Изобретение относится к N-замещенным неароматическим гетероциклическим соединениям. В частности, изобретение относится к N-замещенным неароматическим гетероциклическим соединениям, которые являются антагонистами NMDA/NR2B, пригодными для лечения болезни Паркинсона и боли.
Ионы, такие как глутамат, играют ключевую роль в процессах, связанных с хронической болью и нейротоксичностью, связанной с болью, - действующих, главным образом, через рецепторы N-метил-D-аспартата (NMDA). Следовательно, ингибирование такого действия путем использования ион-канальных антагонистов, особенно антагонистов NMDA, может иметь положительный эффект при лечении и регуляции болезни Паркинсона и боли.
Рецепторы NMDA являются гетеромерными ансамблями субъединиц, из которых два главных семейства субъединиц, обозначаемых NR1 и NR2, были клонированы. Без связи с теорией, обычно считают, что разные функциональные рецепторы NMDA в центральной нервной системе (ЦНС) млекопитающих образуются только комбинациями субъединиц NR1 и NR2, которые обычно выражают сайты узнавания глицина и глутамата. В свою очередь, семейство субъединиц NR2 разделяют на четыре индивидуальных типа субъединиц NR2A, NR2B, NR2C и NR2D. T. Ishii et al., J. Biol. Chem., 268, 2836-2843 (1993) D.J. Laurie et al., Mol. Brain Res., 51, 23-32 (1997) описывают как разные возникающие комбинации дают разные рецепторы NMDA, отличающиеся физиологическими и фармакологическими свойствами, такими как воротный механизм ионных каналов, чувствительность к магнию, фармакологический профиль и анатомическое распределение.
Например, в то время как NR1 находят везде в мозгу, субъединицы NR2 распределяются по-разному. В частности, считается, что карта распределения для NR2В снижает вероятность побочных эффектов при болезни Паркинсона или боли. Таким образом, было бы желательно обеспечить новые антагонисты, нацеленные на рецептор NR2В.
Настоящее изобретение относится к N-замещенным неароматическим гетероциклическим соединениям, представленным формулой (I):
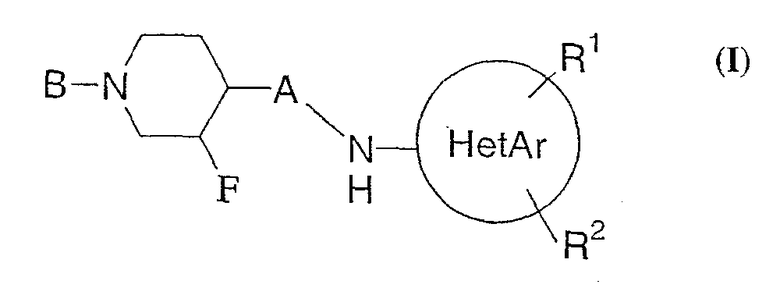
или их фармацевтически приемлемым солям. Настоящее изобретение также относится к фармацевтическим композициям, содержащим такие соединения. Изобретение также относится к способам лечения и предотвращения состояний, включая болезнь Паркинсона, боли, болезнь Альцгеймера и эпилепсия, с использованием настоящих соединений и композиций.
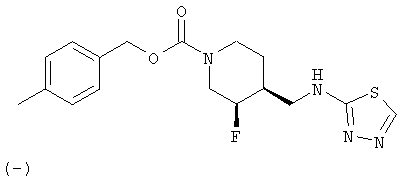
Соединения настоящего изобретения представлены формулой (I)
или их фармацевтически приемлемыми солями, в которой
HetAr представляет 5-6-членное гетероароматическое кольцо, содержащее 1 или 2 атома азота в кольце, или тиазолил, или тиадиазолил;
R1 и R2 представляет, независимо друг от друга, водород, С1-4-алкил, фтор, хлор, бром или иод;
А представляет связь или С1-С2-алкил и
В представляет арил(СН2)0-3-О-С(О)-, инданил(СН2)0-3-О-С(О)-, арил(СН2)1-3-С(О)-, арилциклопропил-С(О)-, арил(СН2)1-3-NH-С(О)-, в которых арил может быть замещен 1-5 заместителями, каждый заместитель независимо друг от друга представляет С1-С4-алкил, фтор или хлор.
В одном аспекте соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой HetAr представляет 6-членное гетероароматическое кольцо, содержащее 1 или 2 атома азота в кольце.
В варианте этого первого аспекта соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой HetAr представляет 6-членное гетероароматическое кольцо, содержащее 1 атом азота в кольце.
В другом варианте этого первого аспекта соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой HetAr представляет 6-членное гетероароматическое кольцо, содержащее 2 атома азота в кольце.
Во втором аспекте соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой HetAr представляет тиазолил или тиадиазолил.
В варианте этого второго аспекта соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой HetAr представляет 1,2,4-иадиазолил.
В другом варианте этого второго аспекта соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой HetAr представляет тиазолил.
В третьем аспекте соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой A представляет связь или -С1-С2-алкил-.
В варианте этого третьего аспекта соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой A представляет связь.
В другом варианте этого третьего аспекта соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой A представляет метилен.
В ином варианте этого третьего аспекта соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой A представляет -C2-алкил-.
В четвертом аспекте соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой В представляет арилциклопропил-С(О)- или арил(СН2)0-3-О-С(О)-, в которой арил может быть замещен С1-С4-алкилом.
В варианте этого четвертого аспекта соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой В представляет арилциклопропил-С(О)-, в которой арил представляет фенил, возможно замещенный С1-4-алкилом.
В другом варианте этого четвертого аспекта соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой В представляет арил(СН2)0-3-О-С(О)-, в которой арил представляет фенил, возможно замещенный С1-С4-алкилом.
В ином варианте этого четвертого аспекта соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой В представляет арил-О-С(О)-, в которой арил представляет фенил, возможно замещенный С1-С4-алкилом.
В другом ином варианте этого четвертого аспекта соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой В представляет арил-(СН2)-О-С(О)-, в которой арил представляет фенил, возможно замещенный С1-С4-алкилом.
В еще одном варианте этого четвертого аспекта соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой В представляет арил-(СН2)-О-С(О)-, в которой арил представляет 4-толил.
В пятом аспекте соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой HetAr представляет 6-членное гетероароматическое кольцо, содержащее 2 атома азота в кольце; А представляет метилен и В представляет арил-(СН2)-О-С(О)-, в которой арил представляет 4-толил.
В шестом аспекте соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой HetAr представляет 1,2,4-тиадиазолил; А представляет метилен и В представляет арил-(СН2)-О-С(О)-, в которой арил представляет 4-толил.
В седьмом аспекте соединения настоящего изобретения представлены формулой (I) или их фармацевтически приемлемыми солями, в которой HetAr представляет 6-членное гетероароматическое кольцо, содержащее 2 атома азота в кольце; А представляет метилен и В представляет арилциклопропил-С(О)-, в которой арил представляет фенил, возможно замещенный С1-С4-алкилом.
"Алкил", а также другие термины, имеющие корень "алк", такие как, например, алкокси, алканоил, алкенил, алкинил и им подобные, обозначает углеродную цепь, которая может быть линейной или разветвленной или их комбинацией. Примеры алкильных групп включают метил, этил, пропил, изопропил, бутил, втор- и трет-бутил, пентил, гексил, гептил и им подобные алкилы.
Термин "арил", если не указано иначе, включает возможно замещенные много- или одноядерные системы, такие как, например, фенил, нафтил и толил.
Термин "HetAr" включает, например, гетероароматические кольца, такие как пиримидин и пиридин.
Термин "(СН2)0" значит, что метил отсутствует. Таким образом, "(СН2)0-3" значит, что метил отсутствует или присутствуют до трех метилов, то есть присутствуют три, два, один или ни одного метила. Когда метильные группы отсутствуют в связывающей алкильной группе, связывание осуществляется прямой связью.
Термин "возможно замещенный" означает как наличие замещения, так и отсутствие замещения. Таким образом, например, возможно замещенный арил может представлять кольцо пентафторфенила или фенила. Далее, замещение может быть произведено на любой группе. Например, замещенный арил(С1-С6)алкил включает замещение как на арильной, так и на алкильной группах.
Соединения, описанные в описании, могут содержать один или больше асимметрических центров и могут, следовательно, давать начало диастереомерам и оптическим изомерам. Настоящее изобретение включает все такие возможные диастереомеры, а также их рацемические смеси, их существенно чистые расщепленные энантиомеры, все возможные геометрические изомеры и их фармацевтически приемлемые соли. Соединения формулы I выше показаны без определенной стереохимии на определенных положениях. Настоящее изобретение включает все стереоизомеры формулы I и их фармацевтически приемлемые соли. Далее, включены смеси стереоизомеров, а также выделенные особые стереоизомеры. В ходе осуществления синтеза таких соединений или при использовании методик рацемизации или эпимеризации, известных специалистам в этой области техники, могут быть получены смесями стереоизомеров.
Термин "фармацевтически приемлемые соли" относится к солям, полученным из фармацевтически приемлемых нетоксичных кислот, включая неорганические и органические кислоты. Такие кислоты включают, например, уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромистоводородную, хлористоводородную, изэтионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, п-олуолсульфоновую и другие кислоты. Особенно предпочтительными являются лимонная, бромистоводородная, хлористоводородная, малеиновая, фосфорная, серная и винная кислоты.
Фармацевтические композиции настоящего изобретения содержат соединение, представленное формулой I (и/или его фармацевтически приемлемую соль (соли)) в качестве активного компонента, фармацевтически приемлемый носитель и, возможно, другие терапевтические компоненты и адъюванты. Настоящие композиции включают композиции, пригодные для перорального, ректального, местного и парентерального (включая подкожное, внутримышечное и внутривенное) введения, хотя наиболее подходящий путь введения в любом данном случае будет зависеть от специфического хозяина и от природы и тяжести состояний, для устранения которых вводится активный компонент. Фармацевтические композиции обычно могут быть представлены в единичной лекарственной форме и изготовлены любым способом, хорошо известным в фармацевтической технике.
На практике соединения, представленные формулой I, или их фармацевтически приемлемые соли, настоящего изобретения в качестве активного компонента могут быть объединены в однородную смесь с фармацевтическим носителем согласно обычным способам фармацевтического компаундирования. Носитель может иметь разнообразные формы, зависящие от формы композиции, желательной для введения, например перорального или парентерального (включая внутривенное). Следовательно, фармацевтические композиции настоящего изобретения могут быть представлены как дискретные единицы, пригодные для перорального введения, такие как капсулы, крахмальные капсулы и таблетки, каждая содержащая предопределенное количество активного компонента. Далее, композиции могут быть представлены в виде порошка, гранул, раствора, суспензии в водной жидкости, неводной жидкости, эмульсии масло-в-воде или жидкой эмульсии вода-в-масле. В дополнение к обычным лекарственным формам, перечисленным выше, соединения, представленные формулой I, и/или их фармацевтически приемлемые соли также могут быть введены с помощью средств регулируемого выделения и/или дозатора. Композиции могут быть получены любым фармацевтическим способом. В общем, такие способы включают стадию объединения активного компонента с носителем, который составляет один или более необходимых компонентов. В целом, композиции получают, тщательно смешивая активный компонент с жидкими носителями, или тщательно измельченными твердыми носителями, или с обоими такими носителями до получения однородной смеси. Затем продукту может быть придана любая форма обычным образом в желательные формы.
Таким образом, фармацевтические композиции настоящего изобретения могут включать фармацевтически приемлемый носитель и соединение или фармацевтически приемлемую соль формулы I. Соединения формулы I или их фармацевтически приемлемые соли также могут быть включены в фармацевтические композиции в комбинации с одним или больше другими терапевтически активными соединениями.
Используемый фармацевтический носитель может быть, например, твердым веществом, жидкостью или газом. Примеры твердых носителей включают лактозу, распыленный гипс, сахарозу, тальк, желатин, агар, пектин, гуммиарабик, стеарат магния и стеариновую кислоту. Примеры жидких носителей включают сахарный сироп, арахисовое масло, оливковое масло и вода. Примеры газообразных носителей включают диоксид углерода и азот.
При получении композиций для пероральных лекарственных форм можно использовать любую подходящую фармацевтическую среду. Например, вода, гликоли, масла, спирты, вкусовые добавки, консерванты, красители и другие средства могут быть использованы, чтобы образовать пероральные жидкие композиции, такие как суспензии, эликсиры и растворы; в то время как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие средства, смазки, связующие, средства для распадения и другие средства могут быть использованы, чтобы образовать пероральные твердые композиции, такие как порошки, капсулы и таблетки. По причине их легкого введения таблетки и капсулы являются предпочтительными пероральными лекарственными единицами, в соответствии с чем и используются твердые фармацевтические носители. Необязательно, таблетки могут быть покрыты стандартными водными и неводными способами.
Таблетка, содержащая композиции настоящего изобретения, может быть получена прессованием или формованием возможно с одним или больше второстепенными компонентами или адъювантами. Прессованные таблетки могут быть получены прессованием, в подходящей машине, активного компонента в свободно-текущей форме, такого как порошок или гранулы, возможно смешанного со связующим, замасливателем, инертным разбавителем, поверхностно-активным или диспергирующим средством. Формованные таблетки могут быть изготовлены формованием в подходящей машине смеси порошкового соединения, увлажненного инертным жидким разбавителем. Каждая таблетка предпочтительно содержит от примерно 0,5 мг до примерно 5 г активного компонента и каждая капсула или крахмальная капсула предпочтительно содержит от примерно 0,5 мг до примерно 5 г активного компонента.
Фармацевтические композиции настоящего изобретения содержат соединение формулы I (или его фармацевтически приемлемую соль) в качестве активного компонента, фармацевтически приемлемый носитель и, необязательно, одно или более дополнительное терапевтическое средство или адъювант. Настоящие композиции включают композиции, пригодные для перорального, ректального, местного и парентерального (включая подкожное, внутримышечное и внутривенное) назначения, хотя наиболее подходящий путь введения в любом данном случае будет зависеть от специфического хозяина и от природы и тяжести состояний, для устранения которых вводится активный компонент. Фармацевтические композиции обычно могут быть представлены в единичной лекарственной форме и изготовлены любым способом, хорошо известным в фармацевтической технике.
Фармацевтические композиции настоящего изобретения, пригодные для парентерального введения, могут быть приготовлены в форме растворов или суспензий активного компонента в воде. Подходящее поверхностно-активное вещество, такое как, например, гидроксипропилцеллюлоза, может быть включено в состав композиции. Также могут быть приготовлены суспензии в глицерине, жидких полиэтиленгликолях и их смеси в маслах. Также могут быть включены консерванты, чтобы предотвратить вредный рост микроорганизмов.
Фармацевтические композиции настоящего изобретения, пригодные для инъекций, включают стерильные водные растворы или дисперсии. Кроме того, композиции могут быть в форме стерильных порошков для незапланированного приготовления таких стерильных инъекционных растворов или дисперсий. Во всех случаях конечная форма для инъекций должна быть стерильной и должна быть эффективно жидкой для легкого введения шприцом. Фармацевтические композиции должны быть стабильны в условиях производства и хранения; следовательно, должны быть сохранены от загрязняющего действия микроорганизмов, таких как бактерии и грибы. Носитель может быть растворителем или дисперсионной средой, содержащей, например, воду, этанол, полиол (например, глицерин, полипропиленгликоль и жидкий полиэтиленгликоль), растительные масла и их подходящие смеси.
Фармацевтические композиции настоящего изобретения могут быть в форме, пригодной для местного применения, такой как аэрозоль, крем, мазь, лосьон, опудривающее средство или другие подобные средства. Далее, композиции могут быть в форме, пригодной для использования в чрескожных методах. Эти композиции могут быть изготовлены обычными способами переработки, используя соединения формулы I настоящего изобретения или их фармацевтически приемлемые соли. Например, крем или мазь изготавливают смешиванием гидрофильного материала и воды с примерно 5 - примерно 10 вес. % соединения, чтобы получить крем или мазь, имеющие желательную консистенцию.
Фармацевтические композиции настоящего изобретения могут быть в форме, пригодной для ректального введения, в которой носителем является твердое вещество. Подходящие носители включают масло какао и другие материалы, обычно используемые в технике. Суппозитории обычно могут быть получены сначала смешиванием композиции с умягчающим или расплавленным носителем с последующим охлаждением и формованием в формах.
В дополнение к вышеуказанным компонентам носителя фармацевтические рецептуры, описанные выше, могут включать один или больше дополнительных компонентов носителя, таких как разбавители, буферные вещества, вкусовые вещества, связующие, поверхностно-активные вещества, загустители, смазочные вещества, консерванты (включая антиоксиданты) и другие вещества. Кроме того, другие адьюванты могут быть включены, чтобы сделать рецептуру изотонической с кровью запланированного реципиента. Композиции, содержащие соединение формулы I и/или его фармацевтически приемлемые соли, также могут быть получены в форме порошка или жидкого концентрата.
Анализы
Основанный на клетках функциональный анализ определения ИК50 антагонистов NR2В
Способность выбранных соединений ингибировать NR1а/NR2B рецептор NMDA, как измерено притоком Са2+, опосредуемым NR1а/NR2B рецептором, оценивали по следующей методике анализа постоянного движения кальция.
L(tk-)-клетки, трансфицированные NR1а/NR2B рецептором, высевали в 96-луночном формате с 3×104 клеток на лунку и выращивали в течение 1-2 дней в нормальной ростовой среде (Dulbeccos MEM с пируватом Na, 4500 мг глюкозы, pen/strep, глутамином, 10% FCS (фетальная телячья сыворотка) и 0,5 мг/мл генетицина). Экспрессию NR1а/NR2B в этих клетках вызывали добавлением 4-20 нМ дексаметазона в присутствии 500 мкМ кетамина в течение 16-24 часов. Растворы антагонистов NR2B получали в ДМСО (диметилсульфоксиде) и серийно разбавляли ДМСО для получения 10 растворов, 3-кратно отличающихся по концентрации. 96-луночный планшет лекарственного средства готовили 250-кратным разбавлением ДМСО раствора в аналитическом буфере HBSS (сбалансированный солевой раствор Хэнкса), не содержащем Mg2+(Gibco #14175-079) и содержащем 20 мМ HEPES (N-2-гидроксиэтилпиперазин-N'-2-этансульфоновая кислота), 2 мМ CaCl2, 0,1% BSA (альбумин бычьей сыворотки) и 250 мкМ Пробенесида (Probenecid, Sigma #P-8761). После индукции клетки дважды промывали (Labsystem cell washer, 3-кратное разбавление, оставляя 100 мкл) аналитическим буфером и загружали 4 мкМ кальциевого индикатора флуоресценции флуо-3 АМ (fluo-3 AM) (молекулярные зонды #Р-1241) в аналитическом буфере, содержащем Плуроник F-127 (Pluronic F-127) (молекулярные зонды # Р-3000) и 10 мкМ кетамина при 37°С за 1 час. Затем клетки промывали восемь раз аналитическим буфером, оставляя 100 мкл буфера в каждой лунке. Интенсивность флуоресценции тотчас измеряли в флуорометрическом сцинцилляционном планшет-ридере (FLIPR), используя возбуждение 488 нм и испускание 530 нм. Через 5 секунд после начала регистрации интенсивности флуоресценции добавляли 50 мкл раствора агониста (40 мкМ глутамат/глицин, конечная концентрация 10 мкМ) и через 1 минуту, после стабилизации сигнала флуоресценции, добавляли 50 мкМ антагониста NR2B и контрольные растворы из планшета лекарственного средства и интенсивность флуоресценции регистрировали еще 30 минут. Величины ИК50 определяли нелинейной аппроксимацией величин конечной флуоресценции по методу наименьших квадратов по уравнению 1 ниже.
Уравнение 1
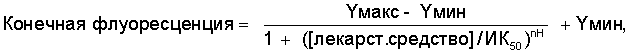
где Yмин - средняя конечная флуоресценция контрольных лунок, содержащих 1 мкМ АМД-2, и Yмакс - средняя конечная флуоресценция лунок, содержащих 0,1% ДМСО в аналитическом буфере.
Связывающий анализ для определении КI антагонистов NR2B
Анализ связывания радиолиганда выполняли при комнатной температуре в 96-луночных титрационных планшетах с конечным аналитическим объемом 1,0 мл в 20 мМ буфере HEPES (рН 7,4), содержащем 150 мМ NaCl. Растворы антагонистов NR2B готовили в ДМСО и серийно разбавляли ДМСО с получением 20 мкл каждого из 10 растворов, отличающихся 3-кратно по концентрации. Неспецифическое связывание (НСС) оценивали, используя АМД-1 (конечная концентрация 10 мкМ), и полное связывание (ПС) измеряли, добавляя ДМСО (конечная концентрация 2%). Во все лунки титрационного планшета добавляли мембраны, экспрессирующие NR1а/NR2B рецепторы (конечная концентрация 40 пМ), и меченный тритием АМД-2 (конечная концентрация 1 нМ). Через 3 часа инкубации при комнатной температуре образцы фильтровали на фильтрах (Packard GF/B), предварительно замоченных в 0,05% ПЭИ (полиэтиленимин Sigma Р-3143), и промывали 10 раз, используя 1 мл холодного буфера HEPES на промывку. После вакуумной сушки фильтровальных пластинок добавляли 40 мкл Паккард Микросцинт-20 (Packard Microscint-20) и связанную радиоактивность определяли в счетчике Packard TopCount. Кажущуюся константу диссоциации (КI), максимальный процент ингибирования (%Имакс), минимальный процент ингибирования (%Имин) и наклон холма (nH) определяли нелинейной аппроксимацией связанной радиоактивности (имп/мин связанная) по методу наименьших квадратов по уравнению 2 ниже:
Уравнение 2
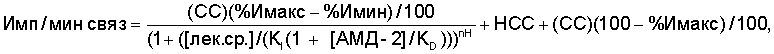 где КD - кажущаяся константа диссоциации радиолиганда для рецептора, как найдено в эксперименте с горячим насыщением, и СС - специфически связанная радиоактивность, определенная как разница ПС и НСС контрольных лунок.
где КD - кажущаяся константа диссоциации радиолиганда для рецептора, как найдено в эксперименте с горячим насыщением, и СС - специфически связанная радиоактивность, определенная как разница ПС и НСС контрольных лунок.
АМД-1
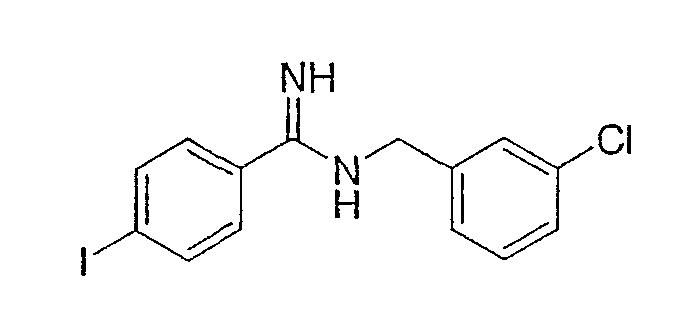
АМД-2
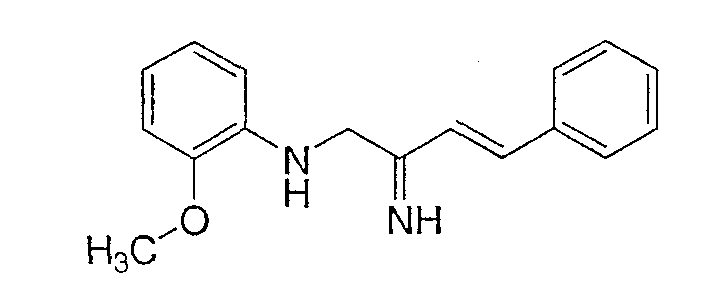
АМД-1 может быть синтезирован по общему способу, описанному C.F. Claiborne et all., Bioorganic and Medchem. Letters, 13, 697-700 (2003).
Предшественник 3 синтеза радиомеченого АМД-1 может быть синтезирован в соответствии со схемой 1а
Схема 1а
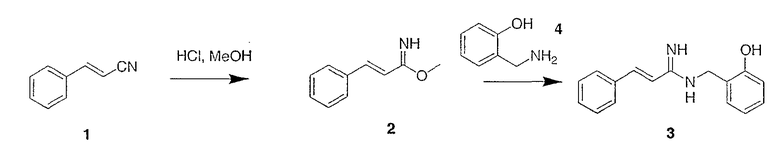
В соответствии со схемой 1а хлористый водород барботируют через раствор циннамонитрила 1 в метаноле при комнатной температуре. Летучие продукты удаляют при пониженном давлении, остаток растирают в эфире и фильтруют, получая промежуточный имидат 2. Имидат 2 растворяют в метаноле при температуре окружающей среды, обрабатывают амином 4 (Acros Chemicals) при температуре окружающей среды и перемешивают под аргоном. Летучие продукты удаляют при пониженном давлении, остаток очищают препаративной ВЭЖХ или растирают с эфиром, чтобы получить амидин 3.
Синтез АМД-2, меченного тритием
Схема 2
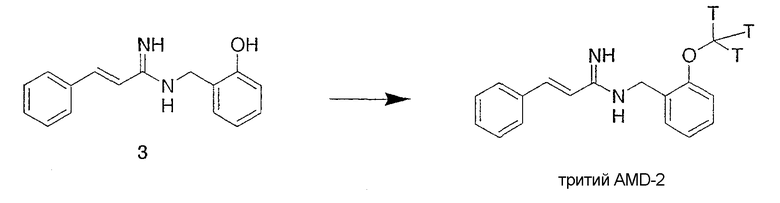
Тритированный АМД-2 получали по методике, показанной в схеме 2. Предшественник 3 (2 мг, 0,008 ммоль) растворяли в диметилформамиде (0,6 мл), содержащем карбонат калия (1,2 мг), в течение 1 часа. При комнатной температуре добавляли тритированный метилиодид (50 мКи, 0,0006 ммоль, в толуоле (1 мл), American Radiolabelled Chemicals) с высокой удельной радиоактивностью и перемешивали 2 часа. Реакционную смесь фильтровали с помощью 0,45-мкм бесшприцевого фильтровального приспособления (Whatman PTFE 0.45-μm syringeless filter device), чтобы удалить нерастворимый карбонат калия, промывали абсолютным этанолом (2 мл, Pharmco) и объединенные фильтраты концентрировали досуха при комнатной температуре, используя роторный испаритель; тем самым непрореагировавший тритированный метилиодид также удалялся. Остаток очищали ВЭЖХ хроматографией на колонке Phenomenx Luna C8 semi-prep column (Luna 5 micro C8(2), 250×10,0 мм), используя градиентную систему от ацетонитрил-вода 20/80 с 0,1% трифторуксусной кислоты до 100% ацетонитрила с 0,1% трифторуксусной кислоты за 20 минут. Полная активность продукта 8 мКи. Дальнейшая очистка проводилась путем адсорбции на колонке Waters C-18 Sep-pak column (Waters Sep-Pak PLUS C-18) и элюирования водой и затем абсолютным этанолом. Продукт разбавляли абсолютным этанолом (10 мл) перед конечным анализом.
Синтез немеченого АМД-2
Схема 1б
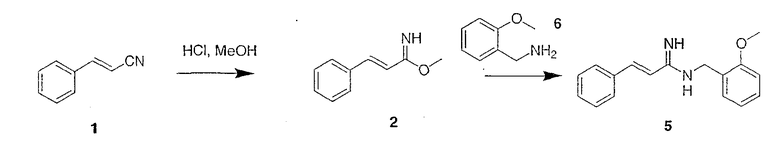
В соответствии со схемой 1б хлористый водород барботируют через раствор циннамонитрила 1 в метаноле при комнатной температуре. Летучие продукты удаляют при пониженном давлении, остаток растирают в эфире и фильтруют, получая промежуточный имидат 2. Имидат 2 растворяют в метаноле при окружающей температуре, обрабатывают амином 6 при температуре окружающей среды и перемешивают под аргоном. Летучие продукты удаляют при пониженном давлении, остаток очищают препаративной ВЭЖХ или растирают с эфиром, чтобы получить амидин 5.
Соединения настоящего изобретения показывают величины ИК50 и К1 меньше, чем 50 мкМ, в функциональном и связывающем анализе соответственно. Полезно, чтобы величины ИК50 и К1 были меньше, чем 5 мкМ, в функциональном и связывающем анализе соответственно. Более полезно, чтобы величины ИК50 и К1 были меньше, чем 1 мкМ, в функциональном и связывающем анализе соответственно. Еще более полезно, чтобы величины ИК50 и К1 были меньше, чем 0,1 мкМ, в функциональном и связывающем анализе соответственно.
Настоящие соединения являются антагонистами NR2B рецепторов NMDA и как таковые полезны для лечения и профилактики заболеваний и нарушений, опосредуемых NR2B рецептором. Такие заболевания и нарушения включают, но ими не ограничиваются, болезнь Паркинсона, нейропатические боли (такие как постгерпетическая невралгия, невралгические раны, "динии", например вульводиния, фантомная боль, авульсии корней, болезненная диабетическая невропатия, болезненная травматическая мононевропатия, болезненная полиневропатия), центральные болевые синдромы (потенциально обусловленные фактически любым патологическим изменением на любом уровне нервной системы) и постхирургические болевые синдромы (например, постмастэктомический болевой синдром, постторакотомический болевой синдром, фантомная боль), костную и суставную боль (остеоартрит), повторяющуюяся боль при движении, зубную боль, раковую боль, миофасциальную боль (мышечные раны, фибромиалгия), периоперационную боль (общая хирургия, гинекология), хроническую боль, дисменорею, а также боль, связанную со стенокардией, и воспалительную боль различной природы (например, остеоартрит, ревматоидный артрит, ревматизм, тендосиновит и подагра), головную боль, мигрень, депрессию, тревогу, шизофрению, "удар", травматическое повреждение мозга, болезнь Альцгеймера, церебральную ишемию, боковой амиотрофический склероз, болезнь Хантингтона, перцептивную тугоухость, шум в ушах, глаукому, неврологическое повреждение, вызванное эпилептическими припадками, или отравлением нейротоксинами, или недостатком глюкозы и/или кислорода в мозгу, потерю зрения, вызванную нейродегенерацией зрительных путей, синдром усталых ног, мультисистемную атрофию, первичную гипералгезию, вторичную гипералгезию, первичную аллодинию, вторичную аллодинию или другую боль, вызванную центральной сенситизацией. Соединения формулы I могут быть использованы, чтобы предотвратить дискинезии, особенно побочные эффекты, сопровождающие нормальные дозы L-ДОФА. Кроме того, соединения формулы I могут быть использованы, чтобы снизить переносимость и/или зависимость от опиоидного лечения боли и для лечения абстинентного синдрома отмены, например, алкоголя, опиоидов и кокаина.
Соединения настоящего изобретения могут быть назначены в профилактически эффективных уровнях доз, чтобы предотвратить состояния, указанные выше, а также чтобы предотвратить другие состояния, опосредованные NR2B рецептором NMDA.
Соединения формулы I могут быть использованы в комбинации с другими лекарственными средствами, которые используют для лечения/предотвращения/подавления или улучшения течения заболеваний или состояний, для которых полезны соединения формулы I. Такие другие лекарственные средства могут вводиться, путем и в количестве, обычно используемыми, одновременно или последовательно, с соединением формулы I. Когда соединения формулы I используют одновременно с одним или более другими лекарственными средствами, предпочтительными являются фармацевтические композиции, содержащие такие другие лекарственные средства в дополнение к соединению формулы I. Соответственно фармацевтические композиции настоящего изобретения включают такие композиции, которые также содержат один или больше активных компонентов в дополнение к соединению формулы I. Примерами других активных компонентов, которые могут быть соединены с соединением формулы I, введены отдельно или в тех же фармацевтических композициях, являются, но ими не ограничиваются, 1) нестероидные противовоспалительные средства; 2) ингибиторы циклооксигеназы 2-го типа, ЦОГ-2 (СОХ-2); 3) антагонисты рецептора брадикинина В1; 4) блокаторы и антагонисты натриевых каналов; 5) антагонисты (оксид азота)-синтазы (NOS); 6) антагонисты сайта глицина; 7) открыватели калиевых каналов; 8) антагонисты рецептора АМРА/каинат; 9) антагонисты кальциевых каналов; 10) модуляторы рецептора ГАМК-А (GABA-A) (например, агонист рецептора ГАМК-А); 11) ингибиторы металлопротеазы матрикса (ММР); 12) тромболитические средства; 13) опиоиды, такие как морфин; 14) нейтрофил-ингибирующий фактор (NIF); 15) L-ДОФА; 16) карбидопа; 17) леводопа/карбидопа; 18) агонисты дофамина, такие как бромкриптин, перголид, прамипексол, ропинирол; 19) антихолинергические средства; 20) амантидин; 21) карбидопа; 22) ингибиторы катехол-О-метилтрансферазы (СОМТ), такие как энтакапон и толкапон; 23) ингибиторы моноаминоксидазы В (МАО-В); 24) агонисты и антагонисты опиатов; 25) агонисты и антагонисты рецепторов 5НТ; 26) агонисты и антагонисты рецепторов NMDA; 27) антагонисты NK1; 28) ингибиторы селективного обратного захвата серотонина (SSRI) и/или ингибиторы селективного обратного захвата серотонина и норэпинефрина (SSNRI); 29) трициклические антидепрессантные лекарственные средства; 30) модуляторы норэпинефрина; 31) литий; 32) вальпроат; 33) нейронтин (габапентин).
Кремы, мази, желе, растворы или суспензии, содержащие настоящие соединения, могут быть использованы для местного применения. Полоскания для ротовой полости и горла включены в композиции местного применения настоящего изобретения.
Препарат, предназначенный для перорального назначения людям, обычно могут содержать от примерно 0,5 мг до примерно 5 г активного компонента, смешанного с подходящим и обычным количеством носителя, которое может изменяться от 5 до 95% от веса композиции. Единичные лекарственные формы могут обычно содержать от примерно 1 мг до примерно 1000 мг активного компонента.
Состояния, указанные в описании, можно лечить или предотвращать введением от примерно 0,01 мг до примерно 140 мг соединений изобретения на килограмм веса тела в день.
Однако конкретный уровень дозы для любого отдельного пациента будет зависеть от многих факторов. Такие факторы включают возраст, вес тела, общее здоровье, пол и питание пациента. Другие факторы включают время и способ введения, скорость выведения, смешивание лекарственного средства и тип, и тяжесть отдельного заболевания, подлежащего терапии. Например, воспалительную боль можно эффективно лечить введением от примерно 0,01 мг до примерно 75 мг соединения изобретения на килограмм веса тела в день или, альтернативно, от примерно 0,5 мг до примерно 3,5 г на пациента в день. Нейропатическую боль можно эффективно лечить введением от примерно 0,01 мг до примерно 125 мг соединения изобретения на килограмм веса тела в день или, альтернативно, от примерно 0,5 мг до примерно 5,5 г на пациента в день.
Сокращения, используемые здесь, если не указано иначе
Способы синтеза
Схема 3
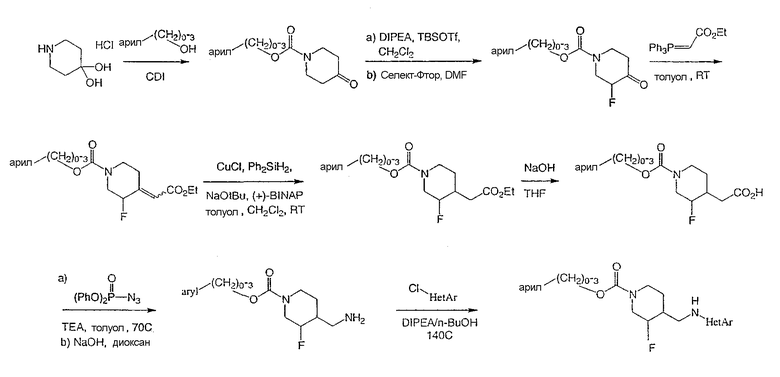
Схема 4
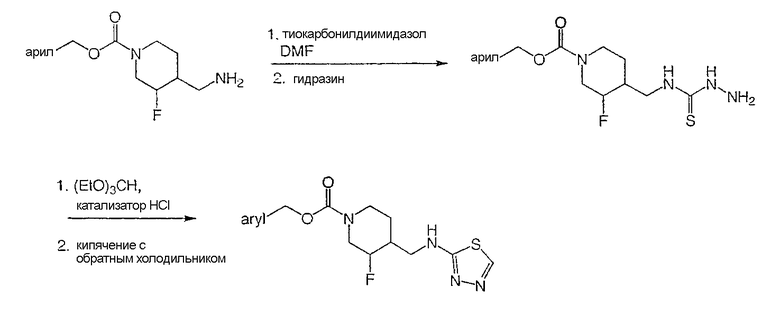
Схема 5
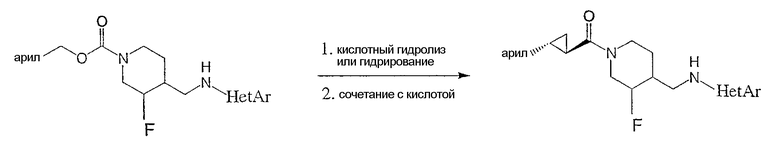
Соединения настоящего изобретения могут быть получены согласно схемам, представленным ниже, а также методикам, представленным в Примерах. Следующие схемы и Примеры далее описывают, но ими не ограничиваются, область действия изобретения.
Примеры 1 и 2
Пример 1
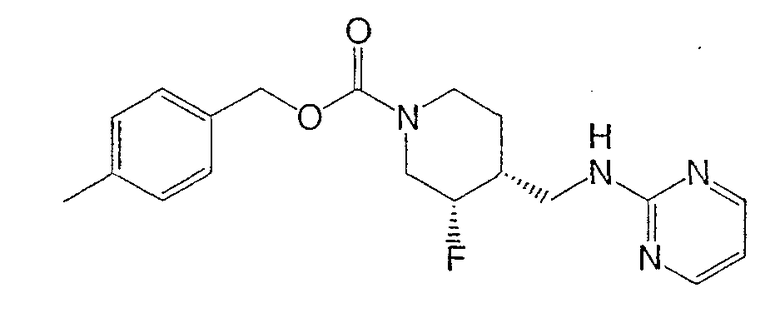
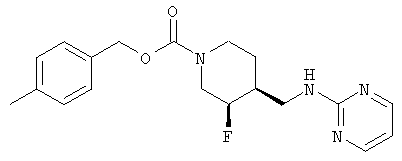
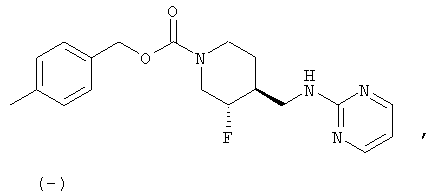
(3S,4R)-4-Метилбензил-3-фтор-4-[(пиримидин-2-иламино)метил]пиперидин-1-карбоксилат
Пример 2
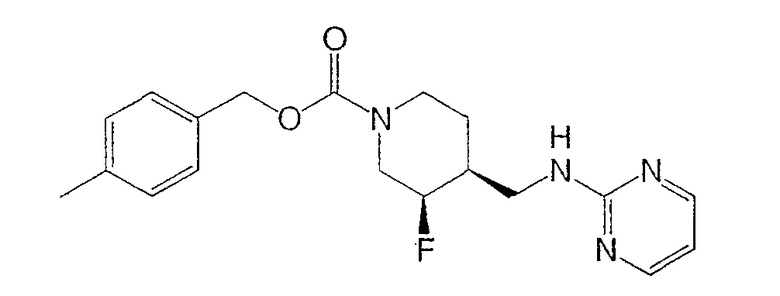
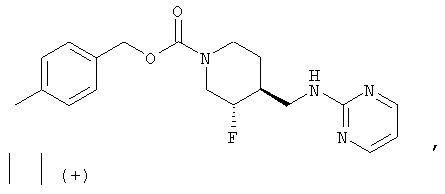
(3R,4S)-4-Метилбензил-3-фтор-4-[(пиримидин-2-иламино)метил]пиперидин-1-карбоксилат
Стадия 1
Получение 4-метилбензил-4-оксопиперидин-1-карбоксилата
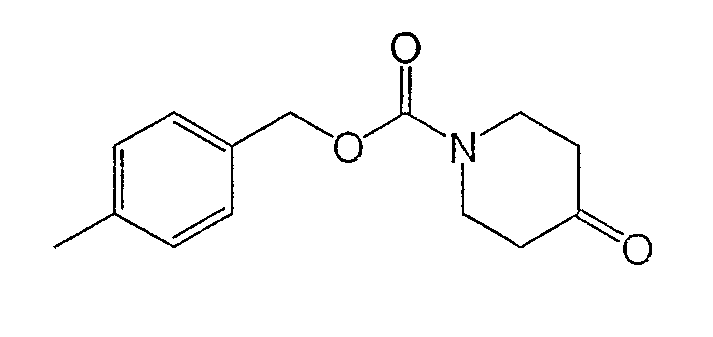
4-Метилбензиловый спирт (37,6 г, 308 ммоль) добавляли к раствору 1,1'-карбонилдиимидазола (50,0 г, 308 ммоль) в ДМФ при комнатной температуре и перемешивали 1 ч. Добавляли гидратгидрохлорид 4-пиперидона (Sigma-Aldrich) (47,0 г, 308 ммоль), образующийся раствор нагревали до 50°С и перемешивали 15 ч. Реакционную смесь разбавляли этилацетатом, промывали 0,1 М HCl, водой (4 раза) и насыщенным раствором NaCl, сушили Na2SO4, фильтровали и концентрировали. Очистка хроматографией на силикагеле (ступенчатое градиентное элюирование: 10, 25 и 50% этилацетата в гексане) дала титульное соединение (42,4 г, 85% выход) в виде прозрачного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,24 (д, 2Н), 7,15 (д, 2Н), 5,08 (с, 2Н), 3,79 (т, 4Н), 2,45 (ушир.с, 4Н), 2,31 (с, 3H) м.д.
Масс-спектроскопия высокого разрешения (электрораспыление) m/z 248,1281 [(M+H)+; рассчитано для C14H18NO3: 248,1287].
Вычислено C14H17NO3: C, 68,03; H, 7,05; N, 5,59. Найдено: C, 68,00; H, 6,93; N, 5,66.
Стадия 2
Получение (±)-4-метилбензил-3-фтор-4-оксопиперидин-1-карбоксилата
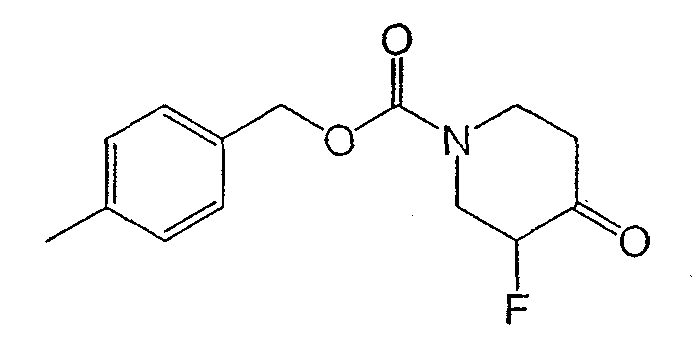
Раствор 4-метилбензил-4-оксопиперидин-1-карбоксилата (21,2 г, 85,7 ммоль) и диизопропилэтиламина (71,3 мл, 428 ммоль) в дихлорметане (425 мл) охлаждали до 0°С и перемешивали. Медленно добавляли TDSOTf (29,5 мл, 129 ммоль), поддерживая температуру внутри ниже 5°С. Добавляли водный NaHCO3 (20 мл) и разделяли слои. Органический слой промывали NaHCO3, водой, насыщенным раствором NaCl, сушили Na2SO4, фильтровали и концентрировали с получением неочищенного TBS эфира енола.
Неочищенный TBS эфир енола растворяли в ДМФ (125 мл) при комнатной температуре. Добавляли реагент Селект-Фтор® (Air Products and Chemicals) (30,4 г, 85,7 ммоль) и реакционную смесь перемешивали 10 мин. Реакционную смесь распределяли между этилацетатом и водой и органический слой промывали водой (три раза). Объединенные водные слои экстрагировали этилацетатом (два раза) и объединенные органические слои сушили Na2SO4, фильтровали и концентрировали. Реакцию повторяли и образовавшиеся продукты реакции соединяли с получением соединения (40 г), которое использовали в следующей стадии без очистки. Данные ЯМР и масс-спектроскопии дают основание считать продукт кетоном, существующим как гидрат.
1H ЯМР (400 МГц, CDCl3) δ 7,24 (м, 2Н), 7,19 (м, 2Н), 5,18 (с, 2Н), 4,81 (ушир.д, 1Н), 4,50 (ушир.д, 1Н), 4,23 (д, 1Н), 3,90 (м, 1Н), 3,60 (м, 1Н), 3,35 (т, 1Н), 2,58 (м, 2Н), 2,35 (с, 3Н) м.д.
Масс-спектроскопия высокого разрешения (электрораспыление) m/z, 284,1292 [(M+H)+; рассчитано для C14H18FNO4: 284,1293].
Вычислено C14H1SFNO4 .1,2 H2O: C, 58,61; H, 6,46; N, 4,88. Найдено: C, 58,28; H, 6,06; N, 4,72.
Стадия 3
Получение
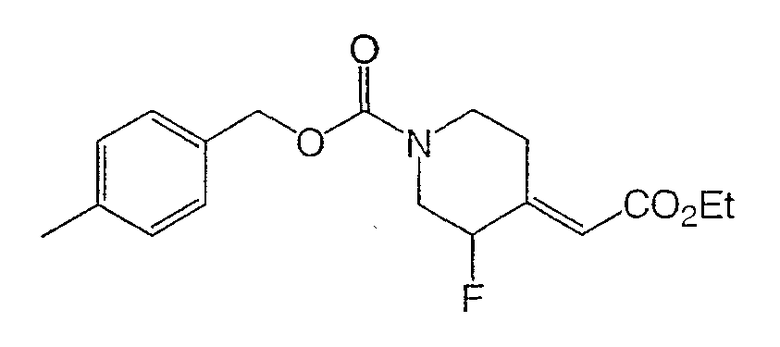
(±)-4-метилбензил-3-фтор-(Е)-4-(2-этокси-2-оксоэтилиден)пиперидин-1-карбоксилата и
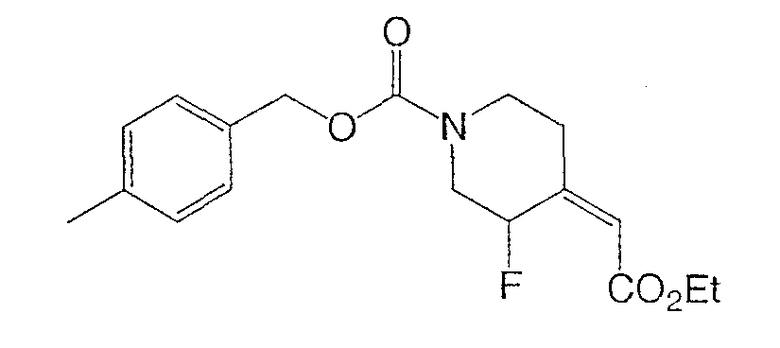
(±)-4-метилбензил-3-фтор-(Z)-4-(2-этокси-2-оксоэтилиден)пиперидин-1-карбоксилата.
К раствору (±)-4-метилбензил-3-фтор-4-оксопиперидин-1-карбоксилата (40 г, 150 ммоль) в толуоле (200 мл) при комнатной температуре добавляли трифенил(карбэтоксиметилен)фосфоран (63,0 г, 181 ммоль) и реакционную смесь перемешивали 1 ч. Реакционную смесь концентрировали и очищали хроматографией на силикагеле (градиентное элюирование: от 10 до 20% этилацетата в гексане) дала (±)-4-метилбензил-3-фтор-(Е)-4-(2-этокси-2-оксоэтилиден)пиперидин-1-карбоксилат и (±)-4-метилбензил-3-фтор-(Z)-4-(2-этокси-2-оксоэтилиден)пиперидин-1-карбоксилат (41,0 г, 78% выход, 3 стадии) в виде смеси E/Z (3:1). Смесь непосредственно применяют в следующей стадии. Маленький образец смеси разделяют хроматографией на силикагеле с целью характеризации.
(±)-4-Метилбензил-3-фтор-(Е)-4-(2-этокси-2-оксоэтилиден)пиперидин-1-карбоксилат: белое твердое вещество
1H ЯМР (400 МГц, CDCl3) δ 7,26 (д, 2Н), 7,17 (д, 2Н), 5,98 (с, 1Н), 5,11 (с, 2Н), 4,85 (м, 1Н), 4,18 (кв., 2Н), 4,08 (ушир.д, 1Н), 3,70 (м, 1Н), 3,55 (м, 1Н) 3,41 (м, 1Н), 3,33, (м, 1Н), 2,63 (ушир.д, 1H), 2,35 (с, 3Н), 1,29 (т, 3Н) м.д.
Масс-спектроскопия высокого разрешения (электрораспыление) m/z 358,1420 [(M+Na)+; рассчитано для C18H22FNO4Na: 358,1425].
Вычислено C18H22FNO4: C, 64,21; H, 6,58; N, 4,27. Найдено: C, 64,46; H, 6,61; N, 4,18.
(±)-4-Метилбензил-3-фтор-(Z)-4-(2-этокси-2-оксоэтилиден)пиперидин-1-карбоксилат: белое твердое вещество
1H ЯМР (400 МГц, CDCl3) δ 7,24 (д, 2Н), 7,15 (д, 2Н), 6,41 (м, 1Н), 5,82 (с, 1Н), 5,11 (д, 2Н), 4,61 (м, 1Н), 4,38 (ушир.д, 1Н), 4,16 (кв., 2Н), 3,05-2,95 (м, 1Н), 2,9-2,75 (м, 2Н), 2,33 (с, 3Н), 2,13 (м, 1Н), 1,27 (т, 3H) м.д.
Масс-спектроскопия высокого разрешения (электрораспыление) m/z 358,1422 [(M+Na)+; рассчитано для C18H22FNO4Na: 358,1425].
Стадия 4
Получение
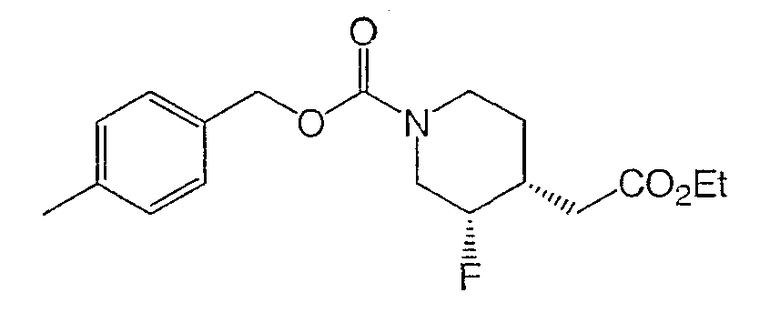
(±)-цис-4-метилбензил-3-фтор-4-(2-этокси-2-оксоэтил)пиперидин-1-карбоксилата и
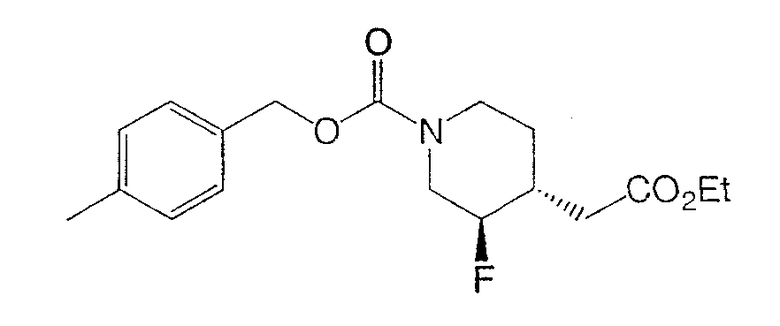
(±)-транс-4-метилбензил-3-фтор-4-(2-этокси-2-оксоэтил)пиперидин-1-карбоксилата.
К раствору смеси олефинов стадии 3 (10,0 г, 29,8 ммоль) в толуоле (160 мл) и дихлорметане (120 мл) добавляли дифенилсилан (5,53 мл, 29,8 ммоль) и (R)-BINAP (1,86 г, 2,98 ммоль). Затем добавляли NaOtBu (0,29 г, 2,98 ммоль) и CuCl (0,30 г, 2,98 ммоль), реакционную смесь защищали от света и перемешивали 15 ч. Добавляли дополнительные порции дифенилсилана (2,76 мл), NaOtBu (0,29 г) и CuCl (0,30 г) и реакционную смесь перемешивали при комнатной температуре 24 ч. Смесь затем фильтровали через целит и концентрировали. Очистка на силикагеле (ступенчатое градиентное элюирование: 5, 10, 15, 25 и 30% этилацетата в гексане) дала регенерированные исходные материалы (3,5 г, 35% выход), (±)-цис-4-метилбензил-3-фтор-4-(2-этокси-2-оксоэтил)пиперидин-1-карбоксилат (5,0 г, 50% выход) и (±)-транс-4-метилбензил-3-фтор-4-(2-этокси-2-оксоэтил)пиперидин-1-карбоксилат (1,2 г, 12%).
(±)-цис-4-метилбензил-3-фтор-4-(2-этокси-2-оксоэтил)пиперидин-1-карбоксилат: прозрачное масло.
1H ЯМР (400 МГц, CDCl3) δ 7,25 (д, 2Н), 7,15 (д, 2Н), 5,10 (с, 2Н), 4,80-4,20 (м, 3Н), 4,15 (кв., 2Н), 3,10-2,73 (м, 2Н), 2,52 (дд, 1Н), 2,35 (с, 3Н), 2,30 (дд, 1Н), 2,10 (м, 1Н), 1,72-1,48 (м, 2Н), 1,29 (т, 3H) м.д.
Масс-спектроскопия высокого разрешения (электрораспыление) m/z 338,1689 [(M+H)+; рассчитано для C18H25FNO4: 338,1762].
(±)-транс-4-метилбензил-3-фтор-4-(2-этокси-2-оксоэтил)пиперидин-1-карбоксилат: прозрачное масло,
1H ЯМР (400 МГц, CDCl3) δ 7,24 (д, 2Н), 7,15 (д, 2Н), 5,08 (с, 2Н), 4,50-3,95 (м, 3Н), 4,15 (кв., 2Н), 2,81 (ушир.т, 2H), 2,70 (ушир.д, 1Н), 2,35 (с, 3Н), 2,17 (м, 2Н), 1,89 (ушир.д, 1Н), 1,25 (м, 1Н), 1,22 (т, 3Н) м.д.
Масс-спектроскопия высокого разрешения (электрораспыление) m/z 338,1699 [(M+H)+; рассчитано для Cl8H25FNO4: 338,1762].
Стадия 5
Получение (±)-цис-3-фтор-1-[(4-метилбензилоксикарбонил)пиперидин-4-ил]уксусной кислоты
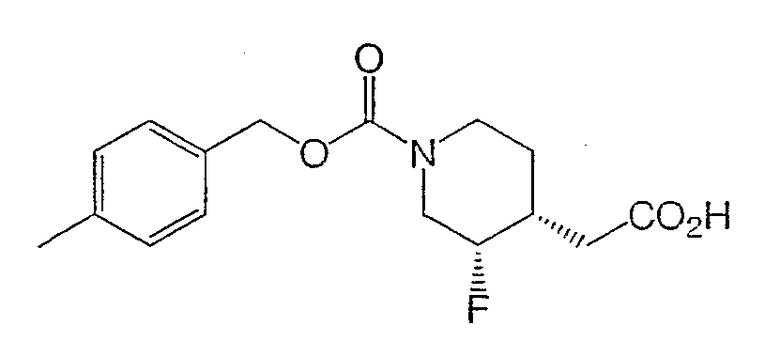
К раствору (±)-цис-4-метилбензил-3-фтор-4-(2-этокси-2-оксоэтил)пиперидин-1-карбоксилата (10,0 г, 29,6 ммоль) в ТГФ (50 мл) добавили водный NaOH (1 М, 50 мл). реакционную смесь перемешивали 5 ч при комнатной температуре и затем разбавляли этилацетатом и 1 М HCl. Слои разделяли, и водный слой дважды экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным раствором NaCl, сушили Na2SO4, фильтровали и концентрировали с получением титульного соединения (9,1 г) в виде белого твердого вещества, которое использовали в следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3) δ 7,24 (д, 2Н), 7,15 (д, 2Н), 5,08 (с, 2Н), 4,79-4,16 (м, 3Н), 3,05-2,75 (м, 2Н), 2,59 (дд, 1Н), 2,36 (дд, 1Н), 2,31 (с, 3Н), 2,20-2,02 (м, 1Н), 1,60 (м, 2Н) м.д.
Масс-спектроскопия высокого разрешения (электрораспыление) m/z 310,1457 [(M+H)+; рассчитано для C16H21FNO4: 310,1449].
Вычислено C16H20FNO4 .0,15 H2O: C, 62,13; H, 6,52; N, 4,53. Найдено: C, 61,55; H, 6,37; N, 4,41.
Стадия 6
Получение (±)-цис-4-метилбензил-3-фтор-4-(аминометил)пиперидин-1-карбоксилата
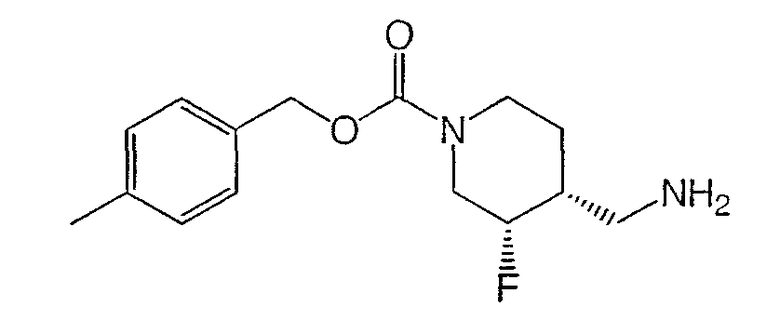
К суспензии неочищенной (±)-цис-3-фтор-1-[(4-метилбензилоксикарбонил)пиперидин-4-ил]уксусной кислоты (9,1 г, 29,4 ммоль) в толуоле добавляли триэтиламин (10,2 мл, 73,5 ммоль) и дифенилфосфорилазид (9,52 мл, 44,1 ммоль). Реакционную смесь нагревали до 70°С и перемешивали 20 мин. Добавляли смесь диоксана (80 мл) и 1 М NaOH (80 мл) и реакционную смесь охлаждали до комнатной температуры. Реакционную смесь упаривали, чтобы удалить диоксан, и 3 раза экстрагировали этилацетатом, сушили Na2SO4, фильтровали и концентрировали. Остаток суспендировали в дихлорметане, перемешивали 30 мин и белый осадок отфильтровывали. Фильтрат концентрировали с получением неочищенного продукта (7,5 г) в виде желтого масла, которое непосредственно использовали в следующей стадии. Аналитический образец очищали хроматографией на силикагеле (градиентное элюирование: от CH2Cl2 до CH2Cl2:MeOH:NH4OH 80:20:2) для характеризации.
1H ЯМР (400 МГц, CDCl3) δ 7,24 (д, 2H), 7,15 (д, 2Н), 5,08 (с, 2Н), 4,90-4,18 (м, 3Н), 2,95-2,75 (м, 2Н), 2,79 (дд, 1Н), 2,70 (дд, 1Н), 2,35 (с, 3Н), 1,59 (м, 3Н) м.д.
Масс-спектроскопия высокого разрешения (электрораспыление) m/z 281,1658 [(М+Н)+; рассчитано для C15H22FN2O2: 281,1660].
Стадия 7
Получение
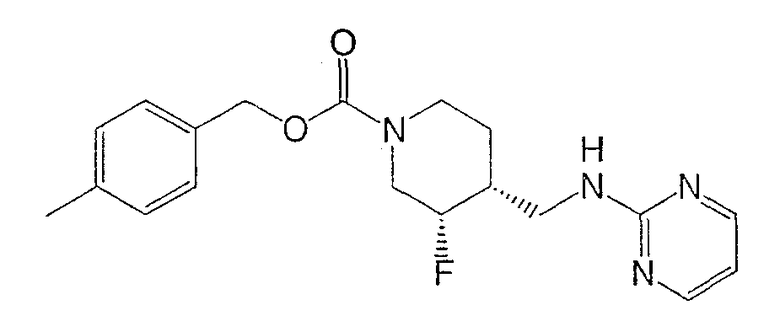
(3S,4R)-4-метилбензил-3-фтор-4-[(пиримидин-2-иламино)метил]пиперидин-1-карбоксилата и
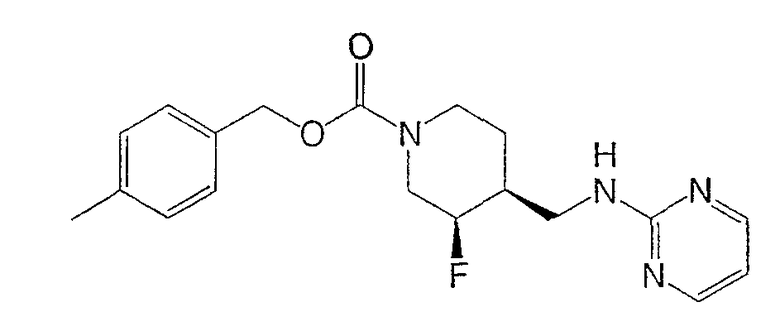
(3R,4S)-4-метилбензил-3-фтор-4-[(пиримидин-2-иламино)метил]пиперидин-1-карбоксилата
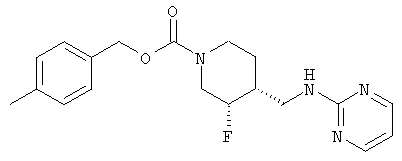
Смесь неочищенного (±)-цис-4-метилбензил-3-фтор-4-(аминометил)пиперидин-1-карбоксилата (стадия 6, 3,7 г, 13,2 ммоль) и 2-хлорпиримидина (1,51 г, 13,2 ммоль) в н-утанолдиизопропилэтиламине (1:1, 13 мл) загружали в две ампулы. Ампулы запаивали, нагревали до 140°С и перемешивали 2 ч. После охлаждения реакционные смеси объединяли и разбавляли этилацетатом и насыщенным NaHCO3. Слои разделяли, органический слой промывали водой и насыщенным раствором NaCl, сушили Na2SO4, фильтровали и концентрировали. Очистка хроматографией на силикагеле (градиентное элюирование: от этилацетат-гексан 1:1 до этилацетата) дала рацемический цис-4-метилбензил-3-фтор-4-[(пиримидин-2-иламино)метил]пиперидин-1-карбоксилат (6,9 г, 65% выход, 3 стадии) в виде белого твердого вещества.
Энантиомеры разделяли препаративной ВЭЖХ на колонке ChiralPak AD (5 см × 50 см, 20 мкМ), используя элюент MeOH-MeCN (15:85, 150 мл/мин). Соль гидрохлорид Примера 1 получали растворением (3S,4R)-цис-4-метилбензил-3-фтор-4-[(пиримидин-2-иламино)метил]пиперидин-1-карбоксилата (6,9 г, 19,3 ммоль) в изопропаноле (100 мл) при 65°С. Добавляли раствор HCl в изопропаноле (1,608 М, 12,6 мл, 20,2 ммоль), раствор медленно охлаждали до комнатной температуры за 15 ч, добавляли Et2O (25 мл), смесь перемешивали 3 ч, охлаждали до 0°С, перемешивали 1 ч и фильтровали, получая гидрохлорид (3S,4R)-4-метилбензил-3-фтор-4-[(пиримидин-2-иламино)метил]пиперидин-1-карбоксилата в форме белого твердого вещества (7,0 г, 92% извлечения).
(3S,4R)-4-метилбензил-3-фтор-4-[(пиримидин-2-иламино)метил]пиперидин-1-карбоксилат·HCl
[α]D -36,4° (с 0,17, MeOH).
Т. пл. 149-150°С.
1H ЯМР (400 МГц, CD3OD) δ 8,58 (ушир.с, 2Н), 7,21 (д, 2Н), 7,17 (д, 2Н), 6,99 (т, 1Н), 5,06 (с, 2Н), 4,79 (м, 1Н), 4,42 (т, 1Н), 4,21 (д, 1Н), 3,60 (дд, 1Н), 3,50 (дд, 1Н), 3,15-2,80 (м, 2Н), 2,30 (с, 3H), 2,10 (м, 1Н), 1,61 (м, 2Н) м.д.
Масс-спектроскопия высокого разрешения (электрораспыление) m/z 359,1879 [(M+H)+; рассчитано для C19H24FN4O2: 359,1878].
Вычислено C19H23FN4O2 .HC1.0,2 H2O: C, 57,27; H, 6,17; N, 14,06. Найдено: C, 57,22; H, 6,37; N, 14,16.
(3R,4S)-4-метилбензил-3-фтор-4-[(пиримидин-2-иламино)метил]пиперидин-1-карбоксилат·HCl
[α]D +34,9° (с 0,18, MeOH).
Т. пл. 149-150°С.
1H ЯМР (400 МГц, CD3OD) δ 8,58 (ушир.с, 2Н), 7,21 (д, 2Н), 7,17 (д, 2Н), 6,99 (т, 1Н), 5,06 (с, 2Н), 4,79 (м, 1Н), 4,42 (т, 1Н), 4,21 (д, 1Н), 3,60 (дд, 1Н), 3,50 (дд, 1Н), 3,15-2,80 (м, 2Н), 2,30 (с, 3H), 2,10 (м, 1Н), 1,61 (м, 2Н) м.д.
Масс-спектроскопия высокого разрешения (электрораспыление) m/z 359,1870 [(M+H)+; рассчитано для C19H24FN4O2: 359,1878].
Вычислено C19H23FN4O2 .HCl.0,5H2O: С, 56,50; H, 6,24; N, 13,87. Найдено: C, 56,68; H, 6,27; N, 13,80.
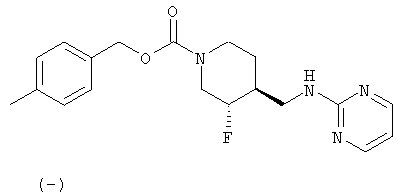
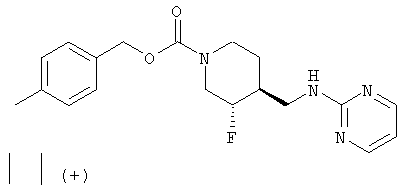
Примеры 3 и 4
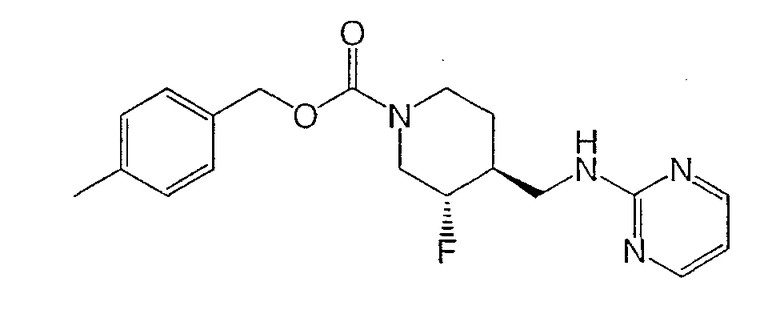
(-)-транс-4-Метилбензил-3-фтор-4-[(пиримидин-2-иламино)метил]пиперидин-1-карбоксилат (пример 3) и (+)-транс-4-метилбензил-3-фтор-4-[(пиримидин-2-иламино)метил]пиперидин-1-карбоксилат (пример 4)
Титульные соединения получали из (±)-транс-4-метилбензил-3-фтор-4-(2-этокси-2-оксоэтил)пиперидин-1-карбоксилат (Примеры 1 и 2, Стадия 4) по методике, описанной в Стадиях 5, 6 и 7 Примеров 1 и 2.
(-)-транс-4-Метилбензил-3-фтор-4-[(пиримидин-2-иламино)метил]пиперидин-1-карбоксилат·HCl
[α]D -11,5° (с 0,22, MeOH).
Т. пл. 113-114°С.
1H ЯМР (400 МГц, CD3OD) δ 8,80-8,39 (м, 2Н), 7,24 (д, 2Н), 7,15 (д, 2Н), 7,00 (т, 1Н), 5,08 (с, 2H), 4,49-4,21 (м, 2Н), 4,00 (д, 1Н), 3,81 (дд, 1H), 3,58 (дд, 1Н), 2,95 (м, 1Н), 2,31 (с, 3Н), 2,12 (м, 1Н), 1,90 (м, 1Н), 1,34 (м, 1Н) м.д.
Масс-спектроскопия высокого разрешения (электрораспыление) m/z 359,1867 [(M+H)+; рассчитано для C19H24FN4O2: 359,1878].
Вычислено C19H23FN4O2 .HCl.H2O: C, 55,27; H, 6,35; N, 13,57. Найдено: C, 55,08; H, 6,11; N, 13,36.
(+)-транс-4-метилбензил-3-фтор-4-[(пиримидин-2-иламино)метил]пиперидин-1-карбоксилат·HCl
[α]D +16,3° (с 0,17, MeOH).
Т. пл. 113-114°С.
1H ЯМР (400 МГц, CD3OD) δ 8,80-8,39 (м, 2Н), 7,24 (д, 2Н), 7,15 (д, 2Н), 7,00 (т, 1Н), 5,08 (с, 2H), 4,49-4,21 (м, 2Н), 4,00 (д, 1Н), 3,81 (дд, 1H), 3,58 (дд, 1Н), 2,95 (м, 1Н), 2,31 (с, 3Н), 2,12 (м, 1Н), 1,90 (м, 1Н), 1,34 (м, 1Н) м.д.
Масс-спектроскопия высокого разрешения (электрораспыление) m/z 359,1873 [(M+H)+; рассчитано для C19H24FN4O2: 359,1878].
Вычислено C19H23FN4O2 .HCl.H2O: C, 55,27; H, 6,35; N, 13,57. Найдено: C, 55,18; H, 6,11; N, 13,38.
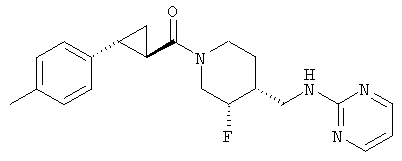
Пример 5
Получение
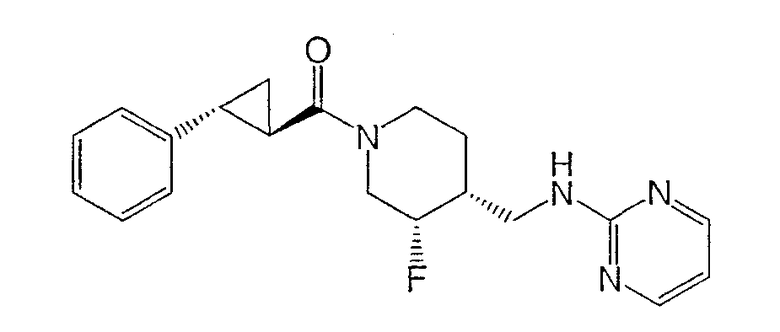
(3S,4R)-N-[(3-цис-фтор-1-{[(1R,2 R)-2-фенилциклопропил]карбонил}иперидин-4-ил)метил]пиримидин-2-амина
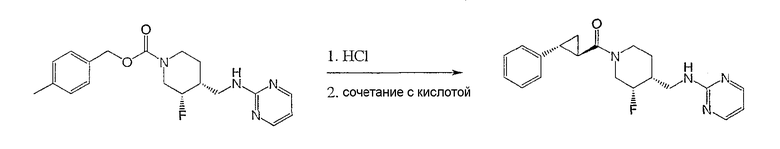
(3S,4R)-4-метилбензил-3-фтор-4-[(пиримидин-2-иламино)метил]пиперидин-1-карбоксилат (Пример 1) (400 мг, 1,16 ммоль) растворяли в 3 н. HCl (4 мл), нагревали до 90°С и перемешивали 1 ч. Реакционную смесь концентрировали и неочищенный гидрохлорид амина растворяли в ДМФ (3 мл) при комнатной температуре и добавляли (1R,2R)-2-фенилциклопропанкарбоновую кислоту (188 мг, 1,16 ммоль) (получение описано T.N. Riley and C.G. Brier, J. Org. Chem., 15, 1187-1188, 1972), EDC (222 мг, 1,16 ммоль), HOAt (158 мг, 1,16 ммоль) и триэтиламин (323 мкл, 2,32 ммоль). Реакционную смесь нагревали до 70°С и перемешивали 10 мин. После охлаждения до комнатной температуры реакционную смесь разбавляли этилацетатом и водой. Слои разделяли, и органический слой промывали водой и насыщенным раствором NaCl, сушили Na2SO4, фильтровали и концентрировали. Очистка хроматографией на силикагеле (градиентное элюирование: от этилацетат-гексан 1:1 до этилацетат/метанол/NH4OH 90:10:1) дала титульное соединение (320 мг, 78% выход) в виде белого твердого вещества. Соединение характеризовали как соль гидрохлорид, которую получали по методике Примера 1, стадия 7.
[α]D -164° (с 0,22, MeOH).
Т. пл. 127-128°С.
1H ЯМР (400 МГц, CD3OD) δ 8,59 (ушир.с, 2Н), 7,25 (м, 2Н), 7,17 (м, 3Н), 7,01 (т, 1Н), 4,85 (м, 2H), 4,65-4,30 (м, 2Н), 3,55 (м, 2Н), 3,30 (м, 1Н), 2,80 (м, 1Н), 2,39 (м, 1Н), 2,30-2,10 (м, 2Н), 1,75 (м, 1Н), 1,55 (м, 2Н), 1,33 (м, 1Н) м.д.
Масс-спектроскопия высокого разрешения (электрораспыление) m/z 355,1925 [(M+H)+; рассчитано для C20H24FN4O: 355,1929].
Вычислено C20H23FN4O.HCl.H2O: C, 58,75; H, 6,41; N, 13,70. Найдено: C, 58,57; H, 6,44; N, 13,54.
Пример 6
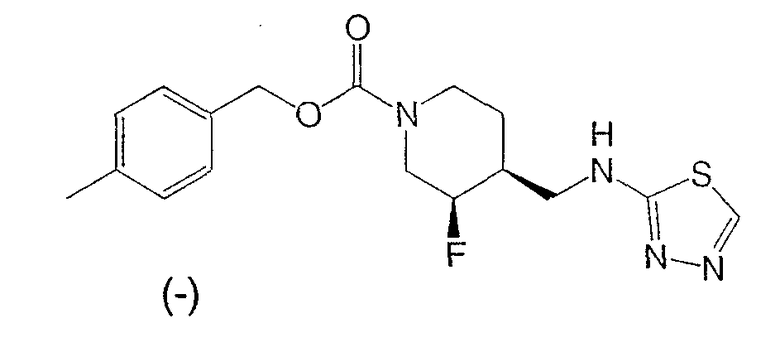
(-)-цис-4-Метилбензил-3-фтор-4-[(1,3,4-тиадиазол-2-иламино)метил]пиперидин-1-карбоксилат
Стадия 1
Получение
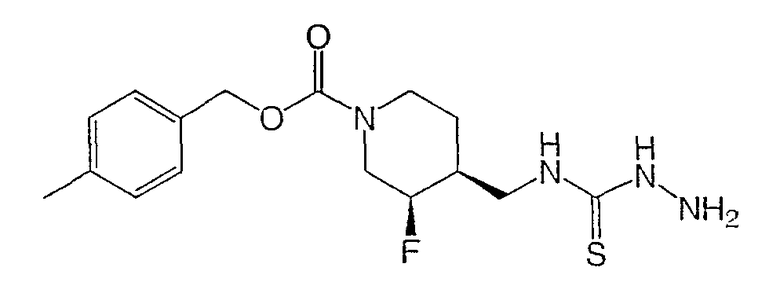
(±)-цис-4-метилбензил-3-фтор-4-{[(гидразинотиокарбонил)амино]метил}иперидин-1-карбоксилата
Раствор (±)-цис-4-метилбензил-3-фтор-4-(аминометил)пиперидин-1-карбоксилата (Пример 1, стадия 6) (200 мг, 0,71 ммоль) в ДМФ (2 мл) добавляли по каплям к перемешиваемому и охлаждаемому (0°С) раствору тиокарбонилдиимидазола (127 мг, 0,71 ммоль) в ДМФ (4 мл). Реакционную смесь перемешивали 30 мин, охлаждающую баню удаляли и реакционную смесь перемешивали 1 ч при комнатной температуре. Затем добавляли гидразин (0,666 мл, 2,14 ммоль) и реакционную смесь перемешивали еще 30 мин, затем выливали в воду и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaHCO3, водой и насыщенным раствором NaCl, сушили Na2SO4, растворитель удаляли в вакууме и получали неочищенный твердый продукт. Твердый продукт растирали с метанолом (10 мл), фильтровали и получали 180 мг титульного соединения.
1H ЯМР (300 МГц, CDCl3) δ 7,67 (ушир.с, 2Н), 7,25 (д, 2Н), 7,19 (д, 2Н), 5,10 (с, 2Н), 4,80-4,20 (ушир.м, 3Н), 3,75 (с, 2Н), 3,80-3,55 (м, 2Н), 3,05-2,70 (м, 2Н), 2,38 (с, 3Н), 2,10 (м, 1Н), 1,60 (м, 2Н) м.д.
Стадия 2
(-)-цис-4-Метилбензил-3-фтор-4-[(1,3,4-тиадиазол-2-иламино)метил]пиперидин-1-карбоксилат
Суспензию (±)-цис-4-метилбензил-3-фтор-4-{[(гидразинотиокарбонил)амино]метил}иперидин-1-карбоксилата (180 мг, 0,51 ммоль) в этаноле (10 мл) обрабатывали триэтилортоформатом (151 мг, 1,02 ммоль) и концентрированной HCl (0,01 мл) и перемешивали при комнатной температуре под азотом 18 ч. После растворения всего твердого вещества реакционную смесь кипятили с обратным холодильником 1,5 ч, охлаждали до комнатной температуры и упаривали летучие продукты. Остаток распределяли между этилацетатом и разбавленным раствором NaHCO3. Органический слой промывали насыщенным раствором NaCl, сушили Na2SO4, растворитель упаривали. Неочищенный продукт очищали хроматографией на силикагеле (градиентное элюирование от 50% этилацетат/гексан до 20% метанол/этилацетат) и получали титульное соединение в виде белого твердого вещества. (-)-Нантиомер отделяют и соль гидрохлорид получают, как описано в Примерах 1 и 2, Стадия 7.
[α]D -47,4° (с 0,17, MeOH).
Т. пл. 115-117°С.
1H ЯМР (400 МГц, CDCl3) δ 8,37 (с, 1Н), 7,25 (д, 2Н), 7,16 (д, 2Н), 6,57 (ушир.с, 1Н), 5,10 (д, 2Н), 4,79 (дд, 1Н), 4,56 (м, 1Н), 4,29 (м, 1Н), 3,44 (д, 2Н), 3,10-2,77 (м, 2Н), 2,35 (с, 3Н), 2,2 (м, 1Н), 1,63 (м, 2Н).
Масс-спектроскопия высокого разрешения (электрораспыление)) m/z 365,1437 [(M+H)+; рассчитано для C17H21FN4O2S: 365,1442].
Вычислено C17H21FN4O2S: C, 56,03; H, 5,81; N, 15,37. Найдено: C, 55,82; H, 5,74; N, 15,11.
Пример 7
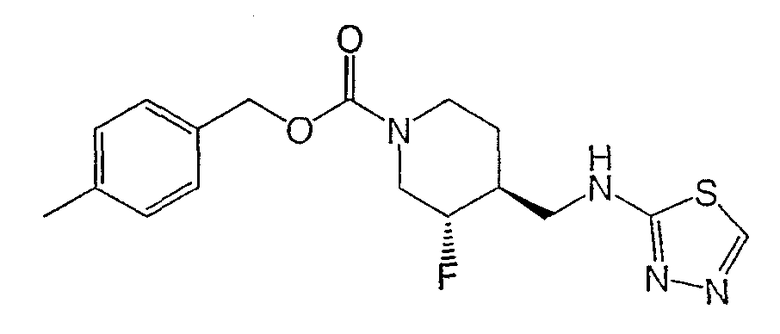
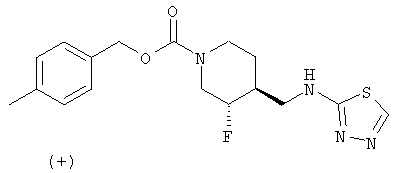
(+)-транс-4-Метилбензил-3-фтор-4-[(1,3,4-тиадиазол-2-иламино)метил]пиперидин-1-карбоксилат
Титульное соединение получали из (±)-транс-4-метилбензил-3-фтор-4-(2-этокси-2-оксоэтил)пиперидин-1-карбоксилата (Примеры 1 и 2, Стадия 4), используя методики, описанные в Примерах 1 и 2, Стадии 5 и 6, с последующим образованием тиадиазола, как описано в Примере 6. (+)-Энантиомер отделяли хиральной ВЭЖХ-хроматографией, как описано в Примерах 1 и 2, Стадия 7.
[α]D +37,1° (с 0,56, MeOH).
Т. пл. 93-95°С.
1H ЯМР (400 МГц, CDCl3) δ 8,36 (с, 1Н), 7,28 (д, 2Н), 7,19 (д, 2Н), 6,56 (ушир.с, 1Н), 5,04 (с, 2Н), 4,45 (м, 2Н), 4,17 (м, 1Н), 3,56 (м, 2Н), 2,80 (м, 2Н), 2,35 (с, 3Н), 2,10 (м, 1Н), 1,96 (м, 1Н), 1,39 (м, 1H) м.д.
Масс-спектроскопия высокого разрешения (электрораспыление)) m/z 365,1432 [(M+H)+; рассчитано для C17H22FN4O2S: 365,1442].
Вычислено C17H22FN4O2S.0,25 H2O: C, 55,34; H, 5,87; N, 15,19. Найдено: C, 55,50; H, 5,61; N, 14,81.
| название | год | авторы | номер документа |
|---|---|---|---|
| 3,3-ДИФТОРПИПЕРИДИНКАРБАМАТНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АНТАГОНИСТОВ NR2B NMDA-РЕЦЕПТОРА | 2016 |
|
RU2735277C2 |
| АЗАГЕТЕРОЦИКЛЫ, ВКЛЮЧАЮЩИЕ ФРАГМЕНТ ПИПЕРИДИН-2-ИЛА-, ФОКУСИРОВАННЫЕ БИБЛИОТЕКИ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2259364C1 |
| ПРОИЗВОДНЫЕ ПИРИДИНА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ БЫСТРО ДИССОЦИИРУЮЩИХСЯ АНТАГОНИСТОВ ДОПАМИНОВЫХ РЕЦЕПТОРОВ 2 | 2008 |
|
RU2480462C2 |
| ЗАМЕЩЕННЫЕ (2R,3R,5R)-3-ГИДРОКСИ-(5-ПИРИМИДИН-1-ИЛ)ТЕТРАГИДРОФУРАН-2-ИЛМЕТИЛ АРИЛ ФОСФОРАМИДАТЫ | 2013 |
|
RU2553996C1 |
| АМИДНЫЕ ПРОИЗВОДНЫЕ КАРБОНОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2001 |
|
RU2271361C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ БЕНЗОПИПЕРАЗИН, В КАЧЕСТВЕ ИНГИБИТОРОВ БРОМОДОМЕНОВ ВЕТ | 2014 |
|
RU2720237C2 |
| ПИРРОЛИДИНОВЫЕ И ПИПЕРИДИНОВЫЕ СОЕДИНЕНИЯ | 2020 |
|
RU2803455C1 |
| ПРОИЗВОДНОЕ ИНДАЗОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2817737C2 |
| КЛАСС КОНДЕНСИРОВАННЫХ КОЛЬЦЕВЫХ СОЕДИНЕНИЙ И ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2021 |
|
RU2831125C1 |
| НОВЫЕ ИНГИБИТОРЫ СЕРИН-ТРЕОНИНОВЫХ КИНАЗ, В ТОМ ЧИСЛЕ ДЛЯ ЛЕЧЕНИЯ ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ И ТУБЕРКУЛЕЗА | 2016 |
|
RU2633032C1 |
Изобретение относится к новым соединениям формулы (1)
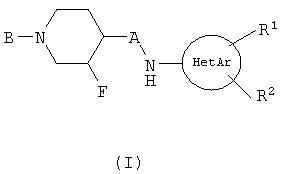
или к его фармацевтически приемлемым солям, в которой HetAr представляет собой пиримидинил или тиадиазолил; R1 и R2 представляют Н; А представляет собой С1-С2-алкил; В представляет арил(СН2)0-3-O-С(O)- или арилциклопропил-С(О)-, в которых арил может быть замещен 1-5 заместителями, каждый заместитель представляет C1-C4-алкил. Изобретение также относится к фармацевтической композиции и к применению соединений по п.1. Технический результат - получение новых соединений, а также фармацевтической композиции, обладающей действием антагонистов NMDA/NR2B. 4 н. и 9 з.п. ф-лы.
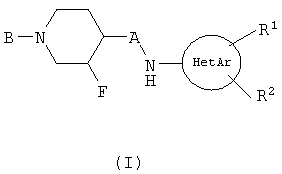
или его фармацевтически приемлемая соль, в которой
HetAr представляет собой пиримидинил или тиадиазолил;
R1 и R2 представляют Н;
А представляет собой С1-С2-алкил;
В представляет арил(СН2)0-3-O-С(O)- или арилциклопропил-С(О)-, в которых арил может быть замещен 1-5 заместителями, каждый заместитель представляет С1-С4-алкил.
или его фармацевтически приемлемые соли.
или его фармацевтически приемлемая соль.
или его фармацевтически приемлемая соль.
или его фармацевтически приемлемая соль.
или его фармацевтически приемлемая соль.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| СОЕДИНЕНИЕ ПИРИМИДИНА(ВАРИАНТЫ) И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ СРОДСТВОМ К ДОПАМИН D3-РЕЦЕПТОРАМ | 1995 |
|
RU2172736C2 |

