Настоящее изобретение касается способа гетерогенного каталитического частичного прямого окисления пропана и/или изо-бутана, по меньшей мере, до одного из таких целевых продуктов, как акриловая кислота, метакриловая кислота, при котором на стадию реакции, которая, за исключением входа для исходной смеси реакционного газа, в случае необходимости, других входов для вспомогательных газов, а также выхода для смеси продукт-газа, герметична для газа, подают пропан и/или изо-бутан, молекулярный кислород и исходную смесь реакционного газа, содержащую, по меньшей мере, один инертный газ-разбавитель, с входным давлением Р1, на указанной стадии реакции путем подачи исходной смеси реакционного газа при повышенной температуре через находящийся в твердом агрегатном состоянии катализатор, содержащийся в исходной смеси реакционного газа пропан и/или изо-бутан, прямым способом частично окисляют и/или аммоокисляют, по меньшей мере, до одного целевого продукта, и смесь реакционного газа в виде содержащей, по меньшей мере, один целевой продукт смеси продукт-газа, выводят из стадии реакции при выходном давлении Р2 и при этом давлении Р2 подают на стадию переработки, которая, за исключением входа для смеси продукт-газа, в случае необходимости, других входов для вспомогательных газов, а также выхода для смеси остаточного продукт-газа, герметична для газа, на стадии переработки из смеси продукт-газа стадии реакции содержащийся в ней целевой продукт грубо отделяют в жидкую фазу, а оставшуюся при этом смесь остаточного продукт-газа, содержащую не только пропан и/или изо-бутан, а также, в случае необходимости, пропен и/или изо-бутен, выводят из стадии переработки при выходном давлении Р3, причем Р3<Р1, и содержащийся в смеси остаточного продукт-газа пропан и/или изо-бутан возвращают на стадию реакции.
Акриловая кислота и метакриловая кислота являются важными промежуточными продуктами, например, для получения полимеров.
Их получение путем гетерогенного каталитического частичного прямого окисления пропана и/или изо-бутана в одну стадию реакции известно (см., например, ЕР-А 529853, ЕР-А 603836, ЕР-А 608838, ЕР-А 895809, DE-A 19835247, DE-А 10051419, DE-A 10122027, ЕР-А 1254707, ЕР-А 1254709, ЕР-А 1192987, ЕР-А 1090684, DE-A 10254279 и цитированную в этих документах литературу).
В качестве окислителя обычно используют молекулярный кислород, который можно добавлять в исходную смесь реакционного газа, например, в чистой форме или в смеси с в основном инертными к частичному окислению газами (например, N2 в воздухе). Инертные газы-разбавители, такие как N2, Н2О, СО, CO2, Не и/или Ar и т.п., поглощают реакционное тепло и поддерживают реакционную смесь за пределами области взрыва. В основном под инертными газами-разбавителями в данной заявке понимают такие газы, которые при одноразовом прохождении смеси реакционного газа через стадию частичного окисления превращаются на менее чем 5 моль.%, предпочтительно на менее чем 3 моль.% и особенно предпочтительно на менее чем 2 моль.%. В качестве катализаторов, как правило, используют находящиеся в твердом агрегатном состоянии многокомпонентные оксиды. Стадия реакции может быть осуществлена на находящихся в твердом агрегатном состоянии многокомпонентных оксидах, а также в стационарном, псевдоожиженном или подвижном (кипящем) слое катализатора.
Рабочее давление на стадии реакции согласно уровню техники может быть как ниже нормального давления (=1 бар), так и выше 1 бара (см., например, DE-A 19835247, ЕР-А 895809 и DE-A 10261186). С целью преодоления сопротивлений потоков на стадии реакции, оно, как правило, составляет чуть больше нормального давления.
Недостатком гетерогенного каталитического частичного прямого окисления пропана и/или изо-бутана до получения, по меньшей мере, одного из таких целевых продуктов, как акриловая кислота, метакриловая кислота, является сравнительно выраженная реакционная инертность пропана и изо-бутана. Она является причиной того, что при одноразовом прохождении смеси реакционного газа через соответствующую стадию реакции даже при повышенных температурах, как правило, достигают лишь частичного превращения пропана и/или изо-бутана.
Поэтому задача гетерогенного каталитического частичного прямого окисления пропана и/или изо-бутана до получения, по меньшей мере, одного из таких целевых продуктов, как акриловая кислота, метакриловая кислота состоит в том, чтобы при максимально низких температурах и одноразовом прохождении смеси реакционного газа через стадию реакции достичь максимально высокого превращения пропана и/или изо-бутана при одновременно максимально высокой селективности получения целевого продукта, то есть достичь максимально высокого показателя объема-времени целевого продукта на выходе при максимально низком потреблении энергии.
Следующее требование для экономичного осуществления гетерогенного каталитического частичного прямого окисления пропана и/или изо-бутана до получения, по меньшей мере, одного из желаемых целевых продуктов состоит в том, чтобы обеспечить возвращение содержащегося в смеси продукт-газа непрореагировавшего пропана и/или изо-бутана на стадию реакции. Для этого в уровне техники предложены следующие решения.
Для получения акриловой кислоты путем гетерогенного каталитического частичного прямого окисления пропана из смеси продукт-газа DE-A 10119933 рекомендует переводить содержащуюся в этой смеси акриловую кислоту путем абсорбции в жидкий абсорбент, а полученную при этом жидкую смесь из абсорбента и акриловой кислоты известным способом ректификативно, экстрактивно и/или кристализационно перерабатывать до получения чистой акриловой кислоты или проводить грубое отделение акриловой кислоты из смеси продукт-газа в жидкую фазу путем фракционной конденсации, как, например, описано в DE-A 19924532, и полученный при этом водный конденсат акриловой кислоты очищать, например, фракционированной кристаллизацией.
Относительно смеси остаточного продукт-газа, оставшегося при таком грубом отделении акриловой кислоты в жидкую фазу, содержащего непрореагировавший пропан, DE-A 10119933 предлагает выделять пропан из смеси остаточного продукт-газа и возвращать его на частичное прямое окисление до акриловой кислоты. В качестве методов выделения используют при этом, например, метод фракционной ректификации под давлением или экстракции с помощью гидрофобного органического растворителя (который способен поглощать пропан) и последующую десорбцию и/или стриппинг воздухом с целью высвобождения пропана.
Точно также при частичном прямом окислении алканов, таких как пропан, ЕР-А 1193240 рекомендует выделять содержащийся в смеси остаточного продукт-газа непрореагировавший алкан (как и в данной публикации уровня техники предпочтительно вместе с образовавшимся в качестве побочного продукта алкеном) из этой смеси (например, абсорбцией или адсорбцией) и повторно использовать его в частичном окислении.
Недостатком при повторном использовании непрореагировавшего алкана и, в случае необходимости, образовавшегося в качестве побочного продукта алкена на стадии реакции согласно рекомендациям документов уровня техники является тот факт, что выделение алкана и, в случае необходимости, алкена из смеси остаточного продукт-газа, в которой непрореагировавший алкан обычно содержится в сильно разбавленном состоянии, является сравнительно дорогостоящим и связано с высокими потерями давления. Последнее свидетельствует о нежелательности использования высоких давлений на стадии реакции, так как при повторном использовании выделенного пропана его необходимо сжимать до таких давлений.
Кроме того, недостаток состоит в том, что компоненты, содержащиеся в смеси остаточного продукт-газа, положительно влияющие на гетерогенное каталитическое частичное прямое окисление, (например, водяной пар, который, как правило, способствует повышению активности и селективности каталитической активной массы, или остаточный кислород, который ненужно сжимать до высоких давлений) не возвращают на стадию реакции (а выпускают) и, в случае необходимости, постоянно подают новые.
Другой причиной предложения возвращать содержащийся в смеси остаточного продукт-газа непрореагировавший углеводород на стадию реакции отдельно, то есть после отделения, может, кроме прочего, быть соображение поддерживать минимальным возвращаемое количество газа (и вместе с ним количества исходной смеси реакционного газа), чтобы таким образом минимизировать производительность подачи и мощность компрессора (циркуляционный газ необходимо сжимать до получения входного давления смеси реакционного газа перед входом на стадию реакции, поскольку этот газ при прохождении стадии реакции, стадии переработки и отделения из смеси остаточного продукт-газа претерпевает потерю давления, которое необходимо для преодоления сопротивления подачи потока, и прежний уровень которого нужно снова достичь), а также необходимых объемов. При этом дальнейшая постановка цели заключается в минимизации потерь используемых веществ.
Таким образом, задача данного изобретения состоит в разработке способа гетерогенного каталитического частичного прямого окисления пропана и/или изо-бутана до, по меньшей мере, одного из таких целевых продуктов, как акриловая кислота, метакриловая кислота, который позволяет выгодным образом минимизировать мощность компрессора и потери используемых веществ и в то же время оптимизировать показатели объема-времени на выходе при максимально низком потреблении энергии.
Соответственно этому был разработан способ гетерогенного каталитического частичного прямого окисления пропана и/или изо-бутана до, по меньшей мере, одного из таких целевых продуктов, как акриловая кислота, метакриловая кислота, при котором на стадию реакции, которая, за исключением входа для исходной смеси реакционного газа, в случае необходимости, других входов для вспомогательных газов, а также выхода для смеси продукт-газа, герметична для газа, подают пропан и/или изо-бутан, молекулярный кислород и исходную смесь реакционного газа, содержащую, по меньшей мере, один инертный газ-разбавитель, при входном давлении Р1, на указанной стадии реакции путем подачи исходной смеси реакционного газа при повышенной температуре через находящийся в твердом агрегатном состоянии катализатор, содержащийся в исходной смеси реакционного газа пропан и/или изо-бутан, прямым способом частично окисляют до, по меньшей мере, одного целевого продукта, и смесь реакционного газа в виде содержащей, по меньшей мере, один целевой продукт смеси продукт-газа, выводят из стадии реакции при выходном давлении Р2 и при том же давлении Р2, подают на стадию переработки, которая за исключением входа для смеси продукт-газа, в случае необходимости, других входов для вспомогательных газов, а также выхода для смеси остаточного продукт-газа, герметична для газа, на стадии переработки из смеси продукт-газа стадии реакции содержащийся в ней целевой продукт грубо отделяют в жидкую фазу, а оставшуюся при этом смесь остаточного продукт-газа, содержащую не только пропан и/или изо-бутан, а также, в случае необходимости, пропен и/или изо-бутен, выводят из стадии переработки при выходном давлении Р3, причем Р3<Р1, содержащийся в смеси остаточного продукт-газа пропан и/или изо-бутан возвращают на стадию реакции, который отличается тем, что Р1 выбирают таким образом, что Р3≥1,5 бар, а смесь остаточного продукт-газа разделяют на две части одинакового состава, при этом одну часть выгружают, а другую часть отводят как циркуляционный газ и в качестве сжатого до исходного давления Р1 компонента исходной смеси реакционного газа повторно подают на стадию реакции. При этом способ согласно изобретению отличается от описанного в ЕР-А 495504 способа тем, что он на и/или после стадии переработки перед выгрузкой не включает стадию каталитического окисления окиси углерода. Кроме того, способ согласно изобретению не требует промывания смеси остаточного продукт-газа от диоксида углерода.
Если на стадии реакции в качестве побочного продукта образуется пропен и/или изо-бутен, то эти соединения на стадии переработки обычно сопровождают пропан и/или изо-бутан и в составе циркуляционного газа совместно возвращаются на стадию реакции.
Все значения давлений в этой заявке, если не указано ничего другого, являются абсолютными.
Обычно смесь остаточного продукт-газа содержит, по меньшей мере, 2 об.%, или, по меньшей мере, 5 об.%, в большинстве случаев, по меньшей мере, 10 об.% компонентов, отличных от пропана и/или изо-бутана, а также пропена и/или изо-бутена, кроме того, в случае необходимости, от кислорода, например, СО, CO2, Н2O и/или N2 и т.д. (как правило, речь при этом идет о компонентах, содержащихся в смеси продукт-газа, более легкокипящих (при нормальном давлении), чем целевой продукт).
В то время как в способе согласно изобретению подлежащее частичному окислению органическое исходное соединение (то есть пропан и/или изо-бутан) на практике часто хранят в жидком состоянии, которое, однако, при нормальном давлении и комнатной температуре переходит в газообразное состояние, достаточно, как правило, простого испарения, чтобы привести органическое исходное соединение к исходному давлению стадии реакции. Используемый, в случае необходимости, в качестве инертного газа-разбавителя водяной пар, имеется из различных источников при достаточном давлении, превышающем атмосферное.
Хотя сказанное выше, как правило, не касается источника кислорода (например, воздуха или обедненного азотом воздуха), в случае необходимости, прочих инертных газов-разбавителей и, прежде всего, содержащего пропан циркуляционного газа (обычно он имеет исходное давление Р1 стадии реакции, за вычетом потери давления при прохождении стадии реакции и стадии переработки, а также разделения на две части; его возвращение на стадию реакции при осуществлении способа согласно изобретению происходит обычно через свободные вводные патрубки без дополнительных высоких потерь давления).
Поэтому на практике следует, по меньшей мере, одну часть (хотя бы циркуляционный газ) компонентов исходной смеси реакционного газа сжимать с помощью компрессора с низкого выходного давления до более высокого конечного давления (исходного давления Р1 на стадии реакции).
При этом сжатие этих компонентов (например, источника кислорода - воздуха, и циркуляционного газа) осуществляют в отдельных компрессорах или в одном компрессоре.
Для осуществления такого сжатия указанных выше газов могут быть использованы компрессоры различного вида, например, компрессор вытеснения (такой как поршневой компрессор, винтовой компрессор и роторно-поршневой компрессор), лопаточный компрессор (например, турбокомпрессор, центробежный компрессор, осевой компрессор и радиальный компрессор), а также струйный компрессор. Согласно изобретению предпочтительно используют радиальные компрессоры, описанные, например, в DE-A 10259023.
Кроме того, согласно изобретению действуют предпочтительно следующим образом: поступающие из отдельных трубопроводов количества исходных смесей реакционного газа разного происхождения, которые в основном приведены к исходному давлению стадии реакции, перемешивают с помощью неподвижной мешалки (устройства с насадками, обладающие повышенной смешивающей способностью по сравнению с порожними трубами), и после этого подают на стадию реакции, в случае необходимости, нагрев до исходной температуры.
Порядок загрузки отдельных газов в подведенный к неподвижной мешалке трубопровод часто устанавливают таким образом, чтобы избежать получения взрывчатой смеси (в случае частичного окисления согласно изобретению этот порядок загрузки может, например, выглядеть так: сначала циркуляционный газ и/или пар, затем воздух и после этого органическое исходное соединение). Естественно содержание водяного пара в исходной смеси реакционного газа можно устанавливать добавлением в нагретую в основном до реакционной температуры путем непрямого теплообмена с потоком продукт-газа предварительную исходную смесь реакционного газа мелкораспыленные капли воды, которые при поглощении тепла испаряются, при этом образуется исходная смесь реакционного газа. Альтернативно изначально подогретую предварительную исходную смесь реакционного газа пропускают через газонасытитель (газовую смесь и воду в параллельном или встречном потоке пропускают над большой поверхностью).
Таким образом, выбранное согласно изобретению выходное давление Р3 в основном влияет на мощность компрессора направленную на сжатие циркуляционного газа и источника кислорода.
На практике давление Р3, при котором смесь остаточного продукт-газа выходит со стадии переработки при осуществлении способа согласно изобретению, как правило, не превышает 30 или 25 бар, часто не превышает 20 бар. Согласно изобретению оптимальное выходное давление Р3 составляет ≥1,5 бар и ≤10 бар, предпочтительно ≥2 бар и ≤8 бар, чаще ≥3 бар и ≤6 бар или ≤5 бар (например, 4 бара).
То есть отличительным признаком способа согласно изобретению является возможность работать при повышенном давлении как на стадии реакции, так и на стадии переработки.
Такой принцип работы является выгодным по указанным ниже причинам:
- неожиданно установили, что гетерогенное каталитическое частичное прямое окисление пропана и/или изо-бутана при повышенном давлении при в остальном одинаковых условия реакции, в расчете на одну стадию, приводит к увеличению объемов превращений, не уменьшая при этом селективности получения целевого продукта;
- проведение стадии переработки при повышенном давлении позволяет подавать увеличенные количества циркуляционного газа в сравнительно небольших необходимых для этого объемах и при сравнительно незначительных потерях давления, поскольку как объем подачи заданного количества газа, так и связанная с его подачей потеря давления при увеличении давления, как правило, уменьшаются; последнее понижает мощность компрессора, необходимую для сжатия циркуляционного газа до входного давления Р1 стадии реакции; одновременно увеличение количества циркуляционного газа по сравнению с количеством на выгрузке уменьшает потери содержащегося в выгрузке непрореагировавшего пропана и/или изо-бутана;
- повторное использование пропана без предварительного выделения его из смеси остаточного продукт-газа предотвращает связанную с таким выделением необходимо наступающую потерю давления и обеспечивает одновременное и энергетически выгодное повторное использование других компонентов, которые, в случае необходимости, входят в состав смеси остаточного продукт-газа, таких, например, как водяной пар и O2.
Таким образом, согласно изобретению посредством повышения давления сравнительно несложными способами удается осуществить все выгодные этапы способа в соответствии с уровнем техники без необходимости проведения требующего больших затрат выделения непрореагировавшего алкана и, в случае необходимости, алкена из смеси остаточного продукт-газа (что дополнительно уменьшает возможность возникновения нежелательных факторов, таких как, например, полная невозможность повторного использования, или требующее больших энергетических затрат повторное использование водяного пара), и одновременно повышение давления обуславливает увеличение объемов превращения эдукта реакции при одноразовом прохождении через стадию реакции без существенного снижения селективности получения целевого продукта.
Понятие “стадия реакции” или “стадия переработки” в данном контексте означает, в частности, одно или несколько последовательно подключенных аппаратных устройств, которые, за исключением входа и выхода, а также, в случае необходимости, других входов для вспомогательных газов, герметичны для газа, что ограничивает потерю давления смеси газа при прохождении через такое аппаратное устройство или через последовательное подключение аппаратных устройств, той потерей, которая необходима смеси газа при преодолении сопротивлений потока.
Под таким аппаративным устройством (или последовательным подключением таких устройств) подразумевают, например, кожухотрубный реактор, реактор с псевдоожиженным слоем, реактор с кипящим слоем, последовательное подключение таких реакторов, абсорбционную колонну, ректификационную колонну, конденсационную колонну или последовательное подключение таких колонн или отдельных стадий резкого охлаждения. Разумеется реактор, подобный вышеописанным реакторам, при осуществлении способа согласно изобретению может иметь возможность при проведении способа согласно изобретению подачи в реактор, например, активатора катализатора, как, например, описано в WO 02/081421. В способе согласно изобретению понятие «вспомогательный газ» включает при последовательном подключении реакторов между реакторами добавлять инертный газ и/или кислород (например, воздух) или на стадии переработки, например, с целью ингибирования полимеризации подавать содержащий молекулярный кислород газ (например, воздух) совместно со смесью продукт-газа через стадию переработки. При этом потери давления при осуществлении способа согласно изобретению на стадии реакции составляют от 0,1 до 3 бар, часто от 0,3 до 1 или 0,5 бар, а на стадии переработки - от 0,5 до 3 бар, часто от 1 до 2 бар.
При осуществлении способа согласно изобретению при особо высоких давлениях потери давления могут быть как на стадии реакции, так и на стадии переработки значительно ниже и составлять, например, до 0,05 бар и ниже.
Входное давление Р1 на входе на стадию реакции в зависимости от дальнейшей стадии переработки при осуществлении способа согласно изобретению превышает выходное давление Р3 со стадии переработки на 0,5 или на 1-4 бар, чаще на 1,5-3,5 бар, еще чаще на 2-3 бар.
При осуществлении способа согласно изобретению в области особо высоких давлений давление Р1 на входе на стадию реакции, как правило, превышает давление
Р3 на выходе со стадии переработки на не более 0,5 бар (например, от 0,1 до 0,01 бар).
Поэтому типичные давления Р1 на входе на стадию реакции составляют от 2,5 до 25 бар. Как правило, давление Р1 на входе на стадию реакции составляет от 3 до 10 бар, предпочтительно, от 4 до 8 бар.
Типичные давления Р2 на входе на стадию переработки составляют от 3 до 25 бар, часто от 3 до 20 бар, или от 3 до 15 бар, в особенности от 3 до 8 бар.
Регулирование соотношений давлений при осуществлении способа согласно изобретению возможно осуществлять, например, с помощью дросселирующего устройства на выводе для выгружаемой части смеси остаточного продукт-газа. Посредством последовательно подсоединенного вместо дросселирующего устройства расширителя (инверсного компрессора, через который осуществляют выгрузку) в способе согласно изобретению дополнительно к описанным выше преимуществам при выгрузке части смеси остаточного продукт-газа путем его контролированной декомпрессии до атмосферного давления часть мощности компрессора, необходимой для сжатия количества смеси остаточного продукт-газа и/или источника кислорода (например, воздуха) до давления на входе Р1, может быть регенерирована.
Количественное соотношение между частью смеси остаточного продукт-газа, которую в способе согласно изобретению повторно используют как циркуляционный газ, и частью смеси остаточного продукт-газа, которую выгружают, в основном зависит от состава исходной смеси реакционного газа. Как правило, V составляет ≥0,5 или ≥1, чаще ≥1,5, предпочтительно ≥2, особенно предпочтительно ≥3. Разумеется в способе согласно изобретению V может также составлять ≥8 или ≥10. Обычно в способе согласно изобретению V составляет ≤30, часто ≤25, чаще ≤20. В большинстве случаев V ≤15 или ≤10, предпочтительно от 2 до 8.
В остальном способ согласно изобретению может быть осуществлен так же, как и известные из уровня техники способы гетерогенного каталитического частичного окисления пропана и/или изо-бутана до, по меньшей мере, одного из целевых продуктов.
Источником молекулярного кислорода, необходимого для осуществления способа в рамках данного изобретения, может быть как воздух, так и обедненный молекулярным азотом воздух (например, ≥90 об.% - O2, ≤10 об.% - N2), a также чистый молекулярный кислород или смеси из молекулярного кислорода и других инертных газов.
В качестве катализаторов при осуществлении способа согласно изобретению используют все катализаторы, рекомендованные в уровне техники для гетерогенного каталитического частичного прямого окисления пропана и/или изо-бутана до получения, по меньшей мере, одного из целевых продуктов.
Такими катализаторами являются, например, катализаторы, описанные в публикациях JP-A 3-170445, ЕР-А 609122 и ЕР-А 747349.
Согласно изобретению существенным является тот факт, что в основном все катализаторы могут быть использованы для каждой из возможных согласно изобретению реакций гетерогенного каталитического частичного прямого окисления.
Под активными массами таких катализаторов, как правило, подразумевают многокомпонентные оксиды, чаще всего полиметаллические оксиды.
Пригодными для осуществления способа согласно изобретению многокомпонентными оксидами являются в особенности полиметаллические оксиды, описанные в публикациях ЕР-А 608838, ЕР-А 529853, DE-A 10254279, DE-A 19835247, ЕР-А 895809, JP-A 7-232071, JP-A 11-169716, DE-A 10261186, ЕР-А 1192987, JP-A 10-57813, JP-A 2000-37623, JP-A 10-36311, WO 00/29105, ЕР-А 767164, DE-A 10029338, JP-A 8-57319, JP-A 10-28862, JP-A 11-43314, JP-A 11-574719, WO 00/29106, JP-A 10-330343, JP-A 11-285637, JP-A 310539, JP-A 11-42434, JP-A 11-343261, JP-A 3423262, WO 99/03825, JP-A 7-53448, JP-A 2000-51693, JP-A 11-263745, DE-A 10046672, DE-A 10118814, DE-A 10119933, JP-A 2000/143244, ЕР-А 318295, ЕР-А 603836, DE-A 19832033, DE-A 19836359, ЕР-А 962253, DE-A 10119933, DE-A 10051419, DE-A 10046672, DE-A 10033121, DE-A 10145958, DE-A 10122027, ЕР-А 1193240 и цитированной в этих публикациях литературе.
Под используемыми активными массами загрузки катализатора в указанных выше случаях подразумевают в основном массы полиметаллических оксидов, которые содержат комбинацию элементов Мо, V, по меньшей мере, одного из элементов Те и Sb и по меньшей мере один из элементов группы, которая включает Mb, Та, W, Ti, Al, Zr, Cr, Mn, Ga, Fe, Ru, Co, Cs, Ca, Sr, Ba, Rh, Ni, Pd, Pt, La, Pb, Cu, Re, Ir, Y, Pr, Nd, Tb, Bi, B, Ce, Sn, Zn, Si, Na, Li, K, Mg, Ag, Au и In.
При этом из последней группы элементов комбинация предпочтительно содержит элементы Nb, Та, W и/или Ti, и особенно предпочтительно, элемент Nb.
Предпочтительно массы полиметаллических оксидов содержат указанные выше комбинации элементов стехиометрической формулы 1

где
М1 означает Те и/или Sb,
М2 означает, по меньшей мере, один из элементов из группы, которая включает Nb, Та, W, Ti, Al, Zr, Cs, Ca, Sr, Ba, Cr, Mn, Ga, Fe, Ru, Co, Rh, Ni, Pd, Pt, La, Bi, Pb, Cu, Re, Ir, Y, Pr, Nd, Tb, Ce, Sn, Zn, Si, Na, Li, K, Mg, Ag, Au и In,
b означает от 0,01 до 1,
с означает от >0 до 1 и
d означает от >0 до 1.
Согласно изобретению М1 предпочтительно означает Те и М2 означает Nb, Та, W и/или Ti. Преимущественно М2 означает Nb.
Стехиометрический коэффициент b предпочтительно составляет от 0,1 до 0,6. Соответственно предпочтительные области значения стехиометрического коэффициента с составляют от 0,01 до 1 или от 0,05 до 0,4, а d преимущественно составляет от 0,001 до 1 или от 0,01 до 0,6.
Согласно изобретению особенно выгодным является одновременное использование стехиометрических коэффициентов b, с и d в указанных выше предпочтительных областях значений.
Сказанное выше имеет силу особенно тогда, когда активная масса загрузки катализатора в отношении ее отличных от кислорода элементов состоит из одной из вышеназванных комбинаций элементов.
Такими являются в особенности активные массы полиметаллических оксидов общей стехиометрической формулы II
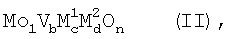
причем переменные имеют указанные в стехиометрической формуле 1 значения, а n означает число, которое определяется валентностью и числом отличных от кислорода элементов в формуле (II).
Предпочтительно массы полиметаллических оксидов содержат указанные вначале комбинации элементов стехиометрической формулы III

где
М4 означает, по меньшей мере, один из элементов группы, которая включает Те и Sb;
М5 означает, по меньшей мере, один из элементов группы, которая включает Mb, Ti, W, Та и Се;
М6 означает, по меньшей мере, один из элементов группы, которая включает Pb, Ni, Co, Bi, Pd, Cs, Ca, Sr, Ва, Ag, Pt, Cu, Au, Ga, Zn, Sn, In, Re, Ir, Sm, Sc, Y, Pr, Nd и Tb;
a' означает от 0,01 до 1;
b' означает от >0 до 1;
с' означает от >0 до 1;
d' означает от 0 до 0,5.
Предпочтительно а' означает от 0,05 до 0,6, особенно предпочтительно от 0,1 до 0,6 или 0,5.
Предпочтительно b' означает от 0,01 до 1, особенно предпочтительно от 0,01 или 0,1 до 0,5 или 0,4.
Предпочтительно с' означает от 0,01 до 1, особенно предпочтительно от 0,01 или 0,1 до 0,5 или 0,4.
Предпочтительно d' означает от 0,00005 или 0,0005 до 0,5, особенно предпочтительно от 0,001 до 0,5, чаще от 0,002 до 0,3 и еще чаще от 0,005 или 0,01 до 0,1.
М4 означает предпочтительно Те.
М5 на, по меньшей мере, 50 моль.%, преимущественно на, по меньшей мере, 75 моль.%, особенно преимущественно на, по меньшей мере, 100 моль.% от своего общего количества состоит предпочтительно из Nb.
М6 означает предпочтительно, по меньшей мере, один элемент из группы, которая включает Ni, Co, Bi, Pd, Ag, Au, Pb и Ga, особенно, по меньшей мере, один элемент из группы, которая включает Ni, Со, Pd и Bi.
Наиболее предпочтительно М5 на, по меньшей мере, 50 моль.%, или на по меньшей мере 75 моль.%, или на, по меньшей мере. 100 моль.% от своего общего количества состоит из Nb, a M6 означает, по меньшей мере, один элемент из группы, которая включает Ni, Со, Pd и Bi.
Согласно изобретению, чрезвычайно предпочтительно, М4 означает Те, М5 означает Nb и М6 означает, по меньшей мере, один элемент из группы, которая включает Ni, Со и Pd.
Сказанное выше имеет силу особенно тогда, когда активная масса загрузки катализатора в отношении ее отличных от кислорода элементов состоит из комбинаций элементов стехиометрической формулы (III). Такими являются в особенности активные массы полиметаллических оксидов общей стехиометрической формулы (IV)

причем переменные имеют указанные в стехиометрической формуле III значения, а n' означает число, которое определяется валентностью и числом отличных от кислорода элементов в формуле (IV).
Кроме того, для осуществления способа согласно изобретению предпочтительно используют такие активные массы полиметаллических оксидов, которые, с одной стороны, содержат одну из указанных выше комбинаций элементов (I), (III) или в отношении отличных от кислорода элементов состоят из такой комбинации и одновременно имеют рентгенодифрактограмму, которая показывает дифракционные рефлексы h и i, пики которых находятся при углах дифракции (2Θ) 22,2±0,5° (h) и 27,3±0,5° (i) (все показатели рентгенодифрактограммы в данном тексте относятся к рентгенодифрактограмме, полученной при использовании в качестве рентгеновского излучения Cu-Кα-излучения (дифрактометр Theta-Theta D-5000 фирмы Siemens, напряжение, приложенное к трубке: 40 кВ, сила тока: 40 миллиампер, апертурная диафрагма V20 (переменная), диафрагма рассеянного излучения V20 (переменная), диафрагма вторичного монохроматора (0,1 мм), диафрагма детектора (0,6 мм), интервал измерения (2Θ):0,02°, время измерения на каждой точке: 2,4 с, детектор: сцинтилляционный счетчик).
Полуширина этих дифракционных рефлексов может при этом быть очень маленькой или очень ярко выраженной.
Выгодными для способа согласно изобретению являются те из указанных выше активных масс полиметаллических оксидов, рентгенодифрактограмма которых, кроме дифракционных рефлексов h и i, показывает также дифракционный рефлекс k, пик которого составляет 28,2±0,5°(k).
Среди последних предпочтение согласно изобретению отдают таким, дифракционные рефлексы h которых в пределах рентгенодифрактограммы являются самыми интенсивными, а полуширина составляет максимум 0,5°, а особенное предпочтение отдают таким, полуширина дифракционного рефлекса i и дифракционного рефлекса k которых одновременно составляют соответственно ≤1°, а интенсивность Pk дифракционного рефлекса k и интенсивность Рi дифракционного рефлекса i удовлетворяют условию 0,2≤R≤0,85, преимущественно 0,3≤R≤0,85, предпочтительно 0,4≤R≤0,85, особенно предпочтительно 0,65≤R≤0,85, наиболее предпочтительно 0,67≤R≤0,75 и абсолютно предпочтительно R=0,70-0,75 или R=0,72, где R означает соотношение интенсивности, которое определяется формулой
R=Рi/(Рi+Рk).
Предпочтительно указанные выше рентгенодифрактограммы не имеют дифракционных рефлексов, пик которых находится при 2Θ 50±0,3°.
Определение упомянутой в данном тексте интенсивности дифракционного рефлекса в рентгенодифрактограмме указано в DE-A 19835247, DE-A 10122027, а также DE-A 10051419 и DE-A 10046672. То же самое касается определения полуширины рефлексов.
Наряду с показателями дифракционного рефлекса h, i и k указанные выше рентгенодифрактограммы предпочтительно используемых согласно изобретению активных масс полиметаллических оксидов имеют и другие дифракционные рефлексы, пики которых находятся при углах дифракции (2Θ):
9,0±0,4° (I)
6,7±0,4° (о) и
7,9±0,4° (р).
Кроме того, предпочтение отдают рентгенодифрактограммам, которые дополнительно имеют дифракционный рефлекс, пик которого находится при угле дифракции (2Θ) 45,2±0,4° (q).
Часто рентгенодифрактограмма имеет также дифракционные рефлексы 29,2±0,4° (m) и 35,4±0,4° (n).
Предпочтение отдают также указанным в формулах (I), (II), (III) и (IV) комбинациям элементов, которые существуют в виде чистой i-фазы. Если каталитически активная оксидная масса содержит еще и k-фазу, то ее рентгенодифрактограмма наряду с описанными выше имеет также другие дифракционные рефлексы, пики которых находятся при следующих углах дифракции (2Θ): 36,2±0,4° и 50±0,4° (понятия i- и k-фаза, используемые в этом тексте, разъяснены в DE-A 10122027 и DE-A 10119933).
Если дифракционному рефлексу П соответствует интенсивность 100, то согласно изобретению предпочтение отдают массам, в которых дифракционные рефлексы i, l, m, n, о, р, q на шкале интенсивности имеют следующие значения интенсивности:
i: от 5 до 95, чаще от 5 до 80, отчасти от 10 до 60;
l: от 1 до 30;
m: от 1 до 40;
n: от 1 до 40;
о: от 1 до 30;
р: от 1 до 30;
q: от 5 до 60.
Если рентгенодифрактограмма содержит упомянутые выше дополнительные дифракционные рефлексы, то их полуширина, как правило, ≤1°.
Удельная поверхность используемых согласно изобретению активных масс полиметаллических оксидов общей формулы (II) или (IV) или активных масс полиметаллических оксидов, которые содержат комбинации элементов общей формулы (I) или (III), составляет часто от 1 до 40 м2/г или от 10 до 30 м2/г (поверхность, определенная методом БЭТ, азот), прежде всего в том случае, когда ее рентгенодифрактограмма имеет описанный выше вид.
Получение таких активных масс полиметаллических оксидов описано в цитированных в уровне техники публикациях. К ним принадлежат, в частности, DE-A 10303526, DE-A 10261186, DE-A 10254279, DE-A 10254278, DE-A 10122027, DE-A 10119933, DE-A 10033121, ЕР-А 1192987, DE-A 10029338, JP-A 2000-143244, ЕР-А 962253, ЕР-А 895809, DE-A 19835247, WO 00/29105, WO 00/29106, ЕР-А 529853 и ЕР-А 608838 (во всех примерах осуществления двух последних публикаций в качестве метода сушки необходимо использовать метод распылительной сушки; например, при температуре на входе от 300 до 350°С и температуре на выходе от 100 до 150°С; встречным или параллельным потоком).
Описанные активные массы полиметаллических оксидов могут быть использованы в способе согласно изобретению как таковые (то есть в форме порошка) или превращены в соответствующие геометрические формы (например, как оболочковые катализаторы в DE-A 10051419 или геометрические способы в DE-A 10122027). Они выгодно подходят для получения акриловой кислоты из пропана, а также для получения метакриловой кислоты из изо-бутана.
Для осуществления способа согласно изобретению могут быть использованы все указанные катализаторы как в неразбавленном, так и в разбавленном инертными частицами и/или формованными изделиями (не содержащими активную массу) состоянии. Пригодным материалом для разбавления является, например, стеатит.
При этом геометрия формованных веществ для разбавления идентична геометрии формованных веществ катализатора.
Как описано в публикациях, касающихся катализаторов активных масс полиметаллических оксидов для осуществления способа согласно изобретению, этот способ может быть проведен как на неподвижном слое катализатора, так и на псевдоожиженном или кипящем слое катализатора. Используемые при этом входные давления стадии реакции были описаны выше.
Реакционные температуры, в частности, при использовании рекомендованных в этом документе катализаторов, могут составлять от 200 до 700°С, предпочтительно от 200 до 550°С или от 230 до 480°С, или от 300 до 440°С.
Нагрузка катализатора пропаном и/или изо-бутаном может составлять от 50 до 3000 нл/л (загрузки катал изатора)/час, или от 80 до 1500 нл/л/час, или от 100 до 1000 нл/л/час, или от 120 до 600 нл/л/час, или от 140 до 300 нл/л/час.
Нагрузка катализатора исходной смесью реакционного газа может при этом составлять от 100 до 10000 нл/л/час, или от 300 до 6000 нл/л/час, или от 300 до 2000 нл/л/час. Средняя длительность пребывания в катализаторе может составлять от 0,01 до 10 с, или от 0,1 до 10 с, или от 2 до 6 с.
В случае получения акриловой кислоты из пропана или метакриловой кислоты из изо-бутана исходная смесь реакционного газа может, например, содержать:
от 0,5 до 15, чаще от 1 до 7 об.% пропана или изо-бутана,
от 10 до 90, чаще от 20 до 50 об.% воздуха,
от 0 до 50 об.% водяного пара и
как остаточное количество циркуляционный газ.
Исходная смесь реакционного газа в случае получения акриловой кислоты из пропана или метакриловой кислоты из изо-бутана при осуществлении способа согласно изобретению может также содержать:
от 0,6 до 1,2 об.% пропана или изо-бутана,
от 65 до 95 об.% воздуха,
от 2 до 30 об.% азота,
от 0,05 до 0,8 об.% СОх и
от 2 до 3 об.% водяного пара.
Предпочтение отдают исходным смесям реакционного газа, которые содержат от 10 до 50 об.% водяного пара (свежего).
В другом возможном составе исходная смесь реакционного газа может содержать, например,
от 70 до 90 об.% пропана или изо-бутана,
от 5 до 25 об.% молекулярного кислорода,
от 0 до 25 об.% водяного пара и
как остаточное количество циркуляционный газ.
При этом предпочтение отдают исходным смесям реакционного газа, которые в общей сложности содержат от 10 до 50 об.% водяного пара. Таким образом, состав исходной смеси реакционного газа при осуществлении способа согласно изобретению в случае частичного окисления пропана или изо-бутана колеблется в следующих пределах (молярные соотношения):
Указанные выше области являются справедливыми в том случае, если как другие газы-разбавители главным образом используют молекулярный азот. Еще другими возможными газами-разбавителями являются, например, Не, Ar, СО и/или CO2 и т.д.
В общем выбирают такой состав исходной смеси реакционного газа, чтобы смесь находилась за пределами области взрыва.
Если способ согласно изобретению осуществляют как частичное окисление, то его можно проводить, например, в однокамерных кожухотрубных реакторах, как, например, описано в ЕР-А 700714 и ЕР-А 700893. Кроме того, его можно осуществлять в многокамерных кожухотрубных реакторах, как, например, описано в DE-A 19927624, DE-A 19948242, DE-A 19948241, DE-A 19910508 и DE-A 19910506. Осуществление способа согласно изобретению в псевдоожиженном слое возможно, как описано, например, в WO 02/0811421.
В зависимости от содержащегося в исходной смеси реакционного газа пропана и/или изо-бутана превращение пропана и/или изо-бутана в способе согласно изобретению при одноразовом прохождении смеси реакционного газа стадии реакции, как правило составляет от 10 или 20 до 70 моль %, часто от 30 до 60 моль % и чаще всего от 40 до 60 моль % или от 45 до 55 моль %. Селективность получения целевого продукта обычно составляет от 40 до 98 или 95, или 90 моль %, часто от 50 до 80 моль %, чаще всего от 60 до 80 моль %.
Грубое отделение, по меньшей мере, одного целевого продукта из смеси продукт-газа, полученной на стадии реакции способа согласно изобретению, может быть осуществлено на стадии переработки известным из уровня техники методом. Кроме того, согласно изобретению используют также способы переработки, известные из уровня техники для выделения тех же целевых продуктов из смесей продукт-газа, как они описаны при получении целевых продуктов путем гетерогенного каталитического частичного окисления пропена и/или изо-бутена.
Как правило, смесь продукт-газа, полученную при осуществлении способа согласно изобретению на стадии реакции, на входе в стадию переработки согласно изобретению подвергают непрямому и/или прямому охлаждению.
С целью грубого отделения содержащегося в смеси продукт-газа целевого продукта в жидкую фазу охлажденную таким образом (в случае акриловой кислоты обычно до 150-250°С) или неохлажденную смесь продукт-газа, например, в абсорбционной колонне противотоком можно подавать в подаваемый сверху вниз жидкий абсорбент, который селективно абсорбирует из смеси продукт-газа по меньшей мере один целевой продукт, как, например описано в JP-A 2001/0026269, ЕР-А 990636, JP-A 2000/327651, ЕР-А 925272, ЕР-А 695736, ЕР-А 778255, DE-A 2136396, DE-A 2449780, DE-A 4308087, ЕР-А 982287, ЕР-А 982289, ЕР-А 982288 и DE-A 19631645 для целевых продуктов и разных абсорбентов.
Как абсорбенты для целевых продуктов используют в основном воду (или водные смеси, например, водный раствор едкого натра или водную акриловую или метакриловую кислоту), используемые для этерификации акриловой и метакриловой кислоты спирты, такие как, например, 2-этилгексанол, а также органические растворители с более высокой температурой кипения. Согласно изобретению температура кипения органического растворителя превышает температуру кипения целевого продукта (акриловой кислоты и/или метакриловой кислоты), подлежащего выведению из смеси продукт-газа, предпочтительно на, по меньшей мере, 20°С, в особенности, на по меньшей мере, 50°С, и особенно предпочтительно, на по меньшей мере, 70°С. Согласно изобретению предпочтительные органические абсорбенты имеют температуру кипения (при атмосферном давлении) от 180 до 400°С, в частности от 220 до 360°С. Согласно изобретению особенно предпочтительными абсорбентами в случае таких целевых продуктов, как акриловая кислота и метакриловая кислота, являются сильно гидрофобные растворители с высокой температурой кипения, которые не содержат взаимодействующих с окружающим пространством боковых полярных групп, такие как алифатические или ароматические углеводороды, например, средние фракции нефти из парафиновой дистилляции, или простые эфиры, содержащие объемные группы у атома О, или их смеси, причем в них предпочтительно добавляют полярный растворитель, например описанный в DE-A 4308087 1,2-диметилфталат. Кроме того, подходящими являются также эфиры бензойной и фталевой кислоты и неразветвленных содержащих 1-8 атомов углерода алканолов, такие как н-бутиловый эфир бензойной кислоты, метиловый эфир бензойной кислоты, этиловый эфир бензойной кислоты, диметиловый эфир фталевой кислоты, диэтиловый эфир фталевой кислоты, а также так называемые масла-теплоносители, такие как дифенил, дифениловый эфир и смеси дифенила и дифенилового эфира или их хлорпроизводные, а также триарилалканы, например, 4-метил-4'-бензилдифенилметан и его изомеры: 2-метил-2'-бензил-дифенилметан, 2-метил-4'-бензилдифенилметан и 4-метил-2'-бензилдифенил-метан и смеси таких изомеров.
Особенно предпочтительным для акриловой кислоты абсорбентом (метакриловая кислота абсорбируется предпочтительно водой) является смесь растворителей, предпочтительно азеотропного состава, состоящая из дифенила и дифенилового эфира, в особенности из приблизительно 25 вес.% дифенила (бифенила) и из приблизительно 75 вес.% дифенилового эфира, в пересчете на 100 вес.% дифенила и дифенилового эфира, например, имеющийся в продаже Diphyl®. Кроме того, эта смесь растворителей предпочтительно содержит полярный растворитель, такой как диметилфталат, в количестве от 0,1 до 25 вес.%, в расчете на всю смесь растворителей. Предпочтительно смесь продукт-газа при использовании в качестве абсорбента органического растворителя с высокой температурой кипения перед абсорбцией охлаждают путем частичного испарения абсорбента в прямом холодильнике или устройстве для резкого охлаждения. Подходящими для этого являются скрубберы Вентури, барботажные колонны или оросительные конденсаторы.
Понятия высококипящие, труднокипящие, среднекипящие и легкокипящие вещества обозначает в данном тексте соединения, которые имеют более высокую температуру кипения, чем целевое соединение (высококипящие), соединения, которые имеют такую же температуру кипения, как и целевое соединение (среднекипящие) или соединения, которые имеют более низкую температуру кипения, чем целевое соединение (легкокипящие). Особенно это касается того случая, когда целевым соединением является акриловая кислота.
В общем, противоточная абсорбция происходит предпочтительно в колонне с упорядоченными или неупорядоченными насадками или в тарельчатой колонне, которая оснащена предпочтительно двухпоточными тарелками и/или клапанными тарелками и в которую сверху загружают растворитель. Смесь продукт-газа (и, в случае необходимости, испарившийся из охлаждающего устройства абсорбент) подают снизу в колонну и после этого охлаждают до температуры абсорбции. Охлаждение осуществляют предпочтительно в режиме циркуляции, то есть нагретый нисходящий в колонне абсорбент выводят из колонны, охлаждают в теплообменниках и снова подают в абсорбционную колонну над местом вывода. После абсорбции в абсорбенте находятся в основном все тяжелокипящие соединения, большая часть целевого соединения (например, акриловой кислоты) и часть легкокипящих соединений. Из содержащего грубо отделенное целевое соединение (например, акриловую кислоту) абсорбата можно далее выделить целевой продукт как описано в уровне техники (например, в цитированной при описании абсорбции литературе) любой чистоты (например, как известно из DE-A 19606877 или DE-A 19838845), а освобожденный от целевого продукта абсорбент снова использовать в абсорбции (например, отделить из органического абсорбата акриловую кислоту от органического абсорбента через верх колонны и дальше очистить ее ректификацией и/или кристаллизацией (например, суспензионной кристаллизацией с выделением кристаллизата в промывной колонне) или выделить ректификативным способом из водного абсорбата воду с помощью органического разделяющего агента, и выделить акриловую кислоту любой чистоты из содержащего акриловую кислоту отстойника ректификацией или кристаллизацией; в последнем случае продукт, выходящий из верха колонны, как правило, разделяют на две фазы (путем охлаждения); органическую фазу возвращают в ректификационную колонну, а водную фазу - в абсорбционную колонну (в верхнюю часть колонны)).
Оставшаяся неабсорбированная смесь остаточного продукт-газа может быть дальше охлаждена с целью выделения легкоконденсируемой части легкокипящих побочных компонентов (например, воды, формальдегида и уксусной кислоты) (как правило, называемых кислой водой). Оставшаяся смесь остаточного продукт-газа согласно изобретению может быть разделена на две порции, и одна из порций может быть повторно использована в качестве циркуляционного газа (на стадии реакции), а другая порция выпущена. Согласно изобретению предпочтительно не осуществляют разделения кислой воды. В особенности при использовании на стадии реакции способа согласно изобретению водяного пара в качестве газа-разбавителя (при использовании масс полиметаллических оксидов (I), (II), (III) или (IV) это является выгодным для селективности получения целевого продукта) согласно изобретению осуществляют переведение содержащегося в смеси продукт-газа стадии реакции целевого продукта в жидкую фазу (независимо от используемого при этом способа выделения, особенно описанных в этом документе) предпочтительно таким образом, что в оставшейся смеси остаточного продукт-газа, по меньшей мере, часть которой (как циркуляционный газ) согласно изобретению повторно используют на стадии реакции, молярное соотношение W содержащегося в ней водяного пара и пропана меньше соответственного молярного соотношения W' в смеси продукт-газа на стадии реакции на не более чем 50%, предпочтительно на не более чем 40 или 30%, еще предпочтительнее на не более чем 20 или 10%, особенно предпочтительно на не более чем 5%. В крайнем случае указанные выше соотношения W и W' в способе согласно изобретению могут быть идентичными.
Попытка оставить в смеси остаточного продукт-газа как можно больше водяного пара преследует цель в как можно больших масштабах отказаться от подачи (соответственно, конденсации и испарения) нового водяного пара в исходную смесь реакционного газа.
Разумеется, при осуществлении способа согласно изобретению выгруженная часть смеси остаточного продукт-газа, содержит, по меньшей мере, такое количество воды, как и на стадии реакции при образовании побочного продукта, с целью предотвращения увеличения содержания воды в смеси реакционного газа (чем более селективно осуществляют реакцию на стадии реакции, тем меньше количество воды, которую выпускают). Это касается также других полученных на стадии реакции побочных компонентов. Если в способе согласно изобретению в качестве источника кислорода используют воздух, то количество выпускаемой смеси остаточного продукт-газа устанавливают таким, чтобы содержащееся в ней количество азота по меньшей мере соответствовало количеству, содержащемуся в притоке воздуха.
Если как абсорбент используют один из высококипящих растворителей, то абсорбцию предпочтительно осуществляют таким образом (особенно в случае абсорбции акриловой кислоты), что сток из абсорбционной колонны является однофазным. Впрочем содержание водяного пара в оставшейся при абсорбции смеси остаточного продукт-газа, независимо от выбранного абсорбента, устанавливают путем соответствующего выбора абсорбционной температуры.
Альтернативно грубому отделению целевого продукта в жидкую фазу путем абсорбции растворителем такое грубое отделение (особенно в случае акриловой кислоты) можно осуществлять также путем конденсации, в частности фракционной конденсации, как описано, например, в DE-A 19924532, DE-A 10200583, DE-A 10053086, DE-A 19627847, DE-A 19740253, DE-A 19740252, DE-A 19740253, DE-A 19814387 и DE-A 10247240.
При этом смесь продукт-газа стадии реакции, в случае необходимости, после прямого и/или косвенного предварительного охлаждения в оснащенной разделительными насадками ректификационной колонне при восхождении подвергают фракционной конденсации, и целевой продукт выводят через боковой отвод ректификационной колонны и, как описано в уровне техники, подвергают последующим стадиям кристаллизационного и/или ректификативного выделения.
В качестве содержащих разделительные насадки конденсационных колонн предпочтительны тарельчатые колонны, которые в качестве разделительных насадок снизу вверх содержат сначала двухпоточные тарелки, а затем гидравлически уплотненные тарелки с перекрестным током, как они описаны в указанном выше уровне техники.
Согласно изобретению указанную выше фракционированную конденсацию осуществляют предпочтительно таким образом, что не происходит практически никакого отделения содержащейся в смеси продукт-газа воды, то есть практически никакого отделения кислой воды.
Выгруженная часть смеси остаточного продукт-газа перед выгрузкой может быть промыта водой с целью избежания потерь метакриловой или акриловой кислоты. Из полученной при этом кислой воды, содержащей метакриловую или акриловую кислоту, акриловая или метакриловая кислота и, в случае необходимости, другие органические ценные продукты могут быть экстрагированы, например, с помощью используемого для абсорбции органического абсорбента и объединены с абсорбатом.
Как абсорбционная колонна, так и колонна для фракционированной конденсации во избежание больших потерь давления могут также быть заменены последовательно подключенными стадиями резкого охлаждения, которые приводятся в действие с помощью абсорбента или конденсата.
В общем, ингибирование полимеризации в рамках грубого отделения в жидкую фазу целевого продукта из смеси продукт-газа происходит, как описано в уровне техники, путем применения соответствующего ингибитора полимеризации.
Согласно изобретению существенным является тот факт, что после выделения целевого продукта смесь остаточного продукт-газа наряду с пропаном и/или изо-бутаном, а также, в случае необходимости, пропеном и/или изо-бутеном обычно содержит по меньшей мере 5 об.%, предпочтительно по меньшей мере 10 об.% компонентов, которые при атмосферном давлении являются более легкокипящими, чем целевой продукт. Такими компонентами являются, в частности, компоненты смеси продукт-газа, более легкокипящие при атмосферном давлении, чем вода (например, N2, СО, СО2), а также предпочтительно сама вода.
Преимущество способа согласно изобретению состоит в том, что циркуляционный газ, как правило, с помощью компрессора низкого давления (при давлении на выходе Р3) может быть сжат до входного давления Р1. При этом под компрессором низкого давления понимают компрессор (обычно осевой компрессор) с низким соотношением давления (конечное давление : исходное давление = 1,1:1-3:1). В отличие от этого используемый как источник кислорода воздух обычно сжимают с помощью центробежного компрессора (приблизительно от давлении окружающей среды) до входного давления Р1. Описанные в этом тексте процессы сжатия могут проходить изотермическим или политропным образом. Предпочтение согласно изобретению отдают последнему.
Часть преимуществ способа согласно изобретению сохраняется также и в том случае, если из подлежащей выгрузке части смеси остаточного продукт-газа, в случае необходимости, после предварительного конденсационного выделения содержащегося в ней количества воды (которая может быть повторно использована на стадии реакции) отделяют содержащийся в ней пропан и/или изо-бутан, а также, в случае необходимости, пропен и/или изо-бутен, сжимают ее до входного давления Р1 и повторно используют на стадии реакции.
Это отделение (которое в уровне техники рекомендуют для общего количества смеси остаточного продукт-газа и в принципе может быть использовано (в особенности как описано ниже)) осуществляют, например, таким образом (см. WO 0196271), что пропан и/или изо-бутан, а также, в случае необходимости, пропен и/или изо-бутен, которые содержатся в подлежащей выгрузке части смеси остаточного продукт-газа, выделяют абсорбцией или адсорбцией и после этого снова высвобождают десорбцией и/или отгонкой. Такое выделение можно, например, осуществлять из абсорбата путем отгонки воздухом, как, например, описано в WO 0196271. После этого воздух сжимают и снова используют на стадии реакции.
Альтернативно часть смеси остаточного продукт-газа, подлежащую выгрузке, подают в реактор окисления, в случае необходимости, после добавления кислорода с целью повышения объемов превращения углеводорода.
Как правило, как было сказано выше, при осуществлении способа согласно изобретению для повторного сжатия циркуляционного газа достаточно использовать компрессор.
Фиг.1 данного документа демонстрирует вариант выполнения способа согласно изобретению.
При этом позиции имеют следующие значения:
1 = стадия реакции
2 = стадия переработки
3 = компрессор циркуляционного газа
4 = расширитель выгрузки
5 = выгрузка
6 = воздух в качестве источника кислорода
7 = компрессор воздуха
8 = свежий пропан и/или изо-бутан, а также, в случае необходимости, водяной пар
9 = повторно сжатый циркуляционный газ
Примеры
1. Получение катализатора, содержащего массу полиметаллических оксидов
Мо1V0,29Те0,13Nb0,13Ох (чистая i-фаза)
87,61 г метаванадата аммония (78,55 вес.% V2O5, фирмы G.f.E., Нюрнберг) при 80°С растворяют в 3040 мл воды (трехгорлая колба с мешалкой, термометр и обратный холодильник). Получают прозрачный желтоватый раствор. Этот раствор охлаждают до 60°С и после этого при поддержании температуры 60°С при перемешивании последовательно добавляют 117,03 г теллуровой кислоты (99 вес.% Н6ТеО6, фирмы Aldrich) и 400,00 г гептамолибдата аммония (82,52 вес.% МоО3, фирмы Starck, Goslar). Полученный темно-красный раствор охлаждают до 30°С (раствор А).
В химическом стакане при 60°С в 500 мл воды отдельно растворяют 112,67 г ниобоксалата аммония (20,8 вес.% Nb, фирмы Starck, Goslar) (раствор В).
Раствор В охлаждают до 30°С и при этой температуре объединяют с раствором А, причем раствор В добавляют в раствор А. Добавление осуществляют непрерывно в течении примерно 5 минут. Получают оранжевую водную суспензию с находящимся во взвешенном состоянии осадком. Затем эту суспензию подвергают распылительной сушке (Тсосуда=30°С, Тна входе=320°С, Тна выходе=110°С, t=1,5 часа, скруббер фирмы Niro типа Atomizer). Полученный порошок имеет также оранжевую окраску и имеет стехиометрию навески Mo1V0,33Te0,22Nb0,11.
2 раза по 100 г полученного порошка обрабатывают термически в шаровой печи согласно фиг.1 в DE-A 10119933 (1 л внутреннего объема) таким образом: сначала в течении 27,5 минут в линейном режиме нагрева потоком воздуха 50 нл/час нагревают от 25°С до 275°С, после этого эту температуру поддерживают в течение 1 часа. После этого в линейном режиме нагрева в течение 32,5 минут нагревают от 275°С до 600°С, причем поток воздуха заменяют потоком азота 50 нл/час. При сохранении потока азота и температуры 600°С продукт выдерживают в течение 2 часов, а затем всю печь оставляют охлаждаться до комнатной температуры. 100 г полученной оксидной массы в течение 7 часов при 70°С перемешивают с 1000 мл 10 вес.%-ного водного раствора HNO3 при нагревании с обратным холодильником, оставшееся твердое вещество отфильтровывают из полученной взвеси и промывают водой до отсутствия в фильтрате нитрат-ионов (25°С). Полученный осадок сушат за ночь в атмосфере воздуха при 110°С в муфельной печи.
Химический анализ полученного твердого вещества показал, что оно имеет такой состав: Mo1V0,29Te0,13Nb0,13Ox. Соответствующая рентгенодифрактограмма показала содержание чистой i-фазы.
После этого высушенный материал измельчают, как описано в DE-A 10119933, в мельнице фирмы Retsch (размер частиц ≤0,12 мм) и согласно примеру А) а) в DE-A 10119933 перерабатывают в оболочковый катализатор:
38 г измельченной активной массы; 150 г шаровидных носителей диаметром от 2,2 до 3,2 мм (материал носителя = стеатит С-220 фирмы CeramTec, DE с шероховатостью поверхности Rz 45 мкм), адгезионное средство = 30 мл смеси глицерина и воды (весовое отношение глицерин: вода = 1:3), продолжительность сушки = 16 часов при 150°С; содержание активной массы полученного оболочкового катализатора 20 вес.% (относительно веса оболочкового катализатора).
2. Гетерогенное каталитическое частичное прямое окисление пропана до акриловой кислоты при различных давлениях
Оболочковым катализатором из пункта 1 загружают реакционную трубу (длина 140 см) из V2A стали (наружный диаметр 60 мм, внутренний диаметр 8,5 мм).
Длина загрузки составляет до 53 см (приблизительно 35,0 г оболочкового катализатора). Предварительная засыпка длиной 30 см из использованных как материал-носитель зерен стеатита служит для позиционирования зоны катализатора. Этими же зернами стеатита наполняют реакционную трубу после зоны катализатора (зона предварительного нагрева для подогрева исходной смеси реакционного газа). Реакционную трубу снаружи по всех длине нагревают с помощью электрических нагревательных матов. Молярный состав исходной смеси реакционного газа пропан: воздух: вода = 1:15:14. Указанная ниже таблица показывает полученную степень превращения пропана при одноразовом прохождении (ППАН, моль %), а также связанную с этим превращением селективность получения акриловой кислоты (САКК моль %) в зависимости от выбранного давления на входе, а также соответствующей температуры нагревательных матов. Продолжительность реакции (относительно объемов загрузки катализатора) во всех случаях составляет 2,4 с. Кроме того, с помощью СПЕН таблица показывает селективность получения побочного продукта пропена.
При более высоком рабочем давлении и при более низких температурах получают более высокие объемы превращения пропана, при этом селективность получения целевого продукт в основном остается неизменной.
Кроме того, во всех случаях смесь продукт-газа содержит малые количества других кислот, таких как, например, уксусная кислота, а также СОх.
3. Мощность компрессора при изотермическом сжатии в способе согласно изобретению в зависимости от выходного давления Р3 при выбранном соотношении циркуляционного газа
Способ согласно вышеприведенному пункту 2 осуществляют в аппаратном устройстве согласно фиг.2. За основу берут степень превращения 40 моль.% и селективность получения акриловой кислоты 70 моль.%. Молярный состав пропан: кислород: вода соответствует составу, указанному в вышеприведенном пункте 2. Подача циркуляционного газа влияет на содержание азота.
При этом позиции имеют следующие значения:
1 = стадия реакции
2 = стадия переработки
3 = выгруженная часть смеси остаточного продукт-газа
4 = компрессор циркуляционного газа
5 = компрессор воздуха
6 = воздух в качестве источника кислорода
7 = свежий пропан и свежий водяной пар
8 = повторно сжатый циркуляционный газ
9 = дросселирующее устройство для регулирования выходного давления Р3
10 = исходная смесь реакционного газа с входным давлением Р1.
Свежий пропан и свежий водяной пар имеют соответственно необходимое входное давление.
Изменение Р3 отражается лишь на мощности компрессора циркуляционного газа и на мощности компрессора воздуха.
С целью упрощения для обоих компрессоров сжатие считают изотермическим сжатием.
Согласно VDI-Warmeatlas, Verlag des Vereins Deutscher Ingenieure, Dusseldorf, 5. Auflage, 1988, Blatt La 1, мощность (VL) компрессора воздуха после изотермического сжатия составляет:
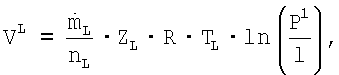
где
 означает массовый поток свежего воздуха;
означает массовый поток свежего воздуха;
nL означает коэффициент полезного действия компрессора воздуха;
ZL означает фактор реального газа для воздуха;
R означает специальную газовую постоянную=идеальная газовая постоянная, разделенная на молярную массу;
TL означает температуру, при которой свежий воздух всасывают из окружающей среды;
l означает атмосферное давление (давление окружающей среды) = 1 бар, при котором всасывают воздух;
P1 означает входное давление стадии реакции, до которого сжимают воздух.
Значит, 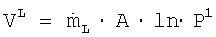 , причем А является постоянной величиной.
, причем А является постоянной величиной.
Соответствующим образом изотермическая мощность компрессора циркуляционного газа VK составляет:
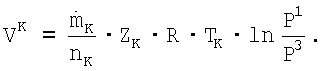
Значит, 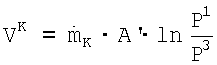 , причем А' является постоянной величиной, и А'≈А.
, причем А' является постоянной величиной, и А'≈А.
Соответственно, общая используемая мощность компрессора Vges=VL+VK составляет:
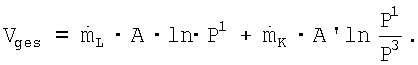
где 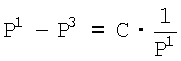 , причем С является характерной для используемого реакционного устройства и устройства переработки постоянной величиной, а
, причем С является характерной для используемого реакционного устройства и устройства переработки постоянной величиной, а 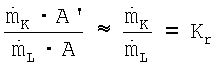 , означает соотношение циркуляционного газа, получают такую формулу
, означает соотношение циркуляционного газа, получают такую формулу

Если соотношение циркуляционного газа выбирают один раз 2,5 и один раз 3,5, и постоянную С определяют как С=(1,5 бар + ΔP1,5)·ΔP1,5,
причем ΔP1,5 означает общую потерю давления на стадии реакции и стадии переработки при выходном давлении (выход из стадии переработки) 1,5 бар, при характерной для способа согласно изобретению потере давления 2 бар дает С=(1,5 бар + 2 бар)·2 бар = 7 бар2, то ( выбирают таким образом, что объемно-временной выход акриловой кислоты в обоих случаях является одинаковым) при Кr=2,5 и массовом потоке циркуляционного газа
выбирают таким образом, что объемно-временной выход акриловой кислоты в обоих случаях является одинаковым) при Кr=2,5 и массовом потоке циркуляционного газа 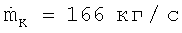 , а также при Кr=3,5 и массовом потоке циркуляционного газа
, а также при Кr=3,5 и массовом потоке циркуляционного газа 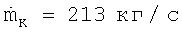 при обоих выбранных соотношениях циркуляционного газа получают показанные на фиг.3 кривые для
при обоих выбранных соотношениях циркуляционного газа получают показанные на фиг.3 кривые для
Vges в зависимости от Р3 (предполагая, что согласно результатам пункта 2 даже при увеличении давления степень превращения и селективность останутся неизменными).
Начиная с Р3=1,5 бар, при увеличении Р3 используемая в общей сложности мощность компрессора в обоих случаях уменьшается, что подтверждает выгодность способа согласно изобретению.
Изобретение относится к усовершенствованному способу гетерогенного каталитического частичного прямого окисления пропана и/или изо-бутана до получения, по меньшей мере, одного из таких целевых продуктов, как акриловая кислота, метакриловая кислота, при котором на стадию реакции, которая за исключением входа для исходной смеси реакционного газа, в случае необходимости, других входов для вспомогательных газов, а также выхода для смеси продукт-газа, герметична для газа, подают пропан и/или изо-бутан, молекулярный кислород и исходную смесь реакционного газа, содержащую, по меньшей мере, один инертный газ-разбавитель, при входном давлении Р1, на указанной стадии реакции путем подачи исходной смеси реакционного газа при повышенной температуре через находящийся в твердом агрегатном состоянии катализатор, содержащийся в исходной смеси реакционного газа пропан и/или изо-бутан, прямым способом частично окисляют до, по меньшей мере, одного целевого продукта, и смесь реакционного газа в виде содержащей, по меньшей мере, один целевой продукт смеси продукт-газа выводят из стадии реакции при выходном давлении
Р2 и при том же давлении Р2 подают на стадию переработки, которая, за исключением входа для смеси продукт-газа, в случае необходимости, других входов для вспомогательных газов, а также выхода для смеси остаточного продукт-газа, герметична для газа, на стадии переработки из смеси продукт-газа стадии реакции, содержащийся в ней, целевой продукт грубо отделяют в жидкую фазу, а оставшуюся при этом смесь остаточного продукт-газа, содержащую не только пропан и/или изо-бутан, а также, в случае необходимости, пропен и/или изо-бутен, выводят со стадии переработки при выходном давлении Р3, причем Р3 меньше Р1, содержащийся в смеси остаточного продукт-газа пропан и/или изо-бутан, возвращают на стадию реакции, где P1 выбирают таким образом, что Р3 больше или равно 1,5 бар, а смесь остаточного продукт-газа разделяют на две части одинакового состава, при этом одну часть выгружают, а другую часть отводят как циркуляционный газ и в качестве сжатого до входного давления Р1 компонента исходной смеси реакционного газа повторно подают на стадию реакции. Задача гетерогенного каталитического частичного прямого окисления состоит в том, чтобы при максимально низких температурах и одноразовом прохождении смеси реакционного газа через стадию реакции достичь максимально высокого превращения пропана и/или изо-бутана при одновременно максимально высокой селективности получения целевого продукта, при максимально низком потреблении энергии. 31 з.п. ф-лы, 1 табл., 3 ил.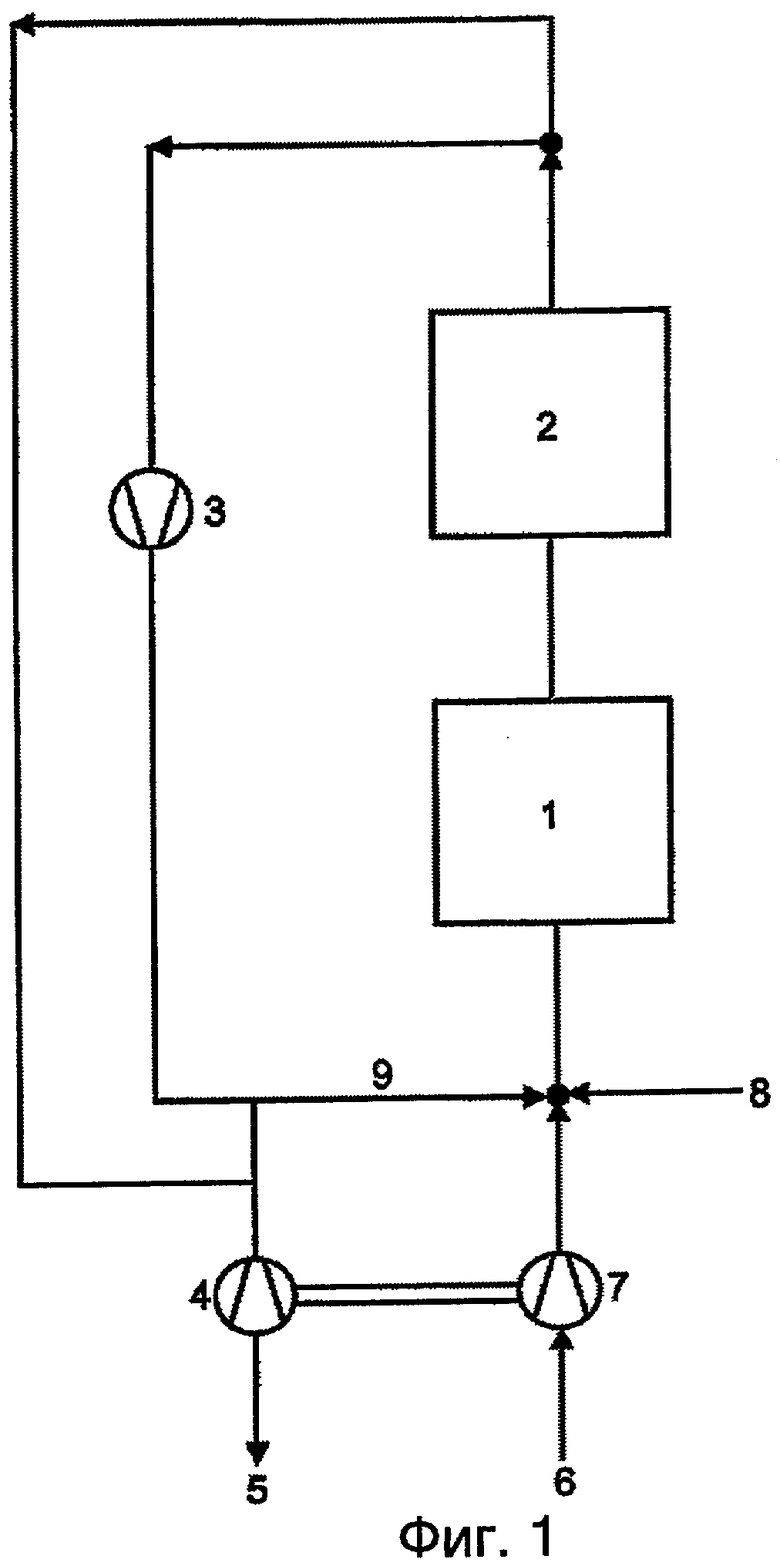
1. Способ гетерогенного каталитического частичного прямого окисления пропана и/или изо-бутана до получения, по меньшей мере, одного из таких целевых продуктов, как акриловая кислота, метакриловая кислота, при котором на стадию реакции, которая за исключением входа для исходной смеси реакционного газа, в случае необходимости, других входов для вспомогательных газов, а также выхода для смеси продукт-газа герметична для газа, подают пропан и/или изо-бутан, молекулярный кислород и исходную смесь реакционного газа, содержащую, по меньшей мере, один инертный газ-разбавитель, при входном давлении Р1, на указанной стадии реакции путем подачи исходной смеси реакционного газа при повышенной температуре через находящийся в твердом агрегатном состоянии катализатор, содержащийся в исходной смеси реакционного газа пропан и/или изо-бутан, прямым способом частично окисляют до, по меньшей мере, одного целевого продукта, и смесь реакционного газа в виде содержащей, по меньшей мере, один целевой продукт смеси продукт-газа выводят из стадии реакции при выходном давлении Р2 и при том же давлении Р2 подают на стадию переработки, которая за исключением входа для смеси продукт-газа, в случае необходимости, других входов для вспомогательных газов, а также выхода для смеси остаточного продукт-газа герметична для газа, на стадии переработки из смеси продукт-газа стадии реакции содержащийся в ней целевой продукт грубо отделяют в жидкую фазу, а оставшуюся при этом смесь остаточного продукт-газа, содержащую не только пропан и/или изо-бутан, а также, в случае необходимости, пропен и/или изо-бутен, выводят со стадии переработки при выходном давлении Р3, причем Р3 меньше Р1, содержащийся в смеси остаточного продукт-газа пропан и/или изо-бутан возвращают на стадию реакции, отличающийся тем, что Р1 выбирают таким образом, что Р3 больше или равно 1,5 бар, а смесь остаточного продукт-газа разделяют на две части одинакового состава, при этом одну часть выгружают, а другую часть отводят как циркуляционный газ и в качестве сжатого до входного давления Р1 компонента исходной смеси реакционного газа повторно подают на стадию реакции.
2. Способ по п.1, отличающийся тем, что смесь остаточного продукт-газа содержит, по меньшей мере, 5 об.% компонентов, отличных от пропана и/или изо-бутана, а также от пропена и/или изо-бутена.
3. Способ по п.1, отличающийся тем, что смесь остаточного продукт-газа содержит, по меньшей мере, 10 об.% компонентов, отличных от пропана и/или изо-бутана, а также от пропена и/или изо-бутена.
4. Способ по п.1, отличающийся тем, что давление Р3 больше или равно 1,5 бар и меньше или равно 25 бар.
5. Способ по п.1, отличающийся тем, что давление Р3 больше или равно 1,5 бар и меньше или равно 20 бар.
6. Способ по п.1, отличающийся тем, что давление Р3 больше или равно 1,5 бар и меньше или равно 10 бар.
7. Способ по п.1, отличающийся тем, что давление Р3 больше или равно 2 бар и меньше или равно 8 бар.
8. Способ по п.1, отличающийся тем, что давление Р1 превышает давление Р3 на 1-4 бар.
9. Способ по п.1, отличающийся тем, что давление Р1 превышает давление Р3 на 1,5-3,5 бар.
10. Способ по п.1, отличающийся тем, что давление Р1 составляет от 3 до 10 бар.
11. Способ по п.1, отличающийся тем, что давление Р1 составляет от 4 до 8 бар.
12. Способ по п.1, отличающийся тем, что выгрузку части смеси остаточного продукт-газа осуществляют через расширитель.
13. Способ по п.1, отличающийся тем, что стадией реакции является загруженный катализатором кожухотрубный реактор или реактор с псевдоожиженным слоем.
14. Способ по п.1, отличающийся тем, что стадией переработки является абсорбционная колонна или колонна для фракционной конденсации или последовательное подключение стадий резкого охлаждения.
15. Способ по п.1, отличающийся тем, что катализатор в качестве активной массы содержит массу полиметаллических оксидов, которая содержит комбинацию элементов Мо, V, по меньшей мере одного из элементов Те и Sb, и по меньшей мере одного из элементов группы, которая включает Nb, Та, W, Ti, Al, Zr, Cr, Mn, Ga, Fe, Ru, Co, Cs, Ca, Sr, Ba, Rh, Ni, Pd, Pt, La, Pb, Cu, Re, Ir, Y, Pr, Nd, Tb, Bi, B, Ce, Sn, Zn, Si, Na, Li, K, Mg, Ag, Au и In.
16. Способ по п.1, отличающийся тем, что катализатор в качестве активной массы содержит массу полиметаллических оксидов, которая содержит комбинацию элементов стехиометрической формулы I
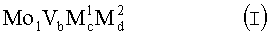
где М1 означает Те и/или Sb,
М2 означает, по меньшей мере, один из элементов из группы, которая включает Nb, Та, W, Ti, Al, Zr, Cs, Ca, Sr, Ва, Cr, Mn, Ga, Fe, Ru, Со, Rh, Ni, Pd, Pt, La, Bi, Pb, Cu, Re, Ir, Y, Pr, Nd, Tb, Се, Sn, Zn, Si, Na, Li, К, Mg, Ag, Au и In,
b означает от 0,01 до 1,
с означает от больше 0 до 1 и
d означает от больше 0 до 1.
17. Способ по п.1, отличающийся тем, что в качестве источника кислорода используют воздух.
18. Способ по п.1, отличающийся тем, что реакционная температура составляет от 200 до 700°С.
19. Способ по п.1, отличающийся тем, что исходная смесь реакционного газа содержит
от 0,5 до 15 об.% пропана или изо-бутана,
от 10 до 90 об.% воздуха,
от 0 до 50 об.% водяного пара и
как остаточное количество циркуляционный газ.
20. Способ по п.1, отличающийся тем, что исходная смесь реакционного газа содержит
от 0,5 до 15 об.% пропана или изо-бутана,
от 10 до 90 об.% воздуха,
от 10 до 50 об.% водяного пара и
как остаточное количество циркуляционный газ.
21. Способ по п.1, отличающийся тем, что исходная смесь реакционного газа содержит
от 70 до 90 об.% пропана или изо-бутана,
от 5 до 25 об.% молекулярного кислорода,
от 0 до 25 об.% водяного пара и
как остаточное количество циркуляционный газ.
22. Способ по п.1, отличающийся тем, что степень превращения пропана и/или изо-бутана при одноразовом прохождении смеси реакционного газа через стадию реакции составляет от 10 до 70 мол.%.
23. Способ по п.22, отличающийся тем, что селективность получения целевого продукта составляет от 40 до 98 мол.%.
24. Способ по п.1, отличающийся тем, что грубое отделение содержащегося в смеси продукт-газа стадии реакции целевого продукта в жидкую фазу осуществляют таким образом, что в оставшейся смеси остаточного продукт-газа молярное соотношение W содержащегося в ней водяного пара и содержащегося в ней пропана является ниже соответственного молярного соотношения W' в смеси продукт-газа стадии реакции на не более чем 50%.
25. Способ по п.1, отличающийся тем, что грубое отделение содержащегося в смеси продукт-газа стадии реакции целевого продукта в жидкую фазу осуществляют в абсорбционной колонне путем абсорбции органическим растворителем таким образом, что сток из абсорбционной колонны является однофазным.
26. Способ по п.1, отличающийся тем, что из выгруженной части смеси остаточного продукт-газа выделяют содержащийся в ней пропан и/или изо-бутан, а также, в случае необходимости, пропен и/или изо-бутен, сжимают до входного давления Р1 и повторно используют на стадии реакции.
27. Способ по п.1, отличающийся тем, что количественное соотношение V части смеси остаточного продукт-газа, которую повторно используют как циркуляционный газ, и части смеси остаточного продукт-газа, которую выгружают, составляет больше или равно 0,5 и меньше или равно 30.
28. Способ по п.1, отличающийся тем, что количественное соотношение V части смеси остаточного продукт-газа, которую повторно используют как циркуляционный газ, и части смеси остаточного продукт-газа, которую выгружают, составляет больше или равно 2 и меньше или равно 25.
29. Способ по п.1, отличающийся тем, что количественное соотношение V части смеси остаточного продукт-газа, которую повторно используют как циркуляционный газ, и части смеси остаточного продукт-газа, которую выгружают, составляет больше или равно 3 и меньше или равно 20.
30. Способ по п.1, отличающийся тем, что циркуляционный газ сжимают при помощи компрессора до входного давления Р1.
31. Способ по п.1, отличающийся тем, что в качестве источника кислорода используют воздух, сжатый при помощи центробежного компрессора до входного давления Р1.
32. Способ по одному из пп.1-31, отличающийся тем, что он является способом частичного прямого окисления пропана до акриловой кислоты.
| US 6448439 А, 10.09.2002 | |||
| US 6433222 A, 13.08.2002 | |||
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| WO 9801415 A1, 15.01.1998 | |||
| RU 96104334 A, 10.06.1998. | |||

