Область техники, к которой относится изобретение
Настоящее изобретение относится к индольным алкалоидам, в частности к способу синтеза десерпидина.
Уровень техники
Резерпин (Ib) впервые был выделен Шлиттером в 1952 г. из экстрактов Rauwolfia serpentine (Muller et al., Experientia 1952, 8, 338) и идентифицирован в качестве основного компонента, обуславливающего гипотензивную активность экстрактов Rauwolfia spp.
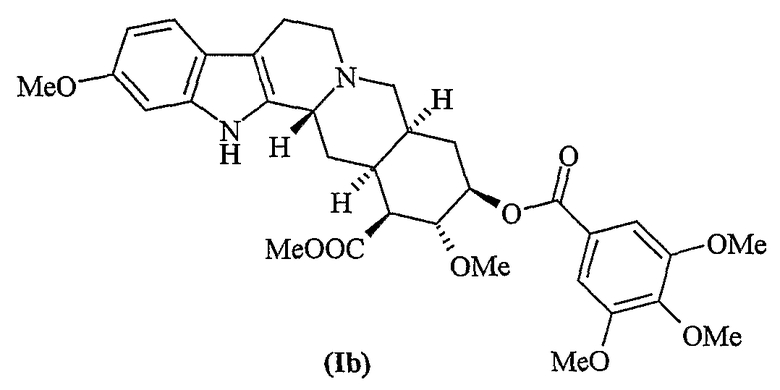
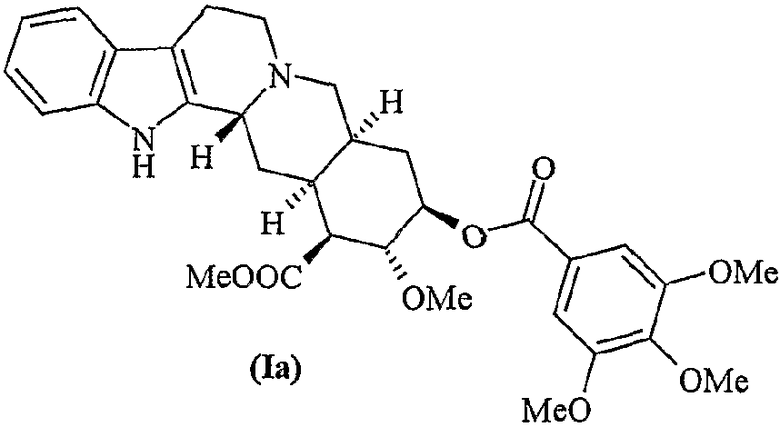
Десерпидин (Ia) впервые был выделен Хофманном в 1955 г. из корней Rauwolfia canescens (Stoll and Hofmann, J. Chem. Soc. 1955, 77, 820).
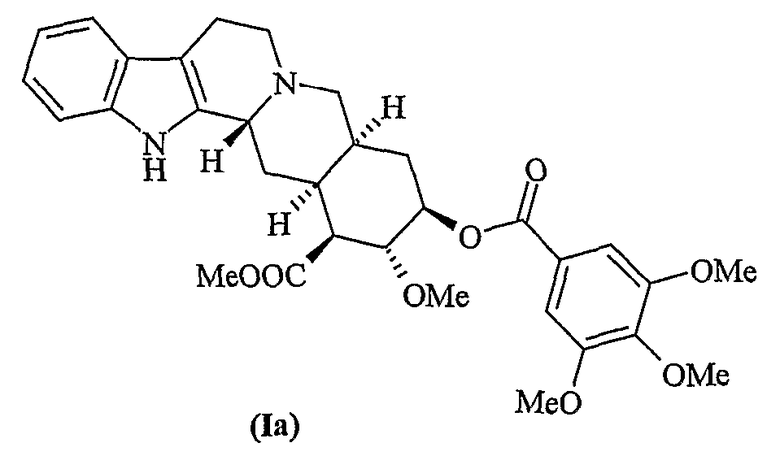
На протяжении многих лет резерпин и родственные индольные алкалоиды, такие как десерпидин, играли важную роль при лечении гипертензии, нервных и психических расстройств. Однако даже в тех случаях, когда десерпидин обладает представляющим интерес фармакологическим профилем, его применение всегда было ограничено по сравнению с резерпином вследствие его малой распространенности в природе. На практике титр десерпидина в корковой части составляет примерно 0,003-0,005%, в то время как титр резерпина составляет примерно 0,1-0,2%.
Десерпидин структурно сходен с резерпином (Ib) и ресциннамином (Ic).
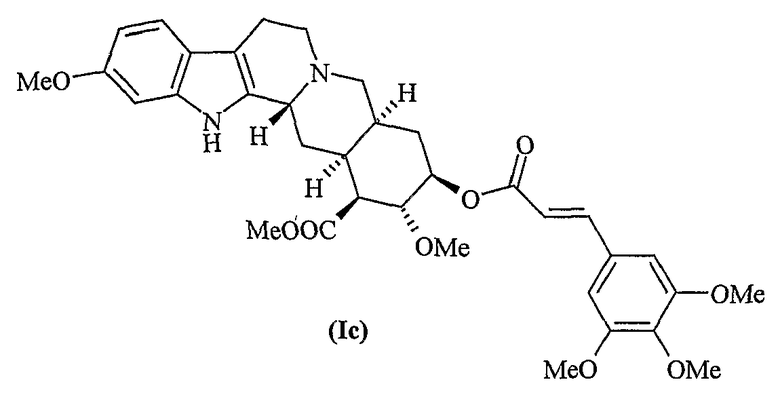
По сравнению с резерпином в десерпидине отсутствует метоксигруппа в положении 11. По сравнению с ресциннамином в десерпидине отсутствует метоксигруппа в положении 11 и положение 18 этерифицировано остатком 3,4,5-триметоксибензойной кислоты вместо остатка 3,4,5-триметоксикоричной кислоты.
Теоретически превращение резерпина в десерпидин могло бы осуществляться через деметоксилирование положения 11. Согласно известным способам органической химии наиболее простым путем было бы либо прямое деметоксилирование резерпина, либо превращение 11-метоксигруппы в гидроксильную группу с последующим восстановлением фенольного кольца до бензольного.
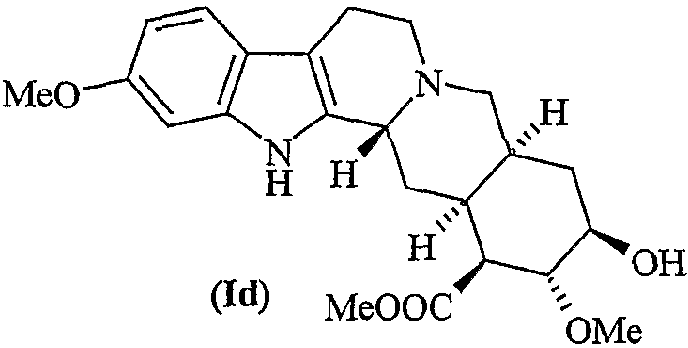
Квалифицированному специалисту в данной области техники известно, что полифункционализация резерпина, ресциннамина и метилрезерпата (Id)
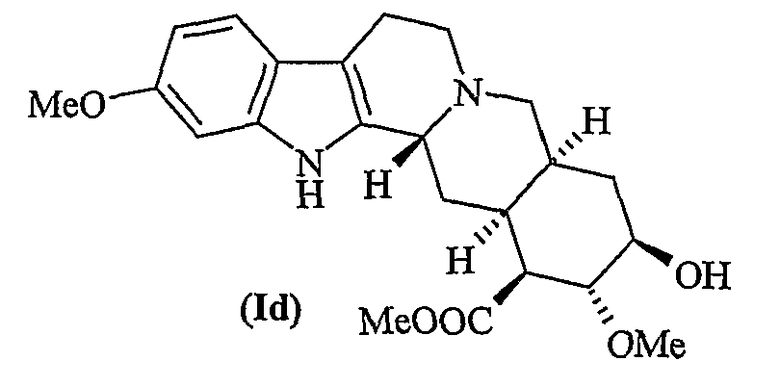
не позволяет селективно О-деметилировать гидроксильную группу в положении 11. Известные способы прямого 11-деметоксилирования или 11-О-деметилирования не обладают региоселективностью и/или химической селективностью.
В настоящее время установлено, что описанные выше проблемы могут быть решены при использовании лактона резерпиновой кислоты в качестве предшественника.
Подробное описание изобретения
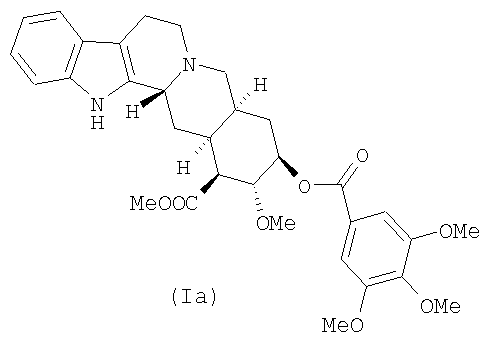
Настоящее изобретение относится к способу синтеза десерпидина, который включает деметилирование лактона резерпиновой кислоты, превращение фенольного кольца в бензольное кольцо и повторную этерификацию 18-гидроксильной группы.
Точнее способ включает следующие стадии:
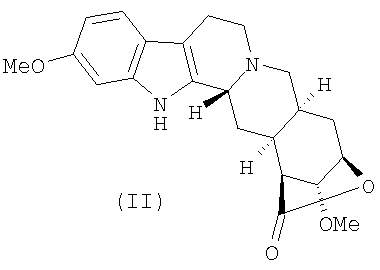
a) деметилирование лактона резерпиновой кислоты (II)
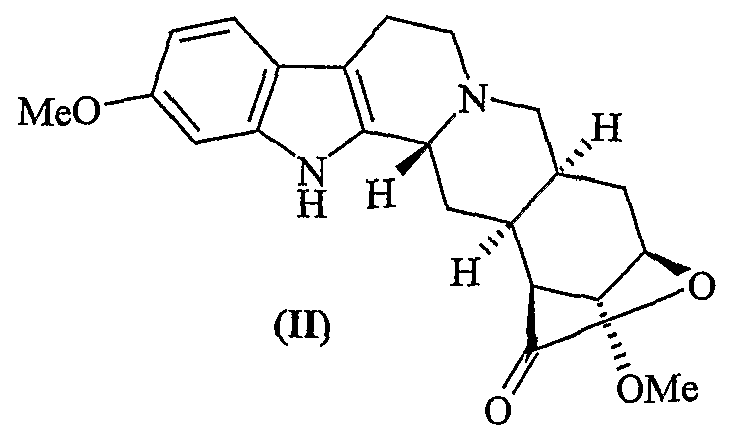
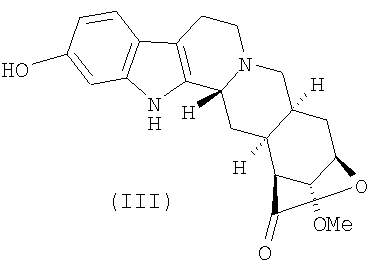
с получением 11-О-деметилированного лактона резерпиновой кислоты (III)
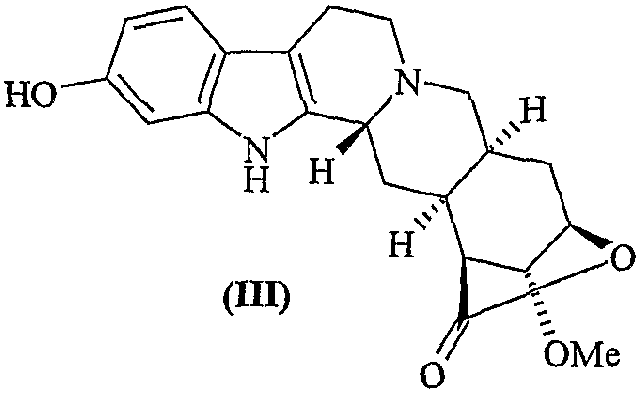
b) превращение соединения (III) в лактон десерпидиновой кислоты (V)
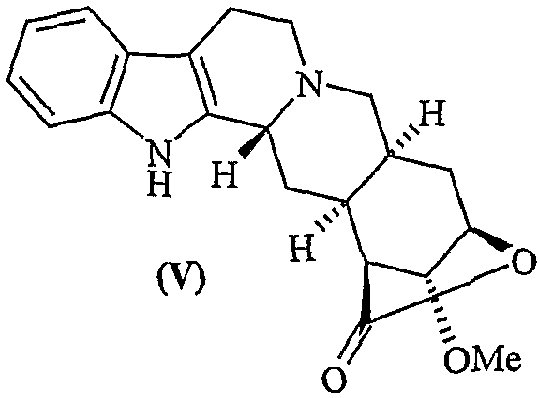
с) гидролиз лактона десерпидиновой кислоты (V) с получением метилдесерпидата (VI)
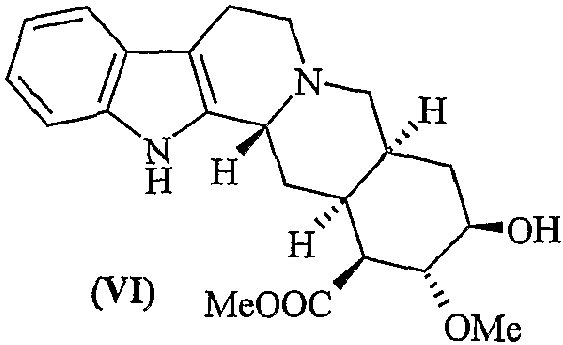
d) этерификацию метилдесерпидата (VI) 3,4,5-триметоксибензойной кислотой с получением десерпидина (Ia)
Лактон резерпиновой кислоты представляет собой известное соединение и обычно может быть получен гидролизом резерпина или ресциннамина, или их смеси метоксидом натрия с получением метилрезерпата (Id)
который далее подвергается циклизации в соответствующий лактон согласно методике, аналогичной описанной в публикации R.B. Woodward et al., Tetrahedron 1958, 2, 1-57. Альтернативно резерпин и ресциннамин могут быть непосредственно превращены в соответствующие лактоны по методикам, описанным в литературе (H.B. MacPhillamy et al., J. Am. Chem. Soc., 1955, 77, 4335-4343).
Селективное деметилирование лактона резерпиновой кислоты (стадия а) может быть проведено с использованием общепринятых агентов деметилирования, предпочтительно выбранных из трибромида бора, йодтриметилсилана и йодистоводородной кислоты, в условиях реакции, которые без труда могут быть оптимизированы квалифицированным химиком, при гарантированной стабильности лактона. Применение трибромида бора особенно предпочтительно, как описано в приведенном примере.
Стадия b) может быть осуществлена способами, подходящими для восстановления фенола в бензол. Предпочтительно данная стадия проводится превращением соединения (III) в соединение формулы (IV)

где R' представляет собой уходящую группу,
и восстановлением (IV).
Предпочтительными уходящими группами являются группы сложных эфиров сульфоновой кислоты, таких как тозилат или мезилат, изоуреидогруппы (в условиях, описанных в публикации E.Vowinkel et al., Chem. Ber. 1974, 107, 907-914), например, группы, полученные обработкой дициклогексилкарбодиимида или диизопропилкарбодиимида, или (5-фенилтетразолил)оксигруппа (полученная обработкой 1-хлор-5-фенилтетразола в условиях, описанных в публикации W.J. Musliner et al., J. Am. Chem. Soc. 1959, 81, 4271-4273). Особенно предпочтительной является группа тозилата, как описано в приведенном примере.
Восстановитель выбирают, например, из никеля Ренея, палладия на угле и платины. Никель Ренея должен применяться для восстановления сложных эфиров сульфоновой кислоты, в то время как палладий на угле предпочтителен для восстановления изомочевин, например, полученных с применением дициклогексилкарбодиимида или диизопропилкарбодиимида.
Гидролиз лактона десерпидиновой кислоты с получением метилдесерпидата (стадия с) может проводиться с использованием метоксида натрия в спиртах, и этерификация метилдесерпидата с получением десерпидина (стадия d) проводится в условиях, аналогичных описанным в литературе (H.B. MacPhillamy et al., J. Am. Chem. Soc., 1955, 77, 4335-4343; M. Lounasmaa et al., Heterocycles 1985, 23, 371-375; R.H. Levin et al., J. Org. Chem. 1973, 38, 1983-1986).
Таким образом, применение лактона резерпиновой кислоты в качестве предшественника позволяет решить проблему регио- и химической селективности известных способов. На практике в лактоне конформация 16-, 17- и 18-заместителей является таковой, что 17-метоксигруппа находится в аксиальном положении, в то время как в предшественниках - в экваториальном положении - аксиальная конформация защищает метоксигруппу от воздействия реагентов деметилирования и позволяет провести селективное деметилирование положения 11 относительно положения 17. Минимальный выход, который обеспечивает способ настоящего изобретения, составляет 40%.
Приведенные далее примеры иллюстрируют изобретение более подробно.
ПРИМЕРЫ
Пример 1. Синтез метилрезерпата
Суспензию резерпина (1 г, 0,16 ммоль) в растворе метоксида натрия (0,150 г, 4,8 ммоль) в метаноле (50 мл) кипятят с обратным холодильником до исчезновения исходного вещества (1 час), затем охлаждают и концентрируют в вакууме до одной трети исходного объема. Раствор разбавляют водой (60 мл) и значение рН доводят до 1 с помощью концентрированной соляной кислоты. Водный раствор затем повторно промывают этиловым эфиром. После этого водную фазу подщелачивают концентрированным раствором гидроксида аммония и экстрагируют метиленхлоридом (4×30 мл). Объединенные органические фазы сушат над сульфатом натрия и концентрируют в вакууме, получая аморфный остаток (0,66 г), который используют на следующей стадии без дополнительной очистки.
Эту же методику используют, исходя из эквивалентных количеств ресциннамина.
Пример 2. Синтез лактона резерпиновой кислоты
А) Из резерпина
Резерпин (4,1 г, 6,74 ммоль) при перемешивании добавляют к раствору изопропоксида алюминия (10,5 г, 51,4 ммоль) в ксилоле (175 мл) и полученную смесь кипятят с обратным холодильником при перемешивании в атмосфере азота в течение 6 часов. Осадок лактона резерпиновой кислоты отфильтровывают и промывают бензолом (3×40 мл), затем этиловым эфиром (4×40 мл). Остаток перекристаллизовывают из CHCl3, получая 2,07 г (5,45 ммоль, 81%) целевого продукта. Такую же методику используют для получения ресциннамина.
В) Из метилрезерпата
Изопероксид алюминия (0,747 г, 3,65 ммоль) растворяют в ксилоле (11,0 мл) в атмосфере азота. Добавляют метилрезерпат (0,200 г, 0,483 ммоль) и реакционную смесь кипятят с обратным холодильником при перемешивании. Сложный эфир быстро растворяется, и спустя 5 минут лактон начинает выпадать в осадок в виде твердого белого вещества. После кипячения с обратным холодильником в течение 2 часов продукт выделяют фильтрованием и промывают ксилолом (3×20 мл) и эфиром (3×20 мл). Остаток перекристаллизовывают из CHCl3, получая 0,168 г (0,440 ммоль, 91%) целевого продукта.
1Н ЯМР (ДМСО-d6, 400 МГц) δ 10,5 (уш.с, 1H, NH), 7,18 (д, 1H, J=8,4 Гц, H-9), 6,77 (д, 1H, J=2,3 Гц, H-12), 6,58 (дд, 1H, J1=8,4 Гц, J2=2,3 Гц, H-10), 4,75 (м, 1H, J=4,3 Гц), 4,10 (т, 1H, J=5 Гц, H-17), 3,73 (с, 3H, OMe), 3,45 (д, 1H, J=12,0 Гц, H-3), 3,35 (с, 3H, OMe), 2,90 (дд, 1H, J1=11,0 Гц, J2=5,2 Гц), 2,76-2,63 (м, 1H), 2,62-2,45 (м, 5H), 2,43-2,24 (м, 3H), 2,00 (м, 1H, J1=15,0 Гц, J2=8,6 Гц), 1,73 (м, 1H), 1,56 (м, 1H, J1=15,0 Гц, J2=4,1 Гц, H-19);
13С ЯМР (ДМСО-d6, 100 МГц) δ 178,3, 155,7, 137,5, 135,2, 121,9, 118,6, 108,5, 106,7, 95,5, 77,7, 77,1, 58,8, 57,1, 55,9, 54,9, 53,2, 45,5, 35,4, 31,3, 27,7, 26,4, 22,2.
Пример 3. Синтез лактона 11-О-деметилрезерпиновой кислоты
Лактон резерпиновой кислоты (0,210 г, 0,550 ммоль) суспендируют в безводном CH2Cl2 (8 мл) в атмосфере аргона и смесь охлаждают до 0°C. Спустя 15 минут добавляют трибромид бора (1,4 мл, 1,37 ммоль, 1,0М раствор в CH2Cl2), и раствор приобретает красно-коричневый цвет. По истечении 5 часов реакцию гасят насыщенным раствором NaHCO3 и экстрагируют CH2Cl2. Водные фазы собирают и снова экстрагируют AcOEt (3×15,0 мл). Органические фазы объединяют и сушат. Водную фазу фильтруют, осадок снова растворяют в смеси 1:1 ТГФ/МеОН и добавляют к полученному ранее органическому раствору. Полученный после фильтрования и концентрирования в вакууме твердый остаток хроматографируют (силикагель, CH2Cl2/МеОН = 15:1, затем 16:1) с получением целевого продукта (0,187 г, 0,51 ммоль, 92%).
1Н ЯМР (ТГФ-d8, 400 МГц) δ 9,38 (уш.с, 1H, NH), 7,54 (уш.с, 1H, OH), 7,07 (д, 1H, J=8,4 Гц, H-9), 6,59 (д, 1H, J=2,2 Гц, H-12), 6,44 (дд, 1H, J1=8,4 Гц, J2= 2,2 Гц, H-10), 4,63 (т, 1H, J=4,3 Гц, H-18), 4,03 (т, 1H, J=5,2 Гц, H-17), 3,61 (д, 1H, H-3), 3,40 (с, 3H, OMe), 2,90 (м, 1H), 2,86-2,46 (м, 7H), 2,38 (м, 1H), 2,22 (дд, 1H, J1=13,5 Гц, J2= 2,0 Гц), 2,11 (м, 1H, J1=14,9 Гц, J2=8,5 Гц), 1,86 (м, 1H), 1,61 (дд, 1H, J1=14,8 Гц, J2=3,9 Гц, H-19);
13С ЯМР (ТГФ-d8, 100 МГц) δ 176,6, 153,2, 138,0, 133,9, 121,3, 117,5, 108,4, 106,9, 96,6, 78,2, 76,7, 58,9, 56,2, 54,6, 53,1, 45,7, 35,8, 31,8, 27,9, 26,2, 22,1.
Пример 4. Синтез лактона 11-О-п-толуолсульфонил-11-О-деметилрезерпиновой кислоты
Лактон 11-О-деметилрезерпиновой кислоты (0,700 г, 1,90 ммоль) в атмосфере азота растворяют в 65 мл безводного ТГФ, затем добавляют триэтиламин (1,86 мл, 13,32 ммоль). Реакционную смесь подвергают взаимодействию в течение 10 минут, затем добавляют п-толуолсульфонилхлорид (1,09 г, 5,71 ммоль) и смесь кипятят с обратным холодильником в течение 42 часов. Реакционную смесь упаривают в вакууме и остаток хроматографируют (силикагель, CH2Cl2/МеОН = 17:1), получая 0,75 г целевого соединения (0,75 г, 1,44 моль, 76%).
1Н ЯМР (ТГФ-d8, 400 МГц) δ 9,99 (уш.с, 1H, NH), 7,63 (2H, Ar), 7,31 (2H, Ar), 7,15 (д, 1H, J=8,4 Гц, H-9), 6,90 (д, 1H, J=2,2 Гц, H-12), 6,47 (дд, 1H, J1=8,4 Гц, J2= 2,2 Гц, H-10), 4,64 (т, 1H, J=4,1 Гц, H-18), 4,03 (т, 1H, J=5,2 Гц, H-17), 3,62 (д, 1H, J=11,8 Гц, H-3), 3,39 (с, 3H, OMe), 2,91 (м, 1H), 2,84-2,43 (м, 7H), 2,38 (м, 4H, 1Me и 1H), 2,22 (дд, 1H, J1=13,5 Гц, J2=2,0 Гц), 2,10 (м, 1H, J1=15,0 Гц, J2= 8,5 Гц), 1,86 (м, 1H), 1,60 (дд, 1H, J1=15,0 Гц, J2=4,0 Гц, H-19);
13С-ЯМР (ТГФ-d8, 100 МГц) δ 176,7, 144,9, 144,8, 136,2, 133,5, 129,6, 128,7, 126,3, 117,4, 113,2, 107,5, 105,0, 78,2, 76,7, 58,8, 56,3, 54,5, 52,9, 45,7, 35,7, 31,7, 29,9, 27,9, 26,2, 21,9, 20,8.
Пример 5. Синтез лактона десерпидиновой кислоты
Ni Ренея, предварительно промытый Н2О (дважды), МеОН (дважды) и EtOH (один раз) вводят (4,86 г, влажный) в реактор гидрирования в атмосфере аргона, затем загружают лактон 11-О-п-толуолсульфонил-11-О-деметилрезерпиновой кислоты (0,300 г, 0,58 ммоль), растворенный в 14 мл безводного ТГФ и 16,0 мл EtOH. Гидрирование проводят при давлении 50 фунтов на кв. дюйм. Спустя 8 часов раствор фильтруют через целит, промывают CHCl3 (6×40 мл) и 100 мл МеОН. Растворитель удаляют в вакууме и остаток очищают хроматографией (силикагель, CH2Cl2/МеОН = 20:1), получая 0,17 г целевого соединения (0,170 г, 0,49 ммоль, 85%).
1Н ЯМР (ТГФ-d8, 400 МГц) δ 9,80 (уш.с, 1H, NH), 7,32 (д, 1H, J=7,8 Гц, H-9), 7,20 (д, 1H, J=7,6 Гц, H-12), 6,98-6,87 (м, 2H, Ar, H-10, H-11), 4,04 (т, 1H, J=5,2 Гц, H-17), 3,66 (д, 1H, J=11,8 Гц, H-3), 3,40 (с, 3H, OMe), 2,94 (м, 1H), 2,84 (м, 1H), 2,78-2,33 (м, 7H), 2,28 (дд, 1H, J1=13,7 Гц, J2=2,0 Гц), 2,12 (м, 1H, J1=15,0 Гц, J2=8,5 Гц), 1,89 (м, 1H), 1,62 (дд, 1H, J1=14,8 Гц, J2=4,0 Гц, H-19);
13С-ЯМР (ТГФ-d8, 100 МГц) δ 176,7, 136,9, 136,1, 127,6, 120,2, 118,3, 117,4, 110,5, 107,2, 78,2, 76,7, 58,9, 56,2, 54,6, 53,1, 45,7, 35,8, 31,7, 27,9, 26,2, 22,1.
Пример 6. Синтез метилдесерпидата
Лактон десерпидиновой кислоты (0,140 г, 0,398 ммоль) растворяют в 27,0 мл безводного МеОН в атмосфере азота. В полученную суспензию добавляют MeONa (0,032 г, 0,597 ммоль), затем реакционную смесь кипятят с обратным холодильником в течение 90 минут. Реакцию гасят добавлением 0,2 мл ледяной уксусной кислоты и растворитель выпаривают в вакууме. Полученный продукт снова растворяют в 0,2М растворе NaOH и экстрагируют CHCl3 (4×25 мл); органическую фазу сушат и фильтруют. Растворитель выпаривают в вакууме и остаток очищают хроматографией (силикагель, CH2Cl2/МеОН = 10:1), получая 0,17 г целевого продукта (0,170 г, 0,38 ммоль, 95%).
1Н ЯМР (CDCl3, 400 МГц) δ 7,80 (уш.с, 1H, NH), 7,46 (д, 1H, J=6,8 Гц), 7,31 (д, 1H, J=8,0 Гц), 7,14 (дт, 1H, J1=6,8 Гц, J2=1,2 Гц), 7,09 (дт, 1H, J1=7,6 Гц, J2=1,2 Гц) 4,46 (уш.с, 1H, H-3), 3,79 (с, 3H, OMe), 3,62-3,48 (м, 5H, 1OMe и 2H), 3,26-3,15 (м, 2H), 3,06-2,91 (м, 2H), 2,59-2,45 (м, 3H), 2,32-2,17 (м, 2H), 2,03-1,91 (м, 1H), 1,89-1,71 (м, 3H);
13С-ЯМР (CDCl3, 100 МГц) δ 173,7, 135,7, 132,1, 127,8, 121,7, 119,7, 118,3, 111,1, 108,3, 81,6, 75,3, 61,2, 53,9, 52,1, 51,5, 51,3, 49,5, 34,7, 33,0, 32,4, 24,5, 16,9.
Пример 7. Синтез десерпидина
Метилдесерпидат (0,5 г, 1,30 ммоль) растворяют в сухом пиридине (4,0 мл) в атмосфере азота. 3,4,5-Триметоксибензилхлорид (0,5 г, 2,17 ммоль) растворяют в бензоле (2 мл), затем медленно по каплям добавляют к реакционной смеси. Реакционную смесь перемешивают в течение 5 дней при 5°C и затем гасят 50 мл воды. В полученный раствор добавляют смесь концентрированного NH3 (2 мл) и 10 мл Н2О. Затем раствор экстрагируют CH2Cl2 (3×25 мл) и органическую фазу сушат и фильтруют. Раствор выпаривают в вакууме и полученный остаток перекристаллизовывают из ацетона, получая 0,168 г (0,440 ммоль, 91%) целевого продукта.
1Н ЯМР (CDCl3, 400 МГц) δ 7,83 (уш.с, 1H, NH), 7,48 (1H), 7,33 (с, 1H), 7,33 (с, 1H), 7,32 (1H), 7,17 (1H), 7,12 (1H), 5,07 (1H), 4,52 (уш.с, 1H, H-3), 3,90 (дд, 1H, J1=12 Гц, J2=9 Гц), 3,89 (с, 3H, OMe), 3,89 (с, 3H, OMe), 3,89 (с, 3H, OMe), 3,80 (с, 3H, OMe), 3,80 (с, 3H, OMe), 3,20 (м, 1H), 3,20 (м, 1H), 3,04 (дд, 1 H, J1=12 Гц, J2=4 Гц), 2,98 (1H), 2,70 (дд, 1H, J1=12 Гц, J2= 5 Гц), 2,54 (1H), 2,47 (дд, 1H, J1=12 Гц, J2=2 Гц), 2,34 (м, 1H), 2,33 (1H), 2,04 (1H), 1,98 (1H), 1,90 (1H), 1,86 (1H);
13С-ЯМР (CDCl3, 100 МГц) δ 176,8, 169,4, 163,8, 161,0, 159,4, 144,0, 141,6, 141,6, 141,4, 140,1, 137,4, 133,1, 133,0, 115,5, 115,5, 86,2, 85,5, 82,3, 75,4, 74,0, 72,1, 67,8, 65,3, 61,8, 57,2, 50,7, 39,8, 27,3, 20,3.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ТАКСАНА, ФУНКЦИОНАЛИЗИРОВАННЫЕ ПО 14-ПОЛОЖЕНИЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2320652C2 |
| СПОСОБЫ СИНТЕЗА АПЛИДИНА И НОВЫХ ПРОТИВООПУХОЛЕВЫХ ПРОИЗВОДНЫХ, СПОСОБЫ ИХ ПРОМЫШЛЕННОГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2001 |
|
RU2299887C2 |
| ИМИДЫ КАК ИНГИБИТОРЫ TNF-АЛЬФА | 1994 |
|
RU2174516C2 |
| 3-ГАЛОГЕН-6-(АРИЛ)-ИМИНОТЕТРАГИДРОПИКОЛИНАТЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ГЕРБИЦИДОВ | 2010 |
|
RU2527954C2 |
| НОВЫЕ ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2006 |
|
RU2434006C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИСПИРОИНДОЛИНОНОВ НА ОСНОВЕ 5-ИНДОЛИДЕН-2-ТИОГИДАНТОИНОВ | 2020 |
|
RU2756463C1 |
| ПОЛУСИНТЕТИЧЕСКИЙ СПОСОБ И НОВЫЕ СОЕДИНЕНИЯ | 2000 |
|
RU2237063C9 |
| НОВЫЕ ПРОИЗВОДНЫЕ ОКСАЗОЛИДИНОНА С ЦИКЛИЧЕСКИМ АМИДОКСИМОМ ИЛИ ЦИКЛИЧЕСКИМ АМИДРАЗОНОМ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2009 |
|
RU2468023C1 |
| Производные аденозина - ингибиторы репродукции вирусов, относящихся к роду Flavivirus | 2023 |
|
RU2828777C1 |
Изобретение относится к улучшенному высокоселективному способу получения десерпидина (Ia) деметилированием 11-метоксигруппы лактона 11-О-метилрезерпиновой кислоты с последующими стадиями восстановления, гидролиза и этерификации полученного метилдесерпидата 3,4,5-триметоксибензойной кислотой. Выход составляет 40%. 3 н. и 3 з.п. ф-лы.
1. Способ получения десерпидина (Ia)
включающий следующие стадии:
а) деметилирование лактона резерпиновой кислоты (II)
с получением лактона 11-O-деметилрезерпиновой кислоты (III)
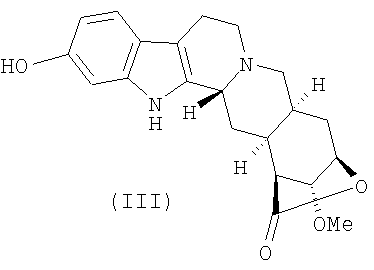 ;
;
b) превращение соединения (III) в соединение формулы (IV)
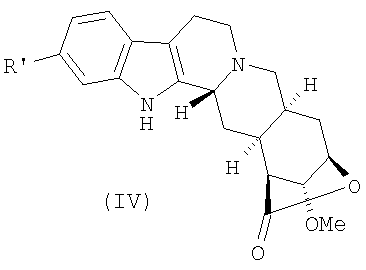
где R' представляет собой уходящую группу, выбранную из сульфоната, и восстановление соединения (IV) с получением лактона десерпидиновой кислоты (V)
 ;
;
с) гидролиз лактона десерпидиновой кислоты (V) до метилдесерпидата (VI)
 ;
;
d) этерификация метилдесерпидата (VI) 3,4,5-триметоксибензойной кислотой с получением десерпидина (Iа).
2. Способ п.1, где в соединении (IV) группа R' представляет собой п-толуолсульфонат или метансульфонат.
3. Способ по п.2, где R' группа представляет собой п-толуолсульфонат.
4. Способ по п.3, где восстановление соединения (IV) проводится с применением никеля Ренея.
5. Соединение формулы (III)
6. Соединение формулы (IV)
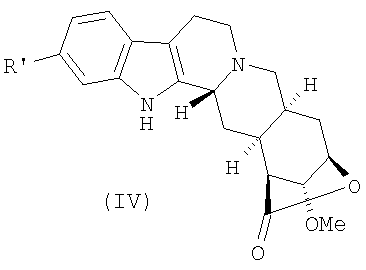
где R' представляет собой п-толуолсульфонат.
| GB 809913 А, 04.03.1959 | |||
| Электроиндукционный пылемер | 1980 |
|
SU868478A1 |
| US 3303195 А, 07.02.1967 | |||
| GB 915449 А, 09.01.1963. | |||

