ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новым производным бензизоселеназолонила, обладающих противоопухолевым, противовоспалительным и антитромботическим действием, а также к их применению. Настоящее изобретение также относится к фармацевтическому составу, содержащему производные бензизоселеназолонила, их использованию в производстве лекарственных средств и способу лечения воспалительных и раковых заболеваний и предотвращению тромбоза.
УРОВЕНЬ ТЕХНИКИ
Темой многих исследований является средство, содержащее селен, так как химический элемент селен выполняет важные функции в биологическом организме. Проблема с неорганическим селеном заключается в том, что он трудно всасывается, срок его жизни в крови небольшой, он обладает низкой активностью и высокой токсичностью. По сравнению с характеристиками неорганического селена характеристики селеноорганических соединений намного улучшились.
Селен является важным микроэлементом. Дефицит селена (<0,1 части на миллион), существующий в течение длительного времени, может вызывать различные заболевания, включая гепатонекроз, поражение сердечной мышцы, рак и ревматические заболевания.
До сих пор было известно, что бензизоселеназолоны (BISA), функционирующие наподобие гломерулостимулирующего гормона (GSH-Px), в лабораторных условиях подавляют липоидную пероксидацию микросомы и оказывают воздействие по предотвращению поражений организма, вызываемых пероксидацией. Наилучшим из соединений, по свойствам напоминающих GSH-Px, с высокой антиокислительной активностью и низкой токсичностью (средняя смертельная доза >6810 мг/кг, у мышей) является 2-фенил-(1,2)-бензизоселеназол-3(2Н)-он (Эбселен), имеющий следующую формулу:
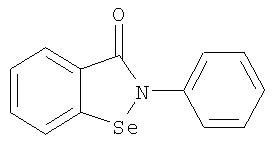
Многие исследования сосредоточены на модификации эбселена для повышения его противоопухолевой активности, но до настоящего времени не сообщалось об основанном на нем успешном противоопухолевом активном соединении. Поэтому целью настоящего исследования является модификация эбселена для образования новых производных бисбензизоселеназолонила, имеющих повышенную противовоспалительную активность, более широкую совместимость и пониженную токсичность. Помимо этого, противоопухолевые селенорганические соединения с характеристикой "регулятора биологической реакции" были получены путем модификации эбселена.
КРАТКОЕ СОДЕРЖАНИЕ ИЗОБРЕТЕНИЯ
В соответствии с одним аспектом изобретения представлены производные бисбензизоселеназолонила, имеющие общие формулы (I)), (II) или (III), и их фармацевтически приемлемые соли:
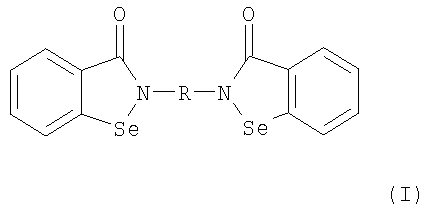
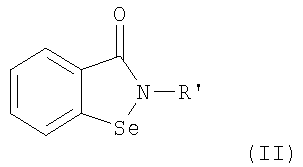
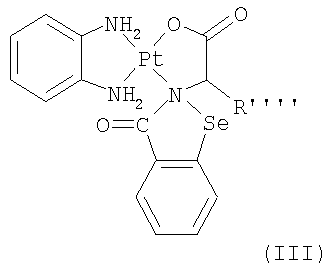
где:
R является C1-6-алкиленом, фенилиденом, бифенилиденом, трифенилиденом или следующей группой:
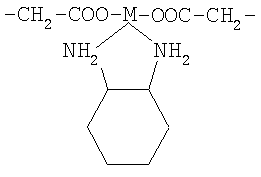
где: М=Pt, Pd или Rh;
R' является полисахаридным остатком или следующей группой:
где: R''=Cl, H2O, ОН, Br или I.
R'''=-Н, -СН2С6Н5ОН, -СН2ОН, -CH2CONH2, -СН2СН2СООН, -СН2(СН2)4NH2, -СН2СООН, -CH2CH2CONH2, -(CH2)3СН, -(CH2)3NHC(NH)NH2, -(СН2)3СНСН2, -СН3, -СН2СН3, -СН2С6Н5, -CH2SH или -CH2CH2SCH3,
R''''=-Н, -СН2С6Н5OH, -СН2OH, -CH2CONH2, -СН2СН2СООН, -СН2(СН2)4NH2, -СН2СООН, -СН2CH2CONH2, -(СН2)3СН,
-(CH2)3NHC(NH)NH2, -(CH2)3CHCH2, -СН3, -СН2СН3, -СН2С6Н5, -CH2SH, или -CH2CH2SCH3.
Согласно еще одному аспекту настоящего изобретения представляется фармацевтический состав, содержащий в качестве активного ингредиента вышеуказанное соединение (I), (II) или (III) или их фармацевтически приемлемые соли и любой фармацевтически приемлемый наполнитель или носитель.
Согласно еще одному аспекту настоящего изобретения предусматривается использование производных бисбензизоселеназолонила с общей формулой (I), (II) или (III) или их фармацевтически приемлемой соли для производства лекарства для лечения рака и воспалительных заболеваний или предотвращения тромбоза.
Согласно еще одному аспекту настоящего изобретения представляется способ лечения воспалительных и раковых заболеваний или предотвращение тромбоза у млекопитающих, включая человека, содержащий стадию назначения терапевтически эффективной дозы производных бисбензизоселеназолонила с общей формулой (I), (II) или (III) или их фармацевтически приемлемой соли пациентам, нуждающимся в лечении.
Согласно еще одному аспекту настоящего изобретения представляется способ лечения воспалительных и раковых заболеваний или предотвращения тромбоза у млекопитающих, включая человека, содержащий стадию назначения терапевтически эффективной дозы производных бисбензизоселеназолонила с общей формулой (I), (II) или (III) или их фармацевтически приемлемой соли в сочетании с другими противовоспалительными или противоопухолевыми средствами пациентам, нуждающимся в лечении.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Производные бензизоселеназолонила согласно настоящему изобретению назначаются с учетом активной фармакокинетики эбселена и усиления функциональной группы. Из-за характеристик этой структуры эти соединения как предмет изобретения имеют множественные цели в биологическом организме и поэтому проявляют многостороннюю биологическую активность. Тот факт, что эти соединения являются противоопухолевыми средствами, функционирующими в качестве модификатора биологической реакции помимо ингибирования рака, делает их новым противоопухолевым средством.
Согласно одному варианту осуществления настоящего изобретения представляются производные бисбензизоселеназолонила с общей формулой (I)), (II) или (III) и их фармацевтически приемлемые соли:
где:
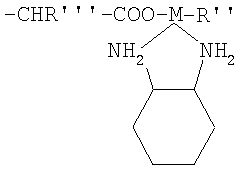
R является C1-6-алкиленом, фенилиденом, бифенилиденом, трифенилиденом или следующей группой:
где: М=Pt, Pd или Rh;
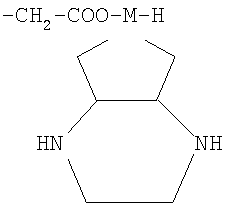
R' является полисахаридным остатком или следующей группой:
где: R'' является Cl, H2O, ОН, Br или I,
R''' является -Н, -СН2С6H5ОН, -СН2ОН, -СН2CONH2, -СН2СН2СООН, -CH2(CH2)4NH2, -СН2СООН, -CH2CH2CONH2, -(СН2)3СН, -(CH2)3NHC(NH)NH2, -(СН2)3СНСН2, -СН3, -СН2СН3, -СН2С6Н5, -CH2SH или -CH2CH2SCH3,
R'''' является -Н, -СН2С6Н5OH, -СН2OH, -CH2CONH2, -СН2СН2СООН, -CH2(CH2)4NH2, -CH2COOH, -CH2CH2CONH2, -(СН2)3СН, -(CH2)3NHC(NH)NH2, -(СН2)3СНСН2, -СН3, -СН2СН3, -СН2С6Н5, -CH2SH или -CH2CH2SCH3.
В данном варианте осуществления предпочтительными соединениями являются производные бисбензизоселеназолонила с общей формулой (I), где R является группой C1-4-алкилена, фенилидена или бифенилидена. Особенно предпочтительными являются соединения, где R является группой С2-алкилена или бифенилидена.
Предпочтительной для R' является группа 1,3,4,6-тетра-O-ацетил-2-деокси -D-глюкопиранозила.
Предпочтительными для R''' и R'''' независимо являются -Н, -СН3, -СН2СН3, -СН2С6Н5, -CH2SH или -CH2CH2SCH3.
Производные бензизоселеназолонила согласно настоящему изобретению могут быть синтезированы любым способом, известным специалистам в данной области техники, или способом, изложенным в описании изобретения к данной заявке. Например, 2-(хлороселено)бензоилхлорид реагирует с соответствующим диамином или аминосахаридом в соответствующем органическом растворителе в холодной атмосфере азота, затем он отделяется с использованием стандартной процедуры, известной специалистам в данной области техники для получения желаемого соединения.
Производные бензизоселеназолонила согласно настоящему изобретению, где R или R' является комплексом металлов, могут быть синтезированы способом, известным специалистам в данной области техники путем использования соединения платины.

Производные бензизоселеназолонила согласно настоящему изобретению или их соли могут назначаться в форме чистого вещества или соответствующего фармацевтического состава, содержащего соединения с общей формулой (I), (II) или (III) в качестве активного ингредиента, который может по выбору назначаться вместе с другими средствами, любым приемлемым способом поступления лекарства в организм. Поэтому изобретение также включает фармацевтический состав, содержащий производные бензизоселеназолонила с общей формулой (I), (II) или (III) или их фармацевтически приемлемые соли и фармацевтически приемлемый наполнитель или носитель, причем этот состав может использоваться для лечения воспалительных и раковых заболеваний или предотвращения тромбоза.
Соединения или состав по настоящему изобретению могут быть назначены для приема различными путями, включая, но без ограничений, пероральный, интраназальный, ректальный, чрескожный или парентеральный, в форме твердого, полутвердого, лиофилизированного порошка или жидкости. Например, состав может использоваться в форме таблеток, свечей, пилюль, капсул из мягкого или твердого желатина, гранул, раствора, суспензии или аэрозоля. Предпочтительной является форма отдельных объектов для обеспечения точности дозировки. Фармацевтический состав включает обычный наполнитель или носитель и одно и более соединений по настоящему изобретению. Состав может также содержать другой лекарственный препарат и т.п.
В общем, в зависимости от способа приема фармацевтически приемлемый состав может содержать от 1 до 99% по массе соединения по настоящему изобретению в качестве активного ингредиента и от 99 до 1% по массе соответствующего фармацевтического наполнителя. Предпочтительный состав содержит примерно 5-75% по массе соединения по настоящему изобретению и остальное как подходящий наполнитель или носитель.
Предпочтительным путем приема является внутривенная инъекция при использовании обычного протокола суточных доз, который может корректироваться с учетом тяжести заболевания. Соединения или их фармацевтически приемлемые соли согласно настоящему изобретению могут быть подготовлены в форме доз для инъекций путем, например, разбавления от примерно 0,5 до 50% по массе соединений по настоящему изобретению в качестве активного ингредиента в жидком наполнителе или носителе, таком как вода, солевой раствор, водный раствор глюкозы, этанол и глицерин для получения раствора или суспензии.
Фармацевтический состав, который может назначаться в форме раствора или суспензии, может быть получен, например, путем растворения или разбавления соединений по настоящему изобретению (в количестве, например, примерно от 0,5 до 20% по массе) и, по выбору, других вспомогательных веществ в носителях, включая, но не ограничиваясь, воду, солевой раствор, водный раствор глюкозы, раствор этанола и глицерина.
Кроме того, если это необходимо, фармацевтический состав согласно настоящему изобретению может включать вспомогательные вещества, такие как увлажняющее средство или эмульгатор, рН-буфер, антиоксидант и т.п.. Конкретными примерами являются лимонная кислота, монолаурат сорбитана, олеат триэтаноламина, гидроксибензол бутила и т.п.
Подготовка состава по настоящему изобретению может быть осуществлена любым способом, известным или очевидным специалистам в данной области (см., например. Remington's Pharmaceutical Sciences, edition 18, Mack Publishing Company, Easton, Pennsylvania, 1990). В любом случае состав по настоящему изобретению включает соединение по настоящему изобретению в количестве, эффективном при лечении соответствующего заболевания.
Согласно еще одному аспекту настоящего изобретения предлагается способ для лечения воспалительных и раковых заболеваний или предотвращения тромбоза у млекопитающих, включая человека, содержащий этап назначения терапевтически эффективной дозы производных бисбензизоселеназолонила с общей формулой (I), (II) или (III) или их фармацевтически приемлемой соли пациентам, нуждающимся в лечении.
Согласно еще одному аспекту настоящего изобретения предлагается способ для лечения воспалительных и раковых заболеваний или предотвращения тромбоза у млекопитающих, включая человека, содержащий этап назначения терапевтически эффективной дозы производных бисбензизоселеназолонила с общей формулой (I), (II) или (III) или их фармацевтически приемлемой соли в сочетании с другими противовоспалительными или противоопухолевыми средствами или антитромботическими средствами пациентам, нуждающимся в лечении.
При применении производных бисбензизоселеназолонила по настоящему изобретению вместе с другими противовоспалительными, противоопухолевыми или антитромботическими средствами они могут назначаться последовательно или одновременно. Например, сначала назначаются производные бисбензизоселеназолонила по настоящему изобретению, а затем другие противовоспалительные, противоопухолевые или антитромботические средства. Альтернативно, другие противовоспалительные, противоопухолевые или антитромботические средства назначаются первыми, а затем назначаются производные бисбензизоселеназолонила по настоящему изобретению.
В одном из предпочтительных вариантов осуществления другим противоопухолевым средством может являться цисплатин, таксол, циклофосфамид, изофосфамид, метотрексат, флуороурацил, эпирубицин, дауномицин, адриамицин, митомицин, пиньянгмицин, карбоплатин, ломустин, кармустин или их сочетания.
В одном из предпочтительных вариантов осуществления другим противовоспалительным средством может являться аспирин, индометацин, цефалоспорины, макроолиды или их сочетания.
В одном из предпочтительных вариантов осуществления другим антитромботическим средством является аспирин.
Дозировка производных бензизоселеназолонила по настоящему изобретению для раковых заболеваний находится в диапазоне 0,05-250 мг на 1 кг массы тела; для воспалительных заболеваний - в диапазоне 1-100 кг на 1 кг массы тела и для предотвращения тромбоза - в диапазоне 1-100 мг на 1 кг массы тела.
В сочетании с другими противовоспалительными, противоопухолевыми или антитромботическими средствами дозировка производных бензизоселеназолонила по настоящему изобретению будет значительно уменьшена, примерно до 1/10-1/2 от дозы, назначаемой при применении их одних.
Ниже приведено более подробное описание настоящего изобретения.
Пример 1
1,2-бис[(1,2)-бензизоселеназол-3(2Н)-онил] этан (Е003)
1 г 2-(хлорселено)бензоилхлорида в тетрагидрофуране добавлялся капельным путем в перемешиваемый раствор 0,14 мл этилендиамина и 1,29 мл триэтиламина в атмосфере азота при охлаждении в ледяной ванне до появляния белого осадка. После перемешивания в течение трех часов образовалась светло-желтая суспензия. Раствор выпаривался в вакууме, и светло-желтый осадок отсасывался, промывался водой и затем повторно кристаллизировался из диметилсульфоксида (ДМСО) для получения вышеназванного соединения. Выход: 0,1 г (11%), температура плавления >320°С.
Электронная масс-спектрометрия: (m/z) (m+) 424; ЯМР для 1Н (ДМСО-d6): 7,37-7,98 (8Н, m, ArH), 4.02 (4Н, s, -CH2СН2-).
Пример 2
4,4'-бис[(1,2)-бензизоселеназол-3(2Н)-онил]-бифенил (Е002)
0,5 г 2-(хлорселено)бензоилхлорида в тетрагидрофуране добавлялось капельным путем в перемешиваемый раствор 0,182 г бифенилдиамина и 0,62 мл триэтиламина в атмосфере азота при охлаждении в ледяной ванне. После перемешивания в течение трех часов образовался белый осадок, который промывался тетрагидрофураном и этанолом. После повторной кристаллизации из ДМСО вышеназванное соединение было получено в форме светло-коричневого осадка. Выход: 0,1 g (18,2%), температура плавления >320°С.
Электронная масс-спектрометрия: (m/z) (m+) 550; ЯМР для 1Н (ДМСО-d6): 7,48-8,12 (m,16H, ArH).
Пример 3
2-(1,3,4,6-тетра-O-ацетил-2-деокси-D-глюкопиранозил)-(1,2)-бензизоселеназол-3(2Н)-он (Е001)
730 мг 1,3,4,6-тетра-O-ацетил-D-глюкозамина были растворены в хлоформе в атмосфере азота при охлаждении в ледяной ванне. Раствор 0,551 г 2-(хлорселено)бензоилхлорида в хлороформе медленно добавлялся при перемешивании. По истечении двух часов реакционный раствор отделялся в колонне с силикагелем с использованием петролекума: этилацетата в соотношении 3:1 в качестве элюента. Вышеназванное соединение было получено в форме светло-желтого твердого вещества. Выход: 200 мг (18,0%), температура плавления 73-75°С.
ИК-спектроскопия 1745(-СО): УФ-спектроскопия (CHCl3) 320 нм, 260 нм (изо-селеназольное кольцо).
Иммунофлуоресцентная масс-спектрометрия (m/z) 566,3 (М+К).
ЯМР для 1Н: δ Н (частей на миллион) 7,24-8,12 (4Н, m, ArH), 6,20(1H, d, аномерный Н в сахаре), 3,97-5,64 (m, 6H, сахар: Н), 1,82-2,16 (12Н, m, -СОСН3).
ЯМР для 13С: δ (частей на миллион) 166,77, 168,66, 169,29, 169,61, 170,44 (-СО), 124,22, 126,28, 128,81, 132,37, 138,30 (углерод в бензоле), 91,43, (углерод в сахаре, C-I), 60,15, 61,35, 68,35, 71,91, 72,35, (углерод в сахаре, С-2, 3,4, 5, 6), 20,34, 20,48, 20,55, 20,78 (-СОСН3).
Пример 4:
Синтез 1,2-диаминоциклогексаноплатин-2-глицин-[(1,2)-бензизоселеназола-3(2Н)-он]
1) Синтез K2PtCl4
10%-й водный раствор гидразина добавлялся капельным путем к перемешиваемому раствору 0,7 г K2PtCl6 (1,44 ммоль) в 7 мл Н2О при 80°С до полного смешивания. Реакция продолжалась до образования темно-красного раствора. Оставшаяся соль K2PtCl6 и металлическая платина отфильтровывались и утилизировались. Фильтрат сгущался до получения K2PtCl4 в форме красных игольчатых кристаллов. Выход: 0,5 г (84%).
2) Синтез 1,2-диаминоциклогексаноплатины (II)
Раствор K2PtCl4 (0,2 г, 0,48 ммоль) в 2 мл H2O перемешивался с раствором KI (0,8 г) в 0,6 г Н2О на ванне кипящей воды при отсутствии освещения. Температура быстро поднималась до 80°С и поддерживалась на этом уровне в течение 30 минут в темноте. К раствору добавлялось 0,05 г твердого 1,2-диаминциклогексана, после чего образовывался желтый осадок. Осадок высасывался и промывался в небольшом количестве ледяной воды, этанола и диэтилового эфира. Выход: 0,21 г (78%).
3) 1,2-диаминциклогексанплатина-2-глицин-[(1,2)-бензизоселеназол-3(2Н)-он]
0,02 г 2-глицинэтилового эфира -[(1,2)-бензизоселеназол-3(2Н)-он] были растворены в 0,5 мл хлороформа. Затем к раствору были добавлены 15 мл NaOH (1 моль/л). Раствор выдерживался в течение 10 часов при температуре 50-60°С. Для полной гидролизации эфира 15 мл раствора NaOH (1 моль/л) добавлялись снова после сбора желтого водного слоя. Для окисления объединенного водного слоя добавлялась HCl (1 моль/л), в результате чего осаждался конечный продукт 2-глицин-[(1,2)-бензизоселеназол-3(2Н)-он]. Нерастворимое твердое вещество высасывалось и высушивалось. Выход: 0,025 г.
0,015 г 1,2-диаминциклогексаноплатины были растворены в 0,15 мл воды для образования желтой пасты. Раствор AgNO3 (0,009 г) в 0,5 мл H2O добавлялся к пасте при перемешивании в течение 4 часов при отсутствии освещения. Образовавшийся желтый осадок AgI утилизировался, и остаток промывался небольшим количеством ледяной воды. В это время не должно появляться белой мутности, если смешать одну каплю фильтрата с одной каплей 1-молярного раствора KCl.
К 0,015 г 2-глицин-[(1,2)-бензизосеназол-3(2Н)-он] было добавлено 0,0036 г КОН и 2 мл воды для получения желтой суспензии. Эта желтая суспензия была смешана при перемешивании с раствором 1,2-диаминциклогексаноплатины в течение 90 минут в темноте, отфильтрована и высушена при пониженном давлении для получения желтых кристаллов. Выход: 25 мг (50%).
Флуоресцентная масс-спектроскопия: m/z (M+1) 566, далекая ИК-спектроскопия 340 см-1 (Pt-O), ИК-спектроскопия 420 см-1 (Pt-N).
Пример 5
Эксперимент по ингибированию соединениями роста раковой клетки
В данном примере была использована проба SRB (плотно прилегающая клетка). Раковые клетки (3-5×104 клеток/мл) были посеяны в чашку с 96 ячейками (180 мкл/ячейка) на воздухе с 5% CO2 и влажностью насыщения при 37°С на 24 часа. В каждую ячейку было добавлено 20 мкл раствора испытываемого соединения с различной концентрацией, и выращивание культуры продолжалось в течение указанного времени на воздухе с 5% CO2 и влажностью насыщения при 37°С. После указанного времени раствор культуры удалялся, затем добавлялись 100 мкл 10%-й трихлоруксусной кислоты и раствор помещался в холодильник с температурой 4°С для фиксации клеток на 1 час. Раствор удалялся, и чашка промывалась дистиллированной водой. После высушивания с помощью центрифуги 50 мкл раствора SRB (0,4% с 1% гиалуроновой кислоты) добавлялись в каждую ячейку и выдерживались при температуре окружающего воздуха в течение 10 минут. После удаления излишнего раствора эритроцитов чашка с 96 ячейками промывалась 5 раз 1%-м раствором ацетата для удаления не связанных эритроцитов. После высушивания с помощью центрифуги чашка подвергалась дальнейшей сушке на воздухе. 100 мкл раствора трис с концентрацией 10 ммоль/л (рН 10,5, щелочной, без буферного раствора) добавлялись в каждую ячейку для того, чтобы полностью растворить связи эритроцитов с клеткой. После гомогенизации значение OD каждой ячейки измерялось при 540 нм считывающим устройством для микрочашек с 96 ячейками (TECAN SUNRISE Magellan, США). Здесь: данные по значению OD= значение OD (МТТ или SRB + клетка) - значение OD (МТТ или SRB, свободная клетка). К параллельным группам относится значение OD±SD. Коэффициент выживаемости клеток и коэффициент ингибирования лекарства рассчитывались по следующим уравнениям:
Коэффициент выживаемости клеток, % = (значение OD испытуемой группы / контрольное значение OD) ×100%
Коэффициент ингибирования лекарства, % = [1-(значение OD испытуемой группы) / контрольное значение OD] ×100%
По вышеизложенному способу SRB соединение Е003 исследовалось на Bel-7402 (раковая клетка печени человека), KB (раковая клетка носоглотки человека) и Hela (клетка цервикальной раковой опухоли человека). Результаты приведены в таблице 1.
Кроме того, средний коэффициент ингибирования (1C) для соединения Е003 определялся на 9 видах линий раковых клеток человека в различное время путем использование вышеизложенного способа. Ингибирующее действие соединения Е003 на рост линий раковых клеток указано в таблице 2.
Пример 6
Влияние соединения на массу опухоли
Ткань легких, пораженных раком Льюиса (Lewis), повторно стимулировалась и пересеивалась по стандартной процедуре. Часть ткани трансплантировалась через кожу спины каждой мыши С 57 для инокуляции и роста. Клетки ткани легких, пораженных раком Льюиса, диспергировались в солевом растворе для образования клеточной суспензии 106/мл. 0,2-0,3 мл этой суспензии инокулировались в каждую мышь С57.
Мыши произвольно распределялись (рандомизировались) на три группы, по 10 мышей в каждой группе. В испытуемой группе мышам вводили внутрибрюшинно соединение Е003 в количестве 50 мг/кг; мышам контрольной группы вводили цисплатин в количестве 2 мг/кг; и мышам отрицательной контрольной группы (группы на растворяющем веществе) вводили 0,5%-й раствор CMC-Na. Общий объем для каждой группы был одинаковым. Соединение Е003 назначался внутрибрюшинно со второго дня трансплантации в течение трех дней до гибели мышей. Мыши С57 дезинфицировались 70%-м спиртом. Легочная ткань, пораженная раком Льюиса, удалялась, фотографировалась и взвешивалась. Затем подкожные опухоли фиксировались формальдегидом для дальнейшего анализа.
Результаты воздействия цисплатина и соединения Е003 на объем опухоли приведены в таблице 3.
Из данных таблицы 3 можно видеть, что ингибирующее действие соединения Е003 на клеточную ткань легких, пораженных раком Льюиса, сильнее, чем действие цисплатина.
Пример 7
Синергическое действие соединения по настоящему изобретению в сочетании с другими противоопухолевыми средствами
В данном примере действие соединения Е003, соответственно в сочетании с таксолом, адриамицином и цисплатином в качестве другого противоопухолевого средства, на рост раковых клеток исследовалось по способу, изложенному в Примере 6. Соединение Е003 назначалось совместно, после или до другого противоопухолевого средства с интервалом 4,0 часа.
Е и А взяты в качестве примера для объяснения порядка применения. Е+А означает, что Е и А вводились в одно и то же время в разных концентрациях, которые указаны в таблице. Е-А означает, что введение Е осуществлялось за 4 часа до введения А. А-Е означает, что введение А осуществлялось за 4 часа до введения Е. Остальные примеры также соответствуют данным пояснениям.
Пример 8
Противовоспалительная активность
Противовоспалительное действие исследуемых соединений Е001, Е002 и Е003 оценивались на мышах с отеком уха, индуцированным ксилолом. Мыши были рандомизированы на группу, не получавшую лекарственных средств, три контрольных группы и три группы, получавшие лекарственные средства. В каждой группе было по 10 мышей. Исследуемые соединения, солевой раствор и контрольные лекарственные средства (индометацин, аспирин и эбселен) вводились перорально. Отек уха был индуцирован путем локального введения ксилола (0,5 мл в ухо) на внутреннюю поверхность правого уха через 1 час после введения лекарственных средств. Спустя 2 часа мышей умерщвляли. Изменения в массе уха в диаметре 8 мм измерялись с помощью прецизионного микрометра. Коэффициент ингибирования отека уха, индуцированного ксилолом, для каждого соединения вычислялся в соответствии с массой уха; результаты приведены в таблице 7.
Можно сделать вывод, что соединения по настоящему изобретению проявляют более высокую противовоспалительную активность, чем аспирин или индометацин.
Пример 9
Действие соединений по настоящему изобретению на тромбоз
Самцы крыс со стандартными отклонениями (300-400 г) были рандомизированы на контрольную группу без лечения (0,25% химиотерапии CMC), контрольную группу ASA (0,25% химиотерапии CMC, несколько Tween-80) и три исследуемые группы (получавшие соединения Е001, Е002 и Е003). В каждой группе было по 5 крыс. Соединения по настоящему изобретению и контрольные лекарственные средства вводились перорально в количестве 30 мг/кг. Спустя 1 час после введения животных анестезировали уретаном. Шейная артерия отделялась хирургическим путем, и измерялось значение ОТ (условия стимуляции: электрический ток - 3 мА; время - 180 секунд). Результаты приведены в таблице 8.
Изобретение относится к производным бензоизоселеназолонила с общей формулой (I) или (II), где R - С1-С6-алкилен, фенилиден, бифенилиден,
R' - полисахаридный остаток или остаток
где М - Pt или Pd.
Соединения обладают противовоспалительными, противовирусными и антитромботическими активностями. 3 н. и 9 з.п. ф-лы, 8 табл.
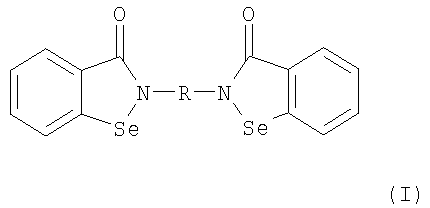
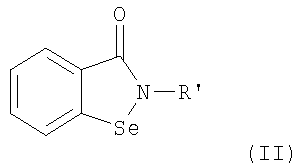
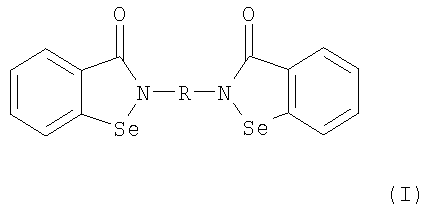
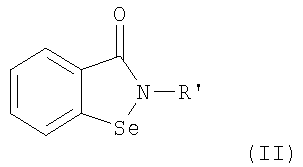
где R является С1-С6-алкиленом, фенилиденом, бифенилиденом,
R' является полисахаридным остатком или остатком
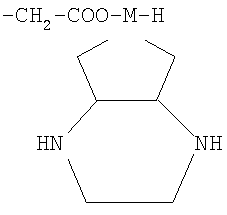
где М - Pt или Pd.
| US 5683863 А, 04.11.1997 | |||
| US 4531001 А, 23.07.1985 | |||
| OSAJDAM et al // Polish Journal of Chem | |||
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |

