Область техники
Настоящее изобретение относится к новому сополимеру, фармацевтической композиции, содержащей указанный сополимер, модификатору белка, содержащему указанный сополимер, комплексу указанного сополимера и белка, способу профилактики или лечения заболеваний с использованием указанного комплекса, применению указанного комплекса для изготовления лекарственного средства, предназначенного для профилактики или лечения заболеваний, и способам синтеза указанных сополимеров и указанных комплексов.
Уровень техники
Модифицирование белков с использованием дополнительных агентов, таких как полимеры, обычно использовали для целей придания улучшенных фармацевтических свойств, например улучшенных стабильности и удерживания в крови и пониженной антигенности [например, см. F. M. Veronese and J. M. Harris, "Peptide and Protein Pegylation", Advanced Drug Delivery Reviews 54(4), 2002].
При модифицировании белков с использованием полимера одна методика, использовавшаяся в прошлом, включает связывание белка и полимерного модификатора при помощи ковалентной связи (например, см. WO-A-97/23614). В других примерах модифицирования белка с использованием полимера, таких как при модифицировании с использованием полимера лекарственного средства, лекарственное средство модифицировали при помощи нековалентной связи. Один такой пример приводится в японской патентной заявке (Kokai) No. Hei 11-302199, которая описывает то, что привитой сополимер, который включает привитую цепь неионного полимера и основную цепь отрицательно заряженного полимера, образует комплекс включения с веществом, способным нести положительный заряд в физиологических условиях, например, с липосомой или поли-L-лизином, несущими положительный заряд, что приводит к улучшению удерживания в крови. Другая альтернатива предлагается в WO-A-99/02131, которая описывает то, что белок и растворимый в воде полимер могут быть перемешаны в специфических условиях в присутствии органического растворителя с получением микрочастиц с контролируемым высвобождением.
К сожалению, по множеству причин особенно успешным оказалось небольшое число данных полимерных модификаторов белка. Один недавний пример полимерного модификатора белка, который демонстрирует некоторые улучшенные свойства, включает сополимер на основе производного полималеиновой кислоты, содержащий в качестве образующего его элементарного звена производное полиоксиалкиленалкилового эфира [см., например, японские патенты с номерами 3035675 и 3271265]. Данные полимерные модификаторы определенно демонстрируют улучшенное связывание с целевыми белками. Однако, для таких сополимеров все еще существуют значительные проблемы. Как было обнаружено, их звено малеинового ангидрида связывается с белками неспецифически. Результатом этого становится получение комплексов между сополимером и белком, демонстрирующих неоднородные свойства в зависимости от условий. В частности, было обнаружено, что данные полимерные модификаторы имеют тенденцию к легкому образованию с белками разупорядоченных сшитых структур, таким образом формируя объемные комплексы, что вызывает избыточное модифицирование структуры белка, а следовательно, и уменьшение желательной активности белка. Кроме того, комплексы между данными полимерными модификаторами белка и белком, как было обнаружено, демонстрируют неудовлетворительное удерживание в крови после приема.
Поэтому существует потребность в полимерном модификаторе, способном привести к получению комплекса, обладающего однородными свойствами, в особенности характеризующегося пониженным образованием неупорядоченных сшитых структур с белком, лучшим сохранением активности белка и превосходным удерживанием белка в крови после приема указанного комплекса.
Краткое изложение изобретения
Поэтому целью настоящего изобретения является получение полимерного модификатора, способного привести к получению комплекса, обладающего однородными свойствами, в особенности характеризующегося пониженным образованием неупорядоченных сшитых структур с белком, лучшим сохранением активности белка и превосходным удерживанием белка в крови после приема указанного комплекса.
Изобретатели настоящего изобретения провели обширные исследования различных модификаторов белка и в результате достигли успеха в получении новых сополимеров, которые способны формировать комплексы с белками, которые обладают однородными свойствами, и значительно улучшать удерживание в крови белков указанных комплексов, что, таким образом, приводит к совершению настоящего изобретения.
Другие цели и преимущества настоящего изобретения станут очевидными по мере прочтения описания.
Таким образом, настоящее изобретение предлагает сополимер или его фармакологически приемлемую соль, которые содержат в качестве образующих их элементарных звеньев
(а) одно или несколько структурных элементарных звеньев, которые могут быть идентичными или отличными друг от друга и которые описываются приведенной ниже формулой (I):
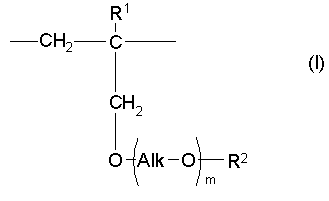
где:
m представляет собой целое число в диапазоне от 3 до 100,
Alk представляет собой алкиленовую группу, содержащую от 1 до 6 атомов углерода, и
R1 и R2 являются идентичными или различными, и каждый из них представляет собой атом водорода или алкильную группу, содержащую от 1 до 6 атомов углерода, которая необязательно может быть замещена, по меньшей мере, одним заместителем, выбираемым из группы, состоящей из гидрокси-групп, атомов галогена и арильных групп, содержащих от 6 до 14 атомов углерода, которые необязательно могут быть замещены заместителями в количестве от 1 до 5, выбираемыми из определенных ниже заместителей А, и
(b) одно или несколько структурных элементарных звеньев, которые могут быть идентичными или отличными друг от друга и которые описываются формулой (II):
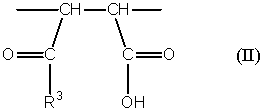
где:
R3 представляет собой
гидроксильную группу,
алкокси-группу, содержащую от 1 до 6 атомов углерода, которая необязательно может быть замещена, по меньшей мере, одним заместителем, выбираемым из группы, состоящей из гидрокси-групп, атомов галогена и арильных групп, содержащих от 6 до 14 атомов углерода, которые необязательно могут быть замещены заместителями в количестве от 1 до 5, выбираемыми из определенных ниже заместителей А,
арилокси-группу, содержащую от 6 до 14 атомов углерода, которая необязательно может быть замещена заместителями в количестве от 1 до 5, выбираемыми из определенных ниже заместителей A, или
группу, описанную формулой -NR4R5, где R4 и R5 являются идентичными или отличными друг от друга, и каждый из них представляет собой атом водорода или алкильную группу, содержащую от 1 до 6 атомов углерода, которая необязательно может быть замещена, по меньшей мере, одним заместителем, выбираемым из группы, состоящей из гидрокси-групп, атомов галогена и арильных групп, содержащих от 6 до 14 атомов углерода, которые необязательно могут быть замещены заместителями в количестве от 1 до 5, выбираемыми из определенных ниже заместителей А;
заместители А выбирают из алкильных групп, содержащих от 1 до 6 атомов углерода, алкокси-групп, содержащих от 1 до 6 атомов углерода, атомов галогена, гидрокси-групп, нитро-групп и карбокси-групп.
Настоящее изобретение дополнительно предлагает сополимер или его фармакологически приемлемую соль, получаемые в результате проведения для одного или нескольких звеньев ангидрида карбоновой кислоты, описанных формулой (III), в сополимере, который содержит в качестве образующих его элементарных звеньев
(а) одно или несколько структурных элементарных звеньев, которые могут быть идентичными или отличными друг от друга и которые описываются приведенной ниже формулой (I):
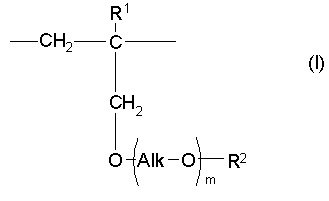
где:
m представляет собой целое число в диапазоне от 3 до 100,
Alk представляет собой алкиленовую группу, содержащую от 1 до 6 атомов углерода, и
R1 и R2 являются идентичными или различными, и каждый из них представляет собой атом водорода или алкильную группу, содержащую от 1 до 6 атомов углерода, которая необязательно может быть замещена, по меньшей мере, одним заместителем, выбираемым из группы, состоящей из гидрокси-групп, атомов галогена и арильных групп, содержащих от 6 до 14 атомов углерода, которые необязательно могут быть замещены заместителями в количестве от 1 до 5, выбираемыми из определенных ниже заместителей А, и
(b) указанные одно или несколько звеньев ангидрида карбоновой кислоты, описанных формулой (III):
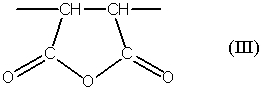
одной или нескольких реакций, выбираемых из группы, состоящей из (i) гидролиза, (ii) аммонолиза, (iii) аминолиза и (iv) алкоголиза;
заместители А выбирают из алкильных групп, содержащих от 1 до 6 атомов углерода, алкокси-групп, содержащих от 1 до 6 атомов углерода, атомов галогена, гидрокси-групп, нитро-групп и карбокси-групп.
Настоящее изобретение также предлагает фармацевтическую композицию, содержащую, по меньшей мере, один сополимер или его фармакологически приемлемую соль настоящего изобретения, описанные выше, в частности, такую композицию, которая также содержит, по меньшей мере, один белок или его аналог или вариант.
Настоящее изобретение также предлагает модификатор, способный модифицировать белок или его аналог или вариант, при этом указанный модификатор содержит сополимер или его фармакологически приемлемую соль настоящего изобретения, описанные выше.
Настоящее изобретение также предлагает комплекс, содержащий, по меньшей мере, один белок или его аналог или вариант, который связывают, по меньшей мере, с одним сополимером или его фармакологически приемлемой солью настоящего изобретения, описанными выше.
Настоящее изобретение также предлагает фармацевтическую композицию, содержащую эффективное количество фармакологически активного вещества совместно с носителем или разбавителем для него, где указанное фармакологически активное вещество представляет собой комплекс, содержащий, по меньшей мере, один белок или его аналог или вариант, которые связывают, по меньшей мере, с одним сополимером или его фармакологически приемлемой солью настоящего изобретения, описанными выше.
Настоящее изобретение также предлагает способ продления времени, в течение которого белок или его аналог или вариант удерживаются в токе крови после приема пациентом, в результате комплексообразования между указанными белком или его аналогом или вариантом и, по меньшей мере, одним сополимером или его фармакологически приемлемой солью настоящего изобретения, описанными выше.
Настоящее изобретение также предлагает способ лечения или профилактики у пациента заболевания, которое восприимчиво к действию белка или его аналога или варианта, включающий прием указанным пациентом эффективного количества комплекса, содержащего указанные белок или его аналог или вариант, которые связывают, по меньшей мере, с одним сополимером или его фармакологически приемлемой солью настоящего изобретения, описанными выше.
Настоящее изобретение также предлагает применение комплекса, содержащего белок или его аналог или вариант, которые связывают, по меньшей мере, с одним сополимером или его фармакологически приемлемой солью настоящего изобретения, описанными выше, для изготовления лекарственного средства, предназначенного для профилактики или лечения заболевания, восприимчивого к действию указанных белка или его аналога или варианта.
Настоящее изобретение также предлагает способ получения сополимера или его фармакологически приемлемой соли, содержащих в качестве образующих их элементарных звеньев
(а) одно или несколько структурных элементарных звеньев, которые могут быть идентичными или отличными друг от друга и которые описываются приведенной ниже формулой (I):
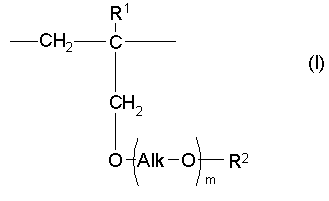
где:
m, Alk, R1 и R2 представляют собой определенное выше, и
(b) одно или несколько структурных элементарных звеньев, которые могут быть идентичными или отличными друг от друга и которые описываются формулой (II):
где:
R3 представляет собой определенное выше;
при этом указанный способ включает проведение для одного или нескольких звеньев ангидрида карбоновой кислоты, описанных формулой (III), в сополимере, который содержит в качестве образующих его элементарных звеньев
(с) одно или несколько структурных элементарных звеньев, которые могут быть идентичными или отличными друг от друга, и которые описываются приведенной выше формулой (I), и
(d) указанные одно или несколько звеньев ангидрида карбоновой кислоты, описанных формулой (III):
одной или нескольких реакций, выбираемых из группы, состоящей из (i) гидролиза, (ii) аммонолиза, (iii) аминолиза и (iv) алкоголиза.
Настоящее изобретение также предлагает способ получения комплекса, содержащего, по меньшей мере, один белок или его аналог или вариант, которые связывают, по меньшей мере, с одним сополимером или его фармакологически приемлемой солью, определяемыми выше, при этом указанный способ включает проведение реакции между указанным сополимером или его фармакологически приемлемой солью и указанными белком или его аналогом или вариантом в условиях, благоприятствующих образованию указанного комплекса.
Краткое описание фигур
Фигура 1 демонстрирует результаты электрофореза в полиакриламидном геле в присутствии SDS (додецилсульфата натрия) для комплексов настоящего изобретения поли(ПЭГ500-MA)a-OCIF (фактор ингибирования остеокластогенеза) в невосстанавливающих условиях, проводимого в приведенном ниже примере испытания 6:
(1) Маркеры молекулярной массы
(2) Комплекс поли(ПЭГ500-MA)a-Na (соединение № 9)-OCIF [OCIF:полимерный модификатор=1:1 (массовое соотношение)]
(3) Комплекс поли(ПЭГ500-MA)a-Na (соединение № 9)-OCIF [OCIF:полимерный модификатор=1:2 (массовое соотношение)]
(4) Комплекс поли(ПЭГ500-MA)a-Na (соединение № 9)-OCIF [OCIF:полимерный модификатор=1:3 (массовое соотношение)]
(5) Комплекс поли(ПЭГ500-MA)a-Na (соединение № 9)-OCIF [OCIF:полимерный модификатор=1:4 (массовое соотношение)]
(6) Комплекс поли(ПЭГ500-MA)a-Na (соединение № 9)-OCIF [OCIF:полимерный модификатор=1:5 (массовое соотношение)]
(7) Немодифицированный OCIF.
Фигура 2 демонстрирует дополнительные результаты электрофореза в полиакриламидном геле в присутствии SDS для комплексов настоящего изобретения поли(ПЭГ500-MA)a-OCIF в невосстанавливающих условиях, проводимого в приведенном ниже примере испытания 6:
(1) Маркер молекулярной массы
(2) Комплекс поли(ПЭГ500-MA)a-Na (соединение № 9)-OCIF [OCIF:полимерный модификатор=1:1 (массовое соотношение), концентрация OCIF во время инкубации: 3,5 мг/мл]
(3) Комплекс поли(ПЭГ500-MA)a-Na (соединение № 9)-OCIF [OCIF:полимерный модификатор=1:1 (массовое соотношение), концентрация OCIF во время инкубации: 1,75 мг/мл]
(4) Комплекс поли(ПЭГ500-MA)a-Na (соединение № 9)-OCIF [OCIF:полимерный модификатор=1:1 (массовое соотношение), концентрация OCIF во время инкубации: 0,875 мг/мл]
(5) Комплекс поли(ПЭГ500-MA)a-Na (соединение № 9)-OCIF [OCIF:полимерный модификатор=1:2 (массовое соотношение), концентрация OCIF во время инкубации: 1,75 мг/мл]
(6) Комплекс поли(ПЭГ500-MA)a-Na (соединение № 9)-OCIF [OCIF:полимерный модификатор=1:4 (массовое соотношение), концентрация OCIF во время инкубации: 0,875 мг/мл]
(7) Немодифицированный OCIF.
Фигура 3 демонстрирует результаты электрофореза в полиакриламидном геле в присутствии SDS для комплексов предшествующего уровня техники поли(ПЭГ500-MA)-OCIF в невосстанавливающих условиях, проводимого в приведенном ниже примере испытания 6:
(1) Маркер молекулярной массы
(2) Комплекс поли(ПЭГ500-MA) (АМ-0530К)-OCIF [OCIF:полимерный модификатор=1:10 (массовое соотношение)]
(3) Комплекс поли(ПЭГ500-MA) (АМ-0530К)-OCIF [OCIF:полимерный модификатор=1:2,5 (массовое соотношение)]
(4) Комплекс поли(ПЭГ500-MA) (АМ-0530К)-OCIF [OCIF:полимерный модификатор=1:1 (массовое соотношение)]
(10) Немодифицированный OCIF.
Подробное описание изобретения
(1) Как отмечалось выше, один аспект настоящего изобретения предлагает сополимер или его фармакологически приемлемую соль, которые содержат в качестве образующих их элементарных звеньев
(а) одно или несколько структурных элементарных звеньев, которые могут быть идентичными или отличными друг от друга и которые описываются приведенной ниже формулой (I):
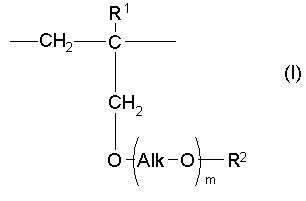
где:
m, Alk, R1 и R2 представляют собой определенное выше, и
(b) одно или несколько структурных элементарных звеньев, которые могут быть идентичными или отличными друг от друга и которые описываются формулой (II):
где R3 представляет собой определенное выше. В числе данных сополимеров и их фармакологически приемлемых солей предпочтительные соединения включают:
(2) сополимер или его фармакологически приемлемую соль, соответствующие позиции (1), где для структурных элементарных звеньев, описанных формулой (I), и структурных элементарных звеньев, описанных формулой (II), получают конфигурацию в виде последовательности с чередованием «голова к голове», последовательности с чередованием «голова к хвосту» или смешанной последовательности с чередованием «голова к голове» и «голова к хвосту»;
(3) сополимер или его фармакологически приемлемую соль, соответствующие позиции (1), где для структурных элементарных звеньев, описанных формулой (I), и структурных элементарных звеньев, описанных формулой (II), получают конфигурацию в виде статистической последовательности;
(4) сополимер или его фармакологически приемлемую соль, соответствующие любой одной позиции от (1) до (3), где Alk представляет собой этиленовую или триметиленовую группу;
(5) сополимер или его фармакологически приемлемую соль, соответствующие позиции (4), где Alk представляет собой этиленовую группу;
(6) сополимер или его фармакологически приемлемую соль, соответствующие любой одной позиции от (1) до (5), где m представляет собой целое число в диапазоне от 3 до 50;
(7) сополимер или его фармакологически приемлемую соль, соответствующие позиции (6), где m представляет собой целое число в диапазоне от 3 до 40;
(8) сополимер или его фармакологически приемлемую соль, соответствующие позиции (7), где m представляет собой целое число в диапазоне от 6 до 16 или в диапазоне от 28 до 38;
(9) сополимер или его фармакологически приемлемую соль, соответствующие позиции (8), где m представляет собой целое число в диапазоне от 6 до 16;
(10) сополимер или его фармакологически приемлемую соль, соответствующие любой одной позиции от (1) до (9), где R1 представляет собой атом водорода или метильную группу;
(11) сополимер или его фармакологически приемлемую соль, соответствующие позиции (10), где R1 представляет собой атом водорода;
(12) сополимер или его фармакологически приемлемую соль, соответствующие любой одной позиции от (1) до (11), где R2 представляет собой атом водорода или метильную группу;
(13) сополимер или его фармакологически приемлемую соль, соответствующие позиции (12), где R2 представляет собой метильную группу;
(14) сополимер или его фармакологически приемлемую соль, соответствующие любой одной позиции от (1) до (13), где R3 представляет собой гидроксильную группу, алкокси-группу, содержащую от 1 до 6 атомов углерода, или группу, описанную формулой -NR4R5, где R4 и R5 являются идентичными или различными, и каждый из них представляет собой атом водорода или алкильную группу, содержащую от 1 до 6 атомов углерода;
(15) сополимер или его фармакологически приемлемую соль, соответствующие позиции (14), где R3 представляет собой гидроксильную группу или алкокси-группу, содержащую от 1 до 6 атомов углерода;
(16) сополимер или его фармакологически приемлемую соль, соответствующие позиции (15), содержащие, по меньшей мере, одно структурное элементарное звено, описанное формулой (II), в которой R3 представляет собой алкокси-группу, содержащую от 1 до 6 атомов углерода, и необязательно, по меньшей мере, одно структурное элементарное звено, описанное формулой (II), в которой R3 представляет собой гидроксильную группу, где соотношение между структурными элементарными звеньями, описанными формулой (II), в которой R3 представляет собой гидрокси-группу, и структурными элементарными звеньями, описанными формулой (II), в которой R3 представляет собой алкокси-группу, содержащую от 1 до 6 атомов углерода, находится в диапазоне от 4:6 до 0:10;
(17) сополимер или его фармакологически приемлемую соль, соответствующие позициям (15) или (16), где R3 представляет собой алкокси-группу, содержащую от 1 до 6 атомов углерода;
(18) сополимер или его фармакологически приемлемую соль, соответствующие любой одной из позиций от (15) до (17), где указанная алкокси-группа, содержащая от 1 до 6 атомов углерода, представляет собой этокси-группу;
(19) сополимер или его фармакологически приемлемую соль, соответствующие позиции (14), где R3 представляет собой гидроксильную группу или группу, описанную формулой -NR4R5, где R4 и R5 являются идентичными или различными, и каждый из них представляет собой атом водорода или алкильную группу, содержащую от 1 до 6 атомов углерода;
(20) сополимер или его фармакологически приемлемую соль, соответствующие позиции (19), содержащие, по меньшей мере, одно структурное элементарное звено, описанное формулой (II), в которой R3 представляет собой группу, описанную формулой -NR4R5, где R4 и R5 являются идентичными или различными, и каждый из них представляет собой атом водорода или алкильную группу, содержащую от 1 до 6 атомов углерода, и необязательно, по меньшей мере, одно структурное элементарное звено, описанное формулой (II), в которой R3 представляет собой гидроксильную группу, где соотношение между структурными элементарными звеньями, описанными формулой (II), в которой R3 представляет собой гидрокси-группу, и структурными элементарными звеньями, описанными формулой (II), в которой R3 представляет собой группу, описанную формулой -NR4R5, находится в диапазоне от 5:5 до 0:10;
(21) сополимер или его фармакологически приемлемую соль, соответствующие позиции (20), где соотношение между структурными элементарными звеньями, описанными формулой (II), в которой R3 представляет собой гидрокси-группу, и структурными элементарными звеньями, описанными формулой (II), в которой R3 представляет собой группу, описанную формулой -NR4R5, находится в диапазоне от 4:6 до 0:10;
(22) сополимер или его фармакологически приемлемую соль, соответствующие любой одной позиции от (19) до (21), где R3 представляет собой группу, описанную формулой -NR4R5, где R4 и R5 являются идентичными или различными, и каждый из них представляет собой атом водорода или алкильную группу, содержащую от 1 до 6 атомов углерода;
(23) сополимер или его фармакологически приемлемую соль, соответствующие любой одной позиции от (19) до (22), где группа, описанная формулой -NR4R5, представляет собой аминогруппу, метиламиногруппу или диметиламиногруппу;
(24) сополимер или его фармакологически приемлемую соль, соответствующие позиции (23), где группа, описанная формулой -NR4R5, представляет собой аминогруппу;
(25) сополимер или его фармакологически приемлемую соль, соответствующие позиции (23), где группа, описанная формулой -NR4R5, представляет собой диметиламиногруппу;
(26) сополимер или его фармакологически приемлемую соль, соответствующие позиции (14), где R3 представляет собой гидроксильную группу;
(27) сополимер или его фармакологически приемлемую соль, соответствующие позиции (14), где R3 представляет собой 1-амино-2-пропанольную группу;
(28) сополимер или его фармакологически приемлемую соль, соответствующие любой одной позиции от (1) до (27), где соотношение между структурным элементарным звеном, описанным формулой (I), и структурным элементарным звеном, описанным формулой (II), находится в диапазоне от 10:1 до 1:10;
(29) сополимер или его фармакологически приемлемую соль, соответствующие позиции (28), где соотношение между структурным элементарным звеном, описанным формулой (I), и структурным элементарным звеном, описанным формулой (II), находится в диапазоне от 3:1 до 1:8;
(30) сополимер или его фармакологически приемлемую соль, соответствующие позиции (28), где соотношение между структурным элементарным звеном, описанным формулой (I), и структурным элементарным звеном, описанным формулой (II), находится в диапазоне от 2:1 до 1:2 или в диапазоне от 1:2 до 1:6;
(31) сополимер или его фармакологически приемлемую соль, соответствующие позиции (28), где соотношение между структурным элементарным звеном, описанным формулой (I), и структурным элементарным звеном, описанным формулой (II), равно 1:1 или находится в диапазоне от 1:2 до 1:4;
(32) сополимер или его фармакологически приемлемую соль, соответствующие любой одной позиции от (1) до (31), где средняя степень полимеризации находится в диапазоне от 5 до 200;
(33) сополимер или его фармакологически приемлемую соль, соответствующие позиции (32), где средняя степень полимеризации находится в диапазоне от 5 до 50;
(34) сополимер или его фармакологически приемлемую соль, соответствующие позиции (33), где средняя степень полимеризации находится в диапазоне от 5 до 20;
(35) сополимер или его фармакологически приемлемую соль, соответствующие позиции (32), где средняя степень полимеризации находится в диапазоне от 20 до 30;
(36) сополимер или его фармакологически приемлемую соль, соответствующие позиции (32), где средняя степень полимеризации находится в диапазоне от 30 до 40;
(37) сополимер или его фармакологически приемлемую соль, соответствующие любой одной позиции от (1) до (31), где их радиус Стокса составляет 9,3 нм или менее;
(38) сополимер или его фармакологически приемлемую соль, соответствующие позиции (37), где их радиус Стокса составляет 7,3 нм или менее;
(39) сополимер или его фармакологически приемлемую соль, соответствующие позиции (38), где их радиус Стокса составляет 6,2 нм или менее;
(40) сополимер или его фармакологически приемлемую соль, соответствующие позиции (39), где их радиус Стокса составляет 4,7 нм или менее;
(41) сополимер или его фармакологически приемлемую соль, соответствующие позиции (40), где их радиус Стокса составляет 3,1 нм или менее;
(42) сополимер или его фармакологически приемлемую соль, соответствующие позиции (37), где их радиус Стокса находится в диапазоне от 1,5 нм до 4,7 нм;
(43) сополимер или его фармакологически приемлемую соль, соответствующие позиции (37), где их радиус Стокса находится в диапазоне от 3,1 нм до 6,2 нм; и
(44) сополимер или его фармакологически приемлемую соль, соответствующие позиции (1), где:
m представляет собой целое число в диапазоне от 3 до 100,
Alk представляет собой алкиленовую группу, содержащую от 1 до 6 атомов углерода,
R1 и R2 являются идентичными или различными, и каждый из них представляет собой атом водорода или алкильную группу, содержащую от 1 до 6 атомов углерода, и
R3 представляет собой гидроксильную группу, алкокси-группу, содержащую от 1 до 6 атомов углерода, которая необязательно может быть замещена одной гидрокси-группой, или группу, описанную формулой -NR4R5, где R4 и R5 являются идентичными или отличными друг от друга, и каждый из них представляет собой атом водорода или алкильную группу, содержащую от 1 до 6 атомов углерода, которая необязательно может быть замещена одной гидрокси-группой;
(45) сополимер или его фармакологически приемлемую соль, соответствующие позиции (1), где Alk представляет собой этиленовую группу, R1 представляет собой атом водорода, R2 представляет собой метильную группу, а m, R3, соотношение между структурными элементарными звеньями, описанными формулами (I) и (II) (соотношение, задаваемое составом) и там, где это будет уместно, соотношение между элементарными звеньями, описанными формулой (II), где R3 представляет собой гидрокси-группу, и элементарными звеньями, описанными формулой (II), где R3 представляет собой группу, отличную от гидрокси (соотношение, задаваемое гидролизом), выбирают из нижеследующего:
(i) m находится в диапазоне от 6 до 16, R3 представляет собой гидрокси-группу, соотношение, задаваемое составом, равно 1:1, а средняя степень полимеризации находится в диапазоне от 30 до 40;
(ii) m находится в диапазоне от 28 до 38, R3 представляет собой гидрокси-группу, соотношение, задаваемое составом, равно 1:1, а средняя степень полимеризации находится в диапазоне от 10 до 15;
(iii) m находится в диапазоне от 6 до 16, R3 представляет собой аминогруппу, соотношение, задаваемое составом, равно 1:1, а средняя степень полимеризации находится в диапазоне от 30 до 40;
(iv) m находится в диапазоне от 6 до 16, R3 представляет собой диметиламиногруппу, соотношение, задаваемое составом, равно 1:1, а средняя степень полимеризации находится в диапазоне от 30 до 40;
(v) m находится в диапазоне от 6 до 16, R3 представляет собой 1-амино-2-пропанольную группу, соотношение, задаваемое составом, равно 1:1, а средняя степень полимеризации находится в диапазоне от 30 до 40;
(vi) m находится в диапазоне от 6 до 16, R3 выбирают из этокси- и гидрокси-групп, соотношение, задаваемое составом, равно 1:1, средняя степень полимеризации находится в диапазоне от 30 до 40, а соотношение, задаваемое гидролизом, равно 4:6;
(vii) m находится в диапазоне от 28 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:1, средняя степень полимеризации находится в диапазоне от 10 до 15, а соотношение, задаваемое гидролизом, равно 4:6;
(viii) m находится в диапазоне от 28 до 38, R3 представляет собой диметиламиногруппу, соотношение, задаваемое составом, равно 1:1, а средняя степень полимеризации находится в диапазоне от 10 до 15;
(ix) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:1, средняя степень полимеризации находится в диапазоне от 30 до 40, соотношение, задаваемое гидролизом, равно 3,1:6,9, а сополимер представляет собой натриевую соль;
(х) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:1, средняя степень полимеризации находится в диапазоне от 30 до 40, а соотношение, задаваемое гидролизом, равно 1,4:8,6;
(xi) m находится в диапазоне от 6 до 16, R3 выбирают из диметиламино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:1, средняя степень полимеризации находится в диапазоне от 30 до 40, соотношение, задаваемое гидролизом, равно 2,9:7,1, а сополимер представляет собой натриевую соль;
(xii) m находится в диапазоне от 6 до 16, R3 представляет собой аминогруппу, соотношение, задаваемое составом, равно 1:2,4, а средняя степень полимеризации находится в диапазоне от 20 до 30;
(xiii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,4:9,6;
(xiv) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 2,9:7,1;
(xv) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,9:9,1;
(xvi) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,5:9,5;
(xvii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 1,3:8,7;
(xviii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 1,9:8,1;
(xix) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 1,0:9,0;
(хх) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,8:9,2;
(xxi) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 4,6:5,4;
(xxii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 1,2:8,8;
(xxiii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 2,0:8,0;
(xxiv) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 1,1:8,9;
(xxv) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 2,4:7,6;
(xxvi) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,9:9,1;
(xxvii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 1,5:8,5;
(xxviii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,7:9,3;
(xxix) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 4,5:5,5;
(xxx) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 1,4:8,6;
(xxxi) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,7:9,3;
(xxxii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,8:9,2;
(xxxiii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 1,4:8,6;
(xxxiv) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:3,1, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,7:9,3;
(xxxv) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,9:9,1;
(xxxvi) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 1,9:8,1;
(xxxvii) m находится в диапазоне от 6 до 16, R3 выбирают из этокси- и гидрокси-групп, соотношение, задаваемое составом, приблизительно равно 1:3, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 3,1:6,9;
(xxxviii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:1, соотношение, задаваемое гидролизом, равно 1,4:8,6, а радиус Стокса составляет 9,3 нм или менее;
(xxxix) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:1, соотношение, задаваемое гидролизом, равно 1,4:8,6, а радиус Стокса находится в диапазоне от 3,1 до 6,2 нм;
(xl) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:1, соотношение, задаваемое гидролизом, равно 1,4:8,6, а радиус Стокса находится в диапазоне от 1,5 до 4,7 нм;
(xli) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:1, соотношение, задаваемое гидролизом, равно 1,4:8,6, а радиус Стокса составляет 3,1 нм или менее;
(xlii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:1, соотношение, задаваемое гидролизом, равно 1,4:8,6, а радиус Стокса составляет 7,8 нм или менее;
(xliii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:1, соотношение, задаваемое гидролизом, равно 1,4:8,6, а радиус Стокса составляет 6,2 нм или менее; и
(xliv) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидрокси-групп, соотношение, задаваемое составом, равно 1:1, соотношение, задаваемое гидролизом, равно 1,4:8,6, а радиус Стокса составляет 4,7 нм или менее.
(46) Как уже отмечалось выше, еще один аспект настоящего изобретения предлагает сополимер или его фармакологически приемлемую соль, получаемые в результате проведения для одного или нескольких звеньев ангидрида карбоновой кислоты, описанных формулой (III), в сополимере, который содержит в качестве образующих его элементарных звеньев
(а) одно или несколько структурных элементарных звеньев, которые могут быть идентичными или отличными друг от друга и которые описываются приведенной ниже формулой (I):
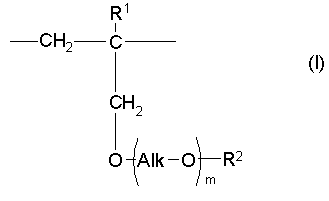
где:
m, Alk, R1 и R2 представляют собой определенное выше, и
(b) указанное структурное элементарное звено, содержащее звено ангидрида карбоновой кислоты, описанное формулой (III):
одной или нескольких реакций, выбираемых из группы, состоящей из (i) гидролиза, (ii) аммонолиза, (iii) аминолиза и (iv) алкоголиза.
В числе данных сополимеров и их фармакологически приемлемых солей предпочтительными соединениями являются:
(47) сополимер или его фармакологически приемлемая соль, соответствующие позиции (46), где для структурного элементарного звена, описанного формулой (I), и структурного элементарного звена, описанного формулой (III), в сополимере получают конфигурацию в виде последовательности с чередованием «голова к голове», последовательности с чередованием «голова к хвосту» или смешанной последовательности с чередованием «голова к голове» и «голова к хвосту»;
(48) сополимер или его фармакологически приемлемая соль, соответствующие позиции (46), где для структурного элементарного звена, описанного формулой (I), и структурного элементарного звена, описанного формулой (III), получают конфигурацию в виде статистической последовательности;
(49) сополимер или его фармакологически приемлемая соль, соответствующие любой одной позиции от (46) до (48), где Alk представляет собой этиленовую или триметиленовую группу;
(50) сополимер или его фармакологически приемлемая соль, соответствующие позиции (49), где Alk представляет собой этиленовую группу;
(51) сополимер или его фармакологически приемлемая соль, соответствующие любой одной позиции от (46) до (50), где m представляет собой целое число в диапазоне от 3 до 50;
(52) сополимер или его фармакологически приемлемая соль, соответствующие позиции (51), где m представляет собой целое число в диапазоне от 3 до 40;
(53) сополимер или его фармакологически приемлемая соль, соответствующие позиции (52), где m представляет собой целое число в диапазоне от 6 до 16 или в диапазоне от 28 до 38;
(54) сополимер или его фармакологически приемлемая соль, соответствующие позиции (53), где m представляет собой целое число в диапазоне от 6 до 16;
(55) сополимер или его фармакологически приемлемая соль, соответствующие любой одной позиции от (46) до (54), где R1 представляет собой атом водорода или метильную группу;
(56) сополимер или его фармакологически приемлемая соль, соответствующие позиции (55), где R1 представляет собой атом водорода;
(57) сополимер или его фармакологически приемлемая соль, соответствующие любой одной позиции от (46) до (56), где R2 представляет собой атом водорода или метильную группу;
(58) сополимер или его фармакологически приемлемая соль, соответствующие позиции (57), где R2 представляет собой метильную группу;
(59) сополимер или его фармакологически приемлемая соль, соответствующие любой одной позиции от (46) до (58), где соотношение между структурным элементарным звеном, описанным формулой (I), и структурным элементарным звеном, полученным в результате проведения для одного или нескольких структурных элементарных звеньев, описанных формулой (III), одной или нескольких реакций, выбираемых из группы, состоящей из (i) гидролиза, (ii) аммонолиза, (iii) аминолиза и (iv) алкоголиза, находится в диапазоне от 10:1 до 1:10;
(60) сополимер или его фармакологически приемлемая соль, соответствующие позиции (59), где соотношение между структурным элементарным звеном, описанным формулой (I), и структурным элементарным звеном, полученным в результате проведения для одного или нескольких структурных элементарных звеньев, описанных формулой (III), одной или нескольких реакций, выбираемых из группы, состоящей из (i) гидролиза, (ii) аммонолиза, (iii) аминолиза и (iv) алкоголиза, находится в диапазоне от 3:1 до 1:8;
(61) сополимер или его фармакологически приемлемая соль, соответствующие позиции (59), где соотношение между структурным элементарным звеном, описанным формулой (I), и структурным элементарным звеном, полученным в результате проведения для одного или нескольких структурных элементарных звеньев, описанных формулой (III), одной или нескольких реакций, выбираемых из группы, состоящей из (i) гидролиза, (ii) аммонолиза, (iii) аминолиза и (iv) алкоголиза, находится в диапазоне от 2:1 до 1:2 или в диапазоне от 1:2 до 1:6;
(62) сополимер или его фармакологически приемлемая соль, соответствующие позиции (59), где соотношение между структурным элементарным звеном, описанным формулой (I), и структурным элементарным звеном, полученным в результате проведения для одного или нескольких структурных элементарных звеньев, описанных формулой (III), одной или нескольких реакций, выбираемых из группы, состоящей из (i) гидролиза, (ii) аммонолиза, (iii) аминолиза и (iv) алкоголиза, равно 1:1 или находится в диапазоне от 1:2 до 1:4;
(63) сополимер или его фармакологически приемлемая соль, соответствующие любой одной позиции от (46) до (62), где средняя степень полимеризации находится в диапазоне от 5 до 200;
(64) сополимер или его фармакологически приемлемая соль, соответствующие позиции (63), где средняя степень полимеризации находится в диапазоне от 5 до 50;
(65) сополимер или его фармакологически приемлемая соль, соответствующие позиции (64), где средняя степень полимеризации находится в диапазоне от 5 до 20;
(66) сополимер или его фармакологически приемлемая соль, соответствующие позиции (63), где средняя степень полимеризации находится в диапазоне от 20 до 30;
(67) сополимер или его фармакологически приемлемая соль, соответствующие позиции (63), где средняя степень полимеризации находится в диапазоне от 30 до 40;
(68) сополимер или его фармакологически приемлемая соль, соответствующие позиции (46), где:
m представляет собой целое число в диапазоне от 3 до 100,
Alk представляет собой алкиленовую группу, содержащую от 1 до 6 атомов углерода, и
R1 и R2 являются идентичными или различными, и каждый из них представляет собой атом водорода или алкильную группу, содержащую от 1 до 6 атомов углерода;
(69) сополимер или его фармакологически приемлемая соль, соответствующие любой одной из позиций от (46) до (68), которые можно получить в результате проведения аммонолиза для звена ангидрида карбоновой кислоты, описанного формулой (III), в сополимере;
(70) сополимер или его фармакологически приемлемая соль, соответствующие позиции (69), которые можно получить в результате проведения аммонолиза с использованием водно-аммиачного раствора;
(71) сополимер или его фармакологически приемлемая соль, соответствующие любой одной из позиций от (46) до (68), которые можно получить в результате проведения аминолиза для звена ангидрида карбоновой кислоты, описанного формулой (III), в сополимере;
(72) сополимер или его фармакологически приемлемая соль, соответствующие позиции (71), которые можно получить в результате проведения аминолиза с использованием водного раствора диметиламина;
(73) сополимер или его фармакологически приемлемая соль, соответствующие любой одной из позиций от (46) до (68), которые можно получить в результате проведения алкоголиза для звена ангидрида карбоновой кислоты, описанного формулой (III), в сополимере; и
(74) сополимер или его фармакологически приемлемая соль, соответствующие позиции (73), которые можно получить в результате проведения алкоголиза с использованием этанола.
Настоящее изобретение также использует сополимеры и их фармакологически приемлемые соли настоящего изобретения с целью создания фармацевтической композиции, модификатора, способного модифицировать белок, комплекса, способа продления времени, в течение которого белок удерживается в токе крови, способа лечения или профилактики заболеваний и применения комплекса изобретения для изготовления лекарственного средства, предназначенного для лечения или профилактики заболеваний. Предпочтительные примеры аспектов данных изобретений включают:
(75) фармацевтическую композицию, содержащую фармацевтически приемлемые разбавитель или носитель и, по меньшей мере, один сополимер или его фармакологически приемлемую соль настоящего изобретения, соответствующие любой одной позиции от (1) до (74);
(76) фармацевтическую композицию, соответствующую позиции (75), где указанная композиция дополнительно содержит, по меньшей мере, один белок или его аналог или вариант;
(77) фармацевтическую композицию, соответствующую позиции (76), где белок или его аналог или вариант представляют собой основной белок;
(78) фармацевтическую композицию, соответствующую позиции (77), где основной белок представляет собой основной фактор роста фибробластов (bFGF), эпидермальный фактор роста (EGF), фактор ингибирования остеокластогенеза (OCIF), тромбоцитарный фактор роста (PDGF), выделенный из мозга нейротрофический фактор (BDNF), фактор роста нервов (NGF), гормон роста человека (HGH), гепатоцитарный фактор роста (HGF) или фактор роста сосудистого эндотелия (VEGF) или их аналоги или варианты;
(79) фармацевтическую композицию, соответствующую позиции (77), где основной белок представляет собой фактор ингибирования остеокластогенеза (OCIF) или его аналог или вариант;
(80) фармацевтическую композицию, соответствующую позиции (79), где указанный OCIF или его аналог или вариант относятся к OCIF природного типа или рекомбинантного типа;
(81) фармацевтическую композицию, соответствующую позиции (79), где указанный OCIF или его аналог или вариант представляют собой мономер или димер;
(82) фармацевтическую композицию, соответствующую позиции (79), где указанный OCIF представляет собой мономер OCIF человека, характеризующийся молекулярной массой, приблизительно равной 60000 согласно измерению методом SDS-PAGE (электрофореза в полиакриламидном геле в присутствии додецилсульфата натрия) в невосстанавливающих условиях, или димер OCIF человека, характеризующийся молекулярной массой, приблизительно равной 120000 согласно измерению методом SDS-PAGE в невосстанавливающих условиях;
(83) фармацевтическую композицию, соответствующую позиции (79), где указанный OCIF содержит аминокислоты от - 21 до + 380 последовательности SEQ ID No. № 1 списка последовательностей;
(84) фармацевтическую композицию, соответствующую позиции (79), где указанный OCIF содержит аминокислоты от + 1 до + 380 последовательности SEQ ID No. № 1 списка последовательностей;
(85) модификатор, способный модифицировать белок или его аналог или вариант, при этом указанный модификатор содержит сополимер или его фармакологически приемлемую соль, соответствующие любой одной позиции от (1) до (74);
(86) модификатор, способный модифицировать белок или его аналог или вариант, соответствующий позиции (85), где белком является основной белок;
(87) модификатор, способный модифицировать белок или его аналог или вариант, соответствующий позиции (86), где основной белок представляет собой основной фактор роста фибробластов (bFGF), эпидермальный фактор роста (EGF), фактор ингибирования остеокластогенеза (OCIF), тромбоцитарный фактор роста (PDGF), выделенный из мозга нейротрофический фактор (BDNF), фактор роста нервов (NGF), гормон роста человека (HGH), гепатоцитарный фактор роста (HGF) или фактор роста сосудистого эндотелия (VEGF) или их аналоги или варианты;
(88) модификатор, способный модифицировать белок или его аналог или вариант, соответствующий позиции (86), где основной белок представляет собой фактор ингибирования остеокластогенеза (OCIF) или его аналог или вариант;
(89) модификатор, способный модифицировать белок или его аналог или вариант, соответствующий позиции (88), где указанный OCIF или его аналог или вариант относятся к OCIF природного типа или рекомбинантного типа;
(90) модификатор, способный модифицировать белок или его аналог или вариант, соответствующий позиции (88), где указанный OCIF или его аналог или вариант представляют собой мономер или димер;
(91) модификатор, способный модифицировать белок или его аналог или вариант, соответствующий позиции (88), где указанный OCIF представляет собой мономер OCIF человека, характеризующийся молекулярной массой, приблизительно равной 60000 согласно измерению методом SDS-PAGE в невосстанавливающих условиях, или димер OCIF человека, характеризующийся молекулярной массой, приблизительно равной 120000 согласно измерению методом SDS-PAGE в невосстанавливающих условиях;
(92) модификатор, способный модифицировать белок или его аналог или вариант, соответствующий позиции (88), где указанный OCIF содержит аминокислоты от - 21 до + 380 последовательности SEQ ID No. № 1 списка последовательностей;
(93) модификатор, способный модифицировать белок или его аналог или вариант, соответствующий позиции (88), где указанный OCIF содержит аминокислоты от + 1 до + 380 последовательности SEQ ID No. № 1 списка последовательностей;
(94) комплекс, содержащий, по меньшей мере, один белок или его аналог или вариант, которые связывают, по меньшей мере, с одним сополимером или его фармакологически приемлемой солью, соответствующими любой одной позиции от (1) до (74);
(95) комплекс, соответствующий позиции (94), где белком является основной белок;
(96) комплекс, соответствующий позиции (95), где основной белок представляет собой основной фактор роста фибробластов (bFGF), эпидермальный фактор роста (EGF), фактор ингибирования остеокластогенеза (OCIF), тромбоцитарный фактор роста (PDGF), выделенный из мозга нейротрофический фактор (BDNF), фактор роста нервов (NGF), гормон роста человека (HGH), гепатоцитарный фактор роста (HGF) или фактор роста сосудистого эндотелия (VEGF) или их аналоги или варианты;
(97) комплекс, соответствующий позиции (95), где основной белок представляет собой фактор ингибирования остеокластогенеза (OCIF) или его аналог или вариант;
(98) комплекс, соответствующий позиции (97), где указанный OCIF или его аналог или вариант относятся к OCIF природного типа или рекомбинантного типа;
(99) комплекс, соответствующий позиции (97), где указанный OCIF или его аналог или вариант представляют собой мономер или димер;
(100) комплекс, соответствующий позиции (97), где указанный OCIF представляет собой мономер OCIF человека, характеризующийся молекулярной массой, приблизительно равной 60000 согласно измерению методом SDS-PAGE в невосстанавливающих условиях, или димер OCIF человека, характеризующийся молекулярной массой, приблизительно равной 120000 согласно измерению методом SDS-PAGE в невосстанавливающих условиях;
(101) комплекс, соответствующий позиции (97), где указанный OCIF содержит аминокислоты от - 21 до + 380 последовательности SEQ ID No. № 1 списка последовательностей;
(102) комплекс, соответствующий позиции (97), где указанный OCIF содержит аминокислоты от + 1 до + 380 последовательности SEQ ID No. № 1 списка последовательностей;
(103) фармацевтическую композицию, содержащую эффективное количество фармакологически активного вещества совместно с носителем или разбавителем для него, где указанное фармакологически активное вещество присутствует в виде комплекса, соответствующего любой одной позиции от (94) до (102);
(104) способ продления времени, в течение которого белок или его аналог или вариант удерживаются в токе крови после приема пациентом в результате комплексообразования между указанным белком или его аналогом или вариантом и, по меньшей мере, одним сополимером или его фармакологически приемлемой солью, соответствующими любой одной позиции от (1) до (74);
(105) способ, соответствующий позиции (104), где белком является основной белок;
(106) способ, соответствующий позиции (105), где основной белок представляет собой основной фактор роста фибробластов (bFGF), эпидермальный фактор роста (EGF), фактор ингибирования остеокластогенеза (OCIF), тромбоцитарный фактор роста (PDGF), выделенный из мозга нейротрофический фактор (BDNF), фактор роста нервов (NGF), гормон роста человека (HGH), гепатоцитарный фактор роста (HGF) или фактор роста сосудистого эндотелия (VEGF) или их аналоги или варианты;
(107) способ, соответствующий позиции (105), где основной белок представляет собой фактор ингибирования остеокластогенеза (OCIF) или его аналог или вариант;
(108) способ, соответствующий позиции (107), где указанный OCIF или его аналог или вариант относятся к OCIF природного типа или рекомбинантного типа;
(109) способ, соответствующий позиции (107), где указанный OCIF или его аналог или вариант представляют собой мономер или димер;
(110) способ, соответствующий позиции (107), где указанный OCIF представляет собой мономер OCIF человека, характеризующийся молекулярной массой, приблизительно равной 60000 согласно измерению методом SDS-PAGE в невосстанавливающих условиях, или димер OCIF человека, характеризующийся молекулярной массой, приблизительно равной 120000 согласно измерению методом SDS-PAGE в невосстанавливающих условиях;
(111) способ, соответствующий позиции (107), где указанный OCIF содержит аминокислоты от - 21 до + 380 последовательности SEQ ID No. № 1 списка последовательностей;
(112) способ, соответствующий позиции (107), где указанный OCIF содержит аминокислоты от + 1 до + 380 последовательности SEQ ID No. № 1 списка последовательностей;
(113) способ лечения или профилактики заболевания у пациента, которое восприимчиво к действию белка или его аналога или варианта, включающий прием указанным пациентом эффективного количества комплекса, содержащего указанный белок или его аналог или вариант, которые связывают, по меньшей мере, с одним сополимером или его фармакологически приемлемой солью, соответствующими любой одной позиции от (1) до (74);
(114) способ, соответствующий позиции (113), где белком является основной белок;
(115) способ, соответствующий позиции (114), где основной белок представляет собой основной фактор роста фибробластов (bFGF), эпидермальный фактор роста (EGF), фактор ингибирования остеокластогенеза (OCIF), тромбоцитарный фактор роста (PDGF), выделенный из мозга нейротрофический фактор (BDNF), фактор роста нервов (NGF), гормон роста человека (HGH), гепатоцитарный фактор роста (HGF) или фактор роста сосудистого эндотелия (VEGF) или их аналоги или варианты;
(116) способ, соответствующий позиции (114), где основной белок представляет собой фактор ингибирования остеокластогенеза (OCIF) или его аналог или вариант;
(117) фармацевтическая композиция, соответствующая позиции (116), где указанный OCIF или его аналог или вариант относятся к OCIF природного типа или рекомбинантного типа;
(118) способ, соответствующий позиции (116), где указанный OCIF или его аналог или вариант представляют собой мономер или димер;
(119) способ, соответствующий позиции (116), где указанный OCIF представляет собой мономер OCIF человека, характеризующийся молекулярной массой, приблизительно равной 60000 согласно измерению методом SDS-PAGE в невосстанавливающих условиях, или димер OCIF человека, характеризующийся молекулярной массой, приблизительно равной 120000 согласно измерению методом SDS-PAGE в невосстанавливающих условиях;
(120) способ, соответствующий позиции (116), где указанный OCIF содержит аминокислоты от - 21 до + 380 последовательности SEQ ID No. № 1 списка последовательностей;
(121) способ, соответствующий позиции (116), где указанный OCIF содержит аминокислоты от + 1 до + 380 последовательности SEQ ID No. № 1 списка последовательностей;
(122) способ, соответствующий любой одной позиции от (116) до (121), где указанным заболеванием является нарушение обмена веществ в костях;
(123) применение комплекса, содержащего белок или его аналог или вариант, который связывают, по меньшей мере, с одним сополимером или его фармакологически приемлемой солью, соответствующими любой одной позиции от (1) до (74), для изготовления лекарственного средства, предназначенного для профилактики или лечения заболевания, восприимчивого к действию указанных белка или его аналога или варианта;
(124) применение, соответствующее позиции (122), где белком является основной белок;
(125) применение, соответствующее позиции (123), где основной белок представляет собой основной фактор роста фибробластов (bFGF), эпидермальный фактор роста (EGF), фактор ингибирования остеокластогенеза (OCIF), тромбоцитарный фактор роста (PDGF), выделенный из мозга нейротрофический фактор (BDNF), фактор роста нервов (NGF), гормон роста человека (HGH), гепатоцитарный фактор роста (HGF) или фактор роста сосудистого эндотелия (VEGF) или их аналоги или варианты;
(126) применение, соответствующее позиции (123), где основной белок представляет собой фактор ингибирования остеокластогенеза (OCIF) или его аналог или вариант;
(127) применение, соответствующее позиции (126), где указанный OCIF или его аналог или вариант относятся к OCIF природного типа или рекомбинантного типа;
(128) применение, соответствующее позиции (126), где указанный OCIF или его аналог или вариант представляют собой мономер или димер;
(129) применение, соответствующее позиции (126), где указанный OCIF представляет собой мономер OCIF человека, характеризующийся молекулярной массой, приблизительно равной 60000 согласно измерению методом SDS-PAGE в невосстанавливающих условиях, или димер OCIF человека, характеризующийся молекулярной массой, приблизительно равной 120000 согласно измерению методом SDS-PAGE в невосстанавливающих условиях;
(130) применение, соответствующее позиции (126), где указанный OCIF содержит аминокислоты от - 21 до + 380 последовательности SEQ ID No. № 1 списка последовательностей;
(131) применение, соответствующее позиции (126), где указанный OCIF содержит аминокислоты от + 1 до + 380 последовательности SEQ ID No. № 1 списка последовательностей; и
(132) применение, соответствующее любой одной позиции от (126) до (131), где указанным заболеванием является нарушение обмена веществ в костях.
«Алкиленовая группа, содержащая от 1 до 6 атомов углерода,» в определении заместителя Alk в приведенной выше формуле (I) представляет собой прямую или разветвленную алкиленовую группу, содержащую от 1 до 6 атомов углерода, такую как метиленовая, метилметиленовая, этиленовая, пропиленовая, триметиленовая, тетраметиленовая, 1-метилтриметиленовая, 2-метилтриметиленовая, 3-метилтриметиленовая, пентаметиленовая или гексаметиленовая группа. В числе данных алкиленовых групп предпочтительными являются прямая или разветвленная алкиленовые группы, содержащие от 1 до 4 атомов углерода, более предпочтительными являются этиленовая или триметиленовая группы, а наиболее предпочтительной является этиленовая группа.
Алкильная группа в «алкильной группе, содержащей от 1 до 6 атомов углерода, которая необязательно может быть замещена, по меньшей мере, одним заместителем, выбираемым из группы, состоящей из гидрокси-групп, атомов галогена и арильных групп, содержащих от 6 до 14 атомов углерода, которые необязательно могут быть замещены заместителями в количестве от 1 до 5, выбираемыми из определенных ниже заместителей А,» в определении заместителей R1, R2, R4, R5 и заместителей А в приведенных выше формулах (I) и (II) представляет собой прямую или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода, такую как метильная, этильная, н-пропильная, изопропильная, н-бутильная, изобутильная, втор-бутильная, трет-бутильная, н-пентильная, изопентильная, 2-метилбутильная, неопентильная, 1-этилпропильная, н-гексильная, изогексильная, 4-метилпентильная, 3-метилпентильная, 2-метилпентильная, 1-метилпентильная, 3,3-диметилбутильная, 2,2-диметилбутильная, 1,1-диметилбутильная, 1,2-диметилбутильная, 1,3-диметилбутильная, 2,3-диметилбутильная или 2-этилбутильная группа. В числе данных алкильных групп предпочтительными являются прямые или разветвленные алкильные группы, содержащие от 1 до 4 атомов углерода, более предпочтительными являются метильная и этильная группы, а наиболее предпочтительной является метильная группа.
Алкокси-группа в «алкокси-группе, содержащей от 1 до 6 атомов углерода, которая необязательно может быть замещена, по меньшей мере, одним заместителем, выбираемым из группы, состоящей из гидрокси-групп, атомов галогена и арильных групп, содержащих от 6 до 14 атомов углерода, которые необязательно могут быть замещены заместителями в количестве от 1 до 5, выбираемыми из определенных ниже заместителей А,» в определении заместителя R3 и заместителей А в приведенных выше формулах (I) и (II) представляет собой заместитель, в котором указанную выше алкильную группу, содержащую от 1 до 6 атомов углерода, связывают с атомом кислорода. Примеры такой алкокси-группы включают прямые или разветвленные алкокси-группы, содержащие от 1 до 6 атомов углерода, такие как метокси-, этокси-, н-пропокси-, изопропокси-, н-бутокси-, изобутокси-, втор-бутокси-, трет-бутокси-, н-пентилокси-, изопентилокси-, 2-метилбутокси-, неопентилокси-, н-гексилокси-, 4-метилпентилокси-, 3-метилпентилокси-, 2-метилпентилокси-, 3,3-диметилбутокси-, 2,2-диметилбутокси-, 1,1-диметилбутокси-, 1,2-диметилбутокси-, 1,3-диметилбутокси- и 2,3-диметилбутокси-группы. В числе данных алкокси-групп более предпочтительными являются прямые или разветвленные алкокси-группы, содержащие от 1 до 4 атомов углерода, а наиболее предпочтительной является этокси-группа.
«Атом галогена», которым является один из приведенных выше «заместителей А», представляет собой необязательный заместитель в «алкильной группе, содержащей от 1 до 6 атомов углерода, которая необязательно может быть замещена, по меньшей мере, одним заместителем, выбираемым из группы, состоящей из гидрокси-групп, атомов галогена и арильных групп, содержащих от 6 до 14 атомов углерода, которые необязательно могут быть замещены заместителями в количестве от 1 до 5, выбираемыми из определенных ниже заместителей А,» в определении заместителей R1, R2, R4 и R5 в приведенных выше формулах (I) и (II) и представляет собой необязательный заместитель в «алкокси-группе, содержащей от 1 до 6 атомов углерода, которая необязательно может быть замещена, по меньшей мере, одним заместителем, выбираемым из группы, состоящей из гидрокси-групп, атомов галогена и арильных групп, содержащих от 6 до 14 атомов углерода, которые необязательно могут быть замещены заместителями в количестве от 1 до 5, выбираемыми из определенных ниже заместителей А,» в определении заместителя R3 в приведенной выше формуле (II), представляет собой атом фтора, атом хлора, атом брома или атом иода; и предпочтительно им является атом фтора или атом хлора.
«Арильная группа, содержащая от 6 до 14 атомов углерода», которая представляет собой необязательный заместитель в «алкильной группе, содержащей от 1 до 6 атомов углерода, которая необязательно может быть замещена, по меньшей мере, одним заместителем, выбираемым из группы, состоящей из гидрокси-групп, атомов галогена и арильных групп, содержащих от 6 до 14 атомов углерода, которые необязательно могут быть замещены заместителями в количестве от 1 до 5, выбираемыми из определенных ниже заместителей А,» в определении заместителей R1, R2, R4 и R5 и необязательный заместитель в «алкокси-группе, содержащей от 1 до 6 атомов углерода, которая необязательно может быть замещена, по меньшей мере, одним заместителем, выбираемым из группы, состоящей из гидрокси-групп, атомов галогена и арильных групп, содержащих от 6 до 14 атомов углерода,» в определении заместителя R3 в приведенной выше формуле (II), представляет собой ароматическую углеводородную группу, содержащую от 6 до 14 атомов углерода, и она может являться, например, фенильной, инденильной, нафтильной, фенантрильной или антрильной группой. Предпочтительно ею является фенильная группа.
«Арилокси-группа, содержащая от 6 до 14 атомов углерода, которая необязательно может быть замещена заместителями в количестве от 1 до 5, выбираемыми из заместителей А,» в определении заместителя R3 в приведенной выше формуле (II) представляет собой арильную группу, определенную выше, которую связывают с атомом кислорода, и ей может являться, например, фенокси-, инденилокси-, нафтилокси-, фенантрилокси- или антрилокси-группа. Предпочтительно ею является фенокси-группа.
«Алкильная группа, содержащая от 1 до 6 атомов углерода, которая необязательно замещена, по меньшей мере, одним атомом галогена,» в определении заместителей R1, R2, R4 и R5 в приведенных выше формулах (I) и (II) представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода, описанную выше, и может являться, например, трифторметильной группой, трихлорметильной группой, дифторметильной группой, дихлорметильной группой, дибромметильной группой, фторметильной группой, 2,2,2-трифторэтильной группой, 2,2,2-трихлорэтильной группой, 2-бромэтильной группой, 2-хлорэтильной группой, 2-фторэтильной группой, 2-иодэтильной группой, 3-хлорпропильной группой, 4-фторбутильной группой, 6-иодгексильной группой, 2,2-дибромэтильной группой или пентафторэтильной группой. Предпочтительно ею являются трифторметильная группа, трихлорметильная группа, дифторметильная группа или пентафторэтильная группа; а наиболее предпочтительно ею является трифторметильная группа.
Примеры «алкильной группы, содержащей от 1 до 6 атомов углерода, которая необязательно замещена, по меньшей мере, одной гидрокси-группой,» в определении заместителей R1, R4 и R5 в приведенных выше формулах (I) и (II) включают гидроксиметильную группу, 1-гидроксиэтильную группу, 1-гидроксипропильную группу и 2-гидроксипропильную группу.
«Алкокси-группа, содержащая от 1 до 6 атомов углерода, которая необязательно замещена, по меньшей мере, одним атомом галогена,» в определениях заместителя R3 в приведенной выше формуле (II) представляет собой алкокси-группу, содержащую от 1 до 6 атомов углерода, описанную выше, которая замещена, по меньшей мере, одним атомом галогена, описанным выше, и может являться, например, трифторметокси-группой, трихлорметокси-группой, дифторметокси-группой, дихлорметокси-группой, дибромметокси-группой, фторметокси-группой, 2,2,2-трифторэтокси-группой, 2,2,2-трихлорэтокси-группой, 2-бромэтокси-группой, 2-хлорэтокси-группой, 2-фторэтокси-группой, 2-иодэтокси-группой, 3-хлорпропокси-группой, 4-фторбутокси-группой, 6-иодгексилокси-группой, 2,2-дибромэтокси-группой или пентафторэтокси-группой; предпочтительно ею является С1-С4 алкокси-группа, замещенная атомами фтора или хлора, такая как трифторметокси-группа, трихлорметокси-группа, дифторметокси-группа или пентафторэтокси-группа. Более предпочтительно ею является трифторметокси-группа.
Примеры «алкокси-группы, содержащей от 1 до 6 атомов углерода, которая необязательно замещена, по меньшей мере, одной гидрокси-группой,» в определении заместителя R3 в приведенной выше формуле (II) включают гидроксиметокси-группу, 1-гидроксиэтокси-группу, 1-гидроксипропокси-группу и 2-гидроксипропокси-группу.
«Алкильная группа, содержащая от 1 до 6 атомов углерода, которая необязательно замещена, по меньшей мере, одной арильной группой, содержащей от 6 до 14 атомов углерода, которая необязательно может быть замещена заместителями в количестве от 1 до 5, выбираемыми из заместителей А,» в определении заместителей R1, R2, R4 и R5 в приведенных выше формулах (I) и (II) может являться, например, бензильной группой, 1-нафтилметильной группой, 2-нафтилметильной группой, инденилметильной группой, 1-фенэтильной группой, 2-фенэтильной группой, 1-нафтилэтильной группой, 2-нафтилэтильной группой, 1-фенилпропильной группой, 2-фенилпропильной группой, 3-фенилпропильной группой, 1-нафтилпропильной группой, 2-нафтилпропильной группой, 3-нафтилпропильной группой, 1-фенилбутильной группой, 2-фенилбутильной группой, 3-фенилбутильной группой, 4-фенилбутильной группой, 1-нафтилбутильной группой, 2-нафтилбутильной группой, 3-нафтилбутильной группой, 4-нафтилбутильной группой, 1-фенилпентильной группой, 2-фенилпентильной группой, 3-фенилпентильной группой, 4-фенилпентильной группой, 5-фенилпентильной группой, 1-фенилгексильной группой, 2-фенилгексильной группой, 3-фенилгексильной группой, 4-фенилгексильной группой, 5-фенилгексильной группой или 6-фенилгексильной группой; предпочтительно ею является алкильная группа, замещенная арильной группой, содержащей от 6 до 10 атомов углерода, такая как бензильная группа, 1-нафтилметильная группа, 2-нафтилметильная группа, 1-фенэтильная группа, 2-фенэтильная группа, 1-нафтилэтильная группа, 2-нафтилэтильная группа, 1-фенилпропильная группа, 2-фенилпропильная группа, 3-фенилпропильная группа или 1-нафтилпропильная группа; а более предпочтительно ею является бензильная группа.
«Алкокси-группой, содержащей от 1 до 6 атомов углерода, которая необязательно замещена, по меньшей мере, одной арильной группой, содержащей от 6 до 14 атомов углерода, которая необязательно может быть замещена заместителями в количестве от 1 до 5, выбираемыми из заместителей А,» в определении заместителя R3 в приведенной выше формуле (II) могут являться, например, бензокси-группа, 1-нафтилметокси-группа, 2-нафтилметокси-группа, инденилметокси-группа, 1-фенэтокси-группа, 2-фенэтокси-группа, 1-нафтилэтокси-группа, 2-нафтилэтокси-группа, 1-фенилпропокси-группа, 2-фенилпропокси-группа, 3-фенилпропокси-группа, 1-нафтилпропокси-группа, 2-нафтилпропокси-группа, 3-нафтилпропокси-группа, 1-фенилбутокси-группа, 2-фенилбутокси-группа, 3-фенилбутокси-группа, 4-фенилбутокси-группа, 1-нафтилбутокси-группа, 2-нафтилбутокси-группа, 3-нафтилбутокси-группа, 4-нафтилбутокси-группа, 1-фенилпентокси-группа, 2-фенилпентокси-группа, 3-фенилпентокси-группа, 4-фенилпентокси-группа, 5-фенилпентокси-группа, 1-фенилгексилокси-группа, 2-фенилгексилокси-группа, 3-фенилгексилокси-группа, 4-фенилгексилокси-группа, 5-фенилгексилокси-группа или 6-фенилгексилокси-группа; предпочтительно ею является алкильная группа, замещенная арильной группой, содержащей от 6 до 10 атомов углерода, такая как бензокси-группа, 1-нафтилметокси-группа, 2-нафтилметокси-группа, 1-фенэтокси-группа, 2-фенэтокси-группа, 1-нафтилэтокси-группа, 2-нафтилэтокси-группа, 1-фенилпропокси-группа, 2-фенилпропокси-группа, 3-фенилпропокси-группа или 1-нафтилпропокси-группа; а более предпочтительно ею является бензокси-группа.
Там, где R3 представляет собой «арилокси-группу, содержащую от 6 до 14 атомов углерода, которая необязательно может быть замещена заместителями в количестве от 1 до 5, выбираемыми из заместителей А,» им предпочтительно является арилокси-группа, содержащая от 6 до 10 атомов углерода, которая необязательно замещена заместителями в количестве от 1 до 3, выбираемыми из группы заместителей А; более предпочтительно им является фенокси-группа, которая необязательно замещена заместителями в количестве от 1 до 3, выбираемыми из группы заместителей А; еще более предпочтительно им является фенокси-группа, необязательно замещенная атомами галогена, алкильными группами, содержащими от 1 до 6 атомов углерода, гидрокси-группами или нитро-группами в количестве от 1 до 3; а наиболее предпочтительно им являются фенокси-группа или п-нитрофенокси-группа.
Там, где сополимер настоящего изобретения содержит основную группу, соединение можно превратить в его фармакологически приемлемую соль в результате проведения реакции между некоторыми или всеми данными основными группами и кислотой. Кроме того, сополимеры настоящего изобретения содержат кислотные карбоксильные группы, и сополимер можно превратить в его фармакологически приемлемую соль в результате проведения реакции между некоторыми или всеми данными карбоксильными группами и основанием.
Предпочтительные примеры фармакологически приемлемых солей, получаемых при содержании в сополимерах настоящего изобретения основной группы, включают соли неорганических кислот, такие как соли галогенводородных кислот (например, гидрохлориды, гидробромиды и гидроиодиды), нитраты, перхлораты, сульфаты и фосфаты; соли органических кислот, такие как низшие алкансульфонаты, в которых их низшее алкильное звено представляет собой определенное выше (например, метансульфонаты, трифторметансульфонаты и этансульфонаты), арилсульфонаты, в которых их арильное звено представляет собой определенное выше (например, бензолсульфонат или п-толуолсульфонат), ацетаты, малаты, фумараты, сукцинаты, цитраты, аскорбаты, тартраты, оксалаты и малеаты; и соли аминокислот, такие как соли глицина, соли лизина, соли аргинина, соли орнитина, глютаминаты и аспартаты. В особенности предпочтительными являются соли галогенводородных кислот.
Предпочтительные примеры фармакологически приемлемых солей, получаемых при содержании в сополимерах настоящего изобретения кислотной карбоксильной группы, включают соли металлов, такие как соли щелочных металлов (например, натриевые соли, калиевые соли и литиевые соли), соли щелочноземельных металлов (например, кальциевые соли и магниевые соли), соли металлов, такие как соли алюминия, соли железа, соли цинка, соли меди, соли никеля и соли кобальта; соли аминов, такие как неорганические соли аминов (например, аммониевые соли) и органические соли аминов (например, соли трет-октиламина, соли дибензиламина, соли морфолина, соли глюкозамина, соли фенилглициналкилового сложного эфира, соли этилендиамина, соли N-метилглюкамина, соли гуанидина, соли диэтиламина, соли триэтиламина, соли дициклогексиламина, соли N,N'-дибензилэтилендиамина, соли хлорпрокаина, соли прокаина, соли диэтаноламина, соли N-бензилфенэтиламина, соли пиперазина, соли тетраметиламмония и соли трис(гидроксиметил)аминометана; и соли аминокислот, такие как соли глицина, соли лизина, соли аргинина, соли орнитина, глютаминаты и аспартаты. В особенности предпочтительными являются соли щелочных металлов и соли щелочноземельных металлов.
В настоящем изобретении «структурное элементарное звено» определяют как минимальное элементарное звено, образующее сополимер изобретения, и его изображают при определении указанного выше сополимера в качестве элементарного звена, описанного формулой (I), или элементарного звена, описанного формулой (II). «Структурное элементарное звено» не является структурой мономерного исходного вещества, использованного в реакции полимеризации при синтезе сополимера настоящего изобретения; точнее говоря, это элементарное звено, которое получают из указанного мономерного исходного вещества и которое присутствует в указанном сополимере настоящего изобретения.
В настоящем изобретении фраза «последовательность «голова к голове»» обозначает то, что для структурных элементарных звеньев, описанных формулами (I) и (II), получают конфигурацию, продемонстрированную в нижеследующей формуле:
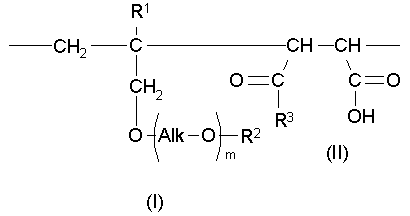
В настоящем изобретении фраза «последовательность «голова к хвосту»» обозначает то, что для структурных элементарных звеньев, описанных формулами (I) и (II), получают конфигурацию, продемонстрированную в нижеследующей формуле:
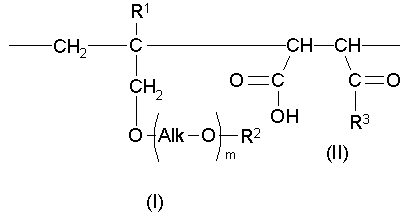
В настоящем изобретении сополимеры и их фармакологически приемлемые соли могут представлять собой чередующиеся сополимеры или статистические сополимеры. Чередующимися сополимерами являются те, в которых соотношение структурных элементарных звеньев, описанных формулой (I), и структурных элементарных звеньев, описанных формулой (II), равно 1:1, и для структурных элементарных звеньев, описанных формулой (I), и структурных элементарных звеньев, описанных формулой (II), получают конфигурацию в виде последовательности с чередованием «голова к голове», последовательности с чередованием «голова к хвосту» или смешанной последовательности с чередованием «голова к голове» и «голова к хвосту». Статистическими сополимерами являются те, в которых для структурных элементарных звеньев, описанных формулой (I), и структурных элементарных звеньев, описанных формулой (II), получают конфигурацию в виде статистической последовательности.
В настоящем изобретении «соотношение, задаваемое составом,» представляет собой среднее соотношение между количеством структурных элементарных звеньев, описанных формулой (I), и количеством структурных элементарных звеньев, описанных формулой (II), в сополимере настоящего изобретения. Если сополимер по существу представляет собой чередующийся сополимер, то тогда соотношение, задаваемое составом, будет равно 1:1. Однако, если сополимер представляет собой статистический сополимер, указанное соотношение может варьироваться. В сополимерах настоящего изобретения на указанное соотношение особенных ограничений не накладывается, и обычно оно может находиться в диапазоне от 10:1 до 1:10; предпочтительно от 3:1 до 1:8, более предпочтительно от 2:1 до 1:2 или от 1:2 до 1:6, а наиболее предпочтительно оно или равно 1:1, или находится в диапазоне от 1:2 до 1:4. Необходимо отметить, что значения соотношения, задаваемого составом, неизбежно в некоторой степени варьируются, что обуславливается наличием незначительных изменений в исходных веществах, условиях полимеризации и тому подобном. В результате приведенные соотношения, задаваемые составом, представляют собой приблизительные значения; вариации для значений соотношений, задаваемых составом, приведенных выше, величиной вплоть до ±30% все еще рассматриваются как попадающие в объем указанных соотношений.
Соотношение, задаваемое составом, для сополимеров настоящего изобретения можно определить с использованием известных аналитических методик. В результате определения содержания карбоксильных групп в сополимере (ммоль/г) методом кондуктометрического титрования [определение, требующее получения соответствующего полностью гидролизованного сополимера (то есть, R3 представляет собой ОН) или в результате гидролиза сополимера, подвергаемого анализу, или в результате независимого синтеза соответствующего гидролизованного сополимера] и, исходя из знания формульных масс для каждого из структурных элементарных звеньев указанного сополимера, можно определить соотношение, задаваемое составом, с использованием нижеследующей формулы:
Rii/Ri=(C×FWi)/(2000-C×FWii),
где Ri представляет собой среднее количество структурных элементарных звеньев (I), Rii представляет собой среднее количество структурных элементарных звеньев (II), С представляет собой содержание карбоксильных групп в сополимере (ммоль/г), FWi представляет собой формульную массу структурного элементарного звена (I), a FWii представляет собой формульную массу структурного элементарного звена (II).
В общем случае молекулярную массу полимера определяют в виде относительной молекулярной массы в приведении к молекулярной массе стандартного соединения, которое включает структуру, подобную структуре указанного полимера, и характеризуется известным значением абсолютной молекулярной массы. Такой способ оценки зачастую используется при определении молекулярной массы новых сополимеров, таких как сополимеры настоящего изобретения.
Средняя молекулярная масса сополимера настоящего изобретения представляет собой значение, измеренное методом гель-фильтрационной хроматографии с использованием в качестве стандартного соединения полимера, характеризующегося известной абсолютной молекулярной массой. Природа геля, условия элюирования и полимер с известной абсолютной молекулярной массой, используемый для сравнения, специалист в соответствующей области может надлежащим образом выбрать с использованием известных методик и общедоступных сведений, например, см. работу "Comprehensive Polymer Science", pub. Pergamon Press (Oxford) 1989. Стандартным соединением, используемым для целей сравнения, предпочтительно является полимер, обладающий подобными структурой и свойствами. В случае сополимеров настоящего изобретения область боковых цепей рассматривается в качестве характеристической структуры. Поэтому стандартное соединение, используемое для сравнения, предпочтительно является полимером, включающим структуру, подобную структуре области боковых цепей сополимера настоящего изобретения, среднюю молекулярную массу которого необходимо измерить. Используемым стандартным соединением более предпочтительно является поли(этиленгликоль).
В настоящем изобретении «средняя степень полимеризации» представляет собой среднее значение степени полимеризации структурных элементарных звеньев в сополимере настоящего изобретения, то есть, это среднее количество структурных элементарных звеньев в указанном сополимере. Его определяют на основе средней молекулярной массы сополимера, формульных масс каждого из структурных элементарных звеньев указанного сополимера и соотношения, задаваемого составом, для структурных элементарных звеньев в указанном сополимере. «Среднюю степень полимеризации» сополимера настоящего изобретения, содержащего структурные элементарные звенья I и II, можно рассчитать с использованием нижеследующей формулы:
Средняя степень полимеризации=Мс/(FWi×Ri+FWii×Rii),
где Мс представляет собой среднюю молекулярную массу сополимера, FWi и FWii представляют собой формульные массы для каждого из структурных элементарных звеньев I и II соответственно, а Ri и Rii представляют собой доли структурных элементарных звеньев I и II в сополимере, вычисленные из соотношения, задаваемого составом (соотношение, задаваемое составом=Ri:Rii; Ri+Rii=1).
В приведенной выше формуле для определения средней степени полимеризации сополимера настоящего изобретения все параметры, такие как значение соотношения, задаваемого составом, для структурных элементарных звеньев, средняя молекулярная масса сополимера и формульные массы структурных элементарных звеньев, можно определить так, как это обсуждается выше. Для того чтобы это сделать, необходимо получить соответствующий сополимер, подвергнутый полному гидролизу, аммонолизу, аминолизу или алкоголизу (предпочтительно соответствующий сополимер, подвергнутый полному гидролизу). В альтернативном варианте, если исходный сополимер, используемый при получении сополимера настоящего изобретения, характеризуется известным составом, данные значения можно определить без необходимого проведения анализа. На среднюю степень полимеризации в сополимерах настоящего изобретения особенных ограничений не накладывается. Обычно она находится в диапазоне от 5 до 200; предпочтительно она находится в диапазоне от 5 до 50; более предпочтительно от 5 до 20 или от 20 до 30 или от 30 до 40.
В сополимерах настоящего изобретения структурные элементарные звенья, описанные формулой (II), могут включать, по меньшей мере, одно структурное элементарное звено, описанное формулой (II), где R3 представляет собой необязательно замещенную алкокси-группу, содержащую от 1 до 6 атомов углерода, необязательно замещенную арилокси-группу или группу, описанную формулой -NR4R5, и необязательно, по меньшей мере, одно структурное элементарное звено, описанное формулой (II), в которой R3 представляет собой гидроксильную группу. Соотношение между структурными элементарными звеньями, описанными формулой (II), в которой R3 представляет собой гидрокси-группу, и структурными элементарными звеньями, описанными формулой (II), в которой R3 представляет собой необязательно замещенную алкокси-группу, необязательно замещенную арилокси-группу или группу, описанную формулой -NR4R5, называют соотношением, задаваемым гидролизом. В результате определения уровня содержания карбоксильных групп в сополимере (ммоль/г) методом кондуктометрического титрования и исходя из знания формульных масс для каждого из структурных элементарных звеньев указанного сополимера и соотношения, задаваемого составом, (см. выше) можно определить соотношение, задаваемое гидролизом, Н:А с использованием нижеследующей формулы:
где Н:А представляет собой соотношение, задаваемое гидролизом, Ri:Rii представляет собой соотношение, задаваемое составом, С представляет собой уровень содержания карбоксильных групп в сополимере (ммоль/г), FWi представляет собой формульную массу структурного элементарного звена (I), FWii представляет собой формульную массу структурного элементарного звена (II), где R3 представляет собой ОН, а FW(A)ii представляет собой формульную массу структурного элементарного звена (II), где R3 представляет собой необязательно замещенную алкокси-группу, необязательно замещенную арилокси-группу или группу, описанную формулой -NR4R5.
Предпочтительные диапазоны для соотношения, задаваемого гидролизом, находятся в интервале от 5:5 до 0:10, от 4:6 до 0:10, от 3:7 до 0:10, от 2:8 до 0:10 и от 1:9 до 0:10. Необходимо отметить, что значения соотношения, задаваемого гидролизом, неизбежно в некоторой степени варьируются, что обуславливается наличием незначительных изменений в исходных веществах, условиях реакции и тому подобном. В результате соотношения, задаваемые гидролизом, приведенные в настоящей заявке, представляют собой приблизительные значения; вариации для значений соотношений, задаваемых гидролизом, приведенных в настоящей заявке, величиной вплоть до ±30% все еще рассматриваются как попадающие в объем указанных соотношений.
Размер молекул сополимеров изобретения можно измерить с использованием известных аналитических методик, таких как эксклюзионная хроматография размеров, с использованием в качестве стандартов белков с известным размером молекулы (приведенные ниже примеры 15 и 16 предлагают примеры применения такой методики). С использованием эксклюзионной хроматографии размеров размер молекулы сополимера получают в виде ее радиуса Стокса. В настоящем изобретении на размер молекулы сополимеров особенных ограничений не накладывается. Обычно радиус Стокса сополимеров настоящего изобретения составляет 9,3 нм или менее, предпочтительно он составляет 7,3 нм или менее, более предпочтительно он составляет 6,2 нм или менее, 4,7 нм или менее или 3,1 нм или менее, или же он находится в диапазоне от 3,1 до 6,2 нм или в диапазоне от 1,5 до 4,7 нм.
Сополимеры и их фармакологически приемлемые соли настоящего изобретения можно получать при проведении подходящей реакции сополимеризациии, известной специалисту в соответствующей области [см., например, работу "Comprehensive Polymer Science" published by Pergamon Press (Oxford) in 1989], с использованием исходных мономеров, описанных нижеследующими формулами (IV) и (V):
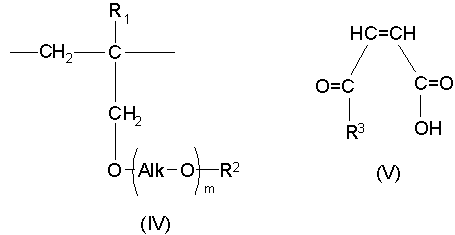
(где m, Alk и R1, R2 и R3 представляют собой определенное выше). Для того чтобы получить сополимер, характеризующийся конкретной желательной средней степенью полимеризации или молекулярной массой, полученный сополимер можно подвергнуть фракционированию с использованием гель-фильтрационной хроматографии.
Мономеры на современном уровне техники известны, или же их легко можно получить в соответствии со способом, хорошо известным специалисту в соответствующей области, [см., например, работу J. M. Harris, "Laboratory synthesis of polyethylene glycol derivatives", Rev. Macromol. Chem. Phys. C25, 326-373 (1985) и японский патент № 2621308]. Мономеры, описанные формулой (V), легко можно получить при проведении для малеинового ангидрида одной или нескольких реакций, выбираемых из группы, состоящей из (а) гидролиза, (b) аммонолиза, (с) аминолиза и (d) алкоголиза.
В альтернативном варианте сополимер или его фармакологически приемлемую соль можно получать в результате проведения для одного или нескольких звеньев ангидрида карбоновой кислоты, описанных формулой (III), в сополимере, который содержит в качестве образующих его элементарных звеньев
(а) одно или несколько структурных элементарных звеньев, которые могут быть идентичными или отличными друг от друга, и которые описываются приведенной ниже формулой (I):
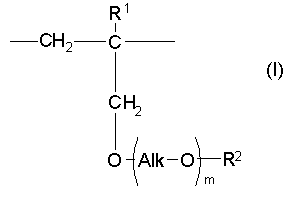
где:
m, Alk, R1 и R2 представляют собой определенное выше, и
(b) указанное структурное элементарное звено, содержащее звено ангидрида карбоновой кислоты, описанное формулой (III):
одной или нескольких реакций, выбираемых из группы, состоящей из (i) гидролиза, (ii) аммонолиза, (iii) аминолиза и (iv) алкоголиза. Исходные сополимеры, содержащие элементарные звенья, описанные формулами (I) и (III), или хорошо известны на современном уровне техники (например, сополимеры, такие как АМ-0530К и АМ-1510К, можно приобрести в компании NOF Corporation), или их легко можно получить в соответствии со способом, хорошо известным специалисту в соответствующей области [см., например, работу Yoshimoto et al., "Polyethylene glycol derivative-modified cholesterol oxidase soluble and active in benzene", Biochem. Biophys. Res. Comm. 148, 876-882 (1987), японский патент № 2621308, публикацию японской патентной заявки № 2003-105040 и публикацию японской патентной заявки № 2003-105003].
Для получения сополимера, характеризующегося желательной средней степенью полимеризации или желательной молекулярной массой, полученный сополимер можно подвергнуть фракционированию способом гель-фильтрационной хроматографии.
В настоящем изобретении сополимеры настоящего изобретения можно получить в результате проведения для одного или нескольких звеньев ангидрида карбоновой кислоты, описанных формулой (III), одной или нескольких реакций, выбираемых из группы, состоящей из (i) гидролиза, (ii) аммонолиза, (iii) аминолиза и (iv) алкоголиза.
В настоящем изобретении «гидролиз» обозначает реакцию с раскрытием цикла для звена ангидрида карбоновой кислоты, описанного формулой (III), под действием воды с получением структурного элементарного звена, описанного формулой (II), где R3 представляет собой гидрокси-группу. На природу фактической реакции гидролиза особенных ограничений не накладывается до тех пор, пока речь идет о способе, обычно используемом специалистами в соответствующей области.
Примеры подходящего растворителя, используемого в реакции гидролиза, включают: воду; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликольдиметиловый эфир; и амиды, такие как формамид, N,N-диметилформамид, N,N-диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон и гексаметилфосфортриамид. В числе данных растворителей предпочтительными являются вода или диоксан.
Используемым реагентом в подходящем случае является вода. Если воду используют в качестве растворителя, то тогда в добавлении дополнительного количества воды необходимости нет. В дополнение к этому, для целей ускорения реакции можно добавлять основание. Примеры такого основания включают: неорганические основания, такие как карбонаты щелочных металлов (например, карбонат натрия, карбонат калия и карбонат лития), гидрокарбонаты щелочных металлов (например, гидрокарбонат натрия, гидрокарбонат калия и гидрокарбонат лития), гидриды щелочных металлов (например, гидрид лития, гидрид натрия и гидрид калия), гидроксиды щелочных металлов (например, гидроксид натрия, гидроксид калия, гидроксид бария и гидроксид лития), фториды щелочных металлов (например, фторид натрия и фторид калия); и органические основания, такие как алкоксиды щелочных металлов (например, метоксид натрия, этоксид натрия, метоксид калия, этоксид калия, трет-бутоксид калия и метоксид лития), N-метилморфолин, триэтиламин, трипропиламин, трибутиламин, диизопропилэтиламин, дициклогексиламин, N-метилпиперидин, пиридин, 4-пирролидинопиридин, пиколин, 4-(N,N-диметиламино)пиридин, 2,6-ди(трет-бутил)-4-метилпиридин, хинолин, N,N-диметиланилин, N,N-диэтиланилин, 1,5-диазабицикло[4.3.0]нон-5-ен (DBN), 1,4-диазабицикло[2.2.2]октан (DABCO) и 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU). В числе данных оснований предпочтительными являются органические основания, а наиболее предпочтительным является пиридин. В данном отношении необходимо отметить, что тогда, когда реакция гидролиза удовлетворительно протекает в отсутствие основания, в добавлении основания необходимости нет.
Температура реакции варьируется в зависимости от исходного соединения и реагента, но обычно она находится в диапазоне от 0 до 100°С, а предпочтительно в диапазоне от 20 до 60°С.
Время реакции варьируется в зависимости от температуры реакции, исходного соединения, реагента и типа использованного растворителя, но обычно оно находится в диапазоне от 10 минут до 3 дней, а предпочтительно в диапазоне от 6 часов до 24 часов.
После завершения реакции желательное соединение реакции гидролиза можно выделить из реакционной смеси с использованием обычного способа, известного специалисту в соответствующей области. Например, получающуюся в результате реакционную смесь можно сконденсировать с использованием ультрафильтрационной мембраны, а после этого подвергнуть лиофильной сушке с получением целевого соединения реакции. В альтернативном варианте желательное соединение можно использовать в виде раствора, без его выделения тогда, когда его необходимо будет использовать для модифицирования белков.
Таким образом, полученное желательное соединение при необходимости можно подвергнуть дополнительной очистке с использованием обычной методики, такой как, например, гель-фильтрационная хроматография. Для получения соединения, характеризующегося конкретной желательной средней степенью полимеризации или желательной молекулярной массой, подвергнутое очистке соединение можно дополнительно фракционировать с использованием гель-фильтрационной хроматографии.
В настоящем изобретении «аммонолиз» обозначает реакцию с раскрытием цикла для звена ангидрида карбоновой кислоты, описанного формулой (III), под действием аммиака с получением звена, описанного формулой (II), где R3 представляет собой аминогруппу. На природу фактической реакции аммонолиза особенных ограничений не накладывается до тех пор, пока речь идет о способе, обычно используемом специалистами в соответствующей области.
Примеры растворителя, подходящего для использования, включают: воду; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликольдиметиловый эфир; и амиды, такие как формамид, N,N-диметилформамид, N,N-диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон и гексаметилфосфортриамид. В числе данных растворителей предпочтительными являются вода или диоксан. Необходимо отметить, что используемый реагент также можно использовать и в качестве растворителя.
Примеры подходящих для использования реагентов включают газообразный аммиак и водно-аммиачный раствор, а предпочтителен аммиак.
Температура реакции варьируется в зависимости от исходного соединения и реагента, но обычно она находится в диапазоне от 0 до 100°С, а предпочтительно в диапазоне от 10 до 40°С.
Время реакции варьируется в зависимости от температуры реакции, исходного соединения, реагента и типа использованного растворителя, но обычно оно находится в диапазоне от 10 минут до 3 дней, а предпочтительно в диапазоне от 6 часов до 24 часов.
После завершения реакции желательное соединение реакции аммонолиза можно выделить из реакционной смеси с использованием обычного способа, известного специалисту в соответствующей области. Для выделения целевого соединения получающуюся в результате реакционную смесь можно, например, подвергнуть обработке следующим образом: (1) диализ через полупроницаемую мембрану с использованием кислоты, такой как водный раствор уксусной кислоты, для удаления избыточного количества аммиака (диализ проводят в таких условиях, при которых раствор реакционной смеси не становится кислым, при добавлении воды по мере надобности) с последующими конденсацией с использованием ультрафильтрационной мембраны, а после этого лиофильной сушкой таким образом полученного конденсата; или (2) добавление водного раствора гидроксида натрия и несмешиваемого органического растворителя, такого как диэтиловый эфир, встряхивание получающейся в результате смеси (встряхивание можно проводить два или более чем два раза по мере надобности), а после этого выделенный водный слой, содержащий целевое соединение, подвергают лиофильной сушке с получением целевого соединения. В альтернативном варианте желательное соединение можно использовать в виде раствора, без его выделения тогда, когда его необходимо будет использовать для модифицирования белков.
Таким образом, полученное желательное соединение при необходимости можно подвергнуть дополнительной очистке с использованием обычной методики, такой как, например, гель-фильтрационная хроматография. Для получения соединения, характеризующегося конкретной желательной средней степенью полимеризации или желательной молекулярной массой, подвергнутое очистке соединение можно дополнительно фракционировать с использованием гель-фильтрационной хроматографии.
Необходимо отметить, что целевое соединение реакции аммонолиза может содержать структурные элементарные звенья, описанные формулой (II), где R3 представляет собой гидрокси-группу, в результате гидролиза некоторых элементарных звеньев, описанных формулой (III), в исходном соединении под действием воды, присутствующей в растворителе или реагенте. Необходимо также отметить, что для улучшения хранения к целевому соединению можно добавлять основание.
В настоящем изобретении «аминолиз» обозначает реакцию с раскрытием цикла для звена ангидрида карбоновой кислоты, описанного формулой (III), под действием амина с получением звена, описанного формулой (II), где R3 представляет собой группу, описанную формулой -NR4R5, где R4 и R5 представляют собой определенное выше. На природу фактической реакции аминолиза особенных ограничений не накладывается до тех пор, пока речь идет о способе, обычно используемом специалистами в соответствующей области.
Примеры растворителя, подходящего для использования, включают: воду; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликольдиметиловый эфир; и амиды, такие как формамид, N,N-диметилформамид, N,N-диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон и гексаметилфосфортриамид. В числе данных растворителей предпочтительными являются вода или диоксан. Необходимо отметить, что используемый реагент также можно использовать и в качестве растворителя.
Используемый реагент с очевидностью зависит от природы целевой группы, описанной формулой -NR4R5. Обычно используемые амины включают метиламин, диметиламин, этиламин, диэтиламин, 2-гидроксиэтиламин, ди-2-гидроксиэтиламин, н-пропиламин, ди-н-пропиламин, изопропиламин, диизопропиламин, 1-амино-2-пропанол и 2-гидроксиизопропиламин и их водные растворы. В числе данных реагентов предпочтительные реагенты включают водный раствор диметиламина и 1-амино-2-пропанол; наиболее предпочтителен водный раствор диметиламина.
Температура реакции варьируется в зависимости от исходного соединения и реагента, но обычно она находится в диапазоне от 0 до 100°С, а предпочтительно в диапазоне от 10 до 40°С.
Время реакции варьируется в зависимости от температуры реакции, исходного соединения, реагента и типа использованного растворителя, но обычно оно находится в диапазоне от 10 минут до 3 дней, а предпочтительно в диапазоне от 6 часов до 36 часов.
После завершения реакции желательное соединение реакции аминолиза можно выделить из реакционной смеси с использованием обычного способа, известного специалисту в соответствующей области. Для выделения целевого соединения получающуюся в результате реакционную смесь можно, например, подвергнуть обработке следующим образом: (1) диализ через полупроницаемую мембрану с использованием кислоты, такой как водный раствор уксусной кислоты, для удаления избыточного количества аммиака (диализ проводят в таких условиях, при которых раствор реакционной смеси не становится кислым, при добавлении воды по мере надобности) с последующей конденсацией с использованием ультрафильтрационной мембраны, а после этого лиофильной сушкой таким образом полученного конденсата; или (2) добавление водного раствора гидроксида натрия и несмешиваемого органического растворителя, такого как диэтиловый эфир, встряхивание получающейся в результате смеси (встряхивание можно проводить два или более чем два раза по мере надобности), а после этого выделенный водный слой, содержащий целевое соединение, подвергают лиофильной сушке с получением целевого соединения. В альтернативном варианте желательное соединение можно использовать в виде раствора, без его выделения тогда, когда его необходимо будет использовать для модифицирования белков.
Таким образом, полученное желательное соединение при необходимости можно подвергнуть дополнительной очистке с использованием обычной методики, такой как, например, гель-фильтрационная хроматография. Для получения соединения, характеризующегося конкретной желательной средней степенью полимеризации или желательной молекулярной массой, подвергнутое очистке соединение можно дополнительно фракционировать с использованием гель-фильтрационной хроматографии.
Необходимо отметить, что целевое соединение реакции аминолиза может содержать структурные элементарные звенья, описанные формулой (II), где R3 представляет собой гидрокси-группу, в результате гидролиза некоторых элементарных звеньев, описанных формулой (III), в исходном соединении под действием воды, присутствующей в растворителе или реагенте. Необходимо также отметить, что для улучшения хранения к целевому соединению можно добавлять основание.
В настоящем изобретении «алкоголиз» обозначает реакцию с раскрытием цикла для звена ангидрида карбоновой кислоты, описанного формулой (III), под действием спирта или арилового спирта с получением звена, описанного формулой (II), где R3 представляет собой алкокси- или арилокси-группу, определенную выше. На природу фактической реакции алкоголиза особенных ограничений не накладывается до тех пор, пока речь идет о способе, обычно используемом специалистами в соответствующей области.
Примеры растворителя, используемого в реакции алкоголиза, включают воду; и амиды, такие как формамид, N,N-диметилформамид, N,N-диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон и гексаметилфосфортриамид. В числе данных растворителей предпочтительной является вода. Необходимо отметить, что используемый реагент также можно использовать и в качестве растворителя.
Используемый реагент с очевидностью зависит от природы целевой алкокси- или арилокси-группы. Типичные примеры спирта, используемого для получения желательной алкокси-группы, включают метанол, этанол, этиленгликоль, н-пропанол, пропиленгликоль, 2-гидрокси-н-пропанол и изопропанол и их водные растворы, в то время как типичные примеры спирта, используемого для получения желательной арилокси-группы, включают фенол и п-нитрофенол. В числе данных реагентов предпочтительным является этанол.
Температура реакции варьируется в зависимости от исходного соединения и реагента, но обычно она находится в диапазоне от 0 до 100°С, а предпочтительно в диапазоне от 10 до 40°С.
Время реакции варьируется в зависимости от температуры реакции, исходного соединения, реагента и типа использованного растворителя, но обычно оно находится в диапазоне от 10 минут до 3 дней, а предпочтительно в диапазоне от 6 часов до 36 часов.
После завершения реакции желательное соединение реакции алкоголиза можно выделить из реакционной смеси с использованием обычного способа, известного специалисту в соответствующей области. Например, для удаления избыточного спирта можно провести конденсирование реакционной смеси с использованием ультрафильтрационной мембраны, добавление воды, а после этого дополнительное конденсирование смеси еще два или более чем два раза с использованием ультрафильтрационной мембраны, а после этого полученный конденсат подвергают лиофильной сушке для получения целевого соединения. В альтернативном варианте желательное соединение можно использовать в виде раствора, без его выделения тогда, когда его необходимо будет использовать для модифицирования белков.
Таким образом, полученное желательное соединение при необходимости можно подвергнуть дополнительной очистке с использованием обычной методики, такой как, например, применение гель-фильтрационной колонки. Для получения соединения, характеризующегося конкретной желательной средней степенью полимеризации или желательной молекулярной массой, подвергнутое очистке соединение можно дополнительно фракционировать с использованием гель-фильтрационной хроматографии.
Необходимо отметить, что целевое соединение реакции алкоголиза может содержать структурные элементарные звенья, описанные формулой (II), где R3 представляет собой гидрокси-группу, в результате гидролиза некоторых элементарных звеньев, описанных формулой (III), в исходном соединении под действием воды, присутствующей в растворителе или реагенте. Необходимо также отметить, что для улучшения хранения к целевому соединению можно добавлять основание.
Тогда, когда желательным сополимером настоящего изобретения является фармакологически приемлемая соль, на способ получения указанной соли особенных ограничений не накладывается до тех пор, пока речь идет о способе, обычно используемом специалистами в соответствующей области. Например, фармакологически приемлемую соль можно получить в результате растворения сополимера изобретения в органическом растворителе, добавления к таким образом полученному раствору основания и после этого сбора получающейся в результате соли, которая выпадает в осадок.
Для регулирования средней молекулярной массы, доли конфигурации с чередованием и соотношения, задаваемого составом, для сополимеров настоящего изобретения возможно использование широкого набора приемов.
Хорошо известно, что мономер малеиновый ангидрид имеет тенденцию к полимеризации с чередованием с сомономерами, в том числе полиоксиэтиленаллилметиловым простым диэфиром [например, см. Comprehensive Polymer Science Vol. 4, Chain polymerization Part II, Ed. by G. C. Eastmond et al., pp. 377-422, Pergamon Press (1989); T. Yoshimoto et al., Biochemical Biophysical Research Communication Vol. 148, 876-882 (1987)]. Среднюю молекулярную массу и конфигурацию полимеров специалист в соответствующей области может регулировать (например, см. работу T. Ohtsu and M. Kinoshita, Koubunshi Gousei no Jikkenhou pp. 125-154, Kagaku-Dojin 1972). В дополнение к этому, несмотря на то, что малеиновый ангидрид имеет тенденцию к полимеризации с чередованием с сомономерами, специалистам в соответствующей области хорошо известна возможность того, что в соотношении, задаваемом составом, доля, образуемая в сополимере элементарными звеньями малеинового ангидрида, будет превышать 50% (мол.) (японские патенты № 2621308, № 2701295, № 2803265, № 3271265, № 3035675 и № 3106265 и публикации японских патентных заявок № 2003-105003 и № 2003-105040).
В общем случае для получения полимеров с повышенной молекулярной массой и большей долей конфигурации с чередованием полимеризацию необходимо проводить в мягких условиях, таких как пониженная температура и пониженная концентрация инициатора. Повышенная концентрация мономера и пониженная концентрация растворителя, что уменьшает относительную концентрацию инициатора, также в результате приводят к получению повышенной молекулярной массы и повышенной доли конфигурации с чередованием.
В альтернативном варианте, для получения полимеров с пониженной молекулярной массой и пониженной долей конфигурации с чередованием благоприятны повышенная температура, повышенная концентрация инициатора, пониженная концентрация мономера и повышенная концентрация растворителя. Кроме того, при таких относительно жестких условиях полимеризации и/или при наличии малеинового ангидрида в количестве, превышающем 50% (мол.) от полного количества мономеров в смеси мономеров, в соотношении, задаваемом составом, у получающегося в результате сополимера доля элементарных звеньев малеинового ангидрида может превышать 50% (мол.).
Некоторые примеры условий полимеризации для полиоксиэтиленаллилметилового простого диэфира и малеинового ангидрида описываются в T. Yoshimoto et al., Biochemical Biophysical Research Communication Vol. 148, 876-882 (1987). В соответствии с данной статьей перемешивали полиоксиэтиленаллилметиловый простой диэфир, малеиновый ангидрид, толуол и бензоилпероксид (инициатор) и проводили полимеризацию при кипячении с обратным холодильником при 80°С в течение 7 часов. Получающийся в результате полимер характеризовался молекулярной массой, равной 13 кДа, и имел 8 цепей ПЭГ и 8 элементарных звеньев малеинового ангидрида. Конфигурация с чередованием для сополимера была предложена исходя из того, что сополимер характеризовался соотношением, задаваемым составом, равным 1:1, и что мономер малеиновый ангидрид имеет тенденцию к сополимеризации с сомономерами с чередованием. Если будет требоваться сополимер, характеризующийся пониженной молекулярной массой и пониженной долей конфигурации с чередованием, то тогда необходимо будет использовать повышенную концентрацию инициатора, пониженную концентрацию мономера и/или повышенную концентрацию растворителя. Например, если в качестве растворителя в полимеризации используют толуол, и полимеризацию проводят при 1 атмосфере, то тогда полимеризацию можно проводить при температуре вплоть до его температуры кипения, равной 110°С. Кроме того, в случае другого растворителя, температура кипения которого превышает температуру кипения толуола, предпочтительной является повышенная температура. В таких случаях подходящим образом необходимо будет выбрать тип инициатора, поскольку каждый инициатор характеризуется своей собственной специфической константой скорости разложения, и при данных повышенных температурах будут разлагаться определенные инициаторы. На современном уровне техники уже известны многочисленные инициаторы с различными скоростями разложения [например, см. работу Polymer Handbook, Third Edition, pp. II/1-II/65, Ed. by J. Brandrup and E. Immergut, John Wiley & Sons (1989)].
В других документах предшествующего уровня техники, таких как публикация японской патентной заявки № 2003-105003 также описываются и альтернативные растворители, инициаторы и условия реакции, приводящие к изменению соотношения, задаваемого составом, молекулярной массы и доли конфигурации с чередованием в реакциях сополимеризации полиоксиэтиленаллилметилового простого диэфира и малеинового ангидрида. Ксилол, например, имеет более высокую температуру кипения при 1 атмосфере (140°С) по сравнению с толуолом. Также известно, что в некоторых случаях реакции сополимеризации можно проводить без использования какого-либо растворителя, что устраняет ограничение для температуры полимеризации, определяемое температурой кипения растворителя. Также описываются и различные альтернативные инициаторы, такие как бензоилпероксид, ди-трет-бутилпероксид и трет-бутилпероксиизобутират и азо-инициаторы, такие как азобисизобутиронитрил. Уменьшить среднюю молекулярную массу сополимера также можно и в результате использования реагентов переноса цепи.
Если в соотношении, задаваемом составом, для сополимера требуется доля элементарных звеньев малеинового ангидрида в полном количестве мономерных элементарных звеньев, по величине превышающая 50%, то тогда в смеси мономеров необходимо будет воспользоваться повышенным процентным содержанием мономера малеинового ангидрида с использованием или без использования таких относительно жестких условий полимеризации.
Тогда, когда получают фармацевтически активный ингредиент с использованием сополимера настоящего изобретения или его фармакологически приемлемой соли и белка или его аналога или варианта, на соотношение между сополимером и белком или его аналогом или вариантом особенных ограничений не накладывается до тех пор, пока получающийся в результате комплекс будет характеризоваться желательной фармацевтической активностью. Обычно массовое соотношение между сополимером настоящего изобретения или его фармакологически приемлемой солью и белком или его аналогом или вариантом находится в диапазоне от 0,01 до 100:1; предпочтительно оно находится в диапазоне от 0,1 до 50:1; еще более предпочтительно оно находится в диапазоне от 1 до 10:1; а наиболее предпочтительно оно находится в диапазоне от 1 до 1,5:1.
При практическом применении фармацевтически активный ингредиент, содержащий сополимер настоящего изобретения или его фармакологически приемлемую соль и белок или его аналог или вариант, обычно получают в виде раствора белка, содержащего сополимер, или подвергнутого лиофильной сушке раствора, содержащего сополимер настоящего изобретения или его фармакологически приемлемую соль и белок или его аналог или вариант. В последнем случае подвергнутую лиофильной сушке форму активного ингредиента растворяют непосредственно перед тем, как ее необходимо будет использовать. В альтернативном варианте фармацевтически активный ингредиент можно получить в форме комплекта. В данном случае сополимер настоящего изобретения или его фармакологически приемлемую соль и белок или его аналог или вариант хранят в различных контейнерах, а после этого перемешивают с получением желательного фармацевтически активного ингредиента непосредственно перед использованием. В числе различных форм наиболее предпочтительной является форма, подвергнутая лиофильной сушке.
Тогда, когда сополимер настоящего изобретения или его фармакологически приемлемую соль используют в качестве модификатора белка, на соотношение между сополимером или его фармакологически приемлемой солью и белком или его аналогом или вариантом никаких особенных ограничений в виде какого-либо специфического значения не накладывается до тех пор, пока добиваются желательного модифицирования белка. Обычно массовое соотношение между сополимером настоящего изобретения или его фармакологически приемлемой солью и белком или его аналогом или вариантом находится в диапазоне от 0,01 до 100:1; предпочтительно оно находится в диапазоне от 0,1 до 50:1; еще более предпочтительно оно находится в диапазоне от 1 до 10:1; а наиболее предпочтительно оно находится в диапазоне от 1 до 1,5:1.
Тогда, когда сополимер настоящего изобретения используют в качестве модификатора белка, на способ модифицирования белка особенных ограничений не накладывается до тех пор, пока речь идет о способе, обычно используемом для модифицирования белка. Например, белок или его аналог или вариант можно модифицировать следующим образом.
Растворитель, используемый в способе модифицирования, представляет собой водный раствор, содержащий электролит, который обычно используют для растворения белка, и на значение его рН ограничений в виде какого-либо специфического значения не накладывается до тех пор, пока оно попадает в диапазон, в котором модификатор белка может быть отрицательно заряжен в результате диссоциации, по меньшей мере, некоторых карбоксильных групп в модификаторе белка настоящего изобретения, а белок или его аналог или вариант могут быть заряжены положительно. Например, значение рН может быть установлено величиной в диапазоне от 3 до изоэлектрической точки белка или его аналога или варианта, и предпочтительно его устанавливают величиной в диапазоне от 4 до 8. В данном отношении необходимо отметить, что изоэлектрическую точку белка или его аналога или варианта можно определить с использованием электрофореза.
Модификатор белка растворяют в описанном выше растворителе, а после этого таким образом полученный раствор добавляют к водному раствору указанного белка или его аналога или варианта. При необходимости полученный раствор можно встряхивать для облегчения протекания реакции. При желании можно провести доводку рН получающейся в результате смеси за счет добавления кислоты и/или основания. На соотношение между используемым модификатором белка настоящего изобретения и указанными белком или его аналогом или вариантом ограничений в виде какого-либо специфического значения не накладывается до тех пор, пока оно приводит к достижению желательного модифицирования белка. Обычно соотношение находится в диапазоне от 0,01 до 100 (мас.); предпочтительно в диапазоне от 0,1 до 50; более предпочтительно в диапазоне от 1 до 10; а наиболее предпочтительно оно находится в диапазоне от 1 до 1,5:1.
Температура реакции для способа модифицирования варьируется в зависимости от используемого соединения и реагента, но обычно она находится в диапазоне от 0 до 100°С, предпочтительно в диапазоне от 4 до 40°С, а наиболее предпочтительно в диапазоне от 30 до 40°С.
Время реакции варьируется в зависимости от температуры реакции, используемого соединения, реагента и типа используемого растворителя, но обычно оно находится в диапазоне от 5 минут до 14 дней, предпочтительно в диапазоне от 1 часа до 12 дней, а более предпочтительно в диапазоне от 5 дней до 10 дней.
Иногда в ходе проведения способа модифицирования белка используемые условия таковы, что некоторые структурные элементарные звенья, описанные формулой (II), в сополимере превращаются в элементарные звенья, описанные формулой (II), имеющие другую сущность. Например, тогда, когда R3 в структурных элементарных звеньях, описанных формулой (II), представляет собой группу, отличную от гидрокси-группы, условия, используемые для способа модифицирования, таковы, что некоторые из указанных групп R3 подвергаются гидролизу с получением структурных элементарных звеньев, описанных формулой (II), где R3 представляет собой гидрокси-группу.
Как отмечалось выше, комплексы настоящего изобретения содержат, по меньшей мере, один белок или его аналог или вариант, которые связывают, по меньшей мере, с одним сополимером настоящего изобретения или его фармакологически приемлемой солью. В указанном комплексе белок или его аналог или вариант и сополимер или его фармакологически приемлемую соль связывают друг с другом при помощи химической связи, такой как ковалентная связь, (например, образование основания Шиффа, образование амидной связи и образование сложноэфирной связи), ионная связь или координационная связь, или же при помощи нехимической связи, такой как гидрофобное взаимодействие, водородная связь, электростатическое взаимодействие или аффинное связывание.
Предпочтительно при проведении эксклюзионной хроматографии размеров или SDS-PAGE в невосстанавливающих условиях комплексов настоящего изобретения в диссоциированной форме в какой-либо значительной степени не обнаруживают. Более предпочтительно комплексов настоящего изобретения в диссоциированной форме в какой-либо значительной степени не обнаруживают при проведении эксклюзионной хроматографии размеров и SDS-PAGE в невосстанавливающих условиях. В дополнение к этому, еще более предпочтительно скорость разрушения, позволяющая обнаруживать белок комплекса методом ELISA (твердофазный иммуноферментный анализ) для комплексов настоящего изобретения, полученных в результате модифицирования структуры белка сополимерами и их фармакологически приемлемыми солями, очень невелика (предпочтительно скорость разрушения составляет 20% или менее, более предпочтительно 15% или менее, а наиболее предпочтительно 10% или менее).
В настоящем изобретении аналог белка определяют как белок, кодируемый молекулой кДНК, клонированной из библиотеки кДНК, полученной из клеток животных, жидкостей организма или тканей в результате гибридизации с использованием кДНК белка в жестких условиях (от 60 до 70°С, 6×SSC).
В настоящем изобретении вариант белка определяют как белок, который получают в результате замещения, удаления, добавления или вставки одной или нескольких аминокислот в исходный белок, и который все еще обнаруживает, по меньшей мере, часть активности исходного белка на детектируемом уровне.
Предпочтительно белок или его аналог или вариант представляют собой основной белок. Более предпочтительно основный белок представляет собой основной фактор роста фибробластов (bFGF), эпидермальный фактор роста (EGF), фактор ингибирования остеокластогенеза (OCIF), тромбоцитарный фактор роста (PDGF), выделенный из мозга нейротрофический фактор (BDNF), фактор роста нервов (NGF), гормон роста человека (HGH), гепатоцитарный фактор роста (HGF) или фактор роста сосудистого эндотелия (VEGF) или его аналог или вариант. Наиболее предпочтительно основной белок представляет собой фактор ингибирования остеокластогенеза (OCIF) или его аналог или вариант.
OCIF, его аналог или его вариант, используемые в настоящем изобретении, могут относиться к естественному типу, или они могут относиться к рекомбинантному типу, и на их происхождение особенных ограничений не накладывается. OCIF природного типа обозначает OCIF, который получают в виде белка, образованного в естественных условиях, в результате экстрагирования, очистки и/или выделения из органа, жидкости организма, клеточной культуры или клеточной среды, полученных из организма человека или животного, не являющегося человеком. OCIF рекомбинантного типа, его аналог или его вариант представляют собой рекомбинантный белок, полученный в результате экстрагирования, очистки и/или выделения указанного белка из тела хозяина, обычно используемого в таких методиках, такого как прокариотическая клетка-хозяин (например, Escherichia coli) или эукариотическая клетка, такая как линия клеток из организма человека или организма, не являющегося им, которые изменяются под действием вектора, содержащего полинуклеотид, который кодирует OCIF, его аналог или его вариант [например, см. рекомбинантные способы, описанные в ЕР-А-0816380 (WO-A-96/26217) и WO-A-97/23614].
На происхождение OCIF, его аналогов и его вариантов, используемых в настоящем изобретении, особенных ограничений не накладывается, и их можно получать из организма человека или животного, не являющегося человеком. Предпочтительно их можно получать из организма млекопитающего, такого как человек, крыса, мышь, кролик, собака, кошка, корова, свинья, овца или коза; или птицы, такой как курица, гусь, цыпленок или индюк. Более предпочтительно их получают из организмов млекопитающих, а наиболее предпочтительно их получают из организма человека.
OCIF или его аналог, используемые в настоящем изобретении, могут относиться к OCIF мономерного типа (например, у человека мономер характеризуется молекулярной массой, приблизительно равной 60000 согласно измерениям методом SDS-PAGE в невосстанавливающих условиях) или димерного типа (например, у человека димер характеризуется молекулярной массой, приблизительно равной 120000 согласно измерениям методом SDS-PAGE в невосстанавливающих условиях) [см. EP-A-0816380 (WO-A-96/26217)]. Предпочтительно им является мономер OCIF человека, характеризующийся молекулярной массой, приблизительно равной 60000 согласно измерениям методом SDS-PAGE в невосстанавливающих условиях, или димер OCIF человека, характеризующийся молекулярной массой, приблизительно равной 120000 согласно измерениям методом SDS-PAGE в невосстанавливающих условиях, а более предпочтительно им является димер OCIF человека, характеризующийся молекулярной массой, приблизительно равной 120000 согласно измерениям методом SDS-PAGE в невосстанавливающих условиях.
Известно, что OCIF транслируется в клетки в виде полипептида, содержащего сигнальный пептид на его амином конце, и что после он подвергается дозреванию в результате процессинга, включающего удаление указанного сигнального пептида, [например, см. рекомбинантные способы, описанные в EP-A-0816380 (WO-A-96/26217) и WO-A-97/23614]. OCIF, его аналог или его вариант, используемые в настоящем изобретении, включают как полипептид, содержащий сигнальный пептид, так и его зрелую форму. Предпочтительные примеры включают OCIF человека, содержащий сигнальный пептид, включающий аминокислоты от -21 до +380 последовательности SEQ ID No. № 1 списка последовательностей, и зрелую форму OCIF человека, не содержащего сигнального пептида, включающего аминокислоты от +1 до +380 последовательности SEQ ID No. № 1 списка последовательностей. В числе данных соединений в особенности предпочтительным является OCIF человека в зрелой форме.
Также известно, что к такой зрелой форме OCIF, его аналога или его варианта можно добавлять метионин, если данная форма экспрессируется в виде рекомбинантного белка в клетке-хозяине, в особенности в прокариотической клетке-хозяине, такой как Escherichia coli. Этого добиваются в результате добавления нуклеотидного триплета, включающего последовательность АТГ (АУГ), к концу 5' полинуклеотида, кодирующего зрелую форму OCIF, его аналога или его варианта, и вставки получающегося в результате полинуклеотида в подходящий экспрессирующий вектор. Желательный зрелый белок, имеющий метионин на своем аминном конце, после этого может экспрессироваться при помощи подходящей клетки-хозяина, которая была изменена благодаря указанному рекомбинантному экспрессирующему вектору. В дополнение к этому, одну или несколько аминокислот можно добавить к указанному белку в положении, соседствующем с метионином на аминном конце, в результате добавления дополнительных нуклеотидных триплетов по соседству с триплетом АТГ, добавленным на конце 5' полинуклеотида, кодирующего зрелую форму OCIF, его аналога или его варианта. Целевую зрелую форму OCIF, его аналога или его варианта, содержащих метионин на аминном конце, можно очистить и выделить из культуры измененной клетки-хозяина в соответствии с обычно используемым способом. В дополнение к этому, одну или несколько аминокислот можно вставить в зрелую форму OCIF, содержащего метионин на аминном конце, его аналога или его варианта в положении, соседствующем с метионином и более близком к карбокси-концу по сравнению с метионином.
В настоящем изобретении аналог OCIF обозначает белок, кодированный полинуклеотидом, который существует в естественных условиях в клетках, жидкости организма и/или органах человека или животного, не являющегося человеком, такого как те, что в качестве примеров приведены выше. Конкретные предпочтительные примеры таких аналогов включают OCIF2, OCIF3, OCIF4 и OCIF5 [см. EP-A-0816380 (WO96/26217)]. Такие аналоги OCIF или их активные фрагменты можно получить способом, таким как следующий: РНК экстрагируют из клетки, органа, ткани или жидкости организма человека или животного, не являющегося человеком; с использованием обратной транскриптазы синтезируют первую цепь кДНК, которая комплиментарна указанной РНК, а после этого с использованием первой цепи в качестве матрицы с использованием ДНК-полимеразы синтезируют вторую цепь указанной кДНК; таким образом полученную двойную цепь кДНК вставляют в подходящий обычно используемый экспрессирующий вектор; подходящую обычно используемую клетку-хозяин после этого изменяют под действием таким образом полученного вектора; хозяина, генерирующего желательный пептид, затем отбирают для использования методики гибридизации, такой как гибридизация тромбоцита или гибридизация фага, с использованием кДНК OCIF или ее фрагмента в качестве зонда в жестких условиях [см. EP-A-0816380 (WO-A-96/26217)]; и после этого, в заключение, желательный аналог OCIF экспрессируется по обычно используемой методике при помощи таким образом полученной клетки-хозяина.
В настоящем изобретении вариант OCIF обозначает белок, который содержит аминокислотную последовательность, где в аминокислотной последовательности OCIF или его аналога замещают, удаляют, добавляют или вставляют один или несколько аминокислотных остатков, и который все еще обладает, по меньшей мере, некоторой активностью OCIF. Такие варианты OCIF можно получать, например, по следующему способу: замещение, удаление, добавление и/или вставка одного нуклеотида или нескольких нуклеотидов в нуклеотидной последовательности, кодирующей OCIF или его аналог, с использованием способа полимеразной цепной реакции (здесь и далее в настоящем документе обозначаемого как ПЦР), способа генетической рекомбинации или способа нуклеазного расщепления с использованием экзонуклеазы или эндонуклеазы, таких как рестрикционный фермент; трансформирование эукариотической клетки-хозяина, такой как клетка животного, или прокариотической клетки-хозяина, такой как Escherichia coli, под действием экспрессирующего вектора, где производят вставку полученного нуклеотида, кодирующего желательный вариант OCIF; а после этого экстрагирование, очистка и/или выделение желательного пептида из белоксодержащей фракции, полученной с использованием клеточной культуры указанного трансформированного хозяина в соответствии со способом, хорошо известным специалисту в соответствующей области.
Процессированные формы OCIF, у которых с карбокси-конца полипептида OCIF удаляют значительную часть аминокислотной последовательности, также известны тем, что они сохраняют, по меньшей мере, некоторую степень активности OCIF [см., например, EP-A-0816380 (WO-A-96/26217) и WO-A-97/23614]. Такие процессированные типы OCIF, сохраняющие, по меньшей мере, некоторую активность полного полипептида OCIF, также включаются в варианты OCIF настоящего изобретения.
Кроме того, OCIF или его процессированная форма, которые подвергают слиянию с иммуноглобулиновым доменом, таким как Fc-домен (например, слитый полипептид, в котором Fc-домен IgG человека присоединяют к карбокси-концу OCIF), и которые сохраняют, по меньшей мере, некоторую активность полного полипептида OCIF, известны (см. работу WO-A-97/23614), и такие слитые белки также включаются в варианты OCIF настоящего изобретения.
Любой образованный в естественных условиях OCIF или его аналог или же рекомбинантный OCIF или его аналог или вариант могут содержать цепь сахара, которую присоединяют к OCIF или его аналогу или варианту на этапе посттрансляции. Образованный в естественных условиях OCIF или его аналог, содержащие цепь сахара, можно получить из клеточных культур, тканей, органов, жидкостей организма или линий клеток, полученных из организма человека или животных, не являющихся человеком, с использованием обычных методик. Рекомбинантный OCIF или его аналог или вариант, содержащие цепь сахара, можно получить из культуры эукариотической клетки-хозяина, трансформированной с использованием вектора, содержащего нуклеотидную последовательность, кодирующую любые OCIF или его аналог или вариант, такие как те, что описываются и приводятся в качестве примеров выше. Примеры подходящих клеток-хозяев, которые можно использовать и которые обладают способностью посттрансляционного модифицирования OCIF или его аналога или варианта с присоединением цепи сахара, включают клетки яичника китайского хомячка и клетки COS [Yasuda, H. et al., Endocrinology, 139, 1329-1337 (1998)]. OCIF или его аналог или вариант, содержащие такую цепь сахара, пригодны для использования при получении комплексов настоящего изобретения. Цепь сахара в OCIF или его аналоге или варианте, содержащих цепь сахара, можно искусственно модифицировать (в частности, под действием ферментов) с использованием полимеров, полисахаридов или модифицированных полисахаридов.
Фармацевтическая композиция настоящего изобретения, которая содержит сополимер настоящего изобретения или его фармакологически приемлемую соль и белок или его аналог, может представлять собой раствор, полученный в соответствии со способом, описанным выше, или композицию, полученную в результате лиофильной сушки раствора. В альтернативном варианте такая фармацевтическая композиция может быть составлена в соответствии с альтернативным способом, или же она может иметь форму комплекта. В последнем случае сополимер настоящего изобретения или его фармакологически приемлемую соль и белок или его аналог или вариант хранят в различных контейнерах, а после этого перемешивают с получением желательной фармацевтической композиции непосредственно перед использованием.
Фармацевтические композиции настоящего изобретения необязательно могут дополнительно содержать основание. На основание никаких ограничений в виде какого-либо специфического основания не накладывается до тех пор, пока им является основание, обычно используемое в фармацевтических композициях. Примеры такого основания включают: неорганические основания, такие как карбонаты щелочных металлов (например, карбонат натрия, карбонат калия и карбонат лития), гидрокарбонаты щелочных металлов (например, гидрокарбонат натрия, гидрокарбонат калия и гидрокарбонат лития), гидриды щелочных металлов (например, гидрид лития, гидрид натрия и гидрид калия), гидроксиды щелочных металлов (например, гидроксид натрия, гидроксид калия, гидроксид бария и гидроксид лития), фториды щелочных металлов (например, фторид натрия и фторид калия); и органические основания, такие как алкоксиды щелочных металлов (например, метоксид натрия, этоксид натрия, метоксид калия, этоксид калия, трет-бутоксид калия и метоксид лития), меркаптаны щелочных металлов (например, метилмеркаптан натрия и этилмеркаптан натрия), N-метилморфолин, триэтиламин, трипропиламин, трибутиламин, диизопропилэтиламин, дициклогексиламин, N-метилпиперидин, пиридин, 4-пирролидинопиридин, пиколин, 4-(N,N-диметиламино)пиридин, 2,6-ди(трет-бутил)-4-метилпиридин, хинолин, N,N-диметиланилин, N,N-диэтиланилин, 1,5-диазабицикло[4.3.0]нон-5-ен (DBN), 1,4-диазабицикло[2.2.2]октан (DABCO) и 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU). В числе данных оснований предпочтительными являются гидроксиды щелочных металлов, а в особенности предпочтительным является гидроксид натрия. В данном отношении необходимо отметить, что часть или все количество оснований могут образовывать соли с сополимером настоящего изобретения.
Примеры фармацевтической композиции по настоящему изобретению включают комплекс между, по меньшей мере, одним веществом, выбираемым из сополимера настоящего изобретения или его фармакологически приемлемой соли, и белком или его аналогом или вариантом, выбираемыми из bFGF (используется для лечения или профилактики ишемического артериопатического заболевания и нерубцующихся кожных язв), EGF (используется для лечения или профилактики язвенных болезней, таких как язвенный колит и длительное нарушение функции эпителия роговицы), PDGF (используется для лечения ран), BDNF и NGF (используется для лечения или профилактики заболеваний центральной нервной системы, таких как болезнь Паркинсона и болезнь Альцгеймера), HGH (используется для лечения или профилактики дефицита гормона роста, синдрома пониженной секреции гормона роста, синдрома Тернера и хрящевой дистрофии), HGF (используется для лечения или профилактики сахарного диабета, артериосклероза, такого как инфаркт головного мозга, и фиброза печени) и VEGF (ишемическое артериопатическое заболевание и окклюзионное поражение периферических артерий).
Один в особенности предпочтительный пример фармацевтической композиции, соответствующей настоящему изобретению, включает комплекс между, по меньшей мере, одним веществом, выбираемым из OCIF, его аналога и варианта, и, по меньшей мере, одним веществом, выбираемым из сополимера настоящего изобретения или его фармакологически приемлемой соли. Такая фармацевтическая композиция в особенности пригодна для профилактики или лечения нарушений обмена веществ в костях. В настоящем изобретении такие нарушения обмена веществ в костях включают любые заболевания, которые характеризуются существенным уменьшением костной массы у пациента, страдающего от них, и при которых необходимо подавить резорбцию костей или скорость резорбции костей для того, чтобы осуществить профилактику или лечение указанных заболеваний.
Нарушения обмена веществ в костях, лечение или профилактику которых можно осуществить с использованием фармацевтической композиции настоящего изобретения, включают: первичный остеопороз (старческий остеопороз, постклимактерический остеопороз и идиопатический ювенильный остеопороз); эндокринный остеопороз (гипертиреоидизм, гиперпаратиреоидизм, гиперадренокортицизм и акромегалию); остеопороз с сопутствующим гипогонадизмом (гипопитуитаризм, синдром Клайнфелтера и синдром Тернера); наследственный и врожденный остеопороз (несовершенный остеогенез, гомоцистинурию, синдром Менкеса и семейную вегетативную дисфункцию); остеопению, обусловленную уменьшением нагрузки от собственного веса или фиксацией и иммобилизацией конечностей; деформирующую остеодистрофию; остеомиелит; инфекционный очаг, обусловленный рарефикацией кости; гиперкальциемию, возникающую в виде последствия солидного рака (рака молочной железы, рака легких, рака почек и рака предстательной железы); гематологические злокачественные заболевания (множественную миелому, лимфому и лейкемию); идиопатическую гиперкальциемию; гиперкальциемию с сопутствующим гипертиреоидизмом или нарушением функции почек; остеопению, возникающую в виде последствия лечения стероидными лекарственными средствами; остеопению, возникающую в виде последствия приема других лекарственных средств (например, иммунодепрессивных препаратов, таких как метотрексат и циклоспорин А, гепарина и противоэпилептических средств); остеопению с сопутствующим нарушением функции почек; остеопению, сопутствующую хирургической операции или заболеваниям пищеварительных органов (например, непроходимости тонкого кишечника, непроходимости толстого кишечника, хронического гепатита, резекции желудка, первичному билиарному циррозу печени и циррозу печени); остеопению, обусловленную различными типами ревматизма, такого как ревматоидный артрит; остеоклазию и разрушение сустава, обусловленные различными типами ревматизма, такого как ревматоидный артрит; ревматоидный артрит, относящийся к типу миелодиспластической трофопатии; остеоартрит; рарефикацию периодонтальной кости; метастазы рака в кости (остеолитические метастазы); некроз кости или омертвление костных клеток, сопутствующие травматическому повреждению, болезни Гоше, серповидноклеточной анемии, системной эритематозной волчанке или нетравматическому повреждению; остеодистрофию, такую как нефрогенная остеодистрофия; остеопению, сопутствующую гипофосфатазии или диабету; остеопению, сопутствующую алиментарной дистрофии или нарушению питания; и другую остеопению. Кроме того, в настоящем изобретении в нарушения обмена веществ в костях также включаются кахексия, обусловленная указанными выше солидным раком, метастазами рака в кости (остеолитическими метастазами) или гематологическими злокачественными заболеваниями (см. японскую патентную заявку (Kokai) № 2000-178200).
Фармацевтическую композицию по настоящему изобретению, описанную выше, человек или животные, отличные от человека, могут безопасно принимать перорально или неперорально. Дозированную форму фармацевтической композиции надлежащим образом можно выбирать в зависимости от типа заболевания, степени заболевания и состояния, возраста, пола и массы пациента. Например, фармацевтическую композицию можно вводить перорально, используя форму таблеток, капсул, порошков, гранул или сиропов; вводить, используя форму внутривенной инъекции, индивидуально или в комбинации с обычным адъювантом, таким как глюкоза, аминокислоты и тому подобное, или же, используя отдельно форму внутримышечной, подкожной, внутрикожной или внутрибрюшинной инъекции; вводить трансдермально, используя форму припарки; вводить трансназально, используя форму капель для носа; вводить трансмукозально или в ротовую полость, используя форму трансмукозального препарата; или вводить интраректально, используя форму суппозитория. Композиции данных препаратов можно составлять обычным образом с использованием хорошо известных адъювантов, которые обычно используются в сфере медицины, таких как наполнители, связующие, дезинтегранты, лубриканты, вкусовые добавки, солюбилизаторы, суспендирующие вещества, красители, регуляторы рН, антисептики, желирующие вещества, поверхностно-активные вещества и вещества для покрытия.
Тогда, когда фармацевтическую композицию получают в виде таблетки, возможно использование различных носителей, известных на современном уровне техники. Примеры таких носителей включают: наполнители, такие как лактоза, сахароза, хлорид натрия, глюкоза, мочевина, крахмал, карбонат кальция, каолин, кристаллическая целлюлоза и кремниевая кислота; связующие, такие как вода, этанол, пропанол, простой сироп, раствор глюкозы, раствор крахмала, раствор желатина, карбоксиметилцеллюлоза, шеллак, метилцеллюлоза, фосфат калия и поливинилпирролидон; дезинтегранты, такие как сухой крахмал, альгинат натрия, порошкообразный агар, порошкообразный ламинаран, гидрокарбонат натрия, карбонат кальция, сложные эфиры, получаемые из полиоксиэтиленсорбитана и жирных кислот, лаурилсульфат натрия, моноглицерид стеариновой кислоты, крахмал и лактоза; ингибиторы дезинтеграции, такие как сахароза, стеарин, масло какао и гидрированное масло; ускорители впитывания, такие как четвертичные аммониевые основания и лаурилсульфат натрия; увлажняющие средства, такие как глицерин и крахмал; абсорбенты, такие как крахмал, лактоза, каолин, бентонит и коллоидальная кремниевая кислота; и лубриканты, такие как очищенный тальк, стеараты, порошкообразная борная кислота и полиэтиленгликоль. В дополнение к этому, при необходимости на такую таблетку можно нанести покрытие в виде обычно используемой оболочки. Примеры такой таблетки, покрытой оболочкой, включают таблетку с сахарным покрытием, таблетку с желатиновым покрытием, таблетку с энтеросолюбильным покрытием, таблетку с пленочным покрытием, двухслойную таблетку и многослойную таблетку.
Тогда, когда фармацевтическую композицию получают в виде пилюли, возможно использование различных носителей, известных на современном уровне техники. Примеры таких носителей включают: наполнители, такие как глюкоза, лактоза, масло какао, крахмал, гидрированное растительное масло, каолин и тальк; связующие, такие как порошкообразная аравийская камедь, порошкообразный трагакант, желатин и этанол; и дезинтегранты, такие как агар на основе ламинарана.
Тогда, когда фармацевтическую композицию получают в виде суппозитория, возможно использование различных носителей, известных на современном уровне техники. Примеры таких носителей включают полиэтиленгликоль, масло какао, высшие спирты, сложные эфиры высших спиртов, желатин и полусинтетические глицериды.
Тогда, когда фармацевтическую композицию получают в виде инъекции, предпочтительно, чтобы композицию в виде раствора или суспензии подвергали стерилизации и делали изотонической по отношению к крови. Тогда, когда такую композицию получают в виде раствора, эмульсии, суспензии или по существу гомогенного водного раствора, возможно использование различных разбавителей, известных на современном уровне техники. Примеры таких разбавителей включают воду, этанол, пропиленгликоль, этоксилированный изостеариловый спирт, полиоксилированный изостеариловый спирт и сложные эфиры, получаемые из полиоксиэтиленсорбитана и жирных кислот. В данном случае фармацевтическая композиция может дополнительно содержать поваренную соль, глюкозу или глицерин в количестве, достаточном для поддержания изотоничности по отношению к крови. Кроме того, фармацевтическая композиция также может содержать и обычно используемые солюбилизаторы, буферные вещества, мягчители, регуляторы рН, стабилизаторы или солюбилизирующие компоненты. Такую инъекцию можно подвергнуть лиофильной сушке.
В дополнение к этому фармацевтическая композиция по изобретению при необходимости также может содержать красители, консерванты, душистые вещества, вкусовые добавки, подсластители или другое лекарственное средство.
На количество комплекса между OCIF или его аналогом или вариантом и сополимером или его фармакологически приемлемой солью, содержащегося в фармацевтической композиции настоящего изобретения, ограничения в виде какого-либо специфического значения не накладывается, но обычно оно находится в диапазоне от 0,1 до 70% (мас.), а предпочтительно в диапазоне от 1 до 30% (мас.).
В настоящем изобретении дозировка комплекса OCIF или его аналога или варианта с сополимером или его фармакологически приемлемой солью, содержащегося в фармацевтической композиции настоящего изобретения, зависит от различных факторов, в том числе от состояния и возраста пациента, способа применения и формы лекарственного средства. В общем случае количество, вводимое взрослому человеку за один прием, находится в диапазоне от верхнего предела в интервале от 1 до 50 мг/кг до нижнего предела в интервале от 0,001 до 0,1 мг/кг, где предпочтительным является диапазон от 0,01 до 1 мг/кг, а более предпочтительным является диапазон от 0,1 до 1 мг/кг.
Фармацевтическую композицию, соответствующую настоящему изобретению, необходимо принимать один раз в несколько месяцев, один раз в месяц, один раз в несколько дней, один раз в день или несколько раз в день в зависимости от типа ингредиента, содержащегося в фармацевтической композиции, способа применения и формы лекарственного средства. Тогда, когда композиция содержит комплекс OCIF или его аналога или варианта с сополимером или его фармакологически приемлемой солью, предназначенный для применения в качестве средства при лечении или профилактике нарушений обмена веществ в костях в соответствии с настоящим изобретением, ее необходимо принимать один раз в несколько месяцев, один раз в месяц, один раз в несколько дней, один раз в день или несколько раз в день в зависимости от типа активного ингредиента, содержащегося в средстве, предназначенном для лечения или профилактики нарушений обмена веществ в костях, способа применения и формы лекарственного средства.
Сополимер настоящего изобретения и его фармакологически приемлемые соли пригодны в качестве полимерного модификатора, способного привести к получению комплекса, обладающего однородными свойствами, в особенности пониженным образованием неупорядоченных сшитых структур с белком, лучшим сохранением активности белка и превосходным удерживанием белка в крови после приема указанного комплекса. Это делает его в особенности подходящим в качестве модификатора при модифицировании фармацевтических свойств белков, обладающих полезной фармацевтической активностью.
Примеры
Нижеследующие примеры, справочные примеры и примеры испытаний предназначены для дополнительного иллюстрирования настоящего изобретения, и они никоим образом не предполагают ограничения объема изобретения. В нижеследующих примерах m и R3 представляют собой то же, что определено выше для формул (I) и (II), "соотн. сост." представляет собой соотношение, задаваемое составом [то есть, соотношение между структурными элементарными звеньями (I) и (II)], "ср. степ. пол." представляет собой среднюю степень полимеризации для полученного сополимера, а "соотн. гидр." для полученного сополимера представляет собой соотношение, задаваемое гидролизом [то есть, среднее соотношение между структурными элементарными звеньями, где R3 представляет собой -ОН, и структурными элементарными звеньями, где R3 отличается от -ОН].
Пример 1
Получение гидролизата поли(ПЭГ500-MA) [поли(ПЭГ500-MA)г], где m=6-16, R3=OH, соотн. сост.=приблизительно 1:1, ср. степ. пол.=30-40, а соотн. гидр.=приблизительно 10:0 (соединение № 1)
В качестве исходного соединения использовали сополимер полиоксиэтиленаллилметилового простого диэфира (m=6-16, Alk=этилен, R1=водород, и R2=метил) и малеинового ангидрида, в котором полиоксиэтиленовая боковая цепь характеризуется средней молекулярной массой, приблизительно равной 500, а средняя степень полимеризации для основной цепи находится в диапазоне от 30 до 40 [AM-0530K, изготовленный в компании NOF Corporation (здесь и далее в настоящем документе обозначаемый как «поли(ПЭГ500-MA)»)]. К 3 г поли(ПЭГ500-MA) для растворения указанного исходного соединения добавляли 50 мл дистиллированной воды и таким образом полученный раствор перемешивали при 40°С в течение 15 часов. Получающийся в результате раствор конденсировали с использованием ультрафильтрационной мембраны, изготовленной из полиэфирсульфона на основе простого эфира (характеризующейся предельной молекулярной массой, равной 10000, изготовленной в компании Millipore Corporation, номер модели PBGC07610), и получающийся в результате конденсат подвергали лиофильной сушке с получением 1,3 г заданного соединения поли(ПЭГ500-MA)г (соединения № 1) в виде маслянистого соединения.
Поли(ПЭГ500-MA)г (соединение № 1) очищали с использованием гель-фильтрации следующим образом. 100 мг поли(ПЭГ500-MA)г (соединения № 1) растворяли в 4 мл 0,001 н. раствора гидроксида натрия. Раствор разделяли на четыре партии и каждую партию в 1 мл наносили на гель-фильтрационную колонку (PD-10, изготовленную в компании Amersham-Pharmacia). Первый 1 мл элюента отбрасывали. После этого на каждую колонку наносили 1,5 мл 0,001 н. раствора гидроксида натрия и в результате элюирования из колонки получали и отбрасывали еще 1,5 мл. Затем на каждую колонку наносили 2,5 мл 0,001 н. раствора гидроксида натрия и в результате элюирования из колонки получали еще 2,5 мл, и именно данная фракция содержала очищенное соединение поли(ПЭГ500-MA)г (соединение № 1). Очищенные фракции из четырех колонок объединяли и получали 10 мл очищенного раствора целевого соединения. Выход после стадии очистки (определенный спектрофотометрически при измерении оптической плотности для поли(ПЭГ500-MA)г в области 210 нм до и после очистки) зафиксировали равным 80% (80 мг полимера), а концентрацию в подвергнутом очистке растворе определили равной 8 мг/мл.
Уровень содержания карбоксильных групп в целевом соединении определяли методом кондуктометрического титрования следующим образом. 7,5 мл раствора очищенного целевого соединения, полученного выше (содержащего 60 мг целевого соединения), разводили до объема 50 мл с использованием дистиллированной воды и рН таким образом полученного раствора доводили до 12 с использованием 1 М водного раствора гидроксида натрия. К раствору порциями по 0,1 мл добавляли 0,1 М хлористоводородную кислоту, при этом после каждого добавления хлористоводородной кислоты проводили измерение рН и проводимости раствора. Затем уровень содержания карбоксильных групп в сополимере рассчитывали из количества 0,1 М хлористоводородной кислоты, добавленной в буферной области проводимости (то есть, в области на графической зависимости проводимости от количества добавленной хлористоводородной кислоты, в которой карбоксильные группы сополимера целевого соединения действуют в качестве буфера, при этом указанная область соответствует диапазону рН приблизительно от 10,5 до 3); мольное количество хлористоводородной кислоты, используемой в буферной области проводимости, равно мольному количеству карбоксильных групп в сополимере целевого соединения. В результате уровень содержания карбоксильных групп в 1 г целевого соединения определили равным 3,22 ммоль. Отсюда соотношение, задаваемое составом, рассчитали равным 1:1,07. Данное численное значение применяли в нижеследующих примерах, где использовали то же самое исходное соединение.
Пример 2
Получение гидролизата поли(ПЭГ1500-MA) [поли(ПЭГ1500-MA)г], где m=28-38, R3=OH, соотн. сост.=приблизительно 1:1, ср. степ. пол.=10-15, а соотн. гидр.=приблизительно 10:0 (соединение № 2)
В качестве исходного соединения использовали сополимер полиоксиэтиленаллилметилового простого диэфира (m=28-38, Alk=этилен, R1=водород, и R2=метил) и малеинового ангидрида, в котором полиоксиэтилен характеризуется средней молекулярной массой, приблизительно равной 1500, а средняя степень полимеризации для основной цепи находится в диапазоне 10-15 (AM-1510K, изготовленный в компании NOF Corporation) [здесь и далее в настоящем документе обозначаемый как «поли(ПЭГ1500-MA)»]. К 1,5 г поли(ПЭГ1500-MA) для растворения указанного исходного соединения добавляли 25 мл дистиллированной воды и таким образом полученный раствор перемешивали при комнатной температуре в течение 20 часов. По окончании данного периода времени раствор конденсировали тем же самым способом, что и описанный в приведенном выше примере 1, с использованием ультрафильтрационной мембраны, изготовленной из полиэфирсульфона на основе простого эфира (характеризующейся предельной молекулярной массой, равной 10000, изготовленной в компании Millipore Corporation, номер модели PBGC07610), и получающийся в результате конденсат подвергали лиофильной сушке с получением 0,8 г целевого соединения поли(ПЭГ1500-MA)г (соединения № 2) в виде маслянистого соединения.
Уровень содержания карбоксильных групп в целевом соединении поли(ПЭГ1500-MA)г (соединении № 2) измеряли тем же самым способом, что и описанный в приведенном выше примере 1, и в результате уровень содержания карбоксильных групп в 1 г целевого соединения определили равным 1,63 ммоль. Отсюда соотношение, задаваемое составом, рассчитали равным 1:1,4. Данное численное значение применяли в нижеследующих примерах, где использовали то же самое исходное соединение.
Пример 3
Получение продукта аммонолиза поли(ПЭГ500-MA) [поли(ПЭГ500-MA)а], где m=6-16, R3=NH2, соотн. сост.=приблизительно 1:1, ср. степ. пол.=30-40, а соотн. гидр.=приблизительно 0:10 (соединение № 3)
К 1 г поли(ПЭГ500-MA) (АМ-0530К, изготовленного в компании NOF Corporation) для растворения указанного исходного соединения добавляли 9,5 г водно-аммиачного раствора (концентрация аммиака: 28% (мас.)) и таким образом полученный раствор перемешивали при комнатной температуре в течение 16 часов. По окончании данного периода времени таким образом полученный раствор подвергали диализу через мембрану из регенерированной целлюлозы (характеризующуюся предельной молекулярной массой в диапазоне от 12000 до 14000, изготовленную в компании Sanko Junyaku Co., Ltd., номер модели UC36-32-100) с использованием в течение одного дня 1 литра водного раствора уксусной кислоты с концентрацией 0,1% (мас.), а затем его дополнительно подвергали диализу с использованием в течение 1 дня 1 литра воды. После обновления 1 литра воды диализ проводили дополнительно в течение еще одного дня. В результате проведения такого диализа из продукта удаляли избыточное количество аммиака. Таким образом полученный раствор конденсировали с использованием ультрафильтрационной мембраны, изготовленной из полиэфирсульфона на основе простого эфира (характеризующейся предельной молекулярной массой, равной 10000, и изготовленной в компании Millipore Corporation, номер модели PBGC07610), и получающийся в результате конденсат подвергали лиофильной сушке с получением 0,99 г целевого соединения поли(ПЭГ500-MA)а (соединения № 3) в виде маслянистого соединения.
Уровень содержания карбоксильных групп в 1 г поли(ПЭГ500-MA)а (соединения № 3) определяли методом кондуктометрического титрования тем же самым способом, что и описанный в приведенном выше примере 1, и в результате уровень содержания карбоксильных групп в 1 г целевого соединения определили равным 1,55 ммоль.
В использованных условиях реакции может оказаться, что остаток малеинового ангидрида будет способен подвергаться реакции с раскрытием цикла в результате протекания не только аммонолиза, но также гидролиза вследствие присутствия в реакционной системе воды. Соотношение между остатками малеинового ангидрида, подвергнутыми аммонолизу, и остатками малеинового ангидрида, подвергнутыми гидролизу, рассчитывали следующим образом.
В примере 1 определяли уровень содержания карбоксильных групп в 1 г поли(ПЭГ500-MA)г (соединения № 1). Отсюда получали численное значение для уровня содержания карбоксильных групп для случая полного гидролиза всех остатков малеиновой кислоты (3,22 ммоль в 1 г полимера). Из данного результата в ходе вычислений определяли массу поли(ПЭГ500-MA)г (соединения № 1) на один моль карбоксильной группы (1/3,22×10-3 г) и массу поли(ПЭГ500-MA)г (соединения № 1) на один моль остатка малеиновой кислоты, полученного при раскрытии цикла [2 × масса поли(ПЭГ500-MA)г (соединения № 1) на один моль остатка малеиновой кислоты, полученного при раскрытии цикла, поскольку имеются 2 карбоксильные группы на один полностью подвергнутый гидролизу остаток малеиновой кислоты], что дает в результате 311 г и 621 г соответственно. Из данных значений определяли массу поли(ПЭГ500-MA) (то есть, массу сополимера перед гидролизом) на один грамм функциональной группы и массу поли(ПЭГ500-MA)а (соединения № 3), полученного в результате добавления к поли(ПЭГ500-MA) аммиака, на один грамм функциональной группы. Говоря конкретно, массу поли(ПЭГ500-MA) (то есть, массу сополимера перед гидролизом) на один моль остатка малеинового ангидрида получали в результате вычитания молекулярной массы молекулы воды (18 г) из массы полностью гидролизованного сополимера, что в результате давало численное значение, равное 603 г. Массу поли(ПЭГ500-MA)а (соединения № 3) на один моль карбоксильной группы получали в результате добавления молекулярной массы молекулы аммиака (17 г) к массе поли(ПЭГ500-MA), что в результате давало численное значение, равное 620 г. Из данной величины в результате вычислений (1 г/620) определяли теоретический уровень содержания карбоксильных групп в 1 г поли(ПЭГ500-MA)а (соединения № 3), в котором все остатки малеинового ангидрида были подвергнуты аммонолизу (то есть, при отсутствии гидролиза), и он был получен равным 1,61 ммоль. Из уровня содержания карбоксильных групп в 1 г поли(ПЭГ500-MA)г (соединения № 1), в котором все остатки малеинового ангидрида были подвергнуты гидролизу, теоретического уровня содержания карбоксильных групп в 1 г поли(ПЭГ500-MA)а (соединения № 3), в котором все остатки малеинового ангидрида были подвергнуты аммонолизу, и фактического уровня содержания карбоксильных групп в 1 г поли(ПЭГ500-MA)а (соединения № 3), измеренного выше (то есть, 1,55 ммоль), рассчитывали соотношение между остатками малеинового ангидрида, подвергнутыми аммонолизу в целевом соединении, и полным количеством остатков малеинового ангидрида в исходном соединении и его определили равным 1,0. Таким образом, было подтверждено, что по существу все остатки малеинового ангидрида были подвергнуты аммонолизу, и что практически никакого гидролиза не протекало.
Пример 4
Получение продукта реакции между диметиламином и поли(ПЭГ500-MA) [поли(ПЭГ500-MA)дма], где m=6-16, R3=NMe2, соотн. сост.=приблизительно 1:1, ср. степ. пол.=30-40, а соотн. гидр.=приблизительно 0:10 (соединение № 4)
К 10 г поли(ПЭГ500-MA) (АМ-0530К, изготовленного в компании NOF Corporation) для его растворения добавляли 71 г водного раствора диметиламина (концентрация диметиламина: 50% (мас.)) и таким образом полученный раствор перемешивали при комнатной температуре в течение 20 часов. По окончании данного периода времени полученный таким образом раствор подвергали диализу через мембрану из регенерированной целлюлозы (характеризующуюся предельной молекулярной массой в диапазоне от 12000 до 14000 и изготовленную в компании Sanko Junyaku Co., Ltd., номер модели UC36-32-100) с использованием в течение одного дня 10 литров водного раствора уксусной кислоты с концентрацией 0,1% (мас.), а затем его дополнительно подвергали диализу с использованием в течение 1 дня 10 литров воды. После обновления 10 литров воды диализ проводили дополнительно в течение еще одного дня. В результате проведения такого диализа из продукта удаляли избыточное количество диметиламина. Таким образом полученный раствор конденсировали с использованием ультрафильтрационной мембраны, изготовленной из полиэфирсульфона на основе простого эфира (характеризующейся предельной молекулярной массой, равной 10000, и изготовленной в компании Millipore Corporation, номер модели PBGC07610), и получающийся в результате конденсат подвергали лиофильной сушке с получением 6,3 г целевого соединения поли(ПЭГ500-MA)дма (соединения № 4) в виде маслянистого соединения.
Уровень содержания карбоксильных групп в 1 г поли(ПЭГ500-MA)дма (соединения № 4) определяли тем же самым способом, что и описанный в приведенном выше примере 1, и он был определен равным 1,53 ммоль. Используя вычисление, подобное тому, что приведено в примере 3 для продукта аммонолиза, в расчете устанавливали теоретический уровень содержания карбоксильных групп в 1 г поли(ПЭГ500-MA)дма (соединения № 4), в котором все остатки малеинового ангидрида были подвергнуты аминолизу, и его определили равным 1,54 ммоль. Из данных результатов рассчитывали соотношение между остатками малеинового ангидрида, подвергнутыми аминолизу в целевом соединении, и полным количеством остатков малеинового ангидрида в исходном соединении и его определили равным 1,0. Таким образом, было подтверждено, что по существу все остатки малеинового ангидрида были подвергнуты аминолизу под действием диметиламина, и что практически никакого гидролиза не протекало.
Пример 5
Получение продукта реакции между 1-амино-2-пропанолом и поли(ПЭГ500-MA) [поли(ПЭГ500-MA)ипа], где m=6-16, R3=NH(CH2CH(OH)CH3), соотн. сост.=приблизительно 1:1, ср. степ. пол.=30-40, а соотн. гидр.=приблизительно 0:10 (соединение № 5)
К 1,5 г поли(ПЭГ500-MA) (АМ-0530К, изготовленного в компании NOF Corporation) для его растворения добавляли 14 г 1-амино-2-пропанола и таким образом полученный раствор перемешивали при комнатной температуре в течение 16 часов. По окончании данного периода времени к раствору добавляли 300 мл дистиллированной воды и для нейтрализации раствора дополнительно добавляли ледяную уксусную кислоту. Таким образом полученный раствор конденсировали с использованием ультрафильтрационной мембраны, изготовленной из полиэфирсульфона на основе простого эфира (характеризующейся предельной молекулярной массой, равной 10000, и изготовленной в компании Millipore Corporation, номер модели PBGC07610), получая 50 мл конденсата. К конденсату добавляли 300 мл дистиллированной воды и получающийся в результате раствор еще раз конденсировали тем же самым способом. Данный цикл разбавления конденсата с использованием дистиллированной воды и последующей повторной конденсации повторяли пять раз для устранения избыточного 1-амино-2-пропанола. Получающийся в результате конденсат подвергали лиофильной сушке с получением 1,3 г целевого соединения поли(ПЭГ500-MA)ипа (соединения № 5) в виде маслянистого соединения.
Уровень содержания карбоксильных групп в 1 г поли(ПЭГ500-MA)ипа (соединения № 5) определяли тем же самым способом, что и описанный в приведенном выше примере 1, и он был определен равным 1,55 ммоль. Используя вычисление, подобное тому, что приведено в примере 3 для продукта аммонолиза, в расчете устанавливали теоретический уровень содержания карбоксильных групп в 1 г поли(ПЭГ500-MA)ипа (соединения № 5), в котором все остатки малеинового ангидрида были подвергнуты аминолизу, и его определили равным 1,47 ммоль. Из данных результатов рассчитывали соотношение между остатками малеинового ангидрида, подвергнутыми аминолизу в целевом соединении, и полным количеством остатков малеинового ангидрида в исходном соединении и его определили равным 1,0. Таким образом, было подтверждено, что по существу все остатки малеинового ангидрида в исходном соединении были подвергнуты аминолизу под действием 1-амино-2-пропанола, и что практически никакого гидролиза не протекало.
Пример 6
Продукт реакции алкоголиза между этанолом и поли(ПЭГ500-MA) [поли(ПЭГ500-MA)эа], где m=6-16, R3=OCH2CH3, соотн. сост.=приблизительно 1:1, ср. степ. пол.=30-40, а соотн. гидр.=приблизительно 4:6 (соединение № 6)
К 1,5 г поли(ПЭГ500-MA) (АМ-0530К, изготовленного в компании NOF Corporation) для его растворения добавляли 25 г абсолютного этанола и таким образом полученный раствор перемешивали при комнатной температуре в течение 16 часов. По окончании данного периода времени добавляли 300 мл воды и получающийся в результате раствор конденсировали с использованием ультрафильтрационной мембраны, изготовленной из полиэфирсульфона на основе простого эфира (характеризующейся предельной молекулярной массой, равной 10000, и изготовленной в компании Millipore Corporation, номер модели PBGC07610), получая 50 мл конденсата. К конденсату добавляли 300 мл дистиллированной воды и получающийся в результате раствор еще раз конденсировали тем же самым способом. Данный цикл разбавления конденсата с использованием дистиллированной воды и последующей повторной конденсации повторяли пять раз для устранения избыточного этанола. Получающийся в результате конденсат подвергали лиофильной сушке с получением 0,8 г целевого соединения поли(ПЭГ500-MA)эа (соединения № 6) в виде маслянистого соединения.
Уровень содержания карбоксильных групп в 1 г поли(ПЭГ500-MA)эа (соединения № 6) определяли тем же самым способом, что и описанный в приведенном выше примере 1, и он был определен равным 2,16 ммоль. Используя вычисление, подобное тому, что приведено в примере 3 для продукта аммонолиза, в расчете устанавливали теоретический уровень содержания карбоксильных групп в 1 г поли(ПЭГ500-MA)эа (соединения № 6), в котором все остатки малеинового ангидрида были подвергнуты алкоголизу, и его определили равным 1,47 ммоль. Из данного результата рассчитывали соотношение между остатками малеинового ангидрида, подвергнутыми алкоголизу в целевом соединении, и полным количеством остатков малеинового ангидрида в исходном соединении и его определили равным 0,6. Таким образом, можно видеть, что 60% остатков малеинового ангидрида в исходном соединении были подвергнуты алкоголизу, а оставшиеся 40% остатков малеинового ангидрида были подвергнуты гидролизу.
Пример 7
Получение продукта аммонолиза поли(ПЭГ1500-MA) [поли(ПЭГ1500-MA)а], где m=28-38, R3=NH2, соотн. сост.=приблизительно 1:1, ср. степ. пол.=10-15, а соотн. гидр.=приблизительно 4:6 (соединение № 7)
К 1,5 г поли(ПЭГ1500-MA) (АМ-1510К, изготовленного в компании NOF Corporation) для его растворения добавляли 14,5 г водно-аммиачного раствора (концентрация аммиака: 28% (мас.)) и таким образом полученный раствор перемешивали при комнатной температуре в течение 20 часов. По окончании данного периода времени к раствору добавляли 300 мл дистиллированной воды и для нейтрализации указанного раствора дополнительно добавляли ледяную уксусную кислоту. Получающийся в результате раствор конденсировали с использованием ультрафильтрационной мембраны, изготовленной из полиэфирсульфона на основе простого эфира (характеризующейся предельной молекулярной массой, равной 10000, и изготовленной в компании Millipore Corporation, номер модели PBGC07610), получая 50 мл конденсата. К конденсату добавляли 300 мл дистиллированной воды и получающийся в результате раствор еще раз конденсировали тем же самым способом. Данный цикл разбавления конденсата с использованием дистиллированной воды и последующей повторной конденсации повторяли пять раз для устранения избыточного аммиака. Получающийся в результате конденсат подвергали лиофильной сушке с получением 0,7 г целевого соединения поли(ПЭГ1500-MA)а (соединения № 7) в виде маслянистого соединения.
Уровень содержания карбоксильных групп в 1 г поли(ПЭГ1500-MA)а определяли тем же самым способом, что и описанный в примере 1, и его определили равным 1,12 ммоль. Как и в случае аммонолиза поли(ПЭГ500-MA)а из примера 3, может оказаться, что остатки малеинового ангидрида в исходном соединении поли(ПЭГ1500-MA) будут способны подвергаться как аммонолизу, так и гидролизу. Поэтому долю остатков малеинового ангидрида, подвергнутых аммонолизу, и долю остатков малеинового ангидрида, подвергнутых гидролизу, рассчитывали следующим образом.
В примере 2 определяли уровень содержания карбоксильных групп в 1 г поли(ПЭГ1500-MA)г (соединения № 2). Отсюда получали численное значение для уровня содержания карбоксильных групп для случая полного гидролиза всех остатков малеиновой кислоты (1,63 ммоль в 1 г полимера). Из данного результата рассчитывали массу поли(ПЭГ1500-MA)г (соединения № 2) на один моль карбоксильной группы (1/1,63×10-3 г) и массу поли(ПЭГ1500-MA)г (соединения № 2) на один моль остатка малеиновой кислоты, полученного при раскрытии цикла [2 × масса поли(ПЭГ1500-MA)г (соединения № 2) на один моль остатка малеиновой кислоты, полученного при раскрытии цикла, поскольку имеются 2 карбоксильные группы на один подвергнутый полному гидролизу остаток малеиновой кислоты], что давало в результате 613 г и 1227 г соответственно. Из полученной таким образом массы поли(ПЭГ1500-MA)г и с использованием подхода, подобного подходу в приведенном выше примере 3, рассчитывали теоретический уровень содержания карбоксильных групп в 1 г поли(ПЭГ1500-MA)а (соединения № 7), в котором все остатки малеинового ангидрида были подвергнуты аммонолизу, и он был получен равным 0,82 ммоль. Из данных результатов и фактического уровня содержания карбоксильных групп в поли(ПЭГ1500-MA)а (соединении № 7), измеренного выше (1,12 ммоль), в результате вычислений устанавливали соотношение между остатками малеинового ангидрида, подвергнутыми аммонолизу в целевом соединении, и полным количеством остатков малеинового ангидрида в исходном соединении и его определили равным 0,6. Таким образом, было подтверждено, что 60% остатков малеинового ангидрида в исходном соединении поли(ПЭГ1500-MA) были подвергнуты аммонолизу, а оставшиеся 40% остатков малеинового ангидрида были подвергнуты гидролизу.
Пример 8
Получение продукта реакции между диметиламином и поли(ПЭГ1500-MA) [поли(ПЭГ1500-MA)дма], где m=28-38, R3=NMe2, соотн. сост.=приблизительно 1:1, ср. степ. пол.=10-15, а соотн. гидр.=приблизительно 0:10 (соединение № 8)
К 1 г поли(ПЭГ1500-MA) (АМ-1510К, изготовленного в компании NOF Corporation) для его растворения добавляли 11 г водного раствора диметиламина (характеризующегося концентрацией 50% (мас.)) и таким образом полученный раствор перемешивали при комнатной температуре в течение 20 часов. По окончании данного периода времени к раствору добавляли 300 мл дистиллированной воды и для нейтрализации раствора дополнительно добавляли ледяную уксусную кислоту. Получающийся в результате раствор конденсировали с использованием ультрафильтрационной мембраны, изготовленной из полиэфирсульфона на основе простого эфира (характеризующейся предельной молекулярной массой, равной 10000, и изготовленной в компании Millipore Corporation, номер модели PBGC07610), получая 50 мл конденсата. К конденсату добавляли 300 мл дистиллированной воды и получающийся в результате раствор еще раз конденсировали тем же самым способом. Данный цикл разбавления конденсата с использованием дистиллированной воды и последующей повторной конденсации повторяли пять раз для устранения избыточного диметиламина. Получающийся в результате конденсат подвергали лиофильной сушке с получением целевого соединения поли(ПЭГ1500-MA)дма (соединения № 8) в виде маслянистого соединения.
Уровень содержания карбоксильных групп в 1 г поли(ПЭГ1500-MA)дма (соединения № 8) определяли тем же самым способом, что и описанный в примере 1, и он был рассчитан равным 0,82 ммоль. Используя подход, подобный тому, что был использован в примере 3, теоретический уровень содержания карбоксильных групп в 1 г поли(ПЭГ1500-MA)дма, в котором все остатки малеинового ангидрида были подвергнуты аминолизу, в ходе вычислений определили равным 0,80 ммоль. Из данных результатов в ходе вычислений устанавливали соотношение между остатками малеинового ангидрида, подвергнутыми аминолизу в целевом соединении, и полным количеством остатков малеинового ангидрида в исходном соединении и его определили равным 1,0. Таким образом, было подтверждено, что по существу все остатки малеинового ангидрида были подвергнуты аминолизу под действием диметиламина.
Пример 9
Получение комплексов между полимерными модификаторами из примеров от 1 до 8 и OCIF
Каждый из полимерных модификаторов, полученных в примерах от 1 до 8, растворяли в физиологическом растворе с фосфатным буфером (PBS) при рН 6,0 (который представляет собой раствор, полученный в результате перемешивания раствора, содержащего 10 мМ гидрофосфата натрия и 150 мМ хлорида натрия, и раствора, содержащего 10 мМ дигидрофосфата натрия и 150 мМ хлорида натрия, при подходящем соотношении с получением буфера, характеризующегося величиной рН, равной 6,0) и получали растворы, характеризующиеся концентрацией модификатора в диапазоне от 1 до 20 мг/мл. Для каждого раствора каждого модификатора, в свою очередь, перемешивали 0,625 мл полученного раствора полимерного модификатора и 0,625 мл раствора, содержащего очищенный, зрелый OCIF человека (OCIF, полученный в соответствии с описанием в WO 96/26217 и ЕР 816380) [концентрация белка: 2 мг/мл, среда: PBS (рН 6,0)], и таким образом полученной смеси давали возможность отстояться при температуре в диапазоне от 4°С до 37°С в течение, по меньшей мере, одного часа с получением серии растворов [среда: PBS (рН 6,0)], содержащих OCIF, модифицированный с использованием полимерных модификаторов из примеров от 1 до 8, в которых соотношение между модификатором и OCIF в каждом растворе определяли концентрация добавленного раствора модификатора и условия реакции. Массовые соотношения модификатор/OCIF для некоторых из полученных комплексов (и условия, в которых их получали) продемонстрированы в таблице 6 в приведенном ниже примере испытания 2. Размер молекулы для каждого из получающихся в результате комплексов измеряли так, как это объясняется в приведенном ниже примере испытания 11. Степень обнаружения OCIF в комплексах методом ELISA измеряли так, как это описывается в приведенном ниже примере испытания 3.
В дополнение к этому, для каждого из полимерных модификаторов точно таким же способом получали раствор OCIF, модифицированного с использованием полимерного модификатора, за исключением того, что в качестве среды использовали PBS с рН 7,4.
Пример 10
Получение натриевой соли продукта аммонолиза поли(ПЭГ500-MA) [поли(ПЭГ500-MA)а-Na], где m=6-16, R3=NH2, соотн. сост.=приблизительно 1:1, ср. степ. пол.=30-40, а соотн. гидр.=приблизительно 3,1:6,9 (соединение № 9)
В качестве исходного соединения использовали сополимер полиоксиэтиленаллилметилового простого диэфира (m=6-16, Alk=этилен, R1=водород, и R2=метил) и малеинового ангидрида, в котором полиоксиэтиленовая боковая цепь характеризуется средней молекулярной массой, приблизительно равной 500, а средняя степень полимеризации для основной цепи находится в диапазоне от 30 до 40 (AM-0530K, изготовленный в компании NOF Corporation, имеющий номер партии М34529) [здесь и далее в настоящем документе обозначаемый как «поли(ПЭГ500-MA)»]. К 10,1 г указанного исходного соединения поли(ПЭГ500-MA) добавляли 61 мл 0,5 М раствора аммиака/1,4-диоксана и получающийся в результате раствор перемешивали при 25°С в течение 20 часов. По окончании данного периода времени к раствору добавляли 200 мл диэтилового эфира и 100 мл 0,2 М водного раствора гидроксида натрия и смесь интенсивно встряхивали в течение приблизительно 3 минут. После протекания фазового разделения органического и водного слоев нижний слой собирали. В отобранный слой добавляли 200 мл диэтилового эфира и 60 мл 1,4-диоксана и таким образом полученный раствор еще раз интенсивно встряхивали. После фазового разделения получающийся в результате нижний слой собирали, а затем подвергали лиофильной сушке и получали 10,1 г целевого соединения натриевой соли поли(ПЭГ500-MA)а (соединения № 9) в виде твердой фазы желтого цвета.
Уровень содержания карбоксильных групп и соотношение, задаваемое гидролизом, для целевого полимера определяли так, как это описывается в приведенном ниже примере испытания 1.
Пример 11
Получение продукта аммонолиза поли(ПЭГ500-MA) [поли(ПЭГ500-MA)а], где m=6-16, R3=NH2, соотн. сост.=приблизительно 1:1, ср. степ. пол.=30-40, а соотн. гидр.=приблизительно 1,4:8,6 (соединение № 10)
К 1 г поли(ПЭГ500-MA) (АМ-0530К, изготовленного в компании NOF Corporation, имеющего номер партии М34529) для его растворения добавляли 9,5 г водно-аммиачного раствора с концентрацией 28% (мас./мас.) и таким образом полученный раствор перемешивали при 25°С в течение 4 часов. По окончании данного периода времени добавляли 250 мл водно-аммиачного раствора с концентрацией 0,28% и получающийся в результате раствор конденсировали с использованием ультрафильтрационной мембраны, изготовленной из полиэфирсульфона на основе простого эфира (характеризующейся предельной молекулярной массой, равной 10000, и изготовленной в компании Millipore Corporation, номер модели PBGC07610) и таким образом полученный конденсат подвергали лиофильной сушке с получением 0,8 г целевого соединения поли(ПЭГ500-MA)а (соединения № 10) в виде маслянистого соединения.
Уровень содержания карбоксильных групп и соотношение, задаваемое гидролизом, для целевого полимера определяли так, как это описывается в приведенном ниже примере испытания 1.
Пример 12
Получение продукта реакции между диметиламином и поли(ПЭГ500-MA) [соль поли(ПЭГ500-MA)дма-Na], где m=6-16, R3=NMe2, соотн. сост.=приблизительно 1:1, ср. степ. пол.=30-40, а соотн. гидр.=приблизительно 2,9:7,1 (соединение № 11)
К 5 г поли(ПЭГ500-MA) (АМ-0530К, номер партии: М34529, полученного в компании NOF Corporation) для его растворения добавляли 35 г водного раствора диметиламина с концентрацией 50% (мас./мас.) и получающийся в результате раствор перемешивали при 25°С в течение 3 часов, после чего дополнительно перемешивали при 4°С в течение 16 часов. По окончании данного периода времени добавляли 100 мл 0,1 М водного раствора гидроксида натрия и таким образом полученный раствор подвергали лиофильной сушке с получением 5,4 г целевого соединения соли поли(ПЭГ500-MA)дма-Na (соединения № 11) в виде твердой фазы желтого цвета.
Уровень содержания карбоксильных групп и соотношение, задаваемое гидролизом, для целевого полимера определяли так, как это описывается в приведенном ниже примере испытания 1.
Пример 13
Получение гидролизата поли(ПЭГ500-MA) [поли(ПЭГ500-MA)г], где m=6-16, R3=ОН, соотн. сост.=приблизительно 1:1, ср. степ. пол.=30-40, а соотн. гидр.=приблизительно 10:0 (соединение № 12)
К 1 г поли(ПЭГ500-MA) (АМ-0530К, изготовленного в компании NOF Corporation, имеющего номер партии М34529) для его растворения добавляли 17 мл дистиллированной воды и получающийся в результате раствор перемешивали при 40°С в течение 4 часов. По окончании данного периода времени сюда добавляли 250 мл водно-аммиачного раствора с концентрацией 0,28% (мас./мас.) и таким образом полученный раствор конденсировали с использованием ультрафильтрационной мембраны, изготовленной из полиэфирсульфона на основе простого эфира (характеризующейся предельной молекулярной массой, равной 10000, и изготовленной в компании Millipore Corporation, номер модели PBGC07610). Таким образом полученный конденсат подвергали лиофильной сушке с получением 0,7 г целевого соединения поли(ПЭГ500-MA)г (соединения № 12) в виде маслянистого соединения.
Уровень содержания карбоксильных групп и соотношение, задаваемое гидролизом, для целевого полимера определяли так, как это описывается в приведенном ниже примере испытания 1.
Пример 14
Получение комплексов между полимерными модификаторами из примеров от 10 до 13 и OCIF
Каждый из полимерных модификаторов, полученных в примерах от 10 до 13, растворяли в физиологическом растворе с фосфатным буфером (PBS) при рН 7,0 (который представляет собой раствор, полученный в результате перемешивания раствора, содержащего 10 мМ гидрофосфата натрия и 150 мМ хлорида натрия, и раствора, содержащего 10 мМ дигидрофосфата натрия и 150 мМ хлорида натрия, при подходящем соотношении с получением буфера, характеризующегося величиной рН, равной 7,0) и получали растворы, характеризующиеся диапазоном концентраций модификатора в интервале от 1,25 до 105 мг/мл. Для каждого полимерного модификатора, в свою очередь, при объемном соотношении 1:1 перемешивали таким образом полученные растворы полимерных модификаторов и раствора, содержащего очищенный, зрелый OCIF человека (OCIF, полученный в соответствии с описанием в WO 96/26217 и ЕР 816380) [концентрация белка: от 0,25 до 14 мг/мл, среда: PBS (рН 6,0)], и получали растворы, характеризующиеся различными массовыми соотношениями между модификатором и OCIF. У таким образом полученных растворов рН доводили до 5,0, 5,5, 6,0, 6,5, 7,0 или 7,4 с использованием 1 М раствора хлористоводородной кислоты или 1 М водного раствора гидроксида натрия. Каждый из таким образом полученных растворов оставляли отстаиваться при 25°С в течение промежутка времени в пределах от 12 часов до одной недели и получали раствор комплекса между полимерным модификатором настоящего изобретения и OCIF. Полученные растворы хранили при 4°С. Размер молекулы у получающихся в результате комплексов измеряли так, как это объясняется в приведенных ниже примерах испытаний 6 и 11. Степень обнаружения OCIF в комплексах методом ELISA измеряли так, как это описывается в приведенном ниже примере испытания 8.
Пример 15
Получение натриевой соли продукта аммонолиза поли(ПЭГ500-MA) [соли поли(ПЭГ500-MA)а-Na], характеризующегося регулируемым размером молекулы, где m=6-16, R3=NH2, соотн. сост.=приблизительно 1:1, ср. степ. пол. продемонстрирована ниже, а соотн. гидр.=приблизительно 1,4:8,6 (соединения №№ 13-19)
100 мг натриевой соли поли(ПЭГ500-MA)а (соединение № 9), полученной в примере 10, растворяли в 1 мл физиологического раствора с фосфатным буфером (PBS, характеризующегося величиной рН, равной 7,4, полученного в результате перемешивания водного раствора, содержащего 10 мМ гидрофосфата натрия и 150 мМ хлорида натрия, и водного раствора, содержащего 10 мМ дигидрофосфата натрия и 150 мМ хлорида натрия, при подходящем соотношении). Таким образом полученный раствор фракционировали способом гель-фильтрационной хроматографии. Использовали два различных нижеследующих набора условий гель-фильтрации:
(1) Способ фракционирования с использованием Superose 6 (здесь и далее в настоящем документе называемый способом SRF)
Колонка: Superose 6 HR 10/30 Amersham Bioscience
Температура колонки: 8°С
Подвижная фаза: PBS (pH 7,4)
Длина волны детектирования: 280 нм
Скорость потока: 0,3 мл/мин
Количество, вводимое в колонку: 100 мкл
(2) Способ фракционирования с использованием Superdex 200 (здесь и далее в настоящем документе называемый способом SDF)
Колонка: Superdex 200 HR 16/60 Amersham Biosciences
Температура колонки: комнатная температура
Подвижная фаза: PBS (pH 7,4)
Длина волны детектирования: 280 нм
Скорость потока: 2 мл/мин
Количество, вводимое в колонку: 5 мкл
Полимерные модификаторы, элюирование которых проводили с использованием данных двух способов фракционирования за время элюирования в диапазоне от х до у минут, определяли как поли(ПЭГ500-MA)a-Na(SRFx-y) и поли(ПЭГ500-MA)a-Na(SDFx-y), соответственно.
Концентрацию полимерного модификатора в каждой фракции (водного раствора) определяли способом высокоэффективной жидкостной хроматографии. Условиями, использованными в высокоэффективной жидкостной хроматографии, являлись нижеследующие:
Колонка: Shodex OHpak SB-806M HQ (приобретенная в компании SHOWA DENKO K. K.)
Защитная колонка: Shodex OHpak SB-G (приобретенная в компании SHOWA DENKO K. K.)
Температура колонки: 40°С
Подвижная фаза: водный раствор 50 мМ гидрофосфата натрия с доведением рН до 7,0 с использованием 1 М хлористоводородной кислоты
Длина волны детектирования: 210 нм
Скорость потока: 0,5 мл/мин
Количество, вводимое в колонку: 50 мкл
Для поли(ПЭГ500-MA)a-Na(SRFx-y) и поли(ПЭГ500-MA)a-Na(SDFx-y), полученных соответственно в результате фракционирования в соответствии со способом SRF и способом SDF, проводили дополнительную гель-фильтрационную хроматографию (способ SRF) с использованием тех же самых условий элюирования, что и описанные выше для первого фракционирования в соответствии со способом SRF, для оценки размера молекулы для каждого из образцов после фракционирования. В качестве стандартных образцов использовали белки, характеризующиеся известными размерами молекул, (приобретаемые в компании Amersham Bioscience). В данном отношении молекулярная масса и размер молекулы для каждого из белков продемонстрированы в соответствующем каталоге, по которому они были приобретены.
Рассчитанный размер молекулы для полимерных модификаторов, полученных в результате фракционирования, продемонстрирован в приведенной ниже таблице 1.
** Данное значение, рассчитанное из калибровочной кривой, полученной с использованием размера молекулы и времени удерживания для каждого из стандартных белков. В тех случаях, когда нижний предел не показан, то есть, тогда, когда радиус Стокса показан в виде «ХХ или менее», можно сказать, что образец содержит низкомолекулярный компонент, который не может быть оценен с использованием гель-фильтрационной хроматографии, проводимой в условиях данного примера.
С использованием данного подхода получали различные полимеры поли(ПЭГ500-МА)а-Na, характеризующиеся неодинаковыми размерами молекул, (соединения №№ 13-19). Среднюю степень полимеризации для соединений №№ 13, 14, 15, 16, 17, 18 и 19 рассчитывали так, как в ранее приведенных примерах, и, как было показана, она составляла <30, <<30, <<<30, <30, <<30, <<<30 и <<<30 соответственно.
Пример 16
Получение продукта аммонолиза поли(ПЭГ1500-MA) [поли(ПЭГ1500-MA)а], характеризующегося регулируемым размером молекулы, где m=28-38, R3=NH2, соотн. сост.=приблизительно 1:1, ср. степ. пол. приведена ниже (соединения №№ 20-22)
Исходя из [поли(ПЭГ1500-МА)а] (соединения № 7), полученного в приведенном выше примере 7, применяемого в качестве исходного соединения, и воспользовавшись тем же самым подходом, что и используемый в приведенном выше примере 15, по тому же самому способу, что и в примере 15, получали поли(ПЭГ1500-МА)а (SRFx-y). Условия элюирования в случае элюирования SRF были теми же самыми, что и в примере 15. Выходы полимерных модификаторов, полученных в результате фракционирования, продемонстрированы в приведенной ниже таблице 2.
Используя данный подход, получали различные полимеры поли(ПЭГ1500-МА)а, характеризующиеся неодинаковыми размерами молекул (соединения №№ 20-22). Среднюю степень полимеризации для соединений №№ 20, 21 и 22 рассчитывали так, как в ранее приведенных примерах, и, как было показана, она составляла <10, <<10 и <<<10 соответственно.
Пример 17
Получение комплексов между полимерными модификаторами из примеров 15 и 16 и OCIF
Комплексы полимер-OCIF настоящего изобретения получали в виде водных растворов с использованием по существу того же самого препаративного подхода, что и в приведенном выше примере 14, используя в качестве исходных компонентов водные растворы соединений №№ от 13 до 22, полученных в приведенных выше примерах 15 и 16 (среда: PBS, характеризующийся величиной рН, равной 7,4), и водный раствор очищенного, зрелого OCIF человека (OCIF, полученный в соответствии с описанием в WO 96/26217 и ЕР 816380) (среда: PBS, характеризующийся величиной рН, равной 6,0). Говоря более конкретно, в случае каждого из модификаторов перемешивали его раствор в PBS (рН 7,4) с концентрацией 0,5 мг/мл (полученный так, как описывается в приведенном выше примере 9) и раствор OCIF в PBS (рН 6,0) с концентрацией 0,5 мг/мл при объемном соотношении 1:1. Получающиеся в результате реакционные смеси оставляли отстаиваться при 25°С в течение 7 дней. Размер молекул у получающихся в результате комплексов измеряли так, как это объясняется в приведенном ниже примере испытания 11.
Пример 18
Получение продукта аммонолиза поли(ПЭГ500-MA) [соли поли(ПЭГ500-MA)а-Na], характеризующегося регулируемым размером молекулы: (а) m=6-16, R3=NH2, соотн. сост.=приблизительно 1:2, а ср. степ. пол.=20-30, (соединение № 27), и (b) m=6-16, R3=NH2, соотн. сост.=приблизительно 1:1, а ср. степ. пол.=приблизительно 15 (соединение № 28)
Два полимера настоящего изобретения поли(ПЭГ500-МА)а (соединение № 27) и поли(ПЭГ500-МА)а (соединение № 28) получали следующим образом. Исходным соединением для первого из них являлся поли(ПЭГ500-МА) (АМ-0510К, изготовленный с использованием методики, подобной той, что описывается в японском патенте № 2621308 и публикациях японских патентных заявок №№ 2003-105040 и 2003-104003) - сополимер полиоксиэтиленаллилметилового простого диэфира (m=6-16, Alk=этилен, R1=водород, и R2=метил) и малеинового ангидрида, в котором полиоксиэтиленовая боковая цепь характеризуется средней молекулярной массой, приблизительно равной 500, а средняя степень полимеризации для основной цепи находится в диапазоне от 20 до 30, при этом соотношение между элементарными звеньями полиоксиэтиленаллилметилового простого диэфира и элементарными звеньями малеинового ангидрида равно 1:2, а средняя молекулярная масса составляет приблизительно 6000 [среднечисленная молекулярная масса составляет приблизительно 6000, а показатель молекулярно-массового распределения (Mw/Mn) приблизительно равен 1,25]. Исходным соединением для второго из них являлся поли(ПЭГ500-МА) (АМ-0510К, изготовленный с использованием методики, подобной той, что описывается в японском патенте № 2621308 и публикациях японских патентных заявок №№ 2003-105040 и 2003-104003) - сополимер полиоксиэтиленаллилметилового простого диэфира (m=6-16, Alk=этилен, R1=водород, и R2=метил) и малеинового ангидрида, в котором полиоксиэтиленовая боковая цепь характеризуется средней молекулярной массой, приблизительно равной 500, средняя степень полимеризации для основной цепи составляет приблизительно 15, а средняя молекулярная масса приблизительно равна 10000. Оба исходных соединения подвергали аммонолизу с использованием условий, по существу идентичных условиям, использованным в примере 10, и получали целевые соединения соль поли(ПЭГ500-MA)а-Na (соединение № 27) и соль поли(ПЭГ500-MA)а-Na (соединение № 28). Уровень содержания карбоксильных групп в целевом полимере определяли так, как это описывается в приведенном ниже примере испытания 1, и, как было обнаружено, он равен 2,73 ммоль/г для соединения № 27 и 2,05 ммоль/г для соединения № 28.
Пример 19
Получение комплексов между полимерными модификаторами из примера 18 и OCIF
Комплексы полимер-OCIF настоящего изобретения получали в виде водных растворов с использованием по существу того же самого препаративного подхода, что и в приведенном выше примере 14, используя в качестве исходных компонентов водные растворы соединений №№ 27 и 28, полученных в приведенном выше примере 18. Говоря более конкретно, в случае каждого из модификаторов перемешивали его раствор в PBS (рН 7,4) с концентрацией 5 мг/мл и раствор зрелого OCIF человека в PBS (рН 6,0) с концентрацией 5 мг/мл при объемном соотношении 1:1. Значение рН у получающейся в результате смеси доводили до 5,5 с использованием 1 М хлористоводородной кислоты и после этого реакционную смесь оставляли отстаиваться при 25°С в течение 7 дней. Размер молекул у получающихся в результате комплексов измеряли так, как это объяснятся в приведенном ниже примере испытания 11.
Пример 20
Оценка композиции поли(ПЭГ500-МА) и получение и оценка поли(ПЭГ500-МА)а (соединений №№ 29-53) при различных условиях
20(1) Определение соотношения, задаваемого составом, для поли(ПЭГ500-МА)
Проводили испытания для нескольких сополимеров полиоксиэтиленаллилметилового простого диэфира и малеинового ангидрида поли(ПЭГ500-МА) с различным составом в целях определения соотношения между элементарными звеньями ПЭГ-аллилметилового простого диэфира и малеинового ангидрида (соотношения, задаваемого составом) в них (различные партии АМ-0510К, изготовленного с использованием методики, подобной той, что описывается в японском патенте № 2621308 и публикациях японских патентных заявок №№ 2003-105040 и 2003-104003) - статистических сополимерах полиоксиэтиленаллилметилового простого диэфира (m=6-16, Alk=этилен, R1=водород, и R2=метил) и малеинового ангидрида, в которых полиоксиэтиленовая боковая цепь характеризуется средней молекулярной массой, приблизительно равной 500, а средняя степень полимеризации для основной цепи находится в диапазоне от 20 до 30. Среднечисленная молекулярная масса (Mn) и показатель молекулярно-массового распределения (Mw/Mn, где Mw представляет собой среднемассовую молекулярную массу) для каждой партии подвергнутых испытанию сополимеров продемонстрированы ниже. Молекулярную массу (MM) для каждой партии поли(ПЭГ500-МА) определяли способом гель-фильтрационной хроматографии с использованием поли(этиленгликоля) с предварительно определенной ММ в качестве стандарта. Следовательно, ММ поли(ПЭГ500-МА), продемонстрированная ниже, не является абсолютной ММ, а представляет собой относительную ММ, измеренную с использованием ПЭГ в качестве стандарта.
М3О538, Mn=6431; Mw/Mn=1,27
М3N549, Mn=6360; Mw/Mn=1,23
М3N550, Mn=5891; Mw/Mn=1,28
М3N569, Mn=5897; Mw/Mn=1,25
Для того чтобы определить соотношение, задаваемое составом, для каждого из сополимеров, необходимо было превратить их в соответствующие натриевые соли гидролизованных сополимеров, то есть, в натриевую соль поли(ПЭГ500-МА)г. Использованные условия гидролиза представляли собой нижеследующее.
К 1 г поли(ПЭГ500-МА) (партия М3О538) добавляли 25 мл 1,4-диоксана, 100 мл простого эфира и 42 мл 0,1 н. водного раствора NaOH. Получающуюся в результате смесь затем интенсивно встряхивали, а после того как ей давали возможность отстояться, собирали таким образом полученный водный слой. Таким образом полученный водный слой перемешивали при 40°С в течение 2 часов после фильтрования через фильтровальную бумагу (704×40 м/м, полученную в компании Nihon Rikagaku-kiki Corporation). По окончании данного периода времени получающийся в результате раствор подвергали лиофильной сушке. Натриевую соль гидролизованного продукта, полученного из исходного соединения, а именно натриевую соль поли(ПЭГ500-МА)г (партия М3О538), получали в виде твердой фазы желтого цвета (выход 100%).
К 1 г поли(ПЭГ500-МА) (партия М3N549) добавляли 12 мл 1,4-диоксана и 9 мл 1 н. водного раствора NaOH. Получающуюся в результате смесь после этого перемешивали при комнатной температуре в течение 24 ч. По окончании данного периода времени добавляли 1 мл 1 н. водного раствора NaOH и получающийся в результате раствор подвергали лиофильной сушке. Натриевую соль гидролизованного продукта, полученного из исходного соединения, а именно натриевую соль поли(ПЭГ500-МА)г (партия М3N549), получали в виде твердой фазы желтого цвета (выход 100%).
К 2 г поли(ПЭГ500-МА) (партия М3N550) добавляли 5 мл 1,4-диоксана и 9 мл 1 н. водного раствора NaOH. Получающуюся в результате смесь после этого перемешивали при 40°С в течение 23 ч. По окончании данного периода времени реакционную смесь подвергали лиофильной сушке. Натриевую соль гидролизованного продукта, полученного из исходного соединения, а именно натриевую соль поли(ПЭГ500-МА)г (партия М3N550), получали в виде твердой фазы желтого цвета (выход 100%).
К 2 г поли(ПЭГ500-МА) (партия М3N569) добавляли 5 мл 1,4-диоксана и 9 мл 1 н. водного раствора NaOH. Получающуюся в результате смесь после этого перемешивали при 40°С в течение 23 ч. По окончании данного периода времени реакционную смесь подвергали лиофильной сушке. Натриевую соль гидролизованного продукта, полученного из исходного соединения, а именно натриевую соль поли(ПЭГ500-МА)г, получали в виде твердой фазы желтого цвета (выход 100%).
Уровень содержания карбоксильных групп для каждой из натриевых солей поли(ПЭГ500-МА)г, полученных таким образом, измеряли методом кондуктометрического титрования так, как это описывается выше. Результаты продемонстрированы в приведенной ниже таблице 3.
Натриевая соль поли(ПЭГ500-МА)г содержит следующие два мономерных элементарных звена:
Если количество мономерных элементарных звеньев натриевой соли малеиновой кислоты на 1 элементарное звено ПЭГ-аллилметилового простого диэфира обозначить как «а», а теоретическое минимальное повторяющееся элементарное звено будет включать одно элементарное звено ПЭГ-аллилметилового простого диэфира и «а» элементарных звеньев малеата натрия, то тогда формульную массу (ФМ) минимального повторяющегося элементарного звена будут получать в соответствии с уравнением 1:
ФМ[(ПЭГ-аллилметиловый простой диэфир)1 + (натриевая соль малеиновой кислоты)а] = ФМ(ПЭГ-аллилметиловый простой диэфир) + а[ФМ(натриевая соль малеиновой кислоты)] = 541 + 160а (уравнение 1)
Количество карбоксильных групп в минимальном повторяющемся элементарном звене натриевой соли поли(ПЭГ500-МА)г равно 2а. Следовательно, уровень содержания карбоксильных групп (С, ммоль/г) в минимальном повторяющемся элементарном звене натриевой соли поли(ПЭГ500-МА)г получается в соответствии с уравнением 2:
С=2а/(541+160а)·1000 (уравнение 2)
Соотношение между количеством элементарных звеньев натриевой соли малеиновой кислоты и количеством элементарных звеньев ПЭГ-аллилметилового простого диэфира в фактических натриевых солях поли(ПЭГ500-МА)г является тем же самым, что и соотношение в минимальных повторяющихся элементарных звеньях. Следовательно, значение С также является одним и тем же в минимальном повторяющемся элементарном звене и в фактических натриевых солях поли(ПЭГ500-МА)г, уровни содержания карбоксильных групп в которых определяли методом кондуктометрического титрования (см. выше). Таким образом, в результате измерения уровня содержания карбоксильных групп и с использованием уравнения 2 можно получить значение соотношения, задаваемого составом, «а», а именно количество элементарных звеньев натриевой соли малеиновой кислоты на 1 элементарное звено ПЭГ-аллилметилового простого диэфира [что соответствует составу мономерной композиции для поли(ПЭГ500-МА)]. Результаты продемонстрированы в приведенной ниже таблице 3.
Соотношение, задаваемое составом, для натриевых солей поли(ПЭГ500-МА)г находилось в диапазоне от 1:2 до 1:3,3. Очевидно, что соотношение, задаваемое составом, является одним и тем же в случаях поли(ПЭГ500-МА) и натриевой соли поли(ПЭГ500-МА)г, так что соотношения, задаваемые составом мономерной смеси, для различных исходных сополимеров поли(ПЭГ500-МА), как было подтверждено, находились в диапазоне от 1:2 до 1:3:3.
20(2) Аммонолиз поли(ПЭГ500-МА) в различных условиях с получением поли(ПЭГ500-МА)а (соединения №№ 29-53) и определение соотношений, задаваемых гидролизом, для указанных соединений
В результате использования соотношения, задаваемого составом, (а) и уровня содержания карбоксильных групп для натриевых солей поли(ПЭГ500-МА)г соотношение, задаваемое гидролизом (то есть, соотношение между элементарными звеньями ангидрида малеиновой кислоты в исходном полимере, подвергнутыми аммонолизу, и данными элементарными звеньями, подвергнутыми гидролизу) можно рассчитать следующим образом. Формула натриевой соли элементарного звена амидированной малеиновой кислоты (натриевой соли полуамида малеиновой кислоты) продемонстрирована ниже:
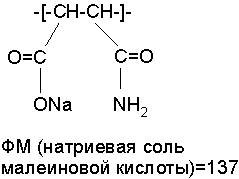
Точно так же, как и в случае указанных выше натриевых солей поли(ПЭГ500-МА)г, можно предположить, что минимальное элементарное звено натриевой соли поли(ПЭГ500-МА)а включает одно элементарное звено ПЭГ-аллилметилового простого диэфира, ах элементарных звеньев натриевой соли полуамида малеиновой кислоты и а(1-х) элементарных звеньев натриевой соли малеиновой кислоты. Формульная масса минимального элементарного звена натриевой соли поли(ПЭГ500-МА)а получается с использованием следующего далее уравнения 3:
ФМ[(ПЭГ-аллилметиловый простой диэфир)1 + (натриевая соль полуамида малеиновой кислоты)ах + (натриевая соль малеиновой кислоты)а(1-х)] = ФМ(ПЭГ-аллилметиловый простой диэфир) + ах[ФМ(натриевая соль полуамида малеиновой кислоты)] + а(1-х)[ФМ(натриевая соль малеиновой кислоты)]=541+137ах+160а(1-х)=541+160а-23ах (уравнение 3)
Поскольку уровень содержания карбоксильных групп в минимальном элементарном звене натриевой соли поли(ПЭГ500-МА)а представляет собой ах+2а(1-х)=2а-ах, уровень содержания карбоксильных групп (С, ммоль/г) в минимальном элементарном звене натриевой соли поли(ПЭГ500-МА)а можно получить из уравнения 4:
С=10000×(2а-ах)/(541+160а-23ах) (уравнение 4)
Таким образом, соотношение, задаваемое гидролизом, (1-х):х можно определить из уравнения 4 с использованием уровня содержания карбоксильных групп (С, ммоль/г), которое можно получить методом кондуктометрического титрования натриевой соли поли(ПЭГ500-МА)а, и значения «а», которое было получено выше.
Некоторые примеры получения поли(ПЭГ500-МА)а и рассчитанного соотношения, задаваемого гидролизом, для продукта поли(ПЭГ500-МА)а продемонстрированы ниже.
(а) К 100 мг поли(ПЭГ500-МА) добавляли 0,5 М раствор аммиака/1,4-диоксана (количества указываются в приведенной ниже таблице 4) и полученный раствор перемешивали при температурах и временах, представленных в приведенной ниже таблице 4. По окончании времени реакции к реакционной смеси добавляли приблизительно 0,4 мл (в диапазоне от 0,3 до 0,5 мл) 1 н. водного раствора NaOH. Получающийся в результате раствор подвергали лиофильной сушке и получали натриевую соль поли(ПЭГ500-МА)а в виде твердой фазы.
«Степень прохождения реакции для аминного компонента» для каждой из натриевых солей поли(ПЭГ500-МА)а [то есть, процентная доля элементарных звеньев ангидрида малеиновой кислоты в исходном полимере, подвергнутых аммонолизу (100·*х)], полученных в данных различных условиях реакции, продемонстрирована в приведенной ниже таблице 4.
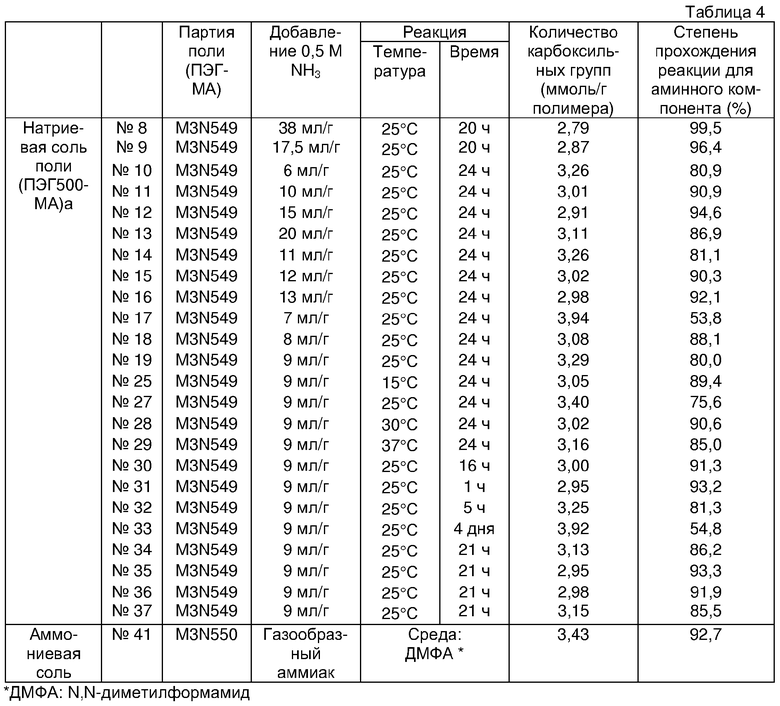
№№ 8-19, 25 и 27-37 представляют собой целевые полимеры поли(ПЭГ500-МА)а (соединения №№ 29-52). Каждый из данных полимеров является статистическим, и m=6-16, R3=NH2, соотношение, задаваемое составом=1:2,4, и средняя степень полимеризации=20-30.
Как продемонстрировано в таблице 4, все продукты реакции поли(ПЭГ500-МА)а (соединения №№ 29-52) характеризуются очень высокими «степенями прохождения реакции для аминного компонента», что свидетельствует о достижении степени превращения для аминогруппы, очень близкой к 100%, с использованием различных условий реакции, описанных выше.
Несколько дополнительных примеров альтернативных способов получения поли(ПЭГ500-МА)а описываются ниже:
(b) 0,44 г поли(ПЭГ500-МА) растворяли в 9 мл диметилформамида. После этого в раствор при - 50°С барботировали газообразный аммиак. Конечное количество аммиака в реакционной смеси составляло 0,67 г. Таким образом полученную реакционную смесь запаивали в стеклянной ампуле в атмосфере азота, а после этого перемешивали при комнатной температуре в течение 24 ч. По окончании данного периода времени газообразный аммиак выпаривали при пониженном давлении после того, как стеклянную ампулу вскрывали. После этого реакционную смесь по каплям добавляли к 90 мл диэтилового эфира. Таким образом полученный осадок собирали и высушивали при пониженном давлении. В виде желтого порошка получали 0,28 г аммониевой соли поли(ПЭГ500-МА)а (соединения № 53) (обозначенного как № 41 в приведенной выше таблице 4). Полимер является статистическим, и m=6-16, R3=NH2, соотношение, задаваемое составом=1:3,10, а средняя степень полимеризации=20-30.
(с) Аммониевую соль поли(ПЭГ500-МА)а получали по тому же самому способу, что и (b), с использованием толуола вместо диметилформамида.
(4) Аммониевую соль поли(ПЭГ500-МА)а получали по тому же самому способу, что и (b), с использованием 1,4-диоксана вместо диметилформамида.
Пример 21
Получение продукта реакции алкоголиза между этанолом и поли(ПЭГ500-MA) [поли(ПЭГ500-MA)эа-Na], где m=6-16, R3=OCH2CH3, соотн. сост.=приблизительно 1:3, ср. степ. пол.=20-30, соотн. гидр.=приблизительно 3,1:6,9 (соединение № 54)
В качестве исходного соединения использовали поли(ПЭГ500-МА) (АМ-0510К, партия M3N550, изготовленный с использованием методики, подобной той, что описывается в японском патенте № 2621308 и публикациях японских патентных заявок №№ 2003-105040 и 2003-104003) - сополимер полиоксиэтиленаллилметилового простого диэфира (m=6-16, Alk=этилен, R1=водород, и R2=метил) и малеинового ангидрида, в котором полиоксиэтиленовая боковая цепь характеризуется средней молекулярной массой, приблизительно равной 500, средняя степень полимеризации для основной цепи находится в диапазоне от 20 до 30, соотношение между элементарными звеньями полиоксиэтиленаллилметилового простого диэфира и элементарными звеньями малеинового ангидрида составляет приблизительно 1:3, среднечисленная молекулярная масса (Mn) равна 5891, а показатель молекулярно-массового распределения (Mn/Mw) составляет 1,28 (см. приведенный выше пример 20). К 100 мг указанного исходного соединения добавляли 1 мл этанола и получающуюся в результате реакционную смесь оставляли отстаиваться в течение 16 часов при 40°С. По окончании данного периода времени добавляли 52 мкл 2,5 н. раствора гидроксида натрия в этаноле. Таким образом полученную смесь концентрировали при 35°С в результате упаривания, а после этого высушивали в вакууме до получения натриевой соли поли(ПЭГ500-МА)эа (соединения № 54) в виде маслянистого вещества. Соотношение, задаваемое гидролизом, рассчитывали с использованием способа, описанного в приведенных выше примерах 3 и 6. А именно, в результате определения уровня содержания карбоксильных групп в натриевой соли поли(ПЭГ500-МА)эа (соединении № 54) и соответствующей натриевой соли поли(ПЭГ500-МА)г рассчитывали соотношение между остатками малеинового ангидрида в целевом соединении, подвергнутыми алкоголизу, и полным количеством остатков малеинового ангидрида в исходном соединении, и оно было определено равным 0,69. Таким образом, можно видеть, что 69% остатков малеинового ангидрида в исходном соединении было подвергнуто алкоголизу, а оставшийся 31% остатков малеинового ангидрида был подвергнут гидролизу.
Пример 22
Получение комплексов между полимерным модификатором из примера 21 и OCIF
Комплексы полимер-OCIF настоящего изобретения получали в виде водных растворов с использованием по существу того же самого препаративного подхода, что и в приведенном выше примере 14, используя водный раствор соединения № 54, полученный в приведенном выше примере 21 [концентрация соединения № 54: 17,2 мг/мл; среда: PBS, рН 7,4 (который представляет собой раствор, полученный в результате перемешивания водного раствора, содержащего 10 мМ гидрофосфата натрия и 150 мМ хлорида натрия, и водного раствора, содержащего 10 мМ дигидрофосфата натрия и 150 мМ хлорида натрия, при подходящем соотношении с получением буфера, характеризующегося величиной рН, равной 7,4)], и водный раствор очищенного, зрелого OCIF человека (OCIF получали так, как это описывается в WO 96/26217 и ЕР 816380) (концентрация OCIF: 4 мг/мл; среда: буфер, содержащий 10 мМ фосфатного иона и 150 мМ NaCl, рН 6,0). Говоря более конкретно, к 1,328 мл указанного раствора OCIF с концентрацией 4 мг/мл добавляли 0,472 мл раствора соединения № 54 с концентрацией 17,2 мг/мл и получали раствор, содержащий 4,5 мг/мл соединения № 54 и 3 мг/мл OCIF. Получающейся в результате реакционной смеси давали возможность отстояться в течение 3 дней при 4, 10 или 25°С до получения водных растворов желательных комплексов настоящего изобретения. Размеры комплексов измеряли в приведенном ниже примере испытания 13.
Пример 23
Получение продукта реакции алкоголиза между этанолом и поли(ПЭГ500-MA) [поли(ПЭГ500-MA)эа], где m=6-16, R3=OCH2CH3, соотн. сост.=приблизительно 1:3, ср. степ. пол.=20-30 (соединение № 55)
В качестве исходного соединения использовали поли(ПЭГ500-МА) (АМ-0510К, партия M3N550, изготовленный с использованием методики, подобной той, что описывается в японском патенте № 2621308 и публикациях японских патентных заявок №№ 2003-105040 и 2003-104003) - статистический сополимер полиоксиэтиленаллилметилового простого диэфира (m=6-16, Alk=этилен, R1=водород, и R2=метил) и малеинового ангидрида, в котором полиоксиэтиленовая боковая цепь характеризуется средней молекулярной массой, приблизительно равной 500, средняя степень полимеризации для основной цепи находится в диапазоне от 20 до 30, соотношение между элементарными звеньями полиоксиэтиленаллилметилового простого диэфира и элементарными звеньями малеинового ангидрида составляет приблизительно 1:3, среднечисленная молекулярная масса (Mn) равна 5891, а показатель молекулярно-массового распределения (Mn/Mw) составляет 1,28 (см. приведенный выше пример 20). К 50 мг указанного исходного соединения добавляли 0,5 мл этанола и получающуюся в результате реакционную смесь оставляли отстаиваться при 37°С в течение 24 часов. Целевое соединение поли(ПЭГ500-МА)эа (соединение № 55) получали в виде раствора в этаноле [концентрация поли(ПЭГ500-МА)эа: 100 мг/мл].
Пример 24
Получение комплексов между полимерным модификатором из примера 23 и OCIF
Комплексы полимер-OCIF настоящего изобретения получали в виде водных растворов с использованием по существу того же самого препаративного подхода, что и в приведенном выше примере 14, используя раствор соединения № 55 в этаноле, полученный в приведенном выше примере 23, и водный раствор очищенного, зрелого OCIF человека (OCIF получали так, как это описывается в WO 96/26217 и ЕР 816380) (концентрация OCIF: 5 мг/мл; среда: буфер, содержащий 10 мМ фосфатного иона и 150 мМ NaCl, рН 6,0). Говоря более конкретно, к 0,5 мл указанного раствора OCIF с концентрацией 5 мг/мл добавляли 37,5 мкл раствора соединения № 55 в этаноле и получающейся в результате реакционной смеси давали возможность отстояться в течение 3 дней при 25°С до получения растворов желательных комплексов настоящего изобретения. Размеры комплексов измеряли в приведенном ниже примере испытания 13.
Сравнительный пример 1
Получение сополимера монометоксиполиэтиленгликоля-метилвинилового эфира-малеиновой кислоты (ПЭГ-ПМВМА)
Привитой сополимер монометоксиполиэтиленгликоля-метилвинилового эфира-малеиновой кислоты (ПЭГ-ПМВМА) получали в соответствии со способом, описанным в примере 2 японской патентной заявки (Kokai) № Hei 11-302199.
Сравнительный пример 2
Получение комплекса, содержащего сополимер монометоксиполиэтиленгликоля-метилвинилового эфира-малеиновой кислоты (ПЭГ-ПМВМА) и белок OCIF
Подвергнутый очистке OCIF человека (OCIF получали так, как это описывается в WO 96/26217 и ЕР 816380), модифицированный с использованием ПЭГ-ПМВА, полученного так, как это описывается в приведенном выше сравнительном примере 1, получали в виде раствора [среда: PBS (pH 6,0)] по существу по тому же самому способу, что и в примере 9. Говоря более конкретно, перемешивали 1 мл раствора OCIF [концентрация OCIF: 2 мг/мл; среда: PBS (рН 6,0)] и 1 мл раствора ПЭГ-ПМВА [концентрация ПЭГ-ПМВА: 2 или 20 мг/мл; среда: PBS (рН 6,0)] и для получения целевого комплекса смесь оставляли отстаиваться в течение 24 часов при 25°С.
Сравнительный пример 3
Получение комплекса, содержащего полимерный модификатор и белок OCIF с использованием поли(ПЭГ500-МА) в качестве полимерного модификатора
К 28,4 мкл водного раствора подвергнутого очистке OCIF человека (OCIF получали так, как это описывается в WO 96/26217 и ЕР 816380) (концентрация полимера: 3,5 мг/мл, среда: 0,5 М водный раствор NaH2PO4, рН которого доводили до 7,6 с использованием 5 М водного раствора NaOH) добавляли 2,2 мкл раствора в диметилсульфоксиде, содержащего поли(ПЭГ500-МА) [АМ-0530К, изготовленный в компании NOF Corporation (здесь и далее в настоящем документе обозначаемый как «поли(ПЭГ500-МА)»] (концентрация полимера: от 35 до 350 мг/мл) и таким образом полученный раствор (концентрация OCIF: 3,2 мг/мл, концентрация поли(ПЭГ500-МА): 2,5 мг/мл или 6,3 мг/мл) встряхивали при 25°С в течение 40 часов. По окончании данного периода времени раствор разбавляли с использованием PBS (рН 7,0) и получали раствор OCIF, модифицированного при помощи полимерного модификатора, с концентрацией OCIF, равной 0,25 мг/мл. Таким образом полученный раствор хранили при 4°С.
Пример испытания 1
Измерение уровня содержания карбоксильных групп для полимеров из примеров от 10 до 13 методом кондуктометрического титрования
Уровень содержания карбоксильных групп для каждого из полимеров [поли(ПЭГ500-МА)а (соединений №№ 9 и 10), поли(ПЭГ500-МА)дма (соединения № 11) и поли(ПЭГ500-МА)г (соединения № 12)], полученных в примерах от 10 до 13, определяли методом кондуктометрического титрования следующим образом.
Во-первых, каждый из полимеров подвергали очистке с использованием гель-фильтрации следующим образом. Для каждого полимера 100 мг полимера растворяли в 4 мл 0,001 н. раствора гидроксида натрия. Раствор разделяли на четыре партии и каждую партию в 1 мл наносили на гель-фильтрационную колонку (PD-10, изготовленную в компании Amersham-Pharmacia). Первый 1 мл элюента отбрасывали. После этого на каждую колонку наносили 1,5 мл 0,001 н. раствора гидроксида натрия и в результате элюирования из колонки получали и отбрасывали еще 1,5 мл. Затем на каждую колонку наносили 2,5 мл 0,001 н. раствора гидроксида натрия и в результате элюирования из колонки получали еще 2,5 мл, и именно данная фракция содержала очищенное соединение. Очищенные фракции из четырех колонок объединяли и получали очищенный раствор целевых соединений. Выход после стадии очистки (определенный спектрофотометрически при измерении оптической плотности для поли(ПЭГ500-MA)г в области 210 нм до и после очистки) зафиксировали равным 80%, а концентрацию в подвергнутом очистке растворе определили равной 8 мг/мл.
Для каждого из растворов подвергнутого очистке полимера аликвоту (от 2,5 до 7,5 мл) доводили до 50 мл с использованием дистиллированной воды или 0,001 М водного раствора гидроксида натрия, а после этого к получающемуся в результате раствору добавляли 1 М водный раствор гидроксида натрия для доведения значения рН до 12. Затем к раствору или порциями по 0,1 мл, или непрерывно со скоростью 0,1 мл/мин добавляли 0,1 М хлористоводородную кислоту. В первом случае рН и проводимость измеряли после каждого добавления, а во втором случае их измерение проводили каждые 15 секунд. После этого уровень содержания карбоксильных групп в полимере рассчитывали из количества 0,1 М хлористоводородной кислоты, добавленной в буферной области проводимости (соответствующей диапазону рН в пределах приблизительно от 10 до 5,5). Результаты продемонстрированы в приведенной ниже таблице 5.
В случае полимеров из примеров от 10 до 12 соотношение между остатками малеинового ангидрида в исходном соединении, подвергнутыми аммонолизу или аминолизу, и остатками малеинового ангидрида, подвергнутыми гидролизу, в каждом случае определяли в результате вычислений следующим образом. Как продемонстрировано в приведенной выше таблице 5, уровень содержания карбоксильных групп в 1 г поли(ПЭГ500-МА)г (соединения № 12) составляет 3,21 ммоль. Исходя из данных величин определяли массу поли(ПЭГ500-МА) (то есть, массу сополимера до проведения гидролиза) на один грамм функциональной группы и массу поли(ПЭГ500-МА)а (соединений №№ 9 и 10), полученных в результате добавления аммиака к поли(ПЭГ500-МА), на один грамм функциональной группы. Говоря конкретно, массу поли(ПЭГ500-МА) (то есть, массу сополимера до проведения гидролиза) на один моль остатка малеинового ангидрида получали в результате вычитания молекулярной массы молекулы воды (18 г) из массы полностью гидролизованного сополимера, что давало в результате численное значение, равное 605 г. Массу поли(ПЭГ500-МА)а (соединений №№ 9 и 10) на один моль карбоксильной группы получали в результате добавления молекулярной массы молекулы аммиака (17 г) к массе поли(ПЭГ500-МА), что давало в результате численное значение, равное 622 г. Исходя из данного значения в результате вычисления (1 г/622) определяли теоретический уровень содержания карбоксильных групп в 1 г поли(ПЭГ500-МА)а (соединениях №№ 9 и 10), в которых все остатки малеинового ангидрида были подвергнуты аммонолизу (то есть, при отсутствии гидролиза), и, как было установлено, он составляет 1,61 ммоль. По такому же способу в результате добавления молекулярной массы молекулы диметиламина к массе поли(ПЭГ500-МА) определяли массу поли(ПЭГ500-МА)дма (соединения № 11), полученного в результате добавления диметиламина к поли(ПЭГ500-МА), на один моль карбоксильной группы, что давало в результате численное значение, равное 650 г. Исходя из данного значения в результате вычисления определяли теоретический уровень содержания карбоксильных групп в 1 г поли(ПЭГ500-МА)дма, в котором все остатки малеинового ангидрида были подвергнуты аминолизу под действием диметиламина, и, как было установлено, он составляет 1,54 ммоль.
Для каждого из полимеров из примеров от 10 до 12 соотношение, задаваемое гидролизом (процентную долю исходного соединения, подвергнутого гидролизу, в сопоставлении с аммонолизом или аминолизом), и степень прохождения реакции аммонолиза или аминолиза (%) определяли из уровня содержания карбоксильных групп в 1 г поли(ПЭГ500-МА)г (соединения № 11), в котором все остатки малеинового ангидрида были подвергнуты гидролизу, теоретического уровня содержания карбоксильных групп в 1 г поли(ПЭГ500-МА)а (соединениях №№ 9 и 10), в которых все остатки малеинового ангидрида были подвергнуты аммонолизу, теоретического уровня содержания карбоксильных групп в 1 г поли(ПЭГ500-МА)дма (соединении № 11), в котором все остатки малеинового ангидрида были подвергнуты аминолизу под действием диметиламина, и фактических измеренных уровней содержания карбоксильных групп в 1 г каждого из полимеров из примеров от 10 до 12 в соответствии с измерениями по описанному выше методу кондуктометрического титрования. Результаты продемонстрированы в приведенной выше таблице 5.
Пример испытания 2
Оценка удерживания комплексов OCIF-модификатор в крови на примере крысы
Каждый из полученных образцов, описанных в примере 9, сравнительном примере 2, и немодифицированный подвергнутый очистке OCIF человека (OCIF получали так, как это описывается в WO 96/26217 и ЕР 816380) надлежащим образом разводили с использованием PBS (рН 6,0) таким образом, что концентрация OCIF составляла 0,25 мг/мл. Каждый из разбавленных образцов, полученных таким образом, вводили через вену в хвост самке крысы Wistar возрастом в пять недель (имеющей массу тела, приблизительно равную 100 г, и которой не давали пищу в течение одного дня) таким образом, чтобы доза OCIF составляла бы 0,5 мг/кг (2 мл/кг по объему). Спустя 6 часов после введения из сердца крысы отбирали 200 мкл крови, а после этого методом ELISA измеряли концентрацию OCIF в сыворотке крови, при этом условиями были те, что описываются в приведенном ниже примере испытания 3.
Концентрация OCIF в сыворотке крови при измерении после введения каждого образца продемонстрирована в приведенной ниже таблице 6.
Концентрация OCIF в сыворотке крови после внутривенного введения каждого образца OCIF
Из приведенной выше таблицы 6 легко можно видеть, что каждый из комплексов между полимерным модификатором и белком настоящего изобретения значительно улучшает удерживание белка в крови в сопоставлении с удерживанием белка при введении только его одного. Из сопоставления комплекса предшествующего уровня техники между ПЭГ-ПМВМА и белком (описанного в выложенном японском патенте № Hei 11-302199 и полученного в приведенном выше сравнительном примере 2) и комплексов настоящего изобретения, характеризующихся тем же самым массовым соотношением между модификатором и белком (1 или 10), также можно видеть, что комплексы между модификатором и белком настоящего изобретения обеспечивают достижение значительно улучшенного удерживания белка в крови в сопоставлении с комплексом предшествующего уровня техники между ПЭГ-ПМВМА и белком.
Пример испытания 3
Оценка степени обнаружения OCIF методом ELISA
Одна из проблем, с которой сталкиваются в случае модификаторов белков предшествующего уровня техники, заключается в том, что связывание модификаторов с белком становится причиной модифицирования структуры белка и/или экранирования белка вследствие образования объемных комплексов. Для того чтобы протестировать комплексы OCIF-модификатор настоящего изобретения, методом ELISA проводили определение степени обнаружения каждого из комплексов, полученных в соответствии с описанием в примере 9 и сравнительном примере 2, на основе немодифицированного, подвергнутого очистке, OCIF человека (OCIF получали так, как это описывается в WO 96/26217 и ЕР 816380). Метод ELISA реализовывали следующим образом.
Моноклональное антитело против OCIF человека OI-19 (полученное в соответствии со способом, описанным в ЕР0974671) растворяли в 0,1 М растворе кислого карбоната натрия (рН 9,6) и получали раствор, характеризующийся концентрацией OI-19, равной 10 мкг/мл. 100 мкл таким образом полученного раствора OI-19 помещали в каждую лунку 96-луночного иммунопланшета (изготовленного в компании Nunc) и планшеты оставляли выдерживаться при 4°С в течение ночи. По окончании данного периода времени в каждую лунку для блокирования добавляли 50%-ный Block Ace (приобретаемый в компании Snow Brand Milk Products Co., Ltd.) и после этого планшеты три раза промывали с использованием PBS (промывающего буфера), содержащего 0,1% Tween 20. Подвергнутый очистке OCIF человека (OCIF получали так, как это описывается в WO 96/26217 и ЕР 816380) растворяли в первичном реакционном буфере (то есть, 0,2 М трис-буферном растворе, включающем хлористоводородную кислоту, (рН 7,4), содержащем 40% Block Ace, 0,1% Tween 20 и 10 мкг/мл IgG мыши) для получения стандартных растворов с различными концентрациями OCIF. 100 мкл каждого из таким образом полученных растворов с различными концентрациями OCIF добавляли в каждую лунку, планшеты встряхивали при комнатной температуре в течение 2 часов, а после этого каждую лунку промывали шесть раз с использованием промывающего буфера. После этого проводили 10000-кратное разбавление POD-OI-4 (то есть, антитела, помеченного пероксидазой и распознающего OCIF, полученного так, как это описывается в ЕР 0974671) с использованием вторичного реакционного буфера (то есть, 0,1 М трис-буферного раствора, включающего хлористоводородную кислоту, (рН 7,4), содержащего 25% Block Ace, 0,1% Tween 20 и 10 мкг/мл IgG мыши), 100 мкл получающегося в результате раствора добавляли в каждую лунку, планшеты встряхивали при комнатной температуре в течение 2 часов, а после этого каждую лунку промывали шесть раз. Как только это осуществляли, в каждую лунку добавляли 100 мкл субстратного раствора (реагента, растворимого в ТМБ, доступного в компании Scytek) и планшеты встряхивали при комнатной температуре в течение промежутка времени продолжительностью от 10 до 15 минут. После этого в каждую лунку добавляли 100 мкл реагента, прекращающего реакцию (содержащего ТМБ стоп-буфера, доступного в компании Scytek), и планшеты подвергали осторожному встряхиванию. По окончании данного периода времени для каждой лунки при помощи микропланшет-ридера (MEML 001, изготовленного в компании Molecular Device corporation) проводили измерение оптической плотности при длине волны в области 450 нм. Исходя из данных результатов, получали калибровочную кривую в виде зависимости оптической плотности от концентрации OCIF. После получения данной калибровочной кривой методику повторяли для каждого из тестируемых комплексов между OCIF и модификатором, при этом в каждую лунку добавляли 100 мкл каждого из растворов, содержащих тестируемый комплекс, реакцию с POD-OI-4 проводили по тому же самому способу, что и указанный выше, и после этого для каждой лунки при помощи микропланшет-ридера измеряли оптическую плотность при длине волны в области 450 нм. Сопоставление с оптическими плотностями, полученными с использованием калибровочной кривой, делало возможным получение концентрации OCIF, детектируемой методом ELISA для каждого из комплексов, для которых проводились измерения.
Для каждого из образцов рассчитывали степень необнаружения OCIF методом ELISA, и результаты продемонстрированы в приведенной ниже таблице 7. Степень необнаружения OCIF методом ELISA определяли с использованием нижеследующего уравнения:
Степень=[1-(концентрация OCIF, измеренная методом ELISA)/(концентрация OCIF, измеренная методом Лоури)]×100
В приведенном выше уравнении метод Лоури для измерения концентрации OCIF в комплексах определяли так, как это описывается в японской патентной заявке № 2002-190407. Это позволяло получить меру полной концентрации OCIF в комплексах. Степень необнаружения OCIF в комплексе методом ELISA представляет собой меру изменения конформации OCIF, вызванную комплексообразованием с модификатором. Низкая степень необнаружения представляет собой свидетельство того, что OCIF в комплексе может легко связываться с антителами против OCIF как OI-19, так и OI-4, что, таким образом, демонстрирует для структуры OCIF в комплексе наличие незначительного модифицирования или его отсутствие.
Степень необнаружения для образца OCIF методом ELISA
Исходя из результатов, продемонстрированных в приведенной выше таблице 7, можно видеть, что в случае комплексов между полимерным модификатором и белком настоящего изобретения степень необнаружения OCIF методом ELISA в результате модифицирования структуры белка значительно уменьшалась в сопоставлении с высокой степенью, которую получали в случае комплекса предшествующего уровня техники между ПЭГ-ПМВМА и белком, полученного в приведенном выше примере испытания 2.
Таким образом, исходя из результатов примеров испытаний 2 и 3 было подтверждено, что понижение чувствительности при обнаружении белка, вызванное избыточным модифицированием белка в случае комплексов между полимерным модификатором и белком настоящего изобретения, очень невелико, и, кроме того, удерживание в крови для белка в указанных комплексах значительно лучше по сравнению с достигаемым в случае комплексов предшествующего уровня техники.
Пример испытания 4
Измерение концентрации OCIF в крови
Каждый из образцов, полученных так, как это описывается в примере 9, и подвергнутый очистке OCIF человека (OCIF получали так, как это описывается в WO 96/26217 и ЕР 816380) надлежащим образом разбавляли с использованием PBS (характеризующегося величиной рН в диапазоне от 6,0 до 7,4) таким образом, чтобы концентрация OCIF находилась бы в диапазоне от 0,1 до 1 мг/мл. Каждый из таким образом полученных разбавленных образцов после этого вводили через подкожную вену ноги или подкожно в спину самке обезьяны рода Cynomolgus возрастом шести или семи лет (имеющей массу тела в диапазоне от 2 до 4 кг, и которой не давали пищу в течение одного дня) таким образом, чтобы ввести дозу OCIF в диапазоне от 0,1 до 1 мг/кг (1 мл/кг по объему). По истечении предварительного заданного периода времени после введения в диапазоне от пяти минут до одного месяца из ее кровеносного сосуда бедра отбирали 500 мкл крови и концентрацию OCIF в сыворотке крови измеряли методом ELISA, описанным в приведенном выше примере испытания 3.
Пример испытания 5
Измерение плотности кости
Каждый из образцов, полученных так, как это описывается в примере 9, и подвергнутый очистке OCIF человека (OCIF получали так, как это описывается в WO 96/26217 и ЕР 816380) надлежащим образом разбавляли с использованием PBS (характеризующегося величиной рН в диапазоне от 6,0 до 7,4) таким образом, чтобы концентрация OCIF находилась бы в диапазоне от 0,7 до 3,5 мг/мл. После этого получали адъювант с использованием убитых клеток Mycobacterium butyricum и жидкого парафина и его вводили в виде инъекции в кожу корня хвоста самке крысы Lewis возрастом от 5 до 10 недель для стимулирования развития у указанной крысы артрита. Спустя две недели после инъекции адъюванта крысе через хвостовую вену или подкожно в спину вводили каждый из полученных образцов таким образом, чтобы доза OCIF находилась бы в диапазоне от 1,4 до 7 мг/кг (2 мл/кг по объему). Спустя три недели после инъекции адъюванта крысу препарировали для извлечения обеих бедренных костей и измеряли плотность ее костей.
Пример испытания 6
Оценка размера молекулы методом электрофореза в полиакриламидном геле в присутствии SDS в невосстанавливающих условиях
Размер молекулы для каждого из комплексов между полимерным модификатором и OCIF, полученным так, как это описывается в примере 14 и сравнительном примере 3, и подвергнутого очистке OCIF человека (OCIF получали так, как это описывается в WO 96/26217 и ЕР 816380) оценивали методом SDS-PAGE в невосстанавливающих условиях следующим образом.
К 10 мкл каждого из тестируемых образцов [которые при необходимости разбавляли с использованием физиологического раствора с фосфатным буфером (PBS (рН 7,0), который представляет собой буферный раствор, полученный в результате перемешивания раствора, содержащего 10 мМ гидрофосфата натрия и 150 мМ хлорида натрия, и раствора, содержащего 10 мМ дигидрофосфата натрия и 150 мМ хлорида натрия, при подходящем объемном соотношении с получением буфера, характеризующегося величиной рН, равной 7,0) таким образом, чтобы концентрация белка составляла бы 250 мкг/мл] добавляли 5 мкл содержащего LDS (додецилсульфат лития) буфера для образцов (4Х) NuPAGE (торговая марка) (полученного в компании Invitrogen Life Technology) и 5 мкл очищенной воды и каждый из получающихся в результате растворов нагревали при 95°С в течение 7 минут. По окончании данного периода времени к полиакриламидному гелю для проведения электрофореза в присутствии SDS (3-8%-ному трис-ацетатному гелю, имеющему толщину 1 мм и изготовленному в компании NOVEX) добавляли полное количество реакционной смеси и к гелю прикладывали напряжение 150 В с использованием блока электропитания (PhoreStar Pro, изготовленного в компании Anatech). После завершения электрофореза белок на геле окрашивали с использованием кумасси голубого в соответствии со способом, хорошо известным специалистам в соответствующей области.
Как можно видеть из фигур 1 и 2, OCIF, модифицированный с использованием полимерного модификатора настоящего изобретения, устойчиво детектировался в качестве вещества, характеризующегося молекулярной массой, превышающей молекулярную массу немодифицированного OCIF (120 кДа) при всех соотношениях компонентов при смешении (первичную полосу в области от 130 до 150 кДа и вторичную полосу в области от 180 до 200 кДа детектировали, основываясь на метках маркеров молекулярной массы), и, кроме того, ни обнаруживали никакого комплекса, характеризующегося молекулярной массой, превышающей 210 кДа. Подобные результаты (не показаны) также получали и для других полимерных модификаторов настоящего изобретения, которые представляли собой продукты аминолиза и алкоголиза. С другой стороны, как можно видеть из фигуры 3, в случае OCIF, модифицированного с использованием обычного полимерного модификатора поли(ПЭГ500-МА), полученного в сравнительном примере 3, количество полученного сырого комплекса значительно увеличивается по мере увеличения доли модификатора.
Данный результат продемонстрировал, что в сравнении со структурно довольно похожим полимерным модификатором поли(ПЭГ500-МА) предшествующего уровня техники использование полимерного модификатора настоящего изобретения может обеспечить значительное подавление образования сырых комплексов, которые не являются предпочтительными с точки зрения фармацевтики, и получение комплекса между полимерным модификатором и белком, который обладает устойчивыми свойствами вне зависимости от соотношения компонентов при смешении.
Пример испытания 7
Оценка активности поли(ПЭГ500-МА)а при образовании ковалентной связи
Определяли реакционную способность для каждого из полимерных модификаторов поли(ПЭГ500-МА)а (соединения № 9) и поли(ПЭГ500-МА)г (соединения № 12) по отношению к тетраметилродаминкадаверину, который представляет собой флуоресцентное вещество, характеризующееся наличием аминогруппы и молекулярной массой, равной 514,62, (доступное в компании Molecular Probes, Inc.) (здесь и далее в настоящем документе обозначаемое как "Rho-NH2") и активность модификаторов при образовании ковалентной связи оценивали в результате сопоставления реакционной способности двух полимерных модификаторов следующим образом.
К 18,9 мкл PBS (рН 6,0), содержащего 1,08 мг/мл Rho-NH2, добавляли 3,8 мкл растворов каждого из поли(ПЭГ500-МА)г (соединения № 12) (полученного так, как описывается в приведенном выше примере 13) и поли(ПЭГ500-МА)а (соединения № 9) [концентрация полимера: 21 мг/мл, среда: PBS (рН доводили до 9,5 с использованием 1 М NaOH)] (полученного так, как описывается в приведенном выше примере 10). Таким образом полученные растворы оставляли отстаиваться при 25°С в течение 3 дней. По окончании данного периода времени реакционные смеси фракционировали методом гель-фильтрационной хроматографии (колонка: PD-10, изготовленная в компании Amersham Biotech, подвижная фаза: очищенная вода) следующим образом. На гель-фильтрационную колонку наносили 0,5 мл реакционной смеси и отбрасывали 0,5 мл стока из колонки. В колонку добавляли 2 мл дистиллированной воды и в результате элюирования получали и отбрасывали еще 2 мл. В колонку добавляли еще 2 мл дистиллированной воды и в результате элюирования получали еще 2 мл, и данная фракция содержала фракцию полимера. Данные по количеству Rho-NH2, содержащегося во фракциях полимера, получали методом флуоресцентной спектрометрии (длина волны возбуждения: 544 нм, длина волны флуоресценции: 571 нм, среда: очищенная вода с доведением значения рН до 3), проводя вычисление соотношения между количеством Rho-NH2, содержащегося в каждом комплексе между полимером и Rho-NH2, и полным количеством Rho-NH2 в реакционной смеси, то есть, степени связывания между Rho-NH2 и полимером.
Результаты продемонстрированы в приведенной ниже таблице 8.
Реакционная способность полимерного модификатора
Из приведенной выше таблицы 8 легко можно видеть, что для реакционной смеси между полимерным модификатором поли(ПЭГ500-МА)а (соединением № 9) и Rho-NH2 имеет место довольно высокий уровень Rho-NH2, детектируемого во фракции полимера. Этим данный случай отличается от реакционной смеси между полимерным модификатором поли(ПЭГ500-МА)г (соединением № 12) и Rho-NH2, для которой имеет место только низкий уровень Rho-NH2, детектируемого во фракции полимера. Если в колонку вводили один только Rho-NH2, то тогда во «фракции полимера» никакого элюирования Rho-NH2 не происходило. Данные результаты свидетельствуют о том, что полимерный модификатор поли(ПЭГ500-МА)а (соединение № 9) настоящего изобретения прочно связывается с аминогруппой Rho-NH2. С другой стороны, степень связывании для полимерного модификатора поли(ПЭГ500-МА)г (соединения № 12) настоящего изобретения невелика, предположительно свидетельствуя о том, что данный полимерный модификатор с аминогруппой Rho-NH2 прочно не связывается.
Пример испытания 8
Оценка степени обнаружения OCIF методом ELISA
Методом ELISA проводили определение степени обнаружения каждого из комплексов между OCIF и полимерным модификатором, полученных в соответствии с описанием в приведенных выше примере 14 и сравнительном примере 3, на основе немодифицированного подвергнутого очистке OCIF человека (OCIF получали так, как это описывается в WO 96/26217 и ЕР 816380). Метод ELISA реализовывали следующим образом.
Моноклональное антитело против OCIF человека OI-19 (полученное в соответствии со способом, описанным в ЕР0974671) растворяли в 0,1 М растворе кислого карбоната натрия (рН 9,6) и получали раствор, характеризующийся концентрацией OI-19, равной 10 мкг/мл. 100 мкл таким образом полученного раствора OI-19 помещали в каждую лунку 96-луночного иммунопланшета (изготовленного в компании Nunc) и планшеты оставляли выдерживаться при 4°С в течение ночи. По окончании данного периода времени в каждую лунку для блокирования добавляли 50%-ный Block Ace (приобретаемый в компании Snow Brand Milk Products Co., Ltd.) и после этого планшеты три раза промывали с использованием PBS (промывающего буфера), содержащего 0,1% Tween 20. Подвергнутый очистке OCIF человека (OCIF получали так, как это описывается в WO 96/26217 и ЕР 816380) растворяли в первичном реакционном буфере (то есть, 0,2 М трис-буферном растворе, включающем хлористоводородную кислоту (рН 7,4), содержащем 40% Block Ace, 0,1% Tween 20 и 10 мкг/мл IgG мыши) для получения стандартных растворов с различными концентрациями OCIF. 100 мкл каждого из таким образом полученных растворов с различными концентрациями OCIF добавляли в каждую лунку, планшеты встряхивали при комнатной температуре в течение 2 часов, а после этого каждую лунку промывали шесть раз с использованием промывающего буфера. После этого проводили 10000-кратное разбавление POD-OI-4 (то есть, антитела, помеченного пероксидазой и распознающего OCIF, полученного так, как это описывается в ЕР 0974671) с использованием вторичного реакционного буфера (то есть, 0,1 М трис-буферного раствора, включающего хлористоводородную кислоту (рН 7,4), содержащего 25% Block Ace, 0,1% Tween 20 и 10 мкг/мл IgG мыши), 100 мкл получающегося в результате раствора добавляли в каждую лунку, планшеты встряхивали при комнатной температуре в течение 2 часов, а после этого каждую лунку промывали шесть раз. Как только это осуществляли, в каждую лунку добавляли 100 мкл субстратного раствора (реагента, растворимого в ТМБ, доступного в компании Scytek) и планшеты встряхивали при комнатной температуре в течение промежутка времени продолжительностью от 10 до 15 минут. После этого в каждую лунку добавляли 100 мкл реагента, прекращающего реакцию (содержащего ТМБ стоп-буфера, доступного в компании Scytek), и планшеты подвергали осторожному встряхиванию. По окончании данного периода времени для каждой лунки при помощи микропланшет-ридера (MEML 001, изготовленного в компании Molecular Device corporation) проводили измерение оптической плотности при длине волны в области 450 нм. Концентрацию OCIF в каждом стандартном образце OCIF рассчитывали, основываясь на калибровочной кривой, полученной с использованием растворов OCIF, имеющих известную концентрацию.
Для каждого из образцов тестируемого комплекса измеряли оптические плотности и в соответствии с объяснениями в приведенном выше примере испытания 3 проводили вычисление концентраций OCIF, рассчитываемых по калибровочной кривой, и степени необнаружения OCIF методом ELISA. Полученные результаты продемонстрированы в приведенной ниже таблице 9.
Степень необнаружения для образца OCIF методом ELISA
Из приведенной выше таблицы 9 легко можно видеть, что для каждого из комплексов между полимерным модификатором и белком настоящего изобретения, полученных в примере 14, степень необнаружения OCIF методом ELISA, обусловленная модифицированием белка, была значительно уменьшена по сравнению с комплексом между полимером и белком предшествующего уровня техники, полученным в сравнительном примере 3, для которого степень необнаружения OCIF методом ELISA была очень большой.
Исходя из данных результатов становится ясно, что комплексы между полимерным модификатором и белком настоящего изобретения вызывают только очень небольшое понижение чувствительности детектирования методом ELISA, которое может быть обусловлено избыточным модифицированием белка и/или образованием объемного комплекса.
Пример испытания 9
Оценка удерживания в крови на примере крысы
Каждый из полученных образцов, описанных в примере 14, и немодифицированный подвергнутый очистке OCIF человека (OCIF получали так, как это описывается в WO 96/26217 и ЕР 816380) надлежащим образом разводили с использованием PBS (рН 7,0) таким образом, что концентрация OCIF составляла 0,25 или 0,025 мг/мл. Каждый из разбавленных образцов, полученных таким образом, вводили через бедренную вену самке крысы Wistar возрастом пять недель (имеющей массу тела, приблизительно равную 100 г, и которой не давали пищу в течение одного дня) таким образом, чтобы доза OCIF составляла бы 0,5 или 0,05 мг/кг (2 мл/кг по объему). Спустя 6 часов после введения из яремной вены крысы отбирали 200 мкл крови, а после этого по описанному выше методу ELISA измеряли концентрацию OCIF в сыворотке крови.
Концентрация OCIF в сыворотке крови при измерении после введения каждого образца продемонстрирована в приведенной ниже таблице 10.
Концентрация OCIF в сыворотке крови после внутривенного введения каждого образца OCIF
Как с очевидностью следует из приведенной выше таблицы 10, комплекс между полимерным модификатором и белком настоящего изобретения обеспечивает значительное улучшение удерживания указанного белка в крови в сопоставлении со случаем введения одного только белка.
Исходя из приведенных выше результатов становится очевидным, что комплекс между полимерным модификатором и белком настоящего изобретения можно устойчиво получать в широком диапазоне условий, и что указанный комплекс обеспечивает значительное улучшение удерживания в крови белка комплекса после введения. Поэтому комплекс, вероятно, является чрезвычайно полезным в сфере фармацевтики и биохимии.
Пример испытания 10
Оценка удерживания в крови на примере крысы
Для каждого из образцов, полученных в примере 17 и примере 19, проводили оценку по тому же самому способу, что и в примере испытания 9. Концентрация OCIF в сыворотке крови при измерении после приема каждого образца продемонстрирована в приведенной ниже таблице 11.
Концентрация OCIF в сыворотке крови после внутривенного введения каждого образца OCIF
Из приведенной выше таблицы 11 легко можно видеть, что все протестированные полимерные модификаторы настоящего изобретения, характеризующиеся различными размерами молекул, обеспечивают значительное улучшение удерживание белка в крови в сопоставлении со случаем введения одного только белка.
Пример испытания 11
Оценка размера молекулы методом эксклюзионной хроматографии по размеру
Размер молекулы для каждого из комплексов между полимерным модификатором и OCIF, полученных в примере 9, примере 14, примере 17 и примере 19, оценивали методом эксклюзионной хроматографии по размеру. Условия проведения испытания продемонстрированы в приведенной ниже таблице 12.
Условия при проведении эксклюзионной хроматографии размеров
Колонка: Superdex 200 HR10/30 (Amersham Biotech)
Подвижная фаза: физиологический раствор с фосфатным буфером (8 мМ Na2HPO4, 15 мМ КН2РО4, 145 мМ NaCl, 0,5 г/л NaN3)
Температура проведения анализа: 4°С
Длина волны детектирования: 280 нм
Скорость потока подвижной фазы: 0,6 мл/мин
Время удерживания нижеследующих стандартных белков в указанных выше условиях продемонстрировано в приведенной ниже таблице 13.
Время удерживания для каждого стандартного белка
Исходя из результатов эксклюзионной хроматографии по размеру, радиус Стокса для незакомплексованного OCIF определили равным 5,63 нм, а радиус Стокса для каждого комплекса полимерный модификатор-OCIF данного изобретения определяли следующим образом:
Комплексы, полученные в примере 9:
Комплексы поли(ПЭГ500-МА)г (соединение № 1)-OCIF (радиусы Стокса в диапазоне от 6,13 нм и 7,32 нм),
Комплексы поли(ПЭГ500-МА)а (соединение № 3)-OCIF (от 6,12 нм и 6,54 нм),
Комплекс поли(ПЭГ500-МА)дма (соединение № 4)-OCIF (6,39 нм),
Комплекс поли(ПЭГ500-МА)ипа (соединение № 5)-OCIF (6,26 нм),
Комплекс поли(ПЭГ500-МА)эа (соединение № 6)-OCIF (6,44 нм),
Комплекс поли(ПЭГ1500-МА)г (соединение № 2)-OCIF (от 6,44 нм до 6,71 нм),
Комплекс поли(ПЭГ1500-МА)а (соединение № 7)-OCIF (от 6,40 нм до 6,47 нм),
Комплекс поли(ПЭГ1500-МА)дма (соединение № 8)-OCIF (6,55 нм).
Исходя из данных результатов становится очевидным то, что каждый из комплексов изобретения характеризуется радиусом Стокса, который приблизительно на 1 нм превышает соответствующую величину для незакомплексованного OCIF в физиологическом растворе с фосфатным буфером, использованном в качестве подвижной фазы. Кроме того, у каждого из образцов пик, получаемый вследствие присутствия немодифицированного OCIF, не фиксировался, что свидетельствует о стабильности комплексов.
Радиусы Стокса для других комплексов определяли в условиях, подобных тем, что описываются выше. Результаты представляли собой нижеследующее:
Комплексы, полученные в примере 14:
Комплексы, полученные в условиях: концентрация OCIF 5 мг/мл; концентрация полимерного модификатора в диапазоне от 1,25 мг/мл до 5 мг/мл; рН 7,4; 25°С; 36 ч; среда в виде физиологического раствора с фосфатным буфером (концентрация фосфата 10 мМ; концентрация NaCl 150 мМ), как было показано, характеризуются радиусами Стокса в диапазоне от 6,2 нм до 6,5 нм. Кроме того, у каждого из образцов пик, получаемый вследствие присутствия немодифицированного OCIF, не фиксировался, что свидетельствует о стабильности комплексов.
Комплексы, полученные в примере 17:
Комплексы, полученные в условиях инкубации: концентрация OCIF 0,5 мг/мл; концентрация полимерного модификатора 0,5 мг/мл; рН 6,0; 25°С; 168 ч; среда в виде физиологического раствора с фосфатным буфером (концентрация фосфата 10 мМ; концентрация NaCl 150 мМ), как было показано, характеризуются радиусами Стокса в диапазоне от 6,1 нм до 6,7 нм. Кроме того, у каждого из образцов пик, получаемый вследствие присутствия немодифицированного OCIF, не фиксировался, что свидетельствует о стабильности комплексов.
Комплексы, полученные в примере 19:
Комплексы, полученные в условиях инкубации: концентрация OCIF 5 мг/мл; концентрация полимерного модификатора 5 мг/мл; рН 5,5; 25°С; 168 ч; среда в виде физиологического раствора с фосфатным буфером (концентрация фосфата 10 мМ; концентрация NaCl 150 мМ), как было показано, характеризуются радиусами Стокса в диапазоне от 6,3 нм до 6,8 нм. Кроме того, у каждого из образцов пик, получаемый вследствие присутствия немодифицированного OCIF, не фиксировался, что свидетельствует о стабильности комплексов.
Увеличение радиусов Стокса для комплексов модификатор-OCIF настоящего изобретения в сопоставлении с незакомплексованным OCIF можно отнести к модифицированию OCIF под действием полимерных модификаторов настоящего изобретения.
Пример испытания 12
Оценка удерживания в крови на примере крысы и оценка степени обнаружения OCIF методом ELISA
Испытания по определению удерживания в крови и степени обнаружения OCIF методом ELISA проводили для комплекса, полученного в примере 22 (комплекса между соединением № 54 и OCIF) (температура инкубации: 25°С) по тому же самому способу, что и описанный в приведенных выше примерах испытаний 2 и 3. Эквивалентная доза для OCIF была установлена на уровне 0,1 мг/кг. Концентрация OCIF в сыворотке, как было обнаружено, составляла 189 нг/мл спустя 6 часов после внутривенной инъекции комплекса. Таким образом, было подтверждено, что комплексы, полученные с использованием натриевой соли поли(ПЭГ500-МА)эа (соединения № 54) в приведенном выше примере 22, демонстрируют превосходный уровень удерживания в крови. Кроме того, понижение чувствительности при обнаружении OCIF методом ELISA, обусловленное избыточным модифицированием белка в комплексе под действием модификатора, как было обнаружено, очень невелико.
Пример испытания 13
Оценка размера комплекса
Размер молекул для комплексов, полученных в примерах 22 (комплекс между соединением № 54 и OCIF) и 24 (комплекс между соединением № 55 и OCIF), оценивали методом гель-фильтрационной хроматографии так, как это описывается в приведенном выше примере испытания 11. Было обнаружено, что размер молекул у данных комплексов демонстрирует высокий уровень однородности. В случае комплексов между соединением № 54 и OCIF, образованных при 4°С, 10°С и 25°С, радиусы Стокса, как было показано, были равны 6,0 нм, 6,0 нм и 6,0 нм. В случае комплексов между соединением № 55 и OCIF радиусы Стокса были равны 6,4 нм. Таким образом, каждый из комплексов, полученных в примере 22 и 24, характеризовался большим радиусом Стокса в сравнении с немодифицированным подвергнутым очистке OCIF человека (радиус Стокса: 5,63 нм).
Пример препарата
Раствор, содержащий комплекс, полученный в стерильных условиях по тому же самому способу, что и описанный в приведенном выше примере 14, подвергали лиофильной сушке и получали лиофильно высушенный препарат.




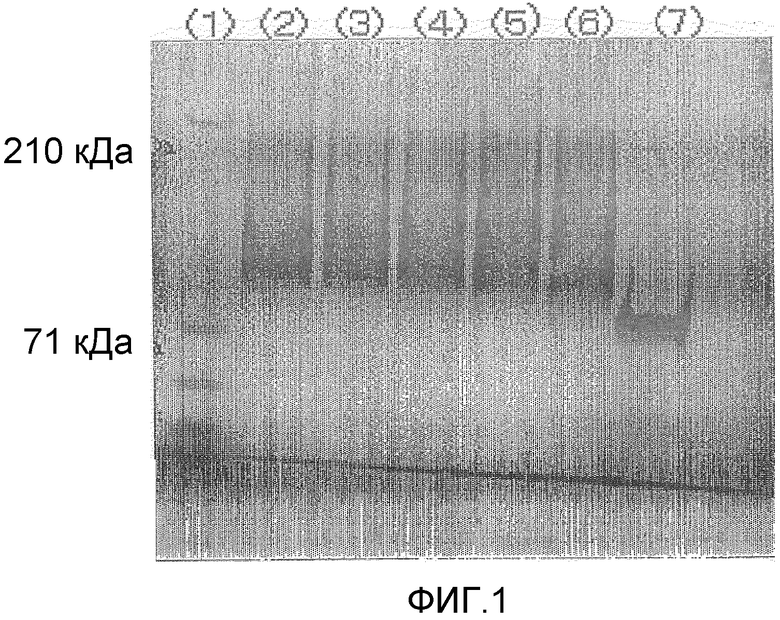
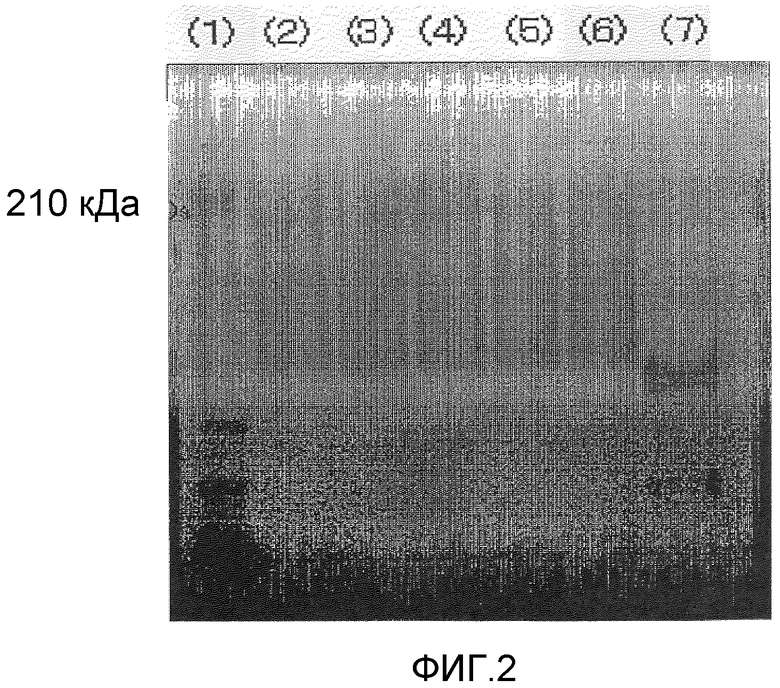
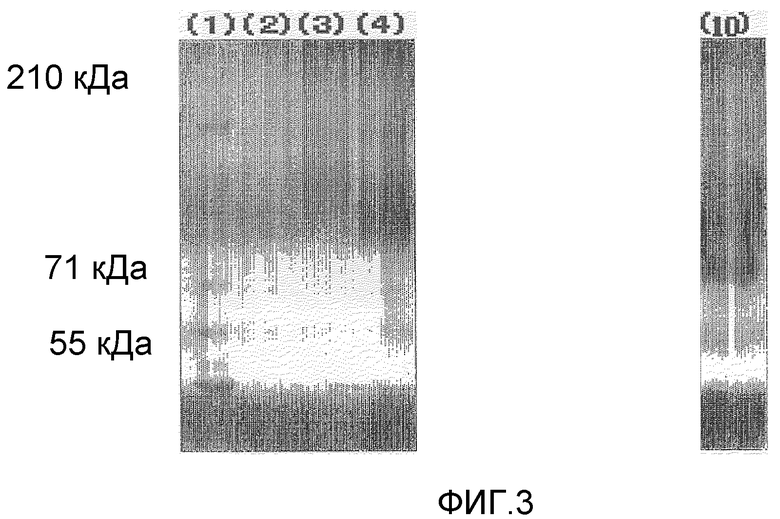
Изобретение относится к сополимеру или его фармакологически приемлемой соли, которые содержат в качестве образующих их элементарных звеньев (а) одно или несколько структурных элементарных звеньев, описываемых формулой (I), и (b) одно или несколько структурных звеньев, описываемых формулой (II), причем расположение структурных звеньев, представленных формулами (I) и (II), выбираются следующих последовательностей: (i) последовательность с чередованием «голова к голове», (ii) последовательность с чередованием «голова к хвосту», (iii) смешанная последовательность с чередованием «голова к голове» и «голова к хвосту», (iv) произвольная последовательность, с учетом того, что соотношение между структурными звеньями формулы (I) и структурными звеньями формулы (II) в указанном сополимере находится в диапазоне от 10:1 до 1:10. Также настоящее изобретение относится к сополимеру или его фармакологически приемлемой соли, полученным в результате введения одного или нескольких звеньев ангидрида карбоновой кислоты, описанных формулой (III), в сополимер, который содержит в качестве образующих его элементарных звеньев (а) одно или несколько структурных элементарных звеньев, описываемых формулой (I), и (b) структурное звено, содержащее звено ангидрида карбоновой кислоты, описанное формулой (III) в одно или несколько реакций, выбираемых из группы, состоящей из (i) гидролиза, (ii) аммонолиза, (iii) аминолиза и (iv) алкоголиза. Данное изобретение также относится к фармацевтической композиции для профилактики или лечения нарушений обмена веществ в костях, содержащей фармацевтически приемлемые разбавитель или носитель, по меньшей мере, один из указанных выше сополимеров или их фармацевтически приемлемых солей и, по меньшей мере, один белок, представляющий собой фактор ингибирования остеокластогенеза (OCIF), или его аналог или вариант. Также изобретение относится к модификатору, содержащему указанные выше сополимеры, к комплексу между одним из указанных выше сополимеров и белком, или его аналогом или вариантом, к фармацевтической композиции, содержащей данный комплекс, к способу продления времени, в течение которого фактор ингибирования остеокластогенеза (OCIF) удерживается в токе крови после приема пациентом комплексообразования между белком и, по меньшей мере, одним из указанных выше сополимеров, а также к способу лечения или профилактики нарушений обмена веществ в костях, включающему прием пациентом эффективного количества комплекса, содержащего комплекс, включающий фактор ингибирования остеокластогенеза (OCIF), или его аналог или вариант, связанные, по меньшей мере, с одним из заявленных сополимеров, и к применению комплекса, содержащего фактор ингибирования остеокластогенеза (OCIF), связанного, по меньшей мере, с одним из заявленных сополимеров, предназначенного для изготовления лекарственного средства, предназначенного для профилактики или лечения нарушения обмена веществ в костях, восприимчивого к действию белка. Модифицирование белка, а именно фактора ингибирования остеокластогенеза (OCIF), заявленными сополимерами приводит к формированию комплекса, обладающего однородными свойствами, в особенности характеризующегося пониженным образованием неупорядоченных сшитых структур с белком, лучшим сохранением активности белка и превосходным удерживанием белка в крови после приема указанного комплекса. 9 н. и 101 з.п. ф-лы, 13 табл., 3 ил.
(а) одно или несколько структурных элементарных звеньев, которые могут быть идентичными или отличными друг от друга, и которые описываются приведенной ниже формулой (I)
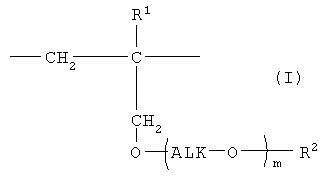
где m представляет собой целое число в диапазоне от 3 до 100,
Alk представляет собой алкиленовую группу, содержащую от 1 до 6 атомов углерода, и
R1 и R2 являются идентичными или различными, и каждый из них представляет собой атом водорода или алкильную группу, содержащую от 1 до 6 атомов углерода, и
(b) одно или несколько структурных звеньев, которые могут быть идентичными или отличными друг от друга, и которые описываются формулой (II)
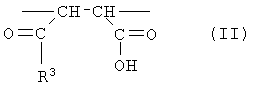
где R3 представляет собой
гидроксильную группу,
алкокси-группу, содержащую от 1 до 6 атомов углерода, которая необязательно может быть замещена арильной группой, содержащей от 6 до 14 атомов углерода,
арилокси-группу, содержащую от 6 до 14 атомов углерода, которая необязательно может быть замещена нитрогруппой, или
группу, описанную формулой -NR4R5, где R4 и R5 являются идентичными или отличными друг от друга, и каждый из них представляет собой атом водорода или алкильную группу, содержащую от 1 до 6 атомов углерода, которая необязательно может быть замещена гидроксигруппой,
причем расположение структурных звеньев, представленных формулой (I), и структурных звеньев, представленных формулой (II), выбирается из следующих вариантов (i)-(iv):
(i) расположение указанных структурных звеньев формулы (I) и формулы (II) в виде последовательности с чередованием «голова к голове»
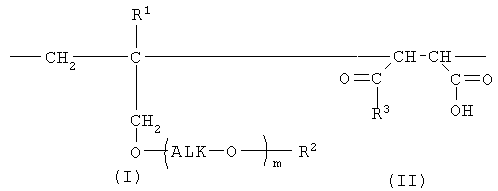
(ii) расположение структурных звеньев формулы (I) и формулы (II) в виде последовательности с чередованием «голова к хвосту»
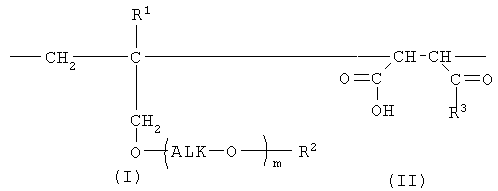
(iii) расположение указанных структурных звеньев формулы (I) и формулы (II) в виде смешанной последовательности с чередованием «голова к голове» и «голова к хвосту»
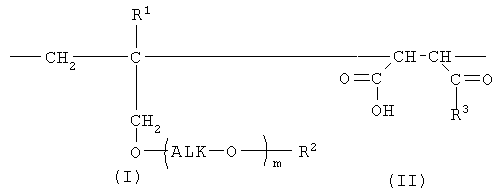
и
(iv) расположение указанных структурных звеньев формулы (I) и формулы (II) в виде произвольной последовательности; и
где соотношение между указанными структурными звеньями, представленными формулой (I), и структурными звеньями, представленными формулой (II), в указанном сополимере находится в диапазоне от 10:1 до 1:10.
(i) m находится в диапазоне от 6 до 16, R3 представляет собой гидроксигруппу, соотношение, задаваемое составом, равно 1:1, а средняя степень полимеризации находится в диапазоне от 30 до 40;
(ii) m находится в диапазоне от 28 до 38, R3 представляет собой гидроксигруппу, соотношение, задаваемое составом, равно 1:1, а средняя степень полимеризации находится в диапазоне от 10 до 15;
(iii) m находится в диапазоне от 6 до 16, R3 представляет собой аминогруппу, соотношение, задаваемое составом, равно 1:1, а средняя степень полимеризации находится в диапазоне от 30 до 40;
(iv) m находится в диапазоне от 6 до 16, R представляет собой диметиламиногруппу, соотношение, задаваемое составом, равно 1:1, а средняя степень полимеризации находится в диапазоне от 30 до 40;
(v) m находится в диапазоне от 6 до 16, R3 представляет собой 1-амино-2-пропанольную группу, соотношение, задаваемое составом, равно 1:1, а средняя степень полимеризации находится в диапазоне от 30 до 40;
(vi) m находится в диапазоне от 6 до 16, R3 выбирают из этокси- и гидроксигрупп, соотношение, задаваемое составом, равно 1:1, средняя степень полимеризации находится в диапазоне от 30 до 40, а соотношение, задаваемое гидролизом, равно 4:6;
(vii) m находится в диапазоне от 28 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:1, средняя степень полимеризации находится в диапазоне от 10 до 15, а соотношение, задаваемое гидролизом, равно 4:6;
(viii) m находится в диапазоне от 28 до 38, R3 представляет собой диметиламиногруппу, соотношение, задаваемое составом, равно 1:1, а средняя степень полимеризации находится в диапазоне от 10 до 15;
(ix) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:1, средняя степень полимеризации находится в диапазоне от 30 до 40, соотношение, задаваемое гидролизом, равно 3,1:6,9, а сополимер представляет собой натриевую соль;
(х) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:1, средняя степень полимеризации находится в диапазоне от 30 до 40, а соотношение, задаваемое гидролизом, равно 1,4:8,6;
(xi) m находится в диапазоне от 6 до 16, R3 выбирают из диметиламино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:1, средняя степень полимеризации находится в диапазоне от 30 до 40, соотношение, задаваемое гидролизом, равно 2,9:7,1, а сополимер представляет собой натриевую соль;
(xii) m находится в диапазоне от 6 до 16, R3 представляет собой аминогруппу, соотношение, задаваемое составом, равно 1:2,4, а средняя степень полимеризации находится в диапазоне от 20 до 30;
(xiii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,4:9,6;
(xiv) m находится в диапазоне от 6 до 16, R выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 2,9:7,1;
(xv) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,9:9,1;
(xvi) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,5:9,5;
(xvii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 1,3:8,7;
(xviii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 1,9:8,1;
(xix) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 1,0:9,0;
(хх) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,8:9,2;
(xxi) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 4,6:5,4;
(xxii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 1,2:8,8;
(xxiii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 2,0:8,0;
(xxiv) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 1,1:8,9;
(xxv) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 2,4:7,6;
(xxvi) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,9:9,1;
(xxvii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 1,5:8,5;
(xxviii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,7:9,3;
(xxix) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 4,5:5,5;
(ххх) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 1,4:8,6;
(xxxi) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,7:9,3;
(xxxii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,8:9,2;
(xxxiii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 1,4:8,6;
(xxxiv) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:3,1, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,7:9,3;
(xxxv) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 0,9:9,1;
(xxxvi) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:2,4, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 1,9:8,1;
(xxxvii) m находится в диапазоне от 6 до 16, R3 выбирают из этокси- и гидроксигрупп, соотношение, задаваемое составом, приблизительно равно 1:3, средняя степень полимеризации находится в диапазоне от 20 до 30, а соотношение, задаваемое гидролизом, равно 3,1:6,9;
(xxxviii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:1, соотношение, задаваемое гидролизом, равно 1,4:8,6, а радиус Стокса составляет 9,3 нм или менее;
(xxxix) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:1, соотношение, задаваемое гидролизом, равно 1,4:8,6, а радиус Стокса находится в диапазоне от 3,1 до 6,2 нм;
(xl) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:1, соотношение, задаваемое гидролизом, равно 1,4:8,6, а радиус Стокса находится в диапазоне от 1,5 до 4,7 нм;
(xli) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:1, соотношение, задаваемое гидролизом, равно 1,4:8,6, а радиус Стокса составляет 3,1 нм или менее;
(xlii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:1, соотношение, задаваемое гидролизом, равно 1,4:8,6, а радиус Стокса составляет 7,8 нм или менее;
(xliii) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:1, соотношение, задаваемое гидролизом, равно 1,4:8,6, а радиус Стокса составляет 6,2 нм или менее; и
(xliv) m находится в диапазоне от 6 до 16, R3 выбирают из амино- и гидроксигрупп, соотношение, задаваемое составом, равно 1:1, соотношение, задаваемое гидролизом, равно 1,4:8,6, а радиус Стокса составляет 4,7 нм или менее.
где m представляет собой целое число в диапазоне от 3 до 100,
Alk представляет собой алкиленовую группу, содержащую от 1 до 6 атомов углерода, и
R1 и R2 являются идентичными или различными, и каждый из них представляет собой атом водорода или алкильную группу, и
(b) указанное структурное элементарное звено, содержащее звено ангидрида карбоновой кислоты, описанное формулой (III)
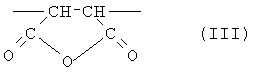
в одной или нескольких реакциях, выбираемых из группы, состоящей из (i) гидролиза, (ii) аммонолиза, (iii) аминолиза и (iv) алкоголиза.
Приоритет по пунктам:
| JP 6233679 А, 23.08.1994 | |||
| НОВЫЕ БЕЛКИ И СПОСОБЫ ПОЛУЧЕНИЯ ТАКИХ БЕЛКОВ | 1996 |
|
RU2194714C2 |
| Литьевая форма для изготовления полимерных изделий с резьбой | 1985 |
|
SU1270015A1 |
| Устройство для дистракции позвоночника | 1982 |
|
SU1127578A1 |
| JP 6205675 А, 26.07.1994. | |||

