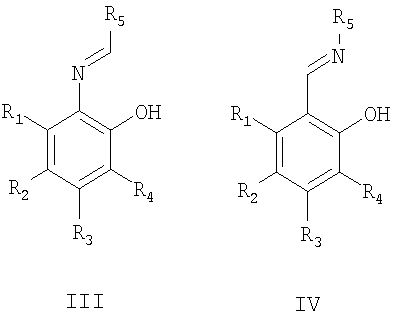
Изобретение относится к области химии лекарственных средств, а именно к получению оптически активного 5-метокси-2-((4-метокси-3,5-диметилпиридин-2-ил)метилсульфинил)-1Н-бензо[d]имидазола общей формулы I, проявляющего противоязвенную активность.
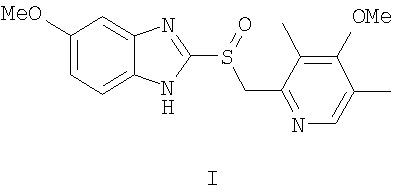
Эзомепразол ((S)-I), современный высокоэффективный противоязвенный препарат, является (S)-энантиомером омепразола и значительно превосходит последний по клиническому эффекту [Лапина Т.Л. Клиническая фармакология и терапия, 2002, 11, №2].
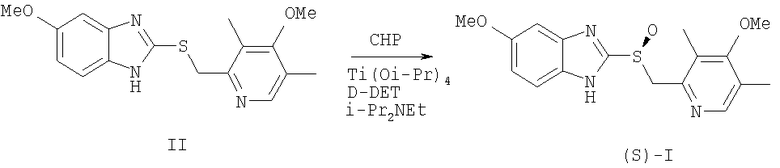
Единственным доступным методом получения эзомепразола ((S)-I) до 1996 г. было разделение рацемической смеси [Erlandsson P., Isaksson R., Lorentzon P., Lindberg Р. J. Chromatogr. 1990, 532, 305; Kohl В., Senn-Bilfinger J. DE 4035455 (1990); Lindberg P., von Unge, S. WO 9427988 (1994); Deng J., Chi Y., Fu F, Cui X., Yu K., Zhu J., Jiang Y. Tetrahedron: Asymmetry, 2000, 11, 1729]. В 1996 г. был предложен способ энантиоселективного окисления 5-метокси-2-((4-метокси-3,5-диметилпиридин-2-ил)метилтио)-1H-бензо[d]имидазола (II) в эзомепразол ((S)-I) [Larsson E.M., Stenhede U.J., Sorensen Н., von Unge Р.О.S., Cotton H.К. WO 9602535 (1996)], включавший в себя приготовление каталитического комплекса изопропилата титана (IV), D-диэтилтартрата (D-DET) и воды в присутствии сульфида (II), выдерживание полученного каталитического комплекса при повышенной температуре (до добавления окислителя), проведение реакции в присутствии аминов, предпочтительно N,N-диизопропилэтиламина, в качестве окислителя использовался гидропероксид кумола (СНР), а растворителя - толуол, катализатор использовался в количестве 30 мольных % (схема 1). После обработки (экстракция водным раствором аммиака, подкисление водного слоя уксусной кислотой, экстракция метилизобутилкетоном), получения натриевой соли и перекристаллизации из ацетонитрила был получен эзомепразол ((S)-I) с выходом до 55%. Аналогично могут быть получены и другие родственные эзомепразолу оптически активные дигетарилсульфиды.
Схема 1.
Дальнейшие исследования в этой области в основном сосредоточились на изменениях в обработке реакционной смеси с целью повышения чистоты получаемого продукта и практически не затрагивали стадии энантиоселективного окисления [Hashimoto H., Maruyama H. WO 2001087874 (2001); Hashimoto H., Urai T. WO 2001083473 (2001)]. В патенте Kawada M., Yamano Т., Тауа N. [WO 2001014366 (2001)] к реакционной смеси были дополнительно добавлены молекулярные сита 3 Å. Замена хирального диола D-DET на монодентантный лиганд - метиловый эфир (S)-(+)-миндальной кислоты, также позволила получить эзомепразол ((S)-I), но со значительно меньшим выходом [Thennati R., Rehani R.B., Soni R.R., Chhabada V.C., Patel V.M. WO 2003089408 (2003)].
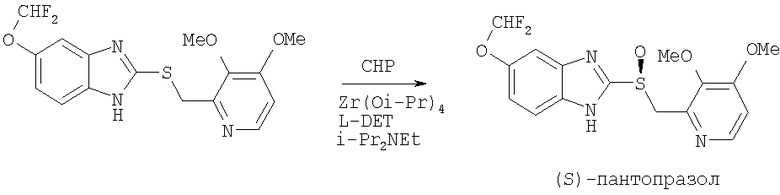
Аналог эзомепразола, (S)-пантопразол (схема 2), был синтезирован асимметрическим окислением соответствующего сульфида с помощью СНР в присутствии каталитического комплекса Zr(Oi-Pr)4 и L-DET, а также N,N-диизопропилэтиламина с выходом 80% [Kohl В., Mueller В., Weingart R.S. WO 2004052881 (2004]).
Схема 2.
Все указанные способы получения эзомепразола ((S)-I) и его аналогов обладают существенными недостатками, стимулирующими поиски новых путей синтеза. Во-первых, использование систем на основе изопропилатов титана и циркония предъявляет повышенные требования к контролю за влажностью среды как при хранении и смешивании реагентов (изопропилаты титана и циркония неустойчивы в присутствии даже следов влаги), так и в процессе реакции, что не только усложняет аппаратное оформление процесса, но и может приводить к плохой воспроизводимости результатов; во-вторых, для воспроизводимого достижения высокой энантиоселективности необходимо использовать значительные количества металлокомплексного катализатора (30 мольных % изопропилата титана, 60 мольных % дорогостоящего D-изомера диэтилтартрата), что не только приводит к необратимому расходу большого количества достаточно дорогих реагентов, но и создает существенные проблемы с обработкой реакционной смеси.
Задачей настоящего изобретения является разработка простого и экономичного способа получения оптически активного 5-метокси-2-((4-метокси-3,5-диметилпиридин-2-ил)метилсульфинил)-1Н-бензо[d]имидазола I.
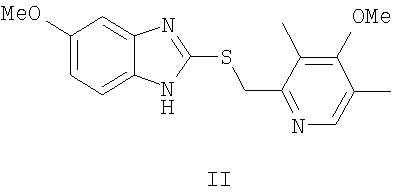
Химическая схема решения поставленной задачи заключается в окислении 5-метокси-2-((4-метокси-3,5-диметилпиридин-2-ил)метилтио)-1H-бензо[d]имидазола формулы II:
органическими пероксидами (предпочтительно гидропероксидом кумола) в присутствии предварительно полученного in situ комплекса соли ванадия (предпочтительно ацетилацетоната ванадила) с хиральным основанием Шиффа, описываемым общими формулами III или IV,
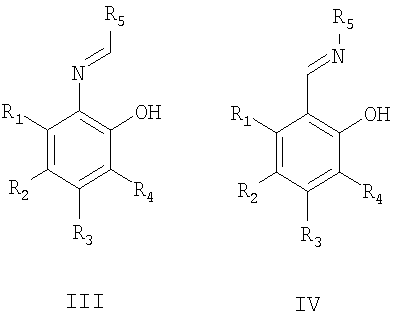
причем заместители R1, R2, R3, R4 могут быть одинаковы или отличаться и могут быть атомом водорода, алкильной группой (в том числе, содержащей атомы галогена), алкоксигруппой (в том числе, содержащей атомы галогена), нитрогруппой, диалкиламиногруппой, галогеном; R5 - оптически активный заместитель;
при пониженной температуре (предпочтительно 0-10°С) в органическом растворителе (предпочтительно толуоле) с возможным присутствием органического основания (предпочтительно N,N-диизопропилэтиламина). Комплекс соли ванадия с хиральным основанием Шиффа может применяться в каталитических количествах (предпочтительно 1 мольный % по отношению к окисляемому сульфиду II). Образующийся в процессе реакции продукт может выделяться фильтрацией или экстракционной методикой.
Хиральные основания Шиффа, описываемые общими формулами III или IV, могут быть синтезированы, например, взаимодействием салицилового альдегида или его производных с оптически активными аминами или взаимодействием 2-аминофенола или его производных с оптически активными альдегидами [Bolm С., Schlingloff G., Bienewald F. J. Mol. Cat. A: Chem. 1997, 117, 347; Хоменко Т.М., Саломатина О.В., Курбакова С.Ю., Ильина И.В., Волчо К.П., Комарова Н.И., Корчагина Д.В., Салахутдинов Н.Ф., Толстиков А.Г. ЖОрХ, 2006, 42, 1666].
Применение приведенной выше окислительной системы, включающей в себя органическую перекись в качестве окислителя, комплекс соли ванадия с хиральным основанием Шиффа и, при необходимости, органическое основание, для получения оптически активных дигетарилсульфоксидов не описано.
Предлагаемый способ получения оптически активного соединения I с использованием в качестве катализатора окисления устойчивого в присутствии воды комплекса соли ванадия с хиральным основанием Шиффа позволяет проводить реакцию без создания специальных условий (определенной влажности, использования инертной атмосферы) на всех стадиях процесса, использовать комплекс соли ванадия с хиральным основанием Шиффа в каталитических количествах, а также упрощает обработку реакционной смеси.
Изобретение иллюстрируется следующими примерами.
Пример 1. Синтез лиганда 2-(((1S,2S,3R,5S)-3-гидроксиметил-2,6,6-триметилбицикло[3.1.1]гептан-2-илимино)метил)фенола V.
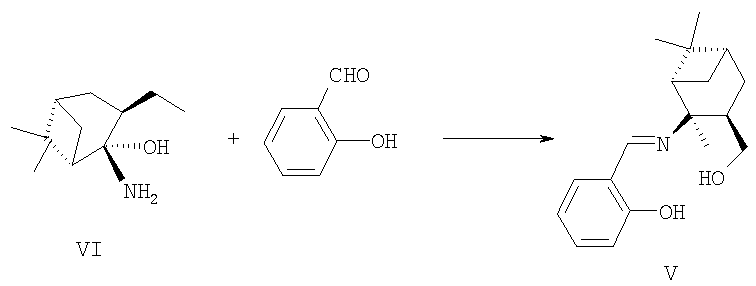
Синтез ((1R,2R,3S,5R)-2-амино-2,2,6-триметилбицикло[3.1.1]гептан-3-ил)метанола VI проводили в соответствии с методикой, опубликованной в работе Szakonyi Z., Martinek Т., Hetenyi A. Tetrahedron [Asymmetry. 2000, 11, 4571].
В стеклянный реактор с магнитной мешалкой поместили раствор 0.050 г (0.27 ммоль) соединения VI в 4 мл абсолютного метанола и при перемешивании прибавили 0.033 г (0.27 ммоль) 2-гидроксибензальдегида в 5 мл метанола. Реакционную смесь перемешивали при комнатной температуре в течение 6 ч. Получили 0.078 г 2-(((1S,2S,3R,5S)-3-гидроксиметил-2,6,6-триметилбицикло[3,1.1]гептан-2-илимино)метил)фенола V (100%), 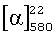 =+2.8 (с 2.8, CHCl3). Спектр ЯМР 1Н, δ, м.д. (J, Гц): 1.13 (с, 3Н, С(9)Ме), 1.19 (д, 1Н, Н(7ан), J7ан, 7 син=10), 1.27 (с, 3Н, С(10)Ме), 1.42 (с, 3Н, С(8)Ме), 1.44 (д.д.д, 1Н, Н(5ан), J5ан,5 син=13, J5ан,4 син=8, Jан,6=2), 1.92 (д.д. 1Н. Н(2), J2,6=5.5, J2,7 син=5.5), 1.95 (м, 1Н, Н(6)), 2.09 (д.д.д.д, 1Н, Н(5 син), J5 син,5ан=13, J5 син,4 син=12, J5 син,6=3.5, J5 син,7 син=2), 2.20 (д.д.д.д, 1Н, Н(7 син), J7 син,7ан=10, J7 син,2=5.5, J7 син,6 =5.5, J7 син,5 син=2), 2.28-2.42 (м, 1Н. Н(4 син)), 3.51 (д.д, 1Н, Н(11), J11,11'=10, J11,4=6.5), 3.67 (д.д, 1Н, Н(11'), J11',11=10, J11',4=8), 6.72 (д.д.д, 1Н, Н(17), J17,18=8, J17,16=7, J17,15=1.2), 6.82 (д.д, 1Н, H(15), J15,16=8.3, J15,17=1.2), 7.14 (д.д, 1Н, H(18), j18,17=8, J18,16=1.5), 7.21 (д.д.д, 1Н, H(16), J16,15=8.3, J16,17=7, J16,18=1.5), 8.20 (с, 1Н. Н(12)), 14.35 (ш.с, 1Н, С(14)-ОН). Спектр ЯМР 13С, δ, м.д.: 39.77 (с, С(1)), 53.69 (д, С(2)), 64.91 (с, С(3)),41.53 (д, С(4)), 31.32 (т, С(5)), 40.07 (д, С(6)), 29.47 (т, С(7)), 28.48 (к, С(8)), 23.64 (к, С(9)), 28.28 (к, С(10)), 66.01 (т, С(11)), 160.93 (д, С(12)), 118.41 (с, С(13)), 163.41 (с, С(14)), 117.92 (д, С(15)), 132.50 (д, С(16)), 117.40 (д, С(17)), 131.63 (д, С(18)). Найдено: M 287.18944.C18H25NO2. Вычислено: M 287.18852.
=+2.8 (с 2.8, CHCl3). Спектр ЯМР 1Н, δ, м.д. (J, Гц): 1.13 (с, 3Н, С(9)Ме), 1.19 (д, 1Н, Н(7ан), J7ан, 7 син=10), 1.27 (с, 3Н, С(10)Ме), 1.42 (с, 3Н, С(8)Ме), 1.44 (д.д.д, 1Н, Н(5ан), J5ан,5 син=13, J5ан,4 син=8, Jан,6=2), 1.92 (д.д. 1Н. Н(2), J2,6=5.5, J2,7 син=5.5), 1.95 (м, 1Н, Н(6)), 2.09 (д.д.д.д, 1Н, Н(5 син), J5 син,5ан=13, J5 син,4 син=12, J5 син,6=3.5, J5 син,7 син=2), 2.20 (д.д.д.д, 1Н, Н(7 син), J7 син,7ан=10, J7 син,2=5.5, J7 син,6 =5.5, J7 син,5 син=2), 2.28-2.42 (м, 1Н. Н(4 син)), 3.51 (д.д, 1Н, Н(11), J11,11'=10, J11,4=6.5), 3.67 (д.д, 1Н, Н(11'), J11',11=10, J11',4=8), 6.72 (д.д.д, 1Н, Н(17), J17,18=8, J17,16=7, J17,15=1.2), 6.82 (д.д, 1Н, H(15), J15,16=8.3, J15,17=1.2), 7.14 (д.д, 1Н, H(18), j18,17=8, J18,16=1.5), 7.21 (д.д.д, 1Н, H(16), J16,15=8.3, J16,17=7, J16,18=1.5), 8.20 (с, 1Н. Н(12)), 14.35 (ш.с, 1Н, С(14)-ОН). Спектр ЯМР 13С, δ, м.д.: 39.77 (с, С(1)), 53.69 (д, С(2)), 64.91 (с, С(3)),41.53 (д, С(4)), 31.32 (т, С(5)), 40.07 (д, С(6)), 29.47 (т, С(7)), 28.48 (к, С(8)), 23.64 (к, С(9)), 28.28 (к, С(10)), 66.01 (т, С(11)), 160.93 (д, С(12)), 118.41 (с, С(13)), 163.41 (с, С(14)), 117.92 (д, С(15)), 132.50 (д, С(16)), 117.40 (д, С(17)), 131.63 (д, С(18)). Найдено: M 287.18944.C18H25NO2. Вычислено: M 287.18852.
Пример 2. Синтез лиганда 4,6-ди-трет-бутил-2-((7,7-диметил-3-оксатрицикло[4.1.1.02,4]окт-2-ил)метиленамино)-фенола VII.
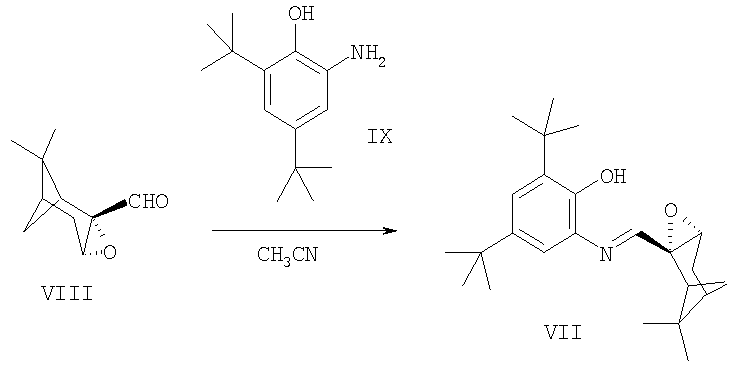
К раствору 0.023 г (0.1 ммоль) 2-амино-4,6-ди-трет-бутилфенола IX в 2 мл ацетонитрила при 25°С и перемешивании в атмосфере аргона прибавили раствор 0.022 г (0.13 ммоль) эпоксида миртеналя VIII, полученного в соответствии с методикой [Хоменко Т.М., Саломатина О.В., Курбакова С.Ю., Ильина И.В., Волчо К.П., Комарова Н.И., Корчагина Д.В., Салахутдинов Н.Ф., Толстиков А.Г. ЖОрХ, 2006, 42, 1666], в 0.8 мл ацетонитрила, перемешивание продолжали 0.5 ч. Растворитель отогнали. Получили 0.037 г (96%) 4,6-ди-трет-бутил-2-((7,7-диметил-3-оксатрицикло[4.1.1.02,4]окт-2-ил)метиленамино)-фенола VII, 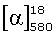 =2.6 (с 2.1, СН3CN). Спектр ЯМР 1H (ацетон-d6), δ, м.д. (J, Гц): 0.91 с (3Н, С16Н3), 1.28 с (9Н, С23Н3, С24Н3, С25Н3), 1.40 с (3Н, С17Н3), 1.41 с (9Н, С19Н3, С20Н3, С21Н3), 1.70 д (1Н, Н7анти, J7анти,7 син 10), 1.81 м (1Н, Н5, J5,7 син 5.5, J5,1 5.5, J5.4 3, J5.3 2), 2.10 м (2Н, Н4), 2.17 д.д.д (1Н, H7 син, J7 син,7анти 10, J7 син.1 5.5, J7 син,5 5.5), 2.94 д.д (1Н, Н1, J1,5 5.5, J1.7 син 5.5), 3.63 м (1Н, Н3, J2-3), 7.23 д и 7.25 д (2Н, Н13, Н15, J 2.2), 7.69 ш.с (1Н, ОН), 7.88 с (1Н, Н8). Спектр ЯМР 13С, δ, м.д.: 40.26 д (С1), 63.47 с (С2), 56.50 д (С3), 27.89 т (С4), 40.87 д (С5), 40.95 с (С6), 26.04 т (С7), 162.43 д (С5), 135.63 с (С10), 149.05 с (С11), 135.36 с (С12), 123.82 д (С13), 142.20 с (С14), 111.80 д (С15), 20.76 к (С16), 26.72 к (С17), 35.12 с (С18), 29.80 к (С19-С21), 35.46 с (С22), 31.87 к (С23-С25). Найдено М 369.26682. C24H35NO2. Вычислено М 369.26676.
=2.6 (с 2.1, СН3CN). Спектр ЯМР 1H (ацетон-d6), δ, м.д. (J, Гц): 0.91 с (3Н, С16Н3), 1.28 с (9Н, С23Н3, С24Н3, С25Н3), 1.40 с (3Н, С17Н3), 1.41 с (9Н, С19Н3, С20Н3, С21Н3), 1.70 д (1Н, Н7анти, J7анти,7 син 10), 1.81 м (1Н, Н5, J5,7 син 5.5, J5,1 5.5, J5.4 3, J5.3 2), 2.10 м (2Н, Н4), 2.17 д.д.д (1Н, H7 син, J7 син,7анти 10, J7 син.1 5.5, J7 син,5 5.5), 2.94 д.д (1Н, Н1, J1,5 5.5, J1.7 син 5.5), 3.63 м (1Н, Н3, J2-3), 7.23 д и 7.25 д (2Н, Н13, Н15, J 2.2), 7.69 ш.с (1Н, ОН), 7.88 с (1Н, Н8). Спектр ЯМР 13С, δ, м.д.: 40.26 д (С1), 63.47 с (С2), 56.50 д (С3), 27.89 т (С4), 40.87 д (С5), 40.95 с (С6), 26.04 т (С7), 162.43 д (С5), 135.63 с (С10), 149.05 с (С11), 135.36 с (С12), 123.82 д (С13), 142.20 с (С14), 111.80 д (С15), 20.76 к (С16), 26.72 к (С17), 35.12 с (С18), 29.80 к (С19-С21), 35.46 с (С22), 31.87 к (С23-С25). Найдено М 369.26682. C24H35NO2. Вычислено М 369.26676.
Пример 3. Синтез оптически активного соединения I.
Общая методика.
Катализатор синтезировали in situ непосредственно перед проведением каталитического синтеза смешением 1 мг (4×10-6 моль) ацетилацетоната ванадила и 6×10-6 моль лиганда (хирального основания Шиффа, описываемого общей формулой III или IV) в 3 мл сухого толуола (технический толуол, осушенный азеотропной перегонкой) в стеклянной колбе емкостью 25 мл при перемешивании на магнитной мешалке (≥500 об/мин-1). Смесь перемешивали 10 мин при 20°С до полного растворения кристаллов ацетилацетоната ванадила, свидетельствующего об образовании каталитического комплекса VO·Лиганд. Добавили, если это предусмотрено условиями опыта, 0.5 мг (4×10-6 моль) диизопропилэтиламина (ДИЭА), перемешивали 10 мин.
К полученному раствору добавили 130 мг (4×10-4 моль) сульфида II, перемешивали 10 мин, охладили суспензию баней с ледяной водой до температуры 0÷5°С, перемешивали 10 мин и прибавили 80 мкл (4×10-4 моль) технического 80%-ного гидропероксида кумола (СНР). После перемешивания в течение 4-6 ч при температуре 0÷5°С добавили 1 мл 10%-ного водного раствора Na2SO3, перемешивали 5 мин при комнатной температуре. Осадок отфильтровали на стеклянном пористом фильтре (размер пор 16-40 мкм), промыли 5 мл гексана, высушили на фильтре. Получили оптически активный 5-метокси-2-((4-метокси-3,5-диметилпиридин-2-ил)метилсульфинил)-1H-бензо[d]имидазол I.
Конверсию исходного сульфида II определяли методом ВЭЖХ (хроматограф Миллихром А-02, колонка Pronto SIL-120-5-C18 AQ: размер - 2.0·75 мм, зерно - 5.0 μм; подвижная фаза - 0.1% трифторуксусная кислота в метаноле; скорость подачи - 150.00 мкл/мин; температура 35°С; давление 3.3 МПа). Оптическую чистоту продукта определяли методом ВЭЖХ (хроматограф Миллихром 01, колонка Kromasil CHI-DMB: размер - 2.0·75 мм, зерно - 5.0 μм; подвижная фаза - 10% изопропанола в гексане; скорость подачи - 100.00 мкл/мин; температура 35°С). Конфигурацию оптически активного сульфоксида I определили сравнением с ВЭЖХ хроматограммой заведомого образца.
В таблице приведены конкретные лиганды, используемые для образования каталитического комплекса, условия и результаты синтеза оптически активного сульфоксида I.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЭЗОМЕПРАЗОЛА | 2007 |
|
RU2339631C1 |
| СПОСОБ ПОЛУЧЕНИЯ СУЛЬФОКСИДОВ | 2010 |
|
RU2448954C1 |
| 5,5-ДИЗАМЕЩЕННЫЕ-2-МЕТИЛ-9,9-ДИОКСО-9-ТИАБИЦИКЛО[4.3.0]НОНАНЫ | 2001 |
|
RU2184732C1 |
| (3aR,4aS,8aR,9aR,E)-3-АРИЛИДЕН-8a-МЕТИЛ-5-МЕТИЛЕН-ДЕКАГИДРОНАФТО[2,3-b]ФУРАН-2(3Н)-ОНЫ, ОБЛАДАЮЩИЕ ПРОТИВОЯЗВЕННОЙ АКТИВНОСТЬЮ | 2009 |
|
RU2413724C1 |
| ЭНАНТИОСЕЛЕКТИВНЫЙ СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ СУЛЬФОКСИДОВ | 2005 |
|
RU2380357C2 |
| ПРОТИВООПУХОЛЕВОЕ СРЕДСТВО ТРИТЕРПЕНОВОЙ ПРИРОДЫ, ПОЛУЧЕННОЕ ПУТЕМ МОДИФИКАЦИИ ГЛИЦИРРЕТОВОЙ КИСЛОТЫ | 2009 |
|
RU2401273C1 |
| ПРОИЗВОДНЫЕ 4,4,7-ТРИМЕТИЛ-2-ФЕНИЛ-4a,5,8,8a-ТЕТРАГИДРО-4Н-БЕНЗО[1,3]ДИОКСИН-8-ОЛА В КАЧЕСТВЕ АНАЛЬГЕЗИРУЮЩИХ СРЕДСТВ | 2009 |
|
RU2409353C1 |
| ПРОИЗВОДНЫЕ 5,7-ДИМЕТИЛ-1,3-ДИАЗААДАМАНТАН-6-ОНА, СОДЕРЖАЩИЕ МОНОТЕРПЕНОВЫЙ ОСТАТОК, НОВЫЕ АНАЛЬГЕЗИРУЮЩИЕ СРЕДСТВА | 2014 |
|
RU2564446C1 |
| МОНОГИДРАТ НАТРИЕВОЙ СОЛИ S-ТЕНАТОПРАЗОЛА И ЕГО ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТОННОГО НАСОСА | 2005 |
|
RU2376304C2 |
| СПОСОБЫ СИНТЕЗА ЗАМЕЩЕННЫХ СУЛЬФОКСИДОВ | 1995 |
|
RU2157806C2 |
Изобретение относится к способу получения оптически активного 5-метокси-2-((4-метокси-3,5-диметилпиридин-2-ил)метилсульфинил)-1Н-бензо[d]имидазола, проявляющего противоязвенную активность, путем энантиоселективного окисления органическими пероксидами 5-метокси-2-((4-метокси-3,5-диметилпиридин-2-ил)метилтио)-1Н-бензо[d]имидазола в присутствии катализатора в органическом растворителе, при этом в качестве катализатора используют предварительно полученный in situ комплекс соли ванадия с хиральным основанием Шиффа, описываемым общими формулами III и IV,
причем заместители R1, R2, R3, R4 могут быть одинаковы или отличаться и могут быть атомом водорода, алкильной группой (в том числе, содержащей атомы галогена), алкоксигруппой (в том числе, содержащей атомы галогена), нитрогруппой, диалкиламиногруппой, галогеном; R5 - оптически активный заместитель; и реакцию проводят при пониженной температуре с возможным присутствием органического основания с дальнейшим выделением продукта обычными способами. 1 табл.
Способ получения оптически активного 5-метокси-2-((4-метокси-3,5-диметилпиридин-2-ил)метилсульфинил)-1Н-бензо[d]имидазола, проявляющего противоязвенную активность, формулы I
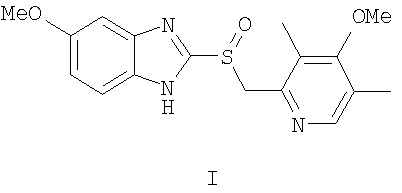
путем энантиоселективного окисления органическими пероксидами (предпочтительно гидропероксидом кумола) 5-метокси-2-((4-метокси-3,5-диметилпиридин-2-ил)метилтио)-1Н-бензо[d]имидазола формулы II
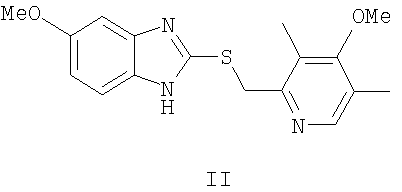
в присутствии катализатора в органическом растворителе (предпочтительно толуоле), отличающийся тем, что в качестве катализатора в каталитическом количестве (предпочтительно 1 мол.% по отношению к окисляемому сульфиду II) используют предварительно полученный in situ комплекс соли ванадия (предпочтительно ацетилацетоната ванадила) с хиральным основанием Шиффа, описываемым общими формулами III и IV
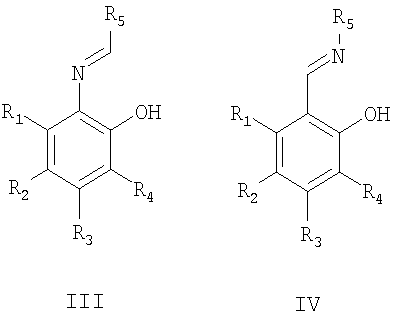
причем заместители R1, R2, R3, R4 могут быть одинаковы или отличаться и могут быть атомом водорода, алкильной группой (в том числе, содержащей атомы галогена), алкоксигруппой (в том числе, содержащей атомы галогена), нитрогруппой, диалкиламиногруппой, галогеном; R5 - оптически активный заместитель;
и реакцию проводят при пониженной температуре (предпочтительно, 0-10°С) с возможным присутствием органического основания (предпочтительно N,N-диизопропилэтиламина) с дальнейшим выделением продукта обычными способами.
| ОПТИЧЕСКИ ЧИСТЫЕ NA, MG, LI, K ИЛИ СА СОЛИ (-)-5-МЕТОКСИ-2[[(4-МЕТОКСИ-3,5-ДИМЕТИЛ-2-ПИРИДИНИЛ)МЕТИЛ] СУЛЬФИНИЛ]-1H-БЕНЗИМИДАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМКОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1994 |
|
RU2137766C1 |
| WO 9602535 A1, 01.02.1996 | |||
| WO 03089408 A2, 30.10.2003 | |||
| WO 2004052881 A2, 24.06.2004. | |||

