ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к области химического синтеза и относится к способу синтеза соединения (I):
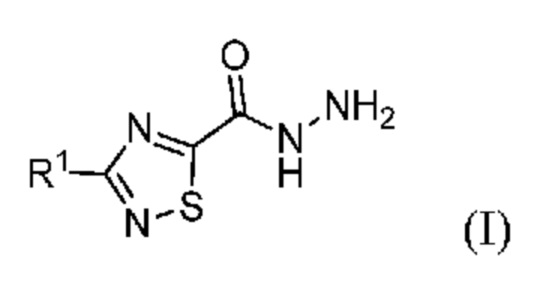
или его соли, где R1 означает метил или метил-d3, что соответствует 3-метил-1,2,4-тиадиазол-5-карбогидразиду или его метил-d3 дейтерированной форме. Эти соединения применимы в качестве ключевых промежуточных продуктов в синтезе фармацевтических соединений, в особенности фезолинетанта и дейтерированного фезолинетанта.
УРОВЕНЬ ТЕХНИКИ
Фезолинетант разработан, как селективный антагонист рецептора NK-3 и применяется в качестве терапевтического соединения, в частности, для лечения и/или предупреждения зависимых от полового гормона заболеваний. Фезолинетант соответствует (R)-(4-фторфенил)-(8-метил-3-(3-метил-1,2,4-тиадиазол-5-ил)-5,6-дигидро-[1,2,4]триазоло[4,3-a]пиразин-7(8H)-ил)метанону и описан в WO2014/154895.
Дейтерированный фезолинетант, (R)-(4-фторфенил)-(8-метил-3-(3-(метил-d3)-1,2,4-тиадиазол-5-ил)-5,6-дигидро-[1,2,4]триазоло[4,3-a]пиразин-7(8H)-ил)метанон, также разработан для тех же целей и описан в WO2019/012033.
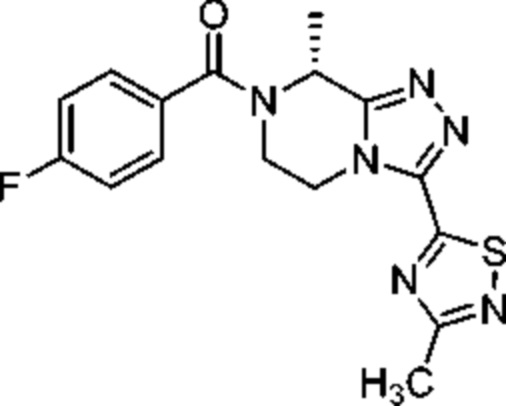
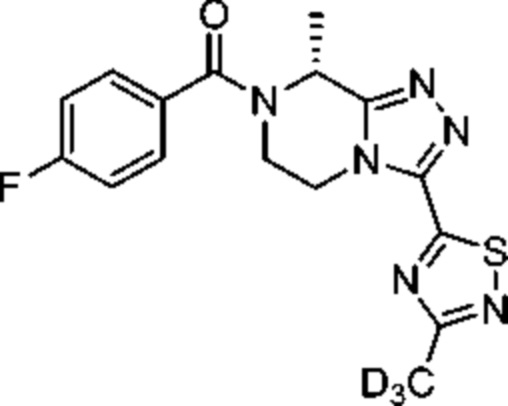
Фезолинетант Дейтерированный фезолинетант
Способы синтеза фезолинетанта и дейтерированного фезолинетанта описаны в WO2014/154895 и WO2019/012033. Эти способы включают 3-метил-1,2,4-тиадиазол-5-карбогидразид или его дейтерированную форму в качестве ключевого промежуточного продукта (I):
где R1 означает метил или метил-d3.
Способ синтеза недейтерированного промежуточного продукта 3-метил-1,2,4-тиадиазол-5-карбогидразида (I-1) раскрыт в WO2013/050424, где его получают из соответствующего метилового эфира (II-1-а), этот последний получают в одну стадию из ацетамида, хлоркарбонилсульфенилхлорида и метилцианоформиата:
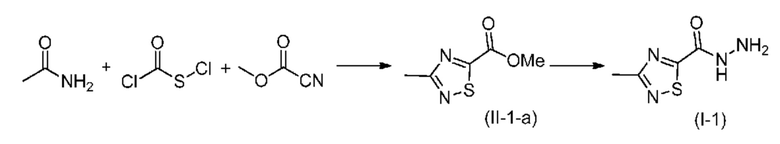
Однако хлоркарбонилсульфенилхлорид и метилцианоформиат являются опасными реагентами, для которых затруднительно снабжение в крупном масштабе и следует избегать увеличения масштаба. Кроме того, этот путь синтеза приводит к загрязнению серой, что вредно влияет на качество конечного фармацевтического продукта. Кроме того, даже после оптимизации общий выход промежуточного продукта (I-1) остается меньшим, чем 30%.
Соответствующий дейтерированный промежуточный продукт 3-(метил-d3)-1,2,4-тиадиазол-5-карбогидразид (I-2) получали по такой же методике, как описанная в WO2019/012033, с теми же недостатками.
Поэтому, необходим более безопасный, надежный, масштабируемый и более эффективный синтез промежуточного продукта (I).
Настоящее изобретение относится к способу синтеза промежуточного продукта (I) из его соответствующего эфира (II), который получают путем алкоксикарбонилирования соответствующего галогенированного промежуточного продукта (III):
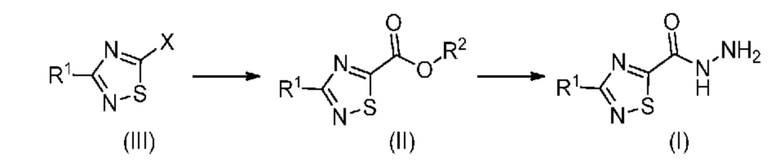
или его солей, где R1 означает метил или метил-d3, R2 означает алкильную или арилалкильную группу и X означает атом галогена.
Этот тип алкоксикарбонилирования описан ранее для более прочных 5-членных гетероциклов, таких как тиофены, фураны, тиазолы, имидазол, оксазолы, пиразолы и индолы. Однако не описана обработка менее прочных гетероциклов (т. е. содержащих более 2 гетероатомов), намного более подверженных раскрытию цикла, например, таких как тиадиазольные кольца, как в настоящем изобретении. В случае образования раскрытых промежуточных 1,2,4-тиадиазолов это может привести к очень реакциоонноспособным промежуточным продуктам (таким как меркаптоамидин) и летучим побочным продуктам (в особенности, если R1 означает метил) (см. аналогию для более прочного оксазола в: Verrier et al., Beilstein J. Org. Chem., 2011, 7, 1584-1601; Bellina et al., Current Org. Chem., 2008, 12(9), 774-790 and Strotman et al., Org. Lett., 2010, 12, 3578-3581).
Способ, предлагаемый в настоящем изобретении, обладает тем преимуществом, что включает не особенно опасные специальные реагенты. Общий выход весьма удовлетворительный (более 67%) даже при масштабировании, что показывает приведенная ниже экспериментальная часть.
Исходное галогенированное соединение формулы (III) можно получить по реакции Зандмейера из амина формулы (IV):
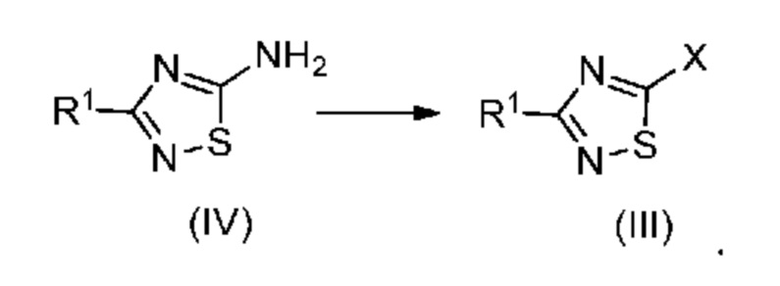
Настоящее изобретение также относится к новому способу синтеза дейтерированного промежуточного продукта (IV). Этот способ включает реакцию Пиннера с дейтерированным ацетонитрилом (VII-2), который сначала превращают в соль Пиннера (VI-2) (стадия a) и затем в дейтерированный ацетамидин (V-2) (стадия b), затем образуется тиадиазольное кольцо (стадия c) с получением соединения (IV-2):
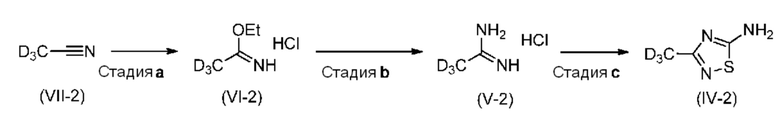
Этот способ обладает тем преимуществом, что обеспечивает весьма высокую изотопную чистоту, что показывает приведенная ниже экспериментальная часть.
Настоящее изобретение также относится к альтернативному новому способу синтеза дейтерированного промежуточного продукта (IV). Этот способ включает образование дейтерированного N-гидроксиацетамидина (IХ-2) из дейтерированного ацетонитрила (VII-2), который затем активируют путем тозилирования с образованием промежуточного продукта (X-2) с последующим образованием тиадиазольного кольца с получением соединения (IV-2):
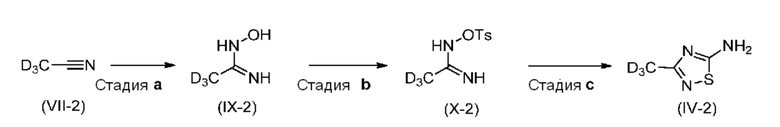
Этот способ также обладает тем преимуществом, что обеспечивает весьма высокую изотопную чистоту.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Таким образом, настоящее изобретение относится к способу получения соединения формулы (I):

или его соли, где R1 означает метил или метил-d3; указанный способ включает следующие стадии: a) проведение реакции алкоксикарбонилирования соединения формулы (III):
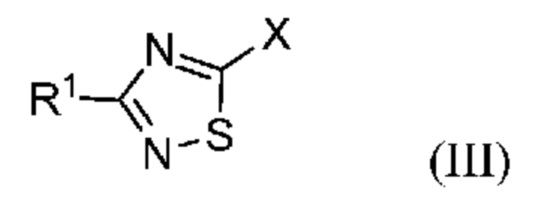
где X означает галоген; и R1 означает метил или метил-d3; с получением соединения формулы (II):
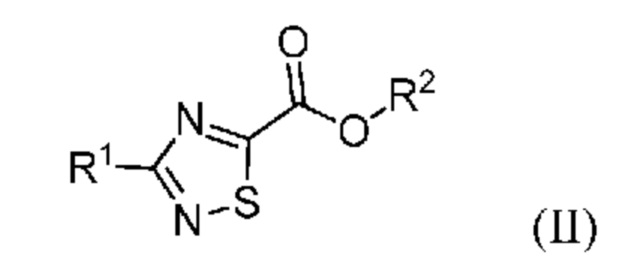
где R1 означает метил или метил-d3; и R2 означает алкил или арилалкил; и
b) получение соединения формулы (I) по реакции соединения формулы (II) с гидразинмоногидратом.
В одном варианте осуществления в способе, предлагаемом в настоящем изобретении X означает бром или йод; предпочтительно, если X означает бром. В одном варианте осуществления в способе, предлагаемом в настоящем изобретении, R2 означает метил или этил; предпочтительно, если R2 означает этил.
В одном варианте осуществления реакцию алкоксикарбонилирования на стадии a) проводят в присутствии монооксида углерода, палладиевого катализатора, основания и спиртового растворителя, и необязательно в присутствии фосфорорганического лиганда.
В одном варианте осуществления палладиевым катализатором является Pd(OAc)2, фосфорорганическим лигандом является 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos), основанием является ацетат натрия и растворитель выбран из группы, включающей этанол, метанол и смеси метил-трет-бутилового эфира с этанолом или метанолом. Предпочтительно, если стадию a) проводят в этаноле в качестве растворителя или в смеси этанола и метил-трет-бутилового эфира. Предпочтительно, если стадию a) проводят при температуре в диапазоне от 50°C до 150°C; предпочтительно от 63°C до 67°C; более предпочтительно примерно при 65°C. В одном варианте осуществления стадию a) проводят в присутствии Fe(CO)5.
В другом варианте осуществления палладиевым катализатором является бис(трифенилфосфин)палладийхлорид (Pd(PPh3)2Cl2), основанием является триэтиламин и растворителем является этанол.
В одном варианте осуществления реакцию алкоксикарбонилирования на стадии a) проводят посредством обмена лития с помощью проводимого сначала взаимодействия соединения формулы (III) с литийорганическим реагентом; с последующим добавлением хлорформиата или цианоформиата. Предпочтительно, если литийорганическим реагентом является н-гексиллитий и хлорформиатом является алкилхлорформиат, предпочтительно этилхлорформиат.
В одном варианте осуществления способ, предлагаемый в настоящем изобретении, включает предварительную стадию реакции Зандмейера соединения формулы (IV):
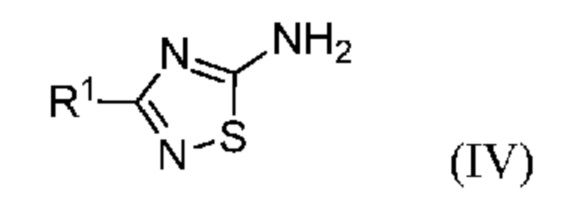
или его соли, где R1 означает метил или метил-d3; с получением соединения формулы (III).
Реакцию Зандмейера можно провести в присутствии нитрита натрия и бромида водорода в водной среде. Альтернативно, реакцию Зандмейера можно провести в присутствии трет-бутилнитрита и йода или в присутствии йодида калия и п-толуолсульфоновой кислоты.
Настоящее изобретение также относится к способу получения 3-(метил-d3)-1,2,4-тиадиазол-5-амина (IV-2):
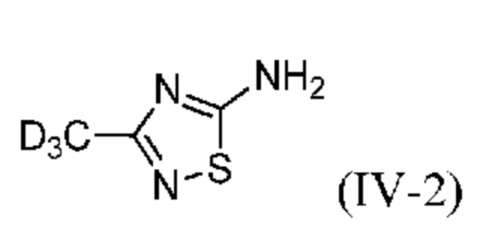
или его соли, включающему следующие стадии:
a) реакция d3-ацетонитрила с этанолом в присутствии HCl с образованием соли Пиннера формулы (VI-2):
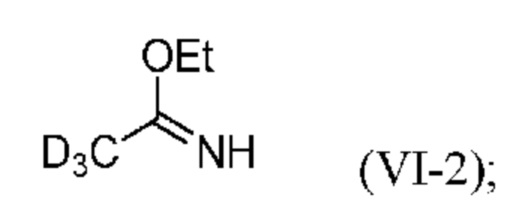
b) реакция соли Пиннера (VI-2) с аммиаком с образованием d3-ацетамидина (V-2) или его соли:
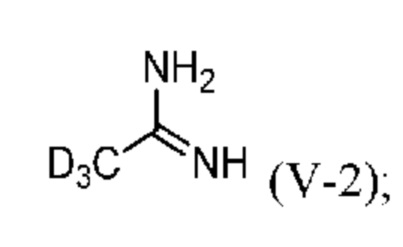
и
c1) реакция d3-ацетамидина (V-2) с бромом, тиоцианатом и метоксидом натрия с образованием соединения формулы (IV-2); или
c2) реакция d3-ацетамидина (V-2) сначала с гипохлоритом натрия (NaOCl) и затем с тиоцианатом с образованием соединения формулы (IV-2).
ОПРЕДЕЛЕНИЯ
В настоящем изобретении указанные ниже термины обладают следующими значениями:
"Примерно" перед числом означает±10% от значения указанного числа.
"Спиртовый растворитель" означает вещество, которое растворяет растворенное вещество (химически иную жидкость, твердое вещество или газ) с образованием раствора и которым является спирт, т. е. органическое соединение, содержащее по меньшей мере одну функциональную гидроксигруппу (-OH), связанную с атомом углерода. Примеры спиртовых растворителей включают метанол, этанол и изопропиловый спирт.
"Реакция алкоксикарбонилирования" означает химическую реакцию образования связи C-C, обеспечивающую введение алкоксикарбонильного фрагмента в каркас. "Алкоксикарбонильный фрагмент означает группу -C(=O)-O-алкил, где алкил является таким, как определено ниже, включая случаи, когда сам алкил является замещенным, например, арильной группой (т. е. образование арилалкильной группы) с получением -C(=O)-O-алкиларила.
"Алкил" означает гидрокарбильный радикал формулы CnH2n+1, где n является числом, большим или равным 1. Обычно алкильные группы в настоящем изобретении содержат от 1 до 6 атомов углерода, предпочтительно от 1 до 4. Алкильные группы могут быть линейными или разветвленными и могут быть замещены, как указано в настоящем изобретении. Примерами алкильных групп являются метил, этил, н-пропил, изопропил, бутил и его изомеры (например, н-бутил, изобутил и трет-бутил); пентил и его изомеры, гексил и его изомеры
"Арил" означает полиненасыщенную ароматическую гидрокарбильную группу, содержащую одно кольцо (т. е. фенил) или несколько ароматических колец, сконденсированных друг с другом (например, нафтил), или связанных ковалентно, обычно содержащее от 5 до 12 атомов; предпочтительно от 6 до 10, где по меньшей мере одно кольцо является ароматическим. Ароматическое кольцо необязательно включать от 1 до 2 дополнительных сконденсированных с ним колец (циклоалкил, гетероциклил или гетероарил). Неограничивающие примеры арила включают фенил, бифенилил, бифениленил, нафталин-1- или -2-ил. В одном варианте осуществления арильной группой является фенил. Необязательно, арильные группы могут быть замещены одной или большим количеством групп, таких как например, алкоксигруппа. В предпочтительном варианте осуществления арильная группа замещена с помощью от 1 до 3 алкоксигрупп, предпочтительно с помощью от 1 до 3 метоксигрупп.
"Арилалкил" означает фрагмент арилалкил, где арил и алкил являются такими, как определено в настоящем изобретении. Примеры арилалкильных групп включают бензил, 4-метоксибензил (PMB), 2,4-диметоксибензил (DMB) и 2,4,6-триметоксибензил (TMB).
"Реакция карбонилирования" означает химическую реакцию образования связи C-C, обеспечивающую введение карбонильного фрагмента в каркас, например, группы -C(=O)-O-R2, где R2 означает, например, алкил или арилалкил.
"Хлорформиат" означает реагент формулы ROC(O)Cl, где R может означать алкил или арилалкил, например, с образованием алкилхлорформиата и арилалкилхлорформиата соответственно.
"Цианоформиат" означает реагент формулы ROC(O)CN, где R может означать алкил или арилалкил, например, с образованием алкилцианоформиата и арилалкилцианоформиата соответственно.
"Галоген" означает фтор, хлор, бром или йод. В настоящем изобретении предпочтительными галогенидными группами являются бром и йод.
"Метил-d3" означает дейтерированный фрагмент -CD3.
"Литийорганический реагент" означает металлоорганическое соединение, которое содержит связи углерод-литий. Примеры литийорганических реагентов включают н-гексиллитий и н-бутиллитий.
"Фосфорорганический лиганд" означает органическое соединение, содержащее фосфор, предпочтительно фосфиновые лиганды, т. е. соединения формулы PR3 где R означает органическое производное. Примеры фосфорорганического лиганда включают трифенилфосфин (PPh3), три-трет-бутилфосфин (РtBu3), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos), 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (BINAP),
1,3-бис(дифенилфосфино)пропан (dppp) и ди(1-адамантил)-н-бутилфосфин (cataCXium® A).
"Палладиевый катализатор" означает комплекс палладия, способный катализировать реакцию. Примеры палладиевых катализаторов включают: ацетат палладия (Pd(OAc)2), хлорид палладия (PdCl2), трис(дибензилиденацетон)дипалладий, (Pd(PPh3)2Cl). Палладиевый катализатор включает прекатализаторы, которые становятся активированными in situ, такие как например, Pd(PPh3)2Cl, который восстанавливается в комплекс Pd(0) или трансметаллируется в арильный комплекс Pd(II) перед участием в каталитическом цикле.
"Соль Пиннера" означает продукт реакции нитрила со спиртом, т. е. соль сложного иминоэфира (соль алкилимидата).
"Реакция Зандмейера" означает химическую реакцию, использующуюся для синтеза арил- или гетероарилгалогенидов из арильных или гетероарильных диазониевых солей, с помощью радикального нуклеофильного ароматического замещения.
"Тиоцианат" означает анион [SCN]". Его можно использовать в виде соли с противоионами, такими как например, калий или натрий.
"Тозилирование" означает реакцию, обеспечивающую введение п-толуолсульфонильной группы (также называющейся тозильной группой) в гидроксильный фрагмент. В предпочтительном варианте осуществления тозилирование проводят с помощью п-толуолсульфонилхлорида.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Синтез соединения (I)
Настоящее изобретение относится к способу получения соединения формулы (I):
или его соли, где R1 означает метил или метил-d3; указанный способ включает следующие стадии: a) проведение реакции алкоксикарбонилирования соединения формулы (III):
где X означает галоген; и R1 означает метил или метил-d3; с получением соединения формулы (II):
где R1 означает метил или метил-d3; и R2 означает алкил или
арилалкил; и
b) получение соединения формулы (I) по реакции соединения формулы (II) с гидразинмоногидратом.
Конечное соединение или промежуточные продукты, использующиеся в способах, предлагаемых в настоящем изобретении, могут находиться в форме солей, включая соли присоединения с кислотой и с основанием. В одном варианте осуществления солями являются фармацевтически приемлемые соли. Подходящими солями присоединения с кислотой являются, например, ацетат, адипат, аспартат, бензоат, безилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камзилат, цитрат, цикламат, эдизилат, эзилат, формиат, фумарат, глюцептат, глюконат, глюкуронат, гексафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидройодид/йодид, изетионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, 2-напзилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, пироглутамат, сахарат, стеарат, сукцинат, таннат, тартрат, тозилат, трифторацетат и ксинофоат. Подходящими солями присоединения с основанием являются, например, соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина, глицина, лизина, магния, меглумина, оламина, калия, натрия, трометамина, 2-(диэтиламино)этанола, этаноламина, морфолина, 4-(2-гидроксиэтил)морфолина и цинка.
Стадия a) - Алкоксикарбонилирование
В одном варианте осуществления в соединении формулы (III) X означает бром или йод. В предпочтительном варианте осуществления в соединении формулы (III) X означает бром. В другом варианте осуществления в соединении формулы (III) X означает йод.
В одном варианте осуществления в соединении формулы (II) R2 означает C1-C4 алкильную группу, предпочтительно метил или этил, более предпочтительно, если R2 означает этил. В другом варианте осуществления в соединении формулы (II) R2 означает арилалкильную группу, предпочтительно бензил, 4-метоксибензил (PMB), 2,4-диметоксибензил (DMB) или 2,4,6-триметоксибензил (TMB).
Алкоксикарбонилирование с помощью CO/Pd
В одном варианте осуществления реакцию алкоксикарбонилирования на стадии a) проводят в присутствии монооксида углерода. В одном варианте осуществления монооксид углерода используют при давлении в диапазоне от 1 до 20 бар, предпочтительно от 3,5 до 8,5 бар, более предпочтительно от 4 до 5 бар.
В одном варианте осуществления реакцию алкоксикарбонилирования на стадии a) проводят в присутствии палладиевого катализатора. В одном варианте осуществления палладиевым катализатором является, например, выбранный из группы, включающей ацетат палладия (Pd(OAc)2), хлорид палладия (PdCl), трис(дибензилиденацетон)дипалладий, бис(дибензилиденацетон)палладий и бис(трифенилфосфин)палладийхлорид (Pd(PPh3)2Cl). В предпочтительном варианте осуществления палладиевым катализатором является ацетат палладия. В предпочтительном варианте осуществления палладиевым катализатором является бис(трифенилфосфин)палладийхлорид.
В одном варианте осуществления реакцию алкоксикарбонилирования на стадии a) проводят в присутствии фосфорорганического лиганда. В одном варианте осуществления фосфорорганический лиганд выбран из группы, включающей трифенилфосфин (PPh3), три-трет-бутилфосфин (РtBu3), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos), 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (BINAP), 1,3-бис(дифенилфосфино)пропан (dppp) и ди(1-адамантил)-н-бутилфосфин (cataCXium® A). В предпочтительном варианте осуществления фосфорорганическим лигандом является 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos). В другом варианте осуществления фосфорорганическим лигандом является трифенилфосфин (PPh3).
Альтернативно, в зависимости от палладиевого катализатора реакцию алкоксикарбонилирования на стадии a) проводят при отсутствии фосфорорганического лиганда. Это так, например, при использовании бис(трифенилфосфин)палладийхлорид (Pd(PPh3)2Cl) в качестве палладиевого катализатора.
В одном варианте осуществления реакцию алкоксикарбонилирования на стадии a) проводят в присутствии основания. В одном варианте осуществления основанием является выбранное из группы, включающей ацетат натрия (NaOAc), N, N-диизопропилэтиламин (DIPEA), лутидин, N-метилморфолин (NMM), трибутиламин, триэтиламин (TEA) и их смеси. В предпочтительном варианте осуществления основанием является ацетат натрия. В одном варианте осуществления основание используют в сухой форме.
В одном варианте осуществления стадию a) проводят в растворителе, приспособленном для введения фрагмента R2. В одном варианте осуществления стадию a) проводят в спирте, предпочтительно в метаноле или этаноле. В одном варианте осуществления стадию a) проводят в смеси спиртового растворителя и метил-трет-бутилового эфира (MTBE), такой как например, смесь MTBE с метанолом или этанолом. В одном варианте осуществления, если R2 означает этил, стадию a) проводят в этаноле или в смеси этанола и MTBE. Растворитель может быть сухим или не сухим. В одном варианте осуществления используют от 2 до 30 объемов растворителя, предпочтительно от 5 до 10 объемов, более предпочтительно примерно 10 объемов.
В одном варианте осуществления соединение формулы (III) используют в концентрации в диапазоне от 0,01 M до 1 M, предпочтительно от 0,1 M до 0,5 M, более предпочтительно от 0,2 M до 0,4 M.
В одном варианте осуществления количество молярных эквивалентов основания (такого как ацетат натрия) находится в диапазоне от 1 до 3 в пересчете на соединение (III); предпочтительно от 1,1 до 2; более предпочтительно от 1,1 до 1,5, более предпочтительно примерно 1,3 молярного эквивалента.
В одном варианте осуществления количество молярных эквивалентов палладиевого катализатора (такого как ацетат палладия) находится в диапазоне от 0,003 до 0,1 в пересчете на соединение (III); предпочтительно от 0,005 до 0,05; более предпочтительно от 0,005 до 0,01, более предпочтительно примерно 0,005 молярного эквивалента.
В одном варианте осуществления количество молярных эквивалентов фосфорорганического лиганда (такого как Xantphos) находится в диапазоне от 0,003 до 0,2 в пересчете на соединение (III); предпочтительно от 0,005 до 0,15; более предпочтительно от 0,005 до 0,05, более предпочтительно примерно 0,005 молярного эквивалента.
В одном варианте осуществления стадию a) проводят при температуре в диапазоне от 50°C до 150°C, предпочтительно от 63°C до 67°C, более предпочтительно примерно при 65°C. Альтернативно, стадию a) можно провести при температуре в диапазоне от 90°C до 120°C, предпочтительно от 100°C до 110°C.
В одном варианте осуществления стадию a) проводят в течение не менее 3 ч, предпочтительно в диапазоне от 10 ч до 48 ч, предпочтительно от 15 до 40 ч. В одном варианте осуществления стадию a) проводят в течение примерно 19 ч. В одном варианте осуществления стадию a) проводят в течение примерно 30 ч. В одном варианте осуществления стадию a) проводят в течение примерно 37 ч. Длительность реакции подбирают в соответствии с количествами, участвующими в реакции.
В одном варианте осуществления после завершения реакции полученный продукт экстрагируют дихлорметаном, трет-бутилметиловым эфиром или метилциклогексаном, предпочтительно метилциклогексаном.
В одном варианте осуществления полученный продукт (II) можно очистить с помощью перегонки.
В одном варианте осуществления реакцию алкоксикарбонилирования на стадии a) проводят в присутствии монооксида углерода, палладиевого катализатора, фосфорорганического лиганда, основания и спиртового растворителя. В одном варианте осуществления палладиевым катализатором является ацетат палладия, фосфорорганическим лигандом является 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos), основанием является ацетат натрия и растворителем является этанол или смесь MTBE и этанола. В другом варианте осуществления палладиевым катализатором является ацетат палладия, фосфорорганическим лигандом является трифенилфосфин (PPh3), основанием является триэтиламин и растворителем является этанол или смесь MTBE и этанола.
В другом варианте осуществления реакцию алкоксикарбонилирования на стадии a) проводят в присутствии монооксида углерода, палладиевого катализатора, основания и спиртового растворителя. В одном варианте осуществления палладиевым катализатором является бис(трифенилфосфин)палладийхлорид (Pd(PPh3)2Cl), основанием является триэтиламин и растворителем является этанол или смесь MTBE и этанола.
Алкоксикарбонилирование с помощью Fe(CO)5
В одном варианте осуществления реакцию алкоксикарбонилирования на стадии a) проводят в присутствии Fe(CO)5.
В одном варианте осуществления реакцию алкоксикарбонилирования на стадии a) проводят в присутствии монооксида углерода, палладиевого катализатора, фосфорорганического лиганда, основания и Fe(CO)5 в спиртовом растворителе. Варианты осуществления, подробно описанные выше для условий алкоксикарбонилирования и особенно для этих компонентов, также применимы для случая использования Fe(CO)5.
В одном варианте осуществления количество молярных эквивалентов Fe(CO)5 находится в диапазоне от 0,01 до 0,5 в пересчете на соединение (III); предпочтительно от 0,05 до 0,2; более предпочтительно примерно 0,01 молярного эквивалента.
Алкоксикарбонилирование с использованием обмена Li
В одном варианте осуществления реакцию алкоксикарбонилирования на стадии a) проводят посредством обмена лития.
В одном варианте осуществления реакцию алкоксикарбонилирования на стадии a) проводят путем проводимого сначала взаимодействия соединения формулы (III) с литийорганическим реагентом; и затем путем добавления хлорформиата или цианоформиата с получением соединения формулы (II).
В одном варианте осуществления литийорганический реагент выбран из группы, включающей н-гексиллитий и н-бутиллитий; предпочтительным литийорганическим реагентом является н-гексиллитий.
В одном варианте осуществления хлорформиат выбран из группы, включающей алкилхлорформиат и арилалкилхлорформиат; предпочтительно, если алкилхлорформиат выбран из группы, включающей этилхлорформиат, метилхлорформиат и сим-бутилхлорформиат, и арилалкилхлорформиатом является, например, бензилхлорформиат; более предпочтительно, если хлорформиатом является алкилхлорформиат, такой как этилхлорформиат.
В одном варианте осуществления цианоформиат выбран из группы, включающей алкилцианоформиат и арилалкилцианоформиат; предпочтительно, если алкилцианоформиат выбран из группы, включающей этилцианоформиат, метилцианоформиат и трет-бутилцианоформиат, и арилалкилцианоформиатом является, например, бензилцианоформиат; более предпочтительно, если цианоформиатом является алкилцианоформиат, такой как этилцианоформиат.
В одном варианте осуществления количество молярных эквивалентов литийорганического реагента (предпочтительно гексиллитий) находится в диапазоне от 1 до 2 в пересчете на соединение (III); предпочтительно от 1 до 1,5 более предпочтительно примерно 1,1 молярного эквивалента.
В одном варианте осуществления количество молярных эквивалентов хлорформиата (предпочтительно этилхлорформиата) находится в диапазоне от 1 до 10 в пересчете на соединение (III); предпочтительно от 4 до 8, более предпочтительно примерно 6 молярных эквивалентов.
В одном варианте осуществления обмен лития проводят в инертной атмосфере.
В одном варианте осуществления обмен лития проводят в сухом растворителе, таком как метилтетрагидрофуран (MeTHF), тетрагидрофуран (THF), трет-бутилметиловый эфир (TBME), н-гексан и их смеси, предпочтительно в метилтетрагидрофуране.
В одном варианте осуществления обмен лития проводят при температуре ниже 0°C, предпочтительно примерно при -65°C.
В одном варианте осуществления полученное соединение формулы (II) можно очистить с помощью перегонки.
Стадия b) - Образование ацилгидразина (стадия b)
На стадии b) соединение формулы (I) получают по реакции соединения формулы (II) с гидразином, предпочтительно гидразинмоногидратом.
В одном варианте осуществления количество молярных эквивалентов гидразина находится в диапазоне от 1 до 2 в пересчете на соединение формулы (II); предпочтительно от 1,1 до 1,2; более предпочтительно примерно 1,14 молярного эквивалента.
В одном варианте осуществления стадию b) проводят в растворителе, выбранном из группы, включающей метанол, этанол и изопропиловый спирт. В предпочтительном варианте осуществления растворителем является изопропиловый спирт. В другом предпочтительном варианте осуществления растворителем является этанол.
В одном варианте осуществления стадию b) проводят при температуре ниже 10°C, предпочтительно в диапазоне от 0°C до 5°C.
В одном варианте осуществления стадию b) проводят в течение не менее 30 мин, предпочтительно не менее 1 ч, предпочтительно в диапазоне от 1 ч до 48 ч, более предпочтительно от 1 ч до 24 ч.
В одном варианте осуществления после завершения реакции соединение формулы (I) выделяют в виде твердого вещества и его можно промыть растворителем, таким как метанол или изопропиловый спирт.
Реакция Зандмейера (предварительная стадия)
В одном варианте осуществления способ, предлагаемый в настоящем изобретении, включает предварительную стадию реакции Зандмейера соединения формулы (IV), приводящую к соединению формулы (III). Например, если X означает Br, соединение (III-1-а) можно получить по методике, описанной в публикации Goerdeler et al. in Chem. Ber, 1956, 89, 1534-1540 или в US2007/0078155.
В одном варианте осуществления галогенид формулы (III) получают по реакции Зандмейера из соединения формулы (IV):
или его соли, где R1 означает метил или метил-d3.
В одном варианте осуществления реакцию Зандмейера проводят путем проводимого сначала образования соли диазония из амина в присутствии азотистой кислоты, образованной in situ, с последующим ее замещением галогенидным анионом в качестве нуклеофила.
В одном варианте осуществления бромирование по реакции Зандмейера проводят в присутствии нитрита натрия (NaNO2) и бромида водорода (HBr), предпочтительно в водной среде. В этом случае в соединении формулы (III) X означает бром.
В одном варианте осуществления количество молярных эквивалентов бромида водорода находится в диапазоне от 1 до 5 в пересчете на соединение (IV); предпочтительно от 2 до 4; более предпочтительно примерно 3 молярных эквивалента.
В одном варианте осуществления количество молярных эквивалентов нитрита натрия находится в диапазоне от 1,15 до 4 в пересчете на соединение (IV); предпочтительно от 1,5 до 2; более предпочтительно примерно 1,5 молярного эквивалента.
В одном варианте осуществления реакцию Зандмейера проводят при температуре в диапазоне от комнатной температуры до 60°C, предпочтительно от 35°C до 50°C, более предпочтительно от примерно 40°C до примерно 45°C.
В одном варианте осуществления реакцию Зандмейера проводят в течение не менее 30 мин, предпочтительно в диапазоне от 30 мин до 22 ч, предпочтительно от 30 мин до 2 ч, более предпочтительно в течение примерно 1 ч.
В одном варианте осуществления реакцию Зандмейера проводят в присутствии 1 молярного эквивалента амина формулы (IV), 3 молярных эквивалентов бромида водорода и 1,5 молярного эквивалента нитрита натрия в воде. Предпочтительно, если реакцию проводят при температуре, равной примерно 40°C. Предпочтительно, если реакцию проводят в 5 объемах воды.
В одном варианте осуществления после завершения реакции продукт выделяют экстрагированием дихлорметаном. Предпочтительно, если органическую фазу нейтрализуют раствором NaOH.
В другом варианте осуществления бромирование по реакции Зандмейера проводят в присутствии трет-бутилнитрита (tBuONO) и CuBr2. В этом случае в соединении формулы (III) X означает бром.
В одном варианте осуществления количество молярных эквивалентов CuBr2 находится в диапазоне от 1 до 3 в пересчете на соединение (IV); предпочтительно от 1 до 1,1; более предпочтительно примерно 1,03 молярного эквивалента.
В одном варианте осуществления количество молярных эквивалентов трет-бутилнитрита находится в диапазоне от 1 до 3 в пересчете на соединение (IV); предпочтительно от 1 до 2; более предпочтительно примерно 1,5 молярного эквивалента.
В одном варианте осуществления реакцию Зандмейера проводят при температуре в диапазоне от 0°C до 40°C, предпочтительно от 0°C до комнатной температуры.
В одном варианте осуществления реакцию Зандмейера проводят в течение не менее 30 мин, предпочтительно в диапазоне от 30 мин до 10 ч, предпочтительно от 30 мин до 5 ч, более предпочтительно от 2 ч до 3 ч.
В одном варианте осуществления реакцию Зандмейера проводят в присутствии 1 молярного эквивалента амина формулы (IV), 1,03 молярного эквивалента CuBr2 и 1,5 молярного эквивалента трет-бутилнитрита в ацетонитриле. Предпочтительно, если реакцию сначала проводят при температуре, равной примерно 0-5°C и затем при комнатной температуре.
В одном варианте осуществления после завершения реакции продукт выделяют экстракцией трет-бутилметиловым эфиром.
В другом варианте осуществления йодирование по реакции Зандмейера проводят в присутствии трет-бутилнитрита (tBuONO) и йода. В этом случае в соединении формулы (III) X означает йод. Предпочтительно, если такую реакцию проводят в растворителе, таком как например, ацетонитрил.
В одном варианте осуществления количество молярных эквивалентов йода находится в диапазоне от 1 до 3 в пересчете на соединение (IV); предпочтительно от 1 до 2; более предпочтительно примерно 1 эквивалент.
В одном варианте осуществления количество молярных эквивалентов трет-бутилнитрита находится в диапазоне от 1 до 5 в пересчете на соединение (IV); предпочтительно от 3 до 5; более предпочтительно примерно 4 молярных эквивалента.
В одном варианте осуществления реакцию Зандмейера проводят при температуре в диапазоне от комнатной температуры до температуры кипения.
В одном варианте осуществления реакцию Зандмейера проводят в течение не менее 30 мин, предпочтительно в диапазоне от 30 мин до 5 ч, предпочтительно от 30 мин до 2 ч, более предпочтительно в течение примерно 1 ч.
В одном варианте осуществления реакцию Зандмейера проводят в присутствии 1 молярного эквивалента амина формулы (IV), 1 молярного эквивалента йода и 4 молярных эквивалентов трет-бутилнитрита.
В одном варианте осуществления после завершения реакции избыток йода нейтрализуют, предпочтительно путем добавления Na2SO3. Затем полученное соединение можно экстрагировать метил-трет-бутиловым эфиром.
В другом варианте осуществления йодирование по реакции Зандмейера проводят в присутствии нитрита натрия (NaNO2), йодида калия (KI) и п-толуолсульфоновой кислоты (TsOH). В этом случае в соединении формулы (III) X означает йод.
В одном варианте осуществления количество молярных эквивалентов йодида калия находится в диапазоне от 1 до 4 в пересчете на соединение (IV); предпочтительно от 2 до 3; более предпочтительно примерно 2,6 эквивалента.
В одном варианте осуществления количество молярных эквивалентов нитрита натрия находится в диапазоне от 1 до 3 в пересчете на соединение (IV); предпочтительно от 1,5 до 2,5; более предпочтительно примерно 2 эквивалента.
В одном варианте осуществления количество молярных эквивалентов п-толуолсульфоновой кислоты находится в диапазоне от 1 до 6 в пересчете на соединение (IV); предпочтительно от 3 до 4; более предпочтительно примерно 3,5 эквивалента.
В одном варианте осуществления реакцию Зандмейера предпочтительно проводят при комнатной температуре.
В одном варианте осуществления реакцию Зандмейера проводят в течение не менее 1 ч, предпочтительно в диапазоне от 1 ч до 24 ч, предпочтительно в течение примерно 12 ч.
В одном варианте осуществления реакцию Зандмейера проводят в присутствии 1 молярного эквивалента амина формулы (IV), 2,6 молярных эквивалентов йодида калия, примерно 2 молярных эквивалентов нитрита натрия и 3,5 молярных эквивалентов п-толуолсульфоновой кислоты.
Синтез дейтерированного соединения (IV-2)
Недейтерированный промежуточный продукт (IV-1), т. е. соединение формулы (IV) где R1 означает метил, также обозначаемый, как AMTD, имеется в продаже или его можно получить по методикам, известным специалисту в данной области техники.
Для соответствующего дейтерированного промежуточного продукта (IV-2), т. е. соединения формулы (IV), где R1 означает метил-d3, на основании путей синтеза недейтерированного продукта, невозможно предсказать, какую изотопную чистоту можно обеспечить. Настоящее изобретение относится к способу синтеза промежуточного продукта (IV-2) с обеспечением очень высокой изотопной чистоты, а именно, общей изотопной чистоты, составляющей более 90%, более предпочтительно более 95%.
Путь с использованием d3-ацетамидина
Таким образом, настоящее изобретение также относится к способу получения 3-(метил-d3)-1,2,4-тиадиазол-5-амина (IV-2):
или его соли, включающему следующие стадии:
a) реакция d3-ацетонитрила с этанолом в присутствии HCl с образованием соли Пиннера формулы (VI-2):
;
b) реакция соли Пиннера (VI-2) с аммиаком с образованием d3-ацетамидина (V-2) или его соли:
и
c1) реакция d3-ацетамидина (V-2) с бромом, тиоцианатом и метоксидом натрия с образованием соединения формулы (IV-2); или
c2) реакция d3-ацетамидина (V-2) сначала с гипохлоритом натрия (NaOCl) и затем с тиоцианатом с получением соединения формулы (IV-2).
В этом способе стадию a) проводят для образования соли Пиннера по реакции Пиннера. "Реакция Пиннера" означает катализируемую кислотой реакцию нитрила (в этом случае d3-ацетонитрила) со спиртом (в этом случае этанолом) с образованием простого иминоэфира соли, также называющейся солью Пиннера. Соли Пиннера сами являются реакционноспособными и вступают в последующее нуклеофильное присоединение, например, с аммиаком с образованием амидина на стадии b).
Последнюю стадию можно провести "путем с использованием брома" (стадия c1) или "путем с использованием гипохлорита" (стадия c2), оба приводят к удовлетворительным результатам.
Как отмечено выше, конечное соединение или промежуточные продукты, использующиеся в способах, предлагаемых в настоящем изобретении, могут находиться в форме солей, включая соль присоединения с кислотой и с основанием.
Стадия a) - Получение соли Пиннера
На стадии a) соль Пиннера формулы (VI-2) получают по реакции d3-ацетонитрила с этанолом в присутствии HCl.
В одном варианте осуществления соль Пиннера (VI-2) образуется на стадии a) путем пропускания газообразного HCl через смесь, содержащую этанол (предпочтительно абсолютный этанол) и d3-ацетонитрил.
В одном варианте осуществления пропускание HCl проводят при температуре ниже 15°C, предпочтительно ниже 10°C.
В одном варианте осуществления газообразный HCl пропускают через смесь в течение не менее 10 ч, предпочтительно не менее 8 ч, более предпочтительно примерно 6 ч.
В одном варианте осуществления после завершения пропускания HCl реакционную смесь перемешивают при комнатной температуре, предпочтительно при температуре в диапазоне от 20°C до 25°C.
В одном варианте осуществления после завершения пропускания HCl реакционную смесь перемешивают в течение не менее 10 ч, предпочтительно в диапазоне от 10 ч до 24 ч, предпочтительно от 15 ч до 20 ч, более предпочтительно в течение примерно 16,5 ч.
В одном варианте осуществления после завершения реакции реакционную смесь обрабатывают трет-бутилметиловым эфиром (TBME), предпочтительно при температуре в диапазоне от 0°C до 5°C, для выделения соли Пиннера (VI-2) в виде твердого вещества.
Стадия b) - Получение дейтерированного ацетамидин
На стадии b) дейтерированный ацетамидин (V-2) получают по реакции соли Пиннера (VI-2) с аммиаком (NH3).
В одном варианте осуществления растворителем, использующимся на стадии b), является спирт, такой как этанол или метанол; предпочтительно этанол; более предпочтительно абсолютный этанол.
В одном варианте осуществления стадию b) проводят при температуре ниже 10°C, предпочтительно при температуре в диапазоне от 0°C до 5°C.
В одном варианте осуществления аммиак (NH3) используют в качестве газа и прямо абсорбируют в реакционной смеси. Предпочтительно, если используют от 3 до 5 молярных эквивалентов аммиака в пересчете на соль Пиннера (VI-2); предпочтительно 4,1 молярных эквивалентов.
В одном варианте осуществления реакцию на стадии b) проводят в течение не менее 1 ч, предпочтительно в диапазоне от 1 до 6 ч, предпочтительно от 2 до 4 ч, более предпочтительно примерно 3 ч.
В одном варианте осуществления после завершения реакции реакционную смесь выпаривают и обрабатывают метилциклогексаном для выделения d3-ацетамидин (V-2) в виде твердого вещества.
Предпочтительно, если d3-ацетамидин (V-2) получают с изотопной чистотой, составляющей не менее 90%, предпочтительно не менее 95%.
Стадия c1) - Циклизация с образованием тиадиазольного кольца "путем с использованием брома"
Последнюю стадию с получением соединения формулы (IV-2) можно провести "путем с использованием брома" (стадия c1).
На стадии c1), соединение формулы (IV-2) получают по реакции d3-ацетамидина (V-2) с бромом, тиоцианатом и метоксидом натрия.
В одном варианте осуществления тиоцианат предпочтительно используют в форме тиоцианата калия.
В одном варианте осуществления метоксид натрия можно получить путем солюбилизации натрия в метаноле.
В одном варианте осуществления количество молярных эквивалентов тиоцианата находится в диапазоне от 1 до 2,6 в пересчете на d3-ацетамидин (V-2); предпочтительно примерно 1,1 эквивалента.
В одном варианте осуществления количество молярных эквивалентов метоксида натрия (NaOMe) находится в диапазоне от 2 до 5 в пересчете на d3-ацетамидин (V-2); предпочтительно от 3 до 3,5; более предпочтительно примерно 3,1 молярных эквивалента.
В одном варианте осуществления количество молярных эквивалентов брома (Br) находится в диапазоне от 1 до 3 в пересчете на d3-ацетамидин (V-2); предпочтительно от 1 до 2; более предпочтительно примерно 1,5 молярного эквивалента.
В одном варианте осуществления растворителем, использующимся на стадии c1), является спирт; предпочтительно метанол. Предпочтительно, если растворитель является сухим, предпочтительно сухим метанолом.
В одном варианте осуществления стадию c1) проводят при температуре ниже 10°C, предпочтительно при температуре в диапазоне от 0°C до 5°C.
В одном варианте осуществления реакцию на стадии c1) проводят в течение не менее 1 ч, предпочтительно в диапазоне от 1 до 6 ч, предпочтительно от 1 до 3 ч, более предпочтительно примерно 2 ч.
В одном варианте осуществления продукт экстрагируют этилацетатом и получают в виде твердого вещества.
Предпочтительно, если тиадиазол (IV-2) получают с изотопной чистотой, составляющей не менее 90%.
Стадия c2) - Циклизация с образованием тиадиазольного кольца "путем с использованием гипохлорита"
Альтернативно, последнюю стадию с получением соединения формулы (IV-2) можно провести "путем с использованием гипохлорита" (стадия c2).
На стадии c2), соединение формулы (IV-2) получают путем проводимого сначала взаимодействия d3-ацетамидина (V-2) с гипохлоритом натрия (NaOCl), с образованием промежуточного продукта (VIII-2) или его соли:
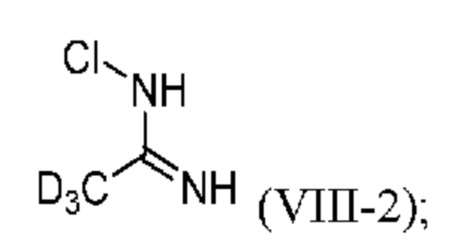
и затем взаимодействия полученного промежуточного продукта (VIII-2) с тиоцианатом.
В одном варианте осуществления гипохлорит натрия (NaOCl) используют в водной форме и его можно получить растворением пентагидрата NaOCl в воде.
В одном варианте осуществления количество молярных эквивалентов гипохлорита натрия (NaOCl) находится в диапазоне от 1 до 5 в пересчете на d3-ацетамидин (V-2); предпочтительно от 1,5 до 2.
В одном варианте осуществления растворителем, использующимся для реакции с гипохлоритом натрия, является вода; предпочтительно деионизированная вода.
В одном варианте осуществления реакцию с гипохлоритом натрия проводят при температуре ниже 10°C, предпочтительно при температуре в диапазоне от 0°C до 5°C.
В одном варианте осуществления реакцию с гипохлоритом натрия проводят в течение не менее 30 мин, предпочтительно в диапазоне от 1 до 6 ч, предпочтительно от 1 до 3 ч, более предпочтительно примерно 2 ч.
В одном варианте осуществления полученный промежуточный продукт (VIII-2) экстрагируют этилацетатом.
В одном варианте осуществления тиоцианат предпочтительно используют в виде тиоцианата натрия.
В одном варианте осуществления количество молярных эквивалентов тиоцианата находится в диапазоне от 1 до 2 в пересчете на промежуточный продукт (VIII-2); предпочтительно от 1 до 1,5; более предпочтительно примерно 1,1 эквивалент.
В одном варианте осуществления растворителем, использующимся для реакции промежуточного продукта (VIII-2) с тиоцианатом является спирт; предпочтительно метанол. Предпочтительно, если растворитель является сухим, предпочтительно сухим метанолом.
В одном варианте осуществления реакцию промежуточного продукта (VIII-2) с тиоцианатом проводят при температуре ниже 10°C, предпочтительно при температуре в диапазоне от 0°C до 5°C.
В одном варианте осуществления реакцию промежуточного продукта (VIII-2) с тиоцианатом проводят в течение не менее 30 мин, предпочтительно в диапазоне от 1 до 6 ч, предпочтительно от 1 до 3 ч, более предпочтительно примерно 1,5 ч.
В одном варианте осуществления продукт экстрагируют этилацетатом и получают в виде твердого вещества.
Предпочтительно, если тиадиазол (IV-2) получают с изотопной чистотой, составляющей не менее 95%.
Путь с использованием d3-гидроксиацетамидина
Настоящее изобретение также относится к другому способу получения 3-(метил-d3)-1,2,4-тиадиазол-5-амин (IV-2):
или его соли,
включающему следующие стадии:
a) реакция d3-ацетонитрил с гидроксиламином (H2N-OH) с образованием d3-гидроксиацетамидина (IХ-2) или его соли:
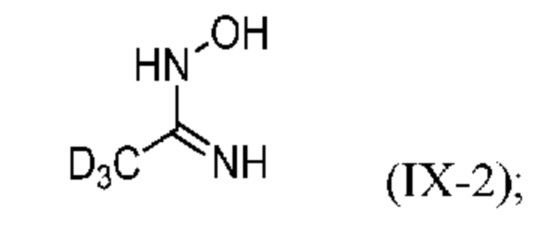
b) проведение тозилирования d3-гидроксиацетамидина (IХ-2) с образованием промежуточного продукта (X-2) или его соли:
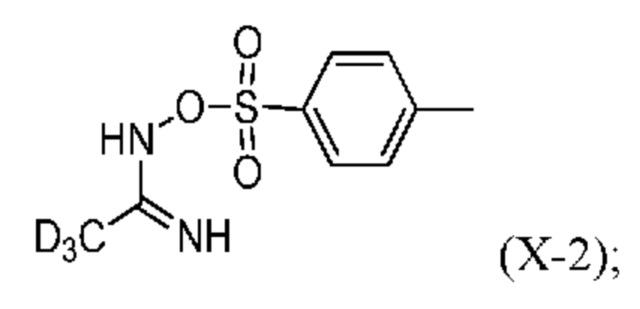
и
c) реакция промежуточного продукта (X-2) с тиоцианатом с образованием соединения формулы (IV-2).
Стадия a) - Получение дейтерированного гидроксиацетамидин
На стадии a) d3-гидроксиацетамидин (IX-2) получают по реакции d3-ацетонитрила с гидроксиламином (H2N-OH).
В одном варианте осуществления гидроксиламин можно использовать в виде водного гидроксиламина или гидроксиламингидрохлорида.
В одном варианте осуществления количество молярных эквивалентов гидроксиламина находится в диапазоне от 1 до 4 в пересчете на d3-ацетонитрил; предпочтительно от 2 до 2,5; более предпочтительно примерно 2,2 эквивалента.
В одном варианте осуществления растворителем, использующимся на стадии a), является спирт; предпочтительно этанол.
В одном варианте осуществления стадию a) проводят при кипячении с обратным холодильником.
В одном варианте осуществления реакцию на стадии a) проводят в течение не менее 1 ч, предпочтительно в диапазоне от 1 до 24 ч, предпочтительно примерно 18 ч.
Предпочтительно, если изотопная чистота сохраняется на стадии a).
Стадия b) - Тозилирование дейтерированного гидроксиацетамидина
На стадии b) реакцию тозилирования проводят с d3-гидроксиацетамидином (IX-2) и получают соответствующий тозилированный промежуточный продукт (X-2). Тозилирование можно провести в присутствии п-толуолсульфонилхлорида и основания.
В одном варианте осуществления основанием является выбранное из группы, включающей триэтиламин (TEA), ацетат натрия (NaOAc), N, N-диизопропилэтиламин (DIPEA), N-метилморфолин (NMM) и их смеси. В предпочтительном варианте осуществления основанием является триэтиламин.
В одном варианте осуществления количество молярных эквивалентов п-толуолсульфонилхлорида находится в диапазоне от 0,8 до 1,5 в пересчете на d3-гидроксиацетамидин (IX-2); предпочтительно от 0,9 до 1,1; более предпочтительно примерно 0,9 эквивалента.
В одном варианте осуществления количество молярных эквивалентов основания (такого как триэтиламин) находится в диапазоне от 1 до 2 в пересчете на d3-гидроксиацетамидин (IX-2); предпочтительно от 1 до 1,5; более предпочтительно примерно 1,3 молярного эквивалента.
В одном варианте осуществления растворителем, использующимся на стадии b), является тетрагидрофуран; предпочтительно сухой тетрагидрофуран.
В одном варианте осуществления добавление п-толуолсульфонилхлорид проводят при температуре ниже 10°C, предпочтительно при температуре в диапазоне от 0°C до 5°C. После завершения добавления п-толуолсульфонилхлорида, реакцию предпочтительно проводят при комнатной температуре.
В одном варианте осуществления реакцию на стадии b) проводят в течение не менее 30 мин, предпочтительно в диапазоне от 30 мин до 3 ч, предпочтительно примерно 1 ч.
В одном варианте осуществления продукт экстрагируют этилацетатом.
Предпочтительно, если изотопная чистота сохраняется на стадии b).
Стадия c) - Циклизация с образованием тиадиазольного кольца
На стадии c) соединение формулы (IV-2) получают по реакции d3-тозилоксиацетамидина (X-2) с тиоцианатом.
В одном варианте осуществления тиоцианат предпочтительно используют в виде тиоцианата калия.
В одном варианте осуществления стадию c) необязательно можно провести в присутствии основания. Если используют основание, его можно выбрать из группы, включающей N,N-диизопропилэтиламин (DIPEA), ацетат натрия (NaOAc), триэтиламин (TEA), N-метилморфолин (NMM) и их смеси.
В одном варианте осуществления количество молярных эквивалентов тиоцианата находится в диапазоне от 1 до 5 в пересчете на d3-тозилоксиацетамидин (X-2); предпочтительно от 2 до 4: более предпочтительно примерно 3 молярных эквивалента.
В одном варианте осуществления количество молярных эквивалентов основания (такого как DIPEA) находится в диапазоне от 0 до 2 в пересчете на d3-тозилоксиацетамидин (X-2); предпочтительно от 1 до 2; более предпочтительно от 1 до 1,5; еще более предпочтительно примерно 1,1 молярного эквивалента.
В одном варианте осуществления растворитель, использующийся на стадии c), выбран из группы, включающей спирт (такой как метанол), диметилформамид (DMF), ацетонитрил, тетрагидрофуран (THF), диметилсульфоксид (DMSO) и их смеси. В одном варианте осуществления растворителем, использующимся на стадии c), является метанол.
В одном варианте осуществления стадию c) проводят при температуре в диапазоне от 20°C до 50°C, предпочтительно примерно при 40°C.
В одном варианте осуществления реакцию на стадии c) проводят в течение не менее 1 ч, предпочтительно в диапазоне от 2 до 20 ч.
Предпочтительно, если тиадиазол (IV-2) получают с изотопной чистотой, составляющей не менее 95%.
ПРИМЕРЫ
Настоящее изобретение дополнительно иллюстрируется следующими примерами. Схемы реакций, описанные в разделе примеров, иллюстрируют в качестве примеров разные возможные подходы.
Материалы и методики
Все приведенные температуры выражены в градусах Цельсия (°C); все реакции проводили при комнатной температуре (rt), если не указано иное.
Методики анализа:
Аналитическую тонкослойную хроматографию (ТСХ) использовали для слежения за реакциями, определения условий для флэш-хроматографии и проверки чистоты промежуточных продуктов или конечных продуктов. Использовали пластины для ТСХ фирмы Merck ТСХ с силикагелем 60 F254 на листе алюминия. Пластины ТСХ проявляли ультрафиолетовым излучением (длина волны=254 нм) при комнатной температуре или проявителем KMnO4 при нагревании при 160°C. Проявитель с KMnO4 для ТСХ получали путем растворения 3 г перманганата калия, 20 г карбоната натрия в 300 мл дистиллированной воды.
Спектры 1H и 13C ЯМР записывали на приборе Bruker Avance 500 МГц. Химические выражали в миллионных долях (м.д., единицы δ). Константы спин-спинового взаимодействия выражали в герцах (Гц). Характеристики расщепления описаны с помощью кажущихся мультиплетностей, как с (синглет), д (дублет), т (триплет), кв (квадруплет), гепт (гептуплет), м (мультиплет), или уш (уширенный).
Исследования посредством ГХ проводили с помощью (условия A) прибора Varian 3900 с использованием пламенного ионизационного детектора (FID) при 260°C; использовали колонку RTX-1301, 30 м×0,32 мм×0,5 мкм; температура инжектора равнялась 200°C, коэффициент деления пробы равнялся 50; инжектируемый объем равнялся 1 мкл; использовали следующую температурную программу: температуру печи устанавливали равной 60°C на 4 мин, линейно повышали со скоростью 20°C/мин до 260°C и поддерживали при 260°C в течение 10 мин; или (условия B) прибора Shimadzu GC-2010 с использованием пламенного ионизационного детектора (FID) при 260°C; температура инжектора равнялась 200°C, коэффициент деления пробы равнялся 50; инжектируемый объем равнялся 1 мкл; использовали колонку Rxi-17Sil MS, 30 м×0,32 мм×0,25 мкм; использовали следующую температурную программу: температуру печи устанавливали равной 60°C на 5 мин, линейно повышали со скоростью 20°C/мин до 300°C и поддерживали при 300°C в течение 8 мин; или (условия C) прибора Varian 3900 с использованием пламенного ионизационного детектора (FID) при 270°C; использовали колонку DB-624, 60 м×0,32 мм×3 мкм; температура инжектора равнялась 200°C, коэффициент деления пробы равнялся 50; инжектируемый объем равнялся 1 мкл; использовали следующую температурную программу: температуру печи устанавливали равной 80°C на 4 мин, линейно повышали со скоростью 20°C/мин до 260°C и поддерживали при 260°C в течение 11 мин.
Спектры ГХ-МС получали на приборе Shimadzu GCMS-QP2010 с использованием детектора с электронной ионизацией (EI) при 250°C. Использовали колонку Phenomenex XB-5MS, 30 м×0,25 мм×0,25 мкм. Скорость потока через колонку равнялась 1,50 мл/мин. Температура инжектора равнялась 250°C, коэффициент деления пробы равнялся 50. Инжектируемый объем равнялся 1 мкл. Использовали следующую температурную программу: в начале выдерживали при 40°C в течение 5 мин, затем повышали до 250°C со скоростью 20°C/мин, затем поддерживали при 250°C в течение 5 мин.
Спектры ВЭЖХ получали на приборе Thermo Scientific Ultimate 3000 HPLC, снабженном УФ детектором.
Условия A: использовали колонку Phenomenex - Kinetex EVO C18 50×4,6 мм×2,6 мкм. Элюентом являлась смесь раствора A (10 мМ HCO2NH4 в H2O) и раствора В (MeCN). Использовали следующий градиентный режим при скорости потока, равной 1,0 мл мин-1: поддерживали начальные условия с использованием 1% раствора В в течение 3 мин, линейно увеличивали до 90% раствора В за 7 мин, поддерживали при 90% в течение 2 мин, возвращались к начальным условиям за 1,0 мин и поддерживали в течение 5 мин. Условия B: использовали колонку Purospher Star RP18e 55 мм×4,6 мм×3,0 мкм. Элюентом являлась смесь раствора A (20 мМ HCO2NH4 в H2O) и раствора В (MeCN). Использовали следующий градиентный режим при скорости потока, равной 1,0 мл мин-1: поддерживали начальные условия с использованием 2% раствора В в течение 1 мин, линейно увеличивали до 90% раствора В за 8 мин, поддерживали при 90% в течение 2 мин, возвращались к начальным условиям за 1,0 мин и поддерживали в течение 3 мин.
Спектры ВЭЖХ-МС получали на приборе Shimadzu LCMS-2020 с использованием УФ детектора при 210 нм и с электронной ионизацией (ESI). Изотопную чистоту определяли путем сопоставления площадей пиков для каждого изотопа в выбранном режиме мониторинга ионов. Условия A: использовали колонку SeQuant ZIC-HILIC 150×4,6 мм×5 мкм. Элюентом являлась смесь 25% раствора A (20 мМ NH4Ac в H2O) и 75% раствора В (MeCN) при скорости потока, равной 1 мл мин-1.
Условия B: использовали колонку YMC-Triart C18 100×3,0 мм×3 мкм. Элюентом являлась смесь раствора A (0,1% HCO2H в H2O) и раствора В (MeCN). Использовали следующий градиентный режим при скорости потока, равной 0,5 мл мин-1: поддерживали начальные условия с использованием 0% раствора В в течение 12 мин, линейно увеличивали до 90% раствора В за 5 мин, поддерживали при 90% в течение 6 мин, возвращались к начальным условиям за 0,1 мин и поддерживали в течение 7 мин. Условия C: использовали колонку Phenomenex - Kinetex EVO C18 100×2,1 мм×2,6 мкм. Элюентом являлась смесь раствора A (0,1% HCO2H в H2O) и раствора В (MeCN). Использовали следующий градиентный режим при скорости потока, равной 0,5 мл мин-1: поддерживали начальные условия с использованием 2% раствора В в течение 1 мин, линейно увеличивали до 90% раствора В за 9 мин, поддерживали при 90% в течение 3 мин, возвращались к начальным условиям за 0,1 мин и поддерживали в течение 5 мин. Условия D: использовали колонку YMC-Triart C18 100×3,0 мм×3 мкм. Элюентом являлась смесь раствора A (0,1% HCO2H в H2O) и раствора В (MeCN). Использовали следующий градиентный режим при скорости потока, равной 0,5 мл мин-1: поддерживали начальные условия с использованием 90% раствора В в течение 23 мин, линейно уменьшали до 1% раствора В за 0,1 мин и поддерживали в течение 7 мин.
Альтернативно, спектры ВЭЖХ-МС получали на приборе Agilent LCMS с использованием ионизации электрораспылением (ESI). Прибор Agilent включает автоматический пробоотборник 1100, бинарный насос 1100, ультрафиолетовый многоволновый детектор 1100 и 6100 масс-спектрометр с одной квадрупольной линзой. Условия D: использовали колонку Sunfire 3,5 мкм, C18, 3,0×50 мм. Элюентом являлась смесь раствора A (0,1% TFA в H2O) и раствора В (0,1% TFA в MeCN). Использовали следующий градиентный режим при скорости потока, равной 1,3 мл мин-1: (для анализа конечных соединений и промежуточных продуктов): поддерживали начальные условия с использованием 5% раствора В в течение 0,2 мин, линейно увеличивали до 95% раствора В за 6 мин, поддерживали при 95% в течение 1,75 мин, возвращались к начальным условиям за 0,25 мин и поддерживали в течение 2,0 мин.
Содержание ионов хлора определяли с помощью Mettler Toledo DL50 или эквивалентного прибора для титрования, DM-141 или эквивалентного электрода, аналитических весов, стандартной лабораторной стеклянной посуды. Использовали раствор для титрования: 0,1 моль/л нитрат серебра (получение 0,1 моль/л нитрата серебра: отвешивают примерно 17 г нитрата серебра (AgNO3) в мерную колбу объемом 1000 мл и растворяют и доводят до объема деионизированной водой. Определяют титр раствора. Приготовление образца: отвешивают 30-500 мг твердого образца на аналитических весах и помещают в сосуд для определения содержания галогенида. Образцы разбавляют до 60 мл растворителем (деионизированная вода, 2-пропанол и т. п.). Расчет результата:
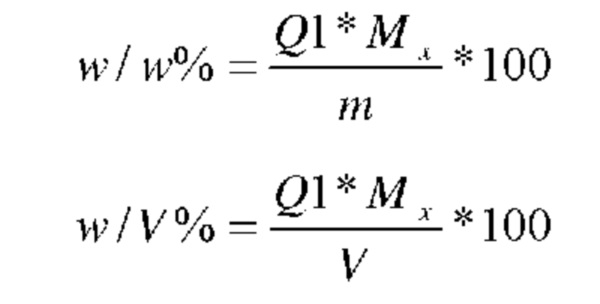
где:
Q1: количество раствора для титрования (в ммолях) для точки эквивалентности,
Mx, молярная масса (мг/ммоль), w масса образца (мг) и V объем образца (мкл)
Результат для содержания хлорид-иона рассчитывали по результату анализа хлорида следующим образом:
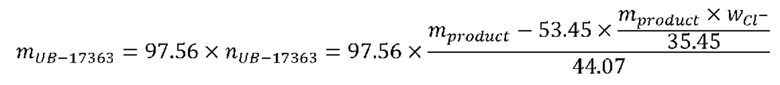
где 97,56 г/моль молекулярная масса соединения (V-2) и 53,45 г/моль молекулярная масса NH4Cl, mproduct масса выделенной соли с HCl и wcl- содержание в мас.% хлорид-иона, определенное титрованием.
Реагенты
Растворители, реагенты и исходные вещества приобретали и использовали в том виде, в котором они получены от поставщиков, если не указано иное.
Используются следующие аббревиатуры:
AMTD: 5-амино-3-метил-1,2,4-тиадиазол
DCM: дихлорметан,
экв.: эквивалент(ы),
EtOAc: этилацетат,
EtOH: этанол,
г: грамм(ы),
ГХ: газовая хроматография,
Гекс: н-гексан,
ВЭЖХ: высокоэффективная жидкостная хроматография,
IPA: изопропиловый спирт,
л: литр(ы),
ЖХМС: жидкостная хроматография с масс-спектрометрией
MeCN: ацетонитрил,
MeOH: метанол,
мл: миллилитр(ы),
мин: минута (минуты),
МС: масс-спектрометрия,
NMM: N-метилморфолин
MW: молекулярная масса,
ЯМР: ядерный магнитный резонанс
rt: комнатная температура,
TBME: трет-бутилметиловый эфир,
TEA: триэтиламин,
THF: тетрагидрофуран,
ТСХ: тонкослойная хроматография, Ts: тозил (т. е. п-толуолсульфонил) об.: объем(ы).
Все соединения, раскрытые в настоящем изобретении, называли с помощью программы ChemDraw Ultra 12®, приобретенной у фирмы CambridgeSoft (Cambridge, MA, USA).
Пример 1A: Синтез дейтерированного d3-AMTD (IV-2) путем с использованием ацетамидина
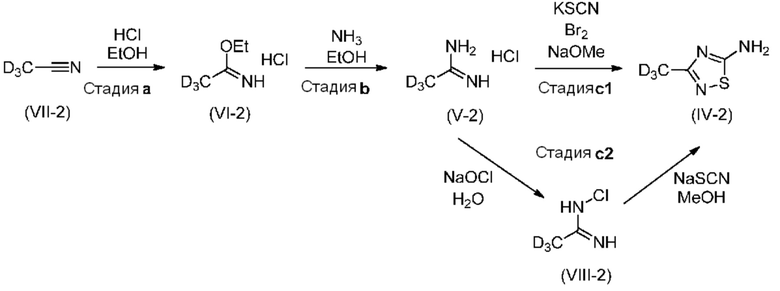
d3-Ацетамидин (V-2) получали по реакции Пиннера через соль Пиннера (VI-2) до проведения циклизации с образованием тиадиазольного кольца соединения (IV-2) "путем с использованием брома" (стадия c1) или "путем с использованием гипохлорита" (стадия c2).
Стадия a: Получение соли Пиннера (VI-2)
В трехгорлую колбу объемом 750 мл, снабженную двумя патрубками для подачи газа, термометром и магнитной мешалкой, при 20-25°C помещали: 74 мл абсолютного EtOH, и 51,2 г (1,161 моля, 1,0 экв.) d3-ацетонитрила (VII-2), затем охлаждали до 0-5°C (охлаждали смесью соли со льдом). При поддержании температуры ниже 10°C через смесь в течение 6 ч пропускали газообразный HCl. Реакционную смесь нагревали до 20-25°C и перемешивали в течение 16,5 ч (смесь превращалась в густую белую суспензию, которую трудно перемешивать). К смеси добавляли 500 мл TBME, затем смесь хорошо перемешивалась и образовывалась белая суспензия. Ее перемешивали в течение 1 ч при 0-5°C, затем фильтровали, промывали с помощью 2×50 мл холодного TBME. Отфильтрованное твердое вещество сушили при 40°C в вакууме и получали 104,3 г белого твердого вещества, соли Пиннера (VI-2), которую использовали без дополнительной очистки на следующей стадии.
Стадия b: Получение d3-ацетамидина (V-2).
В трехгорлую колбу объемом 2 л, снабженную двумя патрубками для подачи газа, термометром и магнитной мешалкой, при 20-25°C помещали: 835 мл (658,8 г) абсолютного EtOH, затем охлаждали до 0-5°C (охлаждали смесью соли со льдом). При этой температуре абсорбировали 57,3 г (3,37 моля, 4,1 экв.) газообразного NH3. Затем при 0-5°C добавляли 104,3 г (823,8 ммоля, 1,0 экв.) соли Пиннера (VI-2) и перемешивали в течение 3 ч. За протеканием реакции следили с помощью ВЭЖХ. Реакционную смесь выпаривали досуха при 30°C в роторном испарителе. К остатку добавляли 200 мл метилциклогексана и перемешивали в течение 15 мин при 0-5°C (охлаждали в бане со льдом). Белый осадок отфильтровывали, промывали с помощью 25 мл холодного метилциклогексана и сушили в вакууме при 45°C. Это давало 77,1 г (790,3 ммоля) белого твердого вещества d3-ацетамидингидрохлорида (V-2). Скорректированный выход: 89,3%. 1H ЯМР (DMSO-d6): δ 9,20 (уш, 1H), 8,75 (уш, 2H), 7,51 (уш, 1H). Изотопная чистота по данным ВЭЖХ-МС (условия A): 95,8%. Содержание хлорид-иона: 38,46 мас./мас.%. Анализ на основании содержания хлорид-иона: 93,1 мас./мас.%
Стадия c1: Синтез d3-AMTD (IV-2) путем с использованием брома
В четырехгорлую колбу объемом 250 мл, снабженную двумя капельными воронками, термометром и магнитной мешалкой, при 20-25°C помещали: 5,8 г (59,45 ммоля, 1,0 экв.) d3-ацетамидингидрохлорида (V-2) (изотопная чистота: 96,5%, анализ на основе содержания хлорида: 64,7 мас./мас.%), 6,35 г (65,4 ммоля, 1,1 экв.) KSCN и 11 мл сухого MeOH (сушили над молекулярными ситами 4A). Эту смесь (суспензию) охлаждали до температуры ниже 10°C при перемешивании. Поддерживая температуру в диапазоне 0-5°C, к смеси в течение 25 мин одновременно добавляли 42,6 мл (184,3 ммоля, 3,1 экв.) 25 мас./мас.% раствора NaOMe в MeOH (54,03 г/моль) и 4,6 мл (14,26 г, 89,16 моля, 1,5 экв.) Br2 (неразбавленного, MW: 159,81 г/моль, d: 3,119 г/мл). Смесь становилась серо-фиолетовой, затем при добавлении образовывалась белая суспензия. После добавления смесь перемешивали в течение 2 ч при температуре 0-10°C и проводили мониторинг с помощью ТСХ (элюент CH2Cl2: MeOH=5:1). К смеси постепенно добавляли 20 мл воды, поддерживая температуру ниже 25°C. Смесь перемешивали в течение 15 мин при температуре окружающей среды, затем отгоняли 40 мл MeOH при пониженном давлении (80-120 мбар, 40°C). Водный остаток (около 40 мл) образовывал суспензию, которую фильтровали. Осадок на фильтре промывали с помощью 3×40 мл EtOAc и получали 6,95 г побочного продукта в виде белого твердого вещества. Каждый фильтрат использовали для экстракции первого водного фильтрата. Водный фильтрат экстрагировали: 3×40 мл EtOAc (т.е. три промывки осадка на фильтре) и еще 3×40 мл EtOAc. Объединенные органические фазы промывали с помощью 50 мл насыщенного раствора NaCl. Экстракт концентрировали при 40°C в вакууме (100-120 мбар). Образовавшуюся взвесь (30 мл) нагревали и кипятили с обратным холодильником в течение 30 мин, затем охлаждали до 0-5°C. После выдерживания при 0-5°C в течение ночи суспензию фильтровали и промывали с помощью 2×5 мл холодного EtOAc. Продукт (IV-2) получали в виде почти белого твердого вещества, 1,62 г. Скорректированный выход: 30%. Изотопная чистота: 93,4% по данным ВЭЖХ-МС (условия B) и 93% по данным 1H ЯМР. Анализ титрованием: 85 мас./мас.%. 1H ЯМР (DMSO-d6): δ 7,77 (уш, 2H), 2,18 (с, 0,07H - приписано сигналу CD2H).
Стадия c2: Синтез d3-AMTD (IV-2) путем с использованием гипохлорита
В четырехгорлую колбу объемом 500 мл, снабженную двумя капельными воронками, термометром и магнитной мешалкой, при 20-25°C помещали: 15,15 г (132,5 ммоля, 1,0 экв.) d3-ацетамидингидрохлорида (V-2) (изотопная чистота: 96,7%, анализ на основании содержания хлорида: 85,7 мас./мас.%), и 75 мл (5 об.) деионизированной воды, затем охлаждали до 0-5°C, поддерживая температуру в диапазоне 0 -5°C, 26,95 г (164 ммоля, 1,24 экв.) NaOCl пентагидрат, растворенный в 160 мл (10,5 об.) воды, добавляли к реакционной смеси в течение 40 мин. За протеканием реакции следили с помощью ТСХ (CH2Cl2:MeOH=5:1). Дополнительное количество реагента добавляли после перемешивания в течение 1 ч, поддерживая температуру в диапазоне 0-5°C, 13,48 г (82 ммоля, 0,62 экв.) добавляли NaOCl пентагидрат, растворенный в 80 мл (5,25 об.) воды. Смесь перемешивали в течение 1 ч при 0 -5°C. Затем к водной смеси добавляли 85 г NaCl, затем экстрагировали с помощью 3×200 мл EtOAc, затем сушили над Na2SO4, фильтровали и выпаривали в вакууме (при 40°C 10 мбар). Промежуточный продукт (VIII-2): 10,0 г красного масла.
Затем в четырехгорлую колбу объемом 500 мл, снабженную двумя капельными воронками, термометром и магнитной мешалкой, при 20-25°C помещали: 10,0 г промежуточного продукта (VIII-2) и 100 мл сухого MeOH (сушили над молекулярными ситами 4A), затем охлаждали до 0-5°C. К этому раствору в течение 20 мин осторожно добавляли 11,75 г (145,5 ммоля, 1,1 экв.) NaSCN. Полученную суспензию перемешивали в течение 1,5 ч, затем реакцию останавливали с помощью 60 мл воды. Затем реакционную смесь фильтровали затем MeOH выпаривали при 40°C в вакууме 100-200 мбар. Остаток (80 мл) фильтровали, осадок промывали с помощью 3×100 мл EtOAc. Маточный раствор экстрагировали каждой жидкостью для промывки, затем с помощью 100 мл EtOAc. Органические фазы (4×100 мл) объединяли и концентрировали до 27 мл. Этот остаток кипятили с обратным холодильником в течение 15 мин, затем охлаждали до 0-5°C, выдерживали в течение 1 ч и отфильтровывали. Отфильтрованный материал являлся продуктом. Неочищенное вещество: 26% (4,1 г) бледно-желтое твердое вещество. Скорректированный выход: 12% (1,93 г). ВЭЖХ(условия B): 47 мас./мас.%. Изотопная чистота: 96,8% по данным ВЭЖХ-МС (условия B). 1H ЯМР (DMSO-d6): δ 7,77 (уш, 2H), 3,46 (уш, 1H), 2,18 (с, 0,03H - приписано сигналу CD2H).
Пример 1B: Синтез дейтерированного d3-AMTD (IV-2) путем с использованием гидроксиацетамидина
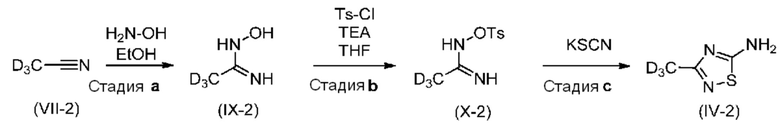
d3-Гидроксиацетамидин (IX-2) получали из дейтерированного ацетонитрила (VII-2). Соответствующий тозилированный промежуточный продукт (X-2) после этого циклизовали в присутствии тиоцианата с образованием тиадиазольного кольца соединения (IV-2).
Стадия a: Получение d3-гидроксиацетамидина (IX-2)
В колбу объемом 100 мл, снабженную обратным холодильником, термометром и магнитной мешалкой, при 20-25°C помещали: 5,5 мл (4,65 г, 105,4 ммоля, 1,0 экв.) d3-ацетонитрила (d: 0,844 г/мл), 33 мл EtOH и 25,1 мл (27,87 г, 422,3 ммоля, 4,0 экв.) водного раствора гидроксиламина (50 мас./мас.%, MW: 33 г/моль, d: 1,11 г/мл). Смесь кипятили с обратным холодильником и перемешивали в течение 5 ч при кипячении с обратным холодильником. Растворитель выпаривали из реакционной смеси в вакууме при 40°C. Продукт (IX-2) получали в виде белого твердого вещества. Выход: 84% (6,84 г). Изотопная чистота: 98,5% по данным ВЭЖХ-МС (условия C). 1H ЯМР (DMSO-d6): δ 8,67 (уш, 1H), 5,33 (уш, 2H). 13C ЯМР (DMSO-d6): δ 149,9, 16,3.
Стадия b: Тозилирование d3-гидроксиацетамидина с образованием соединения (X-2)
В колбу объемом 500 мл, снабженную термометром и магнитной мешалкой, при 20-25°C помещали: 6,3 г (81,74 ммоля 1,0 экв.) d3-гидроксиацетамидина (IX-2), 170 мл (151 г) сухого THF (над молекулярными ситами). Смесь перемешивали в течение 30 мин при температуре окружающей среды, затем 14,8 мл (10,75 г, 106,3 ммоля, 1,3 экв.) TEA добавляли при 20-25°C.
После перемешивания в течение 30 мин добавляли 45 мл сухого THF, но в смеси оставалось некоторое количество нерастворенного вещества. Смесь охлаждали до 0-5°C в бане из соли со льдом, затем к смеси несколькими порциями в течение 15 мин добавляли 14,02 г (73,57 ммоля, 0,9 экв.) п-толуолсульфонилхлорида. После добавления смеси давали нагреться до 20-25°C путем удаления охлаждающей бани и перемешивали в течение 1 ч при 20-25°C. Смесь превращалась в белую суспензию. Мониторинг: ТСХ: CH2Cl2 : MeOH=95:5. После завершения (потребления п-толуолсульфонилхлорида) осадок (9,9 г белое твердое вещество) отфильтровывали, промывали с помощью 2×40 мл THF. Фильтрат выпаривали досуха при 40°C в вакууме. К остатку добавляли 130 мл (118 г) EtOAc, органическую фазу промывали с помощью 1×65 мл воды и 1×65 мл насыщенного водного раствора NaCl. После промывки водой органический слой сушили над Na2SO4, фильтровали, промывали с помощью 2×20 мл EtOAc. Растворитель выпаривали в вакууме при 40°C и получали бесцветное масло, которое затвердевало при выдерживании. Выход: 84% (15,9 г, 68,88 ммоля). Изотопная чистота сохранялась (98,5% по данным ВЭЖХ-МС (условия C)).
Стадия c: Синтез d3-AMTD (IV-2)
В колбу объемом 100 мл, снабженную термометром и магнитной мешалкой, при 20-25°C помещали: 6,39 г (65,76 ммоля, 3,0 экв.) KSCN, 25 мл (19,8 г) сухого MeOH (над молекулярными ситами). Во время перемешивания 5,06 г (21,92 ммоля, 1,0 экв.) d3-N-тозилоксиацетамидина (X-2) добавляли к смеси в виде твердого вещества. После перемешивания в течение 5 мин при 20 -25°C добавляли 4,2 мл (3,12 г, 24,11 ммоля, 1,1 экв.) DIPEA. Реакционную смесь перемешивали при 20 -25°C в течение ночи и она постепенно превращалась в белую суспензию. Мониторинг: ТСХ: CH2Cl2 : MeOH=95:5, визуализация с помощью KMnO4. Реакция не завершалась через 18 ч, затем реакционную смесь нагревали до 40°C и перемешивали в течение 2 ч. Затем осадок (белое твердое вещество) отфильтровывали, промывали с помощью 2×20 мл MeOH. К фильтрату добавляли 30 мл воды, затем MeOH выпаривали при 40°C в вакууме (100-150 мбар). Водный остаток экстрагировали с помощью 7×50 мл EtOAc, объединенные органические фазы сушили над Na2SO4, затем выпаривали. Неочищенный продукт представлял собой 4,2 г масла апельсинового цвета. После обработки с помощью 10 мл CH2Cl2, 700 мг липкого осадка отфильтровывали и обрабатывали с помощью 5 мл iPrOAc и выделяли продукт в виде 173 мг бледно-желтого твердого вещества (выход неочищенного вещества: 7%). Анализ: 63 мас./мас.% с помощью ВЭЖХ(условия B). Изотопная чистота: 73% по данным ВЭЖХ-МС (условия B). Маточные растворы после обработки посредством CH2Cl2 и iPrOAc объединяли и выпаривали в вакууме при 40°C и получали 2,55 г липкого, оранжевого твердого вещества. Анализ: 10 мас./мас.% с помощью ВЭЖХ (условия B). Для реакции в целом скорректированный выход: 14,5% (анализ 0,109 г+0,255 г).
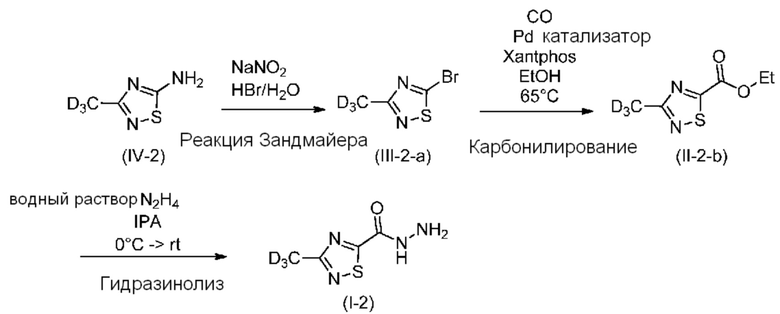
Пример 2: Синтез дейтерированного 3-метил-d3-1,2,4-тиадиазол-5-карбогидразида (I-2)
Изотопная чистота сохранялась на всех стадиях.
Стадия 1: Бромирование по реакции Зандмейера с образованием (III-2-а)
В колбу объемом 25 мл, снабженную патрубком для выхода газа, ведущим к газовой ловушке, заполненной 10 мас./мас.% раствором NaOH, капельной воронкой, термометром и магнитной мешалкой, при 20-25°C помещали: 2,7 мл (4,61 г раствора, содержащего 2,85 г HBr, 35,2 ммоля, 3,0 экв.) 62 мас./мас.% раствора HBr (d: 1,702 г/мл), 2,1 мл воды и 1,39 г (11,76 ммоля, 1 экв.) соединения (IV-2) (изотопная чистота: 93,4%, Анализ титрованием: 85%). Смесь нагревали до 40°C. Затем из капельной воронки добавляли: 1,22 г (17,64 ммоля, 1,5 экв.) NaNO2, растворенного в 2 мл воды со скоростью, обеспечивающей поддержание температуры в диапазоне 40-45°C (в течение 5 мин). Во время добавления образовывался коричневый газ, наблюдалось интенсивное прохождение газа в поглотителе и на дне колбы осаждалась масляная фаза. После добавления смесь перемешивали в течение 1 ч и проводили мониторинг с помощью ТСХ (гексан:EtOAc =1:1) и ВЭЖХ. Реакционную смесь охлаждали до 20-25°C. Двухфазную систему экстрагировали с помощью 2×10 мл дихлорметана. Объединенную органическую фазу (темно-коричневую) промывали с помощью: 5 мл 5 мас./мас.% раствора NaOH. Фазы разделяли. Органическую фазу выпаривали в вакууме (300-100 мбар) при 40°C и получали 1,51 г желтого масла (III-2-a). Выход: 78% (1,51 г). ГХ (условия A): 94,4%. Изотопная чистота по данным ГХ-МС: 93,1%). Не наблюдалось изменение изотопной чистоты.
Стадия 2: Этоксикарбонилирование с образованием (II-2-b)
В инертной атмосфере в автоклав объемом 50 мл при 20-25°C помещали: 1,2 г (6,60 ммоля, 1,0 экв.) неочищенного соединения (III-2-а) (изотопная чистота: 93,1%, ГХ: 94,9%), 12 мл (10 об.) сухого этанола и 706 мг (8,58 ммоля, 1,3 экв.) NaOAc. Затем через смесь пропускали азот, затем добавляли 19 мг (0,033 ммоля, 0,005 моля экв.) Xantphos и 7,5 мг (0,033 ммоля, 0,005 моля экв.) Pd(OAc)2. Колбу закрывали, четырежды продували азотом, затем четырежды продували с помощью CO, затем заполняли с помощью CO до давления в 4 бар. При интенсивном перемешивании смесь нагревали при 65°C. Реакционную смесь выдерживали при 65°C, поддерживая давление в 4 бар до завершения реакции (~ 45 ч). За протеканием реакции следили с помощью ГХ. После удаления CO и продувки азотом растворитель удаляли в вакууме при 40°C, затем добавляли 15 мл метилциклогексана, осадок отфильтровывали и промывали с помощью 5 мл метилциклогексана. Растворитель удаляли в роторном испарителе при 40°C в вакууме (10 мбар). После этой обработки продукт (II-2-b) выделяли в виде зеленого масла. Его использовали на следующей стадии без дополнительной очистки. Выход: 52% (706 мг). ГХ (условия A): 81%. Изотопная чистота по данным ЖХМС (условия C): 93,4%. 1H ЯМР (DMSO-d6): δ 4,43 (кв, J=7,1 Гц, 2H), 1,34 (т, J=7,1 Гц, 3H). 13C ЯМР (DMSO-d6): δ 179,0, 175,9 (д), 158,4, 63,3, 18,2 (гепт), 14,3. Не наблюдалось уменьшение изотопной чистоты.
Стадия 3: Гидразинолиз с образованием (I-2)
В колбу объемом 10 мл, снабженную капельной воронкой, термометром и магнитной мешалкой, при 20-25°C помещали: 4 мл IPA и 177 мкл (181 мг, содержащих 100 мг, 3,11 ммоля, 1,14 экв.) гидразингидрата (55 мас./мас.% в воде, d: 1,027 г/мл). Реакционную смесь охлаждали до 0°C. К смеси из капельной воронки добавляли раствор 595 мг (2,73 ммоля, 1,0 экв.) соединения (II-2-b) (изотопная чистота: 93,4%, ГХ: 81%) в 2 мл IPA со скоростью, обеспечивающей поддержание температуры в диапазоне 0-5°C. После добавления смесь перемешивали при 0-5°C в течение 1 ч, затем в течение ночи при комнатной температуре. За протеканием реакции следили с помощью ВЭЖХ. После завершения суспензию охлаждали до 0-5° С, затем фильтровали, осадок на фильтре промывали с помощью 2×1 мл холодного IPA и сушили в вакууме при 35°C. После этой обработки продукт (1-2) выделяли в виде бледно-желтого твердого вещества. Выход: 72% (321 мг). ВЭЖХ-МС (условия D): 96,4%. Изотопная чистота по данным ВЭЖХ-МС (условия C): 93,4%. 1H ЯМР (DMSO-d6): δ 10,50 (уш, 1H), 4,79 (уш, 2H). 13C ЯМР (DMSO-d6): δ 183,4, 175,0 (д), 157,8 (д), 18,0 (гепт). Не наблюдалось уменьшение изотопной чистоты.
Пример 3: Синтез 3-метил-1,2,4-тиадиазол-5-карбогидразида (I-1)
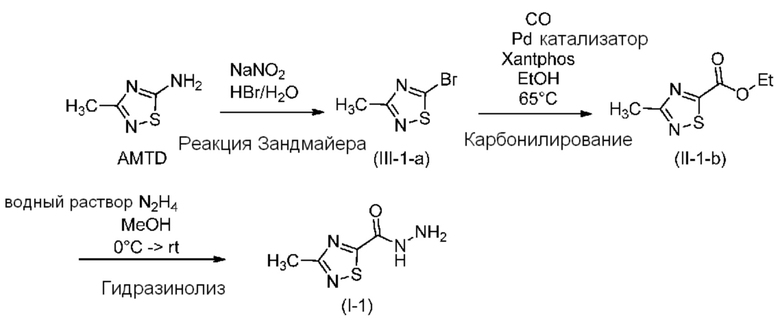
Стадия 1: Бромирование по реакции Зандмейера с образованием (III-1-а)
Процедуру бромирования по реакции Зандмейера успешно проводили с использованием 100 кг 5-амино-3-метил-1,2,4-тиадиазола (AMTD) с выходом 82%: 439,5 кг (295 л) 48% раствора HBr и 100,0 кг AMTD добавляли к 54 кг (л) воды в реакторе. Раствор нагревали максимально до 40°C и перемешивали до растворения всего исходного вещества, что проверяли путем отбора проб. Если исходное вещество полностью не растворялось, было достаточным проводимое после этого перемешивание в течение 2 ч при 40°C. 89,7 кг Нитрита натрия растворяли в 150 кг (л) воды в другом оборудовании, затем раствор помещали в контейнер. Раствор нитрита натрия порциями вводили в реактор, это требовало примерно 6 ч, поддерживая температуру в диапазоне 40-45°C. Образец отбирали через 1 ч после завершения добавления. Критерий завершения: содержание исходного вещества < 1,0%. Если содержание исходного вещества > 1,0%, к реакционной смеси необходимо дополнительно добавить раствор нитрита натрия (10-20%). В случае, если реакционная смесь соответствует критерию завершения, ее охлаждали до 20-25°C и добавляли 399,0 кг (300 л) DCM. После перемешивания в течение 10 мин и осаждения в течение 10 мин нижнюю органическую фазу загружали в контейнер. Водную фазу экстрагировали с помощью 133,0 кг (100 л) DCM в реакторе. Значение pH объединенной органической фазы устанавливали равным 10-11 путем добавления 5% раствора гидроксида натрия. Если pH превышает 11, может образоваться большое количество влаги. Смесь перемешивали в течение 20 мин и после осаждения в течение 30 мин органическую фазу отделяли и помещали в сухое оборудование, и концентрировали в вакууме (примерно -0,9 бар) при температуре не выше 30°C. При отборе образца критерий завершения, для концентрации: максимальное содержание DCM 5,0%. После завершения перегонки необходимо перемешивание в вакууме в течение 4-6 ч для обеспечения равного 5,0% максимального содержания DCM. Содержание DCM, равное 2,0%, может быть благоприятно на следующей стадии. В случае, если вещество соответствует критерию завершения, его помещали в бочку, облицованную с помощью PE (полиэтилен). Получали 126,8 кг соединения (III-1-а); Выход: 81,6%; ГХ чистота (условия C): 97,2%.
Стадия 2: Этоксикарбонилирование с образованием (II-1-b)
Процедуру этоксикарбонилирования успешно проводили с использованием 87,5 кг соединения (III-1-а) с выходом 87%:
87,5 кг Соединения (III-1-а) добавляли к 44 кг абсолютного этанола в бочке, облицованной с помощью PE, и перемешивали до образования прозрачного раствора. 383 кг (484 л) Абсолютного этанола помещали в сухое, стойкое к давлению оборудование. После образования инертной атмосферы с помощью N2 добавляли раствор катализатора, полученный из 0,672 кг ацетата палладия(II) и 1,68 кг Xantphos в 14,5 кг ледяной уксусной кислоты. К смеси добавляли 52,5 кг безводного ацетата натрия. Систему повторно продували с помощью N2 и реакционную смесь нагревали до 63-67°C. Оборудование заполняли с помощью CO до 4-4,5 бар, затем загружали раствор (III-1-а) за 2-3 ч. После добавления 16 кг (20 л) этанола использовали для ополаскивания. Отбор образцов проводили каждые 12 ч, критерий завершения: содержание исходного вещества (III-1-а) максимально 1,0%. Когда реакция отвечала критерию завершения давление сбрасывали с помощью катализатора CATOX и оборудование 6 раз продували с помощью N2. Смесь фильтровали в сухой контейнер с помощью технологического фильтра, промывали с помощью 80 кг (100 л) абсолютного этанола. Фильтрат концентрировали при пониженном давлении при температуре не выше 50°C. Концентрат разбавляли с помощью 120 кг метилциклогексана и перемешивали в течение 30-40 мин. Раствор фильтровали с помощью технологического фильтра, который промывали с помощью 40 кг метилциклогексана. Растворитель удаляли при пониженном давлении при температуре не выше 50°C и продукт выделяли путем фракционированной перегонки в вакууме. Получали 73,5 кг соединения (II-1-b); Выход: 87,0%; ГХ чистота (условия A): 96,9%.
Стадия 3: Гидразинолиз с образованием (1-1)
Процедуру гидразинолиза успешно проводили с использованием 50 кг соединения (II-1-b) с выходом 91%:
50,0 кг Соединения (II-1-b) растворяли в 212,2 кг (270 л) изопропилового спирта. 26,7 кг Гидразингидрата (55% раствор в воде) добавляли к 340 кг (432 л) изопропилового спирта и смесь охлаждали до 0°C. Раствор (II-1-b) порциями добавляли к холодному раствору гидразингидрата, что требовало 1-2 ч, поддерживая температуру в диапазоне 0-5°C. После завершения добавления смесь перемешивают в течение еще 1 ч при 0-5°C. После перемешивания в течение 1 ч проверяли содержание (II-1-b), если содержание (II-1-b) > 1,0%, смесь нагревали до 20-25°C и отбирали образцы для мониторинга протекания реакции, ожидаемое время реакции равно 20 ч, до выполнения критерия завершения: (II-1-b) < 1,0%. В случае, если обеспечивался критерий завершения реакции, суспензию охлаждали до 0-5°C и фильтровали. Отфильтрованное вещество промывали с помощью 31,8 кг (40 л) 0-5°C изопропилового спирта. Получали 37,5 кг соединения (1-1); Выход: 91%; Чистота: 99,9% по данным ВЭЖХ(условия A).
Пример 4: Бромирование по реакции Зандмейера с использованием трет-бутилнитрита с образованием (III-1-а)
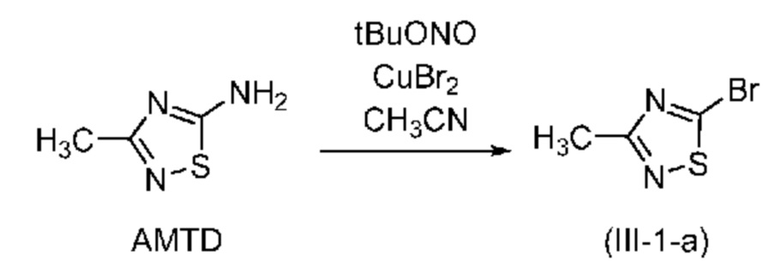
В колбу Шленка помещали CuBr (14 г, 62,68 ммоль, 1,03 экв.), безводный MeCN (110 мл) и трет-бутилнитрит (11 мл, 92,59 ммоля, 1,5 экв.) в атмосфере N2, затем полученную темно-зеленую смесь охлаждали до температуры 0-5°C (в бане со льдом). К этой смеси порциями в течение 1 мин добавляли AMTD (7 г, 60,78 ммоля, 1,0 экв.). Полученную смесь перемешивали при 0-5°C в течение 30 мин и затем ей давали нагреться до rt и перемешивали в течение 2 ч. Смесь разбавляли 1:1 смесью 25% аммиака (50 мл) и 10% Na2CO3 (50 мл) и добавляли TBME (100 мл). Фазы разделяли и водный слой экстрагировали с помощью TBME (2×50 мл). Объединенный органический слой промывали рассолом (3×50 мл), сушили над MgSO4, фильтровали и концентрировали при пониженном давлении (1 мбар, 30°C) досуха и получали соединение (III-1-а) в виде суспензии желтой твердого вещества в оранжевом масле (7,16 г, 39,99 ммоля, 66% выход, > 98% чистота по данным ВЭЖХ-МС (условия D). 1H ЯМР (CDCl3): δ 2,69 (с, 3H).
Пример 5: Йодирование по реакции Зандмейера с использованием трет-бутилнитрита с образованием (III-1-b)
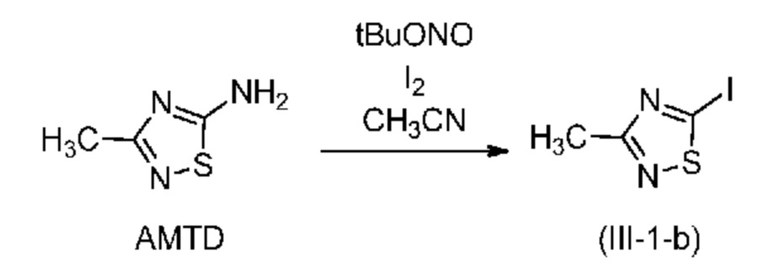
В трехгорлую колбу объемом 1000 мл, снабженную патрубком для подачи газа, капельной воронкой и термометром, помещали 55,0 г (217,1 ммоля 1,0 экв.) йод, 250 мл сухого CH3CN и 102,7 мл (89,54 г, 868,4 ммоля, 4,0 экв.) tBuONO. Реакционную смесь перемешивали в течение 10 мин. При комнатной температуре порциями добавляли 25,0 г (217,1 ммоля, 1,0 экв.) AMTD в виде твердого вещества. Через несколько минут реакционная смесь (коричневая) начинала самопроизвольно кипеть, наблюдалось бурное выделение газа и реакционную смесь охлаждали до комнатной температуры. Этого времени (примерно 40 мин) было достаточно для завершения реакции по данным ТСХ. Избыток йода устраняли путем добавления 450 мл 20 мас./мас.% водного раствора Na2SO3, смесь становилась желтой, но при выдерживании она повторно становилась коричневатой, добавление дополнительного количества водного раствора Na2SO3 (50 мл) делало смесь желтой, но она повторно становилась коричневатой. Фазы разделяли, водную фазу экстрагировали с помощью TBME (4×150 мл). Объединенную органическую фазу промывали с помощью 200 мл рассола, сушили над Na2SO4, фильтровали и выпаривали в вакууме при 30°C. Продукт (III-1-b) получали в виде коричневатого твердого вещества с выходом неочищенного вещества, равным 72,2% (65,7% скорректированный выход) (35,4 г, чистота по данным ВЭЖХ (условия A): 92,3% площади, количественный ЯМР: 91 мас./мас.%). Продукт очищали путем перекристаллизации из кипящего 2-пропанола (2 мл/г неочищенного вещества) с выходом 63% (23,9 г, чистота по данным ВЭЖХ (условия A): 99,3% площади, количественный ЯМР: 99 мас./мас.%). 1H ЯМР (CDCl3): δ 2,73 (с, 3H). 13C ЯМР (CDCl3): δ 175,9, 124,8, 18,9.
Йодированный промежуточный продукт (III-1-b) успешно превращали в соединения (II-1-а) при условиях примера 3 - Стадия 2 (CO, Pd(OAc)2, Xantphos) или при условиях примера 9 (CO, Pd(PPh3)2Cl2).
Пример 6: Йодирование по реакции Зандмейера с использованием нитрита натрия с образованием (III-1-b)
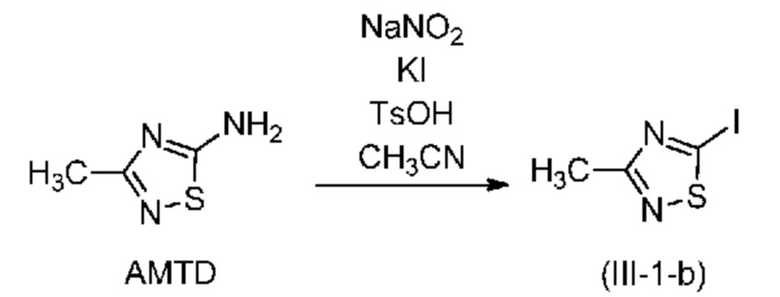
К смеси AMTD (58 мг, 0,50 ммоля) и моногидрата п-толуолсульфоновой кислоты (305 мг, 1,74 ммоля) в безводном MeCN (2 мл) в течение 5 мин добавляли раствор нитрита натрия (70 мг, 1,01 ммоля) и йодид калия (215 мг, 1,3 ммоля) в воде (0,5 мл) при 0°C (бесцветный раствор становился желтым после первой капли и затем превращался в черный сироп, но после завершения добавления становился прозрачным черным раствором). Реакционную смесь перемешивали при КТ в течение ночи. Затем реакционную смесь разбавляли насыщенным раствором NaHCO3 (15 мл) и экстрагировали с помощью EtOAc (3×10 мл). Объединенный органический слой сушили над MgSO4, фильтровали и концентрировали при пониженном давлении (1-2 мбар, 30°C) досуха и получали 149 мг в виде темного масла. ВЭЖХ-МС (условия D): 90% площади. 1H ЯМР (CDCl3): δ 2,73 (с, 3H). 13C ЯМР (CDCl3): δ 175,9, 124,8, 18,9.
Пример 7: Этоксикарбонилирование посредством обмена лития
Введение алкоксикарбонильной группы в тиадиазол также можно провести посредством обмена лития:
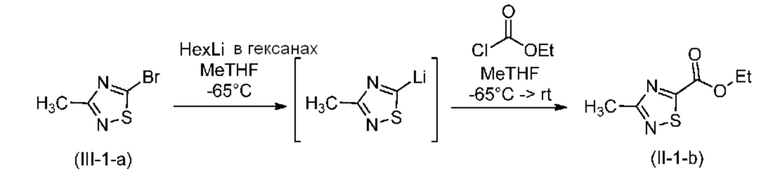
В трехгорлой колбе, снабженной патрубком для подачи газа, капельной воронкой и термометром, 10,0 г (55,87 ммоля, 1 экв.) соединения (III-1-а) растворяли в 100 мл сухого MeTHF в атмосфере аргона. Полученную смесь охлаждали до -65°C. Затем в течение 20 мин по каплям добавляли 15,6 г (22,0 мл, 55,87 ммоля) гексиллития в гексане (33%), поддерживая температуру равной -65°C. После завершения добавления отбирали образец реакционной смеси: реакционная смесь+этилхлорформиат+вода+TBME.
Фазу с TBME анализировали с помощью ВЭЖХ (условия B). С помощью встроенного контроля обнаруживали 5,5% непрореагировавшего исходного вещества, поэтому к реакционной смеси при -65°C добавляли 2,2 мл (5,58 ммоля) гексиллития в гексане. Через 20 мин обнаружено полное израсходование исходного бромида, поэтому реакционную смесь с помощью охлажденной канюли за 15 мин переносили в предварительно охлажденную смесь 32 мл (335,16 ммоля, 6 экв.) этилхлорформиата и 30 мл сухого MeTHF при - 65°. Затем полученной темно-оранжевой реакционной смеси давали нагреваться до комнатной температуры и затем добавляли 200 мл деионизированной воды. Фазы разделяли и органическую фазу сушили над Na2SO4 и концентрировали и получали 13,2 г коричневого неочищенного продукта. 8 г Неочищенного продукта очищали с помощью перегонки в вакууме при 1 мбар и собирали 4 фракции. После перегонки собранные фракции объединяли и получали 1,9 г продукта (выход: 29%, чистота поданным ВЭЖХ (условия B): 92%) в виде бесцветного масла. 1H ЯМР (CDCl3): δ 4,51 (кв, J =1,1 Гц, 2H), 2,77 (с, 3H), 1,44 (т, J=7,1 Гц, 3H). 13C ЯМР (CDCl3): δ 178,8, 175,2, 158,6, 63,4, 19,1, 14,3.
Пример 8: Этоксикарбонилирование с помощью Fe(CO)5
Алкоксикарбонилирование также успешно проводили в присутствии Fe(CO)5:
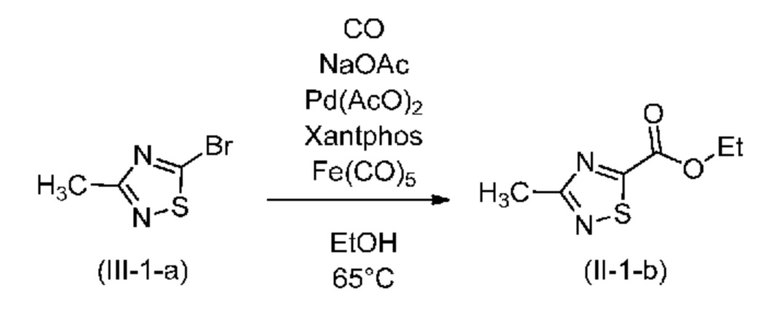
В инертной атмосфере в автоклав объемом 270 мл при 20-25°C помещали: 5,0 г (28,0 ммоля, 1,0 экв.) неочищенного соединения (III-1-а) (чистота: 96 мас./мас.%, содержало 3% дихлорметана), 50,0 мл (10 об.) сухого этанола (содержание воды 256 част./млн) и 3,0 г (36,55 ммоля, 1,3 экв.) NaOAc (содержание воды 256 част./млн). Через эту смесь в течение 5 мин пропускали азот, затем добавляли 80,0 мг (0,14 ммоля, 0,005 мол. экв.) Xantphos, 31,5 мг (0,14 ммоля, 0,005 мол. экв.) Pd(OAc)2 и 376 мкл (560 мг, 2,86 ммоля, 0,1 экв.) Fe(CO)5 (d: 1,45 г/мл). Сосуд закрывали, четырежды продували азотом, затем четырежды продували с помощью CO, затем заполняли посредством CO до 4 бар. При интенсивном перемешивании смесь нагревали до 65°C. Во время нагревания давление повышалось до 5 бар. Реакционную смесь выдерживали при 65°C и давление поддерживали при 4 бар до завершения реакции (~ 24 ч). За протеканием реакции следили с помощью ГХ (условия A), отбор образца: 0,4 мл реакционной смеси разбавляли с помощью 3,6 мл EtOH). Степень превращения по данным ГХ: через 17,5 ч: 59%, через 24 ч: 84,5%.
Пример 9: Этоксикарбонилирование с помощью катализатора Pd(PPh3)2Cl2
Алкоксикарбонилирование также успешно проводили в присутствии Pd(PPh3)2Cl2 в качестве катализатора.
Алкоксикарбонилирование бромированного промежуточного продукта (III-1-а)
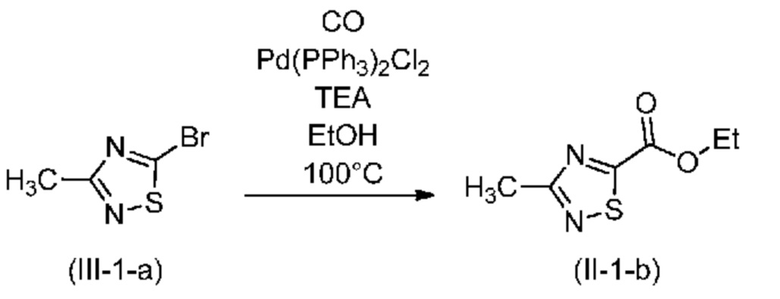
В инертной атмосфере в автоклав объемом 800 мл, снабженный магнитной мешалкой, при 20-25°C помещали: 5,0 г (27,9 ммоля, 1,0 экв.) дистиллированного (III-1-а), 140 мл (28 об.) сухого этанола и 5 мл (3,64 г, 35,9 ммоля, 1,28 экв.) TEA. Эту смесь продували азотом в течение 10 мин, затем добавляли 1,0 г (1,42 ммоля, 0,05 экв.) Pd(PPh3)2Cl2. Сосуд закрывали, трижды продували азотом, затем трижды продували с помощью CO, затем заполняли посредством CO до 5 бар. При интенсивном перемешивании нагревали до 100°C и давление повышалось до 6,5 бар и смесь перемешивали до завершения реакции (~31 ч). За протеканием реакции следили с помощью ЖХМС (условия B) через 8 ч, через 14 ч и через 31 ч (отбор образца: 150 мкл реакционной смеси фильтровали через целит, промывали этанолом (2×150 мкл) и сразу использовали). Затем, реакционную смесь фильтровали через 5 г целита и промывали с помощью 2×50 мл EtOH. EtOH выпаривали из фильтрата в вакууме при 40°C и получали 12,09 г коричневого твердого вещества, которое суспендировали в 150 мл TBME. Затем органическую фазу промывали с помощью 3×50 мл воды, затем выпаривали в вакууме при 40°C и получали желтую смесь масла и твердого вещества. Выход неочищенного вещества: 82,5% (3,96 г), ЖХМС (условия B): 42% площади. 1H ЯМР: по оценке 66 мас./мас.% (без учета PPh3O).
Алкоксикарбонилирование иодированого промежуточного продукта (III-1-b)

В автоклав помещали соединение (III-1-b) (1 г, 4,34 ммоля), TEA (0,75 мл, 5,34 ммоля), дихлорбис(трифенилфосфин)палладий(II) (60 мг, 0,08 ммоля) в безводном этаноле (20 мл). Затем сосуд дважды продували монооксидом углерода и давление повышали до 10 бар и нагревали при 110°C. ВЭЖХ-МС мониторинг (условия D) показывал полное превращение через 4 ч. Смесь фильтровали через целит, твердые вещества промывали с помощью EtOH (20 мл) и фильтрат концентрировали при пониженном давлении (1-2 мбар, 40°C) досуха и получали желтое органическое твердое вещество. Это последнее солюбилизировали в дихлорметане (5 мл) и добавляли TBME (15 мл) до образования осадка. Это почти белое твердое вещество отфильтровывали и промывали с помощью TBME (10 мл) и фильтрат концентрировали в высоком вакууме (2 мбар, 40°C) досуха и получали 854 мг в виде оранжевого твердого вещества. Чистоту оценивали в 59 мас.% нс основании 1H ЯМР (обнаруживали PPh3O и соль TEA-HI), что приводило к скорректированному выходу в 67%.
Пример 10: Этоксикарбонилирование с помощью катализатора Pd(OAc)2 - PPh3
Алкоксикарбонилирование также успешно проводили в присутствии Pd(OAc)2 - PPh3 в качестве катализатора.
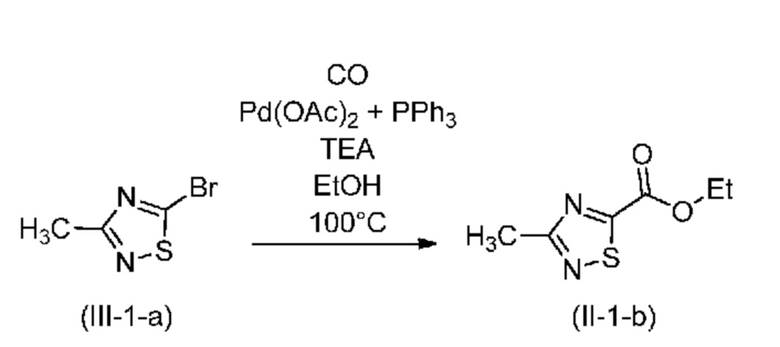
В инертной атмосфере в автоклав объемом 250 мл с механическим перемешиванием при 20-25°C помещали: 3,75 г (20,9 ммоля, 1,0 экв.) дистиллированный (III-1-а), 105 мл (28 об.) сухого этанола и 3,75 мл (2,73 г, 26,99 ммоля, 1,29 экв.) TEA. Эту смесь продували азотом в течение 10 мин, затем добавляли 578 мг (1,05 ммоля, 0,105 экв.) PPh3 и 235 мг (11,17 ммоля, 0,05 экв.) Pd(OAc)2. Сосуд закрывали, трижды продували азотом, затем трижды продували с помощью CO, затем заполняли посредством CO до 5 бар. При интенсивном перемешивании нагревали до 100°C и давление повышалось до 6,5 бар и ее перемешивали до завершения реакции (-17 ч). За протеканием реакции следили с помощью ВЭЖХ(условия B) через 17 ч (отбор образца: 150 мкл реакционной смеси фильтровали через целит, промывали этанолом (2×150 мкл) и сразу использовали). Затем реакционную смесь фильтровали через 5 г целита и промывали с помощью 2×50 мл EtOH. EtOH выпаривали из фильтрата в вакууме при 40°C и получали 7,03 г коричневого твердого вещества, которое суспендировали в 150 мл TBME. Затем органическую фазу промывали с помощью 3×50 мл воды, затем выпаривали в вакууме при 40°C и получали желтую смесь масла и твердого вещества. Выход неочищенного вещества: 84,7% (3,05 г), ЖХМС (условия B): 71% площади. 1H-NMR: оценка 80 мас./мас.% (без учета PPH3O).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ПОЛУЧЕНИЯ МЕТИЛАМИДОВ α-МЕТОКСИИМИНОКАРБОНОВЫХ КИСЛОТ | 1995 |
|
RU2146247C1 |
| ЗАМЕЩЕННЫЕ ФЕНИЛСОДЕРЖАЩИЕ СОЕДИНЕНИЯ | 2012 |
|
RU2592696C2 |
| ПРОИЗВОДНЫЕ РЕЗИКВИМОДА | 2018 |
|
RU2808163C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ЦИТОТОКСИЧЕСКИХ ПРОИЗВОДНЫХ БЕНЗОДИАЗЕПИНА | 2019 |
|
RU2807546C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ (ЦИКЛОПЕНТИЛ[d]ПИРИМИДИН-4-ИЛ)ПИПЕРАЗИНОВЫХ СОЕДИНЕНИЙ | 2015 |
|
RU2732404C2 |
| СПОСОБ СИНТЕЗА 2-ТИОГИСТИДИНА И ЕГО АНАЛОГОВ | 2010 |
|
RU2548153C2 |
| СПОСОБ И СОЕДИНЕНИЕ, ПРИГОДНЫЕ В ПОЛУЧЕНИИ АНТАГОНИСТА РЕЦЕПТОРА ОРЕКСИНА-2, И ЛЕМБОРЕКСАНТ, СОДЕРЖАЩИЙ НЕБОЛЬШОЕ КОЛИЧЕСТВО ПРИМЕСЕЙ | 2020 |
|
RU2823998C2 |
| СПОСОБ ПОЛУЧЕНИЯ СУЛЬФОНИЛКАРБАМАТНЫХ ПРОИЗВОДНЫХ ЖЕЛЧНЫХ КИСЛОТ | 2018 |
|
RU2791682C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ (ЦИКЛОПЕНТИЛ[D]ПИРИМИДИН-4-ИЛ)ПИПЕРАЗИНОВЫХ СОЕДИНЕНИЙ | 2015 |
|
RU2712224C2 |
| ПРОИЗВОДСТВО СОЕДИНЕНИЙ И КОМПОЗИЦИЙ ДЛЯ ПОДАВЛЕНИЯ АКТИВНОСТИ SHP2 | 2019 |
|
RU2797951C2 |
Настоящее изобретение относится к способу синтеза 3-метил-1,2,4-тиадиазол-5-карбогидразида или его метил-d3 дейтерированной формы формулы (I), где R1 означает метил или метил-d3. Указанный способ включает следующие стадии: a) проведение реакции алкоксикарбонилирования соединения формулы (III), где X означает галоген, R1 означает метил или метил-d3, с получением соединения формулы (II), где R1 означает метил или метил-d3, R2 означает алкил или арилалкил; и b) получение соединения формулы (I) по реакции соединения формулы (II) с гидразинмоногидратом. Получаемые заявленным способом соединения применимы в качестве ключевых промежуточных продуктов в синтезе фармацевтических соединений, в особенности фезолинетанта и дейтерированного фезолинетанта. Технический результат изобретения заключается в более безопасном, надежном, масштабируемом и более эффективном синтезе промежуточного соединения формулы (I). 14 з.п. ф-лы, 10 пр.
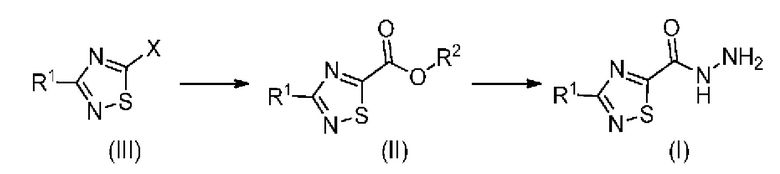
1. Способ получения соединения формулы (I)
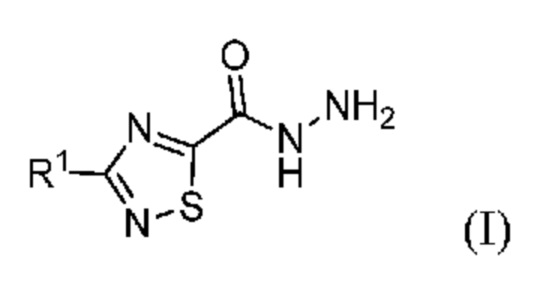
или его соли, где R1 означает метил или метил-d3; указанный способ включает следующие стадии:
a) проведение реакции алкоксикарбонилирования соединения формулы (III)
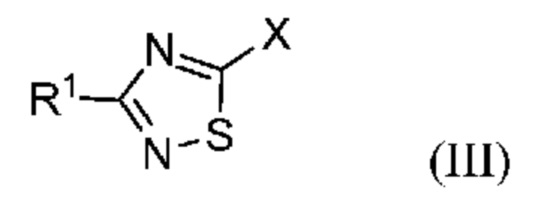 ,
,
где X означает галоген; и R1 означает метил или метил-d3; с получением соединения формулы (II)
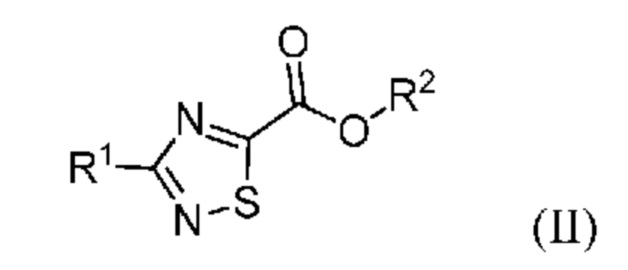 ,
,
где R1 означает метил или метил-d3; и R2 означает алкил или арилалкил; и
b) получение соединения формулы (I) по реакции соединения формулы (II) с гидразинмоногидратом.
2. Способ по п.1, где X означает бром или йод.
3. Способ по п.1 или 2, где R2 означает метил или этил.
4. Способ по любому из пп.1-3, где реакцию алкоксикарбонилирования на стадии a) проводят в присутствии монооксида углерода, палладиевого катализатора, основания и спиртового растворителя и необязательно в присутствии фосфорорганического лиганда.
5. Способ по п.4, где палладиевым катализатором является Pd(OAc)2, фосфорорганическим лигандом является 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos), основанием является ацетат натрия и растворитель выбран из группы, включающей этанол, метанол и смеси метил-трет-бутилового эфира с этанолом или метанолом.
6. Способ по п.4 или 5, где стадию a) проводят в этаноле в качестве растворителя или в смеси этанола и метил-трет-бутилового эфира.
7. Способ по любому из пп.4-6, где стадию a) проводят при температуре в диапазоне от 50 до 150°C.
8. Способ по любому из пп.4-7, где стадию a) проводят в присутствии Fe(CO)5.
9. Способ по п.4, где палладиевым катализатором является бис(трифенилфосфин)палладийхлорид (Pd(PPh3)2Cl2), основанием является триэтиламин и растворителем является этанол.
10. Способ по любому из пп.1-3, где реакцию алкоксикарбонилирования на стадии a) проводят посредством обмена лития с помощью проводимого сначала взаимодействия соединения формулы (III) с литийорганическим реагентом; с последующим добавлением хлорформиата или цианоформиата.
11. Способ по п.10, где литийорганическим реагентом является н-гексиллитий и хлорформиатом является алкилхлорформиат.
12. Способ по любому из пп.1-11, включающий предварительную стадию реакции Зандмейера соединения формулы (IV)
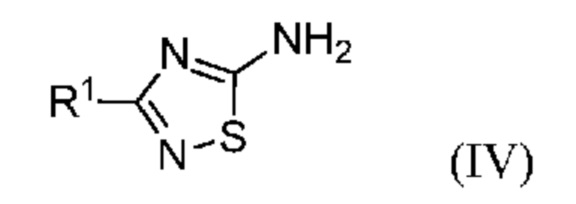
или его соли, где R1 означает метил или метил-d3; с получением соединения формулы (III).
13. Способ по п.12, где реакцию Зандмейера проводят в присутствии нитрита натрия и бромида водорода в водной среде.
14. Способ по п.12, где реакцию Зандмейера проводят в присутствии трет-бутилнитрита и йода или в присутствии йодида калия и п-толуолсульфоновой кислоты.
15. Способ по п.12, где соединение формулы (IV) представляет собой 3-(метил-d3)-1,2,4-тиадиазол-5-амин (IV-2)
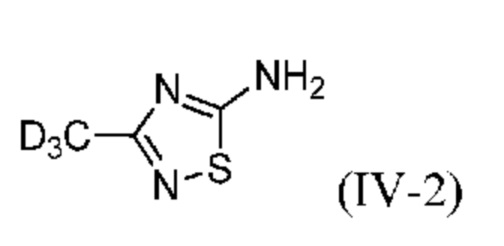 ,
,
и способ включает предварительную стадию получения 3-(метил-d3)-1,2,4-тиадиазол-5-амина (IV-2) или его соли, включающий следующие стадии:
a) реакция d3-ацетонитрила с этанолом в присутствии HCl с образованием соли Пиннера формулы (VI-2):
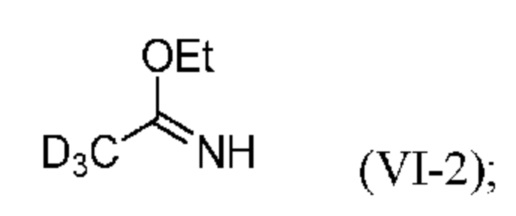
b) реакция соли Пиннера (VI-2) с аммиаком с образованием d3-ацетамидина (V-2) или его соли
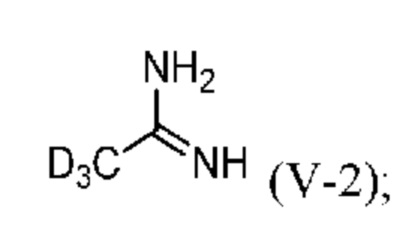
и
c1) реакция d3-ацетамидина (V-2) с бромом, тиоцианатом и метоксидом натрия с образованием соединения формулы (IV-2); или
c2) реакция d3-ацетамидина (V-2) сначала с гипохлоритом натрия (NaOCl) и затем с тиоцианатом с образованием соединения формулы (IV-2).
| WO 2013050424 A1, 11.04.2013 | |||
| GOERDELER J | |||
| et al., Darstellung und Eigenschaften des 1.2.4- und des 1.3.4-Thiodiazols, CHEMISCHE BERICHTE, 1956, vol.89, p.1534-1543 | |||
| EP 1975204 B1, 02.10.2013 | |||
| VERRIER C | |||
| et al., Recent advances in direct C-H arylation: Methodology, selectivity and mechanism in oxazole series, Beilstein J Org Chem, 2011, vol.7, |
