Область техники, к которой относится изобретение
Данное изобретение относится к хромсодержащим композициям и их получению и применению для каталитической переработки углеводородов и/или галогенированных углеводородов.
Предпосылки создания изобретения
Хорошо известно, что α-Cr2O3 и α-Fe2O3 имеют общую структуру α-Al2O3 (корунда) с ионами М+3, занимающими октаэдральные узлы в гексагональной плотно упакованной оксидной решетке. Напротив, СоО имеет решетку, подобную NaCl, тогда как Со3О4 имеет структуру нормальной шпинели. Указанные основные структуры описаны в общеизвестных научных трудах; смотри, например, Structural Inorganic Chemistry by A.F. Wells, 5-th ed. Clarendon Press, Oxford, UK (1986), pp. 538, 543-545, 550. γ-Оксид хрома (CrO2,44) описан в Wilhelmi, Acta Chemica Scandinavica, Vol. 22, pp. 2565-2573 (1968).
Были получены многочисленные смешанные оксиды металлов, у которых катионные узлы решетки заняты различными ионами металлов. Например, известны твердые растворы типа (CrxFe1-x)2O3, где 0<x<1. Указанные материалы были получены по стандартной керамической или золь-гель технологии, как описано в работах Music et al., J. Materials Science, Vol. 31, pp. 4067-4076 (1996) и Bhattacharya et al., J. Materials Science, Vol. 32, pp. 577-560 (1997).
Смешанные Cr-Co-оксиды, имеющие шпинельную структуру, являются известными (смотри, например, Bracconi et al., Ann. Chim. Fr., Vol. 4, pp. 331-338 (1979) и Hanck and Laitinen, J. Inorg. Nucl. Chem., Vol. 33, pp. 63-73 (1971)).
CrCoO3 упоминается в качестве соединительного материала в узле топливного элемента (смотри Chem. Abs. 118:9397). Различные смешанные оксиды металлов также рассматриваются в работах Castiglioni et al., J. Solid State Chemistry, Vol. 152, 526-532 (2000); Nowotny et al., J. Am. Ceram. Soc., Vol. 65, pp. 192-196 (1982); и Zhang et al., Journal of Power Sources, Vol. 83, pp. 121-127 (1999).
Некоторые оксиды металлов используются в качестве катализаторов и/или предшественников катализаторов в получении фторированных углеводородов. Оксид хрома (III), в частности, используется, т.к. было установлено, что он может быть фторирован HF при повышенной температуре с получением смеси частиц фторида хрома и оксифторида хрома, которые являются активными катализаторами превращения C-Cl-связей в C-F-связи в присутствии HF. Данное превращения C-Cl-связей в C-F-связи под действием HF, обычно известное как галогенный обмен, является ключевой стадией во многих способах получения фторированных углеводородов.
Композиции оксида хрома, используемые в качестве предшественников катализаторов, могут быть получены различными способами или могут принимать различные формы. Оксид хрома, подходящий для реакций фторирования в парообразной фазе, может быть получен восстановлением триоксида Cr(VI), дегидратацией гидроксида хрома или осаждением солей Cr(III) основаниями (смотри патент США № 3258500). Другой используемой формой оксида хрома является гидроксид гексагонального оксида хрома с низким содержанием иона щелочного металла, как рассмотрено в патенте США № 3978145. Соединения, такие как MF4 (M=Ti, Th, Ce), MF3 (M=Al, Fe, Y) и MF2 (M=Ca, Mg, Sr, Ba, Zn), были добавлены к гидроксиду гексагонального оксида хрома для увеличения срока службы катализатора, как рассмотрено в патенте США № 3992325. Формой оксида хрома, который является предшественником особенно активного катализатора фторирования, является форма, полученная пиролизом дихромата аммония, как рассмотрено в патенте США № 5036036.
Было рассмотрено введение других соединений (например, других солей металлов) в хромсодержащие катализаторы на носителе и без носителя. Австралийский патент № AU-A-8034/94 рассматривает блочный катализатор или катализатор на носителе, на основе оксида хрома (или оксидов хрома) и, по меньшей мере, одного другого каталитически активного металла (например, Mg, V, Mn, Fe, Co, Ni или Zn), в котором главная часть оксида (оксидов) находится в кристаллическом состоянии (и когда катализатором является блочный катализатор, его удельная поверхность после активации HF составляет не менее 8 м2/г). Рассмотренные кристаллические фазы включают Cr2O3, CrO2, NiCrO3, NiCrO4, NiCr2O4, MgCrO4, ZnCr2O4 и смеси указанных оксидов. Австралийский патент AU-А-29972/92 рассматривает массовый катализатор на основе оксидов хрома и никеля, в которых атомное соотношение Ni:Cr находится между 0,05 и 5. Опубликованная заявка на патент США № US 2001/0011061 А1 рассматривает хромдиоксидсодержащие катализаторы фторирования (необязательно содержащие Mg, Zn, Co и Ni), в которых диоксид хрома является, по меньшей мере, частично кристаллическим. Фторированные катализаторы, содержащие кобальт и хром в комбинации (например, пропитанные на носителе), рассмотрены среди других в патенте США № 5185482. Патент США № 5559069 рассматривает гомогенно диспергированные многофазные каталитические композиции, отличающиеся дисперсными фазами некоторых фторидов двухвалентных металлов (некоторых фторидов Mn, Co, Zn, Mg и/или Cd) и некоторых фторидов трехвалентных металлов (фторидов Al, Ga, V и/или Cr).
Остается потребность в катализаторах галогенного обмена, которые могут использоваться для таких способов, как селективное фторирование и хлорфторирование насыщенных и ненасыщенных углеводородов, хлоруглеводородов, хлорфторуглеводородов и хлорфторуглеродов, фторирование ненасыщенных фторированных углеводородов, изомеризация и диспропорционирование фторированных органических соединений, дегидрофторирование фторуглеводородов и хлордефторирование фторированных углеводородов.
Краткое описание изобретения
Данное изобретение предусматривает кристаллический альфа-оксид хрома, в котором от примерно 0,05 ат.% до примерно 6 ат.% атомов хрома в решетке альфа-оксида хрома замещено атомами трехвалентного кобальта (Со+3), и хромсодержащую каталитическую композицию, содержащую в качестве хромсодержащего компонента указанный кобальтзамещенный альфа-оксид хрома.
Данное изобретение также предусматривает способ получения композиции, содержащей указанный кристаллический кобальтзамещенный альфа-оксид хрома. Способ содержит (а) соосаждение твердого вещества введением гидроксида аммония (водного аммиака) в водный раствор растворимой кобальтовой соли и растворимой соли трехвалентного хрома, который содержит не менее трех молей нитрата (т.е. NO3 -) на моль хрома (т.е. Cr3+) в растворе и имеет концентрацию кобальта от примерно 0,05 мол.% до примерно 6 мол.% общей концентрации кобальта и хрома в растворе; и затем не менее трех молей аммония (т.е. NH4 +) на моль хрома (т.е. Cr3+) в растворе вводят в раствор; (b) собирание соосажденного твердого вещества, образованного на стадии (а); (с) сушку собранного твердого вещества и (d) прокаливание высушенного твердого вещества.
Данное изобретение также предусматривает хромсодержащую каталитическую композицию, содержащую хромсодержащий компонент, полученный обработкой указанного кристаллического кобальтзамещенного альфа-оксида хрома фторирующим агентом (например, фторидом водорода).
Данное изобретение также предусматривает способ изменения распределения фтора (т.е. содержания и/или размещения) в углеводороде или галогенированном углеводороде в присутствии катализатора. Способ характеризуется применением каталитической композиции, содержащей, по меньшей мере, один хромсодержащий компонент, выбранный из группы, состоящей из указанных кристаллических кобальтзамещенных альфа-оксидов хрома и указанных обработанных кобальтзамещенных альфа-оксидов хрома.
Краткое описание чертежей
На фигуре 1 представлен спектр, полученный спектроскопией с рассеянием энергии, образца кобальтзамещенного альфа-оксида хрома, номинально содержащего 2 ат.% кобальта.
На фигуре 2 представлен спектр, полученный спектроскопией с рассеянием энергии, образца альфа-оксида хрома без кобальтового замещения.
На фигуре 3 представлен спектр, полученный спектроскопией с рассеянием энергии, образца кобальт-хромовой шпинели.
На фигуре 4 представлен график функции радиального распределения (т.е. вероятности нахождения атома на некотором расстоянии r от центрального атома), связанной с локальной атомной структурой вокруг (а) центрального атома кобальта в СоО, (b) центрального атома кобальта в Со3О4, (с) центрального атома хрома в Cr2O3, (d) кобальта в образце кобальтзамещенного альфа-оксида хрома, номинально содержащего 2 ат.% кобальта, и (е) кобальта в образце кобальтзамещенного альфа-оксида хрома, номинально содержащего 10 ат.% кобальта.
Подробное описание изобретения
Новые композиции данного изобретения содержат кобальтзамещенный альфа-оксид хрома, содержащий от примерно 0,05 ат.% до примерно 6 ат.% кобальта по отношению к общему содержанию кобальта и хрома в альфа-оксиде хрома, который сохраняет структуру корунда. Данное изобретение включает каталитическую композицию, содержащую указанный кристаллический кобальтзамещенный α-Cr2O3. Кристаллические кобальтзамещенные альфа-оксиды хрома имеют общую формулу α-CoxCr2-xО3, где х = 0,001-0,12.
Композиция настоящего изобретения может быть получена способом, описанным выше, с использованием соосаждения. В обычной технологии соосаждения получают водный раствор солей кобальта (II) или кобальта (III) и солей хрома (III). Относительные концентрации солей кобальта и хрома (III) в водном растворе определяются величиной мольного процентного содержания кобальта относительно хрома, желательного в получаемом катализаторе. Концентрация хрома (III) в водном растворе находится обычно в интервале от 0,3 моль/л до 3 моль/л, причем предпочтительная концентрация составляет 0,75-1,5 моль/л. Солями хрома (III), подходящими для получения водного раствора, являются нитрат, сульфат, ацетат, формиат, оксалат, фосфат, бромид и хлорид и различные гидратированные формы указанных солей. Другие соли хрома (III), которые используются для получения водных растворов, включают гексакоординационные комплексы формулы [CrL6-zAz]+3-z, где каждый L представляет собой нейтральный (т.е. незаряженный) лиганд, выбранный из группы, состоящей из H2O, NH3, первичных, вторичных или третичных органических С1-С4-аминов, С1-С4-алкилнитрилов или пиридина, где каждый А представляет собой анионный лиганд, выбранный из группы, состоящей из фторида, хлорида, бромида, иодида, гидроксида, нитрита и нитрата, и где z имеет значение от 0 до 3 включительно. Включенными являются нейтральные бидентатные лиганды, такие как этилендиамин, которые являются эквивалентными двум L, в которых они могут занимать два координационных узла. Также включенными являются анионные бидентатные лиганды, такие как С1-С4-карбоксилат, которые могут занимать два координационных узла. Также включенными являются дианионные лиганды, такие как сульфат, которые являются эквивалентными двум А-лигандам и могут занимать более одного координационного узла.
Соли, содержащие щелочные металлы, такие как сульфат хрома-калия, не являются предпочтительными, потому что присутствие щелочных металлов может мешать каталитической активности катализатора (смотри патент США 4843181). Хром(VI)содержащие предшественники, такие как CrO3, хотя не являются предпочтительными, могут использоваться, но требуют восстановления до Cr(III) соединением, таким как этанол, перед осаждением.
Нитрат хрома (III) или его гидратированные формы, такие как [Cr(NO3)3(H2О)9], являются наиболее предпочтительной солью хрома (III) для получения указанного водного раствора.
Солями кобальта (II), подходящими для получения водного раствора, являются нитрат, сульфат, формиат, оксалат, бромид и хлорид и различные гидратированные формы указанных солей. Соли, содержащие щелочные металлы, такие как бисульфат кобальта-калия, не являются предпочтительными, потому что присутствие щелочных металлов может мешать каталитической активности. Гидрат нитрата кобальта (II) (например, [Co(NO3)2(H2O)6]) является наиболее предпочтительной солью кобальта (II).
Соли кобальта (III), которые используются для получения водных растворов, включают гексакоординационные комплексы формулы [CoL6-zAz]+3-z, где каждый L представляет собой нейтральный (т.е. незаряженный) лиганд, выбранный из группы, состоящей из H2O, NH3, первичных, вторичных или третичных органических С1-С4-аминов, С1-С4-алкилнитрила или пиридина, где каждый А представляет собой анионный лиганд, выбранный из группы, состоящей из фторида, хлорида, бромида, иодида, гидроксида, нитрита и нитрата, и где z имеет значение от 0 до 3 включительно. Включенными являются нейтральные бидентатные лиганды, такие как этилендиамин, которые являются эквивалентными двум L, в которых они могут занимать два координационных узла. Также включенными являются анионные бидентатные лиганды, такие как С1-С4-карбоксилат, которые могут занимать два координационных узла. Также включенными являются дианионные лиганды, такие как сульфат, которые являются эквивалентными двум А-лигандам и могут занимать более одного координационного узла. Предпочтительными кобальт (III) исходными материалами являются гексаминные соли (например, [Co(NH3)6]+3, где противоионом является хлорид или нитрат. Гексаминкобальт (III) хлорид (например, [Co(NH3)6]Cl3) является наиболее предпочтительной солью кобальта (III).
Водный раствор кобальтовых солей и солей хрома (III) может быть затем выпарен либо в вакууме, либо при повышенной температуре с получением твердого вещества, которое затем прокаливают.
Предпочтительно, однако, водный раствор кобальтовых солей и солей хрома (III) затем обрабатывают основанием, таким как гидроксид аммония (водный аммиак), с осаждением кобальта и хрома в виде гидроксидов. Основания, содержащие щелочные металлы, такие как гидроксид натрия или калия, или карбонаты, могут использоваться, но не являются предпочтительными. Введение гидроксида аммония в водный раствор кобальтовых солей и солей хрома (III) обычно выполняют постепенно за период времени 1-12 ч. рН раствора регулируют в процессе введения основания. Конечный рН обычно находится в интервале 6,0-11,0, предпочтительно от примерно 7,5 до примерно 9,0 и наиболее предпочтительно от примерно 8,0 до 8,7. Осаждение смеси гидроксид кобальта/гидроксид хрома обычно проводят при температуре примерно 15-60°С, предпочтительно от примерно 20°С до примерно 40°С. После введения гидроксида аммония смесь обычно перемешивают в течение до 24 ч.
Известны способы получения, где избыток нитрата аммония (т.е. более трех молей нитрата аммония на моль хрома) присутствует в водном растворе. Например, помимо нитрата аммония, уже присутствующего в результате взаимодействия гидроксида аммония с нитратом хлора, от примерно 0,1 моль до примерно 0,7 моль дополнительного нитрата аммония на моль хрома может быть введено в раствор перед, в процессе или после соосаждения композиций. Неожиданно авторами было найдено, что введение избытка нитрата аммония в осажденную смесь гидроксидов кобальта и хрома до стадии дегидратации может быть использовано для снижения размера частиц α-CoxCr2-xO3-фазы, что, в свою очередь, увеличивает площадь поверхности данной фазы и активность катализатора (смотри примеры получения 15, 17 и 18 и примеры 20, 21, 30 и 31).
После введения нитрата аммония в смесь ее предпочтительно перемешивают в течение примерно 0,5-10 ч (предпочтительно в течение примерно одного-пяти часов) при температуре от примерно 20°С до примерно 60°С. Смесь затем сушат и прокаливают, как указано ниже.
Другие агенты, которые служат указанной цели, включают водный пероксид водорода (1-30% растворы), озон, надкислоты, такие как надуксусная кислота, и персульфат аммония. Такие агенты, как галогены, могут использоваться, но не являются предпочтительными. Агенты, содержащие щелочные металлы, такие как персульфат калия или перборат натрия, могут также использоваться, но не являются предпочтительными.
После того как осаждение смеси гидроксидов кобальта и хрома заканчивается и нитрат аммония или другие агенты введены, если желательно, смесь сушат выпариванием.
Необязательно осажденная смесь гидроксидов кобальта и хрома может быть собрана и, если желательно, промыта деионизованной водой перед сушкой. Это может влиять на активность катализатора (смотри примеры 32 и 33).
После того как смесь гидроксидов кобальта и хрома высушивают, нитратные соли разлагают при нагревании твердого вещества от примерно 250°С до примерно 350°С. Полученное твердое вещество затем прокаливают при температуре от примерно 375°С до примерно 1000°С, предпочтительно от примерно 400°С до примерно 600°С. Температура прокаливания может влиять на активность катализаторов и, в свою очередь, на распределение продукта, когда катализаторы используются для изменения фторораспределения в углеводородах и галогенированных углеводородах (смотри примеры 34 и 35). Более низкие температуры прокаливания могут дать в результате присутствие некоторых остаточных нитратных загрязнений.
Прокаливание предпочтительно проводят в присутствии кислорода, наиболее предпочтительно в присутствии воздуха.
Характеристики композиций оксидов металлов данного изобретения могут быть определены хорошо обоснованными аналитическими методами, включая рентгеновскую абсорбционную спектроскопию ((XAS) (РАС)), дифракцию рентгеновских лучей ((XRD) (ДРЛ)), трансмиссионную электронную микроскопию ((ТЕМ)(ТЭМ)), энергорассеивающую спектроскопию ((EDS)(ЭРС)). ЭРС является аналитическим методом, используемым в сочетании со сканирующей или аналитической ТЭМ.
После прокаливания полученные кобальтзамещенные кристаллиты визуально не отличаются от α-Cr2O3 методом ТЭМ. Кроме того, результаты рентгеновских и электроннодифракционных исследований полностью соответствуют структуре α-Cr2O3 с некоторой решеткой, обратно пропорциональной количеству Co(III), которое замещает Cr(III) в структуре. Поэтому делается вывод, что композиции имеют общую формулу α-СохCr2-хО3, где х=0,001-0,12.
Присутствие кобальта в различных кобальт/хромсодержащих композициях данного изобретения четко подтверждается элементарным анализом с использованием ЭРС. На фигуре 1 представлен ЭРС-спектр образца кобальтзамещенного α-Cr2O3, номинально содержащего 2 ат.% Со. Для сравнения на фигуре 2 представлен ЭРС-спектр α-Cr2O3 без кобальтового замещения, и на фигуре представлен ЭРС-спектр коммерчески доступного CoCr2O4, кобальт-хром-шпинели. На каждой из указанных трех фигур интенсивность рентгеновского излучения I, представляющая тысячи импульсов, изображена в зависимости от уровня энергии Е, представляющей тысячи электрон-Вольт (кэВ). Пики на каждом спектре соответствуют присутствию некоторых элементов. Присутствие кобальта ясно показано на ЭРС-спектре на фигуре 1, тогда как пики кобальта отсутствуют на фигуре 2. При более высоких содержаниях кобальта (например, композиции, имеющие массовый состав кобальта более примерно 6 ат.% кобальта по отношению к общему содержанию металлов) вторая шпинельподобная фаза с номинальным составом (Cr0,5Co0,5)3O4 и с размером кристаллитов в пределах 10 нм может быть легко идентифицирована методом ТЭМ и ЭРС. Относительные высоты 2:1 для Kα пиков Cr (атомная масса 52) и Со (атомная масса 59) соответственно на фигуре 3 указывают, что ЭРС-данные основаны на количественной базе.
РАС и ДРЛ данные были получены для композиций, которые составляли номинально 100% Cr (без введения кобальта), 98% Cr: 2% Со, 95% Cr: 5% Со и 90% Cr: 10% Со. РАС и ДРЛ анализ четко показывает, что кобальт замещает хром в α-Cr2O3. ДРЛ-результаты для 98% Cr: 2% Со показаны в таблице 1. Дифракционные пики, имеющие d-расстояния 3,1368, 1,9211, 1,3581, 1,2464 и 1,1088, обусловлены внутренним эталоном кремния, введенным в образец для калибрования дифрактометра. Все другие дифракционные пики могут быть отнесены к α-Cr2O3-структуре с слегка сниженным объемом элементарной ячейки.
ДРЛ- результаты для композиции Со-замещенного α-Cr2O3, которая номинально содержит 98 ат.% Cr; 2 ат.% Со
Если Со замещает Cr в α-Cr2O3-фазе, это ожидается в октаэдральной координации (N Co-O = 6) и в состоянии окисления 3+. РАС-результаты от Cr-K-края образцов показывают, что весь Cr присутствует как Cr3+ и является октаэдрально координированным. Если кобальт присутствует в шпинельподобной фазе, наблюдаемой электронной микроскопией, часть указанных атомов имеет тетраэдральную координацию и присутствует в состоянии окисления +2.
На фигуре 4 представлена функция радиального распределения ((RDF)(ФРР)) для пяти материалов. Функция радиального распределения представляет вероятность нахождения атома на некотором расстоянии r от центрального атома. Эти вероятности отягощены факторами, которые зависят от типа атома. Таким образом, ФРР является отображением локальной атомной структуры вокруг центрального атома. ФРР получается Фурье-преобразованием данных расширенного поглощения рентгеновских лучей тонкой структурой (EXAFS) и может быть представлена графической зависимостью безразмерной характеристики Фурье-преобразования F от расстояния разделения пары в ангстремах. В упрощенной форме можно видеть пик на ФРР-графике как показатель расстояния, на котором имеется координационная сфера вокруг центрального атома. Ожидается небольшое различие между фактическим расстоянием разделения и "r", показанным на графике, когда коррекция не принимается в расчет для фазового сдвига от обратного рассеяния возбужденных электронов. На фигуре 4 представлена графическая зависимость F от расстояния разделения пары r (показанного в ангстремах, неоткорректированного для фазового сдвига) для каждого из пяти материалов. На фигуре 4 показаны кривая А, представляющая локальную структуру вокруг кобальта в СоО, кривая В, представляющая локальную структуру вокруг кобальта в Со3О4, кривая С, представляющая локальную структуру вокруг хрома в α-Cr2O3. Также на фигуре 4 показаны кривая D, представляющая локальную структуру вокруг кобальта в кобальтзамещенном альфа-оксиде хрома с номинальным составом 98% хрома и 2% кобальта, и кривая Е, представляющая локальную структуру вокруг кобальта в кобальтзамещенном альфа-оксиде хрома с номинальным составом 90% хрома и 10% кобальта. Шпинельная фаза не обнаружена электронной микроскопией в данном образце, так что считается, что весь Со связан с α-Cr2O3-фазой либо как отдельное кобальтоксидное покрытие, либо как заместитель Cr в α-Cr2O3-решетке. Кривая на фигуре 4, представляющая локальную структуру вокруг кобальта в кобальтзамещенном альфа-оксиде хрома с номинальным составом 98% хрома и 2% кобальта, показывает, что локальная атомная структура вокруг Со в данном образце не имеет сходства со структурой ожидаемых общих кобальтоксидных фаз, но очень подобна структуре Cr в α-Cr2O3-фазе.
В таблице 2 приведены средние значения сначала вблизи соседних координационных чисел (N Co-O) и средние состояния окисления Со, полученные РАС-анализом спектров поглощения Со- К-края, а также объемы элементарной ячейки для α-Cr2O3-подобной фазы, полученные методом ДРЛ.
Характеристики металлзамещенного альфа-оксида хрома(III)
ат.%
Снижение объема элементарной ячейки при введении Со может быть понято при рассмотрении ионных радиусов. Если Со+3 замещают в октаэдральном окружающем пространстве Cr+3 в α-Cr2O3, они будут принимать низкоспиновую конфигурацию, как рассмотрено в главе 8 работы Cotton, Chemical Applications of Group Theory, 3rd ed. (New York, Wiley, 1990). При использовании ионных радиусов высокоспинового Cr+3 (62 pm), низкоспинового Со+3 (53 pm) и О-2 (137 pm), как приведено в работе Shannon and Prewitt, Acta Crystallografica, Volume B25, pages 925-945 (1969), сокращение объема элементарной ячейки может быть отнесено к количеству низкоспинового Со+3, заместившего высокоспиновый Cr+3 в α-Cr2O3-решетке. Указанные взаимоотношения обобщены в таблице 3, которая показывает количество кобальта, действительно заместившего хром в α-Cr2O3-фазе, для четырех Со-замещенных композиций на основе координационных чисел, состояний окисления и объемов элементарной ячейки. Данные таблицы 3 находятся в хорошем согласовании и предполагают предел растворимости кобальта в α-Cr2O3 фазе около 6 ат.%. Дополнительное указание ограничения растворимости кобальта для массовой концентрации кобальта выше 6 ат.% предусмотрено кривой на фигуре 4, которая представляет локальную структуру вокруг кобальта в кобальтзамещенном альфа-оксиде хрома с номинальным составом 90% хрома и 10% кобальта. Данная кривая предполагает, что часть кобальта присутствует как Со3О4.
Количество Со, заместившего хром в α-Cr2O3-решетке для различных композиций
N(Co-O)
Другие фазы, такие как хром-кобальт-шпинельные фазы, могут присутствовать в хромоксидных композициях настоящего изобретения. Присутствие указанных фаз, имеющих общую стехиометрию (Co0,5Cr0,5)3O4, было установлено методами ЭРС и ТЭМ. Указанные фазы являются обычно незначительными по отношению к α-Cr2O3 -подобной фазе и обычно имеют меньшие размеры частиц.
Удельная поверхность хромоксидных композиций настоящего изобретения обычно находится в интервале примерно 1-100 м2/г. Фаза α-СохCr2-хO3, присутствующая в композициях, полученных способом данного изобретения, обычно состоит из кристаллитов, имеющих размеры частиц в интервале от примерно 20 до примерно 400 нм, обычно от примерно 40 до примерно 250 нм. В данное изобретение включены микрокристаллические материалы с размерами частиц меньше 20 нм.
Прокаленные хромоксидные композиции настоящего изобретения могут быть формованы в различные формы, такие как таблетки, гранулы и экструдаты, для использования в реакторах с насадками. Они могут также использоваться в порошкообразной форме.
Содержание кобальта в хромоксидных композициях настоящего изобретения влияет на активность катализатора, полученного после фторирования смешанного оксида металлов. Например, в хлорфторировании CCl2=CClF3 до CF3CClFCF3 активность металлоксидных катализаторов для образования CF3CClFCF3 улучшается для композиций, имеющих 2-5 ат.% кобальта в катализаторе по отношению к хромовому катализатору, не содержащему кобальт (смотри таблицу 4). Кроме того, в соответствии с описанием данного изобретения активность композиции, содержащей данное соотношение кобальт:хром, может быть улучшена обработкой исходного раствора нитратов кобальта (II) и хрома (III) таким агентом, как нитрат аммония, до дегидратации и прокаливания.
Активность фторированных оксидов кобальта/хрома для хлорфторирования CCl2=CClF3 до CF3CClFCF3 a)
b) Температура реактора 350°С.
Кобальт-хромовая шпинель не считается эффективным катализатором для реакций фторирования и хлорфторирования (смотри сравнительный пример 26).
Композиции данного изобретения могут дополнительно содержать одну или более добавок в форме соединений металлов, которые изменяют селективность или активность кристаллических кобальтзамещенных альфа-оксидов хрома или фторированных металлоксидных катализаторов, содержащих кобальт и хром. Подходящие добавки могут быть выбраны из группы, состоящей из фторидов, оксидов или оксифторидных соединений Mg, Ca, Sc, Y, La, Ti, Zr, Hf, V, Nb, Ta, Mo, W, Mn, Re, Ru, Co, Rh, Ir, Ni, Pd, Pt, Cu, Ag, Au, Ce и Zn.
Общее содержание добавки (добавок) в композициях настоящего изобретения может быть от примерно 0,05 ат.% до примерно 15 ат.% по отношению к общему содержанию металлов в композициях. Добавки могут быть введены в композиции настоящего изобретения стандартными способами, такими как пропитка.
Обычно прокаленные композиции предварительно обрабатываются фторирующим агентом перед использованием в качестве катализаторов для изменения фторораспредления углеводородов и/или галогенированных углеводородных соединений. Обычно указанным фторирующим агентом является HF, хотя могут использоваться другие материалы, такие как тетрафторид серы, карбонилфторид и фторированные углеводородные соединения, такие как трихлорфторметан, дихлордифторметан, хлордифторметан, трифторметан или 1,1,2-трихлортрифторэтан. Указанная предварительная обработка может быть выполнена, например, при помещении катализатора в подходящий контейнер, которым может быть реактор, используемый для осуществления способа данного изобретения, и затем пропускании HF через высушенный, прокаленный катализатор с тем, чтобы частично насытить катализатор HF. Это удобно выполняется пропусканием HF через катализатор в течение периода времени, например, около 0,1-10 ч при температуре, например, около 200-450°С. Тем не менее указанная предварительная обработка не является важной.
Как отмечено выше, катализаторы, предусмотренные согласно настоящему изобретению, могут использоваться для изменения фторораспределения в углеводородах и/или галогенированных углеводородах. Фторораспределение в углеводороде или галогенированном углеводороде может быть изменено увеличением содержания фтора в углеводороде или галогенированном углеводороде. Фторораспределение галогенированного углеводорода также может быть изменено снижением содержания фтора в галогенированном углеводороде и/или перегруппировкой расположения атомов фтора на углеродных атомах галогенированного углеводорода. Известны способы, где изменяется фторораспределение в галогенированных углеводородах, содержащих от 1 до 12 углеродных атомов, в частности способы, где изменяется фторораспределение в галогенированных углеводородах, содержащих от 1 до 6 углеродных атомов. Также известны способы, где содержание фтора углеводородов, содержащих от 1 до 12 углеродных атомов, увеличивается, в частности способы, где содержание фтора в углеводородах, содержащих 1-6 углеродных атомов, увеличивается. Способы изменения фторораспределения в галогенированных углеводородах включают фторирование, хлорфторирование, изомеризацию, диспропорционирование, дегидрофторирование и хлордефторирование. Способы данного изобретения характеризуются применением каталитической композиции, содержащей, по меньшей мере, один хромсодержащий компонент, выбранный из группы, состоящей из кристаллического кобальтзамещенного альфа-оксида хрома, описанного выше, и указанного кобальтзамещенного альфа-оксида хрома, который обработан фторирующим агентом. Типичными насыщенными галогенированными углеводородами, подходящими для способов фторирования, хлорфторирования, изомеризации, диспропорционирования, дегидрофторирования и хлордефторирования, являются галогенированные углеводороды, имеющие формулу CnHaBrbClcFd, в которой n представляет собой целое число от 1 до 6, а представляет собой целое число от 0 до 12, b представляет собой целое число от 0 до 4, с представляет собой целое число от 0 до 13, d представляет собой целое число от 0 до 13, сумма b, c и d равна не менее 1, и сумма a, b, c и d равна 2n+2, при условии, что n равно не менее 2 для способов изомеризации, диспропорционирования и дегидрофторирования, а равно не менее 1 для способа дегидрофторирования, b равно 0 для способа хлордефторирования, (b+c) равно не менее 1 для способа фторирования и равно 0 для способа дегидрофторирования, (a+b+c) равно не менее 1 для способов фторирования, хлорфторирования, изомеризации, диспропорционирования и дегидрофторирования, и d равно не менее 1 для способов изомеризации, диспропорционирования, дегидрофторирования и хлордефторирования. Типичными ненасыщенными галогенированными углеводородами, подходящими для способов фторирования, хлорфторирования, изомеризации, диспропорционирования и хлордефторирования, являются галогенированные углеводороды, которые имеют формулу CpHeBrfClgFh, в которой p представляет собой целое число от 2 до 6, e представляет собой целое число от 0 до 10, f представляет собой целое число от 0 до 2, g представляет собой целое число от 0 до 12, h представляет собой целое число от 0 до 11, сумма f, g и h равна не менее 1, и сумма е, f, g и h равна 2р, при условии, что f равно 0 для способа хлордефторирования, (e+f+g) равно не менее 1 для способов изомеризации и диспропорционирования, и h равно не менее 1 для способов изомеризации, диспропорционирования и хлордефторирования. Типичными насыщенными углеводородами, подходящими для хлорфторирования, являются насыщенные углеводороды, которые имеют формулу CqHr, где q представляет собой целое число от 1 до 6, и r равно 2q+2. Типичными ненасыщенными углеводородами, подходящими для фторирования и хлорфторирования, являются ненасыщенные углеводороды, которые имеют формулу CiHj, где i представляет собой целое число от 2 до 6, и j равно 2i.
Фторирование
В данное изобретение включен способ увеличения содержания фтора в галогенированном углеводородном соединении или ненасыщенном углеводородном соединении при взаимодействии указанного соединения с фторидом водорода в парообразной фазе в присутствии каталитической композиции, содержащей, по меньшей мере, один хромсодержащий компонент, выбранный из группы, состоящей из кристаллического кобальтзамещенного альфа-оксида хрома, описанного выше, и указанного кобальтзамещенного альфа-оксида хрома, который обработан фторирующим агентом. Каталитическая композиция может необязательно содержать дополнительные компоненты, такие как добавки, для изменения активности и селективности катализатора.
Галогенированные углеводородные соединения, подходящие в качестве исходных материалов для способа фторирования данного изобретения, могут быть насыщенными или ненасыщенными. Насыщенные галогенированные углеводородные соединения, подходящие для способов фторирования данного изобретения, включают насыщенные галогенированные углеводородные соединения общей формулы CnHaBrbClcFd, в которой n представляет собой целое число от 1 до 6, а представляет собой целое число от 0 до 12, b представляет собой целое число от 0 до 4, с представляет собой целое число от 0 до 13, d представляет собой целое число от 0 до 13, и сумма a, b, c и d равна 2n+2, при условии, что (b+c) равно не менее 1. Ненасыщенные галогенированные углеводородные соединения, подходящие для способов фторирования данного изобретения, включают ненасыщенные галогенированные углеводородные соединения общей формулы CpHeBrfClgFh, в которой p представляет собой целое число от 2 до 6, e представляет собой целое число от 0 до 10, f представляет собой целое число от 0 до 2, g представляет собой целое число от 0 до 12, h представляет собой целое число от 0 до 11, сумма f, g и h равна не менее 1, и сумма е, f, g и h равна 2р. Ненасыщенными углеводородами, подходящими для фторирования, являются ненасыщенные углеводороды, которые имеют формулу CiHj, где i представляет собой целое число от 2 до 6, и j равно 2i. Содержание фтора насыщенных соединений формулы CnHaBrbClcFd, ненасыщенных соединений формулы CpHeBrfClgFh и/или ненасыщенных соединений формулы CiHj может быть увеличено при взаимодействии указанных соединений с HF в парообразной фазе в присутствии каталитической композиции, содержащей, по меньшей мере, один хромсодержащий компонент, выбранный из группы, состоящей из кристаллического кобальтзамещенного альфа-оксида хрома, описанного выше, и указанного кобальтзамещенного альфа-оксида хрома, который обработан фторирующим агентом. Такой способ называется здесь реакцией фторирования в парообразной фазе.
Реакции фторирования в парообразной фазе обычно проводят при температурах от примерно 150°С до 500°С. Для насыщенных соединений фторирование обычно проводят при температуре от примерно 175°С до 400°С и более предпочтительно от примерно 200°С до примерно 350°С. Для ненасыщенных соединений фторирование обычно проводят при температуре от примерно 150°С до 350°С и более предпочтительно от примерно 175°С до примерно 300°С.
Реакции фторирования в парообразной фазе обычно проводят при атмосферном давлении и давлении выше атмосферного. В целях удобства в способах разделения с нисходящим потоком (например, дистилляции) могут использоваться давления до примерно 30 атм.
Реакции фторирования в парообразной фазе обычно проводят в трубчатом реакторе. Реактор и его соответствующие линии питания, выпускные линии и связанные установки должны быть сконструированы из материалов, стойких к фториду водорода и хлориду водорода. Типичные материалы конструкции, хорошо известные в технике фторирования, включают нержавеющую сталь, в частности, аустенитичного типа, хорошо известные высоконикелевые сплавы, такие как никель-медные сплавы Monel®, никельсодержащие сплавы Hastelloy® и хром-никелевые сплавы Inconel®, и сталь с медным покрытием.
Время взаимодействия в реакторе обычно составляет от примерно 1 до примерно 120 с и предпочтительно от примерно 5 до примерно 60 с.
Количеством HF, взаимодействовавшего с ненасыщенными углеводородами или галогенированными углеводородными соединениями, должно быть, по меньшей мере, стехиометрическое количество. Стехиометрическое количество основано на числе Br и/или Cl заместителей, замещаемых на F, в дополнение к одному молю HF для насыщения двойной связи углерод-углерод, если присутствует. Обычно мольное отношение HF к указанным соединениям формул CnHaBrbClcFd, CpHeBrfClgFh и CiHj может быть в интервале от примерно 0,5:1 до примерно 100:1, предпочтительно от примерно 2:1 до примерно 50:1 и более предпочтительно от примерно 3:1 до примерно 20:1. В общем случае для данной каталитической композиции чем выше температура и больше время взаимодействия, тем больше превращение во фторированные продукты. Вышеуказанные параметры могут быть сбалансированы друг с другом, так что образование более высокозамещенных продуктов максимизируется.
Примеры насыщенных соединений формулы CnHaBrbClcFd, которые могут взаимодействовать с HF в присутствии катализатора данного изобретения, включают CH2Cl2, CH2Br2, CHCl3, CCl4, C2Cl6, C2BrCl5, C2Cl5F, C2Cl4F2, C2Cl3F3, C2Cl2F4, C2ClF5, C2HCl5, C2HCl4F, C2HCl3F2, C2HCl2F3, C2HClF4, C2HBrF4, C2H2Cl4, C2H2Cl3F, C2H2Cl2F2, C2H2ClF3, C2H3Cl3, C2H3Cl2F, C2H3ClF2, C2H4Cl2, C2H4ClF, C3Cl6F2, C3Cl5F3, C3Cl4F4, C3Cl3F5, C3HCl7, C3HCl6F, C3HCl5F2) C3HCl4F3, C3HCl3F4, C3HCl2F5, C3H2Cl6, C3H2BrCl5, C3H2Cl5F, C3H2Cl4F2, С3Н2Cl3F3, C3H2Cl2F4, C3H2ClF5, C3H3Cl5, C3H3Cl4F, C3H3Cl3F2, C3H3Cl2F3, C3H3ClF4, C3H4Cl4, C4Cl4Cl4, С4С14Cl6, С4H5Cl5, C4H5Cl4F и C5H4Cl8.
Отдельные примеры реакций фторирования насыщенных галогенированных углеводородных соединений, которые могут быть проведены в условиях, описанных выше, с использованием катализаторов данного изобретения, включают превращение CH2Cl2 в CH2F2, превращение CHCl3 в смесь CHCl2F, CHClF2 и CHF3, превращение CH3CHCl2 в смесь CH3CHClF и CH3CHF2, превращение CH2ClCH2Cl в смесь CH3CHClF и CH3CHF2, превращение CH3CCl3 в смесь CH3CCl2F, CH3CClF2 и CH3CF3, превращение CH2ClCF3 в CH2FCF3, превращение CHCl2CF3 в смесь CHClFCF3 и CHF2CF3, превращение CHClFCF3 в CHF2CF3, превращение CHBrFCF3 в CHF2CF3, превращение CCl3CF2CCl3 в смесь CCl2FCF2CClF2 и CClF2CF2CClF2, превращение CCl3CH2CCl3 в CF3CH2CF3, превращение CCl3CH2CHCl2 в смесь CF3CH2CHF2, CF3CH=CHCl и CF3CH=CHF, превращение CF3CCl2CClF2 в смесь CF3CCl2CF3 и CF3ClFCF3, превращение CF3CCl2CF3 в CF3ClFCF3 и превращение смеси, содержащей CF3CF2CHCl2 и CClF2CF2CHClF, в смесь CF3CF2CHClF и CF3CF2CHF2.
Примеры ненасыщенных соединений формул CpHeBrfClgFh и CiHj, которые могут взаимодействовать с HF в присутствии катализаторов данного изобретения, включают C2Cl4, C2BrCl3, C2Cl3F, C2Cl2F2, C2ClF3, C2F4, C2HCl3, C2HBrCl2, C2HCl2F, C2HClF2, C2HF3, C2H2Cl2, C2H2ClF, C2H2F2, C2H3Cl, C2H3F, C2H4, C3H6, C3H5Cl, C3H4Cl2, C3H3Cl3, C3H2Cl4, C3HCl5, C3Cl6, C3Cl5F, C3Cl4F2, C3Cl3F3, C3Cl2F4, C3ClF5, C3HF5, C3H2F4, C3F6, C4Cl8, C4Cl2F6, C4ClF7, C4H2F6 и C4HClF6.
Отдельные примеры реакций фторирования ненасыщенных галогенированных углеводородных соединений, которые могут быть проведены с использованием катализаторов данного изобретения, включают превращение CHCl=CCl2 в смесь CH2ClCF3 и CH2FCF3, превращение CCl2=CCl2 в смесь CHCl2CF3, CHClFCF3 и CHF2CF3, превращение CCl2=CH2 в смесь CH3CCl2F, CH3CClF2 и CH3CF3, превращение CH2=CHCl в смесь CH3CHClF и CH3CHF2, превращение CF2=CH2 в CH3CF3, превращение CCl2=CClCF3 в смесь CF3CHClCClF2, CF3CHClCF3 и/или CF3CCl=CF2, превращение CF3CF=CF2 в CF3CHFCF3, превращение CF3CH=CF2 в CF3CH2CF3 и превращение CF3CH=CHF в CF3CH2CHF2.
Дополнительная информация по получению пентафторэтана содержится в заявке на патент США 60/405223 [CL2109 US PRV], зарегистрированной 22 августа 2002 г и поэтому приведенной здесь в качестве ссылки в ее полноте.
Известен каталитический способ получения 1,1,1,2,2-пентафторэтана (т.е. CHF2CF3, или HFC-125) фторированием галогенэтана формулы CHZ2CZ3, где каждый Z выбран из группы, состоящей из F, Cl и Br, при условии, что не более четырех из Z представляют собой F. Предпочтительные галогенэтаны формулы CHZ2CZ3 включают 2,2-дихлор-1,1,1-трифторэтан (HCFC-123) и 1,2,2-три-хлор-1,1-дифторэтан (HCFC-122). HFC-125 получают взаимодействием соединений CHZ2CZ3 с HF в парообразной фазе в присутствии катализаторов данного изобретения. Реакцию вышеуказанных пентагалогенэтанов с HF в присутствии катализатора данного изобретения предпочтительно проводят при температуре примерно 150-400°С, более предпочтительно от примерно 200°С до примерно 380°С. Количеством HF, подаваемого в реактор, должно быть, по меньшей мере, стехиометрическое количество на основе числа Cl или Br заместителей в CHZ2CZ3 исходном материале (материалах). В случае фторирования HCFC-123 стехиометрическое соотношение HF:HCFC-123 составляет 2:1. Предпочтительные соотношения HF и CHZ2CZ3 исходного материала (материалов) находятся обычно в интервале от примерно стехиометрического соотношения до примерно 20:1. Предпочтительное время взаимодействия составляет от 1 до 60 с. Кислород в виде воздуха или совместного питания с инертным разбавителем, таким как азот, гелий или аргон, может вводиться вместе с реагентами или как отдельная каталитическая обработка, если желательно.
Также известен способ получения 2-хлор-1,1,1,3,3,3-гексафторпропана (т.е. CF3CHClCF3, или HCFC-226da) фторированием гексагалогенпропена формулы C3Cl6-xFx, в которой х равен 0-5. Предпочтительные гексагалогенпропены формулы C3Cl6-xFx включают 1,2,2-трихлор-3,3,3-трифтор-1-пропен (т.е. CCl2=CClCF3, или CFC-1213xa) и гексахлорпропен (т.е. CCl2=CClCCl3). HCFC-226da получают взаимодействием вышеуказанных ненасыщенных соединений с HF в парообразной фазе в присутствии катализаторов данного изобретения при температурах от примерно 150°С до примерно 400°С, предпочтительно примерно 200-350°С.
Количеством HF, подаваемого в реактор, должно быть, по меньшей мере, стехиометрическое количество на основе числа Cl заместителей в C3Cl6-xFx исходном материале (материалах). В случае фторирования CFC-1213ха стехиометрическое соотношение HF:CFC-1213ха составляет 3:1. Предпочтительные соотношения HF и C3Cl6-xFx исходного материала (материалов) находятся обычно в интервале от примерно стехиометрического соотношения до примерно 25:1. Предпочтительное время взаимодействия составляет от 1 до 60 с.
Смеси насыщенных галогенированных углеводородных соединений или смеси ненасыщенных углеводородов и/или галогенированных углеводородных соединений могут также использоваться в реакциях фторирования в парообразной фазе так же, как и смеси, содержащие как ненасыщенные углеводороды, так и галогенированные углеводородные соединения. Отдельные примеры смесей насыщенных галогенированных углеводородных соединений и смесей ненасыщенных углеводородов и галогенированных углеводородных соединений, которые могут быть подвергнуты фторированию в парообразной фазе с использованием катализаторов данного изобретения, включают смесь CH2Cl2 и CCl2=CCl2, смесь CCl2FCClF2 и CCl3CF3, смесь CCl2=CCl2 и CCl2=CClCCl3, смесь СН2=СНСН3 и CH2=CClCH3, смесь CH2Cl2 и CH3CCl3, смесь CHF2CClF2 и CHClFCF3, смесь CHCl2CCl2CH2Cl, смесь CHCl2CH2CCl3 и CCl3CHClCH2Cl, смесь CHCl2CHClCCl3, CCl3CH2CCl3 и CCl3CCl2CH2Cl, смесь CHCl2CH2CCl3 и CCl3CH2CCl3, смесь CF3CH2CCl2F и CF3CH=CCl2 и смесь CF3CH=CHCl и CF3CH=CCl2.
Хлорфторирование
В данное изобретение включен способ увеличения содержания фтора в галогенированном углеводородном соединении или ненасыщенном углеводородном соединении при взаимодействии указанного соединения с фторидом водорода (HF) и хлором (Cl2) в парообразной фазе в присутствии каталитической композиции, содержащей, по меньшей мере, один хромсодержащий компонент, выбранный из группы, состоящей из кристаллического кобальтзамещенного альфа-оксида хрома, описанного выше, и указанного кобальтзамещенного альфа-оксида хрома, который обработан фторирующим агентом. Каталитическая композиция может необязательно содержать дополнительные компоненты, такие как другой каталитически эффективный металл.
Галогенированные углеводородные соединения, подходящие в качестве исходных материалов для способа хлорфторирования данного изобретения, могут быть насыщенными или ненасыщенными. Насыщенные галогенированные углеводородные соединения, подходящие для способов хлорфторирования данного изобретения, включают насыщенные галогенированные углеводородные соединения общей формулы CnHaBrbClcFd, в которой n представляет собой целое число от 1 до 6, а представляет собой целое число от 0 до 12, b представляет собой целое число от 0 до 4, с представляет собой целое число от 0 до 13, d представляет собой целое число от 0 до 13, сумма b, c и d равна не менее 1, и сумма a, b, c и d равна 2n+2, при условии, что (а+b+c) равно не менее 1. Предпочтительные способы хлорфторирования включают указанные насыщенные исходные материалы, где а равно не менее 1. Насыщенными углеводородными соединениями, подходящими для хлорфторирования, являются насыщенные углеводородные соединения, которые имеют формулу CqHr, где q представляет собой целое число от 1 до 6, и r равно 2q+2.
Ненасыщенные галогенированные углеводородные соединения, подходящие для способов хлорфторирования данного изобретения, включают ненасыщенные галогенированные углеводородные соединения общей формулы CpHeBrfClgFh, в которой p представляет собой целое число от 2 до 6, e представляет собой целое число от 0 до 10, f представляет собой целое число от 0 до 2, g представляет собой целое число от 0 до 12, h представляет собой целое число от 0 до 11, сумма f, g и h равна не менее 1, и сумма е, f, g и h равна 2р. Ненасыщенными углеводородными соединениями, подходящими для фторирования, являются ненасыщенные углеводороды, которые имеют формулу CiHj, где i представляет собой целое число от 2 до 6, и j равно 2i. Содержание фтора насыщенных соединений формул CnHaBrbClcFd и CqHr и/или ненасыщенных соединений формул CpHeBrfClgFh и CiHj может быть увеличено при взаимодействии указанных соединений с HF и Cl2 в парообразной фазе в присутствии каталитической композиции, содержащей, по меньшей мере, один хромсодержащий компонент, выбранный из группы, состоящей из кристаллического кобальтзамещенного альфа-оксида хрома, описанного выше, и указанного кобальтзамещенного альфа-оксида хрома, который обработан фторирующим агентом. Такой способ называется здесь реакцией хлорфторирования в парообразной фазе.
Условия реакции хлорфторирования в парообразной фазе являются подобными условиям, описанным выше для реакций фторирования в парообразной фазе в плане температурных интервалов, времени взаимодействия интервалов, давлений и мольных соотношений HF и галогенированных углеводородных соединений. Количество хлора (Cl2), подаваемого в реактор, основано на том, являются ли ненасыщенными галогенированные углеводородные соединения, подаваемые в реактор, и на числе водородов в CnHaBrbClcFd, CqHr, CpHeBrfClgFh и CiHj, которые должно быть замещены хлором и фтором. Один моль Cl2 требуется для насыщения двойной связи углерод-углерод, и моль Cl2 требуется для каждого водорода, замещаемого хлором или фтором. Небольшой избыток хлора по сравнению со стехиометрическим количеством может быть необходим по практическим причинам, но большие избытки хлора будут давать полное хлорфторирование продуктов. Соотношение Cl2 и галогенированного углеводородного соединения составляет обычно от примерно 1:1 до примерно 10:1.
Отдельные примеры реакций хлорфторирования в парообразной фазе насыщенных галогенированных углеводородных соединений общей формулы CnHaBrbClcFd и насыщенных углеводородных соединений общей формулы CqHr, которые могут быть проведены с использованием катализаторов данного изобретения, включают превращение С2Н6 в смесь, содержащую CH2ClCF3, превращение CH2ClCF3 в смесь CHClFCF3 и CHF2CF3, превращение CCl3CH2CH2Cl в смесь CF3CCl2CClF2, CF3CCl2CF3, CF3CClFCClF2 и CF3CClFCF3, превращение CCl3CH2CHCl2 в смесь CF3CCl2CClF2, CF3CCl2CF3, CF3CClFCClF2 и CF3CClFCF3, превращение CCl3CHClCH2Cl в смесь CF3CCl2CClF2, CF3CCl2CF3, CF3CClFCCLF2 и CF3CClFCF3, превращение CHCl2CCl2CCl2CH2Cl в смесь CF3CCl2CClF2, CF3CCl2CF3, CF3CClFCClF2 и CF3CClFCF3, превращение CCl3CH2CH2Cl в смесь CF3CCl2CHF2, CF3CClFCHF2, CF3CClFCClF2 и CF3CCl2CF3 и превращение CCl3CH2CHCl2 в смесь CF3CCl2CHF2, CF3CClFCHF2, CF3CClFCClF2 и CF3CCl2CF3.
Отдельные примеры реакций хлорфторирования в парообразной фазе ненасыщенных галогенированных углеводородных соединений общей формулы CpHeBrfClgFh и ненасыщенных углеводородных соединений общей формулы CiHj, которые могут быть проведены с использованием катализаторов данного изобретения, включают превращение С2Н4 в смесь CCl3CClF2, CCl2FCCl2F, CClF2CCl2F, CCl3CF3, CF3CCl2F и CClF2CClF2, превращение C2Cl4 в смесь CCl3CClF2, CCl2FCCl2F, CClF2CCl2F, CCl3CF3, CF3CCl2F и CClF2CClF2 и превращение С3Н6 или CF3CCl=CCl2 в смесь CF3CCl2CClF2, CF3CCl2CF3, CF3CClFCClF2 и CF3CClFCF3.
Известен каталитический способ получения смеси, содержащей 2-хлор-1,1,1-трифторэтан (т.е. CH2ClCF3 или HCFC-133a), взаимодействием этана с HF и Cl2 в парообразной фазе в присутствии катализаторов данного изобретения. Хлорфторирование этана предпочтительно проводят при температуре примерно 150-450°С, предпочтительно от примерно 300°С до примерно 400°С. Мольное соотношение HF и этана предпочтительно составляет от примерно 3:1 до примерно 15:1, и мольное соотношение хлора и этана - от примерно 2:1 до 5:1. Предпочтительное время взаимодействия составляет от примерно 5 с до примерно 60 с. Кислород в виде воздуха или совместного питания с инертным разбавителем, таким как азот, гелий или аргон, может вводиться вместе с реагентами или как отдельная обработка катализатора, если желательно.
Также известен каталитический способ получения смеси 1,2,2-трихлор-1,1,3,3,3-пентафторпропана (т.е. CClF2CCl2CF3 или CFC-215aa), 2,2-дихлор-1,1,1,3,3,3-гексафторпропана (т.е. CF3CCl2CF3 или CFC-216aa), 1,2-дихлор-1,1,1,3,3,3-гексафторпропана (т.е. CClF2CClFCF3 или CFC-216ba) и 2-хлор-1,1,1,2,3,3,3-гептафторпропана (т.е. CF3CClFCF3 или CFC-217ba) хлорфторированием гексагалогенпропена формулы C3Cl6-xFx, в которой х равен 0-4. Предпочтительные гексагалогенпропены формулы C3Cl6-xFx включают 1,2,2-трихлор-3,3,3-трифтор-1-пропен (т.е. CCl2=CClCF3 или CFC-1213xa) и гексахлорпропен (т.е. CCl2=CClCCl3). Смесь CFC-215aa, CFC-216aa, CFC-216ba и CFC-217ba получают взаимодействием вышеуказанных ненасыщенных соединений с Cl2 и HF в парообразной фазе в присутствии катализаторов данного изобретения при температурах от примерно 150°С до примерно 450°С, предпочтительно от примерно 250°С до 400°С.
Количество HF, подаваемого в реактор, должно быть, по меньшей мере, стехиометрическим количеством относительно числа Cl-заместителей в C3Cl6-xFx исходном материале (материалах) и желаемого состава конечного продукта. В случае хлорфторирования CFC-1213xa до смеси хлорфторпропанов, имеющих среднее число фтор-заместителей шесть, стехиометрическое соотношение HF:CFC-1213xa составляет 3:1. Предпочтительные соотношения HF и C3Cl6-xFx исходного материала (материалов) обычно находятся в интервале от примерно стехиометрического соотношения до примерно 30:1, более предпочтительно от примерно 8:1 до 25:1.
Количеством хлора, подаваемого в реактор, должно быть, по меньшей мере, стехиометрическое количество. Предпочтительные мольные соотношения Cl2:CFC-1213xa составляют от примерно 1:1 до примерно 5:1.
Предпочтительное время взаимодействия составляет от примерно 5 с до примерно 60 с.
Дополнительная информация по хлорфторированию CFC-1213xa содержится в заявке на патент США 60/405222 [CL2108 US PRV], зарегистрированной 22 августа 2002 г. и поэтому приведенной здесь в качестве ссылки в ее полноте.
Смеси насыщенных углеводородных соединений и насыщенных галогенированных углеводородных соединений и смеси ненасыщенных углеводородных соединений и ненасыщенных галогенированных углеводородных соединений, а также смеси, содержащие как насыщенные, так и ненасыщенные соединения, могут хлорфторироваться с использованием катализаторов данного изобретения. Отдельные примеры смесей насыщенных и ненасыщенных углеводородов и галогенированных углеводородов, которые могут использоваться, включают смесь CCl2=CCl2 и CCl2=CClCCl3, смесь CHCl2CCl2CH2Cl и CCl3CHClCH2Cl, смесь CHCl2CH2CCl3 и CCl3CHClCH2Cl, смесь CHCl2CHClCCl3, CCl3CH2CCl3 и CCl3CCl2CH2Cl, смесь CHF2CH2CF3 и CHCl=CHCF3 и смесь СН2=СН2 и СН2=СНСН3.
Изомеризация и диспропорционирование
В изобретение входит способ изменения фторораспределения в галогенированном углеводородном соединении изомеризацией указанного галогенированного углеводородного соединения в присутствии каталитической композиции, содержащей, по меньшей мере, один хромсодержащий компонент, выбранный из группы, состоящей из кристаллического кобальтзамещенного альфа-оксида хрома, описанного выше, и указанного кобальтзамещенного альфа-оксида хрома, который обработан фторирующим агентом.
Также включенным в данное изобретение является способ изменения фторораспределения в галогенированном углеводородном соединении диспропорционированием указанного галогенированного углеводородного соединения в парообразной фазе в присутствии каталитической композиции, содержащей, по меньшей мере, один хромсодержащий компонент, выбранный из группы, состоящей из кристаллического кобальтзамещенного альфа-оксида хрома, описанного выше, и указанного кобальтзамещенного альфа-оксида хрома, который обработан фторирующим агентом.
Галогенированные углеводородные соединения, подходящие в качестве исходных материалов для способов изомеризации и диспропорционирования данного изобретения, могут быть насыщенными или ненасыщенными. Насыщенные галогенированные углеводородные соединения, подходящие для способов изомеризации и диспропорционирования данного изобретения, включают насыщенные галогенированные углеводородные соединения общей формулы CnHaBrbClcFd, в которой n представляет собой целое число от 2 до 6, а представляет собой целое число от 0 до 12, b представляет собой целое число от 0 до 4, с представляет собой целое число от 0 до 13, d представляет собой целое число от 1 до 13, и сумма a, b, c и d равна 2n+2, при условии, что (а+b+c) равно не менее 1. Ненасыщенные галогенированные углеводородные соединения, подходящие для способов изомеризации и диспропорционирования данного изобретения, включают соединения общей формулы CpHeBrfClgFh, в которой p представляет собой целое число от 2 до 6, e представляет собой целое число от 0 до 10, f представляет собой целое число от 0 до 2, g представляет собой целое число от 0 до 12, h представляет собой целое число от 1 до 11, и сумма е, f, g и h равна 2р, при условии, что сумма e+f+g равна не менее 1.
В одном варианте настоящего изобретения фторораспределение галогенированного углеводородного соединения изменяется перегруппировкой заместителей H, Br, Cl и F в молекуле (обычно термодинамически предпочтительной перегруппировкой) при сохранении того же числа заместителей H, Br, Cl и F соответственно. Данный способ называется здесь изомеризацией.
В другом варианте настоящего изобретения фторораспределение галогенированного углеводородного соединения изменяется при замене, по меньшей мере, одного F-заместителя одной молекулы галогенированного углеводородного исходного материала, по меньшей мере, одним заместителем H, Br и/или Cl другой молекулы галогенированного углеводородного исходного материала с образованием в результате одного или более галогенированных углеводородных соединений, имеющих сниженное содержание фтора по сравнению с галогенированным углеводородным исходным материалом, и одного или более галогенированных углеводородных соединений, имеющих увеличенное содержание фтора по сравнению с галогенированным углеводородным исходным материалом. Данный способ называется здесь диспропорционированием.
В другом варианте настоящего изобретения реакции как изомеризации, так и диспропорционирования могут иметь место одновременно.
При проведении изомеризации, диспропорционирования или как изомеризации, так и диспропорционирования фторораспределение насыщенных соединений формулы CnHaBrbClcFd и/или ненасыщенных соединений формулы CpHeBrfClgFh может быть изменено в присутствии каталитической композиции, содержащей, по меньшей мере, один хромсодержащий компонент, выбранный из группы, состоящей из кристаллического кобальтзамещенного альфа-оксида хрома, описанного выше, и указанного кобальтзамещенного альфа-оксида хрома, который обработан фторирующим агентом.
Реакции изомеризации и диспропорционирования обычно проводят при температурах от примерно 150°С до 500°С, предпочтительно от примерно 200°С до примерно 400°С. Время взаимодействия в реакторе составляет обычно от примерно 1 до примерно 120 с и предпочтительно от примерно 5 до примерно 60 с. Реакции изомеризации и диспропорционирования могут осуществляться в присутствии инертного газа, такого как гелий, аргон или азот, хотя это не является предпочтительным. Реакции изомеризации и диспропорционирования могут осуществляться в присутствии HF и HCl, но это не является предпочтительным.
Отдельные примеры реакций изомеризации в парообразной фазе, которые могут осуществляться с использованием катализаторов данного изобретения, включают превращение CClF2CCl2F в CCl3CF3, превращение CClF2CClF2 в CF3CCl2F, превращение CHF2CClF2 в CF3CHClF, превращение CHF2CHF2 в CF3CH2F, превращение CF3CClFCClF2 в CF3CCl2CF3 и превращение CF3CHFCHF2 в CF3CH2CF3.
Отдельные примеры реакций диспропорционирования в парообразной фазе, которые могут осуществляться с использованием катализаторов данного изобретения, включают превращение CClF2CClF2 в смесь CClF2CCl2F, CCl3CF3 и CF3CClF2 и превращение CHClFCF3 в смесь CHCl2CF3 и CHF2CF3.
Известен способ превращения смеси 2-хлор-1,1,2,2-тетра-фторэтана (т.е. CHF2CClF2 или HCFC-124a) и 2-хлор-1,1,1,2-тетрафторэтана (т.е. CF3CHClF или HCFC-124) в смесь, содержащую 2,2-дихлор-1,1,1-трифторэтан (т.е. CHCl2CF3 или HCFC-123) и 1,1,1,2,2-пентафторэтан (т.е. CF3CHF2 или HFC-125) помимо непревращенных исходных материалов. Смесь, содержащая HFC-125 и HCFC-123, может быть получена в парообразной фазе при взаимодействии смеси HCFC-124a и HCFC-124 c катализаторами данного изобретения необязательно в присутствии разбавителя, выбранного из группы, состоящей из HF, HCl, азота, гелия, аргона и диоксида углерода. Диспропорционирование предпочтительно проводят при температуре примерно 150-400°С, более предпочтительно примерно 250-350°С. Если используется, газ-разбавитель может присутствовать в мольном соотношении разбавитель:галогенэтан от примерно 1:1 до примерно 5:1. Предпочтительное время взаимодействия составляет от примерно 10 с до примерно 60 с.
Дегидрофторирование
В данное изобретение включен способ снижения содержания фтора в галогенированном углеводородном соединении дегидрофторированием указанного галогенированного углеводородного соединения в присутствии каталитической композиции, содержащей, по меньшей мере, один хромсодержащий компонент, выбранный из группы, состоящей из кристаллического кобальтзамещенного альфа-оксида хрома, описанного выше, и указанного кобальтзамещенного альфа-оксида хрома, который обработан фторирующим агентом.
Галогенированные углеводородные соединения, подходящие в качестве исходных материалов для способа дегидрофторирования данного изобретения, являются обычно насыщенными. Галогенированные углеводородные соединения, подходящие для способов дегидрофторирования данного изобретения, включают соединения общей формулы CnHaFd, в которой n представляет собой целое число от 2 до 6, а представляет собой целое число от 1 до 12, d представляет собой целое число от 1 до 13, и сумма а и d равна 2n+2. Содержание фтора в насыщенных соединениях формулы CnHaFd может быть снижено в присутствии каталитической композиции, содержащей, по меньшей мере, один хромсодержащий компонент, выбранный из группы, состоящей из кристаллического кобальтзамещенного альфа-оксида хрома, описанного выше, и указанного кобальтзамещенного альфа-оксида хрома, который обработан фторирующим агентом. Указанное снижение содержания фтора обычно связано с удалением фторида водорода (HF) из молекулы и называется здесь дегидрофторированием.
Реакции дегидрофторирования обычно проводят при температурах от примерно 200°С до 500°С, предпочтительно от примерно 300°С до примерно 450°С. Время взаимодействия в реакторе составляет обычно от примерно 1 до примерно 360 с и предпочтительно от примерно 5 до примерно 120 с. Проведение реакций дегидрофторирования в присутствии инертного газа, такого как гелий, аргон или азот, способствует диссоциации фторированного углеводородного соединения, но данная практика может также привести к трудностям в разделении и не является предпочтительной.
Продукт реакции дегидрофторирования состоит из HF и ненасыщенного фторированного углеводородного соединения, образованного в результате потери HF исходным материалом. Отдельные примеры реакций дегидрофторирования в парообразной фазе, которые могут осуществляться с использованием катализаторов данного изобретения, включают превращение CH3CHF2 в CH2=CHF, превращение CH3CF3 в CH2=CF2, превращение CF3CH2F в CF2=CHF, превращение CHF2CH2CF3 в CHF=CHCF3 и превращение CF3CH2CF3 в CF3CH=CF2.
Известен каталитический способ получения фторэтена (т.е. CH2=CHF или винилфторида) дегидрофторированием 1,1-дифторэтана (т.е. CHF2CH3 или HFC-152a). Смесь, содержащая винилфторид и непревращенный HFC-152a, может быть получена в парообразной фазе при взаимодействии HFC-152a c катализаторами данного изобретения необязательно в присутствии разбавителя, выбранного из группы, состоящей из HF, азота, гелия, аргона и диоксида углерода. Дегидрофторирование предпочтительно проводят при температурах от примерно 150°С до примерно 400°С, более предпочтительно от примерно 250°С до примерно 350°С. Если используется, газ-разбавитель присутствует в мольном соотношении разбавитель:галогенэтан от примерно 1:1 до примерно 5:1. Предпочтительное время взаимодействия составляет от примерно 10 с до примерно 60 с.
Хлордефторирование
В данное изобретение включен способ снижения содержания фтора в галогенированном углеводородном соединении при взаимодействии указанного галогенированного углеводородного соединения с хлоридом водорода (HCl) в парообразной фазе в присутствии каталитической композиции, содержащей, по меньшей мере, один хромсодержащий компонент, выбранный из группы, состоящей из кристаллического кобальтзамещенного альфа-оксида хрома, описанного выше, и указанного кобальтзамещенного альфа-оксида хрома, который обработан фторирующим агентом.
Галогенированные углеводородные соединения, подходящие в качестве исходных материалов для способа хлордефторирования данного изобретения, могут быть насыщенными или ненасыщенными. Насыщенные галогенированные углеводородные соединения, подходящие для способов хлордефторирования данного изобретения, включают насыщенные галогенированные углеводородные соединения общей формулы CnHaClcFd, в которой n представляет собой целое число от 1 до 6, а представляет собой целое число от 0 до 12, c представляет собой целое число от 0 до 13, d представляет собой целое число от 1 до 13, и сумма а, с и d равна 2n+2. Ненасыщенные галогенированные углеводородные соединения, подходящие для способов хлордефторирования данного изобретения, включают ненасыщенные галогенированные углеводородные соединения общей формулы CpHeClgFh, в которой р представляет собой целое число от 2 до 6, е представляет собой целое число от 0 до 10, g представляет собой целое число от 0 до 12, h представляет собой целое число от 1 до 11, и сумма e, g и h равна 2р. Содержание фтора в насыщенных соединениях формулы CnHaClcFd и/или ненасыщенных соединений формулы CpHeClgFh может быть снижено при взаимодействии указанных соединений с HCl в парообразной фазе в присутствии каталитической композиции, содержащей, по меньшей мере, один хромсодержащий компонент, выбранный из группы, состоящей из кристаллического кобальтзамещенного альфа-оксида хрома, описанного выше, и указанного кобальтзамещенного альфа-оксида хрома, который обработан фторирующим агентом. Такой способ называется здесь реакцией хлордефторирования в парообразной фазе. Хлордефторирование рассмотрено в патенте США № 5345017 и патенте США № 5763698, и поэтому описания указанных двух патентов приводятся здесь в качестве ссылки.
Реакции хлордефторирования обычно проводят при температурах от примерно 250°С до 450°С, предпочтительно от примерно 300°С до примерно 400°С. Время взаимодействия в реакторе составляет обычно от примерно 1 до примерно 120 с и предпочтительно от примерно 5 до примерно 60 с. Реакции наиболее удобно проводить при атмосферном давлении или давлении выше атмосферного.
Хлордефторирование, включающее насыщенные галогенированные углеводороды, является особенно известным. Мольное соотношение HCl и насыщенного галогенированного углеводородного соединения обычно составляет от примерно 1:1 до примерно 100:1, предпочтительно от примерно 3:1 до примерно 50:1, и наиболее предпочтительно от примерно 4:1 до примерно 30:1. В общем случае для данной каталитической композиции чем выше температура, чем больше время взаимодействия и больше мольное соотношение HCl и насыщенного галогенированного углеводородного соединения, тем больше превращение соединений, имеющих более низкое содержание фтора. Вышеуказанные параметры могут быть сбалансированы друг с другом, так что образование хлорзамещенных продуктов максимизируется.
Продукт реакций хлордефторирования обычно содержит непрореагировавший HCl, HF, непревращенный исходный материал и насыщенные галогенированные углеводородные соединения, имеющие более низкое содержание фтора, чем у исходного материала, в силу замещения одного или более фтор-заместителей хлором. Отдельные примеры реакций хлордефторирования в парообразной фазе, которые могут осуществляться с использованием катализаторов данного изобретения, включают превращение CHF3 в смесь CHCl3, CHCl2F и CHClF2, превращение CClF2CClF2 в смесь CCl3CCl3, CCl3CCl2F, CCl3CClF2, CCl2FCCl2F, CClF2CCl2F и CCl3CF3, превращение CF3CClF2 в смесь CCl3CCl3, CCl3CCl2F, CCl3CClF2, CCl2FCCl2F, CCl3CF3, CClF2CClF2 и CF3CCl2F, превращение CF3CCl2CF3 в смесь CF3CCl2CClF2, CF3CCl2CCl2F, CF3CCl2CCl3 и CClF2CCl2CCl3 и превращение CF3CH2CF3 в смесь CCl2=CHCF3 и CCl2=CClCF3.
Известен каталитический способ получения смеси, содержащей 1,1-дихлор-3,3,3-трифтор-1-пропен (т.е. CCl2=CHCF3 или HCFC-1223za) и 1,1,2-трихлор-3,3,3-трифтор-1-пропен (т.е. CCl2=CClCF3 или CFC-1213xa), хлордефторированием 1,1,1,3,3,3-гексафторпропана (т.е. CF3CH2CF3 или HFC-236fa) при взаимодействии HFC-236fa c HCl в парообразной фазе в присутствии катализаторов данного изобретения. Реакцию предпочтительно проводят при температурах от примерно 275°С до 450°С, более предпочтительно от примерно 300°С до примерно 400°С при мольном соотношении HCl:HFC-236fa, предпочтительно от примерно 3:1 до примерно 20:1. Предпочтительное время взаимодействия составляет от примерно 1 до примерно 40 с. Кислород в виде воздуха или совместного питания с инертным разбавителем, таким как азот, гелий или аргон, может вводиться вместе с реагентами или как отдельная каталитическая обработка, если желательно.
Продукты реакции, полученные способами данного изобретения, могут быть выделены традиционной технологией, такой как комбинации, включающие (но не ограничиваясь этим) использование скрубберов, декантацию или дистилляцию. Некоторые из продуктов различных вариантов данного изобретения могут образовывать одну или более азеотропных смесей друг с другом или с HF. Способы данного изобретения могут быть легко осуществлены с использованием хорошо известной химической технологии.
Применимость
Некоторые реакционные продукты, полученные с использованием катализаторов, рассмотренных здесь, имеют желаемые свойства для прямого промышленного использования. Например, CH2F2 (HFC-32), CHF2CF3 (HFC-125), CHF2CF3 (HFC-125), CH2FCHF2 (HFC-134), CF3CH2CF3 (HFC-236fa) и CF3CH2CHF2 (HFC-245fa) нашли применение в качестве хладагентов, CH2FCF3 (HFC-134a) и CF3CHFCF3 (HFC-227ea) нашли применение в качестве пропеллентов, CH2FCHF2 (HFC-134) и CF3CH2CHF2 (HFC-245fa) нашли применение в качестве вспенивающих агентов, и CHF2CF3 (HFC-125), CF3CH2CF3 (HFC-236fa) и CF3CHFCF3 (HFC-227ea) нашли применение в качестве пламягасителей.
Другие реакционные продукты, полученные с использованием данного изобретения, используются в качестве промежуточных химических веществ для получения полезных продуктов. Например, CCl3CF3 (CFC-113a) может использоваться для получения CFC-114a, который затем может быть превращен в CH2FCF3 (HFC-134a) гидродехлорированием. Аналогично, CF3CCl2CF3 (CFC-216aa) может использоваться для получения CF3CH2CF3 (HFC-236fa) гидродехлорированием, и CF3CCl=CF2 (CFC-1215zc) может использоваться для получения CF3CH2CHF2 (HFC-245fa) гидрированием.
Предполагается, что без дополнительного усовершенствования специалист в данной области техники с использованием данного описания сможет использовать настоящее изобретение в его самой полной степени. Поэтому следующие отдельные варианты истолковываются только как иллюстративные и не ограничивают оставшуюся часть описания совсем никаким образом.
ПРИМЕРЫ
Характеристика катализатора
Энергорассеивающая спектроскопия ((EDS)(ЭРС)) и трансмиссионная электронная микроскопия ((ТЕМ)(ТЭМ))
В данных исследованиях кристаллиты анализируют с использованием трансмиссионного электронного микроскопа высокого разрешения Philips CM-20, работающего при ускоряющем напряжении 200 кВ и спаренного с безоконной ЭРС-системой Oxford c Si (Li) элементарным детектором. В ЭРС-анализах электронопрозрачные тонкие сечения образцов используют для минимизации эффектов толщины образца, таких как флуоресценция. Также благодаря подобию их атомных масс было принято, что рентгеновское поглощение поперечных сечений для Cr, Co и Ni является одинаковым (смотри работу Zaluzek, Introduction to Analytical Electron Microscopy (pp. 121-167), ed. by J.J.Hren, J.I.Goldstein and D.C.Joy (Plenum Press, New York, 1979)). Присутствие меди в ЭРС-спектрах на фигурах 1, 2 и 3 обусловлено ТЭМ-сеткой и фоном в микроскопе.
Рентгеновская абсорбционная спектроскопия ((XAS)(РАС)) и
дифракция рентгеновских лучей ((XRD)(ДРЛ))
ДРЛ-данные получают и анализируют в соответствии с методиками, описанными в Warren, X-Ray Diffraction (Addison-Wesley, Reading, MA, 1969). ДРЛ-данные получают при излучении источника 5MBD, DND-CAT Advanced Photon Source, Argonne National Laboratory. РАС-данные получают и анализируют с использованием методик, описанных в Koningsberger and Prins, X-ray Absorption: Principles, Applications, Techniques of EXAFS, SEXAFS and XANES (John Wiley & Sons, New Jork, 1988). Спектры получают для К-краев Cr, Co и Ni. Cr-края получают в трансмиссионной геометрии, тогда как Со- и Ni-края получают в флуоресцентном варианте благодаря их низким концентрациям.
Данные в таблице 2 представляют средние значения для всех Со-атомов. Координационные числа извлекают из функции радиального распределения, полученной Фурье-преобразованием области расширенного рентгеновского поглощения тонкой структуры (EXAFS) Со-спектра. Состояния окисления получают подгонкой образца вблизи края Со-спектра к эталонам известных состояний окисления.
Использование источника излучения Advanced Photon Source для получения ДРЛ- и РАС-данных подтверждено by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract # W-31-109-Eng-38.
Получение катализатора
Сравнительный пример получения 1
Получение 100% хромового катализатора (400°С)
Раствор 400 г Cr(NO3)3[9(H2O)] (1,0 моль) в 1000 мл деионизованной воды обрабатывают по каплям 477 мл 7,4 М водного аммиака, повышая рН примерно до 8,5. Взвесь перемешивают при комнатной температуре до утра. После повторного доведения аммиаком рН до 8,5 смесь выливают в выпарные чашки и сушат в воздушной среде при 120°С. Полученное твердое вещество затем прокаливают в воздушной среде при 400°С в течение 24 ч.
100% оксид хрома исследуют методами ТЭМ и ЭРС. Продукт состоит из однородных кристаллитов α-Cr2O3 с узким интервалом размеров примерно 20 нм. ЭРС показывает присутствие хрома и кислорода без загрязнений.
Пример получения 2
Получение катализатора 99% хрома/1% кобальта (400°С)
Раствор 792,29 г Cr(NO3)3[9(H2O)] (1,98 моль) и 5,82 г Co(NO3)2[6(H2O)] (0,0200 моль) получают в 2000 мл деионизованной воды. Раствор обрабатывают по каплям 955 мл 7,4 М водного аммиака с увеличением рН до примерно 8,5. Взвесь перемешивают при комнатной температуре до утра. рН доводят до 8,5 на следующий день. Твердое вещество затем собирают с использованием двух фриттованных воронок; полученное твердое вещество в каждой воронке промывают 15-20 л деионизованной воды. Твердое вещество сушат в воздушной среде при 120°С в течение 24 ч и затем прокаливают в воздушной среде при 400°С в течение 24 ч.
Анализ образца методами ТЭМ и ЭРС показывает присутствие кристаллитов α-Cr2O3 в интервале размеров 20-40 нм. Кобальт присутствует в хромоксидной решетке.
Пример получения 3
Получение катализатора 98% хрома/2% кобальта (400°С)
Раствор 784,30 г Cr(NO3)3[9(H2O)] (1,96 моль) и 11,64 г Co(NO3)2[6(H2O)] (0,040 моль) получают в 2000 мл деионизованной воды. Раствор обрабатывают по каплям 950 мл 7,4 М водного аммиака с увеличением рН от примерно 1,8 до примерно 8,5. Взвесь перемешивают при комнатной температуре до утра и затем упаривают досуха в воздушной среде при 110-120°С в течение 48 ч. Высушенный катализатор делят пополам. Одну половину прокаливают в воздушной среде при 400°С в течение 24 ч.
Cr/Co-оксид исследуют методами ТЭМ и ЭРС. Оксиды состоят из кристаллитов в интервале 20-40 нм. ЭРС-спектры показывают однородное распределение кобальта в структуре α-Cr2O3. Анализ образца методами ДРЛ и РАС подтверждает, что кристаллиты имеют α-Cr2O3-структуру и что среднее состояние окисления кобальта составляет 2,94. Количество кобальта, введенного в α-Cr2O3-решетку, находится в интервале 1,7-2,0 ат.%.
Пример получения 4
Получение катализатора 98% хрома/2% кобальта (900°С)
Другую половину высушенного катализатора, полученного в примере получения 3, прокаливают в воздушной среде при 900°С в течение 24 ч.
Пример получения 5
Получение катализатора 98% хрома/2% кобальта (400°С)
Раствор 784,29 г Cr(NO3)3[9(H2O)] (1,98 моль) и 11,64 г Co(NO3)2[6(H2O)] (0,040 моль) получают в 2000 мл деионизованной воды. Раствор обрабатывают по каплям 950 мл 7,4 М водного аммиака до тех пор, пока рН не достигнет примерно 8,5. Взвесь перемешивают при комнатной температуре в течение 24 ч при поддержании рН при 8,5. Взвесь затем упаривают досуха в воздушной среде при 110-120°С с продолжением нагревания при 120°С в течение выходных дней. Высушенный катализатор затем прокаливают в воздушной среде при 400°С в течение 24 ч.
Пример получения 6
Получение катализатора 98% хрома/2% кобальта (550°С)
Раствор 1010 г Cr(NO3)3[9(H2O)] (2,52 моль) и 14,6 г Co(NO3)2[6(H2O)] (0,050 моль) получают в 1000 мл деионизованной воды. Сырой раствор упаривают при температуре примерно 100°С до образования густого черного осадка. Твердое вещество сушат при 300-325°С на нагревательной плитке. Твердое вещество затем переносят в фарфоровую чашку и прокаливают в печи при 550°С в течение 20 ч.
Анализ образца методами ТЭМ и ЭРС показывает наличие кристаллов кобальтзамещенного альфа-оксида хрома только с незначительным увеличением размера частиц благодаря более высокой температуре прокаливания. Размер частиц шпинельной фазы увеличивается до примерно 20-50 нм по сравнению с 10-30 нм, наблюдаемыми в образце, прокаленном при 400°С.
Пример получения 7
Получение катализатора 98% хрома/2% кобальта (400°С)
Cr(NO3)3[9(H2O)] (50,5 г, 0,126 моль) и Co(NO3)2[6(H2O)] (0,73 г, 0,00251 моль) взвешивают в фарфоровом тигле и расплавляют вместе в открытой воздушной среде. Смесь нагревают до разложения и затем прокаливают в печи при 400°С в течение 24 ч.
Анализ образца методами ТЭМ и ЭРС показывает наличие 100-150 нм кристаллов кобальтзамещенного альфа-оксида хрома, а также относительно большого количества шпинельной фазы, имеющей размер частиц примерно 10-30 нм.
Аналогичным образом, как указано выше, получают и прокаливают кобальт/хромоксидные композиции, имеющие массовый состав 0,5 ат.% кобальта/99,5 ат.% хрома, 1,0 ат.% кобальта/99 ат.% хрома, 3 ат.% кобальта/97 ат.% хрома и 4 ат.% кобальта/96 ат.% хрома.
Термогравиметрический анализ указанных пяти композиций кобальт/хром показывает, что прокаливание при 400°С дает в результате неполное разложение нитратных предшественников. Образцы пяти композиций прокаливают повторно при 550°С в течение 12 ч. Анализ этих образцов методом РАС показывает, что среднее состояние окисления кобальта составляет +3,0 - +3,2. Образец, содержащий номинально 0,5 ат.% кобальта, является слишком разбавленным по кобальту для РАС-работы. Среднее состояние окисления хрома составляет +3,2; также присутствует фаза, содержащая небольшое количество Cr+6. ДРЛ- и РАС-данные показывают, что локальная структура вокруг кобальта соответствует его присутствию в α-Cr2O3-решетке. Также присутствует вторая фаза с очень малым размером частиц, которая, как предполагается, является CrCoO3.
Пример получения 8
Получение катализатора 97,8% хрома/2,2% кобальта (550°С)
Cr(NO3)3[9(H2O)] (50,33 г, 0,126 моль) и Co(NO3)2[6(H2O)] (0,82 г, 0,00282 моль) взвешивают в фарфоровом тигле и расплавляют вместе в открытой воздушной среде. Смесь нагревают до разложения и затем прокаливают в печи при 550°С в течение 12 ч.
Аналогичным образом, как указано выше, получают и прокаливают при 550°С кобальт/хромоксидные композиции, имеющие массовый состав 2,4 ат.% кобальта/97,6 ат.% хрома и 2,7 ат.% кобальта/97,3 ат.% хрома.
Анализ указанных трех образцов методом РАС показывает, что среднее состояние окисления кобальта составляет +3,1 - +3,2. Среднее состояние окисления хрома составляет +3,13 - +3,20, причем также присутствует фаза, содержащая небольшое количество Cr+6. ДРЛ- и РАС-данные показывают, что локальная структура вокруг кобальта соответствует его присутствию в α-Cr2O3-решетке. Также присутствует вторая фаза с очень малым размером частиц, которая, как предполагается, является CrCoO3.
Пример получения 9
Получение катализатора 98% хрома/2% кобальта (550°С)
Раствор 1,010 г Cr(NO3)3[9(H2O)] (2,52 моль) и 14,6 г Co(NO3)2[6(H2O)] (0,0502 моль) получают в 1500 мл деионизованной воды. Раствор обрабатывают 500 мл 29% мас. водного аммиака с перемешиванием, обеспечиваемым механической мешалкой. Смесь перемешивают в течение двух часов, и рН стабилизируется при 6,0. Смесь переносят в большую керамическую чашку. Воду удаляют нагреванием. После испарения большей части воды образец нагревают при 250-300°С на нагревательной плитке. Полученное твердое вещество затем переносят в фарфоровую чашку и прокаливают в печи при 550°С в течение 20 ч.
Анализ образца методами ТЭМ и ЭРС показывает, что образование шпинельной фазы является минимальным по технологии осаждения. Около 90% фазы кобальтзамещенного альфа-оксида хрома состоит из кристаллитов размером в интервале 20-40 нм, причем остальные 10% являются очень крупными кристаллитами размером в интервале 200-400 нм.
Пример получения 10
Получение катализатора 97% хрома/3% кобальта (непромытый, 400°С)
Раствор 776,29 г Cr(NO3)3[9(H2O)] (1,94 моль) и 17,46 г Co(NO3)2[6(H2O)] (0,060 моль) получают в 2000 мл деионизованной воды. Раствор обрабатывают по каплям 950 мл 7,4 М водного аммиака до достижения рН примерно 8,5. Взвесь перемешивают при комнатной температуре в течение 24 ч и затем выпаривают досуха в воздушной среде при 110-120°С. Высушенный катализатор измельчают в порошок и затем прокаливают в воздушной среде при 400°С в течение 24 ч. Удельная поверхность прокаленного продукта составляет 30,5 м2/г.
Пример получения 11
Получение катализатора 97% хрома/3% кобальта (промытый, 400°С)
Раствор 776,29 г Cr(NO3)3[9(H2O)] (1,94 моль) и 17,46 г Co(NO3)2[6(H2O)] (0,060 моль) получают в 2000 мл деионизованной воды. Раствор обрабатывают по каплям 955 мл 7,4 М водного аммиака до достижения рН примерно 8,5. Взвесь перемешивают при комнатной температуре до утра и рН доводят до 8,5 на следующий день. Твердое вещество собирают в две 3 л фриттованные воронки, и каждую порцию промывают 15-20 л деионизованной воды. Промытое твердое вещество затем выпаривают досуха в воздушной среде при 120°С в течение 24 ч и затем прокаливают в воздушной среде при 400°С в течение 24 ч. Удельная поверхность прокаленного продукта составляет 17,8 м2/г.
Cr/Co-оксид исследуют методами ТЭМ и ЭРС. Фаза α-Cr2O3 состоит из кристаллитов размером в интервале 100 нм, и ЭРС-спектр показывает наличие кобальта, замещающего хром в решетке. Указанный образец также содержит вторую фазу, состоящую из кристаллитов размером в интервале 10 нм. ЭРС-спектр показывает, что данная фаза содержит сравнительные количества кобальта и хрома и является подобной шпинельной фазе.
Пример получения 12
Получение катализатора 95% хрома/5% кобальта (900°С)
Раствор 380,14 г Cr(NO3)3[9(H2O)] (0,95 моль) и 14,55 г Co(NO3)2[6(H2O)] (0,050 моль) получают в 1000 мл деионизованной воды. Раствор обрабатывают по каплям 450 мл 7,4 М водного аммиака при увеличении рН от примерно 1,7 до 8,4. Взвесь перемешивают при комнатной температуре до утра и затем выпаривают досуха в воздушной среде при 120°С и выдерживают при этой температуре до утра. Высушенный катализатор измельчают в порошок и затем прокаливают в воздушной среде при 900°С в течение 20 ч.
Пример получения 13
Получение катализатора 95% хрома/5% кобальта (400°С)
Раствор 760,28 г Cr(NO3)3[9(H2O)] (1,90 моль) и 29,10 г Co(NO3)2[6(H2O)] (0,10 моль) получают в 2000 мл деионизованной воды. Раствор обрабатывают по каплям 950 мл 7,4 М водного аммиака с достижением рН примерно 8,5. Взвесь перемешивают при комнатной температуре до утра и затем выпаривают досуха в воздушной среде при 110-120°С в течение 48 ч. Высушенный катализатор делят пополам. Одну половину прокаливают в воздушной среде при 400°С в течение 24 ч. Удельная поверхность прокаленного продукта составляет 33,6 м2/г.
Оксид Cr/Co 95/5 исследуют методами ТЭМ и ЭРС. Фаза α-Cr2O3 состоит из кристаллитов размером в интервале 50 нм, и ЭРС-спектр показывает наличие кобальта, замещающего хром в решетке. Данный образец также содержит небольшие кристаллиты шпинельной фазы. Анализ образца методами ДРЛ и РАС подтверждает, что кристаллиты имеют α-Cr2O3-структуру и что среднее состояние окисления кобальта составляет 2,84. Количество кобальта, введенного в α-Cr2O3-решетку, находится в интервале 3,8-4,4 ат.%.
Пример получения 14
Получение катализатора 95% хрома/5% кобальта (900°С)
Другую половину высушенного катализатора, полученного в примере получения 13, прокаливают в воздушной среде при 900°С в течение 24 ч.
Пример получения 15
Получение катализатора 95% хрома/5% кобальта (1,5 экв. избыток NH4NO3, 400°С)
Раствор 760,28 г Cr(NO3)3[9(H2O)] (1,90 моль) и 29,10 г Co(NO3)2[6(H2O)] (0,10 моль) получают в 2000 мл деионизованной воды. Раствор обрабатывают по каплям 950 мл 7,4 М водного аммиака с достижением рН 8,5. Взвесь перемешивают при комнатной температуре в течение 24 ч и затем обрабатывают раствором 240,12 г NH4NO3 (3,0 моль). После перемешивания при комнатной температуре в течение 2 ч смесь выпаривают досуха в воздушной среде при 120°С и выдерживают при этой температуре в течение выходных дней. Высушенный катализатор измельчают в порошок в ступке пестиком и затем прокаливают в воздушной среде при 400°С в течение 24 ч. Удельная поверхность прокаленного продукта составляет 36,5 м2/г.
Пример получения 16
Получение катализатора 90% хрома/10% кобальта (промытый, 400°С)
Раствор 720,27 г Cr(NO3)3[9(H2O)] (1,80 моль) и 58,21 г Co(NO3)2[6(H2O)] (0,20 моль) получают в 2000 мл деионизованной воды. Раствор обрабатывают по каплям 955 мл 7,4 М водного аммиака с увеличением рН от примерно 2,1 до примерно 8,5. Взвесь перемешивают при комнатной температуре до утра. На следующий день рН увеличивают от 8,05 до 8,5 добавлением водного аммиака. Твердое вещество собирают в две 3 л фриттованные воронки и каждую порцию промывают 15-20 л деионизованной воды. Промытое твердое вещество затем выпаривают досуха в воздушной среде при 120°С в течение 24 ч. Высушенный катализатор затем прокаливают в воздушной среде при 400°С в течение 24 ч.
Cr/Co-оксид 90/10 исследуют методами ТЭМ и ЭРС. Образец состоит из двух фаз. Фаза α-Cr2O3 состоит из кристаллитов размером в интервале 100 нм, и ЭРС-спектр показывает наличие кобальта в решетке. Данный образец также содержит относительно большое количество шпинельподобной фазы в виде 10 нм кристаллитов. Анализ образца методами ДРЛ и РАС показывает, что образец имеет, главным образом, α-Cr2O3-структуру и что среднее состояние окисления кобальта составляет 2,63. Количество кобальта, введенного в α-Cr2O3-решетку, находится в интервале 4,5-5,9 ат.%.
Пример получения 17
Получение катализатора 90% хрома/10% кобальта (3,3 экв. избыток NH4NO3; 400°С)
Раствор 72,03 г Cr(NO3)3[9(H2O)] (0,18 моль) и 5,82 г Co(NO3)2[6(H2O)] (0,020 моль) получают в 200 мл деионизованной воды. Раствор доводят до рН 8,5 обработкой 7,4 М водного аммиака с достижением рН 8,5. Взвесь перемешивают при комнатной температуре в течение 24 ч. Смесь затем обрабатывают раствором 48,02 г NH4NO3 (0,60 моль), растворенных в 100 мл воды. Взвесь перемешивают в течение одного часа и затем сушат при 120°С в воздушной среде в течение примерно 90 ч. Высушенное твердое вещество измельчают в порошок и затем помещают в покрытую чашку и прокаливают при 400°С в течение 24 ч в воздушной среде.
Анализ образца методом ДРЛ показывает, что α-Cr2O3-структура главной кристаллической фазы имеет сокращение решетки, связанное с присутствием Со+3, замещающего Cr+3. Интервал размера кристаллитов составляет примерно половину интервала, наблюдаемого в примере получения 16.
Пример получения 18
Получение катализатора 90% хрома/10% кобальта (6,7 экв. избыток NH4NO3; 400°С)
Повторяют получение оксида, указанное выше, за исключением того, что смесь оксид/гидроксиды хрома/кобальта обрабатывают раствором 96,05 г NH4NO3 (1,2 моль), растворенного в 200 мл воды.
Анализ образца методом ДРЛ показывает, что α-Cr2O3-структура главной кристаллической фазы имеет сокращение решетки, связанное с присутствием Со+3, замещающего Cr+3. Интервал размера кристаллитов составляет примерно половину интервала, наблюдаемого в примере получения 16.
Общая методика анализа фторуглеводородного продукта
Следующая общая методика поясняет метод, используемый для анализа продуктов реакций фторуглеводородов. Часть общего потока, выходящего из реактора, отбирается в виде образцов под управлением ЭВМ для анализа органического продукта с использованием газового хроматографа, оборудованного селективным детектором массы (ГХ-МС). Газовую хроматографию выполняют с трубкой длиной 20 фут (6,1 м) и диаметром 1/8 дюйм (0,32 см), содержащей перфторированный полиэфир Krytox® на инертной углеродной подложке. Поток гелия составляет 30 мл/мин. Условиями газовой хроматографии являются 60°С в течение начального периода удерживания три минуты с последующим температурным программированием до 200°С со скоростью 6°С/мин.
Массу потока, выходящего из реактора, содержащего органические продукты, а также неорганические кислоты, такие как HCl и HF, обрабатывают водным каустиком перед сбросом.
Все реакции в парообразной фазе проводят при номинальном давлении одна атмосфера.
Пояснение условных обозначений
Пример 19
Фторирование CF3CHCl2
Кобальтзамещенный оксид хрома (Cr/Co 95/5, 29,04 г, 20 мл, -12 - +20 меш (1,68-0,84 мм)), полученный, как описано в примере получения 15, помещают реакторную трубу диаметром 5/8 дюйм (1,58 см) из никелевого сплава Inconel™, нагреваемую на псевдоожиженной песочной бане. Катализатор нагревают от 77°С до 175°С в токе азота (25 см3/мин, 4,2×10-7 м3/с) в течение примерно 1,2 ч. HF и азот совместно подают в реактор при скорости потока каждого 50 см3/мин (8,3×10-7 м3/с). Через 1,5 ч поток азота снижают до 20 см3/мин (3,3×10-7 м3/с), а поток HF увеличивают до 80 см3/мин (1,3х10-6 м3/с). Температуру реактора постепенно увеличивают до 413°С в течение периода времени 5 ч и поддерживают при 413°С еще 0,6 ч. В конце указанного периода поток HF прекращают и реактор охлаждают до 300°С в токе азота 20 см3/с (3,3×10-7 м3/с).
Катализатор, полученный, как указано выше, помещают в реактор, продутый азотом, и HF при 300°С. Парообразные HF и HCFC-123 подают совместно в реактор в мольном соотношении 6:1 с временем взаимодействия 30 с. Анализ потока, выходящего из реактора, методом ГХ-МС, приведен ниже:
Другие продукты включают CFC-113a, CFC-113, CFC-115.
Пример 20
Фторирование CF3CHClF
Кобальтзамещенный оксид хрома (Cr/Co 90/10, 6,75 г, 4 мл, 12-20 меш (1,68-0,84 мм)), полученный, как описано в примере получения 16, помещают в реакторную трубу диаметром 1/2 дюйм (1,27 см) из никелевого сплава Inconel™, нагретую в печи. Катализатор нагревают от 200°С до 400°С в токе азота (50 см3/мин, 8,3×10-7 м3/с) в течение примерно 25 мин и затем температуру снижают до 300°С при сохранении тока азота в течение еще 80 мин. Поток азота снижают до 35 см3/мин (5,8×10-7 м3/с) и HF затем впускают в реактор при скорости потока 12 см3/мин (2,0×10-7 м3/с). Через 35 мин температуру повышают до 325°С. Через 60 мин ее повышают до 350°С. Через 60 мин ее повышают до 375°С. Через 90 мин ее повышают до 400°С. Через 30 мин ее повышают до 425°С. Через 20 мин поток азота снижают до 15 см3/мин (2,5×10-7 м3/с), а поток HF увеличивают до 28 см3/мин (4,7×10-7 м3/с). Через 20 мин поток азота снижают до 5 см3/мин (8,3×10-8 м3/с), а поток HF увеличивают до 36 см3/мин (6,0×10-7 м3/с). Через 20 мин поток азота перекрывают, а поток HF увеличивают до 40 см3/мин (6,7×10-7 м3/с) и эти условия поддерживают в течение 120 мин. Температуру реактора регулируют до 350°С, парообразные HF и HCFC-124 подают в реактор в мольном соотношении 2:1 с временем взаимодействия 3,3 с. Анализ потока, выходящего из реактора, методом ГХ-МС приводится ниже:
Пример 21
Фторирование CF3CHClF
Кобальтзамещенный оксид хрома, который был предварительно обработан избытком NH4NO3 (Cr/Co 90/10, 5,66 г, 4 мл, 12-20 меш (1,68-0,84 мм)), полученный, как описано в примере получения 18, помещают в реакторную трубу диаметром 1/2 дюйм (1,27 см) из никелевого сплава Inconel™, нагретую в печи. Катализатор нагревают от 200°С до 400°С в токе азота (50 см3/мин, 8,3×10-7 м3/с) в течение примерно 25 мин, и затем температуру снижают до 300°С при сохранении тока азота в течение еще 80 мин. Поток азота снижают до 35 см3/мин (5,8×10-7 м3/с) и HF затем впускают в реактор при скорости потока 12 см3/мин (2,0×10-7 м3/с). Через 35 мин температуру повышают до 325°С. Через 60 мин ее повышают до 350°С. Через 60 мин ее повышают до 375°С. Через 90 мин ее повышают до 400°С. Через 30 мин ее повышают до 425°С. Через 20 мин поток азота снижают до 15 см3/мин (2,5×10-7 м3/с), а поток HF увеличивают до 28 см3/мин (4,7×10-7 м3/с). Через 20 мин поток азота снижают до 5 см3/мин (8,3×10-8 м3/с), а поток HF увеличивают до 36 см3/мин (6,0×10-7 м3/с). Через 20 мин поток азота перекрывают, а поток HF увеличивают до 40 см3/мин (6,7×10-7 м3/с) и эти условия поддерживают в течение 120 мин. Температуру реактора регулируют до 350°С и парообразные HF и HCFC-124 подают в реактор в мольном соотношении 2:1 с временем взаимодействия 3,3 с. Анализ потока, выходящего из реактора, методом ГХ-МС приводится ниже:
Пример 22
Фторирование CCl2=CCl2
Кобальтзамещенный оксид хрома (Cr/Co 90/10, 32,26 г, 20 мл, -12 - +20 меш (1,68-0,84 мм)), полученный, как описано в примере получения 16, помещают в реакторную трубу диаметром 5/8 дюйм (1,58 см) из никелевого сплава Inconel™, нагреваемую на псевдоожиженной песочной бане. Катализатор нагревают от 50°С до 246°С в токе азота (50 см3/мин, 8,3×10-7 м3/с) в течение примерно 1,6 ч. Катализатор продувают 20 см3/мин (3,3×10-7 м3/с) азота до утра при 175°С, затем HF и N2 совместно подают в реактор при скорости потока каждого 50 см3/мин (8,3×10-7 м3/с) при 175°С. Через 1,3 ч поток азота снижают до 20 см3/мин (3,3×10-7 м3/с), поток HF увеличивают до 80 см3/мин (6,3×10-6 м3/с). Температуру реактора постепенно повышают до 375°С в течение периода времени 2 ч, затем проводят обработку 10 см3/мин N2 (1,7×10-7 м3/с) и 90 см3/мин (1,5×10-6 м3/с) HF при 409°С в течение еще 2,3 ч. В конце указанного периода поток HF прекращают и реактор охлаждают до 300°С в токе азота 20 см3/мин (3,3×10-7 м3/с).
Катализатор, полученный как указано выше, помещают в реактор, продутый азотом и HF при 325°С. Температуру реактора регулируют до 375°С и парообразные HF и тетрахлорэтилен подают в реактор в мольном соотношении 6:1 с временем взаимодействия 15 с. Анализ потока, выходящего из реактора, методом ГХ-МС, приведен ниже:
Другие продукты включают CFC-112, CFC-112a, CFC-113a, CFC-113, CFC-114a, HCFC-133a, HFC-23.
Пример 23
Фторирование смеси CF3CF2CHCl2 и CClF2CF2CHClF
Кобальтзамещенный оксид хрома (Cr/Co 97/3, 28,57 г, 20 мл, -12 - +20 меш (1,68-0,84 мм)), полученный, как описано в примере получения 10, помещают в реакторную трубу диаметром 5/8 дюйм (1,58 см) из никелевого сплава Inconel™, нагреваемую на псевдоожиженной песочной бане. Катализатор продувают 25 см3/мин (4,2×10-7 м3/с) азота при 150°С в течение 16 ч. Поток N2 увеличивают до 50 см3/мин (8,3×10-7 м3/с) при 175°С в течение 0,5 ч. Затем HF и N2 совместно подают в реактор при скорости потока каждого 50 см3/мин (8,3×10-7 м3/с). Через 1,2 ч поток азота снижают до 20 см3/мин (3,3×10-7 м3/с), а поток HF увеличивают до 80 см3/мин (1,3×10-6 м3/с). Температуру реактора постепенно увеличивают до 300°С в течение периода времени 5,7 ч. Поток HF перекрывают и реактор продувают 20 см3/мин (3,3×10-7 м3/с) азота в течение примерно 16 ч. Потоки N2 и HF затем устанавливают при 20 см3/мин (3,3×10-7 м3/с) и 80 см3/мин (1,3×10-6 м3/с) соответственно, температуру реактора увеличивают от 298°С до 410°С в течение 3,5 ч и затем поддерживают при 410°С в течение 2,3 ч. В конце указанного периода поток HF останавливают и реактор охлаждают до 300°С в токе азота 20 см3/мин (3,3×10-7 м3/с).
Температуру реактора регулируют до 325°С и парообразные HF и смесь HCFC-225cb/HCFC-225ca (52,8% ГХ поверхности 225cb и 46,8% ГХ поверхности 225са) подают совместно в реактор в мольном соотношении 4:1 с временем взаимодействия 30 с. Анализ потока, выходящего из реактора, методом ГХ-МС, приведен ниже:
Другие продукты включают CFC-217ca, HCFC-226ba, HCFC-226da, HCFC-1224, CFC-216, изомеры HCFC-225, CFC-215ca, HCFC-1223.
Пример 24
Фторирование CF3CCl=CCl2
Кобальтзамещенный оксид хрома (Cr/Co 98/2, 28,04 г, 20 мл, -12 - +20 меш (1,68-0,84 мм)), полученный, как описано в примере получения 5, помещают в реакторную трубу диаметром 5/8 дюйм (1,58 см) из никелевого сплава Inconel™, нагреваемую на псевдоожиженной песочной бане. Катализатор нагревают от 90°С до 177°С в токе азота (50 см3/мин, 8,3×10-7 м3/с) в течение примерно 0,7 ч. Затем HF и N2 совместно подают в реактор при скорости потока каждого 50 см3/мин (8,3×10-7 м3/с). Через 2 ч поток азота снижают до 20 см3/мин (3,3×10-7 м3/с), а поток HF увеличивают до 80 см3/мин (1,3×10-6 м3/с). Температуру реактора постепенно увеличивают до 298°С в течение периода времени 3,5 ч. Поток HF перекрывают и реактор продувают 20 см3/мин (3,3×10-7 м3/с) N2 до утра при 299°С. На следующий день потоки N2 и HF затем устанавливают при 20 см3/мин (3,3×10-7 м3/с) и 80 см3/мин (1,3×10-6 м3/с) соответственно, температуру реактора увеличивают до 400°С в течение 1,7 ч. Реактор поддерживают при 400-410°С в течение еще 1,3 ч. В конце указанного периода поток HF останавливают и реактор охлаждают до 300°С в токе азота 20 см3/мин (3,3×10-7 м3/с). Затем парообразные HF и CFC-1213xa подают совместно в реактор в мольном соотношении 20:1 с временем взаимодействия 15 с. Анализ потока, выходящего из реактора, методом ГХ-МС, при температуре реактора 300°С, приведен ниже:
Другие продукты включают HFC-1225, CFC-217ca, CFC-114, CFC-114a, CFC-113, CFC-215, HCFC-225da.
Пример 25
Фторирование CF3CCl=CCl2
Кобальтзамещенный оксид хрома (Cr/Co 98/2, прокаленный при 550°С, 32,0 г, 20 мл, -12 - +20 меш (1,68-0,84 мм)), полученный, как описано в примере получения 6, помещают в реакторную трубу диаметром 5/8 дюйм (1,58 см) из никелевого сплава Inconel™, нагреваемую на псевдоожиженной песочной бане. Катализатор нагревают от 65°С до 176°С в токе азота (50 см3/мин, 8,3×10-7 м3/с) в течение примерно 0,8 ч. Затем HF и N2 совместно подают в реактор при скорости потока каждого 50 см3/мин (8,3×10-7 м3/с). Через 0,6 ч потоки N2 и HF регулируют до 20 см3/мин (3,3х10-7 м3/с) и до 80 см3/мин (1,3×10-6 м3/с) соответственно, при увеличении температуры реактора до 411°С в течение 3 ч. Катализатор выдерживают при 411°С в течение 0,75 ч. Затем потоки N2 и HF регулируют до 10 см3/мин (1,7×10-7 м3/с) и 50 см3/мин (8,3×10-7 м3/с) соответственно, при поддержании температуры реактора при 411°С в течение еще 2 ч. В конце указанного периода поток HF останавливают и реактор охлаждают до 295°С в токе азота 15 см3/мин (2,5×10-7 м3/с). HF и CFC-1213xa подают совместно в реактор в мольном соотношении 20:1 с временем взаимодействия 15 с. Анализ потока, выходящего из реактора, методом ГХ-МС приведен ниже:
Другие продукты включают ГФП, CFC-216ba, CFC-216cb, CFC-114, CFC-114a, CFC-113, CFC-112.
Сравнительный пример 26
Фторирование CF3CCl=CCl2
Промышленный образец хромита кобальта (CoCr2O4, CAS рег. № [12016-69-2], 40,8 г, 20 мл, -12-+20 меш (1,68-0,84 мм)) гранулируют, просеивают и помещают в реакторную трубу диаметром 5/8 дюйм (1,58 см) из никелевого сплава Inconel™, нагреваемую на псевдоожиженной песочной бане. Катализатор нагревают от 80°С до 174°С в токе азота (50 см3/мин, 8,3×10-7 м3/с) в течение примерно 1,5 ч. Затем HF и N2 совместно подают в реактор при скорости потока каждого 50 см3/мин (8,3×10-7 м3/с). Через 0,2 ч потоки N2 и HF регулируют до 20 см3/мин (3,3×10-7 м3/с) и до 80 см3/мин (1,3×10-6 м3/с) соответственно. Катализатор затем нагревают от 175°С до 401°С в течение 3,2 ч. Затем поток HF прекращают, и реактор охлаждают до 300°С в токе азота 20 см3/мин (3,3×10-7 м3/с). Фторид водорода и CFC-1213xa подают совместно в реактор в мольном соотношении 20:1 с временем взаимодействия 15 с. Анализ потока, выходящего из реактора, методом ГХ-МС, приведен ниже:
Пример 27
Хлорфторирование этилена
Катализатором, использованным в эксперименте, является катализатор, который был использован в примере 24. Температуру реактора регулируют до 250°С и парообразные HF, этилен и хлор подают совместно в реактор в мольном соотношении 12:1:1 со временем взаимодействия 15 с. Анализ методом ГХ-МС потока, выходящего из реактора при 250°С, показан ниже:
Другие продукты включают HFC-143a, COF2, HCFC-142, HCC-1130, HCC-150.
Пример 28
Хлорфторирование этана
Кобальтзамещенный оксид хрома из примера 27 используют в данном эксперименте. Температуру реактора регулируют до 350°С и HF, CFC-1213xa и хлор подают совместно в реактор в мольном соотношении 10:1:4 со временем взаимодействия 15 с. Анализ методом ГХ-МС потока, выходящего из реактора при температуре 350°С, показан ниже:
Другие продукты включают HFC-1141, CFC-115, CFC-114a, HCFC-123a, HCFC-132a.
Пример 29
Хлорфторирование CF3CCl=CCl2
Кобальтзамещенный оксид хрома (Cr/Co 99/1, 29,0 г, 20 мл, -12 - +20 меш (1,68-0,84 мм)), полученный, как описано в примере получения 2, помещают в реакторную трубу диаметром 5/8 дюйм (1,58 см) из никелевого сплава Inconel™, нагреваемую на псевдоожиженной песочной бане. Катализатор нагревают от 52°С до 174°С в токе азота (50 см3/мин, 8,3×10-7 м3/с) в течение примерно одного часа. Затем HF и N2 совместно подают в реактор при скорости потока каждого 50 см3/мин (8,3×10-7 м3/с) в течение 2 ч; в течение этого времени имеется заметная экзотерма. Затем поток азота снижают до 20 см3/мин (3,3×10-7 м3/с), а поток HF увеличивают до 80 см3/мин (1,3×10-6 м3/с). Температуру реактора постепенно повышают до 407°С в течение периода времени 3 ч и выдерживают при 406°С в течение 1,3 ч. Поток HF прекращают и реактор охлаждают до 300°С в токе азота 20 см3/мин (3,3×10-7 м3/с). Хлорфторирование CFC-1213xa начинают при 300°С с HF, CFC-1213xa и хлором, совместно поданными в реактор с временем взаимодействия 15 с. Анализ методом ГХ-МС потока, выходящего из реактора при 320°С с мольным соотношением 20:1:4 (для HF, CFC-1213xa и хлора соответственно) и при 400°С с мольным соотношением 30:1:2, приводится ниже:
Другие продукты включают CFC-1215, CFC-114, CFC-114a, HCFC-225da, CFC-113.
Пример 30
Хлорфторирование CF3CCl=CCl2
Кобальтзамещенный оксид хрома (Cr/Co 95/5, 21,8 г, 15 мл, -12 - +20 меш (1,68-0,84 мм)), полученный, как описано в примере получения 13, помещают в реакторную трубу диаметром 5/8 дюйм (1,58 см) из никелевого сплава Inconel™, нагреваемую на псевдоожиженной песочной бане. Катализатор нагревают от 52°С до 173°С в токе азота (50 см3/мин, 8,3×10-7 м3/с) в течение примерно 1 ч. Затем HF и азот совместно подают в реактор при скорости потока 2 см3/мин (4,2×10-7 м3/с) и 75 см3/мин (1,25×10-6 м3/с) соответственно. Через 2,2 ч скорости потоков азота и HF регулируют до 50 см3/мин (8,3×10-7 м3/с) каждого и температуру реактора постепенно повышают до 299°С в течение 3 ч. Реактор продувают до утра при 299°С азотом 20 см3/мин (3,3×10-7 м3/с). HF и азот совместно подают в реактор при 80 см3/мин (1,3×10-6 м3/с) и 20 см3/мин (3,3×10-7 м3/с) соответственно в течение 0,6 ч. Температуру реактора затем повышают до 400°С в течение 2 ч. Поток азота снижают до 10 см3/мин (1,7×10-7 м3/с), а температуру повышают до 410°С. Через 1 ч температуру регулируют до 280°С. Хлорфторирование CFC-1213xa начинают при 280°С с HF, CFC-1213xa и хлором, совместно поданными в реактор в мольном соотношении 20:1:4 с временем взаимодействия 15 с. Анализ методом ГХ-МС потока, выходящего из реактора при 320°С и 350°С, приводится ниже:
Другие продукты включают CFC-217ca, CFC-1215, CFC-115, CFC-13, CFC-114, CFC-114a, CFC-216cb, CFC-113, CFC-214, CFC-215.
Пример 31
Хлорфторирование CF3CCl=CCl2
Кобальтзамещенный оксид хрома, предварительно обработанный избытком нитрата аммония (Cr/Co 95/5, 21,8 г, 15 мл, -12 - +20 меш (1,68-0,84 мм)), полученный, как описано в примере получения 15, помещают в реакторную трубу диаметром 5/8 дюйм (1,58 см) из никелевого сплава Inconel™, нагреваемую на псевдоожиженной песочной бане. Катализатор нагревают от 77°С до 200°С в токе азота (25 см3/мин, 4,2×10-7 м3/с) в течение примерно 1,3 ч и выдерживают при этой температуре примерно 63 ч. Затем HF и азот совместно подают в реактор при скорости потока 50 см3/мин (8,3×10-7 м3/с) каждого при 177°С. Через 1,6 ч скорости потоков HF и азота регулируют до 80 см3/мин (1,3×10-6 м3/с) и 20 см3/мин (3,3×10-7 м3/с) соответственно и температуру реактора повышают до 413°C в течение 5 ч. Через 0,7 ч при 413°C поток HF прекращают и реактор продувают азотом до утра при 300°С. Хлорфторирование CFC-1213xa начинают при 300°С с HF, CFC-1213xa и хлором, совместно подаваемыми в реактор в мольном соотношении 20:1:4 с временем взаимодействия 15 с. Анализ методом ГХ-МС потока, выходящего из реактора при мольных соотношениях питания 20:1:4 при 320°С и 350°С и при 350°С с мольным соотношением 30:1:2, приводится ниже:
Другие продукты включают CFC-217ca, CFC-1215, CFC-115, CFC-114, CFC-114a, CFC-216cb, CFC-113, CFC-214, CFC-215.
Пример 32
Хлорфторирование CF3CCl=CCl2
Кобальтзамещенный оксид хрома (Cr/Co 97/3, 31,6 г, 20 мл, -12 - +20 меш (1,68-0,84 мм)), полученный, как описано в примере получения 11, помещают в реакторную трубу диаметром 5/8 дюйм (1,58 см) из никелевого сплава Inconel™, нагреваемую на псевдоожиженной песочной бане. Катализатор нагревают от 47°С до 174°С в токе азота (50 см3/мин, 8,3×10-7 м3/с) в течение примерно 0,8 ч. Затем HF и азот совместно подают в реактор при скорости потока 50 см3/мин (8,3×10-7 м3/с) каждого. Через 0,7 ч скорости потоков азота и HF регулируют до 20 см3/мин (3,3×10-7 м3/с) и 80 см3/мин (1,3×10-6 м3/с) соответственно. Через 1,7 ч при 175°С температуру реактора постепенно повышают до 410°С в течение 3,4 ч. Через 1 ч температуру снижают с 410°С до 298, поток HF прекращают и реактор продувают азотом до утра. Хлорфторирование CFC-1213xa начинают при 300°С с парообразными HF, CFC-1213xa и хлором, совместно подаваемыми в реактор в мольном соотношении 20:1:4 с временем взаимодействия 15 с. Анализ методом ГХ-МС потока, выходящего из реактора при мольных соотношениях питания 20:1:4 при 320°С и 350°С и при 400°С с мольными соотношениями питания 30:1:2, приводится ниже:
Другие продукты включают CFC-217ca, CFC-1215, CFC-115, CFC-13, CFC-114, CFC-114a, CFC-216cb, CFC-113, CFC-214, CFC-215.
Пример 33
Хлорфторирование CF3CCl=CCl2
Кобальтзамещенный оксид хрома (Cr/Co 97/3, 28,2 г, 20 мл, -12 - +20 меш (1,68-0,84 мм)), полученный, как описано в примере получения 10, помещают в реакторную трубу диаметром 5/8 дюйм (1,58 см) из никелевого сплава Inconel™, нагреваемую на псевдоожиженной песочной бане. Катализатор продувают 25 см3/мин (8,3×10-7 м3/с) азота при 150°С в течение 16 ч. Поток N2 увеличивают до 50 см3/мин (8,3×10-6 м3/с) при 175°С в течение 0,5 ч. Затем HF и азот совместно подают в реактор при скорости потока 50 см3/мин (8,3×10-7 м3/с) каждого. Через 1,2 ч поток азота снижают до 20 см3/мин (3,3×10-7 м3/с) и поток HF увеличивают до 80 см3/мин. Температуру реактора постепенно повышают до 300°С в течение периода времени 5,7 ч. Поток HF прекращают и реактор продувают 20 см3/мин (3,3×10-7 м3/с) азота при 300°С в течение примерно 16 ч. Потоки N2 и HF затем устанавливают при 20 см3/мин (3,3×10-7 м3/с) и 80 см3/мин (1,3×10-6 м3/с) соответственно и температуру реактора повышают от 298°С до 410°С в течение 3,5 ч и затем удерживают при 410°С в течение 2,3 ч. В конце указанного периода поток HF прекращают и реактор охлаждают до 300°С в токе азота 20 см3/мин. Хлорфторирование CFC-1213xa начинают при 300°С с парообразными HF, CFC-1213xa и хлором, совместно подаваемыми в реактор в мольном соотношении 20:1:4 с временем взаимодействия 15 с. Анализ методом ГХ-МС потока, выходящего из реактора при 320°С и 350°С, приводится ниже:
Другие продукты включают CFC-217ca, CFC-1215, HFC-1225, CFC-115, CFC-13, CFC-114, CFC-114a, CFC-216cb, CFC-113.
Пример 34
Хлорфторирование CF3CCl=CCl2
Кобальтзамещенный оксид хрома (Cr/Co 98/2, прокаленный при 400°С, 21,06 г, 15 мл, -12 - +20 меш (1,68-0,84 мм)), полученный, как описано в примере получения 3, помещают в реакторную трубу диаметром 5/8 дюйм (1,58 см) из никелевого сплава Inconel™, нагреваемую на псевдоожиженной песочной бане. Катализатор нагревают от 77°С до 176°С в токе азота (50 см3/мин, 8,3×10-7 м3/с) в течение примерно 1,7 ч. Затем HF и N2 совместно подают в реактор при скорости потока 50 см3/мин (8,3×10-7 м3/с) каждого. Через 1 ч температуру повышают до 326°С в течение 3 ч при поддержании потоков HF и N2 при 50 см3/мин (8,3×10-7 м3/с). Затем потоки N2 и HF регулируют до 25 см3/мин (4,2×10-7 м3/с) и 50 см3/мин (8,3×10-7 м3/с) соответственно при повышении температуры реактора до 401°С в течение 1 ч. Затем потоки N2 и HF регулируют до 10 см3/мин и 50 см3/мин (8,3×10-7 м3/с) соответственно при поддержании температуры реактора при 401°С в течение 1 ч. В конце указанного периода поток HF прекращают и реактор охлаждают до 280°С в токе азота 20 см3/мин. Хлорфторирование CFC-1213xa начинают при 280°С с парообразными HF, CFC-1213xa и хлором, совместно подаваемыми в реактор в мольном соотношении 20:1:4 с временем взаимодействия 15 с. Анализ методом ГХ-МС потока, выходящего из реактора при 320°С (мольное соотношение питания 20:1:4) и 400°С (мольное соотношение питания 30:1:2, время контактирования = 15 с), приводится ниже:
Другие продукты включают CFC-217ca, CFC-1215, CFC-115, CFC-114, CFC-114a, CFC-113, CFC-1213xa, CFC-112.
Пример 35
Хлорфторирование CF3CCl=CCl2
Кобальтзамещенный оксид хрома (Cr/Co 98/2, прокаленный при 900°С, 27,52 г, 15 мл, -12 - +20 меш (1,68-0,84 мм)), полученный, как описано в примере получения 4, помещают в реакторную трубу диаметром 5/8 дюйм (1,58 см) из никелевого сплава Inconel™, нагреваемую на псевдоожиженной песочной бане. Катализатор нагревают от 96°С до 174°С в токе азота (50 см3/мин, 8,3×10-7 м3/с) в течение примерно 0,5 ч. Затем HF и N2 совместно подают в реактор при скорости потока 50 см3/мин (8,3×10-7 м3/с) каждого. Через 0,3 ч при 175°С потоки HF и N2 регулируют до 80 см3/мин (1,3×10-6 м3/с) и 20 см3/мин (3,3×10-7 м3/с) соответственно и температуру повышают до 400°С в течение 5 ч. В конце указанного периода поток HF прекращают и реактор охлаждают до 300°С в токе азота 20 см3/мин (3,3×10-7 м3/с). Хлорфторирование CFC-1213xa начинают при 300°С с парообразными HF, CFC-1213xa и хлором, совместно подаваемыми в реактор в мольном соотношении 20:1:4 с временем взаимодействия 15 с. Анализ методом ГХ-МС потока, выходящего из реактора при 320°С и 400°С, приводится ниже:
Другие продукты включают CFC-217ca, CFC-1215, CFC-115, CFC-114, CFC-114a, CFC-113, CFC-1213xa, CFC-112.
Пример 36
Хлорфторирование CF3CCl=CCl2
Фторид водорода, CFC-1213xa и хлор подают совместно в реактор, содержащий катализатор из примера 25, с временем взаимодействия 15 с. Анализ методом ГХ-МС потока, выходящего из реактора при 320°С (мольное соотношение подаваемых газов HF:1213xa:Cl2=20:1:4) и при 375°С (мольное соотношение подаваемых газов HF:1213xa:Cl2=30:1:2), приводится ниже:
Другие продукты включают CFC-115, CFC-217ca, CFC-1215, CFC-114, CFC-114a, CFC-1214'ы, CFC-113, CFC-112, CFC-214ab.
Пример 37
Хлорфторирование CF3CCl=CCl2
Кобальтзамещенный оксид хрома (Cr/Co 98/2, прокаленный при 550°С, 29,4 г, 20 мл, -12 - +20 меш (1,68-0,84 мм)), полученный, как описано в примере получения 9, помещают в реакторную трубу диаметром 5/8 дюйм (1,58 см) из никелевого сплава Inconel™, нагреваемую на псевдоожиженной песочной бане. Катализатор продувают 50 см3/мин (8,3×10-7 м3/с) азота при 174°С в течение примерно 72 ч. Затем HF и N2 совместно подают в реактор при скорости потока 50 см3/мин (8,3×10-7 м3/с) каждого. Через 0,8 ч при 175°С потоки HF и N2 регулируют до 80 см3/мин (1,3×10-6 м3/с) и 20 см3/мин (3,3×10-7 м3/с) соответственно и затем температуру повышают до 400°С в течение 4,3 ч. Потоки HF и N2 регулируют до 50 см3/мин (8,3×10-7 м3/с) и 10 см3/мин (1,7×10-7 м3/с) соответственно и реактор выдерживают при 406°С в течение еще 1,7 ч. По окончании этого периода времени поток HF прекращают и реактор охлаждают до 300°С при 20 см3/с (3,3×10-7 м3/с) потоком азота. Хлорфторирование CFC-1213xa начинают при 300°С с парообразными HF, CFC-1213xa и хлором, совместно подаваемыми в реактор в мольном соотношении 20:1:4 с временем взаимодействия 15 с. Анализ методом ГХ-МС потока, выходящего из реактора при 300°С (мольное соотношение подаваемых газов HF:1213xa:Cl2=20:1:4) и при 400°С (мольное соотношение подаваемых газов HF:1213xa:Cl2=30:1:2), приводится ниже:
Другие продукты включают FC-218, CFC-13, CFC-115, CFC-217ca, CFC-1215, CFC-114, CFC-114a, CFC-1214'ы, CFC-113, CFC-214ab.
Пример 38
Хлорфторирование CF3CCl=CCl2
Фторид водорода, CFC-1213xa и хлор подают совместно в реактор, содержащий катализатор из примера 26, с временем взаимодействия 15 с. Анализ методом ГХ-МС потока, выходящего из реактора при 375°С (мольное соотношение подаваемых газов HF:1213xa:Cl2=20:1:4), приводится ниже:
Другие продукты включают CFC-115, CFC-216cb, CFC-114, CFC-114a, CFC-1214'ы, CFC-112, CFC-214ab.
Сравнительный пример 39
Хлорфторирование CF3CCl=CCl2
100% оксид хрома (III) (прокаленный при 400°С, 27,8 г, 20 мл, -12 - +20 меш (1,68-0,84 мм)), полученный, как описано в сравнительном примере получения 1, помещают в реакторную трубу диаметром 5/8 дюйм (1,58 см) из никелевого сплава Inconel™, нагреваемую на псевдоожиженной песочной бане. Катализатор нагревают от 77°С до 200°С в токе азота (50 см3/мин, 8,3×10-7 м3/с) в течение примерно часа. Катализатор продувают до утра азотом (20 см3/мин, 3,3×10-7 м3/с) при 174°С. Затем HF и азот совместно подают в реактор при скорости потока 50 см3/мин (8,3×10-7 м3/с) каждого при 175°С. Через час потоки HF и азота регулируют до 80 см3/мин (1,3×10-6 м3/с) и 20 см3/мин (3,3×10-7 м3/с) соответственно и температуру реактора повышают до 410°С в течение 3,6 ч. Поток HF прекращают и реактор продувают азотом до утра при 300°С. Хлорфторирование CFC-1213xa начинают при 300°С с парообразными HF, CFC-1213xa и хлором, совместно подаваемыми в реактор в мольном соотношении 20:1:4 с временем взаимодействия 15 с. Анализ методом ГХ-МС потока выходящего из реактора при 300°С и при 400°С (мольное соотношение подаваемых газов HF:1213xa:Cl2=30:1:2), приводится ниже:
Другие продукты включают FC-218, CFC-1215, CFC-115, CFC-114, CFC-114a, CFC-216cb, CFC-113, CFC-112.
Пример 40
Изомеризация 1:1 смеси CF3CClFCClF2/CF3CCl2CF3
Катализатор, используемый в примере 34, возвращают в реактор и продувают азотом при 350°С. Смесь CFC-216ba/CFC-216aa (50,2% ГХ поверхности CFC-216ba и 49,8% ГХ поверхности CFC-216aa) и азот одновременно подают в реактор в мольном соотношении 1:4 с временем взаимодействия 30 с. Анализ методом ГХ-МС потока, выходящего из реактора, приводится ниже:
Другие продукты включают CFC-115, FC-218, CFC-217ca, CFC-114, CFC-114a, CFC-216cb, HCFC-226da, HCFC-225da, CFC-1214, CFC-215bb.
Пример 41
Фторирование смеси CF3CHClF/CClF2CHF2
Кобальтзамещенный оксид хрома (Cr/Co 95/5, 29,04 г, 20 мл, -12 - +20 меш (1,68-0,84 мм)), полученный, как описано в примере получения 15, помещают в реакторную трубу диаметром 5/8 дюйм (1,58 см) из никелевого сплава Inconel™, нагреваемую на псевдоожиженной песочной бане. Катализатор нагревают от 77°С до 175°С в токе азота (25 см3/мин, 3,3×10-7 м3/с) в течение примерно 1,2 ч. Затем HF и азот совместно подают в реактор при скорости потока 50 см3/мин (8,3×10-7 м3/с) каждого. Через 1,5 ч поток азота снижают до 20 см3/мин (3,3х10-7 м3/с), а поток HF увеличивают до 80 см3/мин (1,3×10-6 м3/с). Температуру реактора постепенно повышают до 413°С в течение периода времени 5 ч и поддерживают при 413°С еще 0,6 ч. В конце указанного периода поток HF прекращают и реактор охлаждают до 300°С в токе азота 20 см3/мин. Парообразные HF и смесь HCFC-124 и HCFC-124a (52,1% ГХ поверхности HCFC-124a и 44,2% ГХ поверхности HCFC-124) совместно подают в реактор в мольном соотношении 4:1 с временем взаимодействия 10 с. Анализ методом ГХ-МС потока, выходящего из реактора при 300°С, приводится ниже:
Другие продукты включают примеси в исходном материале: HFC-32, HFC-143a, HFC-134a, HFC-134 и побочные продукты: HFC-23, CFC-115, CFC-114a, HCFC-123a, HCFC-123b, CFC-113.
Пример 42
Диспропорционирование смеси CF3CHClF/CClF2CHF2
Азот и смесь HCFC-124 и HCFC-124a (52,1% ГХ поверхности HCFC-124a и 44,2% ГХ поверхности HCFC-124) совместно подают в реактор, содержащий катализатор, используемый в примере 41. Мольное соотношение азота к смеси HCFC-124/HCFC-124a составляет 4:1 и время взаимодействия составляет 10 с. Анализ методом ГХ-МС потока, выходящего из реактора при 250°С и 300°С, приводится ниже:
Другие продукты включают HFC-23, HFC-32, HFC-143a, CFC-115, CFC-114a, HCFC-123b, CFC-113, HCFC-122.
Пример 43
Изомеризация и диспропорционирование смеси CClF2CCl2F/CF3CCl3
Азот и смесь CFC-113 и CFC-113a (52,9% ГХ поверхности CFC-113a и 47,1% ГХ поверхности CFC-113) совместно подают в реактор, содержащий катализатор, используемый в примере 42. Мольное соотношение азота к смеси CFC-113/CFC-113a составляет 4:1, и время взаимодействия составляет 15 с. Анализ методом ГХ-МС потока, выходящего из реактора при 150°С и 300°С, приводится ниже:
Другие продукты включают CFC-13, HCFC-1122, CFC-1113, CFC-1111, CFC-1316.
Пример 44
Диспропорционирование CF3CCl2F
Азот и CFC-114a (99,95 ГХ мол.% CFC-114а) совместно подают в реактор, содержащий катализатор, используемый в примере 43, в мольном соотношении 4:1 с временем взаимодействия 15 с. Анализ методом ГХ-МС потока, выходящего из реактора при 250°С и 350°С, приводится ниже:
Другие продукты включают CFC-13, FC-116, HCFC-1122, CFC-1113, HCFC-123, CFC-1111, CFC-1316.
Пример 45
Диспропорционирование и изомеризация смеси CClF2CClF2/CF3CClF2
Азот и смесь CFC-114 и CFC-114a (87,3 ГХ мол.% CFC-114 и 12,6 ГХ мол.% CFC-114а) совместно подают в реактор, содержащий катализатор, используемый в примере 44. Мольное соотношение азота и смеси CFC-114/CFC-114a составляет 4:1, и время взаимодействия составляет 15 с. Анализ методом ГХ-МС потока, выходящего из реактора при 300°С и 350°С, приводится ниже:
Другие продукты включают HFC-23, CFC-13, FC-116, HCFC-1122, CFC-1113, CFC-1111.
Пример 46
Дегидрофторирование CF3CH2CF3
Азот и HFC-236fa совместно подают в реактор, содержащий катализатор, используемый в примере 45. Мольное соотношение азота и HFC-236fa составляет 4:1, и время взаимодействия составляет 15 с. Анализ методом ГХ-МС потока, выходящего из реактора при 300°С и 350°С, приводится ниже:
Другие продукты включают HFC-143a, CFC-1215, HCFC-226da, HCFC-1224, HCFC-1326, HCFC-1223, CFC-216aa, CFC-217ba.
Пример 47
Дегидрофторирование CH3CHF2
Азот и HFC-152a совместно подают в реактор, содержащий катализатор, используемый в примере 45. Мольное соотношение азота и HFC-152а составляет 4:1, и время взаимодействия составляет 15 с. Анализ методом ГХ-МС потока, выходящего из реактора при 250°С и 350°С, приводится ниже:
Другие продукты включают HCFC-151a, HCC-1140, этилен, метан.
Пример 48
Изомеризация гексафторциклопропана
Катализатор, используемый в примере 34 (15 мл, 21,35 г), возвращают в реактор и продувают азотом при 350°С с последующим HCl. Азот и гексафторциклопропан совместно подают в реактор в мольном соотношении 2:1 с временем взаимодействия 30 с. Анализ методом ГХ-МС потока, выходящего из реактора при 150°С, приводится ниже:
Другие продукты включают HFC-125, CFC-217ba, FC-1318my, ПФИБ, CFC-1215'ы.
Пример 49
Хлордефторирование трифторметана
Катализатор, используемый в примере 33 (5 мл, 7,1 г), возвращают в реактор и продувают азотом при 300°С. Безводный хлорид водорода и трифторметан (HFC-23) совместно подают в реактор в мольном соотношении 20:1 с временем взаимодействия 5 с. Анализ методом ГХ-МС потока, выходящего из реактора при 325°С, приводится ниже:
Пример 50
Хлордефторирование 1,1,1,3,3,3-гексафторпропана
Безводный хлорид водорода и 1,1,1,3,3,3-гексафторпропан (HFC-236fa) совместно подают в мольном соотношении 10:1 в реактор, содержащий катализатор, используемый в примере 49, с временем взаимодействия 10 с. Анализ методом ГХ-МС потока, выходящего из реактора при 325°С, приводится ниже:
Другие продукты включают HFC-143a, CFC-114/114a, CFC-1214, HCC-1120, HCFC-1222, HCC-1110.
Дополнительное получение катализатора
Пример получения 51
Получение 98% хрома/2% кобальта (550°С)
[Cr(NH3)6]Cl3] (16,7684 г, 64,4 ммоль) и [Co(NH3)6]Cl3] (0,3513 г, 1,31 ммоль) растворяют в деионизованной воде. Затем водный гидроксид аммония добавляют к раствору до полного осаждения. Полученный осадок отфильтровывают и сушат в воздушной среде при 110°С в течение 12 ч. Полученный продукт тщательно измельчают в агатовой ступке и затем нагревают при 550°С в воздушной среде в течение 12 ч.
Анализ образца методом ДРЛ показывает преобладающую фазу, имеющую структуру α-Cr2O3; и анализ методами ТЭМ и ЭРС показывает небольшое количество шпинельной фазы, содержащей кобальт и хром. Кобальтсодержащая α-Cr2O3-фаза присутствует в виде 200-400 нм кристаллов. РАС показывает, что кобальт полностью введен в α-Cr2O3-решетку.
| название | год | авторы | номер документа |
|---|---|---|---|
| НИКЕЛЬЗАМЕЩЕННЫЕ И СМЕШАННЫЕ НИКЕЛЬ- И КОБАЛЬТЗАМЕЩЕННЫЕ ХРОМОКСИДНЫЕ КОМПОЗИЦИИ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ КАТАЛИЗАТОРОВ И ПРЕДШЕСТВЕННИКОВ КАТАЛИЗАТОРОВ | 2003 |
|
RU2318595C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ 2-ХЛОР-1,1,1,2,3,3,3-ГЕПТАФТОРПРОПАНА, ГЕКСАФТОРПРОПЕНА И 1,1,1,2,3,3,3-ГЕПТАФТОРПРОПАНА | 2003 |
|
RU2326859C2 |
| СПОСОБ ПОЛУЧЕНИЯ 1,1,1,2,2-ПЕНТАФТОРЭТАНА | 2003 |
|
RU2328482C2 |
| КАТАЛИТИЧЕСКОЕ ГАЗОФАЗНОЕ ФТОРИРОВАНИЕ | 2011 |
|
RU2541541C1 |
| КАТАЛИЗАТОР, СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ И СПОСОБ ФТОРИРОВАНИЯ ГАЛОГЕНИРОВАННЫХ УГЛЕВОДОРОДОВ | 2009 |
|
RU2402378C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕНТАФТОРЭТАНА ДИСМУТАЦИЕЙ ТЕТРАФТОРХЛОРЭТАНА | 2000 |
|
RU2230727C2 |
| КАТАЛИЗАТОР, СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ И АКТИВАЦИИ И СПОСОБ ФТОРИРОВАНИЯ ГАЛОГЕНИРОВАННЫХ УГЛЕВОДОРОДОВ | 2007 |
|
RU2322291C1 |
| КАТАЛИЗАТОР, СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ И СПОСОБ ФТОРИРОВАНИЯ ГАЛОГЕНИРОВАННЫХ УГЛЕВОДОРОДОВ | 2010 |
|
RU2431524C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРИРОВАННЫХ УГЛЕВОДОРОДОВ | 1997 |
|
RU2150454C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ ТЕТРАФТОРПРОПЕНА | 2007 |
|
RU2445302C2 |
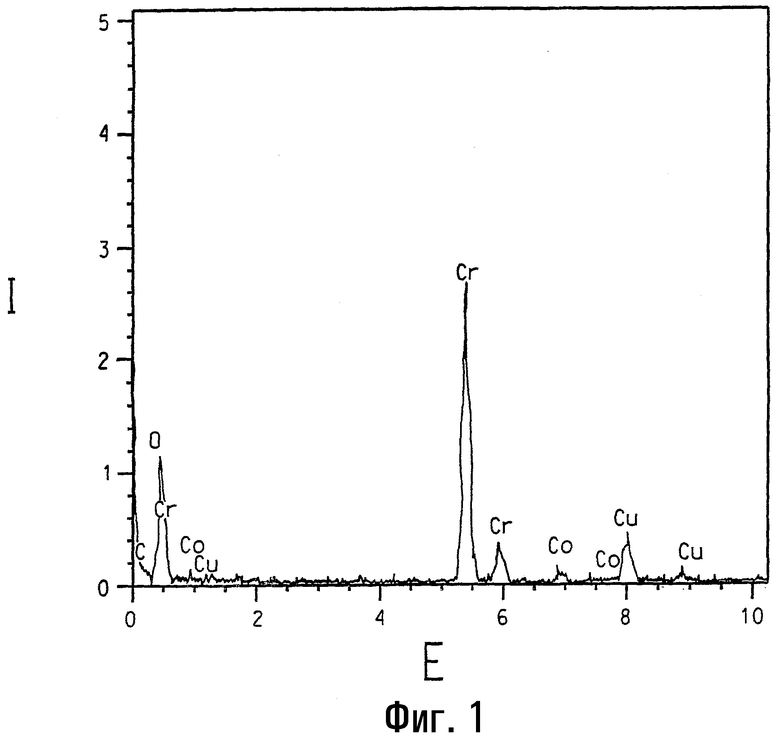
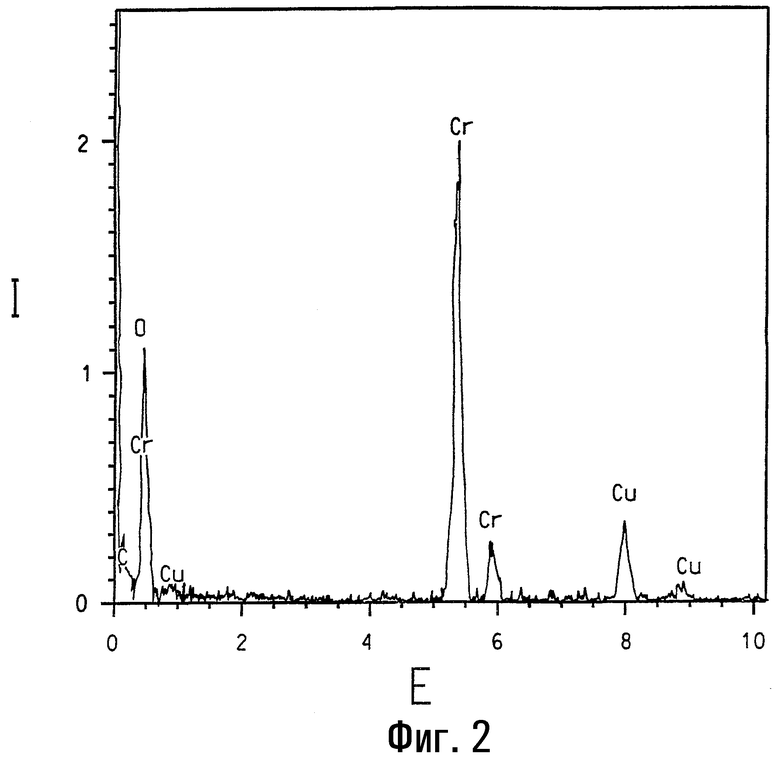
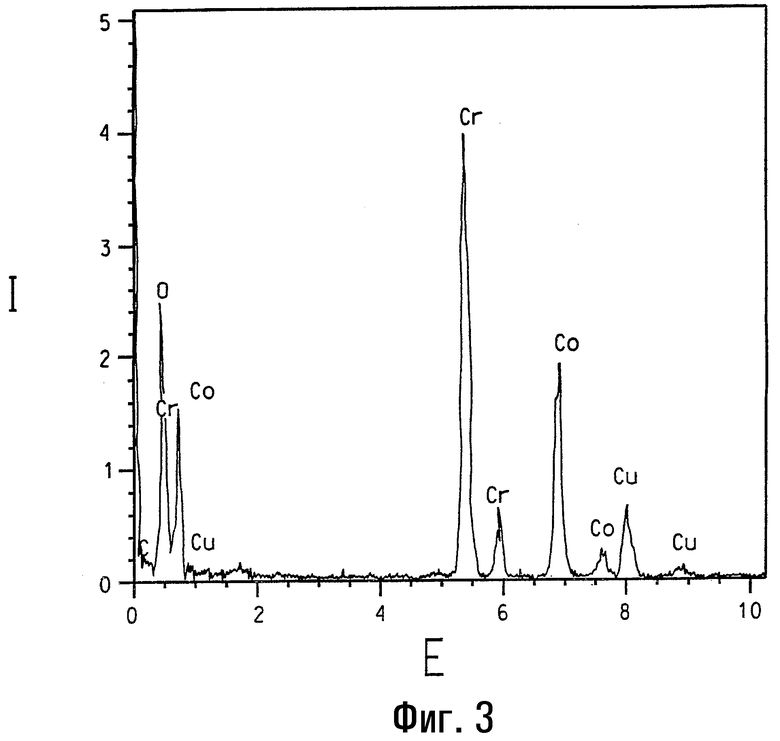
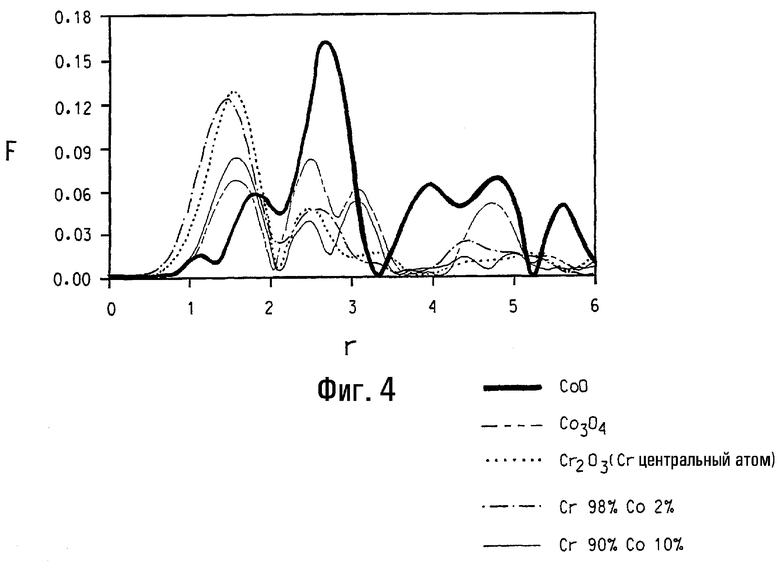
Данное изобретение относится к кристаллическому альфа-оксиду хрома, хромсодержащим каталитическим композициям, способу их получения и к способу изменения фторораспределения в углеводороде и/или галогенированном углеводороде в присутствии указанных каталитических композиций. Заявленный кристаллический альфа-оксид хрома, в котором от примерно 0,05 ат.% до примерно 6 ат.% атомов хрома в решетке альфа-оксида хрома замещено атомами трехвалентного кобальта (Со+3), получают методом соосаждения твердого вещества введением гидроксида аммония, причем указанный метод включает стадию дополнительного введения избытка нитрата аммония в осажденную смесь до стадии дегидратации и стадию прокаливания при температуре от 375°С до 1000°С в присутствии кислорода. Также заявлены хромсодержащие каталитические композиции, содержащие в качестве хромсодержащего компонента кристаллический кобальтзамещенный альфа-оксид хрома, необязательно обработанный фторирующим агентом. Способ получения композиции включает (а) соосаждение твердого вещества введением гидроксида аммония в водный раствор растворимой соли кобальта и растворимой соли трехвалентного хрома, который содержит не менее трех молей нитрата на моль хрома в растворе и имеет концентрацию кобальта от примерно 0,05 мол.% до примерно 6 мол.% от общей концентрации кобальта и хрома в растворе; и последующим введением в раствор не менее трех молей аммония на моль хрома в растворе; (b) сбор соосажденного твердого вещества, образованного на стадии (а); (с) сушку собранного твердого вещества; и (d) прокаливание высушенного твердого вещества при температуре от 375°С до 1000°С в присутствии кислорода. Технический результат - высокая активность каталитической композиции в указанных процессах. 5 н. и 10 з.п. ф-лы, 4 ил., 4 табл.
(а) соосаждение твердого вещества введением гидроксида аммония в водный раствор растворимой соли кобальта и растворимой соли трехвалентного хрома, который содержит не менее трех молей нитрата на моль хрома в растворе и имеет концентрацию кобальта от примерно 0,05 мол.% до примерно 6 мол.% от общей концентрации кобальта и хрома в растворе; и последующим введением в раствор не менее трех молей аммония на моль хрома в растворе;
(b) сбор соосажденного твердого вещества, образованного на стадии (а);
(c) сушку собранного твердого вещества;
(d) прокаливание высушенного твердого вещества при температуре от 375 до 1000°С в присутствии кислорода.
| Устройство для опрессовки сердечников электрических машин при склеивании | 1977 |
|
SU641598A1 |
| US 2001011061 A, 02.08.2001 | |||
| КАТАЛИЗАТОР ДЛЯ ФТОРИРОВАНИЯ НИЗШИХ АЛИФАТИЧЕСКИХ ГАЛОИДУГЛЕВОДОРОДОВ И СПОСОБ ФТОРИРОВАНИЯ НИЗШИХ АЛИФАТИЧЕСКИХ ГАЛОИДУГЛЕВОДОРОДОВ | 1992 |
|
RU2040333C1 |
| RU 2000109577 A, 20.03.2002 | |||
| Кондуктометрический датчик для анализа частиц по объемам | 1981 |
|
SU1038858A1 |
| US 3873471 A, 25.03.1975 | |||
| US 5155082 A, 13.10.1992 | |||
| US 5446215 A, 29.08.1995. | |||

