ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
1. Область изобретения
Изобретение относится к терапевтическим соединениям, фармацевтическим композициям, содержащим эти соединения, способам их изготовления и их применению. В частности, настоящее изобретение относится к соединениям, которые могут быть эффективными в лечении боли, рака, рассеянного склероаз, болезни Паркинсона, хореи Гентингтона, болезни Альцгеймера, тревожных расстройств, желудочно-кишечных расстройств и/или сердечно-сосудистых расстройств.
2. Обсуждение релевантных технических решений
Устранение боли является важной областью исследований в течение многих лет. Общеизвестно, что лиганды каннабиноидных рецепторов (например, рецептора CB1, рецептора СВ2), в том числе агонисты, антагонисты и обратные агонисты, вызывают облегчение боли в различных животных моделях посредством взаимодействия с рецепторами CB1 и/или CB2. Как правило, рецепторы CB1 локализуются преимущественно в центральной нервной системе, а рецепторы СВ2 локализуются, главным образом, на периферии и ограничиваются в основном клетками и тканями, происхождение которых связано с иммунной системой.
Хотя агонисты рецептора CB1, такие как Δ9-тетрагидроканнабинол (Δ9-ТНС) и анадамид, полезны в моделях антиноцицепции у млекопитающих, они имеют тенденцию оказывать нежелательные побочные эффекты на центральную нервную систему (ЦНС), например психоактивные побочные эффекты, возможность злоупотребления, лекарственная зависимость и толерантность и т.д. Известно, что эти нежелательные побочные эффекты опосредованы рецепторами CB1, локализованными в ЦНС. Однако имеются факты, свидетельствующие о том, что агонисты CB1, действующие в периферических сайтах, или с ограниченной ЦНС экспозицией могут устранять боль у людей или животных со значительно улучшенным общим in vivo профилем.
Следовательно, существует потребность в новых лигандах рецептора CB1, таких как агонисты, которые полезны для устранения боли или лечения других связанных с болью симптомов или заболеваний с пониженными или минимальными нежелательными побочными эффектами на ЦНС.
ОПИСАНИЕ ВОПЛОЩЕНИЙ
В настоящем изобретении предложены лиганды рецептора CB1, которые могут быть полезными в лечении боли и/или других родственных симптомов или заболеваний.
Термин "Cm-n" или "Сm-nгруппа", используемый сам по себе или в качестве префикса, относится к любой группе, имеющей от m до n атомов углерода.
Термин "алкил", используемый сам по себе или в качестве суффикса или префикса, относится к одновалентному насыщенному углеводородному радикалу с прямой или разветвленной цепью, содержащему от 1 до примерно 12 атомов углерода. Иллюстративные примеры алкилов включают, без ограничения ими, С1-4алкильные группы, такие как метил, этил, пропил, изопропил, 2-метил-1 -пропил, 2-метил-2-пропил, бутил, изобутил, трет-бутил.
Термин "циклоалкил," используемый сам по себе или в качестве суффикса или префикса, относится к насыщенному одновалентному содержащему кольцо углеводородному радикалу, содержащему по меньшей мере 3 и вплоть до примерно 12 атомов углерода. Примеры циклоалкилов включают, без ограничения ими, С3-7циклоалкильные группы, такие как циклопропил, циклобутил, циклопентил, циклогексил и циклогептил, и насыщенные циклические и бициклические терпены. Циклоалкил может быть незамещенным или может быть замещен одним или двумя подходящими заместителями. Предпочтительно циклоалкил представляет собой моноциклическое или бициклическое кольцо.
Термин "алкокси", используемый сам по себе или в качестве суффикса или префикса, относится к радикалам общей формулы -O-R, где R представляет собой алкил. Примеры алкокси включают метокси, этокси, пропокси, изопропокси, бутокси, трет-бутокси и изобутокси.
Галоген включает фтор, хлор, бром и йод.
"КТ" или "кт" означает комнатную температуру.
В одном аспекте воплощение изобретения представляет собой соединение формулы I, его фармацевтически приемлемую соль, диастереомеры, энантиомеры или их смеси:
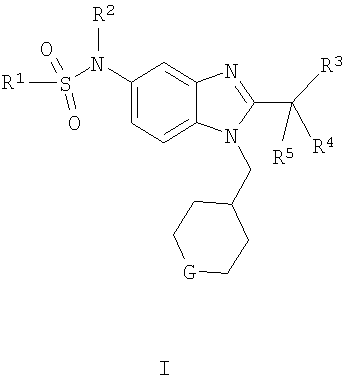
где
G выбран из -O-, -CHF- и -CF2-;
R1 выбран из С1-6алкила и С3-6циклоалкила;
R2 выбран из -Н и метила; и
R3, R4 и R5 независимо выбраны из фторо и метила.
В другом воплощении эти соединения могут представлять собой соединения формулы I, где
G выбран из -О- и -CF2-;
R1 выбран из C1-6алкила и С3-6циклоалкила;
R2 выбран из -Н и метила; и
R3, R4 и R5 независимо выбраны из фторо и метила.
Другое воплощение данного изобретения составляет соединение формулы I, где
G выбран из -О- и -CF2-;
R1 выбран из С1-4алкила и С3-4циклоалкила;
R2 выбран из -Н и метила; и
R3, R4 и R5 независимо выбраны из фторо и метила.
Еще одно воплощение данного изобретения составляет соединение формулы I, где
G представляет собой -O-;
R1 выбран из этила, пропила и циклопропила;
R2 выбран из -Н и метила; и
R3, R4 и R5 независимо выбраны из фторо и метила, причем R3, R4 и R5 являются одинаковыми.
Еще одно воплощение данного изобретения составляет соединение формулы I,
где
G представляет собой -CF2-;
R1 выбран из этила, пропила и циклопропила;
R2 выбран из -Н и метила; и
R3, R4 и R5 независимо выбраны из фторо и метила, причем R3, R4 и R5 являются одинаковыми.
Еще одно воплощение данного изобретения составляет соединение формулы I,
где
G представляет собой -CHF-;
R1 выбран из этила, пропила, трет-бутила и циклопропила;
R2 выбран из -Н и метила; и
R3, R4 и R5 независимо выбраны из фторо и метила, причем R3, R4 и R5 являются одинаковыми.
В еще одном воплощении R1 в формуле I выбран из этила, пропила, трет-бутила и циклопропила.
В еще одном воплощении G в формуле I представляет собой -CHF- или -CF2-.
В еще одном воплощении R3, R4 и R5 в формуле I выбраны из фторо и метила, причем R3, R4 и R5 являются одинаковыми.
В другом воплощении соединение по изобретению может быть выбрано из
N-[2-трет-бутил-1-(тетрагидро-2Н-пиран-4-илметил)-1Н-бензимидазол-5-ил]-N-метилциклопропансульфонамида;
N-[2-трет-бутил-1-(тетрагидро-2Н-пиран-4-илметил)-1Н-бензимидазол-5-ил]-N-метилпропан-1-сульфонамида;
N-[2-трет-бутил-1-(тетрагидро-2H-пиран-4-илметил)-1Н-бензимидазол-5-ил]-N-метилбутан-1-сульфонамида;
N-{2-трет-бутил-1-[(4,4-дифторциклогексил)метил]-1Н-бензимидазол-5-ил}-N-метилбутан-1-сульфонамида;
N-метил-N-[1-(тетрагидро-2Н-пиран-4-илметил)-2-(трифторметил)-1H-бензимидазол-5-ил]пропан-1-сульфонамида;
N-метил-N-[1-(тетрагидро-2Н-пиран-4-илметил)-2-(трифторметил)-1Н-бензимидазол-5-ил]циклопропансульфонамида;
N-[2-трет-бутил-1-(тетрагидро-2Н-пиран-4-илметил)-1H-бензимидазол-5-ил]-N-метилпентан-1-сульфонамида;
N-[2-трет-бутил-1-(тетрагидро-2H-пиран-4-илметил)-1Н-бензимидазол-5-ил]-N-метилэтансульфонамида;
N-[2-трет-бутил-1-(тетрагидро-2Н-пиран-4-илметил)-1Н-бензимидазол-5-ил]-N,2-диметилпропан-2-сульфонамида;
N-{2-трет-бутил-1-[(4,4-дифторциклогексил)метил]-1Н-бензимидазол-5-ил}-N-метилпропан-1-сульфонамида;
N-{2-трет-бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-ил}-N-метилэтансульфонамида;
N-{2-трет-бутил-1-[(4,4-дифторциклогексил)метил]-1Н-бензимидазол-5-ил}пропан-1-сульфонамида;
N-{2-трет-бутил-1-[(4,4-дифторциклогексил)метил]-1Н-бензимидазол-5-ил}метансульфонамида;
N-{2-трет-бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-ил}этансульфонамида;
N-{2-трет-бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-ил}циклопропансульфонамида;
N-{2-трет-бутил-1-[(4,4-дифторциклогексил)метил]-1Н-бензимидазол-5-ил}-N-метилциклопропансульфонамида;
N-{2-трет-бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-ил}-2-метилпропан-2-сульфонамида;
N-[1-[(4,4-дифторциклогексил)метил]-2-(1,1-дифторэтил)-1H-бензимидазол-5-ил]циклопропансульфонамида;
N-[1-[(4,4-дифторциклогексил)метил]-2-(1,1-дифторэтил)-1H-бензимидазол-5-ил]этансульфонамида;
N-[1-[(4,4-дифторциклогексил)метил]-2-(1,1-дифторэтил)-1H-бензимидазол-5-ил]-2-метилпропан-2-сульфонамида;
N-[2-(1,1-дифторэтил)-1-(тетрагидро-2Н-пиран-4-илметил)-1Н-бензимидазол-5-ил]-N-метилэтансульфонамида;
N-[2-(1,1-дифторэтил)-1-(тетрагидро-2Н-пиран-4-илметил)-1H-бензимидазол-5-ил]-N-метилпропан-1-сульфонамида;
N-[2-(1,1-дифторэтил)-1-(тетрагидро-2Н-пиран-4-илметил)-1H-бензимидазол-5-ил]-N-метилциклопропансульфонамида;
N-{2-трет-бутил-1-[(4-фторциклогексил)метил]-1H-бензимидазол-5-ил}этансульфонамида;
N-{2-трет-бутил-1-[(4-фторциклогексил)метил]-1H-бензимидазол-5-ил}циклопропансульфонамида;
N-{2-трет-бутил-1-[(4-фторциклогексил)метил-1Н-бензимидазол-5-ил}-2-метилпропан-2-сульфонамида;
и их фармацевтически приемлемых солей.
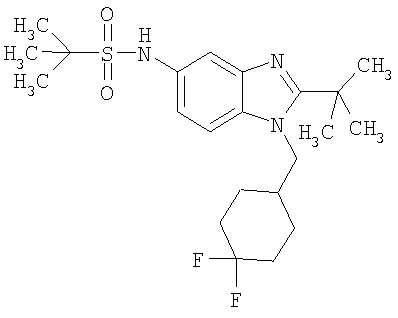
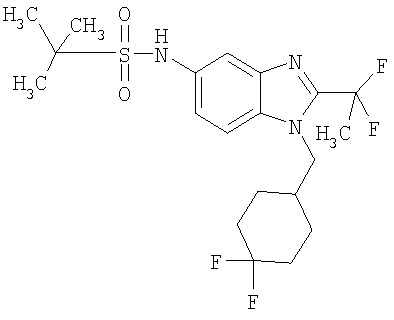
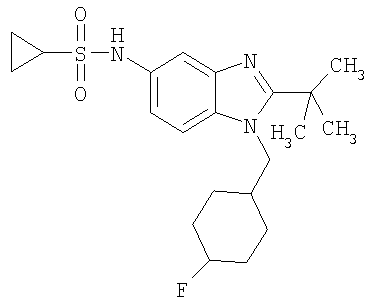
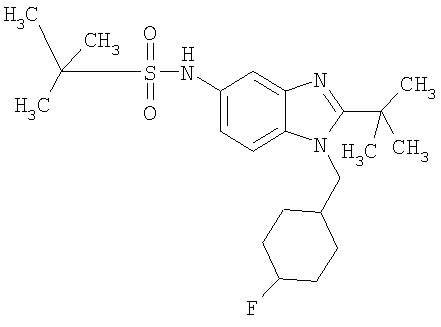
В еще одном воплощении изобретения предложено соединение, выбранное из
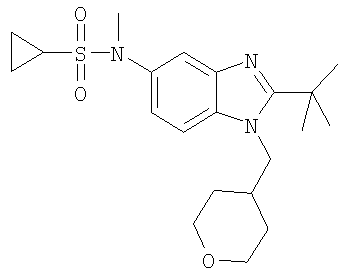
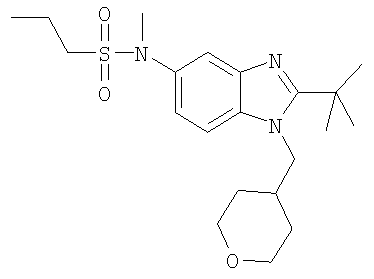
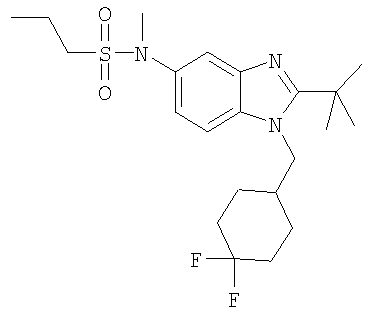
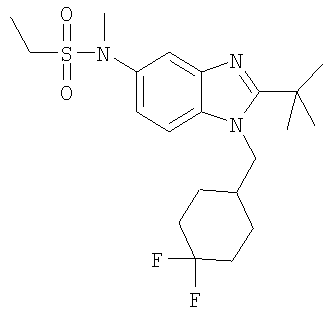
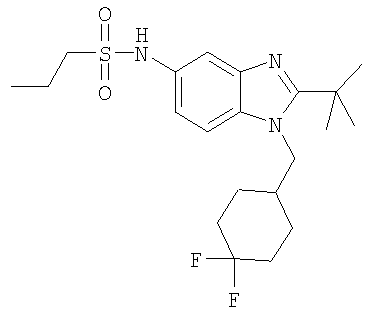
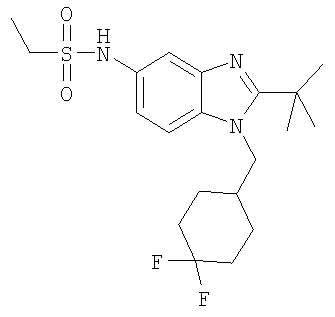
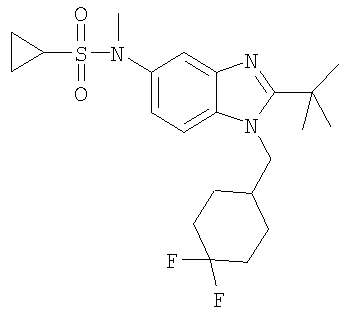
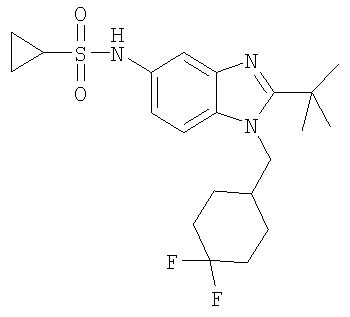
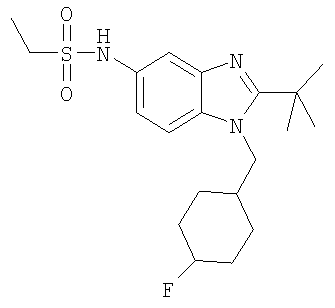
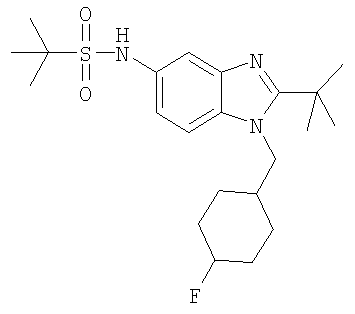
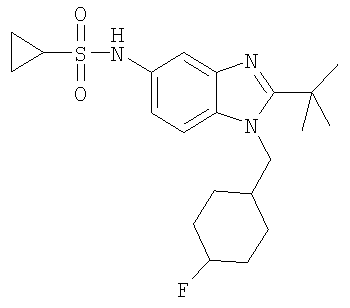
и их фармацевтически приемлемых солей.
Понятно, что когда соединения по настоящему изобретению содержат один или более хиральных центров, соединения по изобретению могут существовать в энантиомерных или диастереомерных формах или в виде рацемической смеси, или могут быть выделенными в виде энантиомерных или диастереомерных форм или в виде рацемической смеси. Настоящее изобретение охватывает любые возможные энантиомеры, диастереомеры, рацематы или их смеси соединения формулы I. Оптически активные формы соединения по изобретению могут быть получены, например, хиральным хроматографическим разделением рацемата, синтезом из оптически активных исходных веществ или асимметрическим синтезом по методикам, описанным ниже.
Также следует принять во внимание, что некоторые соединения по настоящему изобретению могут существовать в виде геометрических изомеров, например Е и Z изомеров алкенов. Настоящее изобретение охватывает любой геометрический изомер соединения формулы I. Следует иметь в виду также, что настоящее изобретение охватывает таутомеры соединений формулы I.
Понятно также, что некоторые соединения по настоящему изобретению могут существовать в сольватированной, например гидратированной, а также в несольватированной формах. Следует иметь в виду также, что настоящее изобретение охватывает все такие сольватированные формы соединений формулы I.
В объем данного изобретения также входят соли соединений формулы I. В общем, фармацевтически приемлемые соли соединений по настоящему изобретению могут быть получены с использованием стандартных методик, общеизвестных в данной области, например путем осуществления взаимодействия достаточно основного соединения, например алкиламина, с подходящей кислотой, например НСl или уксусной кислотой, для предоставления физиологически приемлемого аниона. Возможным может быть также получение соответствующей соли щелочного металла (такого как натрий, калий или литий) или щелочно-земельного металла (такого как кальций) путем обработки соединения по настоящему изобретению, имеющего соответственно кислотный протон, такого как карбоновая кислота или фенол, одним эквивалентом гидроксида или алкоксида (такого как этоксид или метоксид) щелочного металла или щелочно-земельного металла, или соответственно основного органического амина (такого как холин или меглумин) в водной среде с последующей очисткой по общепринятым методикам.
В одном воплощении указанное выше соединение формулы 1 может быть превращено в его фармацевтически приемлемую соль или сольват, в частности в соль присоединения кислоты, такую как гидрохлорид, гидробромид, фосфат, ацетат, фумарат, малеат, тартрат, цитрат, метансульфонат или п-толуолсульфонат.
Авторы изобретения обнаружили, что соединения по изобретению обладают активностью как фармацевтические средства, в частности являются модуляторами или лигандами, такими как агонисты, частичные агонисты, обратные агонисты или антагонисты рецепторов CB1. Более конкретно, соединения по изобретению проявляют избирательную активность как агонисты рецепторов CB1 и полезны в терапии, в частности для облегчения различных болевых состояний, таких как хроническая боль, невропатическая боль, острая боль, боль при раковых заболеваниях, боль, вызванная ревматоидным артритом, мигрень, висцеральная боль и так далее. Однако не следует истолковывать этот список как исчерпывающий. Дополнительно соединения по настоящему изобретению полезны при других болезненных состояниях, при которых имеет место или в которые вовлечена дисфункция рецепторов CB1. Кроме того, соединения по изобретению можно использовать для лечения рака, рассеянного склероза, болезни Паркинсона, хореи Гентингтона, болезни Альцгеймера, тревожных расстройств, желудочно-кишечных расстройств и сердечно-сосудистых расстройств.
Соединения по изобретению полезны в качестве иммунномодуляторов, особенно при аутоиммунных заболеваниях, таких как артрит, при пересадке кожи, трансплантации органов и при сходных хирургических ситуациях, при коллагеновых болезнях, различных аллергиях, для применения в качестве противоопухолевых агентов и противовирусных агентов.
Соединения по изобретению полезны при болезненных состояниях, при которых дегенерация или дисфункция каннабиноидных рецепторов имеет место или вовлечена в этом смысле. Это может включать использование меченных изотопами версий соединений по изобретению в диагностических методах и визуализирующих приложениях, таких как позитронная эмиссионная томография (ПЭТ).
Соединения по изобретению полезны для лечения диареи, депрессии, тревожных и связанных со стрессом расстройств, таких как посттравматические стрессовые расстройства, паническое расстройство, генерализованное тревожное расстройство, социальная фобия и обсессивно-компульсивное расстройство, недержания мочи, преждевременной эякуляции, различных психических болезней, кашля, отека легких, различных желудочно-кишечных расстройств, например запора, функциональных желудочно-кишечных расстройств, таких как синдром раздраженного кишечника или функциональная диспепсия, болезни Паркинсона и других нарушений моторики, травматического повреждения головного мозга, инсульта, для кардиозащиты после инфаркта миокарда, повреждения позвоночника и наркомании, включая лечение злоупотребления алкоголем, никотином, опиоидами и другими наркотиками, и для лечения расстройств симпатической нервной системы, например гипертензии.
Соединения по изобретению полезны в качестве аналгезирующих агентов для использования во время общей анестезии и контролируемой анестезии. Комбинации агентов с различными свойствами часто используют для достижения баланса эффектов, необходимого для поддержания состояния анестезии (например амнезии, аналгезии, мышечной релаксации и седативного эффекта). Включаемыми в эту комбинацию являются ингаляционные анестетики, снотворные средства, анксиолитики, нервно-мышечные блокаторы и опиоиды.
Также в объем изобретения входит применение любого из соединений вышеуказанной формулы 1 для изготовления лекарственного средства для лечения любого состояния, описанного выше.
Еще одним аспектом изобретения является способ лечения субъекта, страдающего любым состоянием, описанным выше, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения вышеуказанной формулы I.
Таким образом, согласно изобретению предложено соединение формулы 1 или его фармацевтически приемлемая соль или сольват, как определено здесь выше, для применения в терапии.
В еще одном аспекте настоящего изобретения предложено применение соединения формулы 1 или его фармацевтически приемлемой соли или сольвата, как определено здесь выше, в изготовлении лекарственного средства для использования в терапии.
В контексте настоящего изобретения термин "терапия" также включает "профилактику", если нет конкретных указаний на противоположное. Термины "терапевтический" и "терапевтически" следует истолковывать соответственно. Термин "терапия" в контексте настоящего изобретения также включает введение эффективного количества соединения по настоящему изобретению для облегчения либо уже существующего болезненного состояния, острого или хронического, либо рецидивирующего состояния. Это определение также охватывает профилактические терапии для предупреждения рецидивирующих состояний и длительную терапию хронических расстройств.
Соединения по настоящему изобретению полезны в терапии, в частности в терапии различных болевых состояний, включая, но не ограничиваясь ими, острую боль, хроническую боль, невропатическую боль, боль в спине, боль при раковых заболеваниях и висцеральную боль.
При применении для терапии у теплокровного животного, например человека, соединение по изобретению может быть введено в форме традиционной фармацевтической композиции любым путем, в том числе перорально, внутримышечно, подкожно, местно, интраназально, интраперитонеально, интраторакально, внутривенно, эпидурально, интратекально, трансдермально, интрацеребровентрикулярно и инъекцией в суставы.
В одном воплощении изобретения путь введения может быть пероральным, внутривенным или внутримышечным.
Дозировка будет зависеть от пути введения, тяжести заболевания, возраста и массы пациента и других факторов, обычно учитываемых лечащим врачом при определении индивидуального режима и уровня дозировки, наиболее подходящих для конкретного пациента.
Для приготовления фармацевтических композиций из соединений по данному изобретению инертные фармацевтически приемлемые носители могут быть твердыми или жидкими. Препараты в твердой форме включают порошки, таблетки, дисперсные гранулы, капсулы, облатки и суппозитории.
Твердым носителем может быть одно или более веществ, которые также могут действовать как разбавители, корригенты, солюбилизаторы, смазывающие вещества, суспендирующие агенты, связывающие вещества или разрыхлители для таблеток. Носителем также может быть инкапсулирующее вещество.
В порошках носитель представляет собой тонкоизмельченное твердое вещество, которое находится в смеси с тонкоизмельченным соединением по изобретению или активным компонентом. В таблетках активный компонент смешивают с носителем, имеющим необходимые связывающие свойства, в подходящих соотношениях и прессуют до желаемой формы желаемого размера.
Для приготовления композиций в форме суппозиториев сначала плавят низкоплавкий воск, например смесь глицеридов жирных кислот и масла какао, и диспергируют в нем активный ингредиент, например путем перемешивания. Расплавленную гомогенную смесь затем вливают в формы соответствующего размера и дают ей охладиться и затвердеть.
Подходящими носителями являются карбонат магния, стеарат магния, тальк, лактоза, сахар, пектин, декстрин, крахмал, трагакант, метилцеллюлоза, натрий-карбоксиметилцеллюлоза, низкоплавкий воск, масло какао и тому подобное.
Подразумевается, что термин "композиция" включает препарат активного компонента с инкапсулирующим веществом в качестве носителя, обеспечивающим получение капсулы, в которой активный компонент (с другими носителями или без) окружен носителем, который, следовательно, находится совместно с ним. Аналогично этот термин охватывает и облатки.
Таблетки, порошки, облатки и капсулы могут быть использованы в твердых дозированных лекарственных формах, подходящих для перорального введения.
Композиции в жидкой форме включают растворы, суспензии и эмульсии. Например, растворы активных соединений в стерильной воде или в водном пропиленгликоле могут быть жидкими препаратами, подходящими для парентерального введения. Жидкие композиции также могут быть приготовлены в виде препарата в водном растворе полиэтиленгликоля.
Водные растворы для перорального введения могут быть приготовлены путем растворения активного компонента в воде и добавления подходящих красителей, корригентов, стабилизаторов и загустителей, если необходимо. Водные суспензии для перорального применения могут быть изготовлены путем диспергирования тонкоизмельченного активного компонента в воде вместе с вязким веществом, таким как природные или синтетические камеди, смолы, метилцеллюлоза, натрий-карбоксиметилцеллюлоза и другие суспендирующие агенты, известные в области приготовления фармацевтических препаратов.
В зависимости от способа введения фармацевтическая композиция предпочтительно содержит от 0,05% до 99% мас.(процент по массе), более предпочтительно от 0,10 до 50% мас. соединения по изобретению, причем все процентные содержания по массе даны в расчете на общую массу композиции.
Терапевтически эффективное количество для практического использования настоящего изобретения может быть определено с использованием известных критериев, включающих возраст, массу и ответную реакцию индивидуального пациента, и интерпретировано в контексте заболевания, которое лечат или которое предупреждают, специалистом в данной области.
В объем данного изобретения входит применение любого соединения формулы I, как оно определено выше, для изготовления лекарственного средства.
Также в объем данного изобретения входит применение любого соединения формулы I для изготовления лекарственного средства для терапии боли.
Кроме того, предложено применение любого соединения формулы I для изготовления лекарственного средства для терапии различных болевых состояний, включая, но не ограничиваясь ими, острую боль, хроническую боль, невропатическую боль, боль в спине, боль при раковых заболеваниях и висцеральную боль.
Еще одним аспектом изобретения является способ лечения субъекта, страдающего любым из состояний, описанных выше, при котором пациенту, нуждающемуся в таком лечении, вводят эффективное количество соединения вышеуказанной формулы I.
Кроме того, предложена фармацевтическая композиция, содержащая соединение формулы I или его фармацевтически приемлемую соль совместно с фармацевтически приемлемым носителем.
В частности, предложена фармацевтическая композиция, содержащая соединение формулы I или его фармацевтически приемлемую соль совместно с фармацевтически приемлемым носителем, для терапии, более конкретно для терапии боли.
Предложена также фармацевтическая композиция, содержащая соединение формулы I или его фармацевтически приемлемую соль совместно с фармацевтически приемлемым носителем, для применения при любом из состояний, описанных выше.
В следующем аспекте настоящего изобретения предложен способ получения соединений по настоящему изобретению.
В одном воплощении изобретения предложен способ получения соединения формулы I:
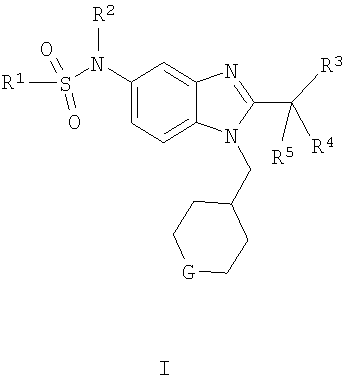
включающий взаимодействие соединения формулы II с соединением формулы III
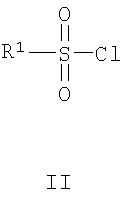
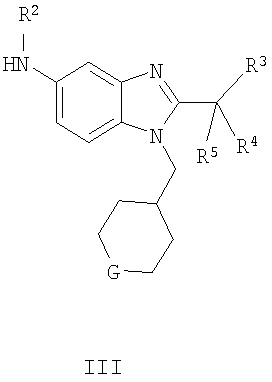
где R1, R2, R3, R4, R5 и G являются такими, как определено выше.
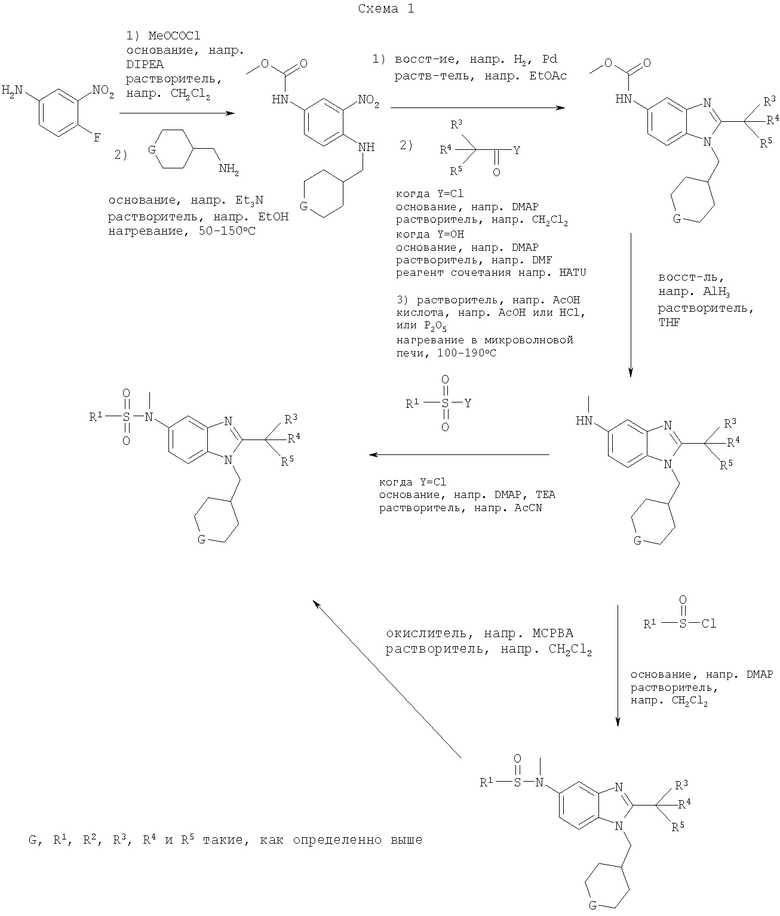
Соединения по настоящему изобретению могут также быть получены согласно путям синтеза, как они представлены на Схемах 1, 2 и 3.
Биологическая оценка
Связывание с рецепторами hCB1 и hCB2
Мембраны, содержащие человеческий рецептор CB1 от Receptor Biology (hCB1) или человеческий рецептор СВ2 от BioSignal (hCB2), оттаивают при 37°С, пропускают 3 раза через иглу с тупым концом 25-го размера, разводят в буфере для связывания каннабиноидов (50 мМ Tris, 2,5 мМ EDTA (этилендиаминтетрауксусная кислота), 5 мМ MgCl2 и 0,5 мг/мл БСА (бычий сывороточный альбумин, не содержащий жирных кислот, рН 7,4) и распределяют аликвоты, содержащие подходящее количество белка, в 96-луночных планшетах. IC50 соединений по изобретению по отношению к hCB1 и hСВ2 оценивают по 10-точечным кривым доза-ответ, построенным для образцов с 3H-CP55,940 при 20000-25000 dpm (распадов в минуту) на лунку (0,17-0,21 нМ) в конечном объеме 300 мкл. Общее и неспецифическое связывание определяют в отсутствие и в присутствии 0,2 мкМ HU210 соответственно. Планшеты встряхивают на вортексе и инкубируют в течение 60 минут при комнатной температуре, фильтруют через Unifilters GF/B (предварительно пропитанные 0,1% полиэтиленимином) на харвестере Tomtec или Packard с использованием 3 мл промывочного буфера (50 мМ Tris, 5 мМ MgCl2, 0,5 мг БСА рН 7,0). Фильтры высушивают в течение 1 ч при 55°С. Радиоактивность (cpm (импульсов в минуту)) подсчитывают в TopCount (Packard) после добавления 65 мкл/лунку сцинтилляционной жидкости MS-20.
Связывание GTPγS hCB1 и hCB2
Мембраны, содержащие человеческий рецептор CB1 от Receptor Biology (hCB1) или человеческий рецептор СВ2 (BioSignal), оттаивают при 37°С, пропускают 3 раза через иглу с тупым концом 25-го размера и разводят в буфере для связывания GTPγS (50 мМ Hepes, 20 мМ NaOH, 100 мМ NaCl, 1 мМ EDTA, 5 мМ MgCl2, pH 7,4, 0,1% БСА). EC50 и Emax соединений по изобретению оценивают по 10-точечным кривым доза-ответ, построенным для образцов в 300 мкл с подходящим количеством мембранного белка и 100000-130000 dpm (число распадов в минуту) GTPg35S на лунку (0,11-0,14 нМ). Исходное и максимальное стимулированное связывание определяют в отсутствие и в присутствии 1 мкМ (hСВ2) или 10 мкМ (hCB1) Win 55212-2 соответственно. Мембраны преинкубируют в течение 5 минут с 56,25 мкМ (hСВ2) или 112,5 мкМ (hCB1) GDP (гуанозиндифосфат), а затем распределяют в планшетах (конечная концентрация GDP 15 мкМ (hСВ2) и 30 мкМ (hСВ1)). Планшеты встряхивают на вортексе и инкубируют в течение 60 минут при комнатной температуре, фильтруют через фильтры Unifilters GF/B (предварительно пропитанные водой) на харвестере Tomtec или Packard с использованием 3 мл промывочного буфера (50 мМ Tris, 5 мМ MgCl2, 50 мМ NaCl, pH 7,0). Фильтры высушивают в течение 1 ч при 55°С. Радиоактивность (cpm) подсчитывают в TopCount (Packard) после добавления 65 мкл/лунку сцинтилляционной жидкости MS-20. Исследования обращения антагониста проводят тем же путем, за исключением того, что (а) кривую доза агониста-ответ получают в присутствии постоянной концентрации антагониста или (б) кривую доза антагониста-ответ получают в присутствии постоянной концентрации агониста.
На основании приведенных выше анализов определяют константу диссоциации (Ki) для конкретного соединения по изобретению в отношении конкретного рецептора, используя следующее уравнение:
Ki=IC50/(1+[rad]/Kd),
где IC50 означает концентрацию соединения по изобретению, при которой наблюдалось 50% замещение;
[rad] означает стандартную или эталонную концентрацию радиоактивного лиганда в данный момент; и
Kd означает константу диссоциации радиоактивного лиганда по отношению к конкретному рецептору.
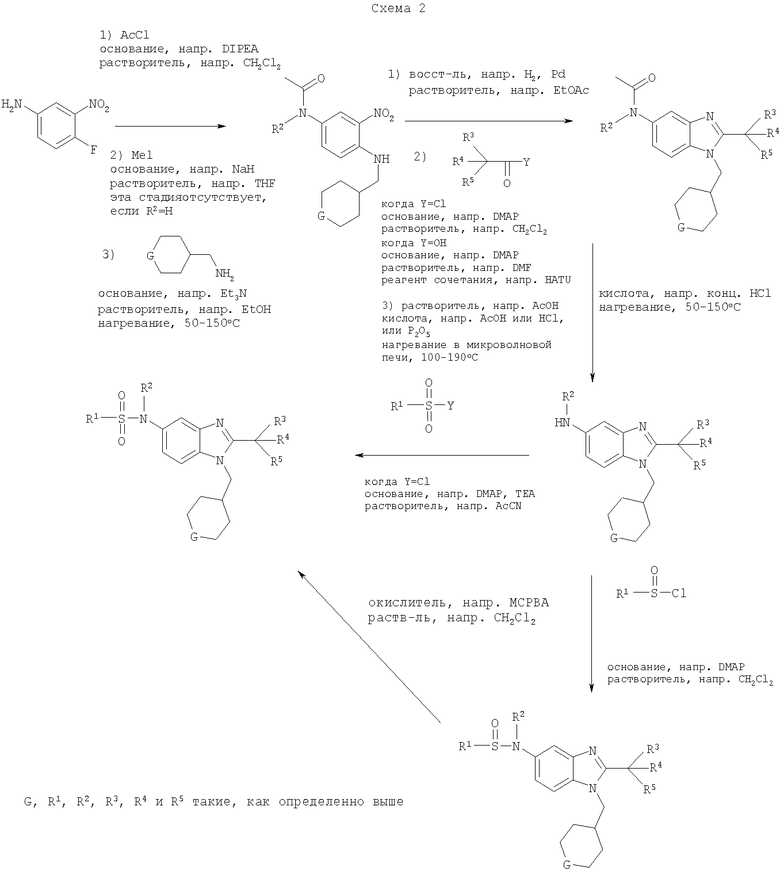
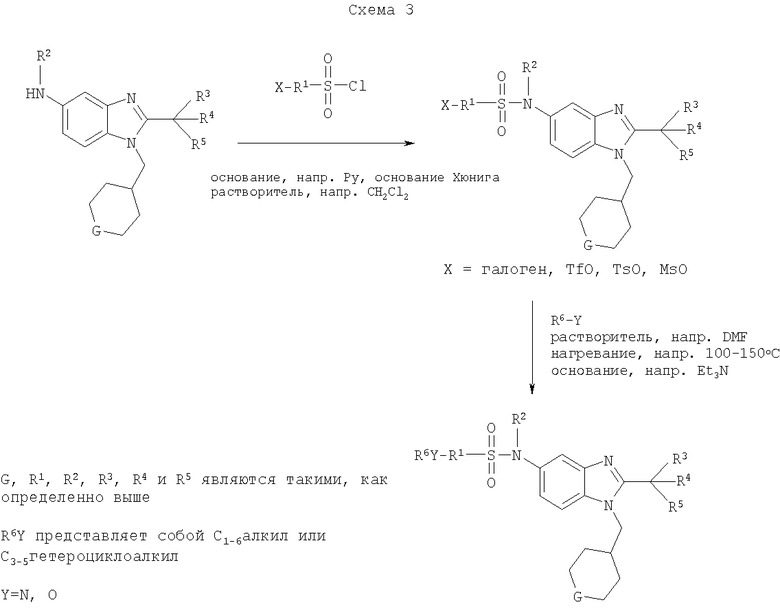
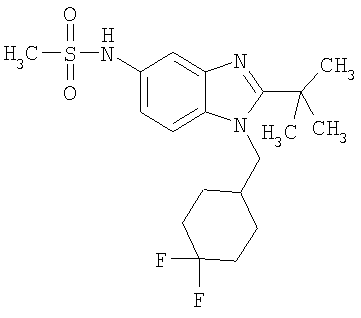
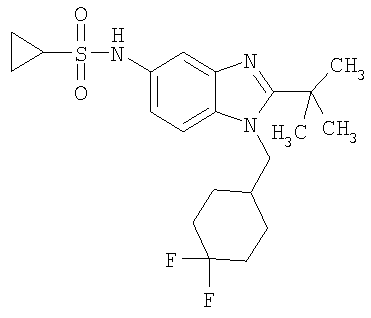
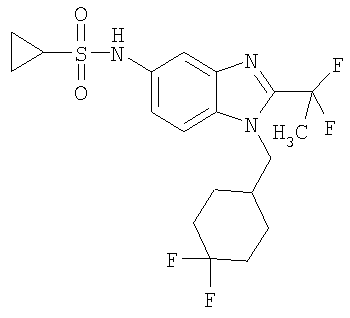
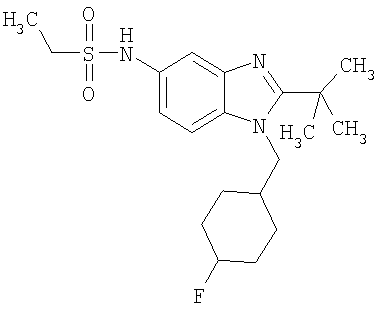
Определенная с использованием описанных выше анализов Ki по отношению к человеческим рецепторам СВ1 для ряда соединений по изобретению имеет значение в диапазоне между 3 нМ и 195 нМ. EC50 для этих соединений лежит в диапазоне между 2,3 нМ и 300 нМ. Emax для этих соединений находится в диапазоне между 109% и 144%. ЕС50 для N-{2-трет-бутил-1-[(4,4-дифторциклогексил)метил]-1Н-бензимидазол-5-ил}этан-сульфонамида составляет 13,5 нМ.
Условия анализа для измерения растворимости
Готовят исходный раствор 30 мМ образца в DMSO (диметилсульфоксид) и затем аликвоты по 25 мкл добавляют в 96-луночный планшет и инкубируют при 40°С в течение 4 часов. К инкубированному соединению добавляют 250 мкл натрий-фосфатного буфера (рН 7,4) и затем смешивают при 1200 об/мин в течение 24 ч, используя Eppendorf Thermomixer, при 25°С. После перемешивания раствор переносят в 96-луночный фильтровальный планшет Whatman GF/B и затем фильтруют в вакууме. Супернатант затем инъецируют в прибор LC/MS (жидкостная хроматография/масс-спектрометрия) для анализа и определение проводят, используя 1-точечную калибровку для представляющего интерес соединения.
Анализ метаболической стабильности в микросомах печени крысы и человека
Раствор 500 мкл 100 мкМ соединения в DMSO инкубируют микросомами печени человека или крысы (843 мкл микросом из 0,5618 мг/мл в 30 мл 0,1 М КН2РO4 буфера рН 7,4) при 37°С в течение 10 мин в 96-луночном глубоком планшете. Для начала реакции добавляли NADPH (никотин-амидаденин-динуклеотидфосфат, 46 мкл) в концентрации 8,33 мг/мл в 100 мМ КН2РО4 буфере с рН 7,4. Реакционные смеси переносили 384-луночный планшет, содержащий ацетонитрил для гашения реакции в моменты 0, 10, 20, 30 минут.Этот 384-луночный планшет центрифугировали в течение 30 мин при 9000 g при 4°С, и образцы из него анализировали посредством LC/MS (модель: XDB Eclipse C18). Три эталона анализировали посредством LC/MS в качестве положительного контроля. Данные обрабатывали, следуя стандартной процедуре. Метаболическую стабильность тестированных соединений выражали как мкл/мин/мг.
Кроме того, с использованием одного или более из описанных выше анализов определяли метаболические стабильности (hClint и rClint) и растворимости (водные) ряда соединений по изобретению. Было установлено, что выбранные соединения обладают улучшенной метаболической стабильностью и/или растворимостью в воде. Метаболическая стабильность и растворимость выбранных соединений проиллюстрированы в Таблице 1 ниже.
ПРИМЕРЫ
Изобретение далее более детально раскрыто в следующих Примерах, которые описывают способы, посредством которых соединения по настоящему изобретению могут быть получены, очищены, проанализированы и подвергнуты биологической оценке, и которые не следует понимать как ограничивающие изобретение.
Пример 1
N-[2-трет-Бутил-1-(тетрагидро-2Н-пиран-4-илметил)-1Н-бензимидазол-5-ил]-N-метилциклопропансульфонамид
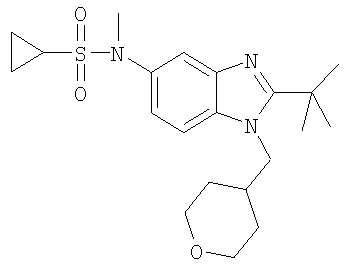
Стадия А: N-[2-трет-Бутил-1-(тетрагидро-2Н-пиран-4-илметил)-1H-бензимидазол-5-ил]-N-метилциклопропансульфонамид
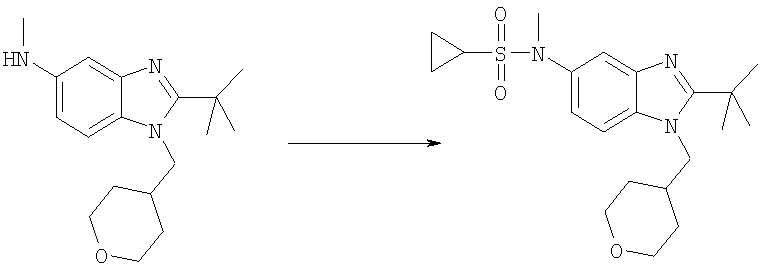
2-трет-Бутил-N-метил-1-(тетрагидро-2Н-пиран-4-илметил)-1Н-бензимидазол-5-амин (для получения смотри следующие далее стадии Б-Е) (50 мг, 0,166 ммоль) и каталитическое количество DMAP (4-диметиламинопиридин) растворяли в 5 мл DCM (дихлорметан). По каплям добавляли циклопропансульфонилхлорид (30 мг, 0,216 ммоль) и раствор перемешивали при кт (комнатная температура) в течение ночи. Раствор промывали насыщенным водным раствором NаНСО3, рассолом и сушили над безводным MgSO4. Продукт очищали посредством HPLC (высокоэффективная жидкостная хроматография) с обращенной фазой, используя смесь 10-70% СН2СN/Н2О, и лиофилизировали с получением указанного в заголовке соединения в виде соотвествующей TFA (трифторуксусная кислота) соли. Выход: 56 мг (65%). 1H ЯМР (400 МГц, МЕТАНОЛ-D4) δ 0.90-0.94 (m, 2 H), 0.97-1.02 (m, 2 H), 1.53-1.59 (m, 2 H), 1.59-1.65 (m, 2 H), 1.69 (s, 9 H), 2.36-2.42 (m, 1 H), 2.60-2.65 (m, 1 H), 3.36 (m, 2 H), 3.43 (s, 3 H), 3.94 (d, J=3,58 Гц, 1 H), 3.96 (d, J=3,07 Гц, 1 H), 4.55 (d, J=7,68 Гц, 2 H), 7.74 (dd, J=8,96, 2,05 Гц, 1 H), 7.81 (d, J=1,54 Гц, 1 H), 7.98 (d, J=8,96 Гц, 1 H); MS (ESI) (масс-спектроскопия с электрораспылительной ионизацией) (М+Н)+ 406,0.
Стадия Б: Метил-(4-фтор-3-нитрофенил)карбамат
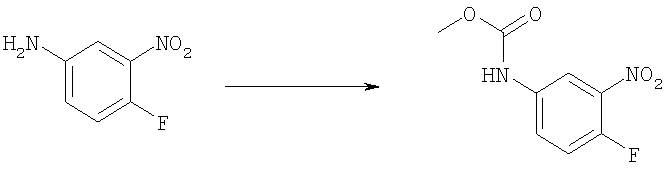
Метилхлорформиат (13,2 мл, 170,2 ммоль) добавляли по каплям к холодному (0°С) дихлорметановому (200 мл) раствору 4-фтор-3-нитроанилина (24,15 г, 154,7 ммоль) и DIPEA (N,N-диизопропилэтиламин, 35 мл, 201 ммоль). Реакционную смесь перемешивали при кт в течение ночи. Раствор затем разбавляли 200 мл дихлорметана и промывали 2 М HCl, рассолом и сушили над безводным МgSO4. Растворитель выпаривали и продукт использовали непосредственно на следующей стадии без дополнительной очистки. Выход: 35,5 г (99%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-D) δ 3.81 (s, 3 H), 7.02 (s, 1 H), 7.23 (m, 1 H), 7.72 (d, J=8,59 Гц, 1 H), 8.17 (dd, J=6,35, 2,64 Гц, 1 H).
Стадия В: Метил-{3-нитро-4-[(тетрагидро-2Н-пиран-4-илметил)амино]фенил}карбамат
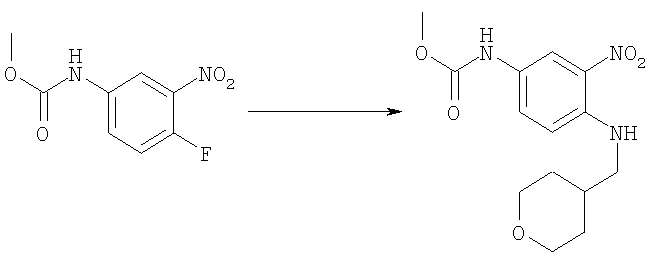
Метил-(4-фтор-3-нитрофенил)карбамат (2,0 г, 9,32 ммоль) и 4-аминометил-тетрагидропиран (1,28 г, 11,2 ммоль) перемешивали в 50 мл ЕtOН, содержащего TEA (триэтиламин, 2,0 мл, 14,0 ммоль) при 75°С в течение 48 ч, растворитель выпаривали. Остаток растворяли в ЕtOАс и промывали водным 5%-ным КНSО4, насыщенным водным раствором NаНСО3, рассолом и сушили над безводным MgSO4. Сырой продукт очищали посредством флэш-хроматографии на силикагеле, используя в качестве элюента смесь 1:1 / гексаны: ЕtOАс. Выход: 2,53 г (88%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-D) δ 1.42 (m, 2 H), 1.73 (d, J=1,76 Гц, 1 H), 1.76 (d, J=1,95 Гц, 1 H), 1.88-2.01 (m, 1 H), 3.22 (dd, J=6,74, 5,57 Гц, 2 H), 3.42 (m, 2 H), 3.78 (s, 3 H), 4.01 (d, J=4,30 Гц, 1 H), 4.04 (d, J=3,51 Гц, 1 H), 6.48 (шир.s, 1H), 6.85 (d, J=9,37 Гц, 1 H), 7.65 (шир.s, 1 H), 8.03-8.09 (m, 2 H).
Стадия Г: Метил-{3-амино-4-[(тетрагидро-2Н-пиран-4-илметил)амино]фенил}карбамат
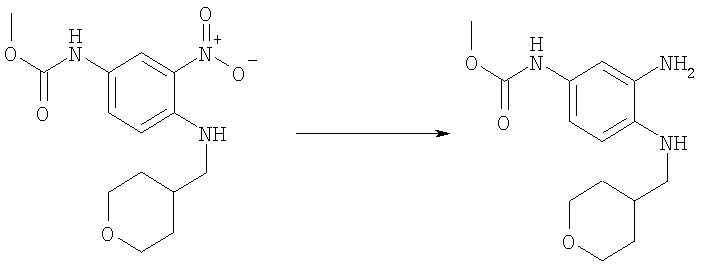
Метил-{3-нитро-4-[(тетрагидро-2Н-пиран-4-илметил)амино]фенил}-карбамат (2,53 г, 8,18 ммоль) растворяли в 50 мл EtOAc, содержащего каталитическое количество 10% Pd/C. Раствор встряхивали в атмосфере H2 (40 фунт-сила на кв. дюйм (276 кПа)), используя аппарат для гидрирования Парра (Parr), в течение ночи при кт. Раствор фильтровали через целит и растворитель выпаривали. Выход: 2,29 г (99%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-D) δ 1.40 (m, 2 H), 1.70-1.74 (m, 1 H), 1.74-1.77 (m, 1 H), 1.81-1.92 (m, 1 H), 2.99 (d, J=6,64 Гц, 2 H), 3.34 (шир.s, 2 H), 3.41 (m, 2 H), 3.74 (s, 3 H), 3.99 (d, J=3,51 Гц, 1 H), 4.02 (d, J=3,51 Гц, 1 H), 6.38 (шир.s, 1 H), 6.55-6.60 (m, 1 H), 6.62-6.68 (m, 1 H), 6.95 (шир.s, 1 H).
Стадия Д: Метил-[2-трет-бутил-1-(тетрагидро-2Н-пиран-4-илметил)-1Н-бензимидазол-5-ил]карбамат
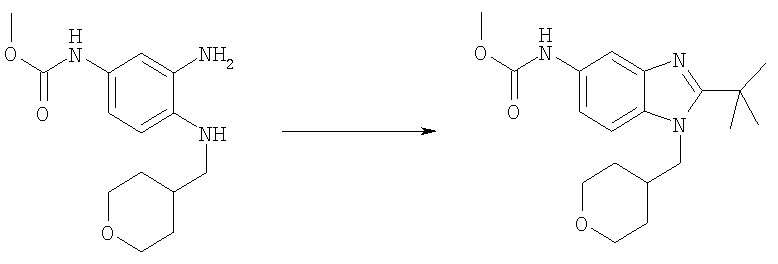
Метил-{3-амино-4-[(тетрагидро-2Н-пиран-4-илметил)амино]фенил}-карбамат (2,29 г, 8,20 ммоль) и DMAP (0,20 г, 1,64 ммоль) растворяли в 75 мл DCM (дихлорметан). Добавляли по каплям триметилацетилхлорид (1,10 мл, 9,02 ммоль) и раствор перемешивали при кт в течение 2 ч. Раствор промывали водным раствором NaHCO3, рассолом и сушили над безводным МgSO4. Остаток растворяли в 25 мл АсОН и нагревали при 125°С в течение 1 ч, используя Personal Chemistry микроволновое устройство. Растворитель выпаривали. Остаток растворяли в EtOAc и промывали водным раствором NaHCO3, рассолом и сушили над безводным MgSO4. Сырой продукт очищали посредством флэш-хроматографии на силикагеле, используя в качестве элюента смесь 4:3 / гексаны:ацетон. Выход: 1,81 г (64%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-D) δ 1.48-1.54 (m, 4 H), 1.56 (s, 9 H), 2.23-2.35 (m, 1 H), 3.27-3.35 (m, 2 H), 3.78 (s, 3 H), 3.96 (t, J=2,93 Гц, 1 H), 3.99 (t, J=3,03 Гц, 1 H), 4.18 (d, J=7,42 Гц, 2 H), 6.63 (шир.s, 1 H), 7.24-7.28 (m, 1 H), 7.41 (шир.s, 1 H), 7.61 (d, J=1,95 Гц, 1 H).
Стадия Е: 2-трет-Бутил-N-метил-1-(тетрагидро-2Н-пиран-4-илметил)-1Н-бензимидазол-5-амин
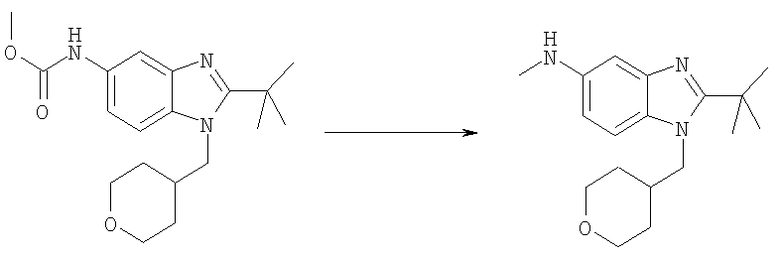
Метил-[2-трет-бутил-1-(тетрагидро-2Н-пиран-4-илметил)-1Н-бензимидазол-5-ил]карбамат (1,80 г, 5,21 ммоль) растворяли в 75 мл THF (тетрагидрофуран) при 0°С. Добавляли по каплям смесь 1М HCl/диэтиловый эфир (7,3 мл, 7,29 ммоль) и раствор перемешивали при 0°С в течение 15 мин. Медленно добавляли LiAlH4 (988 мг, 26,1 ммоль) и раствор перемешивали при кт в течение ночи. Реакционную смесь гасили при 0°С добавлением МеОН (5 мл), а затем воды (10 мл), и раствор оставляли перемешиваться при кт в течение 30 мин. Добавляли безводный Na2SO4 (10 г) и раствор перемешивали при кт в течение еще 30 мин. Раствор фильтровали и растворитель выпаривали. Остаток растворяли в ЕtOАс и промывали водным раствором NаНСО3, рассолом и сушили над безводным МgSО4. Растворитель выпаривали. Выход: 1,54 г (98%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-D) δ 1.49-1.53 (m, 4 H), 1.53-1.57 (m, 9 H), 2,22-2.32 (m, 1 H), 2.87 (s, 3 H), 3.26-3.35 (m, 2 H), 3.95 (t, J=3,03 Гц, 1 H), 3.97-4.00 (m, 1 H), 4.13 (d, J=7,42 Гц, 2 H), 6.61 (dd, J=8,59, 2,15 Гц, 1 H), 6.99 (d, J=1,95 Гц, 1 H), 7.11 (d, J=8,59 Гц, 1 H).
Пример 2
N-[2-трет-Бутил-1-(тетрагидро-2Н-пиран-4-илметил)-1H-бензимидазол-5-ил]-N-метилпропан-1-сульфонамид
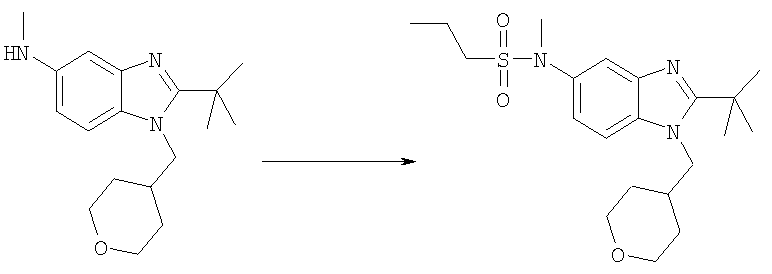
2-трет-Бутил-N-метил-1-(тетрагидро-2Н-пиран-4-илметил)-1H-бензимидазол-5-амин (для получения смотри Стадии Б-Е из Примера 1) (50 мг, 0,166 ммоль) и каталитическое количество DMAP растворяли в 5 мл DCM. Добавляли по каплям 1-пропансульфонилхлорид (0,024 мл, 0,216 ммоль) и раствор перемешивали при кт в течение 3 ч. Раствор промывали насыщенным водным раствором NаНСО3, рассолом и сушили над безводным МgSO4. Продукт очищали посредством HPLC с обращенной фазой, используя смесь 10-70% CH3CN/H2O, и лиофилизировали с получением указанного в заголовке соединения в виде соответствующей TFA соли. Выход: 60 мг (69%); 1H ЯМР (400 МГц, МЕТАНОЛ-D4) δ 1.02 (t, J=7,42 Гц, 3 Н), 1.54-1.59 (m, 2 H), 1.60-1.66 (m,2 H), 1.69(s,9 H), 1.76-1.83 (m, 2 H), 2.36-2.42 (m, 1 H), 3.09-3.13 (m, 2 H), 3.36 (m, 2 H), 3.40 (s, 3 H), 3.94 (d, J=3,58 Гц, 1 H), 3.95 (d, J=3,58 Гц, 1 H), 4.55 (d, J=7,68 Гц, 2 H), 7.70 (dd, J=8,6, 2,05 Гц, 1 H), 7.81 (d, J=1,79 Гц, 1 H), 7.98 (d, J=8,96 Гц, 1 H); MS (ESI) (M+H)+ 408,0.
Пример 3
N-[2-трет-Бутил-1-(тетрагидро-2Н-пиран-4-илметил)-1Н-бензимидазол-5-ил]-N-метилбутан-1-сульфонамид
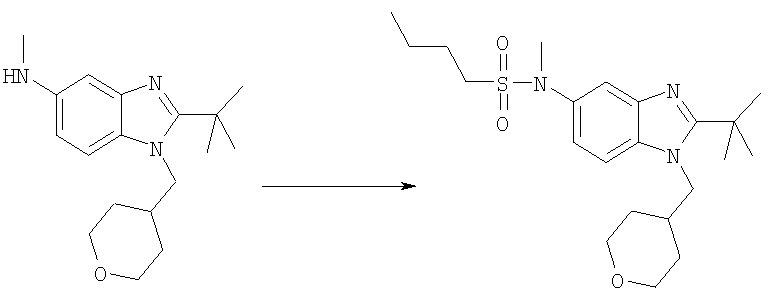
2-трет-Бутил-N-метил-1-(тетрагидро-2Н-пиран-4-илметил)-1Н-бензимидазол-5-амин (для получения смотри Стадии Б, В, Г, Д и Е из Примера 1) (38 мг, 0,126 ммоль) и 1-бутансульфонилхлорид (0,025 мл, 0,189 ммоль) перемешивали в 3 мл DCM, содержащего каталитическое количество DMAP, при кт в течение ночи. Растворитель выпаривали и продукт очищали посредством HPLC с обращенной фазой, используя смесь 10-60% СН3СN/Н2О, и лиофилизировали с получением указанного в заголовке соединения в виде соответствующей TFA соли. Выход: 39 мг (58%). 1H ЯМР (400 МГц, МЕТАНОЛ-D4): δ 0.88-0.94 (m, 3 H), 1.43 (m, 2 H), 1.53-1.59 (m, 2 H,) 1.59-1.66 (m, 2 H), 1.69 (s, 9 H), 1.71-1.77 (m, 2 H), 2.35-2.42 (m, 1 H), 3.10-3.16 (m, 2 H), 3.35 (m, 2 H), 3.40 (s, 3 H), 3.93 (d, J=3,12 Гц, 1 H), 3.96 (d, J=3,71 Гц, 1 H), 4.54 (d, J=7,42 Гц, 2 H), 7.69 (dd, J=8,98, 2,15 Гц, 1 H), 7.81 (d, J=1,56 Гц, 1 H), 7.97 (d, J=8,98 Гц, 1 H); MS (ESI) (M+H)+ 422,2; Аналит. рассч. для С22Н35N3О3S + 1,3 TFA + 1,2H2O: С, 49,96; H, 6,60; N, 7,10. Найдено: С, 49,98; H, 6,67; N, 6,83.
Пример 4
N-{2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-ил}-N-метилбутан-1-сульфонамид
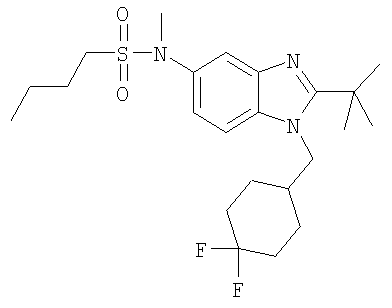
Стадия А: N-{2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1Н-бензимидазол-5-ил}-N-метилбутан-1-сульфонамид
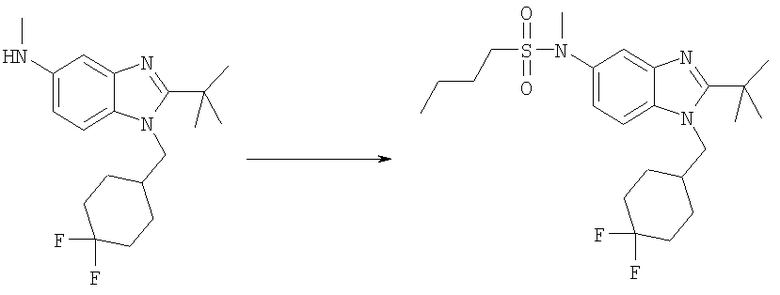
2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-N-метил-1H-бензимидазол-5-амин (для получения смотри следующие далее Стадии Б, В, Г, Д, Е и Ж) (46 мг, 0,137 ммоль) и 1-бутансульфонилхлорид (0,063 мл, 0,411 ммоль) перемешивали в 3 мл DCM, содержащего каталитическое количество DMAP, при кт в течение 6 ч. Растворитель выпаривали и продукт очищали посредством HPLC с обращенной фазой, используя смесь 10-75% СН3СN/H2О, и лиофилизировали с получением указанного в заголовке соединения в виде соответствующей TFA соли. Выход: 48 мг (62%). 1Н ЯМР (400 МГц, МЕТАНОЛ-D4): δ 0.92 (t, J=7,32 Гц, 3 Н), 1.43 (m, 2 H), 1.52-1.63 (m, 2 H), 1.69 (s, 9 H), 1.70-1.76 (m, 4 H), 1.76-1.84 (m, 2 H), 2.02-2.12 (m, 2 H), 2.22-2.31 (m, 1 H), 3.10-3.17 (m, 2 H), 3.41 (s, 3 H), 4.56 (d, J=7,62 Гц, 2 H), 7.69 (dd, J=8,98, 2,15 Гц, 1 H), 7.82 (d, J=1,76 Гц, 1 H), 7.96 (d, J=9,18 Гц, 1 H); MS (ESI) (M+H)+456.
Стадия Б: трет-Бутил-[(4,4-дифторциклогексил)метил]карбамат
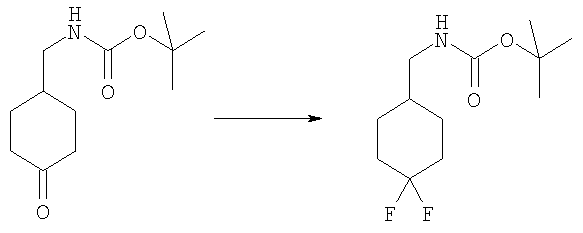
4-N-Вос-аминометилциклогексанон (1,00 г, 4,4 ммоль) растворяли в 30 мл DCM при 0°С. Добавляли по каплям DAST (диэтиламиносернистый трифторид, 1,45 мл, 11,0 ммоль) и раствор перемешивали при кт в течение ночи. Раствор промывали водным 5%-ным раствором KHSO4, насыщенным водным раствором NаНСО3, рассолом и сушили над безводным МgSO4. Сырой продукт очищали посредством флэш-хроматографии на силикагеле, используя в качестве элюента смесь 3:1 / гексаны: ЕtOАс. Выход: 508 мг (46%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-D): δ 1.19-1.36 (m, 2 H), 1.44 (s, 9 H), 1.51-1.56 (m, 1 H), 1.59-1.75 (m, 2 H), 1.75-1.84 (m, 2 H), 2.01-2.16 (m, 2 H), 3.03 (t, J=6,54 Гц, 2 Н), 4.62 (шир.з, 1 H).
Стадия В: [(4,4-Дифторциклогексил)метил]амин гидрохлорид
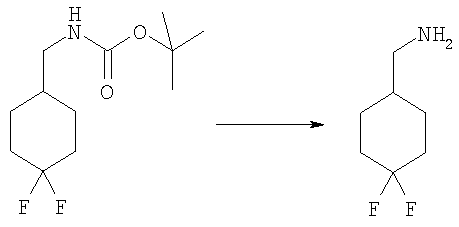
трет-Бутил-[(4,4-дифторциклогексил)метил]карбамат (505 мг, 2,03 ммоль) перемешивали в 5 мл 1М HCl/AcOH при кт в течение 2 ч. Растворитель выпаривали. Остаток промывали диэтиловым эфиром, фильтровали и сушили. Выход: 330 мг (88%). 1H ЯМР (400 МГц, МЕТАНОЛ-D4): δ 1.28-1.40 (m, 2 H), 1.71-1.82 (m, 2 H), 1.84 (d, J=3,12 Гц, 2 H), 1.86-1.89 (m, 1 H), 2.03-2.15 (m, 2 H), 2.85 (d, J=7,03 Гц, 2 H).
Стадия Г: Метил-(4-{[(4,4-дифторциклогексил)метил]амино}-3-нитрофенил)карбамат
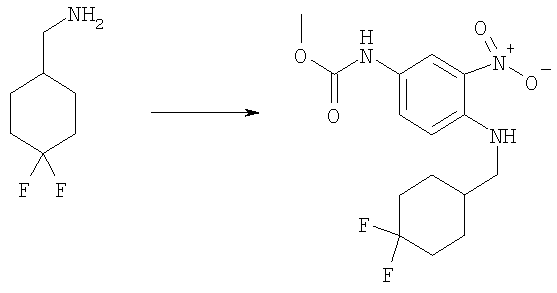
Следовали той же методике, что и на Стадии В из Примера 1, используя [(4,4-дифторциклогексил)метил]амин гидрохлорид (210 мг, 1,12 ммоль), метил-(4-фтор-3-нитрофенил)карбамат (200 мг, 0,934 ммоль) и TEA (0,390 мл, 2,80 ммоль) в 10 мл ЕtOН. Сырой продукт очищали посредством флэш-хроматографии на силикагеле, используя смесь 5% диэтиловый эфир/DCM в качестве элюента. Выход: 200 мг (62%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-D): δ 1.34-1.47 (m, 2 H), 1.65-1.75 (m, 2 H), 1.78-1.85 (m, 1 H), 1.90-1.93 (m, 1 H), 1.94-1.97 (m, 1 H), 2.10-2.21 (m, 2 H), 3.23 (dd, J=6,64, 5,66 Гц, 2 H), 3.78 (s, 3 Н), 6.48 (шир.s, 1 H), 6.83 (d, J=9,18 Гц, 1 H), 7.66 (шир.s, 1 H), 8.05 (шир.s, 1 H), 8.07 (d, J=2,54 Гц, 1 H).
Стадия Д: Метил-(3-амино-4-{[(4,4-дифторциклогексил)-метил]амино}фенил)карбамат
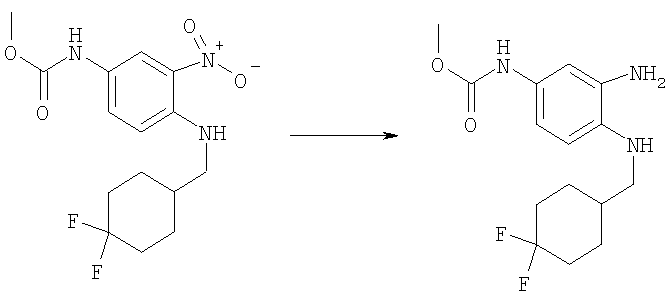
Следовали той же методике, что и на Стадии Г в Примере 1, используя метил-(4-{[(4,4-дифторциклогексил)метил]амино}-3-нитрофенил)карбамат (200 мг, 0,583 ммоль) и каталитическое количество 10% Pd/C в 20 мл ЕtOАс. Выход:
185 мг (99%). MS (ESI) (M+H)+314,29.
Стадия Е: Метил-{2-трет-бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-ил}карбамат
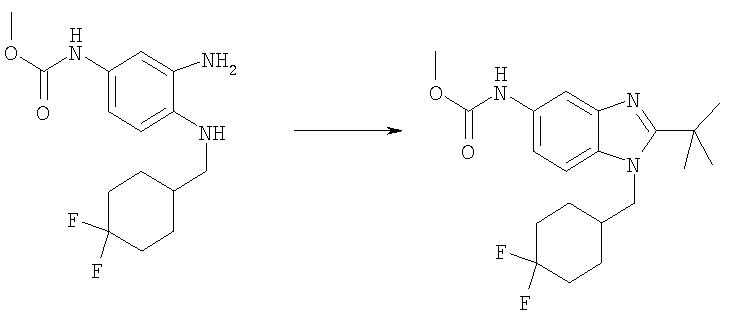
Метил-(3-амино-4-{[(4,4-дифторциклогексил)метил]амино}фенил)-карбамат (185 мг, 0,590 ммоль) и DMAP (15 мг, 0,118 ммоль) растворяли в 10 мл DCM. Добавляли по каплям триметилацетилхлорид (0,080 мл, 0,649 ммоль) и раствор перемешивали при кт в течение 2 ч. Раствор промывали водным раствором NаНСО3, рассолом и сушили над безводным MgSO4. Растворитель выпаривали. Остаток растворяли в 4 мл DCE, добавляли P2O5 (каталитический) и раствор нагревали при 125°С в течение 1 ч, используя Personal Chemistry микроволновое устройство. Раствор промывали водным раствором NaHCО3, рассолом и сушили над безводным MgSO4. Сырой продукт очищали посредством флэш-хроматографии на силикагеле, используя смесь 50-75% ЕtOАс / гексаны. Выход: 122 мг (54%); 1H ЯМР (400 МГц, ХЛОРОФОРМ-D): δ 1.43-1.52 (m, 2 H), 1.55 (s, 9 H), 1.57-1.66 (m, 2 H), 1.67-1.74 (m, 2 H), 2.08-2.18 (m, 3 H), 3.79 (s, 3 H), 4.19 (d, J=7,42 Гц, 2 H), 6.63 (шир.s, 1 H), 7.23 (d, J=8,79 Гц, 1 H), 7.37-7.46 (m, 1 H), 7.62 (d, J=1,76 Гц, 1 H).
Стадия Ж: 2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-N-метил-1H-бензимидазол-5-амин
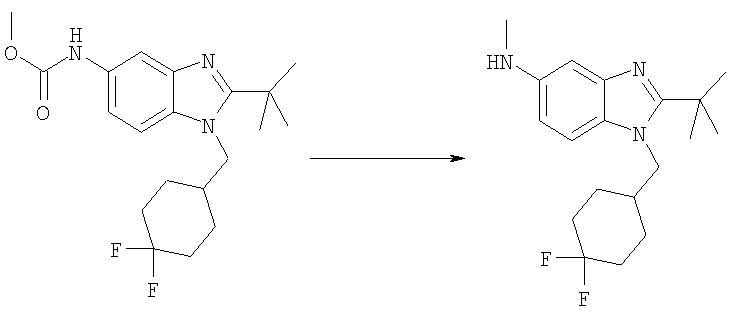
Метил-{2-трет-бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-ил}карбамат (115 мг, 0,303 ммоль) растворяли в 10 мл THF (тетрагидрофуран) при 0°С. Добавляли 1М HCl/диэтиловый эфир (0,425 мл, 0,424 ммоль) и раствор перемешивали при 0°С в течение 15 мин. Медленно добавляли LiAlH4 (57 мг, 1,52 ммоль) и раствор перемешивали при кт в течение ночи. Реакционную смесь гасили при 0°С путем добавления МеОН (1 мл) и воды (2 мл). Добавляли безводный Na2SO4 (5,0 г) и раствор перемешивали при кт в течение 30 мин. Раствор фильтровали и растворитель выпаривали. Остаток растворяли в EtOAc и промывали насыщенным водным раствором NaHCО3, рассолом и сушили над безводным МgSО4. Выход: 95 мг (93%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-D): δ 1.41-1.51 (m, 2 H), 1.54 (s, 9 H), 1.57-1.67 (m, 2 H), 1.68-1.76 (m, 3 H), 2.07-2.17 (m, 3 H), 2.87 (s, 3 H), 4.15 (d, J=7,42 Гц, 2 H), 6.61 (dd, J=8,59, 2,34 Гц, 1 H), 7.01 (d, J=1,95 Гц, 1 H), 7.09 (d, J=8,59 Гц, 1 H).
Пример 5
N-Метил-N-[1-(тетрагидро-2Н-пиран-4-илметил)-2-(трифторметил)-1Н-бензимидазол-5-ил]пропан-1-сульфонамид
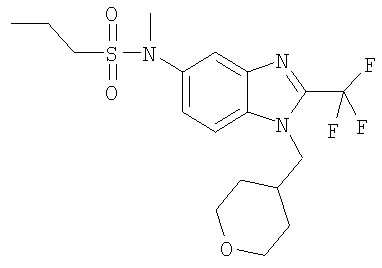
Стадия А: N-Метил-N-[1-(тетрагидро-2Н-пиран-4-илметил)-2-(трифторметил)-1H-бензимидазол-5-ил]пропан-1-сульфонамид
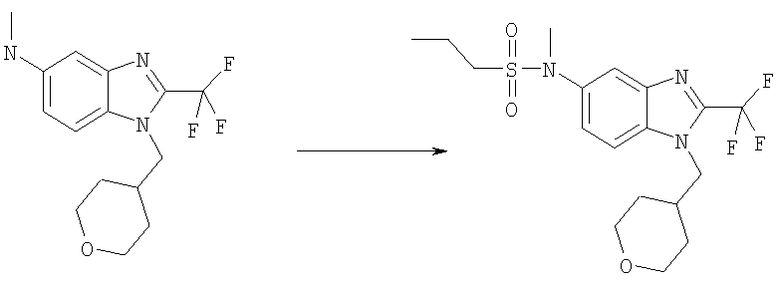
Пропан-1-сульфонилхлорид (27 мкл, 34 мг, 0,24 ммоль) добавляли к раствору N-метил-1-(тетрагидро-2H-пиран-4-илметил)-2-(трифторметил)-1H-бензимидазол-5-амина (63 мг, 0,20 ммоль) (смотри следующие стадии Б, В, Г, Д, Е и Ж для получения), DIPEA (49 мкл, 36 мг, 0,28 ммоль) и DMAP (5 мг, 0,04 ммоль) в DCM (6 мл) при 0°С. Реакционную смесь перемешивали в течение ночи при комнатной температуре, разбавляли DCM (50 мл), промывали насыщенным NаНСО3 (2×10 мл) и сушили над Na2SO4. Сырой продукт очищали посредством MPLC (жидкостная хроматография среднего давления), используя смесь гекс./ЕtOАс (1:1), на силикагеле с получением 40 мг (47%) указанного в заголовке соединения в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-D4): δ 1.00 (t, J=7,42 Гц, 3 Н), 1.38-1.53 (m, 4 H), 1.70-1.88 (m, 2 H), 2.15-2.30 (m, 1 H), 3.01-3.11 (m, 2 H), 3.28-3.33 (m, 2 H), 3.35 (s, 3 H), 3.88-3.91 (m, 2 H), 4.30 (d, J=7,62 Гц, 2 H), 7.55 (dd, J=8,79, 1,76 Гц, 1 H), 7.75 (d, J=8,98 Гц, 1 H), 7.82 (d, J=1,56 Гц, 1 H). MS (ESI) (M+H)+=420,0. Аналит. рассч. для С18Н24F3N3О3S + 0,20 Н2O + 0,30 СН3ОН (432,68): С, 50,80; H, 5,96; N, 9,71; найдено: С, 50,79; H, 5,91; N, 9,69.
Стадия Б. N-(4-фтор-3-нитрофенил)ацетамид
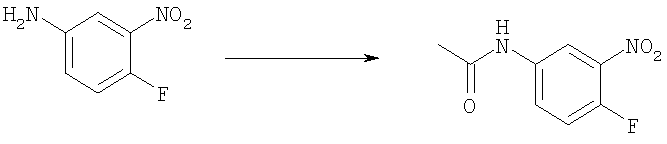
4-Фтор-3-нитро-анилин (45,0 г, 0,288 моль) добавляли порциями к уксусному ангидриду (150 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Белое твердое вещество собирали и сушили в вакууме с получением указанного в заголовке соединения (42,0 г, 70%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-D): δ 2.23 (s, 3 H), 7.26 (m, 1 H), 7.50 (s широкий, 1 H), 7.87 (m, 1 H), 8.23 (dd, J=6,44, 2,73 Гц, 1 H).
Стадия В: N-(4-фтор-3-нитрофенил)-N-метилацетамид
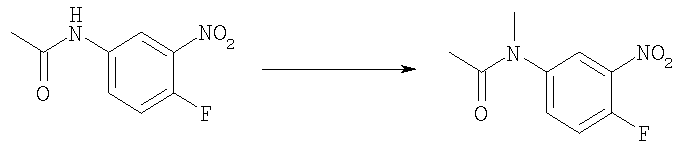
Гидрид натрия (4,22 г, 60%, 106 ммоль) добавляли порциями к раствору N-(4-фтор-3-нитрофенил)ацетамида (13,9 г, 70 ммоль) в THF (200 мл) при 0°С. При перемешивании в течение 20 мин добавляли иодметан (18,5 г, 130 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, гасили насыщенным NаНСО3 (30 мл) и экстрагировали ЕtOАс (3×100 мл). Объединенные органические фазы промывали насыщенным NaCl (2×50 мл). После фильтрования и выпаривания получали 13,1 г (88%) указанного в заголовке соединения в виде желтого твердого вещества. 1H ЯМР (400 МГц, ХЛОРОФОРМ-D): δ 1.92 (s, 3 H), 3.30 (s, 3 H), 7.38 (s, 1 H), 7.52 (s, 1 H), 7.95 (s, 1 H).
Стадия Г. N-метил-N-{3-нитро-4-[(тетрагидро-2H-пиран-4-илметил)амино]фенил}ацетамид
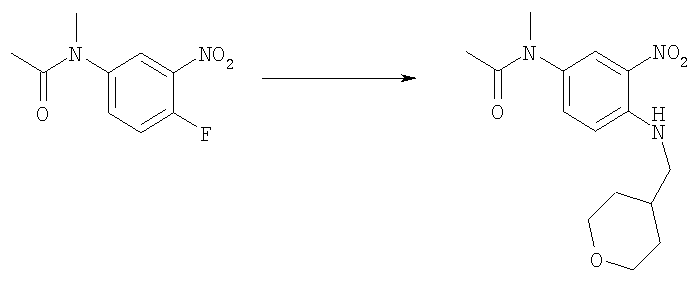
4-Аминометилтетрагидропиран (10,0 г, 86,5 ммоль) добавляли к смеси N-(4-фтор-3-нитрофенил)-N-метилацетамида (15,6 г, 73,3 ммоль) и TEA (триэтиламин, 15,3 мл, 11,1 г, 110 ммоль) в ЕtOН (300 мл) при комнатной температуре. Реакционную смесь нагревали с обратным холодильником в течение 6 ч. После выпаривания этанола остаток растворяли в ЕtOАс (400 мл), промывали Н2О (3×50 мл), насыщенным NaCl (3×50 мл), и сушили над Na2SO4. После фильтрования и выпаривания получали 21,7 г (96%) указанного в заголовке соединения в виде красно-оранжевого твердого вещества. 1H ЯМР (400 МГц, ХЛОРОФОРМ-D): δ 1.38-1.52 (m, 2 H), 1.72-1.81 (m, 2 H), 1.90 (s, 3 H), 1.93-2.02 (m, 1 H), 3.23 (s, 3 H), 3.23-3.27 (m, 2 H), 3.36-3.49 (m, 2 H), 4.01-4.07 (m, 2 H), 6.91 (d, J=9,18 Гц, 1 H), 7.29 (dd, J=9,08, 2.64 Гц, 1 H), 8.05 (d, J=2,34 Гц, 1 H), 8.22 (t, J=5,37 Гц, 1 H). MS (ESI) (M+H)+=309,12.
Стадия Д. N-{3-Амино-4-[(тетрагидро-2H-пиран-4-илметил)амино]фенил}-N-метилацетамид
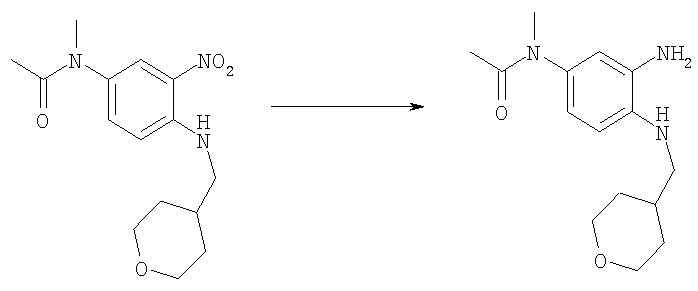
N-Метил-N-{3-нитро-4-[(тетрагидро-2Н-пиран-4-илметил)амино]-фенил}ацетамид (21,7 г, 70,5 ммоль) гидрировали в этилацетате (500 мл) с 10% Pd/C (1,0 г) в качестве катализатора при 30-40 фунт-сила на кв. дюйм (207-276 кПа) Н2 в шейкере Парра в течение 18 ч при комнатной температуре. После фильтрования через целит и выпаривания получали 19,6 г (100%) пурпурного твердого вещества. 1H ЯМР (400 МГц, ХЛОРОФОРМ-D): δ 1.35-1.50 (m, 2 H), 1.67 (s, 1 H), 1.73-1.81 (m, 2 H), 1.88 (s, 3 H), 1.88-1,99 (m, 1 H), 3.04 (d, J=6,64 Гц, 2 H), 3.20 (s, 3 H), 3.33-3.48 (m, 4 H), 3.97-4.08 (m, 2 H), 6.54 (d, J=1,76 Гц, 1 H), 6.60-6.63 (m, 2 H); MS (ESI) (M+H)+: 278,7.
Стадия Е. N-Метил-N-[1-(тетрагидро-2Н-пиран-4-илметил)-2-(трифторметил)-1Н-бензимидазол-5-ил]ацетамид
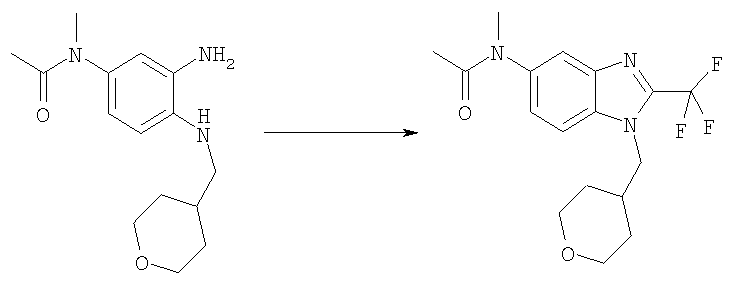
Раствор N-{3-амино-4-[(тетрагидро-2Н-пиран-4-илметил)амино]фенил}-N-метилацетамида гидрохлорида (2,77 г, 10 ммоль) в трифторуксусной кислоте (60 мл) нагревали до кипения в течение 18 ч. После выпаривания растворителя остаток растворяли в ЕtOАс (200 мл), промывали 2 н. NaOH (2×10 мл) и сушили над Na2SO4. Сырой продукт очищали посредством MPLC, используя ЕtOАс на силикагеле, получая 3,18 г (90%) указанного в заголовке соединения в виде белого твердого вещества. MS (ESI) (M+H)+=356,02.
Стадия Ж. N-Метил-1-(тетрагидро-2Н-пиран-4-илметил)-2-(трифторметил)-1H-бензимидазол-5-амин
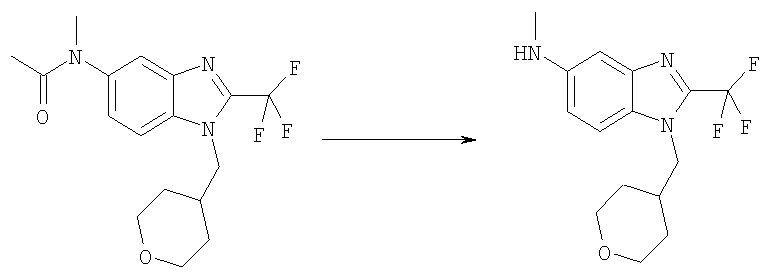
N-Метил-N-[1-(тетрагидро-2Н-пиран-4-илметил)-2-(трифторметил)-1Н-бензимидазол-5-ил]ацетамид (3,18 г, 8,95 ммоль) растворяли в соляной кислоте (37%, 60 мл) и затем нагревали в течение ночи при 95°С. После выпаривания остаток обрабатывали 20 мл 2 н. NaOH, экстрагировали ЕtOАс (4×50 мл). Объединенные органические фазы промывали рассолом (20 мл) и сушили над Na2SO4. После выпаривания получали 2,80 г (100%) указанного в заголовке продукта в виде пурпурно-белого твердого вещества, которое использовали непосредственно на Стадии 3. MS (ESI) (M+H)+=314,20.
Пример 6
N-Метил-N-[1-(тетрагидро-2H-пиран-4-илметил)-2-(трифторметил)-1H-бензимидазол-5-ил]циклопропансульфонамид
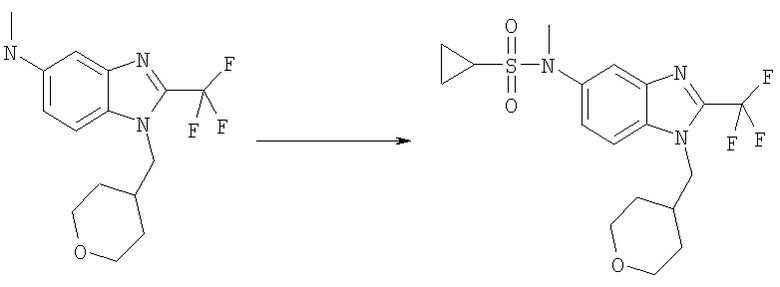
Следовали методике из Примера 5, используя циклопропансульфонилхлорид (34 мг, 0,24 ммоль), N-метил-1-(тетрагидро-2Н-пиран-4-илметил)-2-(трифторметил)-1H-бензимидазол-5-амин (63 мг, 0,20 ммоль) (для получения смотри стадию Ж в примере 1), DIPEA (49 мкл, 36 мг, 0,28 ммоль) и DMAP (5 мг, 0,04 ммоль) в DCM (6 мл) при 0°С. Сырой продукт очищали посредством MPLC, используя смесь гекс./ЕtOАс (1:1) на силикагеле, получая 81 мг (97%) указанного в заголовке соединения в виде белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-D4): δ 0.85-0.92 (m, 2 H), 0.93-1.01 (m, 2 H), 1.37-1.52 (m, 4 Н), 2.18-2.31 (m, 1 H), 2.55-2.65 (m, 1 H), 3.30-3.36 (m, 2 H), 3.38 (s, 3 Н), 3.86-3.95 (m, 2 H), 4.32 (d, J=7,62 Гц, 2 H), 7.58 (dd, J=8,89, 2,05 Гц, 1 H), 7.76 (d, J=8,79 Гц, 1 H) 7.86 (d, J=1,5 Гц, 1 H). MS (ESI) (M+H)+=418,0. Аналит. рассч. для C18H22F3N3O3S + 0,10 Н2O + 0,20 СН3ОН (425,66): С, 51,36; Н, 5,45; N, 9,87; найдено: С, 51,39; Н, 5,49; N, 9,92.
Пример 7
N-[2-трет-Бутил-1-(тетрагидро-2Н-пиран-4-илметил)-1Н-бензимидазол-5-ил]-N-метилпентан-1-сульфонамид
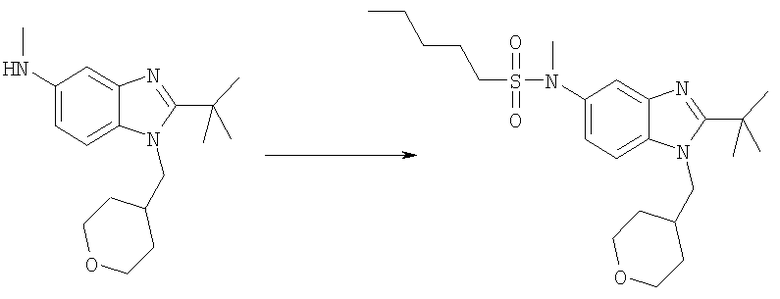
2-трет-Бутил-N-метил-1-(тетрагидро-2Н-пиран-4-илметил)-1H-бензимидазол-5-амин (65 мг, 0,216 ммоль) и каталитическое количество DMAP растворяли в 3 мл DCE. Добавляли н-пентилсульфонилхлорид (44 мг, 0,259 ммоль) и раствор перемешивали при кт в течение 4 ч. Раствор промывали насыщенным водным раствором NаНСО3, рассолом и сушили над безводным MgSO4. Растворитель выпаривали и продукт очищали посредством HPLC с обращенной фазой, используя смесь 10-70% СН3СN/Н2О, и лиофилизировали с получением указанного в заголовке соединения в виде соответствующей TFA соли. Выход: 89 мг (75%). 1H ЯМР (400 МГц, МЕТАНОЛ-
D4) δ 0.89 (t, J=7,13 Гц, 3 Н), 1.26-1.34 (m, 2 H), 1.34-1.43 (m, 2 H), 1.52-1.58 (m, 2 H), 1.58-1.66 (m, 2 H), 1.69(s, 9H), 1.71-1.80(m, 2 H), 2.34-2.43 (m, 1 H), 3.09-3.16 (m, 2 H), 3.36 (td, J=11,47, 2,64 Гц, 2 H), 3.40 (s, 3 H) 3.93 (d, J=3,12 Гц, 1 H), 3.95-3.97 (m, 1 H), 4.55 (d, J=7,62 Гц, 2 H), 7.69 (dd, J=9,08, 2,05 Гц, 1 H), 7.81 (d, J=1,56 Гц, 1 H), 7.97 (d, J=8,59 Гц, 1 H); MS (ESI) (M+H)+ 436,0; Аналит. рассч. (%) для С23H37N3O3S + 1,1 TFA + 0,9 H2O; С, 52,43; H, 6,97; N, 7,28. Найдено: С, 52,39; H, 6,96; N, 7,43.
Пример 8
N-[2-трет-Бутил-1-(тетрагидро-2H-пиран-4-илметил)-1Н-бензимидазол-5-ил]-N-метилэтансульфонамид
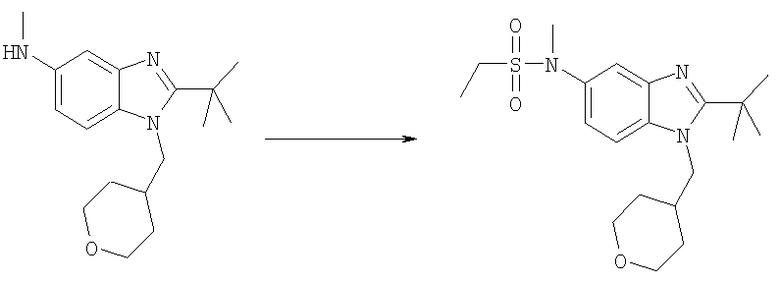
2-трет-Бутил-N-метил-1-(тетрагидро-2Н-пиран-4-илметил)-1H-бензимидазол-5-амин (50 мг, 0,166 ммоль) и каталитическое количество DMAP растворяли в 3 мл DCE. Добавляли этансульфонилхлорид (0,020 мл, 0,215 ммоль) и раствор перемешивали при кт в течение 12 ч. Раствор промывали насыщенным водным раствором NаНСО3, рассолом и сушили над безводным MgSO4. Растворитель выпаривали и продукт очищали посредством HPLC с обращенной фазой, используя смесь 10-70% СН3СN/H2О, и лиофилизировали с получением указанного в заголовке соединения в виде соответствующей TFA соли. Выход: 70 мг (83%). 1H ЯМР (600 МГц, CD3OD) δ 1.31 (t, J=7,30 Гц, 3 H), 1.53-1.58 (m, 2 H), 1.58-1.65 (m, 2 H), 1.69 (s, 9 H), 2.35-2.42 (m, 1 H), 3.16 (m, 2 H), 3.35 (m, 2 H), 3.41 (s, 3 H), 3.94 (d, J=3,84 Гц, 1 H), 3.95 (d, J=3,84 Гц, 1 H), 4.54 (d, J=7,68 Гц, 2 H), 7.69 (dd, J=9,09, 1,92 Гц, 1 H), 7.81 (d, J=1,79 Гц, 1 H), 7.97 (d, J=8,96 Гц, 1 H); MS (ESI) (M+H)+ 394,0; Аналит. рассч. (%) для С20Н31N3О3S + 1,4 TFA: С, 49,50; Н, 5,90; N, 7,60. Найдено: С, 49,51; Н, 6,00; N, 7,24.
Пример 9
N-[2-трет-Бутил-1-(тетрагидро-2Н-пиран-4-илметил)-1Н-бензимидазол-5-ил]-N,2-диметилпропан-2-сульфонамид
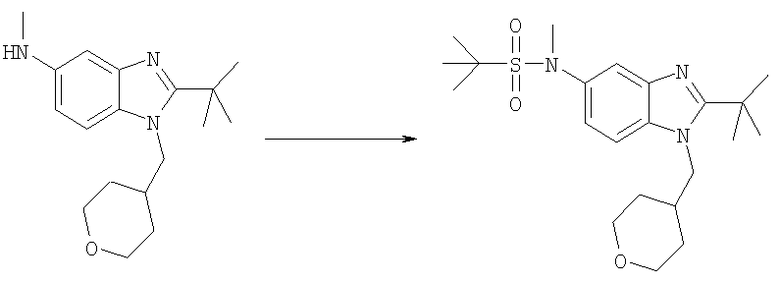
2-трет-Бутил-N-метил-1-(тетрагидро-2Н-пиран-4-илметил)-1H-бензимидазол-5-амин (50 мг, 0,166 ммоль) и DMAP (20 мг, 0,166 ммоль) растворяли в 3 мл DCM. Добавляли трет-бутилсульфинилхлорид (0,027 мл, 0,215 ммоль) и раствор перемешивали при кт в течение 2 ч. Раствор промывали насыщенным водным раствором NaHCO3, рассолом и сушили над безводным MgSO4. Добавляли 3-хлорпероксибензойную кислоту (37 мг, 0,166 ммоль) и раствор перемешивали при кт в течение 1 ч. Раствор промывали насыщенным водным раствором NаНСО3, рассолом и сушили над безводным МgSО4. Продукт очищали посредством HPLC с обращенной фазой, используя смесь 10-70%
СН3СN/Н2О, и лиофилизировали с получением указанного в заголовке соединения в виде соответствующей TFA соли. Выход: 34 мг (38%). 1H ЯМР (400 МГц, МЕТАНОЛ-D4) δ 1.37 (s, 9 Н), 1.52-1.58 (m, 2 H), 1.59-1.66 (m, 2 H), 1.69 (s, 9 H), 2.34-2.44 (m, 1 H), 3.36 (m, 2 H), 3.48 (s, 3 H), 3.93 (d, J=3,32 Гц, 1 H), 3.95-3.97 (m, 1 H), 4.54 (d, J=7,62 Гц, 2 H), 7.78 (dd, J=9,08, 2,05 Гц, 1 H), 7.92 (d, J=2,15 Гц, 1 H), 7.96 (d, J=9,18 Гц, 1 H); MS (ESI) (M+H)+ 422,0.
Пример 10
N-{2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1Н-бензимидазол-5-ил}-N-метилпропан-1-сульфонамид
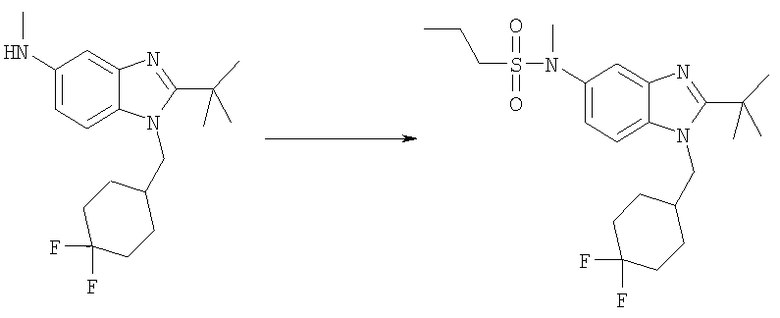
2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-N-метил-1H-бензимидазол-5-амин (45 мг, 0,134 ммоль) и каталитическое количество DMAP растворяли в 3 мл DCE. Добавляли пропансульфонилхлорид (0,020 мл, 0,174 ммоль) и раствор перемешивали при кт в течение 4 ч. Раствор промывали насыщенным водным раствором NаНСО3, рассолом и сушили над безводным МgSО4. Растворитель выпаривали и продукт очищали посредством HPLC с обращенной фазой, используя смесь 10-70% СН3СN/Н2О, и лиофилизировали с получением указанного в заголовке соединения в виде соответствующей TFA соли. Выход: 55 мг (74%). 1H ЯМР (400 МГц, МЕТАНОЛ-
D4) δ 1.00 (t, J=7,42 Гц, 3 Н), 1.51-1.60 (m, 2 H), 1.66 (s, 9 H), 1.68-1.73 (m, 2 H), 1.73-1.81 (m, 4 H), 2.00-2.11 (m, 2 H), 2.18-2.29 (m, 1 H,) 3.06-3.12 (m, 2 H), 3.38 (s, 3 H), 4.54 (d, J=7,62 Гц, 2 H), 7.67 (dd, J=9,08, 2,05 Гц, 1 H), 7.79 (d, J=1,56 Гц, 1 H), 7.94 (d, J=8,98 Гц, 1 H); MS (ESI) (M+H)+ 442,0; Аналит. рассч. (%) для С22Н33N3O2SF2 + 1,0 TFA + 1,6 H2O: С, 49,32; H, 6,42; N, 7,10. Найдено: С, 49,39; H, 6,66; N, 6,71.
Пример 11
N-{2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-ил}-N-метилэтансульфонамид
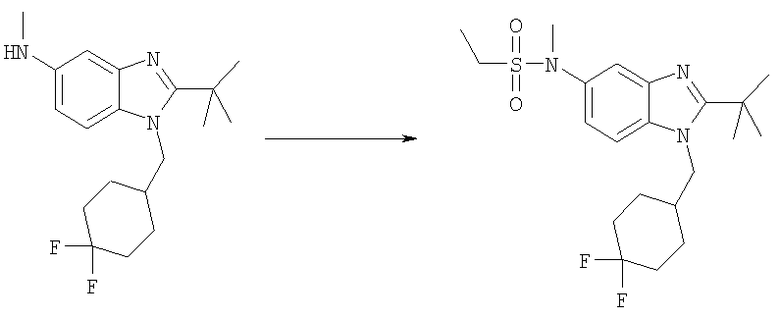
2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-N-метил-1H-бензимидазол-5-амин (49 мг, 0,146 ммоль) и каталитическое количество DMAP растворяли в 3 мл DCM. Добавляли этансульфонилхлорид (0,018 мл, 0,190 ммоль) и раствор перемешивали при кт в течение 12 ч. Раствор промывали насыщенным водным раствором NаНСО3, рассолом и сушили над безводным MgSO4. Растворитель выпаривали и продукт очищали посредством HPLC с обращенной фазой, используя смесь 10-70% СН3СN/H2О, и лиофилизировали с получением указанного в заголовке соединения в виде соответствующей TFA соли. Выход: 58 мг (73%). 1H ЯМР (600 МГц, MeOD) δ 1.31 (t, J=7,42 Гц, 3 H), 1.34-1.41 (m, 2 H), 1.54-1.62 (m, 2 H), 1.69 (s, 9 H), 1.72-1.80 (m, 2 H), 2.03-2.11 (m, 2 H), 2.23-2.30 (m, 1 H), 3.17 (q, J=7,25 Гц, 2 H), 3.41 (s, 3 H), 4.56 (d, J=7,68 Гц, 2 H), 7.70 (dd, J=8,96, 2,05 Гц, 1 H), 7.82 (d, J=2,05 Гц, 1 H), 7.96 (d, J=8,96 Гц, 1 H); MS (ESI) (M+H)+ 428,0.
Пример 12
N-{2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-ил}пропан-1-сульфонамид
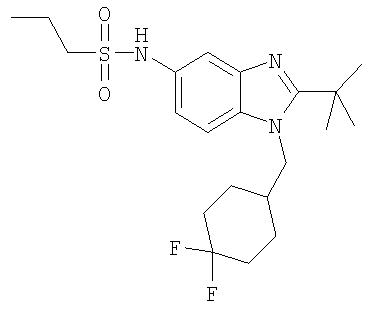
Стадия А: N-{2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-ил}пропан-1-сульфонамид
2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-амин (для получения смотри следующие стадии Б-Д) (45 мг, 0,140 ммоль) и каталитическое количество DMAP растворяли в 3 мл DCM. Добавляли пропансульфонилхлорид (0,020 мл, 0,182 ммоль) и раствор перемешивали при кт в течение 4 ч. Раствор промывали насыщенным водным раствором NaHCO3, рассолом и сушили над безводным MgSO4. Растворитель выпаривали и продукт очищали посредством HPLC с обращенной фазой, используя смесь 10-70% СН3СN/Н2О, и лиофилизировали с получением указанного в заголовке соединения в виде соответствующей TFA соли. Выход: 39 мг (51%). 1H ЯМР (600 МГц, CD3OD) δ 1.00 (t, J=7,55 Гц, 3 H), 1.53-1.61 (m, 2 H), 1.67 (s, 9 H), 1.70-1.77 (m, 3 H), 1.77-1.85 (m, 3 H), 2.02-2.11 (m, 2 H), 2.22-2.29 (m, 1 H), 3.08-3.13 (m, 2 H), 4.53 (d, J=7,42 Гц, 2 H), 7.41 (dd, J=9,09, 1,92 Гц, 1 H), 7.75 (d, J=1,79 Гц, 1 H), 7.89 (d, J=9,22 Гц, 1 H); MS (ESI) (M+H)+428,0.
Стадия Б: N-(4-{[(4,4-дифторциклогексил)метил]амино}-3-нитрофенил)ацетамид
N-(4-Фтор-3-нитрофенил)ацетамид (1,15 г, 5,84 ммоль) и [(4,4-дифторциклогексил)метил]амина гидрохлорид (1,30 г, 7,59 ммоль) перемешивали в 30 мл ЕtOН, содержащего TEA (2,40 мл, 17,5 ммоль), при 80°С в течение 48 ч. Растворитель выпаривали. Остаток растворяли в ЕtOАс и промывали водным 5%-ным раствором КНSО4, насыщенным водным раствором NаНСО3, насыщенным водным раствором NaCl и сушили над безводным Na2SO4. Продукт кристаллизовали из ЕtOАс. Остатки над маточным раствором очищали посредством флэш-хроматографии на силикагеле, используя смесь 2:1 / гексаны:ацетон в качестве элюента. Выход: 1,50 г (78%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-D) δ 1.33-1.47 (m, 2 H), 1.66-1.77 (m, 2 H), 1.77-1.86 (m, 1 H), 1.89-1.93 (m, 1 H), 1.93-1.97 (m, 1 H), 2.10-2.17 (m, 2 H), 2.18 (s, 3 H), 3.23 (dd, J=6,74, 5,76 Гц, 2 H), 6.83 (d, J=9,37 Гц, 1 H), 7.15 (s, 1 H), 7.80 (dd, J=9,18, 2,54 Гц, 1 H), 8.09 (d, J=2,54 Гц, 2 H).
Стадия В: N-(3-Амино-4-{[(4,4-дифторциклогексил)метил]амино}-фенил)ацетамид
N-(4-{[(4,4-дифторциклогексил)метил]амино}-3-нитрофенил)ацетамид (1,48 г, 4,52 ммоль) растворяли в 50 мл ЕtOАс, содержащего каталитическое количество 10% Pd/C. Раствор встряхивали в аппарате для гидрирования Парра в атмосфере Н2 (45 фунт-сила на кв. дюйм, 310 кПа) при кт в течение 24 ч. Раствор фильтровали через целит и растворитель выпаривали. Выход: 1,32 г (98%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-D) δ 1.31-1.43 (m, 2 H), 1.64-1.73 (m, 2 H), 1.74-1.82 (m, 1 H), 1.89-1.93 (m, 1 H), 1.93-1.96 (m, 1 H), 2.08-2.17 (m, 5 H), 3.00 (d, J=6,64 Гц, 2 H), 3.27-3.46 (m, 2 H), 6.55 (d, J=8,40 Гц, 1 H), 6.70 (dd, J=8,40, 2,34 Гц, 1 H), 7.01 (s, 1 H), 7.13 (d, J=2,34 Гц, 1 H).
Стадия Г: N-{2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1Н-бензимидазол-5-ил}ацетамид
N-(3-Амино-4-{[(4,4-дифторциклогексил)метил]амино}фенил)ацетамид (1,32 г, 4,44 ммоль) растворяли в 100 мл DCM, содержащего DMAP (108 мг, 0,89 ммоль). Добавляли по каплям триметилацетилхлорид (0,60 мл, 4,88 ммоль) и раствор перемешивали при кт в течение 2 ч. Раствор промывали насыщенным водным раствором NaHCO3, насыщенным водным раствором NaCl и сушили над безводным
Na2SO4. Часть продукта выпадала в осадок в ходе промывки и фильтрования. Органические фазы выпаривали и объединяли с осадком. Продукт растворяли в 30 мл АсОН и помещали в 6 герметизируемых пробирок (5 мл/пробирка). Каждую пробирку нагревали при 150°С в Personal Chemistry микроволновом устройстве в течение 2,5 ч. Фракции объединяли и растворитель выпаривали. Продукт растворяли в ЕtOАс и промывали водным раствором NаНСО3, насыщенным водным раствором NaCl и сушили над безводным Na2SO4. Продукт очищали посредством флэш-хроматографии на силикагеле, используя смесь 2:1 / ацетон:гексаны в качестве элюента. Выход: 1,11 г (68%). 1H ЯМР (400 МГц, МЕТАНОЛ-D4) δ 1.40-1.49 (m, 2 H), 1.52 (s, 9 H), 1.60-1.65 (m, 2 H), 1.67-1.77 (m, 1 H), 1.96-2.06 (m, 3H),2.11 (s, 3 H), 2.15-2.23 (m, 1 H), 4.28 (d, J=7,62 Гц, 2 H), 7.35-7.39 (m, 1 H), 7.40-7.44 (m, 1 H), 7.85 (d, J=1,76 Гц, 1 H).
Стадия Д: 2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1Н-бензимидазол-5-амин
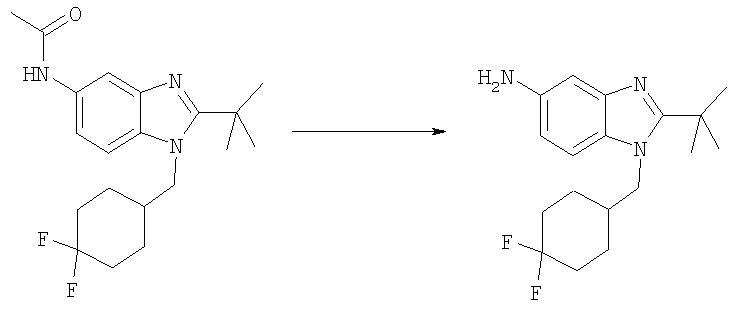
N-{2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1Н-бензимидазол-5-ил}ацетамид (500 мг, 1,37 ммоль) растворяли в 10 мл смеси 1:1 / ЕtOН:2 М HCl. Раствор делили в две герметизируемые пробирки (5 мл/пробирку). Каждую пробирку нагревали при 120°С в Personal Chemistry микроволновом устройстве в течение 1 ч. Фракции объединяли и растворитель выпаривали. Остаток разбавляли 2 М NaOH и экстрагировали (3Х) ЕtOАс. Органическую фазу промывали насыщенным водным раствором NaCl и сушили над безводным Na2SO4. Растворитель выпаривали. Выход: 440 мг (99%). 1Н ЯМР (400 МГц, ХЛОРОФОРМ-D) δ 1.40-1.52 (m, 2 Н), 1.52-1.54 (m, 9 Н), 1.56-1.66 (m, 4 Н), 1.68-1.75 (m, 2 Н), 2.07-2.17 (m, 3 Н), 4.14 (d, J=7,62 Гц, 2 H), 6.65 (dd, J=8,50, 2,25 Гц, 1 H), 7.04-7.09 (m, 2 H).
Пример 13
N-{2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-ил}метансульфонамид
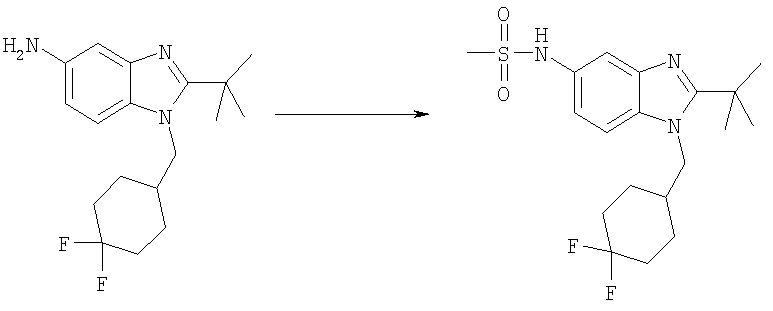
2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1Н-бензимидазол-5-амин (40 мг, 0,124 ммоль) и каталитическое количество DMAP растворяли в 3 мл DCM. Добавляли метансульфонилхлорид (0,012 мл, 0,149 ммоль) и раствор перемешивали при кт в течение 2 ч. Раствор промывали насыщенным водным раствором NaHCO3, рассолом и сушили над безводным МgSО4. Растворитель выпаривали и продукт очищали посредством HPLC с обращенной фазой, используя смесь 10-70% СН3СN/Н2O, и лиофилизировали с получением указанного в заголовке соединения в виде соответствующей TFA соли. Выход: 50 мг (79%). 1H ЯМР (600 МГц, MeOD) δ 1.53-1.61 (m, 2 H), 1.67 (s, 9 H), 1.71-1.76 (m, 3 H), 1.76-1.82 (m, 1 H), 2.04-2.11 (m, 2 H), 2.23-2.29 (m, 1 H), 3.01 (s, 3 H), 4.54 (d, J=7,68 Гц, 2 H), 7.42 (dd, J=9,22, 2,05 Гц, 1 H), 7.75 (d, J=1,79 Гц, 1 H), 7.91 (d, J=8,96 Гц, 1 H); MS (ESI) (M+H)+ 400,0; Аналит. рассч. (%) для
C19H27N3O2SF2 + 1,9 TFA + 0,1 H2O: С, 44,32; H, 4,75; N, 6,80. Найдено: С, 44,34; H, 4,78; N, 6,55.
Пример 14
N-{2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-ил}этансульфонамид
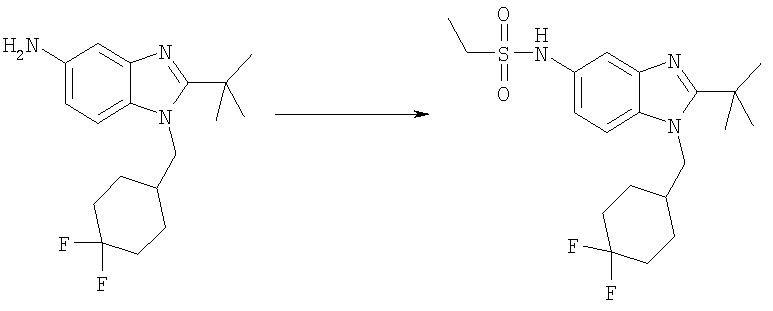
2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-амин (440 мг, 1,37 ммоль) и DMAP (165 мг, 1,37 ммоль) растворяли в 50 мл DCM. Добавляли по каплям этансульфонилхлорид (0,170 мл, 1,78 ммоль) и раствор перемешивали при кт в течение 2,5 ч. Раствор промывали насыщенным водным раствором NаНСО3, насыщенным водным раствором NaCl и сушили над безводным Na2SO4. Продукт очищали посредством флэш-хроматографии на силикагеле, используя ЕtOАс в качестве элюента. Фракции выпаривали и остаток растворяли в 25 мл МеОН. Добавляли по каплям TFA (0,155 мл, 2,06 ммоль) и раствор перемешивали при кт в течение 30 мин. Растворитель выпаривали и продукт осаждали в диэтиловом эфире с получением указанного в заголовке соединения в виде его соответствующей TFA соли. Выход: 565 мг (78%). 1H ЯМР (400 МГц, МЕТАНОЛ-D4) δ 1.29 (t, J=7,42 Гц, 3 H), 1.48-1.60 (m, 2 H), 1.64 (s, 9 H), 1.66-1.72 (m, 2 H), 1.73-1.82 (m, 2 H), 1.99-2.09 (m, 2 H), 2.18-2.28 (m, 1 H), 3.11 (m, 2 H), 4.50 (d, J=7,62 Гц, 2 H), 7.38 (dd, J=9,08, 2,05 Гц, 1 H), 7.72 (d, J=2,15 Гц, 1 H), 7.85 (d, J=8,98 Гц, 1 H); MS (ESI) (M+H)+ 414,0.
Пример 15
N-{2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1Н-бензимидазол-5-ил}циклопропансульфонамид
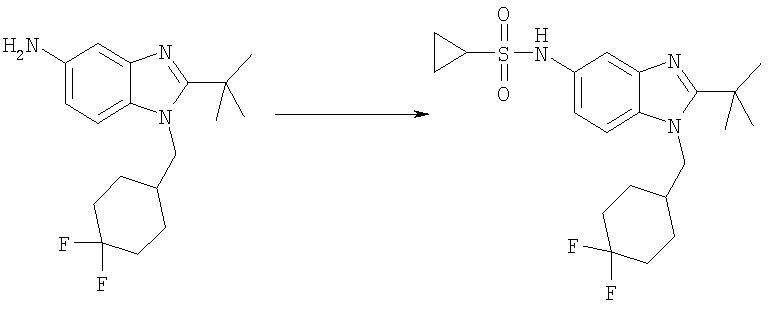
2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-амин (300 мг, 0,934 ммоль) и DMAP (115 мг, 0,934 ммоль) растворяли в 10 мл DCM. Добавляли циклопропансульфонилхлорид (170 мг, 1,21 ммоль) и раствор перемешивали при кт в течение 2 ч. Раствор промывали насыщенным водным раствором NaHCO3, рассолом и сушили над безводным MgSO4. Продукт очищали посредством флэш-хроматографии на силикагеле, используя ЕtOАс в качестве элюента. Фракции выпаривали и остаток растворяли в 25 мл МеОН. Добавляли по каплям TFA (0,143 мл, 1,86 ммоль) и раствор перемешивали при кт в течение 30 мин. Растворитель выпаривали и продукт осаждали в диэтиловом эфире с получением указанного в заголовке соединения в виде его соответствующей TFA соли. Выход: 390 мг (77%). 1H ЯМР (400 МГц, МЕТАНОЛ-D4) δ 0.91-0.97 (m, 2 H), 1.02-1.08 (m, 2 H), 1.48-1.60 (m, 2 H), 1.65 (s, 9 H), 1.67-1.75 (m, 3 H), 1.75-1.82 (m, 1 H), 2.00-2.10 (m, 2 H), 2.18-2.28 (m, 1 H), 2.53-2.61 (m, 1 H), 4.50 (d, J=7,42 Гц, 2 H), 7.42 (dd, J=8,98, 2.15 Гц, 1 H), 7.74 (d, J=1,56 Гц, 1 H), 7.85 (d, J=8,79 Гц, 1 H); MS (ESI) (M+H)+ 426,0; Аналит. рассч. (%) для C21H29N3O2SF2 + 1,0 TFA; С, 51,20; H, 5,60; N, 7,79. Найдено: С, 51,38; H, 5,66; N, 7,56.
Пример 16
N-{2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-ил}-N-метилциклопропансульфонамид
2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-амин (65 мг, 0,202 ммоль) и каталитическое количество DMAP растворяли в 5 мл DCM. Добавляли циклопропансульфонилхлорид (34 мг, 0,242 ммоль) и раствор перемешивали при кт в течение 6 ч. Раствор промывали насыщенным водным раствором NaHCO3, рассолом и сушили над безводным MgSO4. Растворитель выпаривали. Остаток растворяли в 5 мл DMF при 0°С и добавляли NaH (12 мг, 0,303 ммоль). Раствор перемешивали при 0°С в течение 15 мин. Добавляли метилиодид (0,025 мл, 0,404 ммоль) и раствор перемешивали при кт в течение 2 ч. Реакционную смесь гасили насыщенным водным раствором NаНСО3 и растворитель выпаривали. Продукт растворяли в ЕtOАс и промывали водным раствром NаНСО3, насыщенным водным раствором NaCl и сушили над безводным Na2SO4. Растворитель выпаривали и продукт очищали посредством HPLC с обращенной фазой, используя смесь 10-70% СН3СN/Н2O, и лиофилизировали с получением указанного в заголовке соединения в виде соответствующей TFA соли. Выход: 60 мг (54%). 1H ЯМР (600 МГц, СD3OD) δ 0.90-0.94 (m, 2 H), 0.97-1.01 (m, 2 H), 1.54-1.62 (m, 2 H), 1.68 (s, 9 H), 1.73-1.81 (m, 4 H), 2.03-2.11 (m, 2 H), 2.23-2.30 (m, 1 H). 2.59-2.65 (m, 1 H), 3.43 (s, 3 H), 4.56 (d, J=7,68 Гц, 2 H), 7.72 (d, J=9,47 Гц, 1 H), 7.81 (s, 1 H), 7.95 (d, J=8,96 Гц, 1 H); MS (ESI) (M+Н)+ 440,0.
Пример 17
N-{2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-ил}-2-метилпропан-2-сульфонамид
2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-амин (66 мг, 0,205 ммоль) и DMAP (25 мг, 0,205 ммоль) растворяли в 5 мл DCM. Добавляли трет-бутилсульфинилхлорид (0,031 мл, 0,246 ммоль) и раствор перемешивали при кт в течение 2 ч. Раствор промывали насыщенным водным раствором NаНСО3, рассолом и сушили над безводным MgSO4. Добавляли 3-хлорпероксибензойную кислоту (90 мг, 0,410 ммоль) и раствор перемешивали при кт в течение 12 ч. Раствор промывали насыщенным водным раствором NaHCO3, рассолом и сушили над безводным MgSO4. Продукт очищали посредством HPLC с обращенной фазой, используя смесь 10-70%
СН3СN/Н2О, и лиофилизировали с получением указанного в заголовке соединения в виде соответствующей TFA соли. Выход: 55 мг (48%). 1H ЯМР (400 МГц, МЕТАНОЛ-D4) δ 1.35 (s, 9 H), 1.49-1.60 (m, 2 H), 1.64 (s, 9 H), 1.68-1.75 (m, 3 H), 1.76-1.82 (m, 1 H), 2.00-2.09 (m, 2 H), 2.19-2.28 (m, 1 H), 4.50 (d, J=7,42 Гц, 2 H), 7.42 (dd, J=9,08, 2.05 Гц, 1 H), 7.81-7.86 (m, 2 H); MS (ESI) (M+H)+442,0; Аналит. рассч. (%) для С22Н33N3O2SF2 + 1,2 TFA + 0,2 H2O; С, 50,35; H, 5,99; N, 7,22. Найдено: С, 50,36; H, 5,73; N, 7,08.
Пример 18
N-[1-[(4,4-Дифторциклогексил)метил]-2-(1,1-дифторэтил)-1H-бензимидазол-5-ил]циклопропансульфонамид
Стадия А: N-[1-[(4,4-Дифторциклогексил)метил]-2-(1,1-дифторэтил)-1Н-бензимидазол-5-ил]циклопропансульфонамид
N-[1-[(4,4-Дифторциклогексил)метил]-2-(1,1-дифторэтил)-1H-бензимидазол-5-ил]ацетамид (для получения смотри следующую стадию Б) (95 мг, 0,256 ммоль) нагревали в 5 мл смеси 1:1 / 2 M HCl:EtOH при 120°С в течение 1 ч, используя Personal Chemistry микроволновое устройство. Растворитель выпаривали. Остаток подщелачивали с помощью 2 M NaOH и экстрагировали (3Х) ЕtOАс. Органическую фазу промывали насыщенным водным раствором NaCl и сушили над безводным Na2SO4. Растворитель выпаривали. Продукт растворяли в 5 мл DCM, содержащего DMAP (31 мг, 0,256 ммоль), и добавляли циклопропансульфонилхлорид (53 мг, 0,384 ммоль). Раствор перемешивали при кт в течение 3 ч. Раствор промывали насыщенным водным раствором NаНСО3, рассолом и сушили над безводным МgSO4. Растворитель выпаривали и продукт очищали посредством HPLC с обращенной фазой, используя смесь 10-70% СН3СN/Н2О, и лиофилизировали с получением указанного в заголовке соединения в виде соответствующей TFA соли. Выход: 35 мг (25%). 1H ЯМР (400 МГц, МЕТАНОЛ-D4) δ 0.88-0.95 (m, 2 H), 0.98-1.03 (m, 2 H), 1.39-1.51 (m, 2 H), 1.61-1.68 (m, 3 H), 1.70-1.79 (m, 1 H), 2.03 (s, 2 H), 2.15 (s, 1 H), 2.23 (m, 3 H), 2.47-2.55 (m, 1 H), 4.35 (d, J=7,62 Гц, 2 H), 7.39 (dd, J=8,79, 1,95 Гц, 1 H), 7.65 (d, J=8,79 Гц, 1 H), 7.67 (d, J=2,15 Гц, 1 H); MS (ESI) (M+H)+ 43,0; Аналит. рассчитано (%) для С19Н23N3O2SF4 + 0,7 TFA; С, 47,74; H, 4,65; N, 8,19. Найдено: С, 47,88; H, 4,68; N, 8,19.
Стадия Б: N-[1-[(4,4-Дифторциклогексил)метил]-2-(1,1-дифторэтил)-1Н-бензимидазол-5-ил]ацетамид
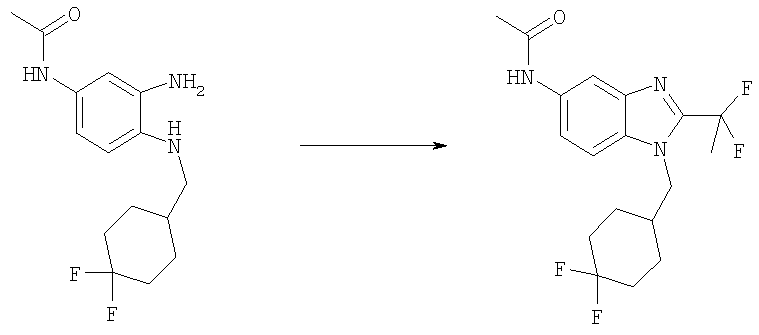
N-(3-Амино-4-{[(4,4-дифторциклогексил)метил]амино}фенил)ацетамид (99 мг, 0,333 ммоль), DIPEA (0,087 мл, 0,500 ммоль), HATU (O-(7-азабензотриазол-1-ил)-N,N,N,N'-тетраметилурония гексафторфосфат, 140 мг, 0,366 ммоль) и 2,2-дифторпропионовую кислоту (40 мг, 0,366 ммоль) перемешивали в 5 мл DMF при кт в течение 1 ч. Растворитель выпаривали. Остаток растворяли в 3 мл ледяной АсОН и нагревали при 80°С в течение 2 ч. Растворитель выпаривали. Продукт растворяли в ЕtOАс и промывали водным раствором NаНСО3, насыщенным водным раствором NaCl и сушили над безводным Na2SO4. Продукт очищали посредством флэш-хроматографии на силикагеле, используя ЕtOАс в качестве элюента. Выход: 100 мг (81%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-D) δ 1.39-1.52 (m, 2 H), 1.57-1.63 (m, 1 H), 1.64-1.71 (m, 3 Н), 2.06-2.16 (m, 3 H), 2.22 (s, 3 H), 2.29 (m, 3 H), 4.25 (d, J=7,42 Гц, 2 H), 7.31 (s, 1 H), 7.35 (d, J=8,79 Гц, 1 H), 7.60 (dd, J=8,89, 1,86 Гц, 1 H), 7.86 (d, J=1,76 Гц, 1 H).
Пример 19
N-[1-[(4,4-Дифторциклогексил)метил]-2-(1,1-дифторэтил)-1H-бензимидазол-5-ил]этансульфонамид
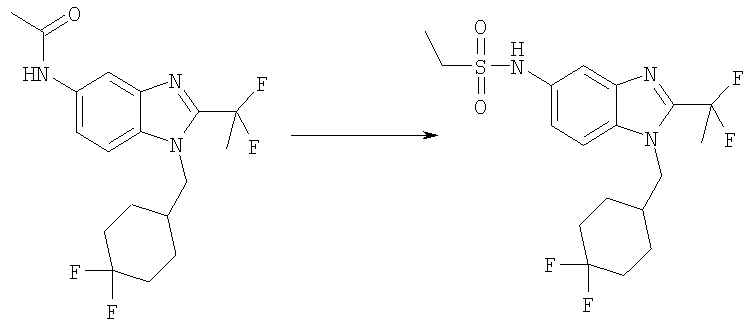
N-[1-[(4,4-Дифторциклогексил)метил]-2-(1,1-дифторэтил)-1H-бензимидазол-5-ил]ацетамид (80 мг, 0,215 ммоль) нагревали в 5 мл смеси 1:1 / 2 M HCl: EtOH при 120°С в течение 1 ч, используя Personal Chemistry микроволновое устройство. Растворитель выпаривали. Остаток подщелачивали с помощью 2 M NaOH и экстрагировали (3Х) ЕtOАс. Органическую фазу промывали насыщенным водным раствором NaCl и сушили над безводным Na2SO4. Растворитель выпаривали. Продукт растворяли в 5 мл DCM, содержащего DMAP (31 мг, 0,256 ммоль), и добавляли этансульфонилхлорид (0,026 мл, 0,280 ммоль). Раствор перемешивали при кт в течение 2 ч. Раствор промывали насыщенным водным раствором NaHCO2, рассолом и сушили над безводным МgSО4. Растворитель выпаривали и продукт очищали посредством HPLC с обращенной фазой, используя смесь 10-70% СН3СN/H2О, и лиофилизировали с получением указанного в заголовке соединения в виде соответствующей TFA соли. Выход: 22 мг (19%). 1H ЯМР (400 МГц, МЕТАНОЛ-D4) δ 1.29 (t, J=7,42 Гц, 3 H), 1.36-1.49 (m, 2 H), 1.58-1.66 (m, 3 H), 1.67-1.78 (m, 1 H), 1.96-2.06 (m, 2 H), 2.11-2.15 (m, 1 H), 2.21 (m, 3 H), 3.04 (m, 2 H), 4.33 (d, J=7,62 Гц, 2 H), 7.34 (dd, J=8,98, 1,95 Гц, 1 H), 7.64 (dd, J=5,47, 3,32 Гц, 2 H); MS (ESI) (М+H)+ 421,9; Аналит. рассчит.(%) для С18Н23N3O2SF4 + 0,8 TFA + 0,1 H2O: С, 45,76; Н, 4,70; N, 8,17. Найдено: С, 45,73; Н, 4,52; N, 7,80.
Пример 20
N-[1-[(4,4-Дифторциклогексил)метил]-2-(1,1-дифторэтил)-1H-бензимидазол-5-ил]-2-метилпропан-2-сульфонамид
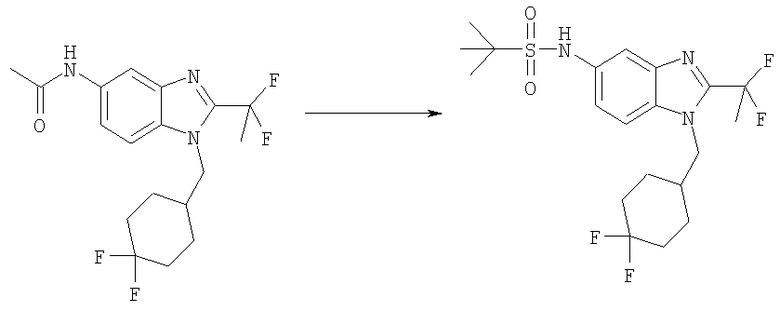
N-[1-[(4,4-Дифторциклогексил)метил]-2-(1,1-дифторэтил)-1Н-бензимидазол-5-ил]ацетамид (185 мг, 0,498 ммоль) нагревали в 5 мл смеси 1:1 / 2М HCl: ЕtOН при 120°С в течение 1 ч, используя a Personal Chemistry микроволновое устройство. Растворитель выпаривали. Остаток подщелачивали с помощью 2 М NaOH и экстрагировали (3Х) ЕtOАс. Органическую фазу промывали насыщенным водным раствором NaCl и сушили над безводным Na2SO4. Растворитель выпаривали. Остаток растворяли в 5 мл DCM и добавляли трет-бутилсульфинилхлорид (0,075 мл, 0,598 ммоль) и DMAP (25 мг, 0,498 ммоль). Раствор перемешивали при кт в течение 1 ч. Раствор промывали насыщенным водным раствором NаНСО3, рассолом и сушили над безводным MgSO4. Добавляли 3-хлорпероксибензойную кислоту (225 мг, 0,996 ммоль) и раствор перемешивали при кт в течение 4 ч. Раствор промывали насыщенным водным раствором NаНСО3, рассолом и сушили над безводным MgSO4. Продукт очищали посредством HPLC с обращенной фазой, используя смесь 10-70% CH3CN/H2O, и лиофилизировали с получением указанного в заголовке соединения в виде соответствующей TFA соли. Выход: 70 мг (25%). 1H ЯМР (400 МГц, МЕТАНОЛ-D4) δ 1.33 (s, 9 Н), 1.37-1.49 (m, 2 H), 1.60-1.65 (m, 3 Н), 1.68-1.78 (m, 1 H), 1.97-2.06 (m, 2 H), 2.11-2.14 (m, 1 H), 2.21 (m, 3 Н), 4.32 (d, J=7,62 Гц, 2 H), 7.40 (dd, J=8,89, 2,05 Гц, 1 H), 7.59 (d, J=8,79 Гц, 1 H), 7.70 (d, J=1,95 Гц, 1 H); MS (ESI) (M+H)+ 449,8.
Пример 21
N-[2-(1,1-Дифторэтил)-1-(тетрагидро-2Н-пиран-4-илметил)-1H-бензимидазол-5-ил]-N-метилэтансульфонамид
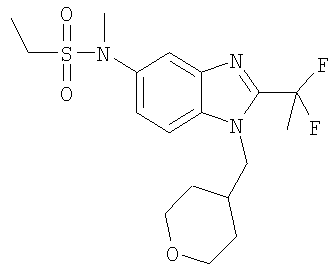
Стадия А. N-[2-(1,1-дифторэтил)-1-(тетрагидро-2Н-пиран-4-илметил)-1Н-бензимидазол-5-ил]-N-метилэтансульфонамид
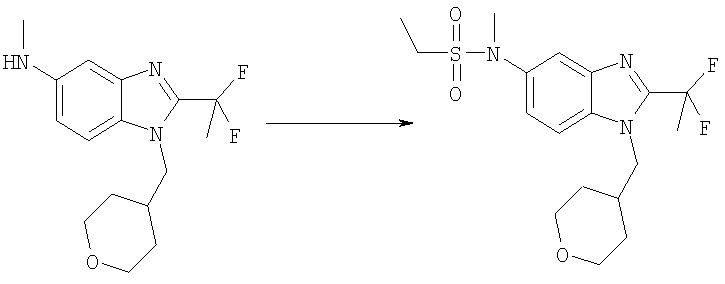
Этансульфонилхлорид (55 мкл, 0,58 ммоль) добавляли к раствору 2-(1,1-дифторэтил)-N-метил-1-(тетрагидро-2H-пиран-4-илметил)-1Н-бензимидазол-5-амина (150 мг, 0,48 ммоль) и DMAP (71 мг, 0,58 ммоль) в DCM (15 мл) при температуре окружающей среды. Реакционную смесь перемешивали в течение ночи и растворитель выпаривали. Продукт очищали посредством препаративной HPLC с обращенной фазой, используя градиент MeCN от 10 до 90% в воде, с получением TFA соли указанного в заголовке соединения в виде белого твердого вещества. Выход: 70 мг (28%); 1H ЯМР (400 МГц, СD3OD) δ 1.24-1.37 (m, 3 Н), 1.36-1.53 (m, 4 Н), 2.12-2.32 (m, 3 Н), 3.05-3.17 (m, 2 H), 3.25-3.31 (m, 2 H), 3.33 (d, J=3,71 Гц, 1 Н), 3.36 (s, 2 H), 3.89 (m, 2 H), 4.33 (d, J=7,42 Гц, 2 H), 7.49 (dd, J=8,79, 1,95 Гц, 1 H), 7.69 (d, J=8,98 Гц, 1 H), 7.77 (d, J=1,76 Гц, 1 H); MS (ESI) (M+H)+ 402,0.
Стадия Б. N-{5-[Ацетил(метил)амино]-2-[(тетрагидро-2Н-пиран-4-илметил)амино]фенил}-2,2-дифторпропанамид
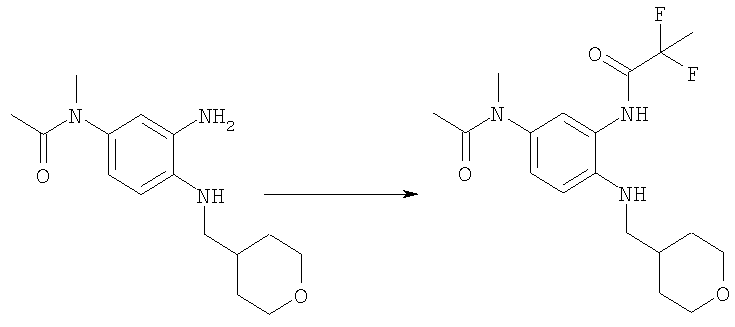
HATU (1,44 г, 3,78 ммоль) и N-{3-амино-4-[(тетрагидро-2Н-пиран-4-илметил)амино]фенил}-N-метилацетамид (1,00 г, 3,60 ммоль) (для получения смотри Пример 1, стадии Б-Д) добавляли к раствору 2,2-дифторпропановой кислоты (0,40 г, 3,60 ммоль) и DIPEA (0,75 мл, 4,32 ммоль) в DMF (100 мл) при комнатной температуре. Реакционную смесь перемешивали в течение ночи. Растворитель выпаривали и сырой продукт выделяли в ЕtOАс. Органическую фазу промывали водой, насыщенным раствором NаНСО3 и рассолом. Органический слой сушили над безводным Nа2SO4 и фильтровали. Растворитель выпаривали с получением указанного в заголовке соединения, которое использовали на следующей стадии без дополнительной очистки. Выход: 1,00 г (75%); MS (ESI) (М+Н)+: 370,2.
Стадия В. N-[2-(1,1-Дифторэтил)-1-(тетрагидро-2H-пиран-4-илметил)-1Н-бензимидазол-5-ил]-N-метилацетамид
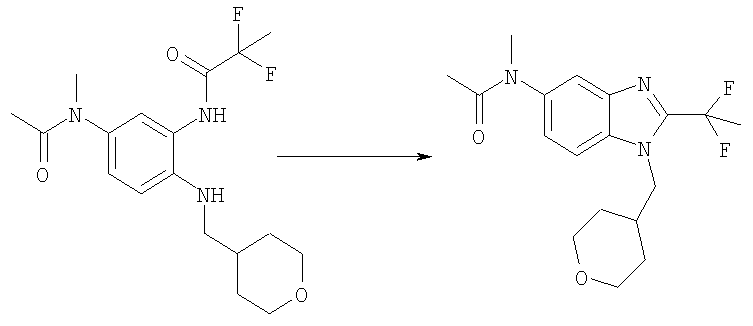
N-{5-[Ацетил(метил)амино]-2-[(тетрагидро-2H-пиран-4-илметил)амино]-фенил}-2,2-дифторпропанамид (1,00 г, 2,70 ммоль) нагревали до 90°С в течение ночи в уксусной кислоте (20 мл). Растворитель выпаривали. Сырой продукт очищали посредством флэш-хроматографии на силикагеле, используя МеОН 3,5% и ацетон 8% в DCM в качестве элюента, получая указанное в заголовке соединение. Выход: 0,48 г (50%); MS (ESI) (M+H)+: 352,0.
Стадия Г. 2-(1,1-дифторэтил)-N-метил-1-(тетрагидро-2Н-пиран-4-илметил)-1H-бензимидазол-5-амин
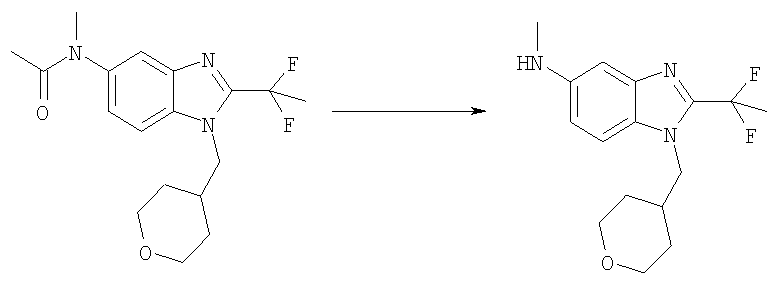
N-[2-(1,1-Дифторэтил)-1-(тетрагидро-2Н-пиран-4-илметил)-1H-бензимидазол-5-ил]-N-метилацетамид (0,48 г, 1,37 ммоль) нагревали до 80°С в течение ночи в концентрированной HCl (80 мл). Реакционную смесь охлаждали до 0°С и доводили до слабо щелочного рН с помощью раствора NaOH. Соединение экстрагировали EtOAc (3X) и объединенные органические слои промывали рассолом, сушили над безводным Na2SO4 и фильтровали. Растворитель выпаривали с получением указанного в заголовке соединения, которое использовали на следующей стадии без дополнительной очистки. Выход: 0,42 г (98%); MS (ESI) (M+H)+: 310,2.
Пример 22
N-[2-(1,1-Дифторэтил)-1-(тетрагидро-2Н-пиран-4-илметил)-1H-бензимидазол-5-ил]-N-метилпропан-1-сульфонамид
Следуя методике стадии А в Примере 21 и используя пропансульфонилхлорид (65 мкл, 0,58 ммоль), получали TFA соль указанного в заголовке соединения в виде белого твердого вещества. Выход: 68 мг (26%); 1H ЯМР (400 МГц, CD3OD) δ 1.02 (t, J=7,42 Гц, 3 Н), 1.40-1.54 (m, 4 H), 1.74-1.87 (m, 1 H), 2.17-2.34 (m, 3 H), 3.05-3.15 (m, 2 H), 3.32-3.37 (m, 2 H), 3.37 (s, 3 H), 3.85-3.97 (m, 2 H), 4.35 (d, J=7,62 Гц, 2 H), 7.50 (dd, J=8,89, 2,05 Гц, 1 H), 7.71 (d, J=8,79 Гц, 1 H), 7.78 (d, J=1,95 Гц, 1 H); MS (ESI) (M+H)+ 416,0; Аналит. рассчитано для C19H27F2N3O3S + 0,1 MeCN: С, 54,96; Н, 6,56; N, 10,35. Найдено: С, 55,02; Н, 6,40; N, 10,24.
Пример 23
N-[2-(1,1-Дифторэтил)-1-(тетрагидро-2Н-пиран-4-илметил)-1H-бензимидазол-5-ил]-N-метилциклопропансульфонамид
Следуя методике стадии А в Примере 21, используя циклопропансульфонилхлорид (81 мкл, 0,58 ммоль) и нагревание до 60°С в течение ночи, получали TFA соль указанного в заголовке соединения в виде белого твердого вещества. Выход: 135 мг (52%); 1H ЯМР (400 МГц, CD3OD) δ 0.85-0.93 (m, 2 H), 0.93-1.03 (m, 2 H), 1.39-1.55 (m, 4H), 2.24 (m, 3H), 2.55-2.66 (m, 1 H), 3.31-3.38 (m, 3 H), 3.39 (s, 3 H), 3.86-3.97 (m, 2 H), 4.36 (d, J=7,42 Гц, 2 H), 7.52 (dd, J=8,79, 2,15 Гц, 1 H), 7.70 (d, J=8,79 Гц, 1 H), 7.81 (d, J=2,15 Гц, 1 H); MS (ESI) (M+H)+ 414,0; Аналит. рассчитано для C19H25F2N3O3S + 0,1 H2O: С, 54,95; H, 6,12; N, 10,12. Найдено: С, 54,91; H, 6,09; N, 9,68.
Пример 24
N-{2-трет-Бутил-1-[(4-фторциклогексил)метил]-1H-бензимидазол-5-ил}этансульфонамид
Стадия А: N-{2-трет-Бутил-1-[(4-фторциклогексил)метил]-1H-бензимидазол-5-ил}этансульфонамид
2-трет-Бутил-1-[(4-фторциклогексил)метил]-1Н-бензимидазол-5-амин (для получения смотри следующие стадии Б-Е) (60 мг, 0,198 ммоль) и DMAP (24 мг, 0,198 ммоль) растворяли в 5 мл DCM. Добавляли этансульфонилхлорид (0,025 мл, 0,257 ммоль) и раствор перемешивали при кт в течение 2 ч. Раствор промывали насыщенным водным раствором NаНСО3, рассолом и сушили над безводным MgSO4. Растворитель выпаривали и продукт очищали посредством HPLC с обращенной фазой, используя смесь 10-70% СН3СN/H2О, и лиофилизировали с получением указанного в заголовке соединения в виде соответствующей TFA соли. Выход: 50 мг (50%). 1H ЯМР (400 МГц, МЕТАНОЛ-D4) δ 1.29 (t, J=7,42 Гц, 3 Н), 1.34-1.41 (m, 2 H), 1.43-1.51 (m, 1 H), 1.53-1.62 (m, 1 H), 1.63-1.66 (m, 9 H), 1.69-1.75 (m, 2 H), 1.96-2.04 (m, 1 H), 2.06-2.12 (m, 2 H), 3.12 (q, J=7,42 Гц, 2 H), 4.44-4.49 (m, 2 H), 7.39 (dd, J=9,08, 2,05 Гц, 1 H), 7.73 (d, J=2,15 Гц, 1 H), 7.85 (d, J=9,18 Гц, 0.7 H), 7.85-7.88 (d, J=9,18 Гц, 0.3 H); MS (ESI) (M+H)+ 396.0; Аналит. рассчитано (%) для С20Н30N3O2SF + 1,3 TFA + 0,5 H2O: С, 49,11; H, 5,89; N, 7,60. Найдено: С, 49,10; H, 5,84; N, 7,52.
Стадия Б: трет-Бутил-[(4-фторциклогекс-3-ен-1-ил)метил]карбамат
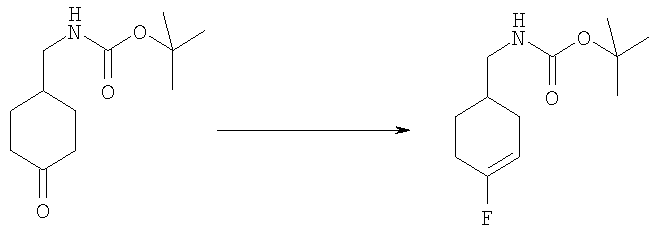
4-N-Вос-аминометилциклогексанон (4,95 г, 21,8 ммоль) растворяли в 80 мл THF. Добавляли по каплям DAST (4,3 мл, 32,7 ммоль) и раствор перемешивали при 50°С в течение 5 ч. Растворитель выпаривали и продукт очищали посредством флэш-хроматографии на силикагеле, используя смесь 3:1 / гексаны:ЕtOАс в качестве элюента. Выход: 1,62 г (30%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-D) δ 1.36-1.42 (m, 1 H), 1.44 (s, 9 H), 1.70-1.80 (m, 2 H), 1.82-1.90 (m, 1 H), 2.09-2.17 (m, 1 H), 2.17-2.29 (m, 2 H), 3.04-3.11 (m, 2 H), 4.61 (s, 1 H), 5.11-5.15 (m, 0.5 H), 5.16-5.19 (m, 0.5 H).
Стадия В: [(4-Фторциклогекс-3-ен-1-ил)метил]амин гидрохлорид
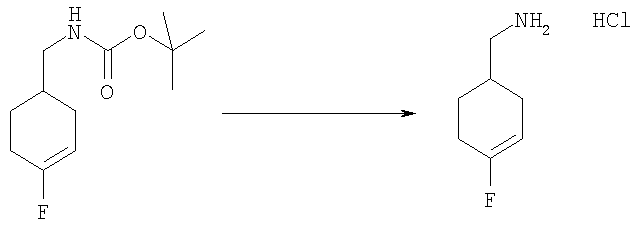
трет-Бутил-[(4-фторциклогекс-3-ен-1-ил)метил]карбамат (1,62 г, 7,06 ммоль) перемешивали в 25 мл 1 М HCl/AcOH при кт в течение 2 ч. Растворитель выпаривали и продукт осаждали в диэтиловом эфире, отфильтровывали и сушили в вакууме. Выход: 1,13 г (97%). 1H ЯМР (400 МГц, МЕТАНОЛ-D4) δ 1.44-1.53 (m, 1 H), 1.80-1.89 (m, 2 H), 1.90-1.98 (m, 1 H), 2.16-2.23 (m, 2 H), 2.26-2.34 (m, 1 H), 2.88 (d, J=6,25 Гц, 2 H), 5.12-5.19 (m, 1 H).
Стадия Г: N-(4-{[(4-Фторциклогекс-3-ен-1-ил)метил]амино}-3-нитрофенил)ацетамид
N-(4-Фтор-3-нитрофенил)ацетамид (460 мг, 2,32 ммоль) и [(4-фторциклогекс-3-ен-1-ил)метил]амин гидрохлорид (350 мг, 2,11 ммоль) перемешивали в 20 мл ЕtOН, содержащего TEA (0,735 мл, 5,28 ммоль), при 75°С в течение 48 ч. Растворитель выпаривали. Остаток растворяли в ЕtOАс и промывали водным 5%-ным KHSO4, насыщенным водным раствором NaHCO3, рассолом и сушили над безводным MgSO4. Сырой продукт очищали посредством флэш-хроматографии на силикагеле, используя смесь 2:1 / гексаны:ацетон в качестве элюента. Выход: 553 мг (85%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-D) δ 1.51-1.61 (m, 1 H), 1.84-1.93 (m, 1 H), 1.96-2.03 (m, 2 H), 2.16-2.18 (m, 3 H), 2.22-2.32 (m, 3 H), 3.26 (m, 2 H), 5.19 (m, 1 H), 6.84 (d, J=9,37 Гц, 1 H), 7.21 (s, 1 H), 7.79 (dd, J=9,18, 2,54 Гц, 1 H), 8.09 (d, J=2,54 Гц, 2 H).
Стадия Д: N-(3-Амино-4-{[(4-фторциклогексил)метил]амино}фенил)ацетамид
N-(4-{[(4-Фторциклогекс-3-ен-1-ил)метил]амино}-3-нитрофенил)ацетамид (340 мг, 1,11 ммоль) растворяли в 25 мл ЕtOАс, содержащего каталитическое количество 10% Pd/C. Раствор втряхивали в атмосфере Н2 (40 фунт-сила на кв. дюйм (275,6 кПа)), используя аппарат для гидрирования Парра, при кт в течение 48 ч. Раствор фильтровали через целит и растворитель выпаривали. Выход: 308 мг (99%). MS (ESI) (M+H)+ 279,95.
Стадия Е: N-{2-трет-Бутил-1-[(4-фторциклогексил)метил]-1H-бензимидазол-5-ил}ацетамид
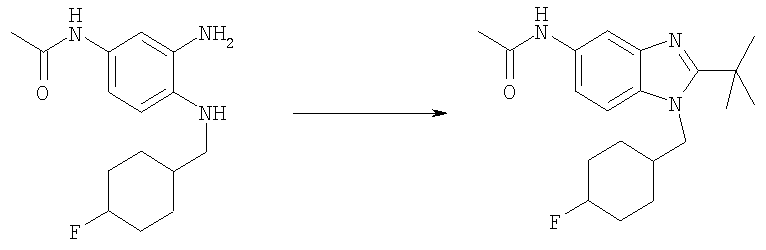
N-(3-Амино-4-{[(4-фторциклогексил)метил]амино}фенил)ацетамид (300 мг, 1,07 ммоль) и DMAP (25 мг, 0,214 ммоль) растворяли в 10 мл DCM. По каплям добавляли триметилацетилхлорид (0,145 мл, 1,18 ммоль) и раствор перемешивали при кт в течение 1 ч. Раствор промывали водным раствором NaHCO3, рассолом и сушили над безводным MgSO4. Остаток растворяли в 5 мл АсОН и нагревали при 150°С в течение 2,5 ч, используя Personal Chemistry микроволновое устройство. Растворитель выпаривали. Остаток растворяли в ЕtOАс и промывали водным раствором NaHCO3, рассолом и сушили над безводным MgSO4. Сырой продукт очищали посредством флэш-хроматографии на силикагеле, используя смесь 2:1 / ацетонтексаны в качестве элюента. Выход: 196 мг (53%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-D) δ 1.14-1.25 (m, 2 H), 1.37-1.45 (m, 1 H), 1.43-1.51 (m, 1 H), 1.54-1.57 (m, 9 H), 1.70-1.78 (m, 2 H), 1.70-1.77 (m, 1 H), 2.02-2.08 (m, 1 H), 2.10-2.17 (m, 1 H), 2.19-2.21 (m, 3H), 4.12-4.19 (m, 2 H), 4.53 (m, 0.3 H), 4.73 (m, 0.3 H), 4.78 (m, 0.2 H), 4.90 (m, 0.2 H), 7.21-7.29 (m, 1 H), 7.30 (s, 1 H), 7.50-7.57 (m, 1 H), 7.64-7.67 (m, 1 H).
Стадия Ж: 2-трет-Бутил-1-[(4-фторциклогексил)метил]-1Н-бензимидазол-5-амин
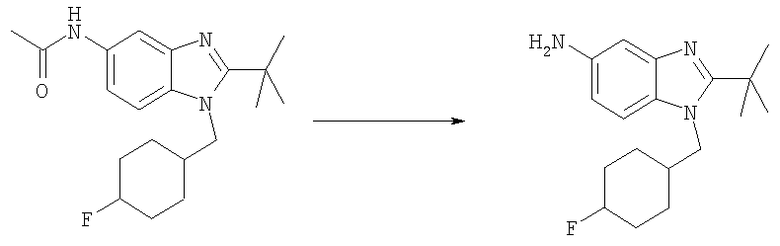
N-{2-трет-Бутил-1-[(4-фторциклогексил)метил]-1H-бензимидазол-5-ил}ацетамид (190 мг, 0,550 ммоль) нагревали в 5 мл смеси 1:1 / 2 М HCl:ЕtOН при 120°С в течение 1 ч, используя Personal Chemistry микроволновое уствройство. Растворитель выпаривали. Остаток подщелачивали с помощью 2 М NaOH и экстрагировали (3Х) ЕtOАс. Органическую фазу промывали насыщенным водным раствором NaCl и сушили над безводным Na2SO4. Растворитель выпаривали. Выход: 154 мг (92%). 1Н ЯМР (400 МГц, МЕТАНОЛ-D4) δ 1.28-1.39 (m, 2 H), 1.41-1.50 (m, 1 H), 1.53-1.59 (m, 1 H), 1.61-1.64 (m, 9 Н), 1.69 (d, J=7,81 Гц, 2 H), 1.95-2.03 (m, 0.7 Н), 2.05-2.11 (m, 2 H), 2.13-2.22 (m, 0.3 Н), 4.37-4.44 (m, 2.7 Н), 4.47-4.56 (m, 0.3 Н), 7.11 (t, J=2,05 Гц, 0.5 Н,) 7.13 (t, J=2,05 Гц, 0.5 Н), 7.15-7.18 (m, 1 H), 7.67-7.73 (m, 1 H).
Пример 25
N-{2-трет-Бутил-1-[(4-фторциклогексил)метил]-1Н-бензимидазол-5-ил}циклопропансульфонамид
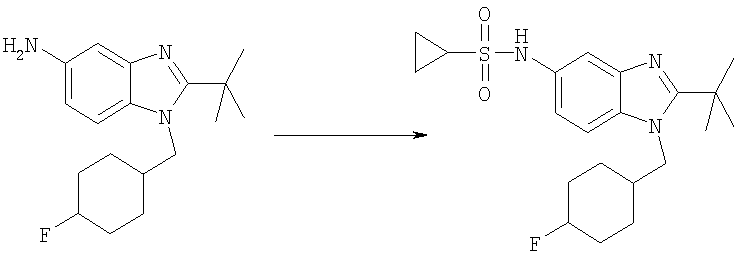
2-трет-Бутил-1-[(4-фторциклогексил)метил]-1H-бензимидазол-5-амин (56 мг, 0,199 ммоль) и DMAP (25 мг, 0,199 ммоль) растворяли в 5 мл DCM. Добавляли циклопропансульфонилхлорид (42 мг, 0,298 ммоль) и раствор перемешивали при кт в течение 3 ч. Раствор промывали насыщенным водным раствором NаНСО3, рассолом и сушили над безводным МgSO4. Растворитель выпаривали и продукт очищали посредством HPLC с обращенной фазой, используя смесь 10-70% СН3СN/H2О, и лиофилизировали с получением указанного в заголовке соединения в виде соответствующей TFA соли. Выход: 58 мг (56%). 1Н ЯМР (400 МГц, МЕТАНОЛ-D4) δ 0.91-0.98 (m, 2 H), 1.03-1.09 (m, 2 H), 1.32-1.43 (m, 2 H), 1.45-1.52 (m, 1 H), 1.54-1.62 (m, 1 H), 1.64-1.67 (m, 9 H), 1.68-1.76 (m, 1 H), 1.97-2.05 (m, 1 H), 2.06-2.13 (m, 2 H), 2.54-2.62 (m, 1 H), 4.44-4.50 (m, 2 H), 4.53 (m, 0.5 H), 4.73 (m, 0.5 H), 7.43 (dd, J=9,08, 2,05 Гц, 1 H), 7.75 (d, J=1,95 Гц, 1 H), 7.83-7.89 (m, 1 H); MS (ESI) (M+H)+ 408,0.
Пример 26
N-{2-трет-Бутил-1-[(4-фторциклогексил)метил]-1Н-бензимидазол-5-ил}-2-метилпропан-2-сульфонамид
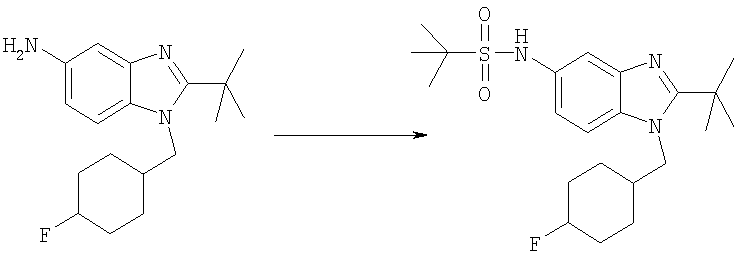
2-трет-Бутил-1-[(4-фторциклогексил)метил]-1H-бензимидазол-5-амин (53 мг, 0,175 ммоль) и DMAP (21 мг, 0,175 ммоль) растворяли в 5 мл DCM. Добавляли трет-бутилсульфинилхлорид (0,026 мл, 0,210 ммоль) и раствор перемешивали при кт в течение 1 ч. Раствор промывали насыщенным водным раствором NаНСО3, рассолом и сушили над безводным MgSO4. Добавляли 3-хлорпероксибензойную кислоту (78 мг, 0,350 ммоль) и раствор перемешивали при кт в течение 2 ч. Раствор промывали насыщенным водным раствором NаНСО3, рассолом и сушили над безводным MgSO4. Продукт очищали посредством HPLC с обращенной фазой, используя смесь 10-70%
СН3СN/Н2О, и лиофилизировали с получением указанного в заголовке соединения в виде соответствующей TFA соли. Выход: 47 мг (50%). 1Н ЯМР (400 МГц, МЕТАНОЛ-D4) δ 1.36 (s, 9 H), 1.38-1.44 (m, 2 H), 1.44-1.51 (m, 1 H), 1.54-1.60 (m, 1 H), 1.63-1.66 (m, 9 H), 1.69-1.75 (m, 2 H), 1.96-2.04 (m, 1 H), 2.06-2.14 (m, 2 H), 4.42-4.48 (m, 2 H), 4.53 (m, 0.5 H), 4.72 (m, 0.5 H), 7.42 (dd, J=8,98, 2,15 Гц, 1 H), 7.79-7.86 (m, 2 H); MS (ESI) (M+H)+ 424,0.
Пример 27
N-{2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1Н-бензимидазол-5-ил}этансульфонамид
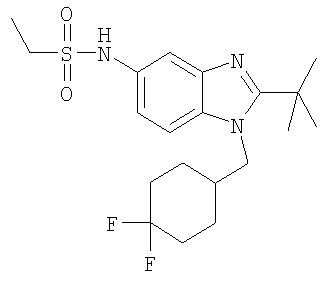
Стадия А. N-{2-трет-Бутил-1-[(4,4-дифторциклогексил)метил]-1Н-бензимидазол-5-ил}этансульфонамид
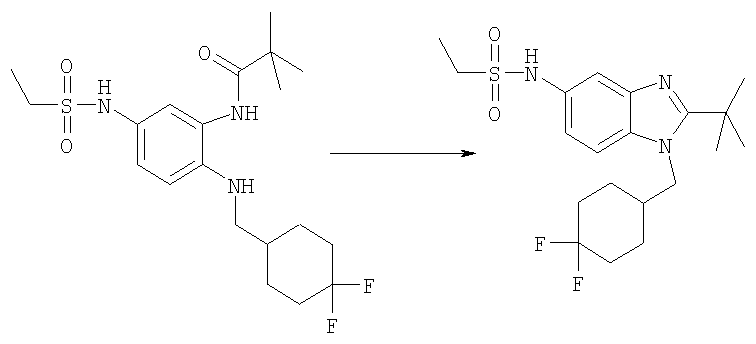
N-{2-{[(4,4-дифторциклогексил)метил]амино}-5-[(этилсульфонил)амино]-фенил}-2,2-диметилпропанамид (22,3 г, 0,051 моль) (для получения смотри следующие стадии Б-Д), PTSA (пара-толуолсульфоновая кислота)×Н2O (10,8 г, 0,057 моль) и DMSO (100 мл) смешивали вместе и нагревали до 120°С в течение ночи. Охлажденную до комнатной температуры реакционную смесь выливали в холодную воду (600 мл). Продукт экстрагировали DCM (5×200 мл). Объединенные органические фазы промывали насыщенным раствором NaHCO3 (4×200 мл), рассолом и сушили над безводным Na2SO4. Растворитель удаляли и сырой продукт очищали на силикагеле (ЕtOАс:гексан 1:1) посредством флэш-хроматографии (и обрабатывали активированным углем) с получением N-{2-трет-бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-ил}этансульфонамида (18,4 г) в виде белого твердого вещества.
Стадия Б. N-(4-Фтор-3-нитрофенил)этансульфонамид
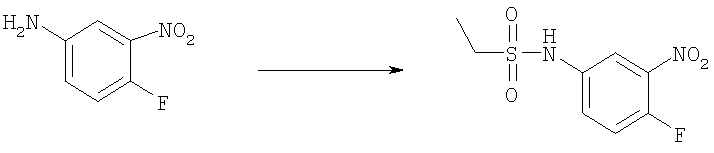
EtSO2Cl (21,5 мл, 0,22 моль) по каплям добавляли к смеси 4-фтор-3-нитроанилина (29,6 г, 0,19 моль) и пиридина (100 мл) при 0°С. Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь разбавляли ЕtOАс (1 л). Полученный раствор промывали 2 н. HCl (4×200 мл), насыщенным раствором NаНСО3 (4×200 мл) и водой (4×200 мл). Органическую фазу сушили над безводным Na2SO4 и удаляли растворитель с получением указанного в заголовке продукта в виде бежевого твердого вещества (46,3 г).
Стадия В. N-(4-{[(4,4-Дифторциклогексил)метил]амино}-3-нитрофенил)этансульфонамид
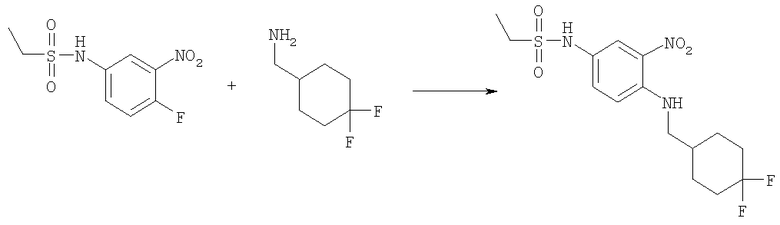
N-(4-Фтор-3-нитрофенил)этансульфонамид (26 г, 0,107 моль), [(4,4-дифторциклогексил)метил]амин (приблизит. 15 г), DIPEA (20 мл) и DMSO (100 мл) смешивали вместе и нагревали до 65°С в течение ночи. Добавляли этаноламин (5 г) и реакционную смесь перемешивали до полного исчезновения N-(4-фтор-3-нитрофенил)этансульфонамида (приблизит. 4-5 часов). Охлажденную до комнатной температуры реакционную смесь выливали в холодную воду (900 мл). Продукт экстрагировали DCM (5×200 мл). Объединенные органические фазы промывали 2 н. HCl (3×200 мл) и сушили над безводным Na2SO4. Растворитель удаляли и сырой продукт очищали на силикагеле посредством флэш-хроматографии (этот материал может быть перекристаллизован с использованием смеси ЕtOАс и гексана) с получением указанного в заголовке продукта (24,2 г) в виде оранжевого твердого вещества.
Стадия Г. N-(3-амино-4-{[(4,4-дифторциклогексил)метил]амино}фенил)этансульфонамид
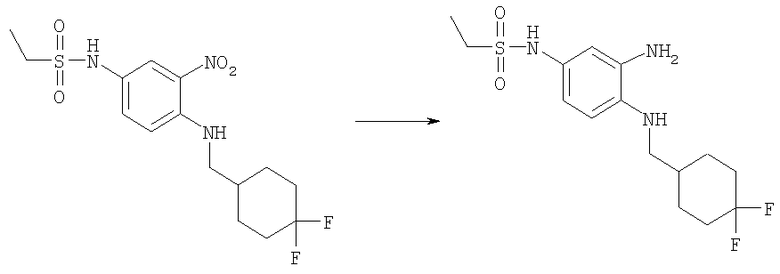
N-(4-{[(4,4-Дифторциклогексил)метил]амино}-3-нитрофенил)этансульфонамид (23,4 г) и Pd/C 10% в ЕtOАс (800 мл) встряхивали вместе в течение ночи в атмосфере H2 (50 фунт-сила на кв. дюйм (344,5 кПа)) в аппарате для гидрирования Парра. Реакционную смесь разбавляли МеОН (400 мл) и фильтровали через слой целита. Растворитель удаляли с получением желаемого указанного в заголовке продукта (22,2 г) в виде бежевого твердого вещества.
Стадия Д. N-{2-{[(4,4-дифторциклогексил)метил]амино}-5-[(этилсульфонил)амино]фенил}-2,2-диметилпропанамид
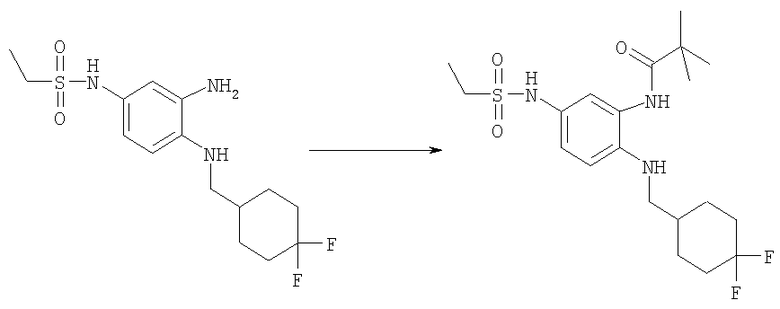
Раствор трет-BuCOCl (7,6 г, 0,063 моль) в DCM (150 мл) медленно добавляли к раствору N-(3-амино-4-{[(4,4-дифторциклогексил)метил]амино}фенил)этансульфонамида (22 г, 0,063 моль) и Et3N (9,7 мл, 0,069 моль) в DCM (500 мл) при 0°С. Реакционную смесь перемешивали в течение 3 часов при 0°С. Добавляли DCM (300 мл) и воду (200 мл). Органический слой отделяли и промывали водой (3×200 мл), рассолом и сушили над безводным Na2SO4. Растворитель удаляли и сырой продукт очищали на силикагеле посредством флэш-хроматографии (ЕtOАс:гексан, 1:1) с получением указанного в заголовке продукта (23,3 г) в виде бежевого твердого вещества.
Пример 28. N-{2-трет-Бутил-1-[(4-фторциклогексил)метил]-1Н-бензимидазол-5-ил}этансульфонамид (изомеры)
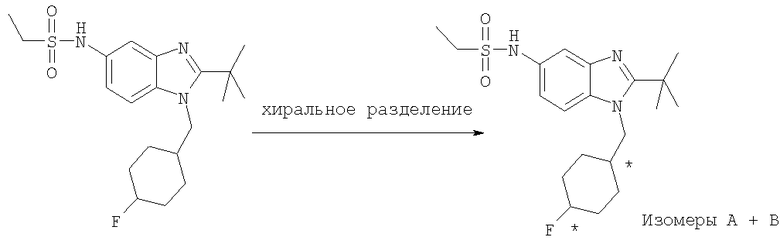
N-{2-трет-Бутил-1-[(4-фторциклогексил)метил]-1H-бензимидазол-5-ил}этансульфонамид (60 мг, TFA соль, 0,117 ммоль) разделяли на хиральной AD колонке, использую смесь 10% ЕtOН / гексаны (0,1% диэтиламин), получая соответственно изомер А (16 мг) и изомер В (31 мг).
Изомер А: 1H ЯМР (400 МГц, МЕТАНОЛ-D4) δ 1.30 (t, J=7,42 Гц, 3 Н), 1.41-1.52 (m, 3 Н), 1.54-1.63 (m, 3 Н), 1.65 (s, 9 Н), 1.97-2.05 (m, 2 H), 2.15-2.24 (m, 1 H), 3.13 (q, J=7,29 Гц, 2 H), 4.47 (d, J=7,62 Гц, 2 H), 4.72 (s, 0.5 Н), 4.85 (s, 0.5 Н), 7.38 (dd, J=8,98, 2,15 Гц, 1 H), 7.73 (d, J=1,95 Гц, 1 H), 7.85 (d, J=8,98 Гц, 1 H); MS (ESI) (M+H)+ 395,8; хиральная AD 15%ЕtOН/гексаны (0,1% DEA) k'=2,97.
Изомер В: 1H ЯМР (400 МГц, МЕТАНОЛ-D4) δ 1.30 (t, J=7,32 Гц, 3 Н), 1.34-1.39 (m, 2 H), 1.39-1.45 (m, 2 H), 1.65 (s, 9 Н), 1.70-1.75 (m, 2 H), 2.06-2.13 (m, 3 Н), 3.13 (q, J=7,42 Гц, 2 H), 4.37-4.43 (m, 0.5 Н), 4.45 (d, J=7,62 Гц, 2 H), 4.49-4.56 (m, 0.5 Н), 7.39 (dd, J=9,08, 2,05 Гц, 1 H), 7.73 (d, J=2,15 Гц, 1 H), 7.84 (d, J=9,18 Гц, 1 H); MS (ESI) (M+H)+395,8; Аналит. рассчитано для C20H30N3O2SF + 1,2 TFA + 0,2 H2O: С, 50,20; Н, 5,94; N, 7,84. Найдено: С, 50,13; Н, 5,81; N, 7,74; хиральная AD 15%ЕtOН/гексаны (0,1% DEA) k'=3,81.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2004 |
|
RU2346938C2 |
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ ПО ОТНОШЕНИЮ К PDE9A, И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2019 |
|
RU2788147C2 |
| Новые производные имидазо[4,5-с]хинолина в качестве ингибиторов LRRK2 | 2018 |
|
RU2773516C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ | 2001 |
|
RU2289581C2 |
| ДИСПИРОПИРРОЛИДИНОВЫЕ ПРОИЗВОДНЫЕ | 2012 |
|
RU2612534C2 |
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ ПО ОТНОШЕНИЮ К PDE9A, И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2019 |
|
RU2788148C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ТЕТРАЗОЛА И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ТУБЕРКУЛЕЗА | 2018 |
|
RU2800930C2 |
| Новые производные имидазо[4,5-c] хинолинов и имидазо[4,5-c][1,5] нафтиридинов в качестве ингибиторов LRRK2 | 2016 |
|
RU2722149C1 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2017 |
|
RU2747318C2 |
| ПРОИЗВОДНЫЕ АЗАИНДАЗОЛА ИЛИ ДИАЗАИНДАЗОЛА В КАЧЕСТВЕ МЕДИКАМЕНТА | 2012 |
|
RU2600976C2 |
Настоящее изобретение относится к соединению формулы:
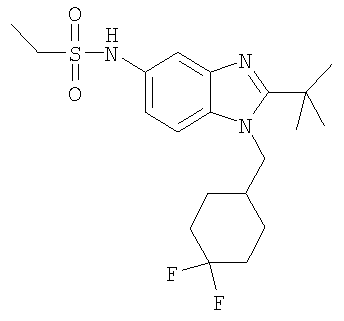
предстаставляющему собой N-{2-трет-бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-ил}этансульфонамид, и его фармацевтически приемлемой соли, его диастереомерам, энантиомерам или их смесям. Также изобретение относится к применению этого соединения для изготовления лекарственного средства, обладающего активностью модулятора CB1 рецепторов; к фармацевтической композиции на основе этого соединения; к способу модулирования CB1 рецепторов, основанному на введении эффективных количеств этого соединения, а также к способу получения вышеуказанного соединения. Технический результат: получено новое производное бензимидазола, обладающего полезной биологической активностью. 7 н. и 1 з.п. ф-лы, 1 табл.
1. Соединение формулы
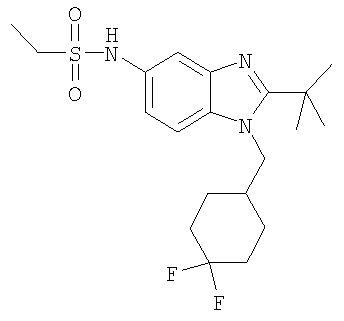
представляющее собой N-{2-трет-бутил-1-[(4,4-дифторциклогексил)метил]-1H-бензимидазол-5-ил}этансульфонамид, и его фармацевтически приемлемая соль, его диастереомеры, энантиомеры или их смеси.
2. Соединение по п.1 для применения в качестве лекарственного средства, обладающего активностью модулятора CB1 рецепторов.
3. Применение соединения по п.1 в изготовлении лекарственного средства для лечения боли.
4. Применение соединения по п.1 в изготовлении лекарственного средства для лечения тревожных расстройств.
5. Применение соединения по п.1 в изготовлении лекарственного средства для лечения рака, рассеянного склероза, болезни Паркинсона, болезни Гентингтона, болезни Альцгеймера, желудочно-кишечных расстройств и сердечно-сосудистых расстройств.
6. Фармацевтическая композиция, обладающая активностью модулятора CB1 рецепторов, содержащая соединение по п.1 и фармацевтически приемлемый носитель.
7. Способ модулирования CB1 рецепторов, включающий стадию введения терапевтически эффективного количества соединения по п.1.
8. Способ получения соединения формулы
включающий взаимодействие соединения формулы II с соединением формулы III
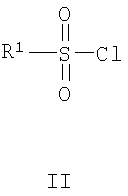
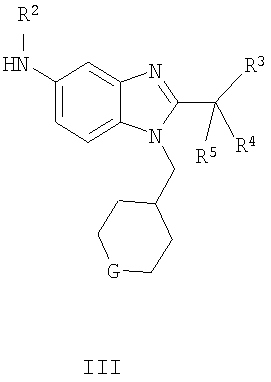
где G представляет собой -СF2-;
R1 представляет собой этил;
R2 представляет собой -Н; и
R3, R4 и R5 независимо представляют собой метил.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ БЕНЗИМИДАЗОЛОВ | 0 |
|
SU237735A1 |
| Устройство для электрохимического снятия заусенцев | 1979 |
|
SU882718A2 |

