Настоящее изобретение относится к новым производным статина. В частности, изобретение относится к нитропроизводным статина, содержащим их фармацевтическим композициям и их применению в качестве холестеринснижающих агентов, в качестве медикаментов, обладающих иммуносупрессивными свойствами, антиоксидантной, антитромботической и противовоспалительной активностью, действующих на эндотелиальную функцию и для лечения и/или предотвращения острых коронарных синдромов, удара, нейродегенеративных заболеваний, таких как болезнь Альцгеймера и болезнь Паркинсона, а также аутоиммунных заболеваний, таких как рассеянный склероз.
Статины, как класс соединений, включают в качестве основных компонентов ловастатин, симвастатин, правастатин, мевастатин, флувастатин, аторвастатин, розувастатин и церивастатин (ривастатин). Они имеют боковую группу, которая структурно подобна HMG-CoA (3-гидрокси-3-метилглутарил-кофермент А). Флувастатин, аторвастатин, розувастатин и церивастатин являются полностью синтетическими соединениями, содержащими в боковой цепи гептановую кислоту, остатки, являющиеся грибковыми метаболитами.
Известно, что различные статины являются ингибиторами HMG-CoA-редуктазы, фермента, который катализирует раннюю, ограничивающую скорость стадию биосинтеза холестеринина, снижает уровень триглицеридов и является индикатором увеличения уровней HDL-C (P.O.Bonetti et al., European Heart Journal (2003), 24, 225-248).
Однако также известно, что статины оказывают неблагоприятные воздействия, такие как, например, гепатопатия, возможный онкогенез, мышечные побочные эффекты и миопатию.
Объектом настоящего изобретения являются новые производные статинов, которые не только способны устранять побочные эффекты, связанные с этими соединениями, но также обладают улучшенной фармакологической активностью. Неожиданно установлено, что нитропроизводные статина обладают улучшенным профилем по сравнению с природными статинами как с точки зрения широты фармакологической активности, так и с точки зрения толерантности. Установлено, что нитропроизводные статина настоящего изобретения проявляют сильную противовоспалительную, антитромботическую и антитромбоцитарную активность и могут использоваться в дальнейшем для снижения уровней холестерина и триглицеридов, для увеличения уровней HDL-C и для лечения и/или предотвращения острых коронарных синдромов, удара, периферических сосудистых расстройств, таких как периферическая ишемия, и нарушений, связанных с дисфункцией эндотелия, таких как сосудистые осложнения при диабете и атеросклерозе. Они также могут использоваться для лечения нейродегенеративных и аутоиммунных заболеваний, таких как болезнь Альцгеймера и болезнь Паркинсона, а также рассеянный склероз.
Объектом настоящего изобретения являются нитропроизводные статина общей формулы (I) и их фармацевтически приемлемые соли или стереоизомеры
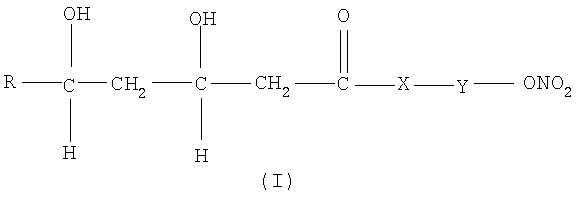
где R остаток статина, представленный ниже, Y означает подходящую связующую группу и Х как указано ниже.
В соответствии с настоящим изобретением остаток статина R в формуле (I) выбирают из группы, состоящей из правастатина, флувастатина, церивастатина, розувастатина и аторвастатина.
В основном в общей формуле (I) R, Х и Y имеют следующие значения: Х означает -О-, -S-, -NH- или -NHR1-, R1 является прямым или разветвленным алкилом с 1-10 углеродными атомами, преимущественно СН3;
R представляет
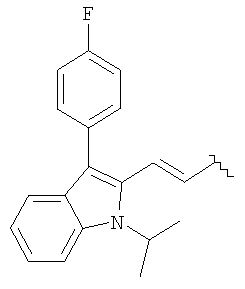
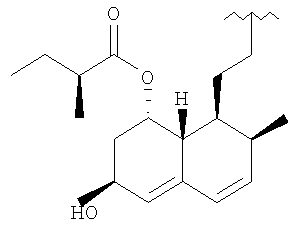
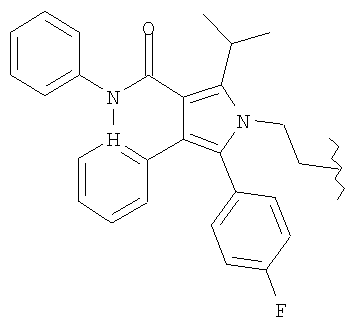
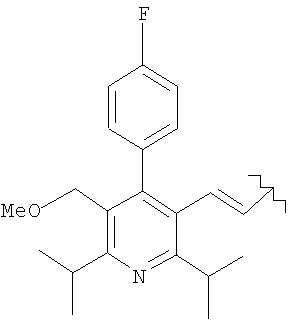
или
Y означает двухвалентный радикал, имеющий следующие значения:
a)
- прямой или разветвленный C1-C20алкилен, преимущественно C1-С10, необязательно замещенный одним или более заместителями, выбранными из группы, состоящей из: атомов галогена, гидрокси, -ONO2 или Т0, где Т0 представляет -CO(O)(C1-C10алкил)-ОNO2 или -O(С1-С10алкил)-ONO2;
- циклоалкилен с 5-7 атомами углерода в циклоалкиленовом кольце, это кольцо возможно замещено боковыми цепями Т, где Т представляет прямой или разветвленный алкил 1-10 атомами углерода, преимущественно СН3;
b)
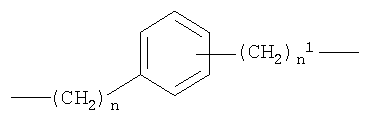
c)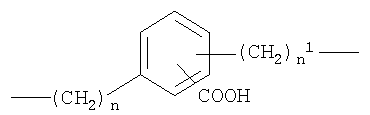
где n от 0 до 20, и n1 от 1 до 20;
d)
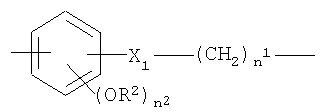
где n1 как указано выше и n2 от 0 до 2;
X1=-ОСО-или -COO- и R2 означает Н или СН3;
e)
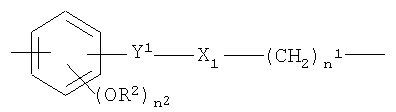
где
n1, n2, R2 и X1 как указано выше;
Y1 означает-СН2-СН2- или -СН=СН-(СН2)n 2-;
f)
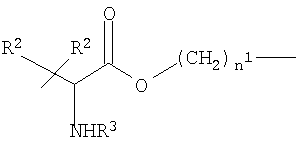
где n1 и R2 как указано выше, R3 означает Н или СОСН3;
при условии, что когда Y выбирают из двухвалентных радикалов, обозначенных под b)-f), группа -ONO2 связана с -(СН2)n1;
g)
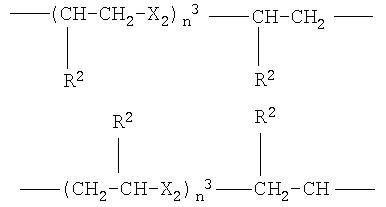
где Х2 означает -О- или -S-, n3 от 1 до 6, преимущественно от 1 до 4, R2 как указано выше;
h)
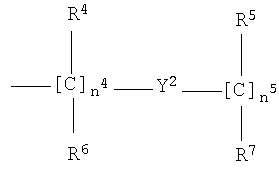
где n4 от 0 до 10;
n5 от 1 до 10;
R4, R5, R6, R7 являются одинаковыми или разными, и означают Н или прямой или разветвленный С1-С4-алкил, предпочтительно R4, R5, R6, R7 означают Н;
где группа -ONO2 связана с
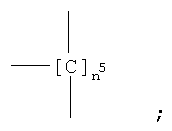
где n5 как указано выше;
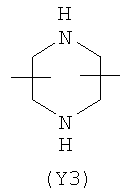
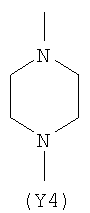
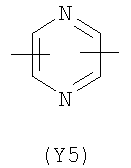
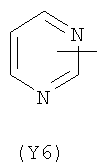
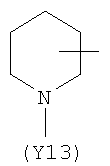
Y2 представляет собой 5-6-членное гетероциклическое, насыщенное, ненасыщенное или ароматическое кольцо, содержащее один или более гетероатомов, выбранных из азота, кислорода, серы, и выбрано из:
 ,
,  ,
,  ,
,  ,
,  ,
,
 ,
, 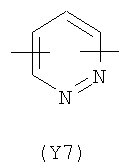 ,
, 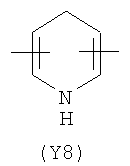 ,
, 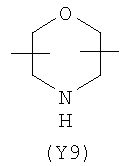 ,
, 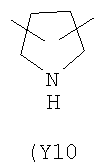 ,
,
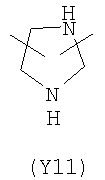 ,
, 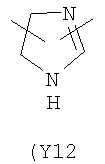 ,
, 
Термин “C1-C20алкилен” означает здесь прямую или разветвленную углеводородную цепь C1-C20, преимущественно имеющую от 1 до 10 атомов углерода, такую как метилен, этилен, пропилен, изопропилен, n-бутилен, пентилен, n-гексилен и тому подобное.
Термин “C1-С10алкил” означает здесь прямые или разветвленные алкильные группы, содержащие от одного до десяти атомов углерода, включая метил, этил, n-пропил, изопропил, n-бутил, изобутил, трет-бутил, пентил, гексил, октил и тому подобное.
Термин “циклоалкилен” означает здесь кольцо, имеющее от 5 до 7 атомов углерода, включая, но не ограничиваясь ими циклопентилен, циклогексилен, необязательно замещенные боковыми цепями, такими как прямой или разветвленный (C1-С10)-алкил, преимущественно СН3.
Термин “гетероциклический” означает здесь насыщенное, ненасыщенное или ароматическое 5-6 членное кольцо, содержащее один или более гетероатомов, выбранных из азота, кислорода, серы, такое как, например, пиридин, пиразин, пиримидин, пирролидин, морфолин, имидазол и тому подобное.
Другим аспектом настоящего изобретения является применение соединений формулы (I) в комбинации с, по крайней мере, соединением, используемым для лечения сердечно-сосудистых заболеваний, выбранным из группы, состоящей из: ингибиторов АТФ, антагонистов рецептора ангиотензина II, бета-адренергических блокаторов, блокаторов кальциевого канала, антитромботических средств, таких как аспирин, нитрозированных ингибиторов АТФ, нитрозированных антагонистов рецептора ангиотензина II, нитрозированных бета-адренергических блокаторов и нитрозированного аспирина.
Подходящими ингибиторами АТФ, антагонистами рецептора ангиотензина II, бета-адренергическими блокаторами, блокаторами кальциевого канала, антитромботическими средствами являются описанные в литературе средства (например, в The Merck Index (13th edition).
Подходящие нитрозированные соединения описаны в WO 98/21193 и WO 97/16405.
Указанные выше соединения можно назначать одновременно или последовательно.
Настоящее изобретение относится также к фармацевтическим наборам, включающим один или более контейнеров, заполненных одним или более веществами и/или композициями настоящего изобретения и одним или более указанными выше веществами, используемыми для лечения сердечнососудистых заболеваний.
Как указано выше изобретение включает также фармацевтически приемлемые соли соединений формулы (I) и их стереоизомеры.
Соединения настоящего изобретения, содержащие в молекуле способный к образованию солей атом азота, могут быть превращены в соответствующие соли реакцией с соответствующими органическими или неорганическими кислотами в органическом растворителе, таком как ацетонитрил или тетрагидрофуран.
Примерами органических кислот являются: щавелевая, винная, малеиновая, янтарная, лимонная кислоты. Примерами неорганических кислот являются: азотная, хлористоводородная, серная, фосфорная кислоты.
Предпочтительными являются соли с азотной кислотой.
Соединения настоящего изобретения, имеющие один или более асимметрических атомов углерода, могут существовать в виде оптически чистых энантиомеров, чистых диастереомеров, энантиомерных смесей, диастереомерных смесей, энантиомерных рацемических смесей, рацематов или рацемических смесей. В объем изобретения входят также все возможные изомеры, стереомеры соединений формулы (I) и их смеси.
Предпочтительными соединениями формулы (I) являются такие, где
Х означает -О- или -S-;
R представляет остаток статина, представленный выше;
Y представляет двухвалентный радикал, имеющий следующие значения:
a)
- прямой или разветвленный C1-С10алкилен, необязательно замещенный Т0, где Т0 имеет указанные выше значения;
b)
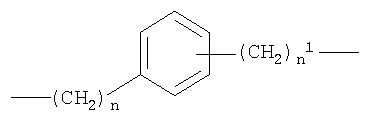
c)
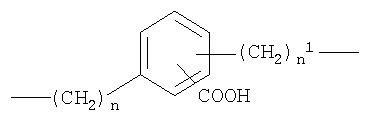
где n от 0 до 5, и n1 от 1 до 5;
d)
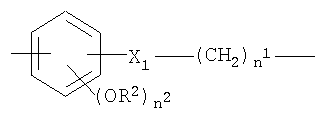
где n1 как указано выше и n2 от 0 до 2;
X1=-ОСО- или -COO- и R2 означает Н или СН3;
e)
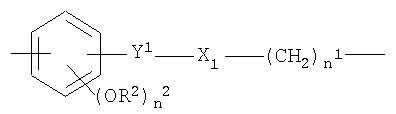
где n1, n2, R2 и X1 как указано выше;
Y1 означает -СН=СН-;
f)
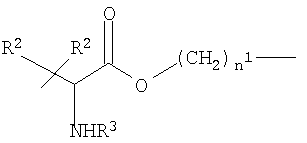
где n1 и R2 означает Н или СН3, R3 означает Н или СОСН3;
при условии, что когда Y выбирают из двухвалентных радикалов, обозначенных под b)-f), группа -ONO2 связана с -(СН2)n 1;
g)
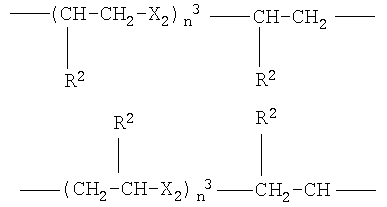
где Х2 означает -О- или -S-, n3 от 1 до 4, преимущественно 1, R2 означает атом водорода или СН3;
h)
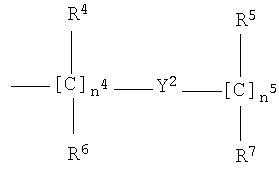
где n4 от 0 до 3;
n5 от 1 до 3;
R4, R5, R6, R7 являются одинаковыми и означают H;
и где группа -ONO2 связана с
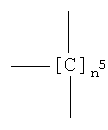 ;
;
Y2 представляет собой 6-членное насыщенное, ненасыщенное или ароматическое гетероциклическое кольцо, содержащее один или более атомов азота и выбранное, например, из
 ,
, 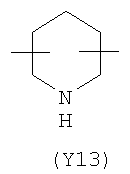 ,
, 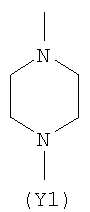 ,
, 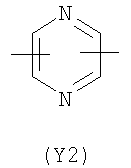 ,
, 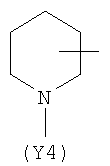 ;
;
В соответствии с настоящим изобретением предпочтительными соединениями являются следующие:
4-(нитроокси)бутиловый эфир флувастатина,
4-(нитрооксиметил)бензиловый эфир флувастатина,
3-(нитрооксиметил)бензиловый эфир флувастатина,
2-(нитрооксиметил)бензиловый эфир флувастатина,
4-(нитрооксиметил)фениловый эфир флувастатина,
3-(нитрооксиметил)фениловый эфир флувастатина,
2-(нитрооксиметил)фениловый эфир флувастатина,
2-[2'-(нитроокси)этилокси] этиловый эфир флувастатина, соответствующий формуле:
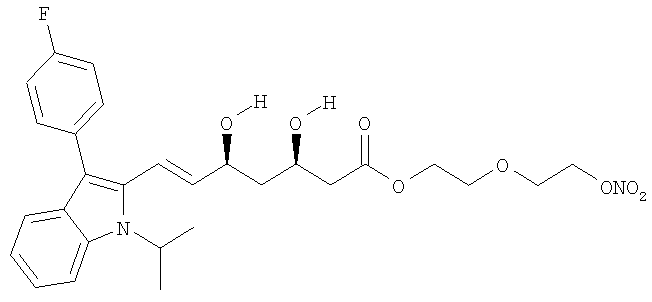
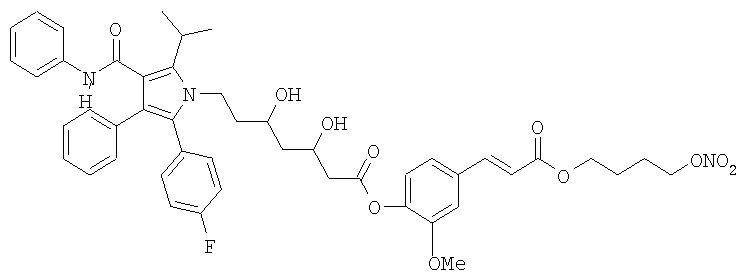
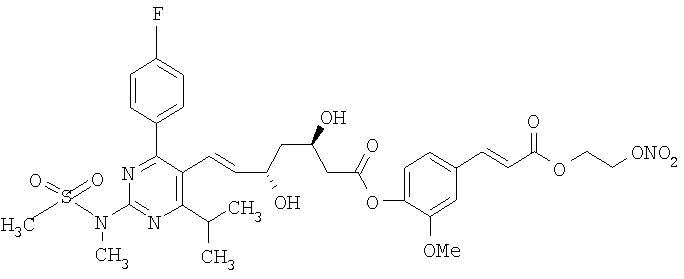
2-метокси-4-[[4'-(нитроокси)бутил]транс-2-пропеноилокси]фениловый эфир флувастатина, соответствующий формуле:
4-(нитроокси)бутиловый эфир правастатина,
4-(нитрооксиметил)бензиловый эфир правастатина,
3-(нитрооксиметил)бензиловый эфир правастатина,
2-(нитрооксиметил)бензиловый эфир правастатина,
4-(нитрооксиметил)фениловый эфир правастатина,
3-(нитрооксиметил)фениловый эфир правастатина,
2-(нитрооксиметил)фениловый эфир правастатина,
2-[2′-(нитроокси)этилокси]этиловый эфир правастатина, соответствующий формуле:
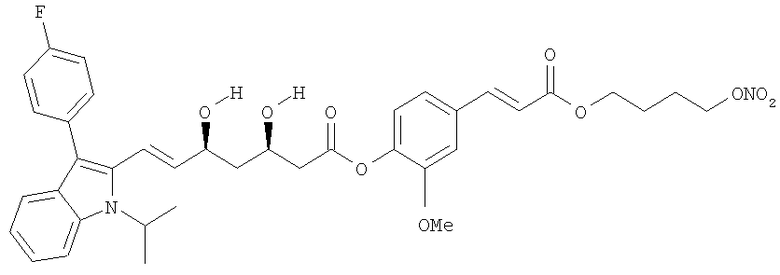
2-метокси-4-[[4′-(нитроокси)бутил]транс-2-пропеноилокси]фениловый эфир правастатина, соответствующий формуле:
4-(нитроокси)бутиловый эфир церивастатина,
4-(нитрооксиметил)бензиловый эфир церивастатина,
3-(нитрооксиметил)бензиловый эфир церивастатина,
2-(нитрооксиметил)бензиловый эфир церивастатина,
4-(нитрооксиметил)фениловый эфир церивастатина,
3-(нитрооксиметил)фениловый эфир церивастатина,
2-(нитрооксиметил)фениловый эфир церивастатина,
2-[2′-(нитроокси)этилокси]этиловый эфир церивастатина, соответствующий формуле:
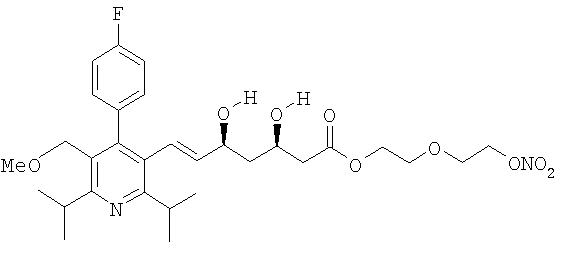
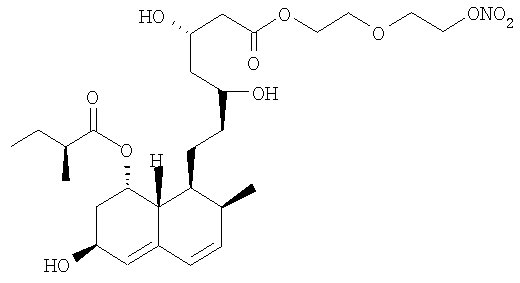
2-метокси-4-[[4′-(нитроокси)бутил]транс-2-пропеноилокси]фениловый эфир церивастатина, соответствующий формуле:
4-(нитроокси)бутиловый эфир аторвастатина,
4-(нитрооксиметил)бензиловый эфир аторвастатина,
3-(нитрооксиметил)бензиловый эфир аторвастатина,
2-(нитрооксиметил)бензиловый эфир аторвастатина,
4-(нитрооксиметил)фениловый эфир аторвастатина,
3-(нитрооксиметил)фениловый эфир аторвастатина,
2-(нитрооксиметил)фениловый эфир аторвастатина,
2-[2′-(нитроокси)этилокси]этиловый эфир аторвастатина, соответствующий формуле:
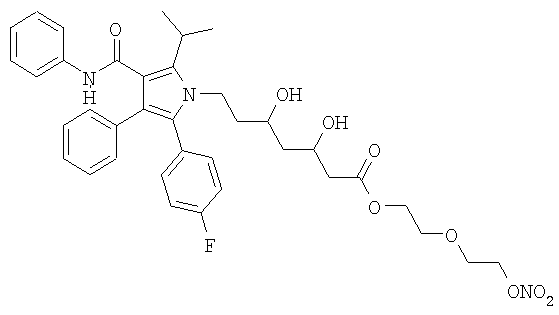
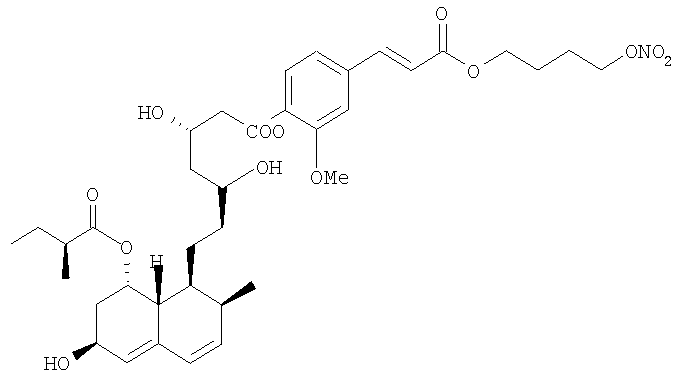
2-метокси-4-[[4'-(нитроокси)бутил]транс-2-пропеноилокси]фениловый эфир аторвастатина, соответствующий формуле:
4-(нитроокси)бутиловый эфир розувастатина,
4-(нитрооксиметил)бензиловый эфир розувастатина,
3-(нитрооксиметил)бензиловый эфир розувастатина,
2-(нитрооксиметил)бензиловый эфир розувастатина,
4-(нитрооксиметил)фениловый эфир розувастатина,
3-(нитрооксиметил)фениловый эфир розувастатина,
2-(нитрооксиметил)фениловый эфир розувастатина,
2-[2′-(нитроокси)этилокси]этиловый эфир розувастатина, соответствующий формуле:
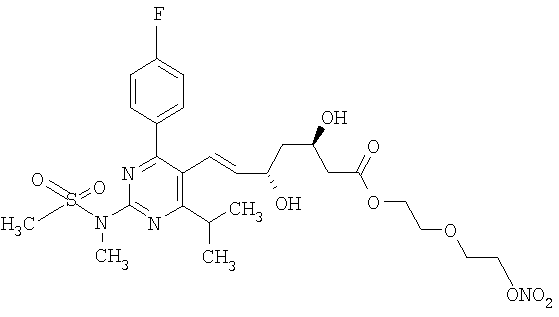
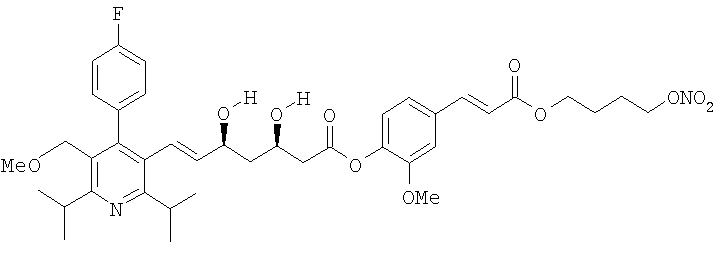
2-метокси-4-[[4′-(нитроокси)бутил]транс-2-пропеноилокси]фениловый эфир розувастатина, соответствующий формуле:
Как указано выше, объектом настоящего изобретения являются также фармацевтические композиции, содержащие, по крайней мере, соединение настоящего изобретения формулы (I) вместе с нетоксичными обычно применяемыми в фармацевтике адъювантами и/или носителями.
Назначаемая дневная доза активного ингредиента может быть единичной дозой или это эффективное количество может быть разделено на несколько небольших доз, которые назначаются в течение дня. Обычно общая дневная может составлять от 50 до 500 мг. Режим дозировки и частота назначения для лечения указанных выше заболеваний соединениями настоящего изобретения и/или фармацевтическими композициями настоящего изобретения следует выбирать в соответствии с различными факторами, включая, например, возраст, вес тела, пол и состояние пациента, также как и тяжесть заболевания, способ назначения, фармакологические обстоятельства и возможную сопутствующую терапию другими лекарственными средствами. В некоторых отдельных случаях уровни доз могут быть ниже или выше вышеуказанного интервала и/или чаще соответствуют ему, определяются врачом и картиной болезни.
Соединения настоящего изобретения могут назначаться орально, парентерально, ректально или местно, путем ингаляции или в виде аэрозоля в составах, по желанию возможно содержащих обычные фармацевтически приемлемые носители, адъюванты и наполнители. Местное назначение может также включать трансдермальные пластыри и повязки или устройства для электофореза. Термин “парентерально” здесь включает подкожные инъекции, внутривенные, внутримышечные, внутригрудинные инъекции или технику вливания.
Инъекционные препараты, например стерильные водные или масляные суспензии, могут формироваться с известными, используемыми в данной области диспергирующими или увлажняющими агентами и суспендирующими агентами. Стерильные инъецируемые препараты могут быть также в виде стерильных инъецируемых растворов или суспензий в нетоксичном, парентерально приемлемом разбавителе или растворителе. Приемлемыми носителями являются вода, раствор Рингера и изотонический хлористый натрий. Кроме того, могут использоваться стерильные, нелетучие масла, обычно используемые в качестве растворяющей или суспендирующей среды. Для этих целей могут использоваться любые легкие нелетучие масла, включая синтетические моно или диглицериды, кроме того, при изготовлении инъецируемых форм находят применение жирные кислоты, такие как олеиновая кислота.
Суппозитории для ректального назначения лекарственного вещества можно приготовить смешиванием активного ингредиента с пригодным, не вызывающим раздражение эксципиентом, таким как масло какао и полиэтиленгликоли.
Твердые лекарственные формы для орального назначения могут включать капсулы, таблетки, пилюли, порошки, гранулы и гели. В таких твердых лекарственных формах активное вещество может быть смешано с по крайней мере одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие лекарственные формы в обычной практике могут также включать дополнительные вещества, иные нежели инертные разбавители, например лубриканты, такие как стеарат магния. В случае капсул, таблеток или пилюль лекарственные формы содержат также буферные агенты.
Жидкие лекарственные формы для орального назначения могут включать фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, содержащие обычно используемые в данной области инертные растворители, такие как вода. Такие композиции могут также содержать адъюванты, такие как увлажняющие агенты, эмульгаторы и суспендирующие агенты, подсластители, ароматизаторы и т.д.
Как уже указывалось выше, новые нитропроизводные статинов сильнее понижают уровень липидов, обладают повышенным противовоспалительным, антитромботическим и антитромбоцитарным действием по сравнению с природными статинами. Более того, они могут быть эффективны при других патологиях, таких как острые коронарные синдромы, удар, периферические сосудистые расстройства, такие как периферическая ишемия, все нарушения, связанные с дисфункцией эндотелия, такие как сосудистые осложнения при диабете и атеросклерозе, нейродегенеративные заболевания, такие как болезнь Альцгеймера (AD) и болезнь Паркинсона (PD), аутоиммунные заболевания, такие как рассеянный склероз.
Изучение сосудистого тонуса.
Способность нитропроизводных статина индуцировать вазорелаксацию по сравнению с природными статинами, исследовали in vitro на изолированных препаратах грудной аорты кролика(Wanstall J.С. et al., Br.J.Pharmacol, 134: 463-472, 2001). Самцов новозеландских кроликов анестезировали тиопенталом натрия (50 мг/кг, в/в), скарифицировали путем обескровливания, затем вскрывали грудную клетку и рассекали аорту. Отдельные кольцевые препараты грудной аорты (4 мм длиной) помещали в небольшие камеры для органов (5 мл) с физиологическим раствором (ФСР) при температуре 37°С. Состав ФСР (мМ): NaCl 130, NaHCO3 14,9,
КH2PO4 1,2, MgSO4 1,2, HEPES 10, CaCl2, аскорбиновая кислота 170 и глюкоза 1,1 (95% O2/5% СО2; рН 7,4). Каждое кольцо помещали в 5 мл камеру для органов под пассивным парциальным давлением 2 g. Изомерное парциальное давление регистрировали с помощью Grass-трансдуктора (Grass FT03), прикрепленного к системе BIOPAC MP150. Препараты оставляли для уравновешивания в течение 1 часа, затем субмаксимально сжимали с помощью норадреналина (NA, 1 мкМ) и, когда сокращение было стабильным, добавляли ацетилхолин (ACh, 10 мкМ). Расслабляющая реакция на ацетилхолин указывает на наличие функционально активного эндотелия. При достижении стабильного предварительного сокращения получали кумулятивную кривую доза-ответ для одного из двух агентов, расслабляющих сосуды, в присутствии функционально активного эндотелия. Временные интервалы между различными концентрациями были основаны на времени, необходимом для достижения полного ответа. Кроме того, действие растворимого ингибитора гуанилил циклазы ODQ (1-Н-(1,2,4)-оксадиазол(4,3-а)хиноксалин-1-он) на реакции расширения, вызванные данными соединениями, проверяли предварительным инкубированием колец аорты с ODQ (10 рМ) в течение 20 минут. Реакции на агенты, расслабляющие сосуды, измеряли от горизонтального участка NA сокращения. IC50 (концентрация, обеспечивающая 50% устранение NA сокращения) интерполировали от точек на кривой “расслабляющий агент-ответ” в зависимости от логарифма молярной концентрации исследуемого соединения.
В течение экспериментального периода плато, полученное с NA, было стабильно без значимого спонтанного уменьшения сокращения в кольцах аорты. При этих экспериментальных условиях, природные статины не приводили к релаксации до 30 мкМ, кривая не отличалась от кривой, полученной в присутствии одного наполнителя.
Как показано в следующей Таблице 1, нитропроизводные по данному изобретению обладали способностью вызывать релаксацию зависимым от концентрации образом. Более того, в экспериментах, проводимых в присутствии ODQ (10 мкМ), сосудорасслабляющие реакции на все тестируемые лекарственные средства были ингибированы.
Эффекты производных нитростатина на каскады воспаления in vitro
Эксперименты проводили с использованием моноцит-макрофагальной клеточной линии RAW264.7. Клетки стимулировали в присутствии LPS (1 мкг/мл) в течение 8 часов (правастатин и производное) или 16 часов (флувастатин, аторвастатин и производные). По окончании инкубации клетки собирали в лизирующий буфер и измеряли содержание белка. Осуществляли вестерн-блот анализ индуцибельных белков iNOS и СОХ2 (показатели протекающего воспалительного процесса). Полученные результаты, представленные в Таблице 2, выражены как % снижения оптической плотности каждого варианта по сравнению с образцами, обработанными LPS. Отрицательные результаты означают повышение по сравнению с одним LPS.
Нитропроизводные флувастатина, правастатина и аторвастатина на клеточной линии RAW264.7 оказывают значительное ингибирующее действие на экспрессию таких белков как СОХ2 и iNOS, которые имеют отношение к процессу воспаления. Напротив, эти эффекты не определяются или являются слабыми у исходных соединений.
Изучение действия нитропроизводного правастатина при заболевании периферических сосудов.
Ангиогенез представляет собой первичный ответ на локальную гипоксию ткани и, вероятно, принимает участие в восстановлении кровотока при ишемических заболеваниях, таких как острые коронарные синдромы и окклюзивное заболевание периферических сосудов. Способность нитропроизводного правастатина (соединение примера 1) ингибировать процесс ангиогенеза оценивали используя in vivo модель ишемии задней конечности и мышей, как описано ранее (Emanueli et al., Circulation 103, 125-132, 2001). Вкратце, самцов мыши CDl (Charles River, Италия) двухмесячного возраста делили на три группы случайным образом (n=20 каждая), корм животных содержал: 1) правастатин (40 мг/кг), 2) нитропроизводное правастатина (48 мг/кг), 3) наполнитель (обычный корм). Через 3 дня мышей анастезировали и вызывали ишемию левой задней конечности путем электрокоагуляции верхней части бедренной артерии. Лечение продолжали на протяжении последующих 2 недель после операции. Измеряли систолическое кровяное давление на 7 и 14 день путем наложения манжеты на хвост. Исход болезни на 14 день определяли подсчетом количества некротизированных или утраченных когтей на лапе, подвергшейся ишемии, а также числа случаев самоампутации. На 14 день после индукции ишемии, задние конечности анестезированной мыши фиксировали перфузией, обе приводящие мышцы кодировали и подвергали гистологическому анализу. Определяли плотность капилляров и артериол ncap/мм2 и nart/мм2).
Как показано в таблице 3, в отличие от исходного соединения, соединение по данному изобретению обладало способностью потенцировать природный репаративный капилляризационный ответ на ишемию. Результаты настоящего исследования показывают, что данное нитропроизводное способствует значительной репаративной неоваскуляризации и улучшает клинический исход у мышей с ишемией задней конечности, вызванной экспериментально.
Влияние нитропроизводного правастатина на адгезию лейкоцитов у нокаут мышей АроЕ.
Взаимодействие между лейкоцитами и эндотелием сосудов является ключевой стадией воспаления в атерогенном процессе. Способность нитропроизводного правастатина (соединение примера 1) ингибировать адгезию лейкоцитов исследовали ex vivo у мышей с атеросклерозом (нокаут АроЕ).
Трем группам нокаут мышей АроЕ определенными дозами ежедневно в течение 5 дней вводили правастатин (40 мг/кг, перорально), нитропроизводное правастатина (эквимолярная доза) или наполнитель, мышей подвергали эвтаназии СO2-асфиксией через 1 час после последней дозы.
Лейкоциты выделяли из селезенки, центрифугировали, эритроциты (RBC) лизировали водой. Готовили сегменты артерий - области аорты & грудной аорты вскрытые продольно и разложенные стороной просвета вверх. Лейкоциты, меченные радиоактивным изотопом (51Сr-лейкоциты) инкубировали на протяжении 30 минут с сегментами, стимулированными тромбином (10 Емл-1 в течение 10 минут). Сегменты отмывали средой и определяли адгезию с помощью гамма-счетчика.
Как показано в Таблице 4, в отличие от исходного соединения, соединение по данному изобретению обладало способностью снижать адгезивное взаимодействие между изолированными лейкоцитами и стенкой кровеносного сосуда, индуцированное тромбином, у обеих пород мышей.
Эти данные демонстрируют, что данное нитропроизводное оказывает противоатерогенные/противовоспалительные эффекты.
Изучение антитромботического действия нитропроизводного правастатина in vivo.
Способность нитропроизводного правастатина ингибировать тромбообразование оценивали у самцов мышей Charles Rivers CD-1 (20-25 г) путем введения правастатина (10 или 20 мг/кг, внутрибрюшинно), соединения примера 1 (эквимолярные дозы) или наполнитель. Через 1-3 часа животным делали инъекцию U46619 (0,2 мг/кг) (стабильный аналог ТхА2) в хвостовую вену. Как описано ранее (Momi et al., Eur. J.Pharmacol. 397: 177-185, 2000), такая доза агониста приводит к смерти в 80-90% случаев в течение 3 минут в контрольной группе. В каждой экспериментальной сессии были обследованы, по крайней мере, 6 животных в каждой группе лечения; группы контроля обследовали в начале и конце каждой экспериментальной сессии. Защита от тромбоэмболии, индуцированной агонистом, выражалась
как (1-Тлекарство/Тфиз.раствор)×100, где Тлекарство представляет собой уровень смертности у мышей, получающих лекарственное средство, а Тфиз.раствор представляет собой число не выживших животных из группы контроля, получавших только тромбин. Результаты выражены как процент защиты от легочной тромбоэмболии, индуцированной U46619, по сравнению с контрольной группой.
Как показано в Таблице 5, в отличие от исходного соединения, соединение по данному изобретению способно ингибировать тромбообразование, индуцированное действием U46619.
Изучение антитромбоцитарного действия нитропроизводного правастатина in vitro.
Тромбоциты являются характерными компонентами тромбов. Способность нитропроизводного правастатина (соединение примера 1) ингибировать агрегацию тромбоцитов оценивали in vitro на тромбоцитах человека. Агрегацию тромбоцитов измеряли в 0,25 мл богатой тромбоцитами плазме (PRP) или в образцах тромбоцитов, пошедших гель-фильтрацию (GFP), как описано ранее (Vezza R, et al., Blood 73: 2006-2013, 1993). Перед добавлением U46619 (агент агрегации, аналог ТхА2) соединения инкубировали 2 минуты при температуре 37°С. Агрегацию проводили в течение 5 минут, и измеряли максимальную амплитуду (см). В качестве носителя использовали DMSO (0,05%). Соединения тестировали при концентрациях в интервале от 50 до 200 мкМ.
Как показано в Таблице 6, в отличие от исходного соединения, нитропроизводное по данному изобретению способно ингибировать агрегацию тромбоцитов, индуцированную действием U46619.
Влияние нитропроизводного правастатина на ингибирование экспрессии тканевого фактора in vitro.
Тканевой фактор (TF) является главным регулятором гомеостаза и тромбообразования. Способность нитропроизводного правастатина (соединение примера 1) ингибировать экспрессию оценивали на изолированных препаратах мононуклеарных клеток (MNC) крови человека. MNC получали из свежей крови центрифугированием в градиенте плотности (Ficoll-Hypaque) и ресуспендированием в бессывороточной среде RPMI в концентрации 2-3×l06/мл.
Нитропроизводное правастатина (3-50 мкМ) или правастатин (50 мкМ) добавляли к клеточным суспензиям перед воздействием LPS (1 мкг/мл). После инкубирования в течение 3 часов при температуре 37°С, экспрессию TF мононуклеарными клетками (MNC) оценивали следующими способами:
1) функциональным анализом интактных клеток (Semeraro et al., 1983. Immunology; 50: 529-35); 2) иммунологическим анализом клеточных экстрактов Тритон Х-100 (Imubind, Instrumentation Laboratory, Милан); 3) RT-PCR (ривертазо-ПЦР) (Rossiello et al. Thromb Haemost 2000; 84: 453-59).
Данные представляют собой среднее значение стандартной ошибки измерения 4-5 экспериментов.
§Условные единицы рассчитывают исходя из стандартной кривой, построенной с релипидированным тканевым фактором человека
**Р<0,01 по сравнению с правастатином и наполнителем.
Как показано в Таблице 7, в отличие от исходного соединения, соединение по данному изобретению заметно ингибировало экспрессию тканевого фактора, индуцированную LPS (активность и антиген). Результаты настоящего исследования показывают, что нитропроизводное проявляет антитромботическую активность.
Следующие примеры дополнительно иллюстрируют изобретение, не ограничивая его.
Общий способ
Соединения общей формулы (I), где Х=О, может быть получено
взаимодействием соединения формулы (II)
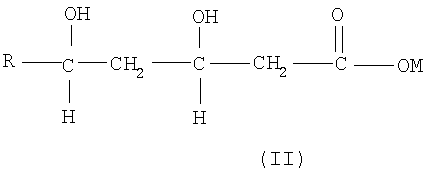
где М означает водород или щелочной или щелочноземельный металл, такой как натрий или кальций, с соединением формулы (III)
 ,
,
где А означает удаляемую группу, такую как хлор, бром, иод, тозил или мезил, В выбирают из хлора, брома, иода или нитроокси группы и Y как указано выше для формулы (I), в инертном органическом растворителе, таком как N,N′-диметилформамид, тетрагидрофуран, бензол, толуол, при температуре от комнатной до 50°С. Реакцию прекращают по прошествии от 30 минут до 24 часов.
Соединения формулы (II) являются известными, имеющимися в продаже соединениями или могут быть получены в соответствии с хорошо известными в литературе методами. Соединения формулы (III), где А как указано выше и В представляет собой Сl, Вr, I, являются коммерчески доступными или могут быть синтезированы в соответствии с хорошо известными из литературы методами.
Соединения формулы (III), где А как указано выше и В представляет ONO2 синтезированы превращением соединения формулы (III), где В представляет собой Сl, Вr, I, в соответствующее нитропроизводное взаимодействием с нитратным источником, таким как нитрат серебра, нитрат лития, нитрат натрия, нитрат калия, нитрат магния, нитрат кальция, нитрат железа, нитрат цинка или нитрат тетраалкиламмония (где алкил означает C1-С10 алкил) в подходящем органическом растворителе, таком как ацетонитрил, тетрагидрофуран, метилэтилкетон, этилацетат, ДМФ, при этом взаимодействие осуществляют в темноте при температуре от комнатной до температуры кипения растворителя. Реакцию прекращают по прошествии от 30 минут до 3 дней. Предпочтительным нитратным источником является нитрат серебра.
Соединения формулы (I), где Х=О, S, NH или NR1, R1 как указано выше, могут быть получены реакцией соединения формулы (II), где М представляет водород, в присутствии дегидратирующего агента с соединением формулы (IV)
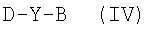
где Y и В как указано выше, и D выбирают из NH2, NHR1, ОН или SH, R1 как указано выше. Предпочтительными дегидратирующими агентами являются N,N′-карбонилдиимидазол, используемый в присутствии каталитического количества этилата натрия, DCC, EDAC. Реакцию осуществляют в инертном органическом растворителе, таком как N,N′-диметилформамид, тетрагидрофуран, бензол, толуол, полигалогенированные алифатические углеводороды, при температуре от -50°С до 50°С от 30 минут до 24 час.
Соединения формулы (IV), где D как указано выше и В представляет Сl, Вr, I, являются коммерчески доступными или могут быть синтезированы в соответствии с хорошо известными из литературы методами.
Например, они могут быть получены из соответствующих гидроксипроизводных формулы (V) D-Y-OH, реакцией с тионил или оксалил хлоридом, галогенидами РIII или РV мезил хлоридо, тозил хлоридом и инертном растворителе, таком как толуол, хлороформ, ДМФ и т.д.
Соединения формулы (IV), где D как указано выше и В представляет ONO2, могут быть получены реакцией соединений формулы (IV), где В представляет Сl, Вr, I с нитратным источником, как указано выше. Кроме того, соединения формулы (IV), где В представляет ONO2 могут быть получены взаимодействием соответствующих гидроксильных соединений формулы (V) с азотной кислотой и уксусным ангидридом при температуре от -50°C до 0°С в соответствии с хорошо известными в литературе методами.
Пример 1
Синтез 4-(нитроокси)бутилового эфира [1S-[1µ(µS*,µS*),2µ,6µ,8µ-(R*),8aµ]]-1,2,6,7,8,8а-гексагидро-µ,µ,6-тригидрокси-2-метил-8-(2-метил-1-оксобутокси)-1-нафталингептановой кислоты (4-(нитроокси)бутилового эфира правастатина).
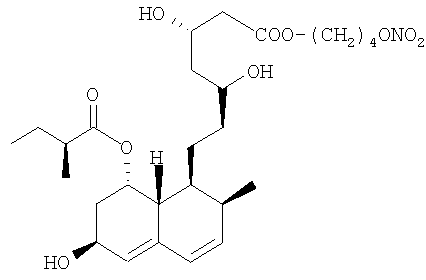
а) 4-Бромбутиловый эфир [1S-[1µ(µS*,µS*),2µ,6µ,8µ-(R*),8аµ]]-1,2,6,7,8,8а-гексагидро-µ,µ,6-тригидрокси-2-метил-8-(2-метил-1-оксобутокси)-1-нафталингептановой кислоты
К раствору правастатин натрия (9,25 г, 21 ммол) в N,N-диметилформамиде (70 мл) прикапывают смесь 1,4-дибромбутана (3,68 мл, 31 ммол) в N,N-диметилформамиде (30 мл). Реакционную смесь перемешивают при комнатной температуре 24 часа. Затем раствор обрабатывают водой и диэтиловым эфиром, органические слои сушат сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают флеш-хроматографией, используя в качестве элюента n-гексан/этилацетат 3/7. Получают указанное в заголовке соединение в виде белого порошка (7,8 г).
b) 4-(Нитроокси)-бутиловый эфир [lS-[1µ(µS*,µS*),2µ,6µ,8µ-(R*),8aµ]]-1,2,6,7,8,8а-гексагидро-µ,µ,6-тригидрокси-2-метил-8-(2-метил-1-оксобутокси)-1-нафталингептановой кислоты
Раствор полученного, как указано в а) соединения (7,7 г, 14 ммол) и нитрата серебра (3,5 г, 21 ммол) в ацетонитриле (90 мл) перемешивают при 40°С в темноте 48 часов. Образовавшийся осадок (серебряной соли) отфильтровывают и растворитель упаривают под вакуумом. Затем остаток очищают флеш-хроматографией, используя в качестве элюента n-гексан/этилацетат 3/7. Получают указанное в заголовке соединение в виде белого порошка (3,7 г), плавящегося при 63°С.
1H-NMR: µ (CDCl3) 6.00 (1H, m); 5.88 (1H, m); 5.53 (1H, s); 5.40 (1H, s); 4.51 (2Н, t); 4.40 (1H, m); 4.26 (1H, m); 4.15 (2H, t); 3.78 (1H, m); 2.60-2.23 (6H, m); 1.8-1.20 (14H, m); 1.10 (3H, d); 0.87 (6H, t).
Пример 2
Синтез 4-(нитрооксиметил)бензилового эфира [1S-[1µ(µS*,µS*),2µ,6µ,8µ-(R*),8aµ]]-1,2,6,7,8,8а-гексагидро-µ,µ,6-тригидрокси-2-метил-8-(2-метил-1-оксобутокси)-1-нафталингептановой кислоты (4-(нитрооксиметил)бензиловый эфир правастатина).
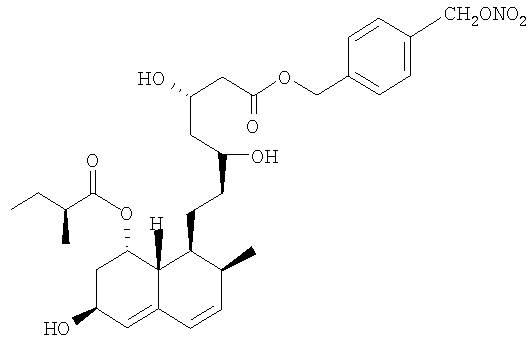
a) 4-(Хлорметил)бензиловый эфир [1S-[1µ(µS*,µS*),2µ,6µ,8µ-(R*),8аµ]]-1,2,6,7,8,8а-гексагидро-µ,µ,6-тригидрокси-2-метил-8-(2-метил-1-оксобутокси)-1-нафталингептановой кислоты
К раствору µ,µ1-дихлор-р-ксилола (5,5 г, 31 ммол) в N,N-диметилформамиде (70 мл) добавляют порциями правастатин натрия (7 г, 15 ммол). Реакционную смесь перемешивают при комнатной температуре 24 часа. Затем полученный раствор обрабатывают водой и диэтилацетатом, органические слои обезвоживают сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают хроматографией на силикагеле, используя в качестве элюента n-гексан/этилацетат 1/1. Получают указанное в заголовке соединение в виде белого порошка (5,5 г), которое используют на следующей стадии без очистки.
b) 4-(Нитрооксиметил)-бензиловый эфир [1S-[1µ(µS*,µS*),2µ,6µ,8µ-(R*),8aµ]]-1,2,6,7,8,8a-гексагидро-µ,µ,6-тригидрокси-2-метил-8-(2-метил-1-оксобутокси)-1-нафталингептановой кислоты
Раствор полученного, как указано, в а) соединения (5,5 г, 7,7 ммол) и нитрата серебра (2,5 г, 15 ммол) в ацетонитриле (60 мл) перемешивают при 60°С в темноте 48 часов. Образовавшийся осадок (серебряной соли) отфильтровывают и растворитель упаривают под вакуумом. Затем остаток очищают флеш-хроматографией, используя в качестве элюента n-гексан/этилацетат 1/1. Получают указанное в заголовке соединение в виде белого порошка (1,6 г), плавящегося при 80-82°С.
1H-NMR  (CDCl3): 7.39 (4H, m); 5.98 (1H, m); 5.86 (1H, m); 5.54 (1H, s); 5.42 (3H, s); 5.16 (2H, s); 4.40 (1H, m); 4.27 (1H; m); 3.80 (1H, m); 2.52 (3H, m); 2.45-2.25 (3H, m); 1.74-1.10 (10H, m); 1.10 (3H, d); 0.87 (6H, t).
(CDCl3): 7.39 (4H, m); 5.98 (1H, m); 5.86 (1H, m); 5.54 (1H, s); 5.42 (3H, s); 5.16 (2H, s); 4.40 (1H, m); 4.27 (1H; m); 3.80 (1H, m); 2.52 (3H, m); 2.45-2.25 (3H, m); 1.74-1.10 (10H, m); 1.10 (3H, d); 0.87 (6H, t).
Пример 3
Синтез 4-(нитроокси)бутилового эфира [R*,S*-(Е)]-7-[3-(4-фторфенил)-1-(1-метилэтил)-1Н-индол-2-ил]-3,5-дигидрокси-6-гептановой кислоты (4-(нитроокси)бутиловый эфир флувастатина)
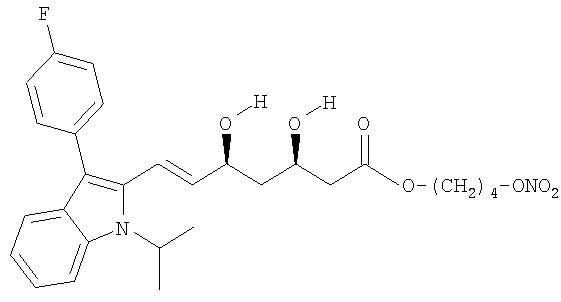
а) 4-Бромбутиловый эфир [R*,S*-(Е)]-7-[3-(4-фторфенил)-1-(1-метилэтил)-1Н-индол-2-ил]-3,5-дигидрокси-6-гептановой кислоты
К раствору 1,4-дибромбутана (4,1 мл, 34 ммол) в N,N-диметилформамиде (60 мл) прикапывают смесь флувастатина натрия (5 г, 11 ммол) в N,N-диметилформамиде (40 мл). Реакционную смесь перемешивают при комнатной температуре 24 часа. Затем полученный раствор обрабатывают водой и диэтилацетатом, органические слои обезвоживают сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают флеш-хроматографией, используя в качестве элюента n-гексан/изопропанол 8,5/1,5. Получают указанное в заголовке соединение в виде желтого масла (4,7 г), которое используют на следующей стадии без очистки.
b) 4-(Нитроокси)бутиловый эфир [R*,S*-(Е)]-7-[3-(4-фторфенил)-1-(1-метилэтил)-1Н-индол-2-ил]-3,5-дигидрокси-6-гептановой кислоты
Раствор полученного как указано в а) соединения (4,7 г, 8,5 ммол) и нитрата серебра (2,9 г, 17 ммол) в ацетонитриле (60 мл) перемешивают при 60°С в темноте 48 часов. Образовавшийся осадок (серебряной соли) отфильтровывают и растворитель упаривают под вакуумом. Затем остаток очищают флеш-хроматографией, используя в качестве элюента n-гексан/этилацетат 1/1. Получают желаемый продукт в виде белого порошка (0,5 г), плавящегося при 112°С.
1H-NMR µ (DMSO):7.69 (1H,d); 7.43 (3H,m); 7.27 (2H,t); 7.25 (1H,t); 7.03 (1H,t); 6.65 (1H,d); 5.75 (1H,dd); 4.98 (1H,d); 4.90 (1H,m); 4.78 (1H,d); 4.50 (2H,t); 4.25 (1H,m); 4.09 (2H,t); 3.9 (1H,m); 2.4 (2H,m); 1.67 (4H,m); 1.61 (6H,d); 1.42 (2H,m).
Пример 4
Синтез 4-(нитрооксиметил)бензилового эфира [R*,S*-(E)]-7-[3-(4-фторфенил)-1-(1-метилэтил)-1Н-индол-2-ил]-3,5-дигидрокси-6-гептановой кислоты (4-(нитрооксиметил)бензиловый эфир флувастатина)
а) 4-(Хлорметил)бензиловый эфир [R*,S*-(Е)]-7-[3-(4-фторфенил)-1-(1-метилэтил)-1Н-индол-2-ил]-3,5-дигидрокси-6-гептановой кислоты
К раствору  -дихлор-р-ксилола (10 г, 57 ммол) в N,N-диметилформамиде (60 мл) прикапывают смесь флувастатина натрия (10 г, 23 ммол) в N,N-диметилформамиде (80 мл). Реакционную смесь перемешивают при комнатной температуре 24 часа. Затем полученный раствор обрабатывают водой и этилацетатом, органические слои обезвоживают сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают флеш-хроматографией, используя в качестве элюента n-гексан/этилацетат 1/1. После кристаллизации из n-гексана получают указанное в заголовке соединение в виде белого порошка (9,4 г).
-дихлор-р-ксилола (10 г, 57 ммол) в N,N-диметилформамиде (60 мл) прикапывают смесь флувастатина натрия (10 г, 23 ммол) в N,N-диметилформамиде (80 мл). Реакционную смесь перемешивают при комнатной температуре 24 часа. Затем полученный раствор обрабатывают водой и этилацетатом, органические слои обезвоживают сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают флеш-хроматографией, используя в качестве элюента n-гексан/этилацетат 1/1. После кристаллизации из n-гексана получают указанное в заголовке соединение в виде белого порошка (9,4 г).
b) 4-(Нитрооксиметил)бензиловый эфир [R*,S*-(Е)]-7-[3-(4-фторфенил)-1-(1-метилэтил)-1Н-индол-2-ил]-3,5-дигидрокси-6-гептановой кислоты
Раствор полученного, как указано, в а) соединения (9,4 г, 17 ммол) и нитрата серебра (10,6 г, 63 ммол) в ацетонитриле (180 мл) перемешивают при 50°С в темноте 24 часа. Образовавшийся осадок (серебряной соли) отфильтровывают и растворитель упаривают под вакуумом. Затем остаток очищают флеш хроматографией, используя в качестве элюента n-гексан/этилацетат 4/6. Получают желаемый продукт в виде белого порошка (4 г), плавящегося при 103-104°С.
1H-NMR (DMSO): 7.65 (1H, d); 7.42 (7H, m); 7.22 (2H, t); 7.15 (1H, t); 7.03 (1H, t); 6.64 (1H, d); 5.75 (1H, dd); 5.51 (2H, s); 5.12 (2H, s); 4.98 (1H, d); 4.88 (1H, t); 4.84 (1H, d); 4.27 (1H, t); 3.94 (1H, t); 2.4-2.6 (2H, m); 1.4-1.7 (2H, M); 1.57 (6H, d).
Пример 5
Синтез 3-(нитрооксиметил)бензилового эфира [R*,S*-(E)]-7-[3-(4-фторфенил)-1-(1-метилэтил)-1Н-индол-2-ил]-3,5-дигидрокси-6-гептановой кислоты (3-(нитрооксиметил)бензиловый эфир флувастатина)
a) 3-(Хлорметил)бензиловый эфир [R*,S*-(Е)]-7-[3-(4-фторфенил)-1-(1-метилэтил)-1Н-индол-2-ил]-3,5-дигидрокси-6-гептановой кислоты
К раствору -дихлор-р-ксилола (0,95 г, 5,4 ммол) в N,N-диметилформамиде (25 мл) прикапывают смесь флувастатина натрия (0,79 г, 1,8 ммол) в N,N-диметилформамиде (25 мл). Реакционную смесь перемешивают при комнатной температуре 24 часа. Затем полученный раствор обрабатывают водой и этилацетатом, органические слои обезвоживают сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают хроматографией на силикагеле, используя в качестве элюента n-гексан/этилацетат 1/1, получая указанное в заголовке соединение (0,41 г).
b) 3-(Нитрооксиметил)бензиловый эфир [R*,S*-(Е)]-7-[3-(4-фторфенил)-1-(1-метилэтил)-1Н-индол-2-ил]-3,5-дигидрокси-6-гептановой кислоты
Раствор полученного как указано в а) соединения (0,4 г, 0,7 ммол) и нитрата серебра (0,36 г, 2,1 ммол) в ацетонитриле (30 мл) перемешивают при 45°С в темноте 24 часа. Образовавшийся осадок (серебряной соли) отфильтровывают и растворитель упаривают под вакуумом. Затем остаток очищают флеш-хроматографией, используя в качестве элюента метиленхлорид/изопропанол 9,5/0,5, получая 0,2 г желаемого продукта.
1H-NMR (DMSO): 7.67 (1H, d); 7.47-7.40 (7H, m); 7.25-7.10 (3H, m); 7.05 (1H, t); 6.62 (1H, d); 5.72 (1H, dd); 5.49 (2H, s); 5.13 (2H, s); 5.00-4.84 (3H, m); 4.27 (1H, m); 3.95 (1H, m); 2.60-2.35 (2H, m); 1.58 (6H, d); 1.70-1.45 (2H, m).
Пример 6
Синтез 2-(нитрооксиметил)бензилового эфира [R*,S*-(Е)]-7-[3-(4-фторфенил)-1-(1-метилэтил)-1Н-индол-2-ил]-3,5-дигидрокси-6-гептановой кислоты (2-(нитрооксиметил)бензиловый эфир флувастатина)
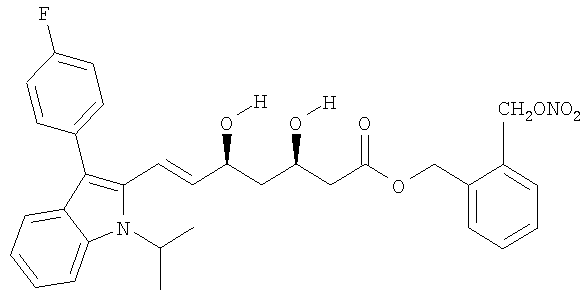
a) 2-(Хлорметил)бензиловый эфир [R*,S*-(Е)]-7-[3-(4-фторфенил)-1-(1-метилэтил)-1H-индол-2-ил]-3,5-дигидрокcи-6-гептановой кислоты
К раствору -дихлор-р-ксилола (1,5 г, 5,4 ммол) в N,N-диметилформамиде (15 мл) прикапывают смесь флувастатина натрия (1,5 г, 8,6 ммол) в N,N-диметилформамиде (15 мл). Реакционную смесь перемешивают при комнатной температуре в темноте 24 часа. Затем полученный раствор обрабатывают водой и этилацетатом, органические слои обезвоживают сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают хроматографией на силикагеле, используя в качестве элюента n-гексан/этилацетат 6/1, получая указанное в заголовке соединение (0,97 г).
b) 2-Нитрооксиметил)бензиловый эфир [R*,S*-(Е)]-7-[3-(4-фторфенил)-1-(1-метилэтил)-1Н-индол-2-ил]-3,5-дигидрокси-6-гептановой кислоты
Раствор полученного как указано в а) соединения (0,97 г, 1,7 ммол) и нитрата серебра (1,3 г, 7,6 ммол) в ацетонитриле (40 мл) перемешивают при 45°С в темноте 24 часа. Образовавшийся осадок (серебряной соли) отфильтровывают и растворитель упаривают под вакуумом. Затем остаток очищают флеш-хроматографией, используя в качестве элюента метиленхлорид/изопропанол 9,5/0,5, получая 0,42 г желаемого продукта в виде желтого порошка.
1H-NMR (DMSO): 6.52 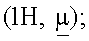 7.47-7.30 (7H, m); 7.24-7.14
7.47-7.30 (7H, m); 7.24-7.14 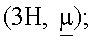 7.03 (1H, t); 6.27 (1H, d); 5.70 (1H, dd); 5.66 (2H, s); 5.22 (2H, s); 4.98 (1H, m); 4.84 (1H, t); 4.82 (1H, d); 4.30 (1H, m); 3.90 2.40 (2H, m); 1.58 (6H, d); 1.40 (2H, m).
7.03 (1H, t); 6.27 (1H, d); 5.70 (1H, dd); 5.66 (2H, s); 5.22 (2H, s); 4.98 (1H, m); 4.84 (1H, t); 4.82 (1H, d); 4.30 (1H, m); 3.90 2.40 (2H, m); 1.58 (6H, d); 1.40 (2H, m).
Пример 7
Синтез 4-(нитроокси)бутилового эфира (µR,µR)-2(4-фторфенил)-µ,µ-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1-Н-пиррол-1-гептановой кислоты (4-(нитроокси)бутиловый эфир аторвастатина).
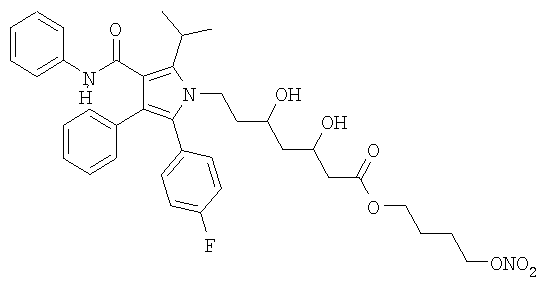
а) 4-(Бромбутиловый эфир (µR,µR)-2(4-фторфенил)-µ,µ-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1-Н-пиррол-1-гептановой кислоты
К раствору 1,4-дибромбутана (0,34 мл, 2,884 ммол) в N,N-диметилформамиде (19 мл) прикапывают смесь аторвастатина кальция (0,83 г, 0,72 ммол) в N,N-диметилформамиде (10 мл). Реакционную смесь перемешивают при комнатной температуре 24 часа. Затем полученный раствор обрабатывают водой и диэтиловым эфиром, органические слои обезвоживают сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают хроматографией на силикагеле, используя в качестве элюента n-гексан/этилацетат 6/4, получая указанное в заголовке соединение (0,97 г) в виде белого твердого остатка.
b) 4-(Нитроокси)бутиловый эфир (µR,µR)-2(4-фторфенил)-µ,µ-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1-Н-пиррол-1-гептановой кислоты
Раствор полученного как указано в а) соединения (0,25 г, 0,36 ммол) и нитрата серебра (0,38 г, 2,2 ммол) в ацетонитриле (10 мл) перемешивают при 50°С в темноте 21 час. Образовавшийся осадок (серебряной соли) отфильтровывают и растворитель упаривают под вакуумом. Затем остаток очищают хроматографией на силикагеле, используя в качестве элюента n-гексан/этилацетат 1/1, получая 0,41 г желаемого продукта в виде желтого порошка.
1H-NMR (DMSO): 9.80 (1Н, s); 7.51 (2Н, d); 7.26-7.19 (6Н, m); 7.09-6.95 (6H, m); 4.73 (1Н, d); 4.61 (1Н, d); 4.54 (2Н, t); 4.04. (2Н, t); 4.00-3.70 (3Н, m); 3.50 (1Н, m); 3.22 (1H, m); 2.40 (1H, dd); 2.25 (1H, dd); 1.80-1.10 (8H, m); 1.38 (6H, d).
Следуя примерами 1-7 исходя из соответствующих реагентов и используя церивастатин и розувастатин вместо правастатина, флувастатина и аторвастатина.
Пример 8
Синтез 2-[2-(нитроокси)этокси]этилового эфира (βR,βR)-2(4-фторфенил)-β,δ-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1Н-пиррол-1-гептановой кислоты 2-[2-(нитроокси)этокси] этиловый эфир аторвастатина).
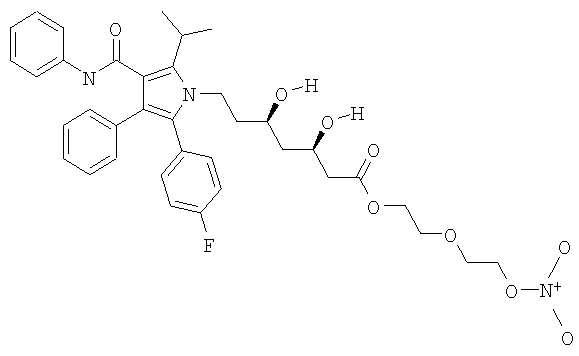
а) 2-[2-(бром)этокси]этилового эфира (βR,δR)-2(4-фторфенил)-β,δ-дигидрокси-5(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1Н-пиррол-1-гептановой кислоты
К раствору 2-бромэтилового эфира (0,65 мл, 5,19 ммол) в N,N-диметилформадиде (8 мл) прикапывают смесь аторвастатина кальция (1,5 г, 1,30 ммол) в N,N-диметилформадиде (10 г). Реакционную смесь перемешивают при комнатной температуре 72 часа. Затем полученный раствор обрабатывают водой этилацетатом, органические слои обезвоживают сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают хромотографией на силикагеле, использую в качестве элюента n-гексан/этилацетат 6/4, получая указанное в заголовке соединение (0,40 г).
в) 2-[2-(нитроокси)этокси]этилового эфира (βR,δR)-2(4-фторфенил)-β,δ-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1Н-пиррол-1-гептановой кислоты
Раствор полученного как указано выше соединения (0,30 г, 0,42 ммол) и нитрата серебра (0,37 г, 2,18 ммол) в ацетонитриле (5 мл) нагревают в микроволновой установке (120°С, 25 минут). Образовавшийся осадок (серебряной соли) отфильтровывают и растворитель упаривают под вакуумом. Затем остаток очищают флэш-хромотографией, использую в качестве элюента n-гексан/этилацетат 4/6, получая (0,08 г) желаемого продукта в виде белого порошка.
1Н-NMR δ (CDCl3): 7.25-7.12 (9Н, m); 7.15-6.94 (5Н, m); 6.88 (1Н, s); 4.63 (2Н, s); 4.41-4.05 (4Н, m); 4.04-3.86 (1Н, m); 3.84-3.68 (5Н, m); 3.67-3.49 (3Н, m); 2.47 (2Н, d); 1.82-1.42 (10Н, m).
Пример 9
Синтез 4-(нитрооксиметил)бензилового эфира (βR,δR)-2(4-фторфенил)-β,δ-дигидрокси-5-(1-метилэтил)-3-[(фениламино)карбонил]-1Н-пиррол-1-гептановой кислоты
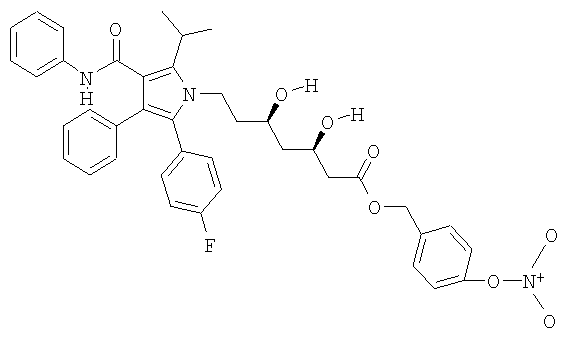
а) 4-(хлорметил)бензилового эфира (βR,δR)-2(4-фторфенил)-β,δ-дигидрокси-5-(1-метилэтил-3-фенил-4-[(фениламино)карбонил]-1Н-пиррол-1-гептановой кислоты
К раствору  ,
, -дихлор-р-ксилена (3,98 г, 22,75 ммол) в N,N-диметилформадиде (40 мл) прикапывают смесь аторвастатина кальция (4,38 г, 3,80 ммол) в диметилформадиде (20 мл). Реакционную смесь перемешивают при комнатной температуре 48 часов. Затем полученный раствор обрабатывают водой и диэтиловым эфиром, органические слои обезвоживают сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают хромотографией на силикагеле, используют в качестве элюента n-гексан/этилацетат 1/1, получая указанное в заголовке соединение (0,80 г) в виде белого твердого остатка.
-дихлор-р-ксилена (3,98 г, 22,75 ммол) в N,N-диметилформадиде (40 мл) прикапывают смесь аторвастатина кальция (4,38 г, 3,80 ммол) в диметилформадиде (20 мл). Реакционную смесь перемешивают при комнатной температуре 48 часов. Затем полученный раствор обрабатывают водой и диэтиловым эфиром, органические слои обезвоживают сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают хромотографией на силикагеле, используют в качестве элюента n-гексан/этилацетат 1/1, получая указанное в заголовке соединение (0,80 г) в виде белого твердого остатка.
в) 4-(нитрооксиметил)бензилового эфира (βR,δR)-2(4-фторфенил)-β,δ-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1Н-пиррол-1-гептановой кислоты
Раствор соединения, полученного выше (0,80 г, 1,15 ммол), и нитрата серебра (0,29 г, 1,72 ммол) ацетонитриле (50 мл), перемешивают при температуре 40°С в темноте 48 часов и затем добавляют дополнительное количество нитрата серебра (0,10 г, 0,57 ммол) и смесь перемешивают при температуре 70°С в темноте 24 часа. Образовавшийся осадок (серебряной соли) отфильтровывают и растворитель упаривают под вакуумом. Затем осадок очищают хромотографией на силикагеле, используют в качестве элюента n-гексан/этил 6/4, получая (0,40 г) желаемого продукта в виде белого порошка.
1Н-NMR δ (DMSO): 9,78 (1H, s); 7,66-7,32 (6Н, m); 7,32-6,82 (12Н, m); 5,57 (2Н, s); 5,12 (2Н, s); 4,79 (1Н, d); 4,63 (1H, d); 4,06-3,64 (3Н, m); 3,54 (1Н, m); 3,23 (1Н, m); 2,51-2,14 (2Н, m); 1,85-1,18 (10Н, m).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРИМЕНЕНИЕ НИТРООКСИПРОИЗВОДНОГО ЛЕКАРСТВЕННОГО СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ МЫШЕЧНОЙ ДИСТРОФИИ | 2007 |
|
RU2429828C2 |
| ВЫДЕЛЯЮЩИЕ ОКСИД АЗОТА СОЕДИНЕНИЯ | 2007 |
|
RU2560144C2 |
| АМОРФНЫЕ ИНГИБИТОРЫ ГМГ-КоА-РЕДУКТАЗЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2309141C2 |
| НИТРООКСИПРОИЗВОДНЫЕ ЛОЗАРТАНА, ВАЛСАРТАНА, КАНДЕСАРТАНА, ТЕЛМИСАРТАНА, ЭПРОСАРТАНА И ОЛМЕСАРТАНА В КАЧЕСТВЕ БЛОКАТОРОВ РЕЦЕПТОРОВ АНГИОТЕНЗИНА II ДЛЯ ЛЕЧЕНИЯ СЕРДЕЧНО-СОСУДИСТЫХ ЗАБОЛЕВАНИЙ | 2004 |
|
RU2374240C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ НАРУШЕНИЙ ЛИПИДНОГО ОБМЕНА | 2001 |
|
RU2246302C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ СТАТИНОВ С ПРЕБИОТИКОМ ДЛЯ ТЕРАПИИ ГИПЕРХОЛЕСТЕРИНЕМИИ И ГИПЕРЛИПИДИМИИ | 2014 |
|
RU2591079C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ VI И VII КАЛЬЦИЕВОЙ СОЛИ АТОРВАСТАТИНА | 2002 |
|
RU2304139C2 |
| СПОСОБ СИНТЕЗА АТОРВАСТАТИНА И ПРОМЕЖУТОЧНЫЙ ФЕНИЛБОРОНАТ (ВАРИАНТЫ) | 2001 |
|
RU2269515C2 |
| НИТРООКСИПРОИЗВОДНЫЕ СТЕРОИДОВ | 2006 |
|
RU2415864C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ МЕВАЛОНОВОЙ КИСЛОТЫ, ИНГИБИРУЮЩИХ ГМГ-СОА РЕДУКТАЗУ | 2003 |
|
RU2335500C2 |
Изобретение относится к новым производным статина формулы:
или его фармацевтически приемлемой соли или стереоизомеру, где
X означает -О-,
R означает остаток статина формулы:
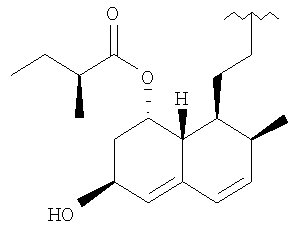
Y означает а) прямой или разветвленный С1-С20алкилен, преимущественно С1-С10, необязательно замещенный одним или более ОН;
b) 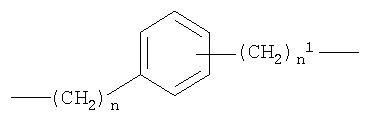
где n равно от 0 до 20, и n1 от 1 до 20;
при условии, что когда Y означает b), группа -ONO2 связана с -(СН2)n 1;
g) 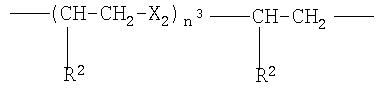
где Х2 означает -О-, n3 от 1 до 6, преимущественно от 1 до 4, R2 означает Н или СН3. Соединения I обладают противовоспалительной, антитромботической и антитромбоцитарной активностью, что позволяет использовать их для приготовления лекарственного препарата для снижения уровней холестерина и триглициридов и/или увеличения уровней HDL-C. 4 н. и 10 з.п. ф-лы, 6 табл.
1. Соединение общей формулы (I) или его фармацевтически приемлемая соль или стереоизомер
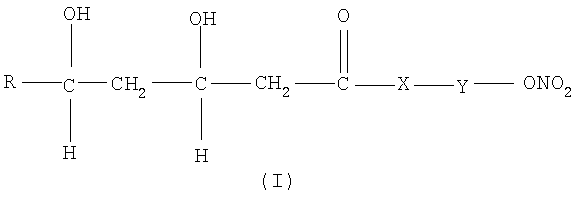
где X означает -О-,
R представляет остаток статина формулы
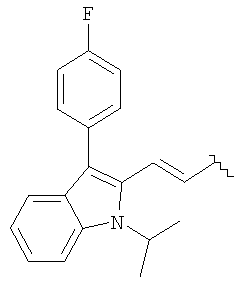
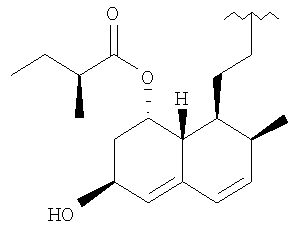
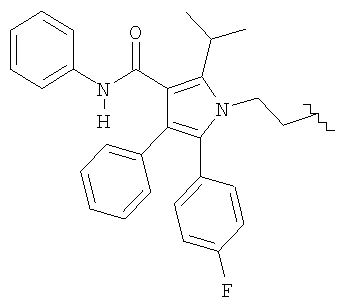
Y означает двухвалентный радикал, имеющий следующие значения:
а) прямой или разветвленный C1-С20алкилен, преимущественно C1-C10, необязательно замещенный одним или более заместителями, выбранными из гидрокси,
b)
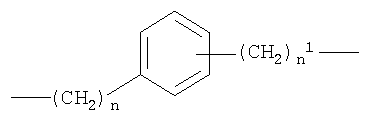
где n от 0 до 20, и n1 от 1 до 20;
при условии, что когда Y выбирают из двухвалентных радикалов, обозначенных под b), группа -ONO2 связана с -(CH2)n 1;
g)
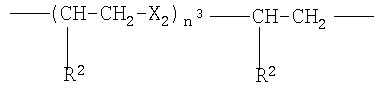
где Х2 означает -О-, n3 от 1 до 6, преимущественно от 1 до 4, R2 означает Н или СН3;
2. Соединение общей формулы (I) и/или его фармацевтически приемлемая соль или стереоизомер по п.1, где
X означает -О-;
R представляет остаток статина по п.1;
Y представляет двухвалентный радикал, имеющий следующие значения:
а) прямой или разветвленный С1-С10алкилен,
b)
где n от 0 до 5, и n1 от 1 до 5;
при условии, что когда Y выбирают из двухвалентных радикалов, обозначенных под b), группа -ONO2 связана с -(СН2)n 1;
g)
где Х2 означает -О-, n3 от 1 до 4, преимущественно 1, R2 как указано в п.1;
3. Соединение формулы (I) по п.1, которое представляет собой 4-(нитроокси)бутиловый эфир правастатина, флувастатина и аторвастатина.
4. Соединение формулы (I) по п.1, которое представляет собой 4-(нитрооксиметил)-бензиловый эфир правастатина, флувастатина и аторвастатина.
5. Соединение формулы (I) по п.1, которое представляет собой 3-(нитрооксиметил)-бензиловый эфир правастатина, флувастатина и аторвастатина.
6. Соединение формулы (I) по п.1, которое представляет собой 2-(нитрооксиметил)-бензиловый эфир правастатина, флувастатина и аторвастатина.
7. Соединение формулы (I) по п.1, которое представляет собой 4-(нитрооксиметил)-фениловый эфир правастатина, флувастатина и аторвастатина.
8. Соединение формулы (I) по п.1, которое представляет собой 3-(нитрооксиметил)-фениловый эфир правастатина, флувастатина и аторвастатина.
9. Соединение формулы (I) по п.1, которое представляет собой 2- (нитрооксиметил)-фениловый эфир правастатина, флувастатина и аторвастатина.
10. Соединение формулы (I) по п.1, которое представляет собой 2-[2′-(нитроокси)этилокси]-этиловый эфир правастатина, флувастатина и аторвастатина.
11. Соединение формулы (I) по п.1 для применения в качестве лекарственного средства, обладающего противовоспалительной, антитромботической и антитромбоцитарной активностью.
12. Применение соединения по п.1 для приготовления лекарственного препарата, обладающего противоспалительной, антитромботической и антитромбоцитарной активностью.
13. Применение соединения по п.1 для приготовления лекарственного препарата для снижения уровней холестерина и триглицеридов и/или увеличения уровней HDL-C.
14. Фармацевтическая композиция, обладающая противовоспалительной, антитромботической и антитромбоцитарной активностью и включающая фармацевтически приемлемый носитель и фармацевтически эффективное количество соединения общей формулы (I) и/или его соль или стереоизомер в соответствии с п.1.
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| IE 904261, 03.06.1992 | |||
| 3-Метокси-4-[1-метил-5-(2-метил-4,4,4-трифторбутилкарбамоил)индол-3-илметил]-N-(2-метилфенилсульфонил)бензамид или его фармацевтически приемлемые соли в качестве антагонистов лейкотриена и полупродукты для их получения | 1990 |
|
RU2002740C1 |

