Данное изобретение относится к объединенному способу синтеза фенола из бензола путем прямого окисления бензола пероксидом водорода в присутствии цеолитного катализатора TS-1 с рециклом побочных продуктов.
Более конкретно, изобретение относится к способу получения фенола, в котором побочные продукты реакции подвергают селективному превращению в фенол и рециклу в производственные потоки.
Фенол является чрезвычайно важным промышленным полупродуктом, который используется, например, в производстве поликарбонатов или других фенольных смол.
Фенол в настоящее время производят в промышленности, исходя из кумола. Однако известны также различные способы получения фенола, включающие прямое окисление бензола пероксидом водорода в присутствии подходящих каталитических систем.
Эти способы в общем случае осуществляют в органическом растворителе, способном улучшить контакт между органическим субстратом и пероксидом водорода, в таком, например, как метанол, этанол или изопропиловый спирт. Для этой цели также пригодны кетоны, такие как ацетон, метилэтилкетон, или же уксусная кислота или ацетонитрил, как описано в патентах US 4396783 и GB 2116974.
В европейской патентной заявке ЕР А 919531 описано использование специфического растворителя, такого как сульфолан, для достижения значительных улучшений в отношении конверсии и селективности этих процессов (ЕР А 919531). В альтернативном варианте улучшения в отношении конверсии и селективности также могут быть достигнуты путем активации катализатора пероксидом водорода и ионами фтора, как описано в европейской патентной заявке ЕР А 958861.
Способы получения фенола путем прямого окисления бензола пероксидом водорода обычно осуществляют в двухфазной реакционной системе (твердый катализатор/органическая фаза) в присутствии подходящих каталитических систем.
В итальянской патентной заявке MI 2001A 002410 описан способ, осуществляемый в трехфазной реакционной системе, состоящей из твердого катализатора/водной фазы/органической фазы (ароматическое соединение + растворитель), которая позволяет повысить производительность процесса окисления бензола без ухудшения селективности.
Однако высокой производительности не удается достичь даже при использовании трехфазной системы. Это является следствием того факта, что процесс необходимо проводить при низких значениях конверсии бензола для ограничения последующих реакций окисления фенола в побочные продукты - пирокатехин и гидрохинон. Например, в указанной выше итальянской патентной заявке MI 2001A 002410 отмечено, что при конверсии бензола 12,2% и селективности по фенолу 90% образуется 111 кг гидрохинона и пирокатехина (при соотношении в смеси 55/45) на каждую тонну фенола. Количество этих побочных продуктов превышает рыночную потребность, и поэтому они подлежат утилизации, что повышает производственные расходы. Кроме того, вследствие низкой производительности необходимо отделить и рециклизовать 20,1 кг растворителей (сульфолан, бензол и вода) на каждый килограмм полученного фенола. Большой объем рециклизуемых продуктов приводит к необходимости иметь в промышленной установке секцию выделения больших размеров.
Теперь установлено, что описанные выше недостатки могут быть частично устранены при использовании способа согласно данному изобретению.
На практике предложенный в изобретении способ предполагает объединение процесса синтеза фенола с секцией гидродеоксигенации побочных продуктов - гидрохинона и пирокатехина, которые подвергают селективному превращению в фенол и подают рециклом в производственные потоки, и работу секции синтеза фенола при определенных технологических условиях.
При осуществлении способа, предложенного в данном изобретении, достигаются следующие преимущества:
- исключение одновременного образования дифенолов (конечная селективность по фенолу после отделения побочных продуктов составляет 99%);
- экономия расходов на утилизацию;
- увеличение производительности процесса, которую можно повысить до 159 г фенола/л реакционной смеси (как в примере 8 данного патента), с соответствующим снижением количества растворителей, подлежащих рециклу (5,3 кг на 1 кг фенола), и капитальных затрат на установку (по отношению к секции выделения).
В соответствии с этим задачей данного изобретения является способ получения фенола, включающий следующие стадии:
1) непрерывное получение фенола путем прямого окисления бензола пероксидом водорода при соотношении Н2O2/бензол от 10 до 70 мол.% в трехфазной реакционной системе, включающей первую жидкую фазу, состоящую из бензола и органического растворителя, вторую жидкую фазу, состоящую из воды, и твердую фазу, состоящую из активированного катализатора, содержащего силикалит титана TS-1;
2) отделение фенола и непрореагировавшего бензола из реакционной смеси стадии (1) окисления путем фракционной перегонки;
3) отделение растворителя и побочных продуктов, содержащих диоксибензолы, от смеси, поступающей из хвостовой фракции перегонки (2), путем экстракции основанием с получением водного раствора диоксибензолов;
4) превращение водного раствора диоксибензолов, полученного на стадии (3), в фенол путем гидродеоксигенации водородом в непрерывных условиях при температуре от 250 до 500°С, давлении 0,1-10 МПа и в присутствии катализатора, содержащего элементы группы VIB или их смеси или элементы группы VIII периодической системы или их смеси и промотор;
5) подачу рециклом фенола, полученного на стадии (4), на перегонную стадию (2).
Объединение процесса окисления бензола в фенол и гидродеоксигенации побочных продуктов в фенол особенно предпочтительно также и потому, что (как описано в итальянской патентной заявке MI 2002A 001187) возможно получать дифенолы, выходящие из секции выделения побочных продуктов, в виде водного раствора, который может быть непосредственно использован в секции гидродеоксигенации без испарения растворителя.
Кроме того, объединение двух процессов позволяет поддерживать молярное соотношение Н2O2/бензол в более широких пределах, чем в процессе, описанном в итальянской патентной заявке MI 2001A 002410, обеспечивая таким образом достижение высоких конверсии бензола и высокой производительности.
В этих условиях происходит значительное образование побочных продуктов, которые, однако, превращают в фенол в секции гидродеоксигенации.
Получение фенола прямым окислением бензола (см. чертеж, секция а) проводят в реакторе, куда подают бензол, растворитель, воду, катализатор и пероксид водорода и где образуется органическая фаза, содержащая растворитель, непрореагировавший бензол, воду, фенол и побочные продукты (пирокатехин, гидрохинон и фенольные смолы).
Трехфазная система образуется при работе с регулируемым количеством воды, которое должно быть таким, чтобы вызывать расслоение жидкой фазы и предотвращать агрегацию катализатора.
Удобно проводить реакцию окисления бензола при концентрации воды в интервале от 5 до 50 мас.%, при этом предпочтительно используют концентрации в интервале от 15 до 40 мас.%.
Органический растворитель можно выбрать из растворителей, обычно используемых в известных процессах окисления, описанных в технике, таких как, например, метанол, этанол, изопропиловый спирт, ацетон, метилэтилкетон, уксусная кислота или ацетонитрил.
Особенно предпочтительными для целей данного изобретения являются растворители, принадлежащие к группе сульфонов, и среди них предпочтителен сульфолан, как описано в европейской патентной заявке ЕР А 919531.
Растворитель используют в количествах, составляющих от 20 до 80 мас.% по отношению к реакционной смеси.
Предпочтительно используют количества, составляющие от 40 до 70%.
Катализаторы, используемые в реакторе окисления согласно изобретению, выбраны из катализаторов, имеющих общую формулу (I)
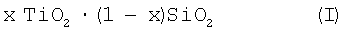
где х составляет от 0,0001 до 0,04, предпочтительно от 0,02 до 0,03.
Указанные силикалиты титана могут быть получены способом, описанным в патенте US 4410501, в котором также определены их структурные характеристики. Силикалиты титана также можно подвергнуть активирующей обработке ионами фтора и пероксидом водорода, как описано в патенте ЕР А 958861. Можно также использовать силикалиты титана, в которых часть титана замещена другими металлами, такими как бор, алюминий, железо или галлий. Эти замещенные силикалиты титана и способы их получения описаны в европейских патентных заявках ЕР 226257, 226258 и 266825.
Катализатор обычно используют в количествах, составляющих от 2 до 60 мас.% по отношению к ароматическому субстрату. Предпочтительно используют количества катализатора, составляющие от 5 до 40 мас.% по отношению к ароматическому субстрату.
Пероксид водорода добавляют в реакционную смесь в количествах, составляющих от 10 до 70 мол.% по отношению к ароматическому субстрату, предпочтительно от 20 до 60 мол.% по отношению к ароматическому субстрату.
Предпочтительно используют растворы пероксида водорода с концентрацией, составляющей от 10 до 60 мас.%, предпочтительно от 15 до 60 мас.%.
Бензол обычно используют в количествах, составляющих от 10 до 80 мас.% по отношению к реакционной смеси. Предпочтительно используют количества бензола, составляющие от 15 до 50 мас.% по отношению к реакционной смеси.
Реакцию окисления проводят при температурах в интервале от 50 до 110°С, предпочтительно от 70 до 100°С. Время реакции, необходимое для полного расходования пероксида водорода, зависит от используемых условий реакции.
Способ окисления, описанный в данном изобретении, можно проводить в реакторах полунепрерывного типа (с подачей пероксида водорода) или типа РНДП (реактора непрерывного действия с перемешиванием) при непрерывной подаче пероксида водорода и смеси бензол/растворитель.
При работе непрерывным способом водную фазу (в которой селективно распределен катализатор) сохраняют внутри реактора, а органическую фазу удаляют из спокойной зоны без перемешивания для осуществления расслоения. Таким образом, на выходе получают только одну фазу, содержащую ароматическое соединение, растворитель, гидроксиароматическое соединение и побочные продукты.
При работе в условиях способа согласно изобретению также возможно работать при 100°С с достижением увеличения каталитической активности без потери селективности, как обычно происходит при работе в двойной фазе.
Отделение и извлечение легких компонентов реакционной смеси, поступающей из секции окисления, проводят путем фракционной перегонки.
В верхней части первой колонны (секция b) получают азеотропную смесь бензол/вода и вместе с тем в хвостовой части получают смесь растворителя, фенола и побочных продуктов (пирокатехины, гидрохинон, фенольные смолы). Эту смесь подают во вторую перегонную колонну (секция с), где в верхней части получают очищенный фенол, а в хвостовой - смесь растворителя и побочных продуктов.
Побочные продукты (пирокатехин, гидрохинон и фенольные смолы) отделяют от реакционного растворителя (предпочтительно сульфолана) путем процедуры, описанной в итальянской патентной заявке MI 2002A 001187 и включающей следующие стадии:
секцию смешивания и разделения (секция d), в которую подают поток из нижней части узла перегонки фенола (секция с), состоящий из сульфолана и побочных продуктов, поток бензола, поступающий из верхней части секции перегонки бензола (секция b), и основной водный раствор. На выходе получают органическую фазу, содержащую сульфолан, бензол и воду (подаваемую рециклом в секцию), и водную фазу, содержащую соли пирокатехина, гидрохинон и смолы;
секцию смешивания (секция е) для подкисления водной фазы, поступающей из секции (d) с неорганической кислотой или CO2 для высвобождения пирокатехина, гидрохинона и смол из их солей;
секцию разделения (секция f), в которую подают поток из секции (е), содержащий воду, соли, пирокатехин, гидрохинон и смолы, а также подают экстракционный растворитель (предпочтительно метилизобутилкетон);
секцию узла перегонки (секция g) для отделения в верхней части экстракционного растворителя, подаваемого рециклом в секцию (f), и в хвостовой части - водного раствора пирокатехина, гидрохинона и смол.
Побочные продукты, полученные в секции выделения (секция g), затем превращают в фенол путем гидродеоксигенации водородом (фиг.1, секция h).
Реакцию осуществляют в газовой фазе при температурах 250-500°С, предпочтительно 300-450°С, при давлении 0,1-10 МПа, предпочтительно 0,3-5 МПа, с объемной скоростью (WHSV = Weight Hourly Space Velocity, часовая объемно-массовая скорость, выражаемая в кг диоксибензолов/час/кг катализатора) 0,1-10 ч-1, предпочтительно 0,5-5 ч-1.
В частности, загрузка реактора состоит из раствора диоксибензолов в воде с концентрацией 5-60 мас.%, предпочтительно 10-40 мас.%, и водорода в мольном соотношении по отношению к диоксибензолам 2-50, предпочтительно 5-30.
Катализатор может быть выбран из катализаторов гидродеоксигенизации, содержащих элементы группы VIB или группы VIII периодической системы.
Если катализатор содержит элементы группы VIB, он может содержать в качестве промоторов элементы, принадлежащие к группе VIII, и фосфор. Элементы группы VIB можно использовать в смеси, и среди них предпочтительными являются молибден и вольфрам. Среди промоторов группы VIII предпочтительными являются никель, кобальт, железо и рутений, и их можно использовать в смеси друг с другом и с фосфором.
Если катализатор содержит элементы группы VIII, он может содержать в качестве промотора цинк, рений, селен, олово, германий и свинец. Элементы группы VIII можно использовать в смеси, и среди них предпочтительными являются кобальт, палладий, никель и платина. Промоторы также можно использовать в смеси друг с другом.
Активную фазу предпочтительно наносят на носитель. Предпочтительными носителями являются неорганические оксиды, такие как оксид алюминия, оксид кремния, диоксид титана, кристаллические или аморфные алюмосиликаты, кристаллические шпинели, имеющие общую формулу F2+R2 3+O4 (где F2+ может представлять собой Mg, Fe, Zn, Mn, Ni и т.д., a R3+ может представлять собой Al, Fe, Cr и т.д.) или их смеси.
Для катализаторов, содержащих элемент группы VIB указанный элемент обычно присутствует на носителе в концентрации, составляющей от 1 до 50 мас.%, предпочтительно от 3 до 30 мас.%. Промоторы этих катализаторов обычно присутствуют в концентрациях от 0,1 до 100% (атомных) по отношению к элементу группы VIB, предпочтительно от 1 до 50%. Без установления каких-либо ограничений по возможным составам или указания каких-либо предпочтений, примерами таких катализаторов являются Мо, W, СоМо, NiMo, NiW, FeMo, RuMo, CoMoP, NiMoP, CoWMo, CoWMoP.
Перед использованием в реакции эти катализаторы могут подвергаться обработке для модификации их химических характеристик, например, сульфированию с помощью H2S, диметилсульфида, диметилдисульфида, сероуглерода или других соединений, пригодных для этой цели.
Для катализаторов, содержащих элемент группы VIII, указанный элемент обычно присутствует на носителе в концентрации в интервале от 0,05 до 20 мас.%, предпочтительно от 0,1 до 10 мас.%. Промоторы этих катализаторов обычно присутствуют в концентрациях в диапазоне от 0,5 до 200% (атомных) по отношению к элементу группы VIII, предпочтительно от 1 до 120%. Не устанавливая каких-либо ограничений для возможных составов и не указывая каких-либо предпочтений, примерами таких катализаторов являются Pt, Pd, Co, Ni, PtZn, PtRe, PtNi, PtSe, PtSn, PtGe, PtPb, PdPb, PdSn.
При наличии наиболее подходящих катализаторов и условий работы можно поддерживать работу реактора в течение времени, составляющего до нескольких сотен часов, при 100% степени превращения диоксибензола и селективности по отношению к фенолу более 95%.
При увеличении времени работы реактора степень превращения имеет тенденцию снижаться, в то время как селективность остается чрезвычайно высокой. Для того чтобы поддерживать желаемую степень превращения, температуру реакции можно постепенно увеличивать в пределах интервала 250-500°С.
Установлено, что катализаторы, которые можно использовать для целей данного изобретения, можно подвергать, без каких-либо особых проблем, периодической регенерации (в соответствии с тем, что известно в данной области техники) для восстановления исходной активности. В частности, весь процесс включает следующие секции:
a) узел окисления, который содержит катализатор и в который подают бензол, сульфолан, воду и пероксид водорода;
b) узел перегонки для отделения в верхней части азеотропной смеси вода/бензол;
c) узел перегонки для отделения в верхней части фенола;
d) узел смешивания и разделения, в который подают поток из нижней части узла перегонки (с), состоящий из сульфолана, дифенолов и фенольных смол, поток бензола из секции (b) и водный раствор основания. На выходе получают органическую фазу, содержащую сульфолан, бензол и воду (в рецикл в секцию а), и водную фазу, содержащую соли дифенолов и смол;
e) узел смешивания для подкисления водной фазы, поступающей из секции (d), неорганической кислотой или CO2 для высвобождения дифенолов и смол из их солей;
f) узел разделения, в который подают поток из секции (е), содержащий воду, соли, дифенолы и смолы, и экстракционный растворитель (например, метилизобутилкетон);
g) секцию перегонки для отделения в верхней части экстракционного растворителя, подаваемого рециклом в секцию (f);
h) узел гидродеоксигенации, который содержит катализатор и в который подают хвостовой поток из секции (g), содержащий дифенолы, смолы и воду, а также подают водород. Выходящий поток, содержащий извлеченный фенол и воду, подают рециклом в секцию (с).
Упрощенная схема объединенного способа представлена на чертеже.
С целью более подробного описания данного изобретения ниже приведены несколько примеров, которые, однако, никак не следует рассматривать как ограничение объема изобретения.
Пример 1
Активация катализатора
В стеклянную колбу вместимостью 100 мл, снабженную механической мешалкой, обратным холодильником термометром и термостатом с циркулирующим маслом, загрузили 3,0 г (1,43 ммоль Ti) катализатора TS-1 (EniChem, Ti=2,29 мас.%) и 0,11 г NH4HF2 (средний титр 92,5%) в 35 мл воды, что соответствует молярному соотношению F/Ti=2,5. Водную суспензию катализатора, поддерживаемую при механическом перемешивании, нагревали до 60°С. Далее добавляли 1,6 мл Н2O2 с концентрацией 30 мас.%, что соответствует молярному отношению H2O2/Ti=11, и суспензию поддерживали при механическом перемешивании при 60°С еще в течение 4 часов. После охлаждения твердое вещество отделили от маточного раствора (рН=4,3) фильтрованием через пористую мембрану, затем многократно промывали деионизированной водой и в завершение - ацетоном. Затем катализатор сушат под вакуумом при 40°С в течение 8 часов и далее подвергают, при скорости нагревания 50°С в час, термообработке на воздухе при 550°С в течение 4 часов. Титр активированного катализатора составляет 1,49% Ti. Растворимый титан составляет 35 мас.%.
Пример 2
Трехфазная система при полупериодических условиях (сравнительный пример MI 2001А 002410)
В реактор AISI 316 (объем = 600 мл) подали азот под давлением 5 атм. Затем загрузили 100 г бензола (1,28 моля), 180 г сульфолана, 43 г воды и 10 г катализатора, активированного, как описано в примере 1 (соответствует 3,1 ммоля Ti). Жидкая часть реакционной смеси в данном случае является трехфазной. Температуру реактора довели до 100°С.
Далее в течение 1 часа подавали 21,75 г водного раствора H2O2 (192 ммоля Н2O2; Н2O2/бензол = 0,15) с концентрацией 30 мас.%.
Затем реакционную смесь охладили до 20°С и катализатор отделили фильтрованием через пористую мембрану.
В конце реакции отделили две фазы при следующих составах:
верхняя органическая фаза (85 мас.%):
бензол/сульфолан/вода = 1/3/96 по массе,
нижняя водная фаза (15 мас.%):
бензол/сульфолан/вода = 61/38/1 по массе.
Органическую фазу анализировали с помощью ВЭЖХ и установили образование следующих продуктов:
фенол - 13,3 г (141,1 ммоля),
гидрохинон - 0,69 г (6,3 ммоля),
пирокатехин - 1,03 г (9,4 ммоля).
Реакционную смесь затем выпаривали при пониженном давлении с получением в кубовом остатке лишь следов полифенольных смол. Характеристики реакции, соответственно, таковы:
- степень превращения бензола (С1) = 12,2% (в молях);
- степень превращения H3O2 (C2) = 98% (в молях);
- селективность по фенолу (S1) = 90% (в молях);
- селективность по Н2О2 (S2) = 75% (в молях);
- концентрация фенола в конечной реакционной смеси (органической фазе) = 4,75% (мас.).
При работе в этих условиях в ходе стадии выделения и очистки продуктов реакции требуется испарить 20,1 кг растворителя (сульфолана и непрореагировавшего бензола) на каждый килограмм фенола.
При этом одновременно образуются побочные продукты в количестве 52,0 кг гидрохинона на 1 кг фенола и 78,0 кг пирокатехина на тонну фенола.
Пример 3
В реактор AISI 316 (объем = 600 мл) подали азот под давлением 5 атм. Затем загрузили 100 г бензола (1,28 моля), 296 г сульфолана, 169 г воды и 10 г катализатора, активированного, как описано в примере 1 (соответствует 3,1 ммолей Ti). Жидкая часть реакционной смеси в данном случае является трехфазной. Температуру реактора довели до 100°С.
Далее в течение 1 часа подавали 29,1 г водного раствора Н2О2 (257 ммолей Н2О2; Н2O2/бензол = 20 мол.%) с концентрацией 30 мас.%.
Затем реакционную смесь охладили до 20°С и катализатор отделили фильтрованием через пористую мембрану.
В конце реакции отделили две фазы при следующих составах:
верхняя органическая фаза (42 мас.%):
бензол/сульфолан/вода = 56/40/4 по массе,
нижняя водная фаза (58 мас.%):
бензол/сульфолан/вода = 49/2/49 по массе.
Органическую фазу анализировали с помощью ВЭЖХ и установили образование следующих продуктов:
фенол - 17,5 г (186,5 ммоля),
гидрохинон - 1,23 г (11,14 ммоля),
пирокатехин - 1,84 г (16,72 ммоля).
Реакционную смесь затем выпаривали при пониженном давлении с получением в кубовом остатке лишь следов полифенольных смол.
Характеристики реакции, соответственно, таковы:
- степень превращения бензола (С1) = 16,7% (в молях);
- степень превращения H2O2 (C2) = 92% (в молях);
- селективность по фенолу (S1) = 87% (в молях);
- селективность по H2O2 (S2) = 79% (в молях);
- концентрация фенола в конечной реакционной смеси (органической фазе) = 7,28% (мас.).
При работе в этих условиях в ходе стадии выделения и очистки продуктов реакции требуется испарить 12,7 кг растворителя (сульфолана и непрореагировавшего бензола) на каждый килограмм фенола.
При этом одновременно образуются побочные продукты в количестве 69,9 кг гидрохинона на 1 кг фенола и 104,9 кг пирокатехина на тонну фенола.
В конце процедуры отделения продуктов реакции, описанной в патенте MI 2002A 001187, указанные побочные продукты получают в водном растворе. Раствор, содержащий пирокатехин (150 г/л) и гидрохинон (100 г/л), затем подают со скоростью, равной 0,14 мл/мин, в трубчатый реактор из стали AISI 316, содержащий 5 г катализатора Angelhard ESCAT™ Н-60 (Со/Мо/Р), при температуре 450°С и давлении 25 бар (2,5 МПа) вместе с потоком водорода, таким, при котором молярное соотношение водород/(пирокатехин + гидрохинон) поддерживается равным 20,5.
При работе в этих условиях достигается 100% степень превращения пирокатехина и гидрохинона с получением 2,49 г (26,5 ммолей), что соответствует степени превращения в фенол, равной 97%.
Общая селективность процесса, рассчитанная как общее количество молей полученного фенола/число молей превращенного бензола × 100, оказывается равной 99%.
Пример 4
Использовали ту же процедуру, что в примере 3, при этом подавали 21,8 г водного раствора H2O2 (384 ммоля H2O2; Н2O2/бензол = 30 мол.%) с концентрацией 60 мас.%.
Органическую фазу анализировали с помощью ВЭЖХ и установили образование следующих продуктов:
фенол - 23,0 г (245,1 ммоля),
гидрохинон - 2,70 г (24,51 ммоля),
пирокатехин - 4,04 г (36,77 ммоля).
Реакционную смесь затем выпаривали при пониженном давлении с получением в кубовом остатке лишь следов полифенольных смол.
Характеристики реакции, соответственно, таковы:
- степень превращения бензола (С1) = 23,89% (в молях);
- степень превращения Н2O2 (С2) = 98% (в молях);
- селективность по фенолу (S1) = 80% (в молях);
- селективность по Н2O2 (S2) = 65% (в молях);
- концентрация фенола в конечной реакционной смеси (органической фазе) = 9,85% (мас.).
При работе в этих условиях в ходе стадии выделения и очистки продуктов реакции требуется испарить 9,2 кг растворителя (сульфолана и непрореагировавшего бензола) на каждый килограмм фенола.
При этом одновременно образуются побочные продукты в количестве 117,0 кг гидрохинона на 1 кг фенола и 175,5 кг пирокатехина на тонну фенола.
При работе, как описано в примере 3, побочные продукты подвергают гидрированию, с достижением 100% степени превращения пирокатехина и гидрохинона, с получением 5,47 г (58,2 ммолей), что соответствует степени превращения в фенол, равной 97%.
Общая селективность процесса, рассчитанная как общее количество молей полученного фенола/число молей превращенного бензола × 100, оказывается равной 99%.
Пример 5
Использовали ту же процедуру, что в примере 3, при этом подавали 29,1 г водного раствора H2O2 (513 ммолей Н2O2; Н2O2/бензол = 40 мол.%) с концентрацией 60 мас.%.
Органическую фазу анализировали с помощью ВЭЖХ и установили образование следующих продуктов:
фенол - 27,4 г (291,63 ммоля),
гидрохинон - 3,83 г (34,84 ммоля),
пирокатехин - 5,75 г (52,27 ммоля).
Реакционную смесь затем выпаривали при пониженном давлении с получением в кубовом остатке лишь следов полифенольных смол.
Характеристики реакции, соответственно, таковы:
- степень превращения бензола (С1) = 29,53% (в молях);
- степень превращения Н2O2 (С2) = 98% (в молях);
- селективность по фенолу (S1) = 77% (в молях);
- селективность по Н2O2 (S2) = 58% (в молях);
- концентрация фенола в конечной реакционной смеси (органической фазе) = 11,73% (мас.).
При работе в этих условиях в ходе стадии выделения и очистки продуктов реакции требуется испарить 7,5 кг растворителя (сульфолана и непрореагировавшего бензола) на каждый килограмм фенола.
При этом одновременно образуются побочные продукты в количестве 139,8 кг гидрохинона на 1 кг фенола и 209,7 кг пирокатехина на тонну фенола.
При работе, как описано в примере 3, побочные продукты подвергают гидрированию, с достижением 100% степени превращения пирокатехина и гидрохинона, с получением 7,78 г (82,7 ммолей), что соответствует степени превращения в фенол, равной 97%.
Общая селективность процесса, рассчитанная как общее количество молей полученного фенола/число молей превращенного бензола × 100, оказывается равной 99%.
Пример 6
В реактор AISI 316 (объем = 600 мл) подали азот под давлением 5 атм. Затем загрузили 100 г бензола (1,28 моля), 216 г сульфолана, 85 г воды и 10 г катализатора, активированного, как описано в примере 1 (соответствует 3,1 ммолей Ti). Жидкая часть реакционной смеси в данном случае является трехфазной. Температуру реактора довели до 100°С.
Далее в течение 1 часа подавали 29,1 г водного раствора H2O2 (257 ммолей Н2O2; Н2O2/бензол = 20 мол.%) с концентрацией 30 мас.%.
Затем реакционную смесь охладили до 20°С, и катализатор отделили фильтрованием через пористую мембрану.
В конце реакции отделили две фазы, при следующих составах:
верхняя органическая фаза (43 мас.%):
бензол/сульфолан/вода = 55/41/4 по массе,
нижняя водная фаза (57 мас.%):
бензол/сульфолан/вода = 49/2/49 по массе.
Органическую фазу анализировали с помощью ВЭЖХ и установили образование следующих продуктов:
фенол - 17,5 г (186,5 ммоля),
гидрохинон - 1,33 г (12,11 ммоля),
пирокатехин - 2,00 г (18,17 ммоля).
Реакционную смесь затем выпаривали при пониженном давлении с получением в кубовом остатке лишь следов полифенольных смол.
Характеристики реакции, соответственно, таковы:
- степень превращения бензола (С1) = 16,9% (в молях);
- степень превращения Н2O2 (С2) = 98% (в молях);
- селективность по фенолу (S1) = 86% (в молях);
- селективность по Н2O2 (S2) = 74% (в молях);
- концентрация фенола в конечной реакционной смеси (органической фазе) = 9,93% (мас.).
При работе в этих условиях в ходе стадии выделения и очистки продуктов реакции требуется испарить 9,1 кг растворителя (сульфолана и непрореагировавшего бензола) на каждый килограмм фенола.
При этом одновременно образуются побочные продукты в количестве 76,2 кг гидрохинона на 1 кг фенола и 114,3 кг пирокатехина на тонну фенола.
При работе, как описано в примере 3, побочные продукты подвергают гидрированию, с достижением 100% степени превращения пирокатехина и гидрохинона, с получением 2,70 г (28,8 ммолей), что соответствует степени превращения в фенол, равной 97%.
Общая селективность процесса, рассчитанная как общее количество молей полученного фенола/число молей превращенного бензола × 100, оказывается равной 99%.
Пример 7
Использовали ту же процедуру, что в примере 6, при этом подавали 21,8 г водного раствора H2O2 (384 ммолей Н2O2; Н2O2/бензол = 30 мол.%) с концентрацией 60 мас.%.
Органическую фазу анализировали с помощью ВЭЖХ и установили образование следующих продуктов:
фенол - 22,69 г (241,1 ммоля),
гидрохинон - 2,65 г (24,14 ммоля),
пирокатехин - 3,98 г (36,20 ммоля).
Реакционную смесь затем выпаривали при пониженном давлении с получением в кубовом остатке лишь следов полифенольных смол.
Характеристики реакции, соответственно, таковы:
- степень превращения бензола (С1) = 23,5% (в молях);
- степень превращения Н2O2 (С2) = 98% (в молях);
- селективность по фенолу (S1) = 80% (в молях);
- селективность по Н2O2 (S2) = 64% (в молях);
- концентрация фенола в конечной реакционной смеси (органической фазе) = 13,41% (мас.).
При работе в этих условиях в ходе стадии выделения и очистки продуктов реакции требуется испарить 6,5 кг растворителя (сульфолана и непрореагировавшего бензола) на каждый килограмм фенола.
При этом одновременно образуются побочные продукты в количестве 117,0 кг гидрохинона на 1 кг фенола и 175,5 кг пирокатехина на тонну фенола.
При работе, как описано в примере 3, побочные продукты подвергают гидрированию, с достижением 100% степени превращения пирокатехина и гидрохинона, с получением 5,38 г (57,3 ммолей), что соответствует степени превращения в фенол, равной 97%.
Общая селективность процесса, рассчитанная как общее количество молей полученного фенола/число молей превращенного бензола × 100, оказывается равной 99%.
Пример 8
Использовали ту же процедуру, что в примере 6, при этом подавали 29,1 г водного раствора Н2O2 (513 ммолей Н2O2; Н2O2/бензол = 40 мол.%) с концентрацией 60 мас.%.
Органическую фазу анализировали с помощью ВЭЖХ и установили образование следующих продуктов:
фенол - 26,9 г (286,6 ммоля),
гидрохинон - 3,98 г (36,20 ммоля),
пирокатехин - 5,97 г (54,30 ммоля).
Реакционную смесь затем выпаривали при пониженном давлении с получением в кубовом остатке лишь следов полифенольных смол.
Характеристики реакции, соответственно, таковы:
- степень превращения бензола (С1) = 29,4% (в молях);
- степень превращения Н2O2 (С2) = 98% (в молях);
- селективность по фенолу (S1) = 76% (в молях);
- селективность по H2O2 (S2) = 57% (в молях);
- концентрация фенола в конечной реакционной смеси (органической фазе) = 15,95% (мас.).
При работе в этих условиях в ходе стадии выделения и очистки продуктов реакции требуется испарить 5,3 кг растворителя (сульфолана и непрореагировавшего бензола) на каждый килограмм фенола.
При этом одновременно образуются побочные продукты в количестве 147,8 кг гидрохинона на 1 кг фенола и 221,7 кг пирокатехина на тонну фенола.
При работе, как описано в примере 3, побочные продукты подвергают гидрированию, с достижением 100% степени превращения пирокатехина и гидрохинона, с получением 8,08 г (85,9 ммолей), что соответствует степени превращения в фенол, равной 97%.
Общая селективность процесса, рассчитанная как общее количество молей полученного фенола/число молей превращенного бензола × 100, оказывается равной 99%.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СОВМЕСТНОГО ПОЛУЧЕНИЯ ПИРОКАТЕХИНА, ГИДРОХИНОНА И ПЛАСТИФИКАТОРА БЕТОНА | 1992 |
|
RU2028288C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФЕНОЛА ПУТЕМ ГИДРОДЕОКСИГЕНАЦИИ ДИОКСИБЕНЗОЛОВ | 2003 |
|
RU2336260C2 |
| СПОСОБ ОКИСЛЕНИЯ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ДО ГИДРОКСИАРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 1998 |
|
RU2185368C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИГИДРОКСИБЕНЗОЛОВ | 2002 |
|
RU2228326C2 |
| Способ получения фенола | 2023 |
|
RU2829821C1 |
| СПОСОБ АКТИВИРОВАНИЯ ТИТАНСОДЕРЖАЩЕГО СИЛИКАЛИТА, ТИТАНСОДЕРЖАЩИЙ СИЛИКАЛИТНЫЙ КАТАЛИЗАТОР И СПОСОБ ОКИСЛЕНИЯ ОРГАНИЧЕСКОГО СУБСТРАТА | 1999 |
|
RU2159675C1 |
| СПОСОБ СОВМЕСТНОГО ПОЛУЧЕНИЯ ПИРОКАТЕХИНА И ГИДРОХИНОНА | 1989 |
|
RU2043331C1 |
| ВОДНЫЙ РАСТВОР ПЕРОКСИДА ВОДОРОДА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ИСПОЛЬЗОВАНИЕ | 2008 |
|
RU2468990C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСИРАНА, УСТАНОВКА ДЛЯ ОСУЩЕСТВЛЕНИЯ СПОСОБА И ОБЪЕДИНЕННЫЙ СПОСОБ ПОЛУЧЕНИЯ ПЕРОКСИДА ВОДОРОДА И ОКСИРАНА | 2001 |
|
RU2247118C2 |
| ПРЯМОЙ СИНТЕЗ ПЕРОКСИДА ВОДОРОДА В МНОГОКОМПОНЕНТНОЙ СИСТЕМЕ РАСТВОРИТЕЛЕЙ | 2002 |
|
RU2270165C2 |
Изобретение относится к объединенному способу синтеза фенола из бензола с рециклом побочных продуктов. Способ включает следующие стадии. На первой стадии непрерывным способом получают фенол путем прямого окисления бензола пероксидом водорода при соотношении Н2O2/бензол от 10 до 70 мол.% в трехфазной реакционной системе, включающей первую жидкую фазу, состоящую из бензола и органического растворителя, вторую жидкую фазу, состоящую из воды, и твердую фазу, состоящую из активированного катализатора, содержащего силикалит титана TS-1. На второй стадии отделяют фенол и непрореагировавший бензол из реакционной смеси путем фракционной перегонки. На третьей стадии отделяют растворитель и побочные продукты, содержащие диоксибензолы, от смеси, поступающей из хвостовой фракции перегонки второй стадии, путем экстракции основанием с получением водного раствора диоксибензолов. На четвертой стадии превращают полученный водный раствор диоксибензолов в фенол путем гидродеоксигенации водородом при работе в непрерывных условиях при температуре от 250 до 500°С, давлении 0,1-10 МПа и в присутствии катализатора, содержащего элемент группы VIB или их смесь или элемент группы VIII периодической системы или их смесь и промотор. На пятой стадии подают рециклом полученный на предыдущей стадии фенол на перегонную стадию. Изобретение позволяет повысить степень превращения бензола и селективность по фенолу, исключить образование дифенолов и снизить количество растворителей. 21 з.п. ф-лы, 1 ил.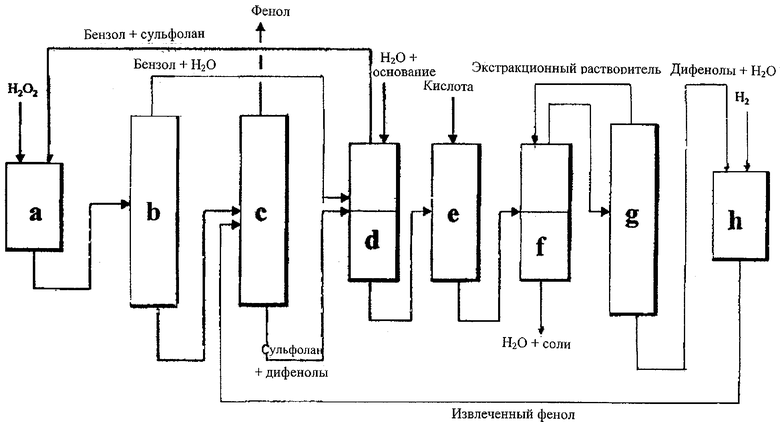
1) непрерывное получение фенола путем прямого окисления бензола пероксидом водорода при соотношении Н2О2/бензол от 10 до 70 мол.% в трехфазной реакционной системе, включающей первую жидкую фазу, состоящую из бензола и органического растворителя, вторую жидкую фазу, состоящую из воды, и твердую фазу, состоящую из активированного катализатора, содержащего силикалит титана TS-1;
2) отделение фенола и непрореагировавшего бензола из реакционной смеси стадии (1) окисления путем фракционной перегонки;
3) отделение растворителя и побочных продуктов, содержащих диоксибензолы, от смеси, поступающей из хвостовой фракции перегонки (2), путем экстракции основанием с получением водного раствора диоксибензолов;
4) превращение водного раствора диоксибензолов, полученного на стадии (3), в фенол путем гидродеоксигенации водородом при работе в непрерывных условиях при температуре от 250 до 500°С, давлении 0,1-10 МПа и в присутствии катализатора, содержащего элемент группы VIB или их смесь или элемент группы VIII периодической системы или их смесь и промотор;
5) подачу рециклом фенола, полученного на стадии (4), на перегонную стадию (2).
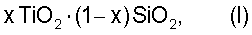
где х составляет от 0,0001 до 0,04, предпочтительно от 0,02 до 0,03, в количествах от 2 до 60 мас.% по отношению к бензолу, активированного путем предварительной обработки ионами фтора и пероксидом водорода.
| US 6288004, В1, 11.09.2001 | |||
| Устройство для моделирования архитектурного освещения застройки | 1979 |
|
SU894783A1 |
| СПОСОБ ОКИСЛЕНИЯ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ДО ГИДРОКСИАРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 1998 |
|
RU2185368C2 |

