Описание
Настоящее изобретение относится к способам синтеза и, в частности, к полусинтетическим способам.
Уровень техники
Европейский патент 309477 имеет отношение к эктеинасцидинам 729, 743, 745, 759А, 759В и 770. Соединения ряда эктеинасцидина, как сообщается, обладают антибактериальными и другими полезными свойствами. Эктеинасцидин 743 в настоящее время проходит клинические испытания в качестве противоопухолевого средства.
Эктеинасцидин 743 имеет сложную трис(тетрагидроизохинолинфенольную) структуру, соответствующую следующей формуле (I):
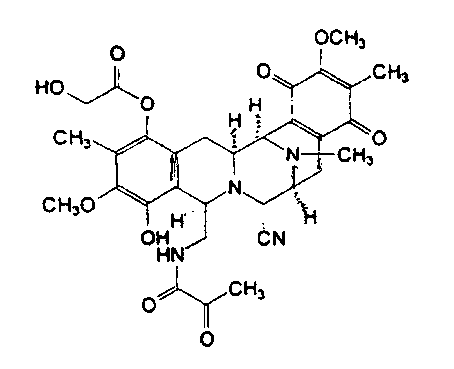
Это соединение в настоящее время получают выделением из экстракта оболочек морских организмов Ecteinascidin turbinata. Выход низкий, в связи с чем пытаются найти альтернативные способы получения.
Синтетический способ получения соединений ряда эктеинасцидина представлен в патенте США 5721362. Описанный способ является длительным и сложным, причем приведено 38 примеров, каждый из которых описывает одну из последовательных стадий синтеза, необходимых для получения эктеинасцидина 743.
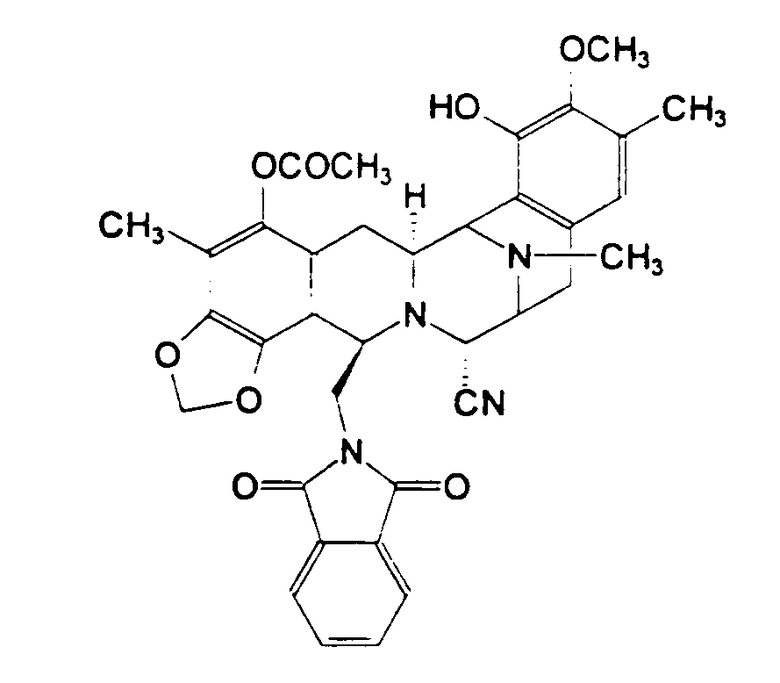
Пункт 25 формулы изобретения, приведенной в патенте США 5721362, относится к промежуточному фенольному соединению, соответствующему формуле (11), которое авторы настоящего изобретения также обозначают как промежуточное соединение 11 или Int-11. Это соединение имеет следующую бис(тетрагидроизохинолинфенольную) структуру (II):

где МОМ представляет метоксиметильный заместитель, а TBDPS представляет 3,5-трет-бутилдифенилсилильный заместитель.
Исходя из промежуточного соединения 11, возможно синтезировать другое, представляющее интерес противоопухолевое средство, фталасцидин, см. Proc. Natl. Acad. Sci. USA, 96, 3496-3501, 1999. Фталасцидин представляет бис(тетрагидроизохинолинфенольное) производное формулы (III):
В эктеинасцидине 743 1,4-мостик имеет структуру, представленную формулой (IV):

Другие известные эктеинасцидины включают соединения с различными мостиковыми циклическими кольцевыми системами, например, с такой, которая присутствует в эктеинасцидинах 722 и 736, где мостик имеет структуру, представленную формулой (V):
а также эктеинасцидины 583 и 597, в которых мостик имеет структуру, представленную формулой (VI):
и эктеинасцидины 594 и 596, в которых мостик имеет структуру, представленную формулой (VII):

Полностью структура этих и родственных им соединений приведена в J. Am. Chem. Soc. (1996) 118, 9017-9023. Эта статья включена в настоящее описание посредством ссылки.
Известны и другие соединения, в которых отсутствует мостиковая циклическая кольцевая система. Такие соединения включают бис(тетрагидроизохинолинхиноновые) противоопухолевые-противомикробные антибиотики - сафрацины и сафрамицины, и вещества из природных морепродуктов - рениэрамицины и ксестомицин, выделеные из культивируемых микроорганизмов или губок. Все они обладают общим димерным тетрагидроизохинолиновым углеродным каркасом. Данные соединения могут быть систематизированы с выделением четырех типов, типы от I
до IV, в зависимости от вида окисления ароматических колец.
Тип I, димерные изохинолинхиноны, представляет систему формулы (VIII), наиболее часто встречающуюся у соединений этого класса, см. приведенную ниже таблицу I.
Таблица I
Структура сафрамициновых антибиотиков типа I
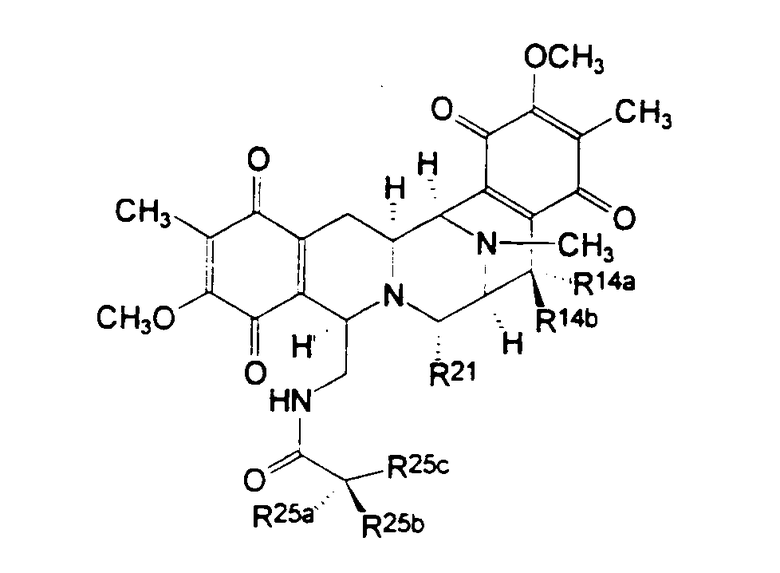
b где группа Q представлена формулой (IX):
Ароматические системы типа I наблюдаются у сафрамицинов типа А, В и С; G и H; и S, выделенных из Streptomyces lavendulae в виде неосновных компонентов. Цианопроизводное сафрамицина А, называемое цианохинонамином, известно из патентов Японии Japanese Kokai JP-A2 59/225189 и 60/084288. Сафрамицины Y3, Yd1, Ad1 и Yd2 продуцирует S. lavendulae путем прямого биосинтеза, при обеспечении подходящей культуральной среды. Сафрамицины Y2b и димеры Y2b-d, образующиеся при связывании азота у С-25 одного из фрагментов с С-14 другого фрагмента, также продуцирует S. lavendulae на подходящей культуральной среде. Сафрамицины AR1 (=АН2), продукт восстановления сафрамицина А в положении С-25 под действием микроорганизмов, продуцируемый Rhodococcus amidophilus, также получают путем нестереоселективного химического восстановления сафрамицина А под действием боргидрида натрия в виде смеси эпимеров 1:1, с последующим хроматографическим разделением [другой изомер АН1 является менее полярным]. Дополнительный продукт восстановления сафрамицин AR3, 21-дециано-25-дигидро-сафрамицин А, (= 25-дигидросафрамицин В) получают тем же самым микробном превращением. Другой тип микробного превращения сафрамицина А, с использованием вида Nocardia, приводит к продуцированию сафрамицина В, а дальнейшее восстановление с использованием вида Mucobacterium приводит к продуцированию сафрамицина АН1Ас. Для биологических исследований 25-О-ацетаты сафрамицина АН2 и АН1 также получают химическим путем.
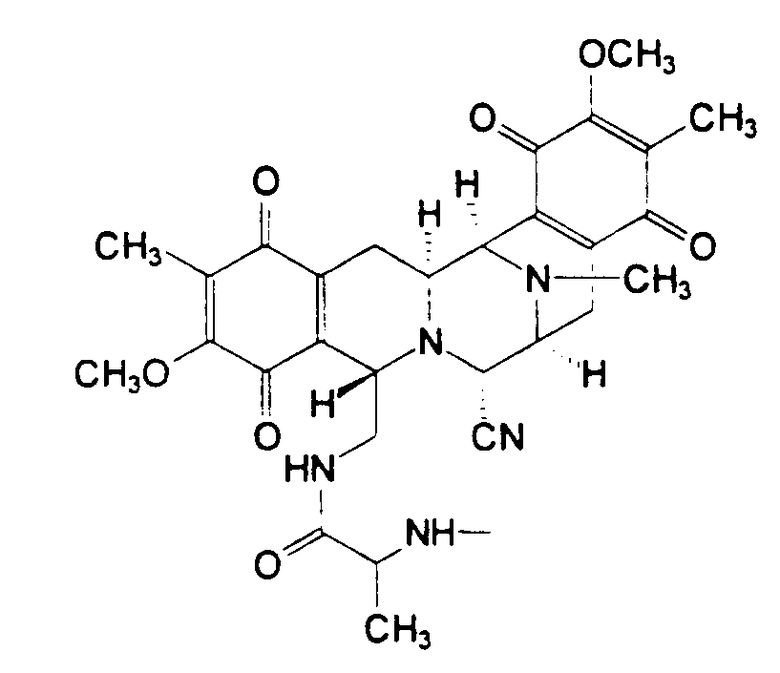
Соединения типа I формулы (X) также были выделены из морских губок, см. таблицу II
Таблица II
Структура соединений типа II из морских губок
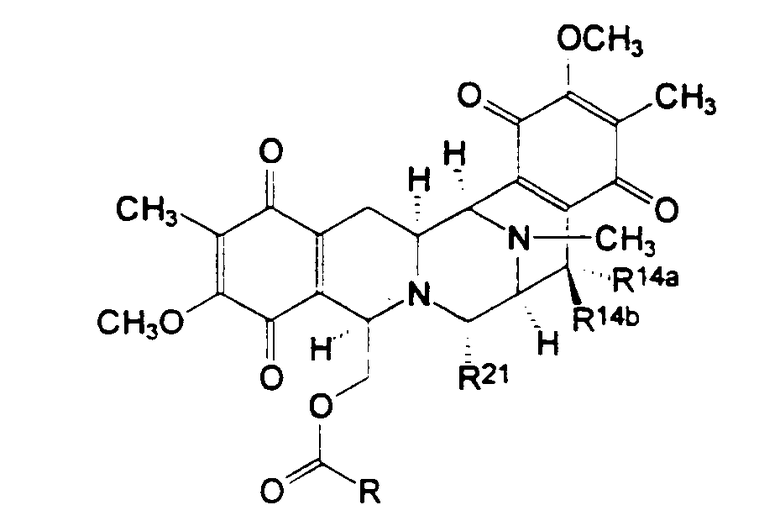
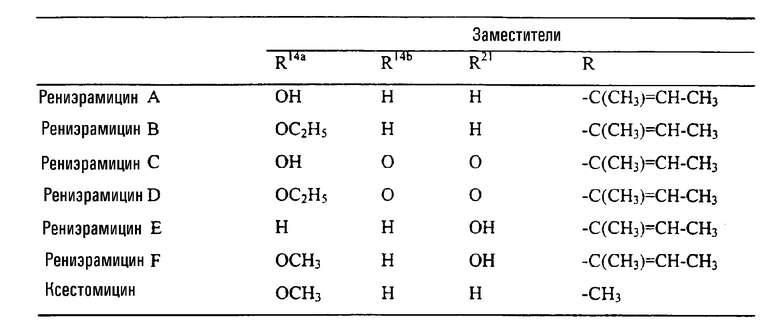
Рениэрамицины А - D выделяли из противомикробного экстракта губок вида Reniera, собранных в Мехико, наряду с родственным по биологическому происхождению рениэроном мономерных изохинолинов и родственными ему соединениями. Структуру рениэрамицина А первоначально связывали с инверсной стереохимией у атомов С-3, С-11 и С-13. Однако тщательное изучение данных 1Н ЯМР, полученных для новых, родственных соединений - рениэрамицинов Е и F, выделенных из тех же самых губок, собранных на островах Палау (Тихий океан), показало, что кольцевое соединение рениэрамицинов идентично тому, которое присуще сафрамицинам. Эти результаты позволили сделать вывод о том, что стереохимия, которую ранее относили к стереохимии рениэрамицинов, от А до D, должна быть той же самой, что и стереохимия сафрамицинов.
Ксестомицин был обнаружен в губках вида Xestospongia, собранных в акватории Шри-Ланка.
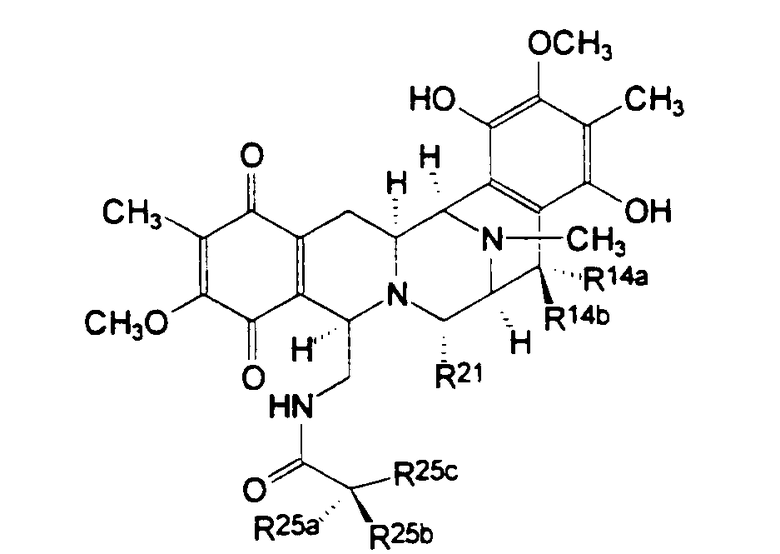
Соединения типа II формулы (XI) с восстановленным гидрохиноновым кольцом включают сафрамицины D и F, выделенные из S. lavendulae, и сафрамицины Мх-1 и Мх-2, выделенные из
Myxococcus xanthus. См. таблицу III.
Таблица III
Соединения типа II
Структура скелета типа III обнаружена у антибиотиков сафрацинов А и В, выделенных из культуры Pseudomonas fluorescens. Эти антибиотики формулы (XII) состоят из тетрагидроизохинолинхиноновой субъединицы и тетрагидроизохинолинфенольной субъединицы:
где R21 представляет Н в случае сафрацина А и ОН в случае сафрацина В.
Сафрамицин R, единственное соединение, классифицированное как соединение, обладающее скелетом типа IV, также было выделено из S. lavendulae. Это соединение формулы (XIII), состоящее из гидрохинонового кольца со сложным гликолевым эфиром в боковой цепи у одного из фенольных кислородов, представляет, вероятно, пролекарство сафрамицина А вследствие его умеренной токсичности.

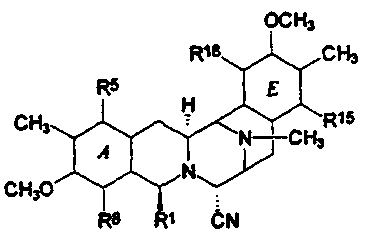
Все эти соединения имеют конденсированную систему из пяти колец от (А) до (Е), как показано в следующей структурной формуле (XIV):

Данные кольца от (А) до (Е) имеют фенольную природу в случае эктеинасцидинов и некоторых других соединений, в то время как в других соединениях, в особенности, в сафрамицинах, кольца А и Е являются хиноновыми. В известных соединениях кольца В и D имеют структуру тетрагидро, в то время как кольцо С представляет структуру пергидро.
Цель изобретения
Имеется необходимость в новых активных соединениях, содержащих конденсированную систему из пяти колец, присущую известным соединениям, и в альтернативных способах синтеза соединений ряда эктеинасцидина и родственных им соединений. Такие синтетические подходы могли бы обеспечить более экономные пути получения известных противоопухолевых средств, а также сделать возможным получение новых активных соединений.
Сущность изобретения
Один из аспектов настоящего изобретения относится к применению известного соединения, сафрацина А, также называемого хинонамином, в полусинтетическом способе синтеза.
В более широком смысле, настоящее изобретение относится к полусинтетическому способу получения промежуточных соединений, производных и структур, родственных эктеинасцидину или другим тетрагидроизохинолинфенольным соединениям, исходя из природных бис(тетрагидроизохинолиновых) алкалоидов. Подходящие исходные вещества для использования в полусинтетическом способе включают соединения класса сафрамициновых и сафрациновых антибиотиков, которые доступны из различных культур, выращиваемых на различных жидких средах, а также соединения класса реинэрамимицина и класса ксестомицина, которые могут быть выделены из морских губок.
Общая структурная формула (XV) исходных соединений является следующей:

где:
R1 представляет амидометиленовую группу, такую, как -CH2-NH-CO-CR25aR25bR25c, где R25a и R25b образуют кетогруппу или один представляет -OH, -NH2 или -OCOCH3, а другой представляет -CH2COCH3, -H, -OH или -OCOCH3, при условии, что в том случае, когда R25a представляет -OH или -NH2, тогда R25b не является -OH и R25c представляет -H, -CH3 или -CH2CH3, или R1 представляет ацилоксиметиленовую группу, такую как -CH2-O-CO-R, где R представляет -C(CH3)=CH-CH3 или -CH3;
R5 и R8 независимо выбирают из -H, -OH или -OCOCH2OH, или R5 и R8 оба являются кето, и кольцо А является п-бензохиноновым кольцом;
R14a и R14b оба представляют -H или один представляет -H, а другой представляет -OH, -OCH3 или -OCH2CH3, или R14a и R14b вместе образуют кетогруппу;
R15 и R18 независимо выбирают из -H и -OH, или R5 и R8 оба являются кето и кольцо А является п-бензохиноновым кольцом; и
R21 представляет -OH или -CN.
Более общая формула для соединений этого класса приведена ниже:

где группы-заместители, определяемые как R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, каждую, независимо, выбирают из ряда, состоящего из H, OH, OCH3, CN, =О, CH3;
где X представляет различные амидные или сложноэфирные функциональные группы, содержащиеся в указанных выше продуктах природного происхождения;
где каждая из пунктирных кольцевых линий представляет одну, две или три необязательно присутствующие двойные связи.
Таким образом, согласно настоящему изобретению, заявитель предлагаются полусинтетические способы получения промежуточных соединений, включая промежуточное соединение 11, и соответственно, способы получения соединений ряда эктеинасцидина, а также фталасцидина и дополнительных соединений. Каждый из полусинтетических способов, являющихся предметом настоящего изобретения, включает ряд стадий превращений, нужных для того, чтобы получить необходимый продукт. Каждая из стадий сама по себе представляет способ согласно настоящему изобретению. Объем изобретения не ограничивается способами, которые проиллюстрированы примерами, и могут быть осуществлены альтернативные варианты, например, при изменении подходящим образом порядка стадий превращений.
В частности, настоящее изобретение включает получение исходного 21-цианосоединения общей формулы (XVI):
в которой R1, R5, R8, R15 и R18 являются такими, как определено выше.
Другие соединения формулы (XVI) с различными заместителями в 21-положении могут также представлять возможные исходные вещества. В общем, любое производное, которое может быть получено при нуклеофильном замещении 21-гидроксигруппы соединения формулы (XV), в котором R21 представляет гидроксигруппу, является потенциальным вариантом. Примеры подходящих 21-заместителей включают, но не ограничиваются, такие группы как:
меркаптогруппа;
алкилтиогруппа (алкильная группа, содержащая от 1 до 6 атомов углерода);
арилтиогруппа (арильная группа, содержащая от 6 до 10 атомов углерода, которая является незамещенной или замещенной
от 1 до 5 заместителями, выбранными, например, из алкильной группы, содержащей от 1 до 6 атомов углерода, алкоксигруппы, содержащей от 1 до 6 атомов углерода, атомов галогена, меркаптогруппы или нитрогруппы);
аминогруппа;
моно- или диалкиламиногруппа (одна или каждая алкильная группа содержит от 1 до 6 атомов углерода);
моно- или диариламиногруппа (одна или каждая арильная группа является такой, как определено выше для арилтиогрупп);
α-карбонилалкильная группа формулы -С(Ra)(Rb)-С(=O)Rc, где Ra и Rb выбирают из атомов водорода, алкильных групп, содержащих от 1 до 20 атомов углерода, арильных групп (таких, как определено выше для арилтиогрупп) и аралкильных групп (в которых алкильная группа, содержащая от 1 до 4 атомов углерода, является замещенной арильной группой, такой как определено выше для арилтиогрупп), при условии, что один из радикалов Ra и Rb представляет атом водорода;
Rc выбирают из атома водорода, алкильной группы, содержащей от 1 до 20 атомов углерода, арильных групп (таких, как определено выше для арилтиогрупп), аралкильной группы (в которой алкильная группа, содержащая от 1 до 4 атомов углерода, является замещенной арильной группой, такой как определено выше для арилтиогрупп), алкоксигруппы, содержащей от 1 до 6 атомов углерода, аминогруппы или моно- или диалкиламиногруппы, как определено выше.
Таким образом, в своем более общем аспекте, настоящее изобретение относится к способам, в которых первая стадия представляет получение 21-производного с использованием нуклеофильного реагента. Такие соединения настоящего изобретения обозначены как 21-Nuc соединения.
Наличие 21-цианогрупы требуется для некоторых конечных продуктов, в особенности, эктеинасцидина 770 и фталасцидина, в то время как в случае других конечных продуктов данная группа служит в качестве защитной группы, которая легко может быть превращена в другие заместители, например, такие как 21-гидроксигруппа эктеинасцидина 743 или 21-гидрокси-фталасцидина. Выбор 21-цианосоединения в качестве исходного вещества приводит к эффективной стабилизации молекулы при осуществлении последующих стадий синтеза, до тех пор, пока данная группа, необязательно, не удалена. Другие 21-Nuc соединения могут обладать этим и другими преимуществами.
В одном из своих важных аспектов настоящее изобретение заключается в применении 21-цианосоединений общей формулы (XVI) для получения бис- и трис(тетрагидроизохинолинфенольных) соединений. Продукты, которые могут быть получены, включают промежуточные соединения, например, такие как промежуточное соединение 11, эктеинасцидины и фталасцидин, а также другие новые и известные соединения родственной структуры.
Предпочтительные исходные вещества включают такие соединения формулы (XV), в которых оба R14a и R14b представляют водород. Предпочтительные исходные вещества также включают соединения формулы (XV) или (XVI), в которых R15 представляет водород. Кроме того, предпочтительные исходные вещества включают такие соединения формулы (XV) или (XVI), в которых кольцо Е представляет фенольное кольцо. Предпочтительные исходные вещества, кроме того, включают такие соединения формулы (XV) или (XVI), в которых, по меньшей мере, один, предпочтительно, по меньшей мере, два или три из R5, R8 R15 и R18 не являются водородом.
Примеры подходящих исходных веществ для использования согласно настоящему изобретению включают сафрамицин А, сафрамицин В, сафрамицин С, сафрамицин G, сафрамицин Н, сафрамицин S, сафрамицин Y3, сафрамицин Yd1, сафрамицин Ad1, сафрамицин Yd2, сафрамицин AH2, сафрамицин AH2Ac, сафрамицин AH1, сафрамицин AH1Ac, сафрамицин AR3, рениэрамицин А, рениэрамицин В, рениэрамицин С, рениэрамицин D, рениэрамицин E, рениэрамицин F, ксестомицин, сафрамицин D, сафрамицин F, сафрамицин Mx-1, сафрамицин Mx-2, сафрацин А, сафрацин В и сафрамицин R. Предпочтительное исходное вещество содержит цианогруппу в 21-положении, в качестве группы R21.
В своем наиболее предпочтительном аспекте настоящее изобретение включает полусинтетический способ, согласно которому стадии превращения применяются к сафрацину В:
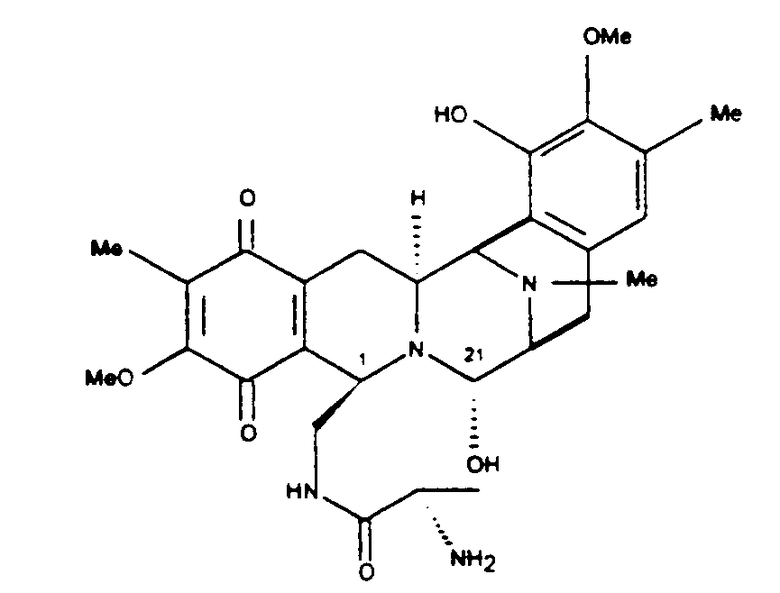
САФРАЦИН В
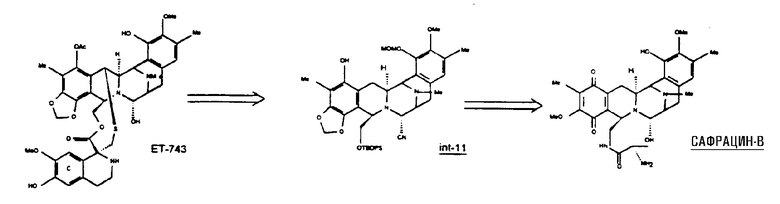
Сафрацин В представляет кольцевую систему, близкую структуре эктеинасцидинов. Данное соединение имеет ту же самую структуру из пяти циклов и тот же характер замещения в правом ароматическом кольце, кольце Е. Кроме того, сафрацин В по своей структуре очень близок одному из синтетических промежуточных соединений, образующихся в полном синтезе ЕТ-743, в частности, к промежуточному соединению 11. Такое промежуточное соединение может быть трансформировано в Et-743 с использованием хорошо отработанных методик. Таким образом, синтетическое превращение сафрацина B в промежуточное соединение 11 предоставит возможность осуществить полусинтетический способ, для того чтобы получить ЕТ-743.
Таким образом, настоящее изобретение предоставляет промежуточное соединение 11, полученное из данного соединения сафрацина В, и соединения, являющиеся производными промежуточного соединения 11, в особенности, производные эктеинасцидина. Дополнительно настоящее изобретение предлагает фталасцидин, полученный из сафрацина В. Изобретение также относится к применению сафрацина В для получения промежуточного соединения 11, фталасцидина, производных эктеинасцидина и других промежуточных соединений настоящего изобретения. Изобретение также относится к описываемым соединениям, являющимся производными других предполагаемых исходных веществ, и применению указанных соединений для получения таких соединений.
Наиболее предпочтительное исходное вещество настоящего изобретения содержит 21-цианогруппу. В настоящее время наиболее предпочтительное соединение, являющееся предметом настоящего изобретения, представляет соединение формулы 2. Данное соединение получают непосредственно из сафрацина В и его рассматривают как ключевое промежуточное соединение при осуществлении полусинтетического способа.
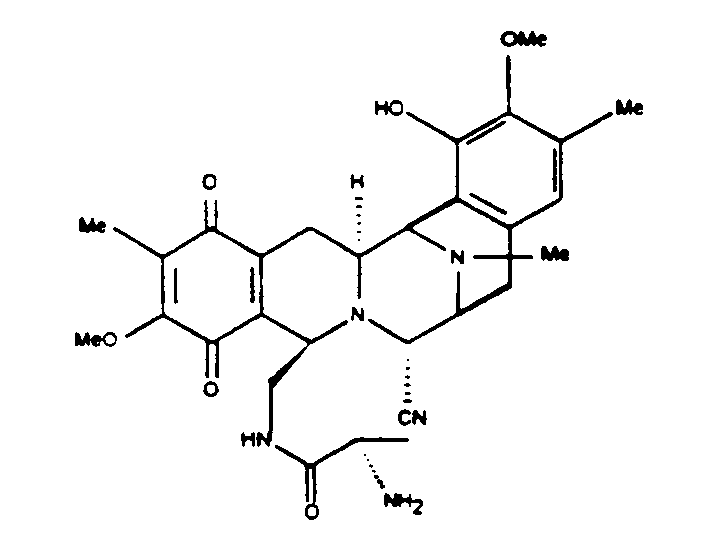
соединение 2
В отношении аспекта настоящего изобретения цианосафрацин В получали ферментацией продуцирующего сафрацин В штамма Pseudomonas fluorescens и обработкой культуральных жидких сред с использованием цианид-иона. Предпочтительный штамм Pseudomonas fluorescens представляет штамм А2-2, FERM BP-14, который используется в соответствии с методикой, описанной в ЕР 055299. Подходящий источник цианид-иона представляет цианид калия. При типичной обработке питательную среду отфильтровывают и добавляют избыток цианид-иона. После соответствующего интервала перемешивания, например, такого как 1 час, значение рН доводят до щелочного, например, рН 9,5, и экстракция органическим веществом дает неочищенный экстракт, который далее может быть очищен для получения цианосафрацина В.
Для устранения сомнений стереохимия, представленная в данном описании, основывается на понимании соответствующей стереохимии продуктов природного происхождения. В той степени, в которой ошибка обнаруживается в указанной стереохимии, эта ошибка нуждается в соответствующей корректировке в формуле, приведенной в настоящем описании. Кроме того, в той степени, в которой синтез может быть подвергнут модификации, данное изобретение распространяется на стереоизомеры.
Продукты, являющиеся предметом настоящего изобретения, обычно представлены формулой (XVIIa):
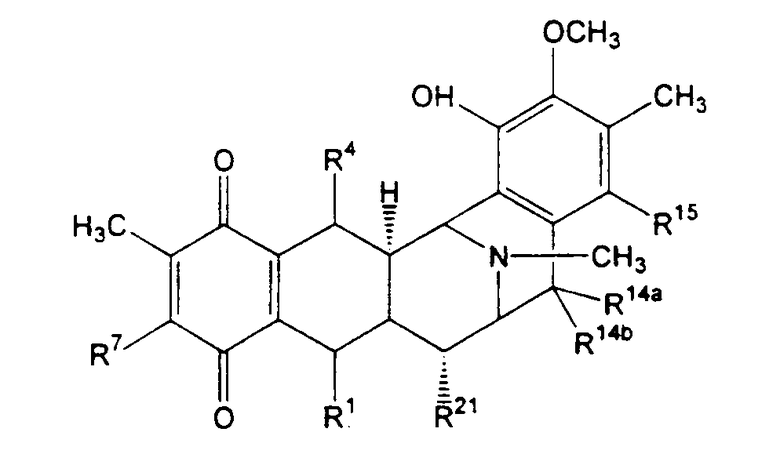
или формулой (XVIIb):
где
R1 представляет необязательно защищенную или модифицированную аминометиленовую группу, необязательно защищенную или модифицированную гидроксиметиленовую группу, например, такую как группа R1, как определено для формулы (XV);
R4 представляет -H; или
R1 и R4 вместе образуют группу формулы (IV), (V), (VI) или (VII):


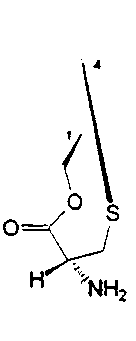 или
или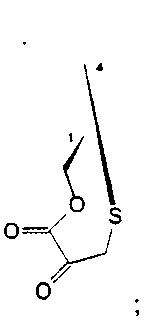
R5 представляет -Н или -ОН;
R7 представляет -ОСН3 и R8 представляет -ОН, или R7 и R8 вместе образуют группу -О-СН2-О-;
R14а и R14b оба представляют -Н или один представляет -Н, а другой представляет -ОН, -ОСН3 или -ОСН2СН3, или R14а и R14b вместе образуют кетогруппу; и
R15 представляет -Н или -ОН;
R21 представляет -Н, -ОН или CN;
и их производные, включая ацилпроизводные, в частности, такие производные, в которых R5 представляет ацетокси или другую ацилоксигруппу, содержащую до 4 атомов углерода, а также включая производные, в которых группа -NCH3- в 12-положении заменена группами -NH- или -NCH2CH3-, и производные, в которых группа -NH2 в соединении формулы (VI) необязательно модифицирована.
В формуле (XVIIa)или (XVIIb) R1 обычно представляет аминометилен, амидометилен, или R1 и R4 вместе образуют группу (IV) или (V). Подходящие амидометиленовые группы включают такие группы формулы -СН2-NH-CO-CHCH3-NH2, которые являются производными аланина, и похожие группы, которые являются производными других аминокислот, в особенности, таких как D, так и L, глицина, валина, лейцина, изолейцина, фенилаланина, тирозина, триптофана, метионина, цистеина, аспартата, аспарагина, глутаминовой кислоты, глутамина, лизина, аргинина, пролина, серина, треонина, гистидина и гидроксипролина. Общая формула группы R1 представляет, в таком случае, -СН2-NH-aa, где aa обозначает ацильную группу аминокислоты.
Группа R1 может быть ацилированной по NH2-группе, и, например, N-ацилпроизводные могут быть образованы из групп -СН2-NH2 и -СН2-NH-aa. Ацилпроизводные могут представлять собой их N-ацил или N-тиоацилпроизводные, также как и циклические амиды. Ацильные группы, для иллюстрации, могут представлять собой алканоил, галоалканоил, арилалканоил, алкеноил, гетероциклилацил, ароил, арилароил, галоароил, нитроароил или другие ацильные группы. Ацильные группы могут представлять собой группы формулы -СО-Ra, где Ra может быть различной группой, например такой, как алкил, алкокси, алкилен, арилалкил, арилалкилен, аминокислотный ацил или гетероциклил, каждая из которых может быть необязательно замещена галогеном, циано, нитро, карбоксиалкилом, алкокси, арилом, арилокси, гетероциклилом, гетероциклилокси, алкилом, амино или замещенной аминогруппой. Другие ацилирующие средства включают изотиоцианаты, такие как арилизотиоцианаты, в особенности, фенилизоцианат. Подходящие алкил, алкокси или алкиленовые группы радикала Ra содержат от 1 до 6 атомов углерода, и могут быть прямыми, разветвленными или циклическими. Арильные группы обычно представляют фенил, бифенил или нафтил. Гетероциклические группы могут быть ароматическими или частично или полностью ненасыщенными и подходящим образом содержат в кольце от 4 до 8 атомов, более предпочтительно, 5 или 6 атомов, с одним или более гетероатомов, выбранных из группы, включающей азот, серу и кислород.
Не являющийся исчерпывающим перечень типичных групп Ra включает алкил, галоалкил, алкоксиалкил, галоалкоксиалкил, арилалкилен, галоалкиларилалкилен, ацил, галоацил, арилалкил, алкенил и остаток аминокислоты. Например, Ra-CO- может представлять собой ацетил, трифторацетил, 2,2,2-трихлорэтоксикарбонил, изовалерилкарбонил, транс-3-(трифторметил)циннамоилкарбонил, гептафторбутирилкарбонил, деканоилкарбонил, транс-циннамоилкарбонил, бутирилкарбонил, 3-хлорпропионилкарбонил, циннамоилкарбонил, 4-метилциннамоилкарбонил, гидроциннамоилкарбонил или транс-гексеноилкарбонил, или аланил, аргинил, аспартил, аспарагил, цистил, глутамил, глутаминил, глицил, гистидил, гидроксипропил, изолейцил, лейцил, лизил, метионил, фенилаланил, пролил, серил, треонил, тиронил, триптофил, тирозил, валил, а также другие ацильные группы менее распространенных аминокислот, также как фталимидо и другие циклические амиды. Другие примеры могут быть найдены в перечне защитных групп.
Соединения, в которых -СО-Ra является производным аминокислоты и включают аминогруппу, могут сами образовывать ацилпроизводные. Подходящие N-ацилсоединения включают дипептиды, которые, в свою очередь, могут образовывать N-ацилпроизводные.
В одном из вариантов, который относится к промежуточным продуктам, кольцо А модифицировано для того, чтобы включить субструктуру, которая показана формулами (XX) или (XXI), что обсуждается ниже.
В другом из вариантов, который относится к промежуточным соединениям, группа R1 может представлять структуру -CH2O-CO-CFu-CH2-S-Prot3, являющуюся производной от соединения формулы (XIX), в котором Prot3 и Fu имеют указанные значения. В таком случае R7 и R8 образуют оксиметиленоксигруппу. Группа R18 обычно защищена. Обычно R21 представляет циано.
Предпочтительно, R14a и R14b представляют водород. Предпочтительно, R15 представляет водород. Подходящие О-ацильные производные представляют алифатические О-ацильные производные, в особенности, ацильные производные, в которых О-ацильный фрагмент содержит от 1 до 4 атомов углерода, и обычно, представляет О-ацетильную группу, в особенности, в 5-положении.
Подходящие защитные группы для фенольных и гидроксигрупп включают простые и сложные эфирные группы, например, такие как алкил, алкоксиалкил, арилоксиалкил, алкоксиалоксиалкил, алкилсилилалкоксиалкил, алкилтиоалкил, арилтиоалкил, азидоалкил, цианоалкил, хлоралкил, гетероциклические группы, арилацил, галоарилацил, циклоалкилалкил, алкенил, циклоалкил, алкилариралкил, алкоксиарилалкил, нитроарилалкил, гало-арилалкил, алкиламинокарбониларилалкил, алкилсульфиниларилалкил, алкилсилил и другие простые эфирные группы, а также арилацил, арилалкилкарбонатные, алифатические карбонатные, алкилсуль-финиларилалкильные карбонатные группы, алкилкарбонатные, арилгалоалкилкарбонатные, арилалкенилкарбонатные, арилкарбаматные, алкилфосфинильные, алкилфосфинотиоильные, арилфосфинотиоильные, арилалкилсульфонатные и другие сложноэфирные группы. Такие группы могут быть необязательно замещены группами, указанными выше при описании R1.
Подходящие защитные группы для аминогрупп включают карбаматные, амидные и другие защитные группы, например, такие как алкил, арилалкил, сульфо- или галоарилалкил, галоалкил, алкилсилилалкил, арилалкил, циклоалкилалкил, алкиларилалкил, гетероциклилалкил, нитроарилалкил, ациламиноалкил, нитроарилдитиоарилалкил, дициклоалкилкарбоксамидоалкил, циклоалкил, алкенил, арилалкенил, нитроарилалкенил, гетероциклилалкенил, гетероциклил, гидроксигетероциклил, алкилдитио, алкокси- или гало- или алкилсульфиниларилалкил, гетероциклилацил и другие карбаматные группы, и алканоил, галоалканоил, арилалканоил, алкеноил, гетероциклилацил, ароил, арилароил, галоароил, нитроароил и другие амидные группы, также как и алкил, алкенил, алкилсилилалкокосиалкил, алкоксиалкил, цианоалкил, гетероциклил, алкоксиариладкил, циклоалкил, нитроарил, арилалкил, алкокси- или гидрокси- арилалкил, и многие другие группы. Такие группы может быть необязательно замещены группами, указанными выше при описании R1.
Примеры таких защитных групп приведены в следующих таблицах.
Сафрацин В содержит боковую аланильную цепь. В одном из вариантов осуществления настоящего изобретения было обнаружено, что защита свободной аминогруппы с использованием Вос-группы может дать большие преимущества.
Некоторые продукты ряда эктеинасцидина, являющиеся предметом настоящего изобретения, включают соединения формулы (XVIII):
где R1 и R4 вместе образуют группу формулы (IV), (V), (VI) или (VII):
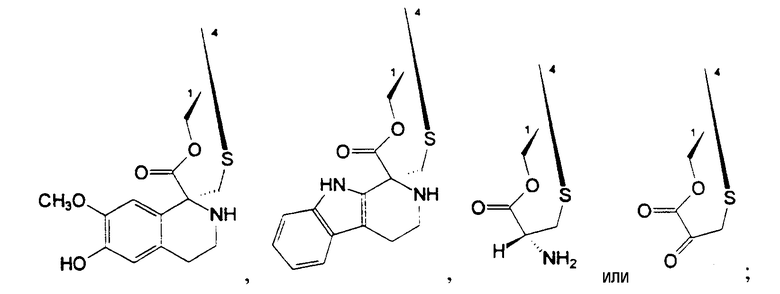
боле предпочтительно, группу формулы (IV) или (V);
R21 представляет -Н, -ОН или -CN, более предпочтительно, -ОН или -CN;
и их ацилпроизводные, боле предпочтительно, 5-ацилпроизводные, включающие 5-ацетилпроизводное.
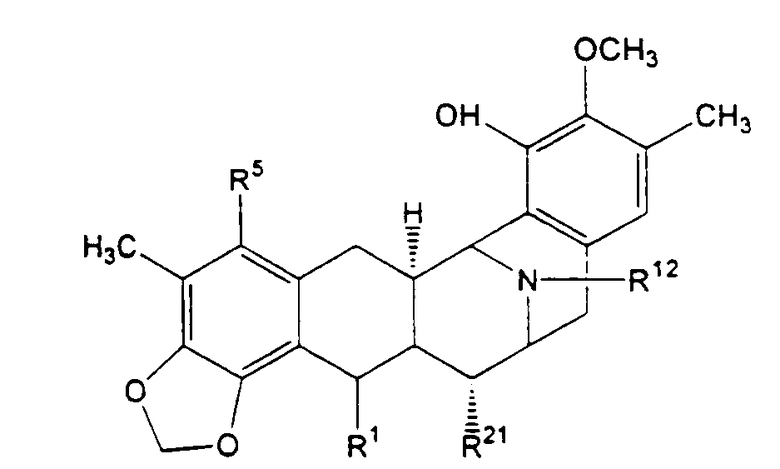
Образование эктеинасцидина 743 и родственных ему соединений
В общем, превращение исходного 21-цианосоединения в продукт - эктеинасцидин, представленный, например, формулой (XVIII), включает:
а) превращение, если необходимо, хиноновой системы кольца Е в фенольную систему;
b) превращение, если необходимо, хиноновой системы кольца А в фенольную систему;
c) превращение фенольной системы кольца А в метилендиоксифенольное кольцо;
d) образование мостиковой спирокольцевой системы формулы (IV), (VI) или (VII), проходящей через 1-положение и 4-положение в кольце В;
е) проведение соответствующей модификации, например, ацилирования.
Стадия (а), превращение, если это необходимо, хиноновой системы кольца Е в фенольную систему, может быть осуществлена с использованием обычных методик восстановления. Подходящая система реагентов представляет водород с катализатором палладий-углерод, хотя могут быть использованы и другие восстанавливающие системы.
Стадия (b), превращение, если это необходимо, хиноновой системы кольца А в фенольную систему, аналогична стадии (а), и нет необходимости в дополнительных деталях.
Стадия (с), превращение фенольной системы кольца А в метилендиоксифенольное кольцо, может быть осуществлена несколькими путями, возможно, вместе со стадией (b). Например, хиноновое кольцо А может быть деметилировано у метоксизаместителя в 7-положении и восстановлено до дигидрохинона и стабилизировано с помощью подходящего электрофильного реагента, например, такого, как CH2Br2, BrCH2Cl или подобного бифункционального реагента, непосредственно с образованием метилендиокси-кольцевой системы, или с бифункциональным реагентом, таким как тиокарбонилдиимидазол, который позволяет получить замещенную метилендиокси-кольцевую систему, которая может быть превращена в необходимое кольцо.
Стадию (d) обычно осуществляют посредством подходящего замещения в 1-положении с использованием участвующего в образовании мостика реагента, который может способствовать образованию нужного мостика, образуя экс-эндо хинонметид в 4-положении с последующим взаимодействием метида с 1-заместителем с образованием мостиковой структуры. Предпочтительные участвующие в образовании мостика реагенты имеют структуру, представленную формулой (XIX)
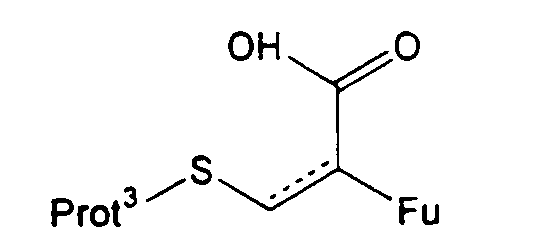
в которой Fu обозначает защищенную функциональную группу, такую как группа -NHProt4a или OProt4b, Prot3 представляет защитную группу, а пунктирная линия показывает необязательно присутствующую двойную связь.
Подходящие метиды образуются введением сначала гидроксигруппы в 10-положение в месте соединения колец А и В с образованием фрагмента формулы (XX):
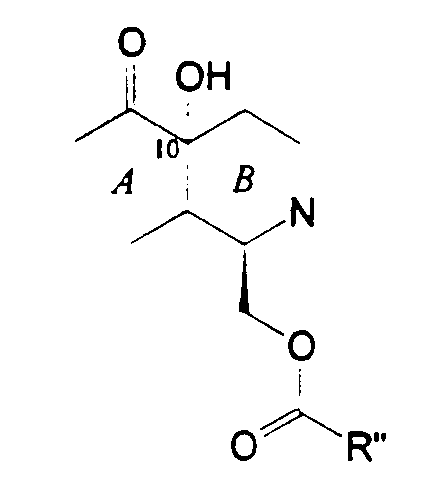
или, более предпочтительно, фрагмента формулы (XXI):

в которой группу R'' выбирают для необходимой группы формулы (IV), (V), (VI) или (VII). Для первых двух таких групп группа R'' обычно имеет структуру -CHFu-CH2-SProt3. Затем защитные группы могут быть удалены, и может быть осуществлена необходимая модификация для получения желаемого соединения.
Типичная методика для осуществления стадии (d) приведена в патенте США 5721362, включенном в настоящее описание посредством ссылки. Особое внимание следует обратить на колонку 8, стадия (1), и пример 33, приведенные в указанном патенте, и соответствующие части описания.
Получение производных на стадии (е) может включать ацилирование, например, для группы Ra-CO-, а также превращение 12-NCH3 группы в 12-NH или 12-NCH2CH3. Такое превращение может быть осуществлено до или после проведения других стадий, с использованием доступных методик.
В качестве иллюстрации в настоящее время вполне возможно превратить соединение цианосафрацин В формулы 2 в ЕТ-743, что дает более короткий и более прямой путь получения ЕТ-743, чем описанные выше способы. Цианосафрацин В может быть превращен в промежуточное соединение 25;
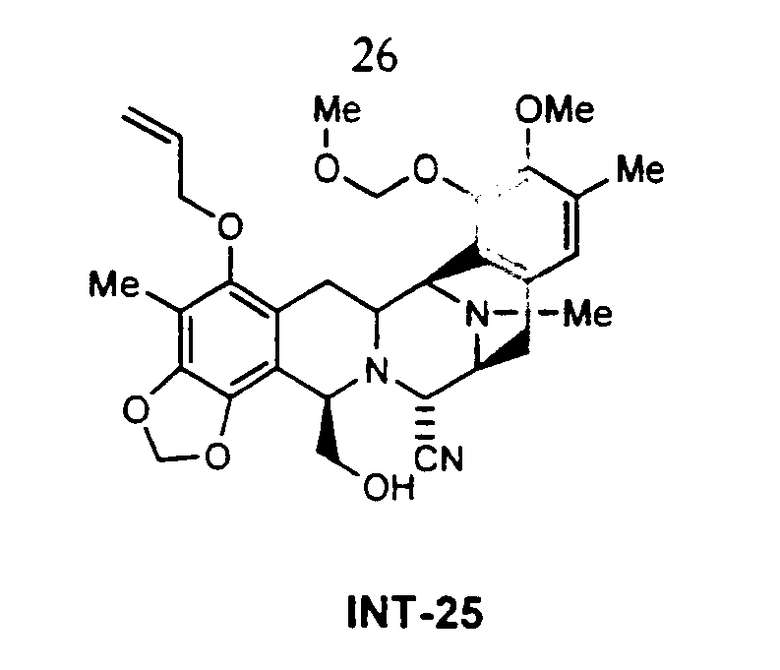
а в это производное возможно ввести ряд цистеиновых производных, которые далее могут быть превращены в Et-743. Предпочтительные цистеиновые производные представлены следующими двумя соединениями:
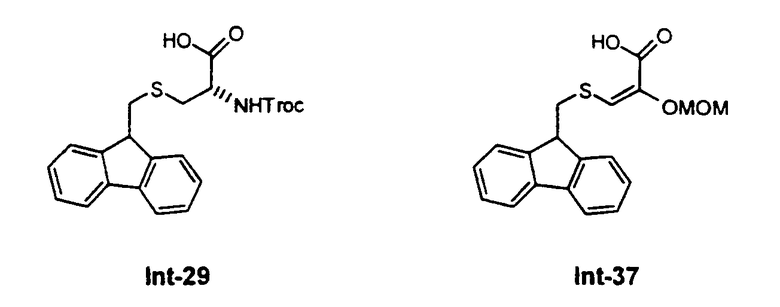
Ретросинтетический анализ получения ЕТ-743 с использованием соединения 29 приведен на схеме I.

Схема I
Следуя приведенной выше схеме I, возможно получить ET-743 за 21 последовательную стадию. Этот метод включает превращение цианосафрацина В в промежуточное соединение 25 путем осуществления последовательности реакций, которые включают, по существу: (1) удаление метоксигруппы, находящейся в кольце А, (2) восстановление кольца А и образование метилендиоксигруппы за одну операцию, (3) гидролиз амидной функциональной группы, расположенной у углерода 1, (4) превращение полученной аминогруппы в гидроксильную группу. Кроме того, при осуществлении способа исключено введение и снятие защиты первичной спиртовой функциональной группы в положении 1 кольца В соединения 25 при использовании непосредственно цистеинового остатка 29 для получения промежуточного соединения 27. Аминогруппу производного цистеина 29 защищают с помощью β,β,β-трихлорэтоксикарбонильной защитной группы, для того чтобы достичь совместимости с имеющимися аллильной и МОМ группами. Промежуточное соединение 27 непосредственно окисляют и циклизуют. Эти обстоятельства, наряду с различной стратегией введения и снятия защиты на поздних стадиях синтеза, позволяют считать способ новым и более пригодным для промышленного применения, чем способ, описанный в патенте США 5721362.
Превращение 2-цианосоединения в промежуточное соединение 25 обычно включает следующие стадии (см. схему II):
образование защищенного соединения формулы 14 взаимодействием соединения 2 с трет-бутоксикарбонил-ангидридом;
превращение соединения 14 в дизащищенное соединение формулы 15 взаимодействием с простым бромметилметиловым эфиром и диизопропилэтиламином в ацетонитриле;
селективное удаление метоксигруппы, находящейся в хиноновой системе соединения 15 для того, чтобы получить соединение формулы 16 путем взаимодействия с метанольным раствором гидроксида натрия;
превращение соединения 16 в метилендиоксисоединение формулы 18, используя следующую предпочтительную последовательность реакций: (1) хиноновую группу соединения 16 восстанавливают, используя 10% Pd/C в атмосфере водорода; (2) гидрохиноновое промежуточное соединение превращают в метилендиоксисоединение формулы 17 взаимодействием с бромхлорметаном и карбонатом цезия в атмосфере водорода; (3) соединение 17 превращают в соединение формулы 18 посредством защиты свободной гидроксильной группы, в виде группы OCH2R. Эту реакцию проводят, используя BrCH2R и карбонат цезия, где R может представлять арил, СН=СН2, OR' и т.д.;
удаление трет-бутоксикарбонильной и метилоксиметильной защитных групп соединения 18 для того, чтобы получить соединение формулы 19 взаимодействием с раствором HCl в диоксане. Данная реакция также протекает при смешивании соединения 18 с раствором трифторуксусной кислоты в дихлорметане;
образование тиомочевины формулы 20 взаимодействием соединения 19 с фенилизотиоцианатом;
превращение соединения формулы 20 в амин формулы 21 взаимодействием с раствором хлористого водорода в диоксане;
превращение соединения формулы 21 в N-Troc-производное 22 взаимодействием с трихлорэтилхлорформиатом и пиридином;
образование защищенного гидроксисоединения формулы 23 взаимодействием соединения 22 с простым бромметилметиловым эфиром и диизопропилэтиламином;
превращение соединения формулы 23 в N-H производное 24 взаимодействием с уксусной кислотой и цинком;
превращение соединения формулы 24 в гидроксисоединение формулы 25 взаимодействием с нитритом натрия в уксусной кислоте. В качестве альтернативы, может быть использован тетраоксид азота в смеси с уксусной кислотой и ацетонитрилом, с последующей обработкой гидроксидом натрия. Также может быть использован нитрит натрия в смеси уксусный ангидрид - уксусная кислота, с последующей обработкой гидроксидом натрия.
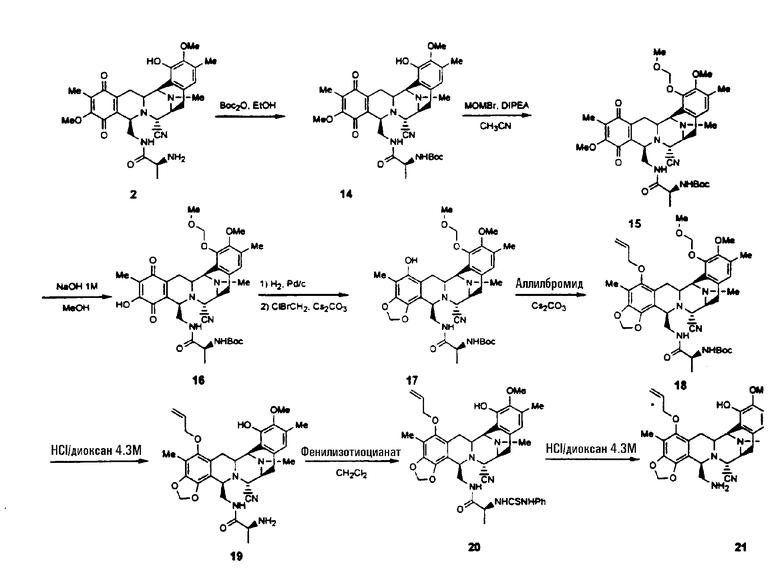
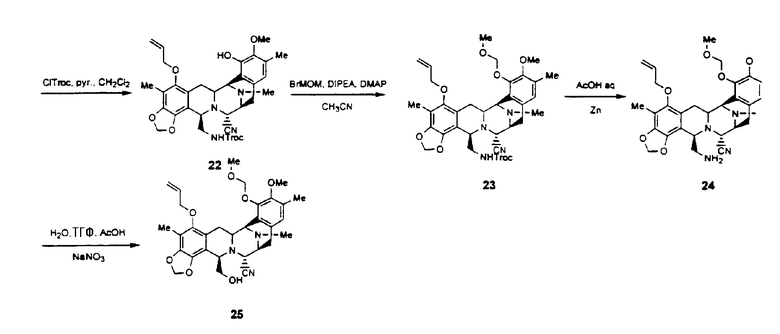
Схема II
Превращение промежуточного соединения 25 в ЕТ-743 с использованием производного цистеина 29 обычно включает следующие стадии (см. схему III):
превращение соединения формулы 24 в производное 30 путем введения защиты первичной гидроксильной функциональной группы с использованием (S)-N-2,2,2-трихлорэтоксикарбонил-S-(9Н-флуорен-9-илметил)цистеина 29;
превращение защищенного соединения формулы 30 в фенольное производное 31 путем отщепления аллильной группы с использованием гидрида трибутилолова и дихлорпалладий-бис(трифенилфосфина);
превращение фенольного соединения формулы 31 в соединение формулы 32 окислением ангидридом бензолселеновой кислоты при низкой температуре;
превращение гидроксисоединения формулы 32 в лактон 33 в результате следующей последовательности реакций: (1) взаимодействие соединения формулы 32 с 2 экв. ангидрида трифторметилсерной кислоты и 5 экв. ДМСО (DMSO), (2) последующее взаимодействие с 8 экв. диизопропилэтиламина, (3) последующее взаимодействие с 4 экв. трет-бутилового спирта, (4) последующее взаимодействие с 7 экв. 2-трет-бутил-1,1,3,3-тетраметилгуанидина, (5) последующее взаимодействие с 10 экв. уксусного ангидрида;
превращение лактона 33 в гидроксилсодержащее соединение 34 путем удаления МОМ-защитной группы с использованием TMSI;
отщепление N-трихлорэтоксикарбонильной группы соединения формулы 34 с получением соединения 35 реакцией с Zn/AcOH;
превращение аминосоединения 35 в соответствующий α-кетолактон 36 взаимодействием с хлоридом N-метилпиридиний-карбоксальдегида и затем с DBU;
образование ET-770 взаимодействием соединения формулы 36 с 3-гидрокси-4-метоксифенилэтиламином;
превращение ET-770 в ET-743 взаимодействием с нитратом серебра в смеси AcN/H2O.

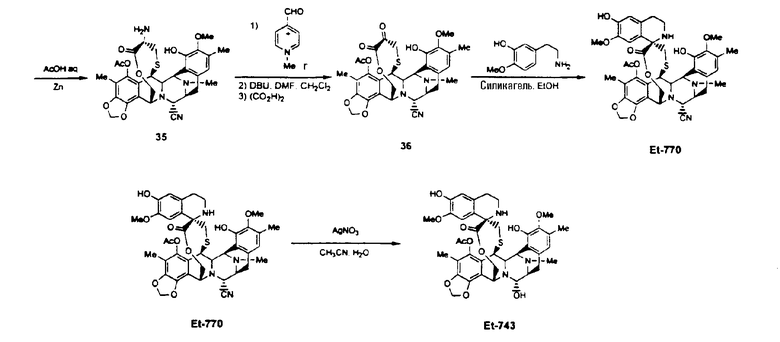
Схема III
Описанный выше путь превращения промежуточного соединения 25 в соединение ЕТ-743 может быть легко модифицирован с использованием других производных цистеина, например, соединения 37, называемого 2-метоксиметилокси-3-(9Н-флуорен-9-илметил)тиопропеновая кислота. В данном соединении уже имеется введенная кетогруппа в форме енольного простого эфира, в то время как в других аналогах цистеина присутствует аминогруппа, которая должна быть превращена далее в кетогруппу путем реакции трансаминирования, протекающей с умеренным выходом - 55-60%. Следовательно, использование соединения 37 возможно для того, чтобы в значительной степени повысить выход постадийного синтеза, поскольку исключается стадия трансаминирования.
Превращение промежуточного соединения 25 в соединение ЕТ-743 с использованием производного цистеина 37 может быть осуществлено аналогичным образом с использованием тех же самых реагентов, что и в случае производного цистеина 29, с исключением стадий превращения (f) и (g). Последовательность реакций иллюстрирует следующая схема (схема IV):
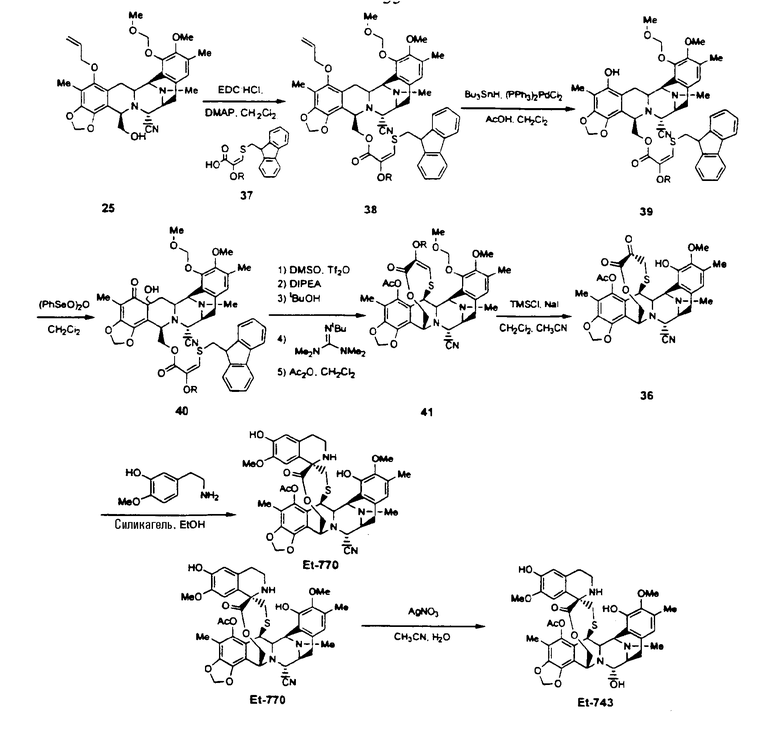
Схема IV
Соединение 38 также может быть получено взаимодействием промежуточного соединения 12, описанного в патенте США 5721362, с промежуточным соединением 37, что является усовершенствованием способа, описанного в указанном патенте.
Образование фталасцидина и родственых ему соединений
Согласно настоящему изобретению ключевой класс продуктов включает фталасцидин и представлен общей формулой (XX):

в которой R1 представляет амидометиленовую группу; R5 представляет небольшую боковую окси-цепь; и R21 представляет цианогруппу или гидроксигруппу. В случае фталасцидина, R1 представляет фталимидометиленовую группу; R5 представляет ацетоксигруппу; и R21 представляет цианогруппу. Другие группы R1 включают моно- и ди-N-замещенные амидометиленовые группы, а также и другие циклические амидометиленовые группы, а другие группы в случае R5 включают, кроме того, С1-С4 ацильные группы, а также С1-С4 алкильные группы.
Превращение 21-цианосоединения во фталасцидин или родственные ему продукты формулы (XX) включает следующие стадии:
а) превращение, если необходимо, хиноновой системы кольца Е в фенольную систему;
b) образование группы -R5 в 5-положении кольца А;
c) образование группы -R1 в 1-положении кольца В;
d) превращение, если необходимо, хиноновой системы кольца A в фенольную систему;
e) превращение, если необходимо, фенольной системы кольца А в метилендиоксифенольное кольцо.
Данные стадии имеют много сходного со стадиями, указанными при получении эктеинасцидинов. Стадия (с) обычно включает образование группы -СН2-NH2 в 1-положении и ее ацилирование.
Фталасцидин может быть получен с использованием промежуточных соединений, описанных при превращении цианосафрацина В в промежуточное соединение 25. Например, промежуточные соединения 21 и 17 являются подходящими исходными веществами для получения фталасцидина.
Как показано далее на схеме V, способ синтеза фталасцидина, исходя из промежуточного соединения 21, включает следующую последовательность стадий:
превращение соединения 21 в соединение формулы 27 взаимодействием со фталевым ангидридом в дихлорметане и с карбонилдиимидазолом;
превращение соединения 27 во фталасцидин по реакции с гидридом трибутилолова и дихлорпалладий-бис(трифенилфосфином) или щелочной средой, с последующим взаимодействием с ацетилхлоридом.
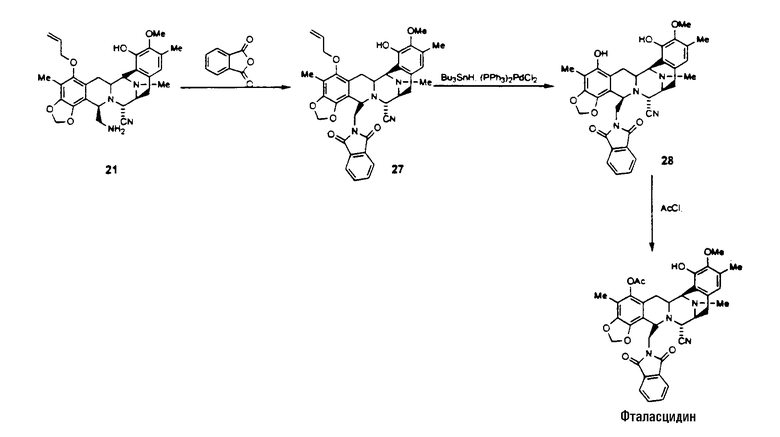
Схема V
Как показано далее на схеме VI, способ синтетического получения фталасцидина, исходя из промежуточного соединения 17, включает следующую последовательность стадий:
ацетилирование гидроксильной группы соединения формулы 17 ацетилхлоридом с использованием пиридина с получением ацетилированного промежуточного соединения формулы 42;
удаление трет-бутоксикарбонильной и метилоксиметильной защитных групп соединения 42 с получением соединения формулы 43 взаимодействием с раствором HCl в диоксане. Данная реакция также может протекать при смешивании соединения 42 с раствором трифторуксусной кислоты в дихлорметане;
образование производного тиомочевины формулы 44 взаимодействием соединения 43 с фенилизотиоцианатом;
превращение соединения формулы 44 в амин формулы 45 взаимодействием с раствором хлористого водорода в диоксане;
превращение соединения 45 во фталасцидин взаимодействием со фталевым ангидридом в дихлорметане с использованием карбонилдиимидазола.

Схема VI
Образование промежуточного соединения 11 и родственных ему промежуточных соединений
Ретросинтетический анализ проиллюстрирован следующей схемой.
Согласно настоящему изобретению класс ключевых промежуточных соединений включает промежуточное соединение 11 и характеризуется общей формулой (XXI):
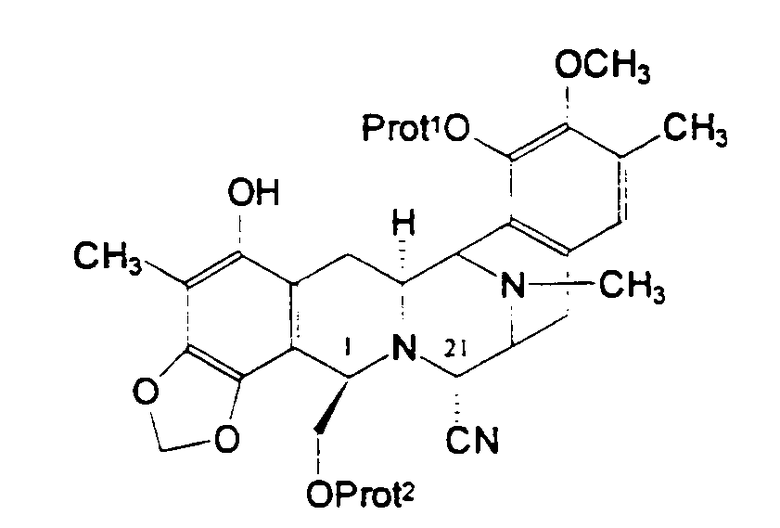
в которой Prot1 и Prot2 представляют защитные группы для гидроксигрупп, предпочтительно различные. Что касается самого промежуточного соединения 11, то группа Prot1 представляет метоксиметильную группу, а Prot2 представляет трет-бутилдифенилсилильную группу.
Превращение 21-цианосоединения в промежуточное соединение 11 или родственное ему промежуточное соединение формулы (XXI) обычно включает следующие стадии:
а) превращение, если необходимо, хиноновой системы кольца Е в фенольную систему;
b) образование группы -OProt1 в 18-положении, в кольце E;
c) образование группы -CH2-OProt2 в 1-положении, в кольце В;
d) превращение, если необходимо, хиноновой системы кольца A в фенольную систему;
e) превращение фенольной системы кольца А в метилендиоксифенольное кольцо.
Стадия (b), образование защитной группы -OProt1 в 18-положении, в кольце E, представляет типичную реакцию введения защиты фенольной группы и не нуждается в специальных комментариях. Подходящие условия выбирают в зависимости от природы защитной группы. Другие стадии аналогичны стадиям других реакций.
Стадию (с), образование группы -CH2-OProt2 в 1-положении кольца В, обычно осуществляют путем образования группы -CH2-NH2 в 1-положении, с последующим превращением аминной функциональной группы в функциональную гидроксигруппу и введением защиты. Таким образом, в тех случаях, когда исходное вещество содержит группу R1, которая представляет -CH2-NH-CO-CR25aR25bR25c, необходимо удаление N-ацильной группы. В тех случаях, когда исходное вещество содержит группу R1, которая представляет -CH2-О-CO-R, при получении продукта - эктеинасцидина, в котором заместитель R1 является тем же самым, нет необходимости в изменениях. В случае других продуктов, возникает задача удаления О-ацильной группы. Для осуществления такого деацетилирования имеются в распоряжении различные методики. В одном из вариантов деацетилирование и модификация функциональной гидроксигруппы осуществляются в одну стадию. Кроме того, гидроксигруппа может быть подвергнута ацилированию или быть модифицирована иным образом для получения подходящей R1-группы.
В патенте США 5721362 описаны методы синтеза, используемые для получения ЕТ-743 путем длительного многостадийного синтеза. Одним из промежуточных соединений, получаемых в этом синтезе, является промежуточное соединение 11. Используя в качестве исходного вещества цианосафрацин В, возможно получить промежуточное соединение 11, используя значительно более короткий путь получения такого промежуточного соединения, усовершенствовав таким образом способ получения ЕТ-743.
Цианосафрацин В может быть превращен в промежуточное соединение 25 по методикам, описанным выше. Исходя из промежуточного соединения 25, возможно получить промежуточное соединение 11, осуществив следующие стадии, см. схему VII:
образование защищенного гидроксисоединения формулы 26 взаимодействием соединения 25 с трет-бутилдифенилсилил-хлоридом в присутствии основания;
заключительное отщепление аллильной группы соединения 26 под действием гидрида трибутилолова и дихлорпалладий-бис(трифенилфосфина), которое приводит к образованию промежуточного соединения 11.

Схема VII
Один из вариантов осуществления синтетического способа согласно настоящему изобретению для превращения сафрацина В в промежуточное соединение 11 представляет модификацию и расширение способа, представленного на схеме VIII, и включает следующую последовательность стадий:
стереоспецифическое превращение соединения формулы 1 (сафрацин В) в соединение формулы 2 путем селективного замещения ОН на CN взаимодействием с KCN в кислой среде;
образование производного тиомочевины формулы 3 взаимодействием соединения формулы 2 с фенилизотиоцианатом;
превращение тиомочевины формулы 3 в ацетамид формулы 5 путем гидролиза в кислой среде с последующим добавлением уксусного ангидрида; промежуточный амин формулы 4 может быть выделен путем гащения реакции гидролиза в кислой среде бикарбонатом натрия, однако данное производное крайне нестабильно и быстро превращается в пятичленный циклический имин, обозначенный как соединение 6;
образование защищенного соединения формулы 7 взаимодействием с бромметилметиловым простым эфиром и диизопропилэтиламином в дихлорметане;
селективное деметилирование метоксигруппы, принадлежащей хиноновой системе соединения формулы 7, с образованием соединения формулы 8 при взаимодействии с метанольным раствором гидроксида натрия;
превращение соединения формулы 8 в метилендиокси-соединение формулы 9, предпочтительно при осуществлении следующей последовательности реакций: (1) хиноновую группу соединения 8 восстанавливают, используя 10% Pd/C в атмосфере водорода; (2) гидрохиноновое промежуточное соединение превращают в метилендиоксисоединение формулы 9 взаимодействием с бромхлорметаном и карбонатом цезия в атмосфере водорода; (3) соединение формулы 9 переводят в соединение формулы 10 введением защиты гидроксильной группы в виде группы ОСН2R посредством взаимодействия с BrCH2R и карбонатом цезия, где R может означать арил, СН=СН2, OR'и т.д.; превращение ацетамидной группы соединения формулы 10 в соответствующую гидроксильную группу соединения формулы 11 взаимодействием с тетраоксидом азота в смеси уксусной кислоты и ацетоуксусного эфира с последующей обработкой гидроксидом натрия; в качестве альтернативы может быть использован нитрит натрия в смеси уксусного ангидрида и уксусной кислоты, с последующей обработкой гидроксидом натрия;
в качестве альтернативы ацетамидная группа соединения формулы 10 может быть превращена в первичную аминогруппу взаимодействием с гидразином или с Boc2O, DMAP после гидразина; такая первичная аминогруппа может быть превращена в соответствующую гидроксильную группу (соединение формулы 11) окислительным превращением первичного амина в соответствующий альдегид под действием 4-формил-1-метилпиридинийбензолсульфоната или другого иона пиридиния, с последующей обработкой DBU или другим основанием и дополнительным гидролизом, и с последующим восстановлением альдегидной группы до соответствующей гидроксильной группы алюмогидридом лития или другим восстанавливающим средством;
образование защищенного соединения формулы 26 взаимодействием с трет-бутилдифенилсилилхлоридом и диметиламинопиридином в дихлорметане;
превращение силилированного соединения формулы 26 в промежуточное соединение 11 путем снятия защиты с ОСН2R защитной группы, взаимодействием в восстановльных или кислотных условиях. Обычные методики представляют использование палладия на углероде в атмосфере азота или обработку водным раствором TFA, или гидридом трибутилолова и дихлор-бис(трифенилфосфинпалладием).
Согласно еще одной предпочтительной модификации цианосоединение формулы 2 может быть превращено в промежуточное соединение 11 с использованием дополнения к схеме II, включающего следующие стадии:
образование защищенного гидроксисоединения формулы 26 взаимодействием соединения 25 с трет-бутилдифенилсилил-хлоридом в присутствии основания;
заключительное отщепление аллильной группы соединения 26 гидридом трибутилолова и дихлорпалладий-бис(трифенилфосфином), что приводит к образованию промежуточного соединения 11.
Образование активных соединений
Возможно превратить сафрацин В в ряд промежуточных соединений и производных с потенциальной противоопухолевой терапевтической активностью. Такие промежуточные соединения могут быть получены из уже описанных соединений, либо с использованием альтернативных подходов.
Описываемые промежуточные соединения, включают соединение 47 и ряд амидных производных, полученных с использованием соединений 45 и 43.
На схеме VIII показано получение соединения 47 с использованием следующей последовательности стадий:
образование производного тиомочевины формулы 3 взаимодействием соединения формулы 2 с фенилизотиоцианатом;
превращение тиомочевины формулы 3 в ацетамид формулы 5 путем гидролиза в кислой среде с последующим добавлением уксусного ангидрида; промежуточный амин формулы 4 может быть выделен гашением реакции гидролиза в кислой среде бикарбонатом натрия, однако данное производное крайне нестабильно и быстро превращается в пятичленный циклический имин, названный соединением 6;
образование защищенного соединения формулы 7 взаимодействием с бромметилметиловым простым эфиром и диизопропилэтиламином в дихлорметане;
селективное деметилирование метоксигруппы, принадлежащей хиноновой системе соединения формулы 7, с образованием соединения формулы 8 взаимодействием с метанольным раствором гидроксида натрия;
превращение соединения формулы 8 в метилендиоксисоединение формулы 10, предпочтительно при осуществлении следующей последовательности реакций: (1) хиноновую группу соединения 8 восстанавливают, используя 10% Pd/C в атмосфере водорода; (2) гидрохиноновое промежуточное соединение превращают в метилендиоксисоединение формулы 9 взаимодействием с бромхлорметаном и карбонатом цезия в атмосфере водорода; (3) соединение формулы 9 переводят в соединение формулы 10 введением защиты гидроксильной группы в виде аллилоксигруппы взаимодействием с аллилбромидом и карбонатом цезия;
превращение соединения формулы 9 в ацетилпроизводное 46 взаимодействием с ацетилхлоридом в пиридине;
превращение соединения формулы 46 в защищенное соединение 47 взаимодействием с хлористоводородной кислотой в диоксане.
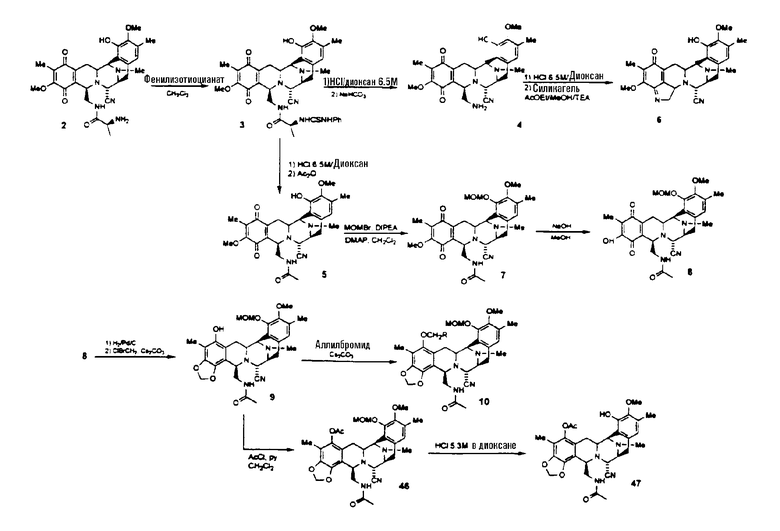
Схема VIII
Другие полезные амидные производные промежуточных соединений могут быть получены, исходя из уже описанного промежуточного соединения 45 по следующей схеме:
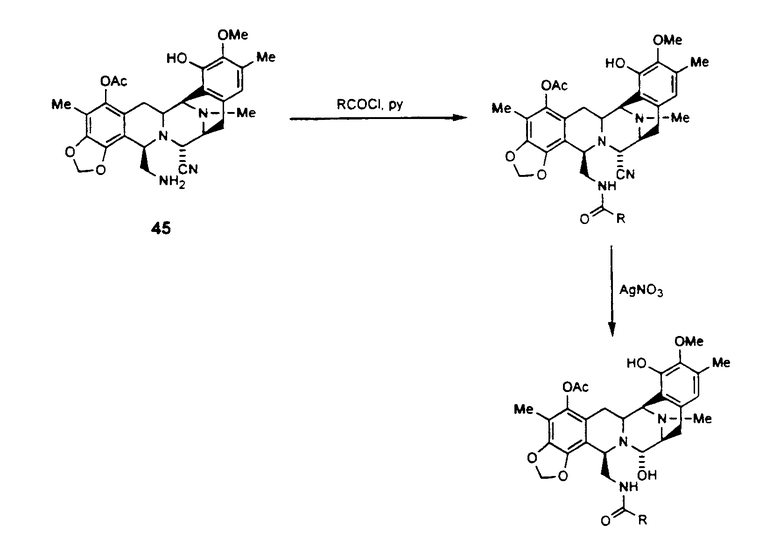
Вторая стадия является необязательной. Данный способ является важной частью настоящего изобретения, в особенности, в том случае, когда группа R представляет группу Ra, определенную выше. Кроме того, схема VIII может быть легко расширена для того, чтобы осуществить синтез соединений формулы (XXIII), путем введения в исходное вещество различных групп в 5-положение либо группы, непосредственно необходимой продукту, либо группы, которая может быть удалена или каким-либо образом модифицирована для получения необходимой группы.
Схема IX
Исходя из соединения 45, может быть получена группа аналогов путем осуществления следующей последовательности операций:
ацилирование аминогруппы соединения формулы 45 с использованием широкого диапазона ацилпроизводных для получения соответствующих амидов, в которых предпочтительные ацильные группы представляют ацетил, циннамоилхлорид, п-трифторциннамоилхлорид, изовалерилхлорид фенилтиоизоцианата или остаток аминокислоты, или другие группы, указанные выше в качестве примеров групп RaCO-.
превращение CN-группы в ОН-группу взаимодействием с нитратом серебра в смеси AcN/H2O.
Другие полезные амидные производные промежуточных соединений могут быть получены, исходя из уже описанного промежуточного соединения 43 по следующей схеме:

Схема X
Из соединения 43 может быть получена другая группа представляющих интерес производных с использованием следующей последовательности операций:
(а) ацилирование аминогруппы соединения формулы 43 с использованием широкого диапазона ацилпроизводных для получения соответствующих амидов, в которых предпочтительные ацильные группы представляют ацетил, циннамоилхлорид, п-трифторциннамоилхлорид, изовалерилхлорид или остаток аминокислоты, или другие группы, указанные выше в качестве примеров групп RaCO-.
(b) превращение CN-группы в ОН-группу взаимодействием с нитратом серебра в смеси AcN/H2O.
Новые промежуточные соединения
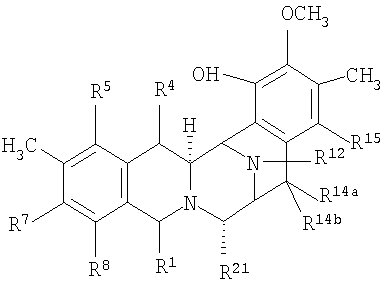
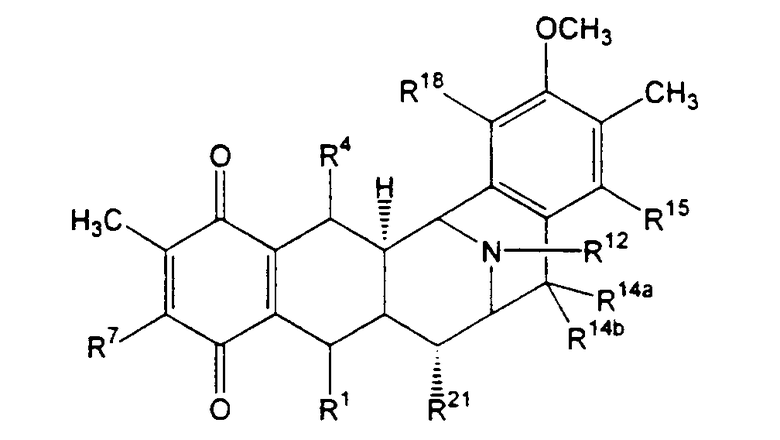
В свете предыдущих разъяснений может быть очевидно, что настоящее изобретение обеспечивает возможность получения новых промежуточных соединений. В зависимости от типа кольца А, промежуточные соединения представлены формулой (XXIIa):
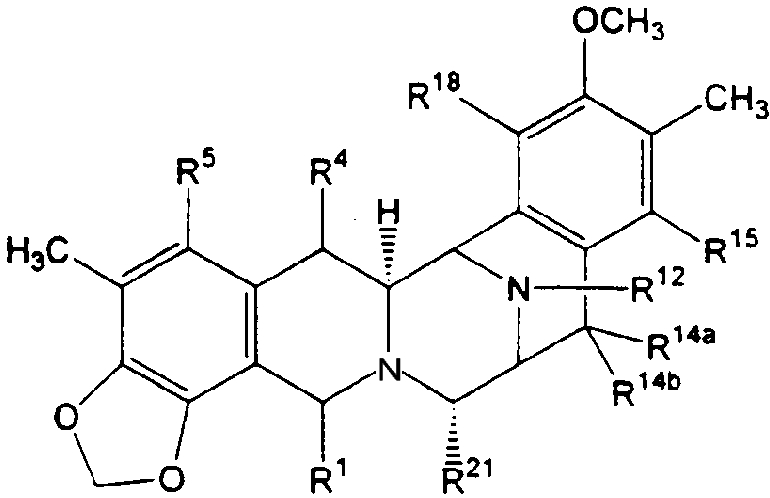
или формулой (XXIIb):
в которых:
R1 представляет -CH2-NH2 или -CH2ОН, или защищенный или модифицированный вариант такой группы, и R4 представляет -Н; или
R1 и R4 вместе образуют группу формул (IV), (VI) или (VII):

R5 представляет -ОН, или защищенный или модифицированный вариант такой группы;
R14a и R14b оба представляют -Н, или один представляет -Н, а другой -ОН, или защищенный или модифицированный вариант такой группы; -ОСН3 или -ОСН2СН3, или R14a и R14b вместе образуют кетогруппу;
R12 представляет -H, -CH3 или -CH2CH3;
R15 представляет -ОН, или защищенный или модифицированный вариант такой группы;
R18 представляет -ОН, или защищенный или модифицированный вариант такой группы; и
R21 представляет -H, -ОН или -CN.
В одном из воплощений, предпочтительно, по меньшей мере, одна из групп R1, R5,R14a , R14b, R15 или R18 представляет защищенный или модифицированный вариант такой группы.
В одном из вариантов осуществления изобретения группа R1 не является 3,5-трет-бутилдифенилсилильнам заместителем и/или группа R18 не является метоксиметильной группой.
Предпочтительно, R1 представляет -CH2-NH2 или -CH2ОН, или защищенный или модифицированный вариант такой группы, и R4 представляет -H; или
R1 и R4 вместе образуют группу:
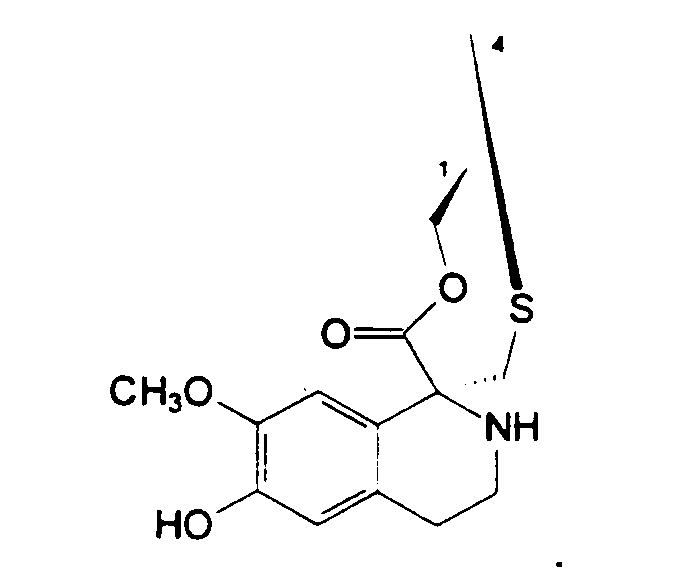
Предпочтительно, R14a и R14b обе представляют -H.
Один из предпочтительных классов промежуточных соединений включает соединение, которое обозначено как соединение 25, формулы:
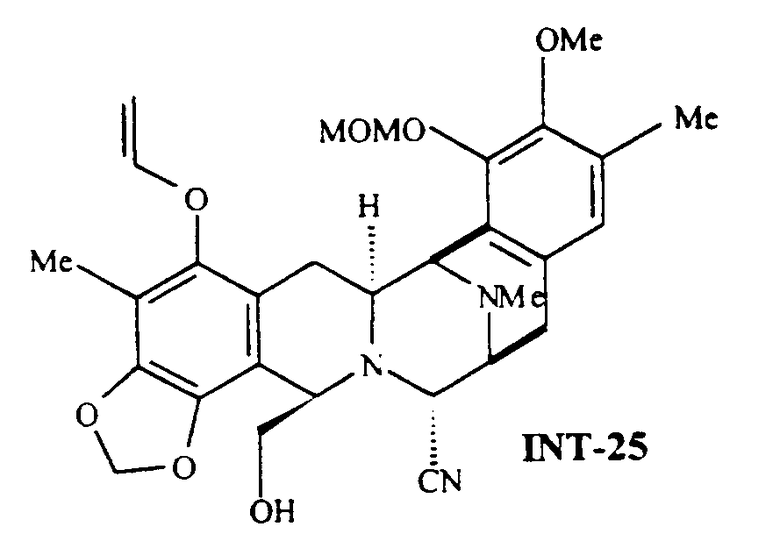
Предпочтительным классом, является класс соединений указанной общей формулы, где группа МОМ заменена любой другой защитной группой.
Другие предпочтительные промежуточные соединения включают соединения, которые обозначены как соединения 45 и 47. Другие N-ацильные производные могут быть легко получены из соединения 45 и представляют важную часть настоящего изобретения. Подходящие ацильные группы включают такие группы, которые были указаны выше. Соответствующие 21-гидроксисоединения также являются полезными и относятся к числу обнаруженных активных соединений.
Новые активные соединения
В дополнение было обнаружено, что некоторые из соединений настоящего изобретения, которые первоначально были получены в качестве промежуточных соединений, обладают исключительной активностью при лечении различных разновидностей рака, например, таких как лейкемия, рак легкого, рак толстой кишки, рак почки и меланома.
Таким образом, настоящее изобретение предоставляет способ лечения какого-либо млекопитающего, в особенности, человека, подверженного раку, который включает введение подверженному болезни субъекту терапевтически эффективного количества соединения настоящего изобретения или его фармацевтической композиции.
Настоящее изобретение также относится к фармацевтическим композициям, которые содержат в качестве активного ингредиента соединение или соединения настоящего изобретения, а также к способам получения таких композиций.
Примеры фармацевтических композиций включают любые твердые (таблетки, пилюли, капсулы, гранулы и т. д.) или жидкие (растворы, суспензии или эмульсии) композиции подходящего состава для перорального, местного или парентерального введения, и могут содержать чисто соединение настоящего изобретения или соединение в сочетании с любым носителем или другим фармакологически активным соединением. Такие композиции могут нуждаться в стерилизации в случае парентерального введения.
Введение соединений или композиций настоящего изобретения может осуществляться любым подходящим способом, например, таким как, внутривенное вливание, использование пероральных препаратов, интраперитонеальное и внутривенное введение. Наиболее предпочтительным является проведение вливания в течение вплоть до 24 часов, предпочтительно, 2-12 часов, наиболее предпочтительно, 2-6 часов. Особенно желательны кратковременные вливания, которые позволяют осуществлять вливание, не оставляя пациента в клинике на ночь. Однако, если требуется, вливание может продолжаться от 12 до 24 часов и даже дольше. Введение может продолжаться, с подходящими интервалами, в течение от 2 до 4 недель. Фармацевтические композиции, содержащие соединения настоящего изобретения, могут быть доставлены с помощью липосом или нанокапсул, с использованием композиций замедленного высвобождения или с использованием других обычных средств доставки.
Правильная дозировка соединений будет варьироваться в зависимости от конкретной композиции, способа применения, а также конкретного местонахождения, «хозяина» и опухоли, которая подлежит лечению. Следует принимать во внимание и другие факторы, такие как возраст, масса тела, пол, режим питания, время введения, скорость выделения, особенности «хозяина», сочетаемость лекарственных препаратов, чувствительность к препарату и степень выраженности симптомов. Введение может осуществляться непрерывно или периодически в пределах максимально допустимой дозы.
Соединения и композиции настоящего изобретения могут быть использованы совместно с другими препаратами для проведения комбинированной терапии. Другие лекарственные средства могут составлять часть той же самой композиции или быть представлены в виде отдельной композиции для введения в то же самое или другое время. Природа других лекарственных средств особенно не ограничена, подходящие варианты лекарственных средств включают:
а) лекарственные средства, обладающие антимитотическим действием, в особенности, такие, мишенью которых являются элементы цитоскелета, включая модуляторы микротрубочек, такие как таксановые лекарственные средства (такие как таксол, паклитаксел, таксотер, доцетаксел), подофилотоксины или винкаалкалоиды (винкристин, винбластин);
b) лекарственные средства - антиметаболиты, такие как 5-фторурацил, цитарабин, гемцитабин, пуриновые аналоги, такие как пентостатин, метотрексат);
c) алкилирующие средства, такие как азотсодержащие производные горчичного масла (такие как циклофосфамид или дифосфамид);
d) лекарственные средства, мишенью которых является ДНК, такие как антрациклиновые лекарственные средства адриамицин, доксорубицин, фарморубицин или эпирубицин;
e) лекарственные средства, мишенью которых являются топоизомеразы, такие как этопозид;
f) гормоны и агонисты гормонов или антагонисты гормонов, такие, как эстрогены, антиэстрогены (тамоксифен и родственные ему соединения) и андрогены, флутамид, лейпрорелин, госерелин, ципротрон или октреотид;
g) лекарственные средства, мишенью которых является система передачи сигнала в опухолевых клетках, включая производные антител, такие как герцептин;
h) алкилирующие лекарственные средства, такие как препараты платины (цисплатин, карбонплатин, оксалиплатин, параплатин) или нитрозомочевины;
i) лекарственные средства, потенциально воздействующие на метастазы опухолей, такие как ингибиторы матричных протеиназ;
j) лекарственные средства для генной терапии и средства, обладающие антисенсивным действием;
k) лекарственные средства для терапии с участием антител;
l) другие биологически активные соединения морского происхождения, в особенности, дидемнины, такие как аплидин;
m) аналоги стероидов, в частности, дексаметазон;
n) противовоспалительные лекарственные средства, в частности, дексаметазон;
o) противорвотные лекарственные средства, в частности, дексаметазон.
Настоящее изобретение также распостраняется на соединения настоящего изобретения для применения в способе лечения, а также на применение данных соединений для получения композиций для лечения рака.
Цитотоксическая активность
Культуры клеток. Использовали клетки в логарифмической фазе роста, в минимальной эссенциальной среде Игла, содержащей добавки поддерживающих солей Эрла, 2,0 мМ L-глутамина, не незаменимые аминокислоты, без бикарбоната натрия (ЕМЕМ/neaa); с добавлением 10% фетальной телячьей сыворотки (FCS), 10-2 М бикарбоната натрия и 0,1 г/л пенициллина-G + сульфата стрептомицина.
Для оценки и сравнения противоопухолевой активности этих соединений осуществляли простую методику скрининга, используя адаптированный вариант метода, описанного Bergeron et al (1984). Используемая линия клеток опухоли представляла собой Р-338 (суспензионная культура лимфоидной неоплазмы мыши линии DBA-2), А-549 (монослойная культура карциномы легкого человека), НТ-29 (монослойная культура карциномы толстой кишки человека) и MEL-28 (монослойная культура меланомы человека).
Клетки Р-338 высевали в 16 мм лунки в количестве 1х104 клеток на лунку в 1 мл аликвоте MEM 5FCS, содержащей определенную концентрацию лекарственного средства. Отдельно высевали набор культур в качестве контроля роста, не используя лекарственное средство, для того, чтобы быть в уверенности в том, что клетки остаются в экспоненциальной фазе роста. Все определения проводили дважды. Через три дня инкубирования при 37°С, 10% СО2 в атмосфере с 98%-ной влажностью определяли приблизительное значение IC50 путем сравнения роста клеток в лунках, содержащих лекарственное средство, и в контрольных лунках.
А-549, НТ-29 и MEL-28 высевали в 16 мм лунки в количестве 2х104 клеток на лунку в 1 мл аликвоте MEM 10FCS, содержащей определенную концентрацию лекарственного средства. Отдельно высевали набор культур в качестве контроля роста, не вводя лекарственное средство, для того, чтобы быть в уверенности в том, что клетки остаются в экспоненциальной фазе роста. Все определения проводили дважды. Через три дня инкубирования при 37°С, 10% СО2 в атмосфере с 98%-ной влажностью лунки окрашивали 0,1%-ным красителем кристаллическим фиолетовым. Приблизительное значение IC50 определяли путем сравнения роста клеток в лунках, содержащих лекарственное средство, и в контрольных лунках.
1. Raymond J.Bergeron. Paul F. Cavanaugh, Jr., Steven J. Kline. Robert G. Hughes, Jr., Gary T. Elliot and Carl W. Porter. Antineoplastic and antiherpetic activity of spermidine catecholamide iron chelators. Biochem. Bioph. Res. Comm. 1984, 121(3), 848-854.
2. Alan C. Schroeder, Robert G. Hughes, Jr., and Alexander Bloch. Effects of Acyclic Pyrimidine Nucleoside Analoges. J.Med.Chem. 1981, 24 1078-1083.
Цитотоксическая активность

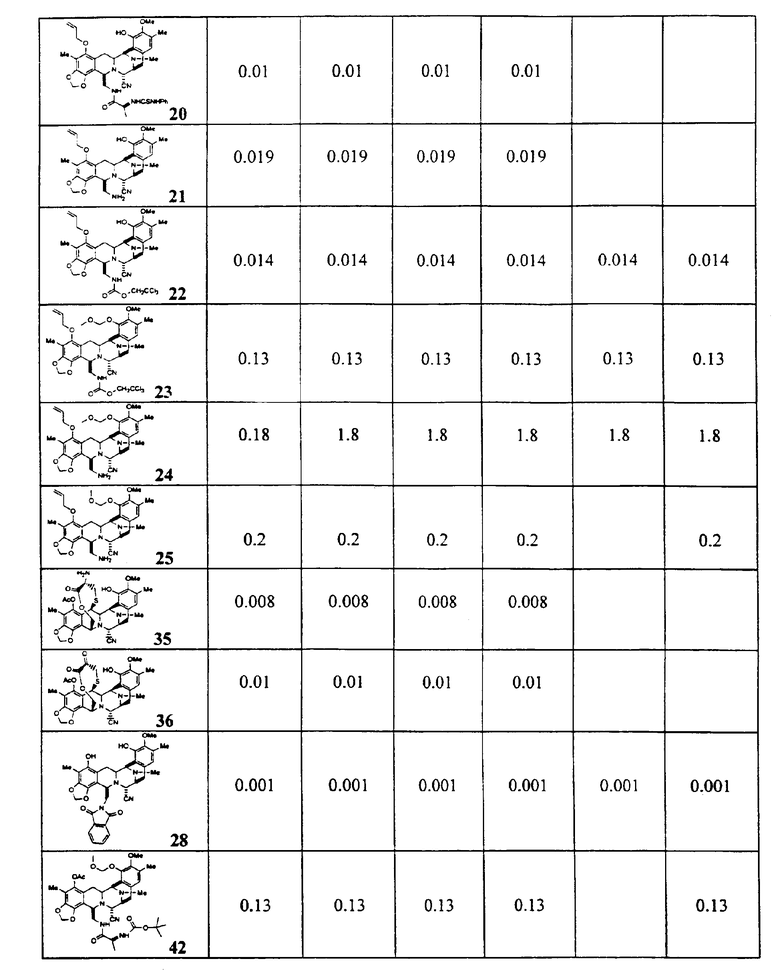
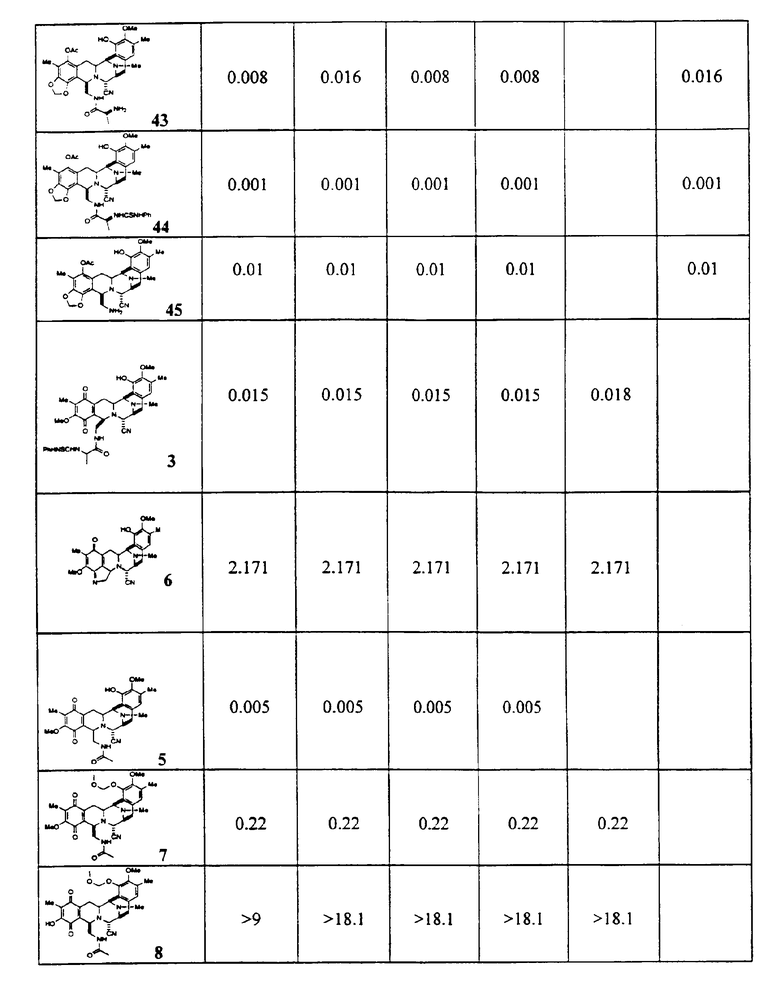

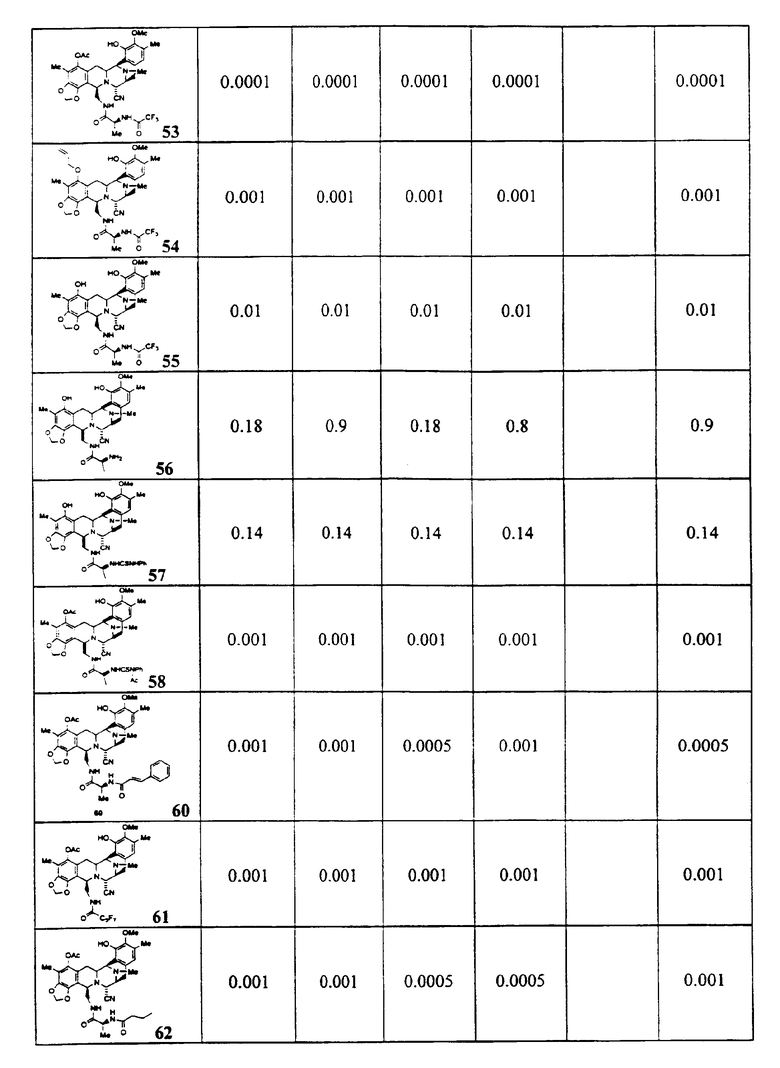

Из этих данных, характеризующих активность, и других анализов может быть видно, что активные соединения, являющиеся предметом настоящего изобретения, включают предпочтительный класс соединений общей формулы (XXIII):
в которой R1 является таким, как определено выше для группы (XVIIb) и предпочтительно представляет производное аминометиленовой группы среднего размера;
R5 является таким, как определено выше для группы (XVIIb) и предпочтительно представляет производное гидроксигруппы небольшого размера;
R12 является таким, как определено выше и предпочтительно представляет -CH3 и
R21 представляет гидрокси или цианогруппу.
R1 представляет подходящую гидрофобную группу, в которой, таким образом, отсутствуют свободные амино, гидрокси и другие гидрофильные функциональные группы. Обычно R1 представляет группу -CH2-NH-CO-Ra, в которой Ra является таким, как определено, но предпочтительно имеет линейную цепь длиной менее, чем 20 атомов, более предпочтительно, менее чем 15 или 10 атомов, где 1,4-фенил определяется как цепь длиной в четыре атома, и аналогичные рассуждения применяются к другим циклическим группам (например, 1,2-циклогексил представляет цепь длиной в два атома), и линейная цепь длиной менее чем 10, 15 или 20 атомов сама может быть замещенной. В частности, данные дают возможность предположить, что должно быть достигнуто некоторое равновесие между отсутствием такой группы, как Ra-CO- и присутствием большой, объемной группы.
В одном из вариантов является предпочтительным, чтобы группа R1 не содержала циклических групп, особенно, ароматических групп. В родственных вариантах настоящее изобретение не относится к получению соединений, которые описаны в статье, опубликованной в: Proc. Natl. Acad. Sci. USA, 96, 3496-3501, 1999, которая включена посредством ссылки. Другие предпочтительные группы для R1, в соответствии с настоящим изобретением, исключают соответствующие заместители CH2R2, приведенные в таблице 1 указанной статьи, в особенности группы А, В, С и D для R2.
R5 предпочтительно представляет ацетильную группу.
В особенно предпочтительных соединениях группа R1 ацилирована по -NH2-группе, и например, N-ацилпроизводные могут быть образованы из групп -CH2-NH2 и -CH2-NH-аа. Ацилпроизводные могут представлять собой их N-ацил или N-тиоацилпроизводные. Ацильные группы могут быть формулы -СО-Ra, где Ra является таким, как определено и выбирается таким образом, чтобы удовлетворять указанным критериям. Подходящие ацильные группы включают аланил, аргинил, аспартил, аспарагил, цистил, глутамил, глутаминил, глицил, гистидил, гидроксипропил, изолейцил, лейцил, лизил, метионил, фенилаланил, пролил, серил, треонил, тиронил, триптофил, тирозил, валил, а также другие ацильные группы аминокислот. Такие ацильные группы аминокислот предпочтительно модифицированы по аминогруппе для достижения гидрофобности.
В одном из вариантов группа R1 представляет модифицированную гидроксиметиленовую группу. Применимы рассуждения, аналогичные рассуждениям относительно модификации аминометиленовой группы.
В свете активности данных соединений важным способом согласно настоящему изобретению является следующий:
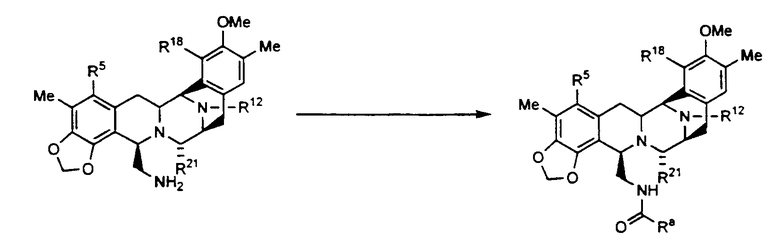
где R5 в конечном продукте является таким, как определено для соединения (XXIII), и может быть другим в исходном веществе и образоваться в результате превращения, составляющего часть способа,
R18 представляет в конечном продукте гидроксигруппу, но может являться защищенной гидроксигруппой в исходном веществе и образоваться в результате превращения, составляющего часть способа,
R12 в конечном продукте может быть таким же, как и в исходном веществе, или может образоваться в результате превращения, составляющего часть способа,
R21 в конечном продукте является таким, как определено, и если является гидроксигруппой, то может образоваться из цианогруппы в результате превращения, составляющего часть способа,
Ra является таким, как определено, и может быть дополнительно ацилирован, что составит часть способа получения конечного продукта с ацилированными группами Ra, как обсуждалось.
R5 в исходном веществе предпочтительно представляет ацетильную или другую небольшую ацильную группу и не изменяется при проведении реакции. R18 в исходном веществе предпочтительно представляет гидроксигруппу и не изменяется при проведении реакции. R12 предпочтительно представляет -CH3 в исходном веществе и не изменяется при проведении реакции. R21 в конечном продукте является такой, как определено, и в том случае, если является гидроксигруппой, то может образоваться из цианогруппы в результате превращения, составляющего часть способа. Ra в конечном продукте является предпочтительно такой, как определено для соединения формулы (XXXIII).
Другой важный способ настоящего изобретения включает реакцию:

Другой важный способ настоящего изобретения включает реакцию:

Другой важный способ настоящего изобретения включает реакцию, согласно которой группа R1, представляющая собой аминометиленовую группу, превращается в гидроксиметиленовую группу.
Другой важный способ настоящего изобретения включает реакцию, согласно которой соединение, содержащее группу R1, представляющую собой гидроксиметиленовую группу, взаимодействует с реагентом формулы (XIX)
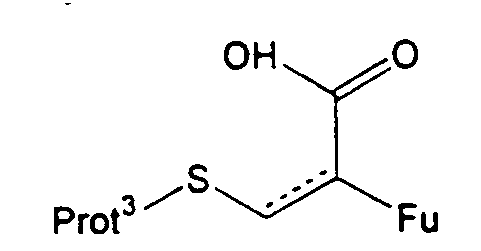
в которой Fu означает защищенную функциональную группу, Prot3 представляет защитную группу, а пунктирная линия показывает необязательную двойную связь
Другой важный способ настоящего изобретения включает реакцию получения 21-цианосоединения формулы (XVI), которая включает взаимодействие соединения формулы (XV):

в которой R1, R5, R8, R14a, R15 и R18 являются такими, как определено, и R21 представляет гидроксигруппу, с источником цианид-иона с получением при этом необходимого 21-цианосоединения.
В дополнение также предусматриваются способы с использованием других нуклеофилсодержащих соединений, для того, чтобы получить соединения, аналогичные соединениям формулы (XVI), в которых 21-положение защищено другой нуклеофильной группой, 21-Nuc группой. Например, 21-Nuc соединение формулы (XVI) с алкиламинозаместителем в 21-положении может быть получено взаимодействием соединения формулы (XV), в котором R21 представляет гидроксигруппу, с подходящим алкиламином. 21-Nuc соединение формулы (XVI) с алкилтиозаместителем в 21-положении может быть получено взаимодействием соединения формулы (XV), в котором R21 представляет гидроксигруппу, с подходящим алкантиолом. В качестве альтернативы, 21-Nuc соединение формулы (XVI) с α-карбонилалкилзаместителем в 21-положении может быть получено взаимодействием соединения формулы (XV), в котором R21 представляет гидроксигруппу, с подходящим карбонильным соединением, как правило, в присутствии основания. Для получения других 21-Nuc соединений доступны другие методики синтеза.
Другая важная реакция настоящего изобретения включает обработку продукта - 21-цианосоединения, являющегося предметом настоящего изобретения, для образования 21-гидроксисоединения. Такие соединения обладают интересными свойствами in vivo.
Примеры
Пример 1

К раствору соединения 2 (21,53 г, 39,17 ммоль) в этаноле (200 ммоль) добавляют трет-бутоксикарбонилангидрид (7,7 г, 35,25 ммоль) и перемешивают смесь в течение 7 час при 23°С. Затем реакционную смесь концентрируют в вакууме и полученный остаток очищают колоночной флэш-хроматографией (SiO2, гексан:этилацетат, 6:4), получая при этом соединение 14 (20,6 г, 81%) в виде желтого твердого вещества.
Rf: 0,52 (этилацетат : CHCl3, 5:2)
1H ЯМР (300 МГц, CDCl3): δ 6,49 (с, 1H), 6,32 (шир.с, 1H), 5,26 (шир.с, 1H), 4,60 (шир.с, 1H), 4,14 (д, J=2,4 Гц, 1H), 4,05 (д, J=2,4 Гц, 1H), 3,94 (с, 3H), 3,81 (д, J=4,8 Гц, 1H), 3,7 (с, 3H), 3,34 (шир. д, J=7,2 Гц, 1H), 3,18-3,00 (м, 5H), 2,44 (д, J=18,3 Гц, 1H), 2,29 (с, 3H), 2,24 (с, 3H), 1,82 (с, 3H), 1,80-1,65 (м, 1H), 1,48 (с, 9H), 0,86 (д, J=5,1 Гц, 3H)
13C ЯМР (75 МГц, CDCl3): δ 185,5, 180,8, 172,7, 155,9, 154,5, 147,3, 143,3, 141,5, 135,3, 130,4, 129,2, 127,5, 120,2, 117,4, 116,9, 80,2, 60,7, 60,3, 58,5, 55,9, 55,8, 54,9, 54,4, 50,0, 41,6, 40,3, 28,0, 25,3, 24,0, 18,1, 15,6, 8,5.
ESI-масс-спектроскопия, m/z: вычислено для C34H43N5O8: 649,7. Найдено (М+Н)+: 650,3
Пример 2

К перемешиваемому раствору соединения 14 (20,6 г, 31,75 ммоль) в СН3CN (159 мл), добавляют при 0°С диизопропилэтиламин (82,96 мл, 476,2 ммоль), метокси-метиленбромид (25,9 мл, 317,5 ммоль) и диметиламинопиридин (155 мг, 1,27 ммоль). Полученную смесь перемешивают при 23°С в течение 24 час. Реакцию гасят добавлением при 0°C водного 0,1 N раствора HCl (750 мл) (рН=5), и экстрагируют, используя СН2Cl2 (2 х 400 мл). Органическую фазу сушат (сульфат натрия) и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией (SiO2, градиент от гексан:этилацетат, 4:1, до гексан:этилацетат, 3:2), получая при этом соединение 15 (17,6 г, 83%) в виде желтого твердого вещества.
Rf: 0,38 (гексан: этилацетат, 3:7)
1H ЯМР (300 МГц, CDCl3): δ 6,73 (с, 1H), 5,35 (шир.с, 1H), 5,13 (с, 2H), 4,50 (шир.с, 1H), 4,25 (д, J=2,7 Гц, 1H), 4,03 (д, J=2,7 Гц, 1H), 3,97 (с, 3H), 3,84 (шир.с, 1H), 3,82-3,65 (м, 1H), 3,69 (с, 3H), 3,56 (с, 3H), 3,39-3,37 (м, 1H), 3,20-3,00 (м, 5H), 2,46 (д, J=18 Гц, 1H), 2,33 (с, 3H), 2,23 (с, 3H), 1,85 (с, 3H), 1,73-1,63 (м, 1H), 1,29 (с, 9H), 0,93 (д, J=5,1 Гц, 3H)
13C ЯМР (75 МГц, CDCl3): δ 185,4, 180,9, 172,4, 155,9, 154,5, 149,0, 148,4, 141,6, 135,1, 131,0, 129,9, 127,6, 124,4, 123,7, 117,3,99,1, 79,3, 60,7, 59,7, 58,4, 57,5, 56,2, 55,9, 55,0, 54,2, 50,0, 41,5, 39,9, 28,0, 25,2, 24,0, 18,1, 15,6, 8,5,
ESI-масс-спектроскопия m/z: вычислено для C36H47N5O9: 693,8. Найдено (М+Н)+: 694,3
Пример 3
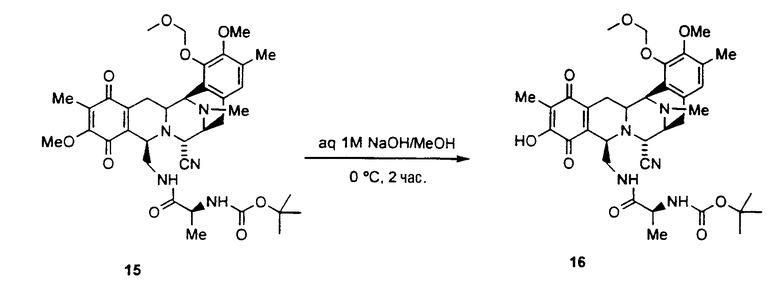
В колбу, в которой находится соединение 15 (8 г, 11,5 ммоль) в метаноле (1,6 л), при 0°С добавляют водный раствор 1 М гидроксида натрия (3,2 л). Реакционную смесь перемешивают в течение 2 час при этой температуре и затем гасят реакцию добавлением 6 М раствора HCl до рН=5. Смесь экстрагируют этилацетатом (3 х 1 л) и объединенные органические слои сушат над сульфатом натрия и концентрируют в вакууме. Полученный остаток очищают колоночной флэш-хроматографией (SiO2, градиент от CHCl3 до смеси гексан:этилацетат, 2:1), получая при этом соединение 16 (5,3 мг, 68%).
Rf: 0,48 (CH3CN: H2O, 7:3, RP-C18)
1H ЯМР (300 МГц, CDCl3): δ 6,73 (с, 1H), 5,43 (шир.с, 1H), 5,16 (с, 2H), 4,54 (шир.с, 1H), 4,26 (д, J=1,8 Гц, 1H), 4,04 (д, J=2,7 Гц 1H), 3,84 (шир.с, 1H), 3,80-3,64 (м, 1H), 3,58 (с, 3H), 3,41-3,39 (м, 1H), 3,22-3,06 (м, 5H), 2,49 (д, J=18,6 Гц 1H), 2,35 (с, 3H), 2,30-2,25 (м, 1H), 2,24 (с, 3H), 1,87 (с, 3H), 1,45-1,33 (м, 1H), 1,19 (с, 9H), 1,00 (шир. д, J=6,6 Гц, 3H)
13C ЯМР (75 МГц, CDCl3): δ 184,9, 180,9, 172,6, 154,7, 151,3, 149,1, 148,6, 144,7, 132,9, 131,3, 129,8, 124,5, 123,7, 117,3, 116,8, 99,1, 79,4, 59,8, 58,6, 57,7, 56,2, 55,6, 54,9, 54,5, 50,1, 41,6, 40,1, 28,0, 25,3, 24,4, 18,1, 15,7, 8,0.
ESI-масс-спектроскопия m/z: вычислено для C35H45N5O9: 679,7. Найдено (М+Н)+: 680,3
Пример 4

К дегазированному раствору соединения 16 (1,8 г, 2,64 ммоль) в DMF (221 мл) добавляют 10% Pd/C (360 мг) и перемешивают в атмосфере Н2 (атмосферное давление) в течение 45 мин. Реакционную смесь отфильтровывают через целит в атмосфере аргона в колбу, содержащую безводный Cs2CO3 (2,58 г, 7,92 ммоль). Затем добавляют бромхлорметан (3,40 мл, 52,8 ммоль), горло колбы герметично закрывают и перемешивают при 100°С в течение 2 час. Реакционную смесь охлаждают, отфильтровывают через слой целита и промывают, используя CH2Cl2. Органический слой концентрируют и сушат (сульфат натрия), получая при этом соединение 17 в виде коричневого масла, которое используют на следующей стадии без дополнительной очистки.
Rf: 0,36 (гексан :этилацетат 1:5, SiO2)
1H ЯМР (300 МГц, CDCl3): δ 6,68 (с, 1H), 6,05 (шир.с, 1H), 5,90 (с, 1H), 5,79 (с, 1H), 5,40 (шир.с, 1H), 5,31-5,24 (м, 2H), 4,67 (д, J=8,1 Гц, 1H), 4,19 (д, J=2,7 Гц, 1H), 4,07 (шир.с, 1H), 4,01 (шир.с, 1H), 3,70 (с, 3H), 3,67 (с, 3H), 3,64-2,96 (м, 5H), 2,65 (д, J=18,3 Гц, 1H), 2,33 (с, 3H), 2,21 (с, 3H), 2,04 (с, 3H), 2,01-1,95 (м, 1H), 1,28 (с, 9H), 0,87 (д, J=6,3 Гц, 3H)
13C ЯМР (75 МГц, CDCl3): δ 172,1, 162,6, 154,9, 149,1, 145,7, 135,9, 130,8, 130,7, 125,1, 123,1, 117,8, 100,8, 99,8, 76,6, 59,8, 59,2, 57,7, 57,0, 56,7, 55,8, 55,2, 49,5, 41,6, 40,1, 36,5, 31,9, 31,6, 29,7, 28,2, 26,3, 25,0, 22,6, 18,2, 15,8, 14,1, 8,8.
ESI-масс-спектроскопия m/z: вычислено для C36H47N5O9: 693,34. Найдено (М+Н)+: 694,3
Пример 5

В колбу, содержащую раствор соединения 17 (1,83 г, 2,65 ммоль), в DMF (13 мл), Cs2CO3 (2,6 г, 7,97 ммоль) при 0°С добавляют аллилбромид (1,15 мл, 13,28 ммоль). Полученную смесь перемешивают при 23°С в течение 1 час. Реакционную смесь отфильтровывают через слой целита и промывают, используя CH2Cl2. Органический слой концентрируют и сушат (сульфат натрия). Полученный остаток очищают колоночной флэш-хроматографией (SiO2, CHCl3:этилацетат, 1:4), получая при этом соединение 18 (1,08 мг, 56%) в виде белого твердого вещества.
Rf: 0,36 (CHCl3:этилацетат, 1:3)
1H ЯМР (300 МГц, CDCl3): δ 6,70 (с, 1H), 6,27-6,02 (м, 1H), 5,94 (с, 1H), 5,83 (с, 1H), 5,37 (дд, J1=1,01 Гц, J2=16,8 Гц, lH), 5,40 (шир.с, 1H), 5,25 (дд, J1=1,0 Гц, J2=10,5 Гц, 1H), 5,10 (с, 2H), 4,91 (шир.с, 1H), 4,25-4,22 (м, 1H), 4,21 (д, J=2,4 Гц, 1H), 4,14-4,10 (м, 1H), 4,08 (д, J=2,4 Гц, 1H), 4,00 (шир.с, 1H), 3,70 (с, 3H), 3,59 (с, 3H), 3,56-3,35 (м, 2H), 3,26-3,20 (м, 2H), 3,05-2,96 (дд, J1=8,1 Гц, J2=18 Гц, 1H), 2,63 (д, J =18 Гц, 1H), 2,30 (с, 3H), 2,21 (с, 3H), 2,09 (с, 3H), 1,91-1,80 (м, 1H), 1,24 (с, 9H), 0,94 (д, J=6,6 Гц, 3H)
13С ЯМР (75 МГц, CDCl3): δ 172,0, 154,8, 148,8, 148,6, 148,4, 144,4, 138,8, 133,7, 130,9, 130,3, 125,1, 124,0, 120,9, 117,8, 117,4, 112,8, 112,6, 101,1, 99,2, 73,9, 59,7, 59,3, 57,7, 56,9, 56,8, 56,2, 55,2, 40,1, 34,6, 31,5, 28,1, 26,4, 25,1, 22,6, 18,5, 15,7, 14,0, 9,2.
ESI-масс-спектроскопия m/z: вычислено для C39H51N5O9: 733,4. Найдено (М+Н)+: 734,4
Пример 6

К раствору соединения 18 (0,1 г, 0,137 ммоль) в диоксане (2 мл) добавляют 4,2 М HCl/диоксан (1,46 мл) и полученную смесь перемешивают в течение 1,2 час при 23°С. Реакцию гасят при 0°С добавлением насыщенного водного раствора бикарбоната натрия (60 мл) и экстрагируют этилацетатом (2х70 мл). Органические слои сушат (сульфат натрия) и концентрируют в вакууме, получая при этом соединение 19 (267 мг, 95%), в виде белого твердого вещества, которое используют в последующих реакциях без дополнительной очистки.
Rf: 0,17 (этилацетат:метанол 10:1, SiO2)
1H ЯМР (300 МГц, CDCl3): δ 6,49 (с, 1H), 6,12-6,00 (м, 1H), 5,94 (с, 1H), 5,86 (с, 1H), 5,34 (дд, J=1,0 Гц, J=17,4 Гц, 1H), 5,25 (дд, J=1,0 Гц, J=10,2 Гц, 1H), 4,18-3,76 (м, 5H), 3,74 (с, 3H), 3,71-3,59 (м, 1H), 3,36-3,20 (м, 4H), 3,01-2,90 (м, 1H), 2,60 (д, J=18,0 Гц, 1H), 2,29 (с, 3H), 2,24 (с, 3H), 2,11 (с, 3H), 1,97-1,86 (м, 1H), 0,93 (д, J=8,7 Гц, 3H)
13C ЯМР (75 МГц, CDCl3): δ 175,5, 148,4, 146,7, 144,4, 142,4, 138,9, 133,7, 131,3, 128,3, 120,8, 117,9, 117,4, 113,8, 112,4, 101,1, 74,2, 60,5, 59,1, 56,5, 56,1, 56,3, 56,0, 55,0, 50,5, 41,6, 39,5, 29,5, 26,4, 24,9, 21,1, 15,5, 9,33.
ESI-масс-спектроскопия, m/z: вычислено для C32H39N5O6: 589. Найдено (М+Н)+: 590.
Пример 7

К раствору соединения 19 (250 мг, 0,42 ммоль) в CH2Cl2 (1,5 мл) добавляют фенилизотиоцианат (0,3 мл, 2,51 ммоль) и полученную смесь перемешивают при 23°С в течение 1 час. Реакционную смесь концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией (SiO2, градиент от гексана до смеси гексан:этилацетат, 5:1), получая при этом соединение 20 (270 мг, 87%) в виде белого твердого вещества.
Rf: 0,56 (CHCl3: этилацетат, 1:4)
1H ЯМР (300 МГц, CDCl3): δ 8,00 (шир.с, 1H), 7,45-6,97 (м, 4H), 6,10 (с, 1H), 6,08-6,00 (м, 1H), 5,92 (с, 1H), 5,89 (с, 1H), 5,82 (с, 1H), 5,40 (дд, J=1,5 Гц, J=17,1 Гц, 1H), 3,38 (шир.с, 1H), 5,23 (дд, J=1,5 Гц, J=10,5 Гц, 1H), 4,42-4,36 (м, 1H), 4,19-4,03 (м, 5H), 3,71 (с, 3H), 3,68-3,17 (м, 4H), 2,90 (дд, J =7,8 Гц, J=18,3 Гц, 1H), 2,57 (д, J=18,3 Гц, 1H), 2,25 (с, 3H), 2,12 (с, 3H), 2,10 (с, 3H), 1,90 (дд, J=12,3 Гц, J=16,5 Гц, 1H), 0,81 (д, J=6,9 Гц, 3H).
13С ЯМР (75 МГц, CDCl3): δ 178,4, 171,6, 148,6, 146,8, 144,3, 142,7, 138,7, 136,2, 133,6, 130,7, 129,8, 126,6, 124,2, 124,1, 120,9, 120,5, 117,7, 117,4, 116,7, 112,6, 112,5, 101,0, 74,0, 60,6, 59,0, 57,0, 56,2, 56,1, 55,0, 53,3, 41,4, 39,7, 26,3, 24,8, 18,3, 15,5, 9,2.
ESI-масс-спектроскопия m/z: вычислено для C39H44N6O6S: 724,8. Найдено (М+Н)+: 725,3
Пример 8
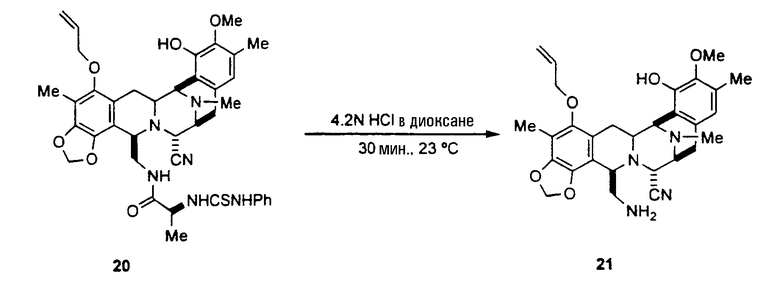
К раствору соединения 20 (270 мг, 0,37 ммоль) в диоксане (1 мл) добавляют 4,2 М HCl/диоксан (3,5 мл) и реакционную смесь перемешивают в течение 30 мин при 23°С. Затем добавляют этилацетат (20 мл) и Н2О (20 мл) и декантируют органический слой. Водную фазу подщелачивают водным раствором бикарбоната натрия (60 мл) (рН=8) при 0°С и затем экстрагируют, используя CH2Cl2 (2 х 50 мл). Объединенные органические экстракты сушат (сульфат натрия) и концентрируют в вакууме. Полученный остаток очищают колоночной флэш-хроматографией (SiO2, этилацетат:метанол, 5:1), получая при этом соединение 21 (158 мг, 82%) в виде белого твердого вещества.
Rf: 0,3 (этилацетат:метанол, 1:1)
1H ЯМР (300 МГц, CDCl3): δ 6,45 (с, 1H), 6,12-6,03 (м, 1H), 5,91 (с, 1H), 5,85 (с, 1H), 5,38 (дд, J1=1,2 Гц, J2=17,1 Гц, 1H), 5,24 (дд, J1=1,2 Гц, J2=10,5 Гц, 1H), 4,23-4,09 (м, 4H), 3,98 (д, J=2,1 Гц, 1H), 3,90 (шир.с, 1H), 3,72 (с, 3H), 3,36-3,02 (м, 5H), 2,72-2,71 (м, 2H), 2,48 (д, J=18,0 Гц, 1H), 2,33 (с, 3H), 2,22 (с, 3H), 2,11 (с, 3H), 1,85 (дд, J,= 11,7 Гц, J=15,6 Гц, 1H).
13C ЯМР (75 МГц, CDCl3): δ 148,4, 146,7, 144,4, 142,8, 138,8, 133,8, 130,5, 128,8, 121,5, 120,8, 118,0, 117,5, 116,9, 113,6, 112,2, 101,1, 74,3, 60,7, 59,9, 58,8, 56,6, 56,5, 55,3, 44,2, 41,8, 29,7, 26,5, 25,7, 15,7, 9,4.
ESI-масс-спектроскопия m/z: вычислено для C29H34N4O5: 518,3. Найдено (М+Н)+: 519,2.
Пример 9

К раствору соединения 21 (0,64 г, 1,22 ммоль) в CH2Cl2 (6,13 мл) при -10°С добавляют пиридин (0,104 мл, 1,28 ммоль) и 2,2,2-трихлорэтилхлорформиат (0,177 мл, 1,28 ммоль). Смесь перемешивают при этой температуре в течение 1 час, после чего реакцию гасят добавлением 0,1 N раствора HCl (10 мл) и экстрагируют, используя CH2Cl2 (2х10 мл). Органический слой сушат над сульфатом натрия и концентрируют в вакууме. Полученный остаток очищают колоночной флэш-хроматографией (SiO2, гексан:этилацетат, 1:2), получая при этом соединение 22 (0,84 г, 98%) в виде белого вспененного твердого вещества.
Rf: 0,57 (этилацетат: метанол, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 6,50 (с, 1H), 6,10-6,00 (м, 1H), 6,94 (д, J=1,5 Гц, 1H), 5,87 (д, J=1,5 Гц, 1H), 5,73 (шир.с, 1H), 5,37 (д.кв., J1=1,5 Гц, J2=17,1 Гц, 1H), 5,26 (д.кв., J1=1,8 Гц, J2=10,2 Гц, 1H), 4,60 (д, J=12 Гц, 1H), 4,22-4,10 (м, 4H), 4,19 (д, J=12 Гц, 1H), 4,02 (м, 2H), 3,75 (с, 3Н), 3,37-3,18 (м, 5Н), 3,04 (дд, J1=8,1 Гц, J2=18 Гц, 1H), 2,63 (д, J=18Гц, 1H), 2,31 (с, 3H), 2,26 (с, 3H), 2,11 (с, 3H), 1,85 (дд, J1=12,3 Гц, J2=15,9 Гц, 1H).
13С ЯМР (75 МГц, CDCl3): δ 154,3, 148,5, 146,7, 144,5, 142,8, 139,0, 133,8, 130,7, 128,7, 121,3, 120,8, 117,8, 117,7, 116,8, 112,7, 101,2, 77,2, 74,3, 60,7, 59,9, 57,0, 56,4, 55,3, 43,3, 41,7, 31,6, 26,4, 25,3, 22,6, 15,9, 14,1, 9,4.
ESI-масс-спектроскопия m/z: вычислено для C32H35Cl3N4O7 694,17. Найдено (М+Н)+: 695,2.
Пример 10

К раствору соединения 22 (0,32 г, 0,46 ммоль) в CH3CN (2,33 мл) при 0°С добавляют диизопропилэтиламин (1,62 мл, 9,34 ммоль), простой бромметилметиловый эфир (0,57 мл, 7,0 ммоль) и диметиламинопиридин (6 мг, 0,046 ммоль). Смесь нагревают при 30°С в течение 10 час. Затем реакционную смесь разбавляют дихлорметаном (30 мл) и выливают в водный раствор HCl при рН=5 (10 мл). Органический слой сушат над сульфатом натрия, растворитель удаляют при пониженном давлении, получая при этом остаток, который очищают колоночной флэш-хроматографией (SiO2, гексан:этилацетат, 2:1), получая при этом соединение 23 (0,304 г, 88%) в виде белого вспененного твердого вещества.
Rf: 0,62 (гексан:этилацетат, 1:3)
1H ЯМР (300 МГц, CDCl3): δ 6,73 (с, 1H), 6,10 (м, 1H), 5,94 (д, J=1,5 Гц, 1H), 5,88 (д, J=1,5 Гц, 1H), 5,39 (д.кв., J1=1,5 Гц, J2=17,1 Гц, 1H), 5,26 (д.кв., J1=1,8 Гц, J2=10,2 Гц, 1H), 5,12 (с, 2H), 4,61 (д, J=12 Гц, 1H), 4,55 (т, J=6,6 Гц, 1H), 4,25 (д, J=12 Гц, 1H), 4,22-4,11 (м, 4H), 4,03 (м, 2H), 3,72 (с, 3H), 3,58 (с, 3H), 3,38-3,21 (м, 5H), 3,05 (дд, J1=8,1 Гц, J2=18 Гц, 1H), 2,65 (д, J=18 Гц, 1H), 2,32 (с, 3H), 2,23 (с, 3H), 2,12 (с, 3H), 1,79 (дд, J1=12,3 Гц, J2=15,9 Гц, 1H).
13С ЯМР (75 МГц, CDCl3): δ 154,3, 148,6, 148,4, 144,5, 139,0, 133,6, 130,6, 130,1, 125,07, 124,7, 124,0, 121,1, 117,7, 112,6, 101,2, 99,2, 77,2, 74,4, 74,1, 59,8, 59,8, 57,7, 57,0, 56,8, 56,68, 55,3, 43,2, 41,5, 26,4, 25,2, 15,9, 9,3.
ESI-масс-спектроскопия m/z: вычислено для C34H39Cl3N4O8 738,20. Найдено (М+Н)+: 739,0.
Пример 11

К суспензии соединения 23 (0,304 г, 0,41 ммоль) в 90%-ной водной уксусной кислоте (4 мл) добавляют порошок цинка (0,2 г, 6,67 ммоль) и реакционную смесь перемешивают при 23°С в течение 7 час. Смесь отфильтровывают через слой целита, который промывают, используя CH2Cl2. Органический слой промывают насыщенным водным раствором бикарбоната натрия (рН=9) (15 мл) и сушат над сульфатом натрия. Растворитель удаляют при пониженном давлении, получая при этом соединение 24 (0,191 г, 83%) в виде белого твердого вещества.
Rf: 0,3 (этилацетат:метанол, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 6,68 (с, 1H), 6,09 (м, 1H), 5,90 (д, J=1,5 Гц, 1H), 5,83 (д, J=1,5 Гц, 1H), 5,39 (д.кв., J1=1,5 Гц, J2=17,1 Гц, 1H), 5,25 (д.кв., J1=1,5 Гц, J2=10,2 Гц, 1H), 5,10 (с, 2H), 4,22-4,09 (м, 3Н), 3,98 (д, J=2,4 Гц, 1H), 3,89 (м, 1H), 3,69 (с, 3Н), 3,57 (с, 3Н), 3,37-3,17 (м, 3Н), 3,07 (дд, J1=8,1 Гц, J2=18 Гц, 1H), 2,71 (м, 2H), 2,48 (д, J=18 Гц, 1H), 2,33 (с, 3Н), 2,19 (с, 3Н), 2,17 (с, 3Н), 1,80 (дд, J1=12,3 Гц, J2=15,9 Гц, 1H)
13С ЯМР (75 МГц, CDCl3): δ 148,5, 148,2, 144,3, 138,7, 133,7, 130,7, 129,9, 125,0, 123,9, 121,3, 117,9, 117,5, 113,6, 112,0, 101,0, 99,2, 74,0, 59,8, 59,7, 58,8, 57,6, 57,0, 56,2, 55,2, 44,2, 41,5, 31,5, 26,4, 25,6, 22,5, 16,7, 14,0, 9,2.
ESI-масс-спектроскопия m/z: вычислено для C31H38N4O6: 562,66. Найдено (М+Н)+: 563,1
Пример 12

К раствору соединения 24 (20 мг, 0,035 ммоль) в H2О (0,7 мл) при 0°С добавляют ТГФ (0,7 мл), NaNO2 (12 мг, 0,17 ммоль) и 90%-ный водный раствор AcOH (0,06 мл) и перемешивают смесь при 0°С в течение 3 час. После разбавления с использованием CH2Cl2 (5 мл) органический слой промывают водой (1 мл), сушат над сульфатом натрия, и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией (SiO2, гексан:этилацетат, 2:1), получая при этом соединение 25 (9,8 мг, 50%) в виде белого твердого вещества.
Rf: 0,34 (гексан: этилацетат, 1:1)
1H ЯМР (300 МГц, CDCl3): δ 6,71 (с, 1H), 6,11 (м, 1H), 5,92 (д, J=1,5 Гц, 1H), 5,87 (д, J=1,5 Гц, 1H), 5,42 (д.кв., J1=1,5 Гц, J2=17,1 Гц, 1H), 5,28 (д.кв., J1=1,5 Гц, J2=10,2 Гц, 1H), 5,12 (с, 2H), 4,26-4,09 (м, 3Н), 4,05 (д, J=2,4 Гц, 1H), 3,97 (т, J=3,0 Гц, 1H), 3,70 (с, 3Н), 3,67-3,32 (м, 4H), 3,58 (с, 3Н), 3,24 (дд, J1=2,7 Гц, J2=15,9 Гц, 1H), 3,12 (дд, J1=8,1 Гц, J2=18,0 Гц, 1H), 2,51 (д, J=18 Гц, 1H), 2,36 (с, 3Н), 2,21 (с, 3Н), 2,12 (с, 3Н), 1,83 (дд, J,= 12,3 Гц, J=15,9 Гц, 1H)
13C ЯМР (75 МГц, CDCl3): δ 148,7, 148,4, 138,9, 133,7, 131,1, 129,4, 125,1, 123,9, 120,7, 117,6, 117,5, 113,2, 112,3, 101,1, 99,2, 74,0, 63,2, 59,8, 59,7, 57,9, 57,7, 57,0, 56,5, 55,2, 41,6, 29,6, 26,1, 25,6, 22,6, 15,7, 9,2.
ESI-масс-спектроскопия, m/z: вычислено для C31H37N3O7 536,64. Найдено (М+Н)+: 564,1.
Пример 13

Исходное вещество (2,0 г, 5,90 ммоль) добавляют к суспензии гидрида натрия (354 мг, 8,86 ммоль) в ТГФ (40 мл) при 23°С, с последующей обработкой суспензии аллилхлорформиатом (1,135 мл, 8,25 ммоль) при 23°С и кипячением с обратным холодильником в течение 3 час. Суспензию охлаждают, отфильтровывают, твердое вещество промывают этилацетатом (100 мл) и концентрируют фильтрат. Неочищенное масло обрабатывают гексаном (100 мл) и выдерживают при 4°С в течение ночи. После этого растворитель декантируют и полученную светло-желтую суспензию обрабатывают, используя CH2Cl2 (20 мл), и осаждают гексаном (100 мл). Через 10 мин растворитель опять декантируют. Операцию повторяют до тех пор, пока не появится белое твердое вещество. Полученное белое твердое вещество отфильтровывают и сушат, получая при этом соединение 29 (1,80 г, 65%) в виде белого твердого вещества.
1H-ЯМР (300 МГц, CDCl3): δ 7,74 (д, J=7,5 Гц, 2H), 7,62 (д, J=6,9 Гц, 2H), 7,33 (т, J=7,5 Гц, 2H), 7,30 (т, J=6,3 Гц, 2H), 5,71 (д, J=7,8 Гц, 1H), 4,73 (д, J=7,8 Гц, 2H), 4,59 (м, 1H), 4,11 (т, J=6,0 Гц, 1H), 3,17 (дд, J=6,0 Гц, J=2,7 Гц, 2H), 3,20 (дд, J=5,4 Гц, J=2,1Гц, 2H).
13C-ЯМР (75 МГц, CDCl3): δ 173,6, 152,7, 144,0, 139,7, 137,8, 126,0, 125,6, 123,4, 118,3, 73,4, 52,4, 45,5, 35,8, 33,7.
ESI-масс-спектроскопия, m/z: вычислено для C20H18Cl3NO4S: 474,8. Найдено (М+Н)+: 564,1.
Пример 14
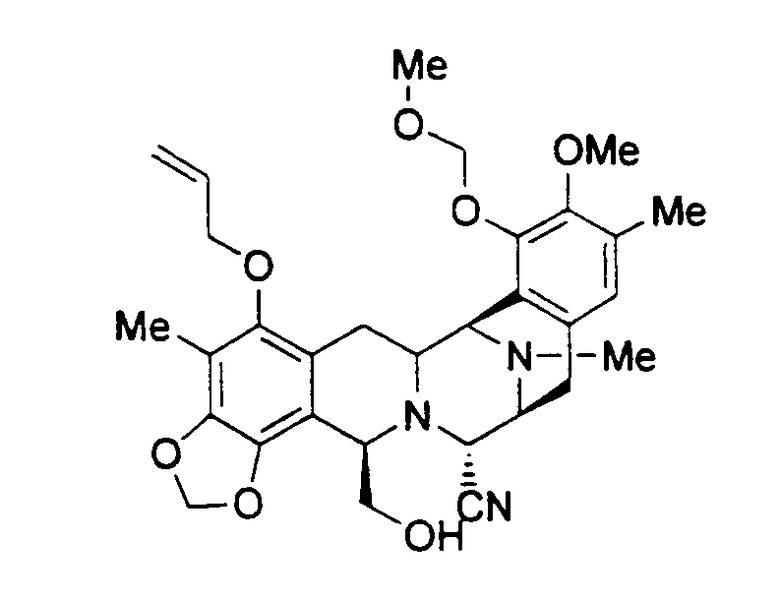
Смесь соединения 25 (585 мг, 1,03 ммоль) и соединения 29 (1,47 г, 3,11 ммоль) подвергают азеотропной перегонке с безводным толуолом (3 х 10 мл). К раствору соединения 25 и соединения 29 в безводном CH2Cl2 (40 мл) добавляют DMAP (633 мг, 5,18 ммоль) и EDC·HCl (994 мг, 5,18 ммоль) при 23°С. Реакционную смесь перемешивают при 23°С в течение 3 час. Полученную смесь распределяют насыщенным раствором бикарбоната натрия (50 мл) и разделяют слои. Водный слой промывают, используя CH2Cl2 (50 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют. Неочищенное вещество очищают колоночной флэш-хроматографией (этилацетат:гексан, 1:3), получая при этом соединение 30 (1,00 г, 95%) в виде светлого желтовато-кремового твердого вещества.
1H-ЯМР (300 МГц, CDCl3): δ 7,72 (м, 2H), 7,52 (м, 2H), 7,38 (м, 2H), 7,28 (м, 2H), 6,65 (с, 1H), 6,03 (м, 1H), 5,92 (д, J=1,5 Гц, 1H), 5,79 (д, J=1,5 Гц, 1H), 5,39 (м, 1H), 5,29 (д.кв., J=10,3 Гц, J=1,5 Гц, 1H), 5,10 (с, 2H), 4,73 (д, J=11,9 Гц, 1H), 4,66 (д, J=11,9 Гц, 1H), 4,53 (м, 1H), 4,36-3,96 (м, 9H), 3,89 (т, J=6,4 Гц, 1H), 3,71 (с, 3H), 3,55 (с, 3H), 3,33 (м, 1H), 3,20 (м, 2H), 2,94 (м, 3H), 2,59 (м, 1H), 2,29 (с, 3H), 2,23 (с, 3H), 2,02 (с, 3H), 1,83 (дд, J=16,0 Гц, J=11,9 Гц, 1H).
13C-ЯМР (75 МГц, CDCl3): δ 169,7, 154,0, 148,8, 148,4, 145,7, 144,5, 140,9, 139,0, 133,7, 130,9, 130,6, 127,6, 127,0, 124,8, 124,6, 124,1, 120,8, 119,9, 118,2, 117,7, 117,3, 112,7, 112,1, 101,3, 99,2, 74,7, 73,9, 64,4, 59,8, 57,7, 57,0, 56,8, 55,4, 53,3, 46,7, 41,4, 36,5, 34,7, 31,5, 26,4, 24,9, 22,6, 15,7, 14,0, 9,1.
ESI-масс-спектроскопия, m/z: вычислено для C51H53Cl3N4О10S: 1020,4. Найдено (М+Н)+: 1021,2
Пример 15
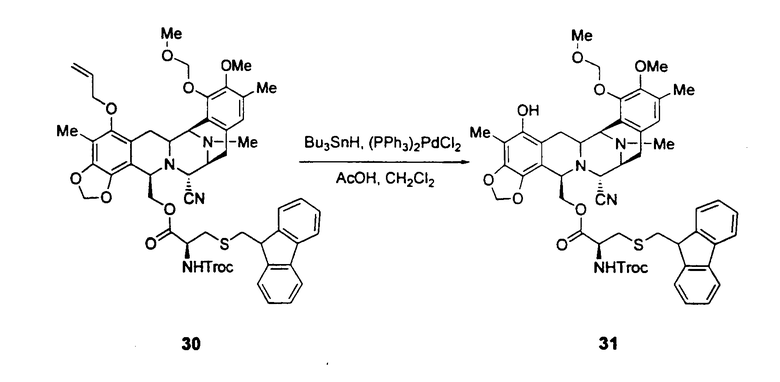
К раствору соединения 30 (845 мг, 0,82 ммоль), уксусной кислоты (500 мг, 8,28 ммоль) и (PPh3)2PdCl2 (29 мг, 0,04 ммоль) в безводном CH2Cl2 (20 мл) добавляют по каплям при 23°С Bu3SnH (650 мг, 2,23 ммоль). Реакционную смесь перемешивают при этой температуре в течение 15 мин, с барботированием. Неочищенный продукт гасят водой (50 мл) и экстрагируют, используя CH2Cl2 (3 х 50 мл). Органические слои сушат над сульфатом натрия, фильтруют и концентрируют. Неочищенное вещество очищают колоночной флэш-хроматографией (этилацетат/гексан, градиент от 1:5 до 1:3), получая при этом соединение 31 (730 мг, 90%) в виде светлого желтовато-кремового твердого вещества.
1H-ЯМР (300 МГц, CDCl3): δ 7,72 (м, 2H), 7,56 (м, 2H), 7,37 (м, 2H), 7,30 (м, 2H), 6,65 (с, 1H), 5,89 (с, 1H), 5,77 (с, 1H), 5,74 (с, 1H), 5,36 (д, J=5,9 Гц, 1H), 5,32 (д, J=5,9 Гц, 1H), 5,20 (д, J=9,0,1H), 4,75 (д, J=12,0 Гц, 1H), 4,73 (м, 1H), 4,48 (д, J=11,9 Гц, 1H), 4,08 (м, 4H), 3,89 (м, 1H), 3,86, (т, J=6,2 Гц, 1H), 3,70 (с, 3H), 3,69 (с, 3H), 3,38 (м, 1H), 3,25 (м, 1H), 3,02-2,89 (м, 4H), 2,67 (с, 1H), 2,61 (с, 1H ), 2,51 (дд, J=14,3 Гц, J=4,5 Гц, 1H), 2,29 (с, 3H), 2,23 (с, 3H), 1,95 (с, 3H), 1,83 (м, 1H).
13C-ЯМР (75 МГц, CDCl3): δ 168,2, 152,5, 148,1, 146,2, 144,4, 144,3, 143,3, 139,6, 134,6, 129,7, 129,6, 126,2, 125,6, 123,4, 123,3, 121,6, 118,5, 116,3, 110,7, 110,2, 105,1, 99,4, 98,5, 75,2, 73,3, 61,7, 58,4, 57,9, 56,3, 56,1, 55,1, 54,7, 53,9, 51,9, 45,2, 40,1, 35,6, 33,3, 24,8, 23,3, 14,5, 7,3.
ESI-масс-спектроскопия, m/z: вычислено для C48H49Cl3N4О10S: 980,3. Найдено (М+Н)+: 981,2
Пример 16
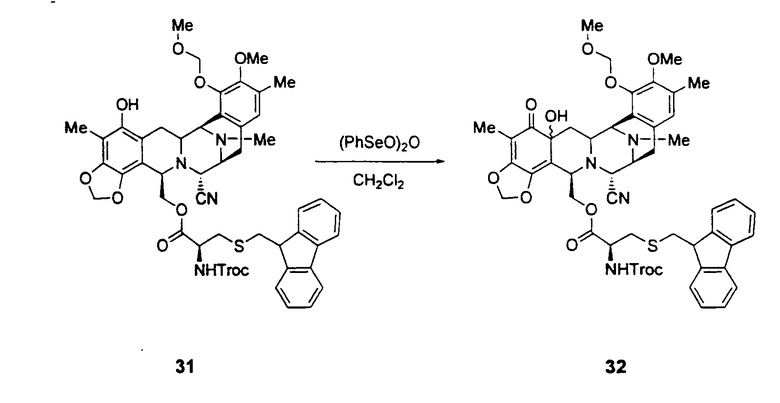
К раствору соединения 31 (310 мг, 0,32 ммоль) в безводном CH2Cl2 (15 мл) при -10°С добавляют с помощью канюли 70%-ный раствор ангидрида бензолселеновой кислоты (165 мг, 0,32 ммоль) в безводном CH2Cl2 (7 мл), поддерживая температуру равной -10°С. Реакционную смесь перемешивают при -10°С в течение 5 мин. Добавляют при этой температуре насыщенный раствор бикарбоната натрия (30 мл). Водный слой промывают большим объемом CH2Cl2 (40 мл). Органические слои сушат над сульфатом натрия, фильтруют и концентрируют. Неочищенное вещество очищают колоночной флэш-хроматографией (этилацетат/гексан, градиент от 1:5 до 1:1), получая при этом соединение 32 (287 мг, 91%, ВЭЖХ: 91,3%) в виде светлого желтовато-кремового твердого вещества, но в виде смеси двух изомеров (65:35), которую используют на следующей стадии.
1H-ЯМР (300 МГц, CDCl3): δ (смесь изомеров) 7,76 (м, 4H), 7,65 (м, 4H), 7,39 (м, 4H), 7,29 (м, 4H), 6,62 (с, 1H), 6,55 (с, 1H), 5,79-5,63 (м, 6H), 5,09 (с, 1H), 5,02 (д, J=6,0 Гц, 1H), 4,99 (д, J=6,0 Гц, 1H), 4,80-4,63 (м, 6H), 4,60 (м, 1H), 4,50 (м, 1H), 4,38 (д, J=12,8 Гц, J =7,5 Гц, 1H), 4,27(дд, J=12,8 Гц, J=7,5 Гц, 1H), 4,16-3,90 (м, 10H), 3,84 (с, 3H), 3,62 (с, 3H), 3,50 (с, 3H), 3,49 (с, 3H), 3,33-2,83 (м, 14H), 2,45-2,18 (м, 2H), 2,21 (с, 6H), 2,17 (с, 6H), 1,77 (с, 6H), 1,67 (м, 2H).
13C-ЯМР (75 МГц, CDCl3): δ (смесь изомеров) 168,6, 168,4, 158,6, 154,8, 152,8, 152,5, 147,3, 147,2, 146,8, 144,1, 144,0, 140,8, 139,7, 137,1, 129,8, 129,3, 128,4, 128,7, 126,5, 125,5, 123,7, 123,6, 123,5, 123,4, 122,2, 121,3, 118,3, 115,8, 115,5, 110,2, 106,9, 103,5, 103,2, 100,1, 99,6, 97,9, 97,7, 93,8, 73,4, 70,9, 69,2, 64,9, 62,5, 59,3, 58,9, 58,4, 56,7, 56,3, 56,2, 55,4, 55,2, 55,1, 54,9, 54,7, 54,3, 54,1, 53,8, 52,8, 45,5, 40,5, 40,0, 39,8, 35,8, 35,5, 33,9, 33,7, 30,1, 28,8, 24,2, 24,1, 21,2, 14,5, 14,4, 12,7, 6,0, 5,7.
ESI-масс-спектроскопия, m/z: вычислено для C48H49Cl3N4О11S: 996,3. Найдено (М+Н)+: 997,2
Пример 17
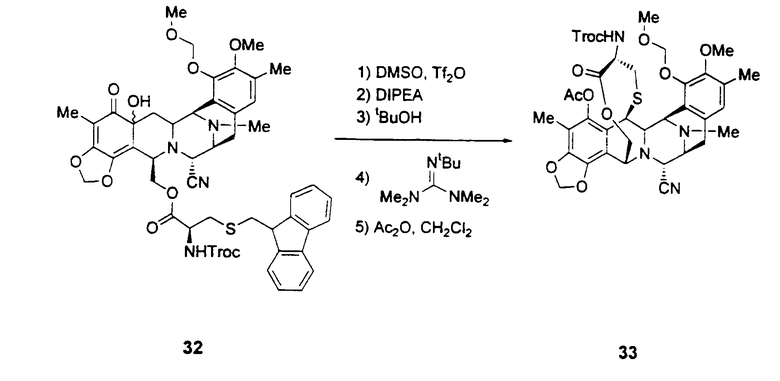
Реакционную колбу дважды обжигают, несколько раз повторяют операцию - откачивание до вакуума/заполнение аргоном и выдерживают в атмосфере аргона в течение реакции. К раствору диметилсульфоксида (DMSO) (39,1 мкл, 0,55 ммоль, 5 эквивалентов) в безводном CH2Cl2 (4,5 мл) добавляют по каплям при -78°С ангидрид трифторметилсерной кислоты (37,3 мкл, 0,22 ммоль, 2 эквивалента). Реакционную смесь перемешивают при -78°С в течение 20 минут, затем при -78°С, с помощью канюли, добавляют раствор соединения 32 (110 мг, 0,11 ммоль, ВЭЖХ: 91,3%) в безводном CH2Cl2 (основное добавление 1 мл и 0,5 мл для промывки). Во время добавления в обеих колбах поддерживают температуру -78°С, окраска изменяется от желтой до коричневой. Реакционную смесь перемешивают при -40°С в течение 35 минут. В течение этого периода времени раствор изменяет свою окраску от желтой до темно-зеленой. После этого добавляют по каплям (изо-Pr)2NEt (153 мкл, 0,88 ммоль, 8 эквивалентов) и выдерживают реакционную смесь при 0°С в течение 45 минут, за это время раствор становится коричневым. Затем добавляют по каплям трет-бутанол (41,6 мкл, 0,44 ммоль, 4 эквивалента) и 2-трет-бутил-1,1,3,3-тетраметилгуанидин (154,6 мкл, 0,77 ммоль, 7 эквивалентов) и перемешивают реакционную смесь при 23°С в течение 40 минут. По истечении этого времени добавляют по каплям уксусный ангидрид (104,3 мкл, 1,10 ммоль, 10 эквивалентов) и выдерживают реакционную смесь при 23°С еще 1 час. Затем реакционную смесь разбавляют, используя CH2Cl2 (20 мл), и промывают насыщенным водным раствором NH4Cl (50 мл), насыщенным водным раствором бикарбоната натрия (50 мл) и насыщенным водным раствором хлорида натрия (50 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют. Полученный остаток очищают колоночной флэш-хроматографией (элюент: этилацетат/гексан, градиент от 1:3 до 1:2), получая при этом соединение 33 (54 мг, 58%) в виде бледно-желтого твердого вещества.
1H-ЯМР (300 МГц, CDCl3): δ 6,85 (с, 1H), 6,09 (с, 1H), 5,99 (с, 1H), 5,20 (д, J=5,8 Гц, 1H), 5,14 (д, J=5,3 Гц, 1H), 5,03 (м, 1H), 4,82 (д, J=12,2, 1H), 4,63 (д, J=12,0 Гц, 1H), 4,52 (м, 1H), 4,35-4,17 (м, 4Н), 3,76 (с, 3Н), 3,56 (с, 3Н), 3,45 (м, 2Н), 2,91 (м, 2H), 2,32 (с, 3Н), 2,28 (с, 3Н), 2,21 (с, 3Н), 2,12 (м, 2H), 2,03 (с, 3Н).
13C-ЯМР (75 МГц, CDCl3): δ 168,5, 167,2, 152,7, 148,1, 147,1, 144,5, 139,6, 139,1, 130,5, 129,0, 123,7, 123,5, 123,3, 118,8, 116,5, 112,1, 100,6, 97,8, 73,3, 60,5, 59,4, 59,2, 58,3, 57,6, 57,4, 56,1, 53,3, 53,1, 40,6, 40,0, 31,0, 22,2, 18,9, 14,4, 8,1.
ESI-масс-спектроскопия, m/z: вычислено для C36H39Cl3N4О11S: 842,1. Найдено (М+Н)+: 843,1
Пример 18

К раствору соединения 33 (12 мг, 0,014 ммоль) в сухом дихлорметане (1,2 мл) и ацетонитриле степени чистоты ВЭЖХ (1,2 мл) добавляют при 23°С иодид натрия (21 мг, 0,14 ммоль) и свежеперегнанный (над гидридом кальция при атмосферном давлении) триметилсилилхлорид (15,4 мг, 0,14 ммоль). Реакционная смесь меняет свою окраску на оранжевую. Через 15 минут раствор разбавляют дихлорметаном (10 мл) и промывают свежим водным насыщенным раствором Na2S2O4 (3 х 10 мл). Органический слой сушат над сульфатом натрия, фильтруют и концентрируют. Получают соединение 34 (13 мг, выход количественный) в виде бледно-желтого твердого вещества, которое используют без дополнительной очистки.
1H-ЯМР (300 МГц, CDCl3): δ 6,85 (с, 1H), 6,09 (с, 1H), 5,99 (с, 1H), 5,27 (д, J=5,8 Гц, 1H), 5,14 (д, J=5,3 Гц, 1H), 5,03 (д, J=11,9 Гц, 1H), 4,82 (д, J=12,2,1H), 4,63 (д, J=13,0 Гц, 1H), 4,52 (м, 1H), 4,34 (м, 1H), 4,27 (шир.с, 1H), 4,18 (м, 2H), 3,76 (с, 3Н), 3,56 (с, 3Н), 3,44 (м, 1H), 3,42 (м, 1H), 2,91 (м, 2H), 2,32 (с, 3Н), 2,28 (с, 3Н), 2,21 (с, 3Н), 2,03 (с, 3H).
ESI-масс-спектроскопия, m/z: вычислено для C34H35N4О10S: 798,1. Найдено (М+Н)+: 799,1
Пример 19

К раствору соединения 34 (13 мг, 0,016 ммоль) в смеси уксусная кислота/Н2О (90:10, 1 мл) добавляют порошок цинка (5,3 мг, 0,081 ммоль) при 23°C. Реакционную смесь нагревают при 70°С в течение 6 часов. После этого реакционную смесь охлаждают до 23°С, разбавляют, используя CH2Cl2 (20 мл), и промывают водным насыщенным раствором бикарбоната натрия (15 мл) и водным раствором Et3N (15 мл). Органический слой сушат над сульфатом натрия, фильтруют и концентрируют. Полученный остаток очищают колоночной флэш-хроматографией на Silica-NH2 (элюент: этилацетат/гексан, градиент от 0:100 до 50:50), получая при этом соединение 35 (6,8 мг, 77% для двух стадий) в виде бледно-желтого твердого вещества.
1H-ЯМР (300 МГц, CDCl3): δ 6,51 (с, 1H), 6,03 (дд, J=1,3 Гц, J=26,5 Гц, 2H), 5,75 (шир.с, 1H), 5,02 (д, J=11,6 Гц, 1H), 4,52 (м, 1H), 4,25 (м, 2H), 4,18 (д, J=2,5 Гц, 1H), 4,12 (дд, J=1,9 Гц, J=11,5 Гц, 1H), 3,77 (с, 3Н), 3,40 (м, 2H), 3,26 (т, J=6,4 Гц, 1H), 2,88 (м, 2H), 2,30-2,10 (м, 2H), 2,30 (с, 3Н), 2,28 (с, 3Н), 2,18 (с, 3Н), 2,02 (с, 3Н).
13C-ЯМР (75 МГц, CDCl3): δ 174,1, 168,4, 147,8, 145,4, 142,9, 140,8, 140,1, 131,7, 130,2, 129,1, 128,3, 120,4, 118,3, 117,9, 113,8, 111,7, 101,7, 61,2, 59,8, 59,2, 58,9, 54,4, 53,8, 54,4, 41,3, 41,5, 34,1, 23,6, 20,3, 15,5, 9,4.
ESI-масс-спектроскопия, m/z: вычислено для C31H34N4О8S: 622,7. Найдено (М+Н)+: 623,2
Пример 20

К раствору соединения 36 (49 мг, 0,08 ммоль) и 2-[3-гидрокси-4-метоксифенил]этиламина (46,2 мг, 0,27 ммоль) в этаноле (2,5 мл) добавляют при 23°С силикагель (105 мг). Реакционную смесь перемешивают при 23°С в течение 14 час. Затем ее разбавляют гексаном и вводят в хроматографическую колонку (этилацетат/гексан, от 1:3 до 1:1), получая при этом соединение Et-770 (55 мг, 90%) в виде бледно-желтого твердого вещества.
1H-ЯМР (300 МГц, CDCl3): δ 6,60 (с, 1H), 6,47 (с, 1H), 6,45 (с, 1H), 6,05 (с, 1H), 5,98 (с, 1H), 5,02 (д, J =11,4 Гц, 1H), 4,57 (шир.с, 1H), 4,32 (шир.с, 1H), 4,28 (д, J=5,3 Гц, 1H), 4,18 (д, J=2,5 Гц, 1H), 4,12 (дд, J=2,1 Гц, J=11,5 Гц, 1H), 3,78 (с, 3H), 3,62 (с, 3H), 3,50 (д, J=5,0 Гц, 1H), 3,42 (м, 1H), 3,10 (ддд, J=4,0 Гц, J=10,0 Гц, J=11,0 Гц, 1H), 2,94 (м, 2H), 2,79 (м, 1H), 2,61 (м, 1H), 2,47 (м, 1H), 2,35 (м, 1H), 2,32 (с, 3H), 2,27 (с, 3H), 2,20 (с, 3H), 2,09 (м, 1H), 2,04 (с, 3H).
ESI-масс-спектроскопия, m/z: вычислено для C40H42N4О10S: 770,7. Найдено (М+Н)+: 771,2
Пример 22
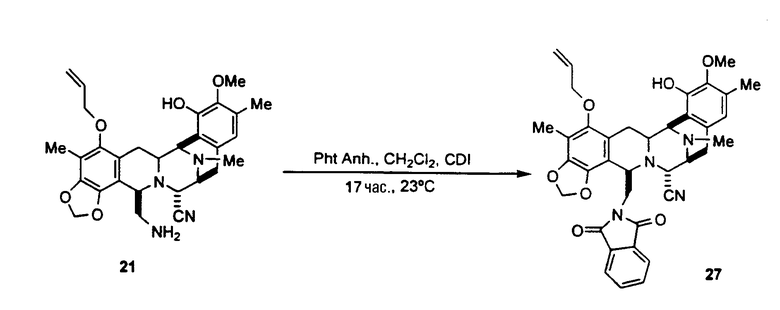
К раствору соединения 21 (22 мг, 0,042 ммоль) в CH2Cl2 (0,8 мл) добавляют фталевый ангидрид (6,44 мг, 0,042 ммоль) и реакционную смесь перемешивают в течение 2 час при 23°С. Затем добавляют карбонилдиимидазол (1 мг, 0,006 ммоль) и полученную смесь перемешивают при 23°С в течение 7 час. Затем добавляют карбонилдиимидазол (5,86 мг, 0,035 ммоль) и реакционную смесь перемешивают при 23°С дополнительно в течение 17 час. Раствор разбавляют, используя CH2Cl2 (15 мл), и промывают 0,1 N раствором HCl (15 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении. Полученный остаток очищают колоночной флэш-хроматографией (SiO2, гексан:этилацетат, 2:1), получая при этом соединение 27 (26,4 мг, 96%) в виде белого твердого вещества.
Rf: 0,58 (этилацетат)
1H ЯМР (300 МГц, CDCl3): δ 7,73-7,64 (м, 4H), 6,40 (с, 1H), 6,12-6,01 (м, 1H), 5,63 (с, 1H), 5,58 (д, J=1,5 Гц, 1H), 5,37 (дд, J1=1,8 Гц, J2=17,4 Гц), 5,23 (дд, J1=1,8 Гц, J2=10,5 Гц, 1H), 5,12 (д, J=1,5 Гц, 1H), 4,22-4,15 (м, 3H), 4,08 (д, J=1,8 Гц, 1H), 3,68 (с, 3H), 3,59-3,55 (м, 2H), 3,35 (д, J=8,1 Гц, 1H), 3,27-3,16 (м, 2H), 3,05 (дд, J1=8,1 Гц, J2=18,3 Гц, 1H), 2,64 (д, J=18,0 Гц, 1H), 2,30 (с, 3H), 2,24 (с, 3H), 2,09 (с, 3H), 1,80 (дд, J1=11,4Гц, J2=15Гц, 1H).
13C ЯМР (75 МГц, CDCl3): δ 167,7, 148,9, 146,4, 144,2, 142,6, 139,5, 134,0, 133,5, 132,0, 131,0, 128,3, 123,0, 121,3, 120,9, 118,1, 117,5, 116,8, 113,6, 112,4, 100,8, 74,5, 60,6, 60,5, 57,7, 56,6, 55,6, 55,5, 42,3, 41,7, 26,6, 25,5, 15,9, 9,46.
ESI-масс-спектроскопия, m/z: вычислено для C37H35N4О7: 648,79. Найдено (М+Н)+: 649,3
Пример 23
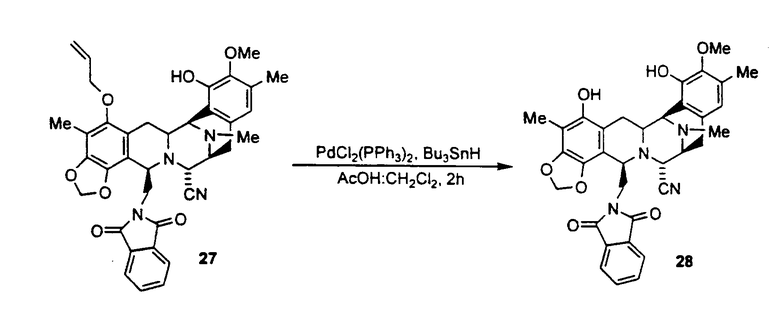
К раствору соединения 27 (26 мг, 0,041 ммоль) в CH2Cl2 (11 мл) добавляют при 23°С уксусную кислоту (11 мл), (PPh3)2PdCl2 (2,36 мг) и Bu3SnH (28 мл, 0,10 ммоль). После перемешивания при этой температуре в течение 2 час реакционную смесь вводят в колонку для флэш-хроматографии (SiO2, градиент от гексана до смеси гексан:этилацетат, 2:1) получая при этом соединение 28 (24,7 мг, 99%) в виде белого твердого вещества.
Rf: 0,33 (гексан:этилацетат, 2:1)
1H ЯМР (300 МГц, CDCl3): δ 7,75-7,70 (м, 2H), 7,69-7,65 (м, 2H), 6,39 (с, 1H), 5,82 (шир.с, 1H), 5,50 (д, J=1,5 Гц, 1H), 5,0 (д, J=1,5 Гц, 1H), 4,45 (шир.с, 1H), 4,23-4,19 (м, 2H), 4,10-4,09 (м, 1H), 3,73 (с, 3Н), 3,60-3,48 (м, 2H), 3,36-3,33 (м, 1H), 3,26-3,20 (м, 1H), 3,14-3,08 (м, 1H), 3,98 (д, J=14,4 Гц, 1H), 2,61 (д, J=18,3 Гц, 1H), 2,30 (с, 3Н), 2,23 (с, 3Н), 2,06 (с, 3Н), 1,85 (дд, J1=12 Гц, J2=15,3 Гц).
13C ЯМР (75 МГц, CDCl3): δ 167,8, 146,4, 145,1, 143,9, 142,7,137,1, 133,5, 131,9, 130,8, 128,4, 122,9, 120,8, 118,0, 116,8, 114,0, 113,4, 106,4, 100,4, 60,6, 60,5, 57,8, 56,6, 55,5, 55,2, 42,6, 41,5, 25,6, 25,5, 15,8, 8,9.
ESI-масс-спектроскопия m/z: вычислено для C34H32N4O7 608,6. Найдено (М+Н)+: 609,2.
Пример 24

К раствору соединения 28 (357 мг, 0,58 ммоль) в CH2Cl2 (3 мл) добавляют при 0°С хлорангидрид уксусной кислоты (41,58 мкл, 0,58 ммоль) и пиридин (47,3 мкл, 0,58 ммоль). Реакционную смесь перемешивают в течение 1 час, после чего раствор разбавляют, используя CH2Cl2 (15 мл), и промывают 0,1 N раствором HCl (15 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении. Полученный остаток очищают колоночной флэш-хроматографией (RP-18, CH3CN:H2O, 60:40), получая при этом фталасцидин (354 мг, 94%) в виде белого твердого вещества.
Rf: 0,37 (CH3CN:H2O, 7:3, RP-18)
1H ЯМР (300 МГц, CDCl3): δ 7,72-7,68 (м, 2H), 7,67-7,63 (м, 2H), 6,38 (с, 1H), 5,69 (д, J=1,2 Гц, 1H), 5,64 (д, J=1,2Гц, 1H), 5,30 (шир.с, 1H), 4,25-4,21 (м, 2H), 4,02 (д, J=2,1 Гц, 1H), 3,64-3,62 (м, 5H), 3,33 (д, J=8,4 Гц, 1H), 3,21-3,16 (м, 1H), 3,02 (дд, J1=8,1 Гц, J2=18 Гц, 1H), 2,76 (дд, J1=1,8 Гц, J2=15,6 Гц, 1H), 2,63 (д, J=17,7 Гц, 1H), 2,29 (с, 3Н), 2,28 (с,3H), 2,21 (с, 3Н), 2,0 (с, 3Н), 1,73 (дд, J1=12,0 Гц, J2=15,3 Гц, 1H).
13C ЯМР (75 МГц, CDCl3): δ 168,5, 167,6, 146,2, 144,2, 142,5, 141,0, 140,5, 133,4, 131,8, 130,7, 128,2, 120,9, 120,8, 117,9, 116,4, 113,6, 101,1, 60,4, 60,0, 57,0, 56,3, 55,6, 55,4, 41,6, 41,5, 26,5, 25,2, 20,2, 15,7, 9,4.
ESI-масс-спектроскопия, m/z: вычислено для C36H34N4О8: 650. Найдено (М+Н)+: 651,2
Пример 25
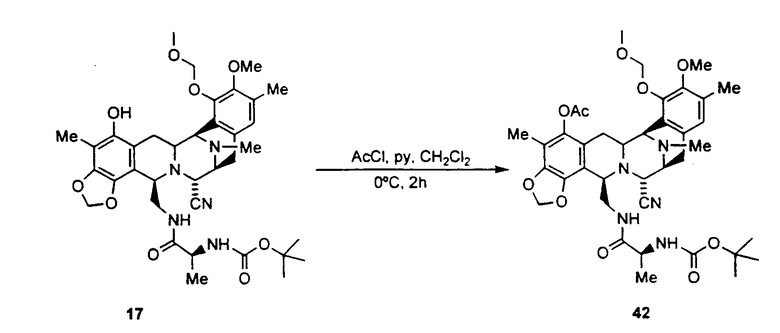
К раствору соединения 17 (300 мг, 0,432 ммоль) в CH2Cl2 (2 мл) добавляют при 0°С хлорангидрид уксусной кислоты (30,7 мкл, 0,432 ммоль) и пиридин (34,9 мкл, 0,432 ммоль). Реакционную смесь перемешивают при этой температуре в течение 2 час, после чего раствор разбавляют, используя CH2Cl2 (15 мл), и промывают 0,1 N раствором HCl (15 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении, получая при этом соединение 42 (318 мг, 100%) в виде белого твердого вещества, которое используют в последующих реакциях без дополнительной очистки.
Rf: 0,5 (этилацетат:метанол, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 6,66 (с, 1H), 5,93 (д, J=1,2 Гц, 1H), 5,83 (д, J=1,2 Гц, 1H), 5,42 (т, J=6,6 Гц, 1H), 5,07 (д, J=5,7 Гц, 1H), 4,98 (д, J=5,7 Гц, 1H), 4,16 (д, J=1,8 Гц, 1H), 4,11 (д, J=2,7 Гц, 1H), 3,98 (шир.с, 1H), 3,73-3,61 (м, 2H), 3,64 (с, 3H), 3,52-3,48 (м, 1H), 3,50 (с, 3H), 3,33 (д, J=9,6 Гц, 1H), 3,17-3,14 (м, 1H), 2,97-2,87 (м, 1H), 2,75-2,70 (д, J=16,8 Гц, 1H), 2,26 (с, 6H), 2,16 (с, 3H), 1,96 (с, 3H), 1,70 (дд, J,= 11,7 Гц, J=15,6 Гц, 1H), 1,33 (с, 9H), 0,59 (д, J =6,0 Гц, 3H).
13C ЯМР (75 МГц, CDCl3): δ 172,0, 168,3, 162,3, 148,2, 144,4, 140,4, 140,2, 130,9, 130,5, 125,3, 123,4, 120,8, 117,6, 112,7, 111,7, 101,4, 99,1, 79,2, 59,5, 58,8, 57,5, 57,4, 56,4, 55,5, 55,0, 41,3, 39,0, 28,2, 26,4, 24,6, 19,9, 18,4, 15,4, 9,1.
ESI-масс-спектроскопия, m/z: вычислено для C38H49N5О10: 735,82. Найдено (М+Н)+: 736,3
Пример 26

К раствору соединения 42 (318 мг, 0,432 ммоль) в CH2Cl2 (2,16 мл) добавляют трифторуксусную кислоту (1,33 мл, 17,30 ммоль) и реакционную смесь перемешивают при 23°С в течение 3,5 час. Реакцию гасят обработкой насыщенным водным раствором бикарбоната натрия (60 мл) и экстрагируют, используя CH2Cl2 (2 х 70 мл). Объединенные органические слои сушат (сульфат натрия) и концентрируют в вакууме. Полученный остаток очищают колоночной флэш-хроматографией (SiO2, этилацетат:метанол, 20:1), получая при этом соединение 43 (154 мг, 60%) в виде белого твердого вещества.
Rf: 0,22 (этилацетат: метанол, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 6,47 (с, 1H), 6,22 (шир.с, 1H), 5,95 (д, J=1,2 Гц, 1H), 5,88 (д, J=1,2 Гц, 1H), 4,08-4,06 (м, 2H), 4,01 (шир.с, 1H), 3,69 (с, 3H), 3,49 (д, J=3,6 Гц, 1H), 3,33 (д, J=8,1 Гц, 1H), 3,26-3,22 (м, 1H), 2,95 (дд, J1=8,1 Гц, J2=18 Гц, 1H), 2,80-2,76 (м, 2H), 2,58 (д, J=18 Гц, 1H), 2,29 (с, 3H), 2,27 (с, 3H), 2,21 (с, 3H), 1,96 (с, 3H), 1,77 (дд, J1=12,3 Гц, J2=15,6 Гц, 1H), 0,90 (д, J=6,9 Гц, 3H).
13C ЯМР (75 МГц, CDCl3): δ 174,8, 169,0, 146,8, 144,4, 142,8, 140,5, 140,2, 131,1, 128,8, 120,8, 120,5, 117,1, 112,9, 111,6, 101,5, 60,3, 59,0, 56,5, 56,3, 55,6, 55,1, 50,2, 41,6, 39,5, 26,8, 26,3, 24,9, 20,2, 15,4, 9,2.
ESI-масс-спектроскопия, m/z: вычислено для C31H37N5О7: 591,65. Найдено (М+Н)+: 592,3
Пример 27
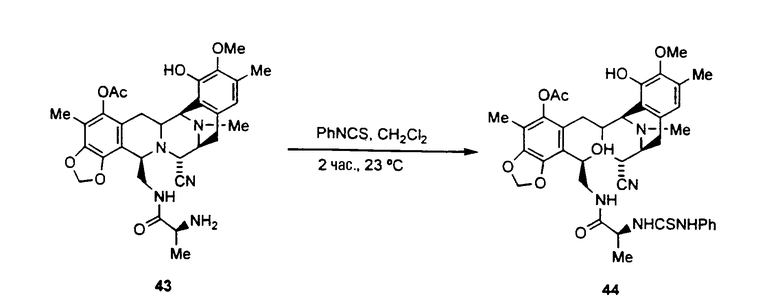
К раствору соединения 43 (254 мг, 0,26 ммоль) в CH2Cl2 (1,3 мл) добавляют фенилизотиоцианат (186 мкл, 1,56 ммоль) и полученную смесь перемешивают при 23°С в течение 2 час. Реакционную смесь концентрируют в вакууме и полученный остаток очищают колоночной флэш-хроматографией (SiO2, градиент от гексана до смеси гексан: этилацетат, 1:1), получая при этом соединение 44 (120 мг, 63%) в виде белого твердого вещества.
Rf: 0,41 (этилацетат: метанол, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 8,17 (с, 1H), 7,49-7,44 (м, 3H), 7,31-7,24 (м, 3H), 7,05 (д, J=6,9 Гц, 1H), 5,98 (д, J=1,2 Гц, 1H), 5,87 (д, J=1,2 Гц, 1H), 5,52 (шир.с, 1H), 4,54 (т, J=6,6 Гц, 1H), 4,15 (д, J=2,1 Гц, 1H), 4,03 (д, J=2,7 Гц, 2H), 3,80 (шир.с, 1H), 3,66 (с, 3H), 3,40 (шир.с, 1H), 3,32 (д, J=7,8 Гц, 1H), 3,16 (д, J=11,7 Гц, 1H), 2,82-2,61 (м, 3H), 2,29 (с, 3H), 2,20 (с, 3H), 2,01 (с, 3H), 1,99 (с, 3H), 1,80 (дд, J1=12,0 Гц, J2=15,9 Гц, 1H), 0,62 (д, J=6,0 Гц, 3Н).
13C ЯМР (75 МГц, CDCl3): δ 178,5, 171,9, 168,7, 146,7, 144,5, 142,6, 140,6, 140,3, 136,3, 131,0, 129,9, 128,9, 126,7, 124,4, 120,9, 120,6, 117,7, 116,6, 112,7, 111,9, 101,4, 60,4, 58,7, 57,5, 56,1, 55,7, 55,1, 53,3, 41,4, 38,8, 26,3, 24,4, 20,2, 18,1, 15,3, 9,2.
ESI-масс-спектроскопия m/z: вычислено для C38H42N6O7S 726,3. Найдено (М+Н)+: 727,3.
Пример 28

К раствору соединения 44 (120 мг, 0,165 ммоль) в диоксане (0,9 мл) добавляют 5,3 N HCl/диоксан (1,8 мл), и реакционную смесь перемешивают при 23°С в течение 2,5 час. Затем к этой реакционной смеси добавляют CH2Cl2 (10 мл) и Н2O (5 мл), и декантируют органический слой. Водную фазу подщелачивают насыщенным водным раствором бикарбоната натрия (20 мл) (рН=8) при 0°С, после чего экстрагируют, используя CH2Cl2 (2 х 15 мл). Объединенные органические слои сушат (сульфат натрия) и концентрируют в вакууме, получая при этом соединение 45 (75 мг, 87%) в виде белого твердого вещества, которое используют в последующих реакциях без дополнительной очистки.
Rf: 0,23 (этилацетат:метанол, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 6,43 (с, 1H), 5,94 (д, J=1,2 Гц, 1H), 5,87 (д, J=1,2 Гц, 1H), 4,10 (д, J=2,1 Гц, 1H), 3,98 (д, J=2,4 Гц, 1H), 3,91 (шир.с, 1H), 3,69 (с, 3Н), 3,34-3,25 (м, 2H), 3,05 (дд, J1=1,8 Гц, J2=8,1 Гц, 1H), 2,80-2,73 (м, 3Н), 2,46 (д, J=18 Гц, 1H), 2,30 (с, 3Н), 2,28 (с, 3H), 2,20 (с, 3Н), 1,98 (с, 3Н), 1,79 (дд, J1=12,6 Гц, J2=16,2 Гц, 1H).
13C ЯМР (75 МГц, CDCl3): δ 168,7, 146,7, 144,4, 142,9, 140,4, 130,4, 128,9, 121,1, 120,8, 117,8, 116,8, 113,6, 111,5, 101,4, 67,6, 60,5, 59,8, 58,4, 56,6, 55,8, 55,3, 43,6, 41,8, 31,3, 25,6, 20,2, 15,6, 9,2.
ESI-масс-спектроскопия, m/z: вычислено для C28H32N4О6: 520,58. Найдено (М+Н)+: 521,3
Пример 29
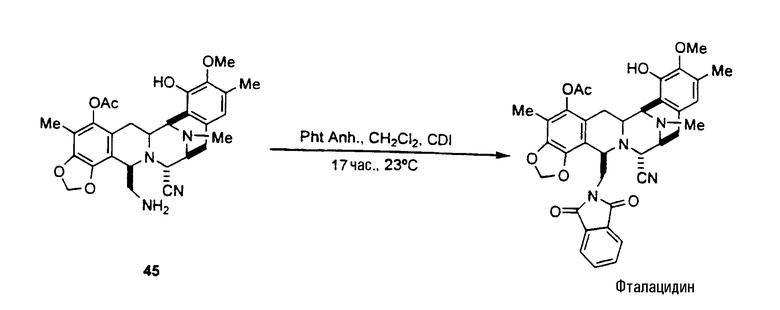
К раствору соединения 45 (10 мг, 0,02 ммоль) в CH2Cl2 (0,4 мл) добавляют фталевый ангидрид (2,84 мг, 0,02 ммоль) и реакционную смесь перемешивают в течение 2 час при 23°С. Затем добавляют карбонилдиимидазол (0,5 мг, 0,003 ммоль) и реакционную смесь перемешивают при 23°С в течение 7 час. После этого добавляют карбонилдиимидазол (2,61 мг, 0,016 ммоль) и реакционную смесь перемешивают при 23°С дополнительно в течение 17 час. Раствор разбавляют CH2Cl2 (10 мл) и промывают 0,1 N раствором HCl (5 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении. Полученный остаток очищают колоночной флэш-хроматографией (RP-18, CH3CN:H2O, 60:40), получая при этом фталасцидин (11,7 мг, 93%) в виде белого твердого вещества.
Rf: 0,37 (CH3CN:H2O, 7:3, RP-18)
1H ЯМР (300 МГц, CDCl3): δ 7,72-7,68 (м, 2h), 7,67-7,63 (м, 2H), 6,38 (с, 1H), 5,69 (д, J =1,2 Гц, lH), 5,64 (д, J =1,2 Гц, 1H), 5,30 (шир.с, 1H), 4,25-4,21 (м, 2H), 4,02 (д, J=2,1 Гц, 1H), 3,64-3,62 (м, 5H), 3,33 (д, J=8,4 Гц, 1H), 3,21-3,16 (м, 1H), 3,02 (дд, J1=8,1 Гц, J2=18 Гц, 1H), 2,76 (дд, J1=1,8 Гц, J2=15,6 Гц, 1H), 2,63 (д, J=17,7 Гц, 1H), 2,29 (с, 3H), 2,28 (с, 3H), 2,21 (с, 3H), 2,0 (с, 3H), 1,73 (дд, J1=12,0 Гц, J2=15,3 Гц, 1H).
13C ЯМР (75 МГц, CDCl3): δ 168,5, 167,6, 146,2, 144,2, 142,5, 141,0, 140,5, 133,4, 131,8, 130,7, 128,2, 120,9, 120,8, 117,9, 116,4, 113,6, 101,1, 60,4, 60,0, 57,0, 56,3, 55,6, 55,4, 41,6, 41,5, 26,5, 25,2, 20,2, 15,7, 9,4.
ESI-масс-спектроскопия, m/z: вычислено для C36H34N4О8: 650. Найдено (М+Н)+: 651,2
Пример 30
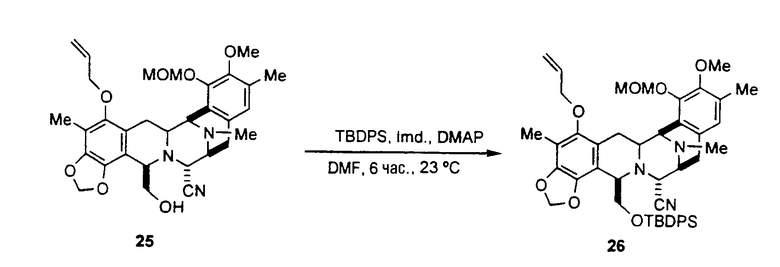
К раствору соединения 25 (18 мг, 0,032 ммоль) в диметилформамиде (DMF) (0,05 мл) добавляют при 0°С кат. DMAP (0,5 мг, 0,004 ммоль), имидазол (5 мг, 0,08 ммоль) и трет-бутилдифенилсилилхлорид (12,5 мкл, 0,048 ммоль) и реакционную смесь перемешивают при 23°С в течение 6 час. Добавляют воду (10 мл) при 0°С и водную фазу экстрагируют смесью гексан: этилацетат, 1:10 (2 х 10 мл). Органический слой сушат (сульфат натрия), фильтруют и удаляют растворитель при пониженном давлении. Неочищенный продукт очищают колоночной флэш-хроматографией (SiO2, гексан: этилацетат, 3:1), получая при этом соединение 26 (27 мг, 88%) в виде белого твердого вещества.
Rf: 0,29 (гексан: этилацетат, 3:1)
1H ЯМР (300 МГц, CDCl3): δ 7,61-7,58 (м, 2H), 7,42-7,28 (м, 8H), 6,71 (с, 1H), 6,19-6,02 (м, 1H), 5,78 (д, J=1,2 Гц, 1H), 5,64 (д, J=1,2 Гц, 1H), 5,40 (дд, J1=1,2 Гц, J2=17,1 Гц, 1H), 5,27 (дд, J1=1,2 Гц, J2=10,2 Гц, 1H), 5,13 (с, 2H), 4,45 (д, J=2,4 Гц, 1H), 4,24 (д, J=2,1 Гц, 1H), 4,17-4,06 (м, 3H), 3,75 (с, 3H), 3,64 (дд, J1=2,4 Гц, J2=9,9 Гц, 1H), 3,59 (с, 3H), 3,42-3,21 (м, 4H), 3,10 (дд, J1=8,1 Гц, J2=17,7 Гц, 1H), 2,70 (д, J=17,7 Гц, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 2,11 (с, 3H), 2,08-1,89 (м, 1H), 0,87 (с, 9H).
13С ЯМР (75 МГц, CDCl3): δ 148,5, 148,3, 148,1, 144,0, 139,0, 135,6, 135,4, 133,8, 133,1, 132,6, 130,5, 130,3, 129,6, 129,4, 127,5, 127,4, 125,1, 124,3, 121,6, 118,5, 117,5, 112,9, 111,7, 100,8, 99,2, 74,0, 67,7, 61,5, 59,6, 59,0, 57,7, 57,1, 55,4, 41,6, 29,6, 26,6, 25,5, 18,8, 15,8, 9,2.
ESI-масс-спектроскопия, m/z: вычислено для C47H55N3O7Si: 801,3. Найдено (М+Н)+:802,3
Пример 31
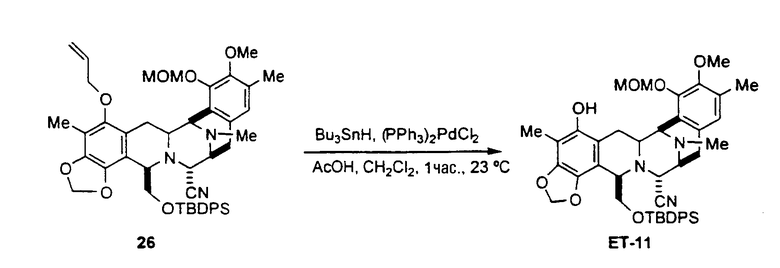
К раствору соединения 26 (7 мг, 0,0087 ммоль) в CH2Cl2 (0,15 мл) добавляют при 23°С уксусную кислоту (2,5 мкл, 0,044 ммоль), (PPh3)2PdCl2 (0,5 мг, 6,96 х 10-4 ммоль) и Bu3SnH (3,5 мкл, 0,013 ммоль). Реакционную смесь перемешивают при этой температуре в течение 1 час. Раствор разбавляют смесью гексан: этилацетат, 5:1 (0,5 мл) и помещают на колонку для флэш-хроматографии (SiO2, градиент гексан: этилацетат, от 5:1 до 1:1), получая при этом соединение ЕТ-11 (5 мг, 75%) в виде белого твердого вещества.
Rf: 0,36 (гексан: этилацетат, 1:5, диоксид кремния)
1H ЯМР (300 МГц, CDCl3): δ 7,56 (м, 2H), 7,41-7,25 (м, 8H), 6,67 (с, 1H), 5,72 (д, J=1,0 Гц, 1H), 5,58 (д, J=1,0 Гц, 1H), 5,51 (с, 1H), 5,38 (д, J=5,75 Гц, 1H), 5,16 (д, J=5,7 Гц, 1H), 4,57 (д, J=2,9 Гц, 1H), 4,21 (м, 1H), 4,09 (м, 1H), 3,72 (с, 3H), 3,71 (с, 3H), 3,68 (дд, J1=2,1 Гц, J2=10,4 Гц, 1H), 3,38-3,26 (м, 3H), 3,11 (дд, J1=2,5 Гц, J2=15,7 Гц, 1H), 3,01 (дд, J1=8,9 Гц, J2=17,9 Гц, 1H), 2,70 (д, J=17,9 Гц, 1H), 2,31 (с, 3H), 2,25 (с, 3H), 2,06 (с, 3H), 1,89 (дд, J1=12,1 Гц, J2=15,7 Гц, 1H), 0,9 (с, 9H),).
13C ЯМР (75 МГц, CDCl3): δ 149,0, 147,4, 145,3, 144,3, 136,3, 135,7, 135,4, 133,2, 130,9, 130,5, 129,6, 129,5, 127,5, 125,0, 118,6, 112,5, 112,1, 105,7, 100,5, 99,8, 68,5, 61,5, 59,7, 58,8, 57,7, 56,9, 56,5, 55,4, 41,7, 26,6, 26,2, 25,5, 18,9, 15,8, 14,2, 8,7.
ESI-масс-спектроскопия m/z: вычислено для C44H51N3O7Si: 761. Найдено (М+Н)+: 762.
Пример 32

Раствор соединения 2 (3,0 г, 5,46 ммоль) и фенилизотиоцианата (3,92 мл, 32,76 ммоль) в CH2Cl2 (27 мл) перемешивают при 23°С в течение 1,5 час. Реакционную смесь распределяют между CH2Cl2 (10 мл) и Н2О (5 мл). Органический слой сушат над сульфатом натрия, фильтруют и концентрируют. Полученный остаток очищают колоночной флэш-хроматографией (SiO2, градиент от гексана до смеси гексан:этилацетат, 2:3) получая при этом соединение 3 (3,29 мг, 88%) в виде желтого твердого вещества.
Rf: 0,27 (ACN: Н2О, 3:2, RP-C18)
1H ЯМР (300 МГц, CDCl3): δ 7,77 (шир.с, 1H), 7,42-7,11 (м, 5H), 6,65 (д, 1H), 6,29 (с, 1H), 5,6-5,5 (м, 1H), 4,19-4,14 (м, 2H), 4,08 (д, 1H), 3,92 (с, 3H), 3,87-3,65 (м, 6H), 3,77 (с, 3Н), 3,37-2,98 (м, 8H), 2,50 (д, 1H), 2,31 (с, 3H), 2,20 (с, 3H), 1,96 (д, 1H), 1,87 (с, 3H), 1,81-1,75 (м, 1H), 0,96 (д, 3H).
13C ЯМР (75 МГц, CDCl3): δ 185,7, 180,9, 178,9, 172,0, 155,7, 147,1, 143,2, 142,4, 136,0, 135,1, 130,5, 129,9, 129,3, 128,5, 126,9, 124,4, 120,2, 117,4, 116,3, 77,1, 60,9, 58,6, 56,2, 55,8, 55,0, 54,6, 53,5, 41,7, 40,3, 25,1, 24,5, 18,4, 15,8, 8,7.
ESI-масс-спектроскопия m/z: вычислено для C36H40N6O6S: 684,8. Найдено (М+Н)+:685,2.
Пример 33

Раствор соединения 3 (0,143 г, 0,208 ммоль) в смеси 6,5 М HCl/диоксан (150 мл) перемешивают в течение 6 час при 23°С. Затем к реакционной смеси добавляют толуол (3 мл) и декантируют органический слой. Остаток распределяют между насыщенным водным раствором бикарбоната натрия (3 мл) и CHCl3 (3 х 3 мл). Органические слои сушат и концентрируют, получая при этом указанное в заглавии соединение в виде смеси соединений 4 и 6 (смесь 4:6, 90:10), которая при стоянии медленно циклизуется с образованием соединения 6.
Rf: 0,4 (этилацетат:метанол 5:1, диоксид кремния)
1H ЯМР (300 МГц, CDCl3): δ 6,45 (с, 1H), 4,16 (м, 1H), 4,02 (д, 1H), 3,96 (с, 3H), 3,79 (м, 2H), 3,75 (с, 3H), 3,35 (м, 1H), 3,20-3,00 (м, 3H), 2,87 (д, 1H), 2,75 (д, 1H), 2,43 (д, 1H), 2,34 (с, 3H), 2,30 (с, 3H), 1,93 (с, 3H), 1,72-1,5 (м, 3H).
ESI-масс-спектроскопия, m/z: вычислено для C26H30N4O5: 478,5. Найдено (М+Н)+:479,2.
Пример 34

Раствор соединения 3 (0,143 г, 0,208 ммоль) в смеси 6,5 М HCl/диоксан (150 мл) перемешивают в течение 1 час при 23°С. После упаривания растворителя полученный остаток очищают колоночной флэш-хроматографией (этилацетат/метанол/триэтиламин, 100:25:0,1), получая при этом соединение 6 (80 мг, 83%) в виде желтого твердого вещества.
Rf: 0,26 (ACN: Н2О, 3:2, RP-C18)
1H ЯМР (500 МГц, CDCl3): δ 6,46 (с, 1H), 5,9 (шир.с, 1H) 4,67 (дд, J=8,3 Гц, J=7,8 Гц, 1H), 4,24 (д, 1Н), 4,16 (с, 3H), 3,93 (д, J =2,7 Гц, 1H), 3,8 (м, 2H), 3,77 (с, 3H), 3,45 (м, 2H), 3,08 (дд, J=17,9 Гц, J=3,6 Гц, 1H), 2,78 (м, 1H), 2,55 (д, 1H), 2,3 (м, 1H), 2,3 (с, 3H), 2,28 (с, 3H), 1,90 (с, 3H).
13С ЯМР (75 МГц, CDCl3): δ 186,2, 162,1, 154,9, 146,9, 145,3, 143,0, 130,1, 129,4, 128,1, 125,0, 121,4, 116,4, 116,2, 66,6, 60,7, 60,7, 60,1, 59,6, 58,8, 55,6, 54,9, 41,9, 25,3, 24,7, 15,7, 8,9.
ESI-масс-спектроскопия m/z: вычислено для C26H28N4O4: 460,5. Найдено (М+Н)+:461,1.
Пример 35

К раствору соединения 3 (2,38 г, 3,47 ммоль) в диоксане (5 мл) добавляют 5,3 М раствор HCl в диоксане (34 мл) и реакционную смесь перемешивают в течение 45 минут при 23°С. Затем добавляют Ас2О (51 мл, 539,5 ммоль) и перемешивают смесь в течение 4 час. Реакционную смесь охлаждают до 0°С и разделяют при этой температуре между насыщенным водным раствором Na2CO3 (300 мл) и этилацетатом (300 мл). Органическую фазу сушат над сульфатом натрия, фильтруют и концентрируют. Полученный остаток очищают колоночной флэш-хроматографией (SiO2, градиент от CH2Cl2 до CH2Cl2:этилацетат, 1:2), получая при этом соединение 5 (1,75 г, 97%) в виде желтого твердого вещества.
Rf: 0,53 (ACN:Н2О, 3:2, RP-C18)
1H ЯМР (300 МГц, CDCl3): δ 6,51 (с, 1 Н), 5,98 (шир.с, 1 Н), 4,84 (дд, 1 Н), 4,17 (д, 1 Н), 4,00 (д, 1H), 3,99 (с, 3H), 3,85 (шир.с, 1H), 3,81 (м, 1H), 3,74 (с, 3H), 3,70 (д, 1H), 3,23 (м, 1H), 3,11 (дд, 1H), 3,09 (м, 1H), 2,93 (м, 2H), 2,44 (д, 1H), 3,67 (с, 3H), 2,25 (с, 3H), 1,70 (с, 3H), 1,60-1,50 (м, 2H), 1,29 (с, 3H).
13C ЯМР (75 МГц, CDCl3): δ 185,9, 180,8, 169,9, 160,2, 156,2, 147,0, 143,1, 140,4, 136,1, 130,6, 129,6, 127,9, 120,4, 117,2, 61,0, 60,7, 58,6, 56,1, 55,7, 55,1, 54,3, 41,8, 41,1, 25,7, 23,9, 22,2, 15,7, 8,7.
ESI-масс-спектроскопия, m/z: вычислено для C28H32N4O6:520,6. Найдено (М+Н)+:521,1.
Пример 36

К раствору соединения 5 (1,75 г, 3,36 ммоль) в CH2Cl2 (17 мл) добавляют при 0°С диизопропилэтиламин (11,71 мл, 67,23 ммоль), DMAP (20 мг, 0,17 ммоль) и простой бромметилметиловый эфир (4,11 мл, 50,42 ммоль). Через 6 час при 23°С реакционную смесь распределяют между CH2Cl2 (50 мл) и насыщенным раствором водным бикарбоната натрия (25 мл). Органический слой сушат над сульфатом натрия и удаляют растворитель при пониженном давлении. Неочищенный продукт очищают колоночной флэш-хроматографией (RP-18, CH3CN/H2O, 1/1), получая при этом соединение 7 (1,32 г, 70%) в виде желтого твердого вещества.
Rf: 0,34 (ACN:Н2О, 2:3, RP-C18)
1H ЯМР (300 МГц, CDCl3): δ 6,74 (с, 1H), 5,14 (с, 2H), 4,82 (м, 1H), 4,22 (д, 1H), 4,00 (с, 3Н), 4,0 (м, 1H), 3,83 (м, 2H), 3,7 (с, 3Н), 3,58 (с, 3Н), 3,4 (м, 1H), 3,2-2,95 (м, 6H), 2,43 (д, 1H), 2,37 (с, 3Н), 2,22 (с, 3Н), 1,89 (с, 3Н), 1,5-1,4 (м, 2H), 1,31 (с, 3Н).
13С ЯМР (75 МГц, CDCl3): δ 185,9, 180,7, 169,6, 156,2, 148,9, 148,5, 140,3, 136,2, 131,3, 130,1, 127,7, 124,6, 123,7, 117,3, 99,5, 99,2, 60,9, 59,7, 58,8, 57,7, 56,4, 55,7, 55,0, 54,2, 51,0, 41,6, 41,0, 40,5, 25,5, 23,9, 22,3, 19,3, 15,6, 14,6, 8,6.
ESI-масс-спектроскопия, m/z: вычислено для C30H36N4O7: 564,6. Найдено (М+Н)+: 565,3.
Пример 37
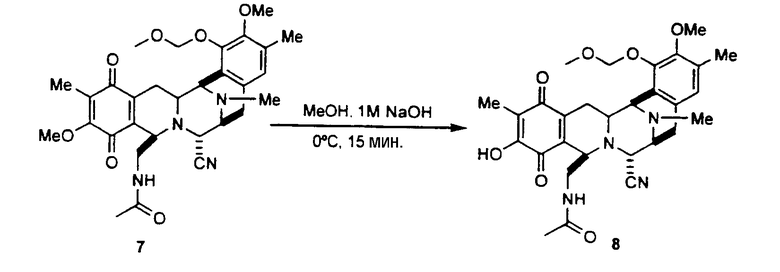
К раствору соединения 7 (0,37 г, 0,65 ммоль) в метаноле (74 мл) при 0°С добавляют 1 М раствор гидроксида натрия (130 мл). Реакционную смесь перемешивают в течение 15 мин и затем реакцию гасят добавлением при 0°С 6 М HCl до рН=5. Смесь экстрагируют этилацетатом (3 х 50 мл) и объединенные органические слои сушат над сульфатом натрия и концентрируют в вакууме. Полученный остаток очищают колоночной флэш-хроматографией (RP-С18, CH3CN:H2O, 1:1), получая при этом соединение 8 (232 мг, 65%) в виде желтого масла.
Rf: 0,5 (ACN:Н2О, 3:2, RP-C18)
1H ЯМР (300 МГц, CDCl3): δ 6,75 (с, 1H), 5,15 (с, 2H), 4,86 (м, 1H), 4,26 (д, 1H),), 4,01 (д, 1H), 3,88-3,81 (м, 2H), 3,70 (с, 3Н), 3,58 (с, 3Н), 3,39 (м, 1H), 3,27-3,21 (м, 1H), 3,18-3,08 (м, 2H), 3,03-2,97 (м, 1H) 2,47 (д, 1H), 2,37 (с, 3Н), 2,22 (с, 3Н), 1,90 (с, 3H), 1,57-1,46 (м, 2H), 1,33 (с, 3H).
13С ЯМР (75 МГц, CDCl3): δ 185,3, 180,6, 175,9, 170,1, 151,5, 148,9, 148,6, 143,3, 133,7, 131,5, 129,9, 124,7, 123,5, 117,1, 117,0, 99,2, 59,8, 58,7, 57,8, 56,3, 55,3, 54,9, 54,3, 41,5, 40,7, 29,6, 25,5, 24,4, 22,2, 20,7, 15,7, 8,0.
ESI-масс-спектроскопия, m/z: вычислено для C29H34N4O7: 550,6. Найдено (М+Н)+: 551,2.
Пример 38

К дегазированному раствору соединения 8 (240 мг, 0,435 ммоль) в диметилформамиде (DMF)(30 мл) добавляют 10% Pd/C (48 мг) и реакционную смесь перемешивают в атмосфере водорода (при атмосферном давлении) в течение 1 час. Реакционную смесь отфильтровывают через слой целита в атмосфере аргона в колбу Шленка, в виде бесцветного раствора, содержащего безводный Cs2CO3 (240 мг, 0,739 ммоль). После этого добавляют бромхлорметан (0,566 мл, 8,71 ммоль). Колбу герметично закрывают и перемешивают при 90°С в течение 3 час. Реакционную смесь охлаждают и фильтруют через целит, затем промывают, используя CH2Cl2. Органический слой концентрируют и сушат (сульфат натрия), получая при этом соединение 9 в виде коричневого масла, которое используют на следующей стадии без дополнительной очистки.
Rf: 0,36 (SiO2, гексан:этилацетат, 1:5)
1H ЯМР (300 МГц, CDCl3): δ 6,71 (с, 3H), 5,89 (д, 1H), 5,81 (д, 1H), 5,63 (шир.с, 1H), 5,33 (д, 1H), 5,17 (д, 1H), 4,97 (м, 1H), 4,20 (д, 1H), 4,09 (м, 1H), 3,99 (м, 1H), 3,68 (м, 1H), 3,65 (с, 6H), 3,59-3,47 (м, 4H), 3,37-3,27 (м, 2H), 3,14-2,97 (м, 2H), 2,62 (д, 1H), 2,32 (с, 3H), 2,20 (с, 3H), 2,08 (с, 3H), 1,72 (м, 1H), 1,36 (с, 3H).
13C ЯМР (75 МГц, CDCl3): δ 169,8, 149,1, 147,4, 145,5, 136,2, 130,9, 130,8, 125,0, 122,9, 117,7, 112,6, 111,8, 106,4, 100,8, 99,8, 59,8, 58,9, 57,7, 56,6, 56,4, 55,5, 55,2, 41,6, 40,1, 29,6, 25,9, 25,0, 22,6, 15,6, 8,8.
ESI-масс-спектроскопия, m/z: вычислено для C30H36SiN4О7: 564,6. Найдено (М+Н)+: 565,3
Пример 39
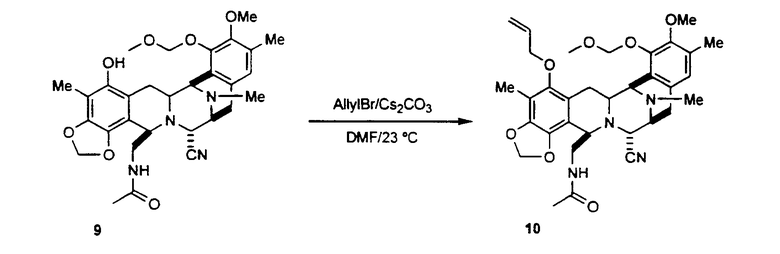
В колбу, содержащую соединение 9 (245 мг, 0,435 ммоль) в диметилформамиде (DMF), (4 мл) добавляют при 0°C карбонат цезия (424 мг, 1,30 ммоль) и аллилбромид (376 мкл, 4,35 ммоль) и перемешивают смесь при 23°С в течение 1 час. Реакционную смесь отфильтровывают через слой целита и затем распределяют между CH2Cl2 (25 мл) и Н2О (10 мл). Органическую фазу сушат (сульфат натрия) и концентрируют при пониженном давлении, получая при этом остаток, который очищают колоночной флэш-хроматографией (SiO2, CHCl3:этилацетат, 1:2), получая при этом соединение 10 (1,75 г, 97%) в виде желтого масла (113 мг, 43%).
Rf: 0,36 (гексан:этилацетат, 1:5)
1H ЯМР (300 МГц, CDCl3): δ 6,74 (с, 1H), 6,3-6,0 (м, 1H), 5,94 (д, 1H), 5,87 (д, 1H), 5,43-5,36 (м, 2H), 5,22 (с, 2H), 5,00 (м, 1H), 4,22 (м, 1H), 4,17-4,01 (м, 1H), 3,98 (м, 2H), 3,71-3,67 (м, 1H), 3,69 (с, 3H), 3,62-3,51 (м, 3H), 3,58 (с, 3H), 3,39-3,37 (м, 1H), 3,31-3,26 (м, 3H), 3,09 (дд, 1H), 2,56 (д, 1H), 2,36 (с, 3H), 2,21 (с, 3H), 2,11 (с, 3H), 2,24-2,10 (м, 1H), 1,82-1,73 (м, 1H), 1,24 (шир.с, 3H)
13C ЯМР (75 МГц, CDCl3): δ 169,4, 148,8, 148,3, 139,1, 133,7, 130,9, 130,3, 125,2, 120,2, 117,7, 113,1, 112,6, 101,3, 99,3, 74,1, 59,7, 59,3, 57,8, 57,0, 56,1, 56,1, 55,2, 41,6, 41,0, 40,9, 29,7, 26,3, 22,5, 15,6, 9,3.
ESI-масс-спектроскопия, m/z: вычислено для C33H40N4O7: 604,7. Найдено (М+Н)+:605,3.
Пример 40
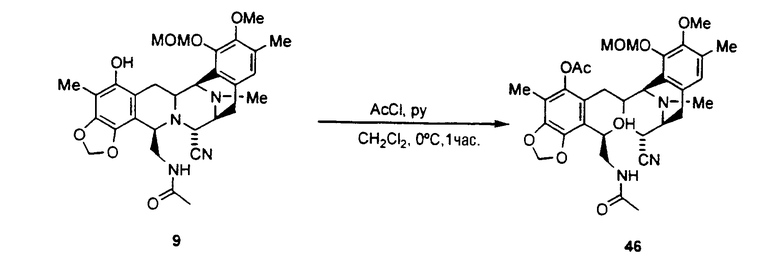
К раствору соединения 9 (22 мг, 0,039 ммоль) в CH2Cl2 (0,2 мл) добавляют при 0°С хлорангидрид уксусной кислоты (2,79 мкл, 0,039 ммоль) и пиридин (3,2 мкл, 0,039 ммоль). Реакционную смесь перемешивают в течение 1 час, после чего раствор разбавляют CH2Cl2 (10 мл) и промывают 0,1 N раствором HCl (5 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении, получая при этом соединение 46 (22 мг, 93%) в виде белого твердого вещества.
Rf: 0,4 (гексан:этилацетат, 1:5)
1H ЯМР (300 МГц, CDCl3): δ 6,74 (с, 1H), 5,97 (д, J=0,9 Гц, 1H), 5,91 (д, J=0,9 Гц, 1H), 5,12 (д, J=5,7 Гц, 2H), 5,04 (д, J=5,7 Гц, 1H) 4,90 (т, J=6 Гц, 1H), 4,17 (д, J=2,7 Гц, 1H), 4,05 (д, J=2,7 Гц, 1H), 4,01 (шир.с, 1H), 3,71 (с, 3H), 3,57 (с, 3Н), 3,50-3,44 (м, 2H), 3,38-3,36 (м, 1H), 3,30-3,26 (м, 1H), 3,00 (дд, J1=7,8 Гц, J2=18,0 Гц, 1H), 2,79 (д, J=12,9 Гц, 1H), 2,60 (д, J=18,0 Гц, 1H), 2,35 (с, 3Н), 2,32 (с, 3Н), 2,21 (с, 3Н), 2,00 (с, 3H), 1,68 (дд, J1=11,7 Гц, J2=15,6 Гц, 1H).
ESI-масс-спектроскопия, m/z: вычислено для C32H38N4О8: 606,7. Найдено (М+Н)+:607,3
Пример 41

К раствору соединения 46 (8 мг, 0,013 ммоль) в диоксане добавляют смесь 5,3 N HCl/диоксан (0,5 мл) и реакционную смесь перемешивают в течение 1 час при 23°С. Затем раствор разбавляют, используя CH2Cl2 (5 мл), и промывают 0,1 N раствором HCl (3 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении, получая при этом соединение 47 (5 мг, 70%) в виде белого твердого вещества.
Rf: 0,4 (гексан:этилацетат, 1:5)
1H ЯМР (300 МГц, CDCl3): δ 6,51 (с, 1H), 5,97 (д, J=1,2 Гц, 1H), 5,91 (д, J=1,2 Гц, 1H), 4,97 (шир.с, 1H), 4,11 (шир.с, 1H), 4,04-4,02 (м, 2H), 3,75 (с, 3Н),), 3,65 (д, J=2,1 Гц, 2H), 3,56-3,30 (м, 2H), 3,04 (дд, J1=7,5 Гц, J2=18 Гц, 1H), 2,80 (д, J=14,4 Гц, 1H), 2,59 (д, J=18,3 Гц, 1H), 2,33 (с, 3Н), 2,24 (с, 3Н), 2,00 (с, 3Н), 1,76 (дд, J1=12,0 Гц, J2 = 15,9 Гц, 1H), 1,33(с, 3H), 1,25 (с, 3H).
= 15,9 Гц, 1H), 1,33(с, 3H), 1,25 (с, 3H).
ESI-масс-спектроскопия, m/z: вычислено для C30H34N4О7: 562,61. Найдено (М+Н)+:563,3
Пример 42

К раствору соединения 45 (10 мг, 0,0192 ммоль) в CH2Cl2 (0,3 мл) добавляют при 0°С изовалерилхлорид (2,34 мкл, 0,0192 ммоль) и пиридин (1,55 мкл, 0,0192 ммоль). Реакционную смесь перемешивают в течение 1 час, после чего раствор разбавляют, используя CH2Cl2 (5 мл), и промывают 0,1 N раствором HCl (3 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении. Остаток очищают колоночной флэш-хроматографией (SiO2, гексан:этилацетат, 1:2), получая при этом соединение 48 (11 мг, 95%) в виде белого твердого вещества.
Rf: 0,12 (гексан:этилацетат, 1:2).
1H ЯМР (300 МГц, CDCl3): δ 6,50 (с, 1H), 5,98 (д, J=1,5Гц, 1H), 5,91 (д, J=1,5 Гц, 1H), 5,75 (с, 1H), 5,02 (т, J=5,4 Гц, 1H), 4,10 (д, J=1,5 Гц, 1H), 4,06 (д, J=2,7 Гц, 1H), 4,02 (д, J=2,7 Гц, 1H), 3,77 (с, 3H), 3,76-3,71 (м, 1H), 3,86-3,28 (м, 3H), 3,04 (дд, J1=8,1 Гц, J2=18,3 Гц, 1H), 2,78 (д, J =15,9 Гц, 1H), 2,55 (д, J=18 Гц, 1H), 2,32 (с, 6H), 2,26 (с, 3H), 1,98 (с, 3H), 1,84-1,68 (м, 2H), 1,36 (д, J=7,2 Гц, 2H), 0,69 (д, J=6,6 Гц, 3H), 0,62 (д, J=6,6 Гц, 3H).
ESI-масс-спектроскопия, m/z: вычислено для C33H40N4О7: 604,69. Найдено (М+Н)+:605,3.
Пример 43

К раствору соединения 45 (10 мг, 0,0192 ммоль) в CH2Cl2 (0,3 мл) добавляют при 0°С деканоилхлорид (3,98 мкл, 0,0192 ммоль) и пиридин (1,55 мкл, 0,0192 ммоль). Реакционную смесь перемешивают в течение 1 час, после чего раствор разбавляют CH2Cl2 (5 мл) и промывают 0,1 N раствором HCl (3 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении. Остаток очищают колоночной флэш-хроматографией (SiO2, гексан: этилацетат, 1:2), получая при этом соединение 49 (12,4 мг, 96%) в виде белого твердого вещества.
Rf: 0,7 (этилацетат: метанол, 10:1)
1H ЯМР (300 МГц, CDCl3): δ 6,50 (с, 1H), 5,98 (д, J=1,5Гц, 1H), 5,91 (д, J=1,5 Гц, 1H), 5,73 (с, 1H), 5,08 (т, J=5,4 Гц, 1H), 4,10 (д, J=1,5 Гц, 1H), 4,05 (м, 1H), 4,01 (м, 1H), 3,76 (с, 3Н), 3,65-3,61 (м, 1H), 3,40-3,27 (м, 3Н), 3,03 (дд, J1=8,1 Гц, J2=18,6 Гц, 1H), 2,78 (д, J=13,2 Гц, 1H), 2,57 (д, J =18,3 Гц, 1H), 2,32 (с,3H), 2,31 (с, 3H), 2,25 (с, 3H), 1,99 (с,3H), 1,79(дд, J1=12,0 Гц, J2=16,5 Гц, 1H), 1,73-1,42 (м, 4Н), 1,33-1,18 (т, 10Н), 1,03 (м, 2H), 0,87 (т, J=6,6 Гц, 3Н).
ESI-масс-спектроскопия, m/z: вычислено для C38H50N4О7: 674,83. Найдено (М+Н)+:675,5
Пример 44
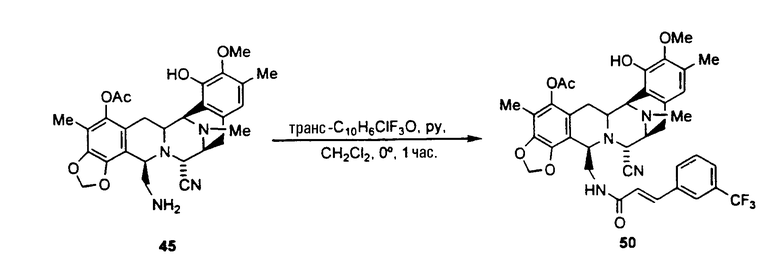
К раствору соединения 45 (14,5 мг, 0,0278 ммоль) в CH2Cl2 (0,3 мл) добавляют при 0°С транс-3-трифторметилциннамоилхлорид (4,76 мкл, 0,0278 ммоль) и пиридин (2,25 мкл, 0,0278 ммоль). Реакционную смесь перемешивают в течение 1 час, после чего раствор разбавляют, используя CH2Cl2 (5 мл), и промывают 0,1 N раствором HCl (3 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении. Остаток очищают колоночной флэш-хроматографией (SiO2, гексан:этилацетат, 1:1), получая при этом соединение 50 (18,7 мг, 94%) в виде белого твердого вещества.
Rf: 0,64 (этилацетат: метанол, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 7,74-7,55 (м, 4Н), 7,23 (д, J=16,0 Гц, 1H), 6,34 (с, 1H), 6,12 (д, J=16,0 Гц, 1H), 6,07 (д, J=0,9 Гц, 1H), 5,96 (д, J=0,9 Гц, 1H), 4,39 (д, J=2,4 Гц, 1H), 4,07-4,05 (м, 1H), 3,81 (шир.с, 1H), 3,46-3,51 (м, 3Н), 3,42 (с, 3Н), 3,09 (шир. д, J=12,0 Гц, 1H), 2,94-2,85 (м, 2H), 2,74 (д, J=18,3 Гц, 1H), 2,38 (с, 3Н), 2,23 (с, 3Н), 2,02 (с, 3Н), 1,80 (с, 3H), 1,84-1,75 (м, 1Н).
13C ЯМР (75 МГц, CDCl3): δ 168,7, 165,3, 146,5, 144,7, 142,6, 140,6, 138,0, 135,9, 131,0, 130,9, 129,1, 128,6, 125,8, 125,7, 124,5, 124,4, 122,7, 121,2, 117,8, 116,5, 113,0, 112,0, 101,7, 60,4, 59,1, 56,5, 56,4, 55,6, 55,3, 41,8, 40,3, 26,6, 25,1, 20,3, 15,4, 9,3.
ESI-масс-спектроскопия, m/z: вычислено для C38H37F3N4О7: 718,72. Найдено (М+Н)+: 719,3
Пример 45
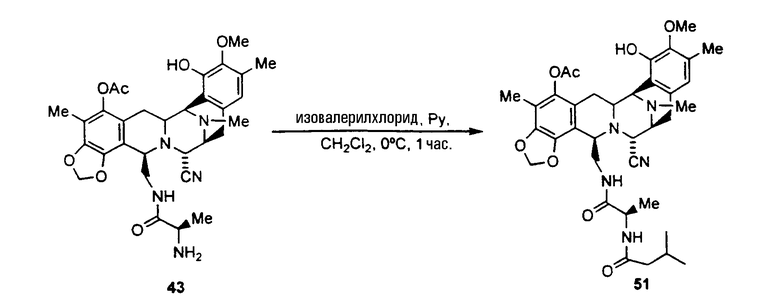
К раствору соединения 43 (33 мг, 0,0557 ммоль) в CH2Cl2 (0,4 мл) добавляют при 0°С изовалерилхлорид (6,79 мкл, 0,0557 ммоль) и пиридин (4,5 мкл, 0,0557 ммоль). Реакционную смесь перемешивают в течение 1 час, после чего раствор разбавляют CH2Cl2 (5 мл) и промывают 0,1 N раствором HCl (3 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении. Остаток очищают колоночной флэш-хроматографией (SiO2, гексан: этилацетат, 1:2), получая при этом соединение 51 (34 мг, 91%) в виде белого твердого вещества.
Rf: 0,09 (гексан:этилацетат, 1:2)
1H ЯМР (300 МГц, CDCl3): δ 6,46 (с, 1H), 6,10 (шир.с, 1H), 5,99 (д, J=0,9 Гц, 1H), 5,90 (д, J=0,9 Гц, 1H), 5,30 (т, J=6,0 Гц, 1H), 4,10-4,05 (м, 3H),3,81 (шир.с, 1H), 3,74 (с, 3H), 3,54 (шир.с, 1H), 3,38-3,36 (м, 1H), 3,29-3,21 (м, 1H), 3,00 (дд, J1=8,0 Гц, J2=18,0 Гц, 1H), 2,25 (с, 3H), 2,20 (с, 3H), 2,00 (с, 3H), 1,95-1,90 (м, 3H), 0,87 (д, J=6,6 Гц, 6H), 0,76 (д, J=6,0 Гц, 3H).
ESI-масс-спектроскопия, m/z: вычислено для C36H45N5О8: 675,77. Найдено (М+Н)+:676,3
Пример 46
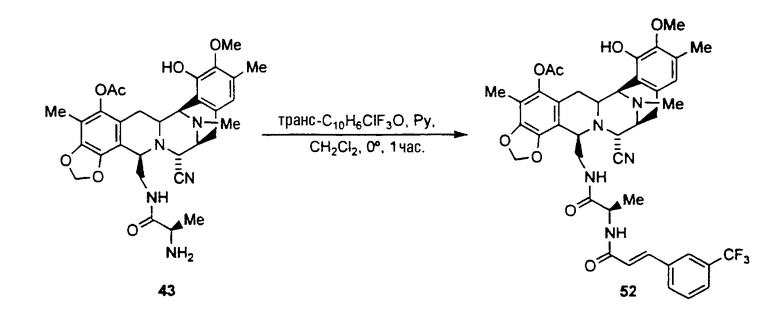
К раствору соединения 43 (33 мг, 0,0557 ммоль) в CH2Cl2 (0,4 мл) добавляют при 0°С транс-3-трифторметилциннамоилхлорид (9,52 мкл, 0,0557 ммоль) и пиридин (4,5 мкл, 0,0557 ммоль). Реакционную смесь перемешивают в течение 1 час, после чего раствор разбавляют, используя CH2Cl2 (5 мл), и промывают 0,1 N раствором HCl (3 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении. Остаток очищают колоночной флэш-хроматографией (SiO2, гексан: этилацетат, 1:1), получая при этом соединение 52 (40 мг, 92%) в виде белого твердого вещества.
Rf: 0,21 (гексан:этилацетат, 1:2)
1H ЯМР (300 МГц, CDCl3): δ 7,74-7,47 (м, 4H), 6,49 (с, 1H), 6,40 (д, J=15,6 Гц, 1H), 6,00 (д, J=1,5 Гц, 1H), 5,90 (д, J=1,5 Гц, 1H), 5,47 (т, J=6 Гц, 1H), 4,12-4,09 (м, 3H), 3,93 (шир.с, 1H), 3,71 (с, 3H), 3,59-3,58 (м, 1H), 3,38 (д, J=7,8 Гц, 1H), 3,29 (д, J=12,0 Гц, 1H), 3,00 (дд, J1=8,1 Гц, J2=18,3 Гц, 1H), 2,79-2,78 (м, 1H), 2,65 (д, J =18,3 Гц, 1H) 2,29 (с, 6H), 2,28 (с, 3H), 2,22 (с, 3H), 1,84-1,80 (м, 1H), 0,85-0,84 (м, 3H).
13С ЯМР (75 МГц, CDCl3): δ 171,9, 168,8, 164,4, 146,9, 144,6, 143,0, 140,5, 140,5, 139,3, 135,7, 131,1, 131,0, 129,4, 129,1, 126,0, 124,1, 124,0, 122,4, 121,1, 120,7, 120,6, 117,7, 116,9, 112,8, 112,0, 101,6, 60,6, 59,3, 57,1, 56,3, 55,9, 55,2, 49,0, 41,7, 49,9, 26,5, 25,1, 20,2, 18,4, 15,7, 9,3.
ESI-масс-спектроскопия, m/z: вычислено для C41H42F3N5О8: 789,8. Найдено (М+Н)+: 790,3
Пример 47
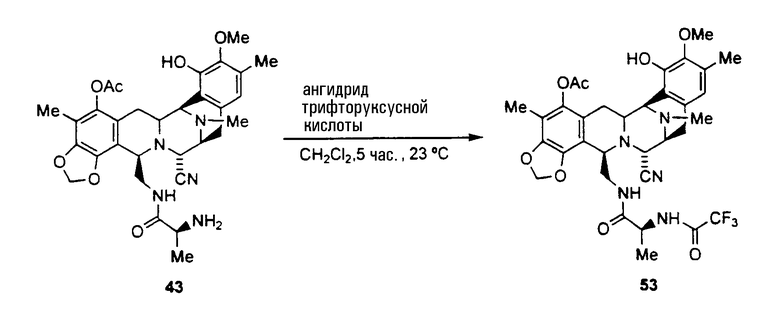
К раствору соединения 43 (10 мг, 0,0169 ммоль) в CH2Cl2 (0,2 мл) добавляют при 23°С ангидрид трифторуксусной кислоты (2,38 мкл, 0,0169 ммоль). Реакционную смесь перемешивают в течение 5 час, после чего раствор разбавляют CH2Cl2 (5 мл) и промывают 0,1 N раствором HCl (3 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении. Остаток очищают колоночной флэш-хроматографией (SiO2, гексан:этилацетат, 3:2), получая при этом соединение 53 (10,7 мг, 93%) в виде белого твердого вещества.
Rf: 0,57 (этилацетат:метанол, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 6,45 (с, 1H), 6,00 (д, J=1,2 Гц, 1H), 5,90 (д, J=1,2 Гц, 1H), 5,87 (шир.с, 1H), 5,32 (шир.с, 1H), 4,12(д, J=2,1 Гц, 1H), 4,08 (д,J=1,8 Гц, 1H), 3,78-3,56 (м, 3H), 3,72 (с, 3H), 3,40 (д, J=8,1 Гц, 1H), 3,25 (д, J=9,3 Гц, 1H), 3,00 (дд, J1=8,4 Гц, J2=18,0 Гц, 1H), 2,77 (дд, J1=2,1 Гц, J2=15,9 Гц, 1H), 2,68 (д, J=18,6 Гц, 1H), 2,30 (с, 3H), 2,28 (с, 3H), 2,22 (с, 3H), 2,00 (с, 3H), 1,75 (дд, J,= 11,4 Гц, JJ=15,9 Гц, 1H), 0,69 (д, J=6,3 Гц, 3H).
13C ЯМР (75 МГц, CDCl3): δ 170,1, 168,6, 156,0, 147,0, 144,6, 143,0, 140,6, 140,4, 131,0, 129,4, 120,9, 120,7, 117,6, 116,8, 112,4, 112,1, 101,6, 60,5, 59,0, 57,1, 56,3, 55,6, 55,2, 48,7, 41,6, 39,4, 26,5, 24,9, 20,2, 17,8, 15,4, 9,2.
ESI-масс-спектроскопия, m/z: вычислено для C33H36F3N5О8: 687,63. Найдено (М+Н)+: 688,66
Пример 48
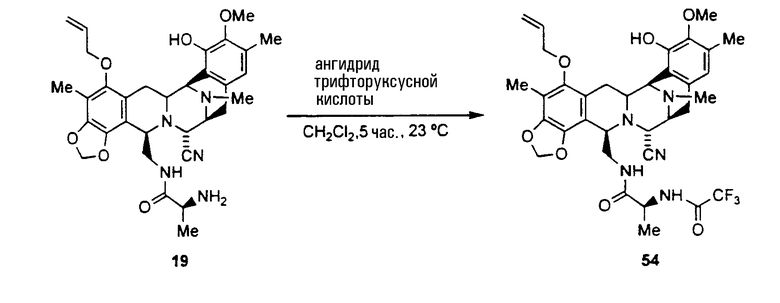
К раствору соединения 19 (11 мг, 0,0169 ммоль) в CH2Cl2 (0,2 мл) добавляют при 23°С ангидрид трифторуксусной кислоты (2,38 мкл, 0,0169 ммоль). Реакционную смесь перемешивают в течение 5 час, после чего раствор разбавляют CH2Cl2 (5 мл) и промывают 0,1 N раствором HCl (3 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении. Остаток очищают колоночной флэш-хроматографией (SiO2, гексан:этилацетат, 3:2), получая при этом соединение 54 (10,7 мг, 93%) в виде белого твердого вещества.
Rf: 0,6 (этилацетат: метанол, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 7,33 (д, J=6,3 Гц, 1H), 6,45 (с, 1H), 6,04 (м, 1H), 5,95 (д, J=1,5 Гц, 1H), 5,84 (д, J=1,5 Гц, 1H), 5,32 (м, 2H), 5,21 (м, 1H), 4,11 (м, 4H), 3,73 (с, 3Н), 3,64 (м, 2H), 3,51 (м, 1H), 3,37 (д, J=7,8 Гц, 1H), 3,22 (м, 2H), 3,03 (дд, 1H, J1=8,1 Гц, J2=18,3 Гц, 1H), 2,60 (д, J=18,3 Гц, 1H), 2,29 (с, 3Н), 2,24 (с, 3Н), 2,08 (с, 3Н), 1,86 (дд, J1=12 Гц, J2=16,2 Гц, 1H), 0,82 (д, J=7,2 Гц, 3Н).
13C ЯМР (75 МГц, CDCl3): δ 170,0, 156,0, 148,4, 147,1, 144,3, 143,0, 138,7, 133,8, 130,5, 129,4, 120,6, 120,4, 117,6, 117,5, 117,0, 113,5, 112,5, 112,4, 101,1, 74,1, 66,8, 60,4, 59,3, 56,9, 56,6, 56,3, 55,4, 48,7, 41,6, 40,1, 26,2, 25,0, 17,6, 15,4, 9,1.
ESI-масс-спектроскопия, m/z: вычислено для C35H39F3N5О7: 685,69. Найдено (М+Н)+: 686,3
Пример 49
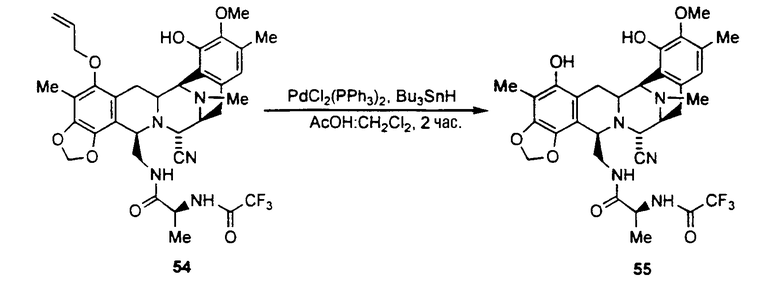
К раствору соединения 54 (100 мг, 0,145 ммоль) в CH2Cl2 (4 мл) добавляют при 23°С уксусную кислоту (40 мл), (PPh3)2PdCl2 (8,4 мг, 0,012 ммоль) и Bu3SnH (151 мл, 0,56 ммоль). После перемешивания при этой температуре в течение 2 час реакционную смесь помещают на колонку для флэш-хроматографии (SiO2, градиент от гексана до смеси гексан: этилацетат, 2:1), получая при этом соединение 55 (90 мг, 96%) в виде белого твердого вещества.
Rf: 0,6 (гексан:этилацетат, 1:2)
1H ЯМР (300 МГц, CDCl3): δ 7,55 (д, J=7,2 Гц, 1H), 6,45 (с, 1H), 5,90 (д, J=1,2 Гц, 1H), 5,82 (д, J=1,2 Гц, 1H), 5,37 (т, J=6,0 Гц, 1H), 4,15 (д, J=2,1 Гц, 1H), 4,04 (д, J=1,8 Гц, 1H), 3,70 (с, 3H), 3,66-3,53 (м, 2H), 3,37-3,31 (м, 2H), 3,19-3,15 (д, J=11,7 Гц, 1H), 3,08-3,00 (м, 2H), 2,56 (д, J=18,3 Гц, 1H), 2,30 (с, 3H), 2,24 (с, 3H), 2,04 (с, 3H), 1,91 (дд, J1=12,0 Гц, J2=15,6 Гц, 1H), 0,84 (д, J=6,9 Гц, 3H).
13C ЯМР (75 МГц, CDCl3): δ 170,1, 156,3, 147,3, 144,9, 144,4, 143,3, 136,7, 130,7, 129,3, 120,6, 117,6, 117,4, 114,4, 112,1, 107,7, 101,0, 85,8, 60,5, 59,3, 56,5, 56,4, 56,2, 55,2, 48,9, 41,6, 40,9, 25,7, 25,3, 18,0, 15,6, 8,7.
ESI-масс-спектроскопия m/z: вычислено для C32H35F3N5О7: 645,63. Найдено (М+Н)+:646,2.
Пример 50

К раствору соединения 17 (200 мг, 0,288 ммоль) в CH2Cl2 (1,44 мл) добавляют трифторуксусную кислоту (888 мкл, 11,53 ммоль) и реакционную смесь перемешивают в течение 4 час при 23°С. Реакцию гасят добавлением при 0°С насыщенного водного раствора бикарбоната натрия (60 мл) и экстрагируют этилацетатом (2 х 70 мл). Объединенные органические слои сушат (сульфат натрия) и концентрируют в вакууме, получая при этом соединение 56 (147 мг, 93%) в виде белого твердого вещества, которое используют в последующих реакциях без дополнительной очистки.
Rf: 0,19 (этилацетат:метанол, 5:1)
1H ЯМР (300 МГц, CD3OD): δ 6,48 (с, 1H), 5,88, д, J=0,9 Гц, 1H), 5,81 (д, J=0,9 Гц, 1H), 4,35 (д, J=2,4 Гц, 1H), 4,15 (д, J=1,8 Гц, 1H), 3,99-3,98 (м, 1H), 3,70 (с, 3H), 3,52-2,96 (м, 7H), 2,68 (д, J=18,3 Гц, 1H), 2,24 (с, 3H), 2,23 (с, 3H), 2,06 (с, 3H), 1,85 (дд, J1=11,7 Гц, J2=15,6 Гц, 1H), 0,91 (д, J=6,6 Гц, 3H).
13C ЯМР (75 МГц, CD3OD): δ 173,2, 149,1, 145,6, 144,9, 138,0, 132,2, 130,6, 121,4, 119,6, 117,4, 114,3, 109,2, 102,5, 82,3, 60,4, 58,4, 58,3, 57,8, 56,6, 50,1, 42,3, 41,6, 27,8, 26,2, 19,5, 15,5, 9,8.
ESI-масс-спектроскопия, m/z: вычислено для C29H35N5О6: 549,62. Найдено (М+Н)+: 550,3
Пример 51
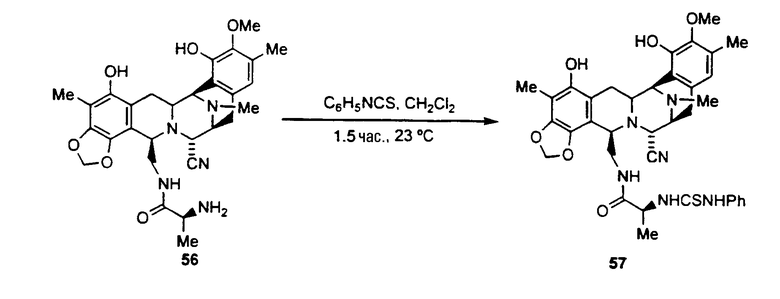
К раствору соединения 56 (10 мг, 0,018 ммоль) в CH2Cl2 (0,4 мл) добавляют фенилизотиоцианат (13 мкл, 0,109 ммоль), и реакционную смесь перемешивают при 23°С в течение 1,5 час. Смесь концентрируют в вакууме и полученный остаток очищают колоночной флэш-хроматографией (SiO2, градиент от гексана до смеси гексан:этилацетат, 1:1), получая при этом соединение 57 (8 мг, 65%) в виде белого твердого вещества.
Rf: 0,57 (этилацетат:метанол, 10:1)
1H ЯМР (300 МГц, CDCl3): δ 7,88 (шир.с, 1H), 7,41-7,36 (м, 2H), 7,27-7,22 (м, 1H), 7,02-7,00 (д, J=7,8 Гц, 2H), 6,71 (д, J=7,2 Гц, 1H), 6,31 (с, 1H), 6,17 (шир.с, 1H), 5,93 (д, J=l2 Гц, 1H), 5,83 (д, J=1,2 Гц, 1H), 5,55 (шир.с, 1H), 5,20-5,17 (м, 1H), 4,16 (д, J=1,8 Гц, 1H), 4,05 (шир.с, 1H), 4,02 (д, J=2,4 Гц, 1H), 3,79 (с, 3H), 3,75-3,71 (м, 1H), 3,35 (д, J=7,8 Гц, 1H), 3,28-3,19 (м, 2H), 3,12-2,97 (м, 2H), 2,50 (д, J=18,3 Гц, 1H), 2,32 (с, 3H), 2,21 (с, 3H), 2,15-2,09 (дд, J1=11,4 Гц, J2=15,9 Гц, 1H), 1,95 (с, 3H), 0,88 (д, J =6,9 Гц, 3H).
13C ЯМР (75 МГц, CDCl3): δ 178,5, 171,7, 147,2, 145,0, 144,3, 143,3, 137,0, 135,7, 130,6, 130,4, 129,6, 127,5, 124,3, 120,6, 117,7, 117,2, 115,3, 112,1, 108,3, 100,9, 60,9, 59,5, 56,7, 56,5, 56,2, 55,2, 54,1, 41,7, 41,1, 26,3, 25,4, 18,5, 15,8, 9,0.
ESI-масс-спектроскопия, m/z: вычислено для C36H40N6О6S: 684,81. Найдено (М+Н)+: 685,3
Пример 52
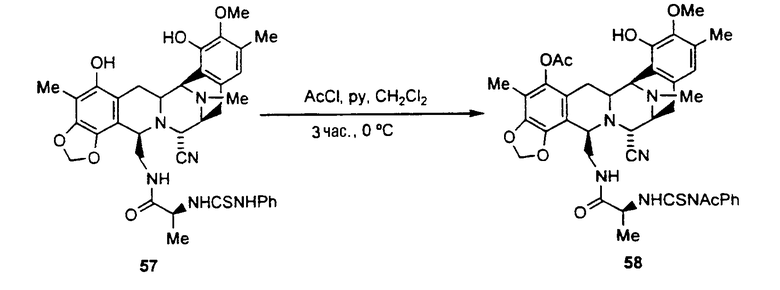
К раствору соединения 57 (45 мг, 0,065 ммоль) в CH2Cl2 (0,5 мл) добавляют при 0°С ацетилхлорид (4,67 мкл, 0,065 ммоль) и пиридин (5,3 мкл, 0,065 ммоль). Реакционную смесь перемешивают в течение 3 час, после чего раствор разбавляют, используя CH2Cl2 (10 мл), и промывают 0,1 N раствором HCl (5 мл). Органический слой сушат над сульфатом натрия, отфильтровывают и удаляют растворитель при пониженном давлении. Остаток очищают колоночной флэш-хроматографией (RP-18, CH3CN:H2O, 40:60), получая при этом соединение 58 (14 мг, 28%) в виде белого твердого вещества.
Rf: 0,34 (CH3CN:H2O, 7:15)
1H ЯМР (300 МГц, CDCl3): δ 11,90 (д, J=6,6 Гц, 1H), 7,45-7,40 (м, 3H), 7,18-7,15 (м, 2H), 6,58 (с, 1H), 6,00 (д, J=1,2 Гц, 1H), 5,89 (д, J=1,2 Гц, 1H), 5,70 (с, 1H), 5,37 (т, J =4,8 Гц, 1H), 4,48 (м, 1H), 4,23 (шир.с, 1H), 4,07 (шир.с, 2H), 3,85-3,75 (м, 1H), 3,70 (с, 3H), 3,46-3,41 (м, 2H), 3,24-3,20 (м, 1H), 3,00-2,95 (м, 1H), 2,87-2,75 (м, 1H), 2,31 (с, 3H), 2,28 (с, 3H), 2,24 (с, 3H), 2,00 (с, 3H), 1,85 (дд, J1=11,4 Гц, J2=15,6 Гц, 1H), 1,66 (с, 3H), 0,82 (д, J=6,0 Гц, 3H).
13С ЯМР (75 МГц, CDCl3): δ 182,6, 174,3, 171,0, 146,6, 144,6, 142,7, 142,3, 140,7, 140,2, 131,3, 129,8, 129,3, 128,9, 128,8, 121,5, 120,4, 117,3, 116,6, 112,8, 112,0, 111,3, 101,5, 60,5, 59,0, 57,6, 56,2, 55,9, 55,3, 55,1, 41,6, 39,4, 27,8, 26,5, 24,8, 20,2, 17,1, 15,5, 9,3.
ESI-масс-спектроскопия, m/z: вычислено для C40H44N6О8S:768,88. Найдено (М+Н)+:769,2.
Пример 53
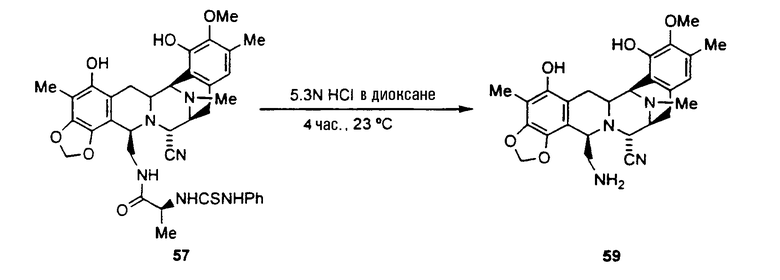
К раствору соединения 57 (130 мг, 0,189 ммоль) в диоксане добавляют смесь 5,3 N HCl/диоксан (1,87 мл) и реакционную смесь перемешивают в течение 4 час при 23°С. Затем к реакционной смеси добавляют CH2Cl2 (15 мл) и Н2О (10 мл) и декантируют органический слой. Водную фазу подщелачивают насыщенным водным раствором бикарбоната натрия (60 мл) (рН=8) при 0°C, после чего экстрагируют этилацетатом (2 х 50 мл). Объединенные органические экстракты сушат (сульфат натрия) и концентрируют в вакууме, получая при этом соединение 59 (63 мг, 70%) в виде белого твердого вещества.
Rf: 0,15 (этилацетат:метанол, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 6,67 (с, 1H), 5,99 (д, J=0,9 Гц, 1H), 5,91 (д, J=1,2 Гц, 1H), 5,10 (шир.с, 1H), 4,32 (д, J=7,2 Гц, 1H), 4,25 (дд, J1=3,6 Гц, J2=9,3 Гц, 1H), 3,7 (с, 3H), 3,71-3,64 (м, 2H), 3,50 (дд, J1=2,4 Гц, J2=15,9 Гц, 1H), 3,42-3,37 (м, 2H), 3,16 (дд, J1=3,6 Гц, J2=12,9 Гц, 1H), 2,57 (дд, J1=9,3 Гц, J2=12,9 Гц, 1H), 2,27 (с, 3H), 2,11 (с, 3H), 1,91 (дд, J1=12,0 Гц, J2=15,9 Гц, 1H).
ESI-масс-спектроскопия, m/z: вычислено для C26H30N4О5: 478,5. Найдено (М+Н)+: 479,3
Пример 54
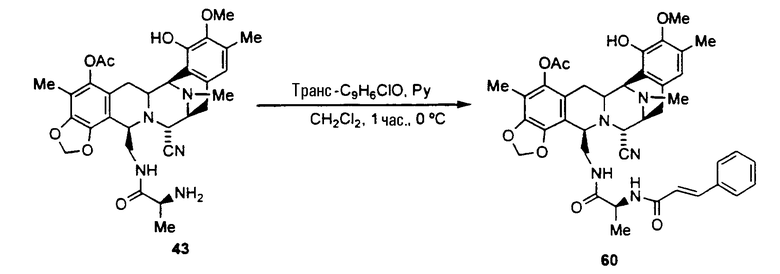
К раствору соединения 43 (20 мг, 0,0338 ммоль) в CH2Cl2 (0,3 мл) добавляют при 0°С циннамоилхлорид (5,63 мг, 0,0338 ммоль) и пиридин (2,73 мл, 0,0338 ммоль). Реакционную смесь перемешивают в течение 1 час, после чего раствор разбавляют, используя CH2Cl2 (10 мл), и промывают 0,1 N раствором HCl (5 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении. Остаток очищают колоночной флэш-хроматографией (SiO2, EtOAc:MeOH, 20:1), получая при этом соединение 60 (22 мг, 90%) в виде белого твердого вещества.
Rf: 0,56 (EtOAc: MeOH, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 7,51 (с, 1H), 7,50-7,47 (м, 2H), 7,36-7,35 (м, 2H), 6,43 (с, 1H), 6,36 (шир.д, J=15,9 Гц, 2H), 6,01 (д, J=1,5 Гц, 1H), 5,90 (шир.д, J=1,5 Гц, 2H), 5,42 (т, J=6,0 Гц 1H), 4,12-4,07 (м, 3H), 3,96-3,95 (м, 1H), 3,73 (шир.с, 3H), 3,58 (шир.с, 2H), 3,39 (д, J=8,7 Гц, 1H), 3,25 (д, J=11,7 Гц, 1H), 3,0 (дд, J1=7,5 Гц, J2=17,7 Гц, 1H), 2,78 (д, J=15,9 Гц, 1H), 2,67 (д, J=16,5 Гц, 1H), 2,29 (с, 6H), 2,23 (с, 3H), 1,99 (с, 3H), 1,82 (дд, J1=11,4 Гц, J2=15,6 Гц, 1H), 0,83 (д, J=6,0 Гц, 3H).
13C ЯМР (75 МГц, CDCl3): δ 172,0, 165,0, 146,9, 144,6, 143,1, 141,0, 140,5, 134,8, 131,0, 129,7, 129,1, 128,8, 127,8, 125,5, 123,8, 123,0, 121,1, 120,5, 117,7, 116,9, 112,8, 112,0, 101,9, 60,6, 59,2, 57,1, 56,4, 55,9, 55,3, 48,8, 41,7, 40,0, 26,5, 25,1, 20,3, 18,5, 15,7, 9,3.
ESI-масс-спектроскопия, m/z: вычислено для C40H43N5О8: 721,8. Найдено (М+Н)+: 722,3
Пример 55
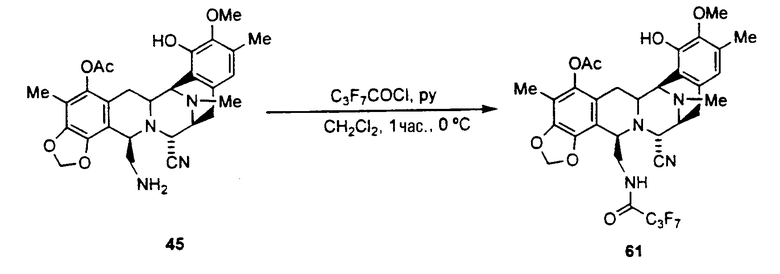
К раствору соединения 45 (19 мг, 0,0364 ммоль) в CH2Cl2 (0,3 мл) добавляют при 0°С гептафторбутирилхлорид (5,44 мкл, 0,0364 ммоль) и пиридин (2,95 мкл, 0,0364 ммоль). Реакционную смесь перемешивают в течение 1 час, после чего раствор разбавляют, используя CH2Cl2 (10 мл), и промывают 0,1 N раствором HCl (5 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении. Остаток очищают колоночной флэш-хроматографией (SiO2, EtOAc:MeOH, 20:1), получая при этом соединение 61 (11,7 мг, 45%) в виде белого твердого вещества.
Rf: 0,76 (EtOAc:MeOH, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 6,46 (с, 1H), 6,12 (шир.с, 1H), 5,98 (д, J=1,2 Гц, 1H), 5,93 (д, J=1,2 Гц, 1H), 5,72 (шир.с, 1H), 4,13-4,11 (м, 2H), 4,0 (д, J=2,4 Гц, 1H), 3,98-3,96 (м, 1H), 3,73 (с, 3H), 3,39 (д, J=7,5 Гц, 1H), 3,39-3,28 (м, 2H), 3,09 (дд, J1=8,1 Гц, J2=18,0 Гц, 1H), 2,80 (д, J=16,2 Гц, 1H), 2,46 (д, J=18,3 Гц, 1H), 2,32 (с, 6H), 2,21 (с, 3H), 1,99 (с, 3H), 1,80 (дд, J1=12,0 Гц, J2=16,2 Гц, 1H).
ESI-масс-спектроскопия, m/z: вычислено для C32H31F7N4О7: 716,6. Найдено (М+Н)+: 717,2
Пример 56

К раствору соединения 43 (24 мг, 0,04 ммоль) в CH2Cl2 (0,3 мл) добавляют при 0°С бутирилхлорид (4,15 мкл, 0,04 ммоль) и пиридин (3,28 мкл, 0,04 ммоль). Реакционную смесь перемешивают в течение 1 час, после чего раствор разбавляют, используя CH2Cl2 (10 мл), и промывают 0,1 N раствором HCl (5 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении. Остаток очищают колоночной флэш-хроматографией (SiO2, EtOAc: MeOH, 20:1), получая при этом соединение 62 (24 мг, 90%) в виде белого твердого вещества.
Rf: 0,35 (EtOAc:MeOH, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 6,47 (с, 1H), 6,10 (д, J=6,5 Гц, 1H), 6,0 (д, J=1,5 Гц, 1H), 5,91 (д, J=1,5 Гц, 1H), 5,86 (шир.с, 1H), 5,31 (д, J=6,9 Гц, 1H), 4,11-4,06 (м, 3Н), 3,85-3,81 (м, 1H), 3,75 (с, 3Н), 3,59-3,53 (м, 2Н), 3,38 (д, J=7,5 Гц, 1H), 3,27-3,22 (м, 1H), 3,0 (дд, j1=7,8 Гц, J2=17,4 Гц, 1H), 2,79(д, J=15,3 Гц, 1H), 2,63 (д, J=17,7 Гц, 1H), 2,31 (с, 3Н), 2,0 (с, 3Н), 1,80 (дд, J1=12,0 Гц, J2=15,9 Гц, 1H), 1,58 (кв., J=7,2 Гц, 2Н), 0,89 (т, J=7,2 Гц, 3Н), 0,76 (д, J=6,6 Гц, 3Н).
ESI-масс-спектроскопия, m/z: вычислено для C35H43N5О8:661,64. Найдено (М+Н)+:662,3
Пример 57

К раствору соединения 43 (19 мг, 0,0364 ммоль) в CH2Cl2 (0,3 мл) добавляют при 0°С циннамоилхлорид (6,06 мг, 0,0364 ммоль) и пиридин (2,95 мкл, 0,0364 ммоль). Реакционную смесь перемешивают в течение 1 час, после чего раствор разбавляют, используя CH2Cl2 (10 мл) и промывают 0,1 N раствором HCl (5 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении. Остаток очищают колоночной флэш-хроматографией (SiO2, EtOAc:MeOH, 20:1), получая при этом соединение 63 (20,1 мг, 85%) в виде белого твердого вещества.
Rf: 0,65 (EtOAc:MeOH, 5:1)
1Н ЯМР (300 МГц, CDCl3): δ 7,39-7,29 (м, 5H), 6,42, (с, 1H), 6,01 (д, J=1,5 Гц, 1H), 5,92 (д, J=1,5 Гц, 1H), 5,73 (шир.с, 1H), 5,24 (т, J=6,8 Гц, 1H), 4,12-4,08 (м, 3H), 3,66-3,64 (м, 2H), 3,58 (шир.с, 3H), 3,36 (д, J=8,7 Гц, 1H), 3,29 (д, J=12,0 Гц, 1H), 2,98 (дд, J1=8,1 Гц, J2=18 Гц, 1H), 2,33 (с, 6Н), 2,29 (с, 3H), 2,01 (с, 3H), 1,84 (дд, J1=12,0 Гц, J2=15,9 Гц, 1H).
ESI-масс-спектроскопия, m/z: вычислено для C37H38N4О7:650,72. Найдено (М+Н)+:651,2
Пример 58
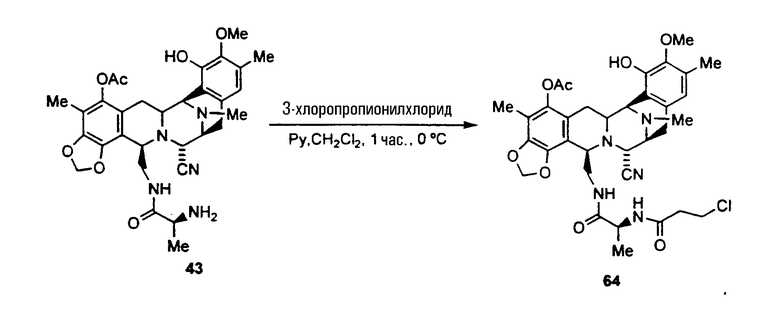
К раствору соединения 43 (20 мг, 0,0338 ммоль) в CH2Cl2 (0,3 мл) добавляют при 0°С 3-хлорпропионилхлорид (3,22 мкл, 0,0338 ммоль) и пиридин (2,73 мкл, 0,0338 ммоль). Реакционную смесь перемешивают в течение 1 час, после чего раствор разбавляют, используя CH2Cl2 (10 мл), и промывают 0,1 N раствором HCl (5 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении. Остаток очищают колоночной флэш-хроматографией (SiO2, EtOAc:MeOH, 20:1), получая при этом соединение 64 (20,5 мг, 89%) в виде белого твердого вещества.
Rf: 0,32 (EtOAc:гексан, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 6,48 (с, 3H), 6,28 (м, 1H), 5,99 (д, J=1,2 Гц, 1H), 5,91 (д, J=1,2 Гц, 1H), 5,86 (шир.с, 1H), 5,31 (м, 1H), 4,08-4,07 (м, 3H), 3,75 (с, 3H), 3,72-3,53 (м, 5Н), 3,39 (д, J=8,1 Гц, 1H), 3,24 (д, J=12,0 Гц, 1H), 3,00 (дд, J1=8,1 Гц, J2=18,0 Гц, 1H), 2,79 (д, J=13,5 Гц, 1H), 2,50 (т, J=6,3 Гц, 2H), 2,32 (с, 3H), 2,28 (с, 3H), 2,25 (с, 3H), 2,0 (с, 3H), 1,79(дд, J1=12,3 Гц, J2=14,8 Гц, 1H), 0,81 (д, J=6,3 Гц, 3H).
Пример 59

К раствору соединения 43 (19 мг, 0,0364 ммоль) в CH2Cl2 (0,3 мл) добавляют при 0°С бутирилхлорид (3,78 мкл, 0,0364 ммоль) и пиридин (2,95 мкл, 0,0364 ммоль). Реакционную смесь перемешивают в течение 1 час, после чего раствор разбавляют, используя CH2Cl2 (10 мл), и промывают 0,1 N раствором HCl (5 мл). Органический слой сушат над сульфатом натрия, фильтруют и удаляют растворитель при пониженном давлении. Остаток очищают колоночной флэш-хроматографией (SiO2, EtOAc:MeOH, 20:1), получая при этом соединение 64 (19 мг, 87%) в виде белого твердого вещества.
Rf: 0,60 (EtOAc: МеОН, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 6,50 (с, 1H), 5,98 (д, J=1,5 Гц, 1H), 5,91 (д, J=1,5 Гц, 1H), 5,75 (с, 1H), 5,01 (т, J=6,4 Гц, 1H), 4,10-4,09 (м, 1H), 4,06 (д, J=2,1 Гц, 1H), 4,03-4,02 (м, 1H), 3,76 (с, 3H), 3,67-3,60 (м, 1H), 3,42-3,35 (м, 2Н), 3,29 (д, J=12,0 Гц, 1H), 3,02 (дд, J1=7,8 Гц, J2=17,7 Гц, 1H), 2,79 (д, J=14,1 Гц, 1H), 2,56 (д, J=18,3 Гц, 1H), 2,32 (с, 3H), 2,31 (с, 3H), 2,25 (с, 3H), 1,78 (дд, J1=12,0 Гц, J2=15,9 Гц, 1H), 1,63 (с, 3H), 1,53-1,46 (м, 2Н), 1,28-1,16 (м, 2Н), 0,68 (т, J=7,2 Гц, 3H).
ESI-масс-спектроскопия, m/z: вычислено для C32H38N4О7: 590,67. Найдено (М+Н)+: 591,2
Пример 60
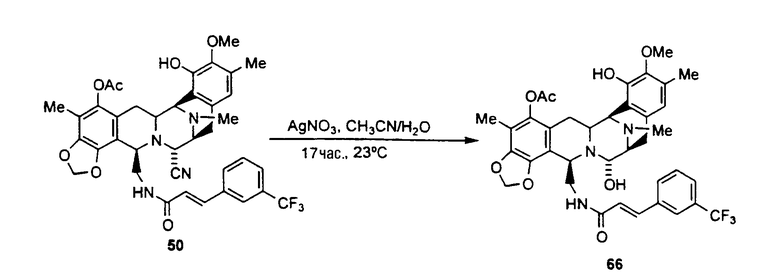
К раствору соединения 50 (31,7 мг, 0,044 ммоль) в CH3CN/H2O (1,5 мл/0,5 мл) добавляют AgNO3 (225 мг, 1,32 ммоль), и реакционную смесь перемешивают при 23°С в течение 17 час. Затем при 0°С добавляют насыщенный раствор соли (10 мл) и насыщенный водный раствор NaNCO3 (10 мл) и полученную смесь перемешивают в течение 15 мин, фильтруют через слой целита и промывают, используя CH2Cl2 (20 мл). Раствор декантируют, органический слой сушат и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией (SiO2, EtOAc:MeOH, 5:1), получая при этом соединение 66 (16 мг, 51%) в виде белого твердого вещества.
Rf: 0,26 (EtOAc:МеОН, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 7,66-7,42 (м, 4H), 7,20 (шир.с, 1H), 6,44 (с, 1H), 5,97 (шир., J=1,2 Гц, 1H), 5,90 (д, J=1,2 Гц, 1H), 5,76 (шир.с, 1H), 5,28 (шир.с, 1H), 4,54 (шир.с, 1H), 4,43 (шир.с, 1H), 4,00 (шир.с, 1H), 3,68-3,57 (м, 4H), 3,47 (д, J=3,3 Гц, 1H), 3,40 (д, J=11,7 Гц, 1H), 3,17 (д, J=6,9 Гц, 1H), 2,92 (дд, j1=8,1 Гц, J2=17,7 Гц, 1H), 2,74 (д, J=17,1 Гц, 1H), 2,48 (д, J=18,6 Гц, 1H), 2,32 (с, 6H), 2,28 (с, 3H), 1,99 (с, 3H), 1,76 (дд, J1=12,0 Гц, J2=16,2 Гц, 1H),
ESI-масс-спектроскопия, m/z: вычислено для C37H38F3N3О8: 709. Найдено (М+-17): 692,3
Пример 61
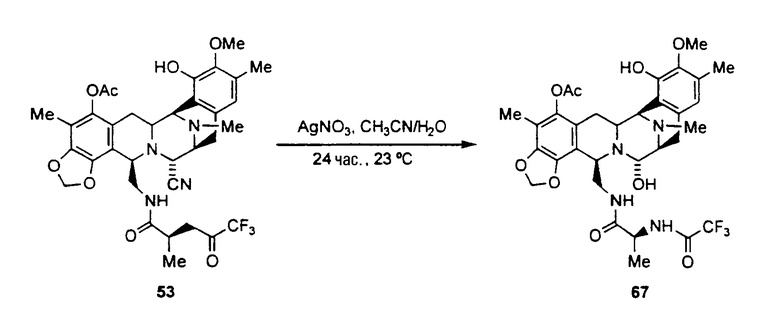
К раствору соединения 53 (57 мг, 0,0828 ммоль) в CH3CN/H2O (1,5 мл/0,5 мл) добавляют AgNO3 (650 мг, 3,81 ммоль) и реакционную смесь перемешивают при 23°С в течение 24 час. Затем при 0°С добавляют насыщенный раствор соли (10 мл) и насыщенный водный раствор NaHCO3 (10 мл) и полученную смесь перемешивают в течение 15 мин, фильтруют через слой целита и промывают, используя CH2Cl2 (20 мл). Раствор декантируют, органический слой сушат и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией (SiO2, EtOAc: MeOH, 5:1), получая при этом соединение 67 (28 мг, 50%) в виде белого твердого вещества.
Rf: 0,28 (EtOAc:МеОН, 10:1)
1H ЯМР (300 МГц, CDCl3): δ 6,47 (с, 1H), 5,97 (с, 1H), 5,88 (с, 1H), 5,35 (шир.с, 1H), 4,51 (шир.с, 1H), 4,41 (шир.с, 1H), 4,12-4,05 (м, 1H), 4,00 (д, J=2,7 Гц, 1H), 3,77 (с, 3H), 3,64 (шир.с, 1H), 3,46 (д, J=3,3 Гц, 1H), 3,34 (д, J=11,4 Гц, 1H), 3,18 (д, J=7,5 Гц, 1H), 2,95 (дд, J1=8,4 Гц, J2=18,3 Гц, 1H), 2,70 (д, J=15,6 Гц, 1H), 2,48 (д, J=17,7 Гц, 1H), 2,28 (с, 3H), 2,27 (с, 3H), 2,26 (с, 3H), 1,98 (с, 3H), 1,68 (дд, J1=12 Гц, J2=15,6 Гц, 1H), 0,86 (д, J=6,3 Гц, 3H).
ESI-масс-спектроскопия, m/z: вычислено для C32H37F3N4О9: 678,66. Найдено (М+-17): 661,2
Пример 62
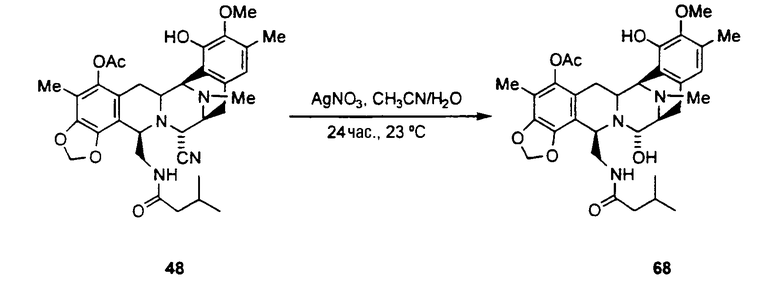
К раствору соединения 48 (32 мг, 0,0529 ммоль) в CH3CN/H2O (1,5 мл/0,5 мл) добавляют AgNO3 (270 мг, 1,58 ммоль), и реакционную смесь перемешивают при 23°С в течение 24 час. Затем при 0°С добавляют насыщенный раствор соли (10 мл) и насыщенный водный раствор NaHCO3 (10 мл), и полученную смесь перемешивают в течение 15 мин, фильтруют через слой целита и промывают CH2Cl2 (20 мл). Раствор декантируют, органический слой сушат и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией (SiO2, EtOAc:MeOH, 5:1), получая при этом соединение 68 (18 мг, 56%) в виде белого твердого вещества.
Rf: 0,40 (EtOAc:МеОН, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 6,50 (с, 1H), 5,95 (д, J=1,2 Гц, 1H), 5,88 (д, J=1,2 Гц, 1H), 5,23 (д, J=6,9 Гц, 1H), 4,45 (д, J=3,3 Гц, 1H), 4,38 (с, 1H), 4,01 (д, J=2,4 Гц, 1H), 3,78 (м, 1H), 3,77 (с, 3H), 3,41-3,37 (м, 1H), 3,17-3,15 (м, 1H), 2,96 (дд, J1=7,8 Гц, J2=18,0 Гц, 1H), 2,70 (д, J=15,3 Гц, 1H), 2,40 (д, J=18,0 Гц, 1H), 2,30 (с, 6H), 2,27 (с, 3H), 1,76-1,65 (м, 1H), 1,35-1,25 (м, 2H), 0,89-0,82 (м, 1H), 0,69 (д, J=6,6 Гц, 3H), 0,58 (д, J=6,6 Гц, 3H).
Пример 63

К раствору соединения 51 (27 мг, 0,04 ммоль) в CH3CN/H2O (1,5 мл/0,5 мл) добавляют AgNO3 (204 мг, 1,19 ммоль) и реакционную смесь перемешивают при 23°С в течение 24 час. Затем при 0°С добавляют насыщенный раствор соли (10 мл) и насыщенный раствор NaHCO3 (10 мл) и полученную смесь перемешивают в течение 15 мин, фильтруют через слой целита и промывают, испльзуя CH2Cl2 (20 мл). Раствор декантируют, органический слой сушат и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией (SiO2, EtOAc: MeOH, 5:1), получая при этом соединение 69 (10 мг, 38%) в виде белого твердого вещества.
Rf: 0,38 (EtOAc:МеОН, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 6,48 (s, 1H), 6,16 (шир.с, 1H), 5,98 (д, J=1,5 Гц, 1H), 5,89 (д, J=1,5 Гц, 1H), 5,33 (т, J=6,0 Гц, 1H), 4,50 (м, 1H), 4,40 (м, 1H), 4,11-4,09 (м, 1H), 4,00 (д, J=2,6 Гц, 1H), 3,78 (с, 3H), 3,41-3,32 (м, 3H), 3,18 (д, J=8,4 Гц, 1H), 2,94 (дд, J1=8,4 Гц, J2=18,3 Гц, 1H), 2,70 (д, J=14,4 Гц, 1H), 4,45 (д, J=18,3 Гц, 1H), 2,31 (с, 3H), 2,28 (с, 3H), 2,27 (с, 3H), 2,04 (с, 3H), 2,00-1,86 (м, 3H), 1,73 (м, 1H), 0,87 (д, J=6,3 Гц, 6H).
Пример 64
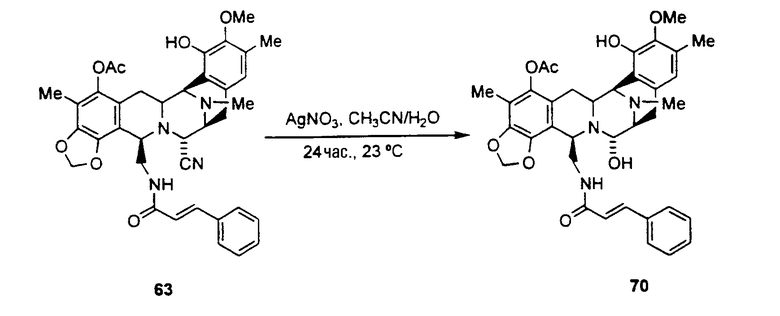
К раствору соединения 63 (15 мг, 0,023 ммоль) в CH3CN/H2O (1,5 мл/0,5 мл) добавляют AgNO3 (118 мг, 0,691 ммоль) и реакционную смесь перемешивают при 23°С в течение 24 час. Затем при 0°С добавляют насыщенный раствор соли (10 мл) и насыщенный водный раствор NaHCO3 (10 мл), полученную смесь перемешивают в течение 15 мин, фильтруют через слой целита и промывают, используя CH2Cl2 (20 мл). Раствор декантируют, органический слой сушат и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией (SiO2, EtOAc: MeOH, 5:1), получая при этом соединение 70 (20,1 мг, 85%) в виде белого твердого вещества.
Rf: 0,43 (EtOAc:МеОН, 5:1)
1H ЯМР (300 МГц, CDCl3): δ 7,38-7,28 (м, 5H), 6,48 (с, 1H), 5,98 (д, J=1,5 Гц, 1H), 5,91 (д, J=1,5 Гц, 1H), 5,75 (шир.с, 1H), 5,38 (шир.д, 1H), 5,30 (шир.с, 1H), 4,53 (м, 1H), 4,42 (м, 1H), 4,02 (д, J=2,7 Гц, 1H), 3,78-3,65 (м, 5H), 3,46-3,40 (м, 2H), 3,17 (д, J=7,8 Гц, 1H), 2,94 (дд, J1 =7,8 Гц, J2=17,7 Гц, 1H), 2,73 (д, J=16,8 Гц, 1H), 2,45 (д, J=18,0 Гц, 1H), 2,31 (с, 6H), 2,28 (с, 3Н), 1,97 (с, 3H), 1,77 (дд, J1=12,0 Гц, J2=15,3 Гц, 1H).
Пример 65
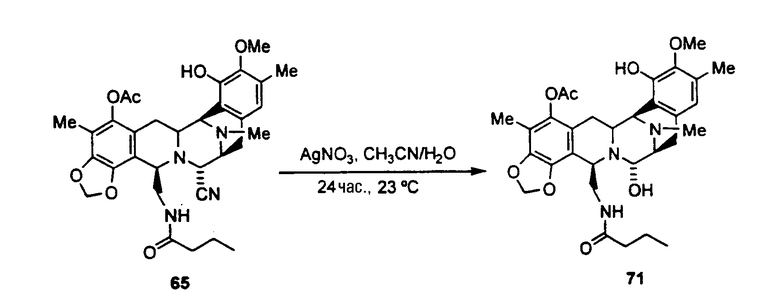
К раствору соединения 65 (25 мг, 0,042 ммоль) в CH3CN/H2O (1,5 мл/0,5 мл) добавляют AgNO3 (215,56 мг, 1,269 ммоль) и реакционную смесь перемешивают при 23°С в течение 24 час. Затем при 0°С добавляют насыщенный раствор соли (10 мл) и насыщенный водный раствор NaHCO3 (10 мл), полученную смесь перемешивают в течение 15 мин, фильтруют через слой целита и промывают, используя CH2Cl2 (20 мл). Раствор декантируют, органический слой сушат и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией (SiO2, EtOAc:MeOH, 5:2), получая при этом соединение 71 (16 мг, 65%) в виде белого твердого вещества.
Rf: 0,5 (EtOAc:МеОН, 5:2)
1H ЯМР (300 МГц, CDCl3): δ 6,50 (с, 1H), 5,95 (д, J=1,5 Гц, 1H), 5,78 (с, 1H), 5,19 (шир.с, 1H), 4,45 (д, J=3,3 Гц, 1H), 4,37(шир.с, 1H), 4,11 (шир.д, 7=4,8 Гц, 1H), 4,01 (д, J=2,1 Гц, 1H), 3,76 (с, 1H), 3,71-3,69 (м, 1H), 3,49-3,35 (м, 1H), 3,24 (д, J=13,5 Гц, 1H), 3,15 (д, J=9,3 Гц, 1H), 2,95 (дд, J1=8,1 Гц, J2=17,7 Гц, 1H), 2,70 (д, J =15,6 Гц, 1H), 2,40 (д, J=18,0 Гц, 1H), 2,31 (с, 3H), 2,29 (с, 3H), 2,26 (с, 3H), 1,96 (с, 3H), 1,75-1,66 (м, 1H), 1,52-1,17 (м, 2H), 0,66 (т, J=7,2 Гц, 3H).
Способы ферментации
Пример А
Среду для посева YMP3, содержащую 1% глюкозы; 0,25% мясного экстракта; 0,5% бактопептона; 0,25% NaCl; 0,8% CaCO3, инокулируют 0,1% маточного замороженного вегетативного микроорганизма, штамм А2-2 Pseudomonas fluorescens, и инкубируют на роторном смесителе (250 об/мин) при 27°С. Через 30 часов инкубирования засевную культуру добавляют в перемешиваемый сосуд-ферментер, содержащий среду для выращивания, состоящую из 2% декстрозы, 4% маннита, 2% сухих пивных дрожжей (Vitalevor® Biolux, Belgium); 1% (NH4)2SO4, 0,04% K2HPO4; 0,8% KCl; 0,001% FeCl3; 0,1% L-Tyr; 0,8% CaCO3; 0,05% PPG-2000; 0,2% антивспенивающего силикона (ASSAF-1000, RHODIA UK). Стерилизацию проводят при 122°C в течение 30 минут. Объем, использованный для инокуляции, составляет 2% (об/об). Температура составляет 27°С (от 0 до 16 часов) и 24°С - от 16 час до окончания процесса (41 час). Давление растворенного кислорода достигает до 25%. Значение рН поддерживают равным 6,0 с помощью разбавленной серной кислоты, после 28 часов с начала процесса и до его окончания. Избыточное давление составляет 0,5 бар. После 16 часов с начала процесса и до его окончания добавляют 1% маннит или сорбит (в течение двух дней) и 2% - для трехдневного процесса ферментации.
По истечении 41 или 64 часов подвергнутую ферментации питательную среду необходимо подвергнуть экстракции для выделения сафрацина В или необходимо обработать осветленную питательную среду добавлением KCN для выделения цианосафрацина В.
Пример В
Получение цианосафрацина В из неочищенного экстракта
Путем осветления или фильтрации подвергнутой ферментации питательной среды при рН 6 удаляют твердые составляющие. Значение рН осветленной питательной среды доводят до рН 9,5 добавлением разбавленного раствора гидроксида натрия и дважды экстрагируют этилацетатом, метиленхлоридом или бутилацетатом, 2:1 (об/об). Экстракцию проводят при перемешивании в сосуде в течение 20 минут, температура смеси поддерживается при 8-10°С. Две фазы разделяют с помощью центрифуги, обеспечивающей разделение жидких фаз. Органическую фазу сушат над безводным сульфатом натрия или замораживают и затем отфильтровывают для удаления льда. Органическую фазу (этилацетатный слой) упаривают до получения неочищенного маслянистого экстракта.
Пример С
Получение цианосафрацина В из осветленной питательной среды
Путем осветления или фильтрации подвергнутой ферментации питательной среды при рН 6 удаляют твердые составляющие. Значение рН осветленной питательной среды доводят до рН 3,9 добавлением концентрированной уксусной кислоты. К осветленной питательной среде добавляют KCN в количестве 0,5 граммов на литр и инкубируют при 20°С в течение 1 часа при перемешивании. Затем температуру понижают до 15°С, доводят рН до 9,5 добавлением разбавленного раствора гидроксида натрия и экстрагируют этилацетатом, 2:1,5 (об/об). Экстракцию проводят при встряхивании в сосуде в течение 20 минут, температура смеси поддерживается при 8-10°С. Две фазы разделяют с помощью центрифуги, обеспечивающей разделение жидких фаз. Органическую фазу сушат над безводным сульфатом натрия. Органическую фазу (этилацетатный слой) упаривают до получения неочищенного масляного экстракта. Этот экстракт очищают колоночной флэш-хроматографией (SiO2, этилацетат:метанол, градиент от 20:1 до 10: до 5:1), получая при этом с количественным выходом соединение 2 в виде светло-желтого твердого вещества.
Rf: 0,55 (этилацетат: метанол, 5:1); tR=19,9 мин [ВЭЖХ, Delta Pack C4, 5 мкм, 300 Å, 150 х 3 мм, λ=215 нм, скорость потока=0,7 мл/мин, температура=50°С, градиент: CH3CN -водн. NaOAc (10 мМ) 85%-70% (20 мин)];
1H ЯМР (300 МГц, CDCl3): δ 6,54 (дд, J1=4,4Гц, J2=8,4 Гц, 1H), 6,44 (с, 1H), 4,12 (д, J=2,4 Гц, 1H), 4,04 (д, J=2,4 Гц, 1H), 4,00 (с, 3H), 3,87 (шир.с, 1H), 3,65 (ддд, J1=1,5 Гц, J2=8,7 Гц, J3=9,9 Гц, 1H), 3,35 (шир.д, J=8,4 Гц, 1H), 3,15-2,96 (м, 4H), 2,92 (кв., J=7,2 Гц, 1H), 2,47 (д, J=18,3 Гц, 1H), 2,29 (с, 3H), 2,18 (с, 3H) 1,83 (с, 3H), 1,64 (ддд, J1=2,7 Гц, J2=11,1 Гц, J3=14,1 Гц, 1H), 0,79 (д, J=7,2 Гц, 3H).
13C ЯМР (75 МГц, CDCl3): δ 186,0 (кв.), 175,9 (кв.), 156,2 (кв.), 146,8 (кв.), 142,8 (кв.), 140,7 (кв.), 136,6 (кв.), 130,5 (кв.), 128,8 (кв.), 127,0 (кв.), 120,5 (с), 117,4 (кв.), 116,5 (кв.), 60,8 (т), 60,4 (с), 58,7 (т), 56,2 (с), 55,7 (с), 54,8 (с), 54,8 (с), 54,4 (с), 50,0 (с), 41,6 (т), 39,8 (д), 25,2 (д), 24,4 (д), 21,2 (т), 15,5 (т), 8,4 (т).
ESI-масс-спектроскопия, m/z: вычислено для C29H35N5О6: 549,6. Найдено (М+Н)+: 572,3
Пример D
Среду (50 л), содержащую декстрозу (2%), маннит (4%), сухие пивные дрожжи (2%), сульфат аммония (1%), вторичный (кислый) фосфат кальция, (0,04%), хлорид калия (0,8%), 6-гидрат хлорида железа (III) (0,001%), L-тирозин (0,1%), карбонат кальция (0,8%), полипропиленгликоль 2000 (0,05%) и антивспенивающее средство ASSAF 1000 (0,2%), выливают в сосуд-ферментер общей емкостью 75 л и, после стерилизации, инокулируют засевной культурой (2%) - штаммом А2-2 (FERM BP-14) и осуществляют культивирование с аэрацией при встряхивании при температуре от 27 до 24°С в течение 64 часов (аэрация со скоростью 75 л в мин и перемешивание, от 350 до 500 об/мин). Значение рН контролируют путем автоматической подачи разбавленного раствора серной кислоты, начиная с 27 часа с начала процесса и до его окончания. Начиная с 16 часа с начала процесса и до его окончания, добавляют 2% раствор маннита. Значение рН полученной таким образом культуральной среды (объем 45 л), после удаления клеток центрифугированием, рН доводят до 9,5 добавлением разбавленного раствора гидроксида натрия, дважды экстрагируя с использованием 25 л этилацетата. Смесь перемешивают во встряхиваемом сосуде в течение 20 минут при 8°С. Две фазы разделяют с помощью центрифуги, обеспечивающей разделение жидких фаз. Органическую фазу замораживают при -20°С и отфильтровывают для удаления льда, после чего упаривают до получения 40 г темного неочищенного масляного экстракта. После введения цианид-группы и очисткполучая при этом 3,0 грамма цианосафрацина В.
Пример Е
Среду (50 л), содержащую декстрозу (2%), маннит (4%), сухие пивные дрожжи (2%), сульфат аммония (1%), вторичный (кислый) фосфат кальция (0,02%), хлорид калия (0,2%), 6-гидрат хлорида железа (III) (0,001%), L-тирозин (0,1%), карбонат кальция (0,8%), полипропиленгликоль 2000 (0,05%) и антивспенивающее средство ASSAF 1000 (0,2%), выливают в сосуд-ферментер общей емкостью 75 л и, после стерилизации, инокулируют засевной культурой (2%) - штаммом А2-2 (FERM BP-14) и осуществляют культивирование с аэрацией при встряхивании при температуре от 27 до 24°С в течение 41 часа (аэрация со скоростью 75 л в мин и перемешивание, от 350 до 500 об/мин). Значение рН контролируют путем автоматической подачи разбавленного раствора серной кислоты, начиная с 28 часа с начала процесса и до его окончания. Начиная с 16 часа от начала процесса и до его окончания, добавляют 1% маннит. Значение рН полученной таким образом культуральной среды (объем 45 л), после удаления клеток центрифугированием, доводят до 3,9 добавлением 200 мл концентрированной уксусной кислоты. Добавляют 25 граммов 97% цианида калия и через 1 час встряхивания при 20°С значение рН доводят до 9,5 добавлением 1500 мл 10%-ного раствора гидроксида натрия. После этого экстрагируют, используя 35 л этилацетата. Смесь перемешивают во встряхиваемом сосуде в течение 20 минут при 8°С. Две фазы разделяют с помощью центрифуги, обеспечивающей разделение жидких фаз. Органическую фазу сушат над безводным сульфатом натрия, после чего упаривают до получения 60 г темного неочищенного масляного экстракта.
После хроматографирования получают 4,9 г цианосафрацина В.
Ссылки
Европейский патент 309477
Патент США 5721362



| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ТАКСАНА, ФУНКЦИОНАЛИЗИРОВАННЫЕ ПО 14-ПОЛОЖЕНИЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2320652C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ИНТЕГРАЗЫ | 2011 |
|
RU2567385C2 |
| ПРОИЗВОДНЫЕ 2-АМИНО-2-ФЕНИЛАЛКАНОЛА, ИХ ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2009 |
|
RU2486174C2 |
| ПРОИЗВОДНЫЕ ЭКТЕИНАСЦИДИНОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРИМЕНЕНИЕ | 2003 |
|
RU2306316C9 |
| ОБЩИЙ СИНТЕЗ МИРИАПОРОНОВ | 2003 |
|
RU2328492C2 |
| ПОЛУСИНТЕТИЧЕСКИЙ СПОСОБ ПОЛУЧЕНИЯ ДЕСЕРПИДИНА | 2005 |
|
RU2372351C2 |
| ПРОИЗВОДНЫЕ КАМПТОТЕЦИНА С ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 2007 |
|
RU2450008C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ОКСАЗОЛИДИНОНА С ЦИКЛИЧЕСКИМ АМИДОКСИМОМ ИЛИ ЦИКЛИЧЕСКИМ АМИДРАЗОНОМ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2009 |
|
RU2468023C1 |
| СПИРОПИПЕРИДИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1993 |
|
RU2168512C2 |
| ИНДОЛЫ, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С АНДРОГЕНОВЫМИ РЕЦЕПТОРАМИ | 2003 |
|
RU2328484C2 |
Изобретение относится к способам получения соединений формулы (XVIIb):
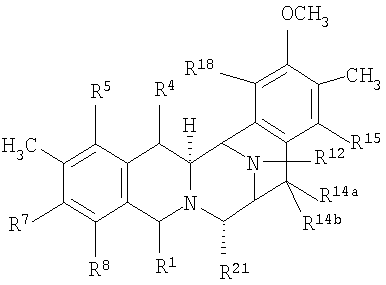
где
R1 представляет необязательно защищенную или модифицированную аминометиленовую группу, необязательно защищенную или модифицированную гидроксиметиленовую группу; и
R4 представляет -Н; или
R1 и R4 вместе образуют группу формулы (IV), (V), (VI) или (VII):


R5 представляет -Н или -ОН;
R7 представляет -ОСН3 и R8 представляет -ОН, или R7 и R8 вместе образуют группу -O-СН2-О-;
R14a и R14b оба представляют -Н или один представляет -Н, а другой представляет -ОН, -ОСН3 или -ОСН2СН3, или R14a и R14b вместе образуют кетогруппу; и R15 представляет -Н или -ОН;
R18 представляет -Н или -ОН;
R21 представляет -Н, -ОН или CN;
и производных, из 21-цианосоединения формулы (XVIb):
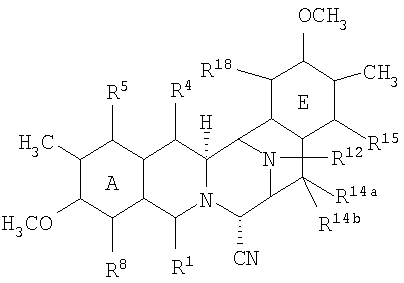
Изобретение также относится к новым соединениям:
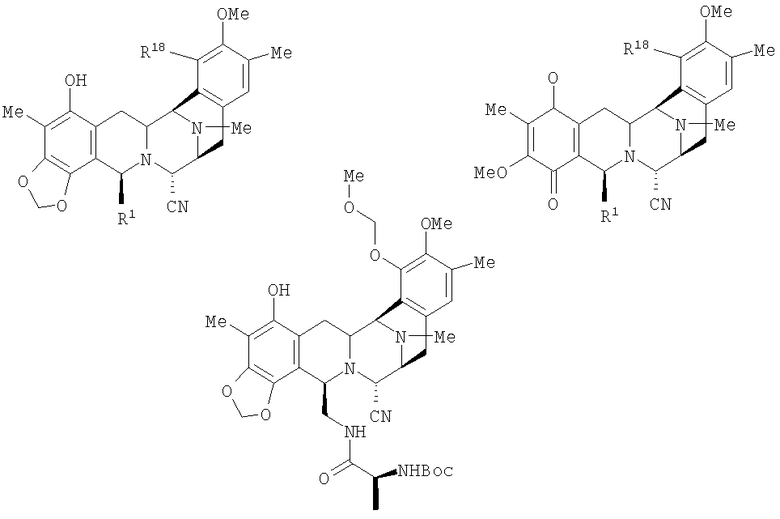
Эти соединения обладают как антибактериальной, так и противоопухолевой активностью. Технический результат - получение новых соединений, содержащих конденсированную систему из пяти колец, обладающих ценными фармакологическими свойствами, и в альтернативных способах синтеза ряда эктеинасцидина и родственным им соединениям, что позволяет обеспечить более экономные пути получения противоопухолевых средств, а также сделать возможным получение новых активных соединений. 5 н. и 47 з.п. ф-лы,4 табл.
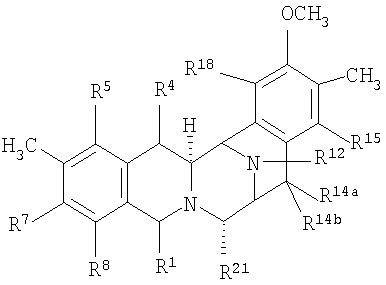
где R1 представляет необязательно защищенную или модифицированную аминометиленовую группу, необязательно защищенную или модифицированную гидроксиметиленовую группу;
R4 представляет -Н или
R1 и R4 вместе образуют группу формулы (IV), (V), (VI) или (VII):

R5 представляет -Н или -ОН;
R7 представляет -ОСН3 и R8 представляет -ОН, или R7 и R8 вместе образуют группу -О-СН2-О-;
R14a и R14b оба представляют -Н или один представляет -Н, а другой представляет -ОН, -ОСН3 или -ОСН2СН3, или R14a и R14b вместе образуют кетогруппу;
R15 представляет -Н или -ОН;
R18 представляет -Н или -ОН;
R21 представляет -Н, -ОН или CN;
и производных, из 21-цианосоединения формулы (XVIb):
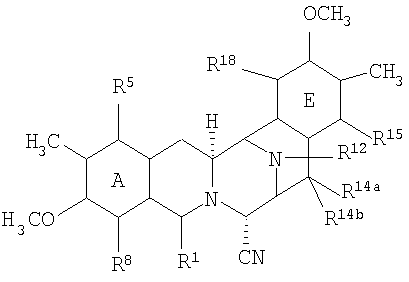
в которой R1 представляет амидометиленовую группу или ацилоксиметиленовую группу;
R5 и R8 независимо выбирают из -Н, -ОН или -ОСОСН2OH, или R5 и R8 оба являются кето, кольцо А является п-бензохиноновым кольцом;
R12 представляет -Н, -СН3 или -СН2СН3;
R14a и R14b оба представляют -Н или один представляет -Н, а другой представляет -ОН, -ОСН3 или -ОСН2СН3, или R14a и R14b вместе образуют кетогруппу;
R15 и R18 независимо выбирают из -Н или -ОН или R15 и R18 оба являются кето, и кольцо Е является п-бензохиноновым кольцом;
при условии, что, по крайней мере, одно из колец А или Е является п-бензохиноновым кольцом, причем реакции способа включают при необходимости: а) превращение хиноновой системы кольца Е в фенольную систему; b) превращение хиноновой системы кольца А в фенольную систему; c) превращение фенольной системы кольца А в метилендиоксифенольное кольцо; d) образование мостиковой спирокольцевой системы формулы (IV), (VI) или (VII), проходящей через 1-положение и 4-положение в кольце В; e) преобразование в производные; с получением требуемого соединения формулы (XVIIb).
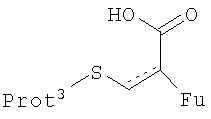
где Fu обозначает защищенную функциональную группу;
Prot3 представляет защитную группу, а пунктирная линия показывает необязательно присутствующую двойную связь.
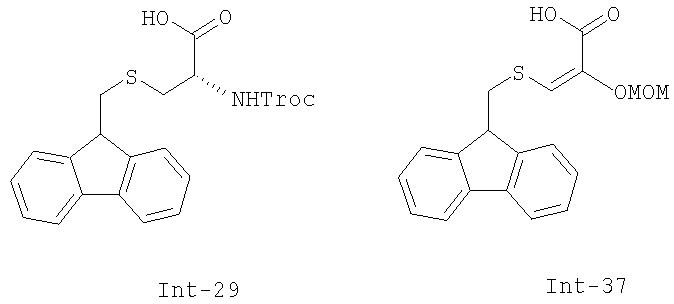
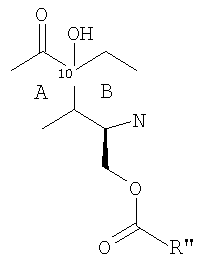
где группу R'' выбирают для образования требуемой группы формулы (IV), (V), (VI) или (VII).
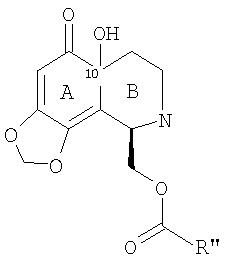
где группа R'' имеет указанные значения.



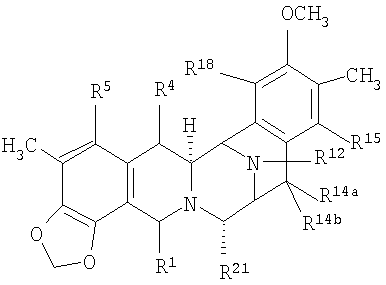
где R1 представляет -CH2-NH2 или -CH2OH, или защищенный или модифицированный вариант такой группы, и R4 представляет -Н;
или R1 и R4 вместе образуют группу формул (IV), (V), (VI) или (VII):

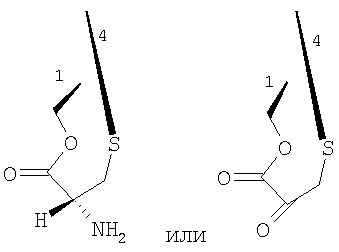
R5 представляет -ОН или защищенный или модифицированный вариант такой группы;
R12 представляет -Н, -СН3 или -СН2СН3;
R14a и R14b оба представляют -Н или один представляет -Н, а другой -ОН, или защищенный или модифицированный вариант такой группы; -ОСН3 или -ОСН2СН3, или R14a и R14b вместе образуют кетогруппу;
R15 представляет Н, -ОН или защищенный или модифицированный вариант такой группы;
R18 представляет -ОН, или защищенный или модифицированный вариант такой группы;
R21 представляет -Н, -ОН или -CN;
из 21-цианосоединения формулы (XVIb)

где R1 представляет амидометиленовую группу или ацилоксиметиленовую группу;
R5 и R8 независимо выбирают из -Н, -OH или -ОСОСН2ОН, или R5 и R8 оба являются кето, кольцо А является п-бензохиноновым кольцом;
R12 представляет -Н, -СН3 или -СН2СН3;
R14a и R14b оба представляют -Н или один представляет -Н, а другой представляет -ОН, -ОСН3 или -ОСН2СН3, или R14a и R14b вместе образуют кетогруппу;
R15 и R18 независимо выбирают из -Н или -ОН, или R15 и R18 оба являются кето, и кольцо Е является п-бензохиноновым кольцом;
при условии, что, по крайней мере, одно из колец А или Е является п-бензохиноновым кольцом, причем реакции способа, включают при необходимости: a) превращение хиноновой системы кольца Е в фенольную систему; b) превращение хиноновой системы кольца А в фенольную систему; c) превращение фенольной системы кольца А в метилендиоксифенольное кольцо; d) образование мостиковой спирокольцевой системы формулы (IV), (VI) или (VII), проходящей через 1-положение и 4-положение в кольце В; e) преобразование в производные; с получением требуемого соединения формулы (XXIIb).


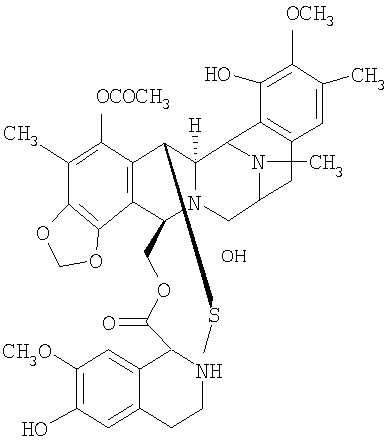
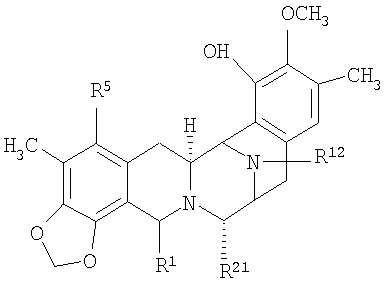
где R1 представляет модифицированную аминометиленовую группу среднего размера;
R5 представляет модифицированную гидроксигруппу небольшого размера;
R12 представляет -СН3;
R21 представляет гидрокси- или цианогруппу.


где R1 и R18 имеют указанные значения.
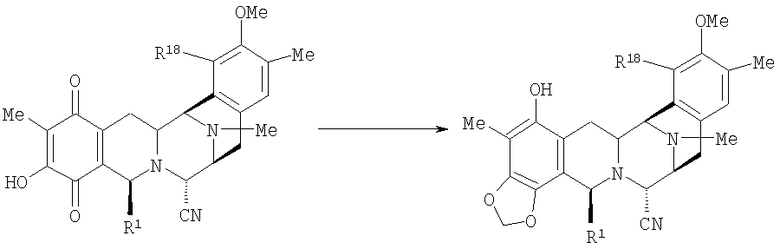
где R1 и R18 имеют указанные значения.
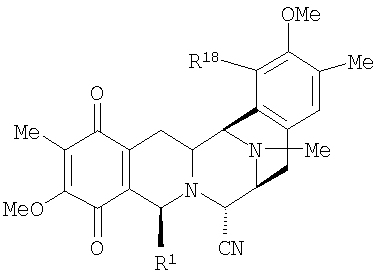
где R1 представляет необязательно защищенную или модифицированную аминометиленовую группу; R18 представляет собой защищенную ОН группу.

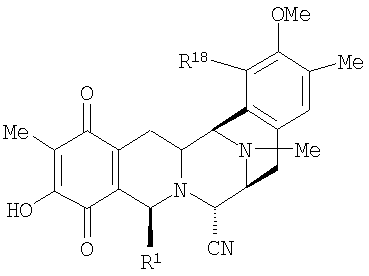
где R1 представляет необязательно защищенную или модифицированную аминометиленовую группу; R18 представляет собой защищенную ОН группу.

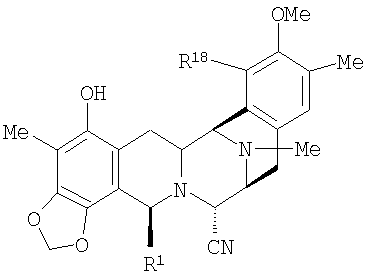
где R1 представляет необязательно защищенную или модифицированную аминометиленовую группу; R18 представляет собой защищенную ОН группу.
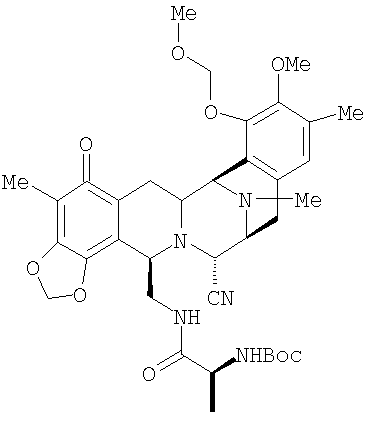
| E.J.Corey et al | |||
| J | |||
| Am | |||
| Chem | |||
| Soc | |||
| Способ использования делительного аппарата ровничных (чесальных) машин, предназначенных для мериносовой шерсти, с целью переработки на них грубых шерстей | 1921 |
|
SU18A1 |
| Fukuyma et al | |||
| J | |||
| Am | |||
| Chem | |||
| Soc | |||
| Способ гальванического снятия позолоты с серебряных изделий без заметного изменения их формы | 1923 |
|
SU12A1 |
| US 5721362 A, 24.02.1998 | |||
| RU 96115394 A, 27.12.1998 | |||
| RU 95112485 A1, 27.12.1998. | |||

