Настоящее изобретение относится к производным гидроксиалкилкрахмала, в частности к производным гидроксиалкилкрахмала, получаемым в соответствии со способом, согласно которому гидроксиалкилкрахмал подвергают реакции с первичной или вторичной аминогруппой в линкерном соединении. В соответствии с особенно предпочтительным вариантом настоящее изобретение относится к производным гидроксиалкилкрахмала, получаемым по способу, в рамках которого гидроксиалкилкрахмал подвергают реакции с первичной или вторичной аминогруппой в линкерном соединении и полученный продукт подвергают реакции с полипептидом, предпочтительно с гликопротеином и, особенно предпочтительно, с эритропоэтином через участие по меньшей мере одной другой реактивной группы в линкерном соединении. Гидроксиалкилкрахмал особенно предпочтительно представляет собой гидроксиэтилкрахмал. Согласно настоящему изобретению гидроксиалкилкрахмал и предпочтительно гидроксиэтилкрахмал подвергают реакции с линкерным соединением по его восстановленному концу, который не окисляют перед проведением данной реакции.
Гидроксиэтилкрахмал (ГЭК) представляет собой производное природного амилопектина, который подвергается деградации в организме под действием α-амилазы. ГЭК представляет собой замещенное производное углеводного полимера амилопектина, который присутствует в кукурузном крахмале в концентрации до 95 мас.%. ГЭК демонстрирует полезные биологические свойства и используется в качестве кровезаменителя и при лечении по методу гемодилюции в клинических условиях (Sommermeyer et al., 1987, Krankenhauspharmazie, 8(8), 271-278, и Weidler et al., 1991, Arzneim.-Forschung/Drug Res., 41, 494-498).
Амилопектин состоит из глюкозных фрагментов, причем в главной цепи имеются альфа-1,4-гликозидные связи, а в сайтах ветвления обнаружены альфа-1,6-гликозидные связи. Физико-химические свойства данной молекулы в основном определяются типом гликозидных связей. Из-за точечных разрывов альфа-1,4-гликозидных связей образуются спиральные структуры с шестью глюкозными мономерами на виток структуры. Физико-химические, а также биохимические свойства полимера могут быть модифицированы путем замещения. Введение гидроксиэтильной группы может быть осуществлено путем щелочного гидроксиэтилирования. При соответствующей адаптации реакционных условий можно использовать различную реакционную способность в отношении гидроксиэтилирования соответствующей гидроксильной группы в незамещенном глюкозном мономере. В этой связи, любой специалист со средним уровнем знаний в данной области может в некотором ограниченном диапазоне оказывать воздействие на характер замещения.
Некоторые способы получения производного гидроксиэтилкрахмала описаны в данной области техники.
В DE 2616086 описан процесс конъюгирования гемоглобина с гидроксиэтилкрахмалом, где на первой стадии сшивающее соединение, например бромциан, связывают с гидроксиэтилкрахмалом и затем гемоглобин связывают с промежуточным продуктом.
Одной важной областью, в которой ГЭК нашел применение, является стабилизация полипептидов, используемых, например, для введения в систему кровообращения с целью достижения определенного физиологического эффекта. Одним конкретным примером таких полипептидов является эритропоэтин, кислый гликопротеин с массой примерно 34000 кДа, который необходим для регулирования уровня эритроцитов в кровотоке.
Хорошо известная в данной области проблема, связанная с применением полипептидов и ферментов, заключается в том, что указанные белки зачастую проявляют неудовлетворительную стабильность. В особенности известно, что эритропоэтин обладает относительно коротким периодом полувыведения из плазмы (Spivak and Hogans, 1989, Blood 73, 90; McMahon et al., 1990, Blood 76, 1718). Данный факт указывает на то, что терапевтические уровни в плазме быстро теряются, и должны проводиться повторные внутривенные введения. Кроме того, в некоторых случаях наблюдается иммунный ответ против вводимых пептидов.
В основном считается, что стабильность полипептидов может быть улучшена и иммунный ответ против указанных полипептидов снижен, если полипептид связать с полимерными молекулами. В WO 94/28024 показано, что физиологически активные полипептиды, модифицированные полиэтиленгликолем (ПЭГ), демонстрируют сниженную иммуногенность и антигенность и циркулируют в кровотоке значительно дольше, чем неконъюгированные белки, то есть имеют более длительный клиренс. Однако конъюгатам ПЭГ-лекарственное средство свойственны некоторые недостатки, определяемые, в частности, тем, что они не имеют природной структуры, которая может распознаваться элементами путей деградации in vivo. В этой связи, кроме ПЭГ-конъюгатов были разработаны другие конъюгаты и белковые полимерные образования. В литературе описано множество методов сшивания различных белков и макромолекул, таких как метод с использованием полимеразы (см. например, Wong, Chemistry of protein conjugation and cross-linking, 1993, CRCS, Inc.).
Известные в настоящее время конъюгаты ГЭК-лекарственное средство имеет недостаток, связанный с тем, что ГЭК не конъюгирует сайт-специфичным образом относительно лекарственного средства. В этой связи, конъюгирование приводит к получению очень гетерогенного продукта, включающего много компонентов, которые могут быть неактивными из-за деструкции трехмерной структуры на стадии конъюгирования. В этой связи, имеется потребность в дальнейшем улучшении конъюгатов ГЭК-полипептиды, которые бы обладали улучшенными стабильностью и/или биологической активностью.
Одним из способов получения таких конъюгатов является использование в качестве исходного материала окисленной формы ГЭК, которую подвергают реакции со сшивающим соединением, после чего полученный продукт подвергают реакции с полипептидом или далее модифицируют и затем подвергают реакции с полипептидом. Основным недостатком такого метода является то, что на первой стадии исходный ГЭК должен быть селективно окислен, в основном по его восстановленному концу, путем окисления терминальной альдегидной группы и/или гемиацетальной группы до лактона, что делает процесс в целом более сложным и дорогостоящим.
В WO 02/08079 А2 описаны соединения, включающие конъюгат активного средства и гидроксиалкилкрахмала, где активное средство и гидроксиалкилкрахмал соединяются либо непосредственно, либо через линкерное соединение. В случае непосредственного соединения реакцию активного средства и гидроксиалкилкрахмала проводят в водной среде, которая включает по меньшей мере 10 мас.% воды. В настоящее время отсутствуют примеры, относящиеся к производному гидроксиалкилкрахмала, получаемому посредством взаимодействия гидроксиалкилкрахмала по его восстановленному концу со сшивающим соединением, включающим структурную единицу -NH-, в водной среде. Все примеры относятся к гидроксиалкилкрахмалу, который вначале окисляют перед проведением другой реакции, так что конкретному описанию, которое приведено в WO 02/08079 А2, также свойственны указанные выше недостатки.
В этой связи, целью по настоящему изобретению является разработка способа получения производного гидроксиалкилкрахмала, который позволял бы осуществлять реакцию гидроксиалкилкрахмала по его восстановленному концу с соответствующим соединением, причем восстановленный конец указанного крахмала при этом не подвергают окислению перед проведением данной реакции.
Другим объектом по настоящему изобретению является разработка способа получения производного гидроксиалкилкрахмала, который позволял бы осуществлять реакцию гидроксиалкилкрахмала по его восстановленному концу с соответствующим соединением, причем восстановленный конец указанного крахмала при этом не подвергают окислению перед проведением данной реакции, указанный способ также характеризуется тем, что продукт взаимодействия гидроксиалкилкрахмала по его восстановленному концу с соответствующим соединением далее подвергают реакции по меньшей мере с одним другим соединением.
Еще одним объектом по настоящему изобретению является разработка описанного выше способа, в соответствии с которым по меньшей мере одно другое соединение представляет собой полипептид, предпочтительно белок и более предпочтительно эритропоэтин.
Еще один объект по настоящему изобретению относится к производному гидроксиалкилкрахмала, получаемому согласно описанному выше способу, который включает реакцию гидроксиалкилкрахмала по его восстановленному концу с соответствующим соединением, причем восстановленный конец крахмала не подвергают окислению перед проведением реакции.
В этой связи, настоящее изобретение относится к способу получения производного гидроксиалкилкрахмала, включающему реакцию гидроксиалкилкрахмала (ГАК) формулы (I)

по его восстановленному концу, который не окисляют перед проведением указанной реакции, с соединением формулы (II)
R'-NH-R", (II)
где R1, R2 и R3 обозначают независимо водород или линейную или разветвленную гидроксиалкильную группу и где любой из R', или R", или R' и R" включают по меньшей мере одну функциональную группу X, способную взаимодействовать по меньшей мере с одним другим соединением до или после реакции (I) и (II).
В контексте настоящего описания термин "гидроксиалкилкрахмал" (ГАК) относится к производным крахмала, которые содержат замещение по меньшей мере одной гидроксиалкильной группой. В этой связи, термин "гидроксиалкилкрахмал" в контексте настоящего описания не ограничивается соединениями, в которых терминальный углеводный фрагмент включает гидроксиалкильные группы R1, R2 и/или R3, как показано, например, для ясности в формуле (I), но также относится к соединениям, в которых по меньшей мере одна гидроксильная группа присутствует в любом месте: на терминальном углеводном фрагменте и/или в оставшейся части молекулы, ГАК', при замещении гидроксиалкильной группой R1, R2 и R3.
В контексте настоящего описания алкильная группа может быть линейной или разветвленной алкильной группой, которая может быть соответствующим образом замещена. Предпочтительно гидроксиалкильная группа содержит от 1 до 10 атомов углерода, более предпочтительно от 1 до 6 атомов углерода, более предпочтительно от 1 до 4 атомов углерода и еще более предпочтительно от 2 до 4 атомов углерода. "Гидроксиалкилкрахмал" также предпочтительно включает гидроксиэтилкрахмал, гидроксипропилкрахмал и гидроксибутилкрахмал, где гидроксиэтилкрахмал и гидроксипропилкрахмал являются особенно предпочтительными.
Гидроксиалкилкрахмал, включающий две или более разных гидроксиалкильных группы, также входит в область по настоящему изобретению.
По меньшей мере одна гидроксиалкильная группа, входящая в ГАК, может содержать две или более гидроксигруппы. В соответствии с предпочтительным вариантом осуществления по настоящему изобретению, по меньшей мере одна гидроксиалкильная группа, входящая в ГАК, содержит одну гидроксигруппу.
Выражение «гидроксиалкилкрахмал» также включает производные, в которых алкильная группа является моно- или полизамещенной группой. В данном контексте предпочтительно, чтобы алкильная группа была замещена галогеном, в особенности атомом фтора, или арильной группой, при условии, что ГАК остается водорастворимым. Кроме того, терминальная гидроксигруппа гидроксиалкила может быть этерифицирована с образованием простого или сложного эфира.
Кроме того, вместо алкила могут также использоваться линейные или разветвленные, замещенные или незамещенные алкеновые группы.
Гидроксиалкилкрахмал представляет собой эфирное производное крахмала. В контексте настоящего изобретения, кроме указанных производных в виде простого эфира, могут также использоваться другие производные крахмала. Например, могут использоваться производные, которые включают этерифицированные гидроксигруппы. Такие производные могут представлять собой, например, производные незамещенных моно- или дикарбоновых кислот, включающих 2-12 атомов углерода, или их замещенных производных. Особенно подходят для использования производные незамещенных монокарбоновых кислот, включающие 2-6 атомов углерода, в особенности производные уксусной кислоты. В контексте по настоящему изобретению предпочтительны ацетилкрахмал, бутилкрахлмал и пропилкрахмал.
Кроме того, предпочтительны производные незамещенных дикарбоновых кислот, включающих 2-6 атомов углерода.
В случае производных дикарбоновых кислот полезно, если вторая карбоксигруппа в дикарбоновой кислоте также этерифицирована. Кроме того, в контексте по настоящему изобретению также пригодны производные сложных моноалкиловых эфиров дикарбоновых кислот.
В случае замещенных моно- или дикарбоновых кислот могут быть предпочтительны те же замещающие группы, что и указанные выше для случая замещенных алкильных остатков.
Методики этерификации известны в данной области техники (см. например, Klemm D. et al., Comprehensive Cellulose Chemistry Vol.2, 1998, Whiley-VCH, Wienheim, New York, см. особенно главу 4.4, Esterification of Celulose (ISBN 3-527-29489-9)).
Гидроксиэтилкрахмал (ГЭК) наиболее предпочтителен для всех вариантов осуществления по настоящему изобретению.
В этой связи, настоящее изобретение также относится к описанному выше способу, в котором гидроксиалкилкрахмал представляет собой гидроксиэтилкрахмал.
ГЭК преимущественно может быть охарактеризован определенным распределением по молекулярному весу и степенью замещения. Имеются две возможности характеристики степени замещения:
1. Степень замещения может быть описана относительно части замещенных глюкозных мономеров в сравнении с соответствующим показателем для всех глюкозных фрагментов (СЗ (DS)).
2. Степень замещения может быть охарактеризована как "молярное замещение" (МЗ (MS)), когда описывают количество гидроксиэтильных групп, приходящихся на фрагмент глюкозы.
Растворы ГЭК присутствуют в виде полидисперсных композиций, где каждая молекула отличается от другой степенью полимеризации, числом и характером сайтов ветвления, а также характером замещения. В этой связи, ГЭК представлен смесью соединений с разной молекулярной массой. Следовательно, конкретный раствор ГЭК может быть определен по средней молекулярной массе с помощью статистических методов. В данном контексте Mn вычисляют как среднее арифметическое значение, зависящее от числа молекул. Альтернативно, MW, как среднее значение молекулярной массы, отражает единицу, которая зависит от массы ГЭК.
В контексте настоящего описания гидроксиэтилкрахмал может иметь значение средней молекулярной массы (средний вес) от 1 до 300 кДа, где средняя молекулярная масса, равная 5-100 кДа, является более предпочтительной. Гидроксиэтилкрахмал может также демонстрировать степень молярного замещения от 0,1 до 0,8 и соотношение между С2:С6-замещением в диапазоне 2-20 относительно гидроксиэтильных групп.
Что касается остатков R1, R2 и R3 в формуле (I), то отсутствуют какие-либо конкретные ограничения, при условии, что соединение (I) сохраняет способность взаимодействовать с соединением формулы (II). В соответствии с предпочтительным вариантом осуществления по настоящему изобретению R1, R2 и R3 обозначают независимо атом водорода или гидроксиалкильную группу, гидроксиарильную группу, гидроксиаралкильную группу или гидроксиалкарильную группу, включающую от 1 до 10 атомов углерода. Водород и гидроксиалкильные группы, включающие от 1 до 6 атомов углерода, являются предпочтительными. Алкильная, арильная, аралкильная и/или алкарильная группа может быть линейной или разветвленной и может быть соответствующим образом замещена.
В этой связи, настоящее изобретение также относится к описанному выше способу, в котором R1, R2 и R3 обозначают независимо атом водорода или линейную или разветвленную гидроксиалкильную группу, включающую от 1 до 6 атомов углерода.
Таким образом, R1, R2 и R3 могут представлять собой гидроксигексил, гидроксипентил, гидроксибутил, гидроксипропил, такой как 1-гидроксипропил, 2-гидроксипропил, 3-гидроксипропил, 1-гидроксиизопропил, 2-гидроксиизопропил, гидроксиэтил, такой как 1-гидроксиэтил, 2-гидроксиэтил, или гидроксиметил. Водород и гидроксиэтильные группы являются предпочтительными, тогда как водород и 2-гидроксиэтильная группа являются особенно предпочтительными.
В этой связи, настоящее изобретение относится также к описанному выше способу, в соответствии с которым R1, R2 и R3 обозначают независимо атом водорода или 2-гидроксиэтильную группу.
В соответствии с настоящим изобретением гидроксиалкилкрахмал подвергают реакции с соединением формулы (II), где соединение (II) может быть подвергнуто реакции с другим соединением перед проведением реакции с соединением (I) с образованием производного гидроксиалкилкрахмала. Что касается соединения (II), то отсутствуют какие-либо конкретные ограничения, при условии, что соединение (II) сохраняет способность взаимодействовать через NH-группу, соединяющую мостиковой связью R' и R", с соединением (I) по восстановленному концу, который не окисляют, с образованием производного гидроксиалкилкрахмала.
Предпочтительные остатки R' в соединении (II) включают водород или алкильный, циклоалкильный, арильный, аралкильный, арилциклоалкильный, алкарильный или циклоалкиларильный остаток, где указанный циклоалкильный, арильный, аралкильный, арилциклоалкильный, алкарильный или циклоалкиларильный остаток может быть присоединен непосредственно к NH-группе, соединяющей мостиковой связью R' и R" в соединении (II), или, в соответствии с другим вариантом осуществления по настоящему изобретению, может быть присоединен через кислородный мостик к NH-группе, соединяющей мостиковой связью R' и R" в соединении (II). Алкильный, арильный, аралкильный или алкарильный остатки могут быть соответствующим образом замещены. В качестве предпочтительных заместителей можно указать галогены, такие как F, Cl или Br. Особенно предпочтительными остатками R' являются водород, алкильные и алкокси группы и еще более предпочтительными являются водород и незамещенные алкильные и алкокси группы.
В этой связи, настоящее изобретение также относится к описанному выше способу, в соответствии с которым R' обозначает водород или линейную или разветвленную алкильную или алкокси группу.
В числе алкильных и алкокси групп предпочтительными являются группы, включающие 1, 2, 3, 4, 5 или 6 атомов С. Более предпочтительными являются метильная, этильная, пропильная, изопропильная, метокси, этокси, пропокси и изопропокси группы. Особенно предпочтительными являются метил, этил, метокси, этокси и особое предпочтение отдается метилу или метоксигруппе.
В этой связи, настоящее изобретение относится к описанному выше способу, в соответствии с которым R' обозначает водород, или метильную, или метоксигруппу.
Кроме функциональной группы X, R" может включать по меньшей мере одну дополнительную функциональную группу W. Указанная по меньшей мере одна дополнительная функциональная группа W в основном может находиться в любом положении R". Предпочтительно W непосредственно присоединяют к NH-группе, связанной с R'.
В основном отсутствуют какие-либо конкретные ограничения относительно функциональной группы W, при условии, что соединение (I) сохраняет способность взаимодействовать с соединением (II). В предпочтительных вариантах осуществления по изобретению функциональная группа W включает структурную единицу -NH- и/или структурную единицу -(C=G)-, где G обозначает O или S и/или структурную единицу -SO2-. В соответствии с более предпочтительными вариантами осуществления по настоящему изобретению функциональную группу W выбирают из группы, состоящей из:

и, в том случае, когда G присутствует дважды, обозначает независимо O или S.
В соответствии с предпочтительными вариантами осуществления по настоящему изобретению, если R' обозначает H и W присоединяют непосредственно к NH-группе, соединяющей мостиковой связью R' и R", то R' и NH-группа, соединяющая мостиковой связью R' и R", образуют вместе с W одну из следующих групп:

Что касается по меньшей мере одной функциональной группы Х, которая включается в R' или R", предпочтительно в R", то отсутствуют какие-либо конкретные ограничения. В основном возможны все функциональные группы, если они позволяют проводить реакцию по меньшей мере с одним другим соединением.
Что касается реакции с другим соединением, то возможны все виды взаимодействий по меньшей мере одной функциональной группы по меньшей мере с одним другим соединением. В качестве возможных, в числе других, можно отметить возможность реакций по меньшей мере одной функциональной группы Х с другим соединением, которые приводят к образованию ковалентной связи, ионной связи и/или к образованию ван-дер-ваальсовых связей, при этом ковалентная связь является особенно предпочтительной.
В числе других можно указать следующие функциональные группы Х:
- С-С-двойные связи или С-С-тройные связи или ароматические С-С-связи;
- тиогруппа или гидроксигруппы;
- гидразид алкилсульфоновой кислоты, гидразид арилсульфоновой кислоты;
- 1,2-диолы;
- 1,2-аминоспирты;
- аминогруппа -NH2 или производные аминогруппы, включающие структурную единицу -NH, такие как аминоалкильные группы, аминоарильная группа, аминоаралкильные группы или алкариламиногруппы;
- гидроксиламиногруппа -О-NH2 или производные гидроксиламиногруппы, включающие структурную единицу -О-NH, такие как гидроксилалкиламиногруппы, гидроксилариламиногруппы, гидроксиларалкиламиногруппы или гидроксилалкариламиногруппы;
- алкоксиаминогруппы, арилоксиаминогруппы, аралкилоксиаминогруппы или алкарилоксиаминогруппы, каждая из которых включает структурную единицу -NH-О;
- остатки, содержащие карбонильную группу, -Q-C(=G)-M, где G обозначает O или S и M обозначает, например,
-OH или -SH;
- алкоксигруппу, арилоксигруппу, аралкилоксигруппу или алкарилоксигруппу;
- алкилтиогруппу, арилтиогруппу, алкарилтиогруппу или аралкилтиогруппу;
- алкилкарбонилоксигруппу, арилкарбонилоксигруппу, аралкилкарбонилоксигруппу, алкарилкарбонилоксигруппу;
- активированные сложные эфиры, такие как сложные эфиры гидроксиламинов, содержащие имидную структуру, такие как N-гдроксисукцинимид, или структурную единицу O-N, где N является частью гетероарильного соединения, или в случае, когда G=O и Q отсутствует, например, арилокси соединения с замещенным арильным остатком, такие как пентафторфенил, паранитрофенил или трихлорфенил;
где Q отсутствует или обозначает NH или гетероатом, такой как S или O;
-NH-NH2 или -NH-NH-;
-NO2;
- нитрильную группу;
- карбонильные группы, такие как альдегидная группа или кетогруппа;
- карбоксигруппу;
-N=C=O группу или -N=C=S группу;
- винилгалогенидные группы, такие как винилиодидная или винилбромидная группа, или винилтрифлат;
-C=C-H;
-(C=NH2Cl)-O-алкил;
- группы -(C=O)-CH2-Hal, где Hal обозначает Cl, Br или I;
- -CH=CH-SO2-;
- дисульфидную группу, включающую структуру -S-S-;
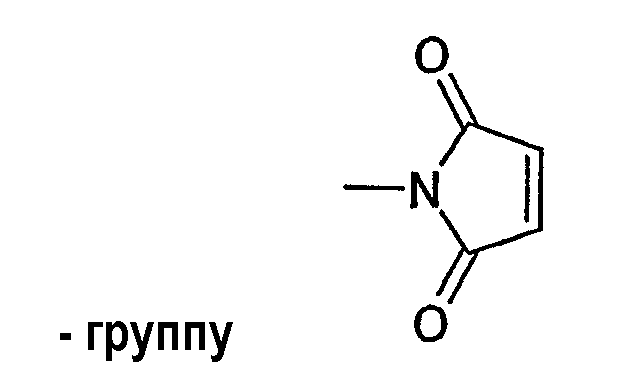
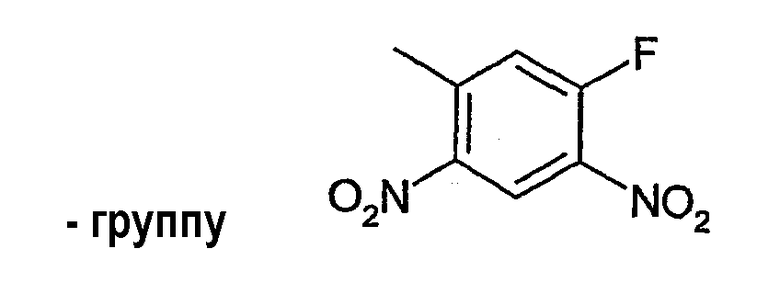
Среди указанных групп особенно предпочтительны тиогруппа, аминогруппа, гидроксиламиногруппа, алкоксиаминогруппы и приведенные ниже группы:
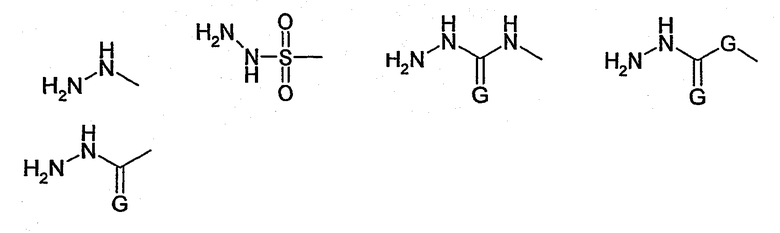
В этой связи, настоящее изобретение также относится к описанному выше способу, в котором по меньшей мере одну функциональную группу Х выбирают из группы, состоящей из -SH, -NN2, -O-NH2, -NH-O-алкила, -(C=G)-NH-NH2, -G-(C=G)-NH-NH2, -NH-(C=G)-NH-NH2 и SO2-NH-NH2, где G обозначает O или S и, если G присутствует дважды, обозначает независимо O или S.
Что касается алкоксиаминогрупп, то особое предпочтение отдается пропоксиаминогруппе, этоксиаминогруппе и метоксиаминогруппе, при этом метоксиаминогруппа -NH-O-CH3 является особенно предпочтительной.
В соответствии с еще одним аспектом по настоящему изобретению по меньшей мере одна функциональная группа Х может быть группой, которая не способна взаимодействовать непосредственно с другим соединением, но которая может быть химически модифицирована для придания ей способности взаимодействовать желательным образом. Указанная модификация функциональной группы Х, включенной в соединение (II), может быть проведена перед реакцией соединения (II) с соединением (I) или после реакции соединения (II) с соединением (I). Если соединение (II) включает по меньшей мере две, необязательно химически разные функциональные группы Х, то возможно провести модификацию по меньшей мере одной функциональной группы Х перед реакцией соединения (II) с соединением (I) и по меньшей мере одной функциональной группой Х после реакции соединения (II) с соединением (I).
Примером функциональной группы, подлежащей модификации перед реакцией с другим соединением, может служить 1,2-аминоспирт или 1,2-диол, которые модифицируют, например, путем окисления с образованием альдегидной или кето группы.
Другим примером функциональной группы Х, подлежащей модификации перед реакцией с другим соединением, является -NH2-группа, которую модифицируют посредством реакции, например, с соединением формулы

с образованием структуры, соответствующей формуле

которая обладает, например, реактивностью в отношении тиогруппы.
Другим примером функциональной группы Х, подлежащей модификации перед реакцией с другим соединением, является -NH2-группа, которую модифицируют посредством реакции, например, с соединением приведенной ниже формулы:

с образованием структуры, соответствующей формуле

которая обладает, например, реактивностью в отношении тиогруппы.
Указанная по меньшей мере одна функциональная группа Х может быть присоединена непосредственно к NH-группе, соединяющей мостиковой связью R' и R". Таким образом, в соответствии с одним вариантом осуществления по настоящему изобретению, функциональная группа Х эквивалентна R". Конкретные примеры соединений, в которых Х непосредственно присоединена к NH группе, соединяющей мостиковой связью R' и R", включают, в числе других
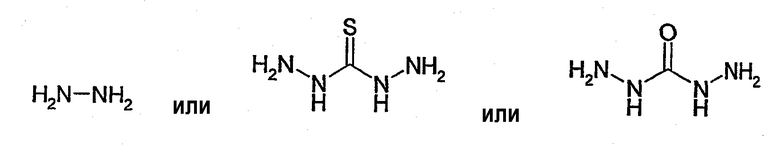
Другим конкретным примером такого соединения, который также входит в область по настоящему изобретению, является NH3.
В соответствии с другим вариантом осуществления по настоящему изобретению NH-группа, соединяющая мостиковой связью R' и R", может быть отделена от по меньшей мере одной функциональной группы Х линейной или разветвленной алкильной, или циклоалкильной, или арильной, или аралкильной, или арилциклоалкильной, или алкарильной, или циклоалкиларильной группой, где указанные группы могут включать по меньшей мере один гетероатом, такой как N, O или S, и где указанные группы могут соответствующим образом замещены. Размер группы, разделяющей NH, соединяющей мостиковой связью R' и R", и по меньшей мере одну функциональную группу Х, может быть адаптирован в соответствии с конкретными потребностями. В основном отделяющая группа включает от 1 до 60, предпочтительно от 1 до 40, более предпочтительно от 1 до 20, более предпочтительно от 1 до 10, более предпочтительно от 1 до 6 и особенно предпочтительно от 1 до 4 атомов углерода. Если присутствуют гетероатомы, то отделяющая группа включает в основном от 1 до 20, предпочтительно от 1 до 8 и особенно предпочтительно от 1 до 4 гетероатомов. В соответствии с особенно предпочтительными вариантами осуществления по настоящему изобретению отделяющая группа включает от 1 до 4 атомов кислорода. Отделяющая группа может включать необязательно разветвленную алкильную цепь или арильную группу или циклоалкильную группу, содержащую, например, от 5 до 7 атомов углерода, или может представлять собой аралкильную группу, алкарильную группу, где алкильная часть может быть линейной и/или циклической алкильной группой. В соответствии с еще более предпочтительным вариантом осуществления по настоящему изобретению отделяющая группа представляет собой алкильную цепь, включающую от 1 до 20, предпочтительно от 1 до 8, более предпочтительно от 1 до 6, более предпочтительно от 1 до 4 и особенно предпочтительно от 2 до 4 атомов углерода. В том случае, если присутствуют гетероатомы, особенно предпочтительна цепь, включающая от 1 до 4 атомов кислорода.
Конкретные примеры соединений (II), в которых группа Х отделена от NH-группы, соединяющей мостиковой связью R' и R", включают, в числе других:













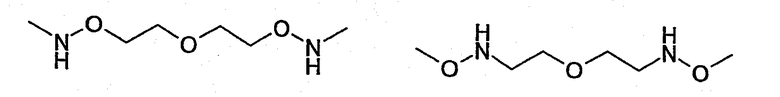

Группа, разделяющая NH, соединяющую мостиковой связью R' и R", и по меньшей мере одну функциональную группу Х, может быть соответствующим образом замещена. Предпочтительные заместители включают, например, галогениды, такие как F, Cl, Br или I.
Группа, разделяющая NH, которая соединяет мостиковой связью R' и R", и по меньшей мере одну функциональную группу Х, может включать один или более сайтов расщепления, таких как
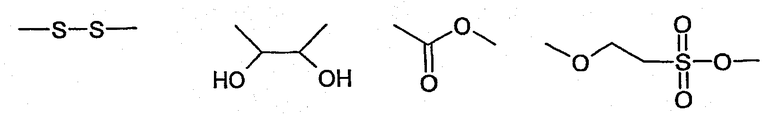
которые позволяют легко расщеплять полученные соединения по заданному сайту.
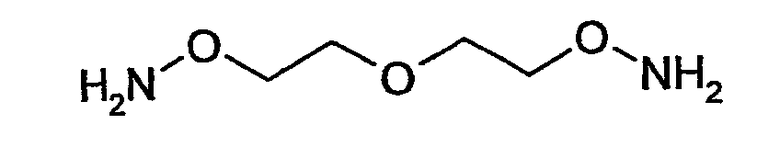
В соответствии с особенно предпочтительным вариантом осуществления по настоящему изобретению соединение (II) представляет собой O-[2-(2-аминооксиэтокси)этил]гидроксиламин
или карбогидразид
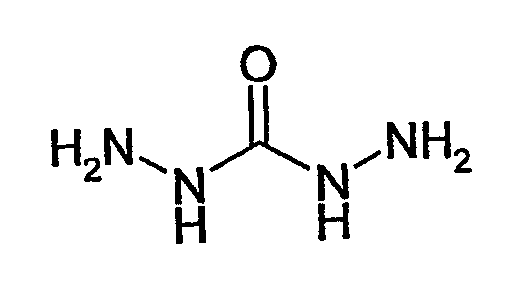
В этой связи, настоящее изобретение относится также к описанному выше способу, в соответствии с которым соединение (II) представляет собой O-[2-(2-аминооксиэтокси)этил]гидроксиламин или карбогидразид.
В том случае, когда соединение (II) включает один или более хиральных центров, указанное соединение (II) может присутствовать в R-конформации, или в S-конформации, или в виде рацемического соединения относительно каждого из хиральных центров.
Как отмечалось выше, соединение (I) может взаимодействовать с соединением (II) как таковым или с соединением (II), которое подвергли реакции по меньшей мере с одним другим соединением перед проведением реакции с соединением (I).
Реакция соединения (I) с соединением (II) может быть проведена по меньшей мере в одном подходящем растворителе. Соответствующий растворитель или смесь двух или более растворителей могут быть подобраны в соответствии с конкретными условиями реакции и в соответствии с химической природой соединения (I) и соединения (II). В соответствии с особенно предпочтительным вариантом осуществления по настоящему изобретению в качестве растворителя используют воду, либо одну, либо в сочетании по меньшей мере с одним другим растворителем. В качестве по меньшей мере одного другого растворителя можно отметить ДМСО, ДМФ, метанол и этанол. Предпочтительные растворители, отличные от воды, включают ДМСО, ДМФ, метанол и этанол.
В этой связи, настоящее изобретение также относится к описанному выше способу, в соответствии с которым реакцию соединения (I) с соединением (II) проводят в водной системе.
Термин "водная система" в контексте настоящего описания относится к растворителю или смеси растворителей, включающих воду в количестве по меньшей мере 10 мас.%, предпочтительно по меньшей мере 50 мас.%, более предпочтительно по меньшей мере 80 мас.%, еще более предпочтительно по меньшей мере 90 мас.% и до 100 мас.% относительно веса используемых растворителей. Предпочтительной реакционной средой являет вода.
Что касается температур, поддерживаемых в ходе данной реакции, то отсутствуют какие-либо ограничения, при условии, что реакция приводит к получению желательного производного гидроксиалкилкрахмала.
В том случае, когда соединение (I) подвергают реакции с соединением (II), и соединение (II) представляет собой гидроксиламин или гидразид, то температуру в реакции поддерживают предпочтительно в диапазоне от 5°С до 45°С, более предпочтительно в диапазоне от 10 до 30°С и особенно предпочтительно в диапазоне от 15 до 25°С.
В том случае, когда соединение (I) подвергают реакции с соединением (II), и указанная реакция является восстановительным аминированием, то температуру предпочтительно поддерживают на уровне 100°С, более предпочтительно в диапазоне от 20°С до 95°С, более предпочтительно в диапазоне от 25°С до 90°С и наиболее предпочтительно в диапазоне от 70°С до 90°С и особенно предпочтительно в диапазоне от 75°С до 85°С.
В этой связи, настоящее изобретение также относится к описанному выше способу, в соответствии с которым реакцию соединения (I) и соединения (II), когда соединение (II) представляет собой гидроксиламин или гидразид, проводят при температуре от 5°С до 45°С.
В этой связи, настоящее изобретение также относится к описанному выше способу, в соответствии с которым реакцию соединения (I) и соединения (II), когда указанная реакция представляет собой восстановительное аминирование, проводят при температуре от 25°С до 90°С.
В ходе реакции температурой можно варьировать, предпочтительно в указанном выше диапазоне, или поддерживать по существу на постоянном уровне.
Длительность реакции соединения (I) с соединением (II) может быть адаптирована к конкретным потребностям и в основном составлять от 1 часа до 7 дней.
В том случае, когда соединение (II) представляет собой гидроксиламин или гидразид, длительность реакции составляет предпочтительно от 1 часа до 3 дней и более предпочтительно от 2 часов до 48 часов.
В том случае, если реакция соединения (I) и соединения (II) представляет собой восстановительное аминирование, длительность реакции предпочтительно составляет от 2 часов до 7 дней.
Величина рН в реакции соединения (I) и соединения (II) может быть адаптирована к конкретным потребностям, таким как химическая природа реагирующих соединений.
В том случае, когда соединение (II) представляет собой гидроксиламин или гидразид, то величина рН составляет предпочтительно от 4,5 до 6,5.
В том случае, когда реакция соединения (1) и соединения (II) представляет собой восстановительное аминирование, то величина рН составляет предпочтительно от 8 до 12.
В этой связи, настоящее изобретение относится к описанному выше способу, в котором реакцию соединения (I) и соединения (II), когда соединение (II) представляет собой гидроксиламин или гидразид, проводят при pH от 4,5 до 6,5.
В этой связи, настоящее изобретение относится к описанному выше способу, в соответствии с которым реакцию соединения (I) и соединения (II), когда указанная реакция представляет собой восстановительное аминирование, проводят при pH от 8 до 12.
Конкретные примеры указанных выше условий реакции включают, например, температуру реакции, составляющую примерно 25°С, и pH примерно 5,5 в том случае, когда указанное соединение представляет собой гидроксиламин, и температуру реакции, составляющую примерно 80°С, и pH примерно 11 в том случае, когда реакция соединения (I) и соединения (II) представляет собой восстановительное аминирование.
Соответствующая величина pH в реакционной системе может быть откорректирована путем добавления по меньшей мере одного приемлемого буфера. Среди предпочтительных буферов следует отметить натрий-ацетатный буфер, фосфатный или боратный буферы.
В соответствии с предпочтительным вариантом осуществления по настоящему изобретению реакционный продукт, получаемый при взаимодействии соединения (I) и соединения (II), подвергают реакции по меньшей мере с одним другим соединением через участие по меньшей мере одной функциональной группы Х.
При необходимости, по меньшей мере одна функциональная группа Х может быть защищена с помощью по меньшей мере одной защитной группы перед проведением реакции соединения (I) с соединением (II). В данном случае возможны для использования все реально доступные защитные группы, которые препятствуют реакции соединения (II) с соединением (I) через по меньшей мере одну функциональную группу Х. В этой связи, защитная группа может быть выбрана в зависимости от химической природы функциональной группы Х, подлежащей защите, включая, например, растворитель, в котором проходит реакция, или рН реакционной смеси. Предпочтительные защитные группы включают, в числе других, бензилоксикарбонильную группу, трет-бутоксикарбонильную группу, метоксифенильную группу, 2,4-диметоксифенильную группу, триарилметильные группы, тритильную группу, монометокситритильную группу, диметокситритильную группу, монометилтритильную группу, диметилтритильную группу, трифторацетильную группу, фталиминовые соединения, 2-(триалкилсилил)этоксикарбонильные соединения, Fmoc-группу, трет-бутильную группу или триалкилсилильные группы.
Если в соединении (II) имеются две или более функциональных групп Х, то по меньшей мере одна группа может быть защищена, тогда как другую группу можно оставить незащищенной.
После реакции соединения (I) и соединения (II) по меньшей мере одна защитная группа может быть оставлена в реакционном продукте или удалена соответствующими способами, такими как традиционные методики, известные специалистам в данной области. Если две разных функциональных группы Х подвергают защите подходящими защитными группами, возможно удалить по меньшей мере одну защитную группу, оставляя, таким образом, по меньшей мере одну функциональную группу Х доступной для последующей реакции по меньшей мере с одним другим соединением и сохраняя по меньшей мере одну другую функциональную группу в защищенном состоянии до реакции продукта взаимодействия соединения (I) и соединения (II) с другим соединением. После этого защитная группа от все еще защищенной функциональной группы может быть удалена, что делает оставшуюся функциональную группу Х доступной для реакции с еще одним соединением.
Использование по меньшей мере одной защитной группы может быть важно для предотвращения реакции, приводящей к образованию производного гидроксиалкилкрахмала, включающего соединение (II), как результат реакции с двумя или более соединениями (I), т.е. соединения (II) с множественным замещением молекулами ГАК. Тот же результат, однако, может быть достигнут посредством реакции соединения (I) с избытком соединения (II). Если в способе по настоящему изобретению используют избыток соединения (II), то молярное соотношение соединения (II) к соединению (I) составляет предпочтительно в диапазоне от 2 до 100.
Образованный при взаимодействии соединения (I) с соединением (II) реакционный продукт может быть выделен из реакционной смеси по меньшей мере одним подходящим способом. При необходимости, продукт реакции может быть осажден перед выделением посредством по меньшей мере одного приемлемого способа.
Если реакционный продукт вначале осаждают, то возможно, например, привести в контакт реакционную смесь по меньшей мере с одним растворителем или смесью растворителей, отличных от растворителя или смеси растворителей, присутствующих в реакции, при определенных температурах. В соответствии с особенно предпочтительным вариантом осуществления по настоящему изобретению, если в качестве растворителя используется вода, то реакционную смесь приводят в контакт со смесью этанола и ацетона, взятых предпочтительно в соотношении 1:1, что означает использование равных объемов указанных соединений при температуре, составляющей предпочтительно от -20 до +50°С, и особенно предпочтительно от 0 до 25°С.
Выделение реакционного продукта может быть осуществлено подходящим способом, который может включать несколько стадий. В соответствии с предпочтительным вариантом осуществления по настоящему изобретению реакционный продукт вначале отделяют от реакционной смеси или смеси реакционной смеси, например, со смесью этанол-ацетон соответствующим способом, таким как центрифугирование или фильтрование. На второй стадии отделенный реакционный продукт может быть подвергнут дополнительной обработке, такой как пост-обработка типа диализа, фильтрования при центрифугировании или фильтрования под давлением, ионообменной хроматографии, ВЭЖХ, ЖХУД (MPLC), гель-фильтрации и/или лиофилизации. В соответствии с еще более предпочтительным вариантом осуществления по настоящему изобретению отделенный реакционный продукт вначале диализуют, предпочтительно против воды, и затем лиофилизируют до того момента, когда содержание растворителя в реакционном продукте станет достаточно низким, в соответствии со спецификациями для данного продукта. Лиофилизация может осуществляться при температуре от 20 до 35°С, предпочтительно от 25 до 30°С.
Выделенные таким образом реакционные продукты, полученные при взаимодействии соединения (I) и соединения (II), могут быть далее подвергнуты реакции по меньшей мере с одним другим соединением через участие по меньшей мере одной функциональной группы Х, включенной в данный реакционный продукт.
В зависимости от химической природы функциональной группы Х может использоваться любое реально доступное соединение, которое способно к образованию химической связи с группой Х. В данной реакции может использоваться один или более подходящих растворителей и все реакционные параметры, такие как температура реакции, длительность реакции, соотношение реагирующих соединений и величина pH реакционной смеси, могут быть соответствующим образом адаптированы к конкретным потребностям.
В соответствии с особенно предпочтительным вариантом осуществления по настоящему изобретению по меньшей мере одно соединение, способное к образованию химической связи по меньшей мере с одной функциональной группой Х, представляет собой полипептид или смесь по меньшей мере двух разных полипептидов.
В этой связи, настоящее изобретение также относится к описанному выше способу, в соответствии с которым реакционный продукт, полученный при взаимодействии соединения (I) и соединения (II), подвергают реакции с полипептидом через функциональную группу Х, включенную в соединение (II).
В соответствии с другим особенно предпочтительным вариантом осуществления по изобретению по меньшей мере одно другое соединение, способное к образованию химической связи по меньшей мере с одной функциональной группой Х, представляет собой сшивающее соединение, которое способно образовать первую химическую связь по меньшей мере с одной функциональной группой Х в реакционном продукте, полученном при взаимодействии соединения (I) и соединения (II), и вторую химическую связь со вторым другим соединением.
В соответствии с еще более предпочтительным вариантом осуществления по настоящему изобретению второе другое соединение представляет собой полипептид или смесь по меньшей мере двух разных полипептидов.
В контексте данного варианта по настоящему изобретению возможно осуществлять реакцию продукта взаимодействия соединения (I) и соединения (II), первого производного гидроксилалкилкрахмала, со сшивающим соединением с образованием второго производного гидроксилалкилкрахмала. Указанное второе производное гидроксилалкилкрахмала может быть впоследствии подвергнуто реакции со вторым другим соединением, предпочтительно полипептидом, с образованием третьего производного гидроксилалкилкрахмала.
Однако возможно провести реакцию продукта, полученного при взаимодействии соединения (I) и соединения (II), первого производного гидроксилалкилкрахмала, с продуктом реакции сшивающего соединения со вторым другим соединением, предпочтительно полипептидом.
В этой связи, настоящее изобретение также относится к описанному выше способу, в соответствии с которым продукт, получаемый при взаимодействии соединения (I) и соединения (II), подвергают реакции с другим соединением, где указанное другое соединение представляет собой сшивающее соединение, посредством реакции функциональной группы V, включенной в сшивающее соединение, и функциональной группы Х, включенной в продукт реакции соединения (I) и соединения (II).
В этой связи, настоящее изобретение также относится к описанному выше способу, в соответствии с которым реакционный продукт, получаемый при взаимодействии соединения (I) и соединения (II), подвергают реакции с другим соединением, где указанное другое соединение представляет собой сшивающее соединение, через реакцию функциональной группы V, включенной в сшивающее соединение, с функциональной группой Х, включенной в продукт реакции соединений (I) и (II), и где указанное сшивающее соединение было подвергнуто реакции со вторым другим соединением перед взаимодействием с продуктом реакции соединений (I) и (II).
В этой связи, настоящее изобретение также относится к описанному выше способу, в соответствии с которым второе другое соединение представляет собой полипептид, предпочтительно эритропоэтин, который подвергают реакции со сшивающим соединением через реакцию функциональной группы Х, включенной в сшивающее соединение.
В этой связи, настоящее изобретение также относится к описанному выше способу, в соответствии с которым соединение (II) подвергают реакции с первым другим соединением, предпочтительно сшивающим соединением, с образованием первого реакционного продукта, и указанный первый реакционный продукт подвергают реакции со вторым другим соединением с образованием второго реакционного продукта, и указанный второй реакционный продукт подвергают реакции с соединением (I).
В этой связи, настоящее изобретение также относится к описанному выше способу, в соответствии с которым первое другое соединение, предпочтительно сшивающее соединение, подвергают реакции со вторым другим соединением, предпочтительно полипептидом, с образованием первого реакционного продукта, и указанный первый реакционный продукт подвергают реакции с соединением (II) с образованием второго реакционного продукта, и указанный второй реакционный продукт подвергают реакции с соединением (I) с образованием производного гидроксиалкилкрахмала.
В соответствии с особенно предпочтительными вариантами осуществления по настоящему изобретению используют сшивающее соединение, позволяющее образовать химическую мостиковую связь между соединением (II) или продуктом реакции соединений (I) и (II) и вторым другим соединением, где функциональная группа во втором другом соединении, которая взаимодействует со сшивающим соединением, представляет собой -SH-группу, или альдегидную группу, или кетогруппу, и функциональная группа соединения (II) или продукта реакции соединений (I) и (II), которая взаимодействует со сшивающим соединением, представляет собой группу, включающую структуру -NH-, особенно предпочтительно -NH2.
В контексте настоящего изобретения термин "сшивающее соединение" относится к химическим соединениям, которые способны образовывать связь между соединением (I) или продуктом реакции соединений (I) и (II) и по меньшей мере одним вторым другим соединением. В зависимости от химической природы второго другого соединения сшивающее соединение включает по меньшей мере одну функциональную группу V, способную взаимодействовать с функциональной группой Х, включенной в соединение (II), или продукт реакции соединения (I) и (II), и по меньшей мере с одной другой функциональной группой, которая способна образовывать химическую связь со вторым другим соединением. Указанная по меньшей мере одна другая функциональная группа, включенная в сшивающее соединение, может представлять собой соединение того же типа, которое обсуждалось выше в контексте функциональной группы Х.
Сшивающее соединение может использоваться для увеличения общей длины химической мостиковой связи между соединением (I) и вторым другим соединением, предпочтительно полипептидом, и/или для воздействия на химическую природу полученного реакционного продукта, при наличии второго другого соединения, либо в его отсутствие, и/или обеспечить возможность создания связи между несколькими вторыми другими соединениями и продуктом реакции соединений (I), (II) и сшивающего соединения, и/или для целей химической модификации функциональной группы Х, включенной в продукт реакции соединений (I) и (II), так чтобы придать указанному реакционному продукту способность взаимодействовать с данным другим соединением.
Таким образом, варианты осуществления по настоящему изобретению, которые обсуждались выше и которые относятся к химической модификации функциональной группы Х, представляющей -NH2-группу, другим соединением, например

с целью обеспечения возможности проведения реакции с -SH группой, включенной во второе другое соединение, предпочтительно полипептид, представляют собой конкретные примеры взаимодействия продукта реакции соединений (I) и (II) со сшивающим соединением.
В соответствии с предпочтительным вариантом осуществления по настоящему изобретению функциональная группа V может представлять собой функциональную группу того же типа, который обсуждался выше в контексте группы Х.
В соответствии с другим предпочтительным вариантом осуществления по настоящему изобретению функциональная группа X или функциональная группа V представляют тиогруппу, и функциональную группу V или функциональную группу Х предпочтительно выбирают из группы, состоящей из

где Gal обозначает Cl, Br или I, предпочтительно Br или I.
В соответствии с еще одним предпочтительным вариантом осуществления по настоящему изобретению функциональную группу X или функциональную группу V выбирают из группы, состоящей из активированного сложного эфира, описанного выше, или из карбоксигруппы, которую необязательно преобразуют в активированный сложный эфир. В указанном конкретном случае функциональная группа V или функциональная группа Х соответственно включает химическую структуру -NH-.
В этой связи, сшивающее соединение представляет собой соединение, включающее по меньшей мере две функциональные группы, которые могут быть одинаковыми или разными. В случае наличия двух функциональных групп сшивающее соединение может быть гомобифункциональным или гетеробифункциональным. Гомобифункциональное сшивающее соединение, например, позволяет сформировать мостиковую связь между продуктом реакции соединений (I) и (II) и вторым другим соединением, продуктом реакции и другим соединением, включающим функциональные группы того же типа. Гетеробифункциональное сшивающее соединение, например, позволяет сформировать мостиковую связь между продуктом реакции соединений (I) и (II) и вторым другим соединением, продуктом реакции и другим соединением, включающим функциональные группы, которые не способны взаимодействовать друг с другом.
По меньше мере две функциональные группы в сшивающем соединении могут быть соединены непосредственно друг с другом или могут быть разделены линейной или разветвленной алкильной, или циклоалкильной, или арильной, или аралкильной, или арилциклоалкильной, или алкарильной, или циклоалкиларильной группами, где указанные группы могут включать по меньшей мере один гетероатом, такой как N, O, S, и где указанные группы могут быть соответствующим образом замещены. Длина группы, разделяющей по меньшей мере две функциональных группы в сшивающем соединении, может быть соответствующим образом адаптирована к конкретным потребностям. В основном, разделяющая группа включает от 1 до 60, предпочтительно от 1 до 40, более предпочтительно от 1 до 20, более предпочтительно от 1 до 10, более предпочтительно от 5 до 10 атомов углерода. Если имеются гетероатомы, то разделяющая группа включает в основном от 1 до 20, предпочтительно от 1 до 8 и особенно предпочтительно от 1 до 4 гетероатомов. В соответствии с еще более предпочтительным вариантом осуществления по настоящему изобретению указанная разделяющая группа представляет собой алкильную или аралкильную цепь, содержащую от 1 до 20 атомов углерода. Кроме того, сшивающее соединение может также включать по меньшей мере один сайт расщепления, как обсуждалось выше в контексте соединения (II).
Другие примеры сшивающих соединений, которые могут быть отмечены в контексте по настоящему изобретению, могут быть отнесены к разным группам в соответствии с прилагаемым перечнем:
соединения
В таблице 1 в конце настоящего описания перечислены некоторые предпочтительные примеры сшивающих соединений.
В том случае, если по меньшей мере одно другое соединение, например сшивающее соединение, включает один или более хиральных центров, то указанное по меньшей мере одно другое соединение может присутствовать в R-конформации или S-конформации или в виде рацемического соединения относительно каждого хирального центра.
Термин «полипептид» в контексте настоящего описания относится к соединению, которое включает по меньшей мере 2 аминокислоты, которые соединены через пептидную связь, т.е. связь структуры -(C=O)-NH-. Полипептид может быть природным или полипептид может быть неприродным, последний вариант включает природную аминокислоту и/или по меньшей мере одну аминокислоту, которая не встречается в природе. Скелет полипептида, полипептидная цепь, может быть далее замещен по меньшей мере одним заместителем, так что он будет иметь по меньшей мере одну боковую цепь. По меньшей мере одна функциональная группа Y может быть частью полипептидного скелета или по меньшей мере одного заместителя в указанном скелете, при этом возможны такие варианты осуществления по настоящему изобретению, в соответствии с которым включается по меньшей мере одна функциональная группа, которая представляет собой часть полипептидного скелета, и по меньшей мере одна функциональная группа, представляющая собой часть по меньшей мере одного заместителя в полипептидном скелете.
Что касается полипептида, то отсутствуют какие-либо ограничения, при условии, что полипептид включает по меньшей мере одну функциональную группу Y. Указанная функциональная группа Y может быть присоединена непосредственно к полипептидному скелету или может представлять собой часть боковой цепи полипептидного скелета. Данная боковая цепь, или функциональная группа Y, или обе они могут быть частью природного полипептида или же они могут быть введены, перед проведением реакции с функциональной группой Х, в природный полипептид или в полипептид, который, по меньшей мере частично, не является природным.
Кроме того, рассматриваемый полипептид может быть, по меньшей мере частично, любого происхождения: из организма человека или животного. В предпочтительном варианте полипептид имеет в качестве источника человеческий организм.
Полипептид может представлять собой цитокин, в особенности эритропоэтин, антитромбин (АТ), такой как АТ III, интерлейкин, в особенности интерлейкин-2, IFN-бета, IFN-альфа, Г-КСФ (G-CSF), КСФ (CSF), интерлейкин-6 и терапевтические антитела.
В соответствии с предпочтительным вариантом осуществления по настоящему изобретению полипептид представляет собой антитромбин (АТ), предпочтительно АТ III (Levy JH, Weisinger A, Ziomek CA, Echelard Y, Recombinant Antithrombin: Production and Role in Cardiovascular Disorder, Seminars in Thrombosis and Hemostasis 27, 4 (2001) 405-416; Edmunds T, Van Patten SM, Pollock J, Hanson E, Bernasconi R, Higgins E, Manavalan P, Ziomek C, Meade H, McPherson J, Cole ES, Transgenically Produced Human Antithrombin: Structural and Functional Comparison to Human Plasma-Derived Antithrombin, Blood 91, 12 (1998) 4661-4671; Minnema MC, Chang ACK, Jansen PM, Lubbers YTP, Pratt BM, Whittaker BG, Taylor FB, Hack CE, Friedman B, Recombinant human antithrombin III improves survival and attenuates inflammatory responses in baboons lethally challenged with Escherichia coli; Blood, 95, 4 (2000) 1117-1123; Van Patten SM, Hanson EH, Bernasconi R, Zhang K, Manavaln P, Cole ES, McPherson JM, Edmunds T, Oxidation of Methionine Residues in Antithrombin, J Biol. Chemistry 274, 15 (1999) 10268-10276).
В соответствии с другим предпочтительным вариантом указанный полипептид представляет собой человеческий IFN-бета, в частности IFN-бета 1а (например, Avonex®, REBIF®) и IFN-бета 1b (например, BETASERON®).
Другой предпочтительный полипептид представляет человеческий Г-КСФ (G-CSF) (гранулоцитарный колониестимулирующий фактор). (См., например, Nagata et al., The chromosomal gene structure and two mRNAs for human granulocyte colony-stimulating factor, EMBO J. 5: 575-581, 1986; Souza et al., Recombinant human granulocyte colony-stimulating factor: effects on normal and leukemic myeloid cells, Science 232 (1986) 61-65; and Herman et al., Characterization, formulation, and stability of Neupogen® (Filgrastim), a recombinant human granulocyte colony-stimulating factor, in: Formulation, characterization, and stability of protein drugs, Rodney Pearlman and Y. John Wang, eds., Plenum Press, New York, 1996, 303-328).
Если используется смесь по меньшей мере двух разных полипептидов, то по меньшей мере два полипептида могут различаться, например, по молекулярной массе, по числу и/или последовательности аминокислот, по разному уровню гликозилирования, по числу и/или химической природе заместителей или по числу полипептидных цепей, присоединяемых посредством соответствующих связей, таких как дисульфидные связи.
В соответствии с предпочтительным вариантом осуществления по настоящему изобретению реакционный продукт, получаемый при взаимодействии соединения (I) и соединения (II), необязательно в присутствии сшивающего соединения, выделяют, предпочтительно с использованием по меньшей мере одного из указанных выше способов, и затем подвергают реакции с полипептидом, включающим по меньшей мере одну функциональную группу Y, способную взаимодействовать по меньшей мере с одной функциональной группой Х в продукте реакции соединения (I) и соединения (II), получаемого в присутствии необязательно сшивающего соединения, с образованием по меньшей мере одной химической связи. Функциональные группы Y в полипептидах, таких как белки, представляют собой, например:
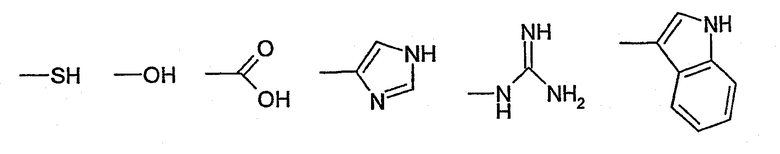

или углеводный фрагмент, который может быть присоединен к полипептиду путем N-гликозилирования или О-гликозилирования.
В контексте настоящего описания термин "углеводный фрагмент" относится к гидроксиальдегидам или гидроксикетонам, а также к их химически модифицированным формам (см. Rompp Chemielexikon, Thieme Verlag Stuttgart, Germany, 9th edition 1990, Volume 9, pp.2281-2285 и цитированную в работе литературу). Кроме того, указанный термин также относится к производным природных углеводных фрагментов типа глюкозы, галактозы, маннозы, сиаловой кислоты и т.п. Указанный термин также включает химически окисленные природные углеводные фрагменты. Структура окисленного углеводного фрагмента может быть линейной или циклической.
Углеводный фрагмент может быть присоединен непосредственно к полипептидному скелету. Предпочтительно углеводный фрагмент представляет собой часть углеводной боковой цепи. Более предпочтительно углеводный фрагмент представляет собой терминальный фрагмент боковой цепи углевода.
В еще более предпочтительном варианте осуществления по настоящему изобретению углеводный фрагмент представляет собой галактозный остаток в боковой цепи углевода, предпочтительно терминальный галактозный остаток в углеводной боковой цепи. Указанный галактозный остаток можно сделать доступным для проведения реакции с реакционным продуктом, получаемым при взаимодействии соединения (I) и соединения (II), за счет удаления терминальных сиаловых кислот с проведением впоследствии окисления, как описано ниже.
В еще одном предпочтительном варианте реакционный продукт, получаемый при взаимодействии соединения (I) и соединения (II), присоединяют к остатку сиаловой кислоты в углеводных боковых цепях, предпочтительно к терминальному остатку сиаловой кислоты в углеводной боковой цепи.
Окисление терминальных углеводных фрагментов может быть осуществлено химическим или ферментативным способом.
Способы химического окисления углеводных фрагментов полипептидов известны в данной области техники и включают обработку периодатом (Chamow et al., 1992, J. Biol. Chem., 267, 15916-15922).
При проведении химического окисления становится принципиально возможным окислить любой углеводный фрагмент, независимо от того, локализован он в концевом положении или нет. Однако при выборе мягких условий обработки (1 мМ периодат, 0°С, тогда как жесткие условия включают: 10 мМ периодат, 1 час при комнатной температуре) достигается возможность окислить предпочтительно терминальный остаток сиаловой кислоты в боковой цепи углевода.
Альтернативно, углеводный фрагмент может быть окислен ферментативным способом. Ферменты, применяемые для окисления отдельных углеводных фрагментов, известны в технике и включают, например, в случае галактозы, фермент галактозооксидазу. В случае проведения окисления терминальных фрагментов галактозы возникает необходимость постепенно удалять терминальные сиаловые кислоты (частично или полностью), если полипептид продуцируется в клетках, способных осуществлять реакцию присоединения сиаловых кислот к углеводным цепям, например в клетках млекопитающих или в клетках, которые были генетически модифицированы, с тем чтобы придать им способность присоединять сиаловые кислоты к углеводным цепям. Химические или ферментативные способы удаления сиаловых кислот известны в данной области техники (Chaplin and Kennedy (eds.), 1986, Carbohydrate Analysis: a practical approach, в особенности Chapter 5 Montreuill, Glycoproteins, pp.175-177; IRL Press Practical approach series (ISBN 0-947946-44-3)).
В этой связи настоящее изобретение относится к описанному выше способу, в соответствии с которым реакционный продукт, получаемый при взаимодействии соединения (I) и соединения (II), подвергают реакции с полипептидом через участие окисленного углеводного фрагмента, входящего в полипептид.
В соответствии с другим предпочтительным вариантом осуществления по настоящему изобретению указанная функциональная группа в полипептиде представляет собой тиогруппу. Соответственно этому, продукт реакции соединения (I) и соединения (II) может быть присоединен к полипептиду через тиоэфирную группу, где атом S может быть получен из любой тиогруппы, входящей в состав полипептида.
Тиогруппа может присутствовать в полипептиде как таковая. Кроме того, возможно встроить тиогруппу в полипептид с помощью подходящего метода. В числе других в качестве подходящих для данной цели можно отметить химические методы. Если в полипептиде имеется дисульфидный мостик, возможно восстановить -S-S-структуру с получением тиогруппы. Возможно также осуществить преобразование аминогруппы, имеющейся в полипептиде, в SH-группу посредством реакции полипептида через его аминогруппу с соединением, который имеет по меньшей мере две разных функциональных группы, одна из которых обладает способностью взаимодействовать с аминогруппой, а вторая представляет собой SH-группу или предшественник SH-группы. Указанную модификацию аминогруппы можно рассматривать как пример способа, когда белок сначала подвергают реакции с соединением (L), которое имеет по меньшей мере две разных функциональных группы, одна из которых обладает способностью взаимодействовать с аминогруппой, а вторая представляет собой SH-группу, и затем полученный реакционный продукт подвергают реакции, например, с производным ГАК, включающим ГАК и соединение (D), где указанное производное включает функциональную группу, способную взаимодействовать с SH-группой. Возможно также встроить SH-группу путем мутации полипептида, такой как введение цистеина или соответствующей SH-фнкциональной аминокислоты в полипептид, или такой как удаление цистеина из полипептида, так что провоцированный таким образом другой цистеин в полипептиде образует дисульфидную связь.
В контексте данного варианта осуществления по изобретению особенно предпочтительно осуществлять реакцию полипептида с реакционным продуктом, получаемым при взаимодействии продукта реакции соединения (I) и соединения (II) со сшивающим соединением.
В этой связи, настоящее изобретение относится к описанному выше способу, в соответствии с которым продукт реакции соединения (I) и соединения (II) подвергают реакции вначале со сшивающим соединением и затем полученный реакционный продукт подвергают реакции с полипептидом через окисленный углеводный фрагмент и/или тиогруппу, включенную в полипептид.
В качестве особенно предпочтительного полипептида используют эритропоэтин (ЭПО).
В этой связи, настоящее изобретение относится в описанному выше способу, в котором указанный полипептид представляет собой эритропоэтин.
ЭПО может быть человеческого происхождения (см., например, Inoue, Wada, Takeuchi, 1994, An improved method for the purification of human erythropoietin with high in vivo activity from the urine of anemic patients, Biol Pharm Bull. 17(2), 180-4; Miyake, Kung, Goldwasser, 1977, Purification of human erythropoietin., J. Biol. Chem., 252(15), 5558-64) или может происходить из другого млекопитающего в качестве источника и может быть получен путем выделения и очистки из природных источников, таких как почка человека, эмбриональная печень человека или животного, предпочтительно из почки обезьяны. Кроме того, выражение "эритропоэтин" или "ЭПО" относится также к варианту ЭПО, где одна или более аминокислот (например, от 1 до 25, предпочтительно от 1 до 10, более предпочтительно от 1 до 5, наиболее предпочтительно 1 или 2) заменены на другую аминокислоту и который проявляет эритропоэтическую активность (см., например, EP 640619 B1). Определение эритропоэтической активности описано в литературе (см., например, описание измерения активности in vitro: Fibi et al., 1991, Blood, 77, 1203 ff; Kitamura et al., 1989, J.Cell Phys., 140, 323-334; описание метода измерения активности ЭПО in vivo см. в Ph. Eur. 2001, 911-917; Ph. Eur. 2000, 1316 Erythropoietini solutio concentrata, 780-785; European Pharmacopoeia (1996/2000); European Pharmacopoeia, 1996, Erythropoietin concentrated solution, Pharmaeuropa, 8, 371-377; Fibi, Hermentin, Pauly, Lauffer, Zettimeissl., 1995, N- and O-glycosylation muteins of recombinant human erythropoietin secreted from BHK-21 cells, Blood, 85(5), 1229-36) (ЭПО и модифицированные формы ЭПО инъецируют самкам мышей NMRI (что эквивалентно по белку 50 нг/мышь) в дни 1, 2 и 3, на 4 день отбирают образцы крови и определяют число ретикулоцитов). Другие публикации, в которых описаны тесты для измерения активности ЭПО, включают: Barbone, Aparicio, Anderson, Natarajan, Ritchie, 1994, Reticulocytes measurements as a bioassay for erythropoietin, J. Pharm. Biomed. Anal., 12(4), 515-22; Bowen, Culligan, Beguin, Kendal, Villis, 1994, Estimation of effective and total erythropoiesis in myelodysplasia using serum transferrin receptor and erythropoietin concentrations, with automated reticulocyte parameters, Leukemi, 8(1), 151-5; Delorme, Lorenzini, Giffin, Martin, Jacobsen, Boone, Elliott, 1992, Role of glycosylation on the secretion and biological activity of erythropoietin, Biochemistry, 31(41), 9871-6; Higuchi, Oheda, Kuboniwa, Tomonoh, Shimonaka, Ochi, 1992; Role of sugar chains in the expression of the biological activity of human erythropoietin, J. Biol. Chem., 267(11), 7703-9; Yamaguchi, Akai, Kawanishi, Ueda, Masuda, Sasaki, 1991, Effects of site-directed removal of N-glycosylation sites in human erythropoietin on its production and biological properties, J. Biol. Chem., 266(30), 20434-9; Takeuchi, Inoue, Strickland, Kubota, Wada, Shimizu, Hoshi, Kozutsumi, Takasaki, Kobata, 1989, Relationship between sugar chain structure and biological activity of recombinant human erythropoietin produced in Chinese hamster ovary cells, Proc. Natl. Acad. Sci. USA, 85(20), 7819-22; Kurtz, Eckardt, 1989, Assay methods for erythropoietin, Nephron., 51(1), 11-4 (German); Zucali, Sulkowski, 1989, Purification of human urinary erythropoietin on controlled-pore glass and silicic acid, Exp. Hematol., 13(3), 833-7; Krystal, 1983, Physical and biological characterization of erythroblast enhancing factor (EEF), a late acting erythropoietin stimulator in serum distinct from erythropoietin, Exp. Hematol., 11(1), 18-31.
Предпочтительно ЭПО получают рекомбинантными способами. Указанный вариант включает продукцию в эукариотических или прокариотических клетках, предпочтительно в клетках млекопитающих, насекомых, дрожжей, в бактериальных клетках или в клетках любого другого типа, которые удобны для получения ЭПО рекомбинантными методами. Кроме того, ЭПО может экспрессироваться в трансгенных животных, например в жидкостях организма (таких как молоко, кровь и т.п.), в яйцах трансгенных птиц, в особенности сельскохозяйственных птиц, предпочтительно кур, или в трансгенных растениях.
Получение полипептидов по рекомбинантной технологии известно специалистам в данной области техники. В целом, такая продукция включает трансфекцию в хозяйскую клетку с помощью соответствующего вектора экспрессии, культивирование хозяйских клеток в условиях, позволяющих осуществлять продукцию полипептида, и выделение и очистку полипептида из хозяйских клеток. Подробная информация содержится в ряде публикаций (см. например, Krystal, Pankratz, Farber, Smart, 1986, Purification of human erythropoietin to homogeneity by a rapid five-step procedure, Blood, 67(1), 71-9; Quelle, Caslake, Burkert, Wojchowski, 1989, High-level expression and purification of a recombinant human erythropoietin produced using a baculovirus vector, Blood, 74(2), 652-7; EP 640619 B1 and EP 668351 B1).
В предпочтительном варианте осуществления настоящего изобретения ЭПО имеет аминокислотную последовательность человеческого ЭПО (см. EP 148605 B2).
ЭПО может включать одну или более углеводных боковых цепей, предпочтительно от 1 до 12, более предпочтительно от 1 до 9, еще более предпочтительно от 1 до 6, в частности, предпочтительно от 1 до 4 и особенно предпочтительно 4 боковых углеводных цепи, присоединяемых к ЭПО через N- и/или O-связанное гликозилирование, т.е. ЭПО гликозилируется. Обычно в случае продукции ЭПО в эукариотических клетках полипептид подвергается пост-трансляционному гликозилированию. Следовательно, углеводные боковые цепи могут присоединяться к ЭПО в процессе биосинтеза в клетках млекопитающего, особенно человека, клетках насекомого или клетках дрожжей. Структура и свойства гликозилированного ЭПО широко исследовались в данной области (см. EP 428267 B1; EP 640619 B1, Rush, Derby, Smith, Merry, Rogers, Rohde, Katta, 1995, Microheterogeneity of erythropoietin carbohydrate structure, Anal Chem., 67(8), 1442-52; Takeuchi, Kobata, 1991, Structure and functional roles of the sugar chains of human erythropoietin, Glycobiology, 1(4), 337-46 (обзор)).
В этой связи, гидроксиалкилкрахмальные производные по настоящему изобретению могут включать по меньшей мере одну, предпочтительно от 1 до 12, более предпочтительно от 1 до 9, еще более предпочтительно от 1 до 6 и особенно предпочтительно от 1 до 4 молекул ГАК на одну молекулу ЭПО. Количество ГАК молекул на одну молекулу ЭПО может быть определено путем количественного анализа углеводного состава с использованием ГХ-МС после гидролиза продукта и дериватизации полученных моносахаридов (см. Chaplin and Kennedy (eds.), 1986, Carbohydrate Analysis: a practical approach, IRL Press Practical approach series (ISBN 0-947946-44-3), в особенности Chapter 1, Monosaccharides, pp.1-36; Chapter 2, Oligosaccharides, pp.37-53, Chapter 3, Neutral Polysaccharides, pp.55-96).
В соответствии с особенно предпочтительным вариантом осуществления по настоящему изобретению углеводный фрагмент, присоединенный к ЭПО, представляет собой часть углеводной боковой цепи. Более предпочтительно углеводный фрагмент представляет собой терминальный фрагмент углеводной боковой цепи. В еще более предпочтительном варианте углеводный фрагмент представляет собой остаток галактозы в углеводной боковой цепи, предпочтительно терминальный остаток галактозы в углеводной боковой цепи. Указанный галактозный остаток можно сделать доступным для проведения реакции с реакционным продуктом, получаемым при взаимодействии соединения (I) и соединения (II), за счет удаления терминальных сиаловых кислот и с проведением впоследствии окисления, как описано ниже. В еще одном предпочтительном варианте осуществления по изобретению реакционный продукт, получаемый при взаимодействии соединения (I) и соединения (II), присоединяют к остатку сиаловой кислоты в боковых углеводных цепях, предпочтительно к терминальному остатку сиаловой кислоты в углеводной боковой цепи. Сиаловую кислоту окисляют, как описано ниже.
В особенно предпочтительном варианте указанный галактозный остаток делают доступным для проведения реакции с реакционным продуктом, получаемым при взаимодействии соединений (I) и (II), или с реакционным продуктом, получаемым при взаимодействии продукта реакции соединений (I) и (II) и сшивающего соединения, через функциональную группу Х за счет удаления терминальной сиаловой кислоты с последующим окислением.
Как указывалось выше, продукт реакции соединения (I) и соединения (II), необязательно при взаимодействии со сшивающим соединением, может быть подвергнут реакции с тиогруппой, входящей в ЭПО.
Возможно также осуществлять реакцию реакционного продукта, получаемого при взаимодействии соединения (I) и соединения (II), необязательно в присутствии сшивающего соединения, с тиогруппой, а также с углеводным фрагментом, каждый из которых входит в состав по меньшей мере одного другого соединения, предпочтительно полипептида, более предпочтительно эритропоэтина.
В соответствии с предпочтительным вариантом указанная SH-группа может быть присоединена к предпочтительно окисленному углеводному фрагменту, например, посредством использования производного гидроксиламина, например гидрохлорида 2-(аминоокси)этилмеркаптана (Bauer L. et al., 1965, J. Org. Chem., 30, 949), или с использованием гидразидного производного, например гидразида тиогликолевой кислоты (Whitesides et al., 1977, J. Org. Chem., 42, 332).
В соответствии с еще одним предпочтительным вариантом осуществления по настоящему изобретению тиогруппу предпочтительно вводят в окисленный углеводный фрагмент ЭПО, более предпочтительно в окисленный углеводный фрагмент, который представляет собой часть боковой углеводной цепи ЭПО.
Предпочтительно тиогруппу получают из природного цистеина или из добавленного цистеина. Более предпочтительно ЭПО имеет аминокислотную последовательность человеческого ЭПО, а природные цистеины представляют собой цистеин 29 и/или 33. В более предпочтительном варианте реакционный продукт, получаемый при взаимодействии соединения (I) и соединения (II), необязательно в присутствии сшивающего соединения, подвергают реакции с цистеином 29, а цистеин 33 замещают другой аминокислотой. Альтернативно реакционный продукт, получаемый при взаимодействии соединения (I) и соединения (II), необязательно в присутствии сшивающего соединения, подвергают реакции с цистеином 33, а цистеин 29 замещают другой аминокислотой.
В контексте настоящего описания термин "добавленные цистеины" указывает на то, что полипептиды, предпочтительно ЭПО, включают цистеиновый остаток, который не присутствует в полипептиде дикого типа.
В контексте данного аспекта по изобретению цистеин может представлять собой дополнительную аминокислоту, добавляемую на N- или С-конце ЭПО.
Кроме того, добавленный цистеин может быть встроен путем замещения природной аминокислоты цистеином или соответствующим образом замещенным цистеином. Предпочтительно, в контексте данного аспекта по изобретению ЭПО представляет собой человеческий ЭПО, и замещенный аминокислотный остаток представляет собой серин 126.
Условия проведения реакции реакционного продукта, получаемого при взаимодействии соединения (I) и соединения (II), необязательно в присутствии сшивающего соединения, по меньшей мере с одним другим соединением, могут быть адаптированы к конкретным потребностям данной реакции, таким как в том случае, когда по меньшей мере одно другое соединение представляет собой полипептид, или в том случае, когда по меньшей мере одно другое соединение представляет собой сшивающее соединение, или в том случае, когда по меньшей мере одно другое соединение представляет собой продукт реакции сшивающего соединения и полипептида. В качестве забуферивающих соединений предпочтительно может использоваться по меньшей мере одно соединение из числа указанных выше. В качестве растворителя или системы растворителей предпочтительно может использоваться по меньшей мере один растворитель из числа указанных выше. Далее может быть проведено выделение или последующая обработка, для осуществления которых выбираются методы из числа обсуждавшихся выше методов.
Если реакционный продукт, получаемый при взаимодействии соединения (I) и соединения (II), например, подвергают далее реакции с полипептидом в качестве другого используемого соединения, предпочтительно с ЭПО, то в качестве растворителя для данной реакции предпочтительно используют воду. Дополнительно, кроме воды, может присутствовать по меньшей мере один другой растворитель. В качестве возможного для использования другого предпочтительного растворителя можно отметить ДМСО, ДМФ, метанол или этанол.
Кроме того, настоящее изобретение относится к описанному выше способу, в соответствии с которым реакцию реакционного продукта, получаемого при взаимодействии соединения (I) и соединения (II), с полипептидом, предпочтительно ЭПО, проводят в водной системе.
В том, что касается температур, поддерживаемых в данной реакции, то отсутствуют какие-либо конкретные ограничения, при условии, что реакция приводит к получению желательного производного гидроксиалкилкрахмала, включающего результат взаимодействия с полипептидом продукта реакции соединения (I) и соединения (II), через участие по меньшей мере одной функциональной группы Х. Температуру реакции поддерживают предпочтительно в диапазоне от 4 до 37°С, более предпочтительно в диапазоне от 10 до 30°С и особенно предпочтительно в диапазоне от 15 до 25°С.
В этой связи, настоящее изобретение относится к описанному выше способу, в соответствии с которым реакцию реакционного продукта, получаемого при взаимодействии соединения (I) и соединения (II), с полипептидом проводят при температуре от 4 до 37°С.
В ходе реакции температура может варьироваться предпочтительно в границах указанного выше диапазона или может поддерживаться по существу на постоянном уровне.
Длительность реакции реакционного продукта, получаемого при взаимодействии соединения (I) и соединения (II), с полипептидом может быть адаптирована к соответствующим потребностям и в основном составляет от 0,5 до 48 часов, предпочтительно от 2 до 24 часов и особенно предпочтительно от 10 до 20 часов.
Уровень рН, поддерживаемый в реакции реакционного продукта, получаемого при взаимодействии соединения (I) и соединения (II), с полипептидом может быть адаптирован к соответствующим потребностям, таким как химическая природа реагентов.
Если, например, реакцию реакционного продукта, получаемого при взаимодействии соединения (I) и соединения (II), проводят с другим соединением посредством взаимодействия функциональной группы Х, которая представляет собой гидроксиламино группу -O-NH2, по меньшей мере с одной альдегидной группой, которая входит в состав полипептида, то рН предпочтительно поддерживают в диапазоне от 4,5 до 6, более предпочтительно на уровне примерно 5,5.
Если реакционный продукт, получаемый при взаимодействии соединения (I) и соединения (II), например, подвергают далее реакции со сшивающим соединением в качестве другого используемого соединения, предпочтительно с ЭПО, то в качестве растворителя для данной реакции предпочтительно используют воду. Дополнительно, кроме воды, может присутствовать по меньшей мере один другой растворитель. В качестве возможного для использования другого предпочтительного растворителя можно отметить ДМСО, ДМФ, метанол или этанол.
В этой связи, настоящее изобретение относится к описанному выше способу, в соответствии с которым реакцию реакционного продукта, получаемого при взаимодействии соединения (I) и соединения (II), со сшивающим соединением проводят в водной системе.
В том, что касается температур, поддерживаемых в данной реакции, то отсутствуют какие-либо конкретные ограничения, при условии, что реакция приводит к получению желательного производного гидроксиалкилкрахмала, включающего результат взаимодействия продукта реакции соединения (I) и соединения (II) со сшивающим соединением, через участие по меньшей мере одной функциональной группы Х. Температуру реакции поддерживают предпочтительно в диапазоне от 4 до 37°С, более предпочтительно в диапазоне от 10 до 30°С и особенно предпочтительно в диапазоне от 15 до 25°С.
В этой связи, настоящее изобретение относится к описанному выше способу, в соответствии с которым реакцию реакционного продукта, получаемого при взаимодействии соединения (I) и соединения (II), со сшивающим соединением проводят при температуре от 4 до 37°С.
В ходе реакции температура может варьироваться предпочтительно в границах указанного выше диапазона или может поддерживаться по существу на постоянном уровне.
Длительность реакции реакционного продукта, получаемого при взаимодействии соединения (I) и соединения (II), со сшивающим соединением может быть адаптирована к соответствующим потребностям и в основном составляет от 10 минут до 10 часов, предпочтительно от 20 минут до 5 часов и более предпочтительно от 30 минут до 2 часов.
Уровень рН, поддерживаемый в реакции реакционного продукта, получаемого при взаимодействии соединения (I) и соединения (II), со сшивающим соединением может быть адаптирован к соответствующим потребностям, таким как химическая природа реагентов.
Если, например, реакционный продукт, получаемый при взаимодействии соединения (I) и соединения (II), подвергают реакции со сшивающим соединением через функциональную группу Х, которая включена в реакционный продукт, получаемый при взаимодействии соединения (I) и соединения (II), и представляет собой аминогруппу -NH2, то рН предпочтительно поддерживают в диапазоне от 7 до 8,5, более предпочтительно на уровне примерно 7,2.
Если реакционный продукт, получаемый при взаимодействии соединения (I) и соединения (II), например, подвергают далее реакции с полипептидом в качестве другого используемого соединения, предпочтительно с ЭПО, то в качестве растворителя для данной реакции предпочтительно используют воду. Дополнительно, кроме воды, может присутствовать по меньшей мере один другой растворитель. В качестве возможного для использования другого предпочтительного растворителя можно отметить ДМСО, ДМФ, метанол или этанол.
В этой связи, настоящее изобретение также относится к описанному выше способу, в соответствии с которым реакцию реакционного продукта, получаемого при взаимодействии соединения (I) и соединения (II) в присутствии сшивающего соединения, с полипептидом проводят в водной системе.
В том, что касается температур, поддерживаемых в данной реакции, то отсутствуют какие-либо конкретные ограничения, при условии, что реакция приводит к получению желательного производного гидроксиалкилкрахмала, включающего продукт взаимодействия продукта реакции соединений (I) и (II) в присутствии сшивающего соединения, с полипептидом через участие по меньшей мере одной функциональной группы Х, входящей в сшивающее соединение. Температуру реакции поддерживают предпочтительно в диапазоне от 4 до 37°С, более предпочтительно в диапазоне от 10 до 30°С и особенно предпочтительно в диапазоне от 15 до 25°С.
В этой связи, настоящее изобретение относится к описанному выше способу, в соответствии с которым реакцию реакционного продукта, получаемого при взаимодействии соединения (I) и соединения (II), в присутствии сшивающего соединения, с полипептидом проводят при температуре от 4 до 37°С.
В ходе реакции температура может варьироваться предпочтительно в границах указанного выше диапазона или может поддерживаться по существу на постоянном уровне.
Длительность реакции реакционного продукта, получаемого при взаимодействии соединения (I) и соединения (II) в присутствии сшивающего соединения, с полипептидом может быть адаптирована к соответствующим потребностям и в основном составляет от 0,5 до 48 часов, предпочтительно от 2 до 24 часов и особенно предпочтительно от 10 до 20 часов.
Уровень рН, поддерживаемый в реакции реакционного продукта, получаемого при взаимодействии соединения (I) и соединения (II) в присутствии сшивающего соединения, с полипептидом может быть адаптирован к соответствующим потребностям, таким как химическая природа реагентов.
Если, например, реакционный продукт, получаемый при взаимодействии соединения (I) и соединения (II) в присутствии сшивающего соединения, подвергают реакции с полипептидом через функциональную группу Х, которая включена в сшивающее соединение и представляет собой аминогруппу -NH2, то рН предпочтительно поддерживают в диапазоне от 7 до 8,5, более предпочтительно на уровне примерно 7,2.
Реакционная смесь может быть доведена до подходящего значения рН путем добавления по меньшей мере одного приемлемого буфера. В числе предпочтительных буферов можно отметить натрий-ацетатный буфер, натрий-фосфатный буфер или боратные буферы.
Реакционный продукт, получаемый при взаимодействии продукта реакции соединения (I) и соединения (II) по меньшей мере с одним другим соединением, где по меньшей мере одно другое соединение представляет собой полипептид или сшивающее соединение, и реакционный продукт, включающий соединения, полученные в ходе реакций соединения (I), соединения (II), сшивающего соединения и полипептида, могут быть выделены из реакционной смеси с помощью подходящего способа и подвергнуты по меньшей мере одной дополнительной обработке, такой как по меньшей мере одна пост-обработка, такая как диализ и/или лиофилизация.
Как только образуется указанный выше реакционный продукт, он может быть выделен из реакционной смеси с помощью по меньшей мере одного подходящего способа.
Выделение реакционного продукта может быть осуществлено с помощью подходящего способа, который может включать одну или более стадий.
В соответствии с предпочтительным вариантом осуществления по настоящему изобретению, при котором реакционный продукт не включает полипептид, реакционный продукт вначале отделяют от реакционной смеси или смеси реакционных смесей, предпочтительно путем фильтрования при центрифугировании. На второй стадии отделенный реакционный продукт может быть подвергнут дополнительной обработке, такой как пост-обработка типа диализа и/или лиофилизации. В соответствии с еще более предпочтительным вариантом осуществления по настоящему изобретению отделенный продукт вначале диализуют, предпочтительно против воды, и затем лиофилизируют до того момента, когда содержание растворителя в реакционном продукте станет достаточно низким, в соответствии со спецификациями для данного продукта.
В соответствии с другим вариантом осуществления по настоящему изобретению при котором реакционный продукт включает полипептид, реакционный продукт предпочтительно выделяют по методу, описанному в примере 7.8.
В соответствии с еще одним вариантом осуществления по настоящему изобретению соединение (II) вначале подвергают реакции с другим соединением перед проведением реакции с соединением (I), например, получают производное соединения (II) посредством взаимодействия соединения (II) через реакцию по меньшей мере одной функциональной группы Х по меньшей мере с одним другим соединением, включающим по меньшей мере одну функциональную группу Y, описанную выше, перед проведением реакции с соединением (I).
Если соединение (II) вначале подвергают реакции с другим соединением, предпочтительно полипептидом, более предпочтительно ЭПО, то в качестве растворителя для данной реакции предпочтительно используют воду. Дополнительно, кроме воды, может присутствовать по меньшей мере один другой растворитель. В качестве возможного для использования другого предпочтительного растворителя можно отметить ДМСО, ДМФ, метанол или этанол.
В этой связи, настоящее изобретение также относится к описанному выше способу, в соответствии с которым реакцию соединения (II), перед осуществлением взаимодействия с соединением (I), с другим соединением, предпочтительно полипептидом, еще более предпочтительно ЭПО, проводят в водной системе.
В том, что касается температур, поддерживаемых в данной реакции, то отсутствуют какие-либо конкретные ограничения, при условии, что реакция приводит к получению желательного производного соединения (II), включающего продукт реакции соединения (II) по меньшей мере с одним другим соединением через участие по меньшей мере одной функциональной группы Х, предпочтительно с полипептидом, более предпочтительно с ЭПО. Температуру реакции поддерживают предпочтительно в диапазоне от 4 до 37°С, более предпочтительно в диапазоне от 10 до 30°С и особенно предпочтительно в диапазоне от 15 до 25°С.
В этой связи, настоящее изобретение относится к описанному выше способу, в соответствии с которым реакцию соединения (II) по меньшей мере с одним другим соединением проводят при температуре от 4 до 37°С.
В ходе реакции температура может варьироваться предпочтительно в границах указанного выше диапазона или может поддерживаться по существу на постоянном уровне.
Длительность реакции и уровень рН в реакции соединения (II) по меньшей мере с одним другим соединением могут быть адаптированы к соответствующим потребностям, таким как химическая природа реагентов. Реакционная смесь может быть доведена до подходящего значения рН путем добавления по меньшей мере одного приемлемого буфера. В числе предпочтительных буферов можно отметить ацетатный, фосфатный или боратные буферы, такие как натрий-ацетатный буфер, натрий-фосфатный буфер или боратные буферы.
Реакционный продукт, получаемый при взаимодействии соединения (II) по меньшей мере с одним другим соединением, может быть выделен из реакционной смеси с помощью подходящего способа и подвергнут по меньшей мере одной дополнительной обработке, такой как по меньшей мере одна пост-обработка, такая как диализ и/или лиофилизация.
Как только образуется продукт взаимодействия соединения (II) по меньшей мере с одним другим соединением, он может быть выделен из реакционной смеси с помощью по меньшей мере одного подходящего способа.
Выделение реакционного продукта может быть осуществлено с помощью подходящего способа, который может включать одну или более стадий, как уже было описано выше.
При желательности и/или необходимости NH-группа, соединяющая мостиковой связью R' и R" в соединении (II), может быть защищена соответствующей защитной группой перед проведением реакции соединения (II) по меньшей мере с одним другим соединением. В качестве защитной группы может использоваться одна из указанных выше защитных групп. Перед проведением реакции продукта взаимодействия соединения (II) и по меньшей мере одного другого соединения, такого как полипептид, предпочтительно ЭПО, с соединением (I) защитную группу удаляют с помощью по меньшей мере одного подходящего для этой цели способа.
Если соединение (I) подвергают вначале реакции со сшивающим соединением или продуктом реакции сшивающего соединения и полипептида, все условия реакции могут быть адаптированы к конкретным потребностям данных реакций. В числе других, для данной цели могут использоваться буферные системы и/или растворители.
На второй стадии продукт реакции соединения (II) по меньшей мере с одним другим соединением подвергают реакции с соединением (I).
В данной реакции все реакционные условия могут быть адаптированы к конкретным потребностям данных реакций. В числе других, для данной цели могут использоваться буферные системы и/или растворители.
Реакционный продукт, получаемый при взаимодействии продукта реакции соединения (II) и по меньшей мере одного другого соединения с соединением (I), может быть выделен из соответствующей реакционной смеси с помощью подходящего способа и подвергнут по меньшей мере одной дополнительной обработке, такой как по меньшей мере одна пост-обработка, такая как диализ и/или лиофилизация. В данном контексте может использоваться любой приемлемый способ из числа описанных выше.
В основном, выделение конъюгата ГАК-полипептид, при наличии сшивающего соединения или в его отсутствие, может быть проведено с использованием известных процедур очистки природных и рекомбинантных полипептидов, таких как вытеснительная хроматография, ионообменная хроматография, ОФ-ВЭЖХ, хроматография на гидроксиапатите, хроматография, основанная на гидрофобном взаимодействии, или сочетание по меньшей мере двух таких методов.
Ковалентное присоединение ГАК к полипептиду может быть подтверждено путем анализа углеводного состава после гидролиза модифицированного белка.
Выявление модификации ГАК по N-присоединенным олигосахаридам в полипептиде может быть осуществлено при удалении ГАК-модифицированных N-гликанов с последующей оценкой наличия предполагаемого сдвига к большей подвижности при проведении электрофореза в ДСН-ПААГ +/- по результатам вестерн-блоттинга.
ГАК-модификация полипептида по цистеиновым остаткам может быть продемонстрирована по неспособности выявить после протеолиза соответствующий Cys-пептид при проведении анализа методом ОФ-ВЭЖХ и MALDI/TOF-MS фрагментов, полученных после протеолиза ГАК-модифицированного продукта (Zhou et al., 1998, Application of capillary electrophoresis, liquid chromatography, electrospray-mass spectrometry and matrix-assisted laserdesorption/ionization - time of flight - mass spectrometry to the characterization of recombinant human erythropoietin. Electrophoresis, 19(13), 2348-55). Выделение ГАК-содержащей фракции после протеолитического расщепления Cys-модифицированного полипептида позволяет провести анализ, подтверждающий наличие искомого продукта в данной фракции соответствующего пептида, в рамках традиционного метода определения аминокислотного состава.
Все описанные выше варианты осуществления по настоящему изобретению в аспекте ГАК-полипептид, которые относятся к свойствам полипептида или ГАК, применимы также к способу получения конъюгата ГАК-полипептид по настоящему изобретению. Кроме того, все описанные выше варианты осуществления по настоящему изобретению в аспекте ГАК-ЭПО или его получения, которые относятся к пептидам в целом или к ГАК, применимы также к способу получения конъюгата ГАК-полипептид по настоящему изобретению.
В соответствии с особенно предпочтительным вариантом осуществления по настоящему изобретению гидроксиэтилкрахмал подвергают реакции с соединением (II), предпочтительно выбранным из гомо- и гетеробифункциональных соединений, описанных выше, и полученный реакционный продукт далее подвергают реакции с гликопротеином, предпочтительно с эритропоэтином, предпочтительно с окисленным терминальным углеводным фрагментом в боковой углеводной цепи ЭПО.
В соответствии с другим особенно предпочтительным вариантом осуществления по настоящему изобретению гидроксиэтилкрахмал подвергают реакции с соединением (II), предпочтительно выбранным из гомо- и гетеробифункциональных соединений, с получением первого производного гидроксиэтилкрахмала. Указанное первое производное гидроксиэтилкрахмала далее подвергают реакции со сшивающим соединением с получением второго производного гидроксиэтилкрахмала. Указанное второе производное гидроксиэтилкрахмала далее подвергают реакции с гликопротеином, предпочтительно с эритропоэтином, предпочтительно с SH-группой, включенной в гликопротеин, с получением третьего производного гидроксиэтилкрахмала. Предпочтительно, сшивающее соединение представляет собой гетеробифункциональное соединение. Более предпочтительно, сшивающее соединение подвергают реакции с функциональной группой, включающей структуру -NH-, которая входит в состав первого производного гидроксиэтилкрахмала. Более предпочтительно, указанная функциональная группа представляет собой -NH2.
Одним преимуществом данного варианта осуществления способа по настоящему изобретению является то, что нет необходимости использовать токсикологически критические растворители по меньшей мере на одной стадии, предпочтительно на всех стадиях реакции, задействованных в данном процессе, так что в этой связи не возникает потребности удалять такие растворители после завершения процесса производства, с тем, чтобы избежать загрязнения продуктов данным растворителем. Кроме того, нет необходимости проводить дополнительные контрольные тесты качества относительно содержания остаточных токсикологически критических растворителей. Если используются органические растворители, предпочтительно в дополнение к воде, то предпочтительно использовать токсикологически не критические растворители, такие как этанол и/или пропиленгликоль.
Другое преимущество осуществления способа по настоящему изобретению заключается в том, что удается избежать необратимых или обратимых структурных изменений на стадиях, где в качестве растворителя используется водная система, которые, в ином случае, индуцируются органическими растворителями. Следовательно, получаемые по способу данного изобретения производные полипептидов отличаются от тех продуктов, которые получают в органических растворителях, таких как ДМСО.
Кроме того, было неожиданно обнаружено, что проведение реакции конъюгирования ГАК с полипептидами, такими как ЭПО, в водном растворе сводит к минимуму или позволяет избежать побочных реакций. Следовательно, данный вариант осуществления способа по настоящему изобретению ведет к получению улучшенных продуктов на основе гидроксиалкилкрахмала с точки зрения их большей чистоты.
В соответствии с другим аспектом настоящее изобретение также относится к производному гидроксиалкилкрахмала, получаемому по способу, включающему реакцию гидроксиалкилкрахмала (ГАК) формулы (I):
(I)
по его восстановленному концу, который не окисляют перед проведением указанной реакции, с соединением формулы (II)
R'-NH-R", (II)
где R1, R2 и R3 обозначают независимо атом водорода или линейную или разветвленную гидроксиалкильную группу, и где R', или R'', или R' и R'' включают по меньшей мере одну функциональную группу Х, способную взаимодействовать по меньшей мере с одним другим соединением до или после реакции (I) и (II).
Как уже отмечалось выше в контексте описания способов по настоящему изобретению, О-[2-(2-аминооксиэтокси)этил]гидроксиламин используют в качестве предпочтительного соединения (II) и гидроксиэтилкрахмал используют в качестве предпочтительного гидроксиалкилкрахмала.
В этой связи, настоящее изобретение также относится к производному гидроксиалкилкрахмала, получаемому по способу, в соответствии с которым гидроксиэтилкрахмал подвергают реакции по его восстановленному концу с О-[2-(2-аминооксиэтокси)этил]гидроксиламином.
В зависимости от соответствующих условий реакции, используемых растворителей или смеси растворителей и/или природы остатков R' и/или R", возможно, чтобы гидроксиалкилкрахмальное производное, получаемое по такому способу или способам, описанным выше, могло иметь указанную ниже структуру (IIIa):

В этой связи, настоящее изобретение также относится к описанным выше гидроксиалкилкрахмальным производным, которые имеют структуру формулы (IIIa).
Возможно также, чтобы, например, в том случае, когда R' обозначает водород, указанное гидроксиалкилкрахмальное производное, получаемое по способу или способам, описанным выше, имело структуру (IIIa) или (IIIb), где (IIIa) и (IIIb) могли бы оба присутствовать в реакционной смеси в состоянии определенного равновесия:
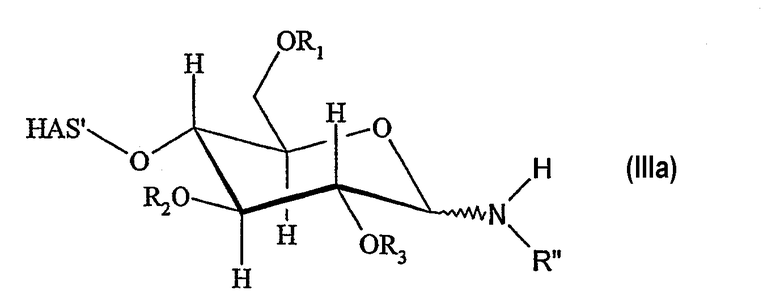
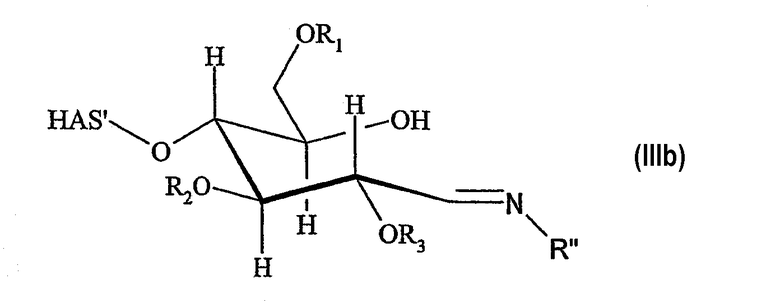
В этой связи, настоящее изобретение также относится к описанному выше производному гидроксиалкилкрахмала формулы (IIIb).
Более того, настоящее изобретение относится также к описанному выше производному гидроксиалкилкрахмала, который присутствует в смеси структур формул (IIIa) и (IIIb).
В зависимости от условий реакции и/или химической природы соединения (II), используемого в реакции, могут присутствовать соединения формулы (IIIa), в которых атом N находится в экваториальном или аксиальном положении, и может присутствовать смесь обеих форм в состоянии их определенного равновесного распределения.
В зависимости от условий реакции и/или химической природы соединения (II), используемого в реакции, могут присутствовать соединения формулы (IIIb), в которых имеется двойная C-N связь в E- или Z-конформации, и может присутствовать смесь обеих форм в состоянии их определенного равновесного распределения.
В некоторых случаях может быть желательно стабилизировать соединение формулы (IIIa). Это особенно справедливо в том случае, когда соединение формулы (IIIa) получают и/или используют в водном растворе. В качестве способа стабилизации особенно предпочтительно проведение ацилирования соединения формулы (IIIa), особенно в том случае, когда R' обозначает водород. В качестве ацилирующего реагента могут использоваться все приемлемые реагенты, которые приводят к получению желательного производного гидроксиалкилкрахмала формулы (IVa)

В соответствии с особенно предпочтительными вариантами осуществления по настоящему изобретению остаток Ra, представляющий собой часть ацилирующего реагента, является метилом. В качестве ацилирующих реагентов предпочтительно используют ангидриды карбоновых кислот, галогенангидриды карбоновых кислот и активированные сложные эфиры карбоновых кислот.
В этой связи, настоящее изобретение также относится к производному гидроксиалкилкрахмала, получаемому по описанному выше способу, где указанное производное имеет структуру формулы (IVa).
Реакцию ацилирования проводят в температурном диапазоне от 0 до 30°С, предпочтительно в диапазоне от 2 до 20°С и особенно предпочтительно в диапазоне от 4 до 10°С.
В других случаях может быть желательно осуществлять стабилизацию соединения формулы (IIIb). Такой подход особенно справедлив в том случае, когда соединение формулы (IIIb) получают и/или используют в водном растворе. В качестве способа стабилизации особенно предпочтительно проведение реакции восстановления соединения формулы (IIIb), особенно в том случае, когда R' обозначает водород. В качестве восстановителя могут использоваться все подходящие реагенты, которые приводят к получению желательного производного гидроксиалкилкрахмала формулы (IVb)

В соответствии с особенно предпочтительными вариантами осуществления по настоящему изобретению в качестве восстановителей используют боргидриды, такие как NaCNBH3 или NaBH4.
В этой связи, настоящее изобретение также относится к производному гидроксиалкилкрахмала, получаемому по описанному выше способу, где указанное производное имеет структуру формулы (IVb).
Реакцию восстановления проводят в температурном диапазоне от 4 до 100°С, предпочтительно в диапазоне от 10 до 90°С и особенно предпочтительно, в диапазоне от 25 до 80°С.
Настоящее изобретение также относится к смесям соединений (IIIa) и (IIIb), (IVa) и (IVb), (IIIa) и (IVa), (IIIa) и (IVb), (IIIb) и (IVa), (IIIb) и (IVb), (IIIa) и (IIIb) и (IVa), (IIIa) и (IIIb) и (IVb), (IVa) и (IVb) и (IIIa) и (IVa) и (IVb) и (IIIb), где (IIIa) и/или (IVa) могут независимо присутствовать в конформации, в которой атом N может находиться в экваториальном или аксиальном положении, и/или где может присутствовать (IIIb), в котором имеется двойная C-N связь в E- или Z- конформации.
В соответствии с одним аспектом по настоящему изобретению соединение (I) подвергают реакции с соединением (II) с получением первого реакционного продукта. Указанный первый реакционный продукт затем необязательно стабилизируют с использованием по меньшей мере одного из описанных выше методов. Первый необязательно стабилизированный реакционный продукт подвергают затем реакции по меньшей мере с одним другим соединением посредством взаимодействия по меньшей мере одной функциональной группы Х, входящей в R" в первом реакционном продукте, по меньшей мере с одной функциональной группой Y, включенной по меньшей мере в одну боковую углеводную цепь, с получением второго реакционного продукта. Указанный второй реакционный продукт затем подвергают необязательной стабилизации с использованием по меньшей мере одного метода из числа описанных выше.
В соответствии с еще одним аспектом по настоящему изобретению по меньшей мере одно другое соединение представляет собой полипептид, или сшивающее, соединение или реакционный продукт, образуемый при взаимодействии сшивающего соединения с полипептидом. В том случае, когда указанное по меньшей мере одно другое соединение представляет собой полипептид, функциональная группа Y включается в полипептид. В том случае, когда указанное по меньшей мере одно другое соединение представляет собой сшивающее соединение, функциональная группа Y включается в сшивающее соединение и необязательно также в полипептид. В том случае, когда указанное по меньшей мере одно другое соединение представляет собой реакционный продукт, образуемый при взаимодействии сшивающего соединения с полипептидом, функциональная группа Y включается в сшивающее соединение.
В соответствии с еще одним аспектом по настоящему изобретению соединение (II) подвергают реакции по меньшей мере с еще одним другим соединением, осуществляемой через взаимодействие по меньшей мере одной функциональной группы Х, включенной в R" в соединении (II), по меньшей мере с одной функциональной группой Y, входящей в состав по меньшей мере одного другого соединения, с получением первого реакционного продукта. По меньшей мере одно другое соединение представляет собой предпочтительно полипептид, или сшивающее соединение, или продукт реакции сшивающего соединения с полипептидом, как обсуждалось выше. Указанный первый реакционный продукт затем подвергают реакции с соединением (I) посредством взаимодействия восстановленного конца соединения (I) с NH-группой первого реакционного продукта, которая соединяет мостиковой связью исходные остатки R' и R" в соединении (II), с получением второго реакционного продукта. Указанный второй реакционный продукт необязательно подвергают стабилизации с использованием по меньшей мере одного метода из числа описанных выше.
В соответствии с особенно предпочтительным вариантом осуществления по настоящему изобретению гидроксиэтилкрахмал используют в качестве соединения (I), О-[2-(2-аминооксиэтокси)этил]гидроксиламин используют в качестве соединения (II) и ЭПО, содержащий окисленный терминальный углеводный фрагмент в боковой углеводной цепи, используют в качестве другого соединения. Более предпочтительно, гидроксиэтилкрахмал подвергают реакции с О-[2-(2-аминооксиэтокси)этил]гидроксиламином с получением первого производного гидроксиэтилкрахмала, и далее указанное первое производное гидроксиэтилкрахмала подвергают реакции с ЭПО, содержащим окисленный терминальный углеводный фрагмент в боковой углеводной цепи, с получением второго производного гидроксиэтилкрахмала. В данном конкретном случае никакой реакции стабилизации не проводится.
В этой связи, настоящее изобретение также относится к производному гидроксиэтилкрахмала, получаемому по способу, согласно которому гидроксиэтилкрахмал подвергают реакции через его восстановленный конец с О-[2-(2-аминооксиэтокси)этил]гидроксиламином и образуемый реакционный продукт подвергают реакции с эритропоэтином через окисленный терминальный углеводный фрагмент в боковой углеводной цепи эритропоэтина.
В соответствии с еще одним особенно предпочтительным вариантом осуществления по изобретению гидроксиэтилкрахмал используют в качестве соединения (I), О-[2-(2-аминооксиэтокси)этил]гидроксиламин используют в качестве соединения (II), используют гетеробифункциональное сшивающее соединение, содержащее малеимидную группу и активную N-гдроксисукцинимидную сложноэфирную группу, а также ЭПО, содержащий по меньшей мере одну -SH-группу (называемый далее как тиоЭПО), используют в качестве полипептида. Более предпочтительно, гидроксиэтилкрахмал подвергают реакции с О-[2-(2-аминооксиэтокси)этил]гидроксиламином с получением первого производного гидроксиэтилкрахмала, указанное первое производное гидроксиэтилкрахмала подвергают реакции с активной N-гдроксисукцинимидной сложноэфирной группой сшивающего соединения с получением второго производного гидроксиэтилкрахмала и далее указанное второе производное подвергают реакции через малеимидную группу с тиоЭПО с получением третьего производного гидроксиэтилкрахмала.
Производное гидроксиалкилкрахмала, которое далее в описании будет обозначаться как конъюгат ГАК-ЭПО и который образуется в результате реакции соединения (I) и соединения (II) и, возможно, также сшивающего соединения и эритропоэтина, обладает преимуществом, связанным с тем, что он демонстрирует повышенную биологическую стабильность в сравнении с эритропоэтином до конъюгирования. Указанное свойство связано главным образом с тем, что данное производное гидроксиалкилкрахмала в меньшей степени или вовсе не распознается системами удаления в печени и почках и, в этой связи, указанный конъюгат присутствует в системе кровообращения в течение более длительного периода времени. Кроме того, поскольку ГАК присоединяется сайт-специфичным образом, минимизируется риск нарушения биологической активности ЭПО in vivo путем конъюгирования ГАК с ЭПО.
Конъюгат ГАК-ЭПО по настоящему изобретению может демонстрировать по существу ту же биологическую активность in vitro, что и рекомбинантный нативный ЭПО, поскольку его биологическая активность in vitro основана на измерении лишь афинности по связыванию с рецептором ЭПО. Способы определения биологической активности in vitro известны в данной области техники.
Кроме того, ГАК-ЭПО демонстрирует более высокую активность in vivo, чем ЭПО, используемый в качестве исходного материала для конъюгирования (неконъюгированный ЭПО). Способы определения биологической активности in vivo известны в технике.
Конъюгат ГАК-ЭПО может демонстрировать активность in vivo, составляющую от 110 до 300%, предпочтительно от 110 до 200%, более предпочтительно от 110% до 180% или от 110% до 150%, наиболее предпочтительно от 110% до 140%, если активность in vivo неконъюгированного ЭПО принять за 100%.
В сравнении с высокосиалилированной формой ЭПО в составе препарата Амген (Amgen) (см. EP 428267 B1), ГАК-ЭПО демонстрирует активность, равную предпочтительно по меньшей мере 50%, более предпочтительно по меньшей мере 70%, еще более предпочтительно по меньшей мере 85% или по меньшей мере 95%, по меньшей мере 150%, по меньшей мере 200% или по меньшей мере 300% от активности высокосиалилированного ЭПО, если активность in vivo высоко сиалилированного ЭПО принять за 100%. Наиболее предпочтительно, указанная форма демонстрирует активность по меньшей мере на уровне 95% от активности in vivo высокосиалилированного ЭПО.
Высокая биологическая активность in vivo конъюгата ГАК-ЭПО по настоящему изобретению в основном связана с тем фактом, что конъюгат ГАК-ЭПО остается дольше в кровотоке, чем неконъюгированный ЭПО, поскольку он хуже распознается системами удаления печени и поскольку снижен почечный клиренс из-за более высокой молекулярной массы. Способы определения in vivo периода полувыведения ЭПО из кровотока известны в данной области техники (Sytkowski, Lunn, Davis, Feldman, Siekman, 1998, Human erythropoietin dimers with markedly enhanced in vivo activity, Proc. Natl. Acad. Sci. USA, 95(3), 1184-8).
В этой связи, большим преимуществом настоящего изобретения является тот факт, что конъюгат ГАК-ЭПО представляет собой форму, которая может вводиться реже, чем препараты ЭПО, коммерчески доступные в настоящее время. В то время как стандартные препараты ЭПО должны вводиться по меньшей мере каждые 3 дня, конъюгат ГАК-ЭПО по настоящему изобретению предпочтительно вводится два раза в неделю, более предпочтительно один раз в неделю.
Кроме того, способ по настоящему изобретению имеет то достоинство, что может быть получено эффективное производное ЭПО со сниженной стоимостью, поскольку данный способ не включает дорогостоящие и длительные по времени стадии очистки, приводящие к низкому конечному выходу, то есть в данном способе нет необходимости очищать продукт от недостаточно сиалилированных форм ЭПО, которые, как известно, характеризуются низкой биологической активностью in vivo или вовсе ее отсутствием.
Кроме того, настоящее изобретение относится к фармацевтической композиции, включающей в терапевтически эффективном количестве конъюгат ГАК-полипептид, предпочтительно конъюгат ГАК-ЭПО, более предпочтительно конъюгат ГЭК-ЭПО по настоящему изобретению. В предпочтительном варианте фармацевтическая композиция включает также по меньшей мере один фармацевтически приемлемый разбавитель, адъювант и/или носитель, используемый в терапии эритропоэтином.
В этой связи, настоящее изобретение относится к фармацевтической композиции, включающей в терапевтически эффективном количестве описанное выше производное гидроксиалкилкрахмала, где продукт взаимодействия соединения (I) с соединением (II) подвергают реакции по меньшей мере через одну функциональную группу Х, входящую в состав соединения (II), по меньшей мере с одним другим соединением, или где соединение (II) подвергают реакции по меньшей мере через одну функциональную группу Х по меньшей мере с одним другим соединением перед проведением реакции с соединением (I) и где по меньшей мере одно другое соединение представляет собой полипептид.
В соответствии с предпочтительными вариантами осуществления по настоящему изобретению полипептид, предпочтительно эритропоэтин, подвергают реакции с соединением (II) или с продуктом реакции соединения (I) и соединения (II) через тиогруппу или окисленный углеводный фрагмент, включенный в полипептид.
В соответствии с еще более предпочтительным вариантом осуществления по настоящему изобретению, полипептид, предпочтительно эритропоэтин, подвергают реакции с соединением (II) или с продуктом реакции соединения (I) и соединения (II) через окисленный углеводный фрагмент, включенный в полипептид.
В этой связи, настоящее изобретение относится к описанной выше фармацевтической композиции, где полипептид подвергают реакции с соединением (II) или с продуктом реакции соединения (I) и соединения (II) через окисленный углеводный фрагмент, включенный в полипептид.
В соответствии с предпочтительными вариантами осуществления по настоящему изобретению, указанный полипептид представляет собой ГКС-Ф (GCS-F), AT III, IFN-бета или эритропоэтин, предпочтительно эритропоэтин.
В этой связи, настоящее изобретение относится к описанной выше фармацевтической композиции, где полипептид представляет собой эритропоэтин.
В соответствии с особенно предпочтительным вариантом осуществления по настоящему изобретению описанную выше фармацевтическую композицию получают посредством реакции гидроксиэтилкрахмала в водной среде с соединением формулы

и посредством взаимодействия реакционного продукта с эритропоэтином.
В соответствии с одним особенно предпочтительным вариантом осуществления по настоящему изобретению эритропоэтин окисляют периодатом натрия перед проведением указанной выше реакции.
В соответствии с другим особенно предпочтительным вариантом осуществления по настоящему изобретению эритропоэтин вначале подвергают частичному десиалилированию и далее окисляют периодатом натрия перед проведением указанной выше реакции.
В соответствии с другим предпочтительным вариантом из настоящего изобретения исключаются фармацевтические композиции, включающие гидроксиалкилкрахмальное производное, который получают, согласно методике примера 5, на основе полностью восстановленного тио-ЭПО.
В соответствии с другим предпочтительным вариантом настоящее изобретение также относится к фармацевтической композиции, включающей в терапевтически эффективном количестве описанное выше производное гидроксиалкилкрахмала, где продукт взаимодействия соединения (I) с соединением (II) подвергают реакции по меньшей мере через одну функциональную группу Х, входящую в состав соединения (II), по меньшей мере с одним другим соединением, или где соединение (II) подвергают реакции по меньшей мере через одну функциональную группу Х по меньшей мере с одним другим соединением перед проведением реакции с соединением (I) и где по меньшей мере одно другое соединение представляет собой сшивающее соединение и реакционный продукт, получаемый при взаимодействии соединения (I) и соединения (II) в присутствии сшивающего соединения, подвергают реакции с полипептидом.
В соответствии с еще одним предпочтительным вариантом, настоящее изобретение относится к фармацевтической композиции, в которой полипептид представляет собой эритропоэтин.
Указанная выше фармацевтическая композиция особенно хорошо подходит для лечения анемических расстройств или гематопоэтических дисфункций или связанных с ними заболеваний.
Термин "терапевтически эффективное количество" в контексте настоящего описания относится к количеству, которое обеспечивает терапевтический эффект в данных условиях и в данном режиме введения. Введение изоформ эритропоэтина предпочтительно проводится парентеральными способами. Конкретный выбираемый способ зависит от состояния пациента, которого предстоит лечить. Введение изоформ эритропоэтина предпочтительно проводят в виде композиции, содержащей соответствующий носитель, такой как человеческий сывороточный альбумин, соответствующий разбавитель, такой как забуференный солевой раствор, и/или соответствующий адъювант. Требуемая дозировка будет представлять собой количества, достаточные для повышения гематокрита у пациента, и будет варьироваться, в зависимости от тяжести состояния, подлежащего лечению, способа введения и т.п.
Объектом лечения фармацевтической композицией по настоящему изобретению предпочтительно является повышение уровня гемоглобина более чем 6,8 ммоль/л в крови. Для этой цели фармацевтическая композиция может вводиться таким способом, чтобы содержание гемоглобина повышалось от 0,6 ммоль/л до 1,6 ммоль/л в неделю. Если содержание гемоглобина превышает уровень 8,7 ммоль/л, то предпочтительно лечение следует прервать до снижения уровня гемоглобина ниже 8,1 ммоль/л.
Композиция по настоящему изобретению предпочтительно используется в составе препарата, пригодного для подкожной, или внутривенной, или парентеральной инъекции. Пригодные для этой цели наполнители и носители включают, например, дигидрофосфат натрия, гидрофосфат динатрия, хлорат натрия, полисорбат 80, ЧСА и воду для инъекций. Указанная композиция может вводиться три раза в неделю, предпочтительно два раза в неделю, более предпочтительно один раз в неделю и наиболее предпочтительно через неделю.
Предпочтительно фармацевтическую композицию вводят в количестве от 0,01 до 10 мкг/кг веса тела пациента, более предпочтительно от 0,1 до 5 мкг/кг, от 0,1 до 1 мкг/кг или от 0,2 до 0,9 мкг/кг, наиболее предпочтительно от 0,3 до 0,7 мкг/кг и еще наиболее предпочтительно от 0,4 до 0,6 мкг/кг веса тела.
В целом, в составе дозы вводят предпочтительно от 10 мкг до 200 мкг, предпочтительно от 15 мкг до 100 мкг.
Настоящее изобретение также относится к применению ГАК-полипептида по настоящему изобретению при получении лекарственного средства, применяемого для лечения анемических расстройств или гематопоэтических дисфункций или связанных с ними заболеваний.
Кроме того, изобретение относится к HAS-полипептиду по настоящему изобретению для применения при лечении человека или животного.
Настоящее изобретение также относится к применению ГАК-ЭПО по настоящему изобретению при получении лекарственного средства, применяемого для лечения анемических расстройств или гематопоэтических дисфункций или связанных с ними заболеваний.
Ниже настоящее изобретение иллюстрируется с помощью поясняющих его примеров, таблиц и чертежей, которые ни в коей мере не следует рассматривать как ограничивающие область по настоящему изобретению.
Краткое описание чертежей
Фиг.1. На фиг.1 показаны результаты анализа в ДСН-ПААГ конъюгата ГЭК-ЭПО, полученного по методике примера 4.1.
Линия А: Белковый маркер Roti®-Mark PRESTAINED (Carl Roth GmbH+Co, Karlsruhe, D); значения молекулярной массы (кД) белкового маркера сверху вниз: 245, 123, 77, 42, 30, 25,4 и 17.
Линия B: Неочищенный продукт, полученный при конъюгировании по методике примера 4.1.
Линия С: Исходный материал ЭПО.
Фиг.2. На фиг.2 показаны результаты анализа в ДСН-ПААГ конъюгата ГЭК-ЭПО, полученного по методике примера 4.3.
Линия А: Неочищенный продукт, полученный при конъюгировании по методике примера 4.3.
Линия В: Исходный материал ЭПО.
Линия С: Белковый маркер Roti®-Mark PRESTAINED (Carl Roth GmbH+Co, Karlsruhe, D); значения молекулярной массы (кД) белкового маркера сверху вниз: 245, 123, 77, 42, 30, 25,4 и 17.
Фиг.3. На фиг.3 показаны результаты анализа в ДСН-ПААГ конъюгатов ГЭК-ЭПО, полученных по методике примеров 6.1 и 6.4.
Линия А: Белковый маркер Roti®-Mark PRESTAINED (Carl Roth GmbH+Co, Karlsruhe, D); значения молекулярной массы (кД) белкового маркера сверху вниз: 245, 123, 77, 42, 30, 25,4 и 17.
Линия B: Неочищенный продукт, полученный при конъюгировании по методике примера 6.1.
Линия С: Неочищенный продукт, полученный при конъюгировании по методике примера 6.4.
Линия D: Исходный материал ЭПО.
Фиг.4. На фиг.4 показаны результаты анализа в ДСН-ПААГ конъюгатов ГЭК-ЭПО, полученных по методике примеров 6.2, 6.3, 6.5 и 6.6.
Линия А: Белковый маркер Roti®-Mark PRESTAINED (Carl Roth GmbH+Co, Karlsruhe, D); значения молекулярной массы (кД) белкового маркера сверху вниз: 245, 123, 77, 42, 30, 25,4 и 17.
Линия B: Неочищенный продукт, полученный при конъюгировании по методике примера 6.6, на основе примера 1.3b).
Линия С: Неочищенный продукт, полученный при конъюгировании по методике примера 6.5, на основе примера 1.1b).
Линия D: Неочищенный продукт, полученный при конъюгировании по методике примера 6.6, на основе примера 1.3а).
Линия Е: Неочищенный продукт, полученный при конъюгировании по методике примера 6.5, на основе примера 1.1а).
Линия F: Неочищенный продукт, полученный при конъюгировании по методике примера 6.2.
Линия G: Неочищенный продукт, полученный при конъюгировании по методике примера 6.3.
Линия К: Исходный материал ЭПО.
Фиг.5. Результаты анализа в ДСН-ПААГ ЕРО-GT-1, подвергнутого мягкой кислотной обработке: в течение 5 минут = линия 2; 10 минут = линия 3; 60 минут = линия 4 и необработанный ЭПО = линия 1; показано смещение мобильности ЭПО после удаления N-гликанов (+PNGASE).
Фиг.6. Результаты анализа в HPAEC-PAD олигосахаридов, выделенных из необработанного ЭПО и из ЭПО, инкубированных в течение 5 минут, 10 минут и 60 минут в условиях мягкого кислотного гидролиза. Римские цифры I-Vуказывают положение при элюировании: I = десиалилированной диантенной структуры, II= трисиалилированных трехантенных структур (два изомера); III = тетрасиалилированной тетраантенной структуры + 2 N-ацетиллактозаминовых повтора; IV= тетрасиалилированной тетраантенной структуры + 1 N-ацетиллактозаминовый повтор; V = тетрасиалилированной тетраантенной структуры без N-аетиллактозаминового повтора. В скобках показана область элюции олигосахаридных структур без сиаловой кислоты и при наличии 1-4 сиаловых кислот.
Фиг.7.Результаты анализа в HPAEC-PAD N-связанных олигосахаридов после десиалилирования; показано положение в ходе элюции N-ацетилнейраминовой кислоты; номера 1-9 указывают положение при элюции стандартных олигосахаридов: 1 = диантенный; 2 = триантенный (2-4 изомера), 3 = триантенный (2-6 изомеров), 4 = тетрантенный; 5 = триантенный + 1 повтор; 6 = тетраантенный + 1 повтор; 7 = триантенный + 2 повтора; 8 = тетраантенный + 2 повтора и 9 = тетраантенный + 3 повтора.
Фиг.8. Результаты анализа в ДСН-ПААГ ЭПО после обработки в мягких условиях и необработанного ЭПО, которые подвергают окислению периодатом по остаткам сиаловой кислоты: 1 = окисление периодатом, но без кислотной обработки; 2 = окисление периодатом + 5 минут кислотной обработки; 3 = окисление периодатом + 10 минут кислотной обработки; 4 = окисление периодатом, но без кислотной обработки; 5 = Стандарт ЭПО BRP без окисления периодатом и без кислотной обработки.
Фиг.9. Результаты анализа в HPAEC-PAD нативных олигосахаридов, выделенных из необработанного ЭПО и из ЭПО, инкубированных в течение 5 минут и 10 минут в условиях мягкого кислотного гидролиза, с последующей обработкой периодатом. В скобках 1-5 показана область элюции олигосахаридных структур без сиаловой кислоты и при наличии 1-4 сиаловых кислот.
Фиг.10. Результаты анализа в ДСН-ПААГ ГЭК-модификации с течением времени ЕРО-GT-1-А: аликвоты по 20 мкл ЕРО-GT-1-А подвергают реакции с производным Х, полученным с использованием модифицированного гидроксиламином ГЭК, в течение 30 минут, 2, 4 и 17 часов. Линия 1 = время реакции 30 минут; линия 2 = время реакции 2 часа; линия 3 = время реакции 4 часа; линия 4 = время реакции 17 часов; линия 5 = ЕРО-GT-1-А без модификации ГЭК. На левом чертеже показано смещение подвижности ЕРО-GT-1-А при увеличении времени инкубации в присутствии модифицированного гидроксиламином ГЭК-производного (скорость течения: 1 мл/мин); Х: линия 1 = время реакции 30 минут; линия 2 = время реакции 2 часа; линия 3 = время реакции 4 часа; линия 4 = время реакции 17 часов; линия 5 = ЕРО-GT-1-А при наличии ГЭК-модификации. Чертеж в правой стороне показывает результат анализа тех же образцов после их обработки N-гликозидазой.
Фиг.11. Результаты анализа в ДСН-ПААГ фракций конъюгатов ГЭК-ЭПО, полученных при элюции с сефарозы (Q-Sepharose). Концентрируют в концентраторе Speed Vac 1% фракции, полученной при прохождении потока через колонку, и 1% фракции, элюируемой при высокой концентрации соли, и затем наносят на гели в буфере для образца. ЭПО белок окрашивают красителем кумасси блю.А = образец I; B = образец II; C = образец III; К = контроль ЕРО-GT-1; А1, В1, С1 и К1 относятся к фракциям, полученным при свободном прохождении потока; А2, В2, С2 и К2 указывают на фракции, элюируемые при высокой концентрации соли.
Фиг.12а. Результаты анализа в ДСН-ПААГ ГЭК-модифицированного образца ЭПО А2 (см. фиг.7), контрольного образца ЭПО К2 и ЕРО-GT-1 препарата ЭПО, расщепленных в присутствии N-гликозидазы, для удаления N-связанных олигосахаридов. Все образцы ЭПО демонстрируют смещение мобильности в сторону форм с низкой молекулярной массой, которые не содержат или содержат О-гликан. Наблюдается сниженный коэффициент соотношения полос для О-гликозированного и негликозилированного белка в случае ГЭК-модифицированного образца ЭПО А2 после де-N-гликозилирования, а также отмечается размытая белковая полоса в области 30 кДа, что преимущественно отражает наличие ГЭК-модификации по сиаловой кислоте О-гиканового остатка (см. стрелку, помеченную звездочкой).
Фиг.12b. Результаты анализа в ДСН-ПААГ ГЭК-модифицированного образца ЭПО А2 после гидролиза в мягких условиях (см. фиг.11), контрольного образца ЭПО К2 и ЕРО-GT-1А, необработанных или расщепленных в присутствии N-гликозидазы для удаления N-связанных олигосахаридов (см. фиг.12а). Как высокомолекулярная форма А2 до, так и форма А после N-гикозидазной обработки (см. участки со скобками, помеченными и не помеченными стрелками), исчезают при кислотной обработке образцов. Стандарт ЭПО BRP, который анализируют для сравнения, не подвергают кислотной обработке в мягких условиях.
Фиг.13. Результаты анализа в HPAEC-PAD N-связанного олигосахаридного материала, высвобождаемого из ГЭК-модифицированного образца А, из ЕРО-GT-1-А и из контрольного образца ЭПО, инкубированного с немодифицированным ГЭК (К). Римские цифры I-V указывают положение при элюции: I = дисиалилированной диантенной структуры, II = трисиалилированных триантенных структур (два изомера); III = тетрасиалилированной тетрантенной структуры + 2 N-ацетиллактозаминовых повтора; IV = тетрасиалилированной тетраантенной структуры + 1 N-аетиллактозаминовый повтор; V= тетрасиалилированной тетраантенной структуры без N-ацетиллактозаминового повтора; в скобках показана область элюции ди-, три- и тетрасиалилированных N-гликанов, как описано в пояснении к фиг.6 и 9.
Фиг.14. Результаты анализа в HPAEC-PAD N-связанного олигосахаридного материала, высвобождаемого из ГЭК-модифицированного образца А, из ЕРО-GT-1А и из контрольного образца ЭПО (К), инкубированного с немодифицированным ГЭК. Показано время удерживания смеси стандартных олигосахаридов: номера 1-9 указывают положение при элюции стандартных олигосахаридов: 1 = диантенного; 2 = триантенного (2-4 изомера); 3 = триантенного (2-6 изомеров); 4 = тетраантенного; 5 = триантенного + 1 повтор; 6 = тетраантенного + 1 повтор; 7 = триантенного+ 2 повтора; 8 = тетраантенного + 2 повтора и 9 = тетраантенного + 3 повтора.
Фиг.15-21. На фиг.15-21 показаны масс-спектры MALDI-TOF энзиматически высвобожденных и химически десиалилированных N-гиканов, выделенных из препаратов ГЭК-модифицированного ЭПО и контрольного ЭПО. Основные сигналы при значениях m/z, равных 1809,7, 2174,8, 2539,9, 2905,0 и 3270,1 ([M+Na]), соответствуют ди-тетраантенным N-гликановым структурам комплексного типа, в отсутствие или при наличии одного или двух N-ацетиллактозаминовых повторов, сопровождаемых слабыми сигналами из-за потери фукозы или галактозы, вызванной условиями кислотной обработки, используемыми для десиалилирования образцов, подвергаемых МС-анализу.
Фиг.15. MALDI-TOF спектр: десиалилированные олигосахариды ГЭК-модифицированного ЭПО А2.
Фиг.16. MALDI-TOF спектр: десиалилированные олигосахариды ЭПО GT-1-A.
Фиг.17. MALDI-TOF спектр: десиалилированные олигосахариды ЭПО К2.
Фиг.18. MALDI-TOF спектр: десиалилированные олигосахариды ЕРО-GT-1.
Фиг.19. MALDI-TOF спектр: десиалилированные олигосахариды ЕРО-GT-1, подвергнутые кислотному гидролизу в течение 5 минут.
Фиг.20. MALDI-TOF спектр: десиалилированные олигосахариды ЕРО-GT-1, подвергнутые кислотному гидролизу в течение 10 минут.
Фиг.21. MALDI-TOF спектр: десиалилированные олигосахариды ЕРО-GT-1, подвергнутые кислотному гидролизу в течение 60 минут.
Примеры
Пример 1
Образование производных гидроксиэтилкрахмала путем восстановительного аминирования
Пример 1.1
Реакция гидроксиэтилкрахмала с 1,3-диамино-2-гидроксипропаном

а) К раствору 200 мг гидроксиэтилкрахмала (ГЭК18/0,4 (М.в. = 18000 Д, СЗ=0,4) в 5 мл воды добавляют 0,83 ммоль 1,3-дамино-2-гидроксипропана и 50 мг цианоборгидрида натрия NaCNBH3. Полученную смесь инкубируют при 80°С в течение 17 часов. Реакционную смесь добавляют к 160 мл охлажденной смеси ацетона и этанола, 1:1 (объем/объем). Осадок собирают центрифугированием, диализуют в течение 4 дней против воды (трубка для диализа SnakeSkin, с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science, Deutschland GmbH, Bonn, Germany) и лиофилизируют.
b) Инкубирование смеси, полученной при добавлении 0,83 ммоль 1,3-диамино-2-гидроксипропана и 50 мг цианоборгидрида натрия NaCNBH3 к раствору 200 мг гидроксиэтилкрахмала, также возможно и проводится при 25°С в течение 3 дней.
Пример 1.2
Реакция гидроксиэтилкрахмала с 1,2-дигидрокси-3-аминопропаном

а) К раствору 200 мг гидроксиэтилкрахмала (ГЭК18/0,4 (М.в. = 18000 Д, СЗ=0,4) в 5 мл воды добавляют 0,83 ммоль 1,2-дгидрокси-3-аминопропана и 50 мг цианоборгидрида натрия NaCNBH3. Полученную смесь инкубируют при 80°С в течение 17 часов. Реакционную смесь добавляют к 160 мл охлажденной смеси ацетона и этанола, 1:1 (объем/объем). Осадок собирают центрифугированием, диализуют в течение 4 дней против воды (трубка для диализа SnakeSkin, с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science, Deutschland GmbH, Bonn, Germany) и лиофилизируют.
b) Инкубирование смеси, полученной при добавлении 0,83 ммоль 1,2-дигидрокси-3-аминопропана и 50 мг цианоборгидрида натрия NaCNBH3 к раствору 200 мг гидроксиэтилкрахмала, также возможно и проводится при 25°С в течение 3 дней.
Осуществление реакции 1,2-дигидрокси-3-аминопропана с ГЭК подтверждается опосредованно при определении количества формальдегида, получаемого при окислительном расщеплении 1,2-дола в реакционном продукте периодатом, как описано в работе Авигада (G. Avigad, Anal. Biochem. 134 (1983) 449-504).
Пример 1.3
Реакция гидроксиэтилкрахмала с 1,4-диаминобутаном

а) К раствору 200 мг гидроксиэтилкрахмала (ГЭК18/0,4 (М.в. = 18000 Д, СЗ=0,4) в 5 мл воды добавляют 0,83 ммоль 1,4-даминобутана и 50 мг цианоборгидрида натрия NaCNBH3. Полученную смесь инкубируют при 80°С в течение 17 часов. Полученную смесь добавляют к 160 мл охлажденной смеси ацетона и этанола, 1:1 (объем/объем). Осадок собирают центрифугированием, диализуют в течение 4 дней против воды (трубка для диализа SnakeSkin, с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science, Deutschland GmbH, Bonn, Germany) и лиофилизируют.
b) Инкубирование смеси, полученной при добавлении 0,83 ммоль 1,4-диаминобутана и 50 мг цианоборгидрида натрия NaCNBH3 к раствору 200 мг гидроксиэтилкрахмала, также возможно и проводится при 25°С в течение 3 дней.
Пример 1.4
Реакция гидроксиэтилкрахмала с 1-меркапто-2-аминоэтаном
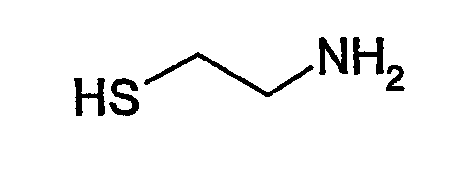
а) К раствору 200 мг гидроксиэтилкрахмала (ГЭК18/0,4 (М.в. = 18000 Д, СЗ=0,4)) в 5 мл воды добавляют 0,83 ммоль 1-мркапто-2-аминоэтана и 50 мг цианоборгидрида натрия NaCNBH3. Полученную смесь инкубируют при 80°С в течение 17 часов. Реакционную смесь добавляют к 160 мл охлажденной смеси ацетона и этанола, 1:1 (объем/объем). Осадок собирают центрифугированием, диализуют в течение 4 дней против воды (трубка для диализа SnakeSkin, с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science, Deutschland GmbH, Bonn, Germany) и лиофилизируют.
b) Инкубирование смеси, полученной при добавлении 0,83 ммоль 1-меркапто-2-аминоэтана и 50 мг цианоборгидрида натрия NaCNBH3 к раствору 200 мг гидроксиэтилкрахмала, также возможно и проводится при 25°С в течение 3 дней.
Пример 2
Образование производных гидроксиэтилкрахмала путем конъюгирования
Пример 2.1
Реакция гидроксиэтилкрахмала с карбогидразидом

0,96 г ГЭК18/0,4 (М.в. = 18000 Д, СЗ=0,4) растворяют в 8 мл водного 0,1 М натрий-ацетатного буфера, pH 5,2 и добавляют 8 ммоль карбогидразида (Sigma, Aldrich, Taufkirchen, D). После перемешивания в течение 18 часов при 25°С реакционную смесь добавляют к 160 мл охлажденной смеси ацетона и этанола, 1:1 (объем/объем). Осажденный продукт собирают центрифугированием, перерастворяют в 40 мл воды, диализуют в течение 3 дней против воды (трубка для диализа SnakeSkin, с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science, Deutschland GmbH, Bonn, Germany) и лиофилизируют.
Пример 2.2
Реакция гидроксиэтилкрахмала с адипиновым дигидразидом
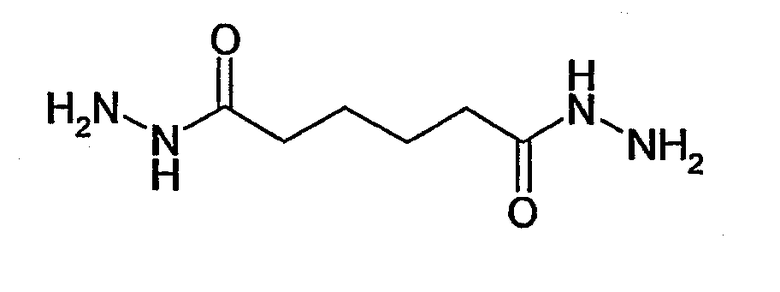
0,96 г ГЭК18/0,4 (М.в. = 18000 Д, СЗ=0,4) растворяют в 8 мл водного 0,1 М натрий-ацетатного буфера, pH 5,2 и добавляют 8 ммоль адипинового дигидразида (Lancaster Synthesis, Frankfurt/Main, D). После перемешивания в течение 18 часов при 25°С реакционную смесь добавляют к 160 мл охлажденной смеси ацетона и этанола, 1:1, (объем/объем). Осажденный продукт собирают центрифугированием, перерастворяют в 40 мл воды, диализуют в течение 3 дней против воды (трубка для диализа SnakeSkin, с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science, Deutschland GmbH, Bonn, Germany) и лиофилизируют.
Пример 2.3
Реакция гидроксиэтилкрахмала с 1,4-фенилен-бис-3-тиосемикарбозидом

0,96 г ГЭК18/0,4 (М.в. = 18000 Д, СЗ=0,4) растворяют в 8 мл водного 0,1 М натрий-ацетатного буфера, pH 5,2 и добавляют 8 ммоль 1,4-фенилен-бис-3-тиосемикарбазида (Lancaster Synthesis, Frankfurt/Main, D). После перемешивания в течение 18 часов при 25°С к реакционной смеси добавляют 8 мл воды и суспензию центрифугируют в течение 15 минут при 4500 об/мин. Прозрачный супернатант декантируют и затем добавляют к 160 мл охлажденной смеси ацетона и этанола, 1:1, (объем/объем). Осажденный продукт собирают центрифугированием, перерастворяют в 40 мл воды и центрифугируют в течение 15 минут при 4500 об/мин. Прозрачный супернатант диализуют в течение 3 дней против воды (трубка для диализа SnakeSkin, с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science, Deutschland GmbH, Bonn, Germany) и лиофилизируют.
Пример 2.4
Реакция гидроксиэтилкрахмала с О-[2-(2-аминоокси-этокси)этил]гидроксиламином
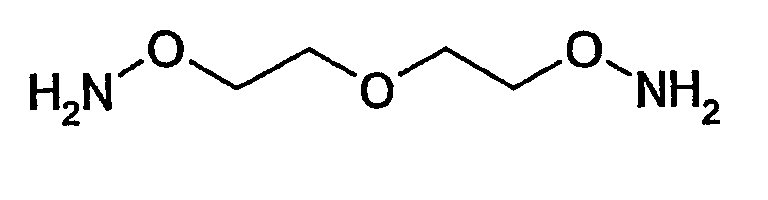
О-[2-(2-аминоокси-этокси)этил]гидроксиламин синтезируют по методу Ботурина с соавт. (Boturyn et al., Tetrahedron 53 (1997) pp.5485-5492) в 2 стадии из коммерчески доступного материала.
0,96 г ГЭК18/0,4 (М.в. = 18000 Д, СЗ=0,4) растворяют в 8 мл водного 0,1 М натрий-ацетатного буфера, pH 5,2 и добавляют 8 ммоль О-[2-(2-аминоокси-этокси)этил]гидроксиламина. После перемешивания в течение 18 часов при 25°С реакционную смесь добавляют к 160 мл охлажденной смеси ацетона и этанола, 1:1 (объем/объем). Осажденный продукт собирают центрифугированием, перерастворяют в 40 мл воды, диализуют в течение 3 дней против воды (трубка для диализа SnakeSkin, с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science, Deutschland GmbH, Bonn, Germany) и лиофилизируют.
Пример 3
Окисление эритропоэтина
Окисленный эритропоэтин получают по методике, описанной в примере 7. В качестве окисленного эритропоэтина используют EPO-GT-1-A, как описано в примере 7.11 (с) (EPO-GT-1 получают без кислотного гидролиза путем обработки в условиях мягкого окисления периодатом).
Пример 4
Конъюгирование гидроксиэтилкрахмальных производных с окисленным эритропоэтином из примера 3
Пример 4.1
Реакция окисленного эритропоэтина с реакционным продуктом из примера 2.1
Окисленный ЭПО (1,055 мкг/мкл) в 20 мМ буфера ФБР доводят до значения pH 5,3 с использованием 5 М натрий-ацетатного буфера, pH 5,2. К 19 мкл раствора ЭПО добавляют 18 мкл раствора производного ГЭК, полученного по процедуре примера 2.1 (M.в. 18 кД; 18,7 мкг/мкл в 0,1 М натрий-ацетатном буфере, pH 5,2), и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт очищают в ДСН-ПААГ с использованием буфера NuPAGE 10% бис-Трис гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях от компании Инвитроген (Invitrogen). Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента проиллюстрирован на фиг.1. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокого молекулярного веса. Расширение полосы связано с распределением по молекулярному весу используемых производных ГЭК и количеством производных ГЭК, присоединенных к белку.
Пример 4.2
Реакция окисленного эритропоэтина с реакционным продуктом из примера 2.3
Окисленный ЭПО (1,055 мкг/мкл) в 20 мМ буфера ФБР доводят до значения pH 5,3 с использованием 5 М натрий-ацетатного буфера, pH 5,2. К 19 мкл раствора ЭПО добавляют 18 мкл раствора производного ГЭК, полученного по процедуре примера 2.3 (M.в. 18 кД; 18,7 мкг/мкл в 0,1 М натрий-ацетатном буфере, pH 5,2) и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом ДСН-ПААГ с использованием буфера NuPAGE 10% бис-Трис гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Инвитроген (Invitrogen, Carlcbad, CA, USA).
Пример 4.3
Реакция окисленного эритропоэтина с реакционным продуктом из примера 2.4
Окисленный ЭПО (1,055 мкг/мкл) в 20 мМ буфера ФБР доводят до значения pH 5,3 с использованием 5 М натрий-ацетатного буфера, pH 5,2. К 19 мкл раствора ЭПО добавляют 18 мкл раствора производного ГЭК, полученного по процедуре примера 2.4 (M.в. 18 кД; 18,7 мкг/мкл в 0,1 М натрий-ацетатном буфере, pH 5,2), и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом ДСН-ПААГ с использованием буфера NuPAGE 10% бис-Трис гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Инвитроген. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.2А. Удачный результат конъюгирования показан в виде перемещения белковой полосы в сторону более высокого молекулярного веса. Расширение полосы связано с распределением по молекулярному весу используемых производных ГЭК и с числом производных ГЭК, присоединенных к белку.
Пример 5
Образование тио-ЭПО путем восстановления эритропоэтина
241,5 мкг эритропоэтина (EPO-GT-1, см. пример 7) в 500 мкл 0,1 М натрий-боратного буфера, 5 мМ ЭДТА, 10 мМ ДТТ (Lancaster, Morcambe, UK), pH 8,3, инкубируют в течение 1 часа при 37°С. ДТТ удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 10 KD MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин, с последующей промывкой 3 раза боратным буфером и дважды фосфатным буфером (0,1 M, 9,15 M NaCl, 50 мМ ЭДТА, pH 7,2).
Пример 6
Конъюгирование производных гидроксиэтилкрахмала с тио-эритропоэтином с использованием сшивающего соединения
В каждом из приведенных ниже примеров в качестве сшивающего соединения используют сложный N-(альфа-малемидоацетокси)сукцинимидный эфир (АМАС; AMAS).

Пример 6.1
Реакция окисленного эритропоэтина с реакционным продуктом из примера 2.1 и сшивающим соединением
К 50 нмоль ГЭК-производного, полученного по методике примера 2.1 и растворенного в 200 мкл 0,1М натрий-фосфатного буфера (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, pH 7,2), добавляют 10 мкл 2,5 мкмоль раствора AMAS (Sigma, Aldrich, Taufkirchen, D) в ДМСО. Прозрачный раствор инкубируют в течение 80 минут при температуре 25°С и 20 минут при 40°С. Оставшийся АМАС удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 5 кД MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с промывкой 4 раза по 30 минут фосфатным буфером.
К оставшемуся раствору добавляют 15 мкг тиоЭПО, полученного по методике примера 5 (1 мкг/мкл в фосфатном буфере), и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом ДСН-ПААГ с использованием буфера NuPAGE 10% бис-Трис гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Инвитроген. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.3. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокого молекулярного веса. Расширение полосы связано с распределением по молекулярному весу используемых производных ГЭК и с числом производных ГЭК, присоединенных к белку.
Пример 6.2
Реакция окисленного эритропоэтина с реакционным продуктом из примера 2.2 и сшивающим соединением
К 50 нмоль ГЭК-производного, полученного по методике примера 2.2 и растворенного в 200 мкл 0,1 М натрий-фосфатного буфера (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, pH 7,2) добавляют 10 мкл раствора 2,5 мкмоль АМАС (Sigma, Aldrich, Taufkirchen, D) в ДМСО. Прозрачный раствор инкубируют в течение 80 минут при температуре 25°С и 20 минут при 40°С. Оставшийся АМАС удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 5 кД MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с промывкой 4 раза в течение 30 минут в фосфатном буфере.
К оставшемуся раствору добавляют 15 мкг тиоЭПО, полученного по методике примера 5 (1 мкг/мкл в фосфатном буфере), и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют в ДСН-ПААГ с использованием буфера NuPAGE 10% бис-Трис гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Инвитроген. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.4. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокого молекулярного веса. Расширение полосы связано с распределением по молекулярному весу используемых производных ГЭК и с числом производных ГЭК, присоединенных к белку.
Пример 6.3
Реакция окисленного эритропоэтина с реакционным продуктом из примера 2.3 и сшивающим соединением
К 50 нмоль ГЭК-производного, полученного по методике примера 2.3 и растворенного в 200 мкл 0,1 М натрий-фосфатного буфера (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, pH 7,2) добавляют 10 мкл раствора 2,5 мкмоль АМАС (Sigma, Aldrich, Taufkirchen, D) в ДМСО. Прозрачный раствор инкубируют в течение 80 минут при температуре 25°С и 20 минут при 40°С. Оставшийся АМАС удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 5 кД MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с промывкой 4 раза по 30 минут в фосфатном буфере.
К оставшемуся раствору добавляют 15 мкг тиоЭПО, полученного по методике примера 5 (1 мкг/мкл в фосфатном буфере), и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом ДСН-ПААГ с использованием буфера NuPAGE 10% бис-Трис гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Инвитроген. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.4. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокого молекулярного веса. Расширение полосы связано с распределением по молекулярному весу используемых производных ГЭК и с числом производных ГЭК, присоединенных к белку.
Пример 6.4
Реакция окисленного эритропоэтина с реакционным продуктом из примера 2.4 и сшивающим соединением
К 50 нмоль ГЭК-производного, полученного по методике примера 2.4 и растворенного в 200 мкл 0,1 М натрий-фосфатного буфера (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, pH 7,2), добавляют 10 мкл раствора 2,5 мкмоль АМАС (Sigma, Aldrich, Taufkirchen, D) в ДМСО. Прозрачный раствор инкубируют в течение 80 минут при температуре 25°С и 20 минут при 40°С. Оставшийся АМАС удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 5 кД MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с промывкой 4 раза по 30 минут в фосфатном буфере.
К оставшемуся раствору добавляют 15 мкг тиоЭПО, полученного по методике примера 5 (1 мкг/мкл в фосфатном буфере), и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом ДСН-ПААГ с использованием буфера NuPAGE 10% бис-Трис гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Инвитроген. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.3. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокой молекулярной массы. Расширение полосы связано с распределением по молекулярному весу используемых производных ГЭК и с числом производных ГЭК, присоединенных к белку.
Пример 6.5
Реакция окисленного эритропоэтина с реакционным продуктом из примера 1.1 и сшивающим соединением
К 50 нмоль ГЭК-производного, полученного по методике примера 1.1, при инкубации в условиях, включающих 80°С в течение 17 часов (пример 1.1а)), а также 25°С в течение 3 дней (пример 1.1b)), и растворенного в 200 мкл 0,1 М натрий-фосфатного буфера (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, pH 7,2) добавляют 10 мкл раствора 2,5 мкмоль АМАС (Sigma, Aldrich, Taufkirchen, D) в ДМСО. Прозрачный раствор инкубируют в течение 80 минут при температуре 25°С и 20 минут при 40°С. Оставшийся АМАС удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 5 кД MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с промывкой 4 раза по 30 минут в фосфатном буфере.
К оставшемуся раствору добавляют 15 мкг тиоЭПО, полученного по методике примера 5 (1 мкг/мкл в фосфатном буфере), и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом ДСН-ПААГ с использованием буфера NuPAGE 10% бис-Трис гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Инвитроген. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.4. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокого молекулярного веса. Расширение полосы связано с распределением по молекулярному весу используемых производных ГЭК и с числом производных ГЭК, присоединенных к белку.
Пример 6.6
Реакция окисленного эритропоэтина с реакционным продуктом из примера 1.3 и сшивающим соединением
К 50 нмоль ГЭК-производного, полученного по методике примера 1.3, при инкубации в условиях, включающих 80°С в течение 17 часов (пример 1.3а)), а также 25°С в течение 3 дней (пример 1.3b)), и растворенного в 200 мкл 0,1 М натрий-фосфатного буфера (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, pH 7,2), добавляют 10 мкл раствора 2,5 мкмоль АМАС (Sigma, Aldrich, Taufkirchen, D) в ДМСО. Прозрачный раствор инкубируют в течение 80 минут при температуре 25°С и 20 минут при 40°С. Оставшийся АМАС удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 5 кД MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с промывкой 4 раза по 30 минут в фосфатном буфере.
К оставшемуся раствору добавляют 15 мкг тиоЭПО, полученного по методике примера 5 (1 мкг/мкл в фосфатном буфере), и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом ДСН-ПААГ с использованием буфера NuPAGE 10% бис-Трис гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Инвитроген. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.4. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокой молекулярной массы. Расширение полосы связано с распределением по молекулярному весу используемых производных ГЭК и с числом производных ГЭК, присоединенных к белку.
Пример 7
Препаративное получение конъюгатов ГЭК-ЭПО
Краткое описание
Конъюгаты ГЭК-ЭПО синтезируют путем связывания ГЭК-производных (среднее значение М.в. составляет 18000 Дальтон; степень замещения гидроксиэтилом составляет 0,4) с частично (в условиях мягкой обработки периодатом) окисленными остатками сиаловой кислоты на олигосахаридных цепях рекомбинантного человеческого ЭПО. На основании структурного анализа углеводов было показано, что введенные модификации не воздействуют на структурную целостность ядерных олигосахаридных цепей, поскольку анализ методом MALDI/TOF-MS ГЭК-модифицированных гликанов в условиях мягкой обработки кислотой выявил наличие интактных нейтральных цепей N-ацетиллактозаминового типа, которые не отличались от соответствующих цепей, наблюдаемых в немодифицированном ЭПО продукте. Полученный результат указывает на то, что по меньшей мере 3 модифицированных ГЭК-остатка присоединяются к молекуле ЭПО в препарате ЭПО, который подвергается модификации без предшествующего частичного удаления сиаловой кислоты. Вариант ЭПО, потерявший примерно 50% остатков сиаловой кислоты относительно предшествующего белка, демонстрирует аналогичную мобильность в ДСН-ПААГ, явно указывающую на высокий молекулярный вес (60-110 кДа против 40 кДа, характерного для стандарта BRP ЭПО). ГЭК-модифицированный ЭПО стабилен в стандартных условиях ионообменной хроматографии при комнатной температуре и pH 3-10.
Биотест на ЭПО в системе нормоцитных мышей указывает на то, что ГЭК-модифицированный ЭПО обладает в 2,5-3,0 раза более высокой удельной активностью (МЕ/мг) в данном тесте в сравнении международным эталонным стандартом BRP ЭПО, что следует из результатов определения белка по величине поглощения в УФ-области, в соответствии с методом Европейской Фармакопеи, и результатов определения белка ЭПО методом ОФ-ВЭЖХ, калиброванным относительно стандартного препарата BRP ЭПО.
Пример 7.1
Материалы и методы
(а) Высвобождение N-связанных олигосахаридов путем расщепления N-гликозидазой
Образцы инкубируют с 25 единицами (в соответствии со спецификацией от производителя, Roche Diagnostics, Germany) рекомбинантной ПНГ-азы F в течение ночи при 37°С. Полноту расщепления отслеживают по специфическому изменению мобильности белка в ДСН-ПААГ. Высвобожденные N-гликаны отделяют от полипептида путем добавления 3 объемов холодного 100% этанола с последующей инкубацией при -20°С в течение по меньшей мере 2 часов (Schroeter S et al., 1999). Осажденный белок удаляют центрифугированием в течение 10 минут при 4°С со скоростью 13000 об/мин. Полученный осадок затем промывают два раза с использованием 500 мкл охлажденного льдом 70% этанола. Олигосахариды в объединенных супернатантах сушат в вакуумной центрифуге (концентратор Speed Vac, Savant Instruments Inc., USA). Образцы гликана перед использованием обессоливают с помощью картриджей Hypercarb (25 мг или 100 мг HyperCarb) следующим образом: колонки промывают 3×500 мкл 80% ацетонитрила (объем/объем) в 0,1% ТФУ с последующими промывками 3·500 мкл воды. Образцы разбавляют водой до конечного объема 300 мкл - 600 мкл перед внесением в картридж, который затем тщательно промывают водой. Олигосахариды элюируют с использованием 1,2 мл (картриджи на 25 мг; берут 1,8 мл в случае картриджей на 100 мг) 25% ацетонитрила в воде, содержащей 0,1% трифторуксусной кислоты (объем/объем). Элюированные олигосахариды нейтрализуют с использованием 2 M NH4OH и сушат в концентраторе Speed Vac. В некоторых случаях обессоливание олигосахаридов, высвобожденных N-гликозидазой, проводят путем абсорбции расщепленной смеси из образцов, содержащих <100 мкг общего (глико)протеина, на картриджах Hypercarb на 100 мг.
(b) Анализ олигосахаридов с помощью лазерной десорбции на основе матричной/ионизационной масс-спектрометрии по времени пролета (MALDI/TOF/TOF-MS)
Используют прибор Bruker ULTRAFLEX для анализа по времени пролета (TOF-TOF): нативные десиалилированные олигосахариды анализируют с использованием 2,5-дигидроксибензойной кислоты в качестве УФ-поглощающего материала, как в варианте положительного, так и отрицательного иона с использованием в обоих случаях рефлектрона. В случае анализов по методу MS-MS выбранные парные ионы подвергают индуцированной лазером диссоциации (LID) и полученные фрагменты ионов разделяют с помощью второго блока TOF прибора (LIFT). Образец растворов по 1 мкл в соответствующей концентрации 1-10 пмоль/мкл смешивают с равными количествами соответствующей матрицы. Указанную смесь наносят капельно на мишень из нержавеющей стали и сушат при комнатной температуре перед проведением анализа.
Пример 7.2
Получение и характеристика рекомбинантного человеческого ЭПО (EPO-GT-1)
ЭПО получают из рекомбинантных CHO клеток при его экспрессии в них, как описано в литературе (Mueller PP et al., 1999, Dorner AJ et al., 1984), и исследуют характеристики полученных препаратов в соответствии с методами Европейской Фармакопеи (Ph. Eur. 4, Monography 01/2002:1316: Erythropoietin concentrated solution). Конечный продукт характеризуется содержанием сиаловой кислоты на уровне 12 нмоль (±1,5 нмоль) на нмоль белка. Определяют структуры N-связанных олигосахаридов методом HPAEC-PAD и MALDI/TOF-MS, как описано в литературе (Nimtz et al., 1999, Grabenhorst, 1999). Полученные ЭПО препараты содержат ди-, три- и тетрасиалилированные олигосахариды (2-12%, 15-28% и 60-80% соответственно, сульфатированные и пентасиалилированные цепи присутствуют в небольших количествах). Было показано, что в целом характеристики гликозилирования препаратов ЭПО аналогичны таковым для международного стандартного препарата BRP ЭПО.
Результаты изоэлектрофокусирования рекомбинантного ЭПО были сравнимы с результатами соответствующего анализа международного стандартного препарата BRP ЭПО, которые демонстрировали соответствующие изоформы. У 25% белка ЭПО отсутствовало О-гикозилирование в положении Ser126 в полипептидой цепи.
Пример 7.3
Получение частично десиалилированных форм ЭПО
Белок EPO GT-1 (2,84 мг/мл) нагревают до 80°С в 20 мМ Na-фсфатном буфере, pH 7,0 и затем добавляют 100 мкл 1N H2SO4 в расчете на 1 мл раствора ЭПО, инкубацию продолжают в течение 5 мин, 10 мин и 60 мин соответственно, что приводит к получению препаратов ЭПО с разной степенью сиалилирования. Количественное определение олигосахаридов с наличием 0-4 сиаловых кислот проводят после высвобождения олигосахаридов N-гликозидазой полипептидов и выделение N-связанных цепей проводят при обессоливании с использованием картриджей Hypercarb (25 мг HyperSep Hypercarb; ThermoHypersil-Keystone, UK). Препараты ЭПО нейтрализуют путем добавления 1N NaOH, затем замораживают в жидком N2 и хранят при температуре -20°С до дальнейшего использования.
Пример 7.4
Окисление периодатом сиалилированных форм ЭПО
К 10 мг необработанного или обработанного кислотой в мягких условиях ЭПО, который растворен в 3,5 мл 20 мМ Na-фосфатного буфера, pH 7,0, добавляют 1,5 мл 0,1 M Na-ацетатного буфера, pH 5,5, и смесь охлаждают до 0°С на ледяной бане; добавляют 500 мкл 10 мМ периодата Na и реакционную смесь выдерживают в темноте в течение 60 минут при 0°C. Затем добавляют 10 мкл глицерина и инкубацию продолжают еще в течение 10 минут в темноте. Частично окисленные формы ЭПО отделяют от реагентов путем обессоливания с использованием концентраторов VIVASPIN (10000 MWCO, PES Vivascience AG, Hannover, Germany), в соответствии с рекомендациями производителя, со скоростью 3000 об/мин в лабораторной центрифуге, снабженной фиксированным угловым ротором. После замораживания в жидком азоте препараты ЭПО хранят в конечном объеме 4 мл при температуре -20°С.
Аликвоты частично окисленного препарата ЭПО по 100 мкг подвергают обработке N-гликозидазой и выделяют олигосахариды с использованием картриджей Hypercarb, как было описано. Олигосахариды подвергают десиалилированию путем мягкой обработки кислотой и анализируют методом HPAEC-PAD, сравнивая полученное значение времени удерживания с соответствующими показателями для аутентичных препаратов стандартных олигосахаридов, как описано в литературе (Nimtz et al., 1990 и 1993).
Пример 7.5
Восстановление дисульфидных связей в ЭПО с использованием дитиоэритрейтола
5 мг EPO-GT-1 инкубируют с 5 мл 0,1 М Трис/HCl буфера, pH 8,1, в присутствии 30 мМ дитиоэритрейтола (ДТТ) при 37°С в течение 60 минут, удаление ДТТ достигается путем использовании концентратора Vivaspin при 4°С с проведением 4 циклов замены буфера. Конечный восстановленный препарат ЭПО замораживают в жидком азоте и хранят при -20°С в 50 мМ Na-ацетатном буфере, pH 5,5.
Пример 7.6
Определение белка в препарате ЭПО
Количественное определение белка в препарате ЭПО проводят путем измерения УФ поглощения при длине волны 280 нм в соответствии с методикой Европейской Фармакопеи (European Pharmacopeia 4, Monography 01/2002; 1316: Erythropoietin concentrated solution) в кювете с длиной пробега 1 см. Кроме того, количественное ЭПО проводят методом ОФ-ВЭЖХ с использованием колонки RP-C4 (Vydac Protein C4, Cat#214TP5410, Grace Vydac, Ca, US); методика ВЭЖХ была откалибрована с использованием эталонного препарата эритропоэтина BRP 1 (European Pharmacopeia, Conseil, de l'Europe B.P. 907-F67029, Strasbourg Cedex 1).
Пример 7.7
Окисление десиалилированного ЭПО галактозооксидазой
4,485 мг полностью десиалилированого ЭПО инкубируют в 20 мМ Na-фосфатном буфере, pH 6,8, в присутствии 16 мкл каталазы (6214 единиц/200 мл) и 80 мкл галактозооксидазы (2250 единиц/мл из Dactylium dendroides (Sigma-Aldrich, Steinheim, Germany)); инкубацию проводят при 37°С в течение ночи; добавляют 2 раза по 20 мкл галактозооксидазы через 4 часа и через 8 часов после начала инкубации.
Пример 7.8
Подготовка образцов ЭПО для биотестирования
Выделение и очистка образцов ЭПО из инкубационных сред, содержащих белковые препараты ЭПО с активированным ГЭК, обработанные периодатом или окисленные галактооксидазой.
Очистку препаратов ЭПО (удаление непрореагировавших производных ГЭК) проводят при комнатной температуре. Смеси для инкубации ЭПО (примерно 5 мг по белку ЭПО) разбавляют 1:10 буфером А (20 мМ N-морфолинпропансульфоновой кислоты [MOPS/NaOH] в бидистиллированной Н2O, pH 8,0) и наносят на колонку, содержащую 3 мл сефарозы (Q-Sepharose HP (Pharmacia Code № 17-1014-03, Lot № 220211)), уравновешенную буфером А в количестве, равном 10 объемам колонки (CV), со скоростью течения 0,5 мл/мин. Колонку промывают 6-8 CV буфера А (скорость течения = 0,8 мл/мин) и проводят элюцию с использованием буфера B (20 мМ морфолинэтансульфоновой кислоты [MES/NaOH], 0,5 M NaCl в бидистиллированной H2O, pH 6,5) со скоростью течения 0,5 мл/мин. ЭПО выявляют по УФ-поглощению при длине волны 280 нм и элюируют примерно в объеме 6 мл. Колонку регенерируют с использованием 3 CV буфера С (20 мМ MES, 1,5 M NaCl в H2O, доведенной до pH 6,5) и подвергают повторному уравновешиванию с использованием 10 CV буфера А (скорость течения = 0,7 мл/мин).
Буферную замену в элюатах ЭПО, полученных на стадии элюции с Q-Sepharose, проводят с помощью концентратора Vivaspin и фосфатно-буферного раствора (ФБР) в течение 3 циклов центрифугирования для каждого образца; объем образцов доводят до 2 мл с помощью ФБР, после чего образцы хранят при-20°С.
Получают лишь <25% частично десиалилированных и впоследствии окисленных в условиях мягкой обработки периодатом форм ЭПО, которые подверглись ГЭК-модификации, из элюата с колонок с сефарозой (Q-Sepharose), поскольку в используемых условиях основные формы ЭПО не связываются с сефарозой (Q-Sepharose) и обнаруживаются в протекающем потоке вместе с непрореагировавшими ГЭК-производными.
Пример 7.9
Анионообменная хроматография при высоком pH с пульсовым амперометрическим обнаружением (HPAEC-PAD)
Очищенные нативные и десиалилированные олигосахариды анализируют с помощью анионообменной хроматографии с высоким pH (HPAE) в системе Dionex BioLC (Dionex, USA), снабженной колонкой CarboPac PA1 (0,4×25 см), в сочетании с системой пульсового амперометрического выявления (PAD) (Schroter et al., 1999; Nimtz et al., 1999). Значения потенциалов детектора (Е) и длительность пульсовой обработки (Т) составляют: Е1: +50 мВ, Т1: 480 мсек; Е2: +500 мВ, Т2: 120 мсек; Е3: -500 мВ, Т3: 60 мсек, а диапазон выхода составляет 500-1500 нА. Затем олигосахариды инъецируют в колонку CarboPac PА1, уравновешенную 100% растворителем А. В случае элюции десиалилированных олигосахаридов (скорость течения: 1 мл/мин) указанную элюцию проводят при использовании линейного градиента (0-20%) растворителя В в течение 40 минут с последующим линейным повышением уровня растворителя В от 20 до 100% в течение 5 минут. Растворитель А представляет 0,2 М NaOH в бидистиллированной Н2О, растворитель В состоит из 0,6 М NaOAc в растворителе А. В случае нативных олигосахаридов колонку уравновешивают 100% растворителем С (0,1 М NaOH в бидистиллированной H2О) и элюцию (скорость течения: 1 мл/мин) проводят с использованием линейного градиента (0-35%) растворителя D в течение 48 минут с последующим линейным повышением уровня растворителя D от 35 до 100% в течение 10 минут. Растворитель D представляет 0,6 М NaAc в растворителе С.
Пример 7.10
Анализ моносахаридного состава N-гликанов, ГЭК-модифицированных N-гликанов и белка ЭПО методом ГХ-МС
Моносахариды анализируют в виде соответствующих метилгликозидов после проведения метанолиза, N-реацетилирования и триметилсиалилирования по методу ГХ/МС [Chaplin, M. F. (1982) A rapid and sensitive method for the analysis of carbohydrate. Anal. Biochem. 123, 336-341]. Анализы проводят в масс-спектрометре, действующем по механизму захвата ионов, Finnigan GCQ (Finnigan MAT corp., San Jose, CA), работающем в режиме положительных ионов EI, который снабжен капиллярной колонкой DB5 длиной 30 м. Прибор работает по следующей температурной программе: 2 минуты изотермический режим при 80°С, затем температуру повышают со скоростью 10 градусов/мин до 300°С.
Моносахариды идентифицируют по значению времени удерживания и показателям характерных фрагментов. Для количественного определения используют некорректированные результаты интеграции электронных пиков. Моносахарид, дающий более чем один пик, в связи с аномерностью и/или присутствием фураноидных и пираноидных форм, количественно определяют путем сложения всех основных пиков. Используют 0,5 мкг миоинозита в качестве внутреннего стандартного соединения.
Пример 7.11
Результаты
Пример 7.11(а)
Характеристика N-гликанов в EPO-GT-1 после его обработки кислотой в мягких условиях (частично десиалилированного)
Препараты EPO-GT-1 подвергают обработке кислотой в мягких условиях в течение 5, 10 или 60 минут и затем анализируют в ДСН-ПААГ до и после высвобождения N-олигосахаридов при инкубации с N-гликозидазой, как показано на фиг.5. N-связанные олигосахариды подвергают картированию олигосахаридов по методу HPAEC-PAD (фиг.6). Необработанный ЕРО-GT-1 содержит более 90% N-связанных олигосахаридов с 3 или 4 остатками сиаловой кислоты, тогда как после 5-минутной инкубации в присутствии слабой кислоты менее 40% углеводных цепей содержат 3 или 4 остатка сиаловой кислоты. Анализ по методу HPAEC-PAD десиалилированных N-гликанов показал, что соотношение нейтральных олигосахаридов, которые были обнаружены в необработанном ЕРО-GT-1, остается стабильным в препаратах, подвергнутых кислотной обработке в течение 5, 10 или 60 минут. Анализ методом MALDI/TOF-MS десиалилированных гликанов показал, что менее 90% проксимальной фукозы присутствует после мягкой кислотной обработки данного белка.
Пример 7.11 (b)
Характеристика ЕРО-GT-1, обработанного периодатом
На фиг.8 приведены данные, показывающие мобильность в ДСН-ПААГ обработанных в мягких условиях периодатом форм ЭПО, которые были предварительно подвергнуты обработке кислотой в течение 5 и 10 минут или которые не обрабатывались. Условия, использованные для периодатного окисления сиаловых кислот, не влияют на поведение препаратов ЭПО в ДСН-ПААГ (см. для сравнения фиг.5). Окисление сиаловых кислот приводит к смещению олигосахаридов в рамках анализа HPAEC-PAD, к более ранним показателям по времени элюирования (для сравнения см. фиг.6и9).
Пример 7.11 (с)
Характеристика ГЭК-модифицированных производных ЭПО
(аа) ГЭК-модификация с течением времени ЕРО-GT-1-А с использованием модифицированного гидроксиламином ГЭК-производного Х, полученного по методике примера 2.4
400 мкг модифицированного гидроксиламином ГЭК-производного Х добавляют к 20 мкг ЕРО-GT-1-А (ЭПО, окисленный в мягких условиях периодатом, но не подвергнутый кислотному гидролизу перед указанным окислением периодатом в мягких условиях) в 20 мкл 0,5 М NaOAc буфера, pH 5,5, и реакцию останавливают через 30 минут, 2, 4, и 17 часов соответственно, путем замораживания образцов в жидком азоте. Впоследствии образцы хранят при -20°С до проведения дальнейшего анализа.
Добавляют буфер для проведения анализа образцов в ДСН-ПААГ и нагревают указанные образцы до 90°С, после чего наносят на гели с ДСН. Как показано на фиг.10, увеличение времени инкубации приводит к более сильному смещению образцов в сторону более высокой молекулярной массы белка. После 17 часов инкубации в присутствии модифицированного гидроксиламином ГЭК-производного Х обнаруживается диффузная окрашиваемая кумасси белковая полоса, мигрирующая в зоне между 60 и 11 кДа, относительно положения стандартных препаратов, анализируемых для определения молекулярного веса (см. правую часть фиг.10). При обработке N-гикозидазой большая часть белка мигрирует в сторону положения де-N-гликозилированного ЭПО (см. фиг.10, правый гель; стрелка А указывает на положение перемещения N-гликозидазы, стрелка В указывает на миграцию де-N-гликозилированного ЭПО; диффузная белковая полоса, видимая на участке между положениями стандартных препаратов для определения молекулярного веса в диапазоне между 28 кДа и 36 кДа, преимущественно отражает те ЭПО формы, которые включают модификации, определяемые ГЭК и сайтом О-гликозилирования молекулы. В связи со специфичностью действия N-гликозидазы, авторы на основании данного эксперимента пришли к выводу, что фактически ГЭК-модификация происходит на окисленных периодатом остатках сиаловой кислоты гликанов в белке ЭПО.
(bb) Характеристика конъюгатов ГЭК-ЭПО
Конъюгаты ГЭК-ЭПО I (получаемые из ЕРО-GT-1 после окисления периодатом в мягких условиях, например, на основе ЕРО-GT-1-А), II (получаемые из ЕРО-GT-1, подвергнутого 5-минутному кислотному гидролизу и окислению периодатом в мягких условиях) и III (получаемые из ЕРО-GT-1, подвергнутого 10-минутному кислотному гидролизу и окислению периодатом в мягких условиях) синтезируют, как было описано ранее. Методика включает вариант контрольной инкубации (К), содержащий немодифицированный ЕРО-GT-1 в том же буфере, к которому добавляют эквивалентное количество немодифицированного ГЭК. Инкубационные смеси подвергают дальнейшей очистке для проведения последующего биохимического анализа производных ГЭК-ЭПО.
Полученные после инкубации конъюгаты ГЭК-ЭПО I, II и III, а также продукт контрольной инкубации К подвергают очистке на сефарозе (Q-Sepharose), как было описано в разделе "Материалы и методы" (пример 7.8), с целью удаления избытка непрореагировавшего ГЭК-реагента, который, как ожидается, выходит с протекающим потоком из ионообменной колонки. В связи с большими количествами основных ЭПО-форм, содержащихся в предварительно обработанных кислотой образцах II и III, авторы ожидали появления в потоке значительных количеств модифицированного ЭПО продукта как результат указанных вариантов инкубации. Как видно на фиг.11, практически весь ЭПО-материал из образцов I удерживается на колонке с сефарозой (Q-Sepharose), тогда как лишь примерно 20-30% образцов II и III выходят во фракции, элюируемой при высокой концентрации соли. Весь белковый материал из инкубационных сред, включающих ГЭК-производное Х, как в протекающем потоке, так и во фракциях, элюируемых при высокой концентрации соли, характеризуется явно более высоким молекулярным весом при анализе в ДСН-ПААГ, в сравнении с контрольным препаратом ЭПО.
С целью более детальной характеристики ГЭК-модифицированного ЭПО в образцах А и К (см. фиг.11) проводят сравнение полученных для них результатов с окисленной периодатом формой ЕРО-GT-1-А. Образцы подвергают обработке N-гликозидазой и, как видно из фиг.12а и 12b, высвобождение N-гликанов приводит к получению двух полос с низким молекулярным весом в положении О-гликозилированной и негликозилированной форм ЭПО стандартного препарата ЭПО. В случае образца А обнаруживается другая полоса, мигрирующая в положение, соответствующее стандарту с М.в. 28 кДа, что, предположительно, указывает на наличие ГЭК-модификации по О-гликану в данном варианте ЭПО (см. пример 7.11(с)(аа)). Указанная полоса (а также высокомолекулярная форма N-гикозилированного ЭПО с высокой степенью ГЭК-модификации, см. фиг.12а и 12b) исчезает после проведения мягкого гидролиза образцов, что согласуется с точкой зрения, согласно которой ГЭК-модификация осуществляется по окисленным периодатом остаткам сиаловой кислоты в эритропоэтине.
Аликвоты инкубационных смесей для N-гликозидазной обработки гидролизуют в условиях, позволяющих полностью удалить остатки сиаловой кислоты (а также производное сиаловой кислоты с присоединенным ГЭК) из олигосахаридов; после нейтрализации смеси сорбируют на небольшие колонки Hypercarb для обессоливания. Колонки тщательно промывают водой и затем проводят элюцию связанных нейтральных олигосахаридов 40% ацетонитрилом в Н2О, содержащей 0,1% трифторуксусную кислоту. Полученные олигосахариды подвергают анализу по методу MALDI/TOF-MS. Спектры фракций десиалилированных олигосахаридов из образца А, ЕРО-GT-1-А и образца К демонстрируют идентичные массы, относительно олигосахаридов комплексного типа, со значениями m/z = 1810 Да (диантенный), 2175 = триантенный, 2540 = тетраантенный, 2906 = тетраантенный + 1N-ацетиллактозаминовый повтор и 3271 = тетраантенный + 2N-ацетиллактозаминовых повтора; обнаруживаются небольшие сигналы, соответствующие отсутствию фукозы (-146) и галактозы (-162), которые определяются условиями кислотного гидролиза, применяемого для удаления сиаловой кислоты (см. MALDI - фиг.15, 16 и 17).
В параллельном эксперименте смесь после расщепления N-гикозидазой сорбируют на картридже RP-C18 на 1 мл (без предварительного кислотного гидролиза олигосахаридов) и проводят элюцию 5% ацетонитрилом в воде, содержащей 0,1% ТФУ; в указанных условиях белок ЭПО полностью удерживается на RP-материале и олигосахариды вымываются из колонки 5% ацетонитрилом в H2O, содержащей 0,1% ТФУ. Белок в де-N-гликозилированном ЭПО элюируют 70% ацетонитрилом в H2O, содержащей 0,1% ТФУ. Олигосахаридные фракции на стадии RP-C18 обработанного N-гликозидазой образца А, ЕРО-GT-1-А и образца К нейтрализуют и подвергают обессоливанию с использованием картриджей Hypercarb, как было описано ранее. Выделенные олигосахариды подвергают картированию по методу анализа HPAEC-PAD до (см. фиг.13) и после обработки кислотой в мягких условиях, позволяющей осуществить количественное удаление сиаловых кислот из гликанов (см. фиг.14).
Профиль, выявляемый при анализе по методу HPAEC-PAD нативного материала, полученного из ГЭК-модифицированного образца А, демонстрирует лишь ничтожные сигналы, характерные для олигосахаридов, тогда как олигосахариды из ЕРО-GT-1-А демонстрируют тот же профиль гликанов, что и вариант, показанный на фиг.9 (образец, обозначенный как ЕРО-GT-1 после обработки периодатом в мягких условиях). Профиль элюции олигосахаридов, полученных из контрольного образца ЭПО (К), соответствует ожидаемым характеристикам (см. для сравнения профиль, показанный на фиг.6). Для сравнения приведен профиль нативного олигосахарида в международном стандартном препарате BRP-ЭПО, выполняющем функцию эталонного стандарта.
После проведения кислотного гидролиза в мягких условиях все олигосахаридные препараты демонстрируют идентичный профиль элюции нейтральных олигосахаридных структур (см. фиг.14) с ожидаемым количественным и качественным составом ди-, три- и тетраантенных углеводных цепей комплексного типа, как описано в методическом разделе относительно препарата ЭПО, который использовался в качестве исходного материала для настоящего исследования. Данный результат показывает, что ГЭК-модификация образца ЭПО приводит к ковалентному связыванию ГЭК-производного, которое отделяется от ЭПО белка с помощью N-гликозидазы и которое является кислотолабильным, поскольку удаляется из N-гиканов в условиях мягкого кислотного гидролиза, приводящего, как известно, к десиалилированию углеводов (см. фиг.12а+b).
(сс) Анализ моносахаридного состава ГЭК-ЭПО и N-гликанов ГЭК-ЭПО по методу ГХ-МС
Для того, чтобы дополнительно подтвердить наличие ГЭК-мдификации ЭПО по N-гликанам в молекуле, образцы ЭПО расщепляют N-гликозидазой и белок ЭПО сорбируют на картриджах RP-C18, тогда как олигосахаридный материал вымывается, как было описано выше. Как показано в таблице 2, глюкоза и гидроксиэтилированные производные глюкозы обнаруживаются только в белке ЭПО, который был подвергнут ГЭК-модификации по цистеиновым остаткам, и в олигосахаридных фракциях ЭПО в образце А2.
Пример 7.11(d)
Тестирование in vivo биологической активности ГЭК-мдифицированного ЭПО
Тестирование ЭПО в системе нормоцитных мышей проводят в соответствии с процедурами, описанными в Европейской Фармакопее; в лаборатории, проводящей ЭПО-тестирование, используется международный стандартный препарат BRP ЭПО. В случае препарата ГЭК-модифицированного ЭПО А2 было определено среднее значение удельной активности, равное 294600 единиц на мг белка ЭПО, указывающее на наличие примерно в 3 раза более высокой удельной активности, чем в международном стандартном препарате BRP ЭПО, который был включен для оценки активности вместе с исследуемыми образцами.
Результаты исследования приведены в таблице 3.
Цитированная литература:
Nimtz M, Noll G, Paques EP, Conradt HS.
Carbohydrate structures of a human tissue plasminogen activator expressed in recombinant Chinese hamster ovary cells.
FEBS Lett. 1990 Oct. 1; 271(1-2):14-8.
Dorner AJ, Wasley LC, Kaufman RJ.
Increased synthesis of secreted proteins induced expression of glucose-regulated proteins in butyrate-treated Chinese hamster ovary cells.
J. Biol. Chem. 1989 Dec 5; 264 (34): 20602-7.
Mueller PP, Schlenke P, Nimtz M, Conradt HS, Hauser H
Recombinant glycoprotein quality in proliferation-controlled BHK-21 cells.
Biotechnol Bioeng. 1999 Dec 5; 65(5): 529-36
Nimtz M, Martin W, Wray V, Kloppel KD, Augustin J, Conradt HS.
Structures of sialylated oligosaccharides of human erythropoietin expressed in recombinant BHK-21 cells.
Eur J Biochem. 1993 Apr. 1; 213(1): 39-56
Hermentin P, Witzel R, Vliegenthart JF, Kamerling JP, Nimtz M, Conradt HS.
A strategy for the mapping of N-glycans by high-ph anion-exchange chromatography with pulsed amperometric detection.
Anal Biochem. 1992 Jun; 203(2): 281-9
Schroter S, Derr P, Conradt HS, Nimtz M, Hale G, Kirchhoff C.
Male specific modification of human CD52.
J Biol Chem. 1999 Oct. 15; 274(42): 29862-73


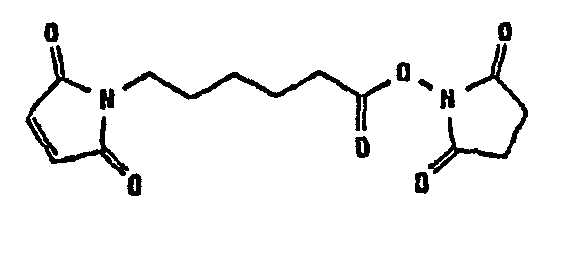
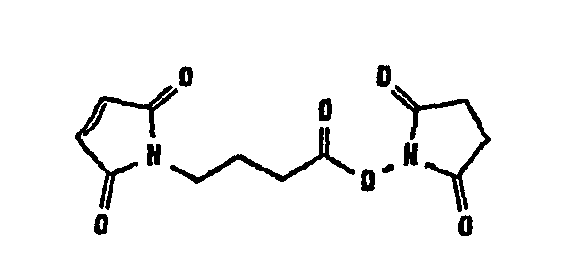
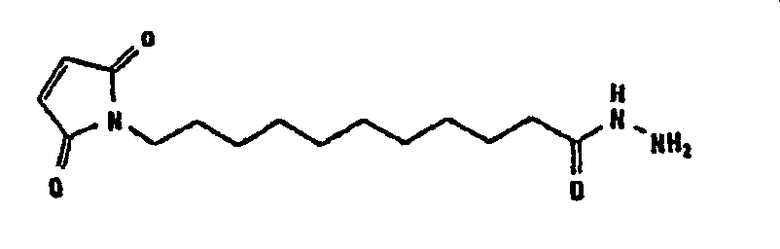
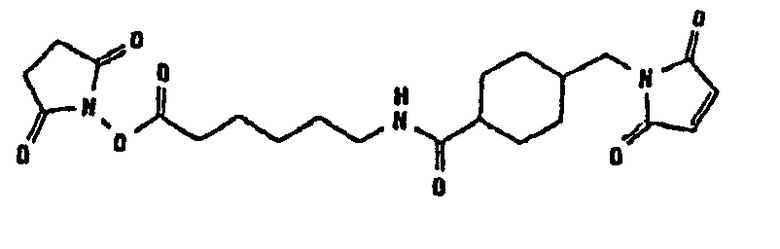







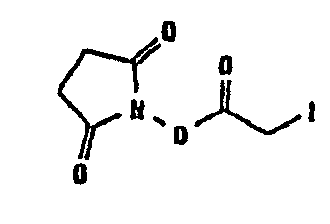



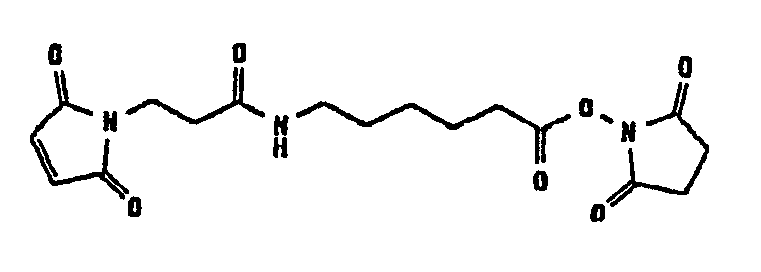



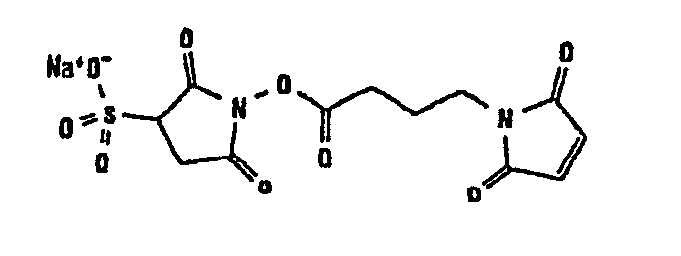

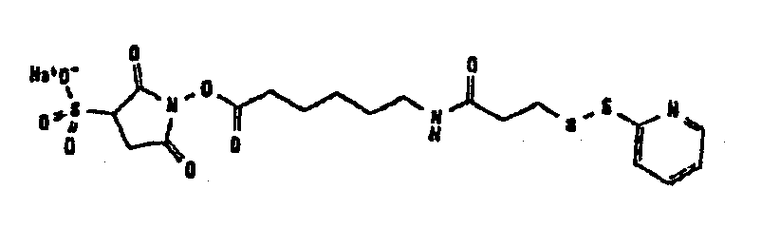
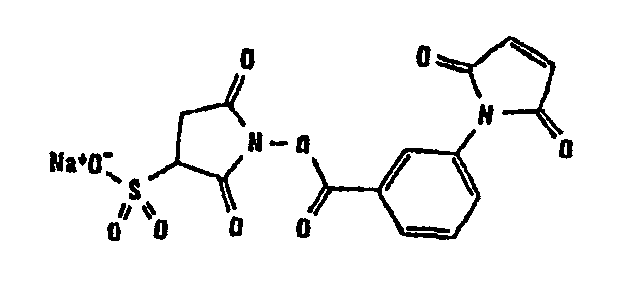


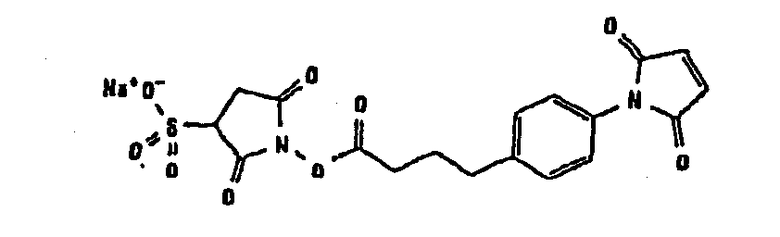

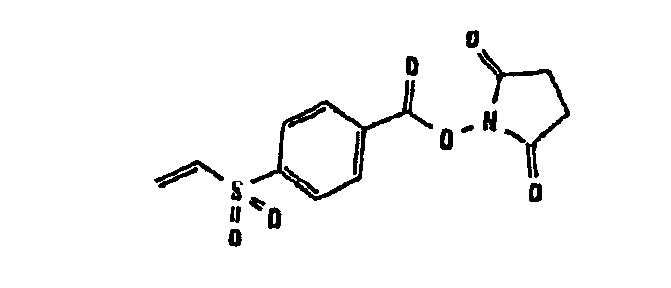
Анализ моносахаридного состава гликанов из ГЭК-модифицированного ЭПО и контрольных образцов
Гликаны
из А2
Гликаны
из EPO-GT-1A
Гликаны
из K2
Гликаны
из A2
Гликаны
из EPO-GT-1A
Гликаны
из K2
ЭПО белок, модифицированный по цистеину*
** Определение количества моносахаридов проводят при однократном пропускании в системе ГХ пертриметилсилилированных метилгликозидов; значения пиков при их электронной интеграции приводятся без коррекции на потери в ходе процедур дериватизации и восстановления каждого соединения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ГИДРОКСИАЛКИЛКРАХМАЛА | 2003 |
|
RU2329274C2 |
| ПОЛИПЕПТИДЫ, ОБРАБОТАННЫЕ HAS, ОСОБЕННО ЭРИТРОПОЭТИН, ОБРАБОТАННЫЙ HAS | 2003 |
|
RU2328505C2 |
| КОНЪЮГАТЫ ГИДРОКСИАЛКИЛКРАХМАЛА И G-CSF | 2004 |
|
RU2370281C2 |
| КОНЪЮГАТЫ ГИДРОКСИАЛКИЛКРАХМАЛА И АЛЛЕРГЕНА | 2003 |
|
RU2325925C2 |
| ОПОСРЕДУЮЩИЕ ТРАНСПОРТ КОЛЛОИДНЫЕ ЛЕКАРСТВЕННЫЕ СОЕДИНЕНИЯ | 2009 |
|
RU2519225C2 |
| ПРОДУКТЫ СЛИЯНИЯ АМИНИРОВАННЫХ ПОЛИСАХАРИДОВ | 2010 |
|
RU2549492C2 |
| ПРИМЕНЕНИЕ ЭРИТРОПОЭТИНА | 2003 |
|
RU2305554C2 |
| КОНЪЮГАТ ГИДРОКСИАЛКИЛКРАХМАЛА И НИЗКОМОЛЕКУЛЯРНОГО ВЕЩЕСТВА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2003 |
|
RU2327702C2 |
| ЛЕЧЕНИЕ НЕЙРОДЕГЕНЕРАТИВНЫХ РАССТРОЙСТВ | 2006 |
|
RU2467761C2 |
| КОНЪЮГАТЫ БЕЛКОВ СВЕРТЫВАНИЯ КРОВИ | 2010 |
|
RU2595442C2 |



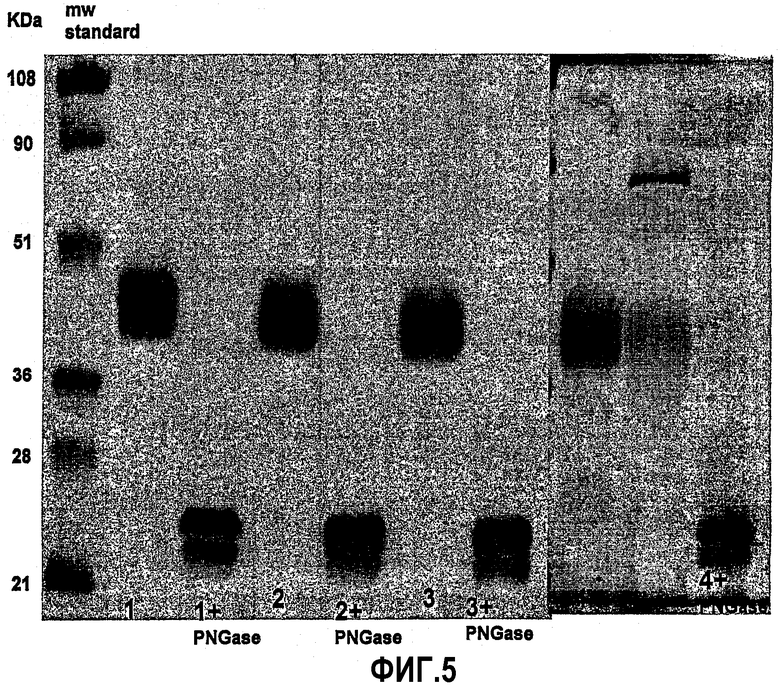
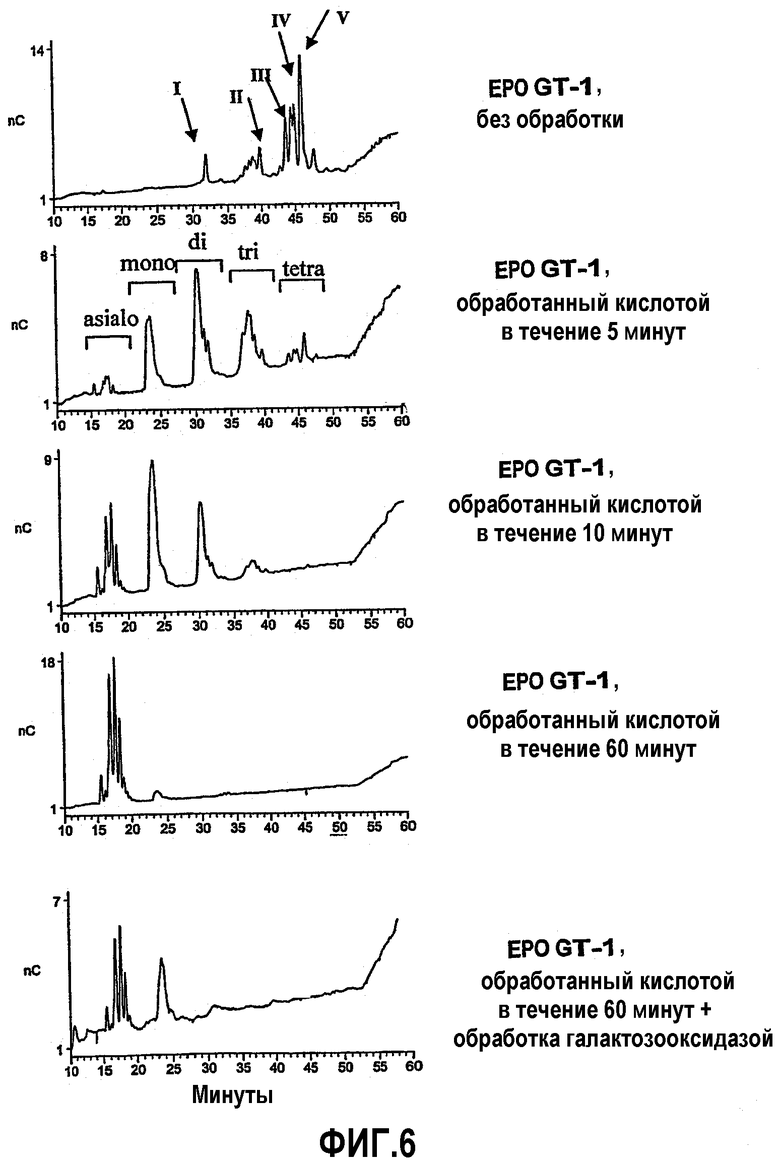

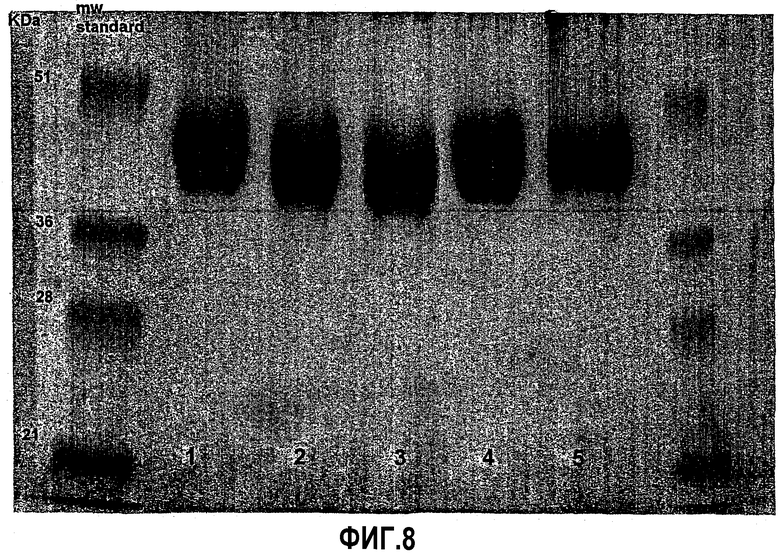

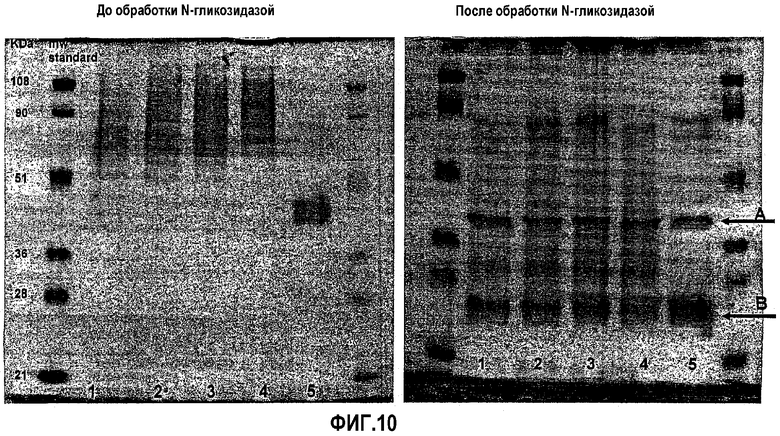
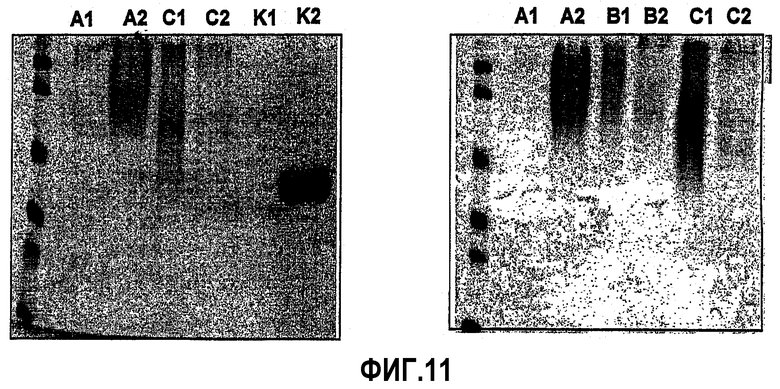

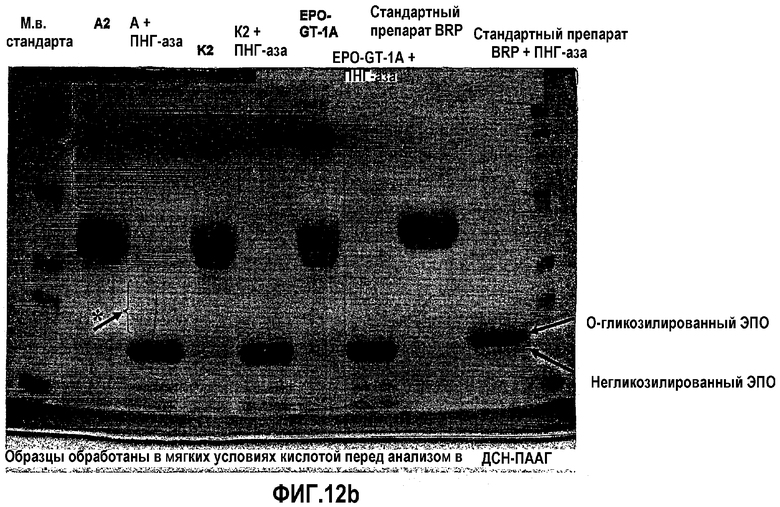

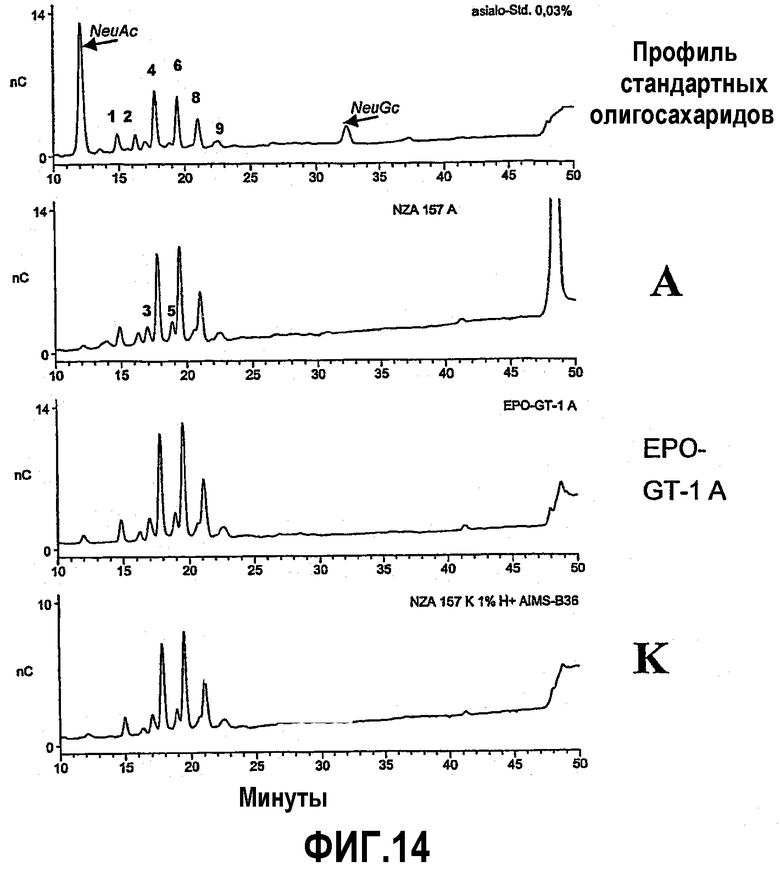


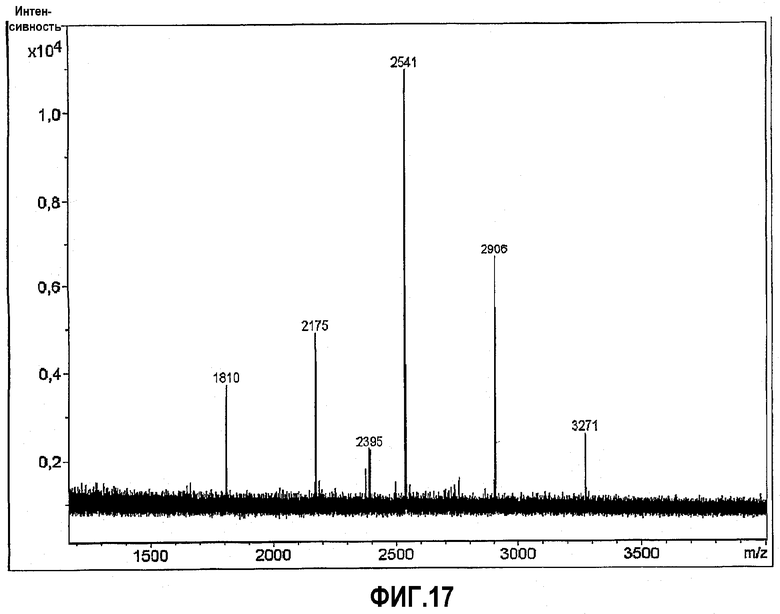
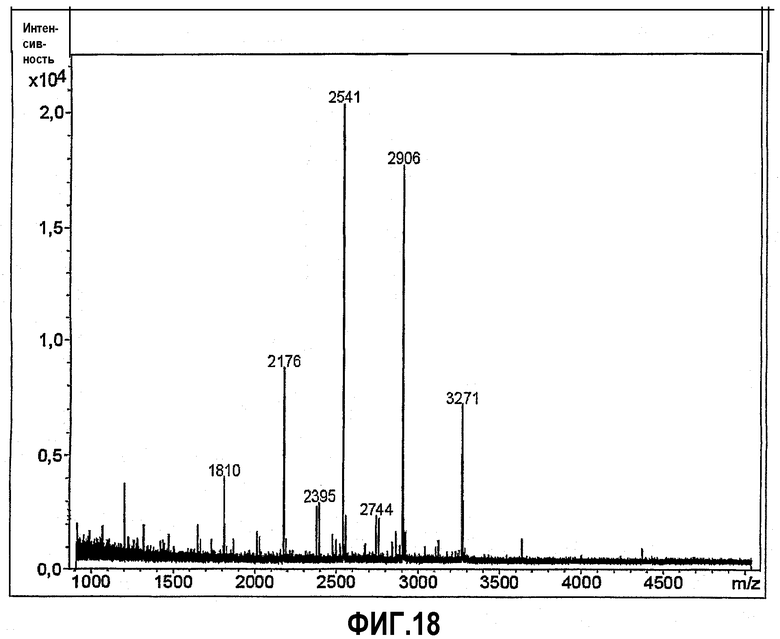

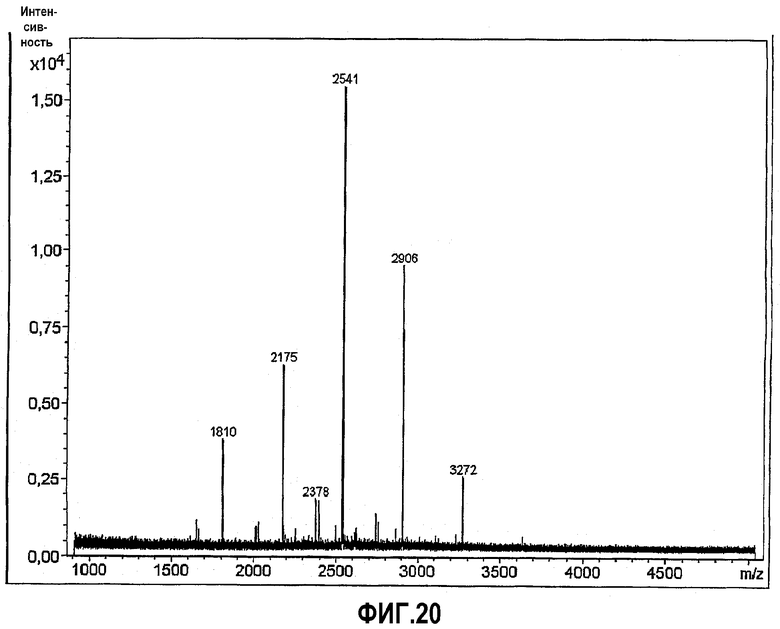
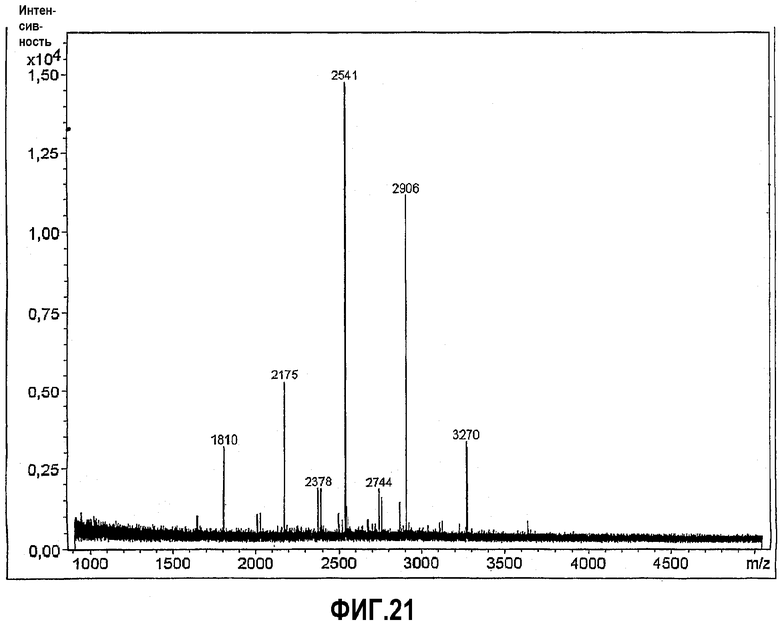
Изобретение относится к способу получения производного гидроксиалкилкрахмала, включающему взаимодействие гидроксиалкилкрахмала формулы (I) по его восстановленному концу, который не окисляют перед проведением данной реакции, с соединением формулы (II) R'-NH-R", где R1, R2 и R3 обозначают независимо водород, или линейную, или разветвленную гидроксиалкильную группу и где R', или R", или R' и R", включают по меньшей мере одну функциональную группу X, способную взаимодействовать по меньшей мере с одним другим соединением до или после реакции (I) и (II), а также к самому производному гидроксиалкилкрахмала, получаемому по данному способу, к способу получения производного гидроксиалкилкрахмала и полипептида, фармацевтической композиции, включающей указанные производные гидроксиалкилкрахмала. 9 н. и 68 з.п. ф-лы, 21 ил., 3 табл.
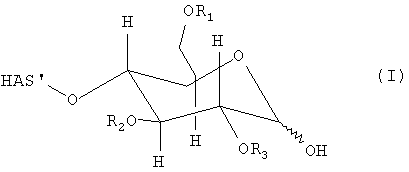
по его восстановленному концу, который не окисляют перед проведением данной реакции, с соединением формулы (II)

где R1, R2 и R3 обозначают независимо водород или линейную или разветвленную гидроксиалкильную группу, и где R' или R" или R' и R" включают по меньшей мере одну функциональную группу X, способную взаимодействовать по меньшей мере с одним полипептидом или сшивающим соединением или продуктом взаимодействия сшивающего соединения или полипептида до или после реакции (I) и (II), и где соединение (II) подвергают реакции через NH-группу, соединяющую мостиковой связью R' и R", с соединением (I) по его восстановленному концу, который не окисляют;
где группа X выбрана из группы, включающей из -SH, -NH2, -O-NH2, -NH-O-алкила, -(C=G)-NH-NH2, -G-(C=G)-NH-NH2, -NH-(C=G)-NH-NH2 и -SO2-NH-NH2, где G обозначает О или S и, если G присутствует дважды, обозначает независимо О или S.
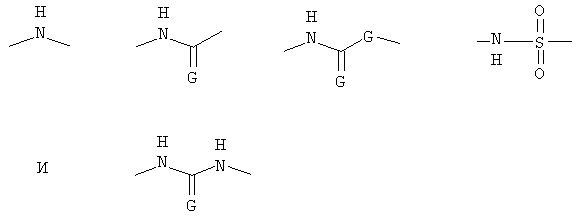
где G обозначает О или S и, если присутствует дважды, G обозначает независимо О или S.

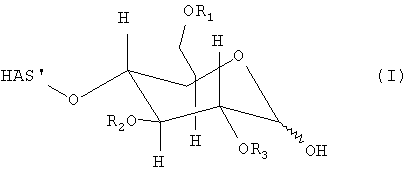
по его восстановленному концу, который не окисляют перед проведением данной реакции в водной среде, с соединением формулы (II)
где R1, R2 и R3 обозначают независимо водород или 2-гидроксиэтильную группу, причем R' представляет собой водород или метильную или метоксигруппу, и где R" включает по меньшей мере одну функциональную группу X, способную взаимодействовать по меньшей мере с одним другим полипептидом или продуктом взаимодействия сшивающего соединения и полипетида до или после реакции (I) и (II),
где группа X выбрана из группы, включающей -SH, -NH2, -O-NH2, -NH-O-алкил, -(C=G)-NH-NH2, -G-(C=G)-NH-NH2, -NH-(C=G)-NH-NH2 и -SO2-NH-NH2, где G обозначает О или S и, если G присутствует дважды, обозначает независимо О или S, и где соединение (II) подвергают реакции через NH-группу, соединяющую мостиковой связью R' и R", с соединением (I) по его восстановленному концу, который не окисляют.

по его восстановленному концу, который не окисляют перед проведением данной реакции, с соединением формулы (II)
где R1, R2 и R3 обозначают, независимо, водород или линейную или разветвленную гидроксиалкильную группу, и где R' или R" или R' и R" включают по меньшей мере одну функциональную группу X, способную взаимодействовать по меньшей мере с одним полипептидом или сшивающим соединением или продуктом взаимодействия сшивающего соединения или полипептида до или после реакции (I) и (II), и где соединение (II) подвергают реакции через NH-группу, соединяющую мостиковой связью R' и R", с соединением (I) по его восстановленному концу, который не окисляют;
где группа X выбрана из группы, состоящей из -SH, -NH2, -O-NH2,
-NH-O-алкила, -(C=G)-NH-NH2, -G-(C=G)-NH-NH2, -NH-(C=G)-NH-NH2 и -SO2-NH-NH2, где G обозначает О или S и, если G присутствует дважды, обозначает независимо О или S.

по его восстановленному концу, который не окисляют перед проведением данной реакции, с соединением формулы (II)
где R1, R2 и R3 обозначают, независимо, водород или 2-гидроксиэтильную группу, и где R' представляет собой водород или метил или метоксигруппу, и где R" включает по меньшей мере одну функциональную группу X, способную взаимодействовать по меньшей мере с одним полипептидом или продуктом взаимодействия сшивающего соединения или полипетида до или после реакции (I) и (II),
где группа X выбрана из группы, включающей -SH, -NH2, -O-NH2,
-NH-O-алкил, -(C=G)-NH-NH2, -G-(C=G)-NH-NH2, -NH-(C=G)-NH-NH2 и -SO2-NH-NH2, где G обозначает О или S и, если G присутствует дважды, обозначает независимо О или S
и где соединение (II) подвергают реакции через NH-группу, соединяющую мостиковой связью R' и R", с соединением (I) по его восстановленному концу, который не окисляют.

где G обозначает О или S и, если присутствует дважды, G обозначает независимо О или S.
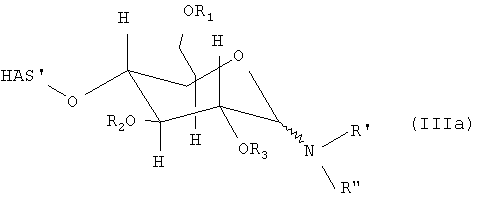
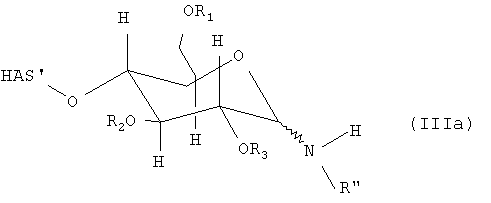


по его восстановленному концу, который не окисляют перед проведением данной реакции, с соединением формулы (II)
где R1, R2 и R3 обозначают независимо водород или линейную или разветвленную гидроксиалкильную группу, и где R' или R" или R' и R" включают по меньшей мере одну функциональную группу X, способную взаимодействовать по меньшей мере с одним полипептидом или сшивающим соединением или продуктом взаимодействия сшивающего соединения и полипептида до или после реакции (I) и (II), и где соединение (II) подвергают взаимодействию через NH-группу соединяющую мостиковой связью R' и R", с соединением (I) по его восстановленному концу, который не окисляют;
где группа X выбрана из группы, включающей из -SH, -NH2, -O-NH2,
-NH-O-алкила, -(C=G)-NH-NH2, -G-(C=G)-NH-NH2, -NH-(C=G)-NH-NH2 и -SO2-H-NH2, где G обозначает О или S и, если G присутствует дважды, обозначает независимо О или S
и последующее взаимодействие с полипептидом.
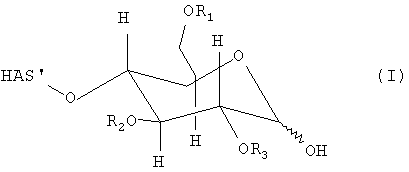
по его восстановленному концу, который не окисляют перед проведением данной реакции, с соединением формулы (II)
где R1, R2 и R3 обозначают, независимо, водород или 2-гидроксиэтильную группу, и где R' представляет собой водород или метил или метокси группу, и где R" включает по меньшей мере одну функциональную группу X, способную взаимодействовать по меньшей мере с одним полипептидом или сшивающим соединением или продуктом взаимодействия сшивающего соединения и полипептида до или после реакции (I) и (II),
где группа X выбрана из группы, включающей из -SH, -NH2, -O-NH2, -NH-O-алкила, -(C=G)-NH-NH2, -G-(C=G)-NH-NH2, -NH-(C=G)-NH-NH2 и -SO2-NH-NH2, где G обозначает О или S и, если G присутствует дважды, обозначает независимо О или S
и где соединение (II) подвергают взаимодействию через NH-группу, соединяющую мостиковой связью R' и R", с соединением (I) по его восстановленному концу, который не окисляют; и где реакционный продукт, полученный при взаимодействии соединения (1) и соединения (11), подвергают реакции по меньшей мере через одну функциональную группу X, включенную в соединение (11), по меньшей мере с одним полипептидом, или где соединение (11) подвергают реакции по меньшей мере через одну функциональную группу X по меньшей мере с полипептидом, перед проведением реакции с соединением (1); и где соединение (11) подвергают реакции через NH-группу, соединяющую мостиковой связью R' и R", с соединением (I) по его восстановленному концу, который не окисляют.

и полученный реакционный продукт подвергают взаимодействию с эритропоэтином.

по его восстановленному концу, который не окисляют перед проведением данной реакции, в водной среде с соединением формулы
где R1, R2 и R3 обозначают, независимо, водород или 2 гидроксиэтильную группу, и где продукт реакции соединения (I) с соединением формулы
подвергают реакции через функциональную группу -O-NH2 в водной среде с эритропоэтином через окисленный углеводный фрагмент, включенный в указанный эритропоэтин.

по его восстановленному концу, который не окисляют перед проведением данной реакции, в водной среде с O-[2-(2-аминооксиэтокси)этил]гидроксиламином, где R1, R2 и R3 обозначают независимо водород или 2-гидроксиэтильную группу, и далее проводят реакцию полученного продукта в водной среде с эритропоэтином через окисленный углеводный фрагмент, входящий в указанный эритропоэтин.
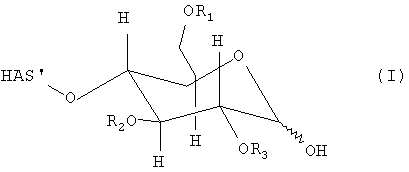
по его восстановленному концу, который не окисляют перед проведением данной реакции, с соединением формулы (II)

где R1, R2 и R3 обозначают независимо водород или линейную или разветвленную гидроксиалкильную группу, и где R' или R" или R' и R" включают по меньшей мере одну функциональную группу X, способную взаимодействовать по меньшей мере с одним полипептидом или сшивающим соединением или продуктом взаимодействия сшивающего соединения или полипептида до или после реакции (I) и (II), и где соединение (11) подвергают реакции через NH-группу, соединяющую мостиковой связью R' и R", с соединением (I) по его восстановленному концу, который не окисляют;
где группа X выбрана из группы, включающей -SH, -NH2, -O-NH2, -NH-O-алкил, -(C=G)-NH-NH2, -G-(C=G)-NH-NH2, -NH-(C=G)-NH-NH2 и -SO2-NH-NH2, где G обозначает О или S и, если G присутствует дважды, обозначает независимо О или S,
и дальнейшую реакцию с полипептидом.

по его восстановленному концу, который не окисляют перед проведением данной реакции в водной среде, с соединением формулы (II)
где R1, R2 и R3 обозначают независимо водород или 2-гидроксиэтильную группу, и где R' представляет собой водород или метильную или метокси группу, R" включает по меньшей мере одну функциональную группу X, способную взаимодействовать по меньшей мере с одним полипептидом или сшивающим соединением или продуктом взаимодействия сшивающего соединения или полипептида до или после реакции (I) и (II), где группа X выбрана из группы, состоящей из -SH, -NH2, -O-NH2, -NH-O-алкила, -(C=G)-NH-NH2, -G-(C=G)-NH-NH2, -NH-(C=G)-NH-NH2 и -SO2-NH-NH2, где G обозначает О или S и, если G присутствует дважды, обозначает, независимо, О или S
и где соединение (II) подвергают реакции через NH-группу, соединяющую мостиковой связью R' и R", с соединением (I) по его восстановленному концу, который не окисляют;
или где соединение (II) подвергают реакции через NH-группу, соединяющую мостиковой связью R' и R", с соединением (I) по его восстановленному концу, который не окисляют; и где продукт реакции соединения (1) с соединением (11) подвергают взаимодействию через по меньшей мере одну функциональную группу X входящую в соединение (11) с по меньшей мере одним полипептидом перед реакцией с соединением (1); и
где соединение (11) подвергают взаимодействию через NH-группу, соединяющую мостиковой связью R' и R", с соединением (I) по его восстановленному концу, который не окисляют.
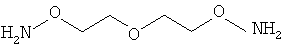
и полученный реакционный продукт подвергают взаимодействию с эритропоэтином.
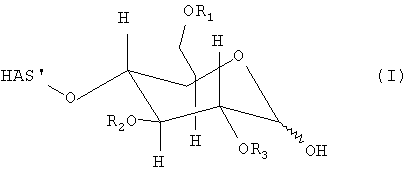
по его восстановленному концу, который не окисляют перед проведением данной реакции, в водной среде с соединением формулы
где R1, R2 и R3 обозначают, независимо, водород или 2-гидроксиэтильную группу, и где продукт реакции соединения (I) с соединением формулы

подвергают реакции через функциональную группу -O-NH2 в водной среде с эритропоэтином через окисленный углеводный фрагмент, включенный в указанный эритропоэтин.
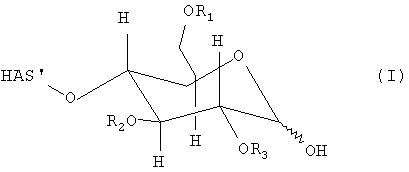
по его восстановленному концу, который не окисляют перед проведением данной реакции в водной среде с O-[2-(2-аминооксиэтокси)этил]гидроксиламином, где R1, R2 и R3 обозначают независимо водород или 2-гидроксиэтильную группу, и далее проводят реакцию полученного продукта в водной среде с эритропоэтином через окисленный углеводный фрагмент, входящий в указанный эритропоэтин.
| Калоризатор для двигателей внутреннего горения | 1927 |
|
SU19403A1 |
| КОМПОЗИЦИИ ДЛИТЕЛЬНОГО ВЫСВОБОЖДЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 1999 |
|
RU2230550C2 |
| US 5217998 А, 08.06.1993 | |||
| Комбинированный дульный тормоз артиллерийского орудия | 2016 |
|
RU2616086C1 |
| СПОСОБ РАДИОЛОКАЦИОННОГО ОПРЕДЕЛЕНИЯ ВЕРТИКАЛЬНОЙ СКОРОСТИ БАЛЛИСТИЧЕСКОГО ОБЪЕКТА И УСТРОЙСТВО ДЛЯ ЕГО РЕАЛИЗАЦИИ | 2015 |
|
RU2646854C2 |

