ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ПРИНАДЛЕЖИТ ИЗОБРЕТЕНИЕ
Настоящее изобретение принадлежит к области медицины и относится к производному индазола, способу его получения и его применению в медицине. В настоящем изобретении раскрыто применение производного индазола в качестве модулятора рецептора эстрогена для предотвращения и/или лечения заболевания или расстройства, опосредованного рецептором эстрогена или зависимого от рецептора эстрогена, где заболевание особенно предпочтительно представляет собой рак молочной железы.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Рак молочной железы является одной из наиболее распространенных злокачественных опухолей у женщин. Согласно статистическим данным интерактивной веб-платформы GLOBALCAN (Global Cancer Observatory) в 2012 г. (СА CANCER J CLIN 2015; 65:87-108), ежегодно в мире отмечается приблизительно 1,7 млн. новых случаев и 520000 смертельных исходов. Независимо от заболеваемости и смертности рак молочной железы занимает первое место среди злокачественных опухолей у женщин. Согласно Ежегодному отчету ракового регистра Китая (2017), выпущенному Национальным онкологическим центром Китая, рак молочной железы занимает первое место по частоте новых случаев злокачественных опухолей у женщин, которая составляет около 279000 новых случаев в год, ежегодно увеличиваясь примерно на 2%.
Около 70% пациентов с раком молочной железы страдают положительным по рецептору эстрогена (ЭР-положительным) раком молочной железы. Среди различных видов терапии для этих пациентов с раком молочной железы важное место занимает эндокринная терапия. Существует три типа эндокринной терапии, а именно: ингибитор ароматазы (ИА), который может ингибировать преобразование андрогенов в эстрогены и снижать уровень эстрогенов в организме; селективный модулятор рецептора эстрогена (СМРЭ), который вызывает антагонистическую активность рецептора эстрогена; и селективный супрессор рецептора эстрогена (ССРЭ), который может не только вызывать антагонистическую активность рецептора эстрогена, но также стимулирует деструкцию рецептора (Pharmacol Then 2017 Dec 28). Хотя эндокринная терапия является наиболее предпочитаемой терапией для положительного по рецептору эстрогена рака молочной железы, примерно у 30% пациентов, получающих адъювантную терапию, будет возникать рецидивирование, и почти у всех пациентов с метастатическим раком молочной железы будет развиваться резистентность и заболевание будет прогрессировать. Существует два основных типа механизмов резистентности к эндокринной терапии. Один тип механизмов фокусируется на самом сигнальном пути рецептора эстрогена, включая активирующую мутацию и амплификацию гена ESR1, кодирующего рецептор эстрогена, слияние кодирующего рецептор эстрогена гена ESR1 с другими генами, нарушение совместно опосредующих факторов рецептора эстрогена и факторов его последующего пути, которые контролируют клеточный цикл, и т.д. Другой тип механизмов включает в себя активацию сигнальных путей, которые перекрестно реагируют с сигнальным путем рецептора эстрогена, таким как путь рецептора фактора роста (Nat Rev Clin Oncol. 2015 Oct; 12(10): 573-83).
В двух исследованиях, проведенных в 2013 г., показано, что мутация в гене ESR1 была обнаружена у 11-55% пациентов с положительным по рецептору эстрогена метастатическим раком молочной железы, которые получили терапию ингибиторами ароматазы. В дополнительных исследованиях обнаружено, что мутантный рецептор может быть фосфорилирован независимо от эстрогена и обеспечивает такое влияние на транскрипцию, при котором после инокуляции эстрогензависимой линией клеток MCF-7 опухоль может расти в организме независимо от эстрогена. Кроме того, мутантный рецептор снизит активность СМРЭ тамоксифена и ССРЭ фулвестранта. Таким образом, мутация в гене ESR1 может представлять собой один из механизмов лекарственной резистентности при положительном по рецептору эстрогена раке молочной железы (Nat Rev Clin Oncol. 2015 Oct; 12(10):573-83 и Nat Genet 2013; 45:1439-45). В последующих исследованиях у пациентов с положительным по рецептору эстрогена раком молочной железы обнаружена некоторая доля мутаций в гене ESR1, которая составляла около 30%. В клиническом исследовании BOLERO-2 мутации в генах ER Y537S и ER D538G были обнаружены в циркулирующей опухолевой ДНК (цоДНК) 29% пациентов с положительным по рецептору эстрогена метастатическим раком молочной железы, который прогрессировал после лечения ингибиторами ароматазы (ИА). В группе, получавшей только эксеместан, беспрогрессивная выживаемость (БПВ) и общая выживаемость (ОВ) пациентов с мутациями была короче, чем у пациентов без мутаций (Nat Genet 2013;45:1446-51).
Таким образом, мутация в гене ESR1 главным образом встречается у пациентов с метастатическим, положительным по рецептору эстрогена раком молочной железы, который прогрессировал после лечения ИА. Данные пациенты были уже нечувствительными к лечению ИА. Таким образом, существует необходимость в разработке антагонистов рецептора эстрогена, которые нацелены на мутацию в гене ESR1.
Наилучший в этом классе препаратов ковалентно связывающий антагонист рецептора эстрогена Н3В-6545, разработанный компанией Eisai, обладает сильным ингибирующим действием на рецептор эстрогена дикого типа и мутантный рецептор и может проявлять пролонгированную эффективность посредством ковалентного связывания с рецептором. В настоящее время он проходит клинические исследования I и II фазы. Опубликованные в настоящее время заявки на патенты, относящиеся к нацеленным на мутацию в гене ESR1 антагонистам рецептора эстрогена, включают WO 2016196346 и WO 2016196342.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Объектом настоящего изобретения является разработка соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли:
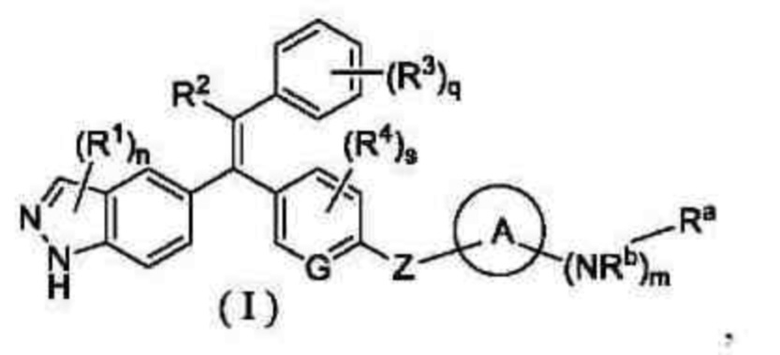
где:
G представляет собой СН или N;
Z выбран из группы, состоящей из химической связи, CR5R6, -О-(CH2)t - и -NR7-(СН2)t-;
кольцо А выбрано из группы, состоящей из циклоалкила и гетероциклила;
Ra выбран из группы, состоящей из -CH2CH=CHC(O)NR8R9, -C(O)CH=CR10R11 и -C(O)C≡CR12;
Rb выбран из группы, состоящей из атома водорода и алкила;
каждый R1 идентичен или отличается, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R2 выбран из группы, состоящей из атома водорода, галогена, алкила, галогеналкила, алкокси, амино, циано, нитро, карбокси, формила, гидрокси, гидроксиалкила, циклоалкила, арила и гетероарила;
каждый R3 идентичен или отличается, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, галогеналкила, алкокси, циано, амино, нитро, карбокси, формила, гидрокси, гидроксиалкила, NR13C(O)R14, C(O)NR13R14, SO2R15, циклоалкила, гетероциклила, арила и гетероарила;
каждый R4 идентичен или отличается, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, галогеналкила, алкокси, циано, амино, нитро, карбокси, формила, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R5 и R6 идентичны или отличаются, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, галогеналкила, алкокси, циано, амино, нитро, карбокси, формила, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R7 выбран из группы, состоящей из атома водорода, алкила, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R8 и R9 идентичны или отличаются, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
или R8 и R9 вместе с атомом азота, к которому они присоединены, образуют гетероциклил, где гетероциклил необязательно содержит от одного до двух идентичных или различных гетероатомов, выбранных из группы, состоящей из N, О и S, в дополнение к указанному атому азота, и указанный гетероциклил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из алкила, алкокси, галогена, амино, циано, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R10 и R11 идентичны или отличаются, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, галогеналкила, алкокси, циано, амино, нитро, карбокси, формила, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R12 выбран из группы, состоящей из атома водорода, алкила, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R13 и R14 идентичны или отличаются, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, галогеналкила, алкокси, циано, амино, нитро, карбокси, формила, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R15 выбран из группы, состоящей из атома водорода, алкила, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
m равно 0 или 1;
n равно 0, 1, 2 или 3;
q равно 0, 1, 2, 3, 4 или 5;
s равно 0, 1, 2 или 3; и
t равно 0, 1, 2, 3, 4, 5 или 6.
В предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смеси, или их фармацевтически приемлемой соли, кольцо А выбрано из группы, состоящей из С3-С6 циклоалкила и 3-6-членного гетероциклила, где гетероциклил содержит от одного до трех гетероатомов, выбранных из группы, состоящей из N, O и S.
В другом предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смеси, или их фармацевтически приемлемой соли, кольцо А выбрано из группы, состоящей из циклопропила, циклопентила, циклогексила, тетрагидропирролила и пиперидинила.
В другом предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смеси, или их фармацевтически приемлемой соли, кольцо А выбрано из группы, состоящей из 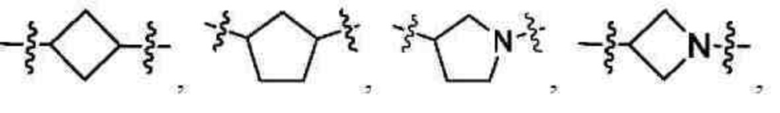
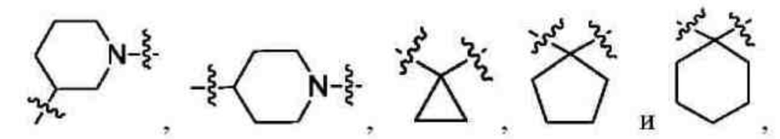 и предпочтительно выбрано из группы, состоящей из
и предпочтительно выбрано из группы, состоящей из 
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль, представляет собой соединение формулы (II) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль:
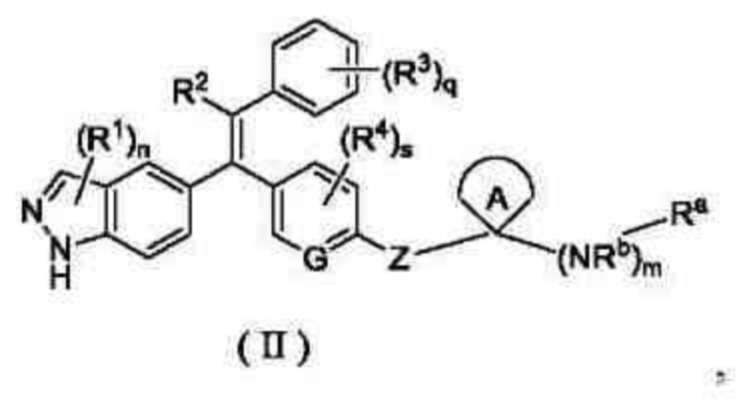
где
кольцо A, G, Z, R1, R2, R3, R4, Ra, Rb, n, m, s и q являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления настоящего изобретения соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль, представляет собой соединение формулы (III) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль:

где: G, Z, R1, R2, R3, R4, Ra, Rb, n, m, s и q являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления настоящего изобретения соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль, представляет собой соединение формулы (IV) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль:

где:
G, Z, R1, R2, R3, R4, R8, R9, Rb, n, s и q являются такими, как определено в формуле (I);
предпочтительно R8 и R9 вместе с атомом азота, к которому они присоединены, образуют гетероциклил, где гетероциклил возможно содержит от одного до двух идентичных или различных гетероатомов, выбранных из группы, состоящей из N, О и S, в дополнение к указанному атому азота, и указанный гетероциклил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из алкила, алкокси, галогена, амино, циано, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила; более предпочтительно гетероциклил необязательно замещен гидрокси или гидроксиалкилом; и еще более предпочтительно гетероциклил необязательно замещен гидрокси.
В другом предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смеси, или его фармацевтически приемлемой соли G представляет собой атом N.
В другом предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смеси, или их фармацевтически приемлемой соли, R1 выбран из группы, состоящей из галогена и атома водорода.
В другом предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смеси, или их фармацевтически приемлемой соли, R2 представляет собой галогеналкил, предпочтительно C1-C6 галогеналкил, и более предпочтительно CH2CF3.
В другом предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смеси, или их фармацевтически приемлемой соли, R3 выбран из группы, состоящей из атома водорода, галогена, алкила, циано, галогеналкила, NR13C(O)R14, C(O)NR13R14 и SO2R15; R13 и R14 идентичны или отличаются, и каждый независимо выбран из группы, состоящей из атома водорода и алкила; и R15 выбран из группы, состоящей из атома водорода и алкила.
В другом предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смеси, или их фармацевтически приемлемой соли, каждый R4 идентичен или отличается, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила и алкокси и предпочтительно атома водорода.
В другом предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смеси, или их фармацевтически приемлемой соли, Z выбран из группы, состоящей из химической связи, -О-,- О-СН2- и -NH-.
В другом предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смеси, или их фармацевтически приемлемой соли Ra выбран из группы, состоящей из -CH2CH=CHC(O)NR8R9, -C(O)CH=CR10R11 и -C(O)C≡CR12; R8 и R9 идентичны или отличаются, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, циклоалкила и гетероциклила; или R8 и R9 вместе с атомом азота, к которому они присоединены, образуют гетероциклил, где гетероциклил необязательно содержит один или два идентичных или различных гетероатома, выбранных из группы, состоящей из N, О и S, в дополнение к одному атому азота, и гетероциклил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из алкила, алкокси, галогена, амино, циано, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила; R10 и R11 идентичны или отличаются, и каждый независимо выбран из группы, состоящей из атома водорода и алкила; и R12 выбран из группы, состоящей из атома водорода и алкила.
В другом предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, или их смеси, или их фармацевтически приемлемой соли, Ra выбран из группы, состоящей из -СН2СН=СНС(O)N(СН3)2, -СН2СН=СНС(O)NH(СН3), -СН2СН=СНС(O)NHC(СН3)3, -С(O)СН=СН2, -С(O)С≡ССН3, 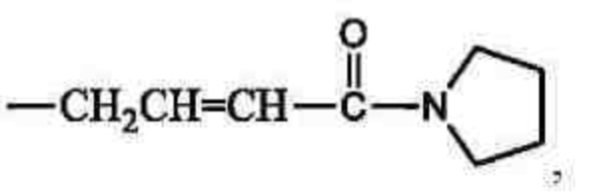

предпочтительно Ra выбран из группы, состоящей из -CH2CH=СНС(O)N(СН3)2, СН2СН=СНС(O)NH(СН3), -СН2СН=СНС(O)NHC(СН3)3, -С(O)СН=СН2, -С(O)С≡ССН3, 
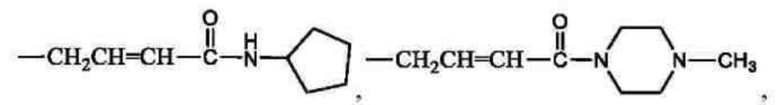

Типовые соединения формулы (I) включают без ограничений:





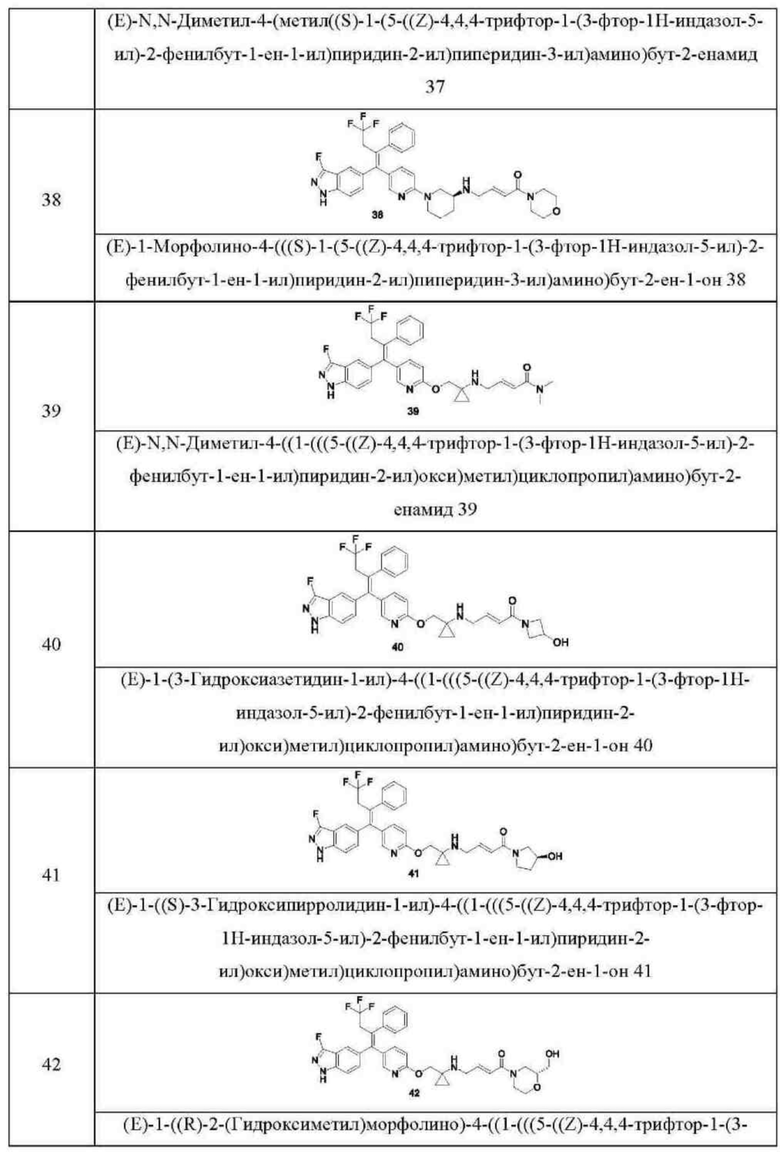

или их таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль.
В другом аспекте настоящее изобретение обеспечивает соединение формулы (IA) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль:

которое является промежуточным соединением для синтеза соединения формулы (I),
где:
Rw представляет собой амино-защитную группу и предпочтительно тетрагидропиранил;
R выбран из группы, состоящей из -CH2CH=CHC(O)NR8R9, -C(O)CH=CR10R11 и -C(O)C≡CR12;
R8 и R9 идентичны или отличаются, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
или R8 и R9 вместе с атомом азота, к которому они присоединены, образуют гетероциклил, где гетероциклил необязятельно содержит от одного до двух идентичных или различных гетероатомов, выбранных из группы, состоящей из N, О и S, в дополнение к одному атому азота, и гетероциклил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из алкила, алкокси, галогена, амино, циано, нитро, гидрокси, гидроксиалкила, COOR16, циклоалкила, гетероциклила, арила и гетероарила;
R10 и R11 идентичны или отличаются, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, галогеналкила, алкокси, циано, амино, нитро, карбокси, формила, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R12 выбран из группы, состоящей из атома водорода, алкила, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R16 выбран из группы, состоящей из атома водорода, алкила, галогеналкила и гидроксиалкила;
кольцо A, G, Z, R1, R2, R3, R4, Rb, n, m, s и q являются такими, как определено в формуле (I).
В другом аспекте настоящее изобретение обеспечивает соединение формулы (IIA) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль:

которое является промежуточным соединением для синтеза соединения формулы (II),
где:
Rw представляет собой амино-защитную группу и предпочтительно тетрагидропиранил;
R является таким, как определено в формуле (IA);
кольцо A, G, Z, R1, R2, R3, R4, Rb, n, m, s и q являются такими, как определено в формуле (II).
В другом аспекте настоящее изобретение обеспечивает соединение формулы (IIIA) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль:

которое является промежуточным соединением для синтеза соединения формулы (III).
где:
Rw представляет собой амино-защитную группу и предпочтительно тетрагидропиранил;
R является таким, как определено в формуле (IA);
G, Z, R1, R2, R3, R4, Rb, n, m, s и q являются такими, как определено в формуле (III).
В другом аспекте настоящее изобретение обеспечивает соединение формулы (IVA) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль:
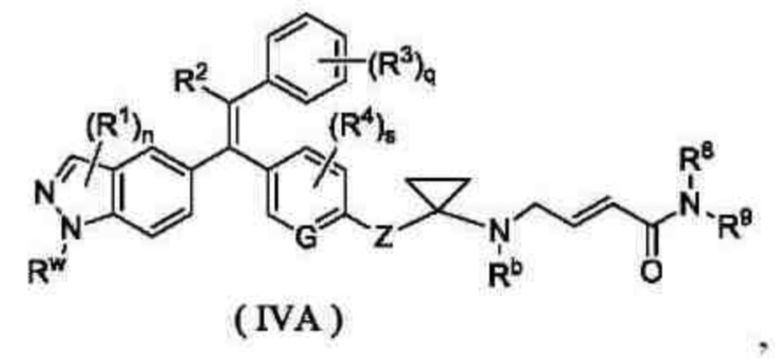
которое является промежуточным соединением для синтеза соединения формулы (IV),
где:
Rw представляет собой амино-защитную группу и предпочтительно тетрагидропиранил;
G, Z, R1, R2, R3, R4, Rb, R8, R9, n, s и q являются такими, как определено в формуле (IV).
Типовые соединения формулы (IA) включают без ограничений:

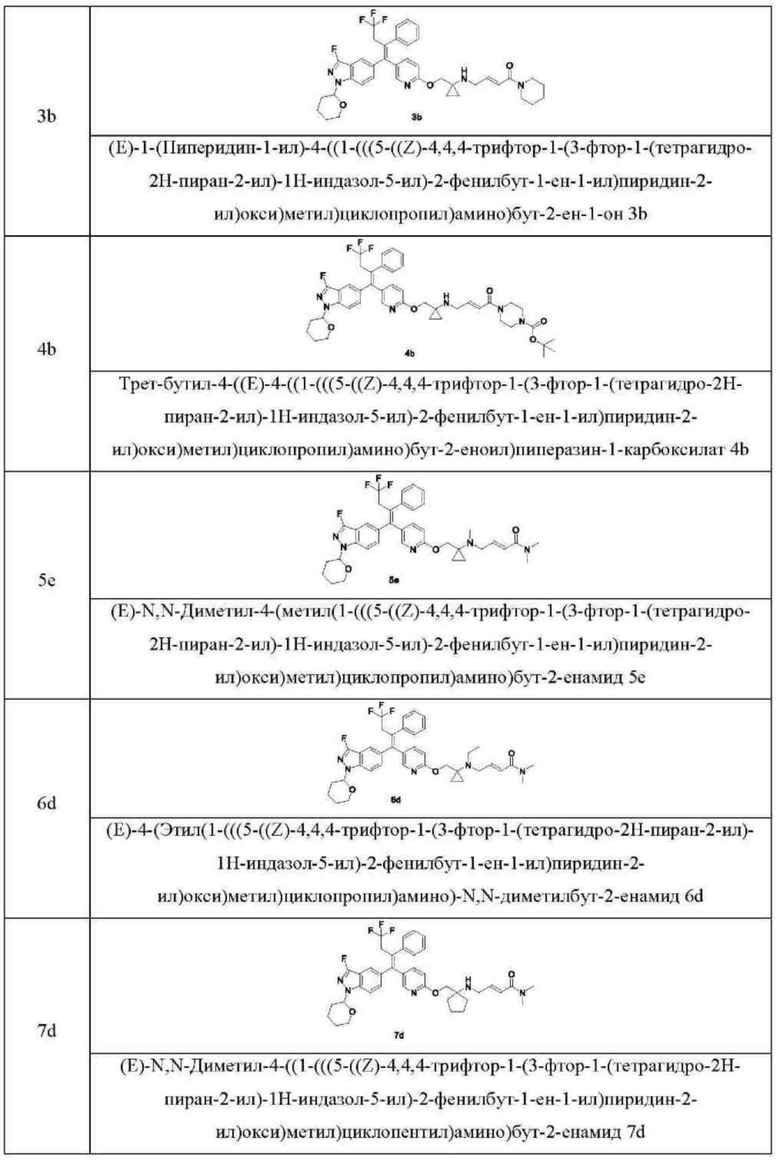





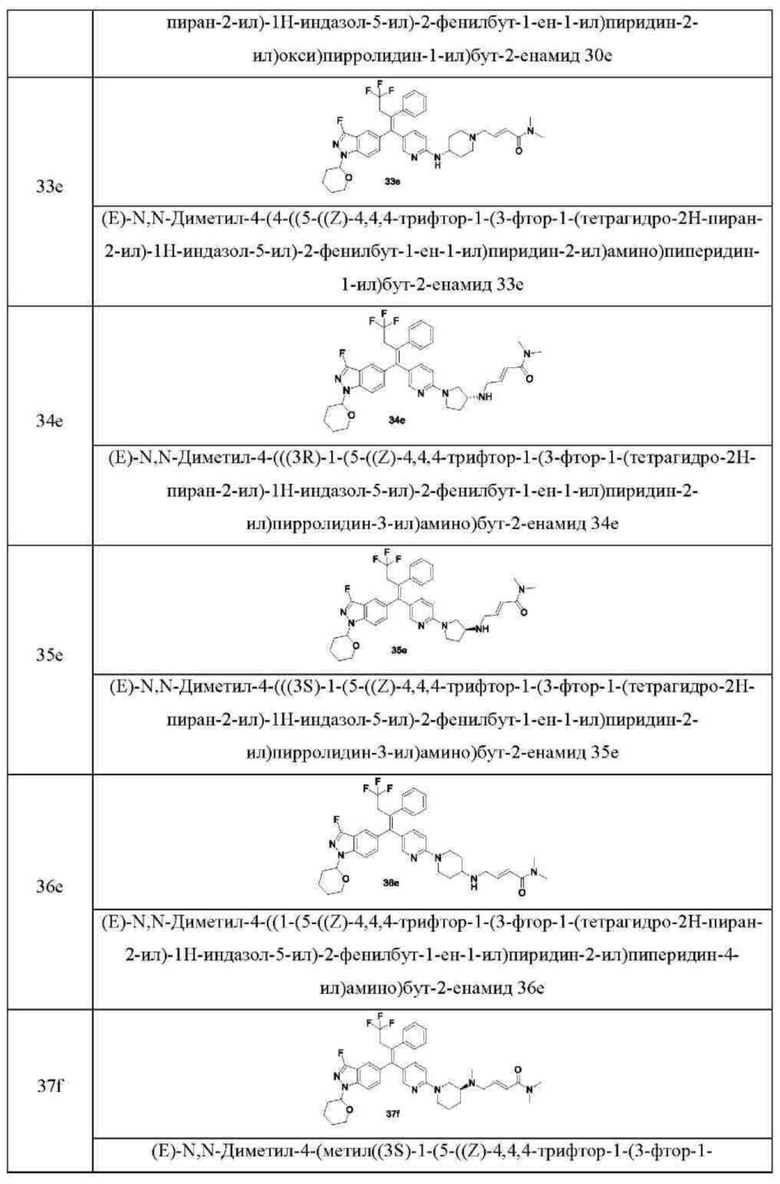


или их таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль.
В другом аспекте настоящее изобретение обеспечивает способ получения соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли, включающий стадию, на которой:
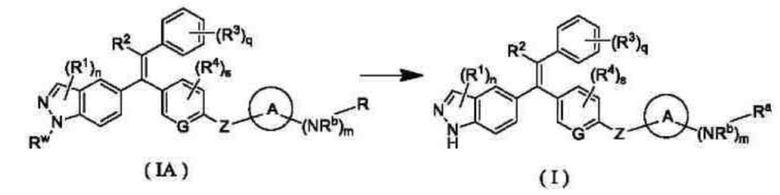
соединение формулы (IA) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль подвергают реакции снятия защиты в кислых условиях с получением соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли;
где:
Rw представляет собой амино-защитную группу и предпочтительно тетрагидропиранил или трет-бутоксикарбонил;
R является таким, как определено в формуле (IA);
кольцо A, G, Z, R1, R2, R3, R4, Ra, Rb, n, m, s и q являются такими, как определено в формуле (I).
В другом аспекте настоящее изобретение обеспечивает способ получения соединения формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли, включающий стадию, на которой:

соединение формулы (IIA), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль подвергают реакции снятия защиты в кислых условиях с получением соединения формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли;
где:
Rw представляет собой амино-защитную группу и предпочтительно тетрагидропиранил или трет-бутоксикарбонил;
R является таким, как определено в формуле (IIA);
кольцо A, G, Z, R1, R2, R3, R4, Ra, Rb, n, m, s и q являются такими, как определено в формуле (II).
В другом аспекте настоящее изобретение обеспечивает способ получения соединения формулы (III) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли, включающий стадию, на которой:

соединение формулы (IIIA), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль подвергают реакции снятия защиты в кислых условиях с получением соединения формулы (III) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли;
где:
Rw представляет собой амино-защитную группу и предпочтительно тетрагидропиранил или трет-бутоксикарбонил;
R является таким, как определено в формуле (IIIA);
G, Z, R1, R2, R3, R4, Ra, Rb, n, s и q являются такими, как определено в формуле (III).
В другом аспекте настоящее изобретение обеспечивает способ получения соединения формулы (IV) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли, включающий стадию, на которой:

соединение формулы (IVA) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль подвергают реакции снятия защиты в кислых условиях с получением соединения формулы (IV) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли;
где:
Rw представляет собой амино-защитную группу и предпочтительно тетрагидропиранил или трет-бутоксикарбонил;
G, Z, R1, R2, R3, R4, R8, R9, Rb, n, s и q являются такими, как определено в формуле (IV).
В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы (I)-(IV), как определено выше, или его таутомера, мезомера, рацемата, энантиомера, или их смеси, или их фармацевтически приемлемой соли и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов. Настоящее изобретение также относится к способу получения фармацевтической композиции, как определено выше, включающему стадию смешивания соединения формулы (I)-(IV) или его таутомера, мезомера, рацемата, энантиомера, или их смеси, или их фармацевтически приемлемой соли с фармацевтически приемлемыми носителями, разбавителями или эксципиентами.
Настоящее изобретение дополнительно относится к применению соединения формулы (I)-(IV) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли, или содержащей их фармацевтической композиции для получения модулятора рецептора эстрогена.
Настоящее изобретение дополнительно относится к применению соединения формулы (I)-(IV) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли, или содержащей их фармацевтической композиции для получения лекарственного средства для предотвращения и/или лечения заболевания или расстройства, опосредованного рецептором эстрогена или зависимого от рецептора эстрогена, где заболевание или расстройство, опосредованное рецептором эстрогена или зависимое от рецептора эстрогена, предпочтительно представляет собой рак, более предпочтительно рак молочной железы, рак яичника, рак эндометрия, рак предстательной железы или рак матки, и еще более предпочтительно рак молочной железы.
Настоящее изобретение дополнительно относится к соединению формулы (I)-(IV) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, или их смеси, или их фармацевтически приемлемой соли, или содержащей их фармацевтической композиции для применения в качестве лекарственного средства.
Настоящее изобретение дополнительно относится к соединению формулы (I)-(IV) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли для применения в качестве лекарственного средства для предотвращения и/или лечения заболевания или расстройства, опосредованного или зависимого от рецептора эстрогена, где заболевание или расстройство, опосредованное или зависимое от рецептора эстрогена, предпочтительно представляет собой рак, более предпочтительно рак молочной железы, рак яичника, рак эндометрия, рак предстательной железы или рак матки, и еще более предпочтительно рак молочной железы.
Настоящее изобретение дополнительно относится к способу лечения заболевания или расстройства, опосредованного или зависимого от рецептора эстрогена, включающему стадию введения нуждающемуся в этом пациенту терапевтически эффективного количества соединения формулы (I)-(IV) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли по настоящему изобретению. Данный способ показывает выраженную эффективность и меньшие побочные эффекты. Заболевание или расстройство, опосредованное или зависимое от рецептора эстрогена, предпочтительно представляет собой рак, более предпочтительно рак молочной железы, рак яичника, рак эндометрия, рак предстательной железы или рак матки, и еще более предпочтительно рак молочной железы.
Активное соединение может быть включено в лекарственную форму, подходящую для введения любым приемлемым путем, и предпочтительно активное соединение находится в форме унифицированной дозы или в форме, в которой пациент может вводить его самостоятельно в однократной дозе. Форма унифицированной дозы соединения или композиции по настоящему изобретению может представлять собой таблетку, капсулу, крахмальную облатку, бутилированное питье, порошок, гранулу, леденец, суппозиторий, восстановленный порошок или жидкий препарат.
Дозировка соединения или композиции, используемая в способе лечения по настоящему изобретению, будет, как правило, варьировать в соответствии с тяжестью заболевания, массой тела пациента и относительной эффективностью соединения. Тем не менее в качестве общей рекомендации подходящая унифицированная доза может составлять от 0,1 до 1000 мг.
В дополнение к активным ингредиентам фармацевтическая композиция по настоящему изобретению может дополнительно содержать одно или более вспомогательных веществ, включая наполнитель (разбавитель), связующее вещество, смачивающий агент, разрыхлитель, эксципиент и т.п. В зависимости от способа введения композиция может содержать от 0,1 до 99% по массе активного соединения.
Фармацевтическая композиция, содержащая активный ингредиент, может принимать форму, подходящую для перорального введения, например, таблетки, троше, леденец, водной или масляной суспензии, диспергируемого порошка или гранул, эмульсии, твердой или мягкой капсулы, сиропа или эликсира. Композицию для перорального применения можно получить в соответствии с любым известным в данной области техники способом получения фармацевтической композиции. Такая композиция может содержать один или более ингредиентов, выбранных из группы, состоящей из подсластителей, вкусоароматических добавок, красителей и консервантов, чтобы обеспечить фармацевтическую форму приятного внешнего вида и вкуса. Таблетка содержит активный ингридиент в смеси с нетоксичными, фармацевтически приемлемыми эксципиентами, подходящими для производства таблеток. Данные эксципиенты могут представлять собой инертные эксципиенты, гранулирующие агенты, вещества для улучшения распадаемости таблеток и смазывающие вещества. Таблетка может быть непокрытой или покрытой оболочкой с помощью известного метода для маскировки вкуса лекарственного препарата или замедления распадения и всасывания активного ингридиента в желудочно-кишечном тракте, в результате чего обеспечивается пролонгированное высвобождение в течение длительного периода времени. Например, можно использовать водорастворимые материалы, маскирующие вкус. Лекарственная форма для перорального применения может быть также обеспечена в виде мягких желатиновых капсул, в которых либо активный ингредиент смешан с инертным твердым разбавителем, либо активный ингредиент смешан с водорастворимым носителем.
Водная суспензия содержит активный ингредиент в смеси с эксципиентами, подходящими для производства водной суспензии. Такие эксципиенты представляют собой суспендирующие агенты, диспергирующие или смачивающие агенты. Активный ингредиент водной суспензии может представлять собой диспергируемые порошки или гранулы. Активный ингредиент смешивают с одним или более диспергирующих агентов или суспендирующих агентов путем добавления воды. Водная суспензия может также содержать один или более консервантов, один или более красителей, одну или более вкусоароматическтих добавок и один или более подсластителей.
Масляную суспензию можно получить путем суспендирования активного ингредиента в растительном или минеральном масле. Масляная суспензия может содержать загуститель. Вышеупомянутые подсластители и вкусоароматические добавки можно добавлять, чтобы обеспечить лекарственную форму приятного вкуса. Эти композиции можно консервировать путем добавления антиоксиданта.
Диспергируемые порошки или гранулы, подходящие для получения водной суспензии, могут обеспечивать активный ингредиент в смеси с диспергирующими агентами или смачивающими агентами, суспендирующим агентом или одним или более консервантами путем добавления воды. Примеры подходящих диспергирующих или смачивающих и суспендирующих агентов представляют собой такие, которые уже упомянутые выше. Можно также добавлять дополнительные эксципиенты, такие как подсластители, вкусоароматические добавки и красители. Данные композиции можно консервировать путем добавления антиоксиданта, такого как аскорбиновая кислота.
Фармацевтическая композиция по настоящему изобретению может быть в форме эмульсии масло-в-воде. Масляная фаза может представлять собой растительное или минеральное масло, или их смесь. Подходящие эмульгирующие агенты могут представлять собой природные фосфолипиды. Может быть использован подсластитель. Такие лекарственные формы могут также содержать мягчительное средство, консервант, краситель и антиоксидант.
Фармацевтическая композиция по настоящему изобретению может быть в форме стерильного водного раствора для инъекций. Среди приемлемых носителей и растворителей, которые могут быть использованы, находятся вода, раствор Рингера или изотонический раствор хлорида натрия. Стерильная лекарственная форма для инъекций может представлять собой стерильную микроэмульсию масло-в-воде для инъекций, в которой активный ингредиент растворен в масляной фазе. Раствор или микроэмульсию для инъекций могут быть введены в кровоток пациента посредством локальной болюсной инъекции. Альтернативно раствор и микроэмульсию предпочтительно вводят таким образом, чтобы поддерживать постоянную концентрацию соединения по настоящему изобретению в кровотоке. Чтобы поддерживать такую постоянную концентрацию, может быть использовано устройство для непрерывной внутривенной доставки. Примером такого устройства является помпа Deltec CADD-PLUS. ТМ. 5400 для внутривенных инъекций.
Фармацевтическая композиция по настоящему изобретению может быть в форме стерильной водной или масляной суспензии для инъекций для внутримышечного и подкожного введения. Такую суспензию можно получить с подходящими диспергирующими или смачивающими и суспендирующими агентами, как описано выше, в соответствии с известными методами. Стерильная лекарственная форма для инъекций может также представлять собой стерильный раствор или суспензию для инъекций, которые получают в нетоксичном парентерально приемлемом разбавителе или растворителе. Кроме того, в качестве растворителя или суспендирующей среды можно использовать стерильные нелетучие масла. В дополнение к этому для получения инъекций могут быть также использованы жирные кислоты.
Соединение по настоящему изобретению может быть введено в форме суппозитория для ректального введения. Данные фармацевтические композиции могут быть получены путем смешивания лекарственного средства с подходящим нераздражающим эксципиентом, который представляет собой твердое вещество при комнатной температуре; но становится жидким в прямой кишке и в результате плавления в прямой кишке для высвобождения лекарственного средства.
Специалистам в данной области техники хорошо известно, что дозировка лекарственного средства зависит от ряда факторов, включая без ограничений следующие факторы: активность конкретного соединения, возраст пациента, масса тела пациента, общее состояние здоровья пациента, поведение пациента, рацион питания пациента, время введения, путь введения, скорость выведения, комбинация лекарственных препаратов и т.п. В дополнение к этому оптимальное лечение, такое как режим лечения, суточная доза соединения формулы (I) или тип его фармацевтически приемлемой соли, может быть верифицирован в соответствии с традиционными схемами лечения.
Определения
Если не указано иное, термины, используемые в описании и формуле изобретения, имеют описанные ниже значения.
Термин «алкил» относится к насыщенной алифатической углеводородной группе, которая представляет собой группу с неразветвленной или разветвленной цепью, содержащую от 1 до 20 атомов углерода, предпочтительно алкил, имеющий от 1 до 12 атомов углерода и более, предпочтительно алкил, имеющий от 1 до 6 атомов углерода. Не имеющие ограничительного характера примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и их различные разветвленные изомеры. Более предпочтительно алкильная группа представляет собой низший алкил, имеющий от 1 до 6 атомов углерода, не имеющие ограничительного характера примеры которого включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и т.п. Алкил может быть замещенным или незамещенным. При замещении группа(-ы) заместителя(-ей) может (могут) быть присоединена(-ы) в любой доступной точке. Группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, оксо, карбокси и алкоксикарбонила.
Термин «алкилен» относится к насыщенной линейной или разветвленной алифатической углеводородной группе, имеющей два остатка, образовавшихся в результате удаления двух атомов водорода от одного и того же атома углерода или от двух различных атомов углерода исходного алкана. Эта группа представляет собой линейный или разветвленный алкилен, имеющий от 1 до 20 атомов углерода, предпочтительно от 1 до 12 атомов углерода и более предпочтительно от 1 до 6 атомов углерода. Не имеющие ограничительного характера примеры алкилена включают без ограничений метилен (-СН2-), 1,1-этилен (-СН(СН3)-), 1,2-этилен (-СН2СН2-), 1,1-пропилен (-СН(СН2СН3)-), 1,2-пропилен (-СН2СН(СН3)-), 1,3-пропилен (-СН2СН2СН2-), 1,4-бутилен (-СН2СН2СН2СН2-) и т.п. Алкилен может быть замещенным или незамещенным. При замещении группа(-ы) заместителя(-ей) может (могут) быть присоединена(-ы) в любой доступной точке. Группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, необязательно независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио и оксо.
Термин «алкенил» относится к алкилу, содержащему в молекуле двойную(-ые) связь(-и) углерод углерод, где определение алкила является таким, как описано выше.
Алкенил может быть замещенным или незамещенным. При замещении группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из атома водорода, алкила, алкокси, галогена, галогеналкила, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, ар ила и гетероарила.
Термин «алкинил» относится к алкилу, содержащему в молекуле тройную(-ые) связь (-и) углерод углерод, где определение алкила является таким, как описано выше. Алкинил может быть замещенным или незамещенным. При замещении группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из атома водорода, алкила, алкокси, галогена, галогеналкила, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, ар ила и гетероарила.
Термин «алкокси» относится к группе -O-(алкил) или -О-(незамещенный циклоалкил), где алкил является таким, как определено выше. Не имеющие ограничительного характера примеры алкокси включают метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси. Группа алкокси необязательно может быть замещенной или незамещенной. При замещении группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, карбокси и алкоксикарбонила.
Термин «циклоалкил» относится к насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной группе заместителя, имеющей от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода (например, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 атомов углерода) и более предпочтительно от 3 до 6 атомов углерода. Не имеющие ограничительного характера примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т.п. Полициклический циклоалкил включает циклоалкил, имеющий спиро-кольцо, конденсированное кольцо или кольцо с внутренним мостиком.
Термин «спиро-циклоалкил» относится к 5-20-членной полициклической группе с отдельными кольцами, соединенными посредством одного общего атома углерода (называемого спиро-атомом), где кольца могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной π-электронной системы.
Спиро-циклоалкил предпочтительно представляет собой 6-14-членный спиро-циклоалкил (например, 6-, 7-, 8-, 9-, 10-, 11-, 12-, 13-, 14-членный спиро-циклоалкил) и более предпочтительно 7-10-членный спиро-циклоалкил. В зависимости от числа общих между кольцами спиро-атомов спиро-циклоалкил можно разделить на моно-спиро-циклоалкил, ди-спиро-циклоалкил или поли-спиро-циклоалкил, и спиро-циклоалкил предпочтительно представляет собой моно-спиро-циклоалкил или ди-спиро-циклоалкил и более предпочтительно 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моно-спиро-циклоалкил. Не имеющие ограничительного характер а примеры спиро-циклоалкила включают:

Термин «конденсированный циклоалкил» относится к 5-20-членной полностью углеродной полициклической группе, где каждое кольцо в системе имеет общую пару соседних атомов углерода с другим кольцом, одно или более колец могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью коньюгированной π-электронной системы. Конденсированный циклоалкил предпочтительно представляет собой 6-14-членный конденсированный циклоалкил и более предпочтительно 7-10-членный конденсированный циклоалкил (например, 7-, 8-, 9- или 10-членный конденсированный циклоалкил). В соответствии с числом колец-членов конденсированный циклоалкил можно разделить на бициклический, трициклический, тетрациклический или полициклический конденсированный циклоалкил, и предпочтительно конденсированный циклоалкил представляет собой бициклический или трициклический конденсированный циклоалкил и более предпочтительно 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный циклоалкил. Не имеющие ограничительного характера примеры конденсированного циклоалкила включают:
Термин «мостиковый циклоалкил» относится к 5-20-членной полностью углеродной полициклической группе, где каждые два кольца в системе имеют два общих несвязанных атома углерода, кольца могут иметь одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной π-электронной системы. Мостиковый циклоалкил предпочтительно представляет собой 6-14-членный мостиковый циклоалкил и более предпочтительно 7-10-членный мостиковый циклоалкил (например, 7-, 8-, 9- или 10-членный мостиковый циклоалкил). В соответствии с числом колец-членов мостиковый циклоалкил можно разделить на бициклический, трициклический, тетрациклический или полициклический мостиковый циклоалкил, и предпочтительно мостиковый циклоалкил представляет собой бициклический, трициклический или тетрациклический мостиковый циклоалкил и более предпочтительно бициклический или трициклический мостиковый циклоалкил. Не имеющие ограничительного характер а примеры мостикового циклоалкила включают:

Циклоалкильное (включая циклоалкильное, спиро-циклоалкильное, конденсированное циклоалкильное и мостиковое циклоалкильное) кольцо может быть конденсировано с кольцом арила, гетероарила или гетероциклила, где кольцо, связанное с исходной структурой, представляет собой циклоалкил. Не имеющие ограничительного характера примеры включают инданил, тетрагидронафтил, бензоциклогептил и т.п. Циклоалкил может быть необязательно замещенным или незамещенным. При замещении группа(-ы) заместителя (-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, оксо, карбокси и алкоксикарбонила.
Термин «гетероциклил» относится к 3-20-членной насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной группе заместителя, где один или более кольцевых атомов выбраны из группы, состоящей из N, О, S, S(O) и S(O)2, но за исключением -O-O-, -O-S- или -S-S- в кольце, при этом остальные кольцевые атомы представляют собой атомы углерода. Предпочтительно гетероциклил имеет от 3 до 12 кольцевых атомов, где от 1 до 4 атомов (например, 1, 2, 3 или 4 атома) представляют собой гетероатомы, более предпочтительно от 3 до 6 кольцевых атомов (например, 3, 4, 5 или 6 кольцевых атомов). Не имеющие ограничительного характера примеры моноциклического гетероциклила включают пирролидинил, имипазолидинил, тетрагидрофуранил, тетрагиротиенил, дигидроимидазолил, дигидрофуранил, дигидропиразолил, дигидропирролил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил и т.п., и предпочтительно пиперидинил, пирролидинил. Полициклический гетероциклил включает гетероциклил, имеющий спиро-кольцо, конденсированное кольцо или кольцо с внутренним мостиком.
Термин «спиро-гетероциклил» относится к 5-20-членной полициклической гетероциклильной группе, 6 которой отдельные кольца соединены через один общий атом углерода (называемый спиро-атомом), где один или более кольцевых атомов представляют собой гетероатомы, выбранные из группы, состоящей из N, О, S, S(O) и S(O)2, при этом остальные кольцевые атомы представляют собой атомы углерода, где кольца могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью коньюгированной π-электронной системы Спиро-гетероциклил предпочтительно представляет собой 6-14-членный спиро-гетероциклил и более предпочтительно 7-10-членный спиро-гетероциклил (например, 7-, 8-, 9- или 10-членный спиро-гетероциклил). В зависимости от числа общих между кольцами спиро-атомов спиро-гетероциклил делится на моно-спиро-гетероциклил, ди-спиро-гетероциклил или поли-спиро-гетероциклил, и спиро-гетероциклил предпочтительно представляет собой моно-спиро-гетероциклил или ди-спиро-гетероциклил и более предпочтительно 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моно-спиро-гетероциклил. Не имеющие ограничительного характера примеры спиро-гетероциклила включают
Термин «конденсированный гетероциклил» относится к 5-20-членной полициклической гетероциклильной группе, где каждое кольцо в системе имеет общую пару соседних атомов углерода с другим кольцом, где одно или более колец содержит одну или более двойных связей, но ни одно из колец не имеет полностью коньюгированной π-электронной системы, и где один или более кольцевых атомов представляют собой гетероатомы, выбранные из группы, состоящей из N, О, S, S(O) и S(O)2, при этом остальные кольцевые атомы представляют собой атомы углерода. Конденсированный гетероциклил предпочтительно представляет собой 6-14-членный конденсированный гетероциклил и более предпочтительно 7-10-членный конденсированный гетероциклил (например, 7-, 8-, 9- или 10-членный конденсированный гетероциклил). В соответствии с числом колец-членов конденсированный гетероциклил можно разделить на бициклический, трициклический, тетрациклический или полициклический конденсированный гетероциклил, и предпочтительно конденсированный гетероциклил представляет собой бициклический или трициклический конденсированный гетероциклил и более предпочтительно 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный гетероциклил. Не имеющие ограничительного характера примеры конденсированного гетероциклила включают:

Термин «мостиковый гетероциклил» относится к 5-14-членной полициклической гетероциклильной группе, где каждые два кольца в системе имеют два общих несвязанных атомов углерода, где кольца могут иметь одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной π-электронной системы, и где один или более кольцевых атомов представляют собой гетероатомы, выбранные из группы, состоящей из N, О, S, S(O) и S(O)2, при этом остальные кольцевые атомы представляют собой атомы углерода. Мостиковый гетероциклил предпочтительно представляет собой 6-14-членный мостиковый гетероциклил и более предпочтительно 7-10-членный мостиковый гетероциклил (например, 7-, 8-, 9- или 10-членный мостиковый гетероциклил). В соответствии с числом колец-членов мостиковый гетероциклил можно разделить на бициклический, трициклический, тетрациклический или полициклический мостиковый гетероциклил, и предпочтительно мостиковый гетероциклил представляет собой бициклический, трициклический или тетрациклический мостиковый гетероциклил и более предпочтительно бициклический или трициклический мостиковый гетероциклил. Не имеющие ограничительного характера примеры мостикового гетероциклила включают:

Гетероциклильное (включая гетероциклил, спиро-гетероциклил, конденсированный гетероциклил и мостиковый гетероциклил) кольцо может быть конденсировано с кольцом арила, гетероарила или цикле алкила, где кольцо, связанное с исходной структурой, представляет собой гетероциклил, и его не имеющие ограничительного характера примеры включают:

Гетероциклил может быть необязательно замещенным или незамещенным. При замещении группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, оксо, карбокси и алкоксикарбонила.
Термин «арил» относится к 6-14-членному полностью углеродному моноциклическому кольцу или полициклическому конденсированному кольцу (т.е. каждое кольцо 6 системе имеет общую пару соседних атомов углерода с другим кольцом в системе), имеющему коньюгированную π-электронную систему, предпочтительно к 6-10-членному арилу, например фенилу и нафтилу Арильное кольцо может быть конденсировано с кольцом гетероарила, гетероциклила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой арильное кольцо, и его не имеющие ограничительного характера примеры включают:

Арил может быть замещенным или незамещенным. При замещении группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, карбокси и алкоксикарбонила и предпочтительно фенила.
Термин «гетероарил» относится к 5-14-членной гетероароматической системе, имеющей от 1 до 4 гетероатомов, выбранных из группы, состоящей из О, S и N. Гетероарил предпочтительно представляет собой 5-10-членный гетероарил (например, 5-, 6-, 7-, 8-, 9- или 10-членный гетероарил), более предпочтительно 5- или 6-членный гетероарил, например имидазолил, фурил, тиенил, тиазолил, пиразолил, оксазолил, пирролил, тетразолил, пиридил, пиримидинил, тиадиазолил, пиразинил и т.п., предпочтительно имидазолил, пиразолил, пиримидинил или тиазолил, и более предпочтительно пиразолил или тиазолил. Гетероарильное кольцо может быть конденсировано с кольцом арила, гетероциклила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой гетероарильное кольцо, и его не имеющие ограничительного характера примеры включают:

Гетероарил может быть необязательно замещенным или незамещенным. При замещении группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, карбокси и алкоксикарбонила.
Термин «гидроксиалкил» относится к алкильной группе, замещенной гидроксигруппой(-ами), где алкил является таким, как определено выше.
Термин «галогеналкил» относится к алкильной группе, замещенной галогеном(-ами), где алкил является таким, как определено выше.
Термин «дейтерированный алкил» относится к алкильной группе, замещенной атомом(-ами) дейтерия, где алкил является таким, как определено выше.
Термин «гидрокси» относится к группе -ОН.
Термин «галоген» относится к фтору, хлору, брому или йоду.
Термин «амино» относится к группе -NH;.
Термин «пиано» относится к группе -CN.
Термин «нитро» относится к группе -NO2.
Термин «карбокси» относится к группе -С(O)ОН.
Термин «формил» относится к группе -СНО.
Термин «алкоксикарбонил» относится к группе -С(O)O(алкила) или -С(O)O(циклоалкил), где алкил и циклоалкил являются такими, как определено выше.
Термин «ацилгалогенид» относится к соединению, содержащему группу -С(O)-галогена.
«Необязательный» или «необязательно» означает, что описанное далее событие или обстоятельство может, но не обязательно, произойти, и такое описание включает ситуацию, в которой это событие или обстоятельство либо происходит, либо не происходит. Например, «гетероциклил, необязательно замещенный алкилом» означает, что алкильная группа может, но не обязательно, присутствовать, и такое описание включает ситуацию, где гетероциклил замещен алкилом и где гетероциклил не замещен алкилом.
«Замещенный» относится к одному или более атомов водорода в группе, предпочтительно в количестве до 5 и более предпочтительно от 1 до 3 атомов водорода, независимо замещенных соответствующим количеством заместителей. Без слов понятно, что заместители могут существовать только в их возможном химическом положении. Специалист в данной области техники способен экспериментальным или теоретическим путем без лишних усилий определить, возможно или невозможно замещение. Например, объединение амино- или гидроксигруппы, имеющей свободные атомы водорода и углерода, имеющие нестабильные связи (такие как олефиновые), может быть нестабильным.
Термин «фармацевтическая композиция» относится к смеси одного или более соединений, описанных в настоящем документе, или их физиологически/фармацевтически приемлемых солей или пролекарств с другими химическими компонентами и другими компонентами, такими как физиологически/фармацевтически приемлемые носители и эксципиенты. Цель фармацевтической композиции состоит в том, чтобы способствовать введению соединения в организм, что создает условия для всасывания активного ингридиента, позволяющего ему проявить биологическую активность.
«Фармацевтически приемлемая соль» относится к соли соединения по настоящему изобретению, которая безопасна и эффективна для млекопитающих и обладает необходимой биологической активностью.
Различные выражения «X выбран из группы, состоящей из А, В или С», «X выбран из группы, состоящей из А, В и С», «X представляет собой А, В или С», «X представляет собой А, В и С» и т.п. все имеют одно и то же значение. Они означают, что Х может представлять собой любое одно или более из А, В и С.
Соединение по настоящему изобретению может также содержать его изотопные производные. Термин «изотопные производные» относится к соединениям, которые отличаются по структуре только в присутствии одного или более атомов, обогащенных изотопами. Например, соединение, имеющее структуру по настоящему изобретению с замещением водорода «дейтерием» или «тритием», или с замещением фтора на метку 18F (изотоп 18F), или с замещением углерода 11С-, 13С- или 14С-обогащенным атомом углерода (метка углерода 11С-, 13С- или 14С- 11С-, 13С-; или 14С-изотоп), входят в объем настоящего изобретения. Такие соединения могут быть использованы, например, в качестве аналитических инструментов или зондов в биологических анализах, или в качестве маркеров для диагностической визуализации заболевания in vivo, или в качестве маркеров для исследований фармакодинамики, фармакокинетики или рецепторов. Дейтерированные соединения могут, как правило, сохранять активность, сопоставимую с активностью не дейтерированных соединений, и при дейтерировании в определенных специфичных участках можно достичь лучшей метаболической стабильности, приводящей в результате к определенным терапевтическим преимуществам (таким как увеличение периода полувыведения in vivo или снижение требуемой дозы).
Для лекарственных средств или фармакологически активных агентов термин «терапевтически эффективное количество» относится к достаточному количеству лекарственного средства или лекарственного препарата, которое нетоксично, но способно к достижению необходимого эффекта. Определение эффективного количества варьирует индивидуально в зависимости от возраста и общего состояния реципиента, а также от конкретной действующей субстанции. Специалисты в данной области техники могут определить приемлемое эффективное количество в каждом случае на основании стандартных экспериментов.
Настоящее изобретение обеспечивает антагонист рецептора эстрогена, имеющий новую структуру формулы (I). Обнаружено, что соединение, имеющее этот вид структуры, показывает великолепную активность in vitro. По сравнению с предшествующим уровнем техники это амидное соединение с α,β-ненасыщенной связью может инактивировать рецептор эстрогена в результате специфичного связывания с цистеином в лиганд-связывающем домене рецептора эстрогена.
Способ синтеза соединения по настоящему изобретению
Для достижения цели настоящего изобретения, настоящее изобретение применяют следующие технические решения:
Схема I
Способ получения соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли в соответствии с настоящим изобретением, включающий следующую стадию, на которой:

соединение формулы (IA) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль подвергают реакции снятия защиты в кислых условиях с получением соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли;
где:
Rw представляет собой амино-защитную группу;
R является таким, как определено в формуле (IA);
кольцо A, G, Z, R1, R2, R3, R4, Ra, Rb, n, m, s и q являются такими, как определено в формуле (I).
Схема II

соединение формулы (IIA) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль подвергают реакции снятия защиты в кислых условиях с получением соединения формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли;
где:
Rw представляет собой амино-защитную группу;
R является таким, как определено в формуле (IIA);
кольцо A, G, Z, R1, R2, R3, R4, Ra, Rb, n, m, s и q являются такими, как определено в формуле (II).
Схема III

соединение формулы (IIIA) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль подвергают реакции снятия защиты в кислых условиях с получением соединения формулы (III) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли;
где:
Rw представляет собой амино-защитную группу;
R является таким, как определено в формуле (IIIA);
G, Z, R1, R2, R3, R4, Ra, Rb, n, s и q являются такими, как определено в формуле (III).
Схема IV

соединение формулы (IVA) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль подвергают реакции снятия защиты в кислых условиях с получением соединения формулы (IV) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли;
где:
Rw представляет собой амино-защитную группу;
G, Z, R1, R2, R3, R4, R8, R9, Rb, n, s и q являются такими, как определено в формуле (IV).
Реагент, который обеспечивает кислые условия в описанных выше схемах, включает без ограничений пиридина гидробромид, трифторуксусную кислоту, муравьиную кислоту уксусную кислоту соляную кислоту, серную кислоту и метансульфоновую кислоту и предпочтительно соляную кислоту.
Амино-защитная группа выбрана из группы, состоящей из тетрагидропиранила, трет-бутоксикарбонила, ацетила; бензила, аллила и п-метоксибензила и предпочтительно тетрагидропиранила.
Описанные выше реакции предпочтительно проводят в растворителе. Используемый растворитель включает без ограничений уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, диметилсульфоксид, 1,4-диоксан, воду, N,N-диметилформамид и их смеси.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение будет приведено дополнительно со ссылкой на следующие ниже примеры, но данные примеры не следует истолковывать как ограничивающие объем настоящего изобретения.
ПРИМЕРЫ
Структуры соединений идентифицировали методом ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (МС). Сдвиги на ЯМР (δ) представлены в 10-6 (млн-1). Спектры ЯМР определяют на устройстве Bruker AVANCE-400. Растворители для определения представляют собой дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD), а внутренний стандарт представлял собой тетраметилсилан (ТМС).
Масс-спектрометрию провоодят с помощью жидкостного хроматографа с масс-спектрометром Agilent 1200 /1290 DAD- 6110/6120 Quadrupole MS (производитель: Agilent, модель МС: 6110/6120 Quadrupole MS), Waters ACQuity UPLC-QD/SQD (производитель: Waters, модель МС: Waters ACQuity Qda Detector/Waters SQ Detector), THERMO Ultimate 3000-Q Exactive (производитель: THERMO, модель МС: THERMO Q Exactive).
Высокоэффективную жидкостную хроматографию (ВЭЖХ) проводят на жидкостном хроматографе высокого давления Agilent HPLC 1200DAD, Agilent HPLC 1200VWD и Waters HPLC e2695-2489.
Хиральную ВЭЖХ проводят на высокоэффективном жидкостном хроматографе Agilent 1260 DAD.
Препаративную высокоэффективную жидкостную хроматографию проводят на препаративных хроматографах Waters 2545-2767, Waters 2767-SQ Detecor2, Shimadzu LC-20AP и Gilson GX-281.
Хиральную препаративную ВЭЖХ проводят на препаративном хроматографе Shimadzu LC-20AP.
Для быстрого получения CombiFlash используют прибор Combiflash Rf200 (TELEDYNE ISCO).
Для тонкослойной хроматографии (ТСХ) используют пластину силикагеля Yantai Huanghai HSGF254 или Qingdao GF254. Размер пластины силикагеля, используемой в ТСХ, составлял от 0,15 мм до 0,2 мм, а размер пластины силикагеля, используемой в очистке продукта, составлял от 0,4 мм до 0,5 мм.
В качестве носителя для колоночной хроматографии на силикагеле, как правило, используют силикагель Yantai Huanghai от 200 до 300 меш.
Средние значения коэффициентов ингибирования киназы и значения IC50 проводят на ридерк для микропланшетов NovoStar (BMG Co., Германия).
Известные исходные материалы по настоящему изобретению могут быть получены известными в данной области техники способами или приобретены у компаний ABCR GmbH & Co. KG, Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc., Dari Chemical Company и т.д.
Если не указано иное, реакции проводят в атмосфере аргона или в атмосфере азота.
«Атмосфера аргона» или «атмосфера азота» означает, что реакционную колбу оснащают баллоном с аргоном или азотом (около 1 л).
«Атмосфера водорода» означает, что реакционную колбу оснащают баллоном с водородом (около 1 л).
Реакцию гидрогенизации под давлением выполняют в гомогенизаторе Parr 3916EKX с генератором водорода Qinglan QL-500 или на приборе для гидрогенизации HC2-SS.
В реакциях гидрогенизации, как правило, из системы откачивают воздух и заполняют водородом, повторяя вышеуказанную операцию три раза.
В микроволновых реакциях используют микроволновой реактор типа СЕМ Discover-S 908860.
Если не указано иное, «раствор» относится к водному раствору.
Если не указано иное, реакции проводят при комнатной температуре от 20°С до 30°С.
Процесс реакции в примерах контролируют методом тонкослойной хроматографии (ТСХ). Используемые в реакции растворитель для проявления, система элюентов колоночной хроматографии и система растворителей для проявления в тонкослойной хроматографии для очистки соединений включали: А: система дихлорметанметанол, В: система н-гексан/этилацетат, и С: система петролейный эфир/этилацетат. Долю объема растворителя регулируют в зависимости от полярности соединений, для чего можно было также добавить небольшое количество щелочного реагента, такого как триэтиламин, или кислого реагента, такого как уксусная кислота.
Пример 1
(E)-1-Морфолино-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 1
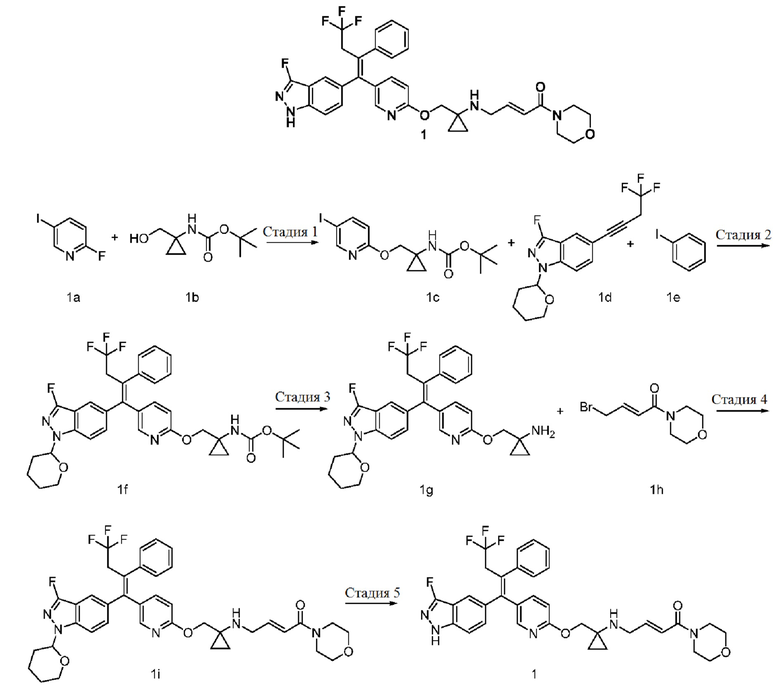
Стадия 1
Трет-бутил-(1-(((5-йодпиридин-2-ил)окси)метил)циклопропил)карбамат 1с
Гидрид натрия (0,4 г, 10,7 ммоль) растворяют в N,N-диметилформамиде (20 мл) с последующим добавлением трет-бутил-(1-(гидроксиметил)циклопропил)карбамата 1b (1,0 г, 5,3 ммоль, получают в соответствии со способом, раскрытым в Journal of Organic Chemistry, 2002, 67(11), 3965-3968) при комнатной температуре. После завершения добавления медленно добавляют 2-фтор-5-йодпиридин 1а (1,8 г, 8,0 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Реакционный раствор концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления В с получением указанного в заголовке продукта 1с (2,4 г, выход: 86%).
MC m/z (ионизация электрораспылением, ИЭР): 391,0 [М+1].
Стадия 2
Трет-бутил-(Z)-(1-(((5-(4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)карбамат 1f
3-Фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-(4,4,4-трифторбут-1-ин-1-ил)-1H-индазол 1d (1,8 г, 5,5 ммоль, получают в соответствии со способом, раскрытым в примере 3 на странице 84 описания патентентной заявки WO 2018098305) растворяют в метилтетрагидрофуране (40 мл) с последующим добавлением бис(пинаколато)дибора (1,7 г, 6,6 ммоль) и тетракис(трифенилфосфин)платины (137 мг, 0,1 ммоль). Реакционный раствор три раза продувают аргоном, подогревают до 85°С и перемешивают в течение 3 часов. Реакционный раствор охлаждают до комнатной температуры, после чего добавляют соединение 1с (2,0 г, 5,2 ммоль), бис(трифенилфосфин)палладия дихлорид (741 мг, 1,1 ммоль), карбонат цезия (3,6 г, 11,0 ммоль) и воду (1 мл). Реакционный раствор перемешивают при комнатной температуре в течение ночи. Добавляют йодбензол 1е (1,2 г, 6,1 ммоль) и гидроксид калия (1,5 г, 27,6 ммоль). Реакционный раствор три раза продувают аргоном, подогревают до 85°С, перемешивают в течение 2 часов и охлаждают до комнатной температуры. Реакционный раствор концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления В с получением указанного в заголовке продукта 1f (3,0 г, выход: 88%).
МС m/z (ИЭР): 667,2 [М+1].
Стадия 3
(Z)-1-(((5-(4,4,4-Трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропан-1-амин 1g
Соединение 1f (1,8 г, 2,7 ммоль) растворяют в дихлорметане (15 мл) с последующим добавлением трифторуксусной кислоты (3 мл). Реакционную смесь перемешивают при комнатной температуре в течение 5 часов. Реакционный раствор концентрируют при пониженном давлении, доводят рН примерно до 8 насыщенным раствором бикарбоната натрия (100 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении с получением неочищенного указанного в заголовке продукта 1g (1,4 г, выход: 89%), который используют непосредственно на следующей стадии без очистки.
Стадия 4
(Е)-1-Морфолино-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 1i
Соединение 1g (1,7 г, 2,8 ммоль) растворяют в N,N-диметилформамиде (20 мл) с последующим добавлением диизопропилэтиламина (1,1 г, 8,5 ммоль) при комнатной температуре. Добавляют (Е)-4-бром-1-морфолинобут-2-ен-1-он 1h (0,7 г, 2,8 ммоль, получают в соответствии со способом, раскрытым в примере 15 на странице 65 описания заявки на патент US 2016347717) и реакционный раствор перемешивают в течение 2 часов. Реакцию останавливают и реакционный раствор охлаждают. Добавляют насыщенный раствор бикарбоната натрия (15 мл) и экстрагируют раствор этилацетатом (2 раза по 50 мл). Органические фазы объединяют, промывают насыщенным раствором хлорида натрия (4 раза по 50 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления А с получением указанного в заголовке продукта 1i (1,3 г, выход: 65%).
МС m/z (ИЭР): 720,2 [М+1].
Стадия 5
(Е)-1-Морфолино-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 1
Соединение 1i (2,0 г, 2,8 ммоль) растворяют в метаноле (5 мл) с последующим добавлением соляной кислоты (12 н.,10 мл). Реакционный раствор перемешивают в течение 3 часов. Реакцию останавливают, и реакционный раствор охлаждают и концентрируют. Добавляют насыщенный раствор бикарбоната натрия (15 мл) и экстрагируют раствор дихлорметаном (4 раза по 50 мл). Органические фазы объединяют, последовательно промывают водой (3 раза по 30 мл) и насыщенным раствором хлорида натрия (50 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления А с получением указанного в заголовке продукта 1 (1,3 г, выход: 73%).
МС m/z (ИЭР (Ионизация электроспреем): 636,2 [М+1].
1H ЯМР (400 МГц, CD3OD) 7,65 (d, 2Н), 7,49 (d, 1H), 7,30-7,22 (m, 7Н), 6,82-6,76 (m, 1H), 6,60-6,52 (m, 2Н), 4,15 (s, 2Н), 3,62-3,39 (m, 12Н), 0,76-0,64 (m, 4Н).
Пример 2
(Е)-1-(Пирролидин-1-ил)-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 2

Стадия 1
(Е)-1-(Пирролидин-1-ил)-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 2b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1h на стадии 4 заменяют (Е)-4-бром-1-(пирролидин-1-ил)бут-2-ен-1-он 2а (получен в соответствии со способом, раскрытым в примере 11 на странице 58 описания патентной заявки US 2016347717) с получением указанного в заголовке соединения 2b (51 мг, выход: 69%).
МС m/z (ИЭР): 704,3 [М+1].
Стадия 2
(Е)-1-(Пирролидин-1-ил)-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 2
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 2b с получением указанного в заголовке соединения 2 (20 мг, выход: 65%).
МС m/z (ИЭР): 620,3 [М+1].
1Н ЯМР (400 МГц, CD3OD) 7,65 (d, 2Н), 7,50 (d, 1Н), 7,31-7,21 (m, 7H), 6,84-6,80 (m, 1H), 6,59 (d, 1H), 6,38 (d, 1H), 4,16 (s, 2H), 3,61-3,39 (m, 8H), 0,92-0,60 (m, 4H), 0,72-0,64 (m, 4H).
Пример 3
(E)-1-(Пиперидин-1-ил)-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 3

Стадия 1
(Е)-1-(Пиперидин-1-ил)-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 3b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1h на стадии 4 заменяют (Е)-4-бром-1-(пиперидин-1-ил)бут-2-ен-1-оном 3а (получен в соответствии со способом, раскрытым в примере 12 на странице 58 описания заявки на патент US 2016347717) с получением указанного в заголовке соединения 3b (40 мг, выход: 74%).
МС m/z (ИЭР): 718,4 [М+1].
Стадия 2
(Е)-1-(Пиперидин-1-ил)-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 3
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 3b с получением указанного в заголовке соединения 3 (15 мг, выход: 61%).
МС m/z (ИЭР): 634,3 [М+1].
1Н ЯМР (400 МГц, CD3OD) 7,65 (d, 2Н), 7,50 (d, 1Н), 7,30-7,21 (m, 7Н), 6,74-6,70 (m, 1Н), 6,60-6,55 (m, 2Н), 5,36-5,35 (m, 1Н), 4,36 (s, 2Н), 3,56-3,53 (m, 5Н), 3,46-3,39 (m, 2Н), 2,05 (s, 1H), 1,67-1,54 (m, 3Н), 0,92-0,68 (m, 6Н).
Пример 4
(Е)-1-(Пиперазин-1-ил)-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 4

Стадия 1
Трет-бутил-4-((Е)-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-еноил)пиперазин-1-карбоксилат 4b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1h на стадии 4 заменяют трет-бутил-(Е)-4-(4-бромбут-2-еноил)пиперазин-1-карбоксилатом 4а (получен в соответствии со способом, раскрытым в примере 32 на странице 129 описания заявки на патент WO 2014160200) с получением указанного в заголовке соединения 4b (61 мг, выход: 72%). МС m/z (ИЭР): 819,4 [М+1].
Стадия 2
(Е)-1-(Пиперазин-1-ил)-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 4
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 4b с получением указанного в заголовке соединения 4 (20 мг, выход: 41%).
МС m/z (ИЭР): 635,3 [М+1].
1H ЯМР (400 МГц, CD3OD) 7,65 (d, 2Н), 7,50 (d, 1Н), 7,30-7,20 (m, 7H), 6,82-6,77 (m, 1H), 6,59-6,53 (m, 2H), 4,15 (s, 2H), 3,69-3,67 (m, 4H), 3,53-3,41 (m, 4H), 2,96-2,94 (m, 4H), 0,72-0,62 (m, 4H).
Пример 5
(Е)-N,N-Диметил-4-(метил(1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-енамид 5
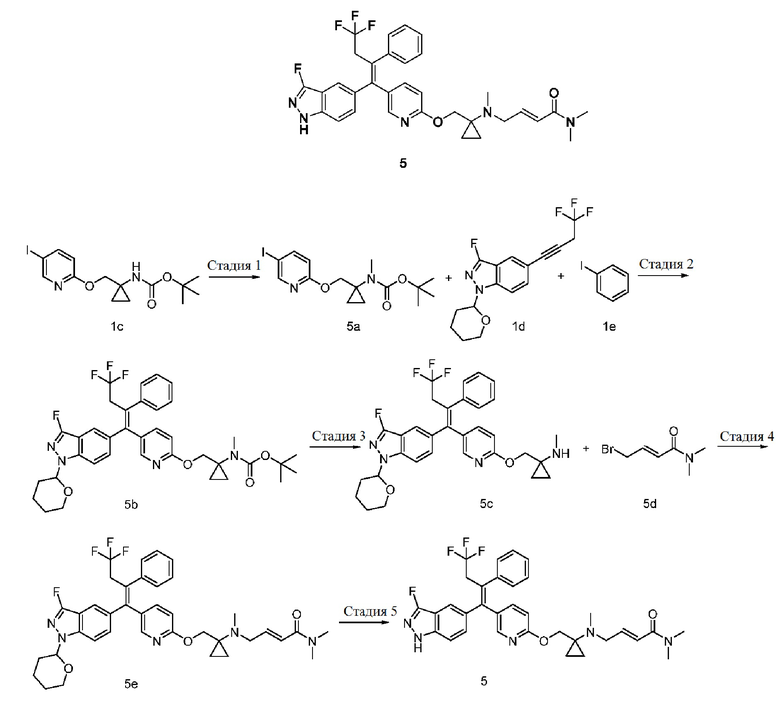
Стадия 1
Трет-бутил-(1-(((5-йодпиридин-2-ил)окси)метил)циклопропил)карбамат 5а
Соединение 1c (0,5 г, 1,3 ммоль) растворяют в N,N-диметилформамиде (25 мл) с последующим добавлением гидрида натрия (0,1 г, 2,6 ммоль) при комнатной температуре. После завершения добавления медленно добавляют йодметан (0,3 г, 1,9 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Реакционный раствор концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления В с получением указанного в заголовке продукта 5а (0,5 г, выход: 87%).
МС m/z (ИЭР): 405,1 [М+1].
Стадия 2
Трет-бутил-(Z)-метил(1-(((5-(4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)карбамат 5b
В соответствии с путем синтеза, описанным в примере 1, исходный материал 1с на стадии 2 заменяют соединением 5а с получением указанного в заголовке соединения 5b (89 мг, выход: 85%).
МС m/z (ИЭР): 681,3 [М+1].
Стадия 3
(Z)-N-метил-1-(((5-(4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропан-1-амин 5с
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1f на стадии 3 заменяют соединением 5b с получением указанного в заголовке соединения 5с (60 мг, выход: 70%).
Стадия 4
(Е)-N,N-Диметил-4-(метил(1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-енамид 5е
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1h на стадии 4 заменяют (Е)-4-бром-N,N-диметилбут-2-енамидом 5d (получен в соответствии со способом, раскрытым в примере 1 на странице 39 описания заявки на патент US 2016347717), а исходный материал 1g заменяют 5с с получением указанного в заголовке соединения 5е (60 мг, выход: 50%).
Стадия 5
(Е)-N,N-Диметил-4-(метил(1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-енамид 5
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 5е с получением указанного в заголовке соединения 5 (15 мг, выход: 28%).
МС m/z (ИЭР): 608,2 [М+1].
1H ЯМР (400 МГц, CDCl3) 9,59 (s, 1H), 7,65 (d, 2Н), 7,33 (d, 1H), 7,28-7,23 (m, 6Н), 7,10 (d, 1Н), 6,79-6,75 (m, 1Н), 6,46 (d, 1H), 6,35 (d, 1H), 4,22 (s, 2Н), 3,48 (d, 2Н), 3,38-3,35 (m, 2Н), 3,05 (d, 6Н), 2,38 (s, 3Н), 0,73-0,63 (m, 4Н).
Пример 6
(Е)-4-(Этил(1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)-N,N-диметилбут-2-енамид 6

Стадия 1
Трет-бутилэтил(1-(((5-йодпиридин-2-ил)окси)метил)циклопропил)карбамат 6а
В соответствии со способом синтеза, описанным в примере 5, реактив йодметан на стадии 1 заменяют йодэтаном с получением указанного в заголовке соединения 6а (58 мг, выход: 88%).
Стадия 2
Трет-бутил-(Z)-этил(1-(((5-(4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)карбамат 6b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1с на стадии 2 заменяют соединением 6а с получением указанного в заголовке соединения 6b (85 мг, выход: 80%).
Стадия 3
(Z)-N-этил-1-(((5-(4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропан-1-амин 6с
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1f на стадии 3 заменяют соединением 6b с получением указанного в заголовке соединения 6с (65 мг, выход: 76%).
Стадия 4
(Е)-4-(Этил(1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)-N,N-диметилбут-2-енамид 6d
В соответствии со способом синтеза, описанным в примере 5, исходный материал 5с на стадии 4 заменяют соединением 6с с получением указанного в заголовке соединения 6d (70 мг, выход: 59%).
МС m/z (ИЭР): 706,3 [М+1].
Стадия 5
(Е)-4-(Этил(1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)-N,N-диметилбут-2-енамид 6
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 6d с получением указанного в заголовке соединения 6 (15 мг, выход: 28%).
МС m/z (ИЭР): 622,3 [М+1].
1Н ЯМР (400 МГц, CDCl3) 9,74 (s, 1H), 7,66 (d, 2Н), 7,31 (d, 1H), 7,28-7,25 (m, 6Н), 7,21 (d, 1H), 6,86-6,82 (m, 1H), 6,46-6,36 (m, 2Н), 4,19 (s, 2Н), 3,53 (d, 2Н), 3,35-3,30 (m, 2Н), 3,06 (d, 6Н), 2,73-2,70 (m, 2Н), 1,05-1,02 (m, 3Н), 0,73-0,61 (m, 4Н).
Пример 7
(Е)-N,N-Диметил-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопентил)амино)бут-2-енамид 7

Стадия 1
1-(((5-Йодпиридин-2-ил)окси)метил)циклопентан-1-амин 7b
Трет-бутоксид калия (0,4 г, 3,5 ммоль) растворяют в тетрагидрофуране (10 мл) с последующим добавлением (1-аминоциклопентил)метанола 7а (0,4 г, 3,5 ммоль, получен в соответствии со способом, раскрытым в Journal of the American Chemical Society, 2004, 126(46), 15195-15201) при комнатной температуре. После завершения добавления медленно добавляют соединение 1а (0,8 г, 3,5 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Реакционный раствор концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления В с получением указанного в заголовке продукта 7b (0,7 г, выход: 66%).
МС m/z (ИЭР): 319,1 [М+1].
Стадия 2
(Е)-4-((1-(((5-Йодпиридин-2-ил)окси)метил)циклопентил)амино)-N,N-диметилбут-2-енамид 7с
Соединение 7b (100 мг, 0,3 ммоль) растворяют в N,N-диметилформамиде (10 мл), после чего последовательно добавляют диизопропилэтиламин (41 мг, 0,3 ммоль) и соединение 5d (60 г, 0,3 ммоль) при комнатной температуре. Реакционный раствор перемешивают в течение 2 часов. Реакцию останавливают и реакционный раствор охлаждают. Добавляют насыщенный раствор бикарбоната натрия (15 мл) и экстрагируют раствор этилацетатом (2 раза по 50 мл). Органические фазы объединяют, промывают насыщенным раствором хлорида натрия (4 раза по 50 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления А с получением указанного в заголовке продукта 7с (86 мг, выход: 67%).
Стадия 3
(Е)-N,N-Диметил-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопентил)амино)бут-2-енамид 7d
Соединение 1d (50 мг, 0,2 ммоль) растворяют в метилтетрагидрофуране (20 мл) с последующим добавлением бис(пинаколато)дибора (47 мг, 0,2 ммоль) и тетракис(трифенилфосфин)палладия (10 mg, 0,01 ммоль). Реакционный раствор три раза продувают аргоном, подогревают до 85°С и перемешивают в течение 3 часов. Реакционный раствор охлаждают до комнатной температуры, после чего добавляют соединение 7с (66 мг, 0,2 ммоль), бис(трифенилфосфин)палладия дихлорид (11 мг, 0,02 ммоль), карбонат цезия (150 мг, 0,5 ммоль) и воду (0,2 мл). Реакционный раствор перемешивают при комнатной температуре в течение ночи. Добавляют соединение 1е (63 мг, 0,3 ммоль) и гидроксид калия (26 мг, 0,5 ммоль). Реакционный раствор три раза продувают аргоном, подогревают до 85°С, перемешивают в течение 2 часов и охлаждают до комнатной температуры. Реакционный раствор концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления В с получением указанного в заголовке продукта 7d (84 мг, выход: 78%).
Стадия 4
(Е)-N,N-Диметил-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопентил)амино)бут-2-енамид 7
Соединение 7d (30 мг, 0,04 ммоль) растворяют в метаноле (1 мл) с последующим добавлением соляной кислоты (12 н., 2 мл). Реакционный раствор перемешивают в течение 3 часов. Реакцию останавливают, реакционный раствор охлаждают и концентрируют. Добавляют насыщенный раствор бикарбоната натрия (15 мл) и экстрагируют раствор дихлорметаном (4 раза по 10 мл). Органические фазы объединяют, последовательно промывают водой (3 раза по 10 мл) и насыщенным раствором хлорида натрия (10 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления А с получением указанного в заголовке продукта 7 (10 мг, выход: 41%).
МС m/z (ИЭР): 622,3 [М+1].
1Н ЯМР (400 МГц, CDCl3) 7,61-7,45 (m, 3Н), 7,29-7,01 (m, 7Н), 6,54-6,42 (m, 3Н), 3,95 (s, 2Н), 3,42-3,39 (m, 1Н), 3,17-3,14 (m, 1Н), 2,91 (s, 1Н), 2,77 (s, 1Н), 2,62 (s, 3Н), 2,26 (s, 3Н), 1,63-1,52 (m, 4Н), 1,49-1,38 (m, 4Н).
Пример 8
(Е)-N,N-Диметил-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклогексил)амино)бут-2-енамид 8

Стадия 1
1-(((5-Йодпиридин-2-ил)окси)метил)циклогексан-1-амин 8b
В соответствии со способом синтеза, описанным в примере 7, исходный материал 7а на стадии 1 заменяют (1-аминоциклогексил)метанолом 8а (получен в соответствии со способом, раскрытым в Journal of Medicinal Chemistry, 2004, 47(18), 4613-4626) с получением указанного в заголовке соединения 8b (650 мг, выход: 68%).
MC m/z (ИЭР): 333,1 [М+1].
Стадия 2
(Е)-4-((1-(((5-Йодпиридин-2-ил)окси)метил)циклогексил)амино)-N,N-диметилбут-2-енамид 8с
В соответствии со способом синтеза, описанным в примере 7, исходный материал 7b на стадии 2 заменяют соединением 8b с получением указанного в заголовке соединения 8с (500 мг, выход: 58%).
MC m/z (ИЭР): 444,2 [М+1].
Стадия 3
(Е)-N,N-Диметил-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклогексил)амино)бут-2-енамид 8d
В соответствии со способом синтеза, описанным в примере 7, исходный материал 7с на стадии 3 заменяют соединением 8с с получением указанного в заголовке соединения 8d (250 мг, выход: 94%).
MC m/z (ИЭР): 720,4 [М+1].
Стадия 4
(Е)-N,N-Диметил-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклогексил)амино)бут-2-енамид 8
В соответствии со способом синтеза, описанным в примере 7, исходный материал 7d на стадии 4 заменяют соединением 8d с получением указанного в заголовке соединения 8 (80 мг, выход: 36%).
MC m/z (ИЭР): 636,2 [М+1].
1Н ЯМР (400 МГц, CD3OD) 7,72 (d, 1Н), 7,65 (s, 1H), 7,49 (dd, 1H), 7,33 (dd, 2Н), 7,31-7,24 (m, 5Н), 6,80 (d, 1H), 6,68-6,65 (m, 2Н), 4,53 (s, 2Н), 3,85 (d, 2Н), 3,46-3,38 (m, 2Н), 3,08 (s, 3Н), 2,98 (s, 3Н), 2,06 (d, 2Н), 1,75-1,64 (m, 3Н), 1,64-1,47 (m, 4Н), 1,31 (m, 1Н).
Пример 9
(Е)-N,N-Диметил-4-(4-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 9

Стадия 1
Трет-бутил-(4-(((5-йодпиридин-2-ил)окси)пиперидин-1-карбоксилат 9b
Гидрид натрия (0,6 г, 14,9 ммоль) растворяют в N,N-диметилформамиде (30 мл) с последующим добавлением трет-бутил-4-гидроксипиперидин-1-карбоксилата 9а (1,0 г, 5,0 ммоль, получают в соответствии со способом, раскрытым в Journal of the American Chemical Society, 2018, 140(1), 155-158) при комнатной температуре. После завершения добавления медленно добавляют соединение 1а (2,2 г, 9,9 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Реакционный раствор концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления В с получением указанного в заголовке продукта 9b (0,6 г, выход: 30%).
Стадия 2
Трет-бутил-(Z)-4-((5-(4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-карбоксилат 9с
Соединение 1d (150 мг, 0,5 ммоль) растворяют в метилтетрагидрофуране (15 мл) с последующим добавлением бис(пинаколато)дибора (140 мг, 0,6 ммоль) и тетракис(трифенилфосфин)палладия (28 мг, 0,02 ммоль). Реакционный раствор три раза продувают аргоном, подогревают до 85°С и перемешивают в течение 3 часов. Реакционный раствор охлаждают до комнатной температуры, после чего добавляют соединение 9b (167 мг, 0,4 ммоль), бис(трифенилфосфин)палладия дихлорид (31 мг, 0,05 ммоль), карбонат цезия (300 мг, 0,9 ммоль) и воду (1 мл). Реакционный раствор перемешивают при комнатной температуре в течение ночи. Добавляют соединение 1е (187 мг, 0,9 ммоль) и гидроксид калия (129 мг, 2,3 ммоль). Реакционный раствор три раза продувают аргоном, подогревают до 85°С, перемешивают в течение 2 часов и охлаждают до комнатной температуры. Реакционный раствор концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления В с получением указанного в заголовке продукта 9с (210 мг, выход: 67%).
МС m/z (ИЭР): 681,3 [М+1].
Стадия 3
(Z)-3-Фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-(4,4,4-трифтор-2-фенил-1-(6-(пиперидин-4-илокси)пиридин-3-ил)бут-1-ен-1-ил)-1Н-имидазол 9d
Соединение 9с (120 мг, 0,2 ммоль) растворяют в дихлорметане (5 мл) с последующим добавлением трифторуксусной кислоты (1 мл). Реакционную смесь перемешивают при комнатной температуре в течение 5 часов. Реакционный раствор концентрируют при пониженном давлении, доводят рН примерно до 8 насыщенным раствором бикарбоната натрия (10 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении с получением неочищенного указанного в заголовке продукта 9d (100 мг, выход: 98%), который используют непосредственно на следующей стадии без очистки.
Стадия 4
(E)-N,N-Диметил-4-(4-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2H-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 9е
Соединение 9d (100 мг, 0,2 ммоль) растворяют в N,N-диметилформамиде (5 мл), после чего последовательно добавляют диизопропилэтиламин (67 мг, 0,5 ммоль) и соединение 5d (31 мг, 0,2 ммоль) при комнатной температуре. Реакционный раствор перемешивают в течение 2 часов. Реакцию останавливают и реакционный раствор охлаждают. Добавляют насыщенный раствор бикарбоната натрия (15 мл) и экстрагируют раствор этилацетатом (2 раза по 20 мл). Органические фазы объединяют, промывают насыщенным раствором хлорида натрия (4 раза по 20 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления А с получением указанного в заголовке продукта 9е (85 мг, выход: 71%).
Стадия 5
(Е)-N,N-Диметил-4-(4-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 9
Соединение 9е (80 мг, 0,1 ммоль) растворяют в метаноле (5 мл) с последующим добавлением слдяной кислоты (12 н., 3 мл). Реакционный раствор перемешивают в течение 3 часов. Реакцию останавливают, и реакционный раствор охлаждают и концентрируют. Добавляют насыщенный раствор бикарбоната натрия (15 мл) и экстрагируют раствор дихлорметаном (4 раза по 10 мл). Органические фазы объединяют, последовательно промывают водой (3 раза по 10 мл) и насыщенным раствором хлорида натрия (10 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления А с получением указанного в заголовке продукта 9 (25 мг, выход: 36%).
MC m/z (ИЭР): 608,4 [М+1].
1Н ЯМР (400 МГц, CDCl3) 9,91 (br, 1H), 7,68 (d, 2Н), 7,45 (d, 1H), 7,33 (s, 1H), 7,24-7,15 (m, 5Н), 6,82 (s, 2Н), 6,47 (d, 1H), 5,31 (s, 1H), 3,86 (s, 2Н), 3,49 (s, 2Н), 3,80-3,31 (m, 2Н), 3,15-3,12 (s, 3Н), 3,11 (s, 3Н), 3,05 (s, 3Н), 2,26-2,18 (m, 4Н).
Пример 10
(Z)-1-(4-((5-(4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-yn-1-он 10

Стадия 1
(Z)-1-(4-((5-(4,4,4-Трифтор-1-(3-фтор-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-yn-1-он 10b
Соединение 9d (60 мг, 0,1 ммоль) растворяют в N,N-диметилформамиде (3 мл), после чего добавляют метиламин (52 мг, 0,5 ммоль), бут-2-иновую кислоту 10а (17 мг, 0,2 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (30 мг, 0,2 ммоль) и 1-гидроксибензотриазол (24 мг, 0,2 ммоль) при комнатной температуре. Реакционный раствор перемешивают в течение 12 часов. Реакционный раствор охлаждают. Добавляют насыщенный раствор бикарбоната натрия (15 мл) и экстрагируют раствор этилацетатом (2 раза по 20 мл). Органические фазы объединяют, промывают насыщенным раствором хлорида натрия (4 раза по 20 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления А с получением указанного в заголовке продукта 10b (45 мг, выход: 67%).
Стадия 2
(Z)-1-(4-((5-(4,4,4-Трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-yn-1-он 10
Соединение 10b (45 мг, 0,07 ммоль) растворяют в метаноле (5 мл) с последующим добавлением соляной кислоты (12 н., 3 мл). Реакционный раствор перемешивают в течение 3 часов. Реакционный раствор охлаждают и концентрируют. Добавляют насыщенный раствор бикарбоната натрия (15 мл) и экстрагируют раствор дихлорметаном (4 раза по 10 мл). Органические фазы объединяют, последовательно промывают водой (3 раза по 10 мл) и насыщенным раствором хлорида натрия (10 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления А с получением указанного в заголовке продукта 10 (15 мг, выход: 38%).
МС m/z (ИЭР): 563,3 [М+1].
1Н ЯМР (400 МГц, CDCl3) 9,67 (br, 1H), 7,65 (d, 2Н), 7,45 (d, 1H), 7,34 (m, 1H), 7,21-7,17 (m, 4Н), 6,50 (d, 1H), 5,11 (s, 1H), 3,94-3,92 (m, 1H), 3,80-3,71 (m, 4Н), 3,37-3,29 (m, 2Н), 2,02 (s, 3Н), 1,94-1,75 (m, 4Н).
Пример 11
(Е)-N-метил-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 11
Стадия 1
(Е)-4-Бром-N-метилбут-2-енамид 11b
(Е)-4-Бромбут-2-еновую кислоту 11а (0,5 г, 3,0 ммоль) растворяют в дихлорметане (5 мл), после чего добавляют триэтиламин (0,3 г, 3,3 ммоль), метиламин (0,1 г, 3,3 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (0,6 г, 3,3 ммоль) и 1-гидроксибензотриазол (0,4 г, 3,3 ммоль) при комнатной температуре. Реакционный раствор перемешивают в течение 12 часов. Реакционный раствор охлаждают. Добавляют насыщенный раствор бикарбоната натрия (15 мл) и экстрагируют раствор этилацетатом (2 раза по 20 мл). Органические фазы объединяют, промывают насыщенным раствором хлорида натрия (4 раза по 20 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления А с получением указанного в заголовке продукта 11b (0,4 г, выход: 77%).
Стадия 2
Трет-бутил-(R)-3-((5-йодпиридин-2-ил)окси)пиперидин-1-карбоксилат 11d
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1b на стадии 1 заменяют трет-бутил-(R)-3-гидроксипиперидин-1-карбоксилатом 11с (получен в соответствии со способом, раскрытым в Tetrahedron, 2011, 67(7), 1485 1500) с получением указанного в заголовке соединения 11d (653 мг, выход: 91%).
Стадия 3
Трет-бутил-(3R)-3-((5-(Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-карбоксилат 11е
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1с на стадии 2 заменяют соединением 11d с получением указанного в заголовке соединения 11е (200 мг, выход: 64%).
МС m/z (ИЭР): 680,9 [М+1].
Стадия 4
3-Фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-((Z)-4,4,4-трифтор-2-фенил-1-(6-(((R)-пиперидин-3-ил)окси)пиридин-3-ил)бут-1-ен-1-ил)-1Н-имидазол 11f
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1f на стадии 3 заменяют соединением 11е с получением указанного в заголовке соединения 11f (170 мг, выход: 100%).
Стадия 5
(Е)-N-Метил-4-((3R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 11g
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют соединением 11f, а исходный материал 1h заменяют 11b с получением указанного в заголовке соединения 11g (50 мг, выход: 86%).
Стадия 6
(Е)-N-метил-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 11
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 11g с получением указанного в заголовке соединения 11 (20 мг, выход: 45%).
МС m/z (ИЭР): 594,2 [М+1].
1Н ЯМР (400 МГц, CD3OD) 7,65 (d, 2Н), 7,49 (d, 1H), 7,31-7,19 (m, 6Н), 6,68-6,62 (m, 2H), 6,30 (d, 1H), 5,39 (s, 1H), 3,88 (s, 2H), 3,67 (s, 1H), 3,48-3,38 (m, 3H), 3,26-3,13 (m, 2H), 3,01 (s, 1H), 2,80 (s, 3H), 2,06 (s, 2H), 1,86-1,73 (m, 2H).
Пример 12
(Е)-1-Морфолино-4-((R)-3-((5-((Z)-454,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-ен-1-он 12

Стадия 1
(Е)-1-Морфолино-4-((3R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-ен-1-он 12а
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют соединением 11f с получением указанного в заголовке соединения 12а (50 мг, выход: 79%).
Стадия 2
(Е)-1-Морфолино-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-ен-1-он 12
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 12а с получением указанного в заголовке соединения 12 (20 мг, выход: 42%).
МС m/z (ИЭР): 650,3 [М+1].
1Н ЯМР (400 МГц, CDCl3) 10,10 (s, 1H), 7,67-7,62 (m, 3Н), 7,50 (d, 1H), 7,27-7,11 (m, 6H), 6,79 (s, 2H), 6,48 (s, 1H), 5,38 (s, 1H), 3,93 (s, 2H), 3,70 (s, 7H), 3,55 (s, 2H), 3,38 331 (m, 3Н), 3,09-2,82 (m, 2H), 1,85-1,65 (m, 4H).
Пример 13
(Е)-N-(Тетрагидро-2Н-пиран-4-ил)-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 13

Стадия 1
(Е)-4-Бром-N-(тетрагидро-2Н-пиран-4-ил)бут-2-енамид 13а
(Е)-4-Бромбут-2-еновую кислоту 11а (1,0 г, 6,1 ммоль) растворяют в дихлорметане (10 мл), после чего добавляют триэтиламин (1,8 г, 18,2 ммоль), тетрагидропиран-4-амин (0,6 г, 6,1 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (1,3 г, 6,7 ммоль) и 1-гидроксибензотриазол (0,9 г, 6,7 ммоль) при комнатной температуре. Реакционный раствор перемешивают в течение 12 часов. Реакционный раствор охлаждают. Добавляют насыщенный раствор бикарбоната натрия (15 мл) и экстрагируют раствор этилацетатом (2 раза по 20 мл). Органические фазы объединяют, промывают насыщенным раствором хлорида натрия (4 раза по 20 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления А с получением указанного в заголовке продукта 13а (1,2 г, выход: 79%).
Стадия 2
(Е)-N-(Тетрагидро-2Н-пиран-4-ил)-4-((3R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 13b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют соединением 11f, а исходный материал 1h заменяют 13а с получением указанного в заголовке соединения 13b (41 мг, выход: 81%).
Стадия 3
(Е)-N-(Тетрагидро-2Н-пиран-4-ил)-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 13
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 13b с получением указанного в заголовке соединения 13 (20 мг, выход: 32%).
МС m/z (ИЭР): 664,3 [М+1].
1Н ЯМР (400 МГц, ДМСО-d6) 12,68 (s, 1H), 7,92-7,90 (d, 1H), 7,60-7,59 (m, 1H), 7,52-7,50 (m, 1H), 7,23-7,14 (m, 6Н), 6,50-6,43 (m, 2Н), 5,97-5,93 (d, 1H), 4,86-4,79 (m, 1Н), 3,79-3,74 (m, 3Н), 3,47-3,38 (m, 2Н), 3,33-3,32 (m, 1Н), 3,00-2,98 (m, 2Н), 2,80-2,78 (m, 1H), 2,55-2,50 (m, 1Н), 2,00-1,24 (m, 12Н).
Пример 14
(Е)-N,N-Диметил-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 14

Стадия 1
(Е)-N,N-Диметил-4-((3R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 14а
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют соединением 11f, а исходный материал 1h заменяют 5d с получением указанного в заголовке соединения 14а (80 мг, выход: 79%).
Стадия 2
(Е)-N,N-Диметил-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 14
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 14а с получением указанного в заголовке соединения 14 (10 мг, выход: 27%).
MC m/z (ИЭР): 608,3 [М+1].
1Н ЯМР (400 МГц, CDCl3) 9,77-7,75 (br, 1Н), 7,68-7,62 (m, 2Н), 7,46-7,43 (m, 1H), 7,36-7,30 (m, 2Н), 7,27-7,18 (d, 5Н), 6,87-6,76 (m, 2Н), 6,80-6,48 (m, 1H), 5,42-5,28 (m, 1Н), 3,97-3,67 (m, 5Н), 3,38-3,31 (m, 2Н), 3,12-3,07 (m, 6Н), 2,31-1,69 (m, 4Н).
Пример 15
(Е)-1-(Пирролидин-1-ил)-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-ен-1-он 15

Стадия 1
(Е)-1-(Пирролидин-1-ил)-4-((3R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-ен-1-он 15а
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют соединением 11f, а исходный материал 1h заменяют 2а с получением указанного в заголовке соединения 15а (49 мг, выход: 72%).
Стадия 2
(Е)-1-(Пирролидин-1-ил)-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-ен-1-он 15
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 15а с получением указанного в заголовке соединения 15 (20 мг, выход: 47%).
МС m/z (ИЭР): 634,3 [М+1].
1Н ЯМР (400 МГц, CD3OD) 7,63 (d, 2Н), 7,49 (dd, 1H), 7,29-7,19 (m, 7Н), 6,79-6,74 (m, 1Н), 6,49-6,43 (m, 2Н), 5,02-4,98 (m, 1H), 3,56-3,54 (m, 2Н), 3,47-3,44 (m, 4Н), 3,25 (d, 2Н), 2,92 (d, 1H), 2,65-2,64 (m, 1H), 2,37-2,35 (m, 2Н), 1,94-1,85 (m, 6Н), 1,64-1,52 (m, 2Н).
Пример 16
(Е)-N-(Трет-бутил)-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 16

Стадия 1
(Е)-4-Бром-N-(трет-бутил)бут-2-енамид 16а
Соединение 11а (0,5 г, 3,0 ммоль) растворяют в дихлорметане (10 мл), после чего добавляют триэтиламин (0,4 г, 3,6 ммоль), трет-бутиламин (0,2 г, 3,0 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (0,7 г, 3,6 ммоль) и 1-гидроксибензотриазол (0,5 г, 3,6 ммоль) при комнатной температуре. Реакционный раствор перемешивают в течение 12 часов. Реакционный раствор охлаждают. Добавляют насыщенный раствор бикарбоната натрия (15 мл) и экстрагируют раствор этилацетатом (2 раза по 20 мл). Органические фазы объединяют, промывают насыщенным раствором хлорида натрия (4 раза по 20 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления А с получением указанного в заголовке продукта 16а (0,3 г, выход: 39%).
Стадия 2
(Е)-N-(Трет-бутил)-4-((3R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 16b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют соединением 11f, а исходный материал 1h заменяют 16а с получением указанного в заголовке соединения 16b (47 мг, выход: 83%).
Стадия 3
(Е)-N-(Трет-бутил)-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 16
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 16b с получением указанного в заголовке соединения 16 (23 мг, выход: 28%).
MC m/z (ИЭР): 636,5 [М+1].
1Н ЯМР (400 МГц, CD3OD) 7,67 (d, 2Н), 7,50 (dd, 1Н), 7,32-7,22 (m, 7Н), 6,64-6,55 (m, 2Н), 6,33 (d, 1Н), 5,40 (s, 1H), 3,88 (s, 2Н), 3,69 (d, 1Н), 3,52-3,41 (m, 3Н), 3,24 (d, 1H), 3,02-2,99 (m, 1H), 2,10-2,07 (m, 2Н), 1,87-1,75 (m, 2Н), 1,36 (s, 9Н).
Пример 17
(Е)-N-Циклопентил-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 17
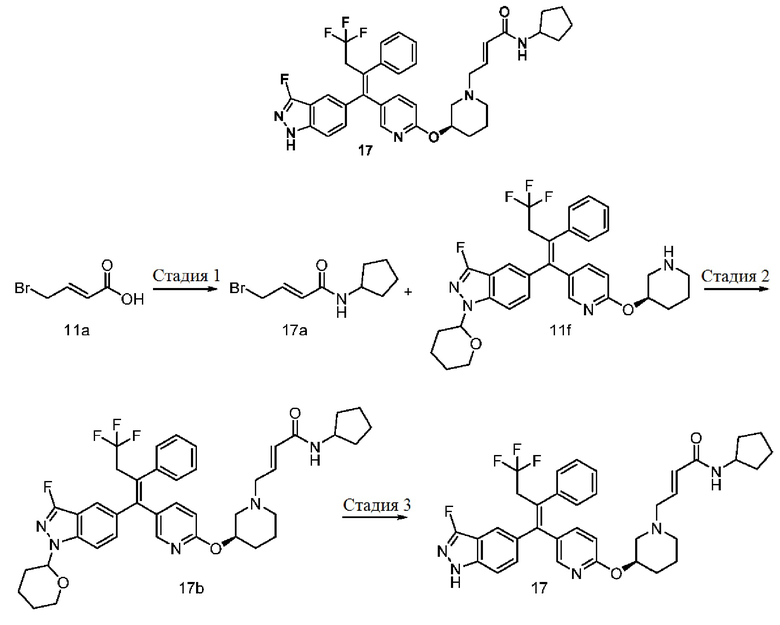
Стадия 1
(Е)-4-Бром-N-циклопентилбут-2-енамид 17а
Соединение 11а (0,5 г, 3,0 ммоль) растворяют в дихлорметане (10 мл), после чего добавляют триэтиламин (0,4 г, 3,6 ммоль), циклопентиламин (0,3 г, 3,0 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (0,7 г, 3,6 ммоль) и 1-гидроксибензотриазол (0,5 г, 3,6 ммоль) при комнатной температуре. Реакционный раствор перемешивают в течение 12 часов. Реакционный раствор охлаждают. Добавляют насыщенный раствор бикарбоната натрия (15 мл) и экстрагируют раствор этилацетатом (2 раза по 20 мл). Органические фазы объединяют, промывают насыщенным раствором хлорида натрия (4 раза по 20 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления А с получением указанного в заголовке продукта 17а (0,3 г, выход: 49%).
Стадия 2
(Е)-N-Циклопентил-4-((3R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 17b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют соединением 11f, а исходный материал 1h заменяют 17а с получением указанного в заголовке соединения 17b (42 мг, выход: 73%).
Стадия 3
(Е)-N-Циклопентил-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 17
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 17b с получением указанного в заголовке соединения 17 (21 мг, выход: 38%).
МС m/z (ИЭР): 648,3 [М+1].
1Н ЯМР (400 МГц, ДМСО-d6) 12,75-12,68 (m, 1H) 7,88-7,86 (d, 1H), 7,61-7,59 (m, 2Н), 7,53-7,50 (m, 1H), 7,23-7,15 (m, 6Н), 6,47-6,41 (m, 2Н), 5,96-5,93 (d, 1H), 4,85-4,79 (m, 1H), 4,02-3,97 (m, 1H), 3,47-3,39 (m, 2Н), 2,99-2,98 (m, 2Н), 2,80-2,79 (m, 1Н), 2,56-2,50 (m, 2Н), 2,30-2,25 (m, 1H), 1,99-1,27 (m, 12Н).
Пример 18
(Е)-1-(Пиперазин-1-ил)-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-ен-1-он 18

Стадия 1
Трет-бутил-4-((Е)-4-((3R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-еноил)пиперазин-1-карбоксилат 18а
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют соединением 11f, а исходный материал 1h заменяют соединением 4а с получением указанного в заголовке соединения 18а (51 мг, выход: 76%).
Стадия 2
(Е)-1-(Пиперазин-1-ил)-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-ен-1-он 18
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 18а с получением указанного в заголовке соединения 18 (21 мг, выход: 31%).
MC m/z (ИЭР): 649,3 [М+1].
1Н ЯМР (400 МГц, CD3OD) 7,67 (d, 2Н), 7,50 (d, 1H), 7,34-7,20 (m, 6Н), 6,92 (d, 1H), 6,78-6,61 (m, 2Н), 5,36 (s, 1H), 3,94-3,88 (m, 5Н), 3,50-3,39 (m, 6Н), 3,28 (s, 2Н), 2,23-1,88 (m, 5Н), 1,34-1,30 (m, 3Н).
Пример 19
(Е)-N,N-Диметил-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-(4-фторфенил)бут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 19

Стадия 1
(R)-5-Йод-2-(пиперидин-3-илокси)пиридин 19а
Соединение 11d (1,0 г, 2,5 ммоль) растворяют в дихлорметане (15 мл) с последующим добавлением трифторуксусной кислоты (3 мл). Реакционную смесь перемешивают при комнатной температуре в течение 5 часов. Реакционный раствор концентрируют при пониженном давлении, доводят рН примерно до 8 насыщенным раствором бикарбоната натрия (100 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении с получением неочищенного указанного в заголовке продукта 19а (0,8 г, выход: 100%), который используют непосредственно на следующей стадии без очистки.
Стадия 2
(R,Е)-4-((3-(((5-Йодпиридин-2-ил)окси)пиперидин-1-ил)-N,N-диметилбут-2-енамид 19b
Соединение 19а (752 мг, 2,5 ммоль) растворяют в N,N-диметилформамиде (20 мл), после чего последовательно добавляют диизопропилэтиламин (1,6 г, 12,4 ммоль) и соединение 5d (470 мг, 2,5 ммоль) при комнатной температуре. Реакционный раствор перемешивают в течение 2 часов. Реакционный раствор охлаждают. Добавляют насыщенный раствор бикарбоната натрия (15 мл) и экстрагируют раствор этилацетатом (2 раза по 50 мл). Органические фазы объединяют, промывают насыщенным раствором хлорида натрия (4 раза по 50 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления А с получением указанного в заголовке продукта 19b (0,7 г, выход: 68%).
МС (ИЭР): m/z 416,1 [М+1].
Стадия 3
(Е)-N,N-Диметил-4-((3R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-(4-фторфенил)бут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 19d
Соединение 1d (50 мг, 0,2 ммоль) растворяют в метилтетрагидрофуране (10 мл) с последующим добавлением бис(пинаколато)дибора (47 мг, 0,2 ммоль) и тетракис(трифенилфосфин)палладия (4 мг, 0,003 ммоль). Реакционный раствор три раза продувают аргоном, подогревают до 85°С и перемешивают в течение 3 часов. Реакционный раствор охлаждают до комнатной температуры, после чего добавляют соединение 19b (57 мг, 0,1 ммоль), бис(трифенилфосфин)палладия дихлорид (20 мг, 0,03 ммоль), карбонат цезия (128 мг, 0,4 ммоль) и воду (0,5 мл). Реакционный раствор перемешивают при комнатной температуре в течение ночи. Добавляют 4-фторйодбензол 19с (68 мг, 0,3 ммоль) и гидроксид калия (43 мг, 0,8 ммоль). Реакционный раствор три раза продувают аргоном, подогревают до 85°С, перемешивают в течение 2 часов и охлаждают до комнатной температуры. Реакционный раствор концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления В с получением указанного в заголовке продукта 19d (50 мг, выход: 46%).
МС m/z (ИЭР): 710,3 [М+1].
Стадия 4
(Е)-N,N-Диметил-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-(4-фторфенил)бут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 19
Соединение 19d (50 мг, 0,07 ммоль) растворяют в метаноле (2 мл) с последующим добавлением соляной кислоты (12 н., 1 мл). Реакционный раствор перемешивают в течение 3 часов. Реакционный раствор охлаждают и концентрируют. Добавляют насыщенный раствор бикарбоната натрия (15 мл) и экстрагируют раствор дихлорметаном (4 раза по 10 мл). Органические фазы объединяют, последовательно промывают водой (3 раза по 10 мл) и насыщенным раствором хлорида натрия (10 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления А с получением указанного в заголовке продукта 19 (15 мг, выход: 34%).
МС m/z (ИЭР): 626,2 [М+1].
1Н ЯМР (400 МГц, CD3OD) 7,70 (s, 1Н), 7,65 (s, 1H), 7,52 (dd, 1H), 7,32-7,26 (m, 4Н), 7,02-6,97 (t, 2Н), 6,91-6,87 (m, 1H), 6,65-6,61 (d, 2Н), 5,44 (s, 1H), 3,94 (s, 3Н), 3,50-3,38 (m, 4Н), 3,14 (s, 3Н), 3,12-3,05 (m, 1H), 3,01 (s, 3Н), 2,11-2,09 (m, 2Н), 1,89-1,77 (m, 2Н).
Пример 20
(Е)-4-((R)-3-((5-((Z)-2-(4-Ацетамидофенил)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)бут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)-N,N-диметилбут-2-енамид 20

Стадия 1
(E)-4-((3R)-3-((5-((Z)-2-(4-Ацетамидофенил)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)бут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)-N,N-диметилбут-2-енамид 20b
В соответствии со способом синтеза, описанным в примере 19, исходный материал 19с на стадии 3 заменяют N-(4-йодфенил)ацетамидом 20а (получен в соответствии со способом, раскрытым в Journal of Organic Chemistry, 2018, 83(2), 930-938) с получением указанного в заголовке соединения 20b (70 мг, выход: 61%).
Стадия 2
(Е)-4-((R)-3-((5-((Z)-2-(4-Ацетамидофенил)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)бут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)-N,N-диметилбут-2-енамид 20
В соответствии со способом синтеза, описанным в примере 19, исходный материал 19d на стадии 4 заменяют соединением 20b с получением указанного в заголовке соединения 20 (22 мг, выход: 41%).
МС m/z (ИЭР): 665,2 [М+1].
1Н ЯМР (400 МГц, CD3OD) 7,67 (d, 2Н), 7,48-7,44 (m, 3Н), 7,28 (dd, 2Н), 7,18 (d, 2Н), 6,82 (d, 1H), 6,67-6,59 (m, 2Н), 5,28 (s, 1H), 3,76 (s, 2Н), 3,49-3,39 (m, 4Н), 3,25 (d, 1Н), 3,10 (s, 3Н), 3,03 (d, 1H), 2,99 (s, 3Н), 2,11 (s, 3Н), 1,99-1,94 (m, 2Н), 1,86-1,74 (m, 2Н).
Пример 21
4-((Z)-1-(6-(((R)-1-((Е)-4-(Диметиламино)-4-оксобут-2-ен-1-ил)пиперидин-3-ил)окси)пиридин-3-ил)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)бут-1-ен-2-ил)-N-метилбензамид 21
Стадия 1
4-((Z)-1-(6-(((R)-1-((Е)-4-(Диметиламино)-4-оксобут-2-ен-1-ил)пиперидин-3-ил)окси)пиридин-3-ил)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)бут-1-ен-2-ил)-N-метилбензамид 21b
В соответствии со способом синтеза, описанным в примере 19, исходный материал 19с на стадии 3 заменяют 4-йод-N-метилбензамидом 21а (получен в соответствии со способом, раскрытым в Journal of the American Chemical Society, 2013, 135(12), 4628-4631) с получением указанного в заголовке соединения 21b (50 мг, выход: 44%).
Стадия 2
4-((Z)-1-(6-(((R)-1-((Е)-4-(Диметиламино)-4-оксобут-2-ен-1-ил)пиперидин-3-ил)окси)пиридин-3-ил)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)бут-1-ен-2-ил)-N-метилбензамид 21
В соответствии со способом синтеза, описанным в примере 19, исходный материал 19d на стадии 4 заменяют соединением 21b с получением указанного в заголовке соединения 21 (10 мг, выход: 32%).
МС m/z (ИЭР): 665,3 [М+1].
1Н ЯМР (400 МГц, CD3OD) 7,71-7,69 (m, 4Н), 7,50 (d, 1Н), 7,36-7,29 (m, 4Н), 6,88 (d, 1H), 6,65-6,58 (m, 2Н), 5,39 (s, 1Н), 3,97-3,91 (m, 2Н), 3,69 (d, 1H), 3,49-3,44 (m, 3Н), 3,06 (s, 3Н), 3,05-3,00 (m, 2Н), 2,99 (s, 3Н), 2,89 (s, 3Н), 2,10-2,03 (m, 2Н), 1,86-1,74 (m, 2Н).
Пример 22
(Е)-N,N-Диметил-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-(4-(трифторметил)фенил)бут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 22

Стадия 1
(Е)-N,N-Диметил-4-((3R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-(4-(трифторметил)фенил)бут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 22b
В соответствии со способом синтеза, описанным в примере 19, исходный материал 19с на стадии 3 заменяют 1-йод-4-(трифторметил)бензолом 22а с получением указанного в заголовке соединения 22b (80 мг, выход: 69%).
Стадия 2
(Е)-N,N-Диметил-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-(4-(трифторметил)фенил)бут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 22
В соответствии со способом синтеза, описанным в примере 19, исходный материал 19d на стадии 4 заменяют соединением 22b с получением указанного в заголовке соединения 22 (20 мг, выход: 31%).
MC m/z (ИЭР): 676,2 [М+1].
1H ЯМР (400 МГц, CD3OD) 7,70 (dd, 2Н), 7,57-7,55 (m, 3Н), 7,47 (d, 2Н), 7,32 (dd, 2Н), 6,88 (d, 1Н), 6,65-6,59 (m, 2Н), 5,37 (s, 1H), 3,91 (d, 2Н), 3,50 (q, 2Н), 3,32 (s, 4Н), 3,12 (s, 3Н), 3,00 (s, 3Н), 2,09-2,04 (m, 2Н), 1,84 (d, 2Н).
Пример 23
(Е)-N,N-Диметил-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-(4-(метилсульфонил)фенил)бут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 23


Стадия 1
(Е)-N,N-Диметил-4-((3R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-(4-(метилсульфонил)фенил)бут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 23b
В соответствии со способом синтеза, описанным в примере 19, исходный материал 19с на стадии 3 заменяют 1-йод-4-(метилсульфонил)бензолом 23а (получен в соответствии со способом, раскрытым в Organic Letters, 2014, 16(9), 2306-2309) с получением указанного в заголовке соединения 23b (60 мг, выход: 51%).
Стадия 2
(Е)-N,N-Диметил-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-(4-(метилсульфонил)фенил)бут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 23
В соответствии со способом синтеза, описанным в примере 19, исходный материал 19d на стадии 4 заменяют соединением 23b с получением указанного в заголовке соединения 23 (10 мг, выход: 28%).
МС m/z (ИЭР): 686,2 [М+1].
1Н ЯМР (400 МГц, CD3OD) 7,83 (d, 2Н), 7,67 (s, 2Н), 7,53-7,50 (m, 3Н), 7,30 (d, 2Н), 6,89 (dd, 1H), 6,66-6,59 (m, 2Н), 5,38 (s, 1H), 3,97-3,91 (m, 3Н), 3,70-3,65 (m, 1H), 3,50 (q, 3Н), 3,28 (d, 1H), 3,11 (s, 6Н), 2,99 (s, 3Н), 2,10-2,01 (m, 2Н), 1,94-1,86 (m, 2Н).
Пример 24
(Е)-4-((R)-3-((5-((Z)-2-(4-Цианофенил)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)бут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)-N,N-диметилбут-2-енамид 24

Стадия 1
(E)-4-((3R)-3-((5-((Z)-2-(4-Цианофенил)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)бут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)-N,N-диметилбут-2-енамид 24b
В соответствии со способом синтеза, описанным в примере 19, исходный материал 19с на стадии 3 заменяют 4-йодбензонитрил 24а с получением указанного в заголовке соединения 24b (80 мг, выход: 73%).
Стадия 2
(Е)-4-((R)-3-((5-((Z)-2-(4-Цианофенил)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)бут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)-N,N-диметилбут-2-енамид 24
В соответствии со способом синтеза, описанным в примере 19, исходный материал 19d на стадии 4 заменяют соединением 24b с получением указанного в заголовке соединения 24 (20 мг, выход: 38%).
MC m/z (ИЭР): 633,2 [М+1].
1Н ЯМР (400 МГц, CD3OD) 7,67-7,61 (m, 4Н), 7,47 (d, 1Н), 7,45 (d, 2Н), 7,33 (d, 2Н), 6,89 (d, 1Н), 6,66-6,61 (m, 2Н), 5,44 (s, 1Н), 3,94-3,91 (m, 2Н), 3,71 (d, 1H), 3,52-3,45 (m, 3Н), 3,25 (d, 1Н), 3,12 (s, 3Н), 3,05 (d, 1Н), 3,01 (s, 3Н), 2,11-2,04 (m, 2Н), 1,90-1,77 (m, 2Н).
Пример 25
(R,Z)-1-(3-((5-(4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-ин-1-он 25

Стадия 1
1-((3R)-3-((5-((Z)-4,4,4-Трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-ин-1-он 25а
В соответствии со способом синтеза, описанным в примере 10, исходный материал 9d на стадии 1 заменяют соединением 11f с получением указанного в заголовке соединения 25а (37 мг, выход: 83%).
Стадия 2
(R,Z)-1-(3-((5-(4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-ин-1-он 25
В соответствии со способом синтеза, описанным в примере 10, исходный материал 10b на стадии 2 заменяют соединением 25а с получением указанного в заголовке соединения 25 (4 мг, выход: 15%).
МС m/z (ИЭР): 563,2 [М+1].
1H ЯМР (400 МГц, CDCl3) 9,38 (s, 1Н), 7,68-7,65 (m, 2H), 7,43-7,12 (m, 8H), 6,46-6,39 (m, 1H), 5,02 (d, 1H), 3,95-3,77 (m, 2H), 3,33-3,00 (m, 4H), 2,13-1,93 (m, 4H), 1,69 (d, 3H).
Пример 26
(R,Z)-1-(3-((5-(4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)проп-2-ен-1-он 26

Стадия 1
1-((3R)-3-((5-((Z)-4,4,4-Трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)проп-2-ен-1-он 26b
В соответствии со способом синтеза, описанным в примере 10, исходный материал 9d на стадии 1 заменяют соединением 11f, а исходный материал 10а заменяют акриловой кислотой 26а с получением указанного в заголовке соединения 26b (30 мг, выход: 73%).
Стадия 2
(R,Z)-1-(3-((5-(4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)проп-2-ен-1-он 26
В соответствии со способом синтеза, описанным в примере 10, исходный материал 10b на стадии 2 заменяют соединением 26b с получением указанного в заголовке соединения 26 (3 мг, выход: 12%).
МС m/z (ИЭР): 551,2 [М+1].
1Н ЯМР (400 МГц, CDCl3) 9,31 (s, 1Н), 7,68 (d, 2Н), 7,43 (d, 2Н), 7,29-7,27 (m, 5Н), 7,20-7,09 (m, 1H), 6,40 (d, 1H), 6,39-6,20 (m, 1H), 5,49-5,37 (m, 2Н), 4,95 (s, 1H), 3,72 (s, 1H), 3,68 (d, 2Н), 3,39 (d, 1H), 3,36-3,31 (m, 2Н), 2,04-1,90 (m, 4Н).
Пример 27
(Е)-N,N-Диметил-4-((S)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 27
Стадия 1
Трет-бутил-(S)-3-((5-йодпиридин-2-ил)окси)пиперидин-1-карбоксилат 27b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1b на стадии 1 заменяют трет-бутил-(S)-3-гидроксипиперидин-1-карбоксилатом 27а (получен в соответствии со способом, раскрытым в Tetrahedron, 2007, 63(2), 331-336) с получением указанного в заголовке соединения 27b (531 мг, выход: 92%).
Стадия 2
Трет-бутил-(3S)-3-((5-(Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2H-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-карбоксилат 27с
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1с на стадии 2 заменяют соединением 27b с получением указанного в заголовке соединения 27с (190 мг, выход: 76%).
MC m/z (ИЭР): 680,9 [М+1].
Стадия 3
3-Фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-((Z)-4,4,4-трифтор-2-фенил-1-(6-(((S)-пиперидин-3-ил)окси)пиридин-3-ил)бут-1-ен-1-ил)-1Н-имидазол 27d
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1f на стадии 3 заменяют соединением 27с с получением указанного в заголовке соединения 27d (85 мг, выход: 100%).
Стадия 4
(Е)-N,N-Диметил-4-((3S)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 27е
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют соединением 27d, а исходный материал 1h заменяют соединением 5d с получением указанного в заголовке соединения 27е (65 мг, выход: 64%).
Стадия 5
(Е)-N,N-Диметил-4-((S)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)бут-2-енамид 27
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 27е с получением указанного в заголовке соединения 27 (15 мг, выход: 43%).
MC m/z (ИЭР): 607,9 [М+1].
1H ЯМР (400 МГц, CDCl3) 9,36 (s, 1H), 7,69-7,67 (m, 1H), 7,64-7,63 (d, 1H), 7,45-7,43 (m, 1Н), 7,35-7,32 (d, 1Н), 7,28-7,19 (m, 4Н), 7,08-7,06 (d, 1H), 6,89-6,82 (m, 1H), 6,46-6,41 (m, 1Н), 5,39-5,36 (m, 1H), 5,08-5,02 (br, 1H), 3,38-3,30 (m, 2Н), 3,22-3,16 (m, 2Н), 3,05 (s, 3Н), 3,10 (s, 3Н), 2,92-2,86 (m, 1H), 2,67-2,61 (m, 1H), 2,34-2,24 (m, 2Н), 2,07-1,77 (m, 4Н), 1,53-1,48 (m, 1H).
Пример 28
(Z)-1-(4-((5-(4,4,4-Трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)проп-2-ен-1-он 28


Стадия 1
(Z)-1-(4-((5-(4,4,4-Трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)проп-2-ен-1-он 28а
В соответствии со способом синтеза, описанным в примере 10, исходный материал 10а на стадии 1 заменяют соединением 26а с получением указанного в заголовке соединения 28а (50 мг, выход: 63%).
Стадия 2
(Z)-1-(4-((5-(4,4,4-Трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пиперидин-1-ил)проп-2-ен-1-он 28
В соответствии со способом синтеза, описанным в примере 10, исходный материал 10b на стадии 2 заменяют соединением 28а с получением указанного в заголовке соединения 28 (10 мг, выход: 23%).
МС m/z (ИЭР): 551,2 [М+1].
1H ЯМР (400 МГц, CDCl3) 9,47 (br, 1H), 7,60 (d, 2Н), 7,35 (d, 1H), 7,28 (d, 1H), 7,26-7,19 (m, 5Н), 6,59 (dd, 1Н), 6,49 (d, 1H), 6,29 (d, 1H), 5,76 (d, 1Н), 5,14 (m, 1H), 3,89-3,53 (m, 5Н), 3,34 (t, 2Н), 1,99-1,94 (m, 2Н), 1,77-1,66 (m, 2Н).
Пример 29
(Е)-N,N-Диметил-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пирролидин-1-ил)бут-2-енамид 29


Стадия 1
Трет-бутил-(R)-3-((5-йодпиридин-2-ил)окси)пирролидин-1-карбоксилат 29b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1b на стадии 1 заменяют трет-бутил-(R)-3-гидроксипирролидин-1-карбоксилатом 29а с получением указанного в заголовке соединения 29b (0,9 г, выход: 86%).
Стадия 2
Трет-бутил-(3R)-3-((5-(Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пирролидин-1-карбоксилат 29с
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1с на стадии 2 заменяют соединением 29b с получением указанного в заголовке соединения 29с (260 мг, выход: 37%).
Стадия 3
3-Фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-((Z)-4,4,4-трифтор-2-фенил-1-(6-(((R)-пирролидин-3-ил)окси)пиридин-3-ил)бут-1-ен-1-ил)-1Н-имидазол 29d
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1f на стадии 3 заменяют соединением 29с с получением указанного в заголовке соединения 29d (93 мг, выход: 99%).
Стадия 4
(Е)-N,N-Диметил-4-((3R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пирролидин-1-ил)бут-2-енамид 29е
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют соединением 29d, а исходный материал 1h заменяют соединением 5d с получением указанного в заголовке соединения 29е (60 мг, выход: 54%).
МС m/z (ИЭР): 678,3 [М+1].
Стадия 5
(Е)-N,N-Диметил-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пирролидин-1-ил)бут-2-енамид 29
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 29е с получением указанного в заголовке соединения 29 (15 мг, выход: 29%).
МС m/z (ИЭР): 594,3 [М+1].
1Н ЯМР (400 МГц, CDCl3) 12,72 (s, 1H), 7,70-7,65 (m, 2Н), 7,60-7,50 (d, 1H), 7,25-7,20 (m, 5Н), 7,19-7,15 (m, 2Н), 6,55-6,45 (m, 3Н), 5,17-5,10 (m, 1H), 3,50-3,35 (m, 2Н), 3,15-3,10 (m, 2Н), 2,95 (s, 3Н), 2,83 (s, 3Н), 2,70-2,60 (m, 2Н), 2,50-2,45 (m, 1H), 2,35-2,25 (m, 1H), 2,15-2,05 (m, 1Н), 1,70-1,60 (m, 1Н).
Пример 30
(Е)-N,N-Диметил-4-((S)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пирролидин-1-ил)бут-2-енамид 30

Стадия 1
Трет-бутил-(S)-3-((5-йодпиридин-2-ил)окси)пирролидин-1-карбоксилат 30b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1b на стадии 1 заменяют трет-бутил-(S)-3-гидроксипирролидин-1-карбоксилатом 30а с получением указанного в заголовке соединения 30b (0,5 г, выход: 50%).
Стадия 2
Трет-бутил-(3S)-3-((5-(Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пирролидин-1-карбоксилат 30с
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1с на стадии 2 заменяют соединением 30b с получением указанного в заголовке соединения 30с (260 мг, выход: 85%).
Стадия 3
3-Фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-((Z)-4,4,4-трифтор-2-фенил-1-(6-(((S)-пирролидин-3-ил)окси)пиридин-3-ил)бут-1-ен-1-ил)-1H-имидазол 30d
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1f на стадии 3 заменяют соединением 30с с получением указанного в заголовке соединения 30d (100 мг, выход: 98%).
Стадия 4
(Е)-N,N-Диметил-4-((3S)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пирролидин-1-ил)бут-2-енамид 30е
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют соединением 30d, а исходный материал 1h заменяют соединением 5d с получением указанного в заголовке соединения 30е (70 мг, выход: 62%).
МС m/z (ИЭР): 678,3 [М+1].
Стадия 5
(Е)-N,N-Диметил-4-((S)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)пирролидин-1-ил)бут-2-енамид 30
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 30е с получением указанного в заголовке соединения 30 (10 мг, выход: 23%).
MC m/z (ИЭР): 593,9 [М+1].
1Н ЯМР (400 МГц, CDCl3) 9,58 (s, 1H), 7,67-7,65 (m, 2Н), 7,46 (d, 1H), 7,34-7,30 (m, 2Н), 7,27-7,16 (m, 4Н), 6,87-6,77 (m, 2Н), 6,59 (d, 1H), 5,53 (s, 1,Н), 4,14 (s, 1H), 3,93 (s, 3Н), 3,39-3,31 (m, 2Н), 3,29-3,03 (m, 7Н), 2,41-2,28 (m, 4Н).
Пример 31
(Е)-N,N-Диметил-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-енамид 31


Стадия 1
Трет-бутил-(R)-3-((5-йодпиридин-2-ил)амино)пиперидин-1-карбоксилат 31b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1b на стадии 1 заменяют трет-бутил-(R)-3-аминопиперидин-1-карбоксилатом 31а с получением указанного в заголовке соединения 31b (0,8 г, выход: 57%).
Стадия 2
Трет-бутил-(3R)-3-((5-(Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)амино)пиперидин-1-карбоксилат 31с
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1с на стадии 2 заменяют соединением 31b с получением указанного в заголовке соединения 31с (210 мг, выход: 75%).
Стадия 3
(R,Z)-N-(Пиперидин-3-ил)-5-(4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-амин 31d
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1f на стадии 3 заменяют соединением 31с с получением указанного в заголовке соединения 31d (42 мг, выход: 92%).
Стадия 4
(Е)-N,N-Диметил-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-енамид 31
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют соединением 31d, а исходный материал 1h заменяют соединением 5d с получением указанного в заголовке соединения 31 (20 мг, выход: 39%).
МС m/z (ИЭР): 607,3 [М+1].
1Н ЯМР (400 МГц, CDCl3) 9,26 (s, 1H), 7,68 (s, 1H), 7,55-7,54 (m, 1H), 7,44-7,41 (m, 1H), 7,36-7,31 (m, 1H), 7,28-7,23 (m, 4Н), 6,92-6,80 (m, 2Н), 6,45-6,41 (m, 1H), 6,15-6,13 (m, 1Н), 5,39-5,37 (m, 1Н), 3,35-3,28 (m, 2Н), 3,17-3,15 (m, 2Н), 3,07 (s, 3Н), 3,02 (s, 3Н), 2,70-2,68 (m, 1Н), 2,47-2,45 (m, 1H), 2,47-2,45 (m, 1H), 2,06-2,04 (m, 2Н), 1,34-1,29 (m, 4Н).
Пример 32
(Е)-1-Морфолино-4-((R)-3-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-ен-1-он 32

В соответствии со способом синтеза, описанным в примере 31, исходный материал 5d на стадии 4 заменяют соединением 1h с получением указанного в заголовке соединения 32 (8 мг, выход: 19%).
МС m/z (ИЭР): 649,2 [М+1].
1Н ЯМР (400 МГц, CDCl3) 9,25 (s, 1H), 7,68 (s, 1H), 7,56-7,52 (m, 1H), 7,45-7,40 (m, 1H), 7,36-7,32 (m, 1H), 7,28-7,21 (m, 4Н), 6,94-6,82 (m, 2Н), 6,44-6,37 (m, 1H), 6,15-6,07 (m, 1H), 5,39-5,35 (m, 1H), 3,76-3,46 (m, 8Н), 3,36-3,26 (m, 2Н), 3,17-3,05 (m, 2Н), 2,99-2,92 (m, 1H), 2,65-2,03 (m, 7Н), 0,95-0,87 (m, 1Н).
Пример 33
(Е)-N,N-Диметил-4-(4-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-енамид 33


Стадия 1
Трет-бутил-4-((5-йодпиридин-2-ил)амино)пиперидин-1-карбоксилат 33b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1b на стадии 1 заменяют трет-бутил-4-аминопиперидин-1-карбоксилатом 33а с получением указанного в заголовке соединения 33b (0,2 г, выход: 20%).
Стадия 2
Трет-бутил-(Z)-4-((5-(4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)амино)пиперидин-1-карбоксилат 33с
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1с на стадии 2 заменяют соединением 33b с получением указанного в заголовке соединения 33с (80 мг, выход: 64%).
МС m/z (ИЭР): 680,3 [М+1].
Стадия 3
(Z)-N-(Пиперидин-4-ил)-5-(4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-амин 33d
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1f на стадии 3 заменяют соединением 33с с получением указанного в заголовке соединения 33d (68 мг, выход: 100%).
Стадия 4
(E)-N,N-Диметил-4-(4-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2H-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-енамид 33е
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют соединением 33d, а исходный материал 1h заменяют соединением 5d с получением указанного в заголовке соединения 33е (70 мг, выход: 86%).
Стадия 5
(Е)-N,N-Диметил-4-(4-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-енамид 33
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 33е с получением указанного в заголовке соединения 33 (15 мг, выход: 24%).
МС m/z (ИЭР): 607,7 [М+1].
1Н ЯМР (400 МГц, CDCl3) 7,63 (s, 1H), 7,59 (s, 1Н), 7,38-7,32 (m, 4Н), 7,24 (d, 2Н), 7,23-7,02 (m, 2Н), 6,77 (s, 2Н), 6,47 (d, 1H), 5,36 (s, 1H), 3,93 (s, 1H), 3,83 (s, 2Н), 3,39-3,29 (m, 6Н), 3,12 (s, 3Н), 3,09 (s, 3Н), 2,42 (m, 2Н), 2,07 (s, 2Н).
Пример 34
(Е)-N,N-Диметил-4-(((R)-1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пирролидин-3-ил)амино)бут-2-енамид 34


Стадия 1
Трет-бутил-(R)-(1-(5-йодпиридин-2-ил)пирролидин-3-ил)карбамат 34b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1b на стадии 1 заменяют трет-бутил-(R)-пирролидин-3-илкарбаматом 34а с получением указанного в заголовке соединения 34b (0,8 г, выход: 70%).
Стадия 2
Трет-бутил-(3R)-1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пирролидин-3-ил)карбамат 34с
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1с на стадии 2 заменяют соединением 34b с получением указанного в заголовке соединения 34с (85 мг, выход: 83%).
МС m/z (ИЭР): 665,9 [М+1].
Стадия 3
(3R)-1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пирролидин-3-амин 34d
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1f на стадии 3 заменяют соединением 34с с получением указанного в заголовке соединения 34d (60 мг, был получен выход: 82%).
Стадия 4
(E)-N,N-Диметил-4-(((3R)-1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пирролидин-3-ил)амино)бут-2-енамид 34е
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют соединением 34d, а исходный материал 1h заменяют соединением 5d с получением указанного в заголовке соединения 34е (45 мг, выход: 63%).
Стадия 5
(Е)-N,N-Диметил-4-(((R)-1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пирролидин-3-ил)амино)бут-2-енамид 34
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 34е с получением указанного в заголовке соединения 34 (9 мг, выход: 23%).
МС m/z (ИЭР): 592,9 [М+1].
1Н ЯМР (400 МГц, CDCl3) 9,53 (s, 1H), 7,67 (s, 2Н), 7,38 (d, 1H), 7,30-7,22 (m, 6Н), 6,95-6,87 (m, 2Н), 6,45 (d, 1H), 6,07 (d, 1H), 3,63-3,59 (m, 1H), 3,54-3,48 (m, 3Н), 3,41-3,28 (m, 3Н), 3,23-3,19 (m, 1H), 3,06 (d, 6Н), 2,27-2,14 (m, 2Н), 1,89-1,81 (m, 2Н).
Пример 35
(Е)-N,N-Диметил-4-(((8)-1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут- 1-ен-1-ил)пиридин-2-ил)пирролидин-3-ил)амино)бут-2-енамид 35


Стадия 1
Трет-бутил-(S)-(1-(5-йодпиридин-2-ил)пирролидин-3-ил)карбамат 35b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1b на стадии 1 заменяют трет-бутил-(S)-пирролидин-3-илкарбаматом 35а с получением указанного в заголовке соединения 35b (0,8 г, выход: 69%).
Стадия 2
Трет-бутил-((3S)-1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2H-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пирролидин-3-ил)карбамат 35с
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1с на стадии 2 заменяют соединением 35b с получением указанного в заголовке соединения 35с (86 мг, выход: 84%).
МС m/z (ИЭР): 666,3 [М+1].
Стадия 3
(3S)-1-(5-((Z)-4,4,4-Трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пирролидин-3-амин 35d
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1f на стадии 3 заменяют соединением 35с с получением указанного в заголовке соединения 35d (55 мг, выход: 80%).
Стадия 4
(Е)-N,N-Диметил-4-(((3S)-1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пирролидин-3-ил)амино)бут-2-енамид 35е
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют соединением 35d, а исходный материал 1h заменяют соединением 5d с получением указанного в заголовке соединения 35е (40 мг, выход: 61%).
Стадия 5
(Е)-N,N-Диметил-4-(((S)-1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пирролидин-3-ил)амино)бут-2-енамид 35
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 35е с получением указанного в заголовке соединения 35 (8 мг, выход: 23%).
МС m/z (ИЭР): 592,9 [М+1].
1Н ЯМР (400 МГц, CDCl3) 9,47 (s, 1H), 7,67 (s, 2Н), 7,39 (dd, 1H), 7,34 (dd, 1Н),7,28-7,22 (m, 5Н), 6,96-6,87 (m, 2Н), 6,45 (dd, 1H), 6,07 (dd, 1H), 3,63-3,59 (m, 1H), 3,54-3,46 (m, 3Н), 3,41-3,28 (m, 3Н), 3,22-3,19 (m, 1Н), 3,07 (d, 6Н), 2,20-2,15 (m, 2Н), 1,88-1,81 (m, 2Н).
Пример 36
(Е)-N,N-Диметил-4-((1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пиперидин-4-ил)амино)бут-2-енамид 36


Стадия 1
Трет-бутил-(1-(5-йодпиридин-2-ил)пиперидин-4-ил)карбамат 36b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1b на стадии 1 заменяют трет-бутил-пиперидин-4-илкарбаматом 36а с получением указанного в заголовке соединения 36b (0,3 г, выход: 28%).
Стадия 2
Трет-бутил-(Z)-(1-(5-(4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2H-пиран-2-ил)-lH-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пиперидин-4-ил)карбамат 36с
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1 с на стадии 2 заменяют соединением 36b с получением указанного в заголовке соединения 36с (120 мг, выход: 74%).
МС m/z (ИЭР): 679,9 [М+1].
Стадия 3
(Z)-1-(5-(4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пиперидин-4-амин 36d
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1f на стадии 3 заменяют соединением 36с с получением указанного в заголовке соединения 36d (68 мг, выход: 100%).
Стадия 4
(E)-N,N-Диметил-4-((1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пиперидин-4-ил)амино)бут-2-енамид 36е
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют соединением 36d, а исходный материал 1h заменяют соединением 5d с получением указанного в заголовке соединения 36е (70 мг, выход: 86%).
Стадия 5
(Е)-N,N-Диметил-4-((1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пиперидин-4-ил)амино)бут-2-енамид 36
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 36е с получением указанного в заголовке соединения 36 (15 мг, выход: 24%).
МС m/z (ИЭР): 607,7 [М+1].
1Н ЯМР (400 МГц, CD3OD) 7,70 (s, 1H), 7,56-7,53 (m, 1H), 7,48-7,42 (m, 1H), 7,35-7,32 (m, 1H), 7,30-7,26 (m, 6Н), 7,00-6,98 (d, 1H), 6,92-6,89 (m, 1H), 6,72-6,64 (m, 1H), 4,25-4,21 (d, 2Н), 3,92-3,90 (d, 2Н), 3,57-3,39 (m, 4Н), 3,17-3,11 (m, 4Н), 3,01 (s, 3Н), 2,24-2,22 (m, 2Н), 1,67-1,67 (m, 2Н).
Пример 37
(Е)-N,N-Диметил-4-(метил((S)-1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пиперидин-3-ил)амино)бут-2-енамид 37


Стадия 1
Трет-бутил-(S)-(1-(5-йодпиридин-2-ил)пиперидин-3-ил)карбамат 37b
Карбонат калия (1,5 г, 20,5 ммоль) растворяют в N,N-диметилформамиде (10 мл) с последующим добавлением трет-бутил-(S)-пиперидин-3-илкарбамат 37а (2,0 г, 10,0 ммоль) при комнатной температуре. После завершения добавления медленно добавляют соединение 1а (2,2 г, 10,0 ммоль). Реакционную смесь нагревают до 100°С и перемешивают в течение 4 часов. Реакционный раствор концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления В с получением указанного в заголовке продукта 37b (3,5 г, выход: 87%).
Стадия 2
Трет-бутил-(S)-(1-(5-йодпиридин-2-ил)пиперидин-3-ил)карбамат 37с
Гидрид натрия (12 мг, 0,5 ммоль) растворяют в N,N-диметилформамиде (10 мл) с последующим добавлением соединения 37b (100 мг, 0,2 ммоль) при комнатной температуре. После завершения добавления медленно добавляют йодметан (28 мг, 0,2 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Реакционный раствор концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления В с получением указанного в заголовке продукта 37с (95 мг, выход: 92%).
МС m/z (ИЭР): 418,1 [М+1].
Стадия 3
Трет-бутилметил-((3S)-1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пиперидин-3-ил)карбамат 37d
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1с на стадии 2 заменяют соединением 37с с получением указанного в заголовке соединения 37d (150 мг, выход: 71%).
МС m/z (ИЭР): 694,4 [М+1].
Стадия 4
(3S)-N-Метил-1-(5-((7)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пиперидин-3-амин 37е
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1f на стадии 3 заменяют соединением 37d с получением указанного в заголовке соединения 37е (80 мг, выход: 93%).
Стадия 5
(Е)-N,N-Диметил-4-(метил((3S)-1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пиперидин-3-ил)амино)бут-2-енамид 37f
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют соединением 37е, а исходный материал 1h заменяют соединением 5d с получением указанного в заголовке соединения 37f (75 мг, выход: 79%).
Стадия 6
(Е)-N,N-Диметил-4-(метил((S)-1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пиперидин-3-ил)амино)бут-2-енамид 37
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 37f с получением указанного в заголовке соединения 37 (22 мг, выход: 25%).
МС m/z (ИЭР): 621,2 [М+1].
1Н ЯМР (400 МГц, CDCl3) 10,51 (s, 1H), 7,71 (s, 1H), 7,62 (s, 1H), 7,74-7,31 (m, 4Н), 7,29-7,21 (m, 4Н), 6,80 (s, 2Н), 6,65 (dd, 1Н), 4,71 (dd, 1Н), 4,04 (d, 2Н), 3,87 (d, 2Н), 3,41 (d, 2Н), 3,35-3,31 (m, 2Н), 3,06 (d, 6Н), 2,84 (s, 3Н), 2,29 (d, 1Н), 2,02-1,92 (m, 2Н), 1,69 (d, 1Н).
Пример 38
(Е)-1-Морфолино-4-(((S)-1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пиперидин-3-ил)амино)бут-2-ен-1-он 38

Стадия 1
Трет-бутилметил-((3S)-1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пиперидин-3-ил)карбамат 38а
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1с на стадии 2 заменяют соединением 37b с получением указанного в заголовке соединения 38а (100 мг, выход: 80%).
МС m/z (ИЭР): 680,4 [М+1].
Стадия 2
(3S)-1-(5-((Z)-4,4,4-Трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пиперидин-3-амин 38b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1f на стадии 3 заменяют соединением 38а с получением указанного в заголовке соединения 38b (35 мг, выход: 82%).
Стадия 3
(Е)-1-Морфолино-4-(((3S)-1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пиперидин-3-ил)амино)бут-2-ен-1-он 38с
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют соединением 38b с получением указанного в заголовке соединения 38с (75 мг, выход: 79%).
Стадия 4
(Е)-1-Морфолино-4-(((S)-1-(5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)пиперидин-3-ил)амино)бут-2-ен-1-он 38
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 38с с получением указанного в заголовке соединения 38 (9 мг, выход: 25%).
МС m/z (ИЭР): 649,3 [М+1].
1Н ЯМР (400 МГц, CDCl3) 9,92 (s, 1H), 7,65 (s, 2Н), 7,37 (d, 1H), 7,30-7,22 (m, 6Н), 6,95-6,88 (m, 2Н), 6,40 (d, 2Н), 4,15 (d, 1H), 3,76-3,49 (m, 7Н), 3,53-3,49 (m, 4Н), 3,32-3,29 (m, 2Н), 3,00-2,95 (m, 1H), 2,86-2,81 (m, 1H), 2,73-2,69 (m, 1H), 1,99-1,94 (m, 1H), 1,78-1,72 (m, 1H), 1,58-1,48 (m, 1H), 1,33-1,27 (m, 2Н).
Пример 39
(Е)-N,N-Диметил-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-енамид 39

Стадия 1
(E)-N,N-Диметил-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-енамид 39а
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1h на стадии 4 заменяют соединением 5d с получением указанного в заголовке соединения 39а (53 мг, выход: 65%).
Стадия 2
(Е)-N,N-Диметил-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-енамид 39
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 39а с получением указанного в заголовке соединения 39 (24 мг, выход: 62%).
МС m/z (ИЭР): 593,9 [М+1].
1Н ЯМР (400 МГц, CD3OD) 7,63 (d, 2Н), 7,41-7,08 (m, 8Н), 6,83-6,76 (m, 1H), 6,48-6,36 (m, 2Н), 4,12 (s, 2Н), 3,49-3,48 (m, 2Н), 3,36-3,31 (m, 2Н), 3,09 (s, 3Н), 3,03 (s, 3Н), 0,67-0,55 (m, 4Н).
Пример 40
(Е)-1-(3-Гидроксиазетидин-1-ил)-4-((1-(((5-(Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 40

Стадия 1
(Е)-4-Бром-1-(3-гидроксиазетидин-1-ил)бут-2-ен-1-он 40а
Соединение 11а (0,5 г, 3,0 ммоль) растворяют в дихлорметане (5 мл), после чего добавляют триэтиламин (0,3 г, 3,3 ммоль), 3-гидроксиазетидин (0,2 г, 3,3 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (0,6 г, 3,3 ммоль) и 1-гидроксибензотриазол (0,4 г, 3,3 ммоль) при комнатной температуре. Реакционный раствор перемешивают в течение 12 часов. Реакционный раствор охлаждают. Добавляют насыщенный раствор бикарбоната натрия (15 мл) и экстрагируют раствор этилацетатом (2 раза по 20 мл). Органические фазы объединяют, промывают насыщенным раствором хлорида натрия (4 раза по 20 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления А с получением указанного в заголовке продукта 40а (0,5 г, выход: 72%).
Стадия 2
(Е)-1-(3-Гидроксиазетидин-1-ил)-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 40b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1h на стадии 4 заменяют соединением 40а с получением указанного в заголовке соединения 40b (43 мг, выход: 51%).
Стадия 3
(Е)-1-(3-Гидроксиазетидин-1-ил)-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 40
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 40b с получением указанного в заголовке соединения 40 (27 мг, выход: 67%).
МС m/z (ИЭР): 622,1 [М+1].
1H ЯМР (400 МГц, CD3OD) 7,71-7,66 (m, 2Н), 7,53-7,51 (m, 1H), 7,35-7,21 (m, 8Н), 6,71-6,65 (m, 2Н), 4,63-4,48 (m, 2Н), 4,43 (s, 2Н), 4,29-4,25 (m, 1Н), 4,05-4,00 (m, 3Н), 3,85-3,81 (m, 1H), 3,48-3,33 (m, 2Н), 1,17-1,11 (m, 4Н).
Пример 41
(Е)-1-((S)-3-Гидроксипирролидин-1-ил)-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 41


Стадия 1
(S,Е)-4-Бром-1-(3-гидроксипирролидин-1-ил)бут-2-ен-1-он 41а
Соединение 11а (0,5 г, 3,0 ммоль) растворяют в дихлорметане (5 мл), после чего добавляют триэтиламин (0,3 г, 3,3 ммоль), (S)-3-гидроксипирролидин (0,2 г, 3,3 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (0,6 г, 3,3 ммоль) и 1-гидроксибензотриазол (0,4 г, 3,3 ммоль) при комнатной температуре. Реакционный раствор перемешивают в течение 12 часов. Реакционный раствор охлаждают. Добавляют насыщенный раствор бикарбоната натрия (15 мл) и экстрагируют раствор этилацетатом (2 раза по 20 мл). Органические фазы объединяют, промывают насыщенным раствором хлорида натрия (4 раза по 20 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления А с получением указанного в заголовке продукта 41а (0,5 г, выход: 73%).
Стадия 2
(Е)-1-((S)-3-Гидроксипирролидин-1-ил)-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 41b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1h на стадии 4 заменяют соединением 41а с получением указанного в заголовке соединения 41b (41 мг, выход: 57%).
Стадия 3
(Е)-1-((S)-3-Гидроксипирролидин-1-ил)-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 41
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 41b с получением указанного в заголовке соединения 41 (37 мг, выход: 69%).
МС m/z (ИЭР): 636,2 [М+1].
1H ЯМР (400 МГц, CD3OD) 7,71-7,66 (m, 2Н), 7,53-7,51 (m, 1Н), 7,35-7,22 (m, 8Н), 6,71-6,66 (m, 2Н), 4,44 (s, 2Н), 4,07-4,00 (m, 2Н), 3,74-3,67 (m, 2Н), 3,55-3,51 (m, 5Н), 2,21-2,19 (m, 2Н), 1,19-1,13 (m, 4Н).
Пример 42
(E)-1-((R)-2-(Гидроксиметил)морфолино)-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил) пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 42

Стадия 1
(S,Е)-4-Бром-1-(2-(гидроксиметил)морфолино)бут-2-ен-1-он 42а
Соединение 11а (0,5 г, 3,0 ммоль) растворяют в дихлорметане (5 мл), после чего добавляют триэтиламин (0,3 г, 3,3 ммоль), (R)-морфолин-2-илметанол (0,4 г, 3,3 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (0,6 г, 3,3 ммоль) и 1-гидроксибензотриазол (0,4 г, 3,3 ммоль) при комнатной температуре. Реакционный раствор перемешивают в течение 12 часов. Реакционный раствор охлаждают. Добавляют насыщенный раствор бикарбоната натрия (15 мл) и экстрагируют раствор этилацетатом (2 раза по 20 мл). Органические фазы объединяют, промывают насыщенным раствором хлорида натрия (4 раза по 20 мл), высушивают над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют при пониженном давлении, и полученные в результате остатки очищают тонкослойной хроматографией с системой проявления А с получением указанного в заголовке продукта 42а (0,5 г, выход: 63%).
Стадия 2
(Е)-1-((R)-2-(Гидроксиметил)морфолино)-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 42b
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1h на стадии 4 заменяют соединением 42а с получением указанного в заголовке соединения 42b (31 мг, выход: 47%).
Стадия 3
(Е)-1-((R)-2-(Гидроксиметил)морфолино)-4-((1-(((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)метил)циклопропил)амино)бут-2-ен-1-он 42
В соответствии со способом синтеза, описанным в примере 1, исходный материал 1i на стадии 5 заменяют соединением 42b с получением указанного в заголовке соединения 42 (27 мг, выход: 59%).
МС m/z (ИЭР): 666,1 [М+1].
1Н ЯМР (400 МГц, CD3OD) 7,67-7,65 (m, 2Н), 7,52-7,50 (m, 1H), 7,31-7,26 (m, 7Н), 6,81-6,77 (m, 1H), 6,61-6,59 (m, 2Н), 4,45-4,42 (m, 1H), 4,30 (s, 2Н), 3,99-3,93 (m, 2Н), 3,61-3,42 (m, 9Н), 3,03-3,00 (m, 1H), 0,81-0,72 (m, 4Н).
Пример 43
(Е)-1-Морфолино-4-((2-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)бут-2-ен-1-он 43

В соответствии со способом синтеза, описанным в примере 1, исходный материал 1g на стадии 4 заменяют 43а (получен в соответствии со способом, раскрытым в примере 1 на странице 77 описания заявки на патент WO 2018098305) с получением указанного в заголовке соединения 43 (21 мг, выход: 42%).
МС m/z (ИЭР): 610,2 [М+1].
1Н ЯМР (400 МГц, CD3OD) 7,69-7,67 (m, 2Н), 7,53-7,50 (m, 1H), 7,32-7,20 (m, 7Н), 6,80-6,59 (m, 3Н), 4,36-4,34 (m, 2Н), 3,66-3,58 (m, 10Н), 3,48-3,40 (m, 3Н), 3,13-3,10 (m, 2Н).
Биологический анализ
Настоящее изобретение будет приведено дополнительно со ссылкой на следующие ниже тестовые примеры, но эти примеры не следует истолковывать как ограничивающие объем настоящего изобретения.
Тестовый пример 1. Ингибирующее действие соединений по настоящему изобретению на активность гена-репортера рецептора эстрогена
1. Цель эксперимента
Цель эксперимента состоит в определении ингибирующего действия соединений по настоящему изобретению на активность гена-репортера рецептора эстрогена и в оценке активности соединений in vitro в соответствии с IC50.
2. Методика эксперимента
Клетки MCF7, линия MCF7/ERE-luc (АТСС, НТВ-22), экспрессирующие ген-репортер люциферазы ERE-luc (синтезирован GENEWIZ, Inc.) под контролем ответного элемента рецептора эстрогена, культивируют в среде MEM (минимальная питательная среда) (GE Healthcare, SH30024.01), содержащей 10% фетальной бычьей сыворотки и 500 мкг/мл G418. В первый день эксперимента клетки MCF7/ERE-luc высеивают в 96-луночный планшет с неполной средой MEM, содержащей 10% фетальной бычьей сыворотки, обработанной активированным углем (BioSun, BS-0004-500), при плотности 30000 клеток/лунка (100 мкл клеточной суспензии на лунку) и инкубируют планшет в термостате для культур клеток при 37°С и 5% СО2 в течение ночи. На второй день в каждую лунку добавляют 10 мкл раствора β-эстрадиола (SIGMA, E2758-250MG), полученного с описанной выше неполной средой, или исследуемого раствора соединения в различных концентрациях, полученного с описанной выше неполной средой, где конечная концентрация β-эстрадиола составляла 0,1 нмоль/л, а конечные концентрации соединения представляют собой девять точек концентрации, полученных путем 10-кратного градиентного разведения, начиная с 10 мкмоль/л; ставят холостой контроль, содержащий 0,5% ДМСО, и планшет инкубируют в термостате для культур клеток при 37°С и 5% СО2 в течение 20 часов. На третий день 96-луночный планшет извлекают и добавляют в каждую лунку 100 мкл системы для анализа люциферазы ONE-Glo™ Luciferase Assay (Promega, E6110) для определения активности люциферазы. Планшет оставляют стоять при комнатной температуре на 3 минуты до полного лизиса клеток и измеряют значения сигнала люциферазы с помощью ридера для микропланшетов с множественными маркерами (PerkinElmer, VICTOR 3). Значения IC50 ингибирующей активности соединений рассчитывают с помощью программного обеспечения Graphpad Prism на основании концентраций и значений люминесцентного сигнала соединений.
3. Результаты эксперимента
Ингибирующее действие соединений по настоящему изобретению на активность гена-репортера рецептора эстрогена определяют с помощью описанного выше теста. График зависимости значений хемилюминесцентного сигнала от логарифмических концентраций соединения строят с помощью Graghpad Prism. Полученные в результате значения IC50 приведены в таблице 1 ниже.


Вывод: соединения по настоящему изобретению обладают значимым ингибирующим действием на ген-репортер рецептора эстрогена.
Тестовый пример 2. Ингибирующее действие соединений по настоящему изобретению на пролиферацию клеток MCF7
1. Цель эксперимента
Цель эксперимента состоит в определении ингибирующего действия соединений по настоящему изобретению на пролиферацию клеток MCF7 и в оценке активности соединений in vitro в соответствии с IC50.
2. Методика эксперимента
Клетки MCF7 (АТСС, НТВ-22) культивируют в полной среде MEM (GE Healthcare, SH30024.01), содержащей 10% фетальной бычьей сыворотки. В первый день эксперимента клетки MCF7 высеивают в 96-луночный планшет с полной средой MEM при плотности 3000 клеток/лунка (100 мкл клеточной суспензии на лунку) и инкубируют планшет в термостате для культур клеток при 37°С и 5% СО2 в течение ночи. На второй день среду в каждой лунке заменяют 135 мкл неполной среды MEM, содержащей 2% фетальной бычьей сыворотки. В каждую лунку добавляют 15 мкл исследуемого раствора соединения в различных концентрациях, полученного с неполной средой, где конечные концентрации соединения представляют собой девять точек концентрации, полученных путем 4-кратного градиентного разведения, начиная со 100 нмоль/л. Ставят холостой контроль, содержащий 0,5% ДМСО. Планшет инкубируют в термостате для культур клеток при 37°С и 5% СО2 в течение 144 часов. На восьмой день 96-луночный планшет извлекают и добавляют в каждую лунку 150 мкл набора реагентов для анализа жизнеспособности клеток CellTiter-Glo® Luminescent Cell Viabi1ity Assay (Promega, G7573). Планшет оставляют стоять при комнатной температуре на 10 минут и измеряют значения сигнала люциферазы с помощью ридера для микропланшетов с множественными маркерами (PerkinElmer, VICTOR 3). Значения IC50 ингибирующей активности соединений рассчитывают на основании концентраций и значений люминесцентного сигнала соединений с помощью программного обеспечения Graphpad Prism.
3. Анализ данных
Для получения значений IC50 строят график зависимости значений хемилюминесцентного сигнала от логарифмических концентраций соединения с помощью Graghpad Prism. Результаты приведены в таблице 2.
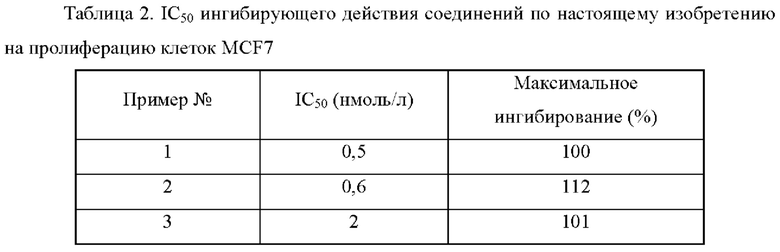


Вывод: соединения по настоящему изобретению обладают значимым ингибирующим действием на пролиферацию клеток MCF7.
Тестовый пример 3. Ингибирующее действие соединений по настоящему изобретению на пролиферацию клеток MCF7, экспрессирующих мутантный ЭР-альфа (ERα)
1. Цель эксперимента
Цель эксперимента состоит в определении ингибирующего действия соединений по настоящему изобретению на пролиферацию клеток MCF7, экспрессирующих мутантный ERα.
2. Методика эксперимента
Сайт-направленный мутагенез и конструирование линии клеток
Мутанты белка рецептора эстрогена α (ERα) человека ERα Y537S и ERα D538G были получены путем сайт-направленного мутагенеза с использованием кДНК гена ESR1 дикого типа (номер по каталогу NM000125) в качестве матрицы для полимеразной цепной реакции (ПЦР) с двумя парами праймеров. Последовательности праймеров, используемые для мутагенеза, приведены ниже (подчеркнутые нуклеотиды представляют собой сайты мутагенеза):

кДНК мутанта ESR1 клонировали в целевом лентивирусном векторе pCDH-CMV-MCS-EF1-Puro (SBI, CD510B-1). Лентивирусную плазмиду с мутантной последовательностью гена ESR1 и лентивирусные упаковочные плазмиды pVSV-G и pCMV-dR8.91 (SBI, LV500A-1) трансфицировали в клетки HEK-293Т (АТСС, CRL-3216) с помощью реагента для трансфекции липофектамина (Lipofectamine 3000 Transfection Reagent, ThermoFisher Scientific, № по каталогу L3000075). Через 48 часов после трансфекции содержащую вирус среду DMEM (минимальная питательная среда, модифицированная по способу Дульбекко среда Игла) с высоким содержанием глюкозы (GE Healthcare, SH30243.01), содержащую 10% фетальной бычьей сыворотки (ФБС), фильтруют и ультрацентрифугируют с получением осадка вируса. Осадок вируса ресуспендируют в соответствующем количестве среды MEM (GE Healthcare, SH30024.01), содержащей 10% ФБС, неэссенциальные аминокислоты (SIGMA, M7145-100ML) и пируват натрия (SIGMA, S8636-100ML), и полученную в результате суспензию добавляют к клеткам MCF7 (АТСС, НТВ-22). Добавляют полибрен (SIGMA, H9268-5G) в конечной концентрации 8 мкг/мл и инкубируют клетки в течение ночи. Через два дня после трансфекции в среду для культивирования клеток добавляют 1 мкг/мл пуромицина (Invitrogen, All 138-03) для скрининга устойчивости. Примерно через две недели получают линию клеток MCF7, способную стабильно экспрессировать мутанты ERαY537S и ERα D538G.
Тест на ингибирование пролиферации клеток
Клетки MCF7, экспрессирующие мутант ERα, культивируют в полной среде MEM (GE Healthcare, SH30024.01), содержащей 10% фетальной бычьей сыворотки. В первый день эксперимента клетки высеивают в 96-луночный планшет с полной средой MEM при плотности 3000 клеток/лунка (100 мкл клеточной суспензии на лунку) и инкубируют планшет в термостате для культур клеток при 37°С и 5% СО2 в течение ночи. На второй день среду в каждой лунке заменяют 135 мкл неполной среды MEM, содержащей 2% фетальной бычьей сыворотки. В каждую лунку добавляют 15 мкл исследуемого раствора соединения в различных концентрациях, полученного с неполной средой, где конечные концентрации соединения представляют собой девять точек концентрации, полученных путем 4-кратного градиентного разведения, начиная со 100 нмоль/л. Ставят холостой контроль, содержащий 0,5% ДМСО. Планшет инкубируют в термостате для культур клеток при 37°С и 5% СО2 в течение 144 часов. На восьмой день 96-луночный планшет извлекают и добавляют в каждую лунку 150 мкл набора реагентов для анализа жизнеспособности клеток CellTiter-Glo® Luminescent Cell Viability Assay (Promega, G7573). Планшет оставляют стоять при комнатной температуре на 10 минут и измеряют значения сигнала люциферазы с помощью ридера для микропланшетов с множественными маркерами (PerkinElmer, VICTOR 3). Значения IC50 ингибирующей активности соединений рассчитывают на основании концентраций и значений люминесцентного сигнала соединений с помощью программного обеспечения Graphpad Prism.


Вывод: соединения по настоящему изобретению обладают значимым ингибирующим действием на пролиферацию клеток MCF7, экспрессирующих мутант ERα.
Тестовый пример 4. Определение ингибирующей активности соединений по настоящему изобретению на NaV1.5
Цель эксперимента состоит в исследовании действия соединений на ионный канал NaV1.5 в эксперименте ex-vivo. Ионный канал NaV1.5 стабильно экспрессируется на клетках HEK293. После того как ток NaV1.5 становится стабильным, сравнивают токи NaV1.5 до и после введения соединения так, чтобы получить действие соединения на ионный канал NaV1.5.
1. Экспериментальные материалы и приборы
1) Усилитель локальной фиксации потенциала: Patch Clamp РС-505В (WARNER Instruments)
2) Устройство для преобразования цифровых величин в непрерывные: Digidata 1440A(Axon CNS)
3) Микроманипулятор: МР-225 (SUTTER Instrument)
4) Микроскоп с инвертированным светом: TL4 (Olympus)
5) Стеклянный оператор микроэлектродов: PC-10 (NARISHIGE)
6) Стеклянный капилляр для микроэлектродов: B12024F (Wuhan Weitan Scientific Instrument Co., Ltd.)
7) Диметилсульфоксид (ДМСО): D2650 (Sigma-Aldrich)
2. Экспериментальные методики
2.1 Получение препаратов соединений
За исключением NaOH и KOH, используемые для титрования кислоты и титрования основания, все агенты для получения препаратов внеклеточной жидкости и внутриклеточной жидкости приобретают у Sigma (Сент-Луис, штат Миссури, США).
Внеклеточная жидкость: NaCl, 137 ммоль/л; KCl, 4 ммоль/л; CaCl2, 1,8 ммоль/л; MgCl2, 1 ммоль/л; ГЭПЭС (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота)), 10; глюкоза, 10 ммоль/л; рН 7,4 (титрование NaOH).
Внутриклеточная жидкость: аспарагиновая кислота, 140 ммоль/л; MgCl2, 2 ммоль/л; этиленгликоль-бис(β-аминоэтиловый эфир)-N,N,N',N'-тетрауксусная кислота (ЭГТА), 11 ммоль/л; ГЭПЭС, 10 ммоль/л; рН 7,2 (титрование CsOH).
Исследуемое соединение растворяют в диметилсульфоксиде (ДМСО) в исходной концентрации 9 нмоль/л. Исходный раствор исследуемого соединения растворяют во внеклеточной жидкости в день теста и получают препарат в необходимой концентрации.
2.2 Процесс тестирования локальной фиксации потенциала вручную
1) Препараты соединений получают в растворах с указанными концентрациями. Растворы добавляют в трубки для доставки, соответственно в порядке от высокой концентрации к низкой, и трубки маркировали.
2) Клетку переносят в резервуар для перфузии и проводят перфузию внеклеточной жидкостью. Внуутриклеточную жидкость хранят небольшими порциями в морозильной камере при -80°С и размораживали в день эксперимента. Электрод извлекают с помощью PC-10 (Narishige, Япония). Регистрацию данных проводят путем локальной фиксации потенциала на целых клетках, и шум отфильтровывали на уровне одной пятой частоты отбора образцов.
3) Перфузию проводят с помощью перфузионной системы, которая использует собственное тяготение. Тестирование каждой концентрации проводят на по меньшей мере двух клетках. После того как ток стабилизировался (или через 5 минут), блокирующее действие соединения рассчитывают путем сравнения изменений тока до и после введения соединения.
4) Резервуар для перфузии очищают. Рефервуар для перфузии промывают растворами лекарственных препаратов в порядке от высокой концентрации к низкой, и продолжительность промывания для каждой концентрации раствора лекарственного препарата составляла 20 секунд. Резервуар для иперфузии окончательно промывают внеклеточной жидкостью в течение 1 минуты.
2.3 Уравнение испытательного напряжения (покоя) и результаты
Локальный потенциал клетки фиксируют при -80 мВ. Деполяризацию клетки проводят до Vhalf при длительности прямоугольной волны 8 миллисекунд, гиперполяризацию проводят до -120 мВ при длительности прямоугольной волны 20 миллисекунд и деполяризацию - при -10 мВ при длительности прямоугольной волны 20 миллисекунд с получением тока NaV1.5. Эту процедуру повторяют раз в 15 секунд. Измеряют максимальный ток, вызванный прямоугольной длиной волны -10 мВ. После того как ток становился стабильным, проводят перфузию исследуемого соединения. После того как ответ становится стабильным, рассчитывают интенсивность блокирования.
3. Анализ данных
Данные для анализа хранились в компьютерной системе. Сбор и анализ данных проводят с помощью программного обеспечения pCLAMP 10 (Molecular Devices, Юнион-Сити, штат Калифорния). Стабильный ток означает, что абсолютное среднее значение пикового тока четырех последовательных сканирований превышает 200 пикоампер (пА), а значение коэффициента вариации (CV) составляет менее 10%, или среднее значение пикового тока четырех последовательных сканирований составляет от 200 пА до 50 пА, а значение CV составляет менее 30%. Для расчета действия соединения при данной концентрации используют среднее значение последних четырех пиков тока после того, как ток стабилизировался.
Ингибирующую активность соединений по настоящему изобретению на NaV1.5 определяют с помощью описанного выше теста, и полученные в результате значения IC50 приведены в таблице 4 ниже.
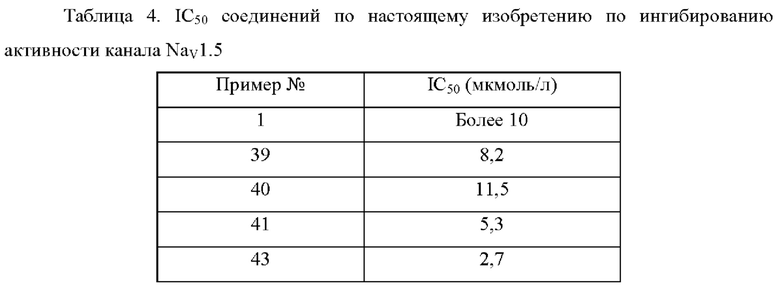
Вывод: ингибирующее действие соединений 1 и 39-41 по настоящему изобретению на канал NaV1.5 слабее по сравнению с соединением примера 43. Видно, что риск кардиотоксичности этих соединений значимо ниже по сравнению с соединением примера 43.
Оценка фармакокинетики
Тестовый пример 5. Анализ фармако кинетики соединений примеров настоящего изобретения у бесшерстных мышей линии BALB/C
1. Аннотация
В качестве экспериментальных животных используют бесшерстных мышей линии BALB/C. Концентрацию лекарственного препарата в плазме крови в различных точках контроля определяют методом жидкостной хроматографии с тандемной масс-спектр ометрией (ЖХ/МС/МС) после внутрижелудочного введения соединений по настоящему изобретению бесшерстным мышам линии BALB/C. У бесшерстных мышей линии BALB/C изучают и оценивают фармакокинетические свойства соединения по настоящему изобретению.
2. Протокол исследования
2.1 Исследуемые соединения и приборы
Соединения примера 1, примера 14, примера 15 и примеров 39-42.
Приборы для ЖХ/МС/МС: Тройной квадрупольный тандемный масс-спектрометр ЖХ/МС/МС API4000 (№3, Applied Biosystems, США), система сверхвысокоэффективной жидкостной хроматографии Shimadzu LC-30AD (Shimadzu, Япония).
2.2 Экспериментальные животные
Шестьдесят три бесшерстных мыши линии BALB/C (самки, равномерно распределенные на семь групп по девять мышей на группу) были приобретены в Jiesijie Laboratory Animal Co., LTD. (Сертификат №: SCXK (Шанхай) 2013-0006).
2.3 Получение исследуемого соединения
Для соединения примера 14 взвешивают навеску соответствующего количества соединения, после чего добавляют 5% ДМСО, 5% Твин 80 и 90% нормального физиологического раствора по объему с получением бесцветного чистого и прозрачного раствора (0,1 мг/мл).
Для соединений примеров 1, 15 и 39-42 взвешивают навеску соответствующего количества соединения, после чего добавляют 9% ПЭГ 400, 0,5% Твин 80 и 90,5% 0,5%-го раствора КМЦ-Na по объему с получением бесцветного чистого и прозрачного раствора (1,5 мг/мл).
2.4 Введение
После ночи натощак исследуемые соединения вводили внутрижелудочно в объеме введения 0,2 мл/10 г соответственно. Вводимая доза соединения 14 составляла 2 мг/кг, а доза введения соединений 1, 15 и 39 42 составляла 30 мг/кг.
3. Способ
После ночи натощак шестидесяти трем самкам бесшерстных мышей линии BALB/C внутрижелудочно вводили исследуемые соединения.
Для соединений примеров 1 и 39 отбирают 0,1 мл крови через 0,25, 0,5, 1,0, 2,0, 4,0, 6,0, 8,0, 11,0 и 24,0 часа после введения (у трех животных на одну точку контроля). Образцы хранят в пробирках для сбора крови, обработанных гепарином (Sinopharm Chemical Reagent Co., Ltd.), и центрифугируют в течение 10 минут при 3500 об/мин для отделения плазмы крови. Плазму крови хранят при -20°С. Определяют содержание исследуемого соединения в плазме крови бесшерстных мышей после внутрижелудочного введения исследуемого соединения. Для каждой точки контроля после введения отбирают 25 мкл плазмы крови бесшерстных мышей, после чего добавляют 30 мкл раствора внутреннего стандарта камптотецина (National Institutes for Food and Drug Control) (100 нг/мл) и 225 мкл ацетонитрила. Полученный в результате раствор перемешивают на вихревой мешалке в течение 5 минут и центрифугируют в течение 10 минут (от 3700 до 4000 об/мин). На анализ ЖХ/МС/МС отбирают от 0,1 до 0,5 мкл супернатанта образцов плазмы крови.
Для соединений примеров 14 и 15 отбирают 0,1 мл крови через 0,5, 1,0, 2,0, 4,0, 6,0, 8,0, 11,0 и 24,0 часа после введения (у трех животных на одну точку контроля). Образцы хранят в гепаринизированных пробирках и центрифугируют в течение 10 минут при 3500 об/мин для отделения плазмы крови. Плазму крови хранят при -20°С. Определяют содержание исследуемого соединения в плазме крови бесшерстных мышей после внутрижелудочного введения исследуемого соединения. Для каждой точки контроля после введения отбирают 25 мкл плазмы крови бесшерстных мышей, после чего добавляют 30 мкл раствора внутреннего стандарта камптотецина (100 нг/мл) и 200 мкл ацетонитрила. Полученный в результате раствор перемешивают на вихревой мешалке в течение 5 минут и центрифугируют в течение 10 минут (от 3700 об/мин). На анализ ЖХ/МС/МС отбирают от 0,25 до 1 мкл супернатанта образцов плазмы крови.
Для соединений примеров 40-42 отбирают 0,1 мл крови через 0,25, 0,5, 1,0, 2,0, 4,0, 6,0, 8,0, 11,0 и 24,0 часа после введения (у трех животных на одну точку контроля). Образцы хранят в пробирках с антикоагулянтом ЭДТА-K2 (Shanghai Titan Scientific Co., Ltd.) и центрифугируют в течение 1 минуты при 10000 об/мин и 4°С. Плазму крови отделяют в течение одного часа и хранят при -20°С. Весь процесс от сбора крови до центрифугирования выполняют в условиях ледяной бани. Определяют содержание исследуемого соединения в плазме крови бесшерстных мышей после внутрижелудочного введения исследуемого соединения. Для каждой точки контроля после введения отбирают 25 мкл плазмы крови бесшерстных мышей, после чего добавляют 50 мкл раствора внутреннего стандарта камптотецина (100 нг/мл) и 200 мкл ацетонитрила. Полученный в результате раствор перемешивают на вихревой мешалке в течение 5 минут и центрифугируют в течение 10 минут (от 3700 об/мин). На анализ ЖХ/МС/МС отбирают 1 мкл супернатанта образцов плазмы крови.
4. Результаты определения фармакокинетических показателей у бесшерстных мышей BALB/C
Фармако кинетические показатели соединений примеров настоящего изобретения представлены в таблице 5 ниже.

Вывод: соединения по настоящему изобретению хорошо всасываются и обладают значимым фармакокинетическим абсорбционным эффектом.
Тестовый пример 6. Биологическая оценка ковалентной модификации рецептора эстрогена ERα дикого типа и ERα мутантного типа Y537S
1. Цель эксперимента
Цель эксперимента состоит в определении влияния ковалентной модификации соединений по настоящему изобретению на рецептор эстрогена ERα дикого типа и ERα мутантного типа Y537S.
2. Методика эксперимента
Лиганд-связывающий домен (LBD, ак 296-554) рецептора эстрогена ERα и ERα мутантного типа Y537S экспрессируют в Е. coli и очищают.2 мкмоль/л белка ERα дикого типа или ERα мутантного типа Y537S и 10 мкмоль/л соединения добавляют в буферный раствор, содержащий 50 ммоль/л Трис-HCl, рН 7,5, 150 ммоль/л NaCl, 1 ммоль/л ТСЕР и 5% глицерина и тщательно перемешивают. Раствор инкубируют при 4°С в течение 24 часов, после чего проводят анализ методом масс-спектрометрии высокого разрешения (Agilent Technologies, Agilent Q-tof 6530). Альтернативно 1 мкмоль/л белка ERα дикого типа или ERα мутантного типа Y537S и 3 мкмоль/л соединения добавляют в буферный раствор, содержащий 50 ммоль/л Трис-HCl, рН 7,5, 150 ммоль/л NaCl, 1 ммоль/л ТСЕР и 5% глицерина и тщательно перемешивают. Раствор инкубируют при 37°С в течение 15 минут, после чего проводят анализ методом масс-спектрометрии высокого разрешения. По результатам анализа методом масс-спектрометрии продуктом ковалентной модификации является пик, молекулярная масса которого представляет собой сумму молекулярной массы белка и соединения. Процент ковалентной модификации получают путем расчета отношения несвязанного белка к общему белку. Результаты приведены в таблице 6 ниже.

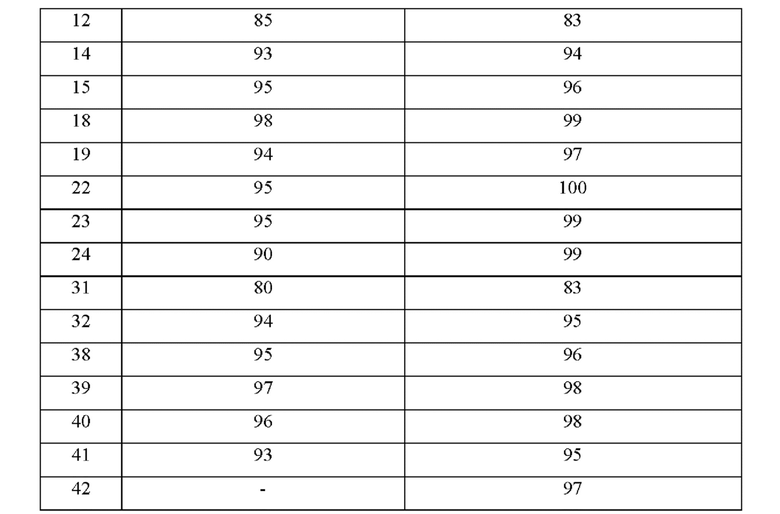
Вывод: исследуемые соединения обладают превосходным влиянием на белок ERα дикого типа или ERα мутантного типа Y537S.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> ЦЗЯНСУ ХЭНЖУЙ МЕДСИН КО., ЛТД.
ШАНХАЙ ХЭНЖУЙ ФАРМАСЬЮТИКАЛ КО., ЛТД.
<120> ПРОИЗВОДНОЕ ИНДАЗОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО
ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ
<130> 702056CPCT
<160> 4
<170> SIPOSequenceListing 1.0
<210> 1
<211> 39
<212> ДНК
<213> Искусственная последовательность
<220>
<221> неопределенный
<223> Y537S-F
<400> 1
aagaacgtgg tgcccctctc tgacctgctg ctggagatg
39
<210> 2
<211> 39
<212> ДНК
<213> Искусственная последовательность
<220>
<221> неопределенный
<223> Y537S-R
<400> 2
catctccagc agcaggtcag agaggggcac cacgttctt
39
<210> 3
<211> 39
<212> ДНК
<213> Искусственная последовательность
<220>
<221> неопределенный
<223> D538G-F
<400> 3
aacgtggtgc ccctctatgg cctgctgctg gagatgctg
39
<210> 4
<211> 39
<212> ДНК
<213> Искусственная последовательность
<220>
<221> неопределенный
<223> D538G-R
<400> 4
2
cagcatctcc agcagcaggc catagagggg caccacgtt
39
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЛИ ПРОИЗВОДНОГО ИНДАЗОЛА И ИХ КРИСТАЛЛЫ | 2017 |
|
RU2747399C2 |
| ТЕТРАЗАМЕЩЕННЫЕ АЛКЕНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2733741C2 |
| ПРОИЗВОДНОЕ ИМИДАЗОИЗОИНДОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2717577C2 |
| ПРОИЗВОДНОЕ 6-ОКСО-1,6-ДИГИДРОПИРИДАЗИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2020 |
|
RU2791534C2 |
| ПРОИЗВОДНОЕ ФЕНИЛПРОПАНАМИДА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2738207C2 |
| ПРОИЗВОДНОЕ ГЕТЕРОАРИЛ[4,3-с]ПИРИМИДИН-5-АМИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКИЕ ПРИМЕНЕНИЯ | 2018 |
|
RU2764655C2 |
| ПРОИЗВОДНОЕ ПИПЕРИДИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2730508C2 |
| ПРОИЗВОДНОЕ БЕНЗОФУРАНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2016 |
|
RU2727198C2 |
| ТРИЦИКЛИЧЕСКОЕ ТЕТРАГИДРОИЗОХИНОЛИНОВОЕ ПРОИЗВОДНОЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2021 |
|
RU2830109C1 |
| ГЕТЕРОАРИЛЬНОЕ ПРОИЗВОДНОЕ ПИПЕРАЗИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2018 |
|
RU2745431C1 |
Изобретение относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, или их смеси, или их фармацевтически приемлемой соли. В формуле (I): G представляет собой N; Z выбран из группы, состоящей из химической связи, -O-(CH2)t- и -NR7-(CH2)t-; кольцо A выбрано из группы, состоящей из C3-C6 циклоалкила и 3-6-членного гетероциклила, содержащего один гетероатом N; Ra выбран из группы, состоящей из -CH2CH=CHC(O)NR8R9, -C(O)CH=CR10R11 и -C(O)C≡CR12; Rb выбран из группы, состоящей из атома водорода и C1-C6 алкила; каждый R1 идентичен или отличается и каждый независимо выбран из группы, состоящей из атома водорода, галогена, C1-C6 алкила и C1-C6 галогеналкила; R2 представляет собой C1-C6 галогеналкил; каждый R3 идентичен или отличается и каждый независимо выбран из группы, состоящей из атома водорода, галогена, C1-C6 алкила, C1-C6 галогеналкила, циано, NR13C(O)R14, C(O)NR13R14, SO2R15; каждый R4 представляет собой атом водорода; R7 представляет собой атом водорода; R8 и R9 идентичны или отличаются и каждый независимо выбран из группы, состоящей из атома водорода, C1-C6 алкила, C1-C6 галогеналкила, C3-C6 циклоалкила и 3-6-членного гетероциклила, содержащего один гетероатом O; или R8 и R9 вместе с атомом азота, к которому они присоединены, образуют 3-8-членный гетероциклил, где 3-8-членный гетероциклил необязательно содержит в дополнение к одному атому азота один гетероатом, выбранный из группы, состоящей из N и O, и 3-8-членный гетероциклил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из C1-C6 алкила, галогена, гидрокси, C1-C6 гидроксиалкила; R10 и R11 идентичны или отличаются и каждый независимо выбран из группы, состоящей из атома водорода, C1-C6 алкила и C1-C6 галогеналкила; R12 выбран из группы, состоящей из атома водорода, C1-C6 алкила и C1-C6 галогеналкила; R13 и R14 идентичны или отличаются и каждый независимо выбран из группы, состоящей из атома водорода, C1-C6 алкила и C1-C6 галогеналкила; R15 выбран из группы, состоящей из атома водорода, C1-C6 алкила и C1-C6 галогеналкила; m равно 0 или 1; n равно 0, 1, 2 или 3; q равно 0, 1, 2, 3, 4 или 5; s равно 0, 1, 2 или 3; и t равно 0, 1, 2, 3, 4, 5 или 6. Также предложено соединение формулы (IA), способ получения соединения (I) из соединения формулы (IA), фармацевтическая композиция и применение соединения формулы (I). Предложенное соединение формулы (I) является антагонистом рецептора эстрогена и может применяться для получения модулятора рецептора эстрогена для предотвращения и/или лечения заболевания или состояния, опосредованного или зависимого от рецептора эстрогена, причем заболеванием особенно предпочтительно является рак молочной железы. 6 н. и 13 з.п. ф-лы, 6 табл., 49 пр.

1. Соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль:
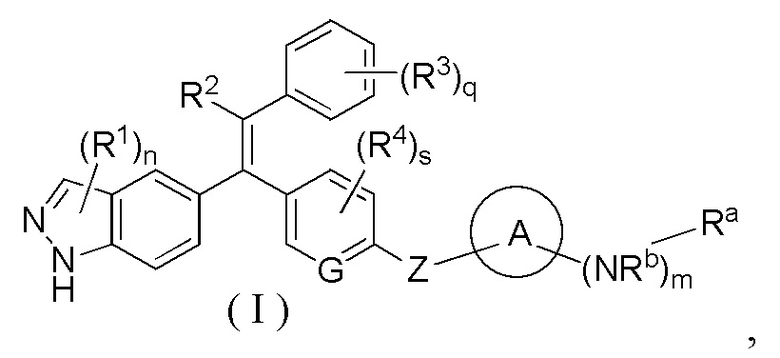
где G представляет собой N;
Z выбран из группы, состоящей из химической связи, -O-(CH2)t- и -NR7-(CH2)t-;
кольцо A выбрано из группы, состоящей из C3-C6 циклоалкила и 3-6-членного гетероциклила, содержащего один гетероатом N;
Ra выбран из группы, состоящей из -CH2CH=CHC(O)NR8R9, -C(O)CH=CR10R11 и -C(O)C≡CR12;
Rb выбран из группы, состоящей из атома водорода и C1-C6 алкила;
каждый R1 идентичен или отличается и каждый независимо выбран из группы, состоящей из атома водорода, галогена, C1-C6 алкила и C1-C6 галогеналкила;
R2 представляет собой C1-C6 галогеналкил;
каждый R3 идентичен или отличается и каждый независимо выбран из группы, состоящей из атома водорода, галогена, C1-C6 алкила, C1-C6 галогеналкила, циано, NR13C(O)R14, C(O)NR13R14, SO2R15;
каждый R4 представляет собой атом водорода;
R7 представляет собой атом водорода;
R8 и R9 идентичны или отличаются и каждый независимо выбран из группы, состоящей из атома водорода, C1-C6 алкила, C1-C6 галогеналкила, C3-C6 циклоалкила и 3-6-членного гетероциклила, содержащего один гетероатом O;
или R8 и R9 вместе с атомом азота, к которому они присоединены, образуют 3-8-членный гетероциклил, где 3-8-членный гетероциклил необязательно содержит в дополнение к одному атому азота один гетероатом, выбранный из группы, состоящей из N и O, и 3-8-членный гетероциклил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из C1-C6 алкила, галогена, гидрокси, C1-C6 гидроксиалкила;
R10 и R11 идентичны или отличаются и каждый независимо выбран из группы, состоящей из атома водорода, C1-C6 алкила и C1-C6 галогеналкила;
R12 выбран из группы, состоящей из атома водорода, C1-C6 алкила и C1-C6 галогеналкила;
R13 и R14 идентичны или отличаются и каждый независимо выбран из группы, состоящей из атома водорода, C1-C6 алкила и C1-C6 галогеналкила;
R15 выбран из группы, состоящей из атома водорода, C1-C6 алкила и C1-C6 галогеналкила;
m равно 0 или 1;
n равно 0, 1, 2 или 3;
q равно 0, 1, 2, 3, 4 или 5;
s равно 0, 1, 2 или 3; и
t равно 0, 1, 2, 3, 4, 5 или 6.
2. Соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль по п. 1, где кольцо A выбрано из группы, состоящей из циклопропила, циклопентила, циклогексила, тетрагидропирролила и пиперидинила.
3. Соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль по п. 1 или 2, где кольцо A выбрано из группы, состоящей из:

4. Соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль по п. 1 или 2, представляющее собой соединение формулы (II) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль:

где кольцо A, G, Z, R1, R2, R3, R4, Ra, Rb, n, m, s и q являются такими, как определено в п. 1.
5. Соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль по любому из пп. 1-4, представляющее собой соединение формулы (III) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль:
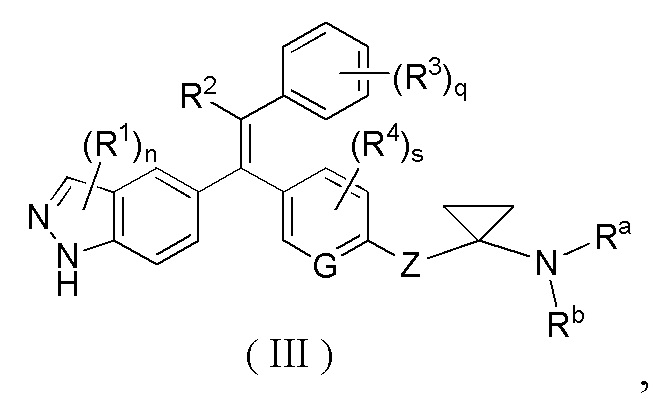
где G, Z, R1, R2, R3, R4, Ra, Rb, n, s и q являются такими, как определено в п. 1.
6. Соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль по любому из пп. 1-5, где R1 выбран из группы, состоящей из галогена и атома водорода.
7. Соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль по любому из пп. 1-6, где R2 представляет собой -CH2CF3.
8. Соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль по любому из пп. 1-7, где R3 выбран из группы, состоящей из атома водорода, циано, галогена, C1-C6 алкила, C1-C6 галогеналкила, NR13C(O)R14, C(O)NR13R14 и SO2R15; R13 и R14 идентичны или отличаются и каждый независимо выбран из группы, состоящей из атома водорода и C1-C6 алкила; и R15 выбран из группы, состоящей из атома водорода и C1-C6 алкила.
9. Соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль по любому из пп. 1-8, где Z выбран из группы, состоящей из химической связи, -O-, -O-CH2- и -NH-.
10. Соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль по любому из пп. 1-9, где Ra выбран из группы, состоящей из -CH2CH=CHC(O)NR8R9, -C(O)CH=CR10R11 и -C(O)C≡CR12;
R8 и R9 идентичны или отличаются и каждый независимо выбран из группы, состоящей из атома водорода, C1-C6 алкила, C3-C6 циклоалкила и 3-6-членного гетероциклила, содержащего один гетероатом O;
или R8 и R9 вместе с атомом азота, к которому они присоединены, образуют 3-8-членный гетероциклил, где 3-8-членный гетероциклил необязательно содержит в дополнение к одному атому азота один гетероатом, выбранный из группы, состоящей из N и O, и 3-8-членный гетероциклил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из C1-C6 алкила, галогена, гидрокси, C1-C6 гидроксиалкила;
R10 и R11 идентичны или отличаются и каждый независимо выбран из группы, состоящей из атома водорода и C1-C6 алкила;
R12 выбран из группы, состоящей из атома водорода и C1-C6 алкила.
11. Соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль по любому из пп. 1-10, где Ra выбран из группы, состоящей из -CH2CH=CHC(O)N(CH3)2, -CH2CH=CHC(O)NH(CH3), -CH2CH=CHC(O)NHC(CH3)3, -C(O)CH=CH2, -C(O)C≡CCH3,

предпочтительно Ra выбран из группы, состоящей из -CH2CH=CHC(O)N(CH3)2, -CH2CH=CHC(O)NH(CH3), -CH2CH=CHC(O)NHC(CH3)3, -C(O)CH=CH2, -C(O)C≡CCH3, 
12. Соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль по любому из пп. 1-11, выбранное из группы, состоящей из:

13. Соединение формулы (IA) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль,

где Rw представляет собой тетрагидропиранил;
R выбран из группы, состоящей из -CH2CH=CHC(O)NR8R9, -C(O)CH=CR10R11 и -C(O)C≡CR12;
R8 и R9 идентичны или отличаются и каждый независимо выбран из группы, состоящей из атома водорода, C1-C6 алкила, C1-C6 галогеналкила, C3-C6 циклоалкила и 3-6-членного гетероциклила, содержащего один гетероатом O;
или R8 и R9 вместе с атомом азота, к которому они присоединены, образуют 3-8-членный гетероциклил, где 3-8-членный гетероциклил необязательно содержит в дополнение к одному атому азота один гетероатом, выбранный из группы, состоящей из N и O, и 3-8-членный гетероциклил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из C1-C6 алкила, галогена, гидрокси, C1-C6 гидроксиалкила и -COOR16-;
R16 выбран из группы, состоящей из атома водорода, C1-C6 алкила, C1-C6 галогеналкила и C1-C6 гидроксиалкила;
кольцо A, G, Z, R1, R2, R3, R4, Rb, R10, R11, R12, n, m, s и q являются такими, как определено в п. 1.
14. Соединение формулы (IA) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль по п. 13, выбранное из группы, состоящей из:


15. Способ получения соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли по любому из пп. 1-12, включающий следующую стадию, на которой соединение формулы (IA), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемую соль подвергают реакции снятия защиты в кислых условиях с получением соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли:
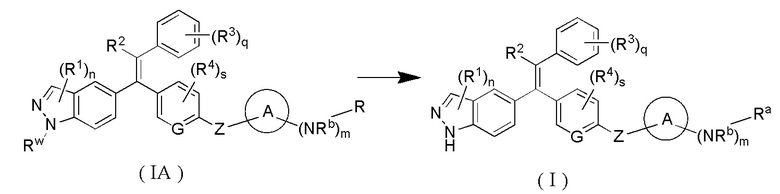 ,
,
где Rw представляет собой тетрагидропиранил;
R является таким, как определено в п. 13;
кольцо A, G, Z, R1, R2, R3, R4, Ra, Rb, n, m, s и q являются такими, как определено в п. 1.
16. Фармацевтическая композиция для использования в качестве модулятора рецептора эстрогена для предотвращения и/или лечения заболевания или расстройства, опосредованного рецептором эстрогена или зависимого от рецептора эстрогена, содержащая терапевтически эффективное количество соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли по любому из пп. 1-12 и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов.
17. Применение соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли по любому из пп. 1-12 или фармацевтической композиции по п. 16 для получения модулятора рецептора эстрогена.
18. Применение соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли по любому из пп. 1-12 или фармацевтической композиции по п. 16 для получения лекарственного средства для предотвращения и/или лечения заболевания или расстройства, опосредованного рецептором эстрогена или зависимого от рецептора эстрогена.
19. Применение по п. 18, где заболевание или расстройство, опосредованное рецептором эстрогена или зависимое от рецептора эстрогена, представляет собой рак, предпочтительно рак молочной железы, рак яичника, рак эндометрия, рак предстательной железы или рак матки и более предпочтительно рак молочной железы.
| WO 2018098305 A1, 31.05.2018 | |||
| US 20160347717 A1, 01.12.2016 | |||
| US 20180141913 A1, 24.05.2018 | |||
| WO 2018098251 A1, 31.05.2018 | |||
| PUYANG X | |||
| et al, Discovery of Selective Estrogen Receptor Covalent Antagonists for the Treatment of ERαWT and ERαMUT Breast Canser, Cancer Discow., 2018, v | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| Ручной станок для испытания материалов на разрыв | 1924 |
|
SU1176A1 |
| GOVEK S.P | |||
| et al, Optimization of an indazole | |||

