Изобретение относится к способам переработки концентратов оксидов природного урана и может быть использовано в технологии получения материалов топливного цикла, в частности, для получения обогащенного урана.
В качестве исходного сырья для обогащения урана могут быть использованы его оксиды в виде технической закиси - окиси (U2O8), получаемой из природного сырья. При этом перед операцией фторирования уран необходимо дополнительно очистить от сопутствующих примесей, имеющихся в рудном концентрате (молибдена, кремния, железа, ванадия и др.). Кроме того, необходима очистка и от примесей, попадающих в уран в процессе переработки природных руд в закись-окись урана (окалина, уголь и т.д.). Для очистки урановых растворов от примесей используют экстракционную технологию переработки азотнокислых растворов урана, содержащих 300-500 г/л урана и 1-3 моль/л HNO3, с применением трибутилфосфата. Перед экстракцией порошок оксидов урана необходимо растворить в азотной кислоте.
Известен способ растворения закиси-окиси урана в азотной кислоте (В.М.Вдовенко. Современная радиохимия. М., 1969, стр.267).
Способ осуществляется по следующей реакции:
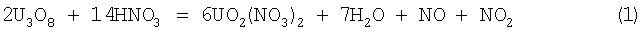
Расход азотной кислоты на растворение по этой реакции составляет 0,62 т на 1 т урана.
Известен способ выщелачивания урана из рудных концентратов, при котором закись-окись растворяется в азотной кислоте по следующим химическим реакциям (Ч.Харрингтон, А.Рюэле. Технология производства урана. Пер. с англ. М.: Госатомиздат, 1961, с.20):
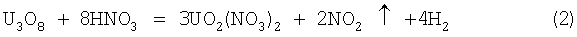
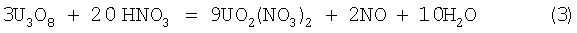
Азотная кислота окисляет уран в растворе из низшего четырехвалентного состояния в шестивалентный уранил-ион с выделением смеси окислов азота NO и NO2. В разбавленной кислоте реакция протекает главным образом с выделением NO, в концентрированной кислоте в основном образуется NO2. Расход азотной кислоты на растворение по реакции (2) составляет 0,7 т на 1 т урана, по реакции (3) - 0,59 т на 1 т урана.
По технической сути и достигаемому техническому результату наиболее близким к заявляемому способу является способ переработки, в котором закись-окись урана выщелачивают концентрированной азотной кислотой по реакции (2) - прототип (Ч.Харрингтон, А.Рюэле. Технология производства урана. Пер. с англ. М.: Госатомиздат, 1961, с.20).
Недостатком известного способа является то, что после растворения концентрата урана и отделения от раствора основной массы нерастворимого остатка азотнокислые растворы урана содержат равномерно распределенную тонкодисперсную твердую фазу, состоящую из соединений железа, кремния и других нерастворимых примесей. Растворы получаются мутными, их дальнейшая экстракционная переработка затруднена, так как экстракционные производства ориентированы на переработку азотнокислых растворов, не содержащих твердой фазы. Фильтрация таких растворов через различные фильтры (полиэфирную, полиамидную ткани, синтепон и др.), а также центрифугирование не позволяют полностью освободиться от взвесей и получить прозрачные растворы.
Задачей изобретения является получение азотнокислых урановых растворов при переработке концентратов оксидов природного урана, легко отделяемых от твердой фазы и не содержащих взвесей, пригодных для экстракционного передела.
Поставленную задачу решают тем, что в способе переработки концентратов оксидов природного урана, включающем выщелачивание урана концентрированной азотной кислотой при повышенной температуре и отделение водной фазы от нерастворимого остатка перед экстракционным переделом, выщелачивание осуществляют при весовом отношении азотной кислоты к урану, составляющем (0,75-0,84):1, при температуре 85-100°С, с получением концентрации урана в водной фазе 700-800 г/л, пульпу в неохлажденном виде разбавляют слабыми азотнокислыми растворами до концентраций урана в водной фазе пульпы 350-500 г/л и азотной кислоты - около 1,0 моль/л, пригодных для водной фазы экстракционного передела, после чего водную фазу отделяют от нерастворимого остатка.
При заявленных на стадии выщелачивания отношении азотной кислоты к урану и содержании урана в водной фазе, при заявленной температуре, по-видимому, некоторые примеси, например железо, растворяются с образованием сложных комплексных соединений, например типа гематита, которые, как правило, хорошо отстаиваются и отделяются от жидкой фазы; с другой стороны, растворение некоторых примесей, в том числе и легкогидролизующихся, образующих взвеси, подавляется вовсе или идет медленнее, чем растворение урана-макрокомпонента.
Разбавление полученной при выщелачивании реакционной смеси в горячем (неохлажденном) состоянии позволяет прекратить растворение примесей, которые еще не успели раствориться, но могли бы раствориться со временем в концентрированных азотнокислых растворах, а также предотвратить выпадение в осадок соединений урана, которое имело бы место при охлаждении высококонцентрированных (пересыщенных) растворов. Разбавление при этом ведут слабыми азотнокислыми растворами, а не водой, чтобы предотвратить гидролиз легкогидролизующихся соединений в момент добавления разбавителя. В качестве слабых азотнокислых растворов (около 0,01 моль/л) можно использовать чистые азотнокислые растворы либо оборотные азотнокислые растворы экстракционного передела, содержащие примеси урана.
Пример.
Раствор азотной кислоты заданного объема и заданной концентрации нагревают до температуры 70-100°С и в него засыпают в течение 10 минут при перемешивании (сжатым воздухом) навеску порошка концентрата оксидов природного урана (25 г). Содержание урана в закиси-окиси составляет 83,19%. Объем и концентрацию азотной кислоты задают из условий обеспечения весового отношения азотной кислоты к урану, составляющего (0,6-0,84):1, и концентрации урана в водной фазе пульпы, полученной при выщелачивании, 660-800 г/л. Перемешивание продолжают в течение 30 минут. Об окончании выщелачивания судят по прекращению газовыделения. Сразу после окончания выщелачивания горячую реакционную смесь разбавляют слабым азотнокислым раствором (0,01 моль/л HNO3) до концентраций урана в водной фазе пульпы 350-500 г/л и азотной кислоты - около 1,0 моль/л. Эти концентрации пригодны (и используются) для экстракционного передела урансодержащих азотнокислых растворов с применением трибутилфосфата. Перед стадией экстракции нерастворимый остаток отделяют. В части опытов отделение осадка от водной фазы проводят фильтрацией через полиэфирную ткань ПЭ-100, при этом на фильтрацию смесь направляют после отстоя в течение нескольких часов. В другой части опытов используют центрифугирование, при этом отстой не производят.
Проводят визуальные наблюдения за состоянием растворов урана после отстаивания и фильтрации через полиэфирную ткань, а также после центрифугирования. Определяют скорость фильтрации раствора через полиэфирную ткань, см3/мин.
Проводят 3 серии опытов.
В первой серии опытов изменяют весовое отношение азотной кислоты к урану (от 0,6 до 0,84 г HNO3 на 1 г U).
Во второй серии опытов при неизменном весовом отношении азотной кислоты к урану создают разную концентрацию урана в водной фазе пульпы - от 660 г/л до 800 г/л. - путем изменения концентрации азотной кислоты в растворе, подаваемом на выщелачивание, т.е. изменения Т:Ж.
В третьей серии изменяют температуру реакционной смеси от 70 до 100°С.
Результаты опытов по переработке концентратов оксидов природного урана приведены в таблице.
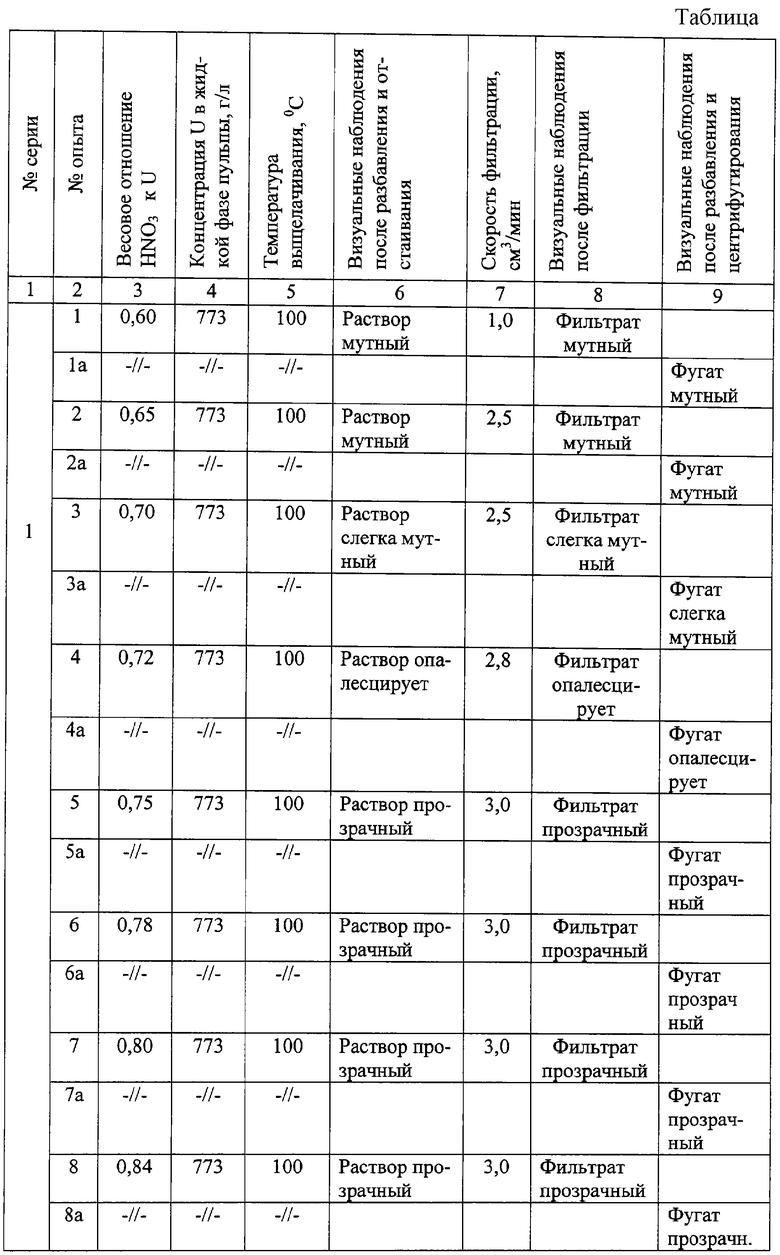

Из результатов таблицы видно, что при весовом отношении азотной кислоты к урану, составляющем (0,75-0,84):1, концентрации урана 700-800 г/л в водной фазе пульпы от выщелачивания, температуре выщелачивания 85-100°С, разбавлении пульпы в неохлажденном виде слабыми азотнокислыми растворами до концентраций урана в водной фазе пульпы 350-500 г/л и азотной кислоты - около 1,0 моль/л, получаются хорошо разделяемые (на фильтре или на центрифуге) смеси, растворы урана до и после отделения осадка - прозрачные (опыты 5-8, 5а-8а первой серии, 1-5, 1a, 5a второй серии, опыты 1-4, 4а третьей серии). При других условиях растворы получаются мутными, скорость фильтрации ниже, тонкодисперсная взвесь забивает фильтр и затрудняет прохождение раствора через фильтр, центрифугирование также не обеспечивает прозрачность растворов.
Таким образом, предлагаемый способ позволяет получать азотнокислые урановые растворы при переработке концентратов оксидов природного урана, легко отделяемые от твердой фазы и не содержащие взвеси, пригодные для экстракционного передела.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПЕРЕРАБОТКИ КРЕМНИЙСОДЕРЖАЩЕГО ХИМИЧЕСКОГО КОНЦЕНТРАТА ПРИРОДНОГО УРАНА | 2013 |
|
RU2517633C1 |
| СПОСОБ ПЕРЕРАБОТКИ КРЕМНИЙСОДЕРЖАЩИХ ОТХОДОВ УРАНОВОГО ПРОИЗВОДСТВА | 2014 |
|
RU2576819C1 |
| СПОСОБ ПЕРЕРАБОТКИ ХИМИЧЕСКОГО КОНЦЕНТРАТА ПРИРОДНОГО УРАНА | 2010 |
|
RU2447168C1 |
| СПОСОБ ЭКСТРАКЦИОННОГО АФФИНАЖА УРАНА | 2005 |
|
RU2295168C1 |
| СПОСОБ ЭКСТРАКЦИОННОГО АФФИНАЖА УРАНА | 2013 |
|
RU2554830C2 |
| СПОСОБ ИЗВЛЕЧЕНИЯ УРАНА ИЗ ТВЕРДЫХ ОТХОДОВ СУБЛИМАТНОГО ПРОИЗВОДСТВА | 2002 |
|
RU2219131C2 |
| СПОСОБ ПЕРЕРАБОТКИ УРАНСОДЕРЖАЩЕГО СЫРЬЯ ПРИРОДНОГО ПРОИСХОЖДЕНИЯ | 2012 |
|
RU2503732C1 |
| СПОСОБ ПЕРЕРАБОТКИ КОНЦЕНТРАТОВ ПРИРОДНОГО УРАНА | 2007 |
|
RU2360988C2 |
| СПОСОБ ПОДГОТОВКИ УРАНСОДЕРЖАЩЕГО СЫРЬЯ К ЭКСТРАКЦИОННОЙ ПЕРЕРАБОТКЕ | 2012 |
|
RU2514557C1 |
| СПОСОБ ПЕРЕРАБОТКИ ХИМИЧЕСКОГО КОНЦЕНТРАТА ПРИРОДНОГО УРАНА | 2010 |
|
RU2444576C1 |
Изобретение относится к способам переработки концентратов оксидов природного урана и может быть использовано в технологии получения материалов топливного цикла, в частности, для получения обогащенного урана. Способ включает выщелачивание урана концентрированной азотной кислотой при повышенной температуре и отделение водной фазы от нерастворимого остатка перед экстракционным переделом. Выщелачивание осуществляют при весовом отношении азотной кислоты к урану, составляющем (0,75-0,84):1, при температуре 85-100°С, с получением концентрации урана в водной фазе 700-800 г/л. Пульпу в неохлажденном виде разбавляют слабыми азотнокислыми растворами до концентрации урана в водной фазе пульпы 350-500 г/л и азотной кислоты - около 1,0 моль/л, пригодных для водной фазы экстракционного передела, после чего водную фазу отделяют от нерастворимого остатка. Способ позволяет получать азотнокислые урановые растворы при переработке концентратов оксидов природного урана, легко отделяемые от твердой фазы и не содержащие взвеси, пригодные для экстракционного передела. 1 табл.
Способ переработки концентратов оксидов природного урана, включающий выщелачивание урана концентрированной азотной кислотой при повышенной температуре и отделение водной фазы от нерастворимого остатка перед экстракционным переделом, отличающийся тем, что выщелачивание осуществляют при весовом отношении азотной кислоты к урану, составляющем (0,75-0,84):1, при температуре 85-100°С с получением концентрации урана в водной фазе 700-800 г/л, пульпу в неохлажденном виде разбавляют слабыми азотнокислыми растворами до концентрации урана в водной фазе пульпы 350-500 г/л и азотной кислоты - около 1,0 моль/л, пригодных для водной фазы экстракционного передела, после чего водную фазу отделяют от нерастворимого остатка.
| ХАРРИНГТОН Ч | |||
| и др., Технология производства урана, Пер | |||
| с англ., Москва, Госатомиздат, 1961, с.20-24 | |||
| СПОСОБ РАСТВОРЕНИЯ ОКСИДОВ УРАНА | 2001 |
|
RU2201398C1 |
| СПОСОБ ИЗГОТОВЛЕНИЯ ТАБЛЕТИРОВАННОГО ЯДЕРНОГО ТОПЛИВА ИЗ ОТХОДОВ ОКИСЛОВ УРАНА И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1998 |
|
RU2152087C1 |
| Способ изготовления ремешковой застежки | 1983 |
|
SU1118337A1 |
| Способ подачи порошков в транспортный трубопровод при продувке металла и устройство для его осуществления | 1982 |
|
SU1041578A1 |
| US 3813464 A, 28.05.1974 | |||
| ВДОВЕНКО В.М., Современная радиохимия, Москва, Атомиздат, 1969, с.267. | |||

