Изобретение относится к области газохроматографического анализа сложных смесей веществ, в частности, для идентификации неизвестных компонентов по собранному банку данных, индексам удерживания веществ и величинам относительных сигналов селективных и универсального детекторов.
Известен способ идентификации веществ в сложных смесях [1] путем измерения в процессе хроматографического анализа времени удерживания или объема удерживания компонента смеси, по которому можно его идентифицировать.
Недостатком этого способа является существенное влияние на характеристику удерживания веществ конкретных условий хроматографического анализа, в результате чего невозможно получить воспроизводимый банк хроматографических данных.
Известен также хроматографический способ идентификации веществ в сложных смесях [2] путем разделения смеси в хроматографической колонке, в которую введены вещества-реперы с регистрацией на выходе из колонки детектором сигналов (пиков) веществ и реперов, и расчета относительного времени или объема удерживания. Индекс удерживания определяется путем логарифмической интерполяции между объемами удерживания двух н-алканов, между которыми находится значение объема удерживания компонента. Индексы удерживания также зависят от температурного режима разделения смеси, кроме того, на них оказывают влияние особо полярные неподвижные фазы и активные твердые носители.
Суть способа состоит в том, что вначале находят условия разделения всех компонентов смеси на кварцевой капиллярной колонке с программированием температуры от То до Тмах до полного разделения компонентов, а затем разделенные компоненты с добавлением в смесь сетки реперных веществ, в качестве которых могут использоваться алканы, направляют во вторую колонку, которая находится при постоянной температуре не ниже Тмах первой колонки с неподвижной фазой той же полярности, что и на разделительной колонке, и выполняет функцию идентификационной колонки. На второй колонке не изменяется порядок выхода компонентов, который был достигнут на первой колонке, но температурный режим - изотермический. Это позволяет определить индексы удерживания и собрать банк воспроизводимых хроматографических данных для последующей идентификации компонентов в сложных смесях.
Способ осуществляют в устройстве, которое содержит источник газа-носителя и последовательно установленные по потоку газа-носителя испаритель с пробовводом, три хроматографические кварцевые капиллярные колонки с неподвижной фазой одной полярности, причем первая колонка расположена в термостате с программированием температуры и соединена через тройник со второй колонкой и первым универсальным детектором УД-1 (пламенно-ионизационный детектор ДИП), вторая колонка расположена во втором по потоку газа-носителя термостате при постоянной температуре, на выходе ее расположен второй универсальный детектор УД-2.
Существующие в настоящее время газохроматографические методы определения содержания токсичных примесей позволяют лишь с определенной долей вероятности идентифицировать примесные соединения, при этом получаемая информация может быть недостаточно достоверной и корректной.
Наиболее близким техническим решением является способ хромато-масс-спектрометрии (3), основанный на сочетаниях капиллярной газовой хроматографии и метода масс-спектрометрии - метода наиболее селективного и чувствительного по детектированию и структурной идентификации. Идентификация вещества проводится по двум аналитическим параметрам:
- времени удерживания и
- масс-спектру.
Разделение проводилось по кварцевой капиллярной колонке HP-FFAP 50 м 0,32 мл, толщина пленки неподвижной фазы 0,52 мкм, температура 60°С (4 мин), объем вводимой пробы 1 мкл. Внутренний стандарт (Вс)-циклогексана - 0,1% в этаноле. К 1 мл пробы добавляли 30 мкл ВС.
Недостатком вышеописанных способов являются ограничения, связанные с близостью времен хроматографического удерживания некоторых присутствующих в нем примесей, а также использование специального и дорогостоящего оборудования.
Задача решается за счет обеспечения разделения примесей, присутствующих в спирте-сырце на колонках с различной фазой, что сэкономит время составления таблиц удерживания разнообразных примесных соединений.
Сущность способа заключается в построении селективных ионных масс-хроматограмм по отдельным ионам, характеризующим определяемые примеси, интерпретацией масс-спектров на основе закономерностей фрагментации молекулярных ионов - представителей различных классов органических соединений с учетом спектроструктурных корреляций, анализе относительных интенсивностей диагностических ионов, применении в качестве маркеров подлинности этилового спирта - алкилпиразинов, применении в качестве маркеров подлинности синтетического спирта - пропеналя (акролеина).
Пример осуществления способа.
На Фиг.1 изображена схема образования диагностических фрагментов соединений алкилпиразинов (181-184) на примере 2,6-диметил-пиразина (183).
На Фиг.2 изображены масс-спектры алкилпиразинов (181-184).
Осуществление технического решения происходит следующим образом: при определении состава примесей в качестве базовой использовалась система хроматограф-масс-спектрометр-ЭВМ, состоящая из газового хроматографа HP 5890А с масс-селективным детектором HP 5972A и системы обработки данных HP ChemStation, содержащей библиотеку 138 тысяч масс-спектров индивидуальных соединений. В ряде случаев использовался хромато-масс-спектрометр HP 5989A. Для получения максимально достоверной и корректной информации о примесях спирта этилового анализ компонентов проводили как минимум на двух капиллярных кварцевых колонках различной полярности из числа НР-1, HP-5, Carbowax 20M, FFAP, INNOWax, Ultra-2. При этом наилучшее разделение исследуемых примесей достигалось на кварцевой колонке с нанесенной жидкой фазой HP-INNOWax с внутренним диаметром 0,53 мм и длиной 30 м с эффективностью по метиловому эфиру ундекановой кислоты не менее 1530 чтт/м и 1360 этт/м, производства фирмы Hewlett Packard. Масс-спектры хроматографических пиков получали при энергии электронов 70 эВ, сканирование масс-спектров от 29 до 300 дальтонов проводили со скоростью 1 спектр/с. Идентификация хроматографических пиков включала анализ с использованием библиотечного поиска масс-спектров (индекс сходства с табличными спектрами превышал 80%), построение селективных ионных масс-хроматограмм по отдельным ионам, характерным для определяемых примесей, интерпретацию масс-спектров на основе закономерностей фрагментации молекулярных ионов представителей различных классов органических соединений с учетом спектроструктурных корреляций.
Условия выполнения анализа на хроматографе Hewlett Packard модель 6890:
Температура испарителя, °С - 210
Температура детектора, °С - 240
Начальная температура термостата, °С - 35
Начальный изотермический участок, мин - 5
Скорость подъема температуры термостата, град./мин - 5
Промежуточная температура термостата, °С - 130
Скорость подъема температуры термостата, град./мин - 20
Конечная температура термостата, °С - 235
Конечный изотермический участок, мин - 7.5
Скорость потока газа-носителя (гелий) через колонку, см3/мин - 4.0
Общий поток гелия, см3/мин - 45.9
Делитель потока гелия в отношении 1:10
Колонка работает в режиме поддержания постоянства потока гелия
Скорость потока водорода, см3/мин - 30.0
Скорость потока воздуха, см3/мин - 450.0
Объем вводимой пробы, мм3 - 1.0
Продолжительность анализа, мин - 36.75
Идентификация индивидуальных соединений, содержащихся в этиловом спирте в микроколичествах, представляет собой нетривиальную задачу, особенно при определении изомерного строения исследуемого соединения.
Поэтому разработанная хромато-масс-спектрометрическая методика идентификации органических микропримесей включает в себя в том числе и интерпретацию масс-спектров на основе знаний особенностей фрагментации представителей каждого класса органических соединений с учетом спектроструктурных корреляций. Для повышения вероятности правильной идентификации участие оператора-аналитика обязательно, особенно при определении изомерных веществ, а также примесей, содержащихся в микроколичествах. В таких случаях идентификация лишь на основании библиотечных данных не может быть однозначной. Например, не все изомерные вещества дают различающиеся масс-спектры. Из этого следует, что идентификацию вещества необходимо проводить как минимум в два этапа. На первом этапе образцы должны подвергаться хромато-масс-спектрометрическому анализу с предварительной идентификацией исследуемых соединений по масс-спектрам с использованием стандартных библиотек, а на втором проводится идентификация по индивидуальным соединениям, сравниваются не только масс-спектры, но и времена удерживания исследуемого соединения и вещества сравнения. Таким образом, в разработанной методике идентификация пиков на хроматограмме проводится путем анализа полученных масс-спектров и времен удерживания веществ (возможно также применение метода добавок индивидуальных соединений). Помимо зависимости времени выхода из хроматографической колонки от степени разветвленности углеродной цепи при интерпретации масс-спектров применяется концепция локализации заряда и радикала, «азотное» правило, «четно-электронное» правило, принцип наименьших структурных изменений при фрагментации. Далее приведен пример, который можно использовать в качестве теста при интерпретации масс-спектров соединений алкилпиразинов.
В масс-спектрах соединений 181-184 (Фиг.2) зарегистрированы интенсивные пики молекулярных ионов М+ с массовыми значениями m/z: 94 (100%) - 181, 108 (96%) - 182, 108 (100%) - 183, 122 (76%) - 184. Анализ относительных интенсивностей пиков М+ и изотопных (М+1)+ позволил рассчитать содержание каждого элемента и установить брутто-формулы соединений алкилпиразинов: 181 - С5Н6N2; 182 - С6Н8N2; 183 - C6H8N2; 184 - С7Н10N2. Хорошо известно, что наличие ароматической системы кратных связей приводит к повышению стабильности М+. Поэтому следует предположить, что соединения 181-184-азотсодержащие ароматические производные (алкилпиразины, или -пиримидины, или -пиридазины).
Интерпретацию масс-спектров проводят исходя из концепции локализации заряда и радикала, «азотного» правила, «четно-электронного» правила, принципа наименьших структурных изменений при фрагментации.
Распад М+ и осколочных ионов всех четырех соединений протекает однотипно: диагностические ионы образуются при выбросе молекулы синильной кислоты (фрагменты А, Д) и ацетонитрила (фрагменты В, С) - Фиг.1. В спектрах соединений 181-183 пики ионов (M-HCN)+ - фрагменты А, Д - имеют большую интенсивность, чем ионов (М-СН3CN)+ - фрагменты В, С. Это говорит, во-первых, о том, что потеря синильной кислоты более выгодный процесс, а во-вторых, что гетероцикл расщепляется по одной из связей C(2)-N (М+-изомеризация - Фиг.1).
Анализ спектров изомеров 2,5- и 2,6-диметилпиразинов показывает, что относительные интенсивности однотипных фрагментов В - m/z 67 - хорошо отражают особенности строения: m/z 67 (10%) для 183 и m/z 67 (0.5%) для 182. Все просто: лимитирующей стадией процесса является раскрытие ароматического цикла по связи C(2)-N (фиг.1). Легко рассеять сомнения о принадлежности спектра 184 триметилпиразину или пропилпиразину. В случае моноалкилпиразина при длине углеводородной цепи, равной 3 С, нужно ожидать характерную перегруппировку через 6-членное переходное состояние с образованием осколочных ионов (М-С2H4)+. Отсутствие пиков ионов (M-C2H4)+ в спектре 184 свидетельствует в пользу структуры триметилпиразина.
Соединения 181-184 не могут быть алкилпиридазинами, поскольку для последних реализовывалось бы отщепление молекулы азота, также не могут быть алкилпиримидинами, поскольку для них характерен последовательный выброс 2-х молекул синильной кислоты.
Таким образом очевидно, что, используя диагностические ионы М+ (m/z 94, 108, 122, 136, ...), m/z 42 (H4С2N)+, m/z 39 (С3Н3)+ в качестве характеристических, определение присутствия алкилпиразинов в неизвестных пробах методом хромато-масс-спектрометрии в режиме селективного ионного детектирования особых сложностей не представляет.
Без предварительной пробоподготовки метод хромато-масс-спектрометрии в режиме селективного ионного детектирования для представителей некоторых классов органических соединений позволяет добиться чувствительности определения порядка 1 мкг/дм3, а с помощью пробоподготовки ее можно повысить еще на один-два порядка. Такая величина чувствительности гарантирует уверенную идентификацию примесей-меток в этанолсодержащих жидкостях независимо от современных способов очистки этанола.
При анализе спиртов различного происхождения выявлены следующие закономерности (таблица 1):
для синтетического этанола характерно наличие ненасыщенных альдегидов, для пищевого - насыщенных; в синтетическом спирте содержатся диалкилкетоны с четным числом атомов углерода, в пищевом - зафиксировано лишь 9 кетонов, причем в виноградном спирте они отсутствуют; в синтетическом спирте идентифицированы только муравьиная и уксусная кислоты, пищевой содержит гомологический ряд карбоновых кислот, причем концентрация их в виноградном спирте больше, чем в зерновом; в синтетическом спирте имеются высшие спирты с четным числом атомов углерода, с гидроксильной группой у второго и третьего атомов углерода, углеводороды, в том числе и ароматические, и практически отсутствуют эфиры карбоновых кислот, в пищевом спирте зафиксированы неразветвленные спирты и этиловые эфиры стеариновой, дикарбоновых и фенилкарбоновых кислот, причем изоамиловые эфиры обнаружены в виноградном спирте, а изобутиловые - в зерновом; из полифункциональных соединений в виноградном этаноле идентифицированы алкилфенолы, гераниол, замещенные ацетали, пировиноградная кислота, замещенные фурфуролы, в зерновом - азот- и серусодержащие соединения и алкоксиалкилфенолы.
Таким образом, разработанный способ позволяет однозначно установить происхождение спирта этилового и идентифицировать сырье, из которого произведен этанол, причем получена чувствительность более чем на два порядка выше, чем при измерении полного ионного тока, что составляет величину, не превышающую 1 мкг/дм3.
Микропримеси, характеризующие вид сырья, из которого изготовлен этиловый спирт
Источники информации
1. Патент RU 2035735, Лейбниц Э. Руководство по газовой хроматографии. М., Мир, 1968 г., с.333-337.
2. Патент RU 2035735, Лейбниц Э. М., Мир, 1988 г., с.241-243, RU 2150699, G01N 33/14.
3. Н.А.Вязьмина, С.А.Савчук, Геохи Ран. Партнеры и конкуренты, №1, 2005 г., стр.32.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОПРЕДЕЛЕНИЯ ФОСФОРОРГАНИЧЕСКИХ ВЕЩЕСТВ | 2006 |
|
RU2313086C2 |
| СПОСОБ ИДЕНТИФИКАЦИИ ПРИМЕСИ ИССЛЕДУЕМОГО ВЕЩЕСТВА, РОДСТВЕННОЙ ЕГО ОСНОВНОМУ КОМПОНЕНТУ | 2017 |
|
RU2669266C1 |
| СПОСОБ ОБНАРУЖЕНИЯ КОМПЛЕКСА КСЕНОБИОТИКОВ В БИОЛОГИЧЕСКОЙ ЖИДКОСТИ ПРИ ДОПИНГОВОМ КОНТРОЛЕ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2011 |
|
RU2473079C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ДЕЛЬТАМЕТРИНА И ЛЯМБДА-ЦИГАЛОТРИНА В БИОЛОГИЧЕСКОМ МАТЕРИАЛЕ | 2012 |
|
RU2478962C1 |
| СПОСОБ ИДЕНТИФИКАЦИИ НАРКОТИЧЕСКИХ И ПСИХОАКТИВНЫХ ВЕЩЕСТВ В БИОЛОГИЧЕСКИХ ЖИДКОСТЯХ | 2009 |
|
RU2390771C1 |
| СПОСОБ ВЫЯВЛЕНИЯ И ОПРЕДЕЛЕНИЯ ПРОИСХОЖДЕНИЯ НЕИЗВЕСТНЫХ ВЕЩЕСТВ В СПИРТНЫХ НАПИТКАХ | 2009 |
|
RU2392616C1 |
| Способ определения фурана и метилфурана в атмосферном воздухе методом капиллярной газовой хроматографии с масс-селективным детектором при использовании метода низкотемпературного концентрирования | 2022 |
|
RU2789634C1 |
| СПОСОБ ИДЕНТИФИКАЦИИ ФОСФОРОРГАНИЧЕСКИХ СОЕДИНЕНИЙ МЕТОДОМ ХРОМАТО-МАСС-СПЕКТРОМЕТРИИ С ЦИЛИНДРИЧЕСКОЙ ИОННОЙ ЛОВУШКОЙ | 2020 |
|
RU2741955C1 |
| ХИМИЧЕСКИЙ МАРКЕР И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2011 |
|
RU2497860C2 |
| Способ определения 2-амино-4,6-динитрофенола в биологическом материале | 2024 |
|
RU2835192C1 |

Изобретение относится к области анализа сложных смесей. Способ заключается в построении селективных ионных масс-хроматограмм по отдельным ионам, характеризующим определенные примеси, интерпретации масс-спектров на основе закономерностей фрагментации молекулярных ионов-представителей различных классов органических соединений с учетом спектро-структурных корреляций, анализе относительных интенсивностей диагностических ионов, применении в качестве маркеров подлинности этилового спирта - алкилпиразинов, а в качестве маркеров подлинности синтетического спирта - пропеналя (акролеина). Технический результат - повышение чувствительности идентификации. 1 табл., 2 ил.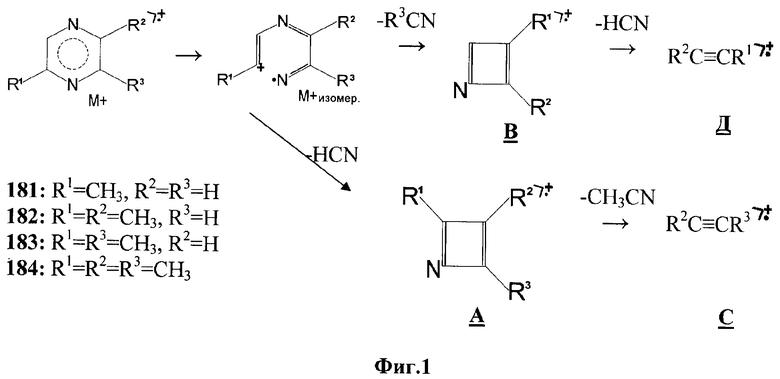
Способ идентификации подлинности этилового спирта и этанолсодержащих жидкостей, характеризующийся построением селективных ионных масс-хроматограмм по отдельным ионам, характеризующим определяемые примеси, интерпретацией масс-спектров на основе закономерностей фрагментации молекулярных ионов-представителей различных классов органических соединений с учетом спектроструктурных корреляций, анализом относительных интенсивностей диагностических ионов, применением в качестве маркеров подлинности этилового спирта - алкилпиразинов, применением в качестве подлинности синтетического спирта - пропеналя (акролеина).
| СПОСОБ ИДЕНТИФИКАЦИИ ПОДЛИННОСТИ СПИРТОСОДЕРЖАЩИХ ЖИДКОСТЕЙ | 1999 |
|
RU2150699C1 |
| СПОСОБ ЭКСПРЕСС КОНТРОЛЯ КАЧЕСТВА СПИРТОВОДОЧНЫХ ИЗДЕЛИЙ ДЛЯ ИХ ИДЕНТИФИКАЦИИ | 1999 |
|
RU2142630C1 |
| ПОБУДИТЕЛЬ РАСХОДА ДЛЯ ХРОМАТОГРАФИИ | 1992 |
|
RU2035736C1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |

