Настоящее изобретение относится к области исследования физико-химических свойств и электронного строения веществ, а именно к области исследования параметров межатомных и межмолекулярных взаимодействий ван-дер-ваальсовых систем, в том числе нано- и мезоструктур.
Описание межатомных и межмолекулярных взаимодействий достигается с помощью так называемых потенциальных функций или межатомных и межмолекулярных потенциалов. Обычно информацию о потенциалах получают на основании данных различных макроскопических свойств вещества (Каплан И.Г. Введение в теорию межмолекулярных взаимодействий. - М.: Наука, 1982, С.245). Данный подход основан на измерении отклонения соответствующей величины свойства реального вещества относительно того же свойства, но относящегося к идеальному состоянию вещества. В качестве измеряемых величин могут быть приняты теплофизические величины, вязкость, диффузия, параметры расстояния частиц и т.п. Потенциальная функция представляет собой зависимость энергии межатомных или межмолекулярных взаимодействий от расстояния между микрочастицами. Однако потенциалы, получаемые по данным одного свойства, могут существенно отличаться от потенциалов, получаемых по данным другого свойства, поскольку разные физические свойства зачастую чувствительны к разным участкам потенциальной кривой по разному. Поэтому для построения корректного потенциала необходима трудоемкая процедура оптимизации максимально возможного числа экспериментальных данных. При этом достоверность получаемой информации неизбежно снижается.
Наиболее близким к предлагаемому способу определения потенциалов межатомных и межмолекулярных взаимодействий являются модельные потенциалы типа потенциала Леннарда-Джонса (Каплан И.Г. Введение в теорию межмолекулярных взаимодействий. - М.: Наука, С.220), в частности потенциал (12-6)

где ε - глубина потенциальной Ямы; параметр σ, соответствующий нулевой энергии u. Положение минимума потенциальной функции соответствует равновесному расстоянию 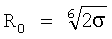 . Здесь притягивательная ветвь функции u(R) описывается энергией дисперсионного взаимодействия, а отталкивательная ветвь - первым членам, характеризующим взаимную непроницаемость атомов.
. Здесь притягивательная ветвь функции u(R) описывается энергией дисперсионного взаимодействия, а отталкивательная ветвь - первым членам, характеризующим взаимную непроницаемость атомов.
Общим недостатком всех модельных потенциалов, в том числе потенциала Леннарда-Джонса, является необходимость введения подгоночных параметров. В уравнении (1) таковыми являются параметры σ и ε. Необходимость подгоночных параметров является следствием неопределенности электронного строения микрочастиц 0 в области связанного состояния микрочастиц.
Сущность предлагаемого изобретения заключается в следующем. Предлагаемая потенциальная функция u(R) сохраняет свой стандартный вид, т.е. включает притягивательную uпр(R) и отталкивательную uот(R) ветви. Притягивательная ветвь uпр(R), как и в известных потенциалах, построена на основе представлений о дисперсионной природе взаимодействия между двумя нейтральными атомами или молекулами [1]. Принципиально новым является построение отталкивательной ветви uот(R) потенциала. В его основе лежат две идеи: конечные и точно заданные размеры атомов, определяемые радиусом аe [2], и конечные заряды q на поверхности атомов. Наличие этих двух параметров предопределяет характер и кулоновскую природу взаимного отталкивания атомов (и молекул), так что
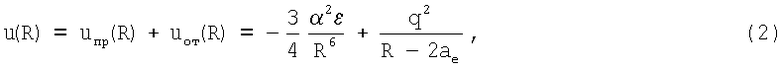
где α - поляризуемость микрочастиц, ε - энергия связи электронов внешней оболочки атомов с их ядром, q - эффективный заряд атомов, а - поляризационный радиус. Поляризационный радиус аe - это атомная константа, определяющая наибольшее расстояние внешних электронов по отношению к ядру; это измеряемая величина [2].
Эффективный заряд q - это реальный заряд на поверхности атома, экспериментально обнаруживаемый в виде энергии сродства к электрону (или протону). Сродство к электрону - это всеобщее свойство атомов, проявляющееся в их способности притягивать к себе электроны (или протоны) [3, С.411]. Конечные заряды q на поверхности атомов можно объяснить эффектом неполного экранирования заряда ядра, как это имеет место у большинства атомов таблицы Менделеева, или эффектом переэкранирования (когда на поверхности образуется небольшой отрицательный заряд), как в случае с атомами II и VIII групп таблицы Менделеева [4, С.20]. Заряд q образуется в соответствии с теоремой Гаусса, согласно которой заряд на оболочке равен сумме всех положительных q+ и отрицательных q- зарядов внутри данной оболочки, так что q=q++q-≠0. Для систем со сферической симметрией, к числу которых в первую очередь относятся атомы, согласно теореме Гаусса такой суммарный заряд эквивалентен точечному заряду q, помещенному в центр атома [5, С.42]. Этот заряд обладает электрическим потенциалом 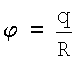 и напряженностью электрического поля
и напряженностью электрического поля 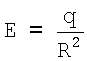 , благодаря которым у атомов и обнаруживается сродство к электрону или протону. Наибольшие значения энергий сродства имеют галогены (порядка 3 эВ), а наименьшие - атомы благородных газов (<0,1 эВ) [4, С.20].
, благодаря которым у атомов и обнаруживается сродство к электрону или протону. Наибольшие значения энергий сродства имеют галогены (порядка 3 эВ), а наименьшие - атомы благородных газов (<0,1 эВ) [4, С.20].
Наличие конечного заряда q на поверхности атомов, как уже отмечалось выше, предопределяет естественный кулоновский характер взаимного отталкивания атомов, представленного с помощью отталкивательной ветви uот(R) потенциала (2). Введение в потенциал (2) поляризационного радиуса ae также имеет принципиальное значение.
Положение электрона по отношению к ядру строго задано и определяется энергией связи ε этого электрона с ядром, так что [2, 6] εae=const=e2, где е - элементарный заряд. Радиус аe имеет смысл радиуса атома, задающего границу максимального удаления электрона от ядра [6]. Внешняя оболочка атома радиусом аe препятствует проникновению внешних электронных полей вовнутрь атома благодаря жесткой связи электронов с ядром, устанавливая его естественную границу. Радиус ae атомов находится по данным измерения их поляризуемостей [2]

Понимание физического смысла радиуса аe атомов позволило по новому подойти и к проблеме построения потенциальной функции. В совокупности с фактом наличия на поверхностях атомов конечных зарядов q введение понятия радиуса ae предопределило возможность построения отталкивательной ветви потенциала u(R) в (2): она представляет энергию кулоновского отталкивания зарядов q атомов, поверхности которых отстоят друг от друга на расстоянии (R-2ae). Действие отталкивательных сил между атомами естественным образом объясняет факт взаимной непроницаемости атомов. При этом по мере сближения атомов и уменьшения расстояния между поверхностями атомов энергия отталкивания быстро увеличивается и при (R-2аe)→0 энергия uот(R)→∝.
Равновесному состоянию простейшей системы из двух атомов, которое описывается парным потенциалом (2), соответствует равновесное расстояние R=R0. Это состояние соответствует экстремуму потенциальной функции, который определяется условием 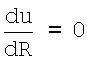 . Выполняя эту операцию дифференцирования (2), получаем
. Выполняя эту операцию дифференцирования (2), получаем

Подстановка величины q, входящей в (4) в выражение для потенциальной функции (2), дает

В целом функция (5) правильно передает поведение энергии взаимодействия между атомами. Вдали от равновесного состояния (при 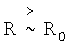 ) слагаемое в квадратных скобках очень мало и энергия взаимодействия между атомами определяется преимущественно дисперсионной энергией притяжения между атомами. Вблизи равновесия R=R0 энергия u(R0) соответствует энергии связи между атомами
) слагаемое в квадратных скобках очень мало и энергия взаимодействия между атомами определяется преимущественно дисперсионной энергией притяжения между атомами. Вблизи равновесия R=R0 энергия u(R0) соответствует энергии связи между атомами
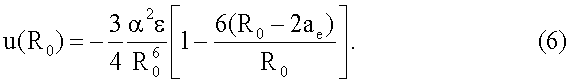
При дальнейшем уменьшении расстояния между атомами, т.е. при R<R0, второе слагаемое в квадратных скобках становится преобладающим в (5) и при R→2ae потенциал u(R) резко возрастает и приобретает преимущественно отталкивательный характер.
В выражении для потенциальной функции u(R) входит равновесное расстояние R0. Экспериментальное определение этой величины затруднено и количественные данные R0 в настоящее время отсутствуют. Оценку данной величины можно сделать, обращаясь к определению энергии сродства к электрону (или протону)

Эта формула представляет собой энергию связи (энергию сродства) атома с электроном (или протоном); по своей сути это закон Кулона, с помощью которого достигается описание взаимодействия эффективного заряда q атома и пробного заряда электрона (протона), находящихся на расстоянии аe.
Из формулы (7) следует

где сделана замена 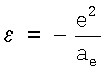 .
.
Подстановка (8) в (4) дает уравнение с одной переменной R0
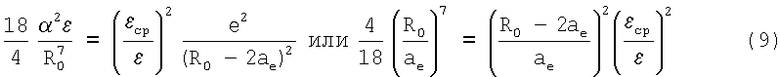
В приближении малости R0-2ae<<R0 это соотношение можно привести к виду

где 2Δа=R0-2ae.
При выводе данного выражения принято приближение 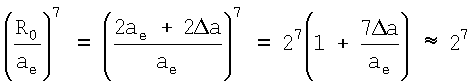 . Для атомов благородных газов это приближение выполняется хорошо. Данные по энергиям сродства εср. приведены в таблице 1.
. Для атомов благородных газов это приближение выполняется хорошо. Данные по энергиям сродства εср. приведены в таблице 1.
В принятом выше приближении энергия связи димеров атомов благородных газов по (6) с учетом (10) принимает вид

Для всех атомов благородных газов второе слагаемое в скобках всегда меньше единицы. Это означает, что для них выполняется условие связывания атомов и образования димеров. Энергия связи димеров невелика, она соизмерима с энергией поступательного движения атомов. Этим можно объяснить неустойчивость димеров и их относительно небольшое время жизни, наблюдаемые в эксперименте [7, С.13].
Существенно, что энергия парного взаимодействия зависит только от входящих в потенциалы (6) и (11) атомных констант (ae, ε, q) и ими полностью определяется. В нем отсутствуют подгоночные параметры и в этом отношении предлагаемый потенциал может быть отнесен к классу ab intio, чем он принципиально отличается от известных модельных потенциалов.
При использовании потенциалов (2) и (5) для практических расчетов следует также учитывать и то, что входящие в них радиус аe и поляризуемость α зависят от расстояния между атомами. В первом приближении радиус аe увеличивается на Δae в результате поляризующего действия атомов друг на друга, так что [2, С.258]
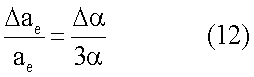
Изменения поляризуемости 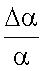 атомов благородных газов достаточно значительны и могут достигать 30-40% [2, С.406]. Поляризуемости атомов в конденсированном состоянии приведены в таблицы 1.
атомов благородных газов достаточно значительны и могут достигать 30-40% [2, С.406]. Поляризуемости атомов в конденсированном состоянии приведены в таблицы 1.
Кроме этого, надо учесть и то, что для парных взаимодействий u(R) приращения по (12) не скомпенсированы, как это имеет место в объеме вещества. Поэтому при определении действительного смещения электронной оболочки в процессе образования димера необходимо смещение по (12) увеличить еще приблизительно в 2 раза.
Подстановка в (10) известных величин ε и εcp (см. таблицу 1) дает для 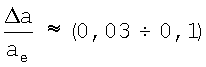 и соответственно для равновесного расстояния
и соответственно для равновесного расстояния 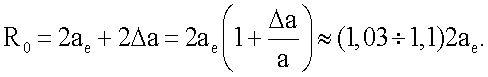 Т.е. при образовании димеров атомов благородных газов равновесное расстояние лишь немного превышает удвоенный радиус ае. В результате энергия связи димеров по (11) оказывается достаточно большой (соизмеримой с энергией связи в конденсированном состоянии) для обеспечения самой возможности образования димеров атомов благородных газов.
Т.е. при образовании димеров атомов благородных газов равновесное расстояние лишь немного превышает удвоенный радиус ае. В результате энергия связи димеров по (11) оказывается достаточно большой (соизмеримой с энергией связи в конденсированном состоянии) для обеспечения самой возможности образования димеров атомов благородных газов.
В принципиальном отношении потенциалы типа (5) и (6) применимы и к молекулярным системам типа Х2. Отличительной особенностью электронного строения молекул является перераспределение электронной плотности вдоль направления химической связи. В результате у молекул всегда имеет место анизотропия поляризуемости и энергии связи, наличие которой предопределяет преимущественную ориентацию молекул относительно друг друга при их сближении. Для молекулярных систем потенциал u(R) принимает вид
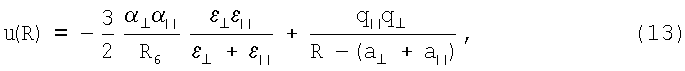
где индексы ⊥ и || соответствуют ⊥-й и ||-й конфигурации молекулы (перпендикулярно оси и вдоль оси молекулы). Здесь первое слагаемое имеет стандартный вид [1]. Поляризуемости а⊥ и а|| молекул и их энергии связи ε⊥ и ε|| могут различаться на 30-40%, хотя характер притягивательной ветви потенциала данное обстоятельство не меняет.
Второе слагаемое в (13) изменяет ситуацию кардинальным образом. Дело в том, что в результате перераспределения заряда вдоль оси молекулы заряды q⊥ и q|| принимают существенно разные величины. Поэтому из всех возможных конфигураций наиболее вероятной является ⊥-конфигурация, соответствующая минимуму энергии за счет взаимного притяжения зарядов q⊥ и q||. Это означает, что потенциал u(R) вырождается в единую притягивательную ветвь, действие которого ограничивает только энергия теплового движения. Отсюда следует, что процессы димеризации и конденсации таких систем протекают одновременно.
Аналогичный вывод можно распространить и на многоатомные молекулы, у которых всегда найдется благоприятная для взаимного притяжения молекул ориентация.
Полученные потенциалы (5), (6) и (11) применимы для определения энергии присоединения (отрыва) атомов к «закрепленным» атомам (или молекулам), имеющим место, например, при нанопроектировании и наноконструировании. Данная ситуация отличается от рассмотренного выше явления димеризации атомов (молекул) тем, что процедура присоединения (отсоединения) атомов происходит в квазистатических условиях, т.е. в условиях закрепленного атома (или группы атомов) и при полном контроле ориентации и положения присоединяемого атома. «Закрепленные» атомы (т.е. атомы в связанном состоянии) в своем большинстве могут быть отнесены к категории ван-дер-ваальсовых систем. Данное обстоятельство существенно расширяет область применения потенциалов типа (5).
В более общем случае присоединения разных атомов первый потенциал имеет вид наподобие (13),
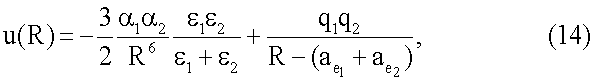
где индексы 1 и 2 относятся к закрепленному атому и присоединяемому атому соответственно.
Как было показано выше, в области механического контакта для ван-дер-ваальсовых систем силы притяжения преобладают над силами отталкивания. Получаемая на основе потенциала (14) энергия связи между атомами имеет вид, аналогичной (11).
Возможность варьирования поляризуемостями α1 и α2 (от 0,2 до нескольких десятков единиц Å3 [2, С.357]) позволяет задавать энергию связи u(R0) в широких пределах, что обеспечивает высокую гибкость проектирования и осуществления операций атомной сборки.
Однако надо иметь в виду то, что у ван-дер-ваальсовых систем электронные оболочки атомов (молекул) не перекрываются (т.е. химическая связь отсутствует). Поэтому связь между атомами весьма неустойчива из-за слабого противодействия тепловым функциям в плоскости, перпендикулярной оси связывания.
Более реалистичен вариант атомной сборки «группа атомов (подложка) - присоединяемый атом». При этом благодаря неоднородности атомной поверхности подложки присоединяемый атом занимает устойчивое положение в лунке, образованной полусферами атомов подложки. Такая лунка может быть образована тремя и большим числом атомов «подложки». В этом случае энергию связи по аналогии с (11) можно записать в виде
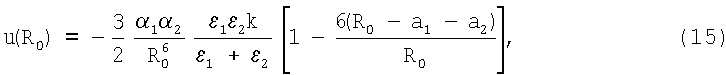
где κ - эффективное число атомов подложки, участвующих в формировании связи. В зависимости от строения подложки величина κ обычно находится в пределах κ=2÷3. Равновесное расстояние R0 может быть найдено на основании данных эффективных зарядов по аналогии с (11).
Очевидно, что в качестве подложки могут выступать также и молекулы (агрегаты молекул), а в качестве элементов сборки - отдельные молекулы. При этом у потенциалов (5) и (6) необходимо дополнительно учесть структурную анизотропию и неоднородность электронной плотности молекул, наподобие (13).
Таким образом, полученные потенциалы (5), (6), (11), (13) и (14) обеспечивают возможность проведения теоретических расчетов, а также проектирование наноизделий. Они могут стать основой для обеспечения нанотехнологии в целом и механосинтеза в частности [8].
Конденсированное состояние вещества отличается кооперативным характером межатомных взаимодействий. Данное обстоятельство количественно выражается в виде суммы энергий uij парных взаимодействий между всеми микрочастицами вещества [1]

Данное общее выражение энергии межатомного взаимодействия вещества можно упростить, если принять во внимание тот факт, что взаимодействия между микрочастицами за пределами первой координационной сферы быстро ослабляются. Для ван-дер-ваальсовых веществ с плотной упаковкой энергия взаимодействия по (15) от атомов второй координационной сферы по сравнению с основным вкладом уменьшается в (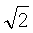 )6=8 раз (здесь принят радиус второй координационной сферы равным
)6=8 раз (здесь принят радиус второй координационной сферы равным 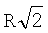 . Соответственно вклад энергии от атомов третьей координационной сферы с радиусом
. Соответственно вклад энергии от атомов третьей координационной сферы с радиусом 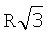 уменьшается в (
уменьшается в (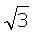 )6=27 раз. Учитывая, что в первой координационной сфере имеется 12 атомов, во второй - 6 и в третьей 24, потенциал в центре ячейки из трех координационных сфер равен
)6=27 раз. Учитывая, что в первой координационной сфере имеется 12 атомов, во второй - 6 и в третьей 24, потенциал в центре ячейки из трех координационных сфер равен
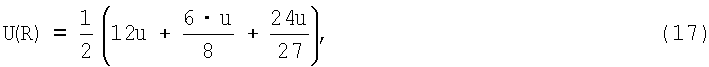
где u - парный потенциал по (15).
Т.е. вклад энергий взаимодействий от второй и третьей координационных сфер в результирующий потенциал (17) не превышает 10%.
Кроме этого, необходимо учитывать и то, что прямое действие атомов третьей и всех последующих координационных сфер фактически исключено, благодаря экранированию атомами первой и второй координационным сфер. Поэтому с хорошим приближением будет применимо выражение для потенциальной функции, учитывающее действие только первой координационной сферы, так что

Данная потенциальная функция применима к описанию конденсированного состояния ван-дер-ваальсовых систем, в первую очередь атомов благородных газов. В отличие от (5) в данном потенциале учтен кооперативный эффект (за счет введения координационного числа η). В первом слагаемом также учтен факт попарного взаимодействия атомов (введением коэффициента 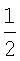 ) как и в общем выражении потенциала (16).
) как и в общем выражении потенциала (16).
Данную потенциальную функцию (18) можно принять за основу при нахождении потенциальной функции наподобие функции (6), так что
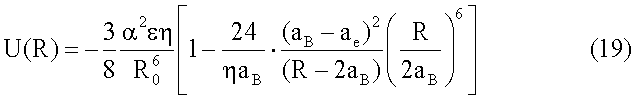
Отличительной особенностью этой функции является использование эмпирического условия равновесия R0=2аB, в котором величина aB представляет собой так называемый ван-дер-ваальсов радиус, который по определению равен половинному расстоянию между ядрами ближайших атомов [9, С.156]. Использование радиуса aB в качестве параметра теории более предпочтительно (по сравнению с эффективным зарядом q атомов), поскольку он может быть измерен с достаточно высокой точностью.
Из (19) следует, что энергия U(R0), соответствующая равновесному состоянию вещества в конденсированном состоянии, равна
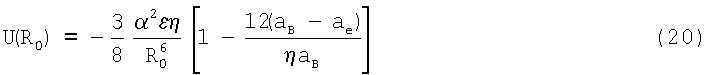
Характерной особенностью потенциалов (19) и (20) является то, что энергия притяжения U(R) увеличивается по сравнению с парной энергией u(R) в η/2 раза. При этом действие дисперсионных сил притяжения проявляется в поляризации (деформации) электронных оболочек атомов. В результате взаимной поляризации атомов их внешнее влияние оболочки расширяются, что приводит к увеличению их радиуса аe. Фактически это означает, что энергия возмущения электронной оболочки атома соответствует дисперсионному вкладу в энергию взаимного притяжения атомов. Другими словами, часть энергии каждого из атомов передается на образование ван-дер-ваальсовой связи между ними. При этом дисперсионные силы межатомного взаимодействия складываются, проявляя кооперативный характер взаимного притяжения атомов.
С другой стороны, кулоновские силы отталкивания проявляются преимущественно в стремлении к смещению атома как целого. В силу сферической симметрии атомов точкой приложения сил отталкивания атомов является центр тяжести (ядро) атома. Поэтому действия окружающих атомов на выделенный атом взаимно компенсируются.
Таким образом, в состоянии равновесия системы все силы притяжения и отталкивания атомов полностью сбалансированы. Сохраняются только дисперсионные взаимодействия, которые аккумулированы в межатомных связях в виде потенциальной энергии поляризации электронных оболочек атомов. Поэтому во втором члене потенциала (19) отсутствует множитель η/2.
Для атомных систем η=12. В этом случае потенциал можно записать в виде
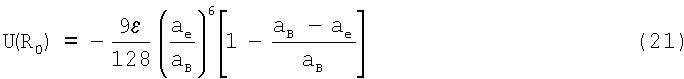
Видно, что в состоянии равновесия отталкивательный вклад в U(R0) относительно невелик (порядка 20-30%) и определяется атомными константами aB и аe (см. таблицу 1).
С другой стороны, в соответствии с (18), по мере приближения aB к аe энергия отталкивания резко возрастает и стремится к ∝ при aB=аe.
Рассчитанные по (21) энергии U(R0) приведены в таблице 1. Здесь же для сравнения приведены экспериментальные данные Uэ [10, С.115]. Согласие рассчитанных и экспериментальных энергий находится в пределах погрешности измеряемых величин Uэ, aB, ε, аe.
Потенциал (19) применим и к молекулярным системам. Для этого входящие в (18) величины α, ε, aB и аe в первом приближении можно рассматривать как некоторые эффективные величины. Для расчета потенциалов ряда типичных молекулярных систем (см. таблицу 1) были приняты: в качестве радиусов аe - средние величины 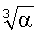 [2]; в качестве ван-дер-ваальсовых радиусов - равновесные расстояния, приведенные в работе [11]; в качестве энергий связи ε - соответствующие им потенциалы ионизации [12]. Результаты расчета равновесного потенциала U(R0) представлены в таблице 1.
[2]; в качестве ван-дер-ваальсовых радиусов - равновесные расстояния, приведенные в работе [11]; в качестве энергий связи ε - соответствующие им потенциалы ионизации [12]. Результаты расчета равновесного потенциала U(R0) представлены в таблице 1.
Здесь же приведены немногочисленные литературные данные Uэ. Имеющееся согласие теоретических (рассчитываемых по (21)) и экспериментальных величин вполне убедительно для установления принципиальной применимости потенциала (19) к молекулярным ван-дер-ваальсовым системам.
Можно ожидать повышения точности описания молекулярных систем, если в выражении (21) учесть анизотропные поляризуемости и энергии связи наподобие потенциалу (13), а также если учесть структурные особенности вещества в конденсированном состоянии. Эти данные в литературе пока отсутствуют.
Очевидно, что при использовании эффективных величин α, ε, аB и ae потенциал (19) может быть применен и к многоатомным ван-дер-ваальсовым системам.
Существенными признаками изобретения является то, что в потенциальную функцию введены атомные константы - поляризационные и ван-дер-ваальсовы радиусы, а также эффективный заряд на поверхности атома, придающие потенциальной функции физический смысл и исключающие необходимость применения подгоночных параметров.
Существенным также представляется то, что энергетические параметры атомных и молекулярных ван-дер-ваальсовых систем могут быть определены непосредственно (прямым методом) по данным измерения атомных констант - поляризационному и ван-дер-ваальсовому радиусам.
Т.о. предложен:
1. Способ определения энергии взаимодействия между атомами ван-дер-ваальсовых систем, основанный на установлении зависимости энергии межатомного взаимодействия от расстояния между атомами, отличающийся тем, что измеряют радиус атома ае, определяющий наибольшее расстояние внешних электронов по отношению к ядру и ван-дер-ваальсов радиус аВ, равный половинному расстоянию между ядрами ближайших атомов вещества в конденсированном состоянии, и рассчитывают потенциальную функцию по формуле
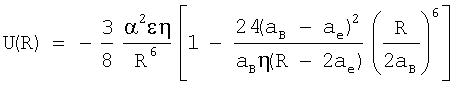
где R - расстояние между центрами атомов, ε - энергия связи, α - поляризуемость, η - координационное число.
2. Способ по п.1, отличающийся тем, что для определения энергии взаимодействия между атомами ван-дер-ваальсовых систем, соответствующей равновесному состоянию вещества в конденсированном состоянии, задают равновесное расстояние R0 равным удвоенному расстоянию 2aB и рассчитывают ее по формуле
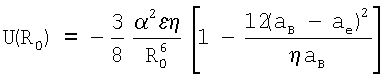
Решаемые задачи:
1) предлагаемые потенциальные функции позволяют производить теоретический расчет энергии связи различных атомных конфигураций и исследовать поведение систем в широком диапазоне межатомных расстояний;
2) предлагаемые потенциалы позволяют проводить инженерные расчеты при проектировании наноизделий, а также могут стать основой теоретического обеспечения нанотехнологии, в том числе механосинтеза;
3) предлагаемый подход к построению потенциальных функций открывает новое направление в области теории межатомных и межмолекулярных взаимодействий.
Осуществление изобретения.
Изобретение осуществляется на основании данных измерения поляризационного и ван-дер-ваальсового радиусов, а также других констант, входящих в потенциальную функцию - энергию связи, энергию сродства к электрону (или протону), поляризуемость. Для каждой из этих величин имеется соответствующая измерительная аппаратура.
Данные по поляризационным радиусам получают на основании данных измерения поляризуемости; обзор методов и средств измерения поляризуемостей приведены в работе [2].
Данные по ван-дер-ваальсовым радиусам получают на основании измерения равновесных расстояний между атомами в конденсированном состоянии [9, 10].
Данные по энергиям связи и энергиям сродства могут быть получены различными способами. Сведения о них можно найти в работах [2, 10, 12, 13].

* аск - радиус атомов с поправкой на поляризационное взаимодействие.
Литература
1. Каплан И.Г. Введение в теорию межмолекулярных взаимодействий. - М.: Наука, 1982.
2. Потапов А.А. Деформационная поляризация: поиск оптимальных моделей. - Новосибирск: Наука, 2004. - 511 с.
3. Химическая энциклопедия, т.4. - М.: Большая Российская энциклопедия, 1985.
4. Свойства неорганических соединений. Справочник. - Л.: Химия, 1983. - 392 с.
5. Парселл Э. Электричество и магнетизм. - М.: Наука, 1975. - 440 с.
6. Потапов А. Абсолютный радиус многоэлектронных атомов по данным их поляризуемостей. // Электронный журнал «Исследовано в России», 49. С.554-470, 2005. http://zhurnal.ape.relarn.ru/articales/2005/049-pdf.
7. Шахпаронов М.И. Межмолекулярные взаимодействия. - М.: Знание. 1983-64 с.
8. Потапов А.А. Состояние и перспективы построения теоретических основ механосинтеза. // Нанотехника, 2005, №4, С.32-46.
9. Физическая энциклопедия, т.1. - М.: Советская энциклопедия, 1988.
10. Киттель Ч. Введение в физику твердого тела. - М.: Наука, 1978. - 792 с.
11. Зефиров Ю.В. Ван-дер-ваальсовы радиусы атомов металлов. // Журнал неорганической химии, 2000. т.45, №10, С.1691-1693.
12. Бацанов С.С. Экспериментальные основы структурной химии. - М.: Изд-во стандартов, 1986. - 240 с.
13. Веденеев В.И. и др. Энергии разрыва химических связей. - М.: Изд-во АН СССР, 1962.
Изобретение относится к области исследования физико-химических свойств и электронного строения веществ, а именно к области исследования параметров межатомных взаимодействий ван-дер-ваальсовых систем. Техническим результатом изобретения является повышение точности описания систем. Способ основан на установлении зависимости энергии межатомного взаимодействия от расстояния между атомами. Измеряют радиус атома, определяющий наибольшее расстояние внешних электронов по отношению к ядру, и ван-дер-ваальсов радиус атома, равный половинному расстоянию между ядрами ближайших атомов вещества в конденсированном состоянии. Рассчитывают зависимость энергии межатомного взаимодействия от расстояния между атомами, представленную потенциальной функцией по определенной формуле. 1 з.п. ф-лы, 1 табл.
1. Способ определения энергии межатомных взаимодействий ван-дер-ваальсовых систем, основанный на установлении зависимости энергии межатомного взаимодействия от расстояния между атомами, отличающийся тем, что измеряют радиус атома аe, определяющий наибольшее расстояние внешних электронов по отношению к ядру, и ван-дер-ваальсов радиус атома аB, равный половинному расстоянию между ядрами ближайших атомов вещества в конденсированном состоянии, и рассчитывают зависимость энергии межатомного взаимодействия от расстояния между атомами, представленную потенциальной функцией по формуле
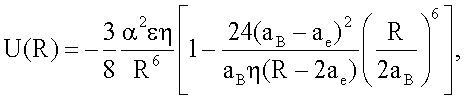
где R - расстояние между центрами атомов, ε - энергия связи, α - поляризуемость, η - координационное число.
2. Способ по п.1, отличающийся тем, что для определения энергии взаимодействия между атомами ван-дер-ваальсовых систем, соответствующей равновесному состоянию вещества в конденсированном состоянии, задают равновесное расстояние
R0 равным расстоянию 2aB и рассчитывают ее по формуле
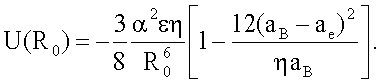
| Каплан И.Г | |||
| Введение в теорию межмолекулярных взаимодействий | |||
| - М.: Наука, с.220 | |||
| Способ исследования надмолекулярной структуры вещества | 1991 |
|
SU1827615A1 |
| СПОСОБЫ МОДЕЛИРОВАНИЯ МЕЖМОЛЕКУЛЯРНОГО ВЗАИМОДЕЙСТВИЯ И ОСНОВАННЫЙ НА НИХ СПОСОБ ПРОГНОЗИРОВАНИЯ СВЯЗЫВАНИЯ МОЛЕКУЛЫ-ЛИГАНДА С МОЛЕКУЛОЙ-МИШЕНЬЮ | 2003 |
|
RU2265243C2 |
| JP 2005233854 A, 02.09.2005. | |||

