Описание
Алкиловые эфиры 2,2-дихлор- или дибромфенилуксусной кислоты применяются в качестве промежуточных соединений, например, для получения пестицидов или в качестве ускорителя вулканизации для эластомеров.
Получение таких соединений проводилось вначале, как, например, описано в патенте EP 0 075 356, реакцией пятихлористого фосфора с эфиром фенилглиоксиловой кислоты, которую получали из бензоилцианида. Бензоилцианид был очень дорогим исходным продуктом, в качестве альтернативы в качестве исходного продукта для получения 2,2-дихлорфенилацетонитрила применяли бензилцианид, например, аналогично патенту EP 0 518 412, который далее согласно патенту EP 0 075 356 переводили в желаемый алкиловый эфир 2,2-дихлорфенилуксусной кислоты.
При этом 2,2-дихлорфенилацетонитрил подвергали взаимодействию с водой и спиртом в присутствии галогеноводорода, предпочтительно газообразного HCl, при температуре от 0 до 80°C, предпочтительно от 15 до 50°C.
Проблемой способа, описанного в патенте EP 0 075 356, является образование побочного продукта 2,2-дихлорфенилацетамида, который затем должен быть отделен от реакционной смеси и который существенно снижает выход желательного целевого соединения. Другой проблемой при применении этанола в качестве спирта и HCl является образование этилхлорида - ядовитого, легковоспламеняющегося побочного продукта, который не должен выделяться. Другими побочными реакциями являются гидролиз конечного продукта в соответствующий алкиловый эфир фенилглиоксиловой кислоты или фенилглиоксиловую кислоту. Чтобы удовлетворить техническим требованиям в отношении дальнейшей переработки, эти побочные продукты, например, фенилглиоксиловая кислота, должны, однако, присутствовать в целевом продукте в чрезвычайно малых количествах.
Кроме того, при получении алкилового эфира 2,2-дихлорфенилуксусной кислоты с использованием описанных в патенте EP 0 075 356 параметров и последовательности стадий желательный целевой продукт получали с выходом только до 75% и с высоким содержанием самых разных побочных продуктов.
Поэтому задачей настоящего изобретения была разработка усовершенствованного способа получения алкиловых эфиров 2,2-дихлор- или дибромфенилуксусной кислоты, обеспечивающего получение желательных продуктов с высокими выходами и с высокой чистотой.
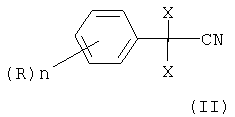
В соответствии с этим объектом изобретения является усовершенствованный способ получения алкилового эфира 2,2-дихлор- или дибромфенилуксусной кислоты формулы
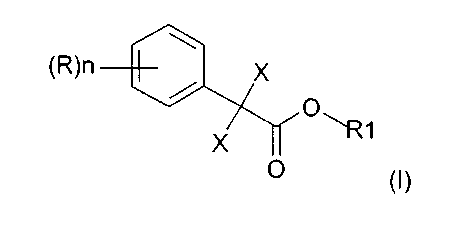
в которой X является Cl или Br, n может быть целым числом от 1 до 5, R означает водород, C1-C8-алкил, арил, гетероарил, C1-C8-алкокси, арилокси или галоген, а R1 означает C1-C8-алкил,
отличающийся тем, что 2,2-дихлор- или дибромфенилацетонитрил формулы
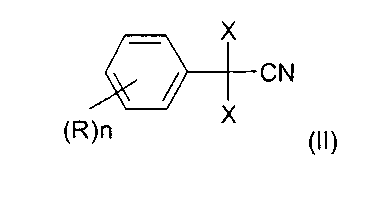
в которой X, n и R определены выше,
подвергают взаимодействию от 0,8 до 2 молей воды на моль нитрила формулы (II), от 1 до 8 молей и спирта формулы
R1OH (III),
в которой R1 определен выше,
на моль нитрила формулы (II),
и в присутствии от 1 до 3 моль HCl или HBr на моль нитрила формулы (II), при необходимости в присутствии растворителя, инертного в условиях реакции, с образованием соответствующего алкилового эфира 2,2-дихлор- или дибромфенилуксусной кислоты формулы (I), причем температура реакции на первой стадии взаимодействия составляет от 30 до 60°C и на второй фазе от 60 до 100°C, затем по окончании взаимодействия реакционную смесь охлаждают до температуры от 20 до 40°C, разбавляют водой и выделяют соответствующий алкиловый эфир 2,2-дихлор- или дибромфенилуксусной кислоты формулы (I).
В способе согласно изобретению 2,2-дихлор- или дибром-фенилацетонитрил формулы (II) с помощью воды, спирта R1OH и HCl или HBr, при необходимости в присутствии растворителя, инертного в условиях реакции, превращают в соответствующий алкиловый эфир 2,2-дихлор- или дибромфенилуксусной кислоты формулы (I).
В формуле (II) X означает хлор или бром, предпочтительно хлор.
n является целым числом от 1 до 5.
При этом R может означать водород, C1-C8-алкил, арил, гетероарил, C1-C8-алкокси, арилокси или галоген.
Под C1-C8-алкилом при этом понимаются линейные, разветвленные или циклически алкильные радикалы с числом С-атомов от 1 до 8, такие как метил, этил, изопропил, н-пропил, циклопропил, н-бутил, трет-бутил, н-пропил, циклопропил, гексил или октил.
Предпочтительны линейные или разветвленные C1-C4-алкильные радикалы.
Под C1-C8-алкокси понимаются алкилоксигруппы с числом С-атомов от 1 до 8, причем алкильная часть может быть линейной или разветвленной, например, метилокси, этилокси, изопропилокси, н-пропилокси, н-бутилокси, трет-бутилокси, н-пропилокси, гексилокси или октилокси.
Предпочтительны линейные или разветвленные C1-C4-алкоксильные остатки.
Алкильные или алкоксильные группы могут в определенных случаях быть одно- или многократно замещены группами, инертными в условиях реакции, например, арильными или гетероарильными группами, возможно, замещенными, галогеном, алкокси, арилокси и т.д.
Под арилом и арилокси понимаются предпочтительно ароматические остатки с числом С-атомов от 6 до 20, как фенил, дифенил, нафтил, инденил, флуоренил, фенокси и т.д.
Предпочтительны как ароматические остатки фенил и фенокси.
Арильные группы могут при желании быть одно- или многократно замещены группами, инертными в условиях реакции, например, арильные или гетероарильные группы, возможно замещенные, галоген, алкокси, арилокси и т.д.
Но арильная группа может также быть аннелированной с фенильным кольцом, так что фенильное кольцо и R, обозначающий арил, образуют конденсированную ароматическую циклическую систему, возможно, замещенную, например, инденил, флуоренил, нафтил и т.д.
Под гетероарилом подразумеваются ароматические остатки, которые содержат по меньшей мере один атом S, O или N в цикле или циклической системе. Такими остатками являются, например, фурил, пиридил, пиримидил, тиенил, изотиазолил, имидазолил, хинолил, бензотиенил, индолил, пирролил и т.д.
Гетероарильные группы могут при желании быть одно- или многократно замещены группами, инертными в условиях реакции, например, арильными или гетероарильными группами, возможно замещенными, галогеном, алкокси, арилокси и т.д.
Но гетероарильная группа может также быть аннелированной с фенильным кольцом, так что фенильное кольцо и R, обозначающий гетероарил, образуют конденсированную циклическую систему, возможно, замещенную, например, хинолинил, индолил, изоиндолил, кумаронил, фтализинил и т.д.
Под галогеном понимается фтор, хлор, бром и иод, причем предпочтительны фтор, бром и хлор.
Если R не является водородом, то n предпочтительно означает целое число от 1 до 3, особенно предпочтительно 1 или 2.
Предпочтительно R означает водород, незамещенный, линейный или разветвленный C1-C4-алкильный или -алкокси радикал, незамещенный фенил или фенокси, или хлор.
Особенно предпочтительно R означат водород.
Исходные соединения формулы (II), применяемые для способа согласно изобретению, являются коммерчески доступными или могут, например, быть получены из бензилцианида, например, согласно патенту EP 0 518 412.
Нитрилы формулы (II) согласно изобретению подвергаются взаимодействию с 0,8 до 2 молей воды на моль нитрила формулы (II), с 1 до 8 молей спирта формулы R1ОH (III) на моль нитрила формулы (II) и с 1 до 3 молей HCl или HBr на моль нитрила формулы (II).
В качестве спиртов формулы (III) подходят такие спирты, в которых R1 означат C1-C8-алкил. Примерами спиртов являются метанол, этанол, н-пропанол, изопропанол, изобутанол, н-пентанол или н-гексанол.
Выбор спирта зависит от желаемого эфира в целевом продукте. Предпочтительно в качестве спирта формулы (III) применяется метанол, этанол или н-бутанол, особенно предпочтителен этанол.
При этом спирт формулы (III) применяется в количестве от 1 до 8 моль на моль нитрила формулы (II), предпочтительно от 3 до 5 моль на моль нитрила формулы (II).
При желании могут применяться и большие количества спирта, однако это нецелесообразно по экономическим причинам.
Если спирт применяется в количестве от 1 до примерно 3 моль на моль нитрила, то предпочтительно используется дополнительный растворитель, инертный в условиях реакции. Подходящими растворителями являются, например, простые эфиры, например, метил-третбутиловый эфир (МТБЭ), диэтиловый эфир, тетрагидрофуран (ТГФ), диоксан или высшие простые эфиры, как, например, диметиловый эфир этиленгликоля и т.д., или возможно, галогенированные углеводороды, как, например, толуол, гексан, гептан, дихлорметан, хлорбензол и т.д.
Воду добавляют в реакционную смесь в количестве от 0,8 до 2 молей на моль нитрила формулы (II), предпочтительно от 0,9 до 1,5 молей на моль нитрила формулы (II).
Кроме того, добавляют от 1 до 3 молей HCl или HBr на моль нитрила формулы (II).
В способе согласно изобретению нитрил формулы (II) может сначала быть растворен в желательном спирте формулы (III), возможно, в растворителе, инертном в условиях реакции, и воде, после чего вводится газообразный HCl или HBr. При этом в качестве газообразного HCl или HBr может вводиться также отходящий газ HCl или HBr, полученный при хлорировании газообразным хлором или, соответственно, бромировании Br2 бензилцианида, возможно, замещенного, для получения желательного нитрила формулы (II), например, согласно патенту EP 0 518 412, в результате чего может быть осуществлено непосредственно объединение получения нитрильного исходного продукта формулы (II) с получением целевого алкилового эфира дихлор- или дибромфенилуксусной кислоты формулы (I).
Соответственно, еще одним объектом изобретения является способ получения алкилового эфира 2,2-дихлор- или дибромфенилуксусной кислоты формулы (I), отличающийся тем, что на первой стадии бензилцианид, возможно, замещенный, формулы
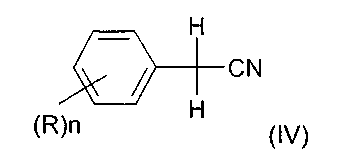
подвергают взаимодействию с хлором в присутствии каталитического количества газообразного хлористого водорода или с бромирующим агентом с получением соответствующего нитрила формулы (II), и образующийся отходящий газ HCl или HBr применяется на второй стадии превращения нитрила формулы (II) в соответствующий алкиловый эфир 2,2-дихлор- или дибромфенилуксусной кислоты формулы (I).
Стадия 1 при этом может быть осуществлена аналогично патенту EP 0 518 412.
В одном предпочтительном варианте для превращения нитрила формулы (II) применяется смесь спирт/вода/HCl или HBr.
Эта тройная смесь может быть получена тем, что в смесь воды и спирта вводят газообразный хлористый водород или HBr.
Особенно предпочтительно смесь спирт/вода/HCl или спирт/вода HBr получают тем, что в смесь воды и спирта вводят отходящий газ HCl или HBr, полученный при галогенировании с помощью Cl2 или Br2 бензилцианида, возможно, замещенного, с получением желательного нитрила формулы (II), например, согласно патенту EP 0 518 412. Преимуществом является то, что получение нитрила формулы (II) не должно быть связано непосредственно с получением алкилового эфира дихлор- или дибромфенилуксусной кислоты формулы (I). HCl или HBr, которые получены в виде отходящего газа со стадии галогенирования, могут тем самым временно храниться в виде водного, спиртового раствора.
Но смесь может также быть получена путем введения HCl или HBr, или соответственно отходящего газа HCl или HBr, в раствор спирта и водного HCl или HBr, или в спирт с последующим разбавлением водой.
Желательные мольные доли в тройной смеси могут быть установлены при желании путем разбавления имеющегося водного, спиртового раствора HCl или HBr спиртом и/или водой.
Если в способе согласно изобретению применяют смесь спирт/вода/HCl или спирт/вода HBr, то сначала может подаваться нитрил формулы (II), а затем добавляется тройная смесь. Однако можно также сначала подавать тройную смесь, а затем добавлять нитрил формулы (II).
В способе согласно изобретению превращение на первой стадии происходит при температуре от 30 до 60°C, предпочтительно от 35 до 55°C.
Для этого, например, сначала подают нитрил формулы (II) или тройную смесь, нагревают до температуры от 30 до 60°C и затем при этой температуре добавляют оставшиеся компоненты реакции. Полученную реакционную смесь затем при этой температуре перемешивают еще от нескольких минут до нескольких часов, предпочтительно от 30 минут до 5 часов.
На второй стадии реакционную смесь нагревают до температуры от 60 до 100°C, предпочтительно от 65 до 80°C, и снова перемешивают при этой температуре от нескольких минут до нескольких часов, предпочтительно от 30 минут до 10 часов.
После проверки полноты превращения реакционную смесь охлаждают до температуры от 20 до 40°C и добавляют столько воды, чтобы выпавший в осадок хлорид или бромид аммония растворился и произошло разделение фаз. Затем водную фазу экстрагируют, возможно, с помощью обычного экстрагента, как, например, гексан, гептан, толуол, простой эфир, или сложные эфиры. Экстракт затем соединяют с органической фазой.
Выделение целевого продукта может проводиться, например, тем, что сначала при нормальном давлении и температуре макс. до 90°C отгоняют возможный экстрагент, воду и спирт, а затем в вакууме отгоняют низкокипящие компоненты или побочные продукты фенилглиоксиловую кислота, этиловый эфир фенилглиоксиловой кислоты, этиловый эфир фенилуксусной кислоты (из-за неполного галогенирования) и монохлор- или монобромфенилацетат, до достижения постоянной температуры кипения, так что соответствующий целевой продукт формулы (I) остается в жидкой фазе. Для дальнейшей очистки продукт может быть отогнан сверху.
После соединения органических фаз можно, однако, также сначала отделить воду на водоотделителе, а затем при нормальном давлении можно отогнать спирт и возможный экстрагент.
Если сырой целевой продукт все еще содержит слишком много органических кислот, которые образуются при гидролизе продукта, то в неочищенный продукт еще раз добавляют указанное выше экстрагент и спирт и снова обрабатывают фракционированием, при этом происходит повторное этерифицирование.
Способом согласно изобретению получен алкиловый эфир 2,2-дихлор- или дибромфенилуксусной кислоты формулы (I) с более высоким выходом и более чистый по сравнению с уровнем техники, при этом происходит существенно меньший выброс газообразных отходов, чем в обычных способах и, кроме того, благодаря утилизации отходящих газов требовалось меньше сырья.
Пример 1: Хлорирование бензилцианида с получением 2,2-дихлор-фенилацетонитрила в лабораторных условиях
1436 г (12,27 моль) бензилцианида загружали в предварительно продутый инертным газом эмалированный автоклав. Затем вводили 239 г (6,55 моль = 0,533 экв.) газообразного хлористого водорода при открытом вентиле для отходящих газов, после чего вентиль закрывали и нагревали до 40°C.
Затем вводили 1830 г (25,77 моль = 2,10 экв.) хлора в течение 6 часов при 60-65°C и давлении 3 бар. При этом в течение первых минут температура резко увеличивалась, а затем в зависимости от скорости введения оставалась приблизительно постоянной. При достижении внутреннего давления 3 бар вентиль для отходящих газов слегка приоткрывали, так что давление оставалось постоянным и равным 3 бар.
В конце реакции температура и давление немного понижались. После окончания ввода хлора вентиль для отходящих газов закрывали и перемешивали 30 минут при 55°C.
Затем давление в автоклаве снижалось и для удаления хлора и газообразного HCl подводили азот.
Газоочиститель отходящих газов заполняли 2823 г (61,3 моль = 5 экв.) этанола и 340 г концентрированной соляной кислоты (12,27 моль = 1 экв., соответствующий 221 г воды, и 3,26 моль = 0,266 экв.= 119 г соляной кислоты) и вводили газ, отходящий с хлорирования, при 10-15°C до 95% (пока в отходящем газе не оставалось хлора (макс. 3%)). После этого включали дополнительный газоочиститель (наполненный 10%-ым раствором едкого натра). Остаточный отходящий газ вводили в 2 подсоединяемых последовательно очистителя с 10%-ным раствором едкого натра.
Выход:
2200 г 2,2-дихлорфенилацетонитрила (96,3% от теор.)
3950 г 24,5%-ного этанольного раствора соляной кислоты (выход HCl при хлорировании: 73% от теор.)
Пример 2: Получение этилового эфира 2,2-дихлорфенилуксусной кислоты из 2,2-дихлорфенилацетонитрила (2-я стадия в лабораторных условиях (вариант I))
70,0 г (0,38 моль) дистиллированного 2,2-дихлорфенилацетонитрила, полученного по примеру 1, нагревали до 40°C.
Затем в течение 30 минут добавляли 123,57 г этанольного раствора соляной кислоты, полученной при хлорировании аналогично примеру 1 (30,22 г = 0,83 моль, соответствующих 2,18 экв. соляной кислоты, 6,77 г = 3,76 моль = 0,99 экв. воды и 86,6 г = 1,88 моль = 4,95 экв. этанола), при 40°C и по окончании добавления перемешивали еще 2 часа при 40°C. Затем нагревали до 75°C и перемешивали еще 3 часа. После проверки полноты превращения реакционную смесь охлаждали до 30°C и добавляли 115 г воды.
Смесь перемешивали до полного растворения твердого вещества (хлорида аммония). Затем органическую фазу отделяли, а оставшуюся водную фазу экстрагировали 18 г гексана. Органический экстракт соединяли с полученной ранее фазой продукта и сначала перегоняли при нормальном давлении для отделения этанола, воды и гексана. Затем при 10 мбар перегоняли для достижения постоянной температуры кипения 128°C.
Оставшаяся жидкая фаза содержала 97,8 вес.% этилового эфира 2,2-дихлорфенилуксусной кислоты.
Выход: 83,6 г этилового эфира 2,2-дихлорфенилуксусной кислоты (95% от теор.) 97,8 вес.%
Пример 3: Получение этилового эфира 2,2-дихлорфенилуксусной кислоты из 2,2-дихлорфенилацетонитрила (2-я стадия в лабораторных условиях (вариант II))
123,57 г этанольного раствора соляной кислоты, полученной при хлорировании аналогично примеру 1 (30,22 г = 0,83 моль = 2,18 экв. соляной кислоты; 6,77 г = 3,76 моль = 0,99 экв. воды; 86,6 г = 1,88 моль = 4,95 экв. этанола) нагревали до 40°C.
Затем в течение 30 минут добавляли 70,0 г (0,38 моль) 2,2-дихлорфенилацетонитрила при 40°C и по окончании добавления перемешивали еще 2 часа при 40°C. Затем нагревали до 75°C и перемешивали еще 3 часа. После проверки полноты превращения реакционную смесь охлаждали до 30°C и добавляли 115 г воды.
Смесь перемешивали до полного растворения твердого вещества (хлорида аммония). Затем органическую фазу отделяли, а оставшуюся водную фазу экстрагировали 18 г гексана. Органический экстракт соединяли с полученной ранее фазой продукта и сначала на водоотделителе удаляли воду. Затем перегоняли при нормальном давлении для отделения этанола и гексана. Затем перегоняли при 10 мбар до достижения постоянной температуры кипения 128°C.
Оставшаяся жидкая фаза содержала 93,9 вес.% этилового эфира 2,2-дихлорфенилуксусной кислоты. Для дальнейшей очистки продукт отгонялся с головы.
Выход: 70 г этилового эфира 2,2-дихлорфенилуксусной кислоты (80% от теор.) 99,4 вес.%.
Пример 4: Хлорирование бензилцианида с получением 2,2-дихлорфенилацетонитрила на пилотной установке
400 кг (3385 моль) бензилцианида загружали в заранее продутый инертным газом эмалированный реактор, затем в течение одного часа вводили при 40-50°C 40 кг (1096 моль = 0,324 экв.) газообразного хлористого водорода при открытом вентиле для отходящих газов, затем вентиль закрывали.
Затем через 15 часов вводили 487 кг (6859 моль = 2,026 экв.) хлора при 55-60°C и 3-3,5 бар.
По окончании ввода хлора вентиль для отходящих газов закрывали и перемешивали 3 часа при 60-63°C. Затем давление в реакторе снижалось и для удаления хлора и газа HCl пропускали азот.
Газоочиститель отходящих газов заполняли 475 кг (10326 моль = 3,05 экв.) этанола и 94 кг концентрированной соляной кислоты (3394 моль = 1 экв. = 61,1 кг воды и 901 моль = 0,266 экв. = 32,9 кг соляной кислоты) и вводили газ, отходящий с хлорирования, при 10-15°C до максимальной концентрации хлора в отходящем газе 3%. После этого включали запасной газоочиститель (наполненный 10%-ным раствором едкого натра).
Остаточный отходящий газ проводили непосредственно в вышеописанный газоочиститель с раствором едкого натра.
Выход:
636 кг 2,2-дихлорфенилацетонитрила (теоретический выход)
825 кг 35,2 %-ой этанольного раствора соляной кислоты (выход HCl при хлорировании: 89% от теор.)
Пример 5: Получение этилового эфира 2,2-дихлорфенилуксусной кислоты из 2,2-дихлорфенилацетонитрила (2-я стадия на пилотной установке (вариант II))
410 кг этанольного раствора соляной кислоты, полученной по примеру 4 (143,5 кг = 3932 моль = 2,31 экв. соляной кислоты; 30,5 кг = 1694 моль = 1 экв. воды; 236 кг = 5130 моль =3,02 экв. этанола) разбавляли 100 кг (2174 моль = 1,28 экв.) этанола и нагревали до 40°C.
Затем в течение 2 часов вводили 316 кг (1699 моль) 2,2-дихлорфенилацетонитрила при 40°C и по окончании добавления перемешивали несколько часов при 40°C. Затем нагревали до 70°C и перемешивали еще 6 часов. После проверки полноты превращения реакционную смесь охлаждали до 30°C.
Полученную суспензию вводили в 570 л воды и перемешивали до полного растворения твердого вещества (хлорида аммония). Затем органическую фазу отделяли, а оставшуюся водную фазу экстрагировали 80 кг гексана. Органический экстракт соединяли с предварительно полученной фазой продукта и сначала перегоняли при нормальном давлении и 90°C (для отделения этанола, воды и гексана), а затем фракционировали при 7 мбар до 135°C.
Выход: 325 кг этилового эфира 2,2-дихлорфенилуксусной кислоты (82% от теор.) 98,8 вес.%
Пример 6: Получение этилового эфира 2,2-дихлорфенилуксусной кислоты из 2,2-дихлорфенилацетонитрила (2-я стадия на пилотной установке (вариант I))
325,3 кг (1749 моль) 2,2-дихлорфенилацетонитрила разбавляли 100 кг (2174 моль = 1,28 экв.) этанола и нагревали до 40°C.
Затем в течение 3 часов добавляли 410 кг этанольного раствора соляной кислоты, полученной по примеру 4 (143,5 кг = 3932 моль = 2,25 экв. соляной кислоты; 30,5 кг = 1694 моль = 0,97 экв. воды; 236 кг = 5130 моль = 2,93 экв. этанола), при 40°C и по окончании добавления перемешивали еще 3 часа при 40°C. Затем нагревали до 70°C и перемешивали еще 6 часов. После проверки полноты превращения реакционную смесь охлаждали до 30°C.
Полученную суспензию вводили в 570 л воды и перемешивали до полного растворения твердого вещества (хлорида аммония). Затем органическую фазу отделяли, а оставшуюся водную фазу экстрагировали 80 кг гексана. Органический экстракт соединяли с предварительно полученной фазой продукта и сначала перегоняли при 20-25°C и небольшом разрежении (для отделения этанола, воды и гексана), а затем фракционировали при 7 мбар до 135°C.
Выход: 346,4 кг этилового эфира 2,2-дихлорфенилуксусной кислоты (85% от теор.) 98,9 вес.%.
Пример 7: Получение этилового эфира 2,2-дихлорфенилуксусной кислоты из 2,2-дихлорфенилацетонитрила (2-я стадия на пилотной установке (вариант II с повторной этерификацией))
375 кг этанольного раствора соляной кислоты, полученной при хлорировании по примеру 4 (136,7 кг = 3745 моль = 2,16 экв. соляной кислоты; 28,2 кг = 1567,5 моль = 0,91 экв. воды; 210,1 кг = 4567 моль = 2,64 экв. этанола) разбавляли 100 кг (2174 моль = 1,26 экв.) этанола и нагревали до 40°C.
Затем в течение 2 часов добавляли 322 кг (1732 моль) 2,2-дихлорфенилацетонитрила при 40°C и по окончании добавления перемешивали еще несколько часов при 40°C. Затем нагревали до 70°C и перемешивали еще 6 часов. После проверки полноты превращения реакционную смесь охлаждали до 30°C.
Полученную суспензию вводили в 570 л воды и перемешивали до полного растворения твердого вещества (хлорида аммония). Затем отделяли органическую фазу, а оставшуюся водную фазу экстрагировали 80 кг гексана. Органический экстракт соединяли с предварительно полученной фазой продукта и затем при нормальном давлении перегоняли при 120°C (для отделения этанола, воды и гексана). Так как сырой продукт содержал слишком много органических кислот (из-за гидролиза продукта), добавляли 40 кг гексана и 20 кг этанола и снова дистиллировали при нормальном давлении до 120°C. Затем сырой продукт фракционировали при 7 мбар до 135°C.
Выход: 329 кг этилового эфира 2,2-дихлорфенилуксусной кислоты (81,5% от теор.) 98,0 вес.%.
Пример 8 Повторная этерификация в лабораторных условиях
11,3 г этилового эфира 2,2-дихлорфенилуксусной кислоты с содержанием 0,13 вес.% фенилглиоксиловой кислоты (продукт гидролиза этилового эфира 2,2-дихлорфенилуксусной кислоты) нагревали с 2 мл гексана и 0,5 мл этанола в течение 2 часов до 70°C. Затем растворители отгоняли.
Результат: содержание фенилглиоксиловой кислоты падает до 0,01 вес.%
Изобретение относится к усовершенствованному способу получения алкилового эфира 2,2-дихлор- или дибромфенилуксусной кислоты формулы (I)
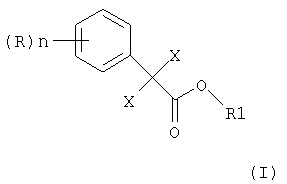
в которой Х является Cl или Br, n может быть целым числом от 1 до 5, R означает водород, C1-C8-алкил, арил, гетероарил, C1-C8-алкокси, арилокси или галоген, и R1 означает C1-C8-алкил, где 2,2-дихлор- или дибромфенилацетонитрил формулы
в которой X, n и R определены выше, подвергают взаимодействию в 0,8 до 2 молей воды на моль нитрила формулы (II), 1 до 8 молей спирта формулы (III): R1OH (III), в которой R1 определен выше, на моль нитрила формулы (II) и в присутствии от 1 до 3 молей HCl или HBr на моль нитрила формулы (II), при необходимости в присутствии растворителя, инертного в условиях реакции, при температуре реакции превращения от 30 до 60°С, затем осуществляют нагревание до 60-100°С и выдерживание при этой температуре, после окончания реакции реакционную смесь охлаждают до температуры от 20 до 40°С и разбавляют водой, и выделяют соответствующий алкиловый эфир 2,2-дихлор- или дибромфенилуксусной кислоты формулы (I). Изобретение относится также к способу получения алкилового эфира 2,2-дихлор- или дибромфенилуксусной кислоты формулы (I)
в которой Х является Cl или Br, n может быть целым числом от 1 до 5, R означает водород, C1-C8-алкил, арил, гетероарил, C1-C8-алкокси, арилокси или галоген и R1 означает C1-C8-алкил, где на первой стадии, возможно, замещенный бензилцианид формулы (IV)
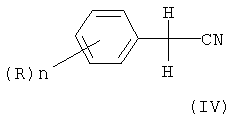
n и R определены выше, подвергают взаимодействию с хлором, в присутствии каталитических количеств газообразного хлористого водорода или с бромирующим агентом с получением соответствующего нитрила формулы (II)
в которой n, Х и R определены выше, и образующийся отходящий газ HCl или HBr используют на второй стадии для превращения нитрила формулы (II) в соответствующий алкиловый эфир 2,2-дихлор- или дибромфенилуксусной кислоты формулы (I), причем превращение в соответствующий алкиловый эфир 2,2-дихлор- или дибромфенилуксусной кислоты формулы (I) на второй стадии происходит в 0,8 до 2 молей воды на моль нитрила формулы (II), 1 до 8 молей спирта формулы (III): R1OH (III), в которой R1 определен выше, на моль нитрила формулы (II) и в присутствии HCl или HBr в виде газа, отходящего со стадии 1, в количестве от 1 до 3 молей на моль нитрила формулы (II), при необходимости в присутствии растворителя, при температуре реакции превращения от 30 до 60°С, затем осуществляют нагревание до 60-100°С и выдерживание при этой температуре, после окончания реакции реакционную смесь охлаждают до температуры от 20 до 40°С и разбавляют водой. Способы позволяют получать целевые продукты с высокими выходами и высокой чистоты. 2 н. и 8 з.п. ф-лы.
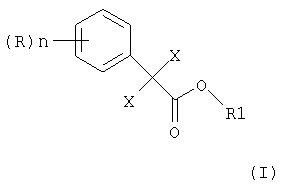
в которой Х является Cl или Br, n может быть целым числом от 1 до 5, R означает водород, C1-C8-алкил, арил, гетероарил, C1-C8-алкокси, арилокси или галоген, и R1 означает C1-C8-алкил, отличающийся тем, что 2,2-дихлор- или дибромфенилацетонитрил формулы
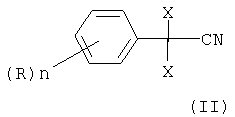
в которой X, n и R определены выше,
подвергают взаимодействию в 0,8 до 2 молей воды на моль нитрила
формулы (II), 1 до 8 молей спирта формулы (III)
R1OH (III)
в которой R1 определен выше,
на моль нитрила формулы (II) и в присутствии от 1 до 3 молей HCl или HBr на моль нитрила формулы (II), при необходимости в присутствии растворителя, инертного в условиях реакции, при температуре реакции превращения от 30 до 60°С, затем осуществляют нагревание до 60-100°С и выдерживание при этой температуре, после окончания реакции реакционную смесь охлаждают до температуры от 20 до 40°С и разбавляют водой, и выделяют соответствующий алкиловый эфир 2,2-дихлор- или дибромфенилуксусной кислоты формулы (I).
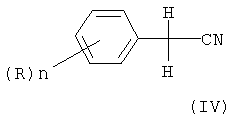
в которой n и R определены как в формуле (I), с хлором, в присутствии каталитических количеств газообразного хлористого водорода, или с бромирующим агентом с образованием соответствующего нитрила формулы (II), за счет чего достигается непосредственное объединение получения нитрила формулы (II) с получением соответствующего алкилового эфира 2,2-дихлор- или дибромфенилуксусной кислоты формулы (I).
в которой Х является Cl или Br, n может быть целым числом от 1 до 5, R означает водород, C1-C8-алкил, арил, гетероарил, C1-C8-алкокси, арилокси или галоген, и R1 означает C1-C8-алкил, отличающийся тем, что на первой стадии, возможно, замещенный бензилцианид формулы (IV)
n и R определены выше,
подвергают взаимодействию с хлором, в присутствии каталитических количеств газообразного хлористого водорода или с бромирующим агентом с получением соответствующего нитрила формулы (II)
в которой n, Х и R определены выше,
и образующийся отходящий газ HCl или HBr используют на второй стадии для превращения нитрила формулы (II) в соответствующий алкиловый эфир 2,2-дихлор- или дибромфенилуксусной кислоты формулы (I)
причем превращение в соответствующий алкиловый эфир 2,2-дихлор- или дибромфенилуксусной кислоты формулы (I) на второй стадии происходит в 0,8 до 2 молей воды на моль нитрила формулы (II), 1 до 8 молей спирта формулы (III)
R1OH (III)
в которой R1 определен выше, на моль нитрила формулы (II) и в присутствии HCl или HBr в виде газа, отходящего со стадии 1, в количестве от 1 до 3 молей на моль нитрила формулы (II), при необходимости в присутствии растворителя, при температуре реакции превращения от 30 до 60°С, затем осуществляют нагревание до 60-100°С и выдерживание при этой температуре, после окончания реакции реакционную смесь охлаждают до температуры от 20 до 40°С и разбавляют водой, и выделяют соответствующий алкиловый эфир 2,2-дихлор- или дибромфенилуксусной кислоты формулы (I).
| Кассета для светокопирования на двухсторонней светочувствительной бумаге и приспособление для зарядки кассеты | 1947 |
|
SU75356A1 |
| СПОСОВ ПОЛУЧЕНИЯ ЭФИРОВ ДИГАЛОИДЗАМЕЩЕННЫХ КАРБОНОВЫХ КИСЛОТ | 0 |
|
SU188951A1 |
| Устройство для швартования судна | 1974 |
|
SU518412A1 |
| ПОРЦИОННАЯ ПРИПРАВА И СПОСОБ ЕЁ ПРИГОТОВЛЕНИЯ | 2023 |
|
RU2827977C1 |

