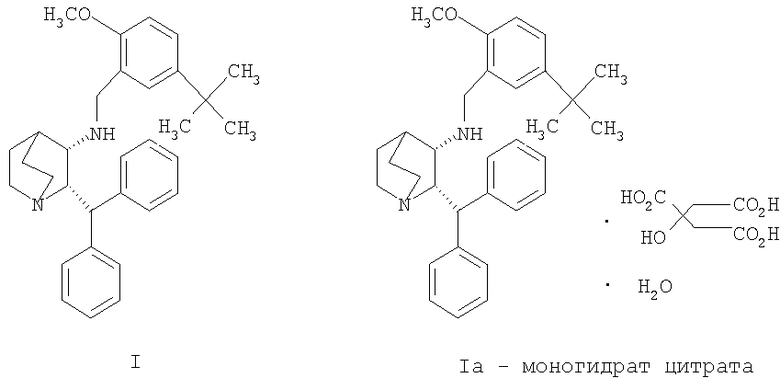
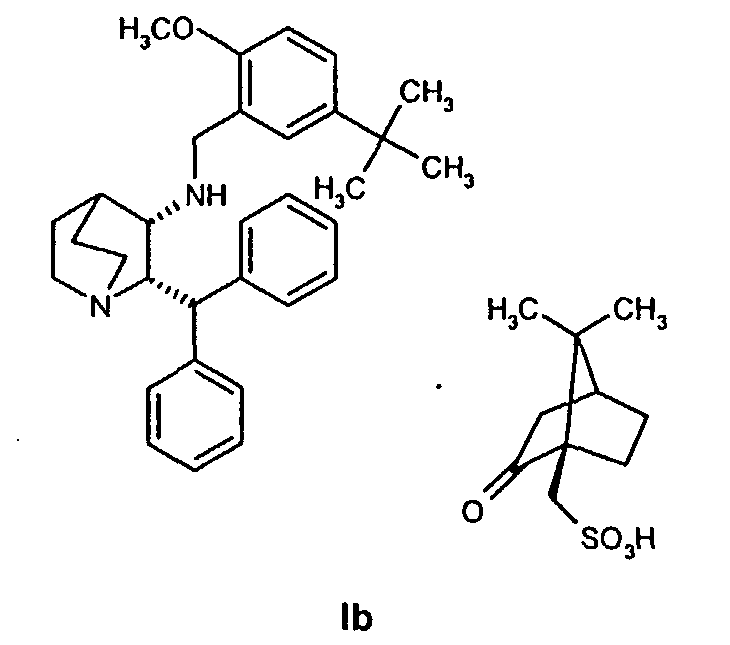
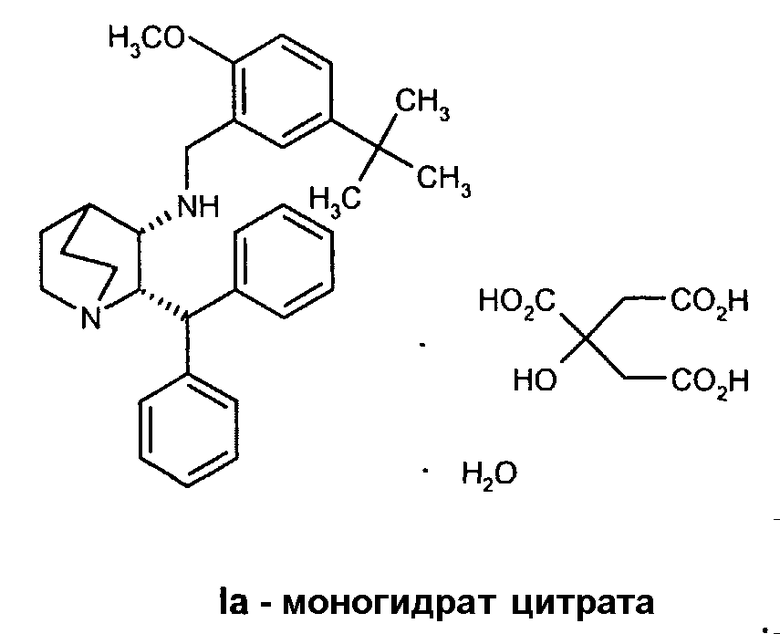
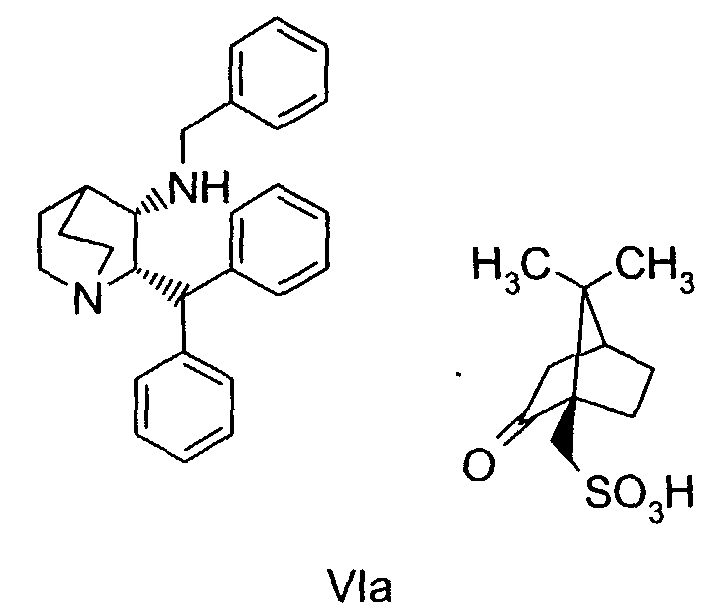
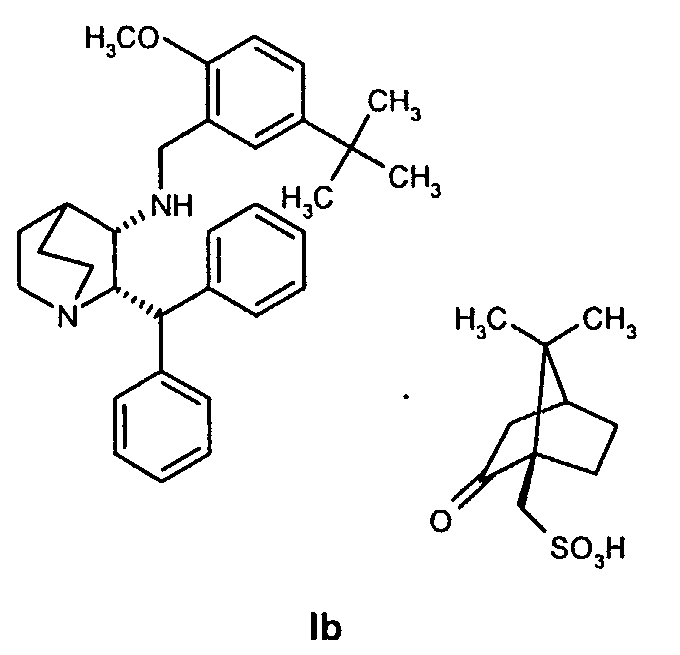
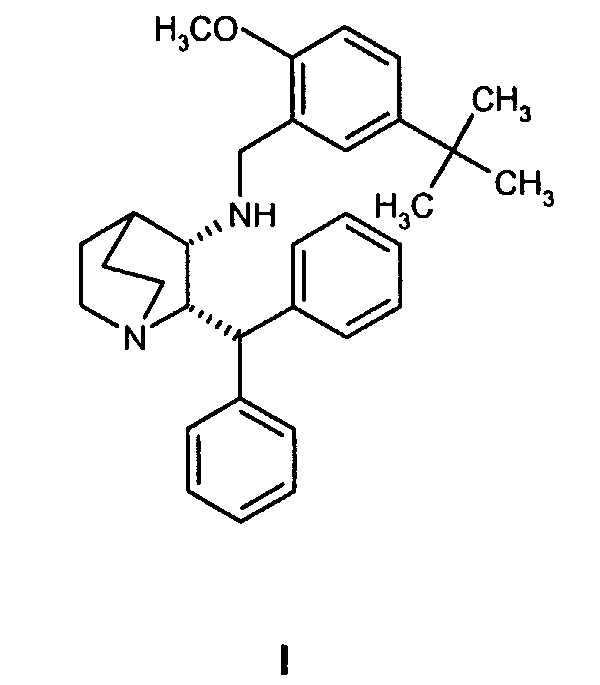
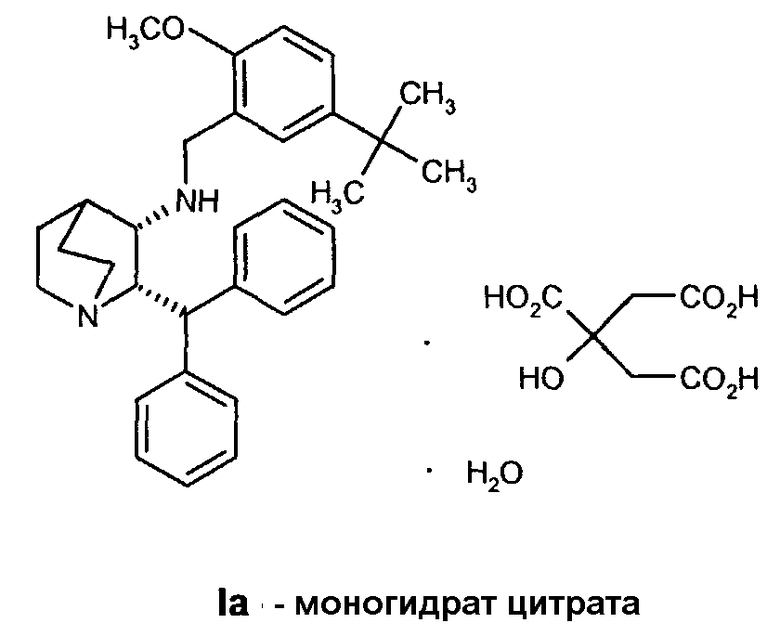
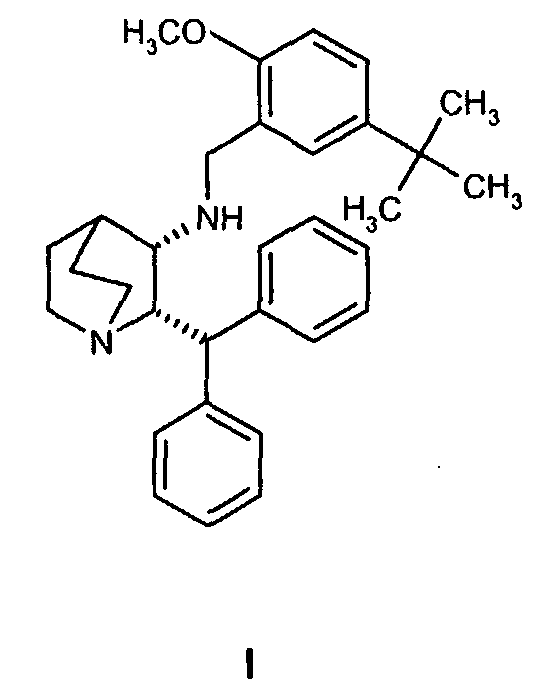
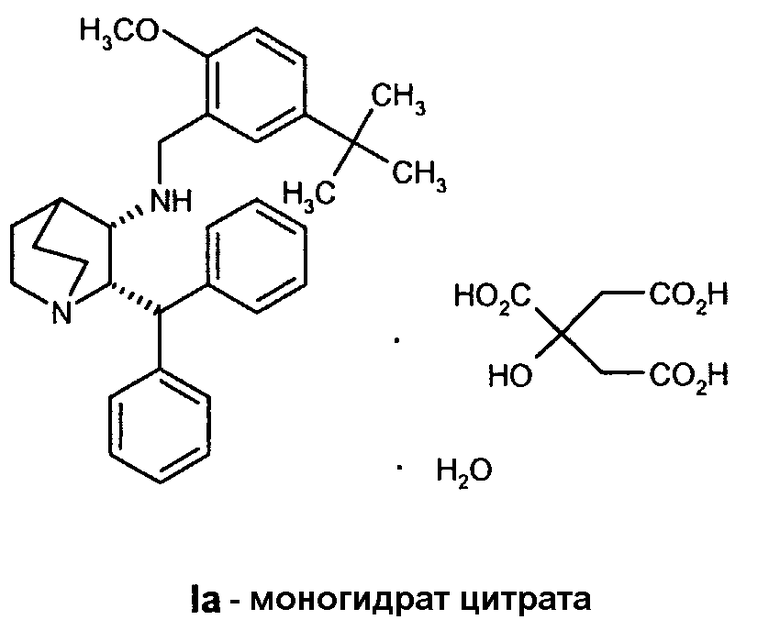
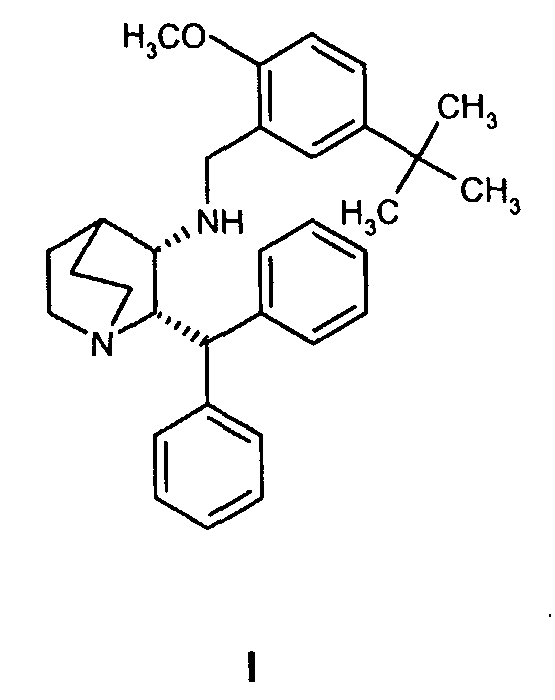
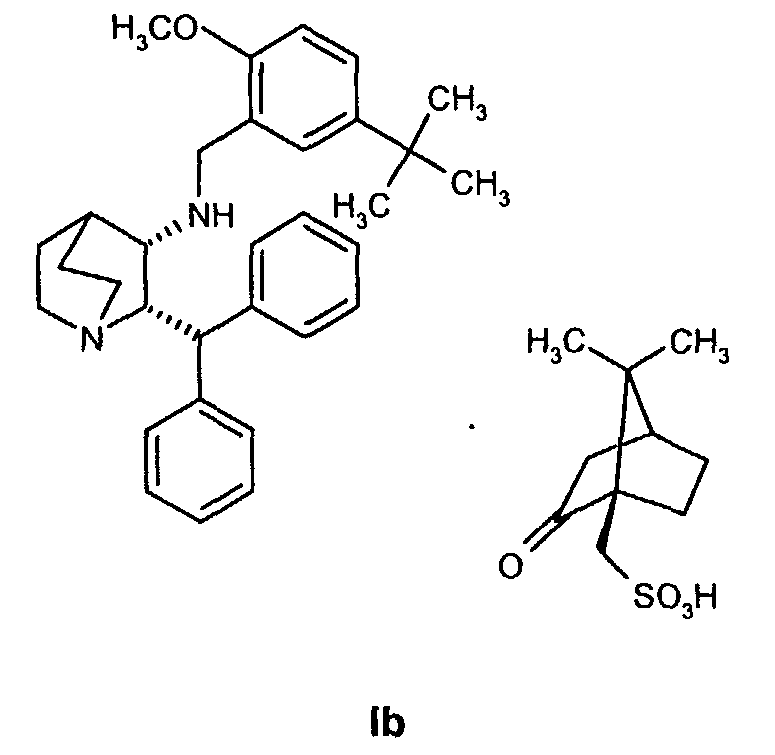
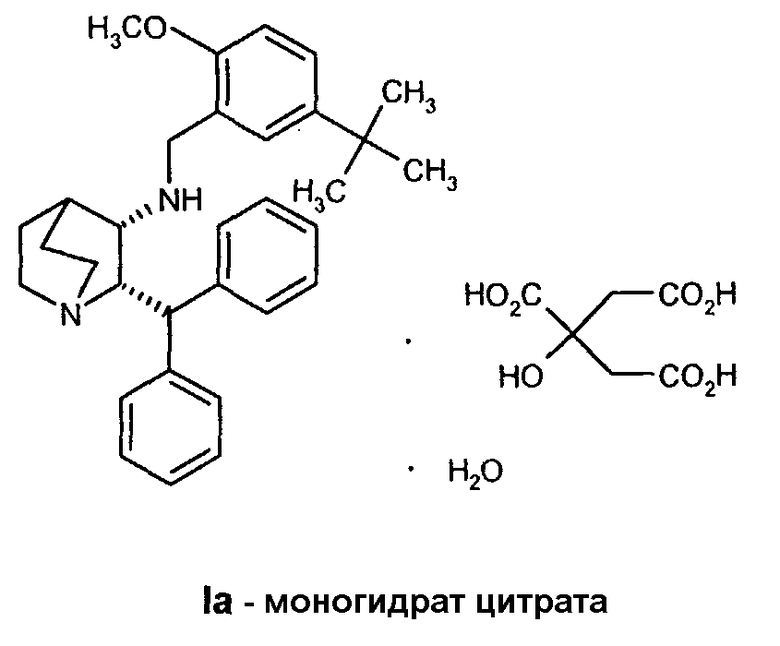
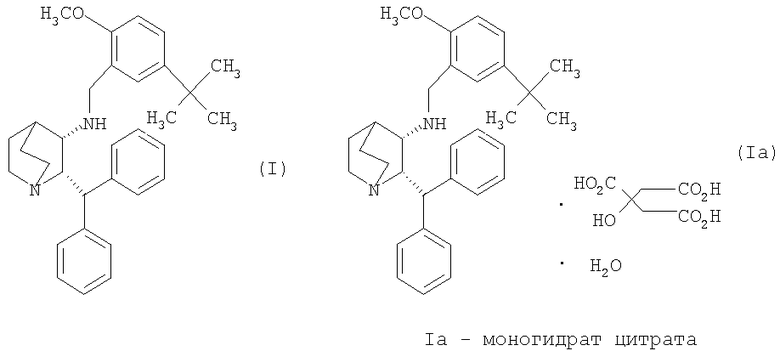
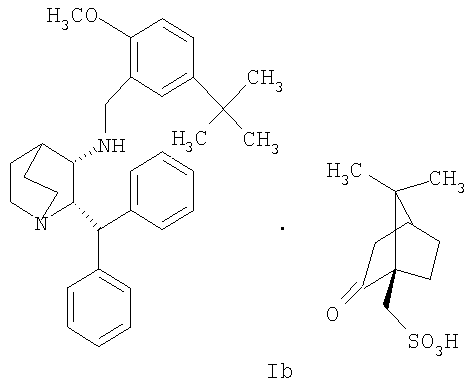
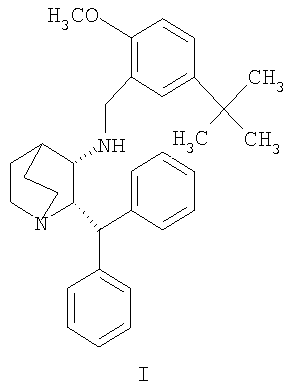
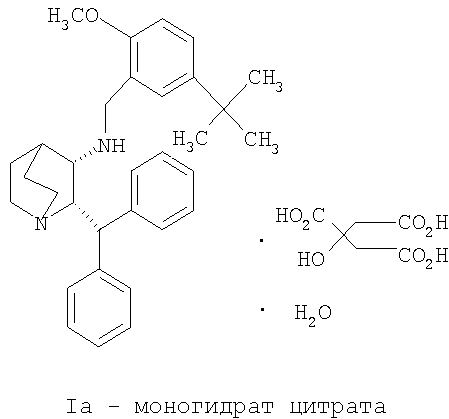
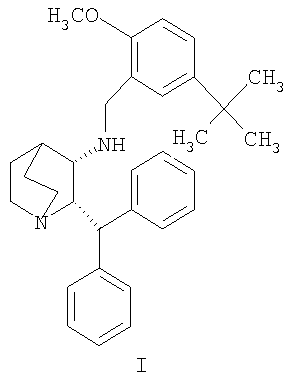
Данное изобретение относится к усовершенствованному способу получения камфорсульфонатной соли (2S,3S)-2-бензгидрил-N-(5-трет-бутил-2-метоксибензил)хинуклидин-3-амина (далее в данном документе «соединение формулы Ib»), свободного основания (2S,3S)-2-бензгидрил-N-(5-трет-бутил-2-метоксибензил)хинуклидин-3-амина (далее в данном документе «соединение формулы I») и его цитратной соли в виде моногидрата формулы Ia.
УРОВЕНЬ ТЕХНИКИ
Соединение формулы I, являющееся антагонистом NK1 рецепторов, эффективно в качестве противорвотного средства для млекопитающих. Соединение формулы I представляет собой объект патентов США №6222038 и №6255320, в которых описано получение соединения формулы I. В патенте США №5393762 также описаны фармацевтические композиции антагонистов рецепторов NK-1 и лечение рвоты такими антагонистами. Композицию соединения формулы I для многократного применения можно вводить парентерально в течение примерно 5 дней в одно и то же место для лечения рвоты или по другим показаниям. В экстренных случаях желательно внутривенное или предпочтительно подкожное введение, так как во время приступов рвоты удерживание и всасывание лекарственной формы при пероральном введении может быть проблематичным. Композиция для многократного применения описана в одновременно рассматриваемой предварительной заявке США № 60/540897, переуступленной компании Pfizer, которая является владельцем данной заявки.
Соединение формулы I также облегчает выход из наркоза у млекопитающих. В одновременно рассматриваемой предварительной заявке США № 60/540697, переуступленной компании Pfizer, которая является владельцем данной заявки, описан метод облегчения выхода из наркоза путем введения NK-1 антагониста до, во время или после введения общего наркоза.
Тексты вышеупомянутых заявок и всех других источников, цитируемых здесь, включены в данную заявку в качестве ссылок в их совокупности.
Некоторые стадии описания способа для синтеза моногидрата цитратной соли соединения формулы I проводят с реагентами, которые нежелательны с точки зрения безопасности, и выходы на некоторых стадиях неудовлетворительны для работы в промышленных масштабах. Настоящее изобретение относится к способу, в котором при проведении химических превращений нет необходимости использовать агрессивные условия для снятия защиты, агрессивные условия для образования оснований Шиффа или агрессивные восстанавливающие агенты, следовательно, повышается качество и увеличивается выход промежуточных соединений и продуктов. Способ в целом совершенствуют за счет применения общеупотребительных растворителей для ключевых стадий химических превращений, уменьшая числа промежуточных продуктов, которые необходимо выделять, что в итоге приводит к повышению общего выхода. Кроме того, эффективность способа достигается возможностью получать исходное соединение (VIa) высокой энантиомерной чистоты, что позволяет избежать последующих стадий очистки. Наконец, условия, используемые на последней стадии производства соединения формулы Ia, оптимизированы для получения нужной морфологической формы моногидрата моноцитратной соли соединения формулы I.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
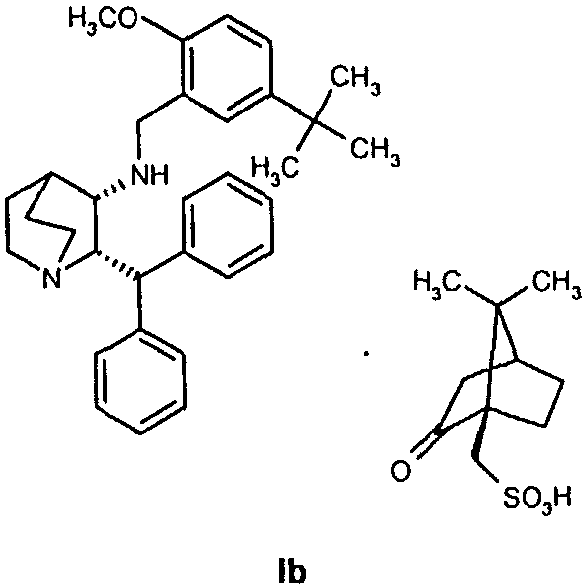
Один аспект изобретения относится к способу получения соединения формулы Ib
включающему
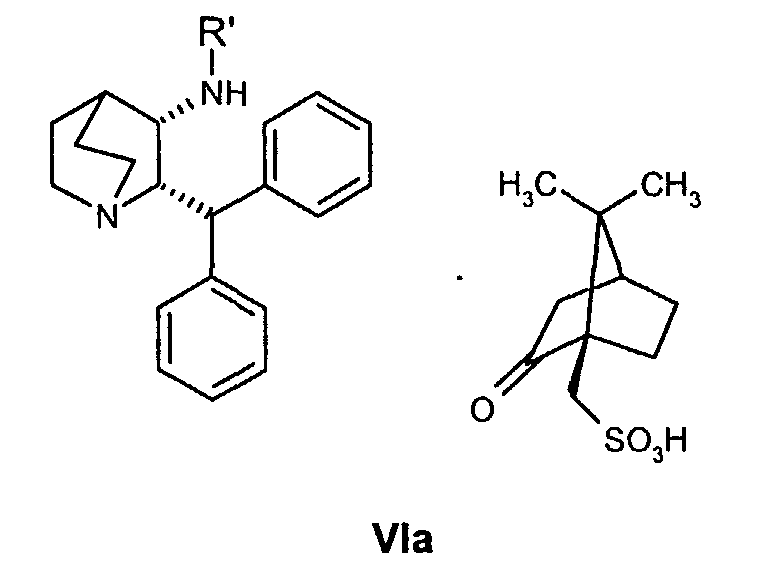
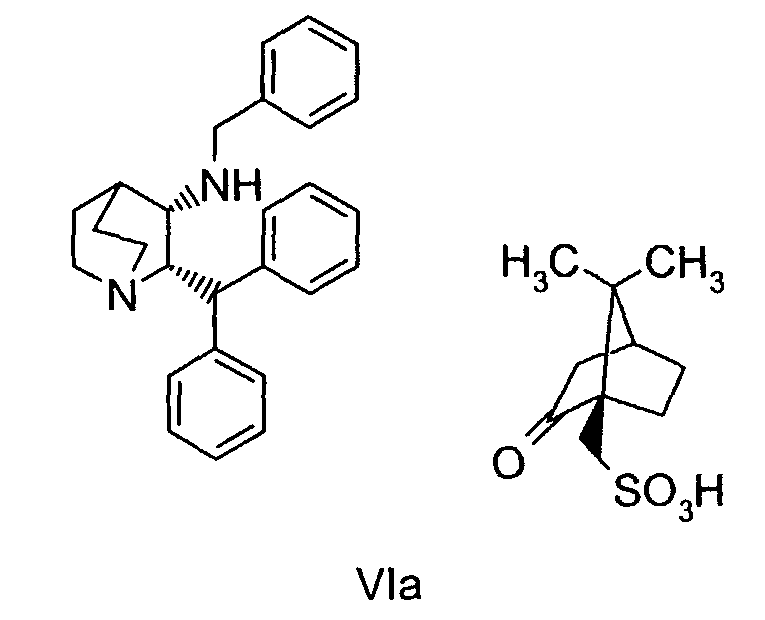
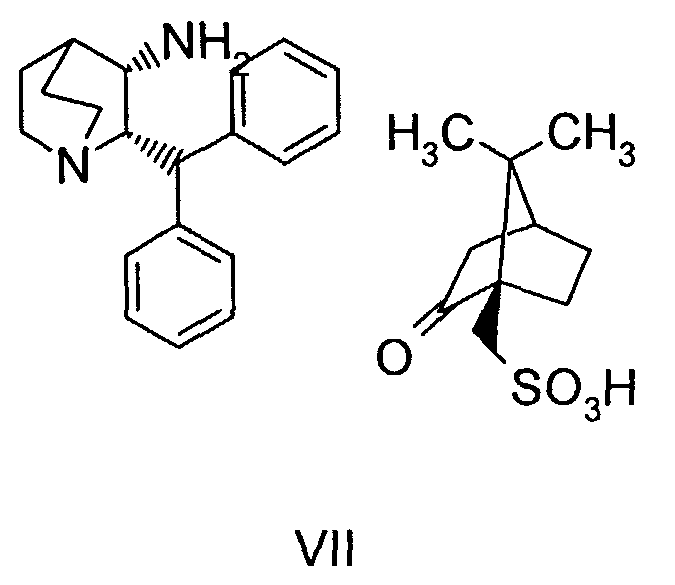
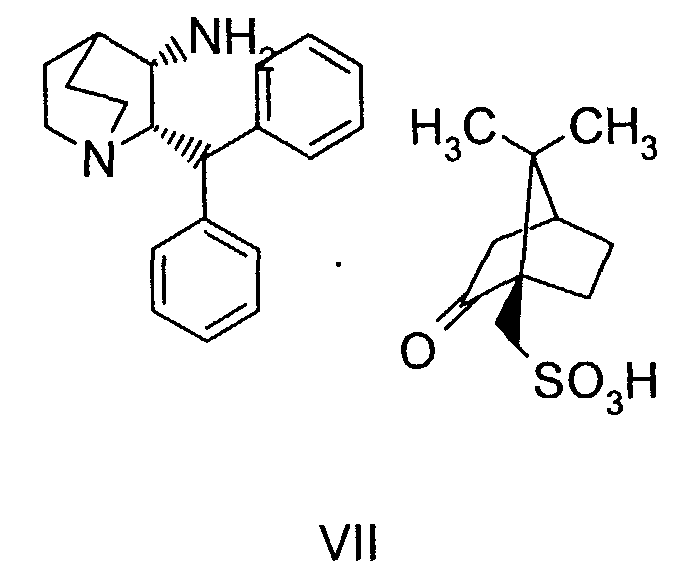
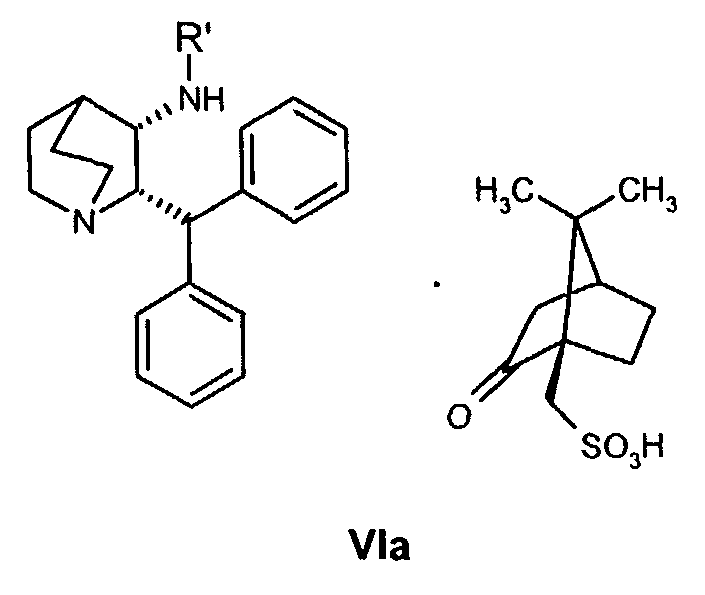
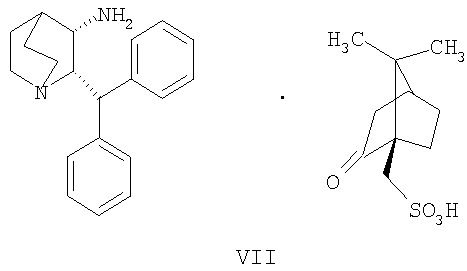
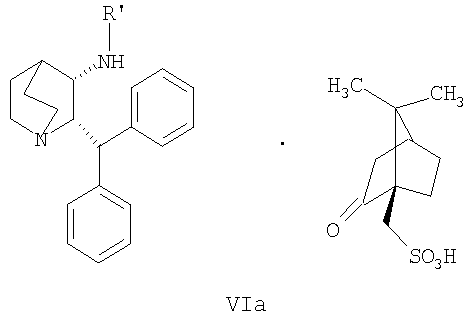
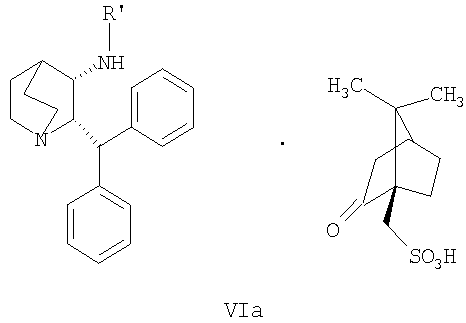
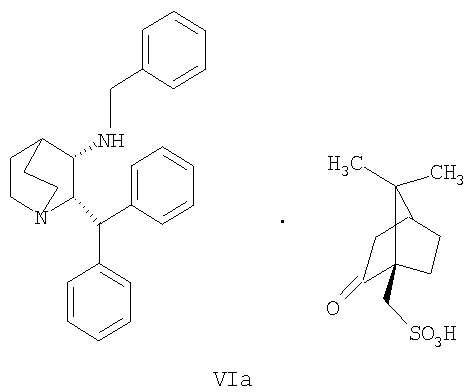
(a) снятие защиты с соединения формулы VIa
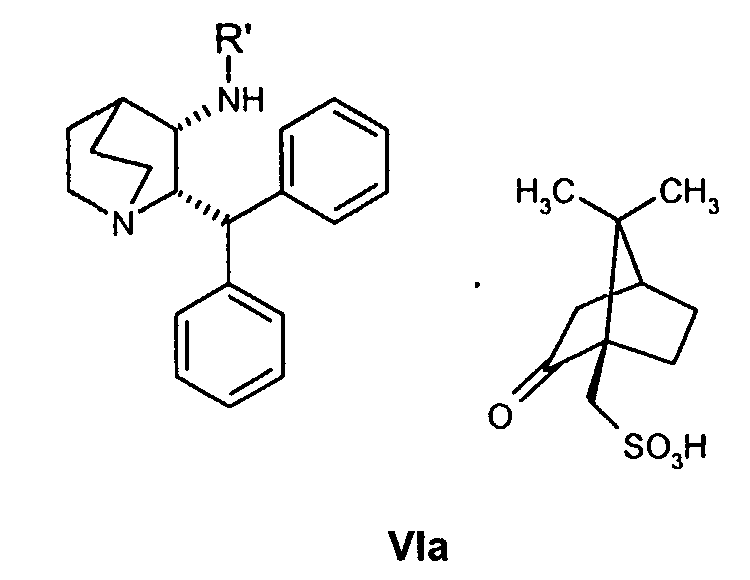
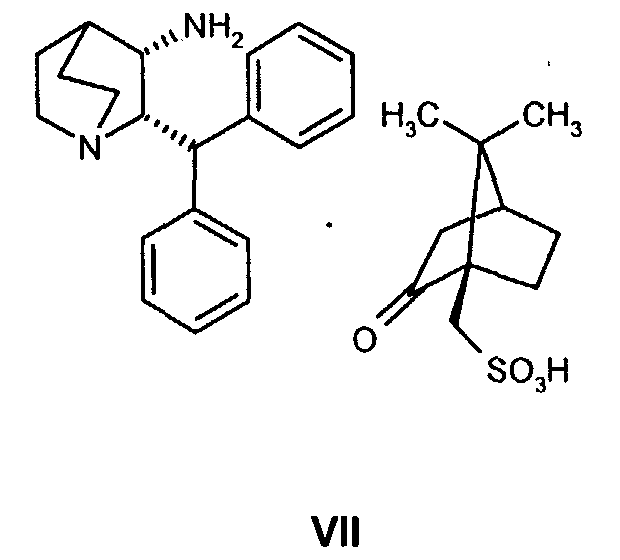
где R' обозначает защитную группу, для получения соединения формулы VII
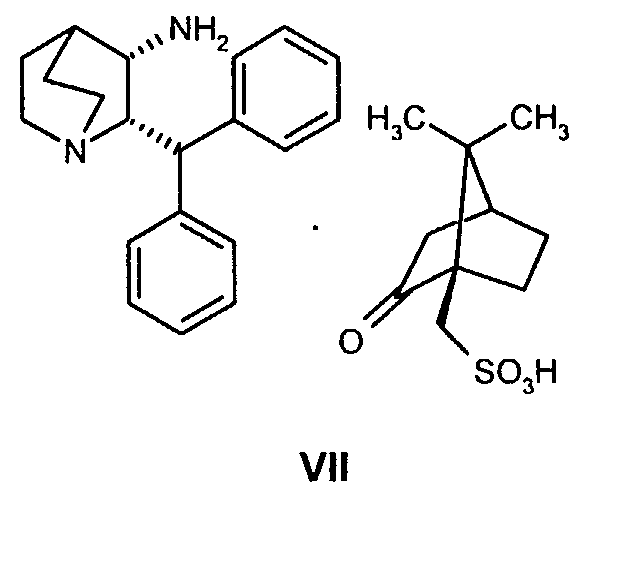
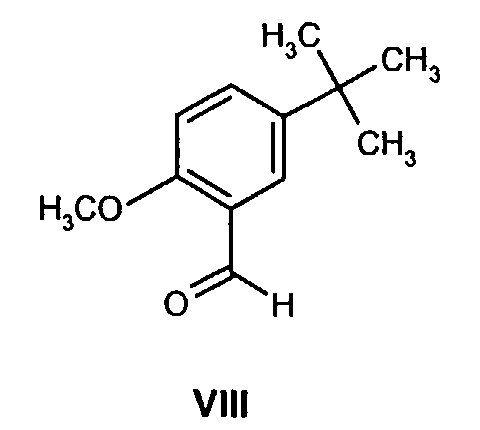
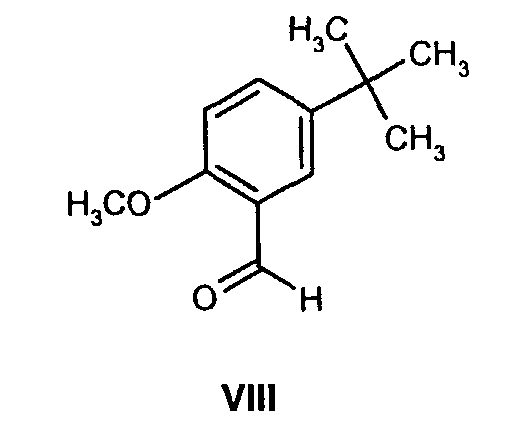
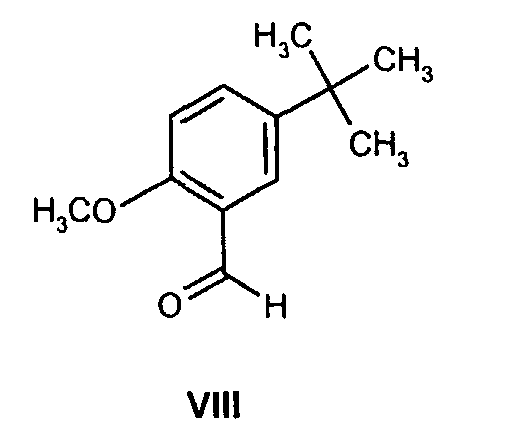
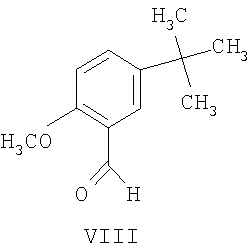
(b) взаимодействие образованного таким образом соединения формулы VII с соединением формулы VIII
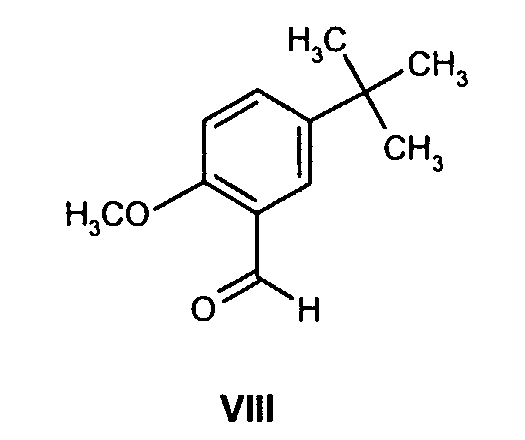
и осуществление восстановительного аминирования для получения соединения формулы Ib
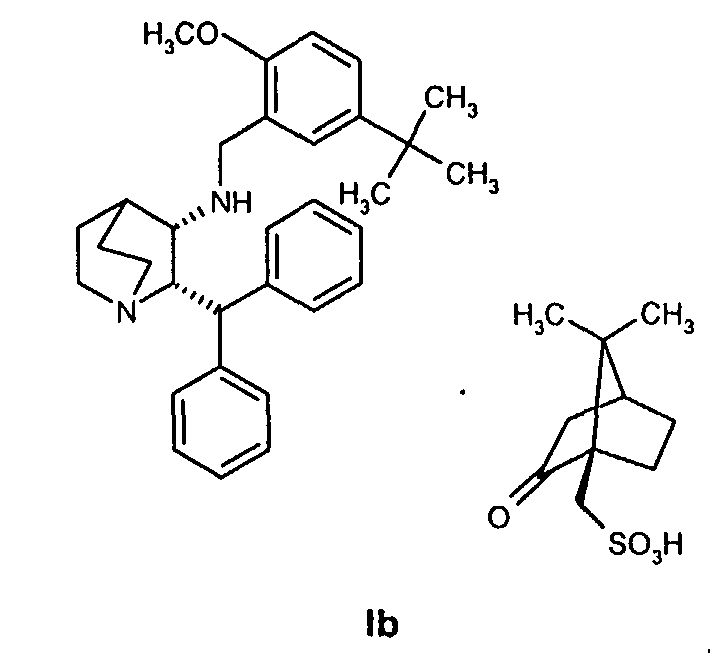
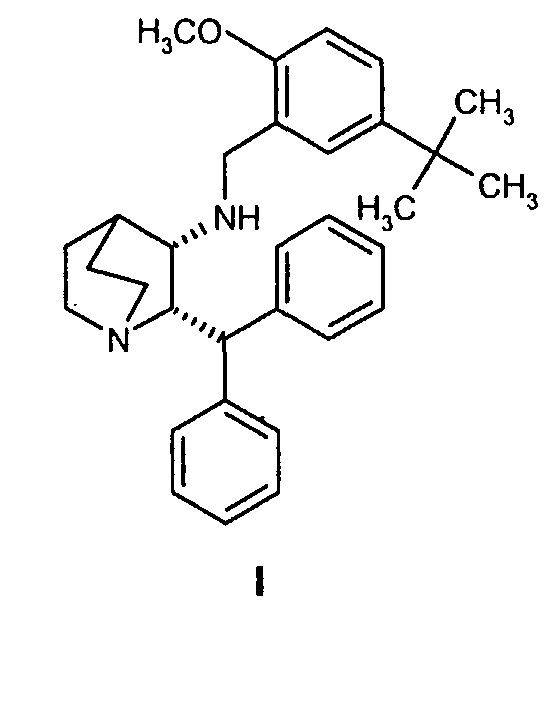
В одном из вариантов осуществления изобретение дополнительно включает удаление камфорсульфонатной соли соединения формулы Ib для получения соединения формулы I
В предпочтительном варианте осуществления изобретения защитная группа представляет собой бензил, 4-метоксибензил, 2,4-диметоксибензил или трифенилметил. Снятие защиты предпочтительно осуществляют каталитическим гидрированием водородом. Катализатор представляет собой предпочтительно палладий на угле, платину на угле, палладий на карбонате кальция или палладий на оксиде алюминия (Al2О3).
В предпочтительном варианте осуществления изобретения восстановительное аминирование осуществляют образованием имина с последующим каталитическим гидрированием. Катализатор гидрирования предпочтительно представляет собой палладий на угле, платину на угле, палладий на карбонате кальция или палладий на оксиде алюминия (Al2О3).
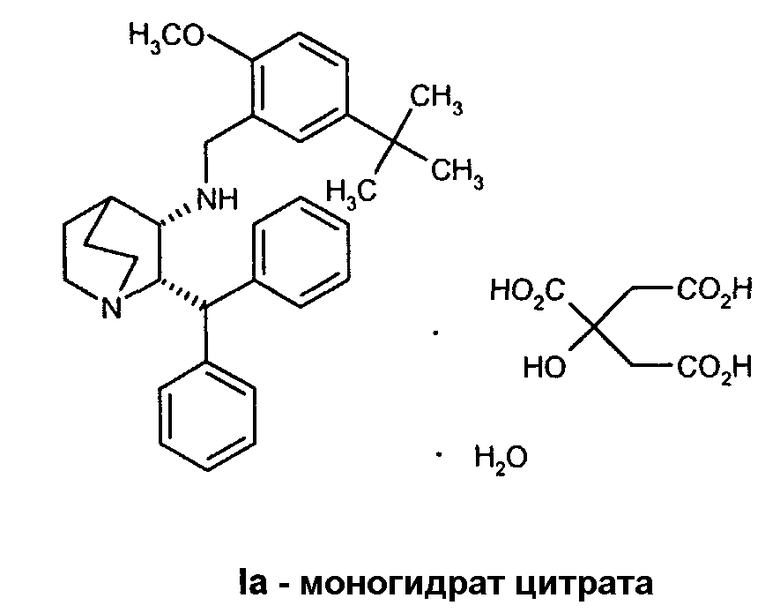
В предпочтительном варианте осуществления изобретения способ дополнительно включает обработку соединения формулы I лимонной кислотой с образованием соединения формулы Ia.
Второй аспект изобретения относится к способу получения соединения формулы I
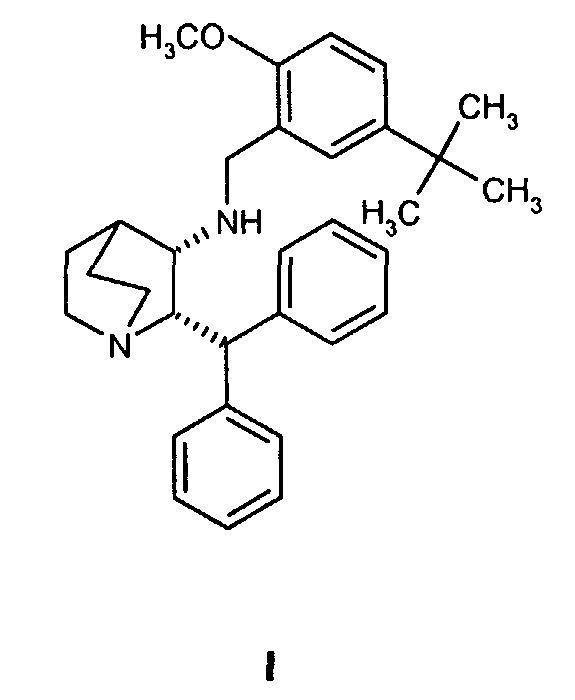
включающему
(а) дебензилирование соединения формулы VIa
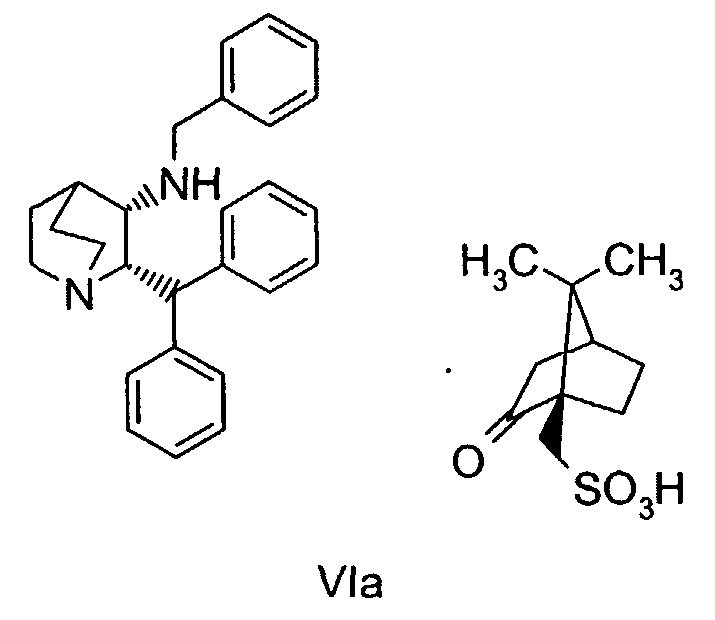
для получения соединения формулы VII
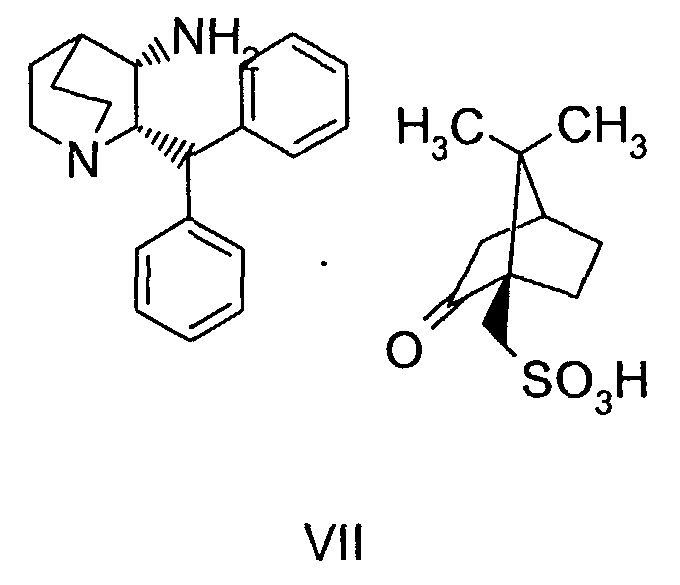
(b) взаимодействие образованного таким образом соединения формулы VII с соединением формулы VIII
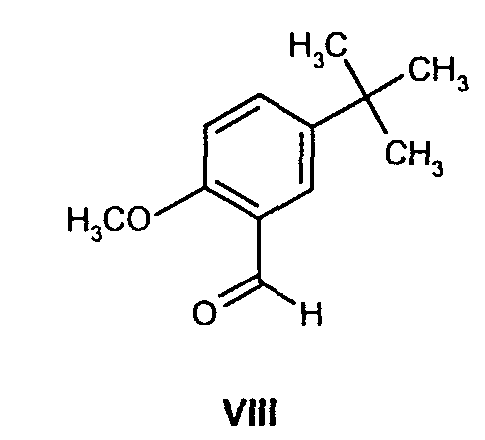
и проведение восстановительного аминирования для получения соединения формулы Ib
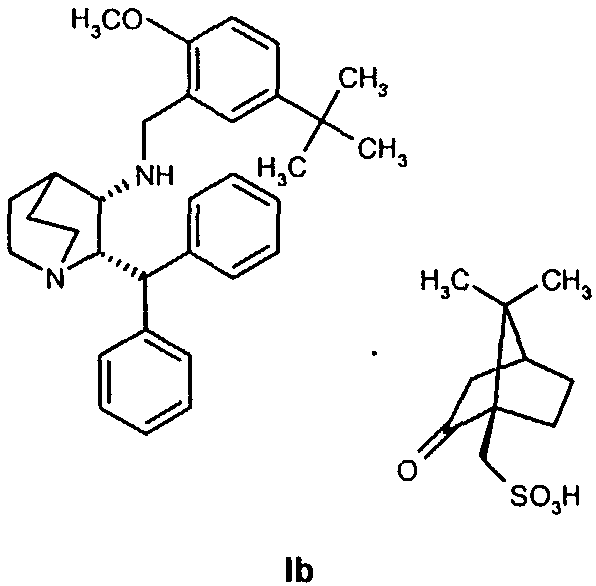
(c) удаление камфорсульфонатной соли соединения Ib для получения соединения формулы I.
В предпочтительном варианте осуществления изобретения дебензилирование проводят каталитическим гидрированием. Катализатор представляет собой предпочтительно палладий на угле, платину на угле, палладий на карбонате кальция или палладий на оксиде алюминия (Al2О3).
В предпочтительном варианте осуществления изобретения способ дополнительно включает восстановительное аминирование на стадии (b), которое проводят каталитическим гидрированием. Катализатор представляет собой предпочтительно палладий на угле, платину на угле, палладий на карбонате кальция или палладий на оксиде алюминия (Al2О3).
В еще одном варианте осуществления изобретения способ дополнительно включает выделение соединения формулы I. Выделение соединения формулы I предпочтительно осуществляется обменом кислотного противоиона или подщелачиванием с последующей селективной кристаллизацией. Кристаллизацию предпочтительно осуществляют в растворителе, выбранном из воды, спиртов, простых эфиров, углеводородов или их смесей. Растворитель предпочтительно представляет собой изопропанол, толуол или воду или их смеси.
В предпочтительном варианте осуществления изобретения подщелачивание осуществляют путем добавления неорганического или органического реагента. Реагент предпочтительно представляет собой гидроксид натрия, карбонат натрия или бикарбонат натрия.
В еще одном варианте осуществления изобретения способ дополнительно включает обработку соединения формулы I лимонной кислотой с образованием соединения формулы Ia.
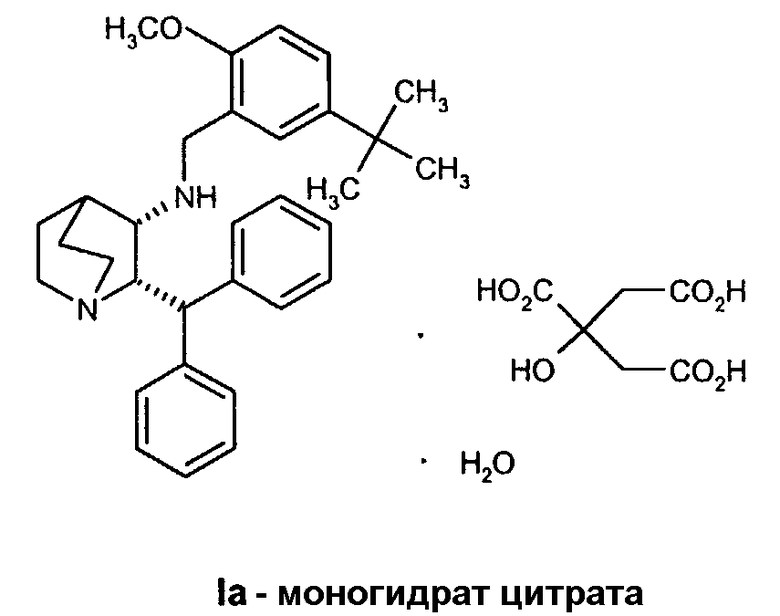
В предпочтительном варианте осуществления изобретения способ дополнительно включает добавление ацетона и воды. Предпочтительно, способ дополнительно включает
a) фильтрование раствора; и
b) добавление фильтрованного растворителя - простого эфира
с получением соединения формулы Ia.
В еще одном варианте осуществления изобретения способ далее включает дополнительную стадию (с) - стадию гранулирования соединения формулы Ia. Простой эфир-растворитель предпочтительно представляет собой трет-бутилметиловый эфир. Предпочтительно, способ дополнительно включает подвод тепла при повышенной температуре во время проведения стадии (b). Кроме того, способ предпочтительно включает добавление затравочных кристаллов соединения формулы Ia во время проведения стадии (b) или после нее. Температура составляет предпочтительно от около 30°С до около 45°С.
В еще одном варианте осуществления изобретения способ дополнительно включает гранулирование соединения формулы I при повышенной температуре. Температура составляет предпочтительно от около 30°С до около 45°С.
Третий аспект изобретения относится к способу получения соединения формулы I
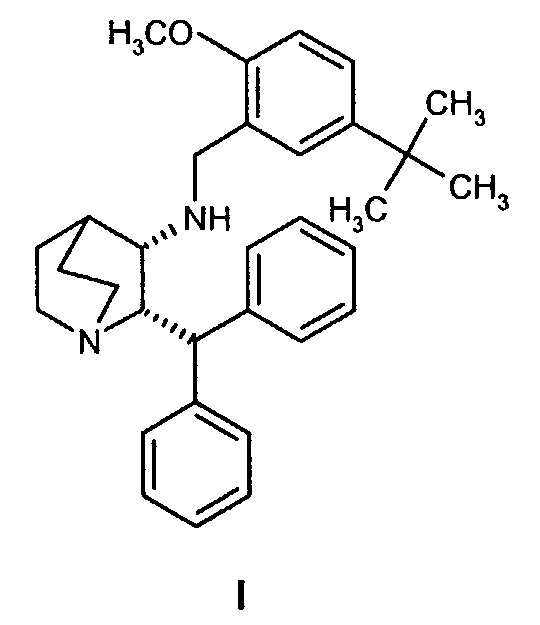
включающему удаление камфорсульфонатной соли соединения Ib
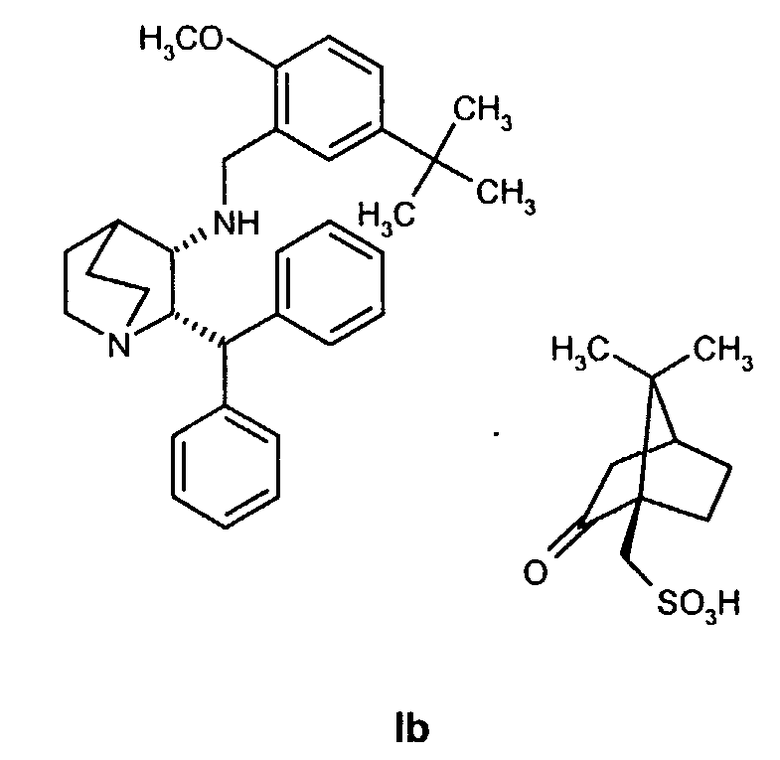
с образованием соединения формулы I.
В предпочтительном варианте осуществления изобретения способ дополнительно включает восстановление соединения IXa
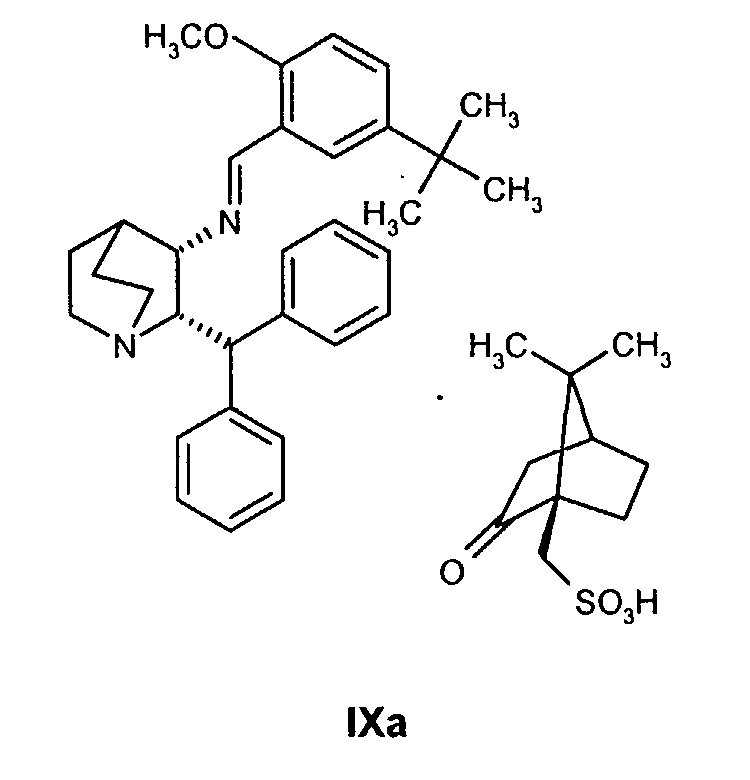
и дает соединение формулы Ib, образовавшееся таким образом.
В предпочтительном варианте осуществления изобретения способ дополнительно включает взаимодействие соединения формулы VII
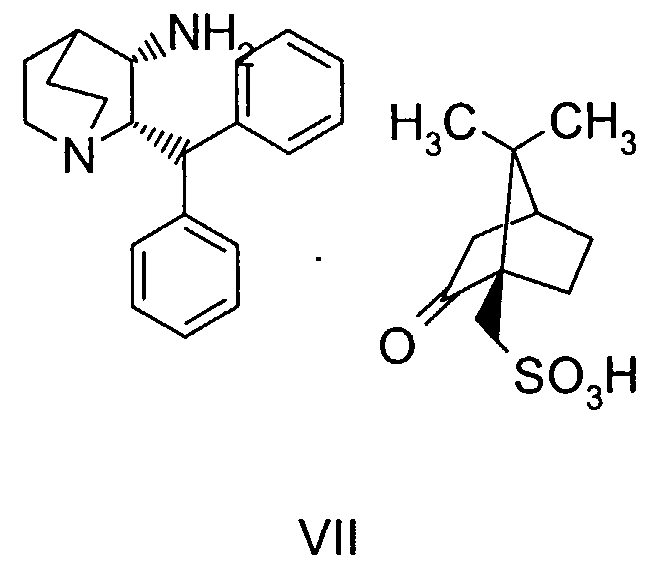
с соединением формулы VIII

и дает соединение формулы IXa, образовавшееся таким образом.
В предпочтительном варианте осуществления изобретения способ дополнительно включает снятие защиты с соединения формулы VIa
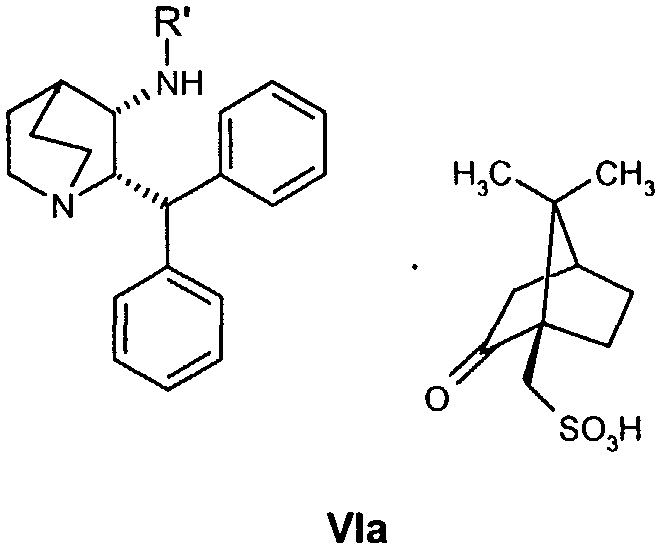
где R′ обозначает защитную группу, выбранную из бензила, 4-метоксибензила, 2,4-диметоксибензила или трифенилметила, и дает соединение формулы VII, образовавшееся таким образом.
В предпочтительном варианте осуществления изобретения способ дополнительно включает обработку соединения формулы I лимонной кислотой с образованием соединения формулы Ia.
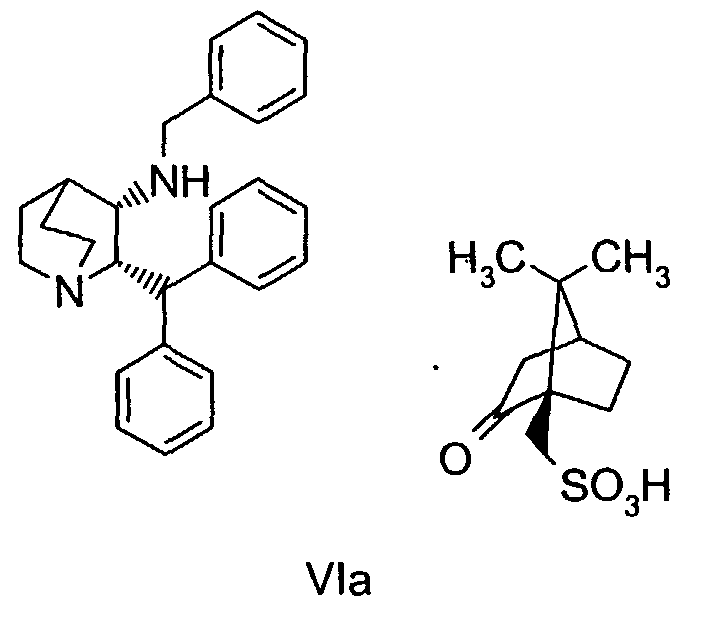
В четвертом аспекте изобретение относится к соединению формулы VIa
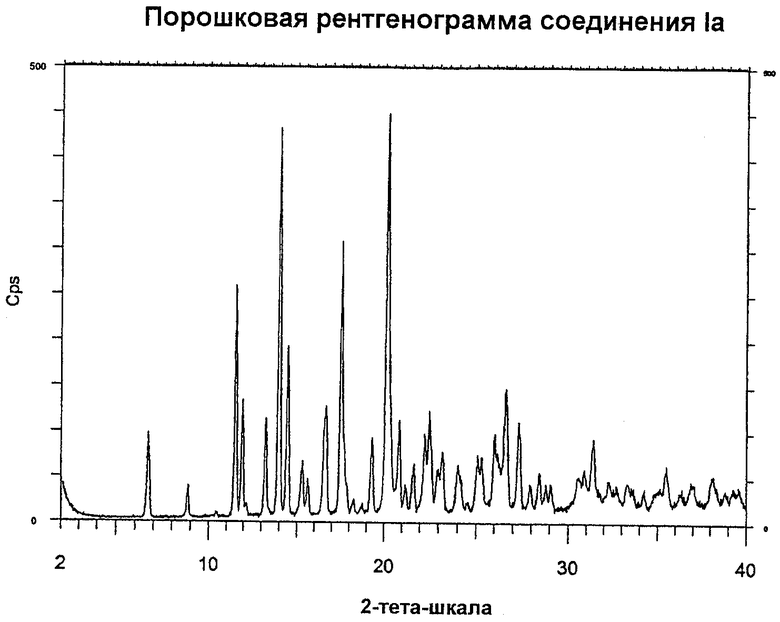
На чертеже показана порошковая рентгенограмма соединения формулы Ia.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Вообще, соединение формулы I можно получить способами, которые включают процессы, известные в химических областях техники, конкретно, с учетом описания данной заявки. Некоторые способы производства соединения формулы I по изобретению иллюстрируются следующими ниже схемами реакций. Некоторые другие способы описаны в экспериментальной части. Некоторые исходные соединения для реакций, описанных в схемах и примерах, получают так, как показано на схемах путей их получения A и B. Все другие исходные соединения можно или приобрести из обычных коммерческих источников, таких как Sigma-Aldrich Corporation, St. Louis, MO, или синтезировать способами, описанными в химической литературе.
Следующие ниже схемы реакций иллюстрируют один возможный путь получения соединения по настоящему изобретению. Специалист в данной области понимает, что для получения вариантов соединения формулы VIa с защитной группой можно использовать также и другие защитные группы, помимо бензила. Другими возможными защитными группами являются, например, 4-метоксибензил, 2,4-диметоксибензил и трифенилметил.
На схемах реакций A и B показаны альтернативные пути получения исходного соединения, соединения формулы VIa, используемого в схемах I и II, в котором защитная группа представляет собой бензил.
ПУТЬ А
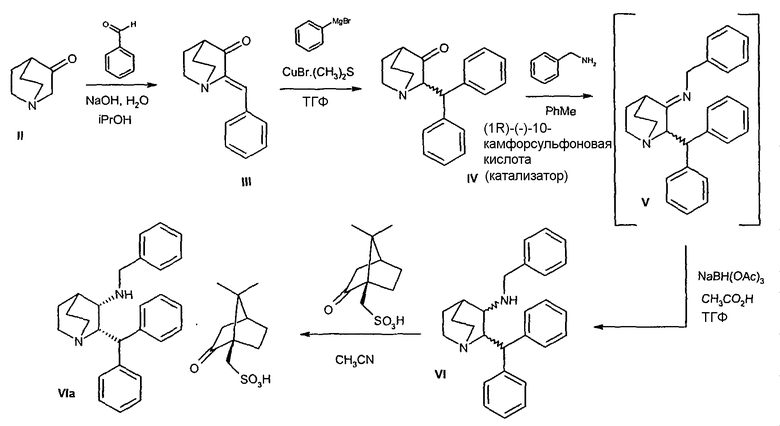
Один из возможных путей синтеза соединения формулы VIa подробно описан выше схемой реакций А. В случае такого пути оптическая чистота соединения формулы VIa достигается селективной кристаллизацией требуемого соединения (цис-2S,3S-формы) в виде (1R)-(-)-10-камфорсульфонатной соли из рацемической смеси соединения VI. Однако при синтезе соединения формулы VIa по указанному пути А получают до 15% нежелательного цис-энантиомера (цис-2R,3R форма, обычно 5-6%) и до 2% нежелательных транс-диастереомеров (транс-2R,3S и транс-2S,3R формы, обычно 1,3%).
Однако при таком конкретном подходе к получению соединения формулы VIa необходимо увеличить как оптическую, так и диастереомерную чистоту, чтобы получить требуемое количество соединения формулы I до его использования в синтезе, показанном на схеме I.
Альтернативный синтез соединения формулы V и в конечном итоге соединения формулы VIa показан на схеме пути B. Путь B является объектом заявки США № 10/679961 (которая не является предварительной), поданной 6 октября 2003. Текст вышеупомянутой заявки настоящим включен полностью в качестве ссылки. Как указано выше, специалист средней квалификации в данной области понимает, что для получения вариантов соединения формулы VIa можно использовать различные защитные группы, помимо бензила. Указанные варианты входят в объем настоящего изобретения.
ПУТЬ В
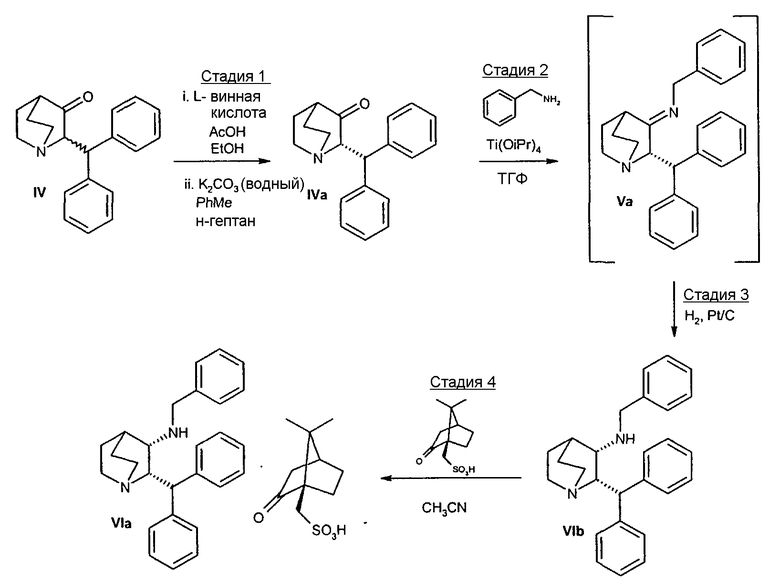
На стадии 1 пути получения B сначала проводят оптическую очистку рацемического кетона формулы IV, причем последний динамически разделяется в виде соли L-винной кислоты, где нежелательный (2R)-энантиомер рацемизуется в условиях реакции и в итоге дает нужный (2S)-энантиомер с выходом более 50%. Оптически чистое соединение формулы IVa (до 98% ee) затем подвергают взаимодействию с бензиламином в условиях образования основания Шиффа и получают промежуточный имин, соединение формулы Va, который каталитически восстанавливают стереоселективно в цис-соединение формулы VIb. Когда соединение VIb превращают в (1R)-(-)-камфорсульфонатную соль, соединение формулы VIa получается с более высокой оптической (энантиомерной) чистотой, при этом устраняется необходимость перекристаллизации соединения VIa для повышения стереохимической чистоты при его синтезе по пути B.
Следующей схемой реакций (схема I) иллюстрируется пример получения соединения формулы Ia из соединения формулы VIa, полученного синтезом по пути A.
СХЕМА I
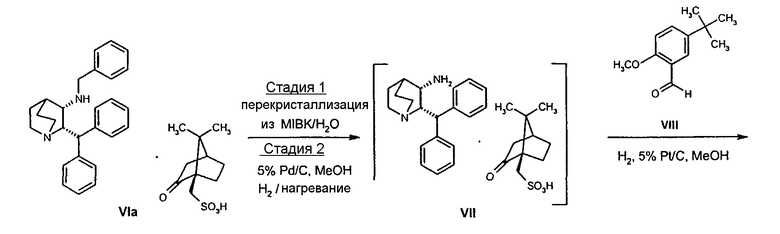
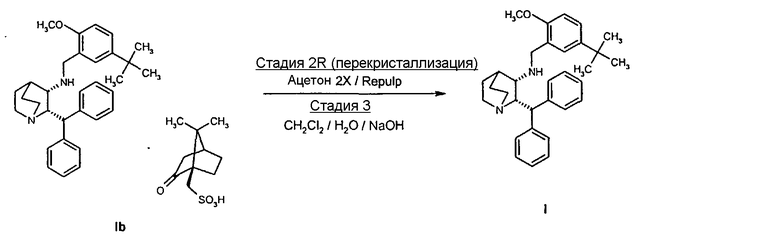
Для использования соединения формулы VIa в производстве соединения формулы I необходима его дополнительная очистка, чтобы свести к минимуму наличие нежелательного (цис-2R-3R)энантиомера. Соответственно на стадии 1 схемы I проводят две последовательные перекристаллизации соединения формулы VIa в 4-метил-2-пентанон ("MIBK").
Соединение VIa (50 г) суспендируют в 10 объемах (500 мл) 10% об./об. раствора вода/MIBK и нагревают до температуры около 88-90°С в течение примерно 2 часов. Раствор охлаждают и продукт отфильтровывают. Влажный продукт, содержащий растворители, повторно суспендируют в 10 объемах водного MIBK и снова нагревают до температуры около 88-90°C в течение 2 часов. Затем раствор охлаждают до температуры примерно 20-25°C и продукт отфильтровывают, промывают 0,5 объемами MIBK и затем сушат и получают соединение VIa высокой энантиомерной чистоты (содержит менее 0,2% ненужного энантиомера) обычно с выходом 83-85%.
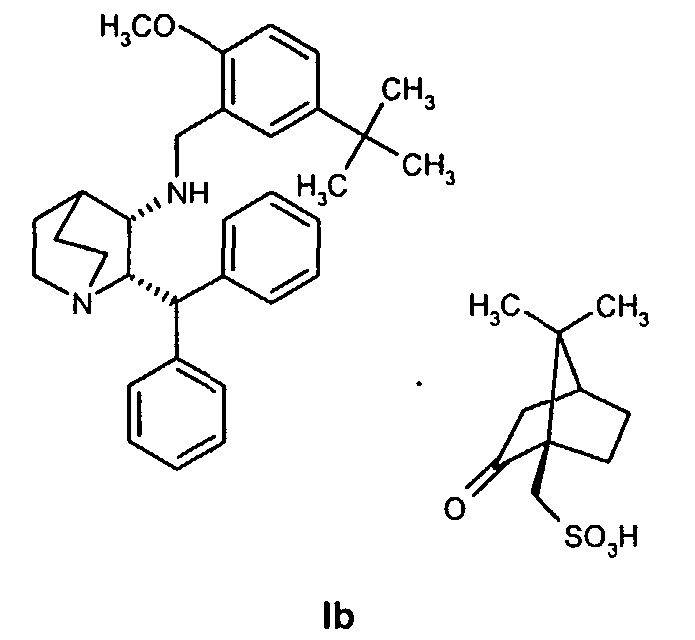
На стадии 2 схемы I снимают защиту соединения формулы VIa в присутствии катализатора, в данном случае соединение формулы VIa дебензилируют, используя подходящий катализатор, такой как палладий на угле, гидроксид палладия на угле, платина на угле, палладий на карбонате кальция или палладий на оксиде алюминия (Al2О3), в растворителе, таком как метанол или изопропанол (пропан-2-ол, "IPA"), и получают соединение формулы VII in situ. В данном конкретном синтезе нет необходимости выделять промежуточное соединение VII. Вместо этого соединение VII подвергают взаимодействию с соединением VIII и водородом в присутствии подходящего катализатора, такого как палладий на угле, гидроксид палладия на угле, платина на угле, палладий на карбонате кальция или палладий на оксиде алюминия (Al2О3) и получают соединение Ib.
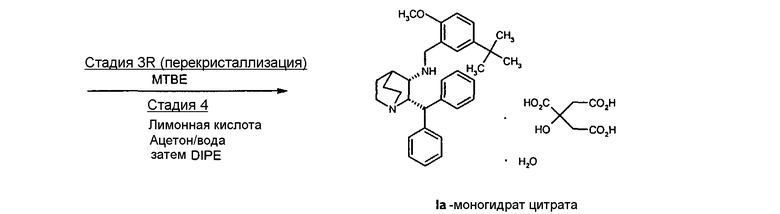
Соединение Ib перекристаллизовывают, используя ацетон в качестве растворителя, и получают очищенное соединение Ib. Затем из соединения Ib получают соединение I подщелачиванием водным гидроксидом натрия и экстракцией дихлорметаном с последующей перекристаллизацией из трет-бутилметилового эфира. Соединение I затем суспендируют в смеси ацетона и воды и добавляют лимонную кислоту, а затем простой диизопропиловый эфир. Образовавшееся твердое вещество отфильтровывают, промывают простым диизопропиловым эфиром, затем сушат и получают соединение Ia.
На следующей ниже схеме II показано альтернативное получение моногидрата цитратной соли соединения формулы I из соединения формулы VIa с выходом, увеличенным от около 68% до около 76%. Кроме того, усовершенствование реакций по схеме II состоит в том, что промежуточные соединения (в квадратных скобках) не требуется выделять при переходе к следующей стадии синтеза.
СХЕМА II
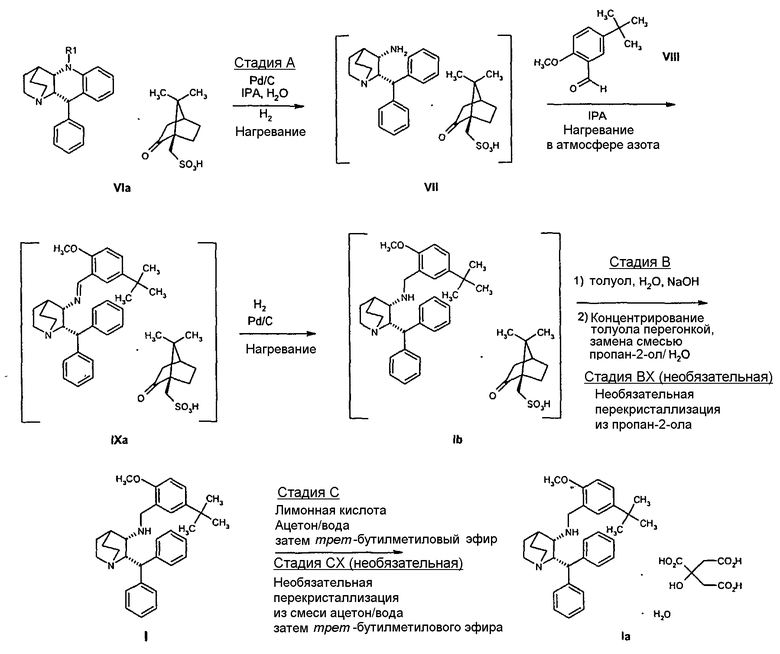
На стадии A схемы II смесь соединения формулы VIa, где R′ обозначает защитную группу (такую как бензил, 4-метоксибензил, 2,4-диметоксибензил или трифенилметил), в спиртовом растворителе, таком как метанол, этанол или н-пропанол, но предпочтительно пропан-2-ол, необязательно в присутствии воды, гидрируют над катализатором (палладий на угле) при повышенной температуре (обычно 75-80°С) и давлении (обычно давление водорода 50 фунт/кв.дюйм). Специалист в данной области понимает, что можно применять и другие катализаторы, такие как палладий на угле, гидроксид палладия на угле, платину на угле, палладий на карбонате кальция или палладий на оксиде алюминия (Al2О3).
Как только образование промежуточного соединения VII завершается (примерно 1 час) соединение формулы VIII, обычно в виде раствора в соответствующем спиртовом растворителе, таком как метанол, этанол и т.п. (предпочтительно в пропан-2-оле (изопропанол, "IPA")), добавляют к реакционной смеси без выделения соединения формулы VII, и смесь перемешивают необязательно при повышенной температуре от около 30 до около 120°С в атмосфере азота. Как только образуется промежуточное соединение IXa, азот вытесняют водородом. Реакционную смесь затем перемешивают необязательно при повышенной температуре (примерно 30-120°С) и повышенном давлении (обычно 50 фунт/кв.дюйм) до тех пор, пока не завершится образование соединения Ib (обычно 18 часов). Затем реакционную смесь охлаждают (примерно до 20-25°C) и газообразный водород выпускают. Катализатор, палладий на угле, отфильтровывают и полученный раствор соединения Ib вводят непосредственно на стадию B.
На стадии B схемы II раствор соединения Ib, обычно в смеси пропан-2-ола и воды, концентрируют дистилляцией с последующим добавлением толуола. Затем смесь опять концентрируют дистилляцией, добавляя дополнительно толуол и воду по мере необходимости во время дистилляции до тех пор, пока достаточное количество изопропанола не будет удалено из смеси и получен соответствующий объем раствора (обычно 2-4 объема на кг соединения Ib). Воду и толуол добавляют по мере необходимости (обычно примерно 3,5 объемов воды и примерно 5 объемов толуола). Специалист в данной области понимает, что, кроме толуола, можно использовать и другие растворители, такие как метиленхлорид, этилацетат, изопропилацетат или трет-бутилметиловый эфир. Доводят рН до соответствующей величины (примерно 11,5-13,5), добавляя при перемешивании водный гидроксид натрия и, если необходимо, водную соляную кислоту.
Как только получают соответствующий рН, водную фазу отделяют. Органическую фазу, содержащую продукт, затем концентрируют дистилляцией. Добавляют смесь пропан-2-ола и воды и смесь опять концентрируют дистилляцией. Добавление воды и пропан-2-ола и последующее концентрирование дистилляцией повторяют по мере необходимости до тех пор, пока из смеси не удаляется достаточное количество толуола (обычно остается менее 3% мас./мас. толуола по данным ГЖХ анализа) и не получается соответствующий объем раствора (около 4 объемов относительно соединения Ib), что в результате приводит к смеси растворителей в конечной взвеси для гранулирования, в которой обычно содержится более 80% мас./мас. пропан-2-ола, менее 20% мас./мас. воды и менее 3% мас./мас. толуола.
Как только удаляют достаточное количество толуола, смесь охлаждают до тех пор, пока не происходит кристаллизация (обычно 70-75°C). Образовавшуюся суспензию охлаждают дополнительно (обычно до 20-25°C), гранулируют в течение некоторого периода времени, прежде чем охлаждать далее до примерно 0-5°С, и перемешивают в течение некоторого периода времени. Твердое вещество отфильтровывают, осадок на фильтре промывают пропан-2-олом, сушат в вакууме при повышенной температуре (обычно 45-55°C) и получают соединение формулы I в виде кристаллического твердого вещества. Специалист в данной области понимает, что можно использовать и другие растворители, кроме пропан-2-ола, такие как метанол, этанол, н-пропанол, ацетонитрил, изопропилацетат, третичный амиловый спирт и 4-метил-2-пентанон.
Как показано на необязательной стадии BX схемы реакций, которая обычно не требуется, соединение I может быть дополнительно очищено. Соединение I суспендируют в пропан-2-оле, смесь кипятят с обратным холодильником и получают раствор. Затем смесь нагревают при повышенной температуре ниже температуры кипения (примерно 70-75°C) в течение примерно 1 часа, в течение которого обычно происходит кристаллизация. Образовавшуюся суспензию выдерживают при такой же температуре в течение примерно 1-2 часов и затем охлаждают (примерно до 20-25°C). После перемешивания при температуре окружающей среды в течение некоторого периода времени (обычно 1-18 часов) твердое вещество отфильтровывают. Осадок на фильтре промывают пропан-2-олом, затем сушат в вакууме при повышенной температуре (примерно 45-55°C) и получают очищенное соединение I в виде твердого кристаллического вещества. Специалист в данной области понимает, что кроме пропан-2-ола можно использовать и другие растворители, такие как метанол, этанол, н-пропанол, ацетонитрил, изопропилацетат, третичный амиловый спирт и 4-метил-2-пентанон.
На стадии C схемы реакций соединение I (1 мольный эквивалент) и безводную лимонную кислоту (обычно 1,1 мольного эквивалента) объединяют в смеси ацетона (обычно около 8-10 объемов) и воды (обычно около 0,4 объема) и фильтруют полученный раствор. Затем опять добавляют ацетон (обычно около 2 объемов, чтобы промыть аппаратуру для подачи. К фильтрату добавляют фильтрованный растворитель - простой эфир, такой как метил-трет-бутиловый эфир (трет-бутилметиловый эфир, "MTBE") или изопропиловый эфир ("IPE") (обычно около 10 объемов), необязательно при повышенной температуре (30-45°C). Как только происходит кристаллизация, которая может быть необязательно инициирована добавлением нескольких затравочных кристаллов, смесь гранулируют в течение некоторого периода времени (обычно 18 часов), обычно при 20-25°C, но необязательно при повышенной температуре (30-45°C) в течение части указанного периода времени. Твердое вещество отфильтровывают. Осадок на фильтре промывают соответствующим фильтрованным растворителем (простым эфиром) и затем сушат при температуре ниже 60°С (при комнатной температуре, если используют изопропиловый эфир) в вакууме необязательно в отсутствие воздуха или при продувании азотом и получают моногидрат цитрата соединения Ia в виде твердого кристаллического вещества. Затем продукт можно необязательно перемалывать или просеивать.
На необязательной стадии CX, можно повысить чистоту соединения Ia, растворяя Ia в смеси ацетона (обычно 7 объемов) и воды (обычно 0,3 объема) при повышенной температуре (примерно 35-50°C). Затем смесь охлаждают (примерно до 20-35°C) и необязательно фильтруют. К полученной смеси затем добавляют фильтрованный растворитель (простой эфир), такой как трет-бутилметиловый эфир или изопропиловый эфир необязательно при повышенной температуре (30-40°C). Как только происходит кристаллизация, которая может быть необязательно инициирована добавлением нескольких затравочных кристаллов, смесь гранулируют в течение некоторого периода времени (обычно 18 часов), обычно при 20-25°C, но необязательно при повышенной температуре (30-45°C) в течение части этого периода времени. Затем твердое вещество отфильтровывают. Осадок на фильтре промывают соответствующим фильтрованным растворителем (простым эфиром) и затем сушат при температуре ниже 60°С (при комнатной температуре, если используют изопропиловый эфир) в вакууме, необязательно в отсутствие воздуха или при продувании азотом и получают моногидрат цитрата соединения Ia в виде твердого кристаллического вещества. Затем продукт можно необязательно перемалывать или просеивать.
Кроме цитрата, можно использовать другие фармацевтически приемлемые соли. Например, такие соли, как малат, малеат, мезилат, лактат и гидрохлориды или их in situ эквиваленты можно получать, добавляя эквимолярное количество соответствующей кислоты к растворам свободного основания соединения I.
ОБЩИЕ МЕТОДИКИ ЭКСПЕРИМЕНТА
Перекристаллизация соединения формулы VIa (Схема I).
В круглодонную колбу объемом 3 л, снабженную механической мешалкой, обратным холодильником, термометром и термостатированной масляной баней, добавляют соединение VIa (200 г), метилизобутилкетон ("MIBK") (1900 мл) и 100 мл воды. Суспензию постепенно нагревают и кипятят при перемешивании в течение примерно 30 мин, выдерживают при 88-90°С в течение примерно 15-30 мин до полного растворения. (Азеотроп MIBK/вода кипит при температуре около 88°C). На указанной стадии смесь является двухфазной с небольшим количеством нерастворенной воды.
Смесь медленно охлаждают до комнатной температуры (около 20-25°С) при перемешивании в течение примерно 2 часов. Продукт начинает выпадать в осадок при температуре около 60°С. Суспензию гранулируют при 20-25°С в течение примерно 2-3 часов, но грануляцию также можно проводить в течение ночи при 20°С; осадок отфильтровывают и промывают примерно 100 мл MIBK.
Влажный слой осадка (примерно 220-230 г) перекристаллизовывают, как описано выше, используя 1700 мл MIBK и 91 мл воды. Суспензию затем опять гранулируют в течение по меньшей мере 3 часов или в течение ночи при 20-25°C. Продукт отфильтровывают и промывают MIBK (100 мл). Очищенное соединение VIa сушат в воздушной лотковой сушилке при 50°С до постоянной массы (в течение примерно 18 часов) и получают твердое белое кристаллическое очищенное вещество, соединение VIa. Выход 85%. Хиральная чистота: (2R-цис)энантиомер 0,3-0,5%.
Получение (2S,3S)-2-бензгидрил-N-(5-трет-бутил-2-метоксибензил)хинуклидин-3-амин(1R)-10-камфорсульфоната, соединение формулы Ib, в виде раствора в смеси пропан-2-ол/вода (Стадия A, Схема II)
К смеси (1R)-10-камфорсульфонатной соли (2S,3S)-2-бензгидрил-N-бензилхинуклидин-3-амина (соединения формулы VIa, 18,0 кг, 29,3 моль) и воды (18,0 кг) в пропан-2-оле (57,9 кг) добавляют 5% палладий на угле (с 50% водной влажностью) (2,88 кг) и полученную смесь гидрируют при давлении водорода 50 фунт/кв.дюйм при 75-80°C в течение 4 часов. Затем смесь охлаждают до 15-20°C, и водород вытесняют азотом (5 фунт/кв.дюйм). К полученной смеси затем добавляют раствор 5-трет-бутил-2-метоксибензальдегида (6,47 кг, 33,7 моль) в пропан-2-оле (6,47 кг). Линию подачи промывают пропан-2-олом (4,24 кг), последний добавляют к реакционной смеси, которую затем перемешивают при 75-80°C в течение 2 часов в атмосфере азота. Полученную смесь затем охлаждают до 30-40°C, и азот вытесняют водородом (50 фунт/кв.дюйм). Затем смесь гидрируют при давлении водорода 50 фунт/кв.дюйм при 75-80°C в течение 3,5 часов; после этого реакционную смесь охлаждают до 25-30°С и уменьшают давление водорода до 10 фунт/кв.дюйм в течение 10 часов для удобства. В реакционную смесь затем повторно подают под давлением водород (50 фунт/кв.дюйм) и нагревают до 75-80°C в течение 11,5 часов, затем реакционную смесь опять охлаждают до 25-30°C и давление водорода уменьшают до 10 фунт/кв.дюйм в течение 10 часов для удобства. В реакционную смесь затем повторно подают под давлением водород (50 фунт/кв.дюйм) и нагревают до 75-80°C в течение 3 часов, чтобы общее время реакции при 75-80°C составляло 18 часов. Реакционную смесь охлаждают до 15-20°C и вытесняют водород азотом. Полученную суспензию затем фильтруют для удаления катализатора, и отфильтрованный осадок промывают пропан-2-олом (19,8 кг). Объединенный фильтрат и промывные воды, содержащие раствор (1R)-10-камфорсульфонатной соли (2S,3S)-2-бензгидрил-N-(5-трет-бутил-2-метоксибензил)хинуклидин-3-амина (соединения формулы Ib) в смеси пропан-2-ол/вода, вводят как таковые в следующую стадию, обозначенную как стадия B.
Получение (2S,3S)-2-бензгидрил-N-(5-трет-бутил-2-метоксибензил)хинуклидин-3-амина, соединения формулы I
(стадия B, Схема 2)
Три раствора (1R)-10-камфорсульфонатной соли (2S,3S)-2-бензгидрил-N-(5-трет-бутил-2-метоксибензил)хинуклидин-3-амина (соединения формулы Ib) в смеси пропан-2-ол/вода, каждый получен из (1R)-10-камфорсульфонатной соли (2S,3S)-2-бензгидрил-N-бензилхинуклидин-3-амина (соединение формулы VIa, 18 кг, 29,3 моль) и 5-трет-бутил-2-метоксибензальдегида (6,47 кг, 33,7 моль) на стадии A (схема II) способа, как описано ранее, объединяют и получают раствор, общий объем которого составляет приблизительно 430 л. Раствор затем концентрируют дистилляцией в вакууме до объема 160 л. Затем добавляют толуол (266 кг) и полученную смесь концентрируют дистилляцией при атмосферном давлении до тех пор, пока объем не станет приблизительно 160 л. Затем добавляют воду (216 кг) и толуол (250 кг) и смесь охлаждают до 20-25°C. Доводят рН водной фазы до 12,5-12,9, добавляя водный гидроксид натрия при перемешивании. Затем удаляют водную фазу и концентрируют органическую фазу до объема приблизительно 160 л дистилляцией в вакууме. Добавляют затем воду (30,8 кг) и пропан-2-ол (218 кг) и полученную смесь затем концентрируют до объема приблизительно 160 л дистилляцией при атмосферном давлении. На данном этапе смесь выдерживают при 25-35°C в течение 18 часов для удобства. Затем добавляют воду (33,8 кг) и пропан-2-ол (218 кг) и смесь концентрируют до объема приблизительно 160 л дистилляцией при атмосферном давлении. Добавляют воду (21 кг) и пропан-2-ол (141 кг) и смесь концентрируют до объема приблизительно 160 л дистилляцией при атмосферном давлении. При поддерживании температуры реакционной смеси выше 75°C медленно добавляют пропан-2-ол (97 кг) и полученную смесь охлаждают до 70°Cв течение 1,5 часов, во время охлаждения происходит кристаллизация. Полученную суспензию затем охлаждают до 20-25°C в течение более 5 часов и перемешивают при указанной температуре 11 часов. Твердое вещество затем отфильтровывают и осадок на фильтре два раза промывают пропан-2-олом (17 кг и 34 кг). Полученное твердое вещество затем сушат в вакууме при 50°С, получая указанное в заголовке соединение (35,3 кг) в виде бесцветного твердого вещества.
1H ЯМР (300 МГц, CDCl3, 30°C) δ: 7,33 (2H, шир.д), 7,27-7,10 (8H, м), 7,10-7,01 (1H, м), 6,92 (1H, д), 6,65 (1H, д), 4,51 (1H, д), 3,73-3,52 (5H, м), 3,26-3,09 (2H, м), 2,77 (1H, дд), 2,82-2,73 (2H, м), 2,59 (1H, шир.т), 2,15-2,06 (1H, м), 2,01-1,87 (1H, м), 1,73-1,60 (1H, м), 1,60-1,43 (1H, м), 1,32-1,19 (10H, м). МС низкого разрешения (LRMS) (химическая ионизация при положительном атмосферном давлении): m/z[MH+] 469.
Необязательная очистка (2S,3S)-2-бензгидрил-N-(5-трет-бутил-2-метоксибензил)хинуклидин-3-амина, соединения формулы I
(стадия BX, Схема II).
Суспензию (2S,3S)-2-бензгидрил-N-(5-трет-бутил-2-метоксибензил)хинуклидин-3-амина (соединения формулы I, 70 г) в пропан-2-оле (350 мл) кипятят с обратным холодильником в течение 1 часа и получают раствор. Полученную смесь затем охлаждают до 70-75°C в течение 2 часов, во время охлаждения происходит кристаллизация и образовавшуюся суспензию охлаждают до 20-25°C в течение приблизительно 4 часов. Затем смесь охлаждают до 0-3°C в течение 0,5 часа и твердое вещество отфильтровывают. Осадок на фильтре дважды промывают пропан-2-олом (по 70 мл каждый раз) и полученное твердое вещество сушат в вакууме при 50°С, получая указанное в заголовке соединение (67,7 кг) в виде бесцветного твердого вещества.
Получение моногидрата цитратной соли (2S,3S)-2-бензгидрил-N-(5-трет-бутил-2-метоксибензил)хинуклидин-3-амина, соединения формулы Ia (стадия C, Схема II).
Раствор (2S,3S)-2-бензгидрил-N-(5-трет-бутил-2-метоксибензил)хинуклидин-3-амина (33,95 кг, 72,4 моль) и безводной лимонной кислоты (15,3 кг, 79,7 моль) в смеси ацетона (215 кг) и воды (13,6 кг) нагревают до 38-42°C. Полученную смесь затем переносят в другой реактор через встроенный фильтр. Линию подачи и фильтр промывают ацетоном (54 кг) и отфильтрованные промывные воды добавляют к раствору. Полученную смесь затем охлаждают до 20-25°C и фильтруют, добавляют порциями трет-бутилметиловый эфир (252 кг) в течение приблизительно 35 минут. Полученную суспензию затем гранулируют при 20-25°C приблизительно 20 часов. Твердое вещество затем отфильтровывают на перемешиваемом фильтре-влагоотделителе и осадок на фильтре дважды промывают фильтрованным трет-бутилметиловым эфиром (по 50 кг каждый раз). Полученное твердое вещество затем сушат при 35°С в вакууме при энергичном перемешивании, получая указанное в заголовке соединение (44,4 кг) в виде бесцветного твердого вещества. Затем продукт размалывают.
1H ЯМР (500 МГц, d4-метанол, 30°C) δ: 7,46 (2H, д), 7,45 (2H, д), 7,37 (4H, м), 7,31 (1H, м), 7,29 (1H, м), 7,24 (1H, дд), 6,95 (1H, д), 6,76 (1H, д), 4,75 (1H, дд), 4,71 (1H, д), 3,76 (1H, м), 3,57 (1H, д), 3,55 (3H, с), 3,37 (1H, м), 3,31 (1H, м), 3,26 (1H, м), 3,24 (1H, д), 3,10 (1H, т), 2,83 (2H, д), 2,75 (2H д), 2,51 (1H, м), 2,35 (1H, м), 2,11 (1H, м), 2,06 (1H, м), 1,85 (1H, м), 1,29 (9H, м). 13C ЯМР (125,7 МГц, d4-метанол, 30°C) δ: 179,4, 175,0, 156,8, 144,0, 141,5, 141,4, 131,1, 130,6, 129,4, 128,9, 128,7, 128,3, 128,2, 127,2, 126,4, 111,0, 74,0, 64,7, 56,1, 54,2, 50,4, 48,5, 48,3, 44,9, 43,8, 34,8, 32,9, 25,3, 22,2, 18,1. МС низкого разрешения (LRMS) (ES+): m/z[MH+] 469.
Твердое соединение формулы Ia, полученное вышеописанным способом, имеет нижеследующие характеристики порошкового рентгеноструктурного анализа.
(2-θ)
(2-θ)
(2-θ)
Особенности получения порошковой рентгенограммы
Порошковую рентгенограмму образца получают с помощью порошкового рентгеновского дифрактометра «SIEMENS D5000», снабженного автоматическим устройством для смены образца, тета-тета гониометром, автоматическими щелями расходимости пучка, вторичным монохроматором и сцинтилляционным счетчиком. Образец для анализа приготавливают, упаковывая порошок в полость диаметром 12 мм и глубиной 0,25 мм, вырезанную в держателе образца из кремниевой пластины. Образец вращают во время облучения Cu K-альфа1 рентгеновскими лучами (длина волны = 1,5406 ангстрем) с помощью рентгеновской трубки при 40 кВ/40 мА. Анализы осуществляют гониометром в режиме пошагового сканирования для импульса 5 секунд на шаг сканирования 0,02° в 2-тета диапазоне от 2° до 40°.
Необязательная очистка моногидрата цитратной соли (2S,3S)-2-бензгидрил-N-(5-трет-бутил-2-метоксибензил)хинуклидин-3-амина, соединения формулы Ia (Стадия CX (Схема II).
Смесь (2S,3S)-2-бензгидрил-N-(5-трет-бутил-2-метоксибензил)хинуклидин-3-амина (38,47 кг, 56,7 моль) и фильтрованную воду (11,5 кг) в фильтрованном ацетоне (213 кг) нагревают до 38-42°С, чтобы получить раствор, который затем охлаждают до 33-37°C. К полученному раствору затем добавляют фильтрованный трет-бутилметиловый эфир (201 кг) в течение приблизительно 35 минут, поддерживая температуру 33-37°C. Полученную суспензию затем охлаждают до 20-25°C и гранулируют при указанной температуре в течение приблизительно 19 часов. Твердое вещество затем отфильтровывают на перемешиваемом фильтре-водоотделителе и осадок на фильтре два раза промывают фильтрованным трет-бутилметиловым эфиром (по 58 кг каждый раз). Полученное твердое вещество затем сушат при 35°C в вакууме при перемешивании, получая указанное в заголовке соединение (32,9 кг) в виде бесцветного твердого вещества. Затем продукт размалывают.
Предпочтительные варианты осуществления изобретения
1. Способ получения соединения формулы Ib
включающий
(а) снятие защиты с соединения формулы VIa
где R′ обозначает защитную группу, для получения соединения формулы VII
(b) взаимодействие полученного таким образом соединения формулы VII с соединением формулы VIII
и осуществление восстановительного аминирования для получения соединения формулы Ib
2. Способ по предпочтительному варианту осуществления изобретения 1, дополнительно включающий удаление камфорсульфонатной соли соединения формулы Ib для получения соединения формулы I
3. Способ по предпочтительному варианту осуществления изобретения 2, в котором защитная группа представляет собой бензил, 4-метоксибензил, 2,4-диметоксибензил или трифенилметил.
4. Способ по предпочтительному варианту осуществления изобретения 3, в котором снятие защиты осуществляют каталитическим гидрированием водородом.
5. Способ по предпочтительному варианту осуществления изобретения 4, в котором катализатор представляет собой палладий на угле, платину на угле, палладий на карбонате кальция или палладий на оксиде алюминия (Al2О3).
6. Способ по п.5, в котором восстановительное аминирование осуществляют путем образования имина с последующим каталитическим гидрированием.
7. Способ по предпочтительному варианту осуществления изобретения 6, в котором катализатор гидрирования представляет собой палладий на угле, платину на угле, палладий на карбонате кальция или палладий на оксиде алюминия (Al2О3).
8. Способ по предпочтительному варианту осуществления изобретения 7, дополнительно включающий обработку соединения формулы I лимонной кислотой, с образованием соединения формулы Ia
9. Способ получения соединения формулы I
включающий
(а) дебензилирование соединения формулы VIa
для получения соединения формулы VII
(b) взаимодействие полученного таким образом соединения формулы VII с соединением формулы VIII
и осуществление восстановительного аминирования для получения соединения формулы Ib
 и
и
(c) удаление камфорсульфонатной соли соединения Ib для получения соединения формулы I.
10. Способ по предпочтительному варианту осуществления изобретения 9, в котором дебензилирование проводят каталитическим гидрированием.
11. Способ по предпочтительному варианту осуществления изобретения 10, в котором катализатор представляет собой предпочтительно палладий на угле, платину на угле, палладий на карбонате кальция или палладий на оксиде алюминия (Al2О3).
12. Способ по предпочтительным вариантам осуществления изобретения 9, 10 или 11, дополнительно включающий восстановительное аминирование (стадия b), которое осуществляют путем каталитического гидрирования.
13. Способ по предпочтительному варианту осуществления изобретения 12, в котором катализатор представляет собой предпочтительно палладий на угле, платину на угле, палладий на карбонате кальция или палладий на оксиде алюминия (Al2О3).
14. Способ по предпочтительному варианту осуществления изобретения 13, дополнительно включающий выделение соединения формулы I.
15. Способ по предпочтительному варианту осуществления изобретения 14, в котором выделение соединения формулы I предпочтительно происходит путем обмена кислотного противоиона или подщелачиванием с последующей селективной кристаллизацией.
16. Способ по предпочтительному варианту осуществления изобретения 15, в котором кристаллизацию проводят в растворителе, выбранном из воды, спиртов, простых эфиров, углеводородов или их смесей.
17. Способ по предпочтительному варианту осуществления изобретения 16, в котором растворитель представляет собой изопропанол, толуол или воду или их смеси.
18. Способ по предпочтительному варианту осуществления изобретения 15, в котором подщелачивание осуществляют добавлением неорганического или органического реагента.
19. Способ по предпочтительному варианту осуществления изобретения 18, в котором реагент представляет собой гидроксид натрия, карбонат натрия или бикарбонат натрия.
20. Способ по предпочтительному варианту осуществления изобретения 9, дополнительно включающий обработку соединения формулы I лимонной кислотой с образованием соединения формулы Ia
21. Способ по предпочтительному варианту осуществления изобретения 20, дополнительно включающий добавление ацетона и воды.
22. Способ по предпочтительному варианту осуществления изобретения 21, дополнительно включающий
a) фильтрование раствора; и
b) добавление фильтрованного растворителя - простого эфира
с получением соединения формулы Ia.
23. Способ по предпочтительному варианту осуществления изобретения 22, включающий дополнительную стадию (с) - стадию гранулирования соединения формулы Ia.
24. Способ по предпочтительному варианту осуществления изобретения 22, в котором простой эфир-растворитель представляет собой трет-бутилметиловый эфир.
25. Способ по предпочтительному варианту осуществления изобретения 22, дополнительно включающий подачу тепла при повышенной температуре во время проведения стадии (b).
26. Способ по предпочтительному варианту осуществления изобретения 22, дополнительно включающий добавление затравочных кристаллов соединения формулы Ia во время проведения стадии (b) или после ее проведения.
27. Способ по предпочтительному варианту осуществления изобретения 25, где температура составляет от около 30 до около 45°С.
28. Способ по предпочтительному варианту осуществления изобретения 23, дополнительно включающий гранулирование соединения формулы I при повышенной температуре.
29. Способ по предпочтительному варианту осуществления изобретения 28, где температура составляет от около 30 до около 45°С.
30. Способ получения соединения формулы I
включающий удаление камфорсульфонатной соли соединения Ib
для получения соединения формулы I.
31. Способ по предпочтительному варианту осуществления изобретения 30, дополнительно включающий восстановление соединения IXa
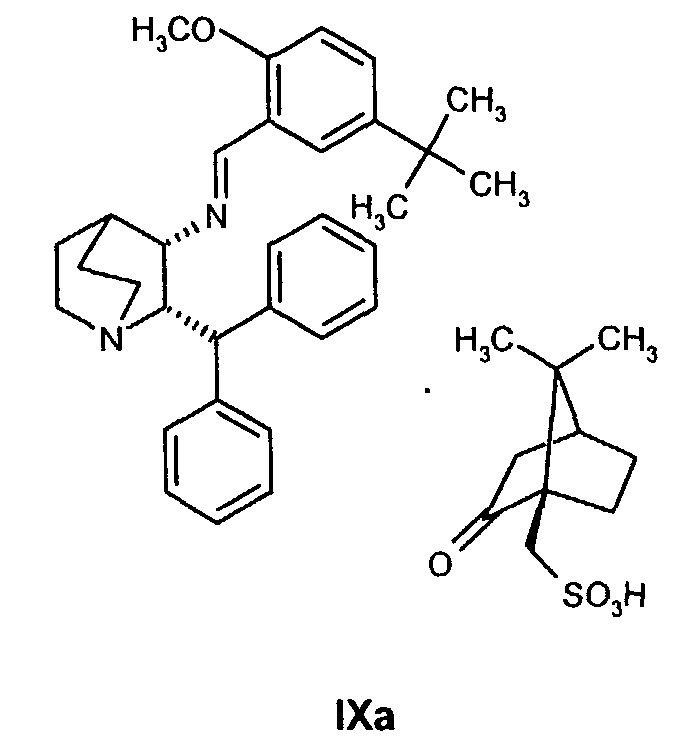
для получения соединения формулы Ib, полученного таким образом.
32. Способ по предпочтительному варианту осуществления изобретения 31, включающий взаимодействие соединения формулы VII
с соединением формулы VIII
для получения соединения формулы IXa, полученного таким образом.
33. Способ по предпочтительному варианту осуществления изобретения 32, дополнительно включающий снятие защиты с соединения формулы VIa
где R′ обозначает защитную группу, выбранную из бензила, 4-метоксибензила, 2,4-диметоксибензила или трифенилметила, для получения соединения формулы VII, образованного таким образом.
34. Способ по предпочтительным вариантам осуществления изобретения 30, 31, 32 и 33, дополнительно включающий обработку соединения формулы I лимонной кислотой с образованием соединения формулы Ia
35. Соединение формулы VIa
Изобретение относится к усовершенствованному процессу получения 1-(2S,3S)-2-бензгидрил-N-(5-трет-бутил-2-метоксибензил)хинуклидин-3-амина (далее именуемого в данной заявке «соединение формулы I») и его фармацевтически приемлемых солей. Конкретно, изобретение относится к усовершенствованному синтезу моногидрата цитратной соли соединения формулы (Ia). 4 н. и 6 з.п. ф-лы, 1 табл., 1 ил.
включающий (с) снятие защиты с соединения формулы VIa
где R' обозначает защитную группу,
для получения соединения формулы VII
(d) взаимодействие соединения формулы VII, полученного таким образом, с соединением формулы VIII
и осуществление восстановительного аминирования для получения соединения формулы Ib
включающий снятие защиты с соединения формулы VIa
где R' обозначает защитную группу, для получения соединения формулы VII
взаимодействие соединения формулы VII, полученного таким образом, с соединением формулы VIII
осуществление восстановительного аминирования для получения соединения формулы Ib
и удаление камфорсульфонатной соли соединения формулы Ib для получения соединения формулы I.
включающий снятие защиты с соединения формулы VIa
где R' обозначает защитную группу, для получения соединения формулы VII
взаимодействие соединения формулы VII, полученного таким образом, с соединением формулы VIII
осуществление восстановительного аминирования для получения соединения формулы Ib
удаление камфорсульфонатной соли соединения формулы Ib для получения соединения формулы I
и обработку соединения формулы I лимонной кислотой для получения соединения формулы Ia.
| РАЗДЕЛЕНИЕ ОПТИЧЕСКИХ ИЗОМЕРОВ 1-АЗАБИЦИКЛО-[2,2,2]ОКТАН-3-АМИН, 2-(ДИФЕНИЛМЕТИЛ)- N-[[2-МЕТОКСИ-5-(1-МЕТИЛЭТИЛ)ФЕНИЛ]МЕТИЛА] | 1996 |
|
RU2136681C1 |
| ПРОИЗВОДНЫЕ ХИНУКЛИДИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2103269C1 |
| US 6255320 B1, 03.07.2001 | |||
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| ПРИСПОСОБЛЕНИЕ ДЛЯ ТОРМОЖЕНИЯ ПОЕЗДА | 1925 |
|
SU3731A1 |
| EA 200101108 A1, 25.04.2002. | |||

