Данное изобретение относится к бициклическим производным, к способам их получения, к содержащим их композициям и к их применению к терапии.
Первый рилизинг-фактор кортикотропина (CRF) был выделен из овечьего гипоталамуса и идентифицирован как пептид, содержащий 41 аминокислоту (Vale et al., Science 213: 1394-1397, 1981). Было обнаружено, что CRF вызывает сильные изменения в функциях эндокринной, нервной и иммунной систем. Полагают, что CRF является основным физиологическим регулятором основного и стрессового выделения адренокортикотропного гормона (АСТН), бендорфина и других проопиомеланокортин (РОМС)-производных пептидов из передней доли гипофиза (Vale et al., Science 213: 1394-1397, 1981).
Кроме того, что он играет роль в стимулировании продуцирования АСТН и РОМС, полагают, что CRF является одним из основных нейротрансмиттеров центральной нервной системы и играет решающую роль в интеграции общей реакции организма на стресс.
Введение CRF непосредственно в мозг вызывает поведенческую, физиологическую и эндокринную реакцию, идентичную той, которую наблюдают у животных, подвергаемых стрессу. Следовательно, клинические данные подтверждают, что антагонисты рецептора CRF могут являться новыми антидепрессивными и/или анксиолитическими лекарственными средствами, которые могут быть полезны при лечении невропатологических и психиатрических расстройств, вызываемых гиперсекрецией CRF.
Первыми антагонистами рецептора CRF были пептиды (см., например, Rivier et al., патент США № 4605642; Rivier et al., Science 224: 889, 1984). Хотя данные пептиды подтверждают, что антагонисты рецепторов CRF могут смягчать фармакологическую реакцию на CRF, пептидные антагонисты рецептора CRF имеют обычные для пептидов недостатки, включая отсутствие стабильности и ограниченную пероральную активность. Сравнительно недавно были описаны небольшие молекулы антагонистов рецептора CRF.
В WO 95/10506 кроме прочего описаны соединения общей формулы (А) с общим антагонистическим действием в отношении CRF,

где Y может быть CR29; V может быть азотом, Z может быть углеродом, R3 может соответствовать производному амида и R4 может совместно с R29 образовывать 5-членное кольцо и является -CH(R28), если R29 является -CH(R30). Конкретные описания соединений, соответствующих этому определению, не даны.
В WO 95/33750 также описаны соединения общей формулы (В), имеющие антагонистическое действие в отношении CRF,

в которой А и Y могут быть азотом и углеродом и В может соответствовать производному амина. Конкретные описания соединений, соответствующих этому определению, не даны.
В WO 98/08846 описаны соединения общей формулы (С), имеющие антагонистическое действие в отношении CRF,

где А может быть углеродом, G может быть азотом или углеродом, В может быть производным амино, и другие группы имеют указанные для них значения.
Из-за физиологической значимости CRF разработка биологически активных малых молекул, обладающих значительным связывающим действием в отношении CRF, и которые могут быть антагонистами рецептора CRF, остается желаемой целью. Такие антагонисты рецептора CRF могут быть полезны при лечении эндокринных, психиатрических и неврологических состояний или заболеваний, включая вообще связанных со стрессом расстройства.
Хотя были достигнуты значительные успехи в регулировании CRF посредством введения антагонистов рецептора CRF, в данной области техники все еще остается потребность в эффективных антагонистах рецептора CRF, имеющих небольшую молекулу. Также существует потребность в фармацевтических композициях, содержащих такие антагонисты рецептора CRF, а также в способах, относящихся к их применению для лечения, например, связанных со стрессом расстройств. Данное изобретение отвечает указанным потребностям и предоставляет другие связанные с ними преимущества.
В частности, данное изобретение относится к новым соединениям, которые являются мощными и специфическими антагонистами рецепторов рилизинг-фактора кортикотропина (CRF).
В данном изобретении представлены соединения формулы (I), включая их стереоизомеры, пролекарства и фармацевтически приемлемые соли и сольваты

где
R является арилом или гетероарилом, каждый из которых может быть замещен 1-4 группами, выбранными из:
галогена, С1-С6алкила, С1-С6алкокси, галоС1-С6алкила, С2-С6алкенила, С2-С6алкинила, галоС1-С6алкокси, -C(O)R5, нитро, -NR6R7, циано и группы R8;
R1 является водородом, С1-С6алкилом, С2-С6алкенилом, С2-С6алкинилом, галоС1-С6алкилом, галоС1-С6алкокси, галогеном, NR6R7 или циано;
R2 является водородом, С3-С7циклоалкилом или группой R9;
R3 является С3-С7циклоалкилом или группой R9; или
R2 и R3 вместе с N образуют 5-14-членный гетероцикл, который может быть замещен 1-3 группами R10;
R4 является водородом, С1-С6алкилом, галогеном или галоС1-С6алкилом;
R5 является С1-С4алкилом, -OR6 или -NR6R7;
R6 является водородом или С1-С6алкилом;
R7 является водородом или С1-С6алкилом;
R8 является 5-6-членным гетероциклом, который может быть насыщен или может содержать от одной до трех двойных связей, и который может быть замещен 1 или более группами R11;
R9 является С1-С6алкилом, который может быть замещен одной или более группами, выбранными из: С3-С7циклоалкила, С1-С6алкокси, галоС1-С6алкокси, гидрокси, галоС1-С6алкилом;
R10 является группой R8, С3-С7циклоалкилом, С1-С6алкилом, С1-С6алкокси, галоС1-С6алкилом, С2-С6алкенилом, С2-С6алкинилом, галоС1-С6алкокси, гидрокси, галогеном, нитро, циано, C(O)NR6R7, фенилом, который может быть замещен 1-4 группами R11;
R11 является С3-С7циклоалкилом, С1-С6алкилом, С1-С6алкокси, галоС1-С6алкилом, С2-С6алкенилом, С2-С6алкинилом, галоС1-С6алкокси, гидрокси, галогеном, нитро, циано или C(O)NR6R7;
Х является углеродом или азотом;
n равно 1 или 2.
Кислотно-аддитивные соли свободных оснований аминосоединений данного изобретения могут быть получены способами, хорошо известными в данной области, и могут быть получены из органических и неорганических кислот. Подходящие органические кислоты включают малеиновую, яблочную, фумаровую, бензойную, аскорбиновую, янтарную, метансульфоновую, п-толуолсульфоновую, уксусную, щавелевую, пропионовую, винную, салициловую, лимонную, глюконовую, молочную, миндальную, коричную, аспарагиновую, стеариновую, пальмитиновую, гликолевую, глутаминовую и бензолсульфоновую кислоты. Подходящие неорганические кислоты включают хлористоводородную, бромистоводородную, серную, фосфорную и азотную кислоты. Таким образом, термин «фармацевтически приемлемая соль» структуры (I) охватывает любую и все приемлемые солевые формы.
Сольваты могут быть, например, гидратами.
Ссылки на соединение в соответствии с данным изобретением включают как соединения формулы (I), так и их фармацевтически приемлемые кислотно-аддитивные соли, а также фармацевтически приемлемые сольваты.
Кроме того, в контекст данного изобретения также включены пролекарства. Пролекарствами являются любые ковалентно связанные носители, которые высвобождают соединение формулы (I) in vivo, если такое пролекарство вводится пациенту. Пролекарства обычно получают модификацией функциональных групп таким образом, что модификация расщепляется, либо обычным преобразованием, либо in vivo, с получением исходного соединения. Пролекарства включают, например, соединения в соответствии с данным изобретением, в которых гидрокси, амино или сульфгидрильная группы связаны с какой-либо группой, которая, при введении пациенту, расщепляется с образованием гидрокси, амино или сульфгидрильной группы. Таким образом, примеры пролекарств включают (но не ограничены ими) ацетат, формиат и бензоат производные спиртовой, сульфгидрильной и амино функциональных групп соединений формулы (I). Кроме того, в случае карбоновой кислоты (-СООН) могут применяться сложные эфиры, такие как метиловые эфиры, этиловые эфиры и подобные.
Относительно стереоизомеров, соединения формулы (I) могут иметь хиральные центры и могут существовать в виде рацематов, рацемических смесей и отдельных энантиомеров или диастереомеров. Все такие изомерные формы включены в объем данного изобретения, включая их смеси. Более того, некоторые кристаллические формы соединений формулы (I) могут существовать в виде полиморфов, которые включены в объем данного изобретения.
Термин С1-С6алкил в данном описании относится к группе или части группы, являющейся линейной или разветвленной алкильной группой, содержащей от 1 до 6 атомов углерода; примеры таких групп включают метил, этил, пропил, изопропил, н-бутил, изобутил, трет-бутил, пентил или гексил.
Термин С3-С7циклоалкильная группа относится к неароматическому моноциклическому углеводородному кольцу, содержащему 3-7 атомов углерода, такому как, например, циклопропил, циклобутил, циклопентил, циклогексил или циклогептил; а ненасыщенные циклоалкилы включают циклопентенил и циклогексенил и подобные.
Термин галоген относится к фтору, хлору, брому или йоду.
Термин галоС1-С6алкил или галоС1-С2 алкил относится к алкильной группе, имеющей один или более атомов углерода и в которой, по крайней мере, один атом водорода замещен галогеном, такой как, например, трифторметильная группа и подобные.
Термин С2-С6алкенил определяет прямой или разветвленный углеводородный радикал, содержащий одну или более двойных связей и имеющий от 2 до 6 атомов углерода, такой как, например, этенил, 2-пропенил, 3-бутенил, 2-бутенил, 2-пентенил, 3-пентенил, 3-метил-2-бутенил или 3-гексенил, и подобные.
Термин С1-С6алкоксигруппа относится к линейной или разветвленной алкоксигруппе, например метокси, этокси, пропокси, проп-2-окси, бутокси, бут-2-окси или метилпроп-2-окси и подобным.
Термин галоС1-С6алкоксируппа относится к С1-С6алкоксигруппе, такой как определено выше, замещенной, по крайней мере, одним галогеном, предпочтительно, фтором, такой как OCHF2 или OCF3.
Термин С2-С6алкинил относится к прямому или разветвленному углеводородному радикалу, содержащему одну или более тройных связей, и имеющему от 2 до 6 атомов углерода, включая ацетиленил, пропинил, 1-бутинил, 1-пентинил, 3-метил-1-бутинил и подобные.
Термин арил относится к ароматической карбоциклической группе, такой как фенил, бифенил или нафтил.
Термин гетероарил относится к 5-10-членному ароматическому гетероциклическому кольцу, имеющему, по крайней мере, один гетероатом, выбранный из азота, кислорода и серы, и содержащему, по крайней мере, 1 атом углерода, включая как моно- так и бициклические системы.
Примеры гетероарилов включают (но не ограничены ими) фурил, бензофуранил, тиофенил, бензотиофенил, пирролил, индолил, изоиндолил, азаиндолил, пиридил, хинолинил, изохинолинил, оксазолил, изоксазолил, бензоксазолил, пиразолил, имидазолил, бензимидазолил, тиазолил, бензотиазолил, изотиазолил, пиридазинил, пиримидинил, пиразинил, триазинил, циннолинил, фталазинил, триазолил, тетразолил и хиназолинил.
Термин 5-14-членный гетероцикл относится к 5-7-членному моноциклу или 7-14-членному полициклическому гетероциклическому кольцу, которое является насыщенным, ненасыщенным или является ароматическим, и которое содержит 1-4 гетероатомов, независимо выбранных из азота, кислорода и серы, и в котором гетероатомы азота или серы могут быть необязательно окислены, и гетероатом азота может быть необязательно кватернизирован, включая бициклические кольца, в которых любой из указанных выше гетероциклов является конденсированным с получением бензольного кольца, а также к трициклическим (и высшим) гетероциклическим кольцам. Гетероциклическое кольцо может быть присоединено через любой гетероатом или атом углерода. Гетероциклы включают гетероарилы, такие как определено выше. Таким образом, кроме ароматических гетероарилов, перечисленных выше, гетероциклы также включают (но не ограничены ими) морфолинил, пирролидинонил, пирролидинил, пиперидинил, гидантоинил, валеролактамил, оксиранил, оксетанил, тетрагидрофуранил, тетрагидропиранил, тетрагидропиридинил, тетрагидропиримидинил, тетрагидротиофенил, тетрагидротиопиранил, тетагидропиримидинил, тетрагидротиофенил, тетрагидротиопиранил и подобные.
Термин 5-6-членный гетероцикл согласно данному выше определению относится к 5-6-членному моноциклическому гетероциклическому кольцу, которое является насыщенным, ненасыщенным или является ароматическим и которое содержит 1-4 гетероатомов, независимо выбранных из азота, кислорода и серы, и в котором гетероатомы азота и серы могут быть необязательно окислены, и гетероатом азота может быть необязательно кватернизирован. Гетероциклы включают гетероарилы, такие как определено выше. Гетероцикл может быть присоединен через любой гетероатом или атом углерода. Таким образом, термин включает (но не ограничен ими) морфолинил, пиридинил, пиразинил, пиразолил, тиазолил, триазолил, имидазолил, оксадиазолил, оксазолил, пирролидинонил, пирролидинил, пиперидинил, гидантоинил, валеролактамил, оксиранил, оксетанил, тетрагидрофуранил, тетрагидропиранил, тетрагидропиридинил, тетрагидропримидинил, тетрагидротиофенил, тетрагидротиопиранил, тетрагидропиримидинил, тетрагидротиофенил, тетрагидротиопиранил и подобные.
Примеры соединений в соответствии с данным изобретением включают следующие формулы (Ia) и (Ib)
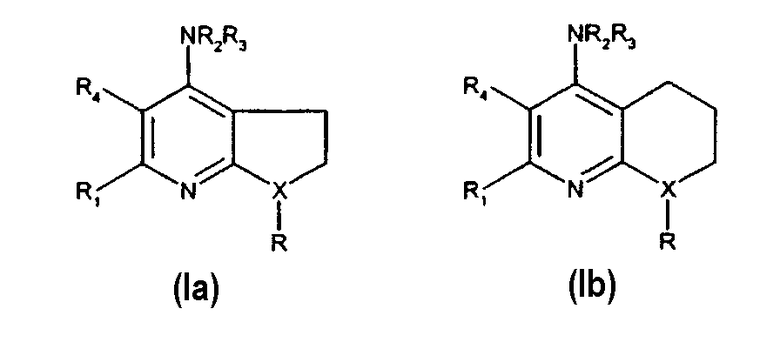
В предпочтительном варианте, в котором n равно 1, согласно определению соединений формулы (I) выше, антагонисты рецептора CRF данного изобретения имеют формулу (Ia) и, если n равно 2, антагонисты рецептора CRF данного изобретения имеют формулу (Ib), где R, R1, R2 и R3 такие, как определено выше.
Другие примеры соединений в соответствии с данным изобретением включают соединения общей формулы (I), в которой
R2 и R3 вместе с N образуют 5-14-членную гетероциклическую группу, которая может быть замещена 1-3 группами R10; где R10 группы такие, как определено выше.
В зависимости от выбора Х, антагонисты рецептора CRF данного изобретения включают соединения формул (IIa) и (IIb), в которых группа NR2R3 представлена 5-6-членной гетероциклической группой, которая может быть замещена 1-3 группами R8.

В частности, включены соединения формул (IIIa) и (IIIb)

в которых R1, R и R8 такие, как определено выше.
Примеры таких соединений представлены в экспериментальной части.
Более предпочтительные варианты данного изобретения включают, но не ограничены ими, соединения формулы (I), (Ia), (Ib), (IIa), (IIb), (IIIa), (IIIb), в которых:
R1 является С1-С3 алкильной группой или галоС1-С3 алкильной группой, предпочтительно, метилом или трифторметилом;
R4 является водородом; и
R является арильной группой, выбранной из 2,4-дихлорфенила, 2-хлор-4-метилфенила, 2-хлор-4-трифторметила,
2-хлор-4-метоксифенила, 2,4,5-триметилфенила, 2,4-диметилфенила,
2-метил-4-метоксифенила, 2-метил-4-хлорфенила,
2-метил-4-трифторметила, 2,4-диметоксифенила,
2-метокси-4-трифторметилфенила, 2-метокси-4-хлорфенила,
3-метокси-4-хлорфенила, 2,5-диметокси-4-хлорфенила,
2-метокси-4-изопропилфенила, 2-метокси-4-трифторметилфенила,
2-метокси-4-изопропилфенила, 2-метокси-4-метилфенила,
2-трифторметил-4-хлорфенила, 2,4-трифторметилфенила,
2-трифторметил-4-метилфенила, 2-трифторметил-4-метоксифенила,
2-бром-4-изопропилфенила, 2-метил-4-цианофенила,
2-хлор-4-цианофенила, 4-метил-6-диметиламинопиридин-3-ила,
3,5-дихлорпиридин-2-ила, 2,6-бисметоксипиридин-3-ила и
3-хлор-5-трихлорметилпиридин-2-ила.
Предпочтительными соединениями в соответствии с данным изобретением являются:
1-(2,4-бис-трифторметилфенил)-6-метил-4-(3-тиазол-2-илпиразол-1-ил)-2,3-дигидро-1Н-пирроло[2,3-b]пиридин;
1-(2,4-бис-трифторметилфенил)-6-метил-4-(3-тиазол-2-илпиразол-1-ил)-2,3-дигидро-1Н-пирроло[2,3-b]пиридин;
3-метил-4-[6-метил-4-(3-тиазол-2-илпиразол-1-ил)-2,3-дигидро-1Н-пирроло[2,3-b]пиридин-1-ил]бензонитрил;
4-[6-метил-4-(3-тиазол-2-илпиразол-1-ил)-2,3-дигидро-1Н-пирроло[2,3-b]пиридин-1-ил]-3-трифторметилбензонитрил;
6-метил-1-(2-метил-4-трифторметоксифенил)-4-(3-тиазол-2-илпиразол-1-ил)-2,3-дигидро-1Н-пирроло[2,3-b]пиридин;
1-(2,4-бис-трифторметилфенил)-7-метил-5-(3-тиазол-2-илпиразол-1-ил)-1,2,3,4-тетрагидро[1,8]нафтиридин;
1-(4-метокси-2-метилфенил)-6-метил-4-(3-тиазол-2-илпиразол-1-ил)-2,3-дигидро-1Н-пирроло[2,3-b]пиридин;
1-(2,4-бис-трифторметилфенил)-6-метил-4-(3-морфолин-4-илпиразол-1-ил)-2,3-дигидро-1Н-пирроло[2,3-b]пиридин;
1-(2,4-бис-трифторметилфенил)-6-метил-4-(3-пиридин-2-илпиразол-1-ил)-2,3-дигидро-1Н-пирроло[2,3-b]пиридин;
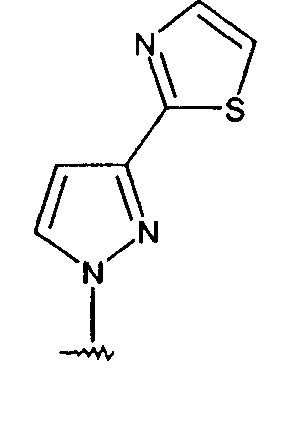
4-[1,3']бипиразолил-1'-ил-1-(2,4-бис-трифторметилфенил)-6-метил-2,3-дигидро-1Н-пирроло[2,3-b]пиридин.
В общем, соединения формулы (I) могут быть получены методами органического синтеза, известными специалистам в данной области, а также методами, представленными в примерах.
Соединения формулы (I) и их соли и сольваты могут быть получены способом, описанным ниже. В представленном ниже описании группы R, R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R11 и n имеют значения, определенные выше для соединений формулы (I), если не указано иначе.
Соединения формулы (IIa) могут быть получены в соответствии со следующей схемой 1:
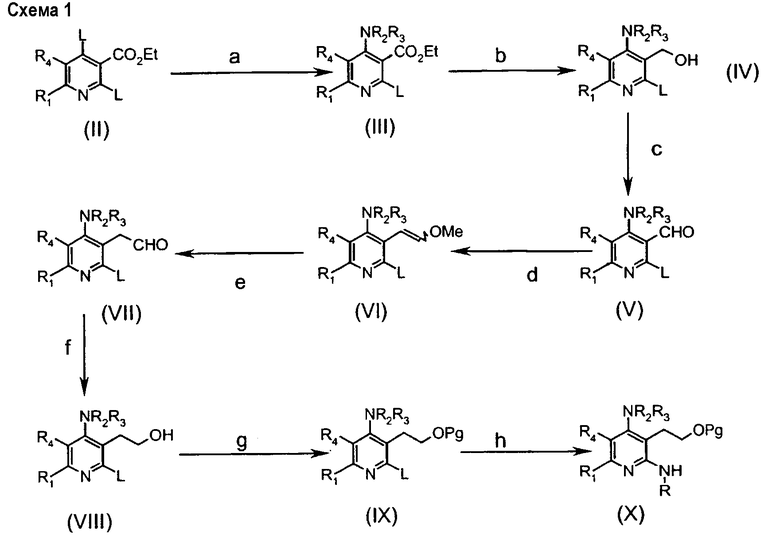
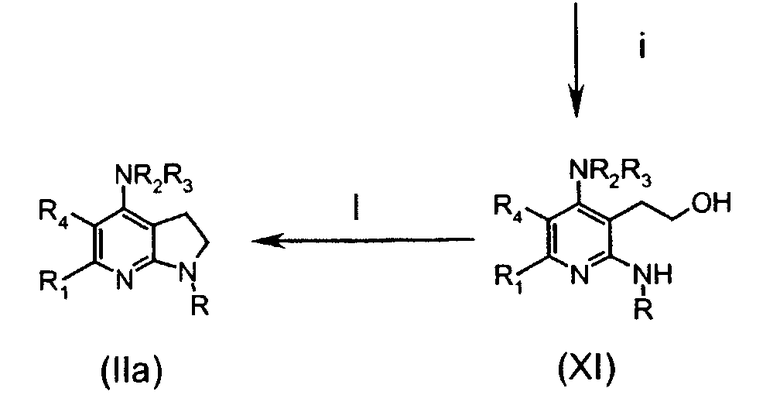
в которой
стадия а включает превращение уходящей группы L, выбранной из группы, включающей галоген или реакционноспособный остаток сульфоновой кислоты (например, мезилат, тозилат), предпочтительно, хлорид, в аминогруппу соединений (III) взаимодействием с подходящим амином NR2R3 в основных условиях;
стадия b включает восстановление сложноэфирной группы подходящим восстанавливающим агентом (таким как DIBAl-H) до гидроксигруппы соединений (IV);
стадия с включает окисление гидроксигруппы подходящим окисляющим агентом (таким как периодинан Десса-Мартина) до альдегидной группы соединения (V);
стадия d включает образование альдегидной группы соединений (VII) по реакции Виттига в обычных условиях через образование енольного эфира с последующим кислотным гидролизом (стадия е);
стадия f включает восстановление альдегидной группы подходящим восстанавливающим агентом (таким как NaBH4) до гидроксигруппы соединений (VIII);
стадия g включает превращение гидроксигруппы в подходящую защитную группу соединений (IX) (такую как TBS: трет-бутилдиметилсилил);
стадия h включает реакцию Бухвальда посредством сочетания с подходящим амином RNH2;
стадия i включает реакцию снятия защиты с получением гидроксильной группы соединений (XI);
стадия l включает внутримолекулярную циклизацию нагреванием после превращения гидроксигруппы соединений (XI) в подходящую уходящую группу (такую как бромид, взаимодействием с CBr4 и PPh3) с получением конечных соединений (IIa).
Альтернативно, соединения формулы (IIa) могут быть получены согласно следующей схеме 2:
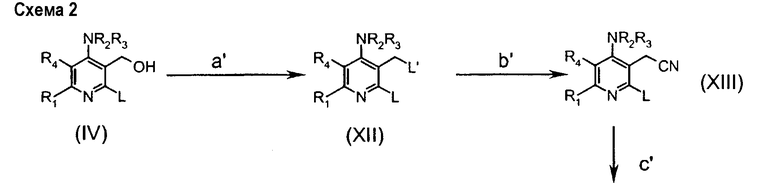
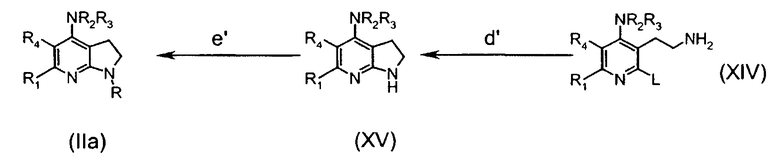
в которой
стадия a' включает превращение гидроксигруппы в подходящую уходящую группу L' соединений (XII), которая, независимо от L, имеет те же определения (например, мезилат, взаимодействием с MsCl в Et3N);
стадия b' включает превращение L' в циано производное соединений (XIII) взаимодействием, например, c KCN в апротонном диполярном растворителе, таком как ДМФА;
стадия c' включает восстановление цианогруппы подходящим восстанавливающим агентом (таким как ВН3-ТГФ) до аминогруппы соединения (XIV);
стадия d' включает внутримолекулярную циклизацию соединения (XIV) нагреванием в подходящем растворителе (таком как NMP) при высокой температуре;
стадия е' соответствует стадии h предыдущей схемы.
Соединения формулы (IIb) могут быть получены в соответствии со следующей схемой 3:
Схема 3
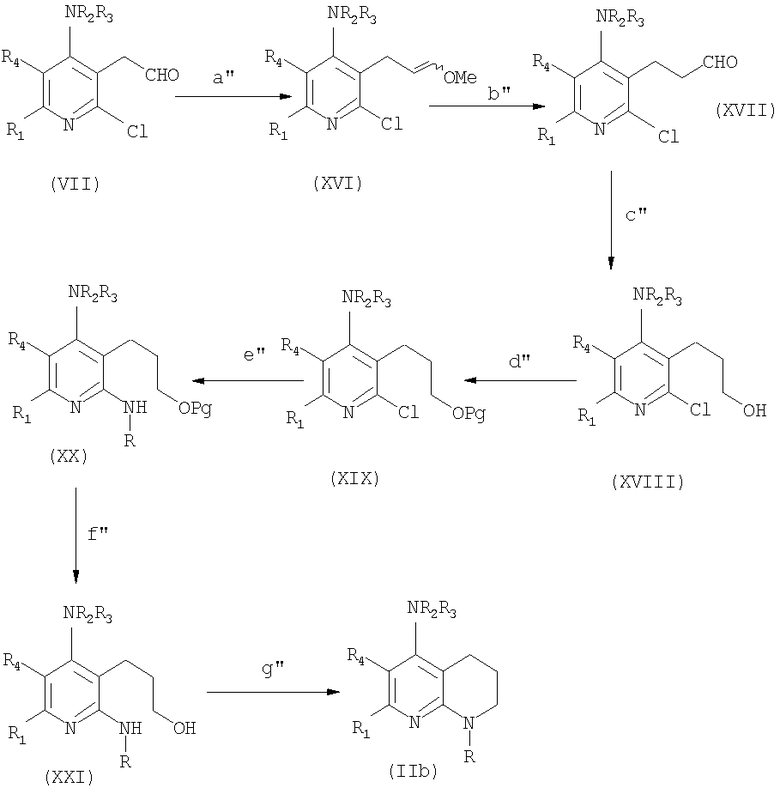
в которой
стадия а" соответствует предыдущей стадии d;
стадия b" соответствует предыдущей стадии е;
стадия с" соответствует предыдущей стадии f;
стадия d'' соответствует предыдущей стадии g;
стадия e'' соответствует предыдущей стадии h;
стадия f'' соответствует предыдущей стадии i;
стадия g'' соответствует предыдущей стадии l.
Соединения формулы (II) являются известными соединениями и могут быть получены способами, описанными в литературе.
Соединения формул (IIIa) и (IIIb) могут быть получены согласно представленным выше схемам 1, 2 и 3, после получения гетероциклического реакционноспособного остатка способами, известными специалистам в данной области.
Примеры подходящих гидроксизащитных групп включают тригидрокарбилсилиловые простые эфиры, такие как триметилсилиловый или трет-бутилдиметилсилиловый эфиры. Гидроксизащитные группы могут быть удалены хорошо известными стандартными способами (такими, как описаны в Protective Groups in Organic Chemistry, стр.46-119, издание J F W McOmie (Plenum Press, 1973)). Например, если защитная группа является трет-бутилдиметилсилильной группой, она может быть удалена обработкой тригидрофторидом триэтиламина.
Фармацевтически приемлемые соли также могут быть получены из других солей, включая другие фармацевтически приемлемые соли соединений формулы (I) с помощью обычных способов.
Соединения формулы (I) легко могут быть выделены в ассоциации с молекулами растворителя кристаллизацией или выпариванием подходящего растворителя с получением соответствующих сольватов.
Если требуется определенный энантиомер соединения общей формулы (I), он может быть получен, например, разделением соответствующей энантиомерной смеси соединения формулы (I) с применением обычных способов. Так, требуемый энантиомер может быть получен из рацемического соединения формулы (I) с применением метода хиральной ВЭЖХ.
Объект данного изобретения также включает меченные изотопами соединения, которые идентичны соединениям, представленным в формулах I и далее, но в которых, фактически, один или более атомов заменены атомами, имеющими атомную массу или массовое число отличающееся от атомной массы или массового числа, обычно существующего в природе. Примеры изотопов, которые могут быть введены в соединения данного изобретения, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора, йода и хлора, такие как 3H, 11C, 14C, 18F, 123I и 125I. Соединения данного изобретения и их фармацевтически приемлемые соли, которые содержат указанные выше изотопы и/или другие изотопы других атомов, включены в объем данного изобретения. Меченные изотопами соединения данного изобретения, например такие, в которые включены радиоактивные изотопы, такие как 3H, 14C, применяются в исследованиях лекарственных средств и/или исследованиях распределения в субстрате ткани. Тритированные, т.е. 3Н и углерод-14, т.е. 14С изотопы особенно предпочтительны благодаря простоте их получения и определяемости. 11С и 8F изотопы особенно полезны в PET (позитронной эмиссионной томографии), и 125I изотопы особенно полезны в SPECT (компьютерной томографии с эмиссией одинарных фотонов), которые применяются для получения изображения мозга. Далее, замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2Н, может дать определенные терапевтические преимущества благодаря большей устойчивости к метаболизму, например, увеличенный период полураспада in vivo и пониженная требуемая дозировка и, следовательно, может быть предпочтительным в определенных обстоятельствах. Меченные изотопами соединения формулы I и другие в соответствии с данным изобретением в основном могут быть получены методами, описанными на схемах и/или представленных ниже примерах, замещением не меченного изотопами реагента на легко доступный меченный изотопами реагент.
Антагонисты рецептора CRF данного изобретения обладают действием на сайт рецептора CRF, включая рецепторы CRF 1 и CRF 2, и могут применяться при лечении состояний, медиированных CRF или рецепторами CRF.
Эффективность соединения как антагониста рецептора CRF может быть определена различными методами исследования. Подходящие антагонисты CRF данного изобретения способны ингибировать специфическое связывание CRF с его рецептором и антагонизировать действия, связанные с CRF. Соединение структуры (I) может оцениваться на предмет его активности в качестве антагониста рецептора CRF с помощью одного или более общепринятых методов исследования для этих целей, включая (но не ограничиваясь ими) исследования, описанные e DeSouza et al., (J. Neuroscience 7: 88, 1987) и Battagcia et al (Synapse 1: 572, 1987).
Исследования связывания рецепторов CRF проводят с применением гомогенного метода сцинтилляционного приближения (SPA). Лиганд связывается с препаратом рекомбинантной мембраны, экспрессирующим рецепторы CRF, которые, в свою очередь, связываются с SPA гранулами, покрытыми агглутинином зародышей пшеницы. Подробное описание экспериментов представлено в экспериментальной части.
Что касается аффинности связывания рецептора CRF, антагонисты рецептора CRF данного изобретения имеют Ki менее 10 мкМ.
Соединения данного изобретения применяются при лечении расстройств центральной нервной системы, в которые вовлечены рецепторы CRF. В частности, при лечении или профилактике основных депрессивных расстройств, включая биполярную депрессию, униполярную депрессию, единичные или повторяющиеся основные депрессивные эпизоды с или без психотических признаков, кататонические признаки, меланхолические признаки, атипичные признаки или послеродовую депрессию, для лечения тревоги и для лечения панических расстройств. Другие расстройства настроения, включенные в термин основные депрессивные расстройства, включают дистимическое расстройство с ранним или поздним появлением (симптомов) и с или без атипичных признаков, невротическую депрессию, посттравматический стресс и социальную фобию; слабоумие типа Альцгеймера, с ранним или поздним появлением (симптомов), при депрессивном настроении; сосудистую деменцию при депрессивном настроении; расстройства настроения, вызванные алкоголем, амфетаминами, кокаином, галлюциногенами, летучими препаратами, опиоидами, фенциклидином, седативными препаратами, снотворными средствами, транквилизаторами и другими веществами; шизоаффективное расстройство депрессивного типа; и регулируемое расстройство с депрессивным настроением. Основные депрессивные расстройства могут также развиваться из общих болезненных состояний, включая, но не ограничиваясь ими, инфаркт миокарда, диабет, выкидыш или аборт и т.д.
Соединения данного изобретения также полезны при лечении или профилактике шизофренических расстройств, включая параноидальную шизофрению, дезорганизованную шизофрению, кататоническую шизофрению, недифференцированную шизофрению, остаточную шизофрению.
Соединения данного изобретения могут применяться в качестве анальгетиков. В частности, они могут применяться при лечении травматической боли, такой как послеоперационная боль; травматической авульсивной боли, такой как в плечевом сплетении; хронической боли, такой как артритная боль, возникающая при остео-, ревматоидном или псориатическом артрите; невропатической боли, такой как пост-герпесная невралгия, невралгия тройничного нерва, сегментная или межреберная невралгия, фибромиалгия, каузалгия, периферийная невропатия, диабетическая невропатия, вызванная химиотерапией невропатия, связанная со СПИД невропатия, затылочная невралгия, коленчатая невралгия, языкоглоточная невралгия, рефлекторная симпатетическая дистрофия, боль в фантомной конечности; различные формы головной боли, такие как мигрень, острая или хроническая тянущая боль, височно-нижнечелюстная боль, боль в верхнечелюстной пазухе, «гистаминовая» головная боль; зубная боль; раковая боль; боль висцерального происхождения; боль в желудочно-кишечном тракте; боль при ущемлении нервов в органах; боль при спортивных повреждениях; дисменорея; боль при менструации; менингиты; арахноидиты; мышечно-скелетная боль; поясничная боль, например позвоночный стеноз; выпадение диска; ущемление седалищного нерва; стенокардия; анкилозирующий спондилоартрит; подагра; ожоги; шрамы; зуд; и травматическая боль, такая как таламическая боль после удара.
Соединения данного изобретения также полезны для лечения расстройства аппетита и приема пищи и для состояний, таких как анорексия, нервная анорексия и булимия.
Соединения данного изобретения также применимы при лечении расстройств сна, включая дисомнию, бессонницу, приступы апноэ во сне, нарколепсию и расстройства циркадных ритмов.
Соединения данного изобретения также применимы для лечения или профилактики расстройств познавательной способности. Расстройства познавательной способности включают деменцию, амнезию и расстройства познавательной способности, не определенные иначе.
Далее, соединения данного изобретения также применяют как средства, улучшающие память и/или познавательную способность у здоровых людей, не страдающих ослаблением познавательной способности и/или памяти.
Соединения данного изобретения также применимы при лечении толерантности к и зависимости от некоторых соединений. Например, их применяют для лечения зависимости от никотина, алкоголя, кофеина, фенциклидина (соединений, подобных фенциклидину) или при лечении толерантности к и зависимости от опиатов (например, марихуаны, героина, морфина) или бензодиазепинов; при лечении пристрастия к лекарственным средствам на основе кокаина, снотворных, амфетаминов или родственными амфетамину соединений (например, декстроамфетамин, метиламфетамин), или при их сочетании.
Соединения данного изобретения также применимы в качестве противовоспалительных агентов. В частности, они применяются при лечении воспаления при астме, гриппе, хронических бронхитов и ревматоидных артритов; при лечении воспалительных заболеваний желудочно-кишечного тракта, таких как болезнь Крона, язвенный колит, воспалительное заболевание кишечника (ВЗК) и повреждений, вызванных приемом нестероидных противовоспалительных средств; воспалительных заболеваний кожи, таких как герпес или экзема; воспалительных заболеваний мочевого пузыря, таких как цистит и недержание мочи; и воспаления глаз и ротовой полости.
Соединения данного изобретения также применимы при лечении аллергических нарушений, в частности, аллергических нарушений кожи, таких как крапивница, и аллергических расстройств дыхательных путей, таких как ринит.
Соединения данного изобретения также применимы при лечении рвоты, т.е. тошноты, рвотных позывов и рвоты. Рвота включает острую рвоту, затяжную рвоту и рецидив рвоты. Соединения данного изобретения применяют при лечении вызванной рвоты. Например, рвота может быть вызвана лекарствами, такими как химиотерапевтические агенты для лечения рака, такие как алкилирующие агенты, например циклофосфамиды, кармустин, ломустин и хлорамбуцил; цитотоксичные антибиотики, например, дактиномицин, доксорубицин, митомицин-С и блеомицин; анти-метаболиты, например, цитарабин, метотрексат и 5-фтороурацил; алкалоиды барвинка, например, этопозид, винбластин и винкристин; и другие, такие как цисплатин, дакарбазин, прокарбазин и гидроксимочевина; и их сочетание; тошнота, вызванная облучением; радиационная терапия, например, облучение грудной клетки или брюшной полости, например, при лечении рака; яды, токсины, такие как токсины, образованные при метаболических расстройствах или инфекции, например, гастритах, или образованные во время бактериальных или вирусных желудочно-кишечных инфекций; беременность; расстройства вестибулярного аппарата, такие как укачивание, головокружение, и болезнь Меньера; тошнота после хирургического вмешательства; кишечная непроходимость; пониженная перистальтика; висцеральная боль, например, при инфаркте миокарда или перитоните; мигрень; повышенное внутричерепное давление; пониженное внутричерепное давление (например, высотная болезнь); опиоидные анальгетики, такие как морфин; и гастроэзофагиальный рефлюкс, изжога, чрезмерное потребление еды и питья, повышенная кислотность желудка, отрыжка, изжога, такая как эпизодическая изжога, ночная изжога и изжога, вызванная перееданием, и диспепсия.
Соединения данного изобретения особенно полезны для лечения желудочно-кишечных расстройств, таких как синдром раздраженного кишечника (СРК); заболевания кожи, такие как псориаз, зуд и солнечная эритема; вазопластические заболевания, такие как ангина, головная боль сосудистого происхождения и болезнь Рейно; церебральная ишемия, такая как церебральный вазоспазм с последующим субарахноидальным кровоизлиянием; фиброзные и коллагеновые заболевания, такие как склеродерма и эозинофагиальный фасциолез; заболевания, связанные с иммунологическим усилением или подавлением, такие как системная волчанка и ревматические заболевания, такие как фиброз; и кашель.
Соединения данного изобретения применяют для лечения нейротоксических повреждений, которые являются следствием церебрального приступа, тромбоэмболического приступа, удара с кровоизлиянием, церебральной ишемии, церебрального вазоспазма, гипогликемии, гипоксии, аноксии, перинатальной асфиксии при остановке сердца.
Данное изобретение представляет соединение формулы (I) или его фармацевтически приемлемую соль или сольват для применения в терапии, в частности в медицине человека.
В другом аспекте данного изобретения также представлено применение соединения формулы (I) или его фармацевтически приемлемой соли или сольвата при изготовлении лекарственного средства для лечения состояний, медиированных CRF.
В альтернативном или другом аспекте данного изобретения представлен способ лечения млекопитающих, включая человека, в частности лечения состояний, медиированных CRF, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли или сольвата.
Должно быть понятно, что термин лечение включает профилактику и облегчение симптомов.
Соединения формулы (I) могут вводиться в виде необработанного химического соединения, но активный ингредиент предпочтительно присутствует в виде фармацевтической композиции.
Следовательно, данное изобретение также представляет фармацевтическую композицию, которая содержит, по крайней мере, одно соединение формулы (I) или его фармацевтически приемлемую соль, и которая предназначена для введения любым удобным способом. Такие композиции предпочтительно имеют форму, адаптированную для применения в медицине, в частности, в медицине человека, и могут быть получены обычными способами с применением одного или более фармацевтически приемлемых носителей или эксципиентов.
Таким образом, соединение формулы (I) может быть составлено в фармацевтическую композицию для перорального, буккального, парентерального, местного (включая офтальмологическое и назальное), депо или ректального введения, или форму, подходящую для введения ингаляцией или инсуффляцией (через рот или нос).
Для перорального введения, фармацевтические композиции могут иметь форму, например, таблеток или капсул, полученных обычными методами, с фармацевтически приемлемыми эксципиентами, такими как связывающие агенты (например, предварительно желатинизированный кукурузный крахмал, поливинилпирролидон или гидроксипропилметилцеллюлоза); наполнители (например, лактоза, микрокристаллическая целлюлоза или гидрофосфат кальция); лубриканты (например, стеарат магния, тальк или двуокись кремния); дезинтеграторы (например, картофельный крахмал или крахмальный гликолят натрия); или смачивающие агенты (например, лаурилсульфат натрия). Таблетки могут быть покрыты оболочкой с применением методов, известных в данной области. Жидкие препараты для перорального применения могут включать, например, растворы, сиропы или суспензии, или они могут быть представлены в виде сухого продукта для разведения водой или другими подходящими носителями перед применением. Такие жидкие препараты могут быть получены обычными методами с фармацевтически приемлемыми добавками, такими как суспендирующие агенты (например, сорбитовый сироп, производные целлюлозы или гидрированные пищевые жиры); эмульгирующие агенты (например, лецитин или аравийская камедь); неводные растворители (например, миндальное масло, масляные сложные эфиры, этиловый спирт или фракционированные растительные масла); и консерванты (например, метил- или пропил-п-гидроксибензоаты или сорбиновая кислота). Препараты также могут содержать буферные соли, вкусовые добавки, красители и подсластители.
Препараты для перорального введения могут обеспечивать контролируемое высвобождение активного соединения.
Для буккального введения композиция может иметь форму таблеток или обычную форму.
Соединения данного изобретения могут быть в виде композиций для парентерального введения, таких как болюсная инъекция или композиция для непрерывного вливания. Композиции для инъекций могут быть представлены в единичной дозированной форме, например, в ампулах, или контейнерах с мультидозами, содержащие консерванты. Композиции могут иметь такие формы, как суспензии, растворы или эмульсии в масляном или водном носителе, и могут содержать агенты для композиции, такие как суспендирующие, стабилизрующие и/или диспергирующие агенты. Альтернативно, активный ингредиент может быть в виде порошка для разбавления подходящим носителем, например, стерильной апирогенной водой, перед применением.
Соединения данного изобретения могут быть составлены в композицию для местного введения, такую как мази, кремы, гели, лосьоны, пессарии, аэрозоли или капли (например, глазные, ушные капли или капли в нос). Мази и кремы могут быть, например, получены на водной или масляной основе с добавлением подходящих загустителей и/или желатинизирующих агентов. Мази для введения в глаза могут быть получены в стерильных условиях с применением стерильных компонентов.
Лосьоны могут быть получены на водной или масляной основе и обычно содержат один или более эмульгирующих агентов, стабилизирующих агентов, диспергирующих агентов, суспендирующих агентов, загустителей или красителей. Капли могут быть получены на водной или не водной основе и также содержать один или более диспергирующих агентов, стабилизирующих агентов, солюбилизирующих агентов или суспендирующих агентов. Также они могут содержать консерванты.
Соединения данного изобретения также могут иметь форму композиций для ректального введения, таких как суппозитории или удерживающая клизма, и содержать, например, обычные основы для суппозиториев, такие как масло какао или другие глицериды.
Соединения данного изобретения также могут быть составлены в композицию как препараты депо. Такие долгодействующие композиции могут вводиться в виде имплантатов (например, подкожно или внутримышечно) или внутримышечной инъекцией. Таким образом, например, соединения данного изобретения могут быть в композиции с подходящими полимерными или гидрофобными материалами (например, в виде эмульсии в приемлемом масле) или ионообменными смолами, или труднорастворимыми производными, например, труднорастворимой солью.
Для интраназального введения, соединения данного изобретения могут иметь форму растворов для введения через подходящие устройства с дозаторами или устройства, содержащие единичную дозу, или, альтернативно, форму порошка для смешивания с подходящим носителем для введения с помощью подходящего устройства доставки.
Предлагаемая доза соединений данного изобретения составляет от 1 до 1000 мг в день. Понятно, что могут быть проведены рутинные изменения дозы, в зависимости от возраста и состояния пациента, и точная дозировка может быть определена лечащим терапевтом или ветеринаром. Дозировка также зависит от способа введения и конкретного выбранного соединения.
Таким образом, для парентерального введения ежедневная доза составляет от 1 до около 100 мг, предпочтительно от 1 до 80 мг в день. Для перорального введения ежедневная доза составляет от 1 до 300 мг, например, от 1 до 100 мг.
Примеры
В примерах промежуточных соединений и примерах соединений, если не указано иначе:
Температуры плавления (т.пл.) определяют на аппарате для измерения т.пл. Gallenkamp и не корректируют. Все температуры даны в °С. Инфракрасный спектр определяют с помощью прибора FT-IR. Ядерный магнитный резонанс (1Н-ЯМР) записывают при 400 МГц, химические сдвиги даны в м.д. по нисходящей (d) относительно Me2Si, применяемого в качестве внутреннего стандарта, и определены в виде синглетов (с), дублетов (д), дублетов дублетов (дд), триплетов (т), квартетов (кв) или мультиплетов (м). Хроматографию на колонке проводят на силикагеле (Merck AG Darmstaadt, Germany). В тексте используют следующие аббревиатуры: EtOAc - этилацетат, cHex - циклогексан, CH2Cl2 - дихлорметан, Et2O - диэтиловый эфир, ДМФА - N,N'-диметилформамид, DIPEA - N,N-диизопропилэтиламин, DME - диметиловый эфир этиленгликоля, МеОН - метанол, Et3N - триэтиламин, ТФУК - трифторуксусная кислота, ТГФ - тетрагидрофуран, DIBAl-Н - гидрид диизобутилалюминия, DMAP - диметиламинопиридин, LHMDS - литийгексаметилдисилазан; ТСХ - тонкослойная хроматография на пластинках с двуокисью кремния и «высушенный» относится к раствору, высушенному над безводным сульфатом натрия; к.т. (КТ) - комнатная температура.
Промежуточное соединение 1
Этиловый эфир 2,4-дихлор-6-метилникотиновой кислоты
Указанное в заголовке соединение получают по известной опубликованной методике: Mittelbach, Martin; Synthesis, 1988, 6, стр.479-80.
Промежуточное соединение 2
Этиловый эфир 2-хлор-6-метил-4-(3-тиазол-2-илпиразол-1-ил)никотиновой кислоты
К раствору 2-(1Н-пиразол-3-ил)-1,3-тиазола (7,71 г 1,05 экв.) в безводном ДМФА (61 мл), при температуре 0°С, в атмосфере N2 добавляют NaH 60% в минеральном масле (2,03 г, 1,05 экв.) и реакционную смесь перемешивают в течение 10 минут при температуре 0°С и затем в течение 1 часа при комнатной температуре. Затем добавляют промежуточное соединение 1 (11,34 г, 48,0 ммоль) в виде раствора в безводном ДМФА (35 мл) при температуре 0°С и полученный раствор нагревают до температуры 110°С в течение 3 часов. Затем реакцию гасят водой, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 7:3) с получением 7,02 г указанного в заголовке соединения в виде белого твердого вещества.
ЯМР (1Н, CDCl3): δ 7,91 (д, 1H), 7,91 (д, 1H), 7,41 (д, 1H), 7,31 (с, 1H), 7,18 (д, 1H), 4,50 (кв, 2H), 2,78 (с, 3H), 1,25 (т, 3H).
МС (m/z): 349 [MH]+.
Промежуточное соединение 3
[2-Хлор-6-метил-4-(3-тиазол-2-илпиразол-1-ил)пиридин-
3-ил]метанол
К раствору промежуточного соединения 2 (1,5 г, 4,3 ммоль) в безводном CH2Cl2 (30 мл) при температуре -78°С в атмосфере N2 добавляют DIBAl-H 1,0M в циклогексане (12,9 мл, 3,0 экв.). Реакционную смесь перемешивают в течение 1 часа при температуре -78°С и затем в течение 1 часа при комнатной температуре. Затем реакцию гасят насыщенным раствором сегнетовой соли, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 1:1) с получением 1,02 г указанного в заголовке соединения в виде белого твердого вещества.
ЯМР (1Н, CDCl3): δ 8,05 (д, 1H), 7,90 (д, 1H), 7,40 (д, 1H), 7,25 (с, 1H), 7,10 (д, 1H), 4,65 (с, 2H), 4,0 (шир.с, 1H), 2,60 (с, 3H).
МС (m/z): 307 [М]+.
Промежуточное соединение 4
2-Хлор-6-метил-4-(3-тиазол-2-илпиразол-1-ил)пиридин-
3-карбальдегид
К раствору промежуточного соединения 3 (150 мг, 0,5 ммоль) в безводном CH2Cl2 (5 мл) при комнатной температуре в атмосфере N2 добавляют периодинан Dess Martin (237 мг, 1,12 экв.) и реакционную смесь перемешивают в течение 1 часа при комнатной температуре. Затем реакцию гасят раствором 0,5 г тиосульфата натрия в насыщенном растворе бикарбоната натрия, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 1:1) с получением 124 мг указанного в заголовке соединения в виде белого твердого вещества.
ЯМР (1Н, CDCl3): δ 10,4 (с, 1H), 8,0-7,9 (2д, 2H), 7,40 (2д, 2H), 7,10 (с, 1H), 2,70 (с, 3H).
МС (m/z): 305 [MH]+.
Промежуточное соединение 5
2-Хлор-3-(2-метоксивинил)-6-метил-4-(3-тиазол-2-илпиразол-
1-ил)пиридин
К раствору хлорида (метоксиметил)трифенилфосфония (4,24 г, 3 экв.) в безводном ТГФ (20 мл) при температуре 0°С, в атмосфере N2 добавляют н-BuLi 1,6M в циклогексане (7,73 мл, 12,37 ммоль), реакционную смесь нагревают до комнатной температуры и затем перемешивают в течение 15 минут. Затем добавляют раствор промежуточного соединения 4 (1,25 г, 4,1 ммоль) в безводном ТГФ (15 мл) и реакционную смесь перемешивают при комнатной температуре в течение 1,5 часов. Затем реакцию гасят водой, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 4:1) с получением 961 мг указанного в заголовке соединения в виде белого твердого вещества (E:Z=3:2 смесь, используется как таковая на следующей стадии).
ЯМР (1Н, CDCl3) основной Е продукт: δ 7,90 (м, 1H), 7,83 (м, 1H), 7,38 (м, 1H), 7,05 (м, 1H), 7,00 (м, 1H), 6,51 (д, 1H), 5,63 (д, 1H), 3,64 (с, 3H), 2,60 (с, 3H).
МС (m/z): 333 [MH]+.
Промежуточное соединение 6
[2-Хлор-6-метил-4-(3-тиазол-2-илпиразол-1-ил)пиридин-
3-ил]ацетальдегид
К раствору промежуточного соединения 5 (936 мг, 2,8 ммоль) в безводном ТГФ (15 мл) добавляют 6N HCl (21 мл, 45 экв.) и реакционную смесь перемешивают при комнатной температуре в течение 15 часов. Затем реакцию гасят насыщенным водным NaHCO3 до рН 7, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме с получением 893 мг указанного в заголовке соединения в виде белого твердого вещества, которое используют на следующей стадии без дальнейшей очистки.
ЯМР (1Н, CDCl3): δ 9,80 (с, 1H), 7,90-7,80 (2д, 2H), 7,70 (д, 1H), 7,20 (д, 1H), 7,0 (с, 1H), 4,25 (с, 2H), 2,70 (с, 3H).
МС (m/z): 319 [MH]+.
Промежуточное соединение 7
2-[2-Хлор-6-метил-4-(3-тиазол-2-илпиразол-1-ил)пиридин-
3-ил]этанол
К раствору промежуточного соединения 6 (903 мг, 2,84 ммоль) в безводном МеОН (10 мл) добавляют CeCl3 (700 мг, 1 экв.) и NaBH4 (107 мг, 1 экв.) и реакционную смесь перемешивают при комнатной температуре в течение 5 минут. Затем реакцию гасят водой, экстрагируют этилацетатом, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме с получением 848 мг указанного в заголовке соединения в виде белого твердого вещества, которое используют на следующей стадии без дальнейшей очистки.
ЯМР (1Н, CDCl3): δ 8,00 (м, 2H), 7,50 (д, 1H), 7,20 (м, 2H), 4,25 (т, 2H), 3,20 (т, 2H), 2,70 (с, 3H).
МС (m/z): 321 [MH]+.
Промежуточное соединение 8
3-[2-(трет-Бутилдиметилсиланилокси)этил]-2-хлор-6-метил-4-(3-тиазол-2-илпиразол-1-ил)пиридин
К раствору промежуточного соединения 7 (840 мг, 2,6 ммоль) в безводном CH2Cl2 (10 мл) добавляют 2,6-лутидин (0,67 мл, 2,2 экв.) и трет-бутилдиметилсилилтрифторацетат (0,89 мл, 1,5 экв.) и реакционную смесь перемешивают при комнатной температуре в течение 15 часов. Затем реакцию гасят водным раствором насыщенного NH4Cl, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 3:2) с получением 950 мг указанного в заголовке соединения в виде бесцветного масла.
ЯМР (1Н, CDCl3): δ 8,20 (д, 1H), 7,75 (д, 1H), 7,35 (д, 1H), 7,00 (м, 2H), 4,00 (т, 2H), 3,05 (т, 2H), 2,55 (с, 3H), 0,80 (с, 9H), -0,10 (с, 6H).
МС (m/z): 435 [MH]+.
Промежуточное соединение 9
(2,4-Бис-трифторметилфенил)-3-[2-(трет-бутилдиметилсиланилокси)этил]-6-метил-4-(3-тиазол-2-илпиразол-1-ил)пиридин-2-ил]амин
К раствору промежуточного соединения 8 (240 мг, 0,553 ммоль) в безводном DME (1 мл) добавляют Pd2(dba)3 (51 мг, 0,1 экв.), 2-(дициклогексилфосфино)-2'-метилбифенил (60 мг, 0,3 экв.), К3РО4 (317 мг, 3 экв.) и 2,4-бис(трифторметил)анилина (0,17 мл, 2 экв.) и реакционную смесь подвергают облучению микроволнами (150 Вт, 100°С, 60 ф/д2) в течение 20 минут. Затем реакцию гасят водным раствором насыщенного NH4Cl, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 9:1) с получением 180 мг указанного в заголовке соединения в виде бесцветного масла.
ЯМР (1Н, CDCl3): δ 8,55 (д, 1H), 8,20 (шир.с, 1H), 7,90 (д, 1H), 7,80 (м, 2H), 7,65 (дд, 1H), 7,40 (д, 1H), 7,05 (д, 1H), 6,85 (с, 1H), 4,20 (т, 2H), 2,90 (т, 2H), 2,60 (с, 3H), 0,80 (с, 9H), 0,10 (с, 6H).
МС (m/z): 628 [MH]+.
Промежуточное соединение 10
2-[2-(2,4-Бис-трифторметилфениламино)-6-метил-4-(3-тиазол-2-илпиразол-1-ил)пиридин-3-ил]этанол
К раствору промежуточного соединения 9 (240 мг, 0,38 ммоль) в безводном ТГФ (5 мл) добавляют Et3N·3HF (0,187 мл, 3 экв.) и реакционную смесь перемешивают в течение 15 часов при комнатной температуре. Затем реакцию гасят водным раствором насыщенного NH4Cl, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 1:1) с получением 180 мг указанного в заголовке соединения в виде бесцветного масла.
ЯМР (1Н, CDCl3): δ 8,45 (шир.с, 1H), 8,20 (д, 1H), 7,85 (д, 1H), 7,85 (2д, 2H), 7,65 (дд, 1H), 7,30 (д, 1H), 7,05 (д, 1H), 6,85 (с, 1H), 4,20 (т, 2H), 2,85 (т, 2H), 2,50 (с, 3H).
МС (m/z): 514 [MH]+.
Промежуточное соединение 11
2-Хлор-6-метил-4-(3-тиазол-2-илпиразол-1-ил)пиридин-3-илметиловый эфир метансульфокислоты
К раствору промежуточного соединения 3 (308 мг, 1,01 ммоль) в безводном CH2Cl2 (2,5 мл) при температуре -25°С, в атмосфере N2 добавляют Et3N (280 мкл, 2 экв.) и CH3SO2Cl (120 мкл, 1,5 экв.). Реакционную смесь перемешивают при температуре -25°С в течение 2 часов и затем при температуре -5°С в течение еще 2 часов. Реакционную смесь разбавляют водой и экстрагируют CH2Cl2. Объединенные органические экстракты сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 6:4 → 1:1) с получением 88 мг указанного в заголовке соединения в виде бесцветного масла.
ЯМР (1Н, CDCl3): δ 7,90 (д, 1H), 7,87 (д, 1H), 7,39 (д, 1H), 7,34 (с, 1H ), 7,14 (д, 1H), 5,5 (с, 2H), 3,0 (с, 3H), 2,78 (с, 3H).
МС (m/z): 385 [MH]+, Cl.
Промежуточное соединение 12
[2-Хлор-6-метил-4-(3-тиазол-2-илпиразол-1-ил)пиридин-3-ил]ацетонитрил
К раствору промежуточного соединения 11 (88 мг, 0,229 ммоль) в безводном ДМФА (2,5 мл) при температуре 0°С в атмосфере N2 добавляют KCN (15 мг, 1 экв.). Реакционную смесь перемешивают при комнатной температуре в течение 5 часов. Реакционную смесь разбавляют водой и 1М NaOH и экстрагируют Et2O. Объединенные органические экстракты сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Указанное в заголовке соединение получают в виде желтого твердого вещества (60 мг) и используют на следующей стадии без дальнейшей очистки.
ЯМР (1Н, CDCl3): δ 7,92 (д, 1H), 7,91 (д, 1H), 7,41 (д, 1H), 7,31 (с, 1H), 7,18 (д, 1H), 3,99 (с, 2H), 2,78 (с, 3H).
МС (m/z): 316 [MH]+, Cl.
Промежуточное соединение 13
2-[2-Хлор-6-метил-4-(3-тиазол-2-илпиразол-1-ил)пиридин-
3-ил]этиламин
К раствору промежуточного соединения 12 (810 мг, 2,571 ммоль) в безводном ТГФ (6 мл) при комнатной температуре, в атмосфере N2 добавляют BH3·ТГФ (10,3 мл, 4 экв.). Реакционную смесь перемешивают при температуре кипения с обратным холодильником в течение 2 часов. Реакционную смесь концентрируют в вакууме и разбавляют МеОН. Добавляют 1М HCl в Et2O (7,7 мкл, 3 экв.) при комнатной температуре и раствор перемешивают при температуре кипения с обратным холодильником в течение 2 часов. Реакционную смесь разбавляют водой и подщелачивают 1М NaOH до рН 12. Неочищенный продукт очищают флэш-хроматографией (силикагель CH2Cl2/MeOH 6:4). Указанное в заголовке соединение получают в виде бледно-желтого твердого вещества (690 мг).
ЯМР (1Н, CDCl3): δ8,42 (д, 1H), 7,94 (д, 1H), 7,79 (д, 1H), 7,48 (с, 1H ), 7,07 (д, 1H), 2,81 (м, 4H), 2,51 (с, 3H), 2,0 (шир.с, 2H).
МС (m/z): 320 [MH]+, Cl.
Промежуточное соединение 14
6-метил-4-(3-тиазол-2-илпиразол-1-ил)-2,3-дигидро-1Н-пирроло[2,3-b]пиридин
К раствору промежуточного соединения 13 (640 мг, 2,01 ммоль) в сухом N-метилпирролидиноне (13 мл) при комнатной температуре в атмосфере N2 добавляют Et3N (1,12 мл, 4 экв.). Реакционную смесь перемешивают при температуре 110°С в течение 7 часов. Реакционную смесь разбавляют водой и экстрагируют EtOAc. Объединенные органические экстракты сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, CH2Cl2/МеОН 98:2). Указанное в заголовке соединение получают в виде белого твердого вещества (187 мг).
ЯМР (1Н, CDCl3): δ7,98 (д, 1H), 7,89 (д, 1H), 7,35 (д, 1H), 7,06 (д, 1H), 4,65 (шир.с, 1H), 3,72 (т, 2Н), 3,48 (т, 2Н), 2,42 (с, 3Н).
МС (m/z): 284 [MH]+.
Промежуточное соединение 15
2-Хлор-3-(3-метоксиаллил)-6-метил-4-(3-тиазол-2-илпиразол-1-ил)пиридин
К перемешиваемой суспензии хлорида (метоксиметил)трифенилфосфония (833 мг, 3 экв.) в безводном ТГФ (4 мл) добавляют по каплям при температуре 0°С в атмосфере N2 н-BuLi в гексане 1,6M (1,50 мл, 3 экв.). Реакционную смесь перемешивают при комнатной температуре в течение 15 минут перед добавлением раствора промежуточного соединения 6 (258 мг, 1 экв.) в безводном ТГФ (3 мл). Реакционную смесь перемешивают в течение 1,5 часов. Смесь гасят водой и экстрагируют EtOAc. Объединенные органические экстракты сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 8:2) с получением 220 мг указанного в заголовке соединения в виде неразделимой смеси транс и цис (60/40) винилового эфира (желтое масло, 78%).
ЯМР (1Н, CDCl3): δ 7,88 (д, 1Н), 7,79 (д, 1H), 7,36 (д, 1H), 7,19 (с, 1H), 7,08 (д, 1H), 6,31 (д, 1H), 4,90 (м, 1H), 3,44 (д, 2H), 3,48 (с, 3H), 2,57 (с, 3H).
МС (m/z): 347 [MH]+, 1Cl.
Промежуточное соединение 16
3-[2-Хлор-6-метил-4-(3-тиазол-2-илпиразол-1-ил)пиридин-3-ил]пропиональдегид
К раствору промежуточного соединения 15 (370 мг, 1,07 ммоль) в ТГФ (5 мл) добавляют 6N HCl (12 мл, 67,5 экв.) и реакционную смесь перемешивают в течение 13 часов при комнатной температуре. К реакционной смеси добавляют раствор NaHCO3 до достижения рН 7 и водную фазу экстрагируют EtOAc. Объединенные органические экстракты сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенное указанное в заголовке соединение (335 мг) используют на следующей стадии без дальнейшей очистки.
ЯМР (1Н, CDCl3): δ 9,84 (с, 1H), 7,84 (д, 1H), 7,75 (д, 1H), 7,30 (д, 1H), 7,06 (д, 1H), 7,05 (с, 1H), 3,10-3,30 (м, 4H), 2,55 (с, 3H).
МС (m/z): 333 [М+1]+, 1Cl.
Промежуточное соединение 17
3-[2-Хлор-6-метил-4-(3-тиазол-2-илпиразол-1-ил)пиридин-3-ил]пропан-1-ол
К раствору промежуточного соединения 16 (335 мг, 1 ммоль) в безводном СН3ОН (5 мл) добавляют CeCl3 (247 мг, 1 экв.) и NaBH4 (38 мг, 1 экв.) при комнатной температуре в атмосфере N2. Реакционную смесь перемешивают в течение 20 минут. Растворитель удаляют в вакууме и остаток повторно растворяют в EtOAc/H2O и слои разделяют. Водный слой экстрагируют EtOAc и объединенные органические экстракты сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Очистка (силикагель, циклогексан/EtOAc 8:2) неочищенного продукта дает 277,4 мг указанного в заголовке соединения в виде прозрачного масла.
ЯМР (1Н, CDCl3): δ 7,90 (д, 1H), 7,76 (д, 1H), 7,36 (д, 1H), 7,12 (с, 1H), 7,07 (д, 1H), 3,70 (м, 2H), 2,90 (т, 2H), 2,58 (с, 3H), 2,20 (шир.т, 1H), 2,04 (м, 2H).
МС (m/z): 335 [М+1]+, 1Cl.
Промежуточное соединение 18
3-[3-трет-Бутилдиметилсиланилокси)пропил]-2-хлор-6-метил-4-
(3-тиазол-2-илпиразол-1-ил)пиридин
К раствору промежуточного соединения 17 (277,4 мг, 0,83 ммоль) в безводном ДМФА (7 мл) добавляют имидазол (621 мг, 11 экв.), трет-бутилдиметилсилил хлорид (350 мг, 2,8 экв.) и каталитическое количество DMAP при температуре 0°С в атмосфере N2. Реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Затем к реакционной смеси добавляют насыщенный водный раствор NH4Cl и экстрагируют EtOAc. Объединенные органические экстракты сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 7:3) с получением 347 мг указанного в заголовке соединения в виде желтого масла.
ЯМР (1Н, CDCl3): δ 7,89 (д, 1H), 7,81 (д, 1H), 7,34 (д, 1H), 7,20 (с, 1H), 7,08 (д, 1H), 3,66 (т, 2H), 2,86 (м, 2H), 2,57 (с, 3H), 1,89 (м, 2H), 0,86 (с, 9H), -0,006 (с, 6H).
МС (m/z): 449 [М]+, 1Cl.
Промежуточное соединение 19
(2,4-Бис-трифторметилфенил)-[3-[3-трет-бутилдиметилсиланилокси)пропил]-6-метил-4-(3-тиазол-2-илпиразол-1-ил)пиридин-2-ил]амин
В пробирку, содержащую Pd(dba)3 (17 мг, 0,1 экв.), 2-(дициклогексилфосфино)-2'-метилбифенил (20 мг, 0,3 экв.) и К3РО4 (103 мг, 2,7 экв.) при комнатной температуре в атмосфере N2 добавляют раствор промежуточного соединения 18 (80 мг, 0,18 ммоль) в безводном DME (0,5 мл) и раствор 2,4-бис(трифторметил)анилина (82 мг, 2 экв.) в сухом DME (0,5 мл). Реакционную смесь пятикратно подвергают облучению микроволнами (3 × 10 мин + 30 мин + 60 мин) при следующих параметрах: Р=110 Вт; Т=110°С, р=18 ф/д2. Раствор разбавляют водой и экстрагируют EtOAc. Объединенные органические экстракты сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 7:3) с получением 49 мг указанного в заголовке соединения в виде желтого масла.
ЯМР (1Н, CDCl3): δ 8,58 (д, 1H), 7,89 (д, 1H), 7,85 (д, 1H), 7,77 (дд, 1H), 7,76 (д, 1H), 7,34 (д, 1H), 7,23 (шир.с, 1H), 7,08 (д, 1H), 6,86 (с, 1H), 3,67 (т, 2H), 2,69 (м, 2H), 2,3 (с, 3H), 1,90 (м, 2H), 0,8 (с, 9H), -0,02 (с, 6H).
МС (m/z): 642 [М+1]+.
Промежуточное соединение 20
3-[2-(2,4-Бис-трифторметилфениламино)-6-метил-4-(3-тиазол-2-илпиразол-1-ил)пиридин-3-ил]пропан-1-ол
К раствору промежуточного соединения 19 (60 мг, 0,094 ммоль) в безводном ТГФ (2 мл) добавляют ТЕА·3HF (0,046 мл, 3 экв.). Реакционную смесь перемешивают при комнатной температуре в течение 12 часов. Затем к реакционной смеси добавляют насыщенный раствор NH4Cl и экстрагируют EtOAc. Объединенные органические экстракты сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 1:1) с получением 34,6 мг указанного в заголовке соединения в виде белого твердого вещества.
ЯМР (1Н, CDCl3): δ 8,62 (д, 1H), 7,90 (д, 1H), 7,85 (шир.с, 1H), 7,76 (дд, 1H), 7,7 (д, 1H), 7,37 (шир.с, 1H), 7,36 (д, 1H), 7,07 (д, 1H), 6,83 (с, 1H), 3,73 (т, 2H), 2,73 (т, 2H), 2,52 (с, 3H), 2,04 (м, 2H).
МС (m/z): 528 [М+1]+.
Промежуточное соединение 21
(2,4-Бис-трифторметилфенил)-[3-(3-бромпропил)-6-метил-4-(3-тиазол-2-илпиразол-1-ил)пиридин-2-ил]амин
К раствору промежуточного соединения 20 (34,6 мг, 0,066 ммоль) в безводном CH2Cl2 (1 мл) добавляют CBr4 (44 мг, 2 экв.) и PPh3 (34 мг, 2 экв.). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Затем к реакционной смеси добавляют насыщенный водный раствор NaHCO3 и экстрагируют EtOAc. Объединенные органические экстракты сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 7:3) с получением 25,7 мг указанного в заголовке соединения в виде белого твердого вещества.
ЯМР (1Н, CDCl3): δ 8,6 (д, 1H), 7,90 (д, 1H), 7,87 (д, 1H), 7,77 (м, 1H), 7,37 (д, 1H), 7,15 (шир.с, 1H), 7,10 (д, 1H), 6,84 (с, 1H), 3,47 (т, 2H), 2,78 (м, 2H), 2,52 (с, 3H), 2,3 (м, 2H).
МС (m/z): 590 [М]+, 1Br; 510 [M-Br]+.
Промежуточное соединение 22
4-[3-[2-(трет-Бутилдиметилсиланилокси)этил]-6-метил-4-(3-тиазол-2-илпиразол-1-ил)-пиридин-2-иламино]-3-метилбензонитрил
К раствору промежуточного соединения 8 (186 мг, 0,43 ммоль) в безводном DME (1 мл) добавляют Pd2(dba)3 (39 мг, 0,1 экв.), 2-(дициклогексилфосфино)-2'-метилбифенил (47 мг, 0,3 экв.), К3РО4 (246 мг, 2,6 экв.) и 3-метил-4-аминобензонитрил (113 мг, 2 экв.) и реакционную смесь облучают микроволнами (150 Вт, 100°С, 60 ф/д2) в течение 20 минут. Реакцию гасят водным раствором насыщенного NH4Cl, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 8:2) с получением 61 мг указанного в заголовке соединения в виде белого твердого вещества.
ЯМР (1Н, CDCl3): δ 8,30 (д, 1H), 8,06 (шир.с, 1H), 7,89 (д, 1H), 7,78 (д, 1H), 7,46 (дд, 1H), 7,44 (д, 1H), 7,36 (д, 1H), 7,09 (д, 1H), 6,81 (с, 1H), 4,34 (м, 2H), 2,82 (т, 2H), 2,56 (с, 3H), 2,36 (с, 3H), 0,85 (с, 9H), 0,02 (с, 6H).
МС (m/z): 531 [МН]+.
Промежуточное соединение 23
4-[3-(2-Гидроксиэтил)-6-метил-4-(3-тиазол-2-илпиразол-1-ил)пиридин-2-иламино]-3-метилбензонитрил
К раствору промежуточного соединения 22 (61 мг, 0,115 ммоль) в безводном ТГФ (2 мл) добавляют Et3N·3HF (0,056 мл, 3 экв.) и реакционную смесь перемешивают в течение 15 часов при комнатной температуре. Затем реакцию гасят водным раствором насыщенного NH4Cl, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 1:1) с получением 46 мг указанного в заголовке соединения в виде белого твердого вещества.
ЯМР (1Н, CDCl3): δ 8,39 (шир.с, 1H), 8,14 (д, 1H), 7,90 (д, 1H), 7,79 (д, 1H), 7,46 (м, 2H), 7,36 (д, 1H), 7,09 (д, 1H), 6,82 (с, 1H), 4,34 (м, 2H), 2,83 (т, 2H), 2,54 (с, 3H), 2,34 (с, 3H).
МС (m/z): 417 [МН]+.
Промежуточное соединение 24
4-[3-(2-Гидроксиэтил)-6-метил-4-(3-тиазол-2-илпиразол-
1-ил)пиридин-2-иламино]-3-трифторметилбензонитрил
К раствору промежуточного соединения 8 (106 мг, 0,244 ммоль) в безводном DME (1 мл) добавляют Pd2(dba)3 (22 мг, 0,1 экв.), 2-(дициклогексилфосфино)-2'-метилбифенил (27 мг, 0,3 экв.), К3РО4 (140 мг, 2,7 экв.) и 3-трифторметил-4-аминобензонитрил (91 мг, 2 экв.) и реакционную смесь облучают микроволнами (150 Вт, 100°С, 60 ф/д2) в течение 20 минут. Реакцию гасят водным раствором насыщенного NH4Cl, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 8:2) и выделенный продукт, содержащий некоторое количество непрореагировавшего анилина, используют на следующей стадии без дальнейшей очистки.
К раствору полученной выше смеси (120 мг) в безводном ТГФ (5 мл) добавляют Et3N·3HF (0,063 мл, 3 экв.) и реакционную смесь перемешивают в течение 15 часов при комнатной температуре. Затем реакцию гасят водным раствором насыщенного NH4Cl, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 1:1) с получением 40 мг указанного в заголовке соединения в виде белого твердого вещества.
ЯМР (1Н, CDCl3): δ 8,81 (шир.с), 8,22 (д, 1Н), 7,90 (д, 1Н), 7,87 (д, 1Н), 7,79 (д, 1Н), 7,68 (дд, 1Н), 7,37 (д, 1Н), 7,17 (д, 1Н), 6,92 (с, 1Н), 4,26 (кв, 2Н), 2,87 (т, 2Н), 2,54 (с, 3Н), 2,63 (т, 1Н).
МС (m/z): 471 [МН]+.
Промежуточное соединение 25
[3-[2-(трет-Бутилдиметилсиланилокси)этил]-6-метил-4-(3-тиазол-2-илпиразол-1-ил)пиридин-2-ил]-(2-метил-4-трифторметоксифенил)амин
К раствору промежуточного соединения 8 (110 мг, 0,253 ммоль) в безводном DME (1 мл) при комнатной температуре в атмосфере N2 добавляют Pd2(dba)3 (23 мг, 0,1 экв.), 2-(дициклогексилфосфино)-2'-метилбифенил (28 мг, 0,3 экв.), К3РО4 (145 мг, 2,7 экв.) и 2-метил-4-трифторметиланилин (97 мг, 2 экв.) и реакционную смесь облучают микроволнами (150 Вт, 100°С, 60 ф/д2) в течение 20 минут. Реакцию гасят водным раствором насыщенного NH4Cl, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 7:3) с получением 80 мг указанного в заголовке соединения в виде желтого масла.
ЯМР (1Н, CDCl3): δ 8,05 (д, 1Н), 7,83 (шир.с, 1Н), 7,78 (д, 1Н), 7,7 (д, 1Н), 7,46 (дд, 1Н), 7,44 (д, 1Н), 7,36 (д, 1Н), 7,09 (д, 1Н), 6,81 (с, 1Н), 4,34 (м, 2Н), 2,82 (т, 2Н), 2,56 (с, 3Н), 2,36 (с, 3Н), 0,85 (с, 9Н), 0,023 (с, 6Н).
МС (m/z): 590 [МН]+.
Промежуточное соединение 26
2-[6-метил-2-(2-метил-4-трифторметоксифениламино)-4-(3-тиазол-
2-илпиразол-1-ил)пиридин-3-ил]этанол
К раствору промежуточного соединения 25 (80 мг, 0,135 ммоль) в безводном ТГФ (2 мл) добавляют Et3N·3HF (66 мкл, 8 экв.) и реакционную смесь перемешивают в течение 15 часов при комнатной температуре. Затем реакцию гасят водным раствором насыщенного NH4Cl, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 1:1) с получением 48 мг указанного в заголовке соединения в виде бесцветного масла.
ЯМР (1Н, CDCl3): δ 7,91 (шир.с, 1Н), 7,85 (д, 1Н), 7,65 (д, 1Н), 7,30 (д, 1Н), 7,15-6,95 (м, 3Н), 6,65 (с, 1Н), 4,34 (м, 2Н), 2,83 (т, 2Н), 2,54 (с, 3Н), 2,34 (с, 3Н).
Пример 1
Синтез типового соединения формулы (IIIa)

Пример 1-1
1-(2,4-Бис-трифторметилфенил)-6-метил-4-(3-тиазол-2-илпиразол-1-ил)-2,3-дигидро-1Н-пирроло[2,3-b]пиридин
К раствору промежуточного соединения 10 (40 мг, 0,078 ммоль) в безводном CH2Cl2 (2 мл) при комнатной температуре в атмосфере N2 добавляют CBr4 (52 мг, 2 экв.) и PPh3 (41 мг, 2 экв.) и реакционную смесь перешивают в течение 3 часов. Затем реакцию гасят водным раствором насыщенного NaHCO3, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 2:1) с получением 18 мг указанного в заголовке соединения в виде твердого белого вещества.
Альтернативно:
Пример 1-1
1-(2,4-Бис-трифторметилфенил)-6-метил-4-(3-тиазол-2-илпиразол-1-ил)-2,3-дигидро-1Н-пирроло[2,3-b]пиридин
К смеси трис(дибензилиденацетон)палладия(0) (3,2 мг, 0,1 экв.), 2-(дициклогексилфосфино)-2'-метилбифенила (3,8 мг, 0,3 экв.) и К3РО4 (20 мг, 2,8 экв.) в пробирке с обжимной крышкой для микроволновой печи добавляют раствор промежуточного соединения 14 (10 мг, 0,035 ммоль) и 2,4-бис(трифторметил)бромбензол (6 мкл, 1 экв.) в безводном DME (1 мл) в атмосфере N2. Реакционную смесь облучают микроволнами в два цикла (2 × 10 мин) при следующих параметрах: Р=150 Вт; Т=100°С, р=60 ф/д2. Затем добавляют воду (1 мл) и продукт экстрагируют EtOAc. Объединенные органические экстракты промывают насыщенным водным NaCl (5 мл) и сушат над безводным Na2SO4. Твердые вещества отфильтровывают и растворитель выпаривают с получением неочищенного продукта, который очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 7:3). Указанное в заголовке соединение получают в виде бесцветного масла (1 мг, 0,002 ммоль).
Пример 1-2
3-Метил-4-[6-метил-4-(3-тиазол-2-илпиразол-1-ил)-2,3-дигидро-1Н-пирроло[2,3-b]пиридин-1-ил]бензонитрил
К раствору промежуточного соединения 23 (44 мг, 0,106 ммоль) в безводном CH2Cl2 (2 мл) добавляют CBr4 (71 мг, 2 экв.) и PPh3 (60 мг, 2 экв.) и реакционную смесь перешивают в течение 3 часов при комнатной температуре. Затем реакцию гасят водным раствором насыщенного NaHCO3, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 4:1) с получением 18 мг указанного в заголовке соединения в виде твердого белого вещества.
Пример 1-3
4-[6-Метил-4-(3-тиазол-2-илпиразол-1-ил)-2,3-дигидро-1Н-пирроло[2,3-b]пиридин-1-ил]-3-трифторметилбензонитрил
К раствору промежуточного соединения 24 (40 мг, 0,085 ммоль) в безводном CH2Cl2 (2 мл) добавляют CBr4 (56 мг, 2 экв.) и PPh3 (45 мг, 2 экв.) и реакционную смесь перешивают в течение 3 часов при комнатной температуре. Затем реакцию гасят водным раствором насыщенного NaHCO3, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 2:1) с получением 13 мг указанного в заголовке соединения в виде твердого белого вещества.
Пример 1-4
6-Метил-1-(2-метил-4-трифторметоксифенил)-4-(3-тиазол-2-илпиразол-1-ил)-2,3-дигидро-1Н-пирроло[2,3-b]пиридин
К раствору промежуточного соединения 26 (48 мг, 0,101 ммоль) в безводном CH2Cl2 (2 мл) добавляют CBr4 (66 мг, 2 экв.) и PPh3 (53 мг, 2 экв.) и реакционную смесь перешивают в течение 3 часов при комнатной температуре. Затем реакцию гасят водным водным раствором насыщенного NaHCO3, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Продукт очищают флэш-хроматографией (силикагель, циклогексан/EtOAc 8:2) с получением 10 мг указанного в заголовке соединения в виде твердого белого вещества.
Пример 1-5
1-(4-Метокси-2-метилфенил)-6-метил-4-(3-тиазол-2-илпиразол-1-ил)-2,3-дигидро-1Н-пирроло[2,3-b]пиридин
К раствору соответствующего промежуточного соединения (31 мг, 0,075 ммоль, 1 экв.) в сухом DCM (5 мл) добавляют CBr4 (53 мг, 0,16 ммоль, 2,1 экв.) и трифенилфосфин (42 мг, 0,16 ммоль, 2,1 экв.) в атмосфере N2. Реакционную смесь перемешивают при комнатной температуре в течение 15 часов. Затем добавляют воду (10 мл) и водную фазу экстрагируют EtOAc (20 мл). Органический слой сушат над Na2SO4, фильтруют и концентрируют в вакууме. Продукт очищают флэш-хроматографией (элюенты: циклогексан/этилацетат 7:3) с получением 5,2 мг указанного в заголовке соединения в виде бесцветного масла.
Пример 1-6
1-(2,4-Бис-трифторметилфенил)-6-метил-4-(3-морфолин-4-илпиразол-1-ил)-2,3-дигидро-1H-пирроло[2,3-b]пиридин
Получают по методике примера 1-1, используя 4-(1H-пиразол-3-ил)морфолин (J. Org. Chem., 1984, 269-276) вместо 2-(1Н-пиразол-3-ил)тиазола при получении промежуточного соединения 2.
Пример 1-7
1-(2,4-Бис-трифторметилфенил)-6-метил-4-(3-пиридин-2-илпиразол-1-ил)-2,3-дигидро-1H-пирроло[2,3-b] пиридин
Получают по методике примера 1-1, используя 2-(1Н-пиразол-3-ил)пиридин (коммерчески доступный) вместо 2-(1Н-пиразол-3-ил)тиазола при получении промежуточного соединения 2.
Пример 1-8
4-[1,3']Бипиразолил-1'-ил-1-(2,4-Бис-трифторметилфенил)-6-метил-2,3-дигидро-1Н-пирроло[2,3-b]пиридин
Получают по методике примера 1-1, используя 1'Н-[1,3']бипиразол (из 1Н-пиразол-3-иламина: J. Heterocycl. Chem., 1983, 1629-1639; затем J. Heterocycl. Chem., 1989, 733-738) вместо 2-(1Н-пиразол-3-ил)тиазола при получении промежуточного соединения 2.
Аналитические данные представлены в таблице 1-1 ниже
МС (m/z): 496 [МН]+.
МС (m/z): 399 [МН]+.
циано
МС (m/z): 453 [MH]+.
МС (m/z): 458,5 [MH]+.
МС (m/z): 404 [M+1]+.
Пример 2
Синтез типового соединения формулы (IIIb)

Аналитические данные представлены в таблице 2-1 ниже.
Пример 2-1
1-(2,4-Бис-трифторметилфенил)-7-метил-5-(3-тиазол-2-илпиразол-1-ил)-1,2,3,4-тетрагидро[1,8]нафтиридин
К раствору промежуточного соединения 21 (24,8 мг, 0,042 ммоль) в безводном N-метилпирролидиноне (2 мл) добавляют Et3N (12 мкл, 2 экв.) в атмосфере N2. Реакционную смесь облучают микроволнами в течение 10 минут при следующих параметрах: Р=90 Вт; Т=99°С, р=6 ф/д2. Затем в реакционную смесь добавляют насыщенный водный раствор NH4Cl и водную фазу экстрагируют EtOAc. Объединенные органические экстракты промывают насыщенным раствором NH4Cl (3х), сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Продукт очищают на SCX Column (элюенты: CH2Cl2, МеОН и раствор концентрированного NH4OH в МеОН (25%) для элюирования желаемого продукта) с получением 18,4 мг указанного в заголовке соединения в виде белой пены.
МС (m/z): 510 [М+1]+.
Пример 3
Активность связывания CRF
Аффинность связывания CRF определяют in vitro по способности соединений замещать 125I-oCRF и 125I-Sauvagine на CRF1 и CRF2 SPA, соответственно, в рекомбинантных CRF рецепторах человека, экспрессированных в мембранах клеток яичников китайского хомячка (СНО). Для получения мембран клетки СНО из конфлюентных Т-колб собирают в SPA буфер (HEPES/KOH 50 мМ; ЭДТК 2 мМ; MgCl2 10 мМ, рН 7,4) в 50 мл пробирки для центрифуги, гомогенизируют с Polytron и центрифугируют (50000 g в течение 5 минут при температуре 4оС: центрифуга Beckman с ротором JA20). Осадок в пробирке повторно суспендируют, гомогенизируют и центрифугируют при тех же условиях.
Эксперимент SPA проводят в Optiplate добавлением 100 мкл смеси реагента к 1 мкл разбавленного соединения (100% раствор в ДМСО) на ячейку. Смесь для исследования получают смешиванием SPA буфера, шариков WGA SPA (2,5 мг/мл), BSA (1 мг/мл) и мембран (50 и 5 мкг белка/мл для CRF1 и CRF2, соответственно) и 50 пМ радиолиганда.
Планшет инкубируют в течение ночи (>18 часов) при комнатной температуре и считывают с помощью Packard Topcount с WGA-SPA 125I по протоколу подсчета.
Пример 4
Функциональное исследование CRF
Соединения данного изобретения характеризуют в функциональном исследовании для определения их ингибирующего действия. Клетки CRF - СНО человека стимулируют CRF и активацию рецептора оценивают измерением аккумулирования сАМФ.
Клетки СНО из конфлюентных Т-колб повторно суспендируют в культуральной среде без G418 и распределяют в 96-луночном планшете, 25000 клеток/лунка, 100 мкл/лунка и инкубируют в течение ночи. После инкубации среду заменяют 100 мкл сАМФ IBMX буфера, нагретого до температуры 37°С (5 мМ KCl, 5 мМ NaHCO3, 154 мМ NaCl, 5 мМ HEPES, 2,3 мМ CaCl2, 1 мМ MgCl2; 1 г/л глюкозы, рН 7,4, с добавлением 1 мг/мл BSA и 1 мМ IBMX) и 1 мкл разбавленного антагониста в чистом ДМСО. Через дополнительных 10 минут инкубирования при температуре 37°С в планшетном инкубаторе без СО2 добавляют 1 мкл разбавленного агониста в чистом ДМСО. Как и раньше, планшет инкубируют в течение 10 минут и затем содержание сАМФ в клетках измеряют с помощью набора Amersham RPA 538.
Все публикации, включая, но не ограничиваясь ими, патенты и заявки на патенты, указанные в данном описании, даны в качестве ссылок, таким образом, что как если бы каждая отдельная публикация была определенно и индивидуально указана в качестве ссылки во всей своей полноте.
Должно быть понятно, что данное изобретение охватывает все комбинации отдельных и предпочтительных групп, описанных выше.
Заявка, частью которой являются представленное описание и формула изобретения, может быть использована как основание для приоритета в отношении любой последующей заявки. Формула изобретения таких последующих заявок может относиться к любому признаку или сочетанию признаков, описанных выше. Они могут иметь форму пунктов на продукт, композицию, способ или применение, и могут включать, например и без ограничений, следующие пункты формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНДЕНСИРОВАННЫЕ N-ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРОВ CRF | 2004 |
|
RU2382785C2 |
| СПОСОБ ЛЕЧЕНИЯ АЛЛЕРГИЙ С ИСПОЛЬЗОВАНИЕМ ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2001 |
|
RU2290179C2 |
| СПОСОБ ЛЕЧЕНИЯ АЛЛЕРГИИ С ИСПОЛЬЗОВАНИЕМ ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2001 |
|
RU2277909C2 |
| СПОСОБ ЛЕЧЕНИЯ АЛЛЕРГИИ С ИСПОЛЬЗОВАНИЕМ ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2001 |
|
RU2259202C2 |
| ИНГИБИТОРЫ РЕПЛИКАЦИИ ВИРУСА ГРИППА, СПОСОБЫ ИХ ПРИМЕНЕНИЯ И ИСПОЛЬЗОВАНИЕ | 2016 |
|
RU2737190C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ПРИМЕНЕНИЕ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ И СПОСОБ ИНГИБИРОВАНИЯ АКТИВНОСТИ КАТЕПСИНА S | 2001 |
|
RU2317988C2 |
| БИАРИЛЬНЫЕ МОНОБАКТАМНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2016 |
|
RU2746129C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ТЕТРАЗОЛА И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ТУБЕРКУЛЕЗА | 2018 |
|
RU2800930C2 |
| МОДУЛЯТОРЫ ПИРУВАТКИНАЗЫ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2797518C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ IRAK И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2743360C2 |
Данное изобретение относится к соединению формулы (I), включая их фармацевтически приемлемые соли или сольваты
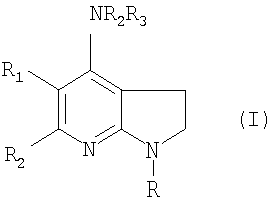
где R является фенилом или пиридинилом, каждый из которых может быть замещен 1-4 группами, выбранными из: галогена, C1-С6алкила, C1-С6алкокси, галоС1-С6алкила, галоС1-С6алкокси, нитро, -NR6R7, циано и группы R8; R1 является водородом или С1-С6алкилом; R2 и R3 вместе с N образуют группу:

R4 является водородом; R6 является водородом или C1-С6алкилом; R7 является водородом или C1-С6алкилом; R8 представляет собой 5-6-членный гетероцикл, который является насыщенным или ароматическим, и который содержит 1-2 гетероатома, независимо выбранных из азота, кислорода и серы. Также описываются способы получения, фармацевтические композиции, содержащие эти соединения, их применение при лечении состояний, медиированных рилизинг-фактором кортикотропина (CRF), и способ лечения млекопитающих с их использованием. Технический результат - получены новые соединения, обладающие полезными биологическими свойствами. 6 н. и 16 з.п. ф-лы, 1 табл.
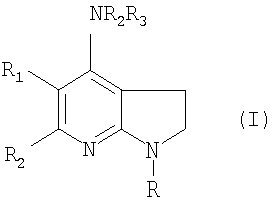
где R является фенилом или пиридинилом, каждый из которых может быть замещен 1-4 группами, выбранными из
галогена, C1-С6алкила, С1-С6алкокси, галоС1-С6алкила, галоС1-С6алкокси, нитро, -NR6R7, циано и группы R8;
R1 является водородом или C1-С6алкилом;
R2 и R3 вместе с N образуют группу

R4 является водородом;
R6 является водородом или C1-С6алкилом;
R7 является водородом или C1-С6алкилом;
R8 представляет собой 5-6-членный гетероцикл, который является насыщенным или ароматическим и который содержит 1-2 гетероатома, независимо выбранных из азота, кислорода и серы.
R является группой, выбранной из 2,4-дихлорфенила,
2-хлор-4-метилфенила, 2-хлор-4-трифторметилфенила,
2-хлор-4-метоксифенила, 2,4,5-триметилфенила, 2,4-диметилфенила,
2-метил-4-метоксифенила, 2-метил-4-хлорфенила,
2-метил-4-трифторметилфенила, 2,4-диметоксифенила,
2-метокси-4-трифторметилфенила, 2-метокси-4-хлорфенила,
3-метокси-4-хлорфенила, 2,5-диметокси-4-хлорфенила,
2-метокси-4-изопропилфенила, 2-метокси-4-трифторметилфенила,
2-метокси-4-изопропилфенила, 2-метокси-4-метилфенила,
2-трифторметил-4-хлорфенила, 2,4-трифторметилфенила,
2-трифторметил-4-метилфенила,
2-трифторметил-4-метоксифенила,
2-бром-4-изопропилфенила, 2-метил-4-цианофенила,
2-хлор-4-цианофенила, 4-метил-6-диметиламинопиридин-3-ила,
3,5-дихлорпиридин-2-ила, 2,6-бисметоксипиридин-3-ила и
3-хлор-5-трихлорметилпиридин-2-ила.
Схема 1

в которой стадия а включает превращение уходящей группы L, выбранной из группы, включающей галоген или реакционноспособный остаток сульфоновой кислоты (например, мезилат, тозилат), предпочтительно хлорид, в аминогруппу соединений (III) взаимодействием с подходящим амином NR2R3 в основных условиях;
стадия b включает восстановление сложноэфирной группы подходящим восстанавливающим агентом (таким как DIBAl-H) до гидроксигруппы соединений (IV);
стадия с включает окисление гидроксигруппы подходящим окисляющим агентом (таким как периодинан Десса-Мартина) до альдегидной группы соединений (V);
стадия d включает образование альдегидной группы соединений (VII) по реакции Виттига в обычных условиях через образование енольного эфира с последующим кислотным гидролизом (стадия е);
стадия f включает восстановление альдегидной группы подходящим восстанавливающим агентом (таким как NaBH4) до гидроксигруппы соединений (VIII);
стадия g включает превращение гидроксигруппы в подходящую защитную группу соединений (IX) (такую как TBS: трет-бутилдиметилсилил);
стадия h включает реакцию Бухвальда посредством сочетания с подходящим амином RNH2;
стадия i включает реакцию снятия защиты с получением гидроксильной группы соединений (XI);
стадия l включает внутримолекулярную циклизацию нагреванием после превращения гидроксигруппы соединений (XI) в подходящую уходящую группу (такую как бромид, взаимодействием с CBr4 и PPh3) с получением конечных соединений (I).
Схема 2
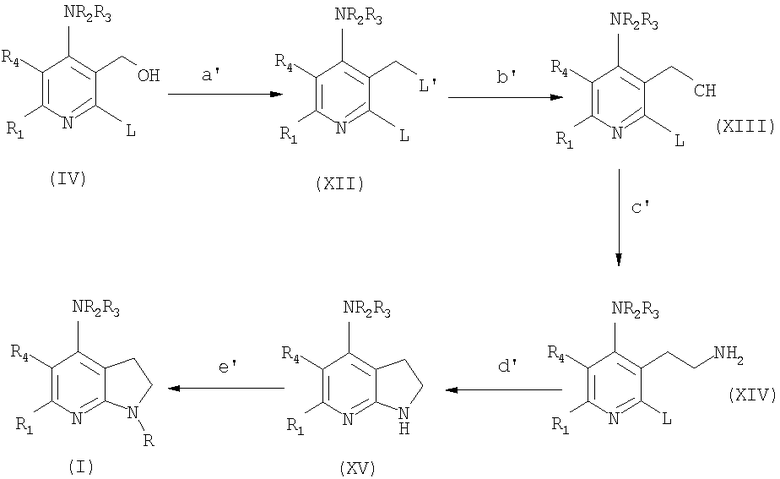
в которой стадия а' включает превращение гидроксигруппы в подходящую уходящую группу L' соединений (XII), которая, независимо от L, имеет те же определения (например, мезилат, взаимодействием с MsCl в Et3N);
стадия b' включает превращение L' в циано производное соединений (XIII) взаимодействием, например, с KCN в апротонном диполярном растворителе, таком как ДМФА;
стадия с' включает восстановление цианогруппы подходящим восстанавливающим агентом (таким как ВН3-ТГФ) до аминогруппы соединения (XIV);
стадия d' включает внутримолекулярную циклизацию соединения (XIV) нагреванием в подходящем растворителе (таком как NMP) при высокой температуре;
стадия е' соответсвует стадии h схемы по п.11.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Способ получения производных пиперазинилбензогетероциклических соединений или их кислотно-аддитивных солей | 1979 |
|
SU993818A3 |

