Область изобретения
Настоящее изобретение относится к ингибиторам образования транстиретиновых амилоидных фибрилл. Более подробно изобретение относится к производным дибензофуранов в качестве ингибиторов образования транстиретиновых амилоидных фибрилл.
Предпосылки создания изобретения
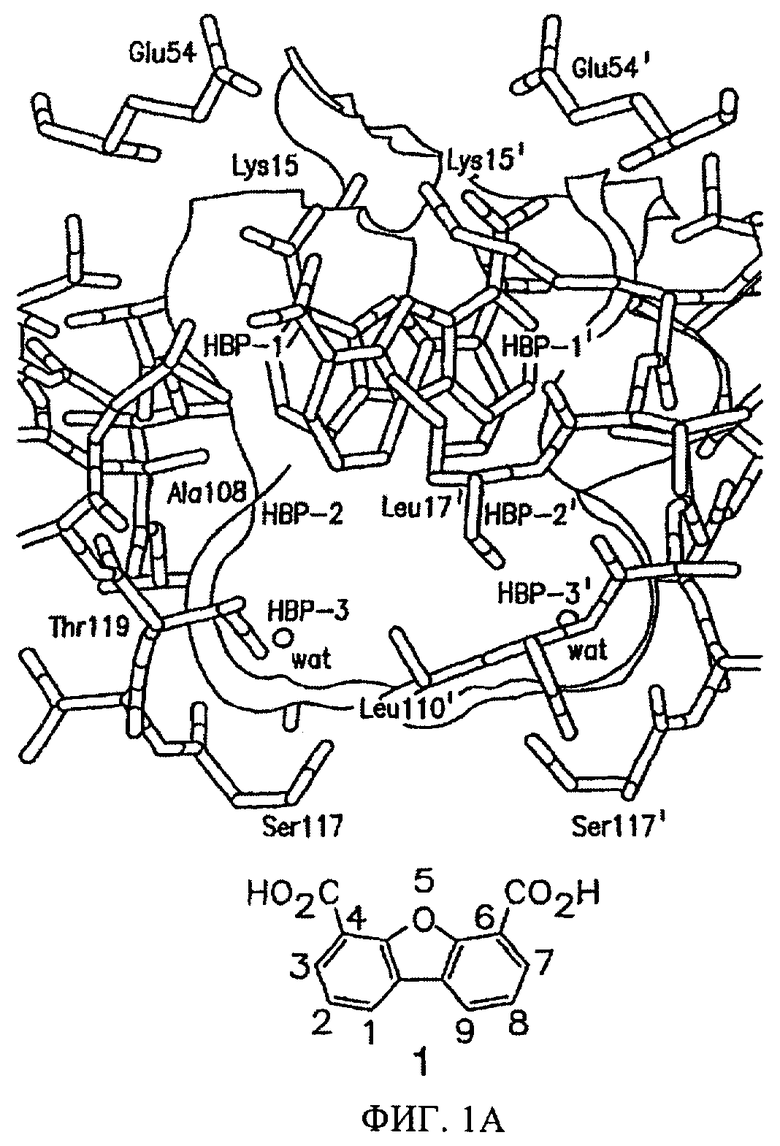
В литературе описано несколько различных структурных классов низкомолекулярных стабилизаторов транстиретина (TTR), из которых наибольший интерес представляет дибензофуран-4,6-дикарбоновая кислота (1), см. фиг.1 (Hammarstrom P. и др., Science, 299, 713-716 (2003); Klabunde Т. и др., Nature Struct. Biol., 7, 312-321 (2000); Razavi H. и др., Angew Chem., 42, 2758-2761 (2003); Miroy G.J. и др., Рrос. Natl. Acad. Sci. USA, 93, 15051-15056 (1996); Peterson S.А. и др., Proc. Natl. Acad. Sci, USA, 95, 12956-12960 (1998); Baures P.W. и др., Bioorg. Med. Chem., 6, 1389-1401 (1998); Baures P.W. и др., Bioorg. Med. Chem., 7, 1339-1347 (1999); Petrassi H.M. и др., J. Am. Chem. Soc., 122, 2178-2192 (2000); McCammon M.G. и др., Structure. 10, 851-863 (2002); Oza V.B. и др., J. Med. Chem., 45, 321-332 (2002); Sacchettini J.С.и др., Nature Rev. Drug Disc., 1, 267-275 (2002); Green N.S. и др., J. Am. Chem. Soc., 125, 13404-13414 (2003); Adamski-Werner S.L. и др., J. Med. Chem., 47, 355-374 (2004); Miller Sean R. и др., Lab. Inv., 84, 545-552 (2004)). Указанный ингибитор (1, 7,2 мкМ) в течение 72 ч уменьшает степень образования амилоидов WT-TTR (3,6 мкМ, рН 4,4) на 90%. По данным рентгеноструктурного анализа сокристаллов TTR·12 установлено, что значительная активность указанного ингибитора связана с тем, что он взаимодействует исключительно с внешней частью каждого участка связывания тироксина (Klabunde Т. и др., Nature Struct. BioL, 7, 312-321 (2000)). Следовательно, существует необходимость в модификации соединения 1 субструктурами, которые способствуют проникновению во внутреннюю полость тироксинсвязывающего кармана, чтобы повысить сродство и селективность связывания производных соединения 1 с TTR в крови человека.
Краткое содержание сущности изобретения
Изобретение относится к производным дибензофуран-4,6-дикарбоновой кислоты, которые получают при введении ароматических заместителей в дибензофурановое кольцо в положение С1 с использованием трех различных типов химических связей. Установлено, что указанные соединения являются чрезвычайно эффективными ингибиторами амилоидогенеза, проявляют повышенную аффинность и значительно повышенную селективность при связывании с TTR по сравнению со всеми остальными плазматическими белками и по сравнению с контрольным соединением 1 (Purkey H.Е. и др., Рrос. Natl. Acad. Sci. USA, 98, 5566-5571 (2001)). Кроме того, функция указанных соединений заключается в повышении кинетической стабильности тетрамерной структуры TTR.
Для процесса транстиретиннового (TTR) амилоидогенеза требуется диссоциация тетрамера и частичная денатурация полученных мономеров, которые ограничивают скорость процесса, при этом образуются аномальные белковые комплексы. Указанный процесс сопровождается повышением мутности, что используют для идентификации ингибиторов транстиретинового амилоидогенеза, включая дибензофуран-4,6-дикарбоновую кислоту (1). По данным рентгеноструктурного анализа сокристаллов TTR·12 установлено, что соединение 1 взаимодействует только с внешней частью двух тироксинсвязывающих карманов, тем самым ингибируя амилоидогенез TTR. Как описано в контексте настоящего изобретения, для повышения сродства к неиспользованной внутренней полости тироксинсвязывающего кармана в дибензофурановое кольцо указанного соединения в положение С1 вводят арильные заместители с использованием трех различных типов химических связей. Получены двадцать восемь высокоэффективных ингибиторов амилоидогенеза, характеризующиеся высокой эффективностью и исключительно высокой селективностью связывания с TTR плазмы крови. Полученные ингибиторы повышают кинетическую стабильность нативной структуры тетрамера TTR, тем самым, исключая возможность амилоидогенеза в физиологических условиях. Так как уже известно, что повышение кинетической стабильности нативного TTR, вызванное межаллельной транс-супрессией, приводит к ослаблению симптомов заболевания, следует полагать, что применение ингибиторов на основе дибензофурана приведет к аналогичному эффекту. Предупреждение развития амилоидогенеза является наиболее консервативной стратегией лечения в клинике, поскольку до настоящего времени не известно, какие промежуточные аномальные агрегаты TTR приводят к токсичности. Указанные ингибиторы обладают чрезвычайно высокой селективностью связывания, что приводит к маскированию тироксинсвязывающих участков TTR в биологической жидкости сложного состава, такой как плазма крови, что и требуется для подавления амилоидогенеза в организме человека. В настоящее время установлено, что ингибиторы амилоидогенеза на основе дибензофурана характеризуются высокой селективностью, аффинностью и эффективностью.
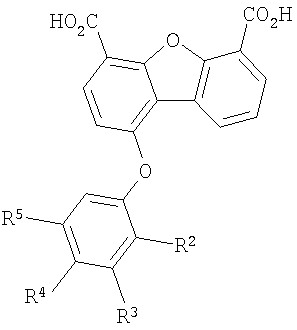
Один объект настоящего изобретения относится к соединению формулы I
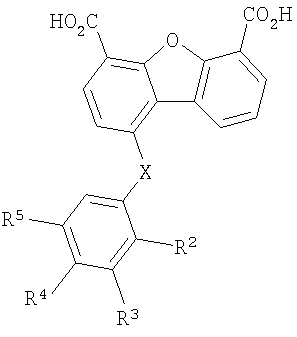
В соединении формулы I заместитель Х отсутствует или означает двухвалентный радикал, который выбирают из группы, включающей -O-, -S- и -NH-, а радикалы R2, R3, R4 и R5 независимо выбирают из группы, включающей -Н, -ОН, -F, -С1, -Вr, -СF3 и -СО2Н. Первая подгруппа первого объекта настоящего изобретения включает соединение формулы II
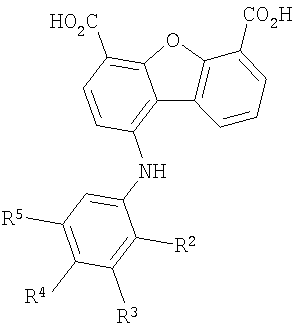
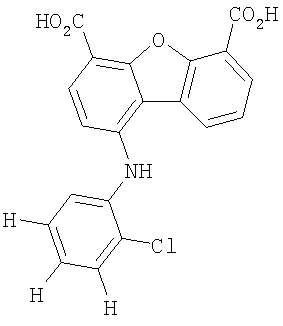
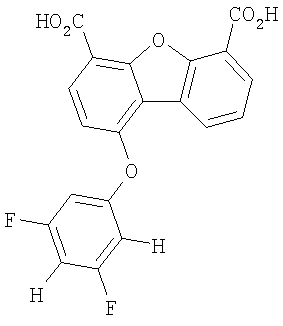
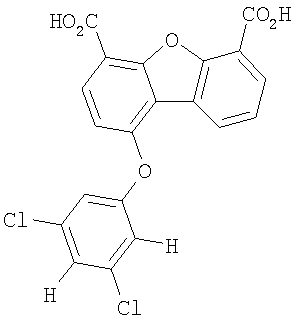
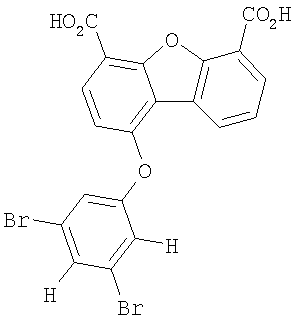
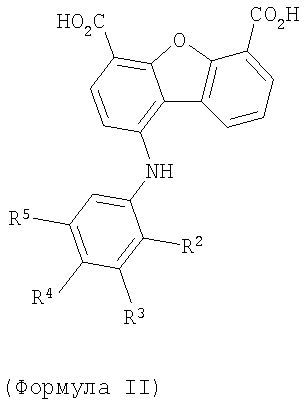
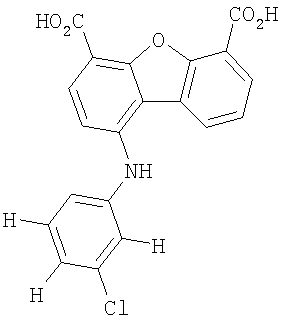
Предпочтительные варианты подгруппы формулы II включают соединения, в которых R2 означает радикал, который выбирают из группы, включающей -Н, -F, -Сl и -СF3, дополнительные предпочтительные варианты включают соединения, в которых R4 означает радикал, который выбирают из группы, включающей -Н, -Сl и -СО2Н, кроме того, предпочтительные варианты включают соединения, в которых R5 означает радикал, который выбирают из группы, включающей -Н, -F и -С1. Предпочтительные варианты подгруппы формулы II включают соединения, которые выбирают из следующих структур:
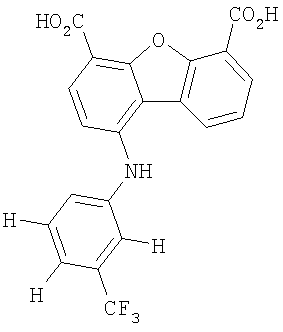
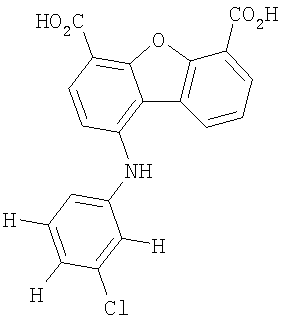
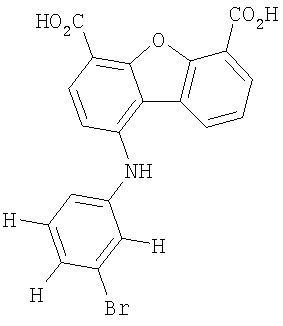
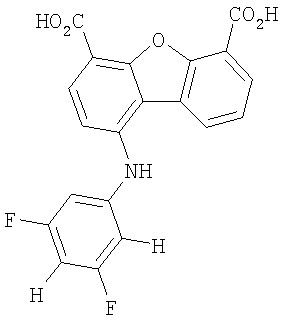
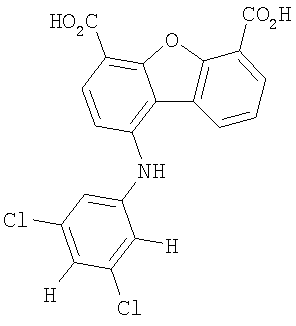
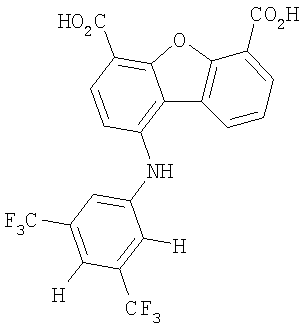
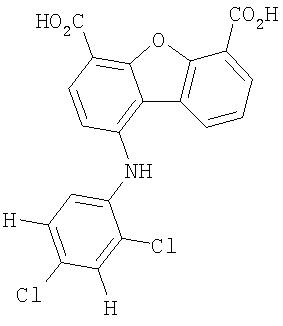
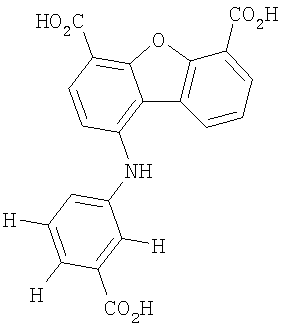
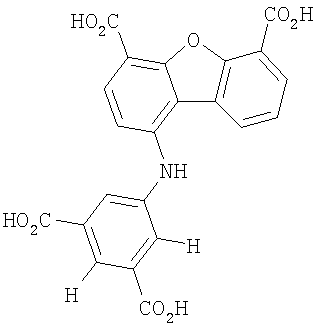
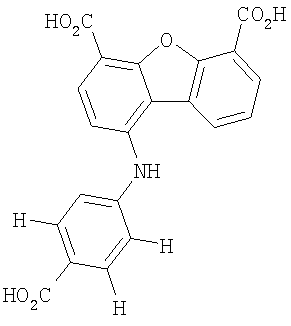
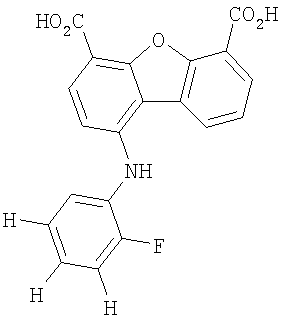
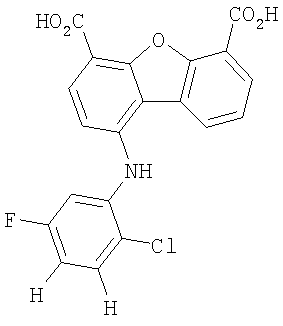
Вторая подгруппа первого объекта настоящего изобретения включает соединение формулы III
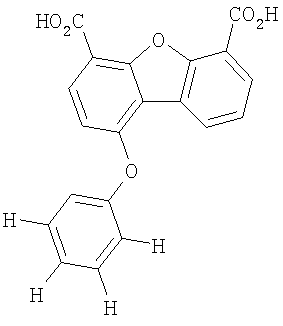
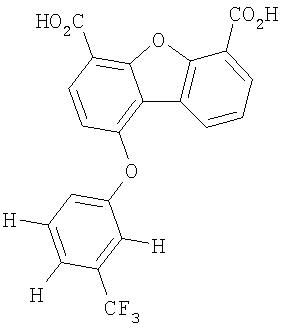
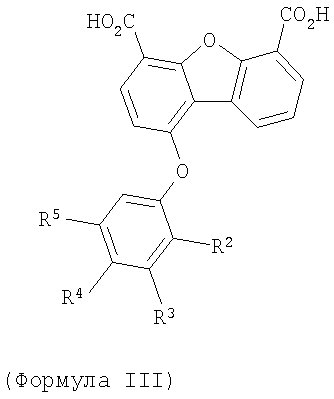
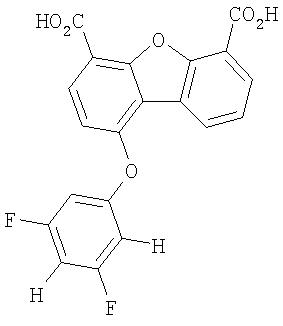
Предпочтительные варианты подгруппы формулы III включают соединения, в которых R3 означает радикал, который выбирают из группы, включающей -Н, -F, -Сl, -Вr и -СF3, дополнительные предпочтительные варианты включают соединения, в которых R5 означает радикал, который выбирают из группы, включающей -Н, -F, -Сl и -Вr. Предпочтительные варианты подгруппы формулы III включают соединения, которые выбирают из следующих структур:
Третья подгуппа первого объекта настоящего изобретения включает соединение формулы IV
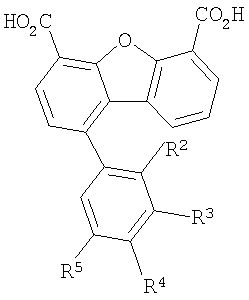
Предпочтительные варианты подгруппы формулы IV включают соединения, в которых R2 означает радикал, который выбирают из группы, включающей -Н, -F и -Сl, и дополнительные варианты включают соединения, в которых R3 означает радикал, который выбирают из группы, включающей -Н, -F, -Сl, -СF3 и -СО2Н, а дополнительные варианты включают соединения, в которых R4 означает радикал, который выбирают из группы, включающей -Н и -СО2Н, дополнительные предпочтительные варианты включают соединения, в которых R5 означает радикал, который выбирают из группы, включающей -Н, -F, -Сl и -СF3.
Предпочтительные варианты подгруппы формулы IV включают соединения, которые выбирают из следующих структур:
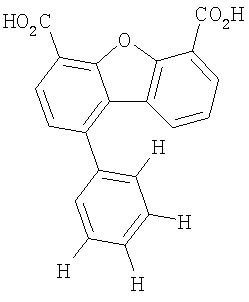
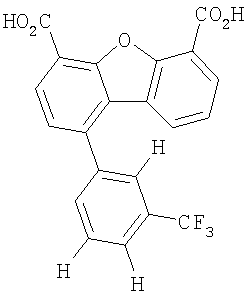
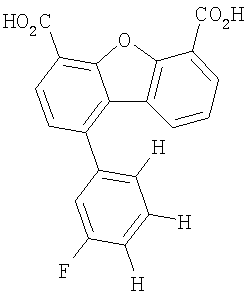
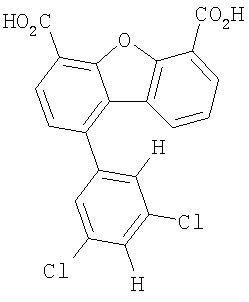
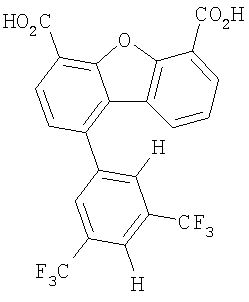
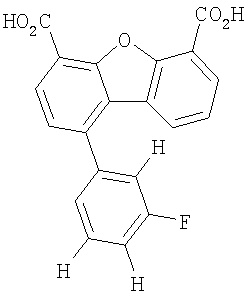
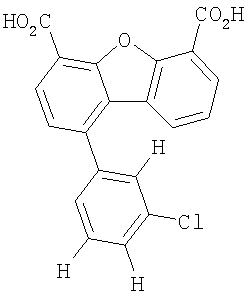
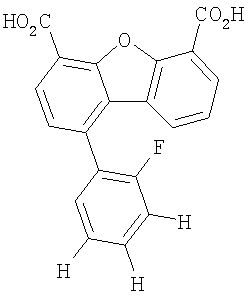
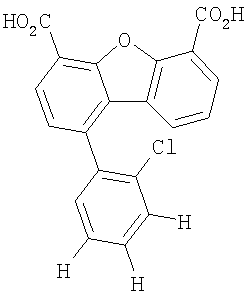
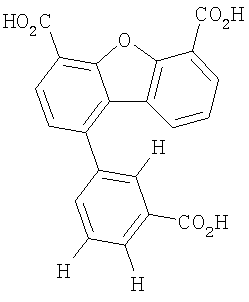
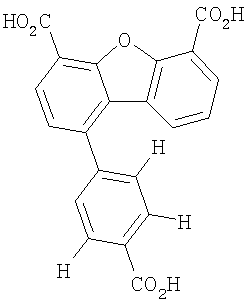
Другим объектом настоящего изобретения является способ, включающий стадию контактирования транстиретина с соединением формул I-IV при концентрации, которой достаточно для ингибирования образования амилоидных транстиретиновых фибрилл.
Краткое описание чертежей
На фиг.1А представлена структура комплекса TTR·12 по данным рентгеноструктурного анализа (Klabunde Т. и др., Nature Struct. Biol, 7, 312-321 (2000)).
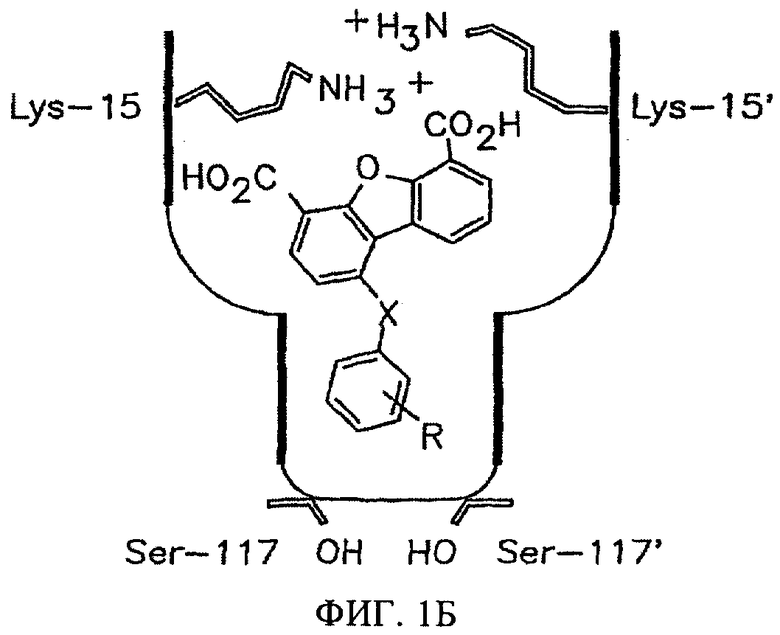
На фиг.1В представлена схема расположения замещенных в положении 1 дибензофуран-4,6-дикарбоновых кислот, в которых Х означает NH, О или прямую Сарил-Сарил связь, во внутреннем тироксинсвязывающем кармане. R означает заместители в составе арильного кольца, которые повышают сродство к внутренней полости связывающего кармана TTR.
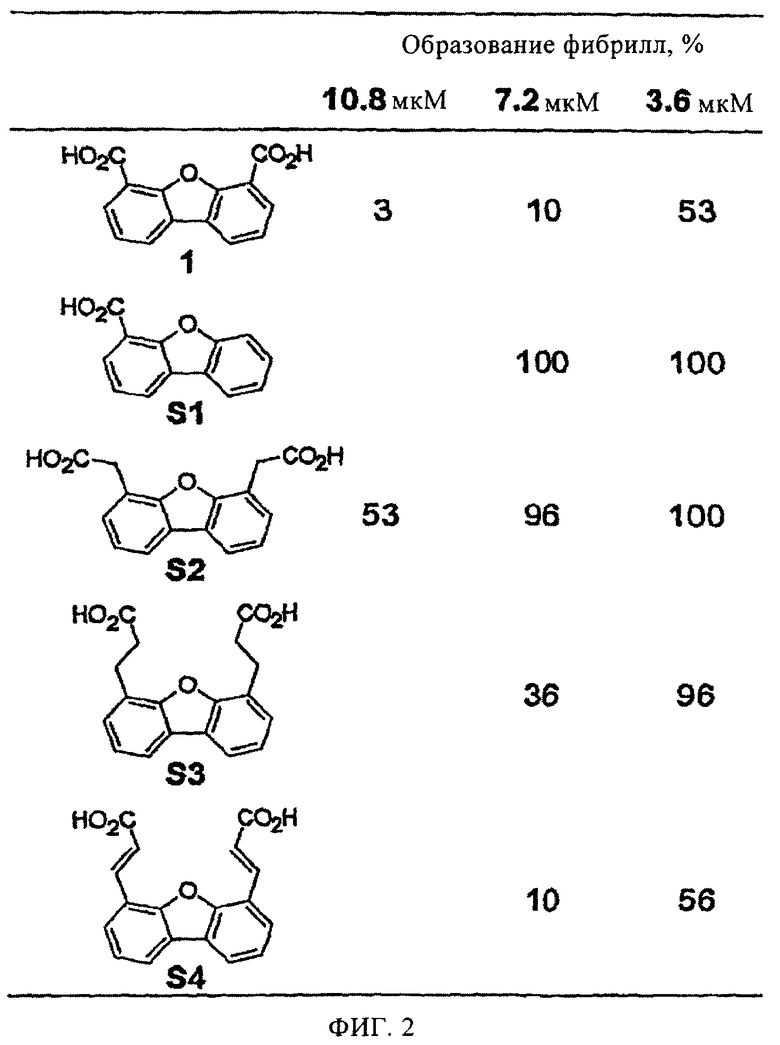
На фиг.2 представлена таблица, в которой показано влияние концентрации производных дибензофурана, замещенного различными кислотами, на активность в отношении образования амилоидных фибрилл WT-TTR (3,6 мкМ) при рН 4,4 (в течение 72 ч).
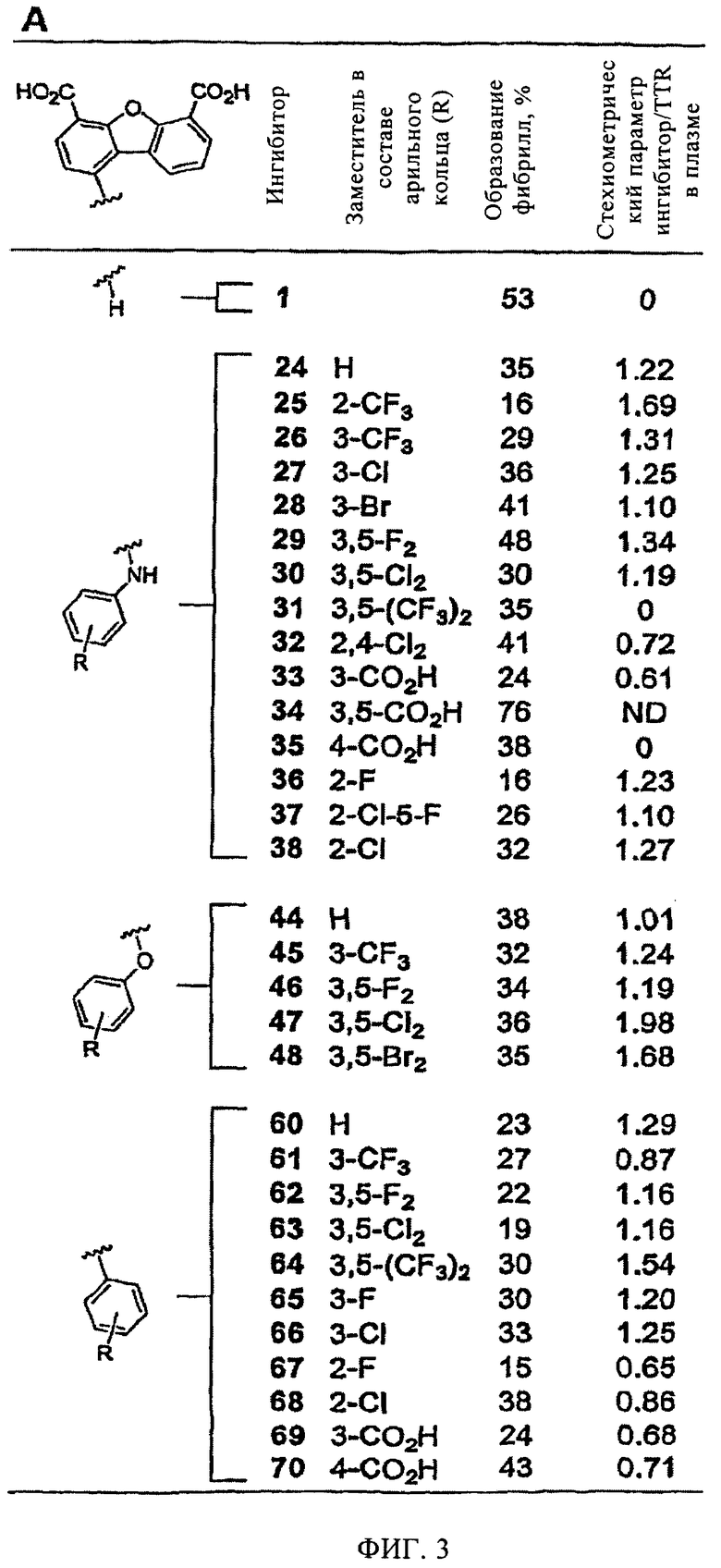
На фиг.3 представлена суммарная таблица, в которой показаны данные по ингибирующей активности производных дибензофурана (3,6 мкМ), в отношении образования фибрилл WT-TTR (рН 4,4, в течение 72 ч, 3,6 мкМ) и стехиометрические параметры связывания указанных соединений с TTR в плазме крови человека.
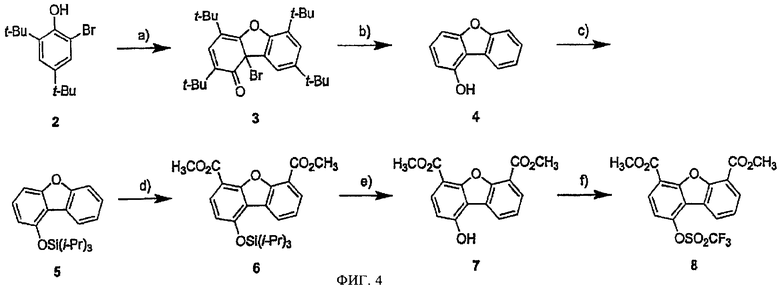
На фиг.4 представлена схема получения диметилового эфира 1-гидроксидибензофуран-4,6-дикарбоновой кислоты и соответствующего трифлата.
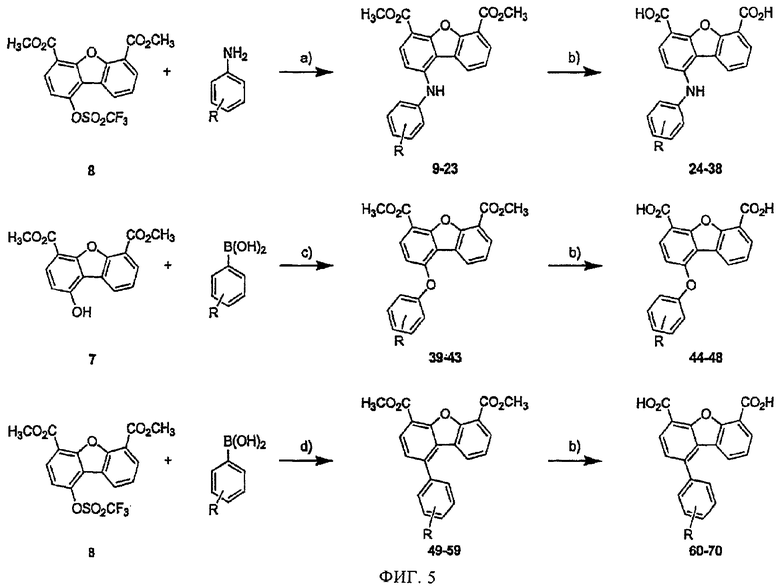
На фиг.5 представлена схема получения диметиловых эфиров 1-фенил-, фенокси- и фениламиндибензофуран-4,6-дикарбоновой кислоты и соответствующих дикарбоновых кислот.
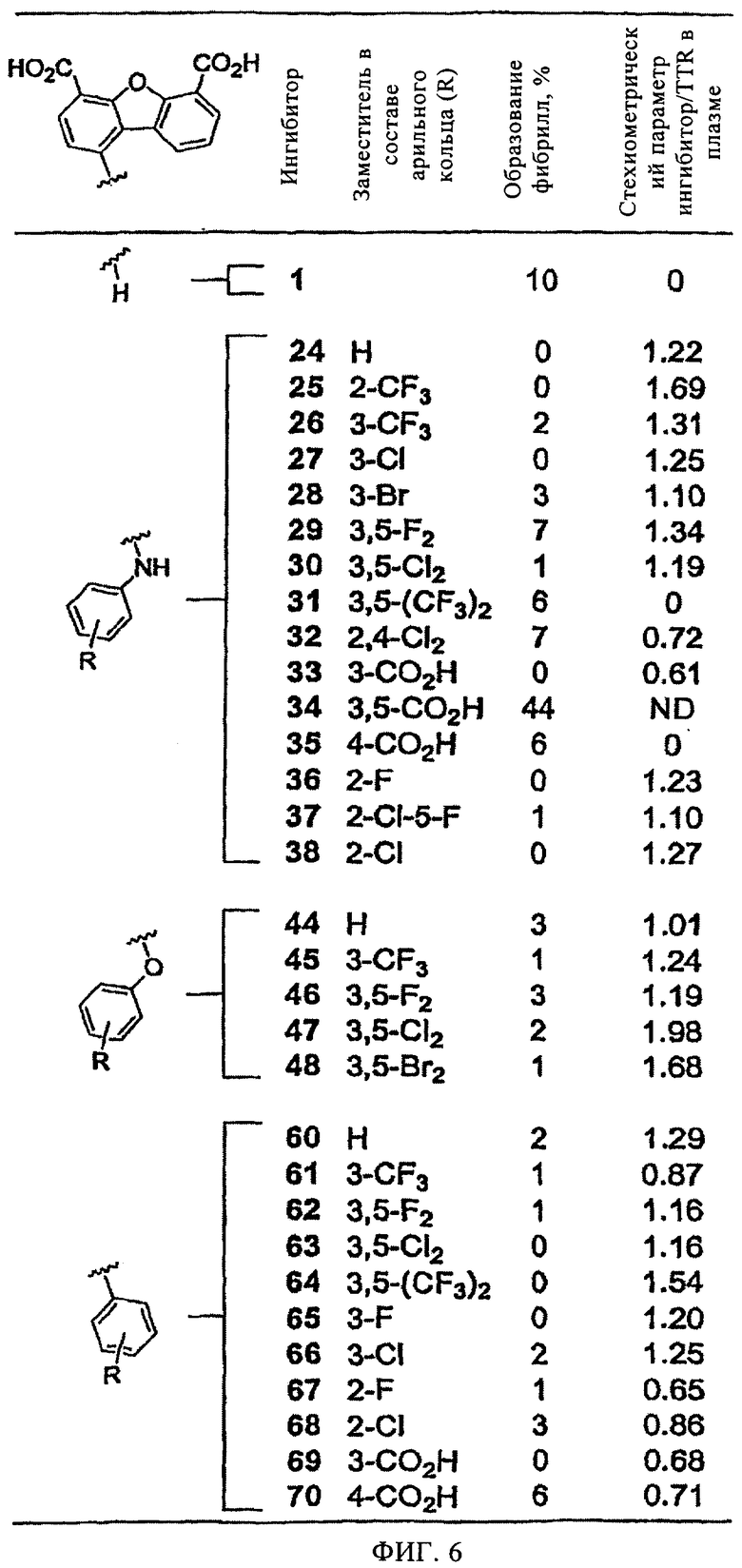
На фиг.6 представлена таблица, в которой показаны данные по ингибирующей активности производных дибензофурана (7,2 мкМ) в отношении образования амилоидных фибрилл WT-TTR (3,6 мкМ) при рН 4,4 (в течение 72 ч).
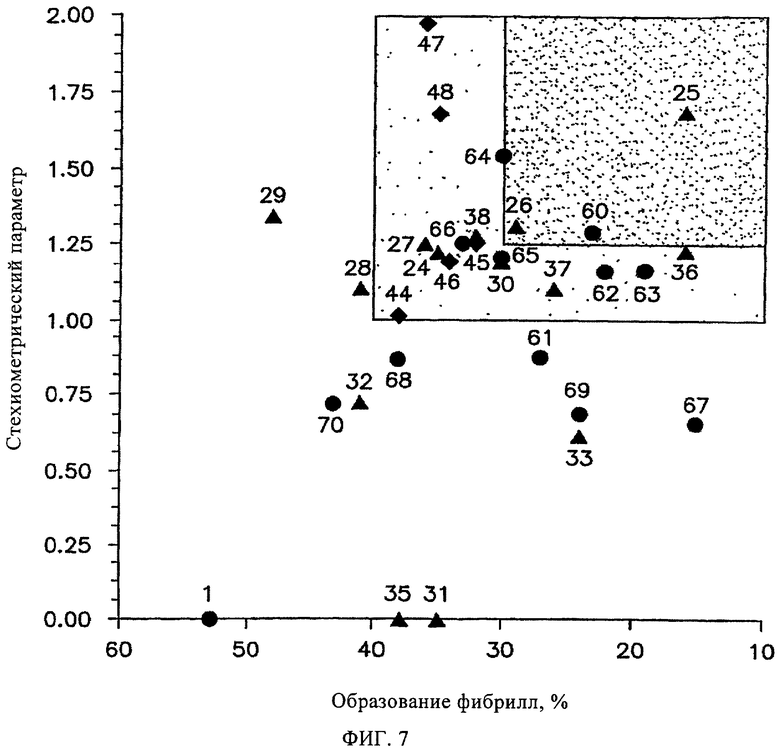
На фиг.7 представлены данные сопоставления эффективности ингибирования образования фибрилл и стехиометрических параметров связывания различных производных дибензофуранов с TTR в плазме крови.
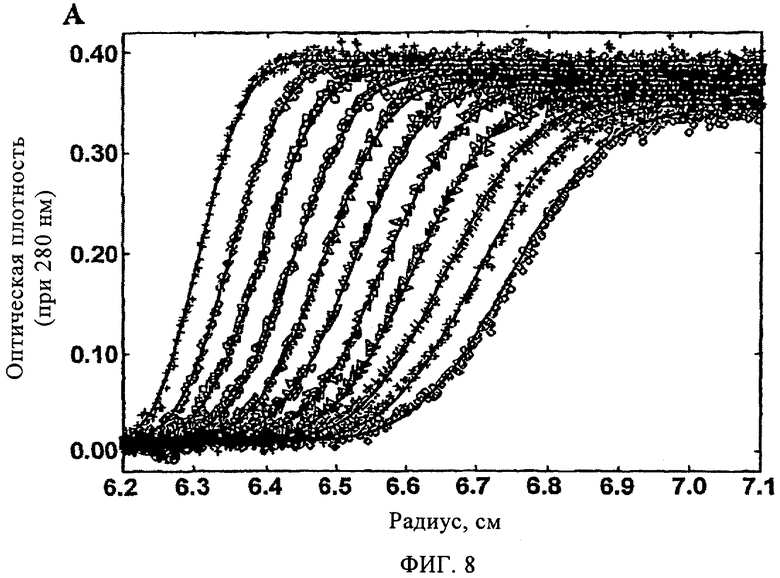
На фиг.8 представлена зависимость оптической плотности при 280 нм от радиуса ротора, полученная методом седиментационного анализа TTR (3,6 мкМ), после предварительной инкубации в присутствии соединения 27 (7,2 мкМ) с последующей инкубацией при рН 4,4 в течение 72 ч (период времени, в течение которого образуется максимальное количество амилоидных фибрилл в отсутствие ингибитора).
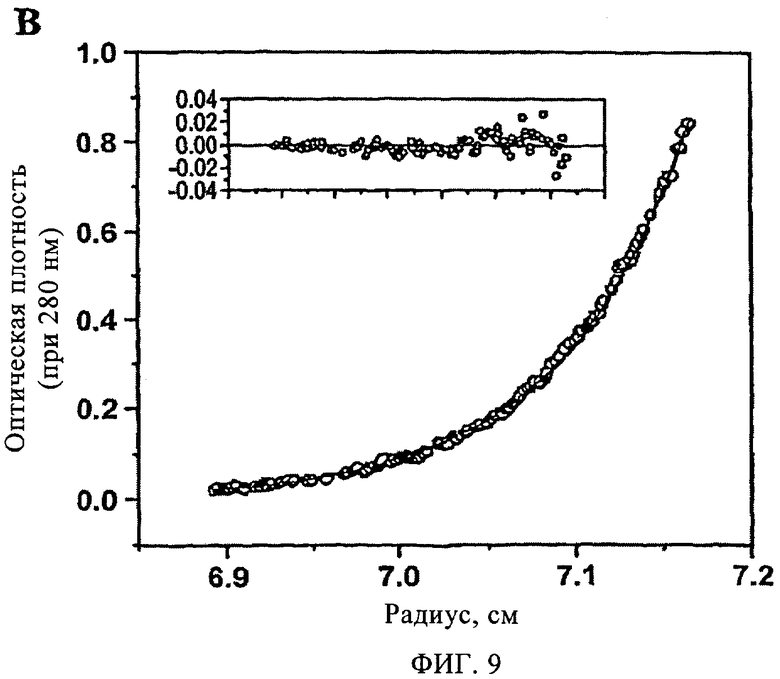
На фиг.9 представлена зависимость оптической плотности при 280 нм от радиуса ротора, полученная методом равновесного ультрацентрифугирования TTR (3,6 мкМ), который предварительно инкубировали в присутствии соединения 27 (7,2 мкМ) с последующей инкубацией при рН 4,4 в течение 72 ч (период времени, в течение которого образуется максимальное количество амилоидных фибрилл в отсутствие ингибитора).
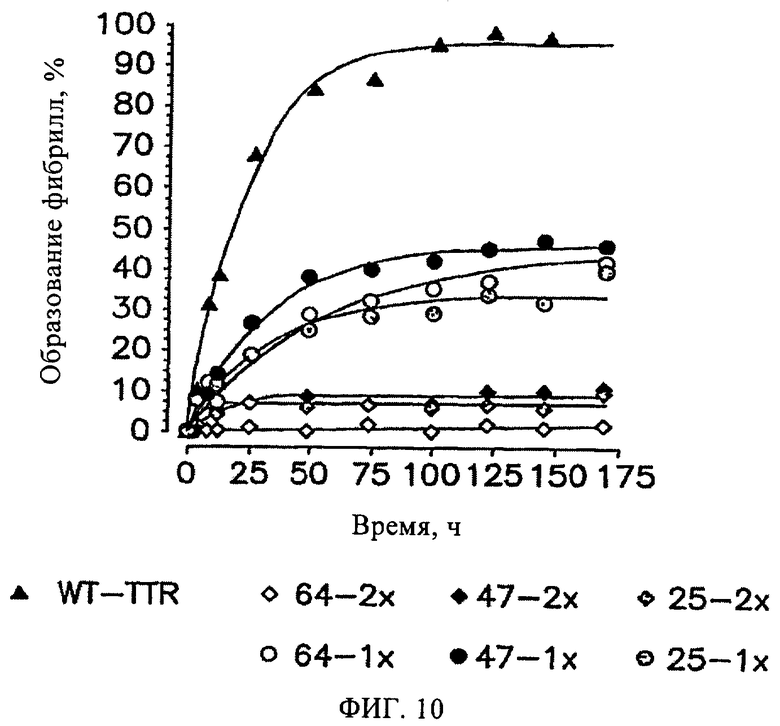
На фиг.10 представлена временная зависимость образования фибрилл WT-TTR (3,6 мкМ), опосредованного частичной денатурацией TTR в кислой среде, в отсутствие (▲) и присутствии 7,2 мкМ (◊) и 3,6 мкМ (○) ингибиторов 25, 47 и 64 (обозначения соответствующих ингибиторов см. на фиг.10). Образование фибрилл определяли по мутности анализируемых растворов при 500 нм.
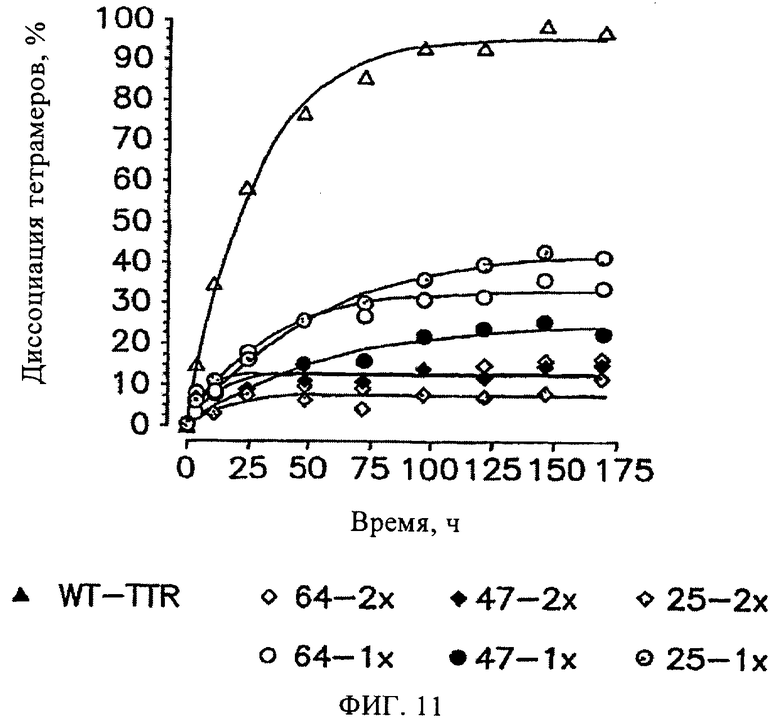
На фиг.11 представлена временная зависимость диссоциации тетрамеров WT-TTR (3,6 мкМ, 6,0 М мочевина) в отсутствие (▲) и присутствии ингибиторов 25, 47 и 64 при концентрации 7,2 мкМ (◊) и 3,6 мкМ (○) (обозначения соответствующих ингибиторов см. на фиг.11).
Подробное описание вариантов осуществления изобретения
Все дибензофураны (7,2 мкМ) кроме одного, содержащие в положении Сl арильные заместители, являются высокоэффективными ингибиторами индуцируемого кислотой (рН 4,4, 37°С) образования фибрилл WT-TTR (3,6 мкМ) in vitro, включая соединения с незамещенными арильными кольцами (фиг.6). Единственным неэффективным ингибитором на основе дибензофурана в отношении амилоидогенеза TTR является соединение 34, которое содержит четыре отрицательных заряда. Так как все соединения (7,2 мкМ) полностью ингибируют образование фибрилл TTR (в пределах ошибки эксперимента ±5%), установить зависимость активности лекарственного средства от структуры (SAR) не удалось. На фиг.3 и 7 представлен диапазон эффективности ингибиторов при концентрации, равной концентрации TTR (3,6 мкМ), что позволяет сделать некоторые выводы по взаимосвязи структуры и активности с учетом полученных 31 аналога и экспериментальной ошибки. Все ингибиторы (кроме соединения 34) характеризуются более высокой эффективностью по сравнению с исходным соединением (1) при 3,6 мкМ (фиг.3). Наиболее важно отметить то, что все тридцать высокоэффективных ингибитора, кроме двух (соединения 31 и 35), характеризуются исключительно высокой селективностью в отношении TTR в плазме крови человека. Таким образом, ингибиторы, содержащие заместитель в положении Сl, являются идеальными агентами для подавления амилоидогенеза TTR в биологических системах сложного состава.
По данным сопоставительного анализа эффективности трех серий ингибиторов, в которых арильное кольцо в положении Сl содержит четыре различных типа заместителей (Н, 3-СF3, 3,5-F2 и 3,5-Сl3), установлено, что ингибирующая активность ингибиторов, в которых арильное кольцо непосредственно присоединено к дибензофурану в положении С1 через простую связь, в дальнейшем называемых диарилами, повышается незначительно по сравнению с их биариламиновыми и биарилэфирными аналогами (фиг.3). Очевидно, это обусловлено структурными различиями, что обеспечивает различное ориентирование колец во внутренней полости тироксинсвязывающего кармана. Однако следует понимать, что при анализе эффективности множества аналогичных соединений помимо диарилов можно выявить другие предпочтительные ингибиторы. Такое предположение подтверждается тем, что из двух других серий ингибиторов можно выявить соединения с наиболее высокой селективностью в отношении TTR в плазме крови. Однако анализ взаимосвязи структуры и активности ингибиторов указанных серий опровергается фактом о том, что более 100 белков конкурируют за связывание с указанными ингибиторами. В то время как известно, что наиболее эффективными ингибиторами являются соединения, содержащие 2-F или 3,5-Cl2 заместители (соединения 36, 63 и 67), в связи с тем, что галоген ориентирован во внутреннюю полость кармана тироксинсвязывающего участка, неожиданно было установлено, что ингибиторы, в которых арил замещен тремя -СО2Н группами (соединение 33 и 69), являются самыми эффективными ингибиторами (несмотря на то, что они характеризуются умеренной селективностью в отношении TTR плазмы). Ранее по данным кристаллографических исследований ингибиторов на основе бифенила и бифениламина было установлено, что предпочтительными ингибиторами являются соединения, в которых арильное кольцо содержит в качестве заместителя карбоксильную группу, которая взаимодействует с внутренней полостью кармана связывания (при этом образуются водородные связи с остатками S117 и Т119) (Klabunde Т. и др. Nature Struct. Biol, 7, 312-321 (2000); Oza V.В. и др., J. Med. Chem., 45, 321-332 (2002); Adamski-Werner S.L. и др., J. Med. Chem., 47, 355-374 (2004)).
Введение в состав соединений (в положение Сl) замещенных арильных групп не только увеличивает эффективность ингибиторов, но что еще более важно, приводит к чрезвычайно значительному повышению их селективности в отношении TTR, по-видимому, за счет повышения аффинности указанных соединений к TTR по сравнению с другими белками плазмы. Чрезвычайно высокая селективность ингибиторов на основе дибензофурана, замещенного в положении С1 арильной группой, в отношении TTR плазмы крови подтверждается тем фактом, что ~2/3 синтезированных соединений взаимодействуют с TTR в стехиометрическом соотношении, равном более единицы. Особый интерес представляет соединение 1 (номер 1 при скрининге), которое взаимодействует только с внешней частью кармана связывания с TTR и не проявляет селективность в отношении TTR в плазме крови. По данным предварительных исследований установлено, что лишь несколько соединений из исследованных ингибиторов амилоидогенеза различной химической структуры взаимодействуют с TTR в стехиометрическом соотношении более 1 (Hammarstrom P. и др., Science, 299, 713-716 (2003); Klabunde Т. и др., Nature Struct. Biol, 7, 312-321 (2000); Razavi H. и др., Angew. Chem., 42, 2758-2761 (2003); Miroy G.J. и др., Proc. Natl. Acad. Sci., USA, 93, 15051-15056 (1996); Peterson S.А. и др., Proc. Natl. Acad. Sci., USA, 95, 12956-12960 (1998); Baures P.W., Peterson S.A., Kelly J.W., Bioorg. Med. Chem., 6, 1389-1401 (1998); Baures P.W. и др., Bioorg. Med. Chem., 7, 1339-1347 (1999); Petrassi H.M. и др., J. Am. Chem. Soc., 122, 2178-2192 (2000); McCammon M. G. и др., Structure, 10, 851-863 (2002); Oza V.В. и др., J. Med. Chem., 45, 321-332 (2002); Sacchettini J.С, Kelly J.W., Nature Rev. Drug Disc., 1, 267-275 (2002); Green N.S. и др., J. Am. Chem. Soc., 125, 13404-13414 (2003); Adamski-Werner S.L. и др., J. Med. Chem., 47. 355-374 (2004); Miller S.R. и др., Lab. Inv., 84, 545-552 (2004)). На фиг.7 соединения на основе дибензофурана, которые удовлетворяют критериям высокой ингибирующей активности in vitro (образование фибрилл <40%) и высокой селективности (стехиометрическое соотношение ингибитор/TTR в плазме крови >1), выделены в отдельный квадрат, который заштрихован серым цветом. Наиболее важным условием является то, что почти все ингибиторы на основе дибензофурана, прежде всего соединения, которые расположены в сером квадрате (фиг.7), характеризуются достаточными активностью и селективностью для кинетической стабилизации TTR в плазме крови, чтобы обеспечивать требуемые профили биодоступности, фармакокинетики и токсичности.
Не удивительно, что величины эффективности ингибитора in vitro (3,6 мкМ) и селективности ингибитора (10,8 мкМ) в отношении TTR в плазме крови не коррелируют друг с другом (фиг.7). Соединения, которые характеризуются наиболее высокой селективностью в отношении TTR, по сравнению со всеми другими плазматическими белками, являются чрезвычайно эффективными ингибиторами образования фибрилл. Однако обратное утверждение не всегда верно. Чрезвычайно эффективные ингибиторы не всегда характеризуются высокой селективностью в отношении TTR плазмы крови. Наиболее пригодными соединениями для лечения человека являются чрезвычайно эффективные ингибиторы, которые характеризуются высокой селективностью в отношении TTR в плазме, поскольку они селективно стабилизируют нативный комплекс TTR и защищают его от переходной диссоциации, таким образом повышая кинетическую стабилизацию TTR в составе биологической жидкости, содержащей высокие концентрации различных белков. Важной характеристикой взаимодействия ингибиторов с TTR является константа связывания, поскольку установлено, что степень кинетической стабилизации пропорциональна величинам констант связывания. Однако не следует всецело полагаться на данные, такие как константы связывания и эффективность, полученные в экспериментах in vitro. Например, если соединения в экспериментах in vitro чрезвычайно эффективно ингибируют амилоидогенез TTR, но взаимодействуют с другими плазматическими белками, то они не пригодны для лечения человека (Purkey Н.Е. и др., Proc. Natl. Acad. Sci., USA, 98, 5566-5571 (2001)). Соединения, характеризующиеся высокой эффективностью in vitro и низкой селективностью в отношении TTR, очевидно взаимодействуют с другими плазматическими белками, например, с альбумином (Purkey Н.Е. и др., Proc. Natl. Acad. Sci., USA, 98, 5566-5571 (2001)). Идеальными объектами для фармакологических испытаний являются ингибиторы на основе дибензофурана, так как они характеризуются чрезвычайно высокой селективностью и ингибирующей активностью по сравнению с различными группами ингибиторов, исследованных ранее (Hammarstrom P. и др., Science, 299, 713-716 (2003); Klabunde Т. и др., Nature Struct. Biol., 7, 312-321 (2000); Razavi Н. и др., Angew. Chem., 42, 2758-2761 (2003); Miroy G.J. и др., Proc. Natl. Acad. Sci., USA, 93, 15051-15056 (1996); Peterson S.А. и др., Proc. Natl. Acad. Sci., USA, 95, 12956-12960 (1998); Baures P.W., Peterson S.A., Kelly, J.W., Bioorg. Med. Chem., 6, 1389-1401 (1998); Baures P.W. и др., Bioorg. Med. Chem., 7, 1339-1347 (1999); Petrassi Н.М. и др., J. Am. Chem. Soc., 122, 2178-2192 (2000); McCammon M. G. и др., Structure, 10, 851-863 (2002); Oza V. В. и др., J. Med. Chem., 45, 321-332 (2002); Sacchettini J.С, Kelly J.W., Nature Rev. Drug Disc., 1, 267-275 (2002); Green N.S. и др., J. Am. Chem. Soc., 125, 13404-13414 (2003); Adamski-Wemer S. L. и др., J. Med. Chem., 47, 355-374 (2004); Miller S.R. и др., Lab. Inv., 84, 545-552 (2004)). Так как TTR является белком-переносчиком Т4 в виде тетрамера, то более 99% участков связывания Т4 не заняты, следовательно взаимодействие указанных ингибиторов с TTR не нарушает гомеостаз Т4.
Соединения на основе замещенных в положении С1 дибензофуранов являются перспективными агентами для перорального введения, так как они ингибируют процесс образования амилоидных фибрилл in vitro, характеризуются чрезвычайно высокой селективностью в отношении TTR в плазме крови, снижают скорость диссоциации TTR (которую можно использовать для оценки высокой селективности в отношении белков плазмы указанным в данном контексте методом), повышают кинетическую стабилизацию тетрамерной структуры TTR, характеризуются химической стабильностью в плазме крови, химически устойчивы при низких значениях рН. Указанные ингибиторы пригодны для лечения TTR-амилоидных заболеваний, включая SSA, FAP и FAC, так как повышение кинетической стабилизации TTR, как известно, приводит к ослаблению симптомов FAP (Hammarstrom P. и др., Science, 299, 713-716 (2003); Coelho Т: и др., J. Rheumatol, 20, 179-179 (1993); Coelho Т. и др., Neuromuscular Disord., 6, 27-27 (1996)).
Описание экспериментов и синтеза
На фиг.1А представлено пространственное изображение соединения 1, которое характеризуется наличием двух симметрично расположенных участков связывания (выделены зеленым и желтым цветом) в составе одного тироксинсвязывающего участка TTR, поверхность которого выделена серым цветом (Klabunde Т. и др., Nature Struct. Biol., 7, 312-321 (2000)). Карбоксильные группы в положении 4 и 6 обеспечивают электростатическое взаимодействие с ε-NН3 +-группами Lys 15 и Lys 15', расположенными во входной области тироксинсвязывающего участка. Установлено, что при удалении одной из карбоксильных групп полученный дибензофуран характеризуется значительно меньшей активностью, аналогично большинству аналогов, в которых карбоксильные группы присоединены к ароматическому кольцу через линкеры (фиг.2). Следует отметить, что дибензофурановое кольцо точно соответствует форме и гидрофобности внешней части тироксинсвязывающего кармана. При анализе кристаллической структуры комплекса TTR·12 (фиг.1А) установлено, что при взаимодействии TTR с соединением 1 большая часть внутреннего объема тироксинсвязывающего участка остается пустой. По данным указанного рентгеноструктурного анализа представляется очевидным, что производные соединения 1, в которых дибензофурановое кольцо в положении С1 содержит заместитель, например арильное кольцо, могут проникать и во внутреннюю полость участка связывания. Как представлено на фиг.1В, указанный заместитель присоединен к дибензофурановому циклу через гетероатом (N или О) или напрямую через связь Сарил-Сарил (на фиг.1В не показано). Ароматические заместители (фиг.3) выбирают таким образом, чтобы они обеспечивали взаимодействие ингибитора с галогенсвязывающими карманами или с субструктурами, которые образуют водородные связи во внутренней полости кармана. При выборе заместителей учитывают теоретические представления ориентации фенильного кольца во внутренней полости связывающего кармана и ранее полученные данные анализа SAR для различных химических соединений, содержащих арильные заместители в аналогичном положении (Hammarstrom P. и др., Science, 299, 713-716 (2003); Klabunde Т. и др., Nature Struct. Biol., 7, 312-321 (2000); Razavi H. и др., Angew Chem., 42, 2758-2761 (2003); Miroy G.J. и др., Proc. Natl. Acad. Sci, USA, 93, 15051-15056 (1996); Peterson S.А. и др., Proc. Natl. Acad. Sci. USA, 95, 12956-12960 (1998); Baures P.W. и др., Bioorg. Med. Chem., 6, 1389-1401 (1998); Baures P.W. и др., Bioorg. Med. Chem., 7, 1339-1347 (1999); Petrassi H.M. и др., J. Am. Chem. Soc., 122, 2178-2192 (2000); McCammon M.G. и др., Structure, 10, 851-863 (2002); Oza V.В. и др., J. Med. Chem., 45, 321-332 (2002); Sacchettini J.С.и др., Nature Rev. Drug Disc., 1, 267-275 (2002); Green N.S. и др., J. Am. Chem. Soc., 125, 13404-13414 (2003); Adamski-Wemer S.L. и др., J. Med. Chem., 47, 355-374 (2004); Miller Sean R. и др., Lab. Inv., 84, 545-552 (2004)).
Ингибиторы на основе дибензофуранов, замещенных в положении С1, получали из 2,4-ди-трет-бутил-6-бромфенола (соединение 2, коммерческий препарат), который на первой стадии конденсировали в условиях реакции радикальной гомоконденсации, с использованием гексацианоферрата (III) калия, при этом получали производное дибензофурана 3 (фиг.4, Tashiro M.Y. и др., Synthesis, 6, 495-496 (1980)). 1-Гидроксидибензофуран (4) получали из указанного тетра-трет-дибензофуранового производного в условиях реакции трансалкилирования в толуоле, общий выход составляет 33% в расчете на количество соединения 2 (альтернативная методика получения указанного промежуточного соединения описана в работах Labiad В., Villemin D., Synthesis, 143-144 (1989); Lee Y.R. и др., Org. Lett., 2, 1387-1389 (2000)). После введения защитных групп в фенол полученный силиловый эфир 5 обрабатывали втор-бутиллитием в условиях реакции орто-металлирования, при этом селективно вводили заместители в положение 4 и 6 (триизопропилсилильная защитная группа в условиях реакции металлирования защищает атом кислорода в положении 1) (Snieckus V., Chem. Rev., 90, 879-933 (1990)). Дианион нейтрализуют газообразным СО2, затем в условиях реакции этерификации получают соединение 6, которое деблокируют обработкой TBAF, затем получают трифлат 8 с высоким выходом.
Соединения 9-23 (аналоги биариламинов на основе дибензофурана, фиг.5) получали при конденсации некоторых анилинов с трифлатом 8 в условиях реакции N-арилирования, в присутствии палладиевого катализатора, как описано в статье Buchwald и Hartwig (фиг.5, Louie J.D. и др., J. Org. Chem., 62, 1268-1273 (1997); Wolfe J.Р.В., Stephen L., Tetrahedron Lett., 38, 6359-6362 (1997)). При взаимодействии фенола 7 с различными фенилбороновыми кислотами в условиях реакции кросс-сочетания, опосредованной соединениями меди по методике, описанной Chan и Evans, получали ариловые эфиры дибензофурана (замещение в положении С1), соединения 39-43 (Chan D.M.Т. и др., Tetrahedron Lett., 39, 2933-2936 (1998); Evans D.А. и др., Tetrahedron Lett., 39, 2933 (1998)). Соединения 49-59 (аналоги биарилов на основе дибензофурана) получали при взаимодействии соединения 8 с различными фенилбороновыми кислотами в условиях реакции конденсации Сузуки (Suzuki A., Modern Arene Chemistry, 53-106 (2002)). При омылении метиловых эфиров указанных предшественников получали требуемые амины (соединения 24-38), простые эфиры (соединения 44-48) и биарилы (соединения 60-70) на основе дибензофуран-4,6-дикарбоновой кислоты и оценивали ингибирующую активность полученных соединений в отношении амилоидогенеза TTR.
Результаты
Наиболее важными характеристиками эффективных низкомолекулярных ингибиторов амилоидогенеза являются высокая аффинность и селективность в отношении TTR в крови, а также стабилизация нативной тетрамерной четвертичной структуры TTR (Hammarstrom P. и др., Science, 299, 713-716 (2003); Razavi H. и др., Angew Chem., 42, 2758-2761 (2003); Purkey H.Е. и др., Рrос. Natl. Acad. Sci., USA, 98, 5566-5571 (2001)). Эффективность ингибирования (соединения 24-38, 44-48 и 60-70) определяли с использованием рекомбинантного TTR в буферном растворе, в котором происходит частичная денатурация белка и в котором ускоряется амилоидогенез (рН 4,4, 37°С). Затем проводили оценку селективности связывания эффективных ингибиторов с TTR по сравнению со всеми другими белками плазмы крови человека.
Оценка ингибирующей способности соединений на основе дибензофурана в отношении амилоидогенеза
Эффективность ингибирования образования амилоидных фибрилл TTR исследовали по описанной ранее методике образования устойчивых фибрилл, при этом частичную денатурацию TTR инициировали подкислением раствора белка (рН 4,4, 37°С, Colon W., Kelly J.W., Biochemistry, 31, 8654-8660 (1992)). Исследуемое соединение (7,2 или 3,6 мкМ) инкубировали с TTR (3,6 мкМ) в течение 30 мин в буферном растворе при рН 7. Амилоидогенез инициировали подкислением полученной смеси до рН 4,4, при этом максимальное количество фибрилл WT-TTR образуется через 72 ч (37°С). Мутность раствора определяли в присутствии потенциального ингибитора (Тиссл) и в отсутствие ингибитора (Тконт). Соединения, полностью подавляющие процесс образования фибрилл (образование фибрилл 0%), относят к чрезвычайно эффективным ингибиторам, соединения, в присутствии которых образуется максимальное количество фибрилл (образование фибрилл 100%) относят к соединениям, не обладающим ингибирующей активностью. Ранее было установлено, что чрезвычайно эффективные ингибиторы при концентрации низкомолекулярного соединения 7,2 мкМ снижают уровень образования фибрилл более чем на 90% (образование фибрилл <10%), а при концентрации, эквивалентной концентрации WT-TTR (3,6 мкМ), более чем на 60% (образование фибрилл <40%) (Hammarstrom P. и др., Science, 299, 713-716 (2003); Klabunde Т. и др., Nature Struct. Biol, 7, 312-321 (2000); Razavi H. и др., Angew Chem., 42, 2758-2761 (2003); Miroy G.J. и др., Proc. Natl. Acad. Sci., USA, 93, 15051-15056 (1996); Peterson S.А. и др., Рrос. Natl. Acad. Sci., USA, 95, 12956-12960 (1998); Baures P.W. и др., Bioorg. Med. Chem., 6, 1389-1401 (1998); Baures P.W. и др., Bioorg. Med. Chem., 7, 1339-1347 (1999); Petrassi H.М. и др., J. Am. Chem. Soc., 122, 2178-2192 (2000); McCammon M.G. и др., Structure, 10, 851-863 (2002); Oza V.В. и др., J. Med. Chem., 45, 321-332 (2002); Sacchettini J.С. и др., Nature Rev. Drug Disc., 1, 267-275 (2002); Green N.S. и др., J. Am. Chem. Soc., 125, 13404-13414 (2003); Adamski-Werner S.L. и др., J. Med. Chem., 47, 355- 374 (2004); Miller Sean R. и др., Lab. Inv., 84, 545-552 (2004)). Установлено, что из 31 исследованных соединений все кроме одного (соединение 34) полностью ингибируют процесс образования фибрилл при концентрации, превышающей концентрацию TTR в два раза (концентрация ингибитора 7,2 мкМ), фиг.6 (при условии, что значения констант диссоциации образуемых комплексов находятся в диапазоне нМ при рН 4,4, таким образом концентрация (7,2 мкМ) является достаточной для связывания исследуемого соединения с двумя участками связывания TTR (ТТR·I2)). Как правило, взаимодействие низкомолекулярных соединений с TTR характеризуется отрицательной кооперативностью, следовательно, значения констант Kd1 и Kd2 различаются на один или два порядка. Поэтому, если в смеси лиганд и TTR присутствуют в равных концентрациях, то содержание комплексов TTR·I, TTR·I2 и свободного TTR определяется величиной констант диссоциации указанных комплексов. К настоящему моменту, на основании данных других независимых исследований, установлено, что достаточным условием стабилизации тетрамерной структуры TTR и подавления амилоидогенеза является взаимодействие ингибитора только с одним из двух участков связывания TTR (Wiseman R.L. и др., J. Am. Chem. Soc. в печати (2005)). Аналогичный эффект подтверждается данными, описанными в настоящем изобретении, т.е. двадцать шесть синтезированных дибензофуранов являются чрезвычайно эффективными ингибиторами амилоидогенеза TTR (при концентрации ингибитора равной концентрации TTR (3,6 мкМ для каждого соединения, фиг.3) образование фибрилл составляет <40%). При изучении типичных низкомолекулярных соединений из трех различных серий синтезированных соединений (соединения 25, 26, 27, 30, 45, 47, 62; 3,6 мкМ) в условиях кислотного амилоидогенеза (рН 4,4, 37°С, 72 ч) в отсутствие TTR установлено, что формирование осадка не происходит.
Оценка селективности связывания ингибиторов на основе дибензофурана с TTR в плазме крови
Для оценки селективности связывания ингибиторов с TTR в плазме крови использовали метод связывания с антителами (Purkey Н.Е. и др., Рrос. Natl. Acad. Sci., USA, 98, 5566-5571 (2001)). Указанный метод включает инкубацию ингибиторов (10,8 мкМ, что ~2-3 раза больше концентрации TTR в плазме крови) в плазме крови человека в течение 24 ч при 37°С. Для удаления всех низкомолекулярных соединений, кроме TTR, в плазму добавляли немодифицированную сефарозу. Затем TTR и комплексы TTR-низкомолекулярный ингибитор связывали с поликлональными антителами к TTR, иммобилизованными на сефарозе, и сефарозу промывали (5×10 мин), комплекс антитела-TTR разрушали при элюции раствором с высоким значением рН, полученный продукт анализировали методом обращено-фазовой ЖХВР. Затем по значениям площади соответствующих пиков ЖХВР, используя калибровочные кривые, рассчитывали стехиометрический параметр: соотношение содержания TTR и ингибитора. В связи с потерями при промывке смолы, ингибиторы, которые характеризуются высокой константой диссоциации, указанный анализ обычно не позволяет определить точный стехиометрический состав комплексов, однако достаточно надежные результаты можно получить при анализе соединений, характеризующихся низкой константой диссоциации. Стехиометрический параметр связывания для двадцати одного ингибитора с TTR составляет более 1 (стехиометрическое соотношение, равное 2, является максимально возможным), девятнадцать из которых более чем на 60% подавляют образование фибрилл при концентрации 3,6 мкМ (фиг.7, заштрихованный квадрат). Высокая селективность связывания с TTR в плазме крови, принимая во внимание отсутствие селективности связывания исходной дибензофуран-4,6-дикарбоновой кислоты (1) с TTR, является доказательством важной роли арильного заместителя (в положении С1) в составе полученных соединений в повышении селективности их взаимодействия с TTR по сравнению со всеми другими белками плазмы.
Стабилизация тетрамерной четвертичной структуры TTR в условиях, индуцирующих амилоидогенез
Способность арилзамещенных в положении С1 дибензофуранов ингибировать образование фибрилл TTR за счет стабилизации его нативной структуры (т.е. стабилизация тетрамерной структуры) оценивали по данным исследования четвертичной структуры TTR методом аналитического ультрацентрифугирования после предварительной инкубации в течение 72 ч в условиях, индуцирующих амилоидогенез (рН 4,4, 37°С). По данным седиментационного анализа (фиг.8) и данным, полученным методом аналитического равновесного ультрацентрифугирования (фиг.9), установлено, что в присутствии соединения 27 (7,2 мкМ) TTR характеризуется гидродинамической молекулярной массой 57,1±0,3 и 55,1±0,4 кДа соответственно (молекулярная масса нативного тетрамера TTR составляет 55,0 кДа). По данным ультрацентрифугирования в отсутствие соединения 27 TTR образовывал олигомеры с чрезвычайно высокой молекулярной массой, которые быстро выпадают в осадок (данные не показаны).
Оценка способности ингибиторов на основе дибензофурана повышать кинетическую стабилизацию TTR
Способность указанных ингибиторов повышать кинетическую стабилизацию тетрамеров TTR оценивали по степени диссоциации тетрамеров TTR (Hammarstrom P. и др., Science, 299, 713-716 (2003); Hammarstrom P. и др., Proc. Natl. Acad. Sci., USA, 99, 16427-16432 (2002)). В кислой среде диссоциация тетрамеров приводит к амилоидогенезу, в то время как в присутствии хаотропного агента (6 М мочевина) диссоциация тетрамеров приводит только к развертыванию полипептидной цепи мономеров. Влияние ингибиторов 25, 47 и 64, относящихся к трем структурно различным классам ингибиторов на основе дибензофурана, на степень диссоциации тетрамеров исследовали как в кислой среде, так и в присутствии мочевины. Установлено, что амилоидогенез TTR, индуцированный частичной денатурацией мономеров в кислой среде, существенно снижается в присутствии соединений 25, 47 и 64 по сравнению с контролем (опыт в отсутствие ингибиторов), и зависит от концентрации указанных ингибиторов (фиг.4А). Степень диссоциации тетрамеров TTR в присутствии 6 М мочевины определяли по переходу от медленных изменений четвертичной структуры белка до быстрых изменений с использованием спектроскопических методов (Hammarstrom P. и др., Proc. Natl. Acad. Sci., USA, 99, 16427-16432 (2002)). По данным спектроскопии кругового дихроизма в дальней области УФ установлено, что значительно замедленная скорость диссоциации тетрамеров TTR (3,6 мкМ) в присутствии 6 М мочевины зависит от концентрации соединений 25, 47 и 64. Полученные результаты согласуются с результатами по различной стабилизации устойчивого состояния по сравнению с переходным состоянием комплекса, который подвергается диссоциации, при связывании с соединениями 25, 47 и 64.
Методики
В экспериментах использовали известные методики получения бактериального рекомбинантного TTR (Lai Z. и др., Biochemistry, 35, 6470-6482 (1996)), количественного анализа образования устойчивых фибрилл (Colon W., Kelly J.W., Biochemistry, 31, 8654-8660 (1992); Lai Z. и др., Biochemistry, 35, 6470-6482 (1996)), определения селективности связывания ингибиторов с белками в плазме крови (Purkey Н.Е. и др., Proc. Natl. Acad. Sci., USA, 98, 5566-5571 (2001)) и аналитического ультрацентрифугирования (Lashuel Н.А. и др., Biochemistry, 37, 17851-17864 (1998)).
Оценка ингибирующей активности соединений 25, 47 и 64 в отношении образования фибрилл WT-TTR в зависимости от времени
Соединения 25, 47 и 64 растворяли в ДМСО, при этом получали первичные исходные растворы с концентрацией 7,2 мМ (10х), которые разбавляли ДМСО в пять и десять раз, при этом получали вторичные исходные растворы с концентрацией 1,44 мМ (2х) и 0,72 мМ (1х) соответственно. В кювету для УФ-спектрофотометра добавляли раствор WT-TTR (495 мкл, 0,4 мг/мл, 7,2 мкМ) в буферном растворе (10 мМ фосфат натрия, 100 мМ KСl и 1 мМ ЭДТА, рН 7,2) и 5 мкл вторичного исходного раствора ингибитора (с концентрацией 1,44 или 0,72 мМ), быстро встряхивали, затем инкубировали в течение 30 мин при 25°С. Смесь подкисляли добавлением кислотного буферного раствора (500 мкл, 100 мМ ацетат, 100 мМ KСl, 1 мМ ЭДТА, рН 4,2) до рН 4,4. Полученный раствор (1 мл) повторно встряхивали и инкубировали в темноте при 37°С без перемешивания. Через 0, 4, 8, 12, 25, 49, 74, 100, 122, 145 и 169 ч (после подкисления раствора) раствор встряхивали и измеряли мутность при 500 нм. В качестве контроля использовали раствор TTR, в который добавляли ДМСО (5 мкл), не содержащий ингибиторы, анализ проводили, как описано выше. Для каждого эксперимента готовили по 10 образцов, содержащих и не содержащих низкомолекулярные ингибиторы, чтобы исключить перемешивание при инкубировании в кювете. После измерения мутности образцы отбрасывали.
Зависимость способности соединений 25, 47 и 64 ингибировать диссоциацию тетрамеров WT-TTR от времени, которую определяли по денатурации мономеров в присутствии мочевины
Соединения 25, 47 и 64 растворяли в ДМСО, при этом получали первичные исходные растворы с концентрацией 10 мМ, которые разбавляли этанолом в десять раз, при этом получали вторичные исходные растворы с концентрацией 1 мМ. В пластиковую пробирку (эппендорф, объемом 2 мл) добавляли WT-TTR (200 мкл, 1,0 мг/мл, 18 мкМ) в буферном растворе (50 мМ фосфат натрия, 100 мМ KСl и 1 мМ ЭДТА, рН 7,2), затем добавляли 7,2 или 3,6 мкл (2х и 1х соответственно) 1 мМ вторичного исходного растворы ингибитора, быстро встряхивали, затем инкубировали в течение 15 мин при 25°С. Растворы TTR-ингибитор (100 мкл) добавляли в буферный раствор (900 мкл) мочевины (6,67 М мочевина, 50 мМ фосфат натрия, 100 мМ KСl, 1 мМ ЭДТА, рН 7,2). Полученные растворы (конечный объем 1 мл) повторно быстро встряхивали и инкубировали в темноте при 25°С без перемешивания. Через 0, 4, 11, 24, 49, 73, 97, 122, 146 и 170 ч (после перемешивания при добавлении мочевины) регистрировали спектры кругового дихроизма (исследуемый интервал 218-215 нм, сканирование через каждые 0,5 нм, усреднение для 5 с). После измерения спектров образцы выливали обратно в соответствующую пластиковую пробирку и снова инкубировали. Для сравнения в качестве контроля использовали раствор ДМСО (7,2 мкл, 10% ДМСО в EtOH) и проводили анализ, как описано выше. Снижение содержания β-складчатой структуры в ходе эксперимента определяли по амплитуде сигнала КД, измеренного в интервале 215-218 нм. Процесс диссоциации тетрамеров TTR сопровождается быстрой денатурацией мономеров (в ~500000 раз быстрее), которую определяют по уменьшению сигнала, соответствующего β-складчатой структуре (Hammarstrom Р. и др., Proc. Natl. Acad. Sci., USA, 99. 16427-16432 (2002)).
Получение ингибиторов
Для синтеза использовали коммерческие реагенты, которые использовались без дополнительной очистки, если не указано иное. Процесс образования требуемых соединений контролировали, используя метод тонкослойной хроматографии на пластинах из алюминиевай фольги со слоем силикагеля 60 F254 (фирмы ЕМ Sciences) или метод аналитической обращено-фазовой жидкостной хроматографии высокого разрешения (ЖХВР). Анализ ЖХВР проводили на хроматографе, оборудованном градиентной системой подачи растворителя Waters 600E, детектором Waters 486 и автосамплером Waters 717 plus. Для обращено-фазовой ЖХВР во всех случаях использовали колонку С 18 фирмы Western Analytical (модель 033-715, размер пор 150 Å, диаметр частиц 3 мкм). В качестве элюента использовали смесь ацетонитрил/вода/трифторуксусная кислота; при формировании градиента использовали раствор А, содержащий указанные растворители в соотношении 4,8%:95%:0,2% соответственно, и раствор В, содержащий указанные растворители в соотношении 95%:4,8%:0,2% соответственно. Хроматографию проводили в следующем режиме: в течение 2 мин промывка в изократическом режиме 100% раствором А, затем в течение 8 мин линейный градиент от 0 до 100% раствора В, скорость потока 1,5 мл/мин. В случае применения метода экспресс-хроматографии во всех случаях использовали колонку с силикагелем 60 (230-400 меш, фирмы ЕМ Sciences). Спектры 1Н- и 13С-ЯМР регистрировали на спектрометре Bruker при частоте 300, 400, 500 или 600 МГц с использованием внутреннего стандарта Me4Si (0,0 част./млн). Величину химического сдвига выражали в част./млн.
(Дибензофуран-1-илокси)триизопропилсилан (5)
В сухую круглодонную колбу (объемом 250 мл) добавляли фенол 4 (Tashiro М.Y. и др., Synthesis, 6, 495-496 (1980)) (492 мг, 2,67 ммоля), помещали перемешивающий стержень и колбу закрывали мембраной. Затем последовательно добавляли CH2Cl2 (5 мл), DMAP (391 мг, 3,2 ммоля) и триизопропилхлорсилан (800 мкл, 3,73 ммоля). Полученный бесцветный раствор инкубировали в течение ночи, при этом получали суспензию белого цвета. Реакционную смесь переносили в делительную воронку (объемом 250 мл) и промывали Н2О (3×10 мл). Водные слои объединяли и экстрагировали CH2Cl2 (3×30 мл). Органические слои объединяли, сушили над MgSO4 и концентрировали при пониженном давлении, при этом получали масло желтого цвета. Масло очищали экспресс-хроматографией на колонке с силикагелем (элюент; 100% гексан), при этом получали соединение 5 (0,70 г, 77%) в виде бесцветного масла.
МС (MALDI-FT) (m/z): для C21H29OSi рассч. 341,1931, найд. 341,1932 (M+H)+.
Диметиловый эфир 1-триизопропилсиланилоксидибензофуран-4,6-дикарбоновой кислоты (6)
Силиловый эфир 5 (654 мг, 1,92 ммоля) добавляли в сухую круглодонную колбу (объемом 50 мл), затем добавляли Et2O (7,4 мл) и TMEDA (0,87 мл, 5,77 ммоля). Колбу в течение 10 мин охлаждали на бане (ацетон/сухой лед) до -78°С, затем в течение 10 мин добавляли втор-бутиллитий (4,44 мл 1,3 М раствора в циклогексане, 5,77 ммоля). Полученную суспензию оранжевого цвета нагревали до комнатной температуры и перемешивали в течение 24 ч. Колбу охлаждали до -78°С, как описано выше, затем через суспензию под давлением (15 фунтов на квадратный дюйм) пропускали поток газообразного СО2 (СО2 пропускали через осушительный патрон, содержащий активированный силикагель). После добавления СО2 охлаждающую баню удаляли, реакционную смесь перемешивали в течение 30 мин и выливали в стакан (объемом 1 л), содержащий ледяную воду (50 мл). Раствор подщелачивали медленным добавлением 0,05 М КОН до рН 9, затем охлаждали до 0°С на ледяной бане (лед/Н2О). Раствор подкисляли добавлением 0,5 М НСl до рН 2, при этом получали осадок твердого вещества белого цвета. Водную суспензию (рН 2) переносили в делительную воронку (объемом 1 л) и экстрагировали EtOAc (5×50 мл). Экстракты объединяли, сушили над MgSO4 и концентрировали при пониженном давлении, при этом получали неочищенную дикислоту в виде масла. В колбу (объемом 100 мл), содержащую неочищенную дикислоту, помещали перемешивающий стержень, закрывали мембраной и вакуумировали. Колбу заполняли аргоном. Затем через шприц добавляли безводный МеОН (2 мл) и бензол квалификации ACS (8 мл). Затем через мембрану из шприца медленно добавляли триметилсилилдиазометан (TMSCHN2, 2,5 мл 2 М раствора в гексане, 5 ммолей). После добавления TMSCHN2 реакционную смесь перемешивали в течение 10 мин и растворитель удаляли при пониженном давлении, при этом получали масло красного цвета. Полученный остаток очищали экспресс-хроматографией на колонке с силикагелем (элюент: 15% EtOAc в гексане), при этом получали соединение 6 в виде твердого вещества белого цвета (0,36 г, 43%).
МС (MALDI-FT) (m/z): для C25H32O6SiNa рассч. 479,1860, найд. 479,1874 (N+Na)+.
Диметиловый эфир 1-гидроксидибензофуран-4,6-дикарбоновой кислоты (7)
Сухую круглодонную колбу (объемом 100 мл), в которую помещали стержень для перемешивания, и содержащую соединение 6 (363 мг, 0,95 ммоля), закрывали мембраной, вакуумировали и заполняли аргоном. Затем в реакционную смесь через шприц добавляли безводный ТГФ (6,3 мл) и фторид тетрабутиламмония (1 М раствор в ТГФ, 1,2 мл, 1,19 ммоля). Реакционную смесь перемешивали в течение 1 ч при комнатной температуре, затем выливали в делительную воронку (объемом 250 мл), содержащую Н2О (30 мл). Водный слой экстрагировали СНСl3 (4×20 мл). Органические слои объединяли, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией на колонке с силикагелем (элюент: 30% EtOAc в гексане), при этом получали соединение 7 (0,23 г, 97%) в виде твердого вещества белого цвета.
ЖХ/МС (m/z): для С16Н12O6 рассч. 301, найд. 301.
Диметиловый эфир 1-трифторметансульфонилоксидибензофуран-4,6-дикарбоновой кислоты (8)
При получении соединения 8 в виде трифлата использовали методику, описанную ранее Стиллом (Echavarren А.М. и др., J. Am. Chem. Soc., 109, 5478-5486 (1987)). Фенол 7 (120 мг, 0,4 ммоля) добавляли в сухую круглодонную колбу (объемом 10 мл), которую затем закрывали мембраной. Растворитель, безводный пиридин (2 мл), добавляли через мембрану из шприца. Реакционную смесь охлаждали до 0°С на бане лед/вода. Реакцию инициировали добавлением ангидрида трифторметансульфоновой кислоты (81 мкл, 12 ммолей) через мембрану из шприца. Ледяную баню удаляли, реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь выливали в стакан (объемом 250 мл), содержащий смесь Н2О и измельченного льда (30 мл) и переносили в делительную воронку (объемом 125 мл). Водный слой экстрагировали Et2O (4×40 мл). Органические слои объединяли, промывали насыщенным раствором СuSО4 (4×20 мл) и солевым раствором (2×20 мл), сушили над MgSO4, затем Et2O удаляли при пониженном давлении, при этом получали твердое вещество светло-желтого цвета, которое очищали экспресс-хроматографией на колонке с силикагелем (элюент: 30% EtOAc в гексане), при этом получали соединение 8 (159 мг, 92%) в виде твердого вещества белого цвета.
МС (FAB (NBA/NaI)) (m/z): для C17H12F3O8S рассч. 433,0205, найд. 433,0215 (М+Н)+.
Типичная методика взаимодействия соединения 8 и замещенных анилинов в условиях реакции кросс-сочетания в присутствии палладиевого катализатора
При получении соединений 9-23 реакцию сочетания арилов проводили по методике, описанной Buchwald и Hartwig. В высушенную в пламени горелки пробирку из боросиликатного стекла (размер 10 мм × 13 см), снабженную стержнем для перемешивания и закрытую мембраной, добавляли соединение 8 (140 мг, 0,324 ммоля), палладийдибензилиденацетон (Pd2(dba)3, 15 мг, 0,016 ммоля), (±)-бинап (15 мг, 0,024 ммоля), Сs2СО3 (147 мг, 0,456 ммоля) и анилин (32 мкл, 0,356 ммоля). После добавления всех реагентов раствор в пробирке продували аргоном в течение 10 мин. Затем через мембрану добавляли безводный толуол (2,4 мл) и реакционную смесь нагревали при 100°С в течение 36 ч на масляной бане. Реакционную смесь фильтровали через слой целлита. Фильтрат упаривали при пониженном давлении. Полученное масло черного цвета очищали экспресс-хроматографией на колонке с силикагелем (элюент: 30% EtOAc в гексане), при этом получали биариламин 17 (0,12 г, 68%) в виде твердого вещества белого цвета. Соединения 10-23 получали аналогично тому, как описано выше, и характеризовали, как описано ниже для соединения 9.
Диметиловый эфир 1-фениламинодибензофуран-4,6-дикарбоновой кислоты (9)
МС (MALDI-FT) (m/z): для C22H17NO5 рассч. 375,1106, найд. 375,1094 (М)+.
Типичная методика получения 1-феноксидибензофуранов 39-43 из фенола 7 и замещенных фенилбороновых кислот по реакции кросс-сочетания в присутствии соединений меди
Для проведения реакции сочетания биариловых эфиров использовали модифицированные методики, описанные Chan и Evans. В сцинтилляционный флакон (объемом 20 мл), оборудованный перемешивающим стержнем для магнитной мешалки, добавляли фенол 7 (150 мг, 0,50 ммоля), ацетат меди (II) (91 мг, 0,5 ммоля), активированные молекулярные сита (4 Å, ~250 мг) и фенилбороновую кислоту (180 мг, 1,5 ммоля). Затем последовательно добавляли дихлорметан (5 мл) и пиридин (201 мкл, 2,5 ммоля), при этом получали бесцветную суспензию. Флакон не плотно закрывали, чтобы реакционная смесь частично контактировала с атмосферой. Завершение реакции определяли методом ТСХ. После завершения реакции в реакционную смесь добавляли силикагель (~6 г), затем растворитель удаляли при пониженном давлении. Реакционную смесь очищали хроматографией на указанном силикагеле (элюент: 30% EtOAc в гексане), при этом получали биариловый эфир 39 (29 мг, 15%) в виде твердого вещества белого цвета. Соединения 40-43 получали и характеризовали аналогично тому, как описано для соединения 39.
Диметиловый эфир 1-феноксидибензофуран-4,6-дикарбоновой кислоты (39)
МС (MALDI-FT) (m/z): для С22Н16О6Nа рассч. 399,0839, найд. 399,0825 (М+Na)+.
Типичная методика взаимодействия трифлата 8 и замещенных фенилбороновых кислот в условиях реакции кросс-сочетания в присутствии палладиевого катализатора
В высушенную в пламени горелки пробирку (размер 10 мм × 13 см), снабженную стержнем для перемешивания, закрытую мембраной, добавляли соединение 8 (100 мг, 0,23 ммоля), Рd (РРh3)4 (14 мг, 0,01 ммоля). LiCl (29 мг, 0,69 ммоля), Nа2СО3 (300 мкл 2 М водного раствора) и толуол (3 мл). Фенилбороновую кислоту (43 мг, 0,35 ммоля) растворяли в EtOH (0,5 мл) и добавляли в реакционную смесь. Все остальные соединения получали при замене EtOH на МеОН, так как в случае EtOH наблюдалась реакция переэтерификации, таким образом, соединение 49 получали в виде диэтилового эфира, а все остальные соединения в виде диметиловых эфиров. После добавления реагентов раствор в пробирке продували аргоном и реакционную смесь нагревали при 100°С в течение 12 ч на масляной бане. Затем реакционную смесь фильтровали через слой целита. Фильтрат упаривали при пониженном давлении, полученный остаток черного цвета очищали экспресс-хроматографией на колонке с силикагелем, при этом получали биарил 49 (52 мг, 63%) в виде твердого вещества белого цвета. Соединения 50-59 получали и характеризовали аналогично тому, как описано для соединения 49.
Диэтиловый эфир 1-фенилдибензофуран-4,6-дикарбоновой кислоты (49) МС (MALDI-FT) (m/z): для C24H20O5Na рассч. 411,1203, найд. 411,1197 (M+Na)+.
Типичная методика получения ингибиторов 24-38, 44-48 и 60-70 гидролизом соответствующих эфиров
В сцинтилляционном флаконе (объемом 20 мл), снабженном перемешивающим стержнем, проводили гидролиз метилового эфира 9 (25 мг, 0,067 ммоля) в ТГФ/МеОН/Н2О, 3:1:1,1 мл. В полученную суспензию добавляли LiOH·H2O (22 мг, 0,53 ммоля) и перемешивали до завершения реакции (как правило, в течение 4 ч), завершение реакции определяли методом ТСХ или методом аналитической обращенно-фазовой ЖХВР. Реакционную смесь разбавляли солевым раствором (2 мл) и подкисляли добавлением 1 М НСl до рН 2 (индикаторные полоски для определения рН), при этом получали двухслойную систему. Верхний слой (ТГФ) отделяли, водный слой экстрагировали ТГФ (3×3 мл). Органические слои объединяли, сушили над MgSO4, затем концентрировали при пониженном давлении, при этом получали дикислоту 24 (21 мг, 92%) в виде твердого вещества белого цвета. Соединения 25-38, 44-48 и 60-70 получали и характеризовали аналогично тому, как описано для соединения 24.
1-Фениламинодибензофуран-4,6-дикарбоновая кислота (24)
МС (MALDI-FT) (m/z): для C20H13NO5 рассч. 347,0788, найд. 347,0794 (M)+.
Кристаллическая структура комплекса TTR·12, полученная рентгеноструктурным методом, представлена на фиг.1А (Klabunde Т. и др., Nature Struct. Biol., 7, 312-321 (2000)). Остатки, выстилающие внутреннюю полость участка связывания, изображены с использованием стержневой модели (атомы кислорода обозначены красным цветом, атомы азота обозначены синим цветом, атомы углерода обозначены серым цветом), белковая поверхность Конноли обозначена серым цветом. На фиг.1А показаны две связанные молекулы соединения 1 в С2 симметрии (обозначены желтым и зеленым цветом). Канал связывания включает 3 типа углублений, так называемые галогенсвязывающие карманы (НВР), так как они взаимодействуют с атомами иода в составе тироксина. Соединение 1 взаимодействует только с внешней частью связывающего кармана и образует связи только с карманами НВР1 и НВР1'. Карбоксильные группы соединения 1 связываются с группами ε-NH3+ остатков K15 и K15'.
На фиг.1Б представлена схема ориентации замещенных в положении 1 дибензофуран-4,6-дикарбоновых кислот (в которых Х означает или NH, О или прямую Сарил-Сарил связь) во внутренней полости тироксинсвязывающего кармана. R означает заместители в составе замещенного арильного кольца, которые вводят для повышения сродства к внутренней полости связывающего кармана TTR.
На фиг.2 представлена таблица, в которой показано влияние концентрации производных дибензофурана, замещенного различными кислотами, на активность в отношении образования амилоидных фибрилл WT-TTR (3,6 мкМ) при рН 4,4 (в течение 72 ч). Степень образования фибрилл используют для оценки эффективности ингибитора по сравнению с количеством фибрилл WT-TTR, образовавшихся в отсутствие ингибиторов (100%). Полное подавление образования фибрилл соответствует отсутствию фибрилл (0%).
На фиг.3 представлена суммарная таблица, в которой показаны данные по ингибирующей активности производных дибензофурана (3,6 мМ) в отношении образования фибрилл WT-TTR (3,6 мМ, рН 4,4, в течение 72 ч, 3,6 мкМ) и стехиометрические параметры связывания указанных соединений с TTR в плазме крови человека.
"Степень образование фибрилл, %", указанная в среднем столбце таблицы, означает количество образовавшихся фибрилл, выраженное в %, которое используют для оценки эффективности ингибитора по сравнению с количеством образовавшихся фибрилл WT-TTR в отсутствие ингибиторов (100%). Полное подавление образования фибрилл соответствует отсутствию фибрилл (0%). В правом столбце приведены значения стехиометрического параметра связывания указанных соединений с TTR, который определяли по связыванию с иммобилизованными антителами (ингибитор использовали в концентрации 10,8 мМ, что приблизительно в 2-3 раза выше концентрации TTR в плазме крови).
На фиг.4 представлена схема получения диметилового эфира 1-гидроксидибензофуран-4,6-дикарбоновой кислоты и соответствующего трифлата.
а) K3[Fе(СN)6], КОН, Н2О, бензол, b) АlСl3, толуол, 33% в расчете на две стадии, с) TIPSCI, DMAP, CH2Cl2, 77%, d) втор-BuLi, Et2O, -78°C, газообразный CO2, TMSCHN2, 43%, e) TBAF, ТГФ, 97%, f) Tf2O, пиридин, 92%.
На фиг.5 представлена схема получения диметиловых эфиров 1-фенил-, фенокси- и фениламинодибензофуран-4,6-дикарбоновой кислоты и соответствующих дикарбоновых кислот.
a) Pd2(DBA)3, (±) бинап, Сs2СО3, толуол 100°С, b) LiOH-H2O, ТГФ/МеОН/Н2O (3:1:1), с) Сu(ОАс)2, пиридин, молекулярные сита, 4 Å, СН2Сl2, d) Pd(PPh3)4, LiCl, водный раствор Nа2СО3, толуол, МеОН, 80°С.
На фиг.6 представлена таблица, в которой показаны данные по ингибирующей активности производных дибензофурана (7,2 мкМ) в отношении образования амилоидных фибрилл WT-TTR (3,6 мкМ) при рН 4,4 в течение 72 ч. Степень образования фибрилл используют для оценки эффективности ингибитора по сравнению с количеством образовавшихся фибрилл WT-TTR в отсутствие ингибиторов (100%). Полное подавление образования фибрилл соответствует отсутствию фибрилл (0%).
На фиг.7 представлены данные сопоставления эффективности ингибирования образования фибрилл и стехиометрических параметров связывания различных производных дибензофуранов с TTR в плазме крови. В слабо заштрихованном квадрате расположены соединения, характеризующиеся высокой ингибирующей активностью и высокой селективностью (образование фибрилл <40%, стехиометрическое соотношение >1), в интенсивно заштрихованном квадрате расположены чрезвычайно эффективные соединения (образование фибрилл <30%, стехиометрическое соотношение >1,25).
Знаком (▲) обозначены соединения, в которых арильный заместитель присоединен через группу NH, (♦) соединения, в которых арильный заместитель присоединен через кислород, (●) соединения, в которых арильный заместитель присоединен через прямую Сарил-Сарил связь. Значения для дибензофуран-4,6-дикарбоновой кислоты (1, (●)) приведены для сравнения.
На фиг.8 представлена зависимость оптической плотности при 280 нм от радиуса ротора, полученная методом седиментационного анализа TTR (3,6 мМ) после предварительной инкубации в присутствии соединения 27 (7,2 мМ) с последующей инкубацией при рН 4,4 в течение 72 ч (период времени, в течение которого образуется максимальное количество амилоидных фибрилл в отсутствие ингибитора). Образцы анализировали после центрифугирования в течение 15 мин при 50000 об/мин. На фиг.8 представлены экспериментальные данные (символы), которые соответствуют единой идеальной модели, с молекулярной массой 57,1±0,2 кДа (обозначены сплошной линией).
На фиг.9 представлена зависимость оптической плотности при 280 нм от радиуса ротора, полученная методом равновесного ультрацентрифугирования TTR (3,6 мМ), который предварительно инкубировали в присутствии соединения 27 (7,2 мМ) с последующей инкубацией при рН 4,4 в течение 72 ч (период времени, в течение которого образуется максимальное количество амилоидных фибрилл в отсутствие ингибитора). Образцы анализировали после центрифугирования в градиенте концентрации в течение 24 ч со скоростью 17000 об/мин, при этом устанавливалось концентрационное равновесие. На фиг.9 представлены экспериментальные данные (о), которые соответствуют идеальной модели, с молекулярной массой 55,0±0,2 кДа (обозначены сплошной линией). Во вставке представлен разброс между экспериментальными и рассчитанными значениями.
На фиг.10 представлена временная зависимость образования фибрилл WT-TTR (3,6 мкМ), опосредованная частичной денатурацией TTR в кислотной среде, в отсутствие  и в присутствии 7,2 мкМ
и в присутствии 7,2 мкМ  и 3,6 (о) ингибиторов 25, 47 и 64 (обозначения соответствующих ингибиторов см. на фиг.10). Образование фибрилл определяли по изменению мутности анализируемых растворов, измеренной при 500 нм. По черно-белым графикам трудно выявить наиболее эффективные соединения. По данным, полученным через 169 ч или при завершении эксперимента, установлено, что эффективность ингибиторов снижается в следующем порядке: соединение 25, соединение 64, соединение 47.
и 3,6 (о) ингибиторов 25, 47 и 64 (обозначения соответствующих ингибиторов см. на фиг.10). Образование фибрилл определяли по изменению мутности анализируемых растворов, измеренной при 500 нм. По черно-белым графикам трудно выявить наиболее эффективные соединения. По данным, полученным через 169 ч или при завершении эксперимента, установлено, что эффективность ингибиторов снижается в следующем порядке: соединение 25, соединение 64, соединение 47.
На фиг.11 представлена временная зависимость диссоциации тетрамеров WT-TTR (3,6 мкМ, 6,0 М мочевина) в отсутствие (▲) и в присутствии ингибиторов 25, 47 и 64 при концентрации 7,2 мкМ (◊) и 3,6 мкМ (○) (обозначения соответствующих ингибиторов см. на фиг.11). Выявить замедленный тип диссоциации тетрамеров методом спектроскопии кругового дихроизма в дальней области УФ не представляется возможным. Однако процесс диссоциации сопровождается быстрой денатурацией мономеров (в ~500000 раз быстрее), которую определяли по уменьшению сигнала, соответствующего β-складчатой структуре (по данным метода спектроскопии кругового дихроизма). По черно-белым графикам трудно выявить наиболее эффективные соединения. По данным, полученным через 169 ч или при завершении эксперимента, установлено, что эффективность ингибиторов снижается в следующем порядке: соединение 25, соединение 64, соединение 47.
Изобретение относится к новым производным дибензофуран-4,6-дикарбоновой кислоты формулы I
где Х отсутствует или означает дирадикал, который выбирают из группы, включающей -O-, -S- и -NH-,
R2 и R4 независимо выбирают из группы, включающей -Н, -ОН, -F, -Сl, -Вr, -СF3 и -СO2Н, а R3 и R5 независимо выбирают из группы, включающей -Н, -ОН, -F, -Сl, -Вr и -СO2Н, которые являются чрезвычайно эффективными ингибиторами амилоидогенеза, проявляют повышенную аффинность и значительно повышенную селективность при связывании транстиретина (TTR) по сравнению со всеми остальными плазматическими белками и по сравнению с контрольным соединением 1. Кроме того, функция указанных соединений заключается в повышении кинетической стабильности тетрамерной структуры TTR. Изобретение также относится к способу ингибирования образования амилоидных фибрилл. 2 н. и 14 з.п. ф-лы, 11 ил.
1. Соединение формулы I
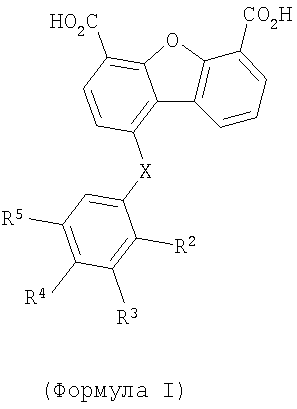
где X отсутствует или означает дирадикал, который выбирают из группы, включающей -O-, -S- и -NH-,
R2 и R4 независимо выбирают из группы, включающей -Н, -ОН, -F, -Сl, -Вr, -СF3 и -СO2Н, а
R3 и R5 независимо выбирают из группы, включающей -Н, -ОН, -F, -Сl, -Вr и -СО2Н.
2. Соединение по п.1 формулы II
где R2-R5 имеют значения, определенные в п.1.
3. Соединение по п.2, в котором
R2 означает радикал, который выбирают из группы, включающей -Н, -F, -Сl и -СF3.
4. Соединение по п.2, в котором
R4 означает радикал, который выбирают из группы, включающей -H, -Сl и -СО3Н.
5. Соединение по п.2, в котором
R5 означает радикал, который выбирают из группы, включающей -Н, -F и -Сl.
6. Соединение по п.2, которое выбирают из группы, включающей следующие структуры:
7. Соединение по п.1 формулы III
где R2-R5 имеют значения, определенные в п.1.
8. Соединение по п.7, в котором
R5 означает радикал, который выбирают из группы, включающей -Н, -F, -Сl и -Вr.
9. Соединение по п.7, которое выбирают из группы, включающей следующие структуры:
10. Соединение по п.1 формулы IV
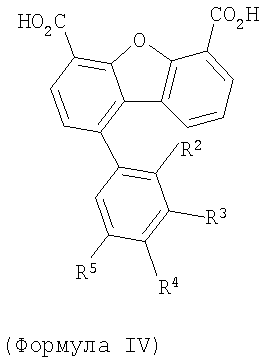
где R2-R5 имеют значения, определенные в п.1.
11. Соединение по п.10, в котором R2 означает радикал, который выбирают из группы, включающей -Н, -F и -Сl.
12. Соединение по п.10, в котором
R3 означает радикал, который выбирают из группы, включающей -Н, -F, -Сl и -СO2Н.
13. Соединение по п.10, в котором
R4 означает радикал, который выбирают из группы, включающей -Н и -СО2Н.
14. Соединение по п.10, в котором
R означает радикал, который выбирают из группы, включающей -H, -F и -Сl.
15. Соединение по п.10, которое выбирают из группы, включающей следующие структуры:
16. Способ ингибирования образования амилоидных фибрилл, включающий контактирование транстиретина с соединением по пп.1-15, при концентрации, достаточной для подавления образования амилоидных фибрилл.
| PURKEY Н.Е | |||
| et al | |||
| Proc | |||
| Nat | |||
| Acad | |||
| Sci | |||
| USA, 2001, v.98, no.10, p.5566-5571 | |||
| BAUSER P.W | |||
| et al | |||
| Bioorg | |||
| Med | |||
| Chem | |||
| Металлический водоудерживающий щит висячей системы | 1922 |
|
SU1999A1 |
| OZA V.B | |||
| et al | |||
| J | |||
| Med | |||
| Chem | |||
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Производные 1-амино-3-(1,2,3,4-тетрагидро-или 1,2,3,4,4 @ ,9 @ -гексагидродибензофуранил-8-окси)-пропанолов-2,обладающие @ -адреноблокирующим,гипотензивным,спазмолитическим,нейротропно-депримирующим и бронхолитическим свойствами | 1980 |
|
SU869278A1 |
| Способ выделения -кето-2-дибензофуранмасляной кислоты | 1973 |
|
SU499810A3 |

