Настоящая заявка испрашивает приоритет на основании китайской патентной заявки №202010287745.6, озаглавленной «АРИЛ-КОНДЕНСИРОВАННОЕ СОЕДИНЕНИЕ ИЗОСЕЛЕНАЗОЛА, СОДЕРЖАЩЕЕ ТЕТРАЗИНОВЫЙ ЗАМЕСТИТЕЛЬ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ», поданной в Национальное управление интеллектуальной собственности Китая 13 апреля 2020 года, содержание которой полностью включено в настоящую заявку посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области фармацевтики и, в частности, относится к способу получения арил-конденсированного соединения изоселеназола, содержащего тетразиновый заместитель, и его применению для получения лекарственного средства для лечения опухоли, в частности глиомы головного мозга.
УРОВЕНЬ ТЕХНИКИ
Глиома головного мозга является распространенной внутричерепной опухолью in situ, на которую приходится более 50% внутричерепных опухолей и 1-3% злокачественных опухолей всего организма по отношению к заболеваемости, а медиана общей выживаемости пациентов составляет всего от 12 до 15 месяцев. Поскольку большинство глиом головного мозга растут инфильтративно и не имеют очевидной гистологической границы с нормальными тканями головного мозга, глиомы головного мозга очень трудно полностью вылечить путем хирургического удаления. Кроме того, лучевая терапия (ЛТ) также является способом лечения, широко применяемым в клинической практике, включая традиционную лучевую терапию, трехмерную конформную лучевую терапию, стереотаксическую лучевую терапию и т.д. Тем не менее, поскольку чувствительность клеток глиомы к лучевой терапии недостаточно высока и варьируется между подтипами, эффект лучевой терапии трудно улучшать, и определенные терапевтические эффекты могут быть достигнуты только путем сочетания фармакотерапии с лучевой терапией. Большинство лекарственных средств не способно эффективно воздействовать на опухолевые участки из-за существования гематоэнцефалического барьера, и поражающее воздействие на опухоли является ограниченным. В качестве алкилирующего агента нового поколения темозоломид (ТМЗ) может быстро абсорбироваться после перорального введения и обладает характеристиками высокой эффективности, низкой токсичности и широкого спектра действия. В физиологических условиях ТМЗ в конечном счете может быть превращен в цитотоксический диазометан, и метилирование в положении О6 гуанина, приводящее к цитотоксичности, является основной причиной его поражающего воздействия. Независимо от того, применяют ли ТМЗ отдельно или в сочетании с лучевой терапией для лечения глиом, ТМЗ обладает относительно хорошим терапевтическим эффектом. В настоящее время основным клиническим протоколом темозоломида является протокол «stupp», т.е. лучевую терапию и адъювантную химиотерапию с применением темозоломида проводят синхронно. Более конкретно, адъювантную ТМЗ химиотерапию применяют через месяц после завершения лучевой терапии, циклами по 28 дней, в которых лекарственное средство применяют один раз в день в течение 5 дней подряд и применение прерывают на 23 дня (150 мг/м2/д × 5 в цикле 1 и 200 мг/м2/д × 5 в циклах от 2 до 6). Медиана общей выживаемости пациентов, получавших лечение по протоколу Stupp, составляет 14,6 месяца, в то время как медиана общей выживаемости пациентов, получавших только лечение путем ЛТ, составляла 12,1 месяца.
Несмотря на некоторые достижения, ТМЗ все еще имеет некоторые недостатки. Во-первых, эффективность не является идеальной. Во-вторых, наблюдается много побочных эффектов, приводящих к неблагоприятным эффектам у пациентов, таким как миелосупрессия, желудочно-кишечная реакция, периферическая нейротоксичность и т.д. В-третьих, также существует проблема резистентности к лекарственному средству, то есть О6-метилгуанин-ДНК-метилтрансфераза (MGMT) может напрямую отрезать метальную группу, что делает ТМЗ неэффективным. Кроме того, также может легко развиться множественная лекарственная резистентность (МЛР). Это основные причины неэффективности ТМЗ химиотерапии при глиомах.
Система тиоредоксина (система TRX) состоит из трех частей: восстановленный никотинамидадениндинуклеотидфосфат (NADPH), тиоредоксин (TRX) и тиоредоксинредуктаза (TrxR), среди которых TrxR представляет собой NADPH-зависимую димерную селеназу, содержащую флавинадениндинуклеотидные (FAD) домены. Существует три подтипа TrxR млекопитающих: TrxR1 в ядрах, TrxR2 в митохондриях и TrxR3, присутствующий в семеннике. Перенос электронов внутри всей системы TRX показан на ФИГ. 1. Полностью окисленный фермент акцептирует электроны из NADPH, а затем образует полностью восстановленный фермент. Наконец, субстрат восстанавливается, при этом С-концевая селенотиоловая пара играет роль в переносе электронов и является основой для функции окислительно-восстановительной регуляции TrxR.
Система TRX может непосредственно противостоять окислению и поддерживать функции других антиоксидантных ферментов, и может способствовать предотвращению превращения нормальных клеток в злокачественные опухоли, путем предотвращения окислительного стресса у экзогенных веществ или канцерогенов. В аспектах прогрессирования и метастазирования опухоли увеличение уровня TrxR/Trx может способствовать пролиферации клеток, противостоять апоптозу и способствовать ангиогенезу, таким образом, стимулируя рост опухолей.
Система TRX играет важную роль в процессе развития, прогрессирования и метастазирования опухолей и является важной системой для поддержания гомеостаза и содействия пролиферации и ангиогенезу в опухолевых клетках.
TrxR экспрессируется на высоком уровне в опухолях головного мозга, и анализ клинических данных по 433 случаям глиобластомы показал, что тиоредоксинредуктаза 1 (TrxR1) повышающе регулировалась более чем в 66% случаев, что было явно связано с большей пролиферативной активностью и более неблагоприятным прогнозом. Таким образом, очень важно изучить лечение глиом головного мозга путем нацеливания на мишень TrxR.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложено арил-конденсированное соединение изоселеназола, содержащее тетразиновый заместитель, представленное формулой (I), или его фармацевтически приемлемая соль:
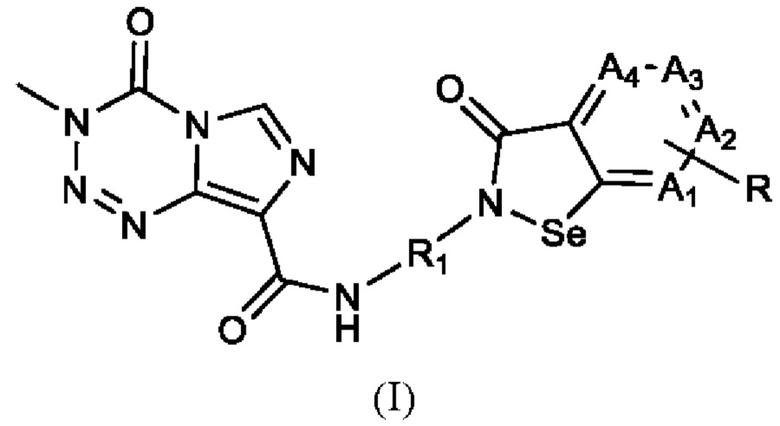
где A1, А2, A3 и А4 являются идентичными или различными и независимо друг от друга выбраны из СН и N, и не больше одного из них представляет собой N; когда A1, А2, А3 или А4 представляет собой СН, атом водорода в нем может быть замещен R, где R выбран из водорода, циано, гидрокси, галогена и нитро, или из амино, сульфгидрила, амидо, алкила, циклоалкила, алкокси, гетероциклила, арила и гетероарила, которые замещены одним или более Ra, где каждый из Ra независимо друг от друга выбран из водорода, циано, гидрокси, галогена, амино, нитро, сульфгидрила, алкила, циклоалкила, алкокси, арила и гетероарила; R1 выбран из алкилена.
Согласно одному варианту реализации настоящего изобретения R выбран из водорода, циано, гидрокси, галогена и нитро, или из амино, сульфгидрила, амидо, С1-10 алкила, С3-10 циклоалкила, С1-10 алкокси, 3-10-членного гетероциклила, С6-14 арила и 5-14-членного гетероарила, которые замещены одним или более Ra, где каждый из Ra независимо друг от друга выбран из водорода, циано, гидрокси, галогена, амино, нитро, сульфгидрила, С1-10 алкила, С3-10 циклоалкила, С1-10 алкокси, 3-10-членного гетероциклила, С6-14 арила и 5-14-членного гетероарила; R1 выбран из С1-10 алкилена.
Согласно одному варианту реализации настоящего изобретения R выбран из водорода и галогена или из С1-6 алкила, С3-6 циклоалкила, C1-6 алкокси, фенила и пиридила, которые замещены одним или более Ra, где каждый из Ra независимо друг от друга выбран из водорода, галогена, C1-6 алкила, С3-6 циклоалкила, C1-6 алкокси, фенила и пиридила; R1 выбран из С1-6 алкилена.
Согласно одному варианту реализации настоящего изобретения R выбран из водорода, галогена, C1-6 алкила и C1-6 алкокси.
Согласно одному варианту реализации настоящего изобретения R выбран из водорода, фтора, хлора, брома, йода, метила, этила, н-пропила, изопропила, н-бутила, изобутила, трет-бутила, н-пентила, изопентила, метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, трет-бутокси, н-пентокси и изопентокси.
Предпочтительно, структура арил-конденсированного соединения изоселеназола, содержащего тетразиновый заместитель, представлена формулой (II):
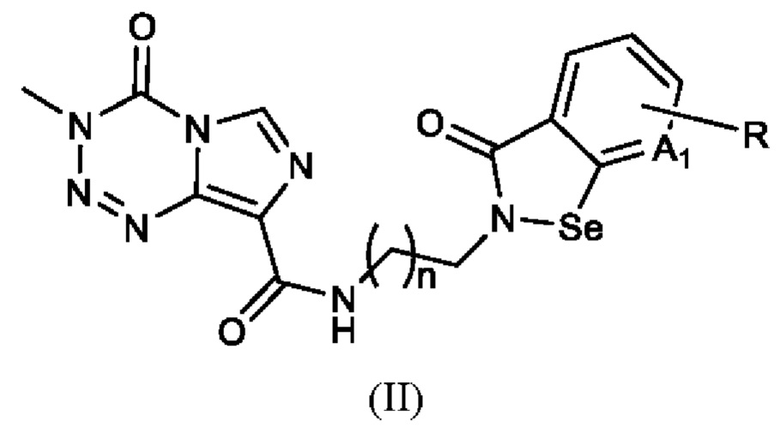
где A1 выбран из СН и N; n равно 1, 2, 3, 4, 5 или 6; R имеет определение, описанное выше.
Согласно одному варианту реализации настоящего изобретения R выбран из водорода, галогена, C1-6 алкила и C1-6 алкокси.
Согласно одному варианту реализации настоящего изобретения R выбран из водорода, фтора, хлора, брома, йода, метила, этила, н-пропила, изопропила, н-бутила, изобутила, трет-бутила, н-пентила, изопентила, метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, трет-бутокси, н-пентокси и изопентокси.
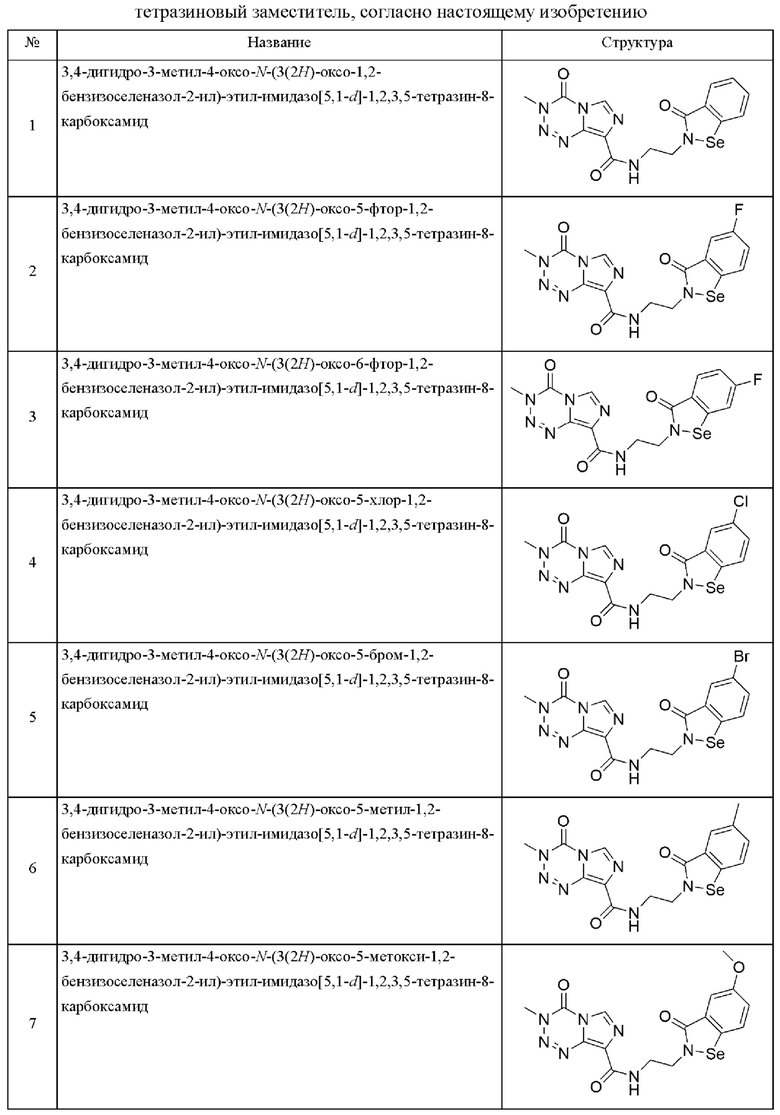
Предпочтительно, арил-конденсированное соединение изоселеназола, содержащее тетразиновый заместитель, выбрано из соединений, представленных ниже в таблице 1:
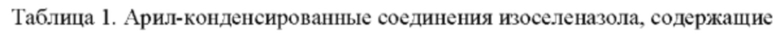

Согласно одному варианту реализации настоящего изобретения фармацевтически приемлемая соль соединения, представленного формулой (I) или формулой (II), включает: соль присоединения кислоты, полученную из соединения и неорганической или органической кислоты; где неорганическая кислота выбрана по меньшей мере из одной из хлористоводородной кислоты, фтористоводородной кислоты, бромистоводородной кислоты, йодистоводородной кислоты, перхлорной кислоты, серной кислоты, пиросерной кислоты, фосфорной кислоты и азотной кислоты; органическая кислота выбрана по меньшей мере из одной из муравьиной кислоты, уксусной кислоты, ацетоуксусной кислоты, пировиноградной кислоты, трифторуксусной кислоты, пропионовой кислоты, масляной кислоты, капроновой кислоты, гептановой кислоты, ундекановой кислоты, лауриновой кислоты, бензойной кислоты, салициловой кислоты, 2-(4-гидроксибензоил)бензойной кислоты, камфорной кислоты, коричной кислоты, циклопентанпропионовой кислоты, диглюконовой кислоты, 3-гидрокси-2-нафтовой кислоты, никотиновой кислоты, памовой кислоты, пектиновой кислоты, надсерной кислоты, 3-фенилпропионовой кислоты, пикриновой кислоты, пивалевой кислоты, 2-гидроксиэтансульфоновой кислоты, итаконовой кислоты, сульфаминовой кислоты, трифторметансульфоновой кислоты, додецилсульфоновой кислоты, этансульфоновой кислоты, бензолсульфоновой кислоты, n-толуолсульфоновой кислоты, метансульфоновой кислоты, 2-нафталинсульфоновой кислоты, нафталиндисульфоновой кислоты, камфорсульфоновой кислоты, лимонной кислоты, винной кислоты, стеариновой кислоты, молочной кислоты, щавелевой кислоты, малоновой кислоты, янтарной кислоты, яблочной кислоты, адипиновой кислоты, альгиновой кислоты, малеиновой кислоты, фумаровой кислоты, D-глюконовой кислоты, миндальной кислоты, аскорбиновой кислоты, глюкогептоновой кислоты, глицерофосфорной кислоты, аспарагиновой кислоты, сульфосалициловой кислоты, гемисерной кислоты и тиоциановой кислоты;
альтернативно, фармацевтически приемлемая соль соединения, представленного формулой (I) или формулой (II), представляет собой соль щелочного металла, соль щелочноземельного металла или аммонийную соль соединения, или соль, полученную из соединения и органического основания, обеспечивающего физиологически приемлемые катионы, например, соль, полученную из соединения и по меньшей мере одного из следующих соединений: ион натрия, ион калия, ион кальция, ион магния, морфолин, пиперидин, триэтиламин, трипропиламин, трибутиламин, диизопропиламин, диизопропилэтилендиамин, пиридин, диметиламин, диэтиламин, N-метилглюкамин, диметилглюкамин, этилглюкамин, лизин, дициклогексиламин, 1,6-гександиамин, этаноламин, глюкозамин, меглюмин, саркозин, серинол, тригидроксиметиламинометан, аминопропандиол и 1-амино-2,3,4-бутантриол.
В настоящем изобретении дополнительно предложен способ получения соединения, представленного формулой (I), который включает следующие стадии:
1) диазотирование аминогруппы в соединении А с последующим взаимодействием с Na2Se2 с получением соединения В:
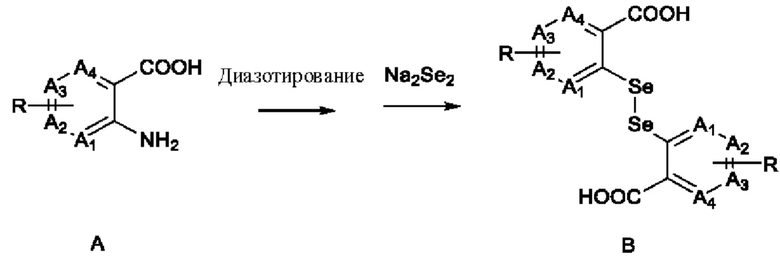 ;
;
где A1, А2, A3, А4, R и R1 имеют определения, описанные выше;
2) взаимодействие соединения В с тионилхлоридом с получением соединения С:
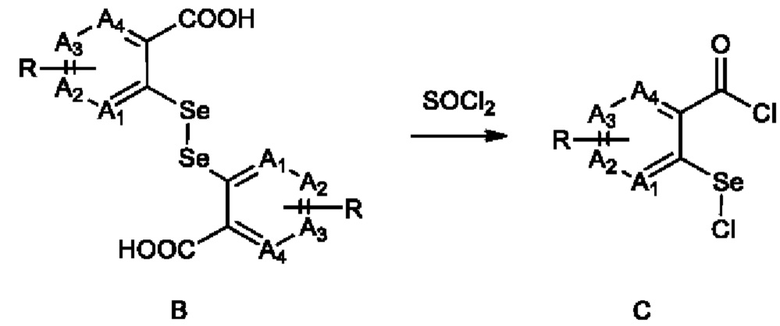 ;
;
3) взаимодействие соединения C с H2N-R1-NHBoc с получением соединения D:
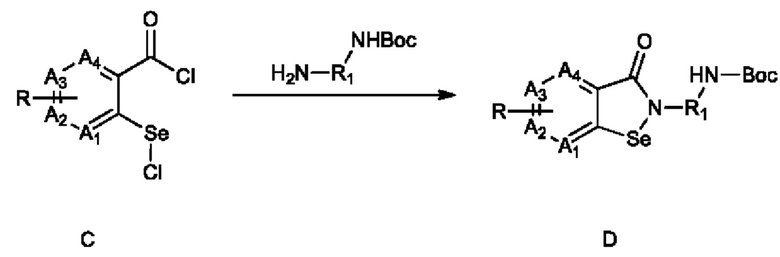 ;
;
где Boc представляет собой трет-бутилоксикарбонил;
4) снятие защиты с Вос-защищенной группы соединения D в присутствии кислоты с получением соединения Е:
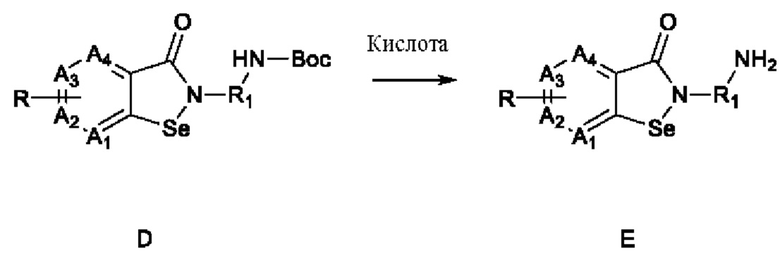 ; и
; и
5) взаимодействие соединения Е с соединением F с получением соединения, представленного формулой (I):
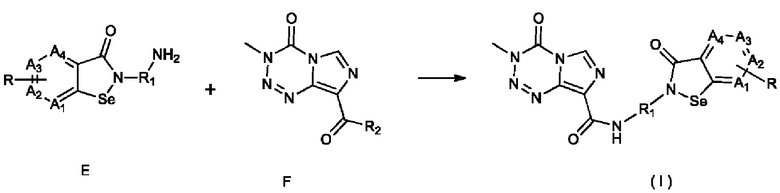 ;
;
где в соединении F R2 выбран из хлора, брома, гидрокси и -OR3, где R3 выбран из C1-6 алкила, сукцинимидила, трет-бутилоксикарбонила, метилсульфонила, п-нитробензолсульфонила, n-толуолсульфонила и изобутилоксикарбонила.
Согласно одному варианту реализации настоящего изобретения Na2Se2 на стадии 1) можно получать из элементарного селена и гидросульфита натрия.
Согласно одному варианту реализации настоящего изобретения стадию 2) можно проводить в присутствии каталитического количества ДМФА. Согласно одному варианту реализации настоящего изобретения кислота на стадии 4) может представлять собой хлористоводородную кислоту. Кроме того, стадию 4) можно проводить в органическом растворителе, таком как этилацетат.
Согласно одному варианту реализации настоящего изобретения соединение F является коммерчески доступным или может быть получено с применением обычного способа, известного в данной области техники, или получено при помощи следующих стадий:
а) взаимодействие соединения ТМЗ (т.е. темозоломида) с серной кислотой и нитритом натрия с получением соединения F-1:
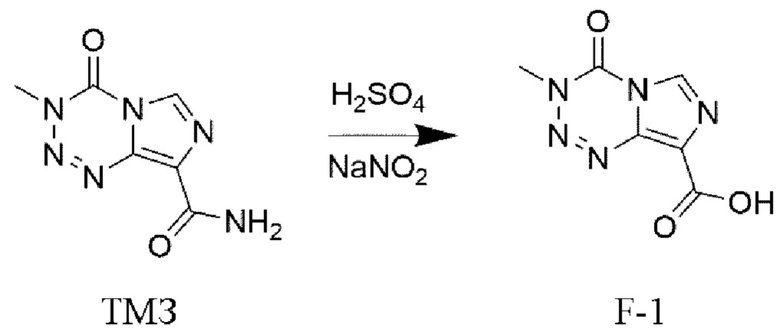 ;
;
и необязательная стадия b): взаимодействие соединения F-1 с тионилхлоридом или дибромсульфоксидом в присутствии каталитического количества ДМФА с получением соединения F-2:
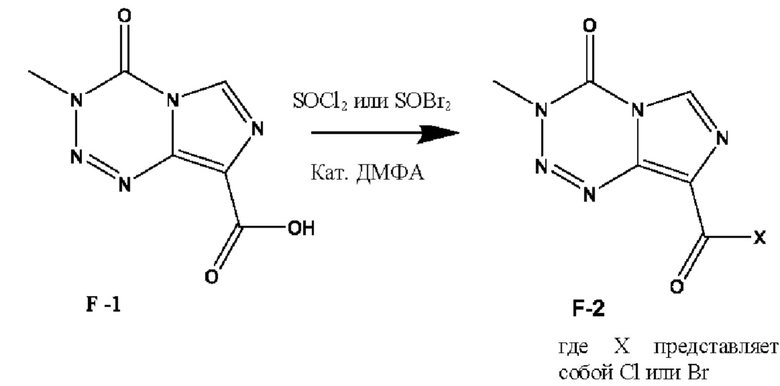 ;
;
или необязательная стадия с): дальнейшее взаимодействие соединения F-1 с получением соединения F, в котором R2 представляет собой -OR3, где R3 представляет собой С1-6 алкил, сукцинимидил, трет-бутоксикарбонил, метансульфонил, n-нитробензолсульфонил, n-толуолсульфонил или изобутилоксикарбонил.
Стадия с) выбрана из стадии с-1), стадии с-2), стадии с-3), стадии с-4) и стадии с-5), представленных ниже:
стадия с-1): взаимодействие соединения F-1 с R4OH под действием кислоты с получением соединения F-3 (т.е. соединения F, в котором R2 представляет собой -OR3, и R3 представляет собой C1-6 алкил):
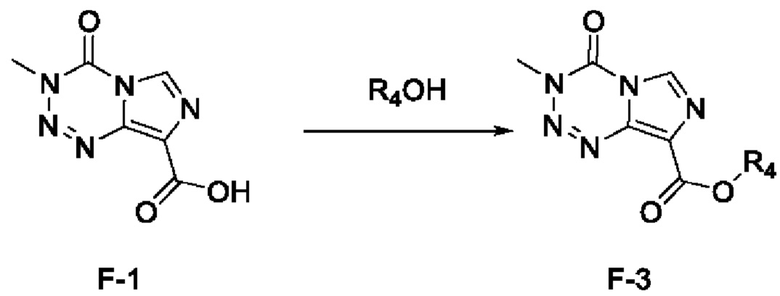 ,
,
где R4 представляет собой C1-6 алкил;
стадия с-2): взаимодействие соединения F-1 с R5Cl под действием основания с получением соединения F-4 (т.е. соединения F, в котором R2 представляет собой -OR3, и R3 представляет собой метансульфонил, n-нитробензолсульфонил или n-толуолсульфонил):
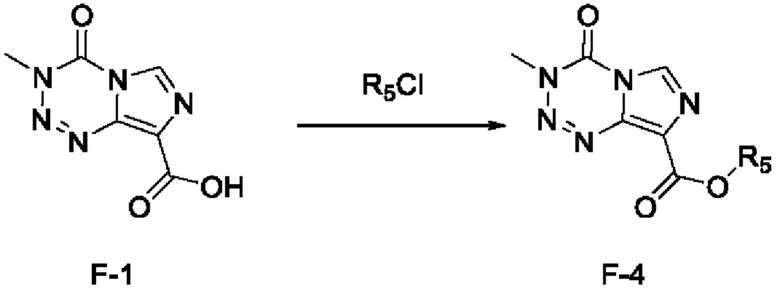 ;
;
где R5 представляет собой метилсульфонил, n-нитробензолсульфонил или n-толуолсульфонил;
стадия с-3): взаимодействие соединения F-1 с N-гидроксисукцинимидом с получением соединения F-5 (т.е. соединения F, в котором R2 представляет собой -OR3, и R3 представляет собой сукцинимидил):
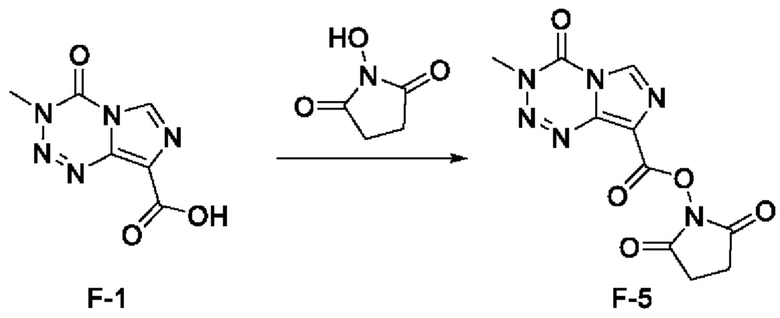 ;
;
стадия с-4): взаимодействие соединения F-1 с ди-трет-бутилдикарбонатом с получением соединения F-6 (т.е. соединения F, в котором R2 представляет собой -OR3, и R3 представляет собой трет-бутоксикарбонил):
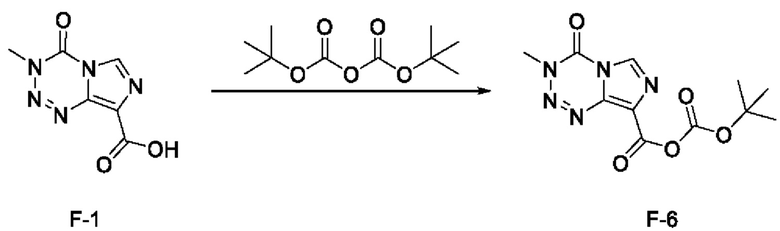 ; и
; и
стадия с-5): взаимодействие соединения F-1 с изобутилхлорформиатом с получением соединения F-7 (т.е. соединения F, в котором R2 представляет собой -OR3, и R3 представляет собой изобутилоксикарбонил):
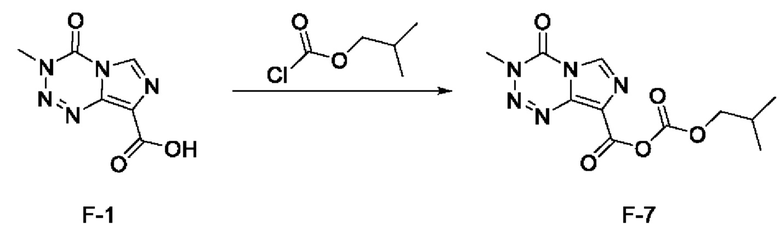 .
.
В настоящем изобретении дополнительно предложена фармацевтическая композиция, которая содержит арил-конденсированное соединение изоселеназола, представленное формулой (I), содержащее тетразиновый заместитель, или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
Согласно одному варианту реализации настоящего изобретения фармацевтическая композиция подходит для энтерального, местного или парентерального введения, например, перорального введения, инъекции, имплантации, местного нанесения, в виде спрея, ингаляции или других способов введения.
Согласно одному варианту реализации настоящего изобретения пероральная фармацевтическая композиция может представлять собой любую форму из таблетки, капсулы, пилюли, жидкого состава для перорального применения, гранул, инъекции, состава для местного нанесения и порошка.
Таблетка может представлять собой обычную таблетку, буккальную таблетку, сублингвальную таблетку, буккальный пластырь, жевательную таблетку, диспергируемую таблетку, растворимую таблетку, шипучую таблетку, вагинальную таблетку, вагинальную шипучую таблетку, таблетку с замедленным высвобождением, таблетку с контролируемым высвобождением, таблетку с кишечнорастворимым покрытием или буккальную таблетку с немедленным высвобождением; капсула может представлять собой твердую капсулу, мягкую капсулу, капсулу с замедленным высвобождением, капсулу с контролируемым высвобождением или капсулу с кишечнорастворимым покрытием; пилюля включает микропилюлю, пилюлю-пустышку или гранулу; жидкий состав для перорального применения может представлять собой сироп, суспензию, раствор для перорального применения, суспензию для перорального применения, эмульсию для перорального применения, сироп, смесь, дистиллят или линимент; гранулы могут представлять собой гранулы суспензии, шипучие гранулы, гранулы с кишечнорастворимым покрытием, гранулы с замедленным высвобождением или гранулы с контролируемым высвобождением. Инъекция может представлять собой любую форму из жидкости для инъекции, стерильного порошка или стерильного блока для инъекции и концентрированного раствора для трансфузии и инъекции. Состав для местного нанесения может представлять собой любую форму из суппозитория, аэрозоля, порошка для ингаляции, спрея, пленки, геля, пластыря, коллоида, медицинского пластыря, гипса, мази, линимента, лосьона, агента для нанесения и желатина.
Согласно одному варианту реализации настоящего изобретения фармацевтическую композицию можно получать при помощи способов получения состава, хорошо известных в данной области техники.
Согласно одному варианту реализации настоящего изобретения фармацевтическая композиция может представлять собой препарат включения или дисперсионный препарат.
Согласно одному варианту реализации настоящего изобретения фармацевтически приемлемый носитель представляет собой обычное вспомогательное вещество или вспомогательный материал, хорошо известные в данной области техники для получения составов, описанных выше, при этом обычное вспомогательное вещество или вспомогательный материал для составов для перорального применения или местного нанесения включает, но не ограничивается ими, наполнитель, разбавитель, смазывающее вещество, скользящее вещество, антиадгезив, диспергатор, увлажняющий агент, связующее вещество, регулятор, солюбилизатор, антиоксидант, бактериостат, эмульгатор и т.д.
Связующее вещество представляет собой, например, сироп, аравийскую камедь, желатин, сорбит, трагакант, целлюлозу или ее производное, желатиновую пасту, сироп, крахмальную пасту или поливинилпирролидон, предпочтительно производное целлюлозы, представляет собой микрокристаллическую целлюлозу, карбоксиметилцеллюлозу натрия, этилцеллюлозу или гидроксипропилметилцеллюлозу; наполнитель представляет собой, например, лактозу, порошкообразный сахар, декстрин, крахмал или его производное, целлюлозу или ее производное, неорганическую соль кальция, сорбит или глицин, при этом неорганическая соль кальция предпочтительно представляет собой сульфат кальция, фосфат кальция, дикальцийфосфат или осажденный карбонат кальция; смазывающее вещество представляет собой, например, коллоидный диоксид кремния, стеарат магния, тальк, гидроксид алюминия, борную кислоту, гидрогенизированное растительное масло или полиэтиленгликоль; разрыхлитель представляет собой, например, крахмал или его производное, поливинилпирролидон или микрокристаллическую целлюлозу, при этом производное крахмала предпочтительно представляет собой карбоксиметилкрахмал натрия, крахмалгликолят натрия, предварительно желатинизированный крахмал, модифицированный крахмал, гидроксипропиловый крахмал или кукурузный крахмал; увлажняющий агент представляет собой, например, додецилсульфат натрия, воду или спирт; вспомогательное вещество предпочтительно представляет собой α-циклодекстрин, β-циклодекстрин, γ-циклодекстрин, Celadon102CG, повидон (PVP) - серия K (включая повидон K30 (PVPK30)), тальк, стеарат магния или этанол и т.д.
Согласно одному варианту реализации настоящего изобретения обычное вспомогательное вещество или вспомогательный материал для инъекции включает: антиоксидант, бактериостат, регулятор рН, эмульгатор или солюбилизатор.
Антиоксидант включает тиосульфат натрия, сульфит натрия, бисульфит натрия, дибутилбензойную кислоту или пиросульфит натрия; бактериостат включает фенол, крезол или хлорбутанол, предпочтительно 0,5% фенол, 0,3% крезол или 0,5% хлорбутанол; регулятор рН включает хлористоводородную кислоту, лимонную кислоту, гидроксид калия, гидроксид натрия, цитрат натрия, дигидрофосфат натрия или гидрофосфат динатрия; эмульгатор включает полисорбат-80, эфир сорбита и жирной кислоты, плуроник F-68, лецитин или фосфолипид сои; солюбилизатор включает Tween-80 или глицерин.
Согласно одному варианту реализации настоящего изобретения соединение, представленное формулой (I), или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель с замедленным/контролируемым высвобождением также могут быть представлены в виде состава с замедленным/контролируемым высвобождением при помощи обычного способа получения составов с замедленным/контролируемым высвобождением. Например, соединение, представленное формулой (I), или его фармацевтически приемлемая соль могут быть покрыты замедлителем или могут быть микроинкапсулированы, а затем представлены в виде гранул, таких как гранулы с замедленным высвобождением или гранулы с контролируемым высвобождением.
Носитель с замедленным/контролируемым высвобождением включает, но не ограничивается ими, жировую добавку, гидрофильный коллоид или покрытие-замедлитель; жировая добавка включает глицерилмоностеарат, гидрогенизированное касторовое масло, минеральное масло, полисилоксан или диметилсилоксан; гидрофильный коллоид включает карбоксиметилцеллюлозу натрия, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, поливинилпирролидон (PVP), аравийскую камедь, трагакант или карбопол; покрытие-замедлитель включает этилцеллюлозу (ЕС), гидроксипропилметилцеллюлозу (НРМС), поливинилпирролидон (PVP), ацетатфталат целлюлозы (САР) или акриловую смолу.
В предпочтительном варианте реализации настоящего изобретения фармацевтическая композиция содержит примерно 1-99 масс. % любого одного или комбинации соединения, представленного формулой (I), и его фармацевтически приемлемой соли и 1-99 масс. % фармацевтически приемлемого носителя, в зависимости от желаемого способа введения.
В настоящем описании дополнительно предложено применение соединения, представленного формулой (I), или его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей указанное соединение, для ингибирования активности тиоредоксинредуктазы (TrxR) или получения ингибитора тиоредоксинредуктазы (TrxR).
В настоящем описании дополнительно предложено применение соединения, представленного формулой (I), или его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей указанное соединение, для лечения или облегчения заболевания или состояния, связанного с повышенной экспрессией или повышенной активностью TrxR, или для получения лекарственного средства для лечения или облегчения заболевания или состояния, связанного с повышенной экспрессией или повышенной активностью TrxR.
В настоящем описании дополнительно предложено применение соединения, представленного формулой (I), или его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей указанное соединение, для лечения опухолей или получения лекарственного средства для лечения опухоли.
В настоящем описании дополнительно предложено соединение, представленное формулой (I), или его фармацевтически приемлемая соль, или фармацевтическая композиция, содержащая указанное соединение, для ингибирования активности тиоредоксинредуктазы (TrxR), для лечения или облегчения заболевания или состояния, связанного с повышенной экспрессией или повышенной активностью TrxR, для лечения опухоли, для применения в способе лечения опухоли или для получения лекарственного средства для лечения опухоли.
В настоящем описании дополнительно предложен способ лечения или облегчения заболевания или состояния, связанного с повышенной экспрессией или повышенной активностью TrxR у субъекта, который включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения, представленного формулой (I), или его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей указанное соединение.
В настоящем описании дополнительно предложен способ лечения опухоли у субъекта, который включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения, представленного формулой (I), или его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей указанное соединение.
Согласно одному варианту реализации настоящего изобретения заболевание или состояние, связанное с повышенной экспрессией или повышенной активностью TrxR, представляет собой опухоль, включая, но не ограничиваясь ими, любую из глиомы головного мозга, рака легкого, рака печени, рака крови, рака кости, рака поджелудочной железы, рака кожи, меланомы, рака матки, рака яичника, рака прямой кишки, рака желудка, рака толстой кишки, рака молочной железы, рака матки, рака фаллопиевой трубы, рака эндометрия, рака шейки матки, рака влагалища, рака вульвы, рака пищевода, рака тонкого кишечника, рака эндокринной системы, саркомы мягких тканей, рака мочеиспускательного канала, рака предстательной железы, лимфоцитомы, рака мочевого пузыря, рака почки, рака мочеточника, опухоли позвоночника, глиомы ствола головного мозга, аденомы гипофиза, рака легкого, рака печени и рака крови, и опухоль предпочтительно представляет собой глиому головного мозга.
Благоприятные эффекты
Арил-конденсированное соединение изоселеназола, содержащее тетразиновый заместитель, согласно настоящему изобретению обладает характеристикой нацеливания на мишень TrxR, и TrxR обладает характеристикой маркера роста опухоли, поскольку TrxR представляет собой фермент, регулирующий рост опухоли. Кроме того, TrxR при глиоме головного мозга являются многократно признанными высокоэкспрессируемыми опухолевыми маркерами, поэтому соединение, подходящее для направленного ингибирования TrxR, будет иметь хороший противоопухолевый эффект в отношении глиом головного мозга. В настоящее время соединение может хорошо ингибировать рост каждой клеточной линии глиомы головного мозга как in vivo, так и in vitro.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
ФИГ. 1 представляет собой схематическую диаграмму переноса электронов внутри системы TRX.
На ФИГ. 2 показано влияние соединения 1 на массу тела мышей с опухолью (U87).
На ФИГ. 3 показано влияние соединения 1 на объем опухоли у мышей с опухолью (U87).
На ФИГ. 4 показано влияние соединения 1 на массу опухоли у мышей с опухолью (U87).
Определения и описание
Термин «алкил» относится к линейной или разветвленной насыщенной одновалентной углеводородной группе, содержащей от 1 до 12 атомов углерода. Например, «С1-10 алкил» относится к линейным и разветвленным алкильным группам, содержащим 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 атомов углерода. Алкил представляет собой, например, метил, этил, пропил, бутил, пентил, гексил, изопропил, изобутил, втор-бутил, трет-бутил, изопентил, 2-метилбутил, 1-метилбутил, 1-этилпропил, 1,2-диметилпропил, неопентил, 1,1-диметилпропил, 4-метилпентил, 3-метилпентил, 2-метилпентил, 1-метилпентил, 2-этилбутил, 1-этилбутил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 2,3-диметилбутил, 1,3-диметилбутил, 1,2-диметилбутил и т.д. или их изомеры.
Термин «алкокси» относится к -О-С1-10 алкилу, где С1-10 алкил имеет определение, описанное выше.
Термин «циклоалкил» относится к насыщенному одновалентному моноциклическому или бициклическому углеводородному кольцу, содержащему от 3 до 20 атомов углерода. Термин «С3-10 циклоалкил» относится к насыщенному одновалентному моноциклическому или бициклическому углеводородному кольцу, содержащему 3, 4, 5, 6, 7, 8, 9 или 10 атомов углерода. С3-10 циклоалкил может представлять собой моноциклическую углеводородную группу, такую как циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил или циклодецил, или бициклическую углеводородную группу, такую как декалиновое кольцо.
Термин «алкилен», применяемый в настоящем описании, относится к двухвалентной алкильной группе; например, С1-10 алкилен относится к двухвалентной С1-10 алкильной группе.
Термин «гетероциклил» относится к насыщенному одновалентному моноциклическому или бициклическому углеводородному кольцу, содержащему от 3 до 20 атомов, которое содержит от 1 до 5 гетероатомов, независимо выбранных из N, О и S, и предпочтительно представляет собой «3-10-членный гетероциклил». Термин «3-10-членный гетероциклил» относится к насыщенному одновалентному моноциклическому или бициклическому углеводородному кольцу, содержащему от 1 до 5 (предпочтительно от 1 до 3) гетероатомов, выбранных из N, О и S. Гетероциклил может быть соединен с остальной частью молекулы через любой из атомов углерода или атома(ов) азота (при наличии). В частности, гетероциклил может включать, но не ограничивается ими: 4-членные кольца, такие как азетидинил и оксетанил; 5-членные кольца, такие как тетрагидрофуранил, диоксолил, пирролидинил, имидазолидинил, пиразолидинил и пирролинил; 6-членные кольца, такие как тетрагидропиранил, пиперидил, морфолинил, дитианил, тиоморфолинил, пиперазинил и тритианил; или 7-членные кольца, такие как диазепанил. Необязательно гетероциклил может являться бензоконденсированным. Гетероциклил может являться бициклическим, например, но не ограничиваясь ими, 5,5-членным кольцом, таким как гексагидроциклопента[с]пиррол-2(1H)-ильное кольцо, или 5,6-членным бициклическим кольцом, таким как гексагидропирроло[1,2-a]пиразин-2(1H)-ильное кольцо. Кольцо, содержащее атомы азота, может являться частично ненасыщенным, т.е. оно может содержать одну или более двойных связей, например, но не ограничиваясь ими, 2,5-дигидро-1H-пирролил, 4H-[1,3,4]тиадиазинил, 4,5-дигидрооксазолил или 4H-[1,4]тиазинил, или оно может являться бензоконденсированным, например, но не ограничиваясь им, дигидроизохинолил. Согласно настоящему изобретению гетероциклил является неароматическим.
Термин «арил» относится к одновалентному ароматическому или частично ароматическому моноциклическому, бициклическому или трициклическому углеводородному кольцу, содержащему от 6 до 20 атомов углерода (предпочтительно от 6 до 14 атомов углерода), и предпочтительно относится к «С6-14 арилу». Термин «С6-10 арил» предпочтительно относится к ароматическому или частично ароматическому одновалентному моноциклическому, бициклическому или трициклическому углеводородному кольцу, содержащему 6, 7, 8, 9, 10, 11, 12, 13 или 14 атомов углерода («С6-14 арил»), в частности, кольцу, содержащему 6 атомов углерода («С6 арил»), такому как фенил или дифенил, кольцу, содержащему 9 атомов углерода («С9 арил»), такому как инданил или инденил, кольцу, содержащему 10 атомов углерода («С10 арил»), такому как тетрагидронафтил, дигидронафтил или нафтил, кольцу, содержащему 13 атомов углерода («С13 арил»), такому как флуоренил, или кольцу, содержащему 14 атомов углерода («С14 арил»), такому как антрил. Когда С6-14 арил является замещенным, он может являться монозамещенным или полизамещенным. Кроме того, место замещения не ограничено и может представлять собой, например, орто-замещение, лора-замещение или мета-замещение.
Термин «гетероарил» относится к одновалентной моноциклической, бициклической или трициклической ароматической кольцевой группе, содержащей от 3 до 20 кольцевых атомов и содержащей от 1 до 5 гетероатомов, независимо выбранных из N, О и S, например, такой как «5-14-членный гетероарил». Термин «5-14-членный гетероарил» относится к одновалентной моноциклической, бициклической или трициклической ароматической кольцевой группе, содержащей 5, 6, 7, 8, 9, 10, 11, 12, 13 или 14 кольцевых атомов (в частности, 5, 6, 9 или 10 атомов углерода) и содержащей от 1 до 5 (предпочтительно от 1 до 3) гетероатомов, независимо выбранных из N, О и S, и может являться бензоконденсированным в каждом случае. В частности, гетероарил выбран из тиенила, фуранила, пирролила, оксазолила, тиазолила, имидазолила, пиразолила, изоксазолила, изотиазолила, оксадиазолила, триазолила, тиадиазолила и т.п. и их бензопроизводных, таких как бензофуранил, бензотиенил, бензоксазолил, бензоизоксазолил, бензимидазолил, бензотриазолил, индазолил, индолил, изоиндолил и т.п.; или пиридила, пиридазинила, пиримидинила, пиразинила, триазинила и т.п. и их бензопроизводных, таких как хинолил, хиназолинил, изохинолил и т.п.; или азоцинила, индолизинила, пуринила и т.п. и их бензопроизводных; или циннолинила, фталазинила, хиназолинила, хиноксалинила, нафтиридинила, птеридинила, карбазолила, акридинила, феназинила, фенотиазинила, феноксазинила и т.п.
Термин «амидо» относится к группе Ra-C(=O)-NH-, где Ra имеет определение, описанное выше.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Технические решения согласно настоящему изобретению будут более подробно проиллюстрированы со ссылкой на следующие конкретные примеры. Следует понимать, что следующие примеры являются лишь иллюстрацией и пояснением настоящего изобретения и не должны рассматриваться, как ограничивающие объем защиты настоящего изобретения. Все способы, реализованные на основе содержания настоящего изобретения, описанного выше, включены в объем защиты настоящего изобретения.
Если не указано иное, исходные материалы и реагенты, применяемые в следующих примерах, являются коммерчески доступными продуктами или могут быть получены при помощи известных способов.
Пример 1
3,4-Дигидро-3-метил-4-оксо-N-(3(2H)-оксо-1,2-бензизоселеназол-2-ил)-этил-мидазо[5,1-d]-1,2,3,5-тетразин-8-карбоксамид (№1)
1) В лабораторный стакан добавляли 28 г 2-аминобензойной кислоты, а затем добавляли 40 мл концентрированной хлористоводородной кислоты и 40 мл воды. Смесь охлаждали на ледяной бане для поддержания температуры реакции ниже 5°С. По каплям медленно добавляли раствор NaNO2 (18 г, растворенных в 40 мл воды). После завершения добавления по каплям смесь оставляли для протекания реакции в течение дополнительных 2 часов с получением раствора соли диазония для последующего применения. 16 г NaOH добавляли к 100 мл воды и растворяли при перемешивании при 50°С, а затем небольшими порциями добавляли 17,6 г гидросульфита натрия. После того, как смесь стала прозрачной, добавляли 16 г порошка селена. После завершения добавления смесь оставляли для протекания реакции в течение дополнительных 3 часов с получением раствора Na2Se2 для последующего применения. По каплям при перемешивании добавляли раствор соли диазония к раствору Na2Se2, при этом температуру реакции поддерживали ниже 5°С. После завершения добавления по каплям раствор оставляли для протекания реакции в течение дополнительных 2 часов, при этом раствор оставался щелочным. После завершения реакции смесь подкисляли хлористоводородной кислотой и фильтровали с получением твердого вещества и твердое вещество промывали водой, а затем сушили в сушильном шкафу с получением 2,2''-диселандиилдибензойной кислоты (25 г, 63%).
2) К 2,2''-диселандиилдибензойной кислоте (4 г, 10 ммоль) добавляли тионилхлорид (20 мл) и 1-2 капли ДМФА. Смесь перемешивали при температуре обратной конденсации в течение 5 ч и концентрировали при пониженном давлении для выпаривания тионилхлорида. Остаток перекристаллизовывали из петролейного эфира (100 мл) и смесь фильтровали через слой целита. Фильтрат хранили в холодильнике с получением желтого игольчатого кристалла, т.е. 2-селенохлорбензоилхлорид (3 г, 59%).
3) Вос-этилендиамин (1,60 г, 10 ммоль) растворяли в дихлорметане (10 мл) и добавляли 1,39 мл триэтиламина. К смеси медленно по каплям добавляли раствор 2-селенохлорбензоилхлорида (2,54 г, 10 ммоль) в дихлорметане (10 мл) при охлаждении на ледяной бане. Смесь нагревали до комнатной температуры, оставляли для протекания реакции в течение 3 ч и концентрировали при пониженном давлении для выпаривания растворителя. Добавляли диэтиловый эфир, а затем перемешивали и получали осадок в виде белого твердого вещества. Твердое вещество последовательно промывали водой и петролейным эфиром, сушили и очищали путем колоночной хроматографии (петролейный эфир : дихлорметан: метанол = 200:200:1) с получением желтого твердого вещества, т.е. трет-бутил-(3(2H)-оксо-1,2-бензизоселеназол-2-ил)-этил-1-карбамата (1,4 г, 41%).
4) В 50 мл одногорлую колбу добавляли трет-бутил-(3(2H)-оксо-1,2-бензизоселеназол-2-ил)этил-1-карбамат (0,500 г, 1,47 ммоль), а затем раствор 2 н. хлористоводородной кислоты в этилацетате (5 мл). Раствор оставляли для протекания реакции в течение 1 ч, при этом раствор превращался из прозрачного в мутный, и образовывался белый осадок. Смесь подвергали фильтрованию с отсасыванием и остаток на фильтре промывали этилацетатом с получением белого твердого вещества, т.е. 2-аминоэтил-[1,2-бензизоселеназол-3(2H)-он]гидрохлорида (0,405 г, 90%).
5) ТМЗ (0,97 г, 5,0 ммоль) добавляли порциями к концентрированной серной кислоте при перемешивании при комнатной температуре. После завершения добавления добавляли по каплям 8 мл водного раствора NaNO2 (1,3 г, 18,8 ммоль) при охлаждении на ледяной бане для поддержания температуры при 0°С. После завершения добавления раствор смеси медленно перемешивали при комнатной температуре, при этом раствор превращался из коричневато-желтой жидкости в зеленую смолу и в светло-желтую смолу. Смолу вносили в измельченный лед и энергично перемешивали до превращения раствора в белую суспензию. Суспензию подвергали фильтрованию с отсасыванием и остаток на фильтре промывали ледяной водой и сушили с получением белого аморфного твердого вещества, т.е. 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоновой кислоты (0,75 г, 75%).
6) 3,4-Дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоновую кислоту (0,585 г, 3 ммоль) подвергали взаимодействию с 30 мл тионилхлорида и 2 каплями ДМФА в течение 5 часов и выпаривали растворитель при пониженном давлении. Добавляли небольшое количество толуола и выпаривали толуол при пониженном давлении с получением 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбонилхлорида (0,545 г, 85%).
7) 2-Аминоэтил-[1,2-бензизоселеназол-3(2H)-он]гидрохлорид (0,611 г, 2,2 ммоль) подвергали взаимодействию с 1 мл триэтиламина в дихлорметане в течение 1 часа и добавляли раствор 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбонилхлорида (0,363 г, 1,7 ммоль) в дихлорметане при охлаждении на ледяной бане. После завершения добавления смесь нагревали до комнатной температуры, оставляли для протекания реакции в течение ночи и концентрировали при пониженном давлении для выпаривания растворителя. Твердое вещество промывали дихлорметаном, водой и ацетоном и сушили с получением целевого соединения в виде желтого твердого вещества (441 мг, 62%). 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,84 (s, 1H), 8,67 (s, 1H), 8,04 (d, J=7,9 Гц, 1H), 7,82 (d, J=7,5 Гц, 1H), 7,59 (t, J=7,2 Гц, 1H), 7,40 (t, J=7,2 Гц, 1H), 3,94 (s, 2Н), 3,86 (s, 3Н), 3,66-3,56 (m, 2Н). 13С-ЯМР (101 МГц, ДМСО-d6) δ 166,55, 159,92, 139,62, 139,11, 134,44, 131,37, 130,19, 128,43, 127,73, 127,27, 125,85, 125,64, 42,75, 39,41, 36,13.
Пример 2
3,4-Дигидро-3-метил-4-оксо-N-(3(2H)-оксо-5-фтор-1,2-бензизоселеназол-2-ил)-этил-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоксамид (№2)
1) В лабораторный стакан добавляли 7,75 г 2-амино-5-фторбензойной кислоты, а затем жидкую смесь из 15 мл концентрированной хлористоводородной кислоты и 15 мл воды. Смесь охлаждали на ледяной бане для поддержания температуры реакции ниже 5°С. По каплям медленно добавляли раствор NaNO2 (4,35 г, растворенных в 10 мл воды). После завершения добавления по каплям смесь оставляли для протекания реакции в течение дополнительных 2 часов с получением раствора соли диазония для последующего применения. К 30 мл воды добавляли 4 г NaOH и растворяли при перемешивании при 50°С, а затем небольшими порциями добавляли 4,35 г гидросульфита натрия. После того, как смесь стала прозрачной, добавляли 4 г порошка селена. После завершения добавления смесь оставляли для протекания реакции в течение дополнительных 2-3 ч с получением раствора Na2Se2 для последующего применения. По каплям при перемешивании добавляли раствор соли диазония к раствору Na2Se2, при этом температуру реакции поддерживали ниже 5°С. После завершения добавления по каплям раствор оставляли для протекания реакции в течение дополнительных 2 часов, при этом раствор оставался щелочным. После завершения реакции смесь подкисляли хлористоводородной кислотой и фильтровали с получением твердого вещества и твердое вещество промывали водой, а затем сушили в сушильном шкафу с получением 5,5''-дифтор-2,2''-диселандиилдибензойной кислоты (6 г, 55%).
2) К 5,5'-дифтор-2,2''-диселандиилдибензойной кислоте (4,36 г, 10 ммоль) добавляли тионилхлорид (20 мл) и 1-2 капли ДМФА. Смесь перемешивали при температуре обратной конденсации в течение 5 ч и концентрировали при пониженном давлении для выпаривания тионилхлорида. Остаток перекристаллизовывали из петролейного эфира (100 мл) и смесь фильтровали через слой целита. Фильтрат хранили в холодильнике с получением желтого игольчатого кристалла, т.е. 5-фтор-2-селенохлорбензоилхлорида (3,25 г, 60%).
3) Boc-этилендиамин (1,60 г, 10 ммоль) растворяли в дихлорметане (10 мл) и добавляли 1,39 мл триэтиламина. К смеси медленно по каплям добавляли раствор 5-фтор-2-селенохлорбензоилхлорида (2,72 г, 10 ммоль) в дихлорметане (10 мл) при охлаждении на ледяной бане. Смесь нагревали до комнатной температуры, оставляли для протекания реакции в течение 3 ч и концентрировали при пониженном давлении для выпаривания растворителя. Добавляли диэтиловый эфир, а затем перемешивали и получали осадок в виде белого твердого вещества. Твердое вещество последовательно промывали водой и петролейным эфиром, сушили и очищали путем колоночной хроматографии (петролейный эфир : дихлорметан : метанол = 200:200:1) с получением желтого твердого вещества, т.е. трет-бутил-(3(2H)-оксо-5-фтор-1,2-бензизоселеназол-2-ил)-этил-1-карбамата (1,22 г, 34%).
4) В 25 мл одногорлую колбу добавляли трет-бутил-(3(2H)-оксо-5-фтор-1,2-бензизоселеназол-2-ил)-этил-1-карбамат (0,359 г, 1,00 ммоль), а затем раствор 2 н. хлористоводородной кислоты в этилацетате (5 мл). Раствор оставляли для протекания реакции в течение 1 ч, при этом раствор превращался из прозрачного в мутный, и образовывался белый осадок. Смесь подвергали фильтрованию с отсасыванием и остаток на фильтре промывали этилацетатом с получением белого твердого вещества, т.е. 2-(2-аминоэтил)-5-фтор-1,2-бензизоселеназол-3(2H)-она гидрохлорида (0,251 г, 85%).
5) ТМЗ (0,97 г, 5,0 ммоль) добавляли порциями к концентрированной серной кислоте при перемешивании при комнатной температуре. После завершения добавления добавляли по каплям 8 мл водного раствора NaNO2 (1,3 г, 18,8 ммоль) при охлаждении на ледяной бане для поддержания температуры при 0°С. После завершения добавления раствор смеси медленно перемешивали при комнатной температуре, при этом раствор превращался из коричневато-желтой жидкости в зеленую смолу и в светло-желтую смолу. Смолу вносили в измельченный лед и энергично перемешивали до превращения раствора в белую суспензию. Суспензию подвергали фильтрованию с отсасыванием и остаток на фильтре промывали ледяной водой и сушили с получением белого аморфного твердого вещества, т.е. 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоновой кислоты (0,75 г, 75%).
6) 3,4-Дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоновую кислоту (0,585 г, 3 ммоль) подвергали взаимодействию с 30 мл тионилхлорида и 2 каплями ДМФА в течение 5 часов и выпаривали растворитель при пониженном давлении. Добавляли небольшое количество толуола и выпаривали толуол при пониженном давлении с получением 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбонилхлорида (0,545 г, 85%).
7) 2-(2-Аминоэтил)-5-фтор-1,2-бензизоселеназол-3(2H)-она гидрохлорид (222 мг, 0,75 ммоль) подвергали взаимодействию с триэтиламином в дихлорметане в течение 1 ч и добавляли раствор 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбонилхлорида (107 мг, 0,5 ммоль) в дихлорметане при охлаждении на ледяной бане. После завершения добавления смесь нагревали до комнатной температуры, оставляли для протекания реакции в течение ночи и концентрировали при пониженном давлении для выпаривания растворителя. Твердое вещество промывали дихлорметаном, водой и ацетоном и сушили с получением целевого соединения в виде белого твердого вещества (50 мг, 23%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,84 (s, 1H), 8,67 (s, 1H), 8,04 (dd, J=8,6, 4,9 Гц, 1H), 7,63-7,44 (m, 2Н), 3,94 (t, J=5,4 Гц, 2Н), 3,86 (s, 3Н), 3,66-3,55 (т, 2Н). 13С-ЯМР (101 МГц, ДМСО-d6) δ 165,62, 165,59, 162,25, 159,95, 159,84, 139,13, 134,61, 134,46, 130,17, 129,40, 129,33, 128,46, 127,88, 127,81, 119,63, 119,39, 113,07, 112,84, 43,01, 38,98, 36,15.
Пример 3
3,4-Дигидро-3-метил-4-оксо-N-(3(2H)-оксо-6-фтор-1,2-бензизоселеназол-2-ил)-этил-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоксамид (№3)
1) В лабораторный стакан добавляли 7,75 г 2-амино-6-фторбензойной кислоты, а затем жидкую смесь из 15 мл концентрированной хлористоводородной кислоты и 15 мл воды. Смесь охлаждали на ледяной бане для поддержания температуры реакции ниже 5°С. По каплям медленно добавляли раствор NaNO2 (4,35 г, растворенных в 10 мл воды). После завершения добавления по каплям смесь оставляли для протекания реакции в течение дополнительных 2 часов с получением раствора соли диазония для последующего применения. К 30 мл воды добавляли 4 г NaOH и растворяли при перемешивании при 50°С, а затем небольшими порциями добавляли 4,35 г гидросульфита натрия. После того, как смесь стала прозрачной, добавляли 4 г порошка селена. После завершения добавления смесь оставляли для протекания реакции в течение дополнительных 3 часов с получением раствора Na2Se2 для последующего применения. По каплям при перемешивании добавляли раствор соли диазония к раствору Na2Se2, при этом температуру реакции поддерживали ниже 5°С. После завершения добавления по каплям раствор оставляли для протекания реакции в течение дополнительных 2 часов, при этом раствор оставался щелочным. После завершения реакции смесь подкисляли хлористоводородной кислотой и фильтровали с получением твердого вещества и твердое вещество промывали водой, а затем сушили в сушильном шкафу с получением 4,4'-дифтор-2,2'-диселандиилдибензойной кислоты (5,5 г, 50%).
2) К 4,4'-дифтор-2,2''-диселандиилдибензойной кислоте (4,36 г, 10 ммоль) добавляли тионилхлорид (20 мл) и 1-2 капли ДМФА. Смесь перемешивали при температуре обратной конденсации в течение 5 ч и концентрировали при пониженном давлении для выпаривания тионилхлорида. Остаток перекристаллизовывали из петролейного эфира (100 мл) и смесь фильтровали через слой целита. Фильтрат хранили в холодильнике с получением желтого игольчатого кристалла, т.е. 4-фтор-2-селенохлорбензоилхлорида (3,00 г, 55%).
3) Boc-этилендиамин (1,60 г, 10 ммоль) растворяли в дихлорметане (10 мл) и добавляли 1,39 мл триэтиламина. К смеси медленно по каплям добавляли раствор 4-фтор-2-селенохлорбензоилхлорида (2,72 г, 10 ммоль) в дихлорметане (10 мл) при охлаждении на ледяной бане. Смесь нагревали до комнатной температуры, оставляли для протекания реакции в течение 3 ч и концентрировали при пониженном давлении для выпаривания растворителя. Добавляли диэтиловый эфир, а затем перемешивали и получали осадок в виде белого твердого вещества. Твердое вещество последовательно промывали водой и петролейным эфиром, сушили и очищали путем колоночной хроматографии (петролейный эфир : дихлорметан : метанол = 200:200:1) с получением желтого твердого вещества, т.е. трет-бутил-(3(2H)-оксо-6-фтор-1,2-бензизоселеназол-2-ил)-этил-1-карбамата (1,07 г, 30%).
4) В 25 мл одногорлую колбу добавляли трет-бутил-(3(2H)-оксо-6-фтор-1,2-бензизоселеназол-2-ил)-этил-1-карбамат (0,359 г, 1,00 ммоль), а затем раствор 2 н. хлористоводородной кислоты в этилацетате (5 мл). Раствор оставляли для протекания реакции в течение 1 ч, при этом раствор превращался из прозрачного в мутный, и образовывался белый осадок. Смесь подвергали фильтрованию с отсасыванием и остаток на фильтре промывали этилацетатом с получением белого твердого вещества, т.е. 2-(2-аминоэтил)-6-фтор-1,2-бензизоселеназол-3(2H)-она гидрохлорида (0,275 г, 93%).
5) ТМЗ (0,97 г, 5,0 ммоль) добавляли порциями к концентрированной серной кислоте при перемешивании при комнатной температуре. После завершения добавления добавляли по каплям 8 мл водного раствора NaNO2 (1,3 г, 18,8 ммоль) при охлаждении на ледяной бане для поддержания температуры при 0°С. После завершения добавления раствор смеси медленно перемешивали при комнатной температуре, при этом раствор превращался из коричневато-желтой жидкости в зеленую смолу и в светло-желтую смолу. Смолу вносили в измельченный лед и энергично перемешивали до превращения раствора в белую суспензию. Суспензию подвергали фильтрованию с отсасыванием и остаток на фильтре промывали ледяной водой и сушили с получением белого аморфного твердого вещества, т.е. 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоновой кислоты (0,75 г, 80%).
6) 3,4-Дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоновую кислоту (0,585 г, 3 ммоль) подвергали взаимодействию с 30 мл тионилхлорида и 2 каплями ДМФА в течение 5 часов и выпаривали растворитель при пониженном давлении. Добавляли небольшое количество толуола и выпаривали толуол при пониженном давлении с получением 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбонилхлорида (0,545 г, 85%).
7) 2-(2-Аминоэтил)-6-фтор-1,2-бензизоселеназол-3(2H)-она гидрохлорид (222 мг, 0,75 ммоль) подвергали взаимодействию с триэтиламином в дихлорметане в течение 1 ч и добавляли раствор 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбонилхлорида (107 мг, 0,5 ммоль) в дихлорметане при охлаждении на ледяной бане. После завершения добавления смесь нагревали до комнатной температуры, оставляли для протекания реакции в течение ночи и концентрировали при пониженном давлении для выпаривания растворителя. Твердое вещество промывали дихлорметаном, водой и ацетоном и сушили с получением целевого соединения в виде белого твердого вещества (120 мг, 55%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,84 (s, 1H), 8,68 (t, J=5,5 Гц, 1H), 7,82 (ddd, J=16,4, 8,8, 3,8 Гц, 2Н), 7,26 (td, J=8,7, 2,1 Гц, 1H), 3,93 (t, J=5,7 Гц, 2Н), 3,86 (s, 3Н), 3,64-3,53 (m, 2Н). 13С-ЯМР (101 МГц, ДМСО-d6) δ 165,71, 165,25, 162,77, 160,01, 141,75, 141,65, 139,17, 134,50, 130,18, 129,49, 129,40, 128,50, 124,55, 114,16, 113,92, 112,45, 112,18, 42,85, 38,97, 36,19.
Пример 4
3,4-Дигидро-3-метил-4-оксо-N-(3(2H)-оксо-5-хлор-1,2-бензизоселеназол-2-ил)-этил-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоксамид (№4)
1) В лабораторный стакан добавляли 8,85 г 2-амино-5-хлорбензойной кислоты, а затем жидкую смесь из 15 мл концентрированной хлористоводородной кислоты и 15 мл воды. Смесь охлаждали на ледяной бане для поддержания температуры реакции ниже 5°С. По каплям медленно добавляли раствор NaNO2 (4,35 г, растворенных в 10 мл воды). После завершения добавления по каплям смесь оставляли для протекания реакции в течение дополнительных 2 часов с получением раствора соли диазония для последующего применения. К 30 мл воды добавляли 4 г NaOH и растворяли при перемешивании при 50°С, а затем небольшими порциями добавляли 4,35 г гидросульфита натрия. После того, как смесь стала прозрачной, добавляли 4 г порошка селена. После завершения добавления смесь оставляли для протекания реакции в течение дополнительных 3 часов с получением раствора Na2Se2 для последующего применения. По каплям при перемешивании добавляли раствор соли диазония к раствору Na2Se2, при этом температуру реакции поддерживали ниже 5°С. После завершения добавления по каплям раствор оставляли для протекания реакции в течение дополнительных 2 часов, при этом раствор оставался щелочным. После завершения реакции смесь подкисляли хлористоводородной кислотой и фильтровали с получением твердого вещества и твердое вещество промывали водой, а затем сушили в сушильном шкафу с получением 5,5''-дихлор-2,2''-диселандиилдибензойной кислоты (5,98 г, 51%).
2) К 5,5''-дихлор-2,2''-диселандиилдибензойной кислоте (4,69 г, 10 ммоль) добавляли тионилхлорид (20 мл) и 1-2 капли ДМФА. Смесь перемешивали при температуре обратной конденсации в течение 5 ч и концентрировали при пониженном давлении для выпаривания тионилхлорида. Остаток перекристаллизовывали из петролейного эфира (100 мл) и смесь фильтровали через слой целита. Фильтрат хранили в холодильнике с получением желтого игольчатого кристалла, т.е. 5-хлор-2-селенохлорбензоилхлорида (3,63 г, 63%).
3) Boc-этилендиамин (1,60 г, 10 ммоль) растворяли в дихлорметане (10 мл) и добавляли 1,39 мл триэтиламина. К смеси медленно по каплям добавляли раствор 5-хлор-2-селенохлорбензоилхлорида (2,88 г, 10 ммоль) в дихлорметане (10 мл) при охлаждении на ледяной бане. Смесь нагревали до комнатной температуры, оставляли для протекания реакции в течение 3 ч и концентрировали при пониженном давлении для выпаривания растворителя. Добавляли диэтиловый эфир, а затем перемешивали и получали осадок в виде белого твердого вещества. Твердое вещество последовательно промывали водой и петролейным эфиром, сушили и очищали путем колоночной хроматографии (петролейный эфир : дихлорметан : метанол = 200:200:1) с получением желтого твердого вещества, т.е. трет-бутил-(3(2H)-оксо-5-хлор-1,2-бензизоселеназол-2-ил)-этил-1-карбамата (1,12 г, 30%).
4) В 25 мл одногорлую колбу добавляли трет-бутил-(3(2H)-оксо-5-хлор-1,2-бензизоселеназол-2-ил)-этил-1-карбамат (0,375 г, 1,00 ммоль), а затем раствор 2 н. хлористоводородной кислоты в этилацетате (5 мл). Раствор оставляли для протекания реакции в течение 1 ч, при этом раствор превращался из прозрачного в мутный, и образовывался белый осадок. Смесь подвергали фильтрованию с отсасыванием и остаток на фильтре промывали этилацетатом с получением белого твердого вещества, т.е. 2-(2-аминоэтил)-5-хлор-1,2-бензизоселеназол-3(2H)-она гидрохлорида (0,284 г, 91%).
5) ТМЗ (0,93 г, 4,8 ммоль) добавляли порциями к концентрированной серной кислоте при перемешивании при комнатной температуре. После завершения добавления добавляли по каплям 8 мл водного раствора NaNO2 (1,3 г, 18,8 ммоль) при охлаждении на ледяной бане для поддержания температуры при 0°С. После завершения добавления раствор смеси медленно перемешивали при комнатной температуре, при этом раствор превращался из коричневато-желтой жидкости в зеленую смолу и в светло-желтую смолу. Смолу вносили в измельченный лед и энергично перемешивали до превращения раствора в белую суспензию. Суспензию подвергали фильтрованию с отсасыванием и остаток на фильтре промывали ледяной водой и сушили с получением белого аморфного твердого вещества, т.е. 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоновой кислоты (0,75 г, 80%).
6) 3,4-Дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоновую кислоту (0,585 г, 3 ммоль) подвергали взаимодействию с 30 мл тионилхлорида и 2 каплями ДМФА в течение 5 часов и выпаривали растворитель при пониженном давлении. Добавляли небольшое количество толуола и выпаривали толуол при пониженном давлении с получением 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбонилхлорида (0,545 г, 85%).
7) 2-(2-Аминоэтил)-5-хлор-1,2-бензизоселеназол-3(2H)-она гидрохлорид (234 мг, 0,75 ммоль) подвергали взаимодействию с триэтиламином в дихлорметане в течение 1 часа и добавляли раствор 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбонилхлорида (147 мг, 0,5 ммоль) в дихлорметане при охлаждении на ледяной бане. После завершения добавления смесь нагревали до комнатной температуры, оставляли для протекания реакции в течение ночи и концентрировали при пониженном давлении для выпаривания растворителя. Твердое вещество промывали дихлорметаном, водой и ацетоном и сушили с получением целевого соединения в виде белого твердого вещества (147 мг, 65%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,84 (s, 1H), 8,67 (t, J=5,1 Гц, 1H), 8,03 (d, J=8,6 Гц, 1H), 7,77 (s, 1H), 7,64 (d, J=8,4 Гц, 1H), 3,94 (t, J=5,3 Гц, 2Н), 3,86 (s, 3Н), 3,60 (d, J=5,6 Гц, 2Н). 13С-ЯМР (101 МГц, ДМСО-d6) δ 165,35, 159,98, 139,14, 138,29, 134,46, 131,31, 130,89, 130,18, 129,47, 128,47, 127,77, 126,46, 42,97, 38,98, 36,16.
Пример 5
3,4-Дигидро-3-метил-4-оксо-N-(3(2H)-оксо-5-бром-1,2-бензизоселеназол-2-ил)-этил-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоксамид (№5)
1) В лабораторный стакан добавляли 10,80 г 2-амино-5-бромбензойной кислоты, а затем жидкую смесь из 15 мл концентрированной хлорбензойной кислоты и 15 мл воды. Смесь охлаждали на ледяной бане для поддержания температуры реакции ниже 5°С. По каплям медленно добавляли раствор NaNO2 (4,35 г, растворенных в 10 мл воды). После завершения добавления по каплям смесь оставляли для протекания реакции в течение дополнительных 2 часов с получением раствора соли диазония для последующего применения. К 30 мл воды добавляли 4 г NaOH и растворяли при перемешивании при 50°С, а затем небольшими порциями добавляли 4,35 г гидросульфита натрия. После того, как смесь стала прозрачной, добавляли 4 г порошка селена. После завершения добавления смесь оставляли для протекания реакции в течение дополнительных 3 часов с получением раствора Na2Se2 для последующего применения. По каплям при перемешивании добавляли раствор соли диазония к раствору Na2Se2, при этом температуру реакции поддерживали ниже 5°С. После завершения добавления по каплям раствор оставляли для протекания реакции в течение дополнительных 2 часов, при этом раствор оставался щелочным. После завершения реакции смесь подкисляли хлористоводородной кислотой и фильтровали с получением твердого вещества и твердое вещество промывали водой, а затем сушили в сушильном шкафу с получением 5,5''-дибром-2,2''-диселандиилдибензойной кислоты (8,37 г, 60%).
2) К 5,5'-дибром-2,2''-диселандиилдибензойной кислоте (5,58 г, 10 ммоль) добавляли тионилхлорид (20 мл) и 1-2 капли ДМФА. Смесь перемешивали при температуре обратной конденсации в течение 5 ч и концентрировали при пониженном давлении для выпаривания тионилхлорида. Остаток перекристаллизовывали из петролейного эфира (100 мл) и смесь фильтровали через слой целита. Фильтрат хранили в холодильнике с получением желтого игольчатого кристалла, т.е. 5-бром-2-селенохлорбензоилхлорида (4,19 г, 63%).
3) Boc-этилендиамин (1,60 г, 10 ммоль) растворяли в дихлорметане (10 мл) и добавляли 1,39 мл триэтиламина. К смеси медленно по каплям добавляли раствор 5-бром-2-селенохлорбензоилхлорида (3,33 г, 10 ммоль) в дихлорметане (10 мл) при охлаждении на ледяной бане. Смесь нагревали до комнатной температуры, оставляли для протекания реакции в течение 3 ч и концентрировали при пониженном давлении для выпаривания растворителя. Добавляли диэтиловый эфир, а затем перемешивали и получали осадок в виде белого твердого вещества. Твердое вещество последовательно промывали водой и петролейным эфиром, сушили и очищали путем колоночной хроматографии (петролейный эфир : дихлорметан : метанол = 200:200:1) с получением желтого твердого вещества, т.е. трет-бутил-(3(2H) -оксо-5-бром-1,2-бензизоселеназол-2-ил)-этил-1-карбамата (1,72 г, 41%).
4) В 25 мл одногорлую колбу добавляли трет-бутил-(3(2H)-оксо-5-бром-1,2-бензизоселеназол-2-ил)-этил-1-карбамат (0,420 г, 1,00 ммоль), а затем раствор 2 н. хлористоводородной кислоты в этилацетате (5 мл). Раствор оставляли для протекания реакции в течение 1 ч, при этом раствор превращался из прозрачного в мутный, и образовывался белый осадок. Смесь подвергали фильтрованию с отсасыванием и остаток на фильтре промывали этилацетатом с получением белого твердого вещества, т.е. 2-(2-аминоэтил)-5-бром-1,2-бензизоселеназол-3(2H)-она гидрохлорида (0,338 г, 95%).
5) ТМЗ (0,97 г, 5,0 ммоль) добавляли порциями к концентрированной серной кислоте при перемешивании при комнатной температуре. После завершения добавления добавляли по каплям 8 мл водного раствора NaNO2 (1,3 г, 18,8 ммоль) при охлаждении на ледяной бане для поддержания температуры при 0°С. После завершения добавления раствор медленно перемешивали при комнатной температуре, при этом раствор превращался из коричневато-желтой жидкости в зеленую смолу и в светло-желтую смолу. Смолу вносили в измельченный лед и энергично перемешивали до превращения раствора в белую суспензию. Суспензию подвергали фильтрованию с отсасыванием и остаток на фильтре промывали ледяной водой и сушили с получением белого аморфного твердого вещества, т.е. 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоновой кислоты (0,75 г, 80%).
6) 3,4-Дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоновую кислоту (0,585 г, 3 ммоль) подвергали взаимодействию с 30 мл тионилхлорида и 2 каплями ДМФА в течение 5 часов и выпаривали растворитель при пониженном давлении. Добавляли небольшое количество толуола и выпаривали толуол при пониженном давлении с получением 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбонилхлорида (0,545 г, 85%).
7) 2-(2-Аминоэтил)-5-бром-1,2-бензизоселеназол-3(2H)-она гидрохлорид (267 мг, 0,75 ммоль) подвергали взаимодействию с триэтиламином в дихлорметане в течение 1 часа и добавляли раствор 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбонилхлорида (107 мг, 0,5 ммоль) в дихлорметане при охлаждении на ледяной бане. После завершения добавления смесь нагревали до комнатной температуры, оставляли для протекания реакции в течение ночи и концентрировали при пониженном давлении для выпаривания растворителя. Твердое вещество промывали дихлорметаном, водой и ацетоном и сушили с получением целевого соединения в виде белого твердого вещества (151 мг, 61%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,84 (s, 1H), 8,66 (t, J=5,7 Гц, 1H), 8,12 (d, J=8,6 Гц, 1H), 7,92-7,84 (m, 1Н), 7,74 (dd, J=8,5 Гц, 1H), 3,92 (t, J=5,8 Гц, 2Н), 3,86 (s, 3Н), 3,59 (q, J=5,8 Гц, 2Н). 13С-ЯМР(101 МГц, ДМСО-d6) δ 165,18, 159,93, 139,25, 139,13, 134,45, 130,21, 130,16, 129,25, 129,06, 128,57, 128,45, 118,74, 42,76, 39,05, 36,16.
Пример 6
3,4-Дигидро-3-метил-4-оксо-N-(3(2H)-оксо-5-метил-1,2-бензизоселеназол-2-ил)-этил-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоксамид (№6)
1) В лабораторный стакан добавляли 7,75 г 2-амино-5-метилбензойной кислоты, а затем жидкую смесь из 15 мл концентрированной хлористоводородной кислоты и 15 мл воды. Смесь охлаждали на ледяной бане для поддержания температуры реакции ниже 5°С. По каплям медленно добавляли раствор NaNO2 (4,35 г, растворенных в 10 мл воды). После завершения добавления по каплям смесь оставляли для протекания реакции в течение дополнительных 2 часов с получением раствора соли диазония для последующего применения. К 30 мл воды добавляли 4 г NaOH и растворяли при перемешивании при 50°С, а затем небольшими порциями добавляли 4,35 г гидросульфита натрия. После того, как смесь стала прозрачной, добавляли 4 г порошка селена. После завершения добавления смесь оставляли для протекания реакции в течение дополнительных 3 часов с получением раствора Na2Se2 для последующего применения. По каплям при перемешивании добавляли раствор соли диазония к раствору Na2Se2, при этом температуру реакции поддерживали ниже 5°С. После завершения добавления по каплям раствор оставляли для протекания реакции в течение дополнительных 2 часов, при этом раствор оставался щелочным. После завершения реакции смесь подкисляли хлористоводородной кислотой и фильтровали с получением твердого вещества и твердое вещество промывали водой, а затем сушили в сушильном шкафу с получением 5,5''-диметил-2,2''-диселандиилдибензойной кислоты (5,88 г, 54%).
2) К 5,5'-диметил-2,2''-диселандиилдибензойной кислоте (4,28 г, 10 ммоль) добавляли тионилхлорид (20 мл) и 1-2 капли ДМФА. Смесь перемешивали при температуре обратной конденсации в течение 5 ч и концентрировали при пониженном давлении для выпаривания тионилхлорида. Остаток перекристаллизовывали из петролейного эфира (100 мл) и смесь фильтровали через слой целита. Фильтрат хранили в холодильнике с получением желтого игольчатого кристалла, т.е. 5-метил-2-селенохлорбензоилхлорида (2,84 г, 53%).
3) Вос-этилендиамин (1,60 г, 10 ммоль) растворяли в дихлорметане (10 мл) и добавляли 1,39 мл триэтиламина. К смеси медленно по каплям добавляли раствор 5-метил-2-селенохлорбензоилхлорида (2,68 г, 10 ммоль) в дихлорметане (10 мл) при охлаждении на ледяной бане. Смесь нагревали до комнатной температуры, оставляли для протекания реакции в течение 3 ч и концентрировали при пониженном давлении для выпаривания растворителя. Добавляли диэтиловый эфир, а затем перемешивали и получали осадок в виде белого твердого вещества. Твердое вещество последовательно промывали водой и петролейным эфиром, сушили и очищали путем колоночной хроматографии (петролейный эфир : дихлорметан : метанол = 200:200:1) с получением желтого твердого вещества, т.е. трет-бутил-(3(2H)-оксо-5-метил-1,2-бензизоселеназол-2-ил)-этил-1-карбамата (1,31 г, 37%).
4) В 25 мл одногорлую колбу добавляли трет-бутил-(3(2H)-оксо-5-метил-1,2-бензизоселеназол-2-ил)-этил-1-карбамат (0,355 г, 1,00 ммоль), а затем раствор 2 н. хлористоводородной кислоты в этилацетате (5 мл). Раствор оставляли для протекания реакции в течение 1 ч, при этом раствор превращался из прозрачного в мутный, и образовывался белый осадок. Смесь подвергали фильтрованию с отсасыванием и остаток на фильтре промывали этилацетатом с получением белого твердого вещества, т.е. 2-(2-аминоэтил)-5-метил-1,2-бензизоселеназол-3(2H)-она гидрохлорида (0,262 г, 90%).
5) ТМЗ (0,97 г, 5,0 ммоль) добавляли порциями к концентрированной серной кислоте при перемешивании при комнатной температуре. После завершения добавления добавляли по каплям 8 мл водного раствора NaNCO2 (1,3 г, 18,8 ммоль) при охлаждении на ледяной бане для поддержания температуры при 0°С. После завершения добавления раствор медленно перемешивали при комнатной температуре, при этом раствор превращался из коричневато-желтой жидкости в зеленую смолу и в светло-желтую смолу. Смолу вносили в измельченный лед и энергично перемешивали до превращения раствора в белую суспензию. Суспензию подвергали фильтрованию с отсасыванием и остаток на фильтре промывали ледяной водой и сушили с получением белого аморфного твердого вещества, т.е. 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоновой кислоты (0,75 г, 75%).
6) 3,4-Дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоновую кислоту (0,585 г, 3 ммоль) подвергали взаимодействию с 30 мл тионилхлорида и 2 каплями ДМФА в течение 5 часов и выпаривали растворитель при пониженном давлении. Добавляли небольшое количество толуола и выпаривали толуол при пониженном давлении с получением 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбонилхлорида (0,545 г, 85%).
7) 2-(2-Аминоэтил)-5-метил-1,2-бензизоселеназол-3(2H)-она гидрохлорид (231 мг, 0,75 ммоль) подвергали взаимодействию с триэтиламином в дихлорметане в течение 1 ч и добавляли раствор 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбонилхлорида (107 мг, 0,5 ммоль) в дихлорметане при охлаждении на ледяной бане. После завершения добавления смесь нагревали до комнатной температуры, оставляли для протекания реакции в течение ночи и концентрировали при пониженном давлении для выпаривания растворителя. Твердое вещество промывали дихлорметаном, водой и ацетоном и сушили с получением целевого соединения в виде белого твердого вещества (147 мг, 68%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,84 (s, 1H), 8,66 (t, J=5,4 Гц, 1H), 7,88 (d, J=8,2 Гц, 1Н), 7,64 (s, 1H), 7,42 (d, J=8,2 Гц, 1H), 3,92 (t, J=6,1 Гц, 2H), 3,86 (s, 3H), 3,59 (q, J=6,1 Гц, 2H), 2,39 (s, 3Н). 13С-ЯМР (101 МГц, ДМСО-d6) δ 166,56, 159,93, 139,14, 136,28, 135,22, 134,46, 132,69, 130,18, 128,46, 127,73, 127,32, 125,56, 42,80, 39,22, 36,16, 20,47.
Пример 7
3,4-Дигидро-3-метил-4-оксо-N-(3(2H)-оксо-5-метокси-1,2-бензизоселеназол-2-ил)-этил-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоксамид (№7)
1) В лабораторный стакан добавляли 8,35 г 2-амино-5-метоксибензойной кислоты, а затем жидкую смесь из 15 мл концентрированной хлористоводородной кислоты и 15 мл воды. Смесь охлаждали на ледяной бане для поддержания температуры реакции ниже 5°С. По каплям медленно добавляли раствор NaNCO2 (4,35 г, растворенных в 10 мл воды). После завершения добавления по каплям смесь оставляли для протекания реакции в течение дополнительных 2 часов с получением раствора соли диазония для последующего применения. К 30 мл воды добавляли 4 г NaOH и растворяли при перемешивании при 50°С, а затем небольшими порциями добавляли 4,35 г гидросульфита натрия. После того, как смесь стала прозрачной, добавляли 4 г порошка селена. После завершения добавления смесь оставляли для протекания реакции в течение дополнительных 3 часов с получением раствора Na2Se2 для последующего применения. По каплям при перемешивании добавляли раствор соли диазония к раствору Na2Se2, при этом температуру реакции поддерживали ниже 5°С. После завершения добавления по каплям раствор оставляли для протекания реакции в течение дополнительных 2 часов, при этом раствор оставался щелочным. После завершения реакции смесь подкисляли хлористоводородной кислотой и фильтровали с получением твердого вещества и твердое вещество промывали водой, а затем сушили в сушильном шкафу с получением 5,5''-диметокси-2,2''-диселандиилдибензойной кислоты (5,17 г, 45%).
2) К 5,5'-диметокси-2,2''-диселандиилдибензойной кислоте (4,60 г, 10 ммоль) добавляли тионилхлорид (20 мл) и 1-2 капли ДМФА. Смесь перемешивали при температуре обратной конденсации в течение 5 ч и концентрировали при пониженном давлении для выпаривания тионилхлорида. Остаток перекристаллизовывали из петролейного эфира (100 мл) и смесь фильтровали через слой целита. Фильтрат хранили в холодильнике с получением желтого игольчатого кристалла, т.е. 5-метокси-2-селенохлорбензоилхлорида (2,67 г, 47%).
3) Boc-этилендиамин (1,60 г, 10 ммоль) растворяли в дихлорметане (10 мл) и добавляли 1,39 мл триэтиламина. К смеси медленно по каплям добавляли раствор 5-метокси-2-селенохлорбензоилхлорида (2,84 г, 10 ммоль) в дихлорметане (10 мл) при охлаждении на ледяной бане. Смесь нагревали до комнатной температуры, оставляли для протекания реакции в течение 3 ч и концентрировали при пониженном давлении для выпаривания растворителя. Добавляли диэтиловый эфир, а затем перемешивали и получали осадок в виде белого твердого вещества. Твердое вещество последовательно промывали водой и петролейным эфиром, сушили и очищали путем колоночной хроматографии (петролейный эфир : дихлорметан : метанол = 200:200:1) с получением желтого твердого вещества, т.е. трет-бутил-(3(2H)-оксо-5-метокси-1,2-бензизоселеназол-2-ил)-этил-1-карбамата (1,67 г, 45%).
4) В 25 мл одногорлую колбу добавляли трет-бутил-(3(2H)-оксо-5-метокси-1,2-бензизоселеназол-2-ил)-этил-1-карбамат (0,371 г, 1,00 ммоль), а затем раствор 2 н. хлористоводородной кислоты в этилацетате (5 мл). Раствор оставляли для протекания реакции в течение 1 ч, при этом раствор превращался из прозрачного в мутный, и образовывался белый осадок. Смесь подвергали фильтрованию с отсасыванием и остаток на фильтре промывали этилацетатом с получением белого твердого вещества, т.е. 2-(2-аминоэтил)-5-метоксибензо[d][1,2]селеноазо-3(2H)-она гидрохлорида (0,271 г, 88%).
5) ТМЗ (0,97 г, 5,0 ммоль) добавляли порциями к концентрированной серной кислоте при перемешивании при комнатной температуре. После завершения добавления добавляли по каплям 8 мл водного раствора NaNO2 (1,3 г, 18,8 ммоль) при охлаждении на ледяной бане для поддержания температуры при 0°С. После завершения добавления раствор смеси медленно перемешивали при комнатной температуре, при этом раствор превращался из коричневато-желтой жидкости в зеленую смолу и в светло-желтую смолу. Смолу вносили в измельченный лед и энергично перемешивали до превращения раствора в белую суспензию. Суспензию подвергали фильтрованию с отсасыванием и остаток на фильтре промывали ледяной водой и сушили с получением белого аморфного твердого вещества, т.е. 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоновой кислоты (0,75 г, 80%).
6) 3,4-Дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоновую кислоту (0,585 г, 3 ммоль) подвергали взаимодействию с 30 мл тионилхлорида и 2 каплями ДМФА в течение 5 часов и выпаривали растворитель при пониженном давлении. Добавляли небольшое количество толуола и выпаривали толуол при пониженном давлении с получением 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбонилхлорида (0,545 г, 85%).
7) 2-(2-Аминоэтил)-5-метокси-1,2-бензизоселеназол-3(2H)-она гидрохлорид (231 мг, 0,75 ммоль) подвергали взаимодействию с триэтиламином в дихлорметане в течение 1 ч и добавляли раствор 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбонилхлорида (107 мг, 0,5 ммоль) в дихлорметане при охлаждении на ледяной бане. После завершения добавления смесь нагревали до комнатной температуры, оставляли для протекания реакции в течение ночи и концентрировали при пониженном давлении для выпаривания растворителя. Твердое вещество промывали дихлорметаном, водой и ацетоном и сушили с получением целевого соединения в виде белого твердого вещества (159 мг, 71%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,84 (s, 1H), 8,67 (t, J=5,7 Гц, 1H), 7,91 (d, J=8,8 Гц, 1H), 7,32 (d, J=2,7 Гц, 1H), 7,23 (dd, J=8,8, 2,7 Гц, 1H), 3,93 (t, J=6,0 Гц, 2Н), 3,87 (s, 3Н), 3,82 (s, 3Н), 3,60 (q, J=6,0 Гц, 2Н). 13С-ЯМР (101 МГц, ДМСО-d6) δ 166,36, 159,93, 158,14, 139,14, 134,46, 130,43, 130,19, 128,72, 128,46, 126,80, 120,46, 109,69, 55,46, 42,96, 39,23, 36,16.
Пример 8
3,4-Дигидро-3-метил-4-оксо-N-(3(2H)-оксо-6-метил-1,2-бензизоселеназол-2-ил)-этил-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоксамид (№8)
1) В лабораторный стакан добавляли 7,75 г 2-амино-6-метилбензойной кислоты, а затем жидкую смесь из 15 мл концентрированной хлористоводородной кислоты и 15 мл воды. Смесь охлаждали на ледяной бане для поддержания температуры реакции ниже 5°С. По каплям медленно добавляли раствор NaNO2 (4,35 г, растворенных в 10 мл воды). После завершения добавления по каплям смесь оставляли для протекания реакции в течение дополнительных 2 часов с получением раствора соли диазония для последующего применения. К 30 мл воды добавляли 4 г NaOH и растворяли при перемешивании при 50°С, а затем небольшими порциями добавляли 4,35 г гидросульфита натрия. После того, как смесь стала прозрачной, добавляли 4 г порошка селена. После завершения добавления по каплям смесь оставляли для протекания реакции в течение дополнительных 3 часов с получением раствора Na2Se2 для последующего применения. По каплям при перемешивании добавляли раствор соли диазония к раствору Na2Se2, при этом температуру реакции поддерживали ниже 5°С. После завершения добавления раствор оставляли для протекания реакции в течение дополнительных 2 ч, при этом раствор оставался щелочным. После завершения реакции смесь подкисляли хлористоводородной кислотой и фильтровали с получением твердого вещества и твердое вещество промывали водой, а затем сушили в сушильном шкафу с получением 4,4''-диметил-2,2''-диселандиилдибензойной кислоты (5,88 г, 55%).
2) К 4,4'-диметил-2,2''-диселандиилдибензойной кислоте (4,28 г, 10 ммоль) добавляли тионилхлорид (20 мл) и 1-2 капли ДМФА. Смесь перемешивали при температуре обратной конденсации в течение 5 ч и концентрировали при пониженном давлении для выпаривания тионилхлорида. Остаток перекристаллизовывали из петролейного эфира (100 мл) и смесь фильтровали через слой целита. Фильтрат хранили в холодильнике с получением желтого игольчатого кристалла, т.е. 4-метил-2-селенохлорбензоилхлорида (2,84 г, 53%).
3) Boc-этилендиамин (1,60 г, 10 ммоль) растворяли в дихлорметане (10 мл) и добавляли 1,39 мл триэтиламина. К смеси медленно по каплям добавляли раствор 4-метил-2-селенохлорбензоилхлорида (2,68 г, 10 ммоль) в дихлорметане (10 мл) при охлаждении на ледяной бане. Смесь нагревали до комнатной температуры, оставляли для протекания реакции в течение 3 ч и концентрировали при пониженном давлении для выпаривания растворителя. Добавляли диэтиловый эфир, а затем перемешивали и получали осадок в виде белого твердого вещества. Твердое вещество последовательно промывали водой и петролейным эфиром, сушили и очищали путем колоночной хроматографии (петролейный эфир : дихлорметан : метанол = 200:200:1) с получением желтого твердого вещества, т.е. трет-бутил-(3(2H)-оксо-6-метил-1,2-бензизоселеназол-2-ил)-этил-1-карбамата (1,85 г, 52%).
4) В 25 мл одногорлую колбу добавляли трет-бутил-(3(2H)-оксо-6-метил-1,2-бензизоселеназол-2-ил)-этил-1-карбамат (0,355 г, 1,00 ммоль), а затем раствор 2 н. хлористоводородной кислоты в этилацетате (5 мл). Раствор оставляли для протекания реакции в течение 1 ч, при этом раствор превращался из прозрачного в мутный, и образовывался белый осадок. Смесь подвергали фильтрованию с отсасыванием и остаток на фильтре промывали этилацетатом с получением белого твердого вещества, т.е. 2-(2-аминоэтил)-6-метил-1,2-бензизоселеназол-3(2H)-она гидрохлорида (0,265 г, 91%).
5) ТМЗ (0,97 г, 5,0 ммоль) добавляли порциями к концентрированной серной кислоте при перемешивании при комнатной температуре. После завершения добавления добавляли по каплям 8 мл водного раствора NaNO2 (1,3 г, 18,8 ммоль) при охлаждении на ледяной бане для поддержания температуры при 0°С. После завершения добавления раствор смеси медленно перемешивали при комнатной температуре, при этом раствор превращался из коричневато-желтой жидкости в зеленую смолу и в светло-желтую смолу. Смолу вносили в измельченный лед и энергично перемешивали до превращения раствора в белую суспензию. Суспензию подвергали фильтрованию с отсасыванием и остаток на фильтре промывали ледяной водой и сушили с получением белого аморфного твердого вещества, т.е. 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоновой кислоты (0,75 г, 80%).
6) 3,4-Дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоновую кислоту (0,585 г, 3 ммоль) подвергали взаимодействию с 30 мл тионилхлорида и 2 каплями ДМФА в течение 5 часов и выпаривали растворитель при пониженном давлении. Добавляли небольшое количество толуола и выпаривали толуол при пониженном давлении с получением 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбонилхлорида (0,545 г, 85%).
7) 2-(2-Аминоэтил)-6-метил-1,2-бензизоселеназол-3(2H)-она гидрохлорид (219 мг, 0,75 ммоль) подвергали взаимодействию с триэтиламином в дихлорметане в течение 1 часа и добавляли раствор 3,4-дигидро-3-метил-4-оксо-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбонилхлорида (107 мг, 0,5 ммоль) в дихлорметане при охлаждении на ледяной бане. После завершения добавления смесь нагревали до комнатной температуры, оставляли для протекания реакции в течение ночи и концентрировали при пониженном давлении для выпаривания растворителя. Твердое вещество промывали дихлорметаном, водой и ацетоном и сушили с получением целевого соединения в виде белого твердого вещества (153 мг, 71%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,84 (s, 1H), 8,67 (t, J=5,7 Гц, 1H), 7,84 (s, 1H), 7,70 (d, J=7,9 Гц, 1H), 7,22 (d, J=7,5 Гц, 1H), 3,92 (t, J=6,0 Гц, 2Н), 3,86 (s, 3Н), 3,59 (q, J=5,9 Гц, 2Н), 2,40 (s, 3Н). 13С-ЯМР (101 МГц, ДМСО) *5 166,55, 159,92, 141,55, 139,78, 139,13, 134,45, 130,18, 128,45, 127,05, 126,98, 125,68, 125,43, 42,70, 38,96, 36,15, 21,46.
Пример 9: Исследование противоопухолевой активности in vitro соединений согласно настоящему изобретению
Ингибирующую рост активность in vitro соединений 1-8 согласно настоящему изобретению в отношении клеток глиомы человека (U87 и LN229), клеток гепатомы человека (SMMC-7721) и клеток карциномы поджелудочной железы человека (Panc 1) исследовали при помощи метода МТТ.
Клетки U87, LN229, SMMC-7721 и Panc 1, растущие в логарифмической фазе, отдельно инокупировали в 96-луночный планшет с плотностью 5×104 клеток/мл, 180 мкл/лунка. После прикрепления клеток к стенке в каждую лунку добавляли по 20 мкл жидкого лекарственного средства, так что конечные концентрации лекарственного средства составляли 0 мкМ, 5 мкМ, 10 мкМ, 20 мкМ и 50 мкМ. Через 48 ч воздействия добавляли 5 мг/мл раствора МТТ (20 мкл/лунка). После инкубирования клеток в СО2 инкубаторе в течение 3-4 часов надосадочную жидкость осторожно удаляли. После высушивания остаточной жидкости на воздухе добавляли подкисленный изопропанол (50 мкл концентрированной хлористоводородной кислоты, растворенной в 500 мл изопропанола) в количестве 200 мл/лунка и планшет встряхивали на шейкере в течение 30 мин. После полного растворения кристаллов измеряли значения оптической плотности при помощи считывающего устройства для микропланшетов при 570 нм. Результаты представлены в таблице 2.
Жизнеспособность клеток % = (ОП обработанных лекарственным средством клеток - ОП холостой группы)/(ОП контрольной клетки - ОП холостой группы) × 100
Скорость уничтожения клеток % = 1 жизнеспособность клеток %
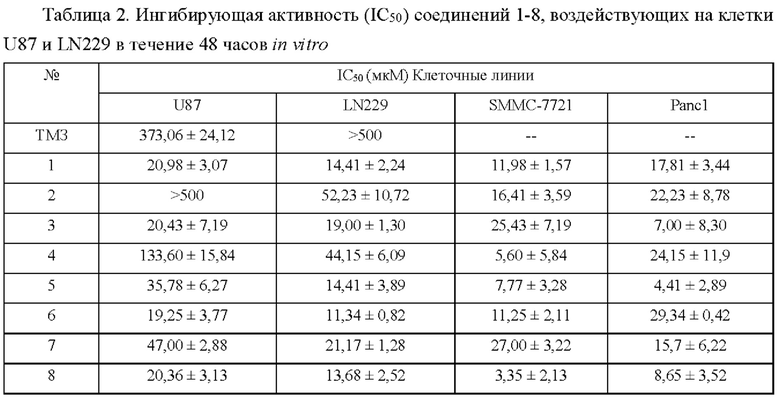
Экспериментальные результаты в таблице 2 свидетельствуют о том, что значения IC50 для соединения 1 и соединений 3-8 согласно настоящему изобретению значительно ниже, чем значения для ТМЗ в клеточной линии глиомы человека U87. В клеточной линии глиомы человека LN229 все соединения согласно настоящему изобретению проявляли хорошую ингибирующую активность; в частности, значение IC50 для соединения 6 значительно ниже, чем значение для ТМЗ, и значения IC50 для соединений 3, 5 и 8 сопоставимы со значениями для ТМЗ. В клеточной линии гепатомы человека SMMC-7721 все соединения согласно настоящему изобретению проявляли хорошую ингибирующую активность; в частности, значения IC50 для соединений 4, 5 и 8 значительно ниже, чем значения для ТМЗ, и значения IC50 для соединений 2 и 6 сопоставимы со значениями для ТМЗ. В клеточной линии карциномы поджелудочной железы человека Panc 1 все соединения согласно настоящему изобретению проявляли хорошую ингибирующую активность; в частности, значения IC50 для соединений 3, 5 и 8 значительно ниже, чем значения для ТМЗ, и значения IC50 для других соединений сопоставимы со значениями для ТМЗ.
Пример 10: Исследование противоопухолевой активности in vivo соединения 1 согласно настоящему изобретению
В данном эксперименте изучали противоопухолевую активность in vivo соединения 1 (т.е. 3,4-дигидро-3-метил-4-оксо-N-(3(2H)-оксо-бензизоселеназол-2-ил)-этил-имидазо[5,1-d]-1,2,3,5-тетразин-8-карбоксамида) (№0409 в эксперименте на животных).
В данном исследовании 4-недельных здоровых бестимусных самок мышей Balb/c массой 12-17 г случайным образом распределяли на группы, по 8 мышей в каждой контрольной группе и группе, получавшей темозоломид, и по 4 мыши в каждой группе, получавшей низкую дозу 0409, и группе, получавшей высокую дозу 0409. Суспензию клеток глиомы человека U87 получали в концентрации 2×107/мл. Каждой бестимусной мыши инокулировали 0,1 мл суспензии в правую подмышку, т.е. количество для инокуляции составляло 2×106 клеток/мышь. Частота возникновения опухоли составляла 100%. Дозирование начинали в день 12 после инокуляции и проводили в течение 11 последовательных дней. Дозы являлись следующими: холостая контрольная группа (5%о карбоксиметилцеллюлоза натрия, вводимая перорально один раз в день), группа, получавшая темозоломид (30 мг⋅кг-1, в/ж, один раз в день), группа, получавшая низкую дозу 0409 (45 мг⋅кг-1, в/ж, один раз в день), и группа, получавшая высокую дозу 0409 (90 мг⋅кг-1, в/ж, один раз в день). Растворителем для группы, получавшей темозоломид, группы, получавшей низкую дозу 0409, и группы, получавшей высокую дозу 0409, являлся буферный раствор на основе уксусной кислоты и ацетата натрия (рН 4,5). Т.е. взвешивали 18 г ацетата натрия и добавляли 9,8 мл ледяной уксусной кислоты; а затем в смесь добавляли воду для разбавления до 1000 мл. Растворитель заново готовили каждый день по мере необходимости. За состоянием мышей наблюдали ежедневно в течение 22 последовательных дней и записывали массу тела и объемы опухолей. Объемы опухолей 100 мм3 использовали в качестве критерия для определения того, возникали ли опухоли у мышей или нет. Результаты представлены в таблице 3 и на ФИГ. 2, 3 и 4.
Таблица 3. Объемы опухолей и масса тела бестимусных мышей в каждой группе в конце эксперимента и степень ингибирования опухоли
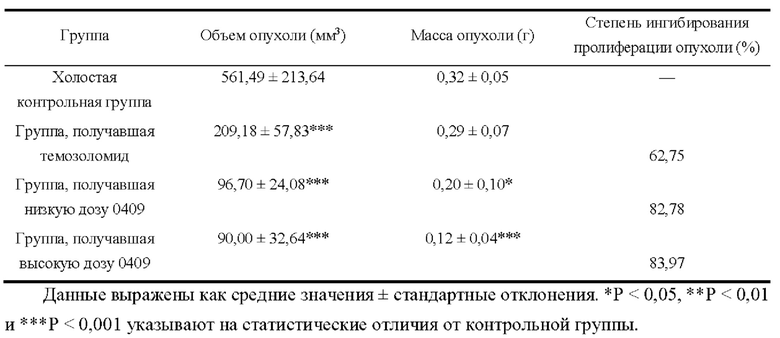
Вывод
На ФИГ. 2 представлены изменения массы тела мышей в каждой группе в день 11 после введения по сравнению с массами тела до введения. Масса тела мышей в контрольной группе, группе, получавшей темозоломид (ТМЗ), группе, получавшей низкую дозу 0409 (0409L), и группе, получавшей высокую дозу 0409 (0409Н), в день 11 после введения была в 1,01 раза, 0,91 раза, 1,02 раза и 1,02 раза больше, чем у мышей перед введением, соответственно. Масса тела группы, получавшей ТМЗ, значительно снижалась из-за токсической реакции, вызванной ТМЗ, а лекарственные средства в других группах не оказывали значительного токсического и побочного действия на мышей.
Объемы опухолей и массы опухолей бестимусных мышей анализировали статистически. Результаты представлены в таблице 3, на ФИГ. 3 и ФИГ. 4. Объемы опухолей и массы опухолей в каждой группе введения статистически отличались от результатов в контрольной группе. Во-первых, группа, получавшая ТМЗ, имела объем опухоли 209,18±57,83 мм3 и массу опухоли 0,29±0,07 г; группа, получавшая низкую дозу 0409, имела объем опухоли 96,70±24,08 мм3 и массу опухоли 0,20±0,10 г; группа, получавшая высокую дозу 0409, имела объем опухоли 90,00±32,64 мм3 и массу опухоли 0,12±0,04 г. По сравнению с темозоломидом 0409 обладает значительно улучшенной эффективностью. Во-вторых, объемы опухолей и массы опухолей в группе, получавшей низкую дозу 0409, и группе, получавшей высокую дозу 0409, последовательно снижались, и степень ингибирования опухоли в группе, получавшей низкую дозу 0409, и группе, получавшей высокую дозу 0409, составляла 82,78% и 83,97%, соответственно, что указывает на тот факт, что эффект ингибирования роста опухоли в случае 0409 в определенной степени зависел от концентрации.
Кроме того, для анализа синхронно применяли низкие дозы 0409 (20 мг⋅кг-1, в/в) и ТМЗ (10 мг⋅кг-1, в/в) для определения степени ингибирования объема опухоли. Результаты представлены в таблице 4.
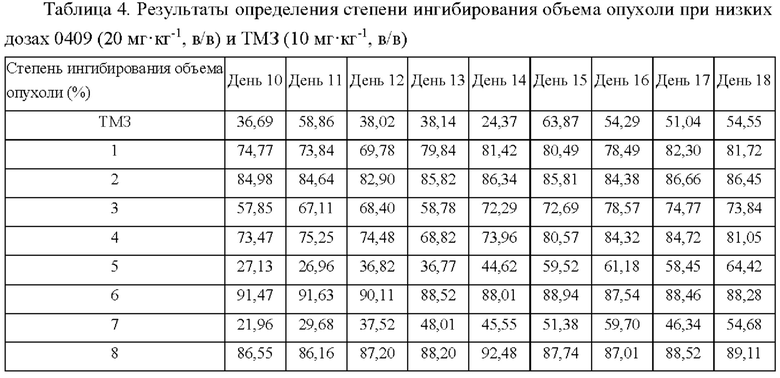
Пример 11: Фармакодинамическое исследование соединений 1, 3, 6 и 8 в подкожных моделях клеток глиомы человека U87, полученных с использованием бестимусных мышей Balb/C
При помощи тех же экспериментальной модели и рабочего процесса, что и в примере 10, одновременно исследовали ингибирующие эффекты соединения 1, соединения 3, соединения 6, соединения 8 и ТМЗ, вводимых отдельно, на рост опухоли в подкожной модели клеток глиомы человека U87, полученных с использованием бестимусных мышей Balb/c. Способ введения являлся внутрижелудочным (в/б, один раз в день × 14 дней). Результаты являлись следующими:
1) Соединение 1 (20 мг/кг), соединение 3 (20 мг/кг), соединение 6 (20 мг/кг) и соединение 8 (20 мг/кг) не приводили к потере массы при дозировках данного исследования. Это указывает на тот факт, что данный тип лекарственных средств не оказывает значительного токсического и побочного воздействия на бестимусных мышей.
Начиная с дня 4 после введения, степень ингибирования опухоли в группе, получавшей соединение 6, являлась значительно более высокой по сравнению с другими группами введения, и в день 7 после введения степень ингибирования опухоли в группах, получавших соединение 3, соединение 6 и соединение 8, составляла 80% или больше. В конце эксперимента степени ингибирования объема опухоли в группах введения, т.е. группе, получавшей ТМЗ, группе, получавшей соединение 1, группе, получавшей соединение 3, группе, получавшей соединение 6, и группе, получавшей соединение 8, составляли 14,26%, 81,42%, 88,01%, 92,48% и 82,68%, соответственно. Результаты представлены в таблице 5:
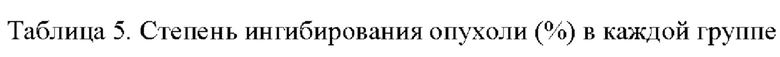
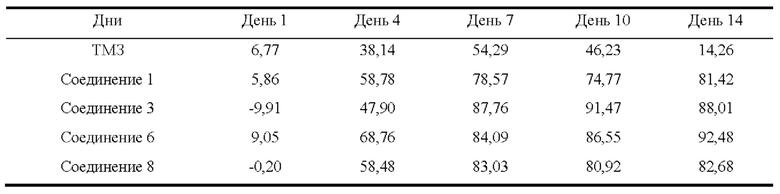
Поскольку тиоредоксинредуктаза широко и высоко экспрессируется в опухолевых тканях и может быть селективно распознана арил-конденсированным соединением изоселеназола согласно настоящему изобретению, арил-конденсированное соединение изоселеназола согласно настоящему изобретению демонстрировало хорошее направленное действие. Лекарственные средства согласно настоящему изобретению могут хорошо ингибировать рост клеток глиомы как in vitro, так и in vivo, значительно расширяя диапазон применения существующих арил-конденсированных соединений изоселеназола.
Пример 12: Исследование противоопухолевой активности in vitro соединений согласно настоящему изобретению
Ссылаясь на способ из примера 9, были выбраны две линии опухолевых клеток, клетки глиомы человека U251MG и U251TR, и темозоломид и соединения согласно настоящему изобретению были подвергнуты скринингу на противораковую активность in vitro с применением способа SRB. Значение IC50 (мкМ) каждого соединения представлено ниже в таблице 6:
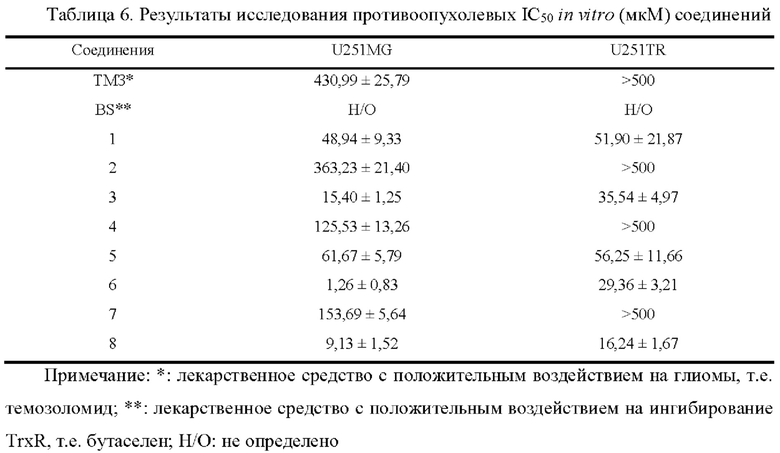
Экспериментальные результаты в таблице 6 показывают, что все из соединений 1, 3, 6 и 8 согласно настоящему изобретению демонстрировали значения ниже 5 мкМ для ингибирования TrxR, и ингибирующая активность соединения 1 в отношении TrxR среди указанных соединений являлась приблизительно такой же, как у BS, лекарственного средства с положительным воздействием на ингибирование TrxR. В клеточных линиях глиомы человека U251MG и U251TR значения IC50 соединений согласно настоящему изобретению были лучше по сравнению с ТМЗ, что указывает на тот факт, что арил-конденсированные соединения изоселеназола, содержащие тетразиновый заместитель, проявляли хорошую активность в отношении клеточных линий глиомы. Соединения 1, 3, 5, 6 и 8 согласно настоящему изобретению имели очень низкие значения IC50 в обеих клеточных линиях, в частности, для лекарственно-устойчивого штамма U251TR, демонстрируя очень сильную ингибирующую активность.
Пример 13: Определение активности соединений согласно настоящему изобретению в отношении тиоредоксинредуктазы (TrxR)
Способ определения являлся следующим:
1. Экстрагировали общий клеточный белок (т.е. тиоредоксинредуктазу) и определяли концентрацию. 30 мкг образца белка добавляли в 96-луночный планшет (3 повторные лунки на концентрацию) и объем доводили до 80 мкл при помощи 0,1 М натрий-фосфатного буфера. Образцы белка инкубировали в печи при 37°С в течение 30 мин.
2. Добавляли 20 мкл 5 мМ NADPH (к контрольной группе добавляли эквивалентный объем 0,1 мМ раствора натрий-фосфатного буфера), а затем добавляли 100 мкл 10 мМ DTNB и испытываемых соединений (IC50 обычно определяли с использованием 5 или более концентраций). Немедленно измеряли поглощение при 405 нм при помощи считывающего устройства для микропланшетов (FlexStation 3, Molecular Devices). Перед первым считыванием проводили встряхивание в течение 10 сек и проводили определение каждые 15 сек, суммарно 30 раз. В качестве показателя ферментативной активности брали максимальную скорость реакции. Эксперимент независимо повторяли три раза.
Результаты определения представлены ниже в таблице 7:
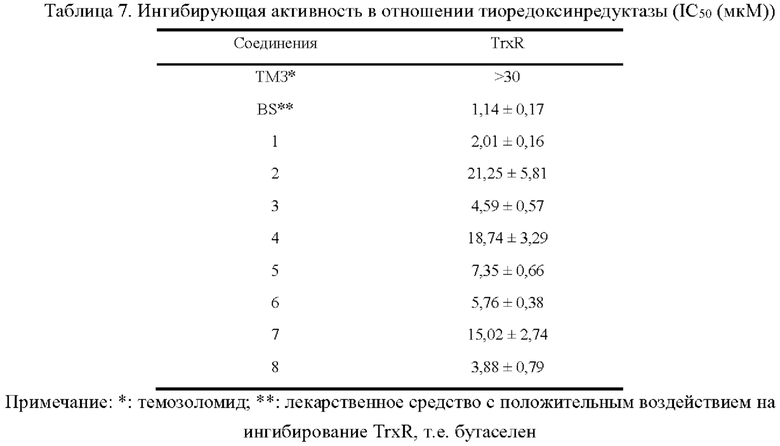
Результаты определения показывают, что соединения согласно настоящему изобретению обладают хорошей ингибирующей активностью в отношении тиоредоксинредуктазы (TrxR); в частности, значения IC50 соединений 1, 3, 5, 6 и 8 являлись того же порядка, что значения для бутаселена, лекарственного средства с положительным воздействием на ингибирование TrxR, демонстрируя превосходную ингибирующую активность в отношении TrxR. Ожидается, что арил-конденсированные соединения изоселеназола согласно настоящему изобретению можно применять в качестве ингибитора TrxR для лечения или облегчения заболевания или состояния, связанного с повышенной экспрессией или повышенной активностью TrxR.
Выше были описаны примеры согласно настоящему изобретению. Тем не менее, настоящее изобретение не ограничивается ими. Любая модификация, эквивалент, усовершенствование и т.п., выполненные без отступления от сущности и принципа настоящего изобретения, попадают под объем защиты настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЭФИР ТЕМОЗОЛОМИДА | 2005 |
|
RU2393160C2 |
| ПРОИЗВОДНОЕ ФТАЛАЗИНОНКЕТОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2011 |
|
RU2564527C2 |
| СОЕДИНЕНИЯ ДЛЯ ИНГИБИРОВАНИЯ КИНАЗЫ EGFR, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2020 |
|
RU2832925C2 |
| ИМИДАЗОПИРИМИДИНЫ КАК ИНГИБИТОРЫ EED И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2836176C2 |
| ПРОИЗВОДНОЕ ИМИДАЗОИЗОИНДОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2717577C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ИМИДАЗОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2686314C1 |
| СОЕДИНЕНИЯ ГЕТЕРОАРИЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ТКБ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2742122C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |
| ИНГИБИТОР, СОДЕРЖАЩИЙ ТРИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2800543C2 |
| Макроциклический ингибитор тирозинкиназы и его применение | 2019 |
|
RU2798231C2 |
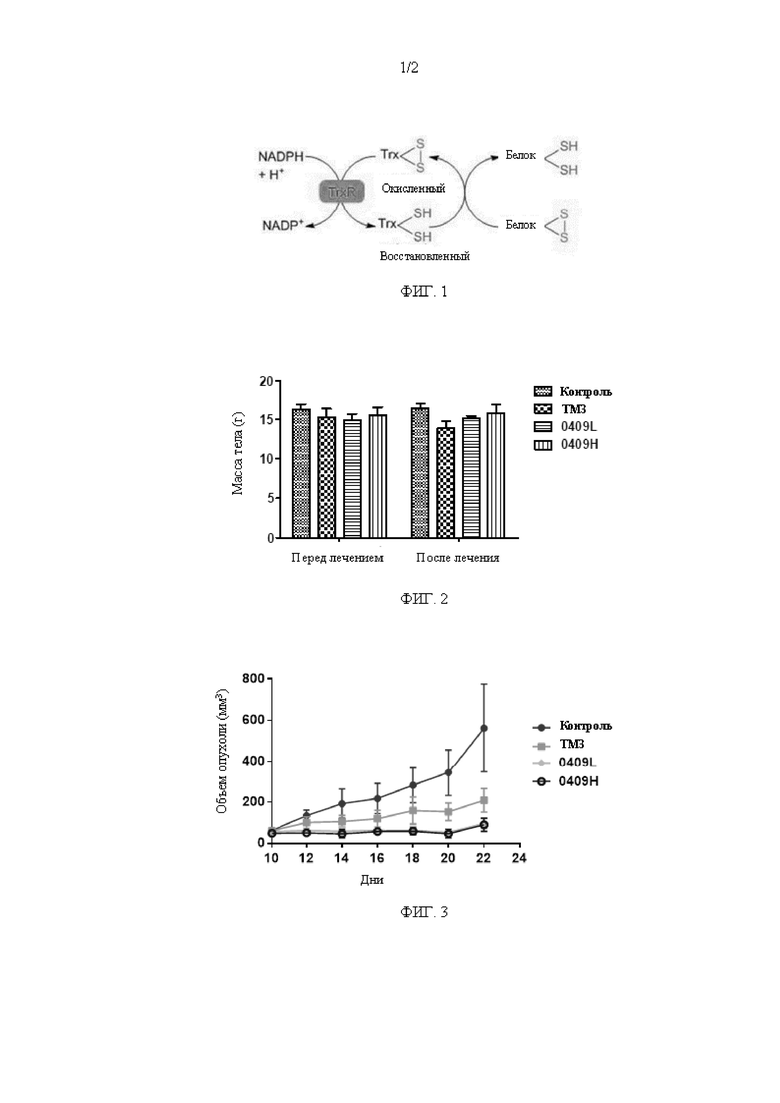
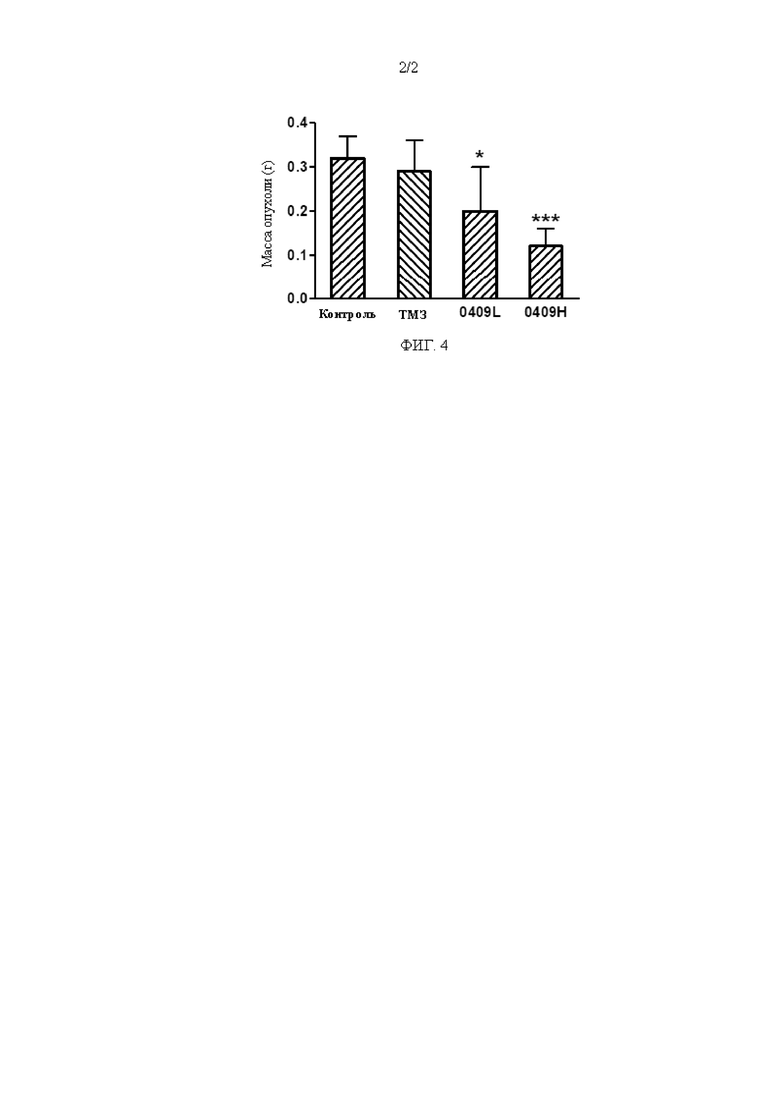
Группа изобретений относится к органической химии и включает арил-конденсированное соединение изоселеназола формулы (I), способ его получения и применение, фармацевтическую композицию, его содержащую, и способ лечения. В формуле (I) A1, A2, A3 и A4 представляют собой CH, R1 представляет собой C1-6 алкилен, R выбран из водорода, галогена, C1-6 алкила или C1-6 алкокси. Технический результат – соединение формулы (I), обладающее ингибирующей активностью в отношении тиоредоксинредуктазы (TrxR). 5 н. и 16 з.п. ф-лы, 4 ил., 7 табл., 13 пр.
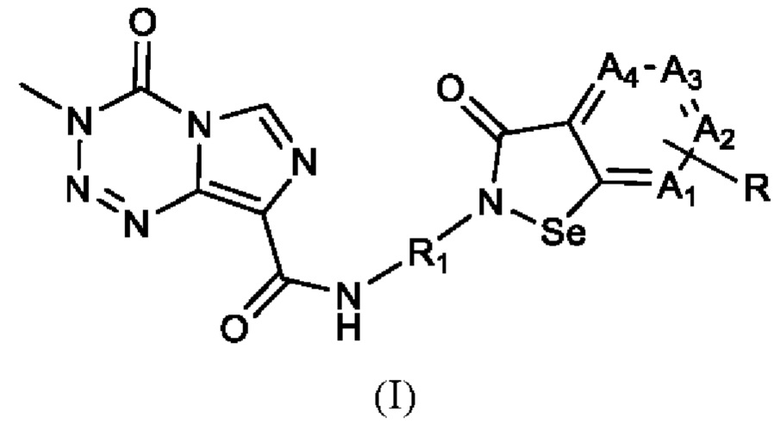
1. Арил-конденсированное соединение изоселеназола, содержащее тетразиновый заместитель, представленное формулой (I), или его фармацевтически приемлемая соль:
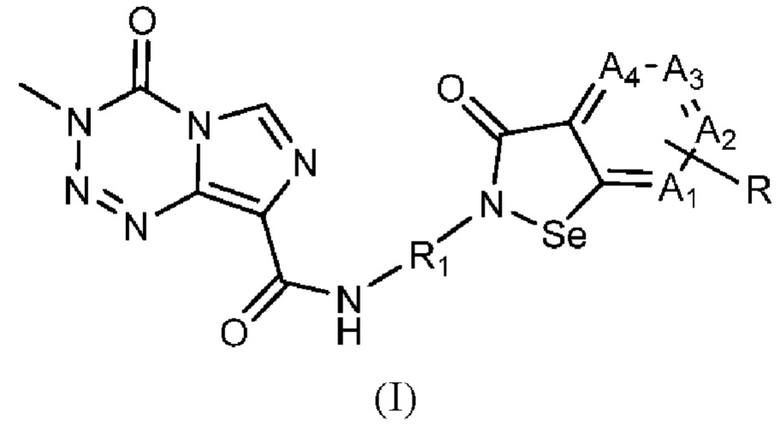
где A1, А2, А3 и А4 представляют собой СН;
R1 выбран из C1-6 алкилена;
R выбран из водорода, галогена, C1-6 алкила или C1-6 алкокси.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где R выбран из водорода, фтора, хлора, брома, йода, метила, этила, н-пропила, изопропила, н-бутила, изобутила, трет-бутила, н-пентила, изопентила, метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, трет-бутокси, н-пентокси и изопентокси.
3. Соединение или его фармацевтически приемлемая соль по п. 1, где арил-конденсированное соединение изоселеназола, содержащее тетразиновый заместитель, выбрано из следующих соединений:
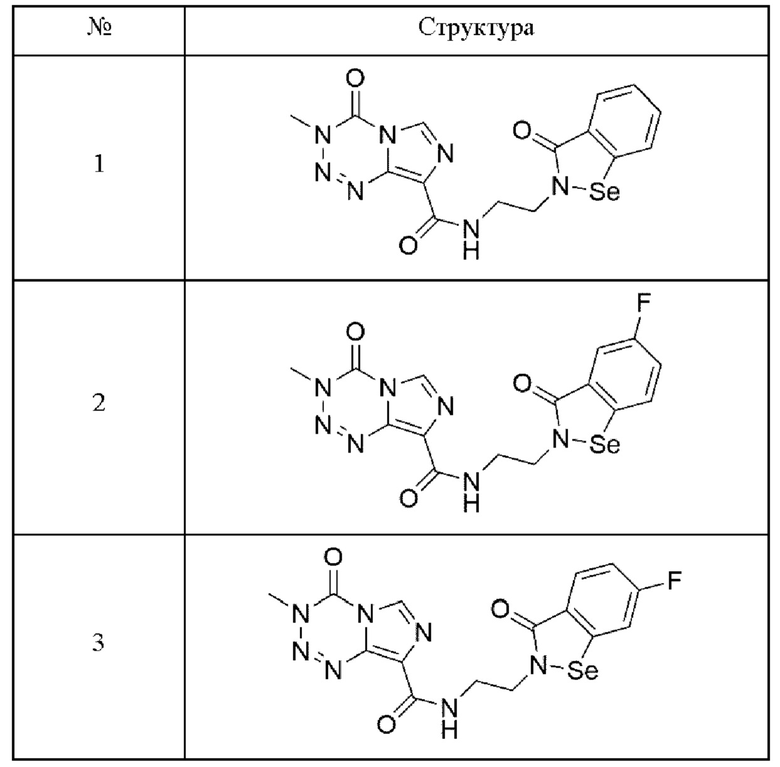
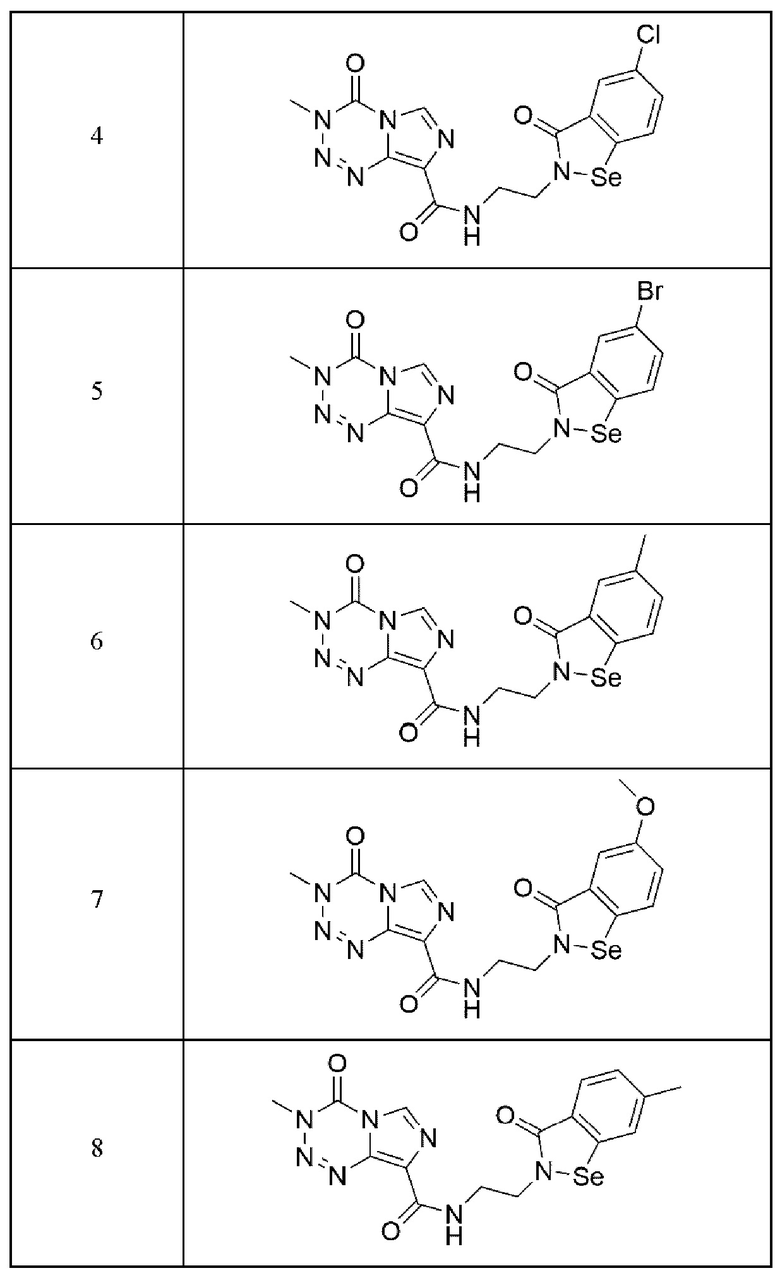
4. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-3, где фармацевтически приемлемая соль включает: соль присоединения кислоты, полученную из соединения и неорганической или органической кислоты; где неорганическая кислота выбрана по меньшей мере из одной из хлористоводородной кислоты, фтористоводородной кислоты, бромистоводородной кислоты, йодистоводородной кислоты, перхлорной кислоты, серной кислоты, пиросерной кислоты, фосфорной кислоты и азотной кислоты; органическая кислота выбрана по меньшей мере из одной из муравьиной кислоты, уксусной кислоты, ацетоуксусной кислоты, пировиноградной кислоты, трифторуксусной кислоты, пропионовой кислоты, масляной кислоты, капроновой кислоты, гептановой кислоты, ундекановой кислоты, лауриновой кислоты, бензойной кислоты, салициловой кислоты, 2-(4-гидроксибензоил)бензойной кислоты, камфорной кислоты, коричной кислоты, циклопентанпропионовой кислоты, диглюконовой кислоты, 3-гидрокси-2-нафтовой кислоты, никотиновой кислоты, памовой кислоты, пектиновой кислоты, надсерной кислоты, 3-фенилпропионовой кислоты, пикриновой кислоты, пивалевой кислоты, 2-гидроксиэтансульфоновой кислоты, итаконовой кислоты, сульфаминовой кислоты, трифторметансульфоновой кислоты, додецилсульфоновой кислоты, этансульфоновой кислоты, бензолсульфоновой кислоты, п-толуолсульфоновой кислоты, метансульфоновой кислоты, 2-нафталинсульфоновой кислоты, нафталиндисульфоновой кислоты, камфорсульфоновой кислоты, лимонной кислоты, винной кислоты, стеариновой кислоты, молочной кислоты, щавелевой кислоты, малоновой кислоты, янтарной кислоты, яблочной кислоты, адипиновой кислоты, альгиновой кислоты, малеиновой кислоты, фумаровой кислоты, D-глюконовой кислоты, миндальной кислоты, аскорбиновой кислоты, глюкогептоновой кислоты, глицерофосфорной кислоты, аспарагиновой кислоты, сульфосалициловой кислоты, гемисерной кислоты и тиоциановой кислоты;
альтернативно фармацевтически приемлемая соль представляет собой соль щелочного металла, соль щелочноземельного металла или аммонийную соль соединения, или соль, полученную из соединения и органического основания, обеспечивающего физиологически приемлемые катионы.
5. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-4, где указанная соль представляет собой соль, полученную из соединения и по меньшей мере одного из следующих соединений: ион натрия, ион калия, ион кальция, ион магния, морфолин, пиперидин, триэтиламин, трипропиламин, трибутиламин, диизопропиламин, диизопропилэтилендиамин, пиридин, диметиламин, диэтиламин, N-метилглюкамин, диметилглюкамин, этилглюкамин, лизин, дициклогексиламин, 1,6-гександиамин, этаноламин, глюкозамин, меглюмин, саркозин, серинол, тригидроксиметиламинометан, аминопропандиол и 1-амино-2,3,4-бутантриол.
6. Способ получения соединения по любому из пп. 1-5, включающий следующие стадии:
1) диазотирование аминогруппы в соединении А с последующим взаимодействием с Na2Se2 с получением соединения В:
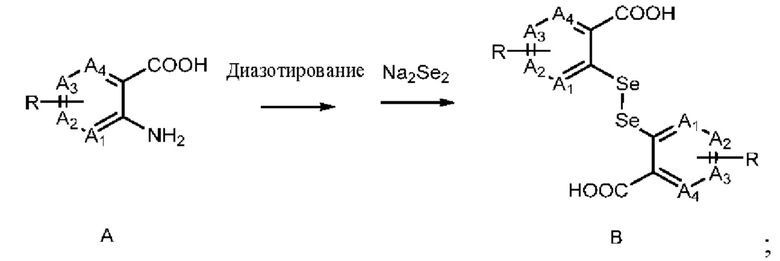
где A1, А2, А3, А4, R и R1 имеют определения, описанные в любом из пп. 1-5;
2) взаимодействие соединения В с тионилхлоридом с получением соединения С:
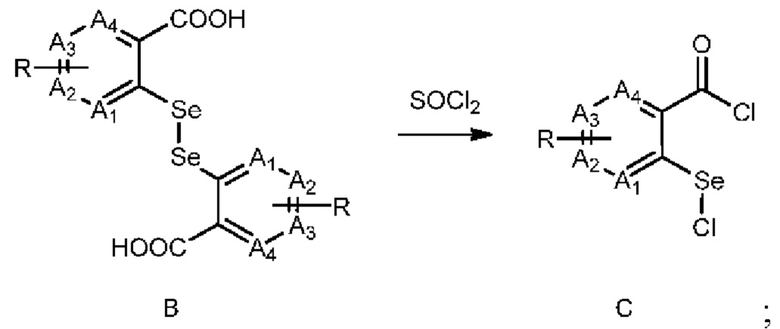
3) взаимодействие соединения С с H2N-R1-NHBoc с получением соединения D:
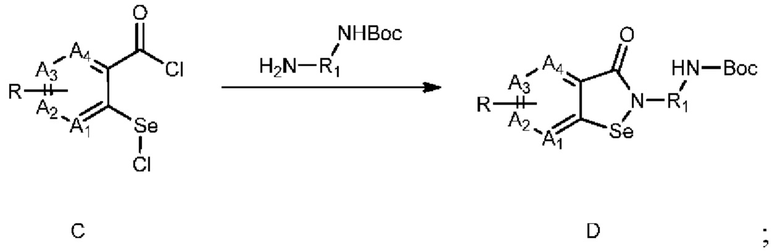
где Вос представляет собой трет-бутилоксикарбонил;
4) снятие защиты с Вос-защищенной группы соединения D в присутствии кислоты с получением соединения Е:
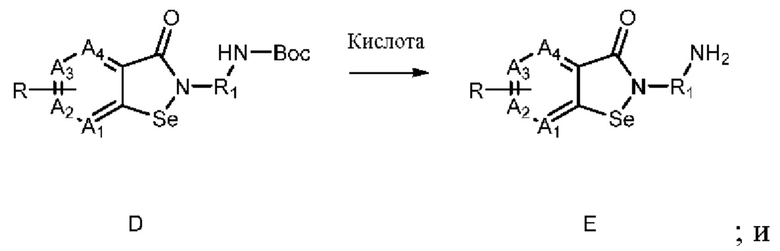
5) взаимодействие соединения Е с соединением F с получением соединения, представленного формулой (I):
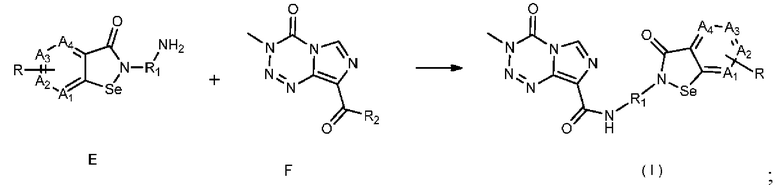
где в соединении F R2 выбран из хлора, брома, гидрокси и -OR3, где R3 выбран из С1-6 алкила, сукцинимидила, трет-бутилоксикарбонила, метилсульфонила, п-нитробензолсульфонила, п-толуолсульфонила и изобутилоксикарбонила.
7. Способ получения по п. 6, где
Na2Se2 на стадии 1) получают из элементарного селена и гидросульфита натрия;
стадию 2) проводят в присутствии каталитического количества ДМФА; кислота на стадии 4) представляет собой хлористоводородную кислоту; стадию 4) проводят в органическом растворителе.
8. Способ получения по п. 6 или 7, где указанное соединение F является коммерчески доступным или может быть получено с применением обычного способа в данной области техники, или получено при помощи следующих стадий:
а) взаимодействие соединения ТМЗ (т.е. темозоломида) с серной кислотой и нитритом натрия с получением соединения F-1:
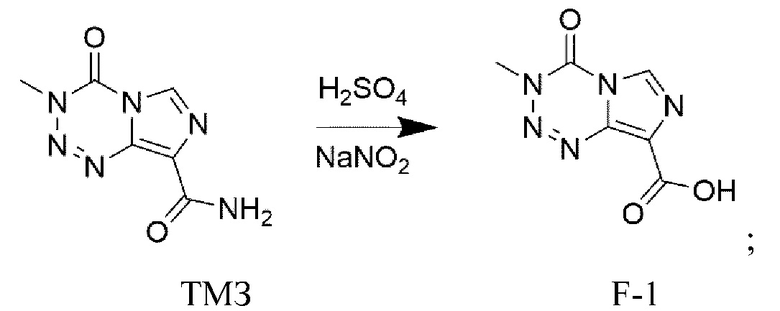
и необязательная стадия b): взаимодействие соединения F-1 с тионилхлоридом или дибромсульфоксидом в присутствии каталитического количества ДМФА с получением соединения F-2:
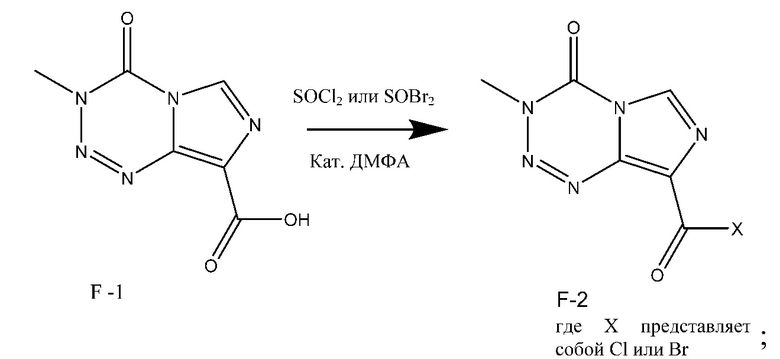
или необязательная стадия с): дальнейшее взаимодействие соединения F-1 с получением соединения F, в котором R2 представляет собой -OR3, где R3 представляет собой C1-6 алкил, сукцинимидил, трет-бутоксикарбонил, метансульфонил, п-нитробензолсульфонил, п-толуолсульфонил или изобутилоксикарбонил;
стадия с) выбрана из стадии с-1), стадии с-2), стадии с-3), стадии с-4) и стадии с-5), представленных ниже;
стадия с-1): взаимодействие соединения F-1 с R4OH под действием кислоты с получением соединения F-3:
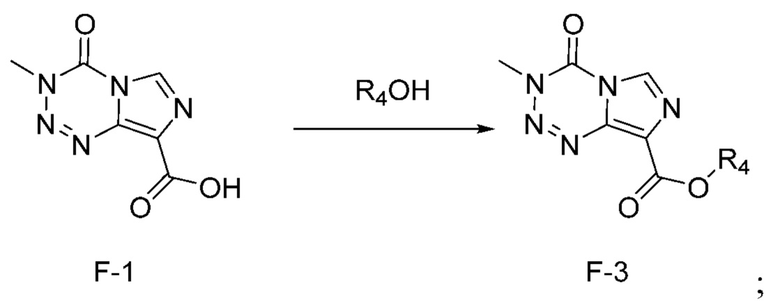
где R4 представляет собой С1-6 алкил;
стадия с-2): взаимодействие соединения F-1 с R5Cl под действием основания с получением соединения F-4:
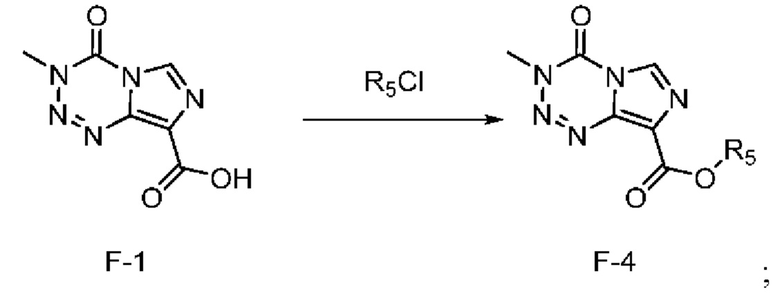
где R5 представляет собой метилсульфонил, n-нитробензолсульфонил или n-толуолсульфонил;
стадия с-3): взаимодействие соединения F-1 с N-гидроксисукцинимидом с получением соединения F-5:
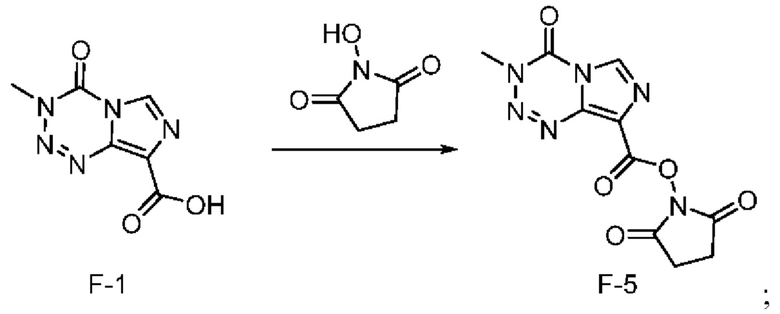
стадия с-4): взаимодействие соединения F-1 с ди-трет-бутилдикарбонатом с получением соединения F-6
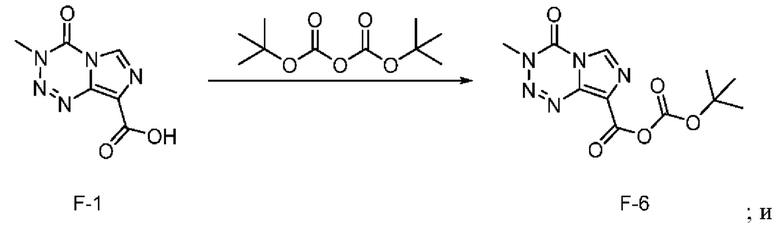
стадия с-5): взаимодействие соединения F-1 с изобутилхлорформиатом с получением соединения F-7:
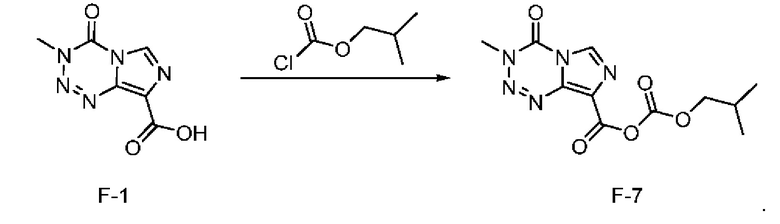
9. Фармацевтическая композиция для ингибирования активности тиоредоксинредуктазы (TrxR), где фармацевтическая композиция содержит соединение или его фармацевтически приемлемую соль по любому из пп. 1-5 в эффективном количестве и фармацевтически приемлемый носитель.
10. Фармацевтическая композиция по п. 9, где указанная фармацевтическая композиция подходит для энтерального, местного или парентерального введения.
11. Фармацевтическая композиция по п. 10, где указанная фармацевтическая композиция подходит для следующих способов введения: пероральное введение, инъекция, имплантация, местное нанесение, в виде спрея или ингаляции.
12. Фармацевтическая композиция по п. 11, где указанная фармацевтическая композиция для перорального введения представляет собой любую форму из таблетки, капсулы, пилюли, жидкого состава для перорального применения, гранул, инъекции, состава для местного нанесения или порошка.
13. Фармацевтическая композиция по п. 12, где таблетка представляет собой обычную таблетку, буккальную таблетку, сублингвальную таблетку, буккальный пластырь, жевательную таблетку, диспергируемую таблетку, растворимую таблетку, шипучую таблетку, вагинальную таблетку, вагинальную шипучую таблетку, таблетку с замедленным высвобождением, таблетку с контролируемым высвобождением, таблетку с кишечнорастворимым покрытием или буккальную таблетку с немедленным высвобождением; капсула представляет собой твердую капсулу, мягкую капсулу, капсулу с замедленным высвобождением, капсулу с контролируемым высвобождением или капсулу с кишечнорастворимым покрытием; пилюля включает микропилюлю, пилюлю-пустышку или гранулу; жидкий состав для перорального применения представляет собой сироп, суспензию, раствор для перорального применения, суспензию для перорального применения, эмульсию для перорального применения, сироп, смесь, дистиллят или линимент; гранулы представляют собой гранулы суспензии, шипучие гранулы, гранулы с кишечнорастворимым покрытием, гранулы с замедленным высвобождением или гранулы с контролируемым высвобождением; инъекция представляет собой любую форму из жидкости для инъекции, стерильного порошка или стерильного блока для инъекции и концентрированного раствора для трансфузии и инъекции; состав для местного нанесения представляет собой любую форму из суппозитория, аэрозоля, порошка для ингаляции, спрея, пленки, геля, пластыря, коллоида, медицинского пластыря, гипса, мази, линимента, лосьона, агента для нанесения и желатина.
14. Фармацевтическая композиция по любому из пп. 9-13, где указанная фармацевтическая композиция представляет собой препарат включения или дисперсионный препарат.
15. Фармацевтическая композиция по любому из пп. 9-14, где соединение или его фармацевтически приемлемая соль по любому из пп. 1-5 и фармацевтически приемлемый носитель с замедленным/контролируемым высвобождением могут быть представлены в виде состава с замедленным/контролируемым высвобождением при помощи обычного способа получения составов с замедленным/контролируемым высвобождением.
16. Фармацевтическая композиция по п. 15, где соединение или его фармацевтически приемлемая соль по любому из пп. 1-5 могут быть покрыты замедлителем или могут быть микроинкапсулированы, а затем представлены в виде гранул.
17. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-5 или фармацевтической композиции по любому из пп. 9-16 для
ингибирования активности тиоредоксинредуктазы (TrxR) или получения ингибитора тиоредоксинредуктазы (TrxR);
лечения или облегчения заболевания или состояния, связанного с повышенной экспрессией или повышенной активностью TrxR, или получения лекарственного средства для лечения или облегчения заболевания или состояния, связанного с повышенной экспрессией или повышенной активностью TrxR;
или
лечения опухоли, связанной с повышенной экспрессией или повышенной активностью TrxR, или получения лекарственного средства для лечения опухоли, связанной с повышенной экспрессией или повышенной активностью TrxR.
18. Применение по п. 17, где опухоль выбрана из любой из глиомы головного мозга, рака легкого, рака печени, рака крови, рака кости, рака поджелудочной железы, рака кожи, меланомы, рака матки, рака яичника, рака прямой кишки, рака желудка, рака толстой кишки, рака молочной железы, рака матки, рака фаллопиевых труб, рака эндометрия, рака шейки матки, рака влагалища, рака вульвы, рака пищевода, рака тонкого кишечника, рака эндокринной системы, саркомы мягких тканей, рака мочеиспускательного канала, рака предстательной железы, лимфоцитомы, рака мочевого пузыря, рака почки, рака мочеточника, опухоли позвоночника, глиомы ствола головного мозга, аденомы гипофиза, рака легкого, рака печени и рака крови.
19. Применение по п. 18, где опухоль представляет собой глиому головного мозга.
20. Способ лечения опухоли, связанной с повышенной экспрессией или повышенной активностью TrxR, у субъекта, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по любому из пп. 1-5 или фармацевтической композиции по любому из пп. 9-16, причем опухоль выбрана из любой из глиомы головного мозга, рака легкого, рака печени, рака крови, рака кости, рака поджелудочной железы, рака кожи, меланомы, рака матки, рака яичника, рака прямой кишки, рака желудка, рака толстой кишки, рака молочной железы, рака матки, рака фаллопиевых труб, рака эндометрия, рака шейки матки, рака влагалища, рака вульвы, рака пищевода, рака тонкого кишечника, рака эндокринной системы, саркомы мягких тканей, рака мочеиспускательного канала, рака предстательной железы, лимфоцитомы, рака мочевого пузыря, рака почки, рака мочеточника, опухоли позвоночника, глиомы ствола головного мозга, аденомы гипофиза, рака легкого, рака печени и рака крови.
21. Способ по п. 20, где опухоль представляет собой глиому головного мозга.
| WO 2017084598 А1, 26.05.2017 | |||
| CN 0108503607 A, 07.09.2018 | |||
| CN 0102234254 A, 09.11.2011 | |||
| CN 0106977472 A, 25.07.2017 | |||
| Yan Jun et al., Design, synthesis and biological evaluation of benzoselenazole-stilbene hybrids as multi-target-directed anti-cancer agents | |||
| European journal of medicinal mhemistry, 2015, vol.95, p.220-229 | |||
| Mao Fei et al., Novel |

