Область техники, к которой относится изобретение
Данное изобретение относится к композиции с солифенацином или его солью для использования в твердой лекарственной форме, причем композиция содержит кристаллическое вещество солифенацина или его соли при содержании аморфного вещества в интервале, не оказывающем влияния на стабильность получающегося продукта, а также способу ее получения. Кроме того, данное изобретение относится к фармацевтической композиции, содержащей солифенацин или его соль и ингибитор образования аморфного препарата.
Уровень техники
Солифенацин представляет следующая формула (I):
[Химическая формула]
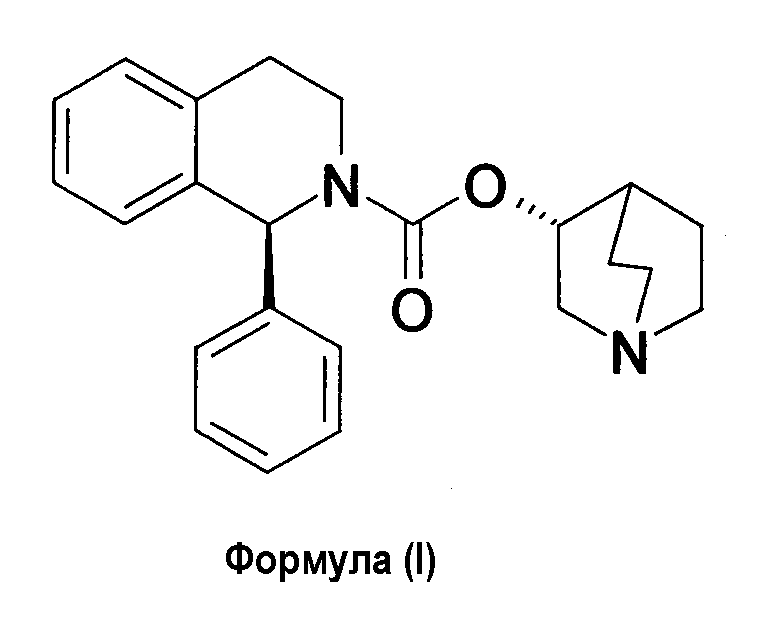
и он имеет химическое название: (1R,3′R)-3′-хинуклидинил-1-фенил-1,2,3,4-тетрагидро-2-изохинолинкарбоксилат.
Сообщают, что серии хинуклидиновых производных, включая солифенацин или его соли, обладают превосходным селективным антагонистическим действием против мускариновых М3 рецепторов и пригодны в качестве профилактических или терапевтических средств при заболеваниях мочевыводящей системы, таких как нервная поллакиурия, нейрогенный мочевой пузырь, ночной энурез, нестабильный мочевой пузырь, контрактура мочевого пузыря и хронический цистит, а также респираторные заболевания, такие как хронические окклюзионные заболевания легких, хронический бронхит, астма и ринит (см. патентный документ 1).
Процесс производства гидрохлорида солифенацина описан в примере 8 патентного документа 1, где кристаллическое вещество, получаемое при кристаллизации в смешанном растворителе из ацетонитрила и диэтилового эфира, имеет температуру плавления 212-214°С и имеет удельное вращение ([α]25 D=98,1 (с=1,00, EtOH).
Однако патентный документ 1 не включает описания или предположения о значительном разложении с течением времени аморфного солифенацина или его аморфной соли, или сукцината солифенацина как активного фармацевтического ингредиента в препарате, когда продукт солифенацина сукцината перерабатывают в лекарственную форму по обычному процессу фармацевтического производства.
Непатентный документ 1, опубликованный Японским Министерством Здравоохранения, Труда и Социального Обеспечения в июне 2003 г., включает описание по спецификации лекарственных продуктов, а именно общее представление о продуктах разложения (примеси) в новых лекарственных продуктах, что наблюдали при испытаниях на стабильность. В соответствии с этим документом, порог для продуктов разложения (деградации), требуемый ограничивающим условием безопасности для лекарственного продукта, ниже в любом случае 1,0%, как процента продукта разложения (деградации), содержащегося в лекарственном веществе, или равен 50 мкг, в виде общего суточного приема продукта разложения, когда количество лекарственного вещества, которое нужно вводить в сутки, составляет менее 10 мг. Когда количество лекарственного вещества, которое нужно вводить в сутки, составляет 10 мг или более до 100 мг или менее, порог продукта разложения, требуемый ограничивающим условием безопасности для лекарственных продуктов, в любом случае ниже 0,5%, как процента продукта разложения, содержащегося в лекарственном веществе, или равен 200 мкг, в виде общего суточного приема продукта разложения. Поэтому технические требования по продукту разложения, которые могут быть определены без условий какого-либо ограничения по безопасности продукта разложения, представляют обычно 1,0% или менее в виде процента продукта разложения, содержащегося в лекарственном веществе, когда препарат представлен, например, с 5-мг содержанием лекарственного вещества. Когда препарат представлен, например, с 10-мг содержанием лекарственного вещества, процент продукта разложения, содержащегося в лекарственном веществе, составляет 0,5% или менее.
Препараты с солифенацином, планируемые в настоящее время к выпуску на рынок по результатам клинических испытаний, представляют собой таблетки с содержанием 2,5 мг, 5 мг и 10 мг. Что касается этих препаратов, которые имеют стабильность, описанную в непатентном документе 1, считают, что количество основного продукта разложения (далее обозначаемое аббревиатурой F1) сукцината солифенацина к общему количеству сукцината солифенацина и продукты его разложения должны быть ограничены 0,5% или менее, и что данное количество должно поддерживаться на уровне 0,4% или менее, с учетом колебания от партии к партии и ошибок испытания.
Патентный документ 1: описание ЕР 801067.
Непатентный документ 1: Сообщение № 0624001, опубликованное Японским комитетом по делам фармации (Japanese Committee of Pharmaceutical Affairs), «Пересмотр указаний по примесям в новых лекарственных продуктах, содержащих новые активные фармацевтические ингредиенты» (“Revision of Guideline about Impurities in New Drug Products containing Novel Active Pharmaceutical Ingredients”).
Описание изобретения
Проблемы, которые изобретение должно разрешить
По данному изобретению сукцинат солифенацина гранулировали способом гранулирования в псевдоожиженном слое при обычных условиях, известных специалистам в данной области, и производили таблетки для разработки сукцината солифенацина в качестве превосходного терапевтического средства при поллакурии и недержании мочи. Затем проводили предварительные испытания стабильности полученных таблеток в течение 6 месяцев при испытании с ускоренным старением (при 40°С и 75% ОВ (относительной влажности) с использованием условий герметично закрытого сосуда) как одном из общих испытаний на стабильность. В результате наблюдали снижение остаточного отношения сукцината солифенацина, так что количество образовавшегося F1 по отношению к общему количеству сукцината солифенацина и его продуктов разложения превышало 0,4% (см. таблицу 2, ниже, более подробно). Стало понятно, что трудно получить его препарат с фармацевтически достаточной стабильностью с помощью такого общепринятого способа фармацевтического производства.
При получении твердого препарата солифенацина или его соли как превосходного терапевтического средства при поллакурии и недержания мочи, другими словами, очень желательна разработка рецептуры твердого препарата солифенацина или его соли, стабильных с течением времени, что может подавлять образуемое количество F1 по отношению к общему количеству солифенацина или его соли и продуктов их разложения, до 0,4% или менее.
Средства решения проблем
Разложение лекарственного вещества в препарате обычно включает, например, окислительно-восстановительную реакцию, реакцию гидролиза, рацемизацию, фоторазложение и полимеризующее разрушение. Было описано, что эти реакции коррелируют с нагреванием, присутствием кислорода, освещением, присутствием воды и взаимодействиями с другими компонентами. Как описано выше, нужно учитывать многие причины в связи с разложением лекарственного средства, чтобы получить стабильные лекарственные продукты. При таком состоянии уровня техники авторами изобретения были проведены исследования по стабилизации продуктов солифенацина. Неожиданно было выяснено, что аморфный сукцинат солифенацина, образующийся в процессе производства лекарственных продуктов, был основной причиной разложения активного фармацевтического ингредиента с течением времени.
Кроме того, авторами было обнаружено, что содержание аморфного вещества в лекарственных продуктах может быть подавлено регулировкой содержания влаги в лекарственном продукте во время процесса производства, когда лекарственные продукты получали с помощью процесса влажного гранулирования с использованием водных растворов обычных связующих средств или путем нагревания и/или процесса увлажнения полученной композиции после производственного процесса получения. Обнаружено, что стабильный твердый препарат солифенацина или его соли, в котором его разложение с течением времени могло быть подавлено, мог быть получен, когда отношение аморфного солифенацина к кристаллическому и аморфному солифенацину имело данное или меньшее значение.
Кроме того, авторами обнаружено, что препарат солифенацина, в котором разложение солифенацина с течением времени могло быть подавлено, мог быть получен с использованием полиэтиленгликоля (под другим названием Macrogol; далее в виде аббревиатуры ПЭГ) в качестве связующего средства, независимо от процесса его производства, хотя сам ПЭГ представляет собой вещество, которое можно использовать обычно в целях получения лекарственных средств в аморфном состоянии. Таким образом, было создано данное изобретение, отличающееся от способов стабилизации, описанных выше.
Другими словами, данное изобретение относится к описанному ниже.
1) Композиция с солифенацином или его солью для использования в твердом препарате, причем композиция содержит кристаллический солифенацин или его соль, где содержание аморфного вещества находится в интервале, не оказывающем влияния на стабильность лекарственного продукта.
2) Композиция с солифенацином или его солью для использования в твердом препарате, которая описана в абзаце 1), в которой содержание аморфного вещества составляет 77% или менее.
3) Композиция для использования в твердом препарате, которая описана выше в абзацах 1) или 2), которая получена способом производства, включающим стадию смешивания солифенацина или его соли с наполнителями без использования растворителя с последующим формованием посредством прессования.
4) Композиция для использования в твердом препарате, как описано выше в абзацах 1) и 2), которая получена способом производства, включающим стадию добавления растворителя к солифенацину или к его соли, причем количество солифенацина или его соли, которое можно растворить в 1 мл растворителя, составляет менее 0,1 мг.
5) Композиция для использования в твердом препарате, которая описана выше в абзаце 4), в которой растворитель, добавленный к солифенацину или к его соли, является ацетоном или гексаном, или их смесью.
6) Композиция для использования в твердом препарате, которая описана выше в абзаце 1) или 2), которая получена способом производства, включающим стадию добавления растворителя для получения солифенацина или его соли в аморфном состоянии, где количество солифенацина или его соли, которое должно быть растворено в 1 мл растворителя, составляет 10 мг или более.
7) Композиция для использования в твердом препарате, которая описана выше в абзаце 6), в которой растворителем для получения солифенацина или его соли в аморфном состоянии является вода, метанол или этанол, или их смесь.
8) Композиция для использования в твердом препарате, которая описана выше в абзацах с 1) по 7), которая получена способом производства, включающим стадию стимуляции кристаллизации аморфного солифенацина или его аморфной соли.
9) Смесь солифенацина или его соли, причем смесь содержит аморфный и кристаллический солифенацин или его аморфную или кристаллическую соль, где содержание аморфных солифенацина или его соли находится в интервале, не оказывающем влияния на стабильность продукта.
10) Фармацевтическая композиция для использования в твердом препарате, где композиция содержит кристаллический и аморфный солифенацин или его кристаллическую и аморфную соль вместе с ингибитором образования аморфной формы препарата.
11) Фармацевтическая композиция, описанная выше в абзаце 10), в которой ингибитор аморфного формирования является веществом, имеющим этиленоксидную цепь.
12) Фармацевтическая композиция, описанная выше в абзаце 11), где вещество, имеющее этиленоксидную цепь, является полиэтиленгликолем.
При прессовании в соответствии с изготовлением препарата в смеси с добавками, известны методики, включая методику стабилизации (Е)-1-[4-(2-диметиламино)этокси]фенил-2-(4-изопропилфенил)-1-(4-фосфоноокси)фенил-1-бутеном, который обладает таким свойством, что продукты его разложения повышают ускоряющим образом с течением времени под влиянием таких условий, как влага, содержащаяся в такой добавке, повышение контакта внутри таблетки с добавкой при формующем прессовании, и снижение кристалличности при сдавливании, и обладающего эффективностью в качестве терапевтического средства от рака молочных желез, при снижении влажности (Chemical & Pharmaceutical Bulletin, 42(12), 2582(1994)), а также методику стабилизации композиции, содержащей данное соединение, посредством производственного процесса грануляции с расплавлением (JP-A-Hei 9-110698), например методику стабилизации (JP-A-Hei 10-007547) путем по существу безводного процесса производства твердого препарата анилидных соединений в таблетированной форме для применения при рассеянном склерозе, что связано с большими трудностями в точности введения анилидных соединений в качестве главного компонента, из-за того, что при хранении в твердом препарате образуется от 6 до 9% других соединений, помимо главного соединения.
Однако эти документы уровня техники никогда не включали какого-либо описания солифенацина или его соли со строением, полностью отличным от структур описанных соединений, и с физико-химическими или фармакологическими свойствами, полностью отличными от свойств описанных соединений, и никогда не включали какого-либо описания или предположения о проблеме разложения твердого препарата, содержащего аморфную форму, с течением времени или о его стабилизации путем регуляции содержания аморфного вещества ниже соответствующего количества в получаемом твердом препарате.
JP-A-Hei-194218, опубликованный в официальном бюллетене, раскрывает методику стабилизации азотосодержащих гетероциклических алкилфенильных производных с действием против ангиотензина II, у которых снижение содержания ускоряется путем деформации кристаллов из-за перемешивания в ходе изготовления, давления, растирания, нагревания и т.п., происходящих при грануляции или формующем прессовании, причем методика включает стадию смешивания маслянистого вещества с низкой температурой плавления, такого как ПЭГ, с такими алкилфенильными производными для стабилизации получаемого перорального препарата, когда алкилфенильные производные перерабатываются в соответствии с рецептурой в смеси с другими компонентами. В этом случае механизм стабилизации с помощью вещества с низкой температурой плавления осуществляется путем подавления термического разложения активного фармацевтического ингредиента при смешивании маслянистого вещества с низкой температурой плавления до однородности с активным фармацевтическим ингредиентом. Нет описаний о содействии вещества с низкой температурой плавления кристалличности активного фармацевтического ингредиента. Этот механизм стабилизации полностью отличается от механизма стабилизации по данному изобретению.
Кроме того, в International Journal of Pharmaceutics, 216 (2001) 43-49, сообщается о том, что при совместном растворении и кристаллизации лактозы с ПЭГ, выпадающая в осадок лактоза находится в кристаллическом состоянии. Альтернативно в International Journal of Pharmaceutics, 127(1996) 261-272 и International Journal of Pharmaceutics, 262(2003) 125-137, сообщается о том, что при совместном растворении и кристаллизации лекарственного вещества с ПЭГ, лекарственное вещество находится в аморфном состоянии. В случае совместного растворения и кристаллизации активного фармацевтического ингредиента с полимером, таким как ПЭГ, получаемый активный фармацевтический ингредиент часто находится в аморфном состоянии, хотя это зависит от свойств активного фармацевтического ингредиента. Исследовательские работы по смешиванию в целях получения аморфного препарата путем солюбилизации низкорастворимых лекарственных веществ и т.п., в большинстве известны. Все соединения, описанные в этих технических патентах, имеют химические структуры, полностью отличающиеся от структуры солифенацина. Эти источники не включают какое-либо описание солифенацина или его соли с разными физико-химическими и фармакологическими свойствами или какое-либо предположение о данных, на основании которых можно ожидать кристаллизацию или получение аморфного препарата солифенацина путем смешивания с ПЭГ. Даже в отношении стабилизации в известных документах не описан такой состав, или не высказано предположение о таком составе, в котором разложение активного фармацевтического ингредиента с течением времени может быть подавлено путем использования кристаллизации с помощью полимеров, таких как ПЭГ.
Композиция по данному изобретению подробно описана ниже.
«Соль солифенацина» для использования по данному изобретению включает кислотно-аддитивные соли солифенацина с минеральными кислотами, такими как соляная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, органическими кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, лимонная кислота, виннокаменная кислота, угольная кислота, пикриновая кислота, метансульфоновая кислота, этансульфоновая кислота и глютаминовая кислота, и его соли четвертичного аммония, которые описаны в патентном документе 1. В частности, предпочтительным для получения фармацевтического продукта является сукцинат солифенацина. При его использовании достигается значительный стабилизационный эффект в соответствии с данным изобретением. Таким образом, сукцинатная соль является особенно предпочтительным выбором.
«Солифенацин или его соль» для использования согласно данному изобретению легко получить способами, описанными в патентном документе 1, или обычными методами. Количество солифенацина или его соли, которое нужно смешать в данной композиции для использования в твердом препарате в соответствии с данным изобретением, является удовлетворительным, если содержит его активное количество на единицу дозы препарата. Данное количество предпочтительно составляет от 0,001% по массе до 97% по массе, более предпочтительно от примерно 0,05% по массе до 50% по массе, еще предпочтительнее от 0,05% по массе до 20% по массе, наиболее предпочтительно от 0,05% по массе до 10% по массе. Если фармацевтическая композиция по данному изобретению представляет собой частицы, такие как гранулы, количество лекарственного вещества, содержащегося в фармацевтической композиции, состоящей из частиц, выбирают соответственно, обычно в зависимости от типов лекарственного вещества и его фармацевтического применения (показания). Терапевтически активное его количество или его профилактически активное количество является удовлетворительным без конкретного ограничения.
Кроме того, суточная доза солифенацина или его соли предпочтительно составляет от 0,01 мг до 100 мг, более предпочтительно от 0,5 мг до 50 мг, еще предпочтительнее от 0,5 мг до 20 мг, наиболее предпочтительно от 0,5 мг до 10 мг.
«Кристаллы» или «кристаллическое вещество солифенацина или его соли» означает вещество солифенацина или его соли с кристаллической структурой, причем значение этого термина соответствует его значению, используемому в области кристаллографии. В соответствии с данным изобретением понятия «кристаллы» или «кристаллическое» характеризуют вещество с меньшим разложением солифенацина с течением времени. Термины «кристаллы» или «кристаллическое» характеризуют вещество, отличающееся от аморфного вещества со значительным разложением солифенацина с течением времени, при этом данное вещество присутствует в количестве, находящемся за пределами интервала, не оказывающего влияния на стабильность лекарственного продукта в препарате.
В соответствии с данным изобретением, термин «аморфный» солифенацин или его соль, или «аморфное вещество» солифенацина или его соль означает вещество с кристаллографически аморфной структурой. В соответствии с данным изобретением, термин «аморфный солифенацин или его аморфная соль» или «аморфное вещество солифенацина или его соль» означает вещество со значительным разложением солифенацина, когда оно присутствует в количестве, находящемся за пределами интервала, не оказывающего влияния на стабильность лекарственного продукта и, кроме того, означает вещество, отличное от «кристаллов» или «кристаллического вещества» с меньшим разложением солифенацина с течением времени.
Дополнительно, в соответствии с данным изобретением, «содержание аморфного вещества» означает отношение аморфного вещества к общему количеству аморфного и кристаллического солифенацина или его аморфной и кристаллической соли в целом.
В соответствии с данным изобретением, фраза «в интервале, не оказывающем влияния на стабильность лекарственного продукта» означает, что продукт солифенацина или его соли является стабильным в жестких условиях в процессе сбыта коммерческого продукта. В частности, количество основного образуемого продукта разложения солифенацина по отношению к солифенацину или его соли и продуктам их разложения в целом может быть снижено до 0,4% или менее при предварительном испытании на стабильность с использованием условий герметично закрытого сосуда при 40°С и 75% ОВ в течение 6 месяцев.
Таким образом, в соответствии с данным изобретением, специфическое содержание аморфной формы вещества в интервале, не оказывающем влияния на стабильность продукта, составляет 77% или менее, предпочтительно 73% или менее, более предпочтительно 71% или менее, наиболее предпочтительно 63% или менее по отношению к общему количеству аморфного и кристаллического солифенацина, или его аморфной и кристаллической соли, в случае измерения с помощью ближней инфракрасной спектрометрии. Кроме того, солифенацин или его соль с первоначальным содержанием аморфного вещества, превышающим интервал, который не оказывает влияния на стабильность продукта, сразу после получения, но с развивающейся кристаллизацией с течением времени таким образом, что содержание аморфного вещества снижается до пределов данного интервала без влияния на стабильность продукта, также включены в объем данного изобретения. Таким образом, время измерения содержания аморфного вещества не имеет конкретных ограничений. Принимая во внимание, что содержание аморфного вещества должно гарантировать стабильность продукта в ходе его сбыта, предпочтительно, когда содержание аморфного вещества определяют во время начала сбыта продукта или в подходящий срок после него.
В целом, способ оценки содержания аморфной формы солифенацина или его соли в соответствии с данным изобретением, может представлять собой любой метод для идентификации кристаллической структуры солифенацина или его соли в композиции, включая, например, метод порошковой рентгеновской дифракции, метод ДСК, ЯМР в твердом состоянии и ближней инфракрасной спектрометрии. Для определения кристаллической структуры лекарственного вещества при низком содержании в композиции в смеси с другими компонентами кристаллическую структуру предпочтительно определяют ЯМР в твердом состоянии или ближней инфракрасной спектрометрией. Более простым способом определения структуры является ближняя инфракрасная спектрометрия.
В качестве метода определения содержания аморфного вещества сукцината солифенацина использовали, например, метод ближней инфракрасной спектрометрии путем определения спектра при помощи спектрометра для ближней инфракрасной спектрометрии с преобразованием Фурье (Vector 22/N, Bruker Optik GmbH, Германия) (интервал определения: 10000 см-1 до 4000 см-1, разрешение: 2 см-1, число сканирований: 126) и вторичным дифференцированием полученного спектра (конволюционный метод Savitzky-Gollay) для анализа с помощью программного обеспечения, предназначенного для анализа ближнего инфракрасного спектра (например, OPUS, Bruker Optik GmbH, Германия). Перед спектральным определением вещества в таблетке анализировали спектры продуктов, полученных путем смешивания в разном соотношении кристаллического и аморфного сукцината солифенацина, предварительно изготовленных методом распылительной сушки водного раствора сукцината солифенацина, с использованием регрессионного анализа частичным методом наименьших квадратов, чтобы получить стандартную кривую. Посредством наложения спектра таблетки на стандартную кривую можно определить содержание аморфной формы сукцината солифенацина.
В качестве метода измерения содержания аморфной формы сукцината солифенацина ЯМР в твердом состоянии, например, спектр таблетки определяют с помощью прибора для ЯМР в твердом состоянии (например, CMX-300, производимый Chemagnetics, США) (например, использовали пробу: изготовлен из керамики, 7,5 мм проба, время контакта: 9 мсек, время повтора импульса: 38 сек, число обращения образца: 5 кГц). Полученный спектр представляют с обработкой данных (например, функциональное окно показателя, фактор уширения: 30 Гц, трапецеидальное окно, t1=0, t2=0, t3=0,5, t4=0,6). Кроме того, кристаллический и аморфный сукцинат солифенацина, предварительно полученные методом распылительной сушки водного раствора сукцината солифенацина, смешивают вместе в разных соотношениях. Затем, используя лактозу в качестве внутреннего стандарта, определяют пропорцию высоты пика кристаллического сукцината солифенацина для построения стандартной кривой. Помещая отношение пик/высота кристаллического сукцината солифенацина, полученного от таблетки спектра, на стандартную кривую, можно определить содержание кристаллического сукцината солифенацина и содержание его аморфной формы.
«Композиция для использования в твердом препарате» в соответствии с данным изобретением является любой фармацевтической композицией для использования в твердом препарате без конкретного ограничения, причем разложение солифенацина или его соли с течением времени может быть задержано, потому что содержание аморфной формы находится в интервале, не оказывающем влияния на стабильность продукта. Этот термин означает композиции для перорального и парентерального введения, такие как таблетки, пилюли, порошки, гранулы и капсулы.
Фраза «смесь солифенацина или его соли, содержащая аморфный и кристаллический солифенацин или его аморфную и кристаллическую соль, причем содержание аморфной формы находится в интервале, не оказывая влияния на стабильность продукта» означает смесь аморфного и кристаллического солифенацина или его аморфной и кристаллической соли, причем разложение солифенацина или его соли подавляется с течением времени, и смесь по существу содержит аморфный солифенацин или его аморфную соль с содержанием аморфной формы в таком интервале, который не оказывает влияния на стабильность продукта.
В отношении количества солифенацина или его соли, которое нужно смешать в композиции для использования в твердом препарате в соответствии с данным изобретением, удовлетворительно, когда композиция содержит активное количество солифенацина или его соли на стандартную дозу препарата.
Способ производства «композиции с солифенацином или его солью для использования в твердом препарате, причем композиция содержит кристаллический солифенацин или его кристаллическую соль, в которой содержание аморфного вещества солифенацина или его соли находится в интервале, не оказывающем влияния на стабильность продукта» является любым способом с использованием любого растворителя для получения солифенацина или его соли в аморфном состоянии; или способом, включающим стадию снижения контакта солифенацина или его соли с растворителем в ходе растворения солифенацина или его соли в растворителе, для получения солифенацина или его соли в аморфном состоянии для образования аморфного вещества, причем содержание аморфного вещества находится в интервале без влияния на стабильность продукта; или способом, включающим стадию нагревания и/или увлажнения композиции с содержанием аморфного вещества за пределами интервала, не оказывающего влияния на стабильность продукта во время производства или после получения, чтобы привести содержание аморфного вещества в рамки интервала, не оказывающего влияния на стабильность продукта, без конкретного ограничения аппаратуры или средств производства.
В качестве производственных условий для приведения содержания аморфной формы солифенацина или его соли в рамки интервала, который не оказывает влияния на стабильность продукта, можно предложить разные условия. В частности, одним из вариантов производственных условий является типичный процесс производства без использования какого-либо растворителя для получения солифенацина или его соли в аморфном состоянии. Что касается фразы «процесс производства без использования какого-либо растворителя для получения солифенацина или его соли в аморфном состоянии», такой процесс производства включает прямое таблетирование, которое включает смешивание солифенацина или его соли с соответствующими вспомогательными веществами без использования какого-либо растворителя и формующее прессование получаемой смеси, если необходимо получить таблетку. В случае, когда способ включает стадию добавления растворителя, данный способ включает использование растворителя, почти не превращающего солифенацин или его соль в аморфное состояние, причем количество солифенацина или его соли, которое должно растворяться в 1 мл растворителя, составляет менее 0,1 мг, например, ацетона, гексана или их смеси, для влажной грануляции.
В случае способа производства для получения солифенацина или его соли в аморфном состоянии, на стадии добавления растворителя в процессе производства с получением солифенацина или его соли в аморфном состоянии, стабильная композиция с солифенацином или его солью для использования в твердом препарате может быть получена в условиях производства для снижения количества и скорости добавления соответствующих растворителей, таких как вода, которые нужно использовать на данной стадии производства, и в условиях производства для надежного достижения заданного качества получаемых гранул, с приведением содержания аморфной формы в рамки интервала, не оказывающего влияния на стабильность продукта. Растворитель для получения солифенацина или его соли в аморфном состоянии, как упомянуто здесь, означает растворитель, для которого количество солифенацина или его соли, которое можно растворить в 1 мл растворителя, составляет 10 мг или более, например, воду, метанол или этанол, или их смесь, который более предпочтительно является водой. Конкретно, на стадии распыления водного раствора с растворенным в нем в качестве связующего раствора на порошок, содержащий солифенацин или его соль, при производстве композиции для использования в твердом препарате, данный продукт удовлетворительно производится путем получения гранул с содержанием влаги, доведенным до заданного значения или менее при распылении связующего раствора. Содержание влаги в гранулах во время и после распыления связующего раствора доводят до предпочтительно 9% или менее, более предпочтительно до 6% или менее, особенно предпочтительно 5% или менее, наиболее предпочтительно 4% или менее.
Даже когда композицию с содержанием аморфной формы солифенацина или его соли, равным 77% или более, получают не способом производства, описанным выше, а обычным методом влажной грануляции, происходит активизация процесса кристаллизации данной композиции с получением композиции с содержанием аморфной формы солифенацина или его соли в интервале, не оказывающем влияния на стабильность продукта. Активизация процесса кристаллизации, на который ссылаются здесь, может быть осуществлена согласно любому производственному процессу активизации кристаллизации аморфного солифенацина или его соли без конкретного ограничения. Способ производства включает, например, нагревание и/или процесс увлажнения, процесс микроволнового облучения, процесс низкочастотного облучения, процесс ультразвукового облучения и процесс термоэлектронного облучения. Процесс нагревания и/или увлажнения включает производственный процесс выдерживания одного вещества в термостате при постоянной влажности, например, в условиях 25°С и 75% ОВ в течение одной недели с последующей повторной сушкой. Удовлетворительным может быть любой производственный процесс нагревания и/или увлажнения композиции до однородности без конкретного ограничения по аппаратам и средствам производства. Что касается процесса микроволнового облучения, можно использовать, например, длину волны от 10 МГц до 25 ГГц. Кроме того, время обработки зависит от исходной степени кристаллизации и выбранного субстрата. Длины волн, описанные выше, используют для облучения, например, в течение от 10 секунд до 60 минут. Облучение можно выполнять непрерывно или прерывисто. Продолжительность осуществления этих процессов активизации кристаллизации может быть любой, при условии, что получают стабильную композицию с солифенацином или его солью для использования в твердом препарате без конкретного ограничения, при производстве гранул солифенацина или его соли, или после получения композиции для использования в твердом препарате.
Данный процесс производства включает, например, процесс прямого таблетирования, состоящий из смешивания солифенацина или его соли с соответствующей добавкой и формующего прессования смеси, если необходимо получить таблетку, процесс влажной грануляции, состоящий из смешивания солифенацина или его соли с соответствующей добавкой, а затем распыления связующего раствора на получаемую смесь с получением гранул, и процесс грануляции плавлением, состоящий из смешивания солифенацина или его соли с соответствующим веществом с низкой температурой плавления и нагревания и грануляции смеси. Так как солифенацин или его соль обладают свойством сильной агрегации, трудно надежно поддерживать однородность содержания, при этом смесь налипает на пуансоны во время прессования при процессе прямого таблетирования, и очень трудно регулировать количество вещества с низкой температурой плавления, которое нужно растворить при процессе грануляции плавлением, поэтому процесс влажной грануляции является предпочтительным в качестве производственного процесса в соответствии с данным изобретением.
Процесс влажной грануляции включает, например, процесс распыления солифенацина или его соли с помощью распыляющего механизма, затем смешивания получаемого порошка с фармацевтически приемлемыми добавками, такими как наполнители и разрыхлители, распыления связующего раствора на смесь для получения гранул, смешивания улучшающего скольжение вещества с гранулами и прессования смесей в таблетки. В соответствии с данным процессом понятно, что кристаллический солифенацин или его кристаллическую соль растворяют в распыляемом связующем растворе на стадии распыления связующего раствора во время грануляции и затем сушки получаемых гранул для образования аморфного продукта. Путем снижения расхода связующего раствора во время грануляции, что снижает общее количество связующего раствора, или повышения температуры подаваемого воздуха и тому подобного, чтобы снизить растворение солифенацина или его соли в связующем растворе с последующим восстановлением образуемой аморфной формы, может быть представлена фармацевтическая композиция в виде твердого препарата.
Предпочтительный расход связующего раствора зависит от процесса производства и масштаба производства. Расход предпочтительно составляет от 40 до 100 г/мин, более предпочтительно 50-80 г/мин при производстве в 5-кг масштабе при процессе грануляции в псевдоожиженном слое. Предпочтительное общее количество связующего раствора зависит от производственного процесса или масштаба производства. Для производства 5-кг масштаба путем процесса в псевдоожиженном слое его общее количество предпочтительно составляет от 1000 кг до 2500 кг, более предпочтительно от 1500 кг до 2200 кг. Предпочтительная температура подаваемого воздуха меняется в зависимости от процесса производства и масштаба производства. Для производства при 5-кг масштабе путем процесса с псевдоожиженным слоем, однако, температура предпочтительно составляет от 50 до 80°С, более предпочтительно от 60 до 80°С.
Механизм, превращающий в порошок, включает, например, молотковую мельницу, шаровую мельницу, струйную мельницу и коллоидную мельницу. В основном, любой способ фармацевтического превращения в порошок может быть удовлетворительным без конкретного ограничения аппаратуры или средств производства.
Аппарат для смешивания отдельных компонентов, который непрерывно используют для получения порошков, включает, например, смеситель типа V, ленточный смеситель, контейнерный смеситель и устройство для высокоскоростного перемешивания. В основном, может быть удовлетворительным любой процесс, при котором возможно смешивание отдельных компонентов до фармацевтически однородного состояния без конкретных ограничений в аппаратуре или средствах осуществления.
Аппарат для грануляции (процесс) включает, например, высокоскоростной процесс грануляции со смешиванием, способ грануляции в псевдоожиженном слое, экструзионный способ грануляции и способ грануляции вращением. Может быть удовлетворительным любой способ грануляции с использованием связующего раствора, без конкретного ограничения аппаратуры и средств осуществления.
Аппарат для таблетирования включает, например, роторную таблетирующую машину и таблетирующую машину с одним пуансоном. В основном, может быть удовлетворительным способ, при котором возможно получить продукты формующим прессованием (предпочтительно таблетки) без конкретного ограничения аппаратуры и средств осуществления.
Связующее вещество для использования при способе влажной грануляции включает, например, гидроксипропилметилцеллюлозу и поливинилпирролидон. В основном, может быть удовлетворительным любое связующее вещество с фармацевтически приемлемой связующей порошок способностью, без конкретного ограничения.
Обычно количество такого связующего вещества, которое нужно использовать, может быть его количеством для получения фармацевтически приемлемого продукта грануляции, без конкретного ограничения. Обычно это количество составляет от 0,5 до 50% по массе на единицу дозы, предпочтительно от 0,5 до 10% по массе на единицу дозы, более предпочтительно от 2 до 5% по массе на стандартную дозу.
В такой фармацевтической композиции, предназначенной для использования в твердом препарате в соответствии с данным изобретением, кроме того, для рецептуры можно соответственно использовать различные фармацевтические вспомогательные вещества. Такие фармацевтические вспомогательные вещества могут быть любыми фармацевтически приемлемыми и фармакологически приемлемыми вспомогательными веществами без особого ограничения. Например, можно использовать связующие вещества, разрыхляющие, подкисляющие вещества, пенообразующие вещества, искусственные подсластители, вкусо-ароматические добавки, смазывающие вещества, красители, стабилизаторы, буферные вещества, антиоксиданты и поверхностно-активные вещества. Для примера, связующие вещества включают гидроксипропилметилцеллюлозу и аравийскую камедь. Разрыхляющие средства включают, например, кукурузный крахмал, картофельный крахмал, кальцийкармеллозу и натрийкармеллозу. Подкисляющие вещества включают, например, лимонную кислоту, виннокаменную кислоту и яблочную кислоту. Пенообразующие вещества включают, например, бикарбонат натрия. Искусственные подсластители включают, например, сахарин-натрий, двухзамещенный калийглицирризин, аспартам, стевию и соматин. Вкусо-ароматические добавки включают, например, лимонную, лайм-лимонную, апельсиновую и ментоловую. Смазывающие вещества включают, например, стеарат магния, стеарат кальция, сложный эфир сахарозы и жирной кислоты, полиэтиленгликоль, тальк и стеариновую кислоту. Подкрашивающие вещества включают, например, желтый сесквиоксид железа, красный сесквиоксид железа, пищевые желтые №№ 4 и 5, пищевые красные №№ 3 и 102 и пищевой синий № 3. Буферы включают, например, лимонную кислоту, янтарную кислоту, фумаровую кислоту, виннокаменную кислоту, аскорбиновую кислоту или их соли, глютаминовую кислоту, глютамин, глицин, аспарагиновую кислоту, аланин, аргинин или их соли, оксид магния, оксид цинка, гидроксид магния, фосфорную кислоту, борную кислоту или их соли. Антиоксиданты включают, например, аскорбиновую кислоту, дибутилгидрокситолуол, пропилгаллат. Поверхностно-активные вещества включают, например, полисорбат 80, лаурилсульфат натрия и отвержденное полиоксиэтиленом касторовое масло. Один вид или два вида, или более таких вспомогательных веществ можно соответственно добавлять в сочетании в подходящем количестве.
Кроме того, «ингибитор аморфного формирования» (ингибитор формирования аморфной формы) означает вещество, подавляющее образование аморфного солифенацина или его аморфной соли, когда солифенацин или его соль растворяют в растворителе, а затем превращают в твердое состояние путем сушки и тому подобного при изготовлении композиции с солифенацином или его солью для использования в твердом препарате с использованием растворителя.
Ингибитор аморфного формирования предпочтительно является веществом с этиленоксидной цепью. Данное вещество с этиленоксидной цепью, которое упомянуто здесь, является любым веществом с этиленоксидной цепью без конкретного ограничения. Поскольку задача подавления аморфного формирования солифенацина или его соли в соответствии с данным изобретением достигается добавлением вещества, причем данное вещество может быть любым веществом с любым видом молекул или может иметь любую молекулярную массу или любую степень полимеризации без конкретного ограничения. Молекулярная масса предпочтительно находится в интервале средней молекулярной массы от 400 до 1000000, более предпочтительно в интервале средней молекулярной массы от 2000 до 200000. В качестве вещества с этиленоксидной цепью в смеси можно использовать два или более видов веществ. В соответствии с данным изобретением, в частности, вещество с этиленоксидной цепью включает, например, ПЭГ, полиэтиленоксид, блоксополимер полиоксиэтилена и полиоксипропилена, отвержденное полиоксиэтиленом касторовое масло (далее обозначаемое аббревиатурой ТКМ) и сложный эфир полиэтиленгликоля и жирной кислоты. Из них предпочтительны, в частности, ПЭГ, блок-сополимер полиоксиэтилена и полиоксипропилена или ТКМ. Более предпочтителен ПЭГ.
Блок-сополимер полиоксиэтилена и полиоксипропилена в соответствии с данным изобретением может быть сополимером пропиленоксида и этиленоксида. В зависимости от соотношения в композиции, существуют различные такие сополимеры. Может быть пригодным любой такой сополимер со свойством подавлять аморфное формирование солифенацина или его соли в составе композиции. В частности, например, можно использовать полиоксиэтилен-(105)-полиоксипропилен-(5)-гликоль и полиоксиэтилен-(160)-полиоксипропилен-(30)-гликоль (под другим названием, Pluronic F68).
Ингибитор аморфного препарата используют в количестве, равном предпочтительно от 0,1 до 90% по массе, более предпочтительно от 1 до 60% по массе по отношению к общей массе препарата. Когда ПЭГ используют в качестве связующего вещества для использования в процессе влажной грануляции путем растворения ПЭГ в дистиллированной воде, его количество предпочтительно составляет от 3 до 20% по массе, более предпочтительно от 4 до 10% по массе от общей массы порошка, который нужно гранулировать. Когда количество ингибитора аморфного формирования определяли на одну массовую часть кристаллического и аморфного солифенацина или его соли, причем данное количество предпочтительно представлено в отношении 0,001-100000% по массе, более предпочтительно в отношении от 0,1 до 1000% по массе, еще более предпочтительно в отношении от 10 до 600% по массе.
В соответствии с данным изобретением, фраза «содержащий» означает солифенацин или его соль в качестве активного фармацевтического ингредиента находится в смеси с ингибитором аморфного формирования. Предпочтительно солифенацин или его соль находится в контакте с ингибитором аморфного формирования, так что солифенацин или его соль распределены в структуре смеси. Как в случае использования фармацевтической композиции в качестве покрывающего средства для препарата с солифенацином, где активный фармацевтический ингредиент, представляющий собой солифенацин или его соль, не находится в контакте с или в смеси с таким ингибитором аморфного формирования, таким образом, что он существует в локализованном состоянии (например, ингибитор аморфного формирования в соответствии с данным изобретением (ПЭГ)), фармацевтические препараты, например, с такой структурой, что солифенацин или его соль не находятся в физическом контакте с ингибитором аморфного формирования в промежуточном слое с использованием других добавок и тому подобного не допускаются.
Фармацевтическая композиция солифенацина или его соли для использования в твердом препарате в соответствии с данным изобретением далее описывается подробно. В следующих примерах и сравнительных примерах данное изобретение описано более подробно. Однако данное изобретение не должно рассматриваться как ограниченное данными примерами.
[Справочный пример 1]
60 частей сукцината солифенацина растворяли в 140 частях воды для сушки распылением в распылительной сушилке (DL-41, производимой Yamato Science) с получением высушенного распылением продукта.
Кристалличность получаемого высушенного распылением продукта сукцината солифенацина определяли с помощью аппарата для рентгеновской дифракции (RINT 1400, производимый Rigaku Denki). Наблюдали картину гало, показывающую, что продукт был аморфным.
<Стабильность при хранении кристаллического и аморфного продукта>
Результаты по стабильности кристаллического продукта перед сушкой распылением и аморфного продукта представлены в таблице 1. Количество продуктов разложения с течением времени при хранении определяли высокоэффективной жидкостной хроматографией. Представлено максимальное количество отдельных продуктов разложения. Через короткий период времени после начала хранения образовывались продукты разложения аморфного продукта сукцината солифенацина, и стабильность его была хуже, чем стабильность кристаллического продукта. Поэтому главной причиной разложения активного фармацевтического ингредиента с течением времени был преимущественно аморфный сукцинат солифенацина, образованный в процессе производства препарата.
Результаты стабильности кристаллического продукта и аморфного продукта сукцината солифенацина
Условия хранения: 40°С и 75% ОВ
Форма упаковки: стеклянный сосуд
Предметы испытания: родственные вещества (индивидуальные максимальные значения)
продукт
закрытый
стеклянный сосуд
стеклянный сосуд
Пример 1
204 части гидроксипропилметилцеллюлозы 2910 растворяли и перемешивали в 1836 частях воды с помощью мешалки с пневматическим двигателем (AM-GC-1, производимой Chuo-Rika Machine), с получением связующего раствора (с концентрацией 10,0 в/о %). Затем смешивали 340 частей сукцината солифенацина и
1360 частей лактозы. Затем полученную смесь превращали в порошок в молотковой мельнице (мельница для образцов АР-S с использованием 1-мм сита, производимая Hosokawa Micron). К смешанному и превращенному в порошок продукту добавляли 2125 частей лактозы и 1020 частей кукурузного крахмала и затем загружали в гранулятор в псевдоожиженном слое (WSG-5, производимый Powlec) для распылении связующего раствора при температуре подаваемого воздуха, равной 65°С, расходе воздуха 4 м3/мин, расходе связующего раствора 75 г/мин, давлении воздуха для распыления, равном 1,5 кг/см2, и цикле распыления/перемешивания в 30 секунд/10 секунд во время грануляции. Содержание влаги в гранулах после распыления всего объема связующего раствора составляла 3,9%. После грануляции гранулы сушили при температуре подаваемого воздуха 50°С в течение 10 минут с получением гранул данного изобретения. Добавляли 12 частей стеарата магния к 1188 частям высушенного гранулированного продукта при смешивании с помощью смесителя (типа DC, производимого Yamanouchi). Затем полученную смесь прессовали с помощью роторной таблетирующей машины (НТ Р-22, производимой Hata Tekkosho) с пуансонами диаметром 7,5 мм при таблетирующем давлении примерно 700 кгс/пуансон до веса таблетки 150 мг. Кроме того, на 800 частей полученных таблеток покрывали распыляемым раствором, изготовленным растворением/диспергированием 84,3 части гидроксипропилметилцеллюлозы, 15,8 частей макрогола 6000, 25,3 части талька, 10,5 части оксида титана и 0,03 части красного сесквиоксида железа в 1223 частях с использованием вентилируемой машины для нанесения покрытия (машина для нанесения высококачественного покрытия НСТ-30, производимая Freund Industry Corporation) при температуре подаваемого воздуха 60°С, скорости вращения чаши 13 об/мин и расходе жидкого покрытия 5 г/мин до пропорции 2,7% покрывающего средства по отношению к весу таблетки с получением покрытых пленкой таблеток данного изобретения.
Пример 2
Грануляцию производили распылением связующего раствора в следующих условиях грануляции с помощью машины для грануляции в псевдоожиженном слое: при температуре подаваемого воздуха составляла 65°С; расход воздуха составлял 4 м3/мин; расход связующего раствора составлял 75 г/мин; давление подаваемого воздуха составляло 0,7 кг/см2; и цикл распыления/перемешивания составлял 30 секунд/10 секунд. Содержание влаги в гранулах составляло 5,5% после распыления всего связующего раствора. После грануляции получали покрытые пленкой таблетки данного изобретения методом, описанным в примере 1.
Пример 3
Грануляцию производили путем распыления связующего раствора в следующих условиях грануляции с помощью машины для грануляции в псевдоожиженном слое: температура подаваемого воздуха составляла 65°С; расход воздуха составлял 4 м3/мин; расход связующего раствора составлял 95 г/мин; давление подаваемого воздуха составляло 1,5 кг/см2; и цикл распыления/перемешивания составлял 30 секунд/10 секунд. Содержание влаги в гранулах составляло 5,7% после распыления всего связующего раствора. После грануляции получали покрытые пленкой таблетки данного изобретения методом, описанным в примере 1.
Пример 4
Грануляцию производили путем распыления связующего раствора в следующих условиях грануляции с помощью машины для грануляции в псевдоожиженном слое: температура подаваемого воздуха составляла 55°С; расход воздуха составлял 4 м3/мин; расход связующего раствора составлял 75 г/мин; давление подаваемого воздуха составляло 1,5 кг/см2; и цикл распыления/перемешивания составлял 30 секунд/10 секунд. Содержание влаги в гранулах составляло 8,4% после распыления всего связующего раствора. После грануляции получали покрытые пленкой таблетки данного изобретения методом, описанным в примере 1.
[Сравнительный пример 1]
Грануляцию производили путем распыления связующего раствора в следующих условиях грануляции с помощью машины для грануляции в псевдоожиженном слое: температура подаваемого воздуха составляла 65°С; расход воздуха составлял 4 м3/мин; расход связующего раствора составлял 115 г/мин; давление подаваемого воздуха составляло 1,5 кг/см2; и цикл распыления/перемешивания составлял 30 секунд/10 секунд. Содержание влаги в гранулах составляло 10,6% после распыления всего связующего раствора. После грануляции получали покрытые пленкой таблетки данного изобретения методом, описанным в примере 1.
[Сравнительный пример 2]
Грануляцию производили путем распыления связующего раствора в следующих условиях грануляции с помощью машины для грануляции в псевдоожиженном слое: температура подаваемого воздуха составляла 65°С; расход воздуха составлял 3 м3/мин; расход связующего раствора составлял 75 г/мин; давление подаваемого воздуха составляло 1,5 кг/см2; и цикл распыления/перемешивания составлял 30 секунд/10 секунд. Содержание влаги в гранулах составляло 10,6% после распыления всего связующего раствора. После грануляции получали покрытые пленкой таблетки данного изобретения методом, описанным в примере 1.
[Сравнительный пример 3]
Грануляцию производили путем распыления связующего раствора в следующих условиях грануляции с помощью машины для грануляции в псевдоожиженном слое: температура подаваемого воздуха составляла 45°С; расход воздуха составлял 4 м3/мин; расход связующего раствора составлял 75 г/мин; давление подаваемого воздуха составляло 1,5 кг/см2; и цикл распыления/перемешивания составлял 30 секунд/10 секунд. Содержание влаги в гранулах составляло 10,8% после распыления всего связующего раствора. После грануляции получали покрытые пленкой таблетки данного изобретения методом, описанным в примере 1.
<Определение содержания влаги в гранулах во время грануляции, содержания аморфного сукцината солифенацина в таблетке и количества продуктов разложения после хранения с течением времени>
Результаты содержания влаги в гранулах после распыления связующего раствора, содержания аморфного сукцината солифенацина и предварительные результаты по стабильности при использовании условий в герметично закрытом сосуде при 40°С и 75% ОВ в течение 6 месяцев представлены в таблице 2 при модификации условий производства во время грануляции. Содержание влаги в гранулах после распыления связующего раствора показано как значение, определяемое по потере по методу высушивания (80°С, 2 часа), тогда как содержание аморфного сукцината солифенацина показано как значение, определяемое спектрометрией ближнего инфракрасного излучения. Спектрометрию ближнего инфракрасного излучения выполняли путем определения спектра спектрометром в ближней инфракрасной области с преобразованием Фурье (Vector 22/N, Bruker Optik GmbH, Germany) (интервал измерения 10000 см-1 - 4000 см-1, разрешение: 2 см-1, число сканирования: 126). Полученный спектр был вторичной производной (конволюционный метод Savitzky-Gollay) и проанализированным с помощью программного обеспечения для анализа спектра в ближней инфракрасной области (OPUS, Bruker Optik GmbH, Германия). Перед определением спектра таблетки спектры продуктов, полученных смешиванием кристаллического и аморфного сукцината солифенацина при разных соотношениях, предварительно изготовленные путем распылительной сушки водного раствора сукцината солифенацина, подвергали регрессионному анализу частичным методом наименьших квадратов для получения стандартной кривой. Спектр таблетки помещали на стандартную кривую для определения количества аморфного сукцината солифенацина. Дополнительно определяли количество продуктов разложения высокоэффективной жидкостной хроматографией после хранения в течение 6 месяцев, применяя условия в герметично закрытом сосуде при 40°С и 75% ОВ. Среди количеств продуктов разложения, определенных таким образом, показано количество образуемого главного продукта разложения (F1), по отношению к общему количеству сукцината солифенацина и продуктов его разложения. Используя отношение образуемого F1 как показатель, оценивали стабильность сукцината солифенацина.
<Содержание влаги в гранулах во время грануляции в 10 мг таблетке сукцината солифенацина, содержание аморфного сукцината солифенацина в таблетке и предварительное испытание на стабильность (в течение 6 месяцев)>
испытания
1
2
3
4
пример 1
пример 2
пример 3
*2: пропорция главного продукта разложения сукцината солифенацина по отношению к общему количеству сукцината солифенацина и продуктов его разложения
Как показано в таблице 2, 10-мг таблетки, полученные при конкретных условиях производства, имеют разное содержание влаги при грануляции. Так как содержание влаги в гранулах было ниже, обычно содержание аморфной формы в таблетках было ниже.
В сравнительных примерах с 1 по 3, как в основных производственных процессах содержание влаги в гранулах после распыления связующих растворов было больше, чем в примерах. Поэтому содержание аморфного солифенацина было таким большим, как 90% и более. Кроме того, количество главного продукта разложения F1 по отношению к сукцинату солифенацина и продуктам его разложения в целом превышает 0,4%. Это показывает, что существует серьезная проблема при получении композиции с солифенацином или его солью, которая является стабильной с течением времени для клинической практики.
В случае удерживания содержания влаги в гранулах на наименьшем возможном уровне, как в примерах с 1 по 4, альтернативно содержание аморфного вещества под контролем содержания влаги составляло 77% или менее, тогда как количество главного продукта разложения F1 по отношению к сукцинату солифенацина и продуктам его разложения в целом составляло 0,4% или менее.
Таким образом, препарат с солифенацином, стабильный во времени, мог быть получен путем удерживания содержания аморфной формы до 77% или менее в препарате, содержащем солифенацин или его соль.
Пример 5
270 частей ПЭГ (под торговым названием Macrogol 6000, производимый Sanyo Chemical) растворяли в 1080 частях воды и перемешивали мешалкой с пневматическим двигателем (AM-GC-1, производимая Chuo Rika) для получения связующего раствора (при концентрации 20,0 в/о %). Затем смешивали 90 частей сукцината солифенацина и 360 частей лактозы (под торговым названием лактоза 200М, производимой DMV). Затем полученную смесь превращали в порошок с помощью молотковой мельницы (мельница для образца АР-S с использованием 1 мм сита, производимая Hosokawa Micron). К смешанному и превращенному в порошок продукту добавляли 3906 частей лактозы и кристаллической целлюлозы (под торговым названием Avicel PH 102, производимой Asahi Chemical), и затем загружали в машину для грануляции в псевдоожиженном слое (WSG-5, производимую Powlec) для распыления связующего раствора при температуре подаваемого воздуха 70°С, расходе связующего раствора 100 г/мин, давлении распыляющего воздуха 1,5 кг/см2 и цикле распыления/перемешивания 30 секунд/10 секунд для грануляции. После грануляции гранулы сушили при температуре подаваемого воздуха 70°С в течение 10 минут с получением гранул данного изобретения. К 1188 частям высушенного гранулированного продукта добавляли 12 частей стеарата магния (производимого NOF) для смешивания с помощью смесителя (типа DC, производимого Yamanouchi). Затем получаемую смесь прессовали с помощью роторной таблетирующей машины (НТ Р-22, производимой Hata Tekkosho) с пуансонами диаметром 5,5 мм при давлении прессования примерно 500 кгс/пуансон с получением таблеток весом 60 мг. Далее на 900 частей получаемых таблеток распыляли раствор, полученный растворением/диспергированием 18,6 частей ГПМЦ 2910 (под торговым названием ТС-5R, производимой Shin-estu Chemical), 3,5 части ПЭГ (под торговым названием Macrogol 6000, производимого Sanyo Chemical), 5,6 части талька (производимого Kihara Chemical), 2,3 части оксида титана (производимого Freund Industry Corporation) и 0,05 части красного сесквиоксида железа в 270 частях воды, и создавали покрытие, используя вентилируемую машину для нанесения покрытия (устройство для нанесения высококачественного покрытия НСТ-30, производимое Freund Industry Corporation) при температуре подаваемого воздуха 60°С, при скорости вращения чаши 13 об/мин, и расходе покрывающей жидкости 5 г/мин до 3,3%-ной пропорции покрывающего компонента в весе таблетки с получением таблеток данного изобретения с покрытием.
[Сравнительный пример 4]
Растворяли 180 частей ГПМЦ 2910 (под торговым названием ТС-5R, производимой Shin-estu Chemical) в 1620 частях воды и перемешивали с помощью мешалки с пневматическим двигателем (АМ-GC-1, производимой Chuo Rika) с получением раствора связующего вещества (при концентрации 10,0 в/о%). Затем смешивали 75 частей сукцината солифенацина и 300 частей лактозы. Затем полученную смесь превращали в порошок с помощью молотковой мельницы (мельница для образца АР-S с использованием 1 мм сита, производимая Hosokawa Micron). К смешанному и превращенному в порошок продукту добавляли 2700 частей лактозы и 900 частей кукурузного крахмала (производимого Nihon Shokuhin) и затем загружали в аппарат для грануляции в псевдоожиженном слое (WSG-5, производимый Powlec) для распыления связующего раствора при температуре подаваемого воздуха 60°С, степени распыления связующего раствора 75 г/мин, давлении распыляющего воздуха 1,5 кг/см2 и цикле распыления/перемешивания 30 секунд/10 секунд для грануляции. После грануляции гранулы сушили при температуре подаваемого воздуха 60°С в течение 10 минут с получением гранул данного изобретения. К 1188 частям высушенных гранул добавляли 12 частей стеарата магния (производимого NOF) для смешивания с помощью смесителя (типа DC, производимого Yamanouchi). Затем полученную смесь прессовали с помощью роторной таблетирующей машины (НТ Р-22, производимой Hata Tekkosho) c пуансонами диаметром 5,5 мм при давлении прессования примерно 500 кгс/пуансон с получением таблеток весом 60 мг. Далее, 900 частей полученных таблеток покрывали способом, показанным в примере 5 для получения покрытых пленкой таблеток данного изобретения.
[Сравнительный пример 5]
108 частей картофельного крахмала (производимого Nihon Shokuhin) добавляли к 2592 частям воды и затем растворяли в ней при нагревании до 80°С. Затем полученный раствор охлаждали до комнатной температуры для получения связующего раствора. Затем смешивали 90 частей сукцината солифенацина и 360 частей лактозы. Затем полученную смесь превращали в порошок с помощью молотковой мельницы (мельница для образца АР-S с использованием 1 мм сита, производимая Hosokawa Micron). К смешанному и превращенному в порошок продукту добавляли 3708 частей лактозы и 1080 частей кукурузного крахмала и затем загружали в аппарат для грануляции в псевдоожиженном слое (WSG-5, производимый Powlec) для распыления связующего раствора при температуре подаваемого воздуха 70°С, степени распыления связующего раствора 90 г/мин, давлении распыляющего воздуха 1,5 кг/см2 и цикле распыления/перемешивания 30 секунд/10 секунд для грануляции. После грануляции гранулы сушили при температуре подаваемого воздуха 70°С в течение 10 минут с получением гранул данного изобретения. К 129 частям высушенного гранулированного продукта добавляли 13 частей стеарата магния для смешивания с помощью смесителя (типа DC, производимого Yamanouchi). Затем полученную смесь прессовали с помощью роторной таблетирующей машины (НТ Р-22, производимой Hata Tekkosho) c пуансонами диаметром 5,5 мм при давлении прессования примерно 500 кгс/пуансон с получением таблеток весом 60 мг. Далее, 800 частей полученных таблеток опрыскивали и покрывали способом, показанным в примере 5 для получения покрытых пленкой таблеток данного изобретения.
<Результаты предварительного испытания стабильности препарата солифенацина, который получен способом влажной грануляции>
Было выполнено предварительное испытание стабильности таблеток сукцината солифенацина, произведенных со связующим раствором, отличающимся от растворов, использованных при данной грануляции (в условиях 25°С и 60% ОВ). Результаты представлены в таблице 3.
Таблетки из сравнительного примера 4, которые получали, используя ГПМЦ, не могли быть достаточно стабилизированы. Даже когда оценивали другие виды связующего вещества, крахмал не мог улучшить стабильность, как показано в сравнительном примере 5. Как показано в примере 5, между тем использование ПЭГ могло улучшать стабильность. Было показано, что даже в более жестких условиях температуры и влажности, чем условия 25°С и 60% ОВ, стабильность препарата солифенацина могла сохраняться.
Результаты предварительного испытания стабильности таблеток сукцината солифенацина
Условия хранения: 25°С и 60% ОВ
Форма упаковки: упакованы во флаконы из ПЭВП с металлической крышкой
Объекты испытания: родственные вещества (количество главного образуемого продукта разложения F1)
Возможность промышленного применения
Техническая сущность данного изобретения состоит в выяснении причины разложения активного фармацевтического ингредиента в препарате, содержащем солифенацин или его соль, с течением времени, который представлял собой аморфный солифенацин или его аморфную соль. Путем изготовления такого препарата при регулировке содержания аморфной формы в нем до заданного значения или менее может быть впервые получен стабильный твердый препарат с солифенацином или его солью, который обладает большими преимуществами для промышленности. В препарате, содержащем солифенацин или его соль, дополнительно содержится ингибитор образования аморфного вещества, чтобы сделать возможным получение стабильной фармацевтической композиции для использования в твердом виде, которая представляет большие преимущества для промышленности.
Таким образом, данное изобретение пригодно в качестве способа, делающего возможным получение стабильной композиции солифенацина или его соли для использования в твердом препарате, из которого была очень желательна разработка фармацевтического продукта для использования при поллакурии и недержании мочи.
| название | год | авторы | номер документа |
|---|---|---|---|
| СТАБИЛЬНАЯ, СОСТОЯЩАЯ ИЗ ЧАСТИЦ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ СОЛИФЕНАЦИН ИЛИ ЕГО СОЛЬ | 2005 |
|
RU2397767C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2018 |
|
RU2767872C2 |
| СТАБИЛЬНАЯ КОМПОЗИЦИЯ АМОРФНЫХ СОЛЕЙ ПЕРИНДОПРИЛА, СПОСОБ ЕЕ ПОЛУЧЕНИЯ, В ЧАСТНОСТИ ПРОМЫШЛЕННОГО ПОЛУЧЕНИЯ, И ЕЕ ПРИМЕНЕНИЕ В ТЕРАПИИ ГИПЕРТЕНЗИИ | 2006 |
|
RU2429878C2 |
| КОМПОЗИЦИЯ НЕНУКЛЕОЗИДНОГО ИНГИБИТОРА ОБРАТНОЙ ТРАНСКРИПТАЗЫ | 2014 |
|
RU2661399C1 |
| ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2017 |
|
RU2764849C2 |
| ТВЕРДАЯ МОЛЕКУЛЯРНАЯ ДИСПЕРСИЯ | 2012 |
|
RU2600816C2 |
| ТВЕРДАЯ ДИСПЕРСИЯ ТОЛВАПТАНА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2011 |
|
RU2579745C2 |
| ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ | 2018 |
|
RU2809144C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ ДЛЯ ЛЕЧЕНИЯ ОПОСРЕДОВАННЫХ CFTR ЗАБОЛЕВАНИЙ | 2014 |
|
RU2718044C2 |
| ТВЕРДАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ) И СПОСОБ КОНТРОЛЯ КОНЦЕНТРАЦИИ ГЛЮКОЗЫ С ЕЕ ПОМОЩЬЮ, СПОСОБ ПОЛУЧЕНИЯ ТВЕРДОЙ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ (ВАРИАНТЫ), ТАБЛЕТКА (ВАРИАНТЫ) И СПОСОБ ПОЛУЧЕНИЯ АМФОРНЫХ ЧАСТИЦ | 2008 |
|
RU2453332C2 |
Изобретение относится к лекарственным средствам и касается способа получения композиции солифенацина или его соли для использования в твердом препарате, который включает по меньшей мере одну стадию, выбранную из группы, состоящей из (i) стадии влажной грануляции с использованием растворителя для солифенацина или его соли, причем количество солифенацина или его соли, которое должно растворяться в 1 мл растворителя, составляет менее 0,1 мг, (ii) стадии снижения количества или скорости добавления растворителя, если растворитель переводит солифенацин или его соль в аморфное состояние, причем количество солифенацина или его соли, которое должно растворяться в 1 мл растворителя, составляет 10 мг или более, и (iii) стадии активизации процесса кристаллизации композиции, полученной с помощью обычного способа влажной грануляции. Также раскрыта фармацевтическая композиция для использования в твердом препарате, проявляющая селективное антагонистическое действие против мускариновых М3 рецепторов. Предложенный способ позволяет повысить стабильность композиций, содержащих солифенацин или его соль. 2 н. и 10 з.п. ф-лы, 3 табл.
1. Способ получения композиции солифенацина или его соли для использования в твердом препарате, который включает по меньшей мере одну стадию, выбранную из группы, состоящей из (i) стадии влажной грануляции с использованием растворителя для солифенацина или его соли, причем количество солифенацина или его соли, которое должно растворяться в 1 мл растворителя, составляет менее 0,1 мг, (ii) стадии снижения количества или скорости добавления растворителя, если растворитель переводит солифенацин или его соль в аморфное состояние, причем количество солифенацина или его соли, которое должно растворяться в 1 мл растворителя, составляет 10 мг или более, и (iii) стадии активизации процесса кристаллизации композиции, полученной с помощью обычного способа влажной грануляции.
2. Способ получения композиции солифенацина или его соли для использования в твердом препарате по п.1, где содержание аморфной формы составляет 77% или менее.
3. Способ получения композиции солифенацина или его соли для использования в твердом препарате по п.1 или 2, который включает стадию прямого прессования с получением гранулированной композиции для использования в твердом препарате.
4. Способ получения композиции солифенацина или его соли для использования в твердом препарате по п.1 или 2, в котором растворитель представляет собой ацетон, гексан или их смесь, причем количество солифенацина или его соли, которое должно растворяться в 1 мл растворителя, составляет менее 0,1 мг.
5. Способ получения композиции солифенацина или его соли для использования в твердом препарате по п.1 или 2, в котором растворитель представляет собой воду, метанол, этанол или их смесь, причем количество солифенацина или его соли, которое должно растворяться в 1 мл растворителя, составляет 10 мг или более.
6. Способ получения композиции солифенацина или его соли для использования в твердом препарате по п.1, в котором стадия снижения количества или скорости добавления растворителя предназначена для доведения содержания влаги гранул в связующем растворе до заданного значения.
7. Способ получения композиции солифенацина или его соли для использования в твердом препарате по п.6, в котором содержание влаги в гранулах составляет 9% или менее.
8. Способ получения композиции солифенацина или его соли для использования в твердом препарате по п.1, в котором стадия (iii) активизации процесса кристаллизации представляет собой по меньшей мере одну стадию, выбранную из группы, состоящей из процесса нагревания и/или процесса увлажнения, процесса микроволнового облучения, процесса низкочастотного облучения, процесса ультразвукового облучения и процесса термоэлектронного облучения.
9. Способ получения композиции солифенацина или его соли для использования в твердом препарате по п.8, в котором процессы нагревания и/или увлажнения проводятся при условиях 25°С и относительной влажности 75%.
10. Фармацевтическая композиция для использования в твердом препарате, где композиция проявляет селективное антагонистическое действие против мускариновых М3 рецепторов, содержащая кристаллический или аморфный солифенацин или его кристаллическую или аморфную соль вместе с ингибитором аморфного формирования, причем ингибитором аморфного формирования является вещество, имеющее этиленоксидную цепь.
11. Фармацевтическая композиция по п.10, в которой вещество с этиленоксидной цепью включает, например, полиэтиленгликоль, полиэтиленоксид, блоксополимер полиоксиэтилена и полиоксипропилена, отвержденное полиоксиэтиленом касторовое масло или сложный эфир полиэтиленгликоля и жирной кислоты.
12. Фармацевтическая композиция по п.11, в которой вещество, имеющее этиленоксидную цепь, представляет собой полиэтиленгликоль.
Приоритет по пунктам:
Пункты 1-9 по US 60/556025 и US 60638388;
Пункты 10-12 по US 60/556025.
| WO 9620194 A1, 04.07.1996 | |||
| КРИСТАЛЛИЧЕСКАЯ СВОБОДНАЯ ЦЕФТИОФУРОВАЯ КИСЛОТА, КОМПОЗИЦИЯ НА ЕЕ ОСНОВЕ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1994 |
|
RU2136685C1 |
| КРИСТАЛЛИЧЕСКИЙ ПИВАЛОИЛОКСИМЕТИЛ (IR, 5S, 6S)-[(4R)-2-ОКСО-4-ПИРРОЛИДИНИЛТИО]-6-[(IR)-1-ГИДРОКСИЭТИЛ]-1-МЕТИЛ-1-КАРБАПЕН-2-ЕМ-3-КАРБОКСИЛАТ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1993 |
|
RU2090567C1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| WO 03099268 A1, 04.12.2003 | |||
| CLAES AHLNECK "The molecular basis of moisture effects on the physical and chemical stability of drugs in the solid state" International journal of Pharmaceutics, 1990, 62, p.87-95. | |||

