Область техники
Настоящее изобретение относится к соединениям класса 2-тиабензоазуленов, к их фармакологически приемлемым солям и сольватам, к способам и промежуточным продуктам для их получения, а также к их противовоспалительным эффектам, в особенности к ингибированию продуцирования фактора некроза опухоли-(ФНО-) и к ингибированию продуцирования интерлейкина-1 (ИЛ-1), а также к их болеутоляющему действию.
Предшествующий уровень
К настоящему времени в литературе были раскрыты производные 1-тиадибензоазуленов, замещенные в положении 2 метилом, метилкетоном, нитрогруппой или карбоксильной группой (Cagniant PG, C.R. Hebd. Sceances Acad. Sci., 1976, 283:683-686). Некоторые производные 1,3-диазадибензоазуленов и их соли известны как новый класс соединений, обладающих противовоспалительным действием (патенты США № 3711489, 4198421 и Канады № 967573). Производные 1-тиадибензоазуленов, имеющие алкилоксизаместители в положении 2 (WO 01/878990), также обладают сильным противовоспалительным действием. Однако в соответствии с имеющимися у авторов сведениями и доступными литературными данными соединения класса 2-тиабензоазуленов до настоящего времени в целом не были известны, поэтому не были получены производные данной структуры и не было известно их противовоспалительное действие в качестве ингибиторов секреции ФНО-и ингибиторов ИЛ-1 или их болеутоляющее действие.
В 1975 г. ФНО- был определен как сывороточный фактор, индуцированный эндотоксином и вызывающий некроз опухоли in vitro и in vivo (Carswell EA et al., Proc. Natl. Acad. Sci. USA, 1975, 72:3666-3670). Кроме противоопухолевого действия ФНО- обладает также другими многочисленными биологическими действиями, являющимися важными в гомеостазе организмов и при патофизиологических состояниях. Основные источники ФНО- представляют собой моноциты-макрофаги, Т-лимфоциты и лаброциты.
Обнаружение того, что антитела анти-ФНО-(сA2) обладают действием при лечении больных ревматоидным артритом (РА) (Elliott M et al., Lancet, 1994, 344:1105-1110), привело к повышенной заинтересованности в нахождении новых ингибиторов ФНО- как возможных сильнодействующих лекарственных средств для РА. Ревматоидный артрит является аутоиммунным хроническим воспалительным заболеванием, характеризующимся необратимыми патологическими изменениями в суставах. Кроме использования при РА, антагонисты ФНО- могут быть использованы также при многочисленных патологических состояниях и заболеваниях, таких как спондилит, остеоартрит, подагра и другие артритные состояния, сепсис, септический шок, токсический шок, атопический дерматит, контактный дерматит, псориаз, гломерулонефрит, красная волчанка, склеродермия, астма, кахексия, хроническое обструктивное заболевание легких, остановка сердца, инсулинорезистентность, фиброз легких, рассеянный склероз, болезнь Крона, язвенный колит, вирусные инфекции и AIDS (СПИД).
Доказательство биологической важности ФНО- было получено в испытаниях in vivo, осуществляемых на мышах, в которых инактивировали гены мышей, отвечающие за ФНО- или его рецептор. Такие животные были резистентны к вызванному коллагеном артриту (Mori L et al., J. Immunol., 1996, 157:3178-3182) и к вызванному эндотоксином шоку (Pfeffer K. et al., Cell, 1993, 73:457-467). Когда в испытаниях на животных уровень ФНО- повышался, проявлялся хронический воспалительный полиартрит (Georgopoulos S et al., J. Inflamm., 1996, 46:86-97; Keffer J. et al., EMBO J., 1991, 10:4025-4031) и его патологическая картина смягчалась ингибиторами продуцирования ФНО-. Лечение таких воспалительных и патологических состояний обычно включает употребление нестероидных противовоспалительных лекарственных средств, а в более тяжелых случаях вводят соли золота, D-пенициллинамин или метотрексат. Данные лекарственные средства действуют симптоматически, но они не останавливают патологический процесс. Новые подходы в терапии ревматоидного артрита основаны на лекарственных средствах, таких как тенидап, лефлуномид, циклоспорин, FK-506, и на биомолекулах, нейтрализующих действие ФНО-. В настоящее время имеются коммерчески доступный этанерсепт (Enbrel, Immunex/Wyeth), слитый белок растворимого рецептора ФНО-, и инфликсимаб (Remicade, Centocor) - химерное моноклональное антитело человека и мыши. Кроме использования в терапии РА, этанерсепт и инфликсимаб зарегистрированы, так же как лекарственные средства для терапии болезни Крона (Exp. Opin. Invest. Drugs, 2000, 9:103).
Кроме ингибирования секреции ФНО-, в терапии РА очень важно также ингибирование секреции ИЛ-1, поскольку ИЛ-1 является важным цитокином в регуляции и иммунорегуляции клеток, а также в патофизиологических состояниях, таких как воспаление (Dinarello CA et al., Rev. Infect. Disease, 1984, 6:51). Хорошо известные виды биологической активности ИЛ-1 представляют собой: активацию Т-лимфоцитов, индуцирование повышенной температуры, стимуляцию секреции простагландина или коллагеназы, хемотаксис нейтрофилов и уменьшение уровня железа в плазме (Dinarello CA, J. Clinical Immunology, 1985, 5:287). Два рецептора, с которыми может быть связан ИЛ-1, хорошо известны: ИЛ-1RI и ИЛ-1RII. В то время как ИЛ-1RI передает сигнал внутриклеточно, ИЛ-1RII расположен на поверхности клеток и не передает сигнал внутрь клеток. Так как ИЛ-1RII связывает ИЛ-1, а также ИЛ-1RI, он может действовать как отрицательный регулятор действия ИЛ-1. Кроме указанного механизма регуляции передачи сигнала, в клетках присутствует другой природный антагонист рецептора ИЛ-1 (ИЛ-1ra). Данный белок связывается с ИЛ-1RI, но не передает какой-либо сигнал. Однако его действенность в прекращении передачи сигнала является невысокой и его концентрация для достижения разрыва в передаче сигнала должна быть в 500 раз больше концентрации ИЛ-1. Рекомбинантный человеческий ИЛ-1ra (Amgen) был клинически испытан (Bresnihan B et al., Arthrit. Rheum., 1996, 39:73) и полученные результаты показали улучшение клинической картины у 472 больных РА по сравнению с плацебо. Полученные результаты показывают важность ингибирования действия ИЛ-1 в лечении заболеваний, таких как РА, при которых нарушено продуцирование ИЛ-1. Поскольку существует синергическое действие ФНО- и ИЛ-1, 2-тиадибензоазулены могут быть использованы при лечении состояний и заболеваний, связанных с повышенной секрецией ФНО- и ИЛ-1.
Сущность изобретения
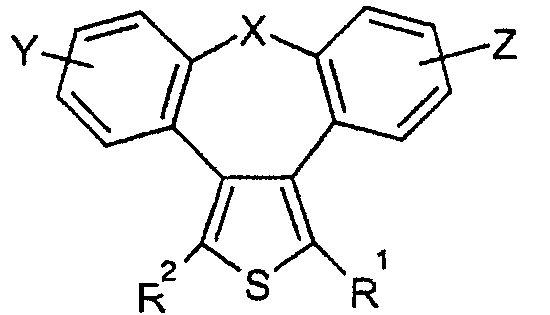
Настоящее изобретение относится к 2-тиадибензоазуленам формулы I
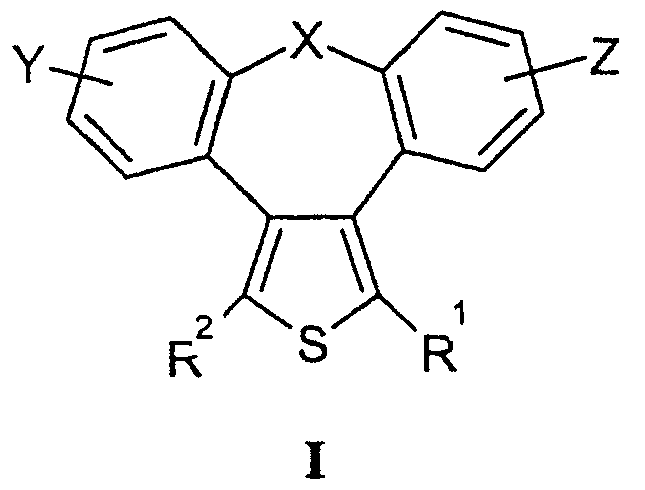
где
Х может представлять собой СН2 или гетероатом, такой как О, S, S(=O), S(=O)2 или NRa, где Ra представляет собой водород или защитную группу;
Y и Z независимо друг от друга означают один или несколько идентичных или различных заместителей, связанных с любым доступным атомом углерода, и могут представлять собой галоген, С1-С4-алкил, С2-С4-алкенил, С2-С4-алкинил, трифторметил, галоген-С1-С4-алкил, гидрокси, С1-С4-алкокси, трифторметокси, С1-С4-алканоил, амино, амино-С1-С4-алкил, С1-С4-алкиламино, N-(С1-С4-алкил)амино, N,N-ди(С1-С4-алкил)амино, тиол, С1-С4-алкилтио, сульфонил, С1-С4-алкилсульфонил, сульфинил, С1-С4-алкилсульфинил, карбокси, С1-С4-алкоксикарбонил, нитро;
R1 может представлять собой водород, галоген, необязательно замещенный С1-С7-алкил или С2-С7-алкенил, С2-С7-алкинил, необязательно замещенный арил или гетероарил и гетероцикл, гидрокси, гидрокси-С2-С7-алкенил, гидрокси-С2-С7-алкинил, С1-С7-алкокси, тиол, тио-С2-С7-алкенил, тио-С2-С7-алкинил, С1-С7-алкилтио, амино, N-(С1-С7-алкил)амино, N,N-ди(С1-С7-алкил)амино, С1-С7-алкиламино, амино-С2-С7-алкенил, амино-С2-С7-алкинил, амино-С1-С7-алкокси, С1-С7-алканоил, ароил, оксо-С1-С7-алкил, С1-С7-алканоилокси, карбокси, необязательно замещенный С1-С7-алкилоксикарбонил или арилоксикарбонил, карбамоил, N-(С1-С7-алкил)карбамоил, N,N-ди(С1-С7-алкил)карбамоил, циано, циано-С1-С7-алкил, сульфонил, С1-С7-алкилсульфонил, сульфинил, С1-С7-алкилсульфинил, нитро,
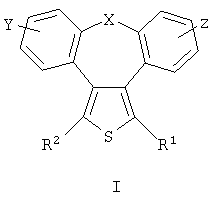
или заместитель формулы II
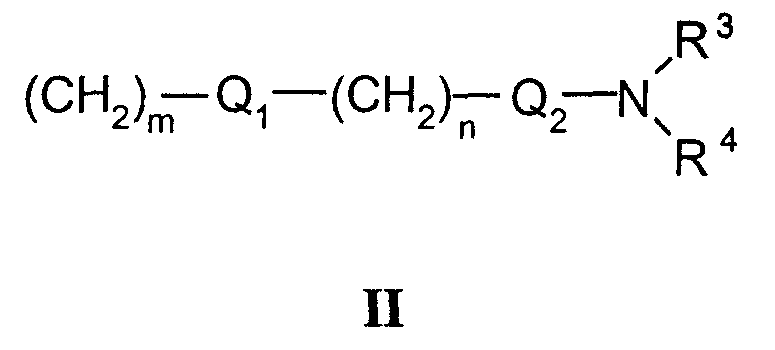
где
R3 и R4 одновременно или независимо друг от друга могут представлять собой водород, C1-C4-алкил, арил или вместе с N означают необязательно замещенный гетероцикл или гетероарил;
m и n представляют собой целое число от 0 до 3;
Q1 и Q2 независимо друг от друга представляют собой кислород, серу или группы:
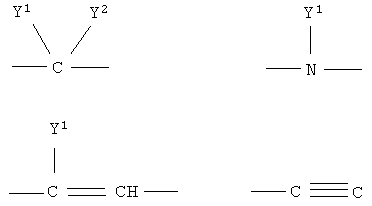
в которых заместители
Y1 и Y2 независимо друг от друга могут представлять собой водород, галоген, необязательно замещенный C1-C4-алкил или арил, гидрокси, C1-C4-алкокси, C1-C4-алканоил, тиол, C1-C4-алкилтио, сульфонил, C1-C4-алкилсульфонил, сульфинил, C1-C4-алкилсульфинил, циано, нитро или вместе образуют карбонильную или иминогруппу;
R2 может представлять собой водород, карбокси или алкилоксикарбонил;
а также к их фармакологически приемлемым солям и сольватам.
Термин "гало", "гал" или "галоген" относится к атому галогена, который может представлять собой фтор, хлор, бром или иод.
Термин "алкил" относится к алкильным группам со значением алканов, из которых произведены радикалы, которые могут быть прямыми, разветвленными или циклическими или могут представлять собой комбинацию прямого и циклического радикала и разветвленного и циклического радикала. Предпочтительными прямыми или разветвленными алкилами являются, например, метил, этил, пропил, изопропил, бутил, втор-бутил и трет-бутил. Предпочтительными циклическими алкилами являются, например, циклопентил или циклогексил.
Термин "галогеналкил" относится к алкильным группам, которые должны быть замещены, по меньшей мере, одним атомом галогена. Наиболее часто галогеналкилы представляют собой, например, хлорметил, дихлорметил, трифторметил или 1,2-дихлорпропил.
Термин "алкенил" относится к алкенильным группам, имеющим значение углеводородных радикалов, которые могут быть прямыми, разветвленными или циклическими или могут представлять собой комбинацию прямого и циклического радикала или разветвленного и циклического радикала, но имеющим, при этом, по меньшей мере, одну углерод-углеродную двойную связь. Наиболее часто алкенилы представляют собой этенил, пропенил, бутенил или циклогексенил.
Термин "алкинил" относится к алкинильным группам, имеющим значение углеводородных радикалов, которые являются прямыми или разветвленными и содержат, по меньшей мере, одну и не более чем две углерод-углеродные тройные связи. Наиболее часто алкинилы представляют собой, например, этинил, пропинил или бутинил.
Термин "алкокси" относится к прямым или разветвленным цепям алкоксильной группы. Примерами таких групп являются метокси, пропокси, пропил-2-окси, бутокси, бутил-2-окси или метилпропил-2-окси.
Термин "арил" относится к группам, имеющим значение ароматического кольца, например к фенилу, а также к конденсированным ароматическим кольцам. Арил содержит одно кольцо, по меньшей мере, с 6 атомами углерода или два кольца, содержащих в целом 10 атомов углерода, при этом кольца имеют чередующиеся двойные (резонансные) связи между атомами углерода. Наиболее часто используемые арилы представляют собой, например, фенил или нафтил. Арильные группы могут быть обычно связаны с остальной молекулой любым доступным атомом углерода через простую связь или через С1-С4-алкиленовую группу, такую как метилен или этилен.
Термин "гетероарил" относится к группам, имеющим значение ароматической и частично ароматической группы моноциклического или бициклического кольца с 4-12 атомами, по меньшей мере, один из которых представляет собой гетероатом, такой как O, S или N, и доступный атом азота или атом углерода является местом связывания группы с остальной молекулой или через простую связь или через определенную выше С1-С4-алкиленовую группу. Примерами групп указанного типа являются тиофенил, пирролил, имидазолил, пиридинил, оксазолил, тиазолил, пиразолил, тетразолил, пиримидинил, пиразинил, хинолинил или триазинил.
Термин "гетероцикл" относится к пятичленным или шестичленным полностью насыщенным или частично ненасыщенным гетероциклическим группам, содержащим, по меньшей мере, один гетероатом, такой как O, S или N, и доступный атом азота или атом углерода является местом связывания группы с остальной молекулой или через простую связь или через определенную выше С1-С4-алкиленовую группу. Наиболее часто встречающимися примерами таких групп являются морфолинил, пиперидинил, пиперазинил, пирролидинил, пиразинил или имидазолил.
Термин "алканоильная" группа относится к прямым цепям ацильной группы, такой как формильная, ацетильная или пропаноильная.
Термин "ароильная" группа относится к ароматическим ацильным группам, таким как бензоильная.
Термин "необязательно замещенный алкил" относится к алкильным группам, которые могут быть необязательно дополнительно замещены одним, двумя, тремя или более заместителями. Такие заместители могут представлять собой атом галогена (предпочтительно фтор или хлор), гидрокси, С1-С4-алкокси (предпочтительно метокси или этокси), тиол, С1-С4-алкилтио (предпочтительно метилтио или этилтио), амино, N-(C1-C4)алкиламино (предпочтительно N-метиламино или N-этиламино), N,N-ди(С1-С4-алкил)амино (предпочтительно диметиламино или диэтиламино), сульфонил, С1-С4-алкилсульфонил (предпочтительно метилсульфонил или этилсульфонил), сульфинил, С1-С4-алкилсульфинил (предпочтительно метилсульфинил).
Термин "необязательно замещенный алкенил" относится к алкенильным группам, необязательно дополнительно замещенным одним, двумя или тремя атомами галогена. Такие заместители могут представлять собой, например, 2-хлорэтенил, 1,2-дихлорэтенил или 2-бромпропен-1-ил.
Термин "необязательно замещенный арил, гетероарил или гетероцикл" относится к арильной, гетероарильной или гетероциклической группам, которые могут быть необязательно дополнительно замещены одним или двумя заместителями. Заместители могут представлять собой галоген (предпочтительно хлор или фтор), С1-С4-алкил (предпочтительно метил, этил или изопропил), циано, нитро, гидрокси, С1-С4-алкокси (предпочтительно метокси или этокси), тиол, С1-С4-алкилтио (предпочтительно метилтио или этилтио), амино, N-(C1-C4)алкиламино (предпочтительно N-метиламино или N-этиламино), N,N-ди(С1-С4-алкил)амино (предпочтительно N,N-диметиламино или N,N-диэтиламино), сульфонил, С1-С4-алкилсульфонил (предпочтительно метилсульфонил или этилсульфонил), сульфинил, С1-С4-алкилсульфинил (предпочтительно метилсульфинил).
Когда Х означает NRa и Ra означает защитную группу, тогда Ra относится к таким группам, как алкильная (предпочтительно метильная или этильная), алканоильная (предпочтительно ацетильная), алкоксикарбонильная (предпочтительно метоксикарбонильная или трет-бутоксикарбонильная), арилметоксикарбонильная (предпочтительно бензилоксикарбонильная), ароильная (предпочтительно бензоильная), арилалкильная (предпочтительно бензильная), алкилсилильная (предпочтительно триметилсилильная) или алкилсилилалкоксиалкильная (предпочтительно триметилсилилэтоксиметильная).
Когда R2 и R3 вместе с N означают гетероарил или гетероцикл, это означает, что такие гетероарилы или гетероциклы имеют, по меньшей мере, один атом углерода, замещенный атомом азота через группы, которые связаны с остальной молекулой. Примерами таких групп являются морфолин-4-ил, пиперидин-1-ил, пирролидин-1-ил, имидазол-1-ил или пиперазин-1-ил.
Термин "фармацевтически приемлемые соли" относится к солям соединений формулы I и включает, например, соли с С1-С4-алкилгалогенидами (предпочтительно с метилбромидом, метилхлоридом) (соли четвертичного аммония), с неорганическими кислотами (такими как хлористоводородная, бромистоводородная, фосфорная, метафосфорная, азотная или серная кислота) или с органическими кислотами (такими как винная, уксусная, лимонная, малеиновая, молочная, фумаровая, бензойная, янтарная, метансульфоновая или п-толуолсульфоновая кислота).
Некоторые соединения формулы I могут образовывать соли с органическими или неорганическими кислотами или основаниями и они также включены в настоящее изобретение.
Предметом настоящего изобретения являются также сольваты (наиболее часто гидраты), которые могут образовывать соединения формулы I или их соли.
В зависимости от природы конкретных заместителей соединения формулы I могут иметь геометрические изомеры и один или несколько хиральных центров, так что могут существовать энантиомеры или диастереоизомеры. Настоящее изобретение относится также к таким изомерам и их смесям, включающим рацематы.
Настоящее изобретение относится также ко всем возможным таутомерным формам конкретных соединений формулы I.
Следующая цель настоящего изобретения состоит в получении соединений формулы I в соответствии со способами, включающими:
а) для получения соединений формулы I, где R1 и R2 независимо друг от друга представляют собой карбоксильную группу, С1-С6-алкилоксикарбонил, арилоксикарбонил или арилалкилоксикарбонил, циклизацию -дикетонов формулы III
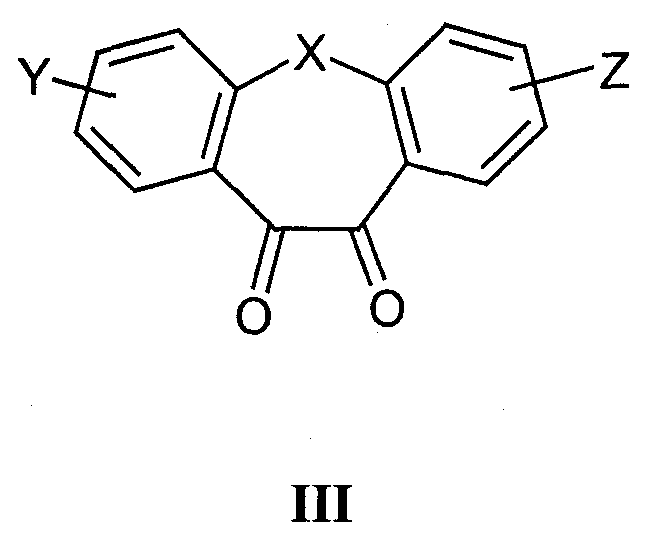
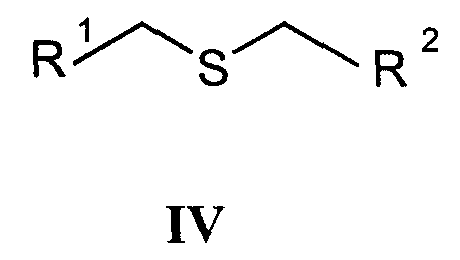
с соединениями формулы IV
b) для получения соединений формулы I, где Q1 означает -О-,
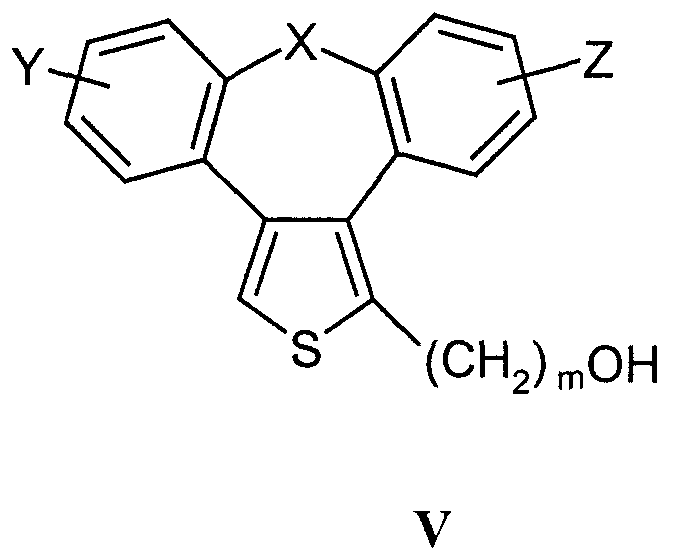
взаимодействие спиртов формулы V
с соединениями формулы VI
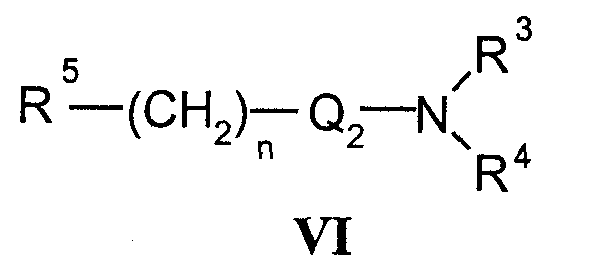
где R5 означает удаляемую группу;
с) для получения соединений формулы I, где Q1 означает -О-, -NH-, -S- или -С С-,
С-,
взаимодействие соединений формулы Va
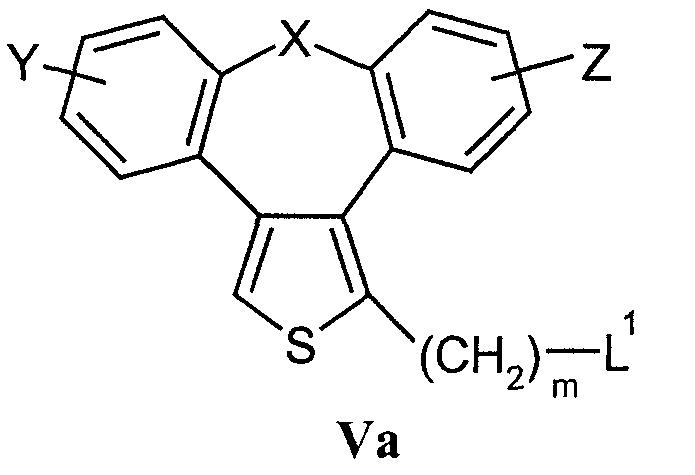
где L1 означает удаляемую группу,
с соединениями формулы VIa
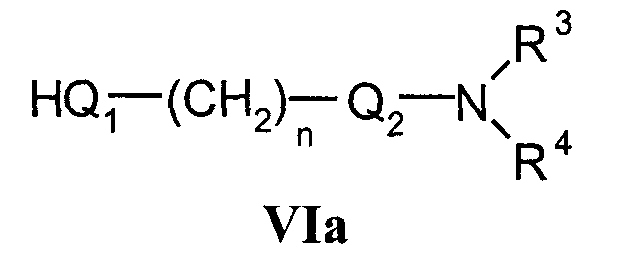
d) для получения соединений формулы I, где Q1 означает -О-, -NH- или -S-,
взаимодействие соединений формулы Vb
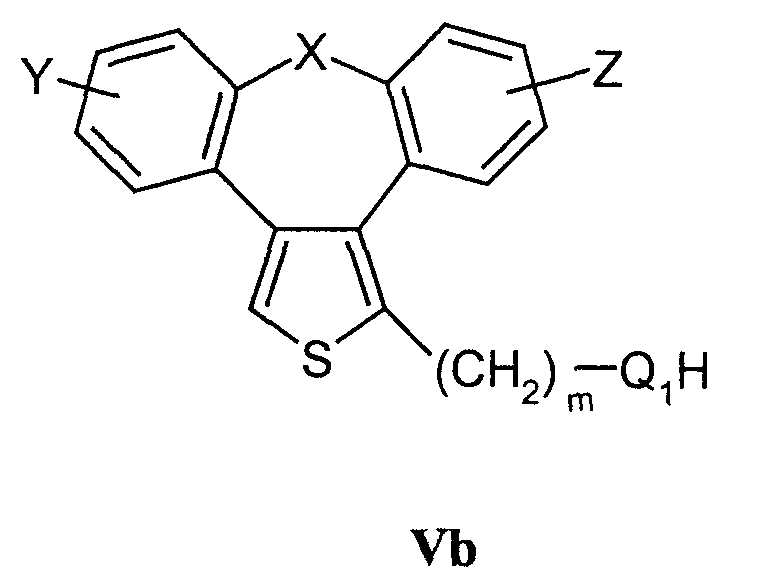
с соединениями формулы VI, где R5 означает удаляемую группу;
е) для получения соединений формулы I, где Q1 означает -С=С-,
взаимодействие соединений формулы Vb, где Q1 означает карбонил, с фосфористыми илидами.
Способы получения:
а) Циклизацию -дикетонов формулы III и соединений формулы IV, где R1 и R2 одновременно или независимо друг от друга представляют собой С1-С6-алкилоксикарбонил, арилоксикарбонил или арилалкилоксикарбонил, осуществляют способами, раскрытыми для получения аналогичных соединений (Chadwick DJ et al., J. Chem. Soc. Perkin Trans. I., 1972, 2079-81). Реакцию циклизации осуществляют в спиртах (наиболее часто в трет-бутаноле) в присутствии алкоголятов (предпочтительно в присутствии трет-бутилата калия).
Исходные соединения для данной реакции уже известны или их получают способами, раскрытыми для получения аналогичных соединений: получение -дикетонов формулы III раскрыто, например, в: Leonard N.J. et al., J.Am. Chem. Soc., 1995, 77:5078, патенте США №3711489 или в: Lombardino J.G., J. Heterocyclic Chem., 1974, 11:17-21; или получение простых тиоэфиров формулы IV раскрыто, например, в: Overberger C.G. et al., J. Am. Chem. Soc., 1950, 72:4958-61. Полученные таким образом соединения могут быть очищены, выделены и охарактеризованы или могут быть подвергнуты последующему превращению без выделения.
b) Соединения формулы I в соответствии с настоящим способом могут быть получены взаимодействием спиртов формулы V и соединений формулы VI, где R5 означает удаляемую группу, которая может представлять собой атом галогена (наиболее часто бром, иод или хлор) или сульфонилоксигруппу (наиболее часто трифторметилсульфонилокси или п-толуолсульфонилокси). Реакция конденсации может быть осуществлена в соответствии со способами, раскрытыми для получения аналогичных соединений (Menozzi G et al., J. Heterocyclic Chem., 1997, 34:963-968 или WO 01/87890). Реакцию осуществляют при температуре от 20 до 100°С в течение времени от 1 до 24-х часов в двухфазной системе (предпочтительно с использованием смеси 50% NaOH/толуол) в присутствии межфазного катализатора (предпочтительно бензилтриэтиламмонийхлорида, бензилтриэтиламмонийбромида, цетилтриметилбромида). После обработки реакционной смеси образованные продукты выделяют перекристаллизацией или хроматографией на колонке с силикагелем.
Исходные соединения для получения спиртов формулы V представляют собой соединения формулы I, в которой R1 и R2 независимо друг от друга означают карбоксильную или сложноэфирную группу (этилоксикарбонил, метилоксикарбонил), которые при декарбоксилировании дают соединения формулы I, где R2 означает водород и R1 означает сложноэфирную группу, которые при восстановлении дают спирты формулы V. Декарбоксилирование осуществляют пиролизом при 250-300°С в присутствии металлов, предпочтительно меди. Реакцию восстановления осуществляют с использованием гидридов металлов, таких как алюмогидрид лития или боргидрид натрия. Кроме того, спирты формулы V могут быть получены гидролизом соответствующих сложных эфиров (в щелочной или кислой среде).
Исходные соединения формулы VI уже известны или их получают в соответствии со способами, раскрытыми для получения аналогичных соединений.
с) Соединения формулы I в соответствии с настоящим способом могут быть получены взаимодействием соединений формулы Va, в которой L1 означает удаляемую группу, определенную выше для R5, и соединений формулы VIa, в которой Q1 означает кислород, азот, серу или -С С-. Наиболее подходящие реакции конденсации представляют собой реакции нуклеофильного замещения при насыщенном атоме углерода, раскрытые в литературе.
С-. Наиболее подходящие реакции конденсации представляют собой реакции нуклеофильного замещения при насыщенном атоме углерода, раскрытые в литературе.
Исходные соединения формулы Va (наиболее часто галогениды) могут быть получены галогенированием (например, бромированием или хлорированием) спиртов формулы V обычными галогенирующими агентами (например, бромистоводородной кислотой, PBr3, SOCl2 или PCl5) способами, раскрытыми в литературе. Полученные соединения могут быть выделены или могут быть использованы без выделения в качестве промежуточных продуктов, подходящих для получения соединений формулы I.
Исходные соединения формулы VIa уже известны или их получают в соответствии со способами, раскрытыми для получения аналогичных соединений.
d) Соединения формулы I, в которой Q означает гетероатом -О-, -NH- или -S-, могут быть получены конденсацией соединений формулы Vb и соединений формулы VI, в которой R5 означает удаляемую группу, определенную выше. Реакция может быть осуществлена в условиях реакции, раскрытых в способе b), или в условиях реакций нуклеофильного замещения, раскрытых в литературе. Исходные спирты, амины и тиолы могут быть получены взаимодействием воды, аммиака или сероводорода с соединениями Va в соответствии со способами, раскрытыми в литературе.
е) Спирты структурной формулы V могут быть окислены в соответствующие соединения формулы Vb, где Q1 означает карбонил, в результате последующего взаимодействия которых с соответствующими илидными реагентами происходит удлинение цепи и образование алкенильного заместителя с карбонильной или сложноэфирной группой, как раскрыто в заявке на патент Хорватии № 20000310.
Соединения формулы I, кроме вышеуказанных реакций, могут быть получены превращением других соединений формулы I, и следует понимать, что настоящее изобретение также включает такие соединения и способы. Специальный пример изменения функциональной группы представляет реакция альдегидной группы с выбранными фосфористыми илидами, приводящая к удлинению цепи и образованию алкенильного заместителя с карбонильной или сложноэфирной группой, которая раскрыта в заявке на патент Хорватии № 20000310. Указанные реакции осуществляют в растворителях, таких как бензол, толуол или гексан, при повышенной температуре (наиболее часто при температуре кипения).
Соединения формулы I, в которой Q1 представляет собой -СС-, получают взаимодействием соединений формулы Va с 1-алкином в щелочной среде (такой как амид натрия в аммиаке). Условия реакции данного способа раскрыты в литературе. При подобных условиях реакции (нуклеофильного замещения) могут быть получены различные производные простого эфира, простого тиоэфира и амина.
Формилирование соединений формулы I такими способами, как, например, ацилирование по Вильсмайеру или реакция н-BuLi и диметилформамида, является дополнительным общим примером превращения. Условия реакций в данных способах хорошо известны в литературе.
В результате гидролиза соединений формулы I, имеющих нитрильные, амидные или сложноэфирные группы, могут быть получены соединения с карбоксильной группой, которые являются подходящими промежуточными продуктами для получения других соединений с новыми функциональными группами, таких как, например, сложные эфиры, амиды, галогениды, ангидриды, спирты или амины.
Реакции окисления или восстановления являются дополнительной возможностью изменения заместителей в соединениях формулы I. Наиболее часто используемые окислители представляют собой пероксиды (пероксид водорода, м-хлорпербензойная кислота или бензоилпероксид) или ионы перманганата, хромата или перхлората. Таким образом, в результате, например, окисления спиртовой группы пиридинилдихроматом или пиридинилхлорхроматом образуется альдегидная группа, которая может быть превращена в карбоксильную группу дополнительным окислением. Окислением соединений формулы I, в которой R1 означает алкил, тетраацетатом свинца в уксусной кислоте или N-бромсукцинимидом с использованием каталитического количества бензоилпероксида получают соответствующее карбонильное производное.
Селективным окислением алкилтиогруппы могут быть получены алкилсульфинильная или алкилсульфонильная группы.
В результате восстановления соединений нитрогруппой возможно получение аминосоединений. Реакцию осуществляют в обычных условиях каталитического гидрирования или электрохимически. Каталитическим гидрированием с использованием палладия на угле алкенильные заместители могут быть превращены в алкильные или нитрильная группа может быть превращена в аминоалкил.
В соединения формулы I стандартными реакциями замещения или обычными заменами отдельных функциональных групп могут быть введены различные заместители ароматической структуры. Примерами таких реакций являются ароматическое замещение, алкилирование, галогенирование, гидроксилирование, а также окисление или восстановление заместителей. Реагенты и условия реакций известны из литературы. Так например, ароматическим замещением в присутствии концентрированной азотной кислоты и серной кислоты вводится нитрогруппа. С использованием ацилгалогенидов или алкилгалогенидов возможно введение ацильной группы или алкильной группы. Реакцию осуществляют в присутствии кислот Льюиса, таких как трихлорид алюминия или железа, в условиях реакции Фриделя-Крафтса. Восстановлением нитрогруппы получают аминогруппу, которую с помощью реакции диазотирования превращают в подходящую исходную группу, которая может быть замещена одной из следующих групп: Н, CN, OH, галоген.
Для предотвращения нежелательного взаимодействия в химических реакциях часто необходимо защитить определенные группы, такие как, например, гидрокси, амино, тио или карбокси. Для этой цели может быть использовано большое число защитных групп [Green T. W, Wuts PGH, Protective Groups in Organic Synthesis, John Wiley and Sons, 1999] и их выбор, использование и удаление осуществляют с применением методов, общепринятых в химическом синтезе.
Традиционной защитой для амино- или алкиламиногрупп являются, например, такие группы как алканоильная (ацетильная), алкоксикарбонильная (метоксикарбонильная, этоксикарбонильная или трет-бутоксикарбонильная); арилметоксикарбонильная (бензилоксикарбонильная), ароильная (бензоильная) или алкилсилильная (триметилсилильная или триметилсилилэтоксиметильная) группы. Условия удаления защитной группы зависят от выбора и свойств этой группы. Так, например, ацильные группы, такие как алканоильная, алкоксикарбонильная или ароильная, могут быть удалены гидролизом в присутствии основания (гидроксида натрия или гидроксида калия), трет-бутоксикарбонильная или алкилсилильная (триметилсилильная) группы могут быть удалены обработкой подходящей кислотой (хлористоводородной, серной, фосфорной или трифторуксусной кислотой), тогда как арилметоксикарбонильная группа (бензилоксикарбонильная) может быть удалена гидрированием с использованием катализатора, такого как палладий на угле.
Соли соединений формулы I могут быть получены общеизвестными способами, например, такими как взаимодействие соединений формулы I с соответствующим основанием или кислотой в подходящем растворителе или растворяющей смеси, например в простых эфирах (диэтиловый эфир) или спиртах (этанол, пропанол или изопропанол).
Другой целью настоящего изобретения является применение заявленных соединений в терапии воспалительных заболеваний и состояний, в особенности всех заболеваний и состояний, вызванных избыточной секрецией ФНО- и ИЛ-1.
Ингибиторы продуцирования цитокинов или медиаторов воспаления, являющиеся объектом настоящего изобретения, или их фармацевтически приемлемые соли могут быть использованы в производстве лекарственных средств для лечения и профилактики патологического состояния или заболевания, вызванного избыточным нерегулированным продуцированием цитокинов или медиаторов воспаления, при этом лекарственные средства должны содержать эффективную дозу данных ингибиторов.
Настоящее изобретение в особенности относится к эффективной дозе ингибитора ФНО-, которая может быть определена обычными методами.
Настоящее изобретение относится также к лекарственному препарату, содержащему эффективные нетоксичные дозы соединений настоящего изобретения и фармацевтически приемлемые носители или растворители.
Приготовление лекарственных препаратов может включать смешивание, гранулирование, таблетирование и растворение ингредиентов. Химические носители могут быть твердыми или жидкими. Твердые носители могут представлять собой лактозу, сахарозу, тальк, желатин, агар, пектин, стеарат магния, жирные кислоты и т.д. Жидкие носители могут представлять собой сиропы, масла, такие как оливковое масло, подсолнечное масло или соевое масло, воду и т.д. Аналогично, носитель может также содержать компонент для пролонгированного высвобождения активного компонента, такой как глицерилмоностеарат или глицерилдистеарат. Могут быть использованы различные формы лекарственных препаратов. Таким образом, если используется твердый носитель, указанные формы могут представлять собой таблетки, твердые желатиновые капсулы, порошок или гранулы, которые могут быть введены в капсулах перорально. Количество твердого носителя может изменяться, но оно обычно составляет от 25 мг до 1 г. Если используется жидкий носитель, препарат может быть в форме сиропа, эмульсии, мягких желатиновых капсул, стерильных инъецируемых растворов, таких как ампулы или неводные жидкие суспензии.
Соединения в соответствии с настоящим изобретением могут быть употреблены перорально, парентерально, местно, внутриназально, внутриректально и интравагинально. Парентеральный путь в данном описании означает внутривенное, внутримышечное и подкожное применение. Соответствующие препараты настоящих соединений могут быть использованы для профилактики, а также в лечении различных заболеваний и патологических воспалительных состояний, вызванных избыточным нерегулированным продуцированием цитокинов или медиаторов воспаления, в особенности ФНО-. Они включают ревматоидный артрит, ревматоидный спондилит, остеоартрит и другие артритные патологические состояния и заболевания, экзему, псориаз и другие воспалительные состояния кожи, такие как ожоги, вызванные ультрафиолетовым излучением (солнечные лучи и подобные источники УФ), воспалительные глазные заболевания, болезнь Крона, язвенный колит и астму.
Ингибирующее действие соединений настоящего изобретения на секрецию ФНО- и ИЛ-1 было определено следующими испытаниями in vitro и in vivo.
Определение in vivo секреции ФНО- и ИЛ-1 в одноядерных клетках человеческой периферийной крови
Одноядерные клетки человеческой периферийной крови (РВМС) получали из гепаринизованной цельной крови после отделения РВМС на Ficoll-Paque™ Plus (Amersham-Pharmacia). Для определения уровня ФНО- 3,5-5×104 клеток общим объемом 200 мкл выращивали в течение времени от 18 до 24-х часов в планшетах для микротитрования с плоским дном (96 лунок, Falcon) в среде RPMI 1640, в которую добавляли 10% FBS (фетальная (эмбриональная) бычья сыворотка, Biowhittaker), предварительно инактивированной при 54°С/30 мин, 100 ед./мл пенициллина, 100 мг/мл стрептомицина и 20 мМ HEPES (N-2-гидроксиэтилпиперазин-N'-2-этансульфоновая кислота) (GIBCO). Клетки инкубировали при 37°С в атмосфере, содержащей 5% CO2, имеющей влажность 90%. При отрицательном контроле клетки выращивали только в среде (NC), тогда как при положительном контроле секрецию ФНО- возбуждали добавлением 1 нг/мл липополисахаридов (ЛПС, серологический тип E. coli 0111:В4, SIGMA)(PC). Действие испытуемых соединений на секрецию ФНО- исследовали после добавления их в культуры клеток, стимулированных ЛПС (TS). Уровень ФНО- в клеточном супернатанте определяли методикой ТИФА (твердофазный иммуноферментный анализ) (ELISA) в соответствии с рекомендациями производителя (R&D Systems). Чувствительность испытаний составляла <3 пг/мл ФНО-. Уровень ИЛ-1 определяли в анализе, осуществляемом при таких же условиях и с таким же числом клеток и такой же концентрацией стимула, методикой ТИФА (R&D Systems). Процент ингибирования продуцирования ФНО- или ИЛ-1 вычисляли в соответствии с уравнением:
% ингибирования = [1-(TS-NC)/(PC-NC)]×100
Значение IC-50 определяли в виде концентрации вещества, при которой происходило 50% ингибирование выработки ФНО-.
Соединения, имеющие значение IC-50 при концентрациях 20 мкМ или более низких, являются активными.
Определение in vitro секреции ФНО- и ИЛ-1 в брюшинных макрофагах мышей
Для получения брюшинных макрофагов мышам-самцам линии Balb/C возрастом от 8 до 12 недель впрыскивали внутрибрюшинно 300 мкг зимосана (SIGMA), растворенного в фосфатном буфере (PBS), общий объем которого составлял 0,1 мл/мышь. Через 24 часа мышей умерщвляли в соответствии с Актом о защите лабораторных животных (Laboratory Animal Welfare Act). Брюшинную полость промывали стерильным физиологическим раствором (5 мл). Полученные брюшинные макрофаги дважды промывали стерильным физиологическим раствором и после последнего центрифугирования (350 g/10 мин) повторно суспендировали в RPMI 1640, в которую добавляли 10% порцию FBS. Для определения секреции ФНО- 5×104 клеток/лунку общим объемом 200 мкл выращивали в течение времени от 18 до 24-х часов в планшете для титрования с плоским дном (96 лунок, Falcon) в среде RPMI 1640, в которую прибавляли 10% FBS (фетальная бычья сыворотка, Biowhittaker), инактивированную нагревом, 100 ед./мл пенициллина, 100 мг/мл стрептомицина, 20 мМ HEPES и 50 мкМ 2-меркаптоэтанола (все от GIBCO). Клетки инкубировали при 37°С в атмосфере, содержащей 5% CO2, имеющей влажность 90%. При отрицательном контроле клетки выращивали только в среде (NC), тогда как при положительном контроле возбуждали секрецию ФНО- добавлением 10 нг/мл липополисахаридов (ЛПС, серологический тип E. coli 0111:В4, SIGMA)(PC). Действие веществ на секрецию ФНО- исследовали после добавления их в культуры клеток, стимулированных ЛПС (TS). Уровень ФНО- в клеточном супернатанте определяли методикой ТИФА (R&D Systems, Biosource). Уровень клеток определяли в анализе, идентичном анализу на ФНО-, осуществляемому методикой ТИФА (R&D Systems). Процент ингибирования продуцирования ФНО- или ИЛ-1 вычисляли в соответствии с уравнением:
% ингибирования = [1-(TS-NC)/(PC-NC)]×100.
Значение IC-50 определяли в виде концентрации вещества, при которой происходило 50% ингибирование выработки ФНО-.
Соединения, имеющие значения IC-50 при концентрациях 10 мкМ или более низких, являются активными.
Модель избыточной секреции ФНО- или ИЛ-1 у мышей, вызванной ЛПС in vivo
Секрецию ФНО- или ИЛ-1 у мышей индуцировали в соответствии с уже известным методом (Badger AM et al., J.Pharmac. Env. Therap., 1996, 279:1453-1461). Использовали самцов Balb/C возрастом от 8 до 12 недель группами, состоящими из 6-10 животных. Животных обрабатывали перорально или только растворителем (при отрицательном или при положительном контролях) или растворами веществ за 30 минут до внутрибрюшинной обработки ЛПС (серологический тип E. Coli 0111:B4, Sigma) в дозе 25 мкг/животное. Через 2 часа животных умерщвляли посредством внутрибрюшинной инъекции румпана (Bayer) и кетанеста (Parke-Davis). Отбирали пробу крови каждого животного в микроцентрифужную пробирку (Becton Dickinson) и отделяли плазму в соответствии с инструкциями производителя. Уровень ФНО- в плазме определяли методикой ТИФА (Biosource, R&D Systems) в соответствии с инструкциями производителя. Чувствительность испытаний составляла <3 пг/мл ФНО-. Уровень ИЛ-1 определяли методикой ТИФА (R&D Systems). Процент ингибирования продуцирования ФНО- или ИЛ-1 вычисляли в соответствии с уравнением:
% ингибирования = [1-(TS-NC)/(PC-NC)]×100.
Соединения, проявляющие 30% или большее ингибирование продуцирования ФНО- при дозе 10 мг/кг, являются активными.
Анализ анальгезирующей активности по числу корчей (судорог)
В данном анализе вызывали боль инъекцией раздражающего средства, наиболее часто уксусной кислоты, в брюшинную полость мышей. Животные реагировали с характерными корчами (судорогами), которые дали название анализу (Collier HOJ et al., Pharmac. Chemother., 1968, 32:295-310; Fukawa K et al., J. Pharmacol. Meth., 1980, 4:251-259; Schweizer A et al., Agents Actions, 1988, 23:29-31). Анализ является удобным для определения активности соединений. Методика: использовали мышей-самцов линии Balb/C (Charles River, Италия) возрастом от 8 до 12 недель. Контрольная группа получала перорально метилцеллюлозу за 30 минут до внутрибрюшинного введения уксусной кислоты с концентрацией 0,6%, тогда как экспериментальные группы получали перорально стандартные (ацетилсалициловая кислота) или испытуемые вещества в метилцеллюлозе за 30 минут до внутрибрюшинного введения 0,6% уксусной кислоты (объем 0,1 мл/10 г). Мышей помещали отдельно под стеклянные воронки и в течение 20 минут регистрировали число корчей (судорог) у каждого животного. Процент ингибирования корчей (судорог) вычисляли в соответствии с уравнением:
% ингибирования = (среднее значение числа корчей (судорог) в контрольной группе - число корчей (судорог) в экспериментальной группе)/число корчей (судорог) в контрольной группе × 100.
Соединения, показывающие такую же анальгезирующую активность, как ацетилсалициловая кислота или более высокую, являются активными.
Модель шока у мышей, вызванного ЛПС in vivo
Использовали мышей-самцов Balb/C (Charles River, Италия) возрастом от 8 до 12 недель. Выделенные из Serratie marcessans ЛПС (Sigma, L-6136) разбавляли стерильным физиологическим раствором. Первую инъекцию ЛПС осуществляли внутрикожно дозой 4 мкг/мышь. Через 18-24 часа ЛПС вводили внутривенно дозой 200 мкг/мышь. Контрольная группа получала две инъекции ЛПС, как указано выше. Экспериментальные группы получали вещества перорально за полчаса до каждого введения ЛПС. Через 24 часа наблюдали выживаемость животных.
Вещества, в присутствии которых выживаемость при дозах 30 мг/кг составляла 40% или более, являются активными.
Соединения из примера 14 показывают активность, по меньшей мере, в двух исследовательских анализах, хотя данные результаты представляют только иллюстрацию биологической активности соединений и не ограничивают изобретение каким-либо образом.
Способы получения с примерами
Настоящее изобретение проиллюстрировано следующими примерами, которые никоим образом его не ограничивают.
Пример 1
Моноэтиловый эфир 8-окса-2-тиадибензо[е,h]азулен-1,3-дикарбоновой кислоты (1)
К раствору трет-бутилата калия (0,013 моль) в 5 мл трет-бутанола (10 мл), нагретому до 60°С, добавляли раствор дибензо[b,f]оксепин-10,11-диона (III; X=О; Y=Z=H) (0,004 моль) и простой тиоэфир (IV; R1, R2=Et) (0,008 моль) в трет-бутаноле. После перемешивания в течение 30 минут при 60°С реакционную смесь охлаждали и подкисляли 5М водным раствором HCl (10 мл), после чего выпаривали большую часть растворителя при температуре 30°С и давлении 30 гПа. К остатку добавляли диэтиловый эфир (20 мл) и раствор затем экстрагировали 2М раствором NH4OH (10 мл). Объединенные экстракты подкисляли разбавленной HCl до кислой реакции и получали дикарбоксилат в форме коричневых кристаллов.
Пример 2
1-Метиловый эфир 5-хлор-8-окса-2-тиадибензо[е,h]азулен-1,3-дикарбоновой кислоты (2)
3-Метиловый эфир 5-хлор-8-окса-2-тиадибензо[е,h]азулен-1,3-дикарбоновой кислоты (3)
Следуя методике примера 1, с использованием в качестве исходных продуктов 2-хлордибензо[b,f]оксепин-10,11-диона (III; X=О; Y=2-Cl; Z=H) и простого тиоэфира (IV; R1, R2=Me) получали смесь дикарбоксилатов в форме коричневого масла.
Пример 3
Моноэтиловый эфир 2,8-дитиадибензо[е,h]азулен-1,3-дикарбоновой кислоты (4)
Следуя методике примера 1, с использованием в качестве исходных продуктов дибензо[b,f]тиепин-10,11-диона (III; X=S; Y=Z=H) и простого тиоэфира (IV; R5=Et) получали дикарбоксилат в форме коричневых кристаллов.
Пример 4
Этиловый эфир 8-окса-2-тиадибензо[е,h]азулен-1-карбоновой кислоты (5)
8-окса-2-тиадибензо[е,h]азулен (9)
Гомогенную смесь дикарбоксилата 1 (200 мг) и меди (150 мг) нагревали в течение 2-х часов при 300°С. После охлаждения реакционной смеси к ней добавляли диэтиловый эфир и отфильтровывали нерастворенный оксид меди. Фильтрат выпаривали при пониженном давлении и полученную смесь продуктов разделяли хроматографией на колонке. Выделяли соединения 5 и 9 в кристаллической форме.
Пример 5
Метиловый эфир 5-хлор-8-окса-2-тиадибензо[е,h]азулен-1-карбоновой кислоты (6)
Метиловый эфир 11-хлор-8-окса-2-тиадибензо[е,h]азулен-1-карбоновой кислоты (7)
5-Хлор-8-окса-2-тиадибензо[е,h]азулен (10)
Следуя методике примера 4, с использованием в качестве исходных продуктов смеси дикарбоксилатов 2 и 3 получали смесь двух монокарбоксилатов 6 и 7 и соединения 10. Соединение 10 выделяли из смеси монокарбоксилатов хроматографией на колонке. Карбоксилаты 6 и 7 отделяли и исследовали ГХ-МС (GC-MC) в виде двух близко расположенных пиков с m/z = 314 (MH+).
Пример 6
Этиловый эфир 2,8-дитиадибензо[е,h]азулен-1-карбоновой кислоты (8)
2,8-Дитиадибензо[е,h]азулен (11)
Следуя методике примера 4, с использованием в качестве исходного продукта дикарбоксилата 4 получали соединения 8 и 11. Смесь соединений разделяли хроматографией на колонке с получением обоих продуктов в кристаллической форме.
Пример 7
(8-Окса-2-тиадибензо[е,h]азулен-1-ил)метанол (12)
К суспензии LiAlH4 в сухом эфире (10 ммоль в 15 мл сухого эфира) добавляли по каплям эфирный раствор сложного эфира 5 (2 ммоль в 15 мл сухого эфира). Реакционную смесь перемешивали при комнатной температуре в течение 4-х часов. После завершения взаимодействия всего количества сложного эфира (за ходом реакции следили с помощью тонкослойной хроматографии) разлагали избыток LiAlH4 добавлением диэтилового эфира и воды. Отфильтровывали полученный белый осадок и после сушки над безводным Na2SO4 выпаривали фильтрат при пониженном давлении. Неочищенный продукт очищали хроматографией на колонке с получением чистого продукта в форме желтоватых кристаллов.
Пример 8
(5-Хлор-8-окса-2-тиадибензо[е,h]азулен-1-ил)метанол (13)
(11-Хлор-8-окса-2-тиадибензо[е,h]азулен-1-ил)метанол (14)
Следуя методике примера 8, с использованием в качестве исходных продуктов смеси сложных эфиров 6 и 7 получали смесь указанных в заголовке спиртов, которую разделяли колоночной хроматографией с получением чистых веществ в форме желтоватых кристаллов.
Пример 9
(2,8-Дитиадибензо[е,h]азулен-1-ил)метанол (15)
Следуя методике примера 8, с использованием в качестве исходного продукта соответствующего сложного эфира 8 получали спирт в форме коричневых кристаллов.
Таблица 1
Пример 10
а) Диметил-[3-(8-окса-2-тиадибензо[е,h]азулен-1-илметокси)пропил]амин
К раствору гидрохлорида 3-диметиламинопропилхлорида (2,5 ммоль) в 50% гидроксиде натрия (3 мл) добавляли хлорид бензилтриэтиламмония (0,3 ммоль) и толуоловый раствор спирта 12 (0,25 ммоль). Реакционную смесь кипятили при интенсивном перемешивании с обратным холодильником в течение 4-х часов. Затем ее охлаждали до комнатной температуры, разбавляли водой и экстрагировали дихлорметаном. После очистки колоночной хроматографией выделяли маслянистый продукт.
1H ЯМР (м.д., CDCl3): 2,08 (м, 2H); 2,58 (с, 6H); 2,84 (м, 2H); 3,69 (м, 2H); 4,75 (шир.д, 2H); 7,16-7,36 (м, 6H), 7,46 (с, 1H); 7,47-7,56 (м, 2H).
b) Диметил-[2-(8-окса-2-тиадибензо[е,h]азулен-1-илметокси)этил]амин
С использованием в качестве исходных продуктов спирта 12 (0,25 ммоль) и гидрохлорида 2-диметиламиноэтилхлорида (2,5 ммоль) получали маслянистый продукт.
1H ЯМР (м.д., CDCl3): 2,52 (с, 6H); 2,86 (шир.с, 2H); 3,85 (шир.с, 2H); 4,80 (шир.д, 2H); 7,16-7,36 (м, 6H); 7,46 (с, 1H); 7,49-7,56 (м, 2H).
с) 3-(8-Окса-2-тиадибензо [е,h]азулен-1-илметокси)пропиламин
С использованием в качестве исходных продуктов спирта 12 (0,25 ммоль) и гидрохлорида 3-хлорпропиламина (2,5 ммоль) получали маслянистый продукт.
1H ЯМР (м.д., CDCl3): 1,99 (м, 2H); 3,05 (т, 2H); 3,70 (шир.с, 2H); 4,3-4,5 (шир., 2H); 4,72 (шир.с, 2H); 7,15-7,60 (м, 9H).
Пример 11
а) 3-(5-Хлор-8-окса-2-тиадибензо[е,h]азулен-1-илметокси)пропиламин
К раствору гидрохлорида 3-хлорпропиламина (2,2 ммоль) в 50% гидроксиде натрия (3 мл) добавляли хлорид бензилтриэтиламмония (0,3 ммоль) и толуоловый раствор спирта 13 (0,22 ммоль). Реакционную смесь кипятили при интенсивном перемешивании с обратным холодильником в течение 5-ти часов. Затем ее охлаждали до комнатной температуры, разбавляли водой и экстрагировали дихлорметаном. После очистки колоночной хроматографией выделяли маслянистый продукт.
МС (m/z): 372 (MH+).
b) [2-(5-Хлор-8-окса-2-тиадибензо[е,h]азулен-1-илметокси)этил]диметиламин
С использованием в качестве исходных продуктов спирта 13 (0,29 ммоль) и гидрохлорида 2-диметиламиноэтилхлорида (2,9 ммоль) получали маслянистый продукт.
МС (m/z): 386 (MH+).
с) [3-(5-Хлор-8-окса-2-тиадибензо[е,h]азулен-1-илметокси)пропил]диметиламин
С использованием в качестве исходных продуктов спирта 13 (0,22 ммоль) и гидрохлорида 3-диметиламинопропилхлорида (2,2 ммоль) получали маслянистый продукт.
МС (m/z): 400 (MH+).
Пример 12
а) [2-(11-Хлор-8-окса-2-тиадибензо[е,h]азулен-1-илметокси)этил]диметиламин
К раствору гидрохлорида 2-диметиламиноэтилхлорида (1,8 ммоль) в 50% гидроксиде натрия (3 мл) добавляли хлорид бензилтриэтиламмония (0,3 ммоль) и толуоловый раствор спирта 14 (0,18 ммоль). Реакционную смесь кипятили при интенсивном перемешивании с обратным холодильником в течение 5-ти часов. Затем ее охлаждали до комнатной температуры, разбавляли водой и экстрагировали дихлорметаном. После очистки колоночной хроматографией выделяли маслянистый продукт.
МС (m/z): 386 (MH+).
b) 3-(11-Хлор-8-окса-2-тиадибензо[е,h]азулен-1-илметокси)пропиламин
С использованием в качестве исходных продуктов спирта 14 (0,18 ммоль) и гидрохлорида 3-хлорпропиламина (1,8 ммоль) получали маслянистый продукт.
1H ЯМР (м.д., CD3COCD3): 1,82 (с, 2H); 1,97 (т, 2H); 3,36 (т, 2H); 3,76 (шир.с, 2H); 4,74 (с, 2H); 7,26-7,82 (м, 8H);
МС (м/z): 372 (MH+).
Пример 13
а) [3-(2,8-Дитиадибензо[е,h]азулен-1-илметокси)пропил]диметиламин
К раствору гидрохлорида 3-диметиламинопропилхлорида (6,7 ммоль) в 50% гидроксиде натрия (5 мл) добавляли хлорид бензилтриэтиламмония (0,88 ммоль) и толуоловый раствор спирта 15 (0,67 ммоль). Реакционную смесь кипятили при интенсивном перемешивании с обратным холодильником в течение 5-ти часов. Затем ее охлаждали до комнатной температуры, разбавляли водой и экстрагировали дихлорметаном. После очистки колоночной хроматографией выделяли маслянистый продукт.
1H ЯМР (м.д., CDCl3): 2,04 (п, 2H); 2,57 (с, 6H); 2,82 (шир.с, 2H); 3,61 (м, 2H); 4,67 (м, 2H); 7,27-7,71 (м, 8H); 7,40 (с, 1H).
b) [2-(2,8-Дитиадибензо[е,h]азулен-1-илметокси)этил]диметиламин
С использованием в качестве исходных продуктов спирта 15 (0,67 ммоль) и гидрохлорида 2-диметиламиноэтилхлорида (6,7 ммоль) получали маслянистый продукт.
1H ЯМР (м.д., CDCl3): 2,49 (с, 6H); 2,86 (шир.с, 2H); 3,78 (м, 2H); 4,72 (м, 2H); 7,23-7,70 (м, 8H); 7,40 (с, 1H).
с) 3-(2,8-Дитиадибензо[е,h]азулен-1-илметокси)пропиламин
С использованием в качестве исходных продуктов спирта 15 (0,27 ммоль) и гидрохлорида 3-хлорпропиламина (2,7 ммоль) получали маслянистый продукт.
МС (m/z): 354 (MH+).
Пример 14
2,8-Дитиадибензо [е,h]азулен-1-карбальдегид
К дихлорметановому раствору спирта 15 (3,0 ммоль в 40 мл) добавляли смесь дипиридина и оксида хрома (VI) (пиридинилдихромат, ПДХ (PDC), 0,006 моль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. К реакционной смеси добавляли диэтиловый эфир (50 мл) и разбавленную таким образом реакционную смесь очищали на колонке с Флорисилом (Florisil) с получением желтого кристаллического продукта.
1H ЯМР (м.д., CDCl3): 7,29-7,45 (м, 5H); 7,53-7,56 (м, 1H); 7,65-7,68 (м, 1H); 7,72-7,75 (м, 1H); 7,81 (д, 1H); 9,84 (с, 1H).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ БЕНЗОНАФТОАЗУЛЕНА, ПРИМЕНЕНИЕ ИХ В КАЧЕСТВЕ ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ И ДЛЯ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ, ПРЕДНАЗНАЧЕННЫХ ДЛЯ ИНГИБИРОВАНИЯ ПРОДУЦИРОВАНИЯ ФНО-α | 2003 |
|
RU2318827C2 |
| 1-ОКСА-3-АЗАДИБЕНЗОАЗУЛЕНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОДУЦИРОВАНИЯ ФАКТОРА НЕКРОЗА ОПУХОЛИ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2323222C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ЗАБОЛЕВАНИЙ | 2001 |
|
RU2270832C2 |
| ЗАМЕЩЕННЫЕ АЗОЛЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2047604C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2678305C1 |
| Производные тетрагидротриазолопиримидина в качестве ингибиторов нейтрофильной эластазы человека | 2012 |
|
RU2622643C2 |
| НОВЫЕ АНТИГЕЛЬМИНТНЫЕ СОЕДИНЕНИЯ | 2019 |
|
RU2828007C2 |
| ЗАМЕЩЕННЫЕ ДИГИДРОПИРАЗОЛОНЫ ДЛЯ ЛЕЧЕНИЯ КАРДИОВАСКУЛЯРНЫХ И ГЕМАТОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ, ИХ ПРИМЕНЕНИЕ, ЛЕКАРСТВЕННОЕ СРЕДСТВО И СПОСОБ ЛЕЧЕНИЯ И/ИЛИ ПРОФИЛАКТИКИ | 2012 |
|
RU2611012C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРНЫХ ФОРМ ПРОИЗВОДНЫХ 1,3-ЦИКЛОГЕКСАНДИОЛА В ЦИС-КОНФИГУРАЦИИ | 2004 |
|
RU2372319C2 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА И ПИРАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2686117C1 |
Настоящее изобретение относится к соединениям формулы I, их фармакологически приемлемым солям и сольватам
где X представляет собой О или S; Y и Z независимо друг от друга означают водород или галоген; R1 - гидрокси-С1-С7-алкил, карбокси, С1-С7-алкилоксикарбонил, или заместитель формулы II
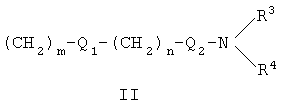
R2 - водород, карбокси или алкилоксикарбонил. Соединения могут быть использованы в качестве ингибиторов продуцирования цитокинов или медиаторов воспаления. Описаны также промежуточные соединения для получения соединений I и применение соединений I. 4 н. и 8 з.п. ф-лы, 1 табл.
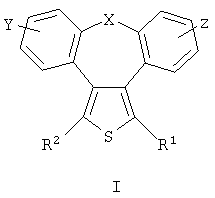
отличающееся тем, что
Х представляет собой О или S;
Y и Z независимо друг от друга означают водород или галоген;
R1 может представлять собой гидрокси-С1-С7-алкил, карбокси, C1-C7-алкилоксикарбонил,
или заместитель формулы II
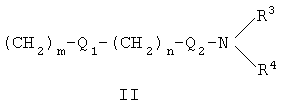
где R3 и R4 одновременно или независимо друг от друга могут представлять собой водород, С1-С4-алкил;
m и n представляют собой целое число от 1 до 3;
Q1 и Q2 независимо друг от друга представляют собой кислород или группу;
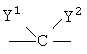
в которой заместители Y1 и Y2 независимо друг от друга могут представлять собой водород;
R2 может представлять собой водород, карбокси или алкилоксикарбонил,
а также его фармакологически приемлемые соли и сольваты.
8-окса-2-тиадибензо[е,h]азулен;
2,8-дитиадибензо[е,h]азулен;
5-хлор-8-окса-2-тиадибензо[е,h]азулен;
моноэтиловый эфир 8-окса-2-тиадибензо[е,h]азулен-1,3-дикарбоновой кислоты;
1-метиловый эфир 5-хлор-8-окса-2-тиадибензо[е,h]азулен-1,3-дикарбоновой кислоты;
3-метиловый эфир 5-хлор-8-окса-2-тиадибензо[е,h]азулен-1,3-дикарбоновой кислоты;
моноэтиловый эфир 2,8-дитиадибензо[е,h]азулен-1,3-дикарбоновой кислоты.
этиловый эфир 8-окса-2-тиадибензо[е,h]азулен-1-карбоновой кислоты;
метиловый эфир 5-хлор-8-окса-2-тиадибензо[е,h]азулен-1-карбоновой кислоты;
метиловый эфир 11-хлор-8-окса-2-тиадибензо[е,h]азулен-1-карбоновой кислоты;
этиловый эфир 2,8-дитиадибензо[е,h]азулен-1-карбоновой кислоты;
2,8-дитиадибензо[е,h]азулен-1-карбальдегид;
(8-окса-2-тиадибензо[е,h]азулен-1-ил)метанол;
(5-хлор-8-окса-2-тиадибензо[е,h]азулен-1-ил)метанол;
(11-хлор-8-окса-2-тиадибензо[е,h]азулен-1-ил)метанол;
(2,8-дитиадибензо[е,h]азулен-1-ил)метанол.
диметил-[3-(8-окса-2-тиадибензо[е,h]азулен-1-илметокси)пропил]амин;
диметил-[2-(8-окса-2-тиадибензо[е,h]азулен-1-илметокси)этил]амин;
3-(8-окса-2-тиадибензо[e,h]азулен-1-илметокси)пропиламин;
3-(5-хлор-8-окса-2-тиадибензо[e,h]азулен-1-илметокси)пропиламин;
[2-(5-хлор-8-окса-2-тиадибензо[e,h]азулен-1-илметокси)этил]диметиламин;
[3-(5-хлор-8-окса-2-тиадибензо[e,h]азулен-1-илметокси)пропил]диметиламин;
[2-(11-хлор-8-окса-2-тиадибензо[е,h]азулен-1-илметокси)этил]диметиламин;
3-(11-хлор-8-окса-2-тиадибензо[е,h]азулен-1-илметокси)пропиламин;
[3-(2,8-дитиадибензо[е,h]азулен-1-илметокси)пропил]диметиламин;
[2-(2,8-дитиадибензо[е,h]азулен-1-илметокси)этил]диметиламин;
3-(2,8-дитиадибензо[е,h]азулен-1-илметокси)пропиламин.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US 3711489, 16.01.1973. | |||

