Область техники, к которой относится изобретение
Изобретение относится к соединениям, композициям, содержащим их, и их применению в терапии, например, в лечении микобактериальных инфекций или в лечении заболеваний, вызванных микобактериями, таких как туберкулез (также известный как ТБ).
Уровень техники
Согласно отчету, опубликованному Всемирной организацией здравоохранения в 2016 г., примерно 10 млн. человек ежегодно заражаются туберкулезом (TБ), что приводит к 1,4 млн. смертей в год и еще 0,4 млн. смертей от туберкулеза среди людей, зараженных вирусом иммунодефицита человека (ВИЧ). Несмотря на доступные методы лечения туберкулеза, глобальное бремя заболевания остается серьезной проблемой за счет Mycobacterium tuberculosis, бактерии-возбудителя туберкулеза, который становится резистентным ко многим видам лечения.
В попытке предупредить повышение резистентности к имеющимся в настоящее время лекарственным препаратам и открытым в будущем лекарственным препаратам, туберкулез лечат с использованием комбинированной терапии из трех или более препаратов. Кроме того, для лечения ТБ часто требуется терапия несколькими препаратами. Стандартное лечение, используемое в настоящее время для лекарственно-чувствительного TB, представляет собой комбинацию изониазида, рифампицина, пиразинамида и этамбутола, которую пациенты должны принимать в течение двух месяцев, с последующим приемом только изониазида и рифампицина в течение еще четырех месяцев.
Туберкулез с множественной лекарственной устойчивостью (МЛУ-ТБ) определяется как наличие устойчивости, по меньшей мере, к изониазиду и рифампицину, двум наиболее эффективным противотуберкулезным препаратам первого ряда, и туберкулез с широкой лекарственной устойчивостью (ШЛУ-ТБ) является формой МЛУ-ТБ, которая также резистентна, по меньшей мере, к одному фторхинолону и любому из противотуберкулезных препаратов второго ряда (т.е. амикацину, канамицину или капреомицину), двум наиболее важным классам лекарственных препаратов в схеме лечения МЛУ-ТБ. Для лечения туберкулеза М(Ш)ЛУ необходимо назначать схему из четырех или более препаратов второго ряда.
Распространенность ТБ инфекции на протяжении всей истории данного заболевания в значительной степени обусловлена способностью Mycobacterium tuberculosis сохраняться в организме хозяина в течение длительных периодов времени и вызывать заболевание даже в условиях хорошо сформированного иммунного ответа хозяина (Flynn J.L. & Chan J. (2001) Annu. Rev. Immunol., 19, 93-129). Наличие такой необычной способности предполагает, что микобактерии могут использовать уникальные патогенные механизмы.
Результаты исследований по идентификации мишени указывают на ингибирование триптофансинтазы в качестве возможной мишени для лечения TБ. Триптофансинтаза (TS) представляет пиридоксаль-5-фосфат-зависимый комплекс α2β2, катализирующий последние две стадии биосинтеза триптофана в бактериях, растениях и грибах. Физиологической реакцией TS является превращение индол-3-глицеролфосфата (IGP) и L-серина в L-триптофан и D-глицеральдегид-3-фосфат (G3P). Mycobacterium tuberculosis могут синтезировать свой собственный триптофан, так что, в отличие от некоторых других внутриклеточных патогенов, они способны пережить внутриклеточное «голодание» триптофана, вызванное интерфероном [Zhang et al., Cell, 2013, 155, 1296-1308]. Кроме того, биосинтез триптофана отсутствует у млекопитающих, что указывает на возможность селективного ингибирования.
В связи с постоянно растущим появлением штаммов Mycobacterium tuberculosis с множественной лекарственной устойчивостью и сохраняющейся высокой заболеваемостью туберкулезом, существует настоятельная необходимость в обеспечении дополнительных лекарственных препаратов для лечения туберкулеза, предпочтительно с новым механизмом действия.
Сущность изобретения
В первом аспекте изобретения обеспечивается соединение формулы (I) или его фармацевтически приемлемая соль:
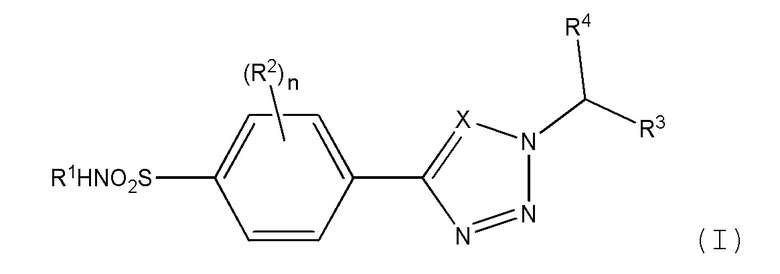
где:
Х является СН или N;
n равно 0, 1 или 2;
R1 представляет метил, этил, цианометил, С-связанный ацетамидо, метилацетат, 2-гидроксиэтил, 2-гидрокси-1-пропил, 1,3-дигидрокси-2-пропил или 1,2-дигидрокси-3-пропил;
R2 независимо выбран из атома галогена, амино, гидроксиметила, C1-2 алкила, необязательно замещенного не более чем тремя атомами фтора, или C1-2 алкокси, необязательно замещенного не более чем тремя атомами фтора;
R3 представляет фенил, пиридил, пиримидинил, пиразинил или пиридазинил, где каждая из этих групп может быть необязательно замещена одним или двумя заместителями, выбранными из атома галогена, циано, C1-2 алкила, необязательно замещенного не более чем тремя атомами фтора, и C1-2 алкокси, необязательно замещенного не более чем тремя атомами фтора, где заместители могут быть одинаковыми или различными; или
R3 представляет циклогексил, который может быть необязательно замещен одним или двумя атомами фтора или хлора, где каждый заместитель может быть присоединен к одному и тому же атому углерода и каждый заместитель может быть одинаковым или различным; или
R3 представляет тетрагидропиран; и
R4 является Н или метилом.
Во втором аспекте изобретения обеспечивается соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии.
В третьем аспекте изобретения обеспечивается соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении туберкулеза.
В четвертом аспекте изобретения обеспечивается соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении микобактериальной инфекции или для применения в лечении заболевания, вызванного микобактериями.
В пятом аспекте изобретения обеспечивается способ лечения микобактериальной инфекции у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В шестом аспекте изобретения обеспечивается способ лечения заболевания, вызванного микобактериями у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В седьмом аспекте изобретения обеспечивается применение соединения формулы (I) или его фармацевтически приемлемой соли в производстве лекарственного средства для применения в лечении микобактериальной инфекции или заболевания, вызванного микобактериями.
В восьмом аспекте изобретения обеспечивается фармацевтическая композиция, содержащая (а) соединение формулы (I) или его фармацевтически приемлемую соль; и (b) фармацевтически приемлемый эксципиент.
В девятом аспекте изобретения обеспечивается комбинация (а) соединения формулы (I) или его фармацевтически приемлемой соли; и (b) по меньшей мере, одного другого противомикобактериального агента.
Подробное описание изобретения
Как описано выше, в одном аспекте изобретения обеспечивается соединение формулы (I) или его фармацевтически приемлемая соль:
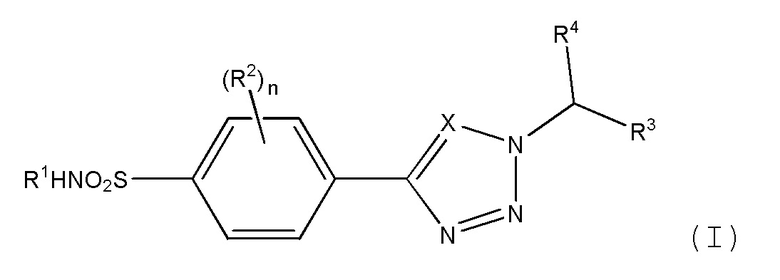
где:
Х является СН или N;
n равно 0, 1 или 2;
R1 представляет метил, этил, цианометил, С-связанный ацетамидо, метилацетат, 2-гидроксиэтил, 2-гидрокси-1-пропил, 1,3-дигидрокси-2-пропил или 1,2-дигидрокси-3-пропил;
R2 независимо выбран из атома галогена, амино, гидроксиметила, C1-2 алкила, необязательно замещенного не более чем тремя атомами фтора, или C1-2 алкокси, необязательно замещенного не более чем тремя атомами фтора;
R3 представляет фенил, пиридил, пиримидинил, пиразинил или пиридазинил, где каждая из этих групп может быть необязательно замещена одним или двумя заместителями, выбранными из атома галогена, циано, C1-2 алкила, необязательно замещенного не более чем тремя атомами фтора, и C1-2 алкокси, необязательно замещенного не более чем тремя атомами фтора, где заместители могут быть одинаковыми или различными; или
R3 представляет циклогексил, который может быть необязательно замещен одним или двумя атомами фтора или хлора, где каждый заместитель может быть присоединен к одному и тому же атому углерода и каждый заместитель может быть одинаковым или различным; или
R3 представляет тетрагидропиран.
В одном варианте осуществления изобретение относится к соединению формулы (I), как определено выше.
В одном варианте осуществления X представляет N.
В одном варианте осуществления R4 представляет H. Когда R4 представляет H, то формула (I) может быть определена в соответствии с формулой (Ia):
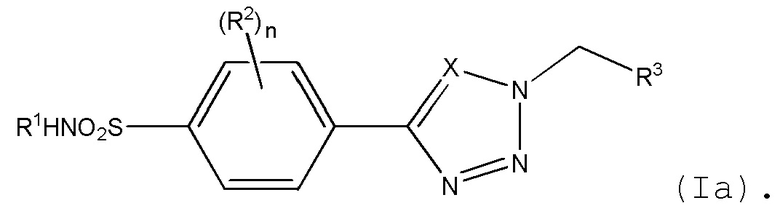
В одном варианте осуществления, когда R4 представляет метил, то он представляет (S)-метил.
В одном варианте осуществления n равно 0 или 1. В конкретном варианте осуществления n равно 0.
Как определено выше, R1 представляет метил, этил, цианометил, С-связанный ацетамидо, метилацетат, 2-гидроксиэтил, 2-гидрокси-1-пропил, 1,3-дигидрокси-2-пропил или 1,2-дигидрокси-3-пропил. Другими словами, R1 выбран из одной из следующих групп, которые соответствуют приведенным ниже:
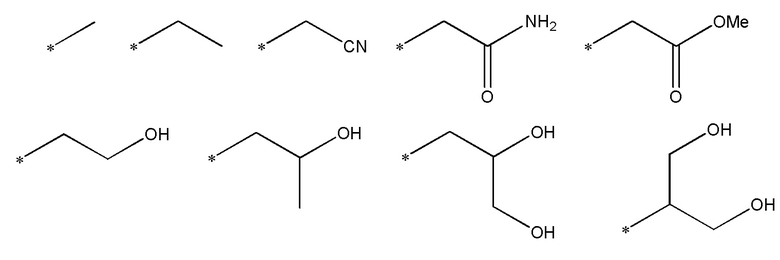
где * представляет место присоединения.
В одном варианте осуществления R1 представляет метил, этил, цианометил, C-связанный ацетамидо, метилацетат, 2-гидроксиэтил, (R)-2-гидрокси-1-пропил, (S)-2-гидрокси-1-пропил, 1,3-дигидрокси-2-пропил или 1,2-дигидрокси-3-пропил.
В одном конкретном варианте осуществления R1 представляет метил, цианометил, C-связанный ацетамидо, 2-гидроксиэтил, (R)-2-гидрокси-1-пропил или (S)-2-гидрокси-1-пропил.
В одном варианте осуществления R1 представляет (R)-2-гидрокси-1-пропил, 2-гидроксиэтил, цианометил или C-связанный ацетамидо.
В одном варианте осуществления R1 представляет 2-гидроксиэтил, цианометил или С-связанный ацетамидо.
В одном варианте осуществления R1 представляет 2-гидроксиэтил или С-связанный ацетамидо.
В одном варианте осуществления R1 представляет 2-гидроксиэтил.
Как определено выше, R2 независимо выбран из атома галогена, амино, гидроксиметила, C1-2 алкила, необязательно замещенного не более чем тремя атомами фтора, или C1-2 алкокси, необязательно замещенного не более чем тремя атомами фтора.
В одном варианте осуществления, когда n равно 1 или 2, то R2 представляет атом галогена, метокси или метил. В частности, R2 представляет атом галогена или метокси. В одном варианте осуществления, когда R2 представляет атом галогена, то он представляет атом фтора или хлора. В конкретном варианте осуществления R2 представляет атом хлора или фтора.
В одном варианте осуществления, когда n равно 2, то R2 представляет атом галогена, который предпочтительно представляет собой атом фтора.
В одном варианте осуществления R2 присоединен к одному из атомов углерода фенильного кольца, который является смежным с -SO2NHR1. Другими словами, R2 присоединен в орто-положении относительно -SO2NHR1. В альтернативном варианте осуществления R2 присоединен в мета-положении относительно -SO2NHR1.
Как определено выше, R3 представляет фенил, пиридил, пиримидинил, пиразинил или пиридазинил, где каждая из этих групп может быть необязательно замещена одним или двумя заместителями, выбранными из атома галогена, циано, C1-2 алкила, необязательно замещенного не более чем тремя атомами фтора, и C1-2 алкокси, необязательно замещенного не более чем тремя атомами фтора, где заместители могут быть одинаковыми или разными
В одном варианте осуществления R3 представляет фенил, пиридил или пиримидинил, где каждая из этих групп может быть необязательно замещена одним или двумя заместителями, выбранными из атома галогена, циано, C1-2 алкила, необязательно замещенного не более чем тремя атомами фтора, C1-2 алкокси, необязательно замещенного не более чем тремя атомами фтора, где заместители могут быть одинаковыми или различными; или
R3 представляет циклогексил, который может быть необязательно замещен одним или двумя атомами фтора или хлора, где каждый заместитель может быть присоединен к одному и тому же атому углерода, и каждый заместитель может быть одинаковым или различным; или
R3 представляет тетрагидропиран.
В одном варианте осуществления R3 представляет фенил, пиридил, пиразинил или пиримидинил, где каждая из этих групп может быть необязательно замещена одним заместителем, выбранным из атома галогена, циано, C1-2 алкила, необязательно замещенного не более чем тремя атомами фтора, и C1-2 алкокси, необязательно замещенного не более чем тремя атомами фтора.
В одном конкретном варианте осуществления R3 представляет фенил или пиридил, где каждая из этих групп может быть необязательно замещена одним или двумя заместителями, выбранными из атома галогена, циано, C1-2 алкила, необязательно замещенного не более чем тремя атомами фтора, и C1-2 алкокси, необязательно замещенного не более чем тремя атомами фтора, где заместители могут быть одинаковыми или различными.
В одном варианте осуществления R3 представляет фенил или пиридил, где каждая из этих групп может быть необязательно замещена одним заместителем, выбранным из атома галогена, циано, C1-2 алкила, необязательно замещенного не более чем тремя атомами фтора, и C1-2 алкокси, необязательно замещенного не более чем тремя атомами фтора.
В одном варианте осуществления R3 представляет фенил, необязательно замещенный одним заместителем, выбранным из атома фтора, метокси, этокси, циано, этила, -CHF2, -OCF3 и -CF3.
В одном варианте осуществления R3 представляет фенил, необязательно замещенный одним заместителем, выбранным из атома фтора, метокси, циано, этила и -CHF2.
В еще одном варианте R3 представляет пиридил, необязательно замещенный одним заместителем, выбранным из атома фтора, хлора, метила, метокси и циано.
В одном варианте осуществления R3 представляет фенил, пиридил, пиримидинил, пиразинил или пиридазинил, где каждая из этих групп может быть необязательно замещена одним или двумя заместителями, выбранными из атома фтора, хлора, циано, метила, этилдифторметила (-CHF2), трифторметила, метокси, этокси и трифторметокси (-OCF3)
В одном варианте осуществления R3 представляет фенил, пиридил или пиримидинил, где каждая из этих групп может быть необязательно замещена одним или двумя заместителями, выбранными из атома фтора, хлора, циано, метила, этилдифторметила (-CHF2), трифторметила, метокси и трифторметокси (-OCF3).
В одном варианте осуществления, когда R3 представляет пиридил, то он представляет 2-пиридил, 3-пиридил или 4-пиридил, в частности, 2-пиридил, необязательно замещенный любой из групп, определенных выше.
В одном варианте осуществления, когда R3 представляет циклогексил, то он замещен двумя атомами фтора, которые присоединены к одному и тому же атому углерода. В конкретном варианте осуществления два атома фтора присоединены в положении 4, так что R3 представляет, например, 4,4-дифторциклогексил.
В одном варианте осуществления R3 представляет пиридил, необязательно замещенный одним заместителем, выбранным из атома фтора и хлора. В конкретном варианте осуществления R3 представляет 2-пиридил, необязательно замещенный атомом фтора. В одном варианте осуществления R3 представляет 2-пиридил, замещенный атомом фтора.
В одном варианте осуществления, когда R3 представляет тетрагидропиран, то он может представлять одно из следующего:
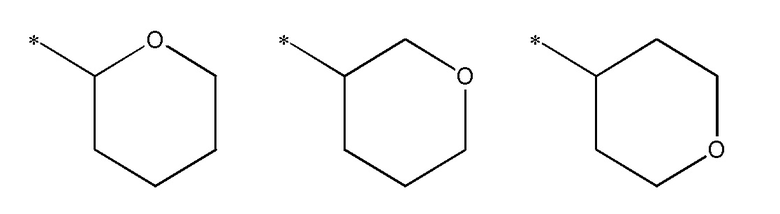
где * представляет место присоединения.
В одном варианте осуществления, когда R3 представляет тетрагидропиран, то он представляет:
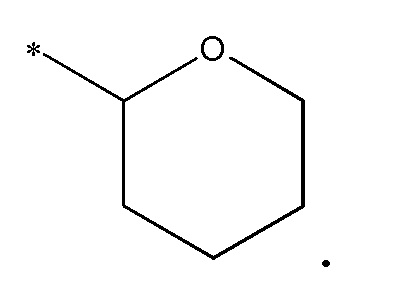
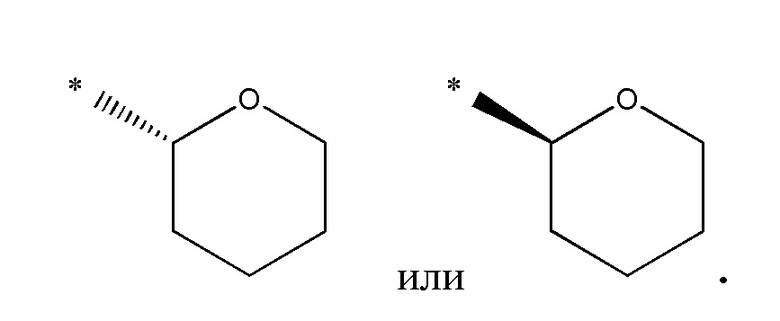
В одном варианте осуществления, когда R3 представляет тетрагидропиран, то он относится к конкретному энантиомеру, т.е. (R)- или (S)-энантиомеру. Примеры включают следующее:
В одном варианте осуществления, когда R1 представляет 2-гидроксиэтил, то:
R3 представляет фенил, необязательно замещенный одним заместителем, выбранным из атома фтора, этила, метокси, этокси, циано, трифторметила, -OCF3 и -CHF2;
R3 представляет пиридил, необязательно замещенный одним или двумя заместителями, выбранными из атома фтора, хлора, циано, метила и метокси, где заместители могут быть одинаковыми или различными; пиримидинил, необязательно замещенный одной метильной группой;
R3 представляет незамещенный пиридазинил;
R3 представляет пиразинил, необязательно замещенный одним заместителем, выбранным из метила и метокси;
R3 представляет циклогексил, который может быть необязательно замещен одним или двумя атомами фтора, где каждый заместитель может быть присоединен к одному и тому же атому углерода; или
R3 представляет тетрагидропиран.
В одном варианте осуществления, когда R1 представляет 2-гидроксиэтил, то R3 представляет фенил, необязательно замещенный одним заместителем, выбранным из атома фтора, этила, метокси, этокси, циано, трифторметила, -OCF3 и -CHF2; пиридил, необязательно замещенный одним или двумя заместителями, выбранными из атома фтора, хлора, циано, метила и метокси, где заместители могут быть одинаковыми или различными; пиримидинил, необязательно замещенный одной метильной группой; незамещенный пиридазинил; или пиразинил, необязательно замещенный одним заместителем, выбранным из метила и метокси.
В одном варианте осуществления, когда R1 представляет 2-гидроксиэтил, то R3 представляет фенил, необязательно замещенный одним заместителем, выбранным из атома фтора, этила, метокси, этокси, циано, трифторметила, -OCF3 и -CHF2; пиридил, необязательно замещенный одним или двумя заместителями, выбранными из атома фтора, хлора, циано, метила и метокси, где заместители могут быть одинаковыми или различными; пиримидинил, необязательно замещенный одной метильной группой; или незамещенный пиридазинил.
В одном варианте осуществления X представляет N; n равно 0; R1 представляет 2-гидроксиэтил; и R3 представляет пиридил, необязательно замещенный атомом фтора или хлора, предпочтительно атомом фтора.
В одном варианте осуществления соединение формулы (I) выбрано из списка, включающего:
(4-(2-(4-фторбензил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид);
N-(2-гидроксиэтил)-4-(2-(пиримидин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
4-(2-(4-фторбензил)-2H-тетразол-5-ил)-N-метилбензолсульфонамид;
N-метил-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
N-этил-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-(4-метоксибензил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
4-(2-(4-хлорбензол)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(2-(4-цианобензол)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(2-(4-(дифторметил)бензил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(2-((4,4-дифторциклогексил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
(R)-N-(2-гидроксипропил)-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид
(R)-4-(2-((4,4-дифторциклогексил)метил)-2H-тетразол-5-ил)-N-(2-гидроксипропил)бензолсульфонамид;
(S)-4-(2-(4-фторбензил)-2H-тетразол-5-ил)-N-(2-гидроксипропил)бензолсульфонамид;
N-(1,3-дигидроксипропан-2-ил)-4-(2-(4-фторбензил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(1,3-дигидроксипропан-2-ил)-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2,3-дигидроксипропил)-4-(2-(4-фторбензил)-2H-тетразол-5-ил)бензолсульфонамид;
4-(2-((5-хлорпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(2-(4-этилбензил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(2-((5-цианопиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(2-((3,5-дифторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-((6-метилпиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-((2-метилпиримидин-4-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
(R)-4-(2-(4-фторбензил)-2H-тетразол-5-ил)-N-(2-гидроксипропил)бензолсульфонамид;
(R)-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксипропил)бензолсульфонамид;
(R)-N-(2-гидроксипропил)-4-(2-((5-метоксипиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
(R)-N-(2-гидроксипропил)-4-(2-((5-метилпиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
(R)-N-(2-гидроксипропил)-4-(2-(4-метоксибензил)-2H-тетразол-5-ил)бензолсульфонамид;
4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(2-(3-фтор-4-метоксибензил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-((5-метоксипиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
(N-(2-гидроксиэтил)-4-(2-((5-метилпиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид);
N-(2-гидроксиэтил)-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-((2-метилпиридин-4-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
2-(4-(2-(4-метоксибензил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
2-(4-(2-(4-метилбензил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
2-(4-(2-(3,4-дифторбензил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
2-(4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
2-(4-(2-((4,4-дифторциклогексил)метил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
2-(4-(2-((5-метилпиридин-2-ил)метил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
2-(4-(2-((5-хлорпиридин-2-ил)метил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
2-(4-(2-((2-метилпиридин-4-ил)метил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
Метил 2-(4-(2-(4-фторбензил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетат;
Метил 2-(4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетат;
2-(4-(2-(4-фторбензил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
2-(4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
(4-(2-(4-фторбензил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)-2-метоксибензолсульфонамид);
(N-(2-гидроксиэтил)-2-метокси-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид);
(4-(2-(4-цианобензил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)-2-метоксибензолсульфонамид);
(4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)-2-метоксибензолсульфонамид);
(2-метокси-N-метил-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид);
(N-(цианометил)-4-(2-(4-фторбензил)-2H-тетразол-5-ил)бензолсульфонамид);
2-(4-(2-(4-фторбензил)-2H-тетразол-5-ил)-2-метоксифенилсульфонамидо)ацетамид;
2-(4-(2-(4-цианобензил)-2H-тетразол-5-ил)-2-метоксифенилсульфонамидо)ацетамид;
2-(2-метокси-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
(N-(цианометил)-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид);
(N-(цианометил)-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид);
(N-(цианометил)-4-(2-(пиримидин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
(N-(цианометил)-2-метокси-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид);
(N-(цианометил)-4-(1-(пиридин-4-илметил)-1H-1,2,3-триазол-4-ил)бензолсульфонамид);
(N-(цианометил)-4-(1-((5-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензолсульфонамид);
(4-(1-((5-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-N-(2-гидроксиэтил)бензолсульфонамидо);
(N-(2-гидроксиэтил)-4-(1-(4-метоксибензил)-1H-1,2,3-триазол-4-ил)бензолсульфонамид);
(4-(1-(4-фторбензил)-1H-1,2,3-триазол-4-ил)-N-(2-гидроксиэтил)бензолсульфонамид);
(N-(2-гидроксиэтил)-4-(1-(пиридин-4-илметил)-1H-1,2,3-триазол-4-ил)бензолсульфонамид);
(4-(1-(4-цианобензил)-1H-1,2,3-триазол-4-ил)-N-(2-гидроксиэтил)бензолсульфонамид);
(N-(2-гидроксиэтил)-4-(1-((5-метилпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензолсульфонамид);
(2-(4-(1-(4-фторбензил)-1H-1,2,3-триазол-4-ил)фенилсульфонамидо)ацетамид);
(2-(4-(1-((4,4-дифторциклогексил)метил)-1H-1,2,3-триазол-4-ил)фенилсульфонамидо)ацетамид);
(R)-4-(1-(4-фторбензил)-1H-1,2,3-триазол-4-ил)-N-(2-гидроксипропил)бензолсульфонамид;
4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)-2-метилбензолсульфонамид;
N-(2-гидроксиэтил)-2-метил-4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)-3-метилбензолсульфонамид;
N-(2-гидроксиэтил)-3-метил-4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
2-фтор-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил) бензолсульфонамид;
2-фтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
2-фтор-4-(2-(4-фторбензил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
2-фтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-4-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
4-(2-(циклогексилметил)-2H-тетразол-5-ил)-2-фтор-N-(2-гидроксиэтил)бензолсульфонамид;
2-хлор-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
3-хлор-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
2-(2-хлор-4-(2-(4-фторбензил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
3-фтор-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
3-фтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
2,3-дифтор-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
2,3-дифтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
2,6-дифтор-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
(R)-2,6-дифтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид
N-(цианометил)-4-(2-((тетрагидро-2H-пиран-4-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(цианометил)-4-(2-(пиразин-2-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(цианометил)-4-(2-((5-метилпиразин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(цианометил)-4-(2-((5-метоксипиразин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(цианометил)-4-(2-((6-метоксипиридин-3-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(цианометил)-4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
2-((4-(2-((4,4-дифторциклогексил)метил)-2H-тетразол-5-ил)-2-метоксифенил)сульфонамидо)ацетамид;
2-((4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)фенил)сульфонамидо)ацетамид;
N-(2-гидроксиэтил)-4-(2-(4-(трифторметокси)бензил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-(4-(трифторметил)бензил)-2H-тетразол-5-ил)бензолсульфонамид;
4-(2-(4-этоксибензил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(2-(циклогексилметил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-((6-метилпиридин-3-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-(пиридин-3-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
(N-(2-гидроксиэтил)-4-(2-((5-метоксипиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид);
N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-3-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-4-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-(пиридазин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-(пиридазин-3-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
4-(2-(4-цианобензил)-2H-тетразол-5-ил)-N-метилбензолсульфонамид;
N-(2-гидроксиэтил)-2-метокси-4-(2-((5-метоксипиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
2-фтор-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)-5-метилбензолсульфонамид;
(4-(2-(1-(5-фторпиридин-2-ил)этил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(2-(1-(5-фторпиридин-2-ил)этил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(1-(1-(4-фторфенил)этил)-1H-1,2,3-триазол-4-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
2-(4-(2-(1-(4-фторфенил)этил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид; и
2-(4-(2-(1-(4-фторфенил)этил)-2H-тетразол-5-ил)-2-метоксифенилсульфонамидо)ацетамид.
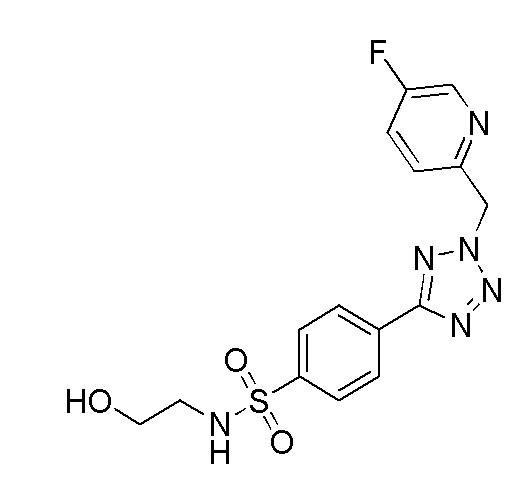
В частности, соединение формулы (I) представляет 4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил) бензолсульфонамид.
Кроме того, следует понимать, что соединение формулы (I) может существовать в разных таутомерных формах. Предполагается, что все возможные таутомеры входят в объем настоящего изобретения.
Соединения формулы (I) или их фармацевтически приемлемые соли могут находиться в кристаллической или аморфной форме. Кроме того, некоторые кристаллические формы соединений по изобретению могут существовать в виде полиморфов, которые все включаются в объем настоящего изобретения. Наиболее термодинамически стабильные полиморфные формы или формы соединений по изобретению представляют особый интерес. В одном аспекте изобретения соединение формулы (I) или его фармацевтически приемлемая соль являются кристаллическими.
Полиморфные формы соединений по изобретению могут быть охарактеризованы и дифференцированы с использованием ряда обычных аналитических методов, включая, не ограничиваясь этим, порошковую рентгеновскую дифракцию (XRPD), инфракрасную спектроскопию (IR), рамановскую спектроскопию, дифференциальную сканирующую калориметрию (DSC), термогравиметрический анализ (TGA) и твердотельный ядерный магнитный резонанс (ssNMR).
Термины и определения
Как здесь используется, термин «C1-2 алкил, необязательно замещенный не более чем тремя атомами фтора» относится к метилу или этилу, каждый из которых может быть замещен не более чем тремя атомами фтора. Таким образом, неограничивающими примерами групп, содержащих атомы фтора, включенных в это определение, являются -CF3, -CHF2, -CH2F и -CH2CF3.
Как здесь используется, термин «C1-2 алкокси, необязательно замещенный не более чем тремя атомами фтора» относится к метокси (т.е. OCH3) или этокси (т.е. OCH2CH3), каждый из которых может быть замещен не более чем тремя атомами фтора. Таким образом, неограничивающими примерами таких групп, содержащих атомы фтора, включенных в это определение, являются -OCF3, -OCHF2, -OCH2F и -OCH2CF3.
Как здесь используется, термин «атом галогена» относится к атому галогена, выбранному из атомов F, Cl и Br, предпочтительно F и Cl.
Как здесь используется, термин «амино» относится к -NH2.
Как здесь используется, термин «гидроксиметил» относится к метильной группе, в которой один из атомов водорода замещен на ОН группу, т.е. гидроксильную группу.
Как здесь используется, термин «цианометил» относится к метильной группе, в которой один из атомов водорода замещен на группу СN-, т.е. цианогруппу.
Как здесь используется, термин «циано» относится к группе CN-.
Как здесь используется, термин «соединения по изобретению» означает соединение формулы (I) или его фармацевтически приемлемую соль. Термин «соединение по изобретению» означает любое из соединений по изобретению, как определено выше.
Кроме того, следует понимать, что выражения, такие как «соединение формулы (I) или его фармацевтически приемлемая соль» или «соединения по изобретению» предназначены для включения соединения формулы (I), фармацевтически приемлемой соли или сольвата соединения формулы (I) или любой их фармацевтически приемлемой комбинации. Таким образом, в качестве неограничивающего примера, используемого здесь для иллюстративной цели, «соединение формулы (I) или его фармацевтически приемлемая соль» включает фармацевтически приемлемую соль соединения формулы (I), которая находится в виде сольвата, и это выражение также включает смесь соединения формулы (I) и фармацевтически приемлемой соли соединения формулы (I).
Следует понимать, что ссылки в настоящем документе на соединение формулы (I) или его фармацевтически приемлемую соль включают соединение формулы (I) в виде свободного основания или в виде его фармацевтически приемлемой соли. Таким образом, в одном варианте осуществления изобретение относится к соединению формулы (I). В еще одном варианте осуществления изобретение относится к фармацевтически приемлемой соли соединения формулы (I).
Термин «фармацевтически приемлемый» относится к таким соединениям (включая соли), веществам, композициям и лекарственным формам, которые в рамках медицинского заключения пригодны для применения в контакте с тканями людей и животных без проявления чрезмерной токсичности, раздражения или другой проблемы или осложнения, соразмерных с разумным соотношением польза/риск.
Фармацевтически приемлемые соли включают, среди прочего, соли, описанные Berge, J. Pharm. Sci., 1977, 66, 1-19, или перечисленные в PH Stahl и CG Wermuth, editors, Handbook of Pharmaceutical Salts; editors, Handbook of Pharmaceutical Salts; Properties, Selection and Use, Second Edition Stahl/Wermuth:: Wiley-VCH/VHCA, 2011 (см. Http://www.wiley.com/WileyCDA/WileyTitle/productCd3906390519.html).
Когда функциональные свойства соединения позволяют, то можно получить подходящие фармацевтически приемлемые соли соединения формулы (I), которые включают аддитивные соли кислоты или основания. Кислотно-аддитивные соли можно получить взаимодействием с подходящей кислотой, необязательно в подходящем растворителе, таком как органический растворитель, с получением соли, которую можно выделить кристаллизацией и фильтрацией. Основно-аддитивные соли можно получить взаимодействием с подходящим основанием, необязательно в подходящем растворителе, таком как органический растворитель, с получением соли, которую можно выделить кристаллизацией и фильтрацией.
Репрезентативные фармацевтически приемлемые кислотно-аддитивные соли включают, не ограничиваясь этим, 4-ацетамидобензоат, ацетат, адипат, альгинат, аскорбат, аспартат, бензолсульфонат (бесилат), бензоат, бисульфат, битартрат, бутират, эдетат кальция, камфорат, камфорсульфонат (камсилат), капрат (деканоат), капроат (гексаноат), каприлат (октаноат), циннамат, цитрат, цикламат, биглюконат, 2,5-дигидроксибензоат, дисукцинат, додецилсульфат (эстолат), эдетат (этилендиаминтетраацетат), эстолат (лаурилсульфат), этан-1,2-дисульфонат (эдисилат), этансульфонат (эсилат), формиат, фумарат, галактарат (мукат), гентизат (2,5-дигидроксибензоат), глюкогептонат (глюцептат), глюконат, глюкуронат, глутамат, глутарат, глицерофосфат, гликолят, гексилрезорцинат, гиппурат, гидрабамин (N, N'-ди(дегидроабиэтил)этилендиамин), гидробромид, гидрохлорид, гидроиодид, гидроксинафтоат, изобутират, лактат, лактобионат, лаурат, малат, малеат, малонат, манделат, метансульфонат (мезилат), метилсульфат, мукат, нафталин-1,5-дисульфонат (нападизилат), нафталин-2-сульфонат (напсилат), никотинат, нитрат, олеат, пальмитат, п-аминобензолсульфонат, п-аминосалицилат, памоат (эмбонат), пантотенат, пектинат, персульфат, фенилацетат, фенилэтилбарбитурат, фосфат, полигалактуронат, пропионат, п-толуолсульфонат (тозилат), пироглутамат, пируват, салицилат, себакат, стеарат, субацетат, сукцинат, сульфамат, сульфат, таннат, тартрат, теоклат (8-хлортеофиллинат), тиоцианат, триэтиодид, ундеканоат, ундециленат и валерат.
Репрезентативные фармацевтически приемлемые основно-аддитивные соли включают, не ограничиваясь этим, соли алюминия, 2-амино-2-(гидроксиметил)-1,3-пропандиола (ТРИС, трометамина), аргинина, бенетамина (N-бензилфенэтиламина), бензатина (N, N'-дибензилэтилендиамина), бис-(2-гидроксиэтил)амина, висмута, кальция, хлорпрокаина, холина, клемизола (1-п-хлорбензил-2-пирролилдин-1'-илметилбензимидазола), циклогексиламина, дибензилэтилендиамина, диэтиламина, диэтилтриамина, диметиламина, диметилэтаноламина, дофамина, этаноламина, этилендиамина, L-гистидина, железа, изохинолина, лепидина, лития, лизина, магния, меглюмина (N-метилглюкамина), пиперазина, пиперидина, калия, прокаина, хинина, хинолина, натрия, стронция, трет-бутиламина и цинка.
Как здесь используется, термин «терапевтически эффективное количество» означает любое количество, которое по сравнению с соответствующим субъектом, который не получил такого количества, приводит к повышению эффективности лечения, заживления, профилактике или ослаблению заболевания, расстройства или побочного эффекта, или снижению скорости развития заболевания или расстройства.
Подходящее «терапевтически эффективное количество» будет зависеть от ряда факторов, включая, например, возраст и массу тела субъекта, конкретное патологическое состояние, требующее лечения, и его тяжесть, природу композиции и путь введения, и в конечном итоге будет находиться на усмотрении лечащего врача.
Специалистам в данной области будет понятно, что ссылки в данном описании на лечение относятся к лечению установленных патологических состояний, включая, например, микобактериальную инфекцию или заболевание, возникающее в результате микобактериальной инфекции, такое как туберкулез. Однако соединения формулы (I) или их фармацевтически приемлемые соли могут, в зависимости от патологического состояния, также быть пригодными для профилактики микобактериальной инфекции или заболевания, возникающего в результате микобактериальной инфекции, такого как туберкулез. Таким образом, в одном варианте осуществления обеспечивается лечение или профилактика заболевания. В еще одном варианте осуществления обеспечивается лечение заболевания. В дополнительном варианте осуществления обеспечивается профилактика заболевания.
Соединения формулы (I) могут содержать один или более асимметричных центров (также называемых хиральными центрами) и, следовательно, могут находиться в виде отдельных энантиомеров, диастереоизомеров или других стереоизомерных форм или в виде их смесей. Хиральные центры, такие как хиральные атомы углерода, также могут присутствовать в заместителе, таком как алкильная группа. Когда стереохимия хирального центра, присутствующего в формуле (I) или в любой химической структуре, показанной здесь, не указана, то предполагается, что структура охватывает любой стереоизомер и все их смеси. Таким образом, соединения формулы (I), содержащие один или более хиральных центров, можно использовать в виде рацемических модификаций, включая рацемические смеси и рацематы, энантиомерно обогащенные смеси или в виде энантиомерно чистых индивидуальных стереоизомеров.
Получение соединений
Соединения по изобретению можно получить различными способами, включая обычную химию. Любая ранее определенная переменная будет иметь ранее определенное значение, если не указано иное. Иллюстративные общие способы синтеза приведены на следующих схемах реакций, и их можно легко адаптировать для получения других соединений по изобретению. Конкретные соединения по изобретению можно получить в соответствии с экспериментальными процедурами, раскрытыми в разделе «Примеры».
Общие процедуры, используемые для синтеза соединений формулы (I), показаны на схемах реакций 1-4 ниже и иллюстрированы в примерах.
Получение соединений формулы (I)
Соединения формулы (Ia), которые представляют производные тетразола формулы (I), где X=N, и R2 имеет значения, определенные в п.1, (т.е. n равно 1 или 2), в частности, F или Cl или OMe, когда они являются смежными с сульфонилхлоридом или где R2 отсутствует (т.е. n равно 0), можно получить, следуя общей процедуре, схематично показанной на схеме реакций 1.
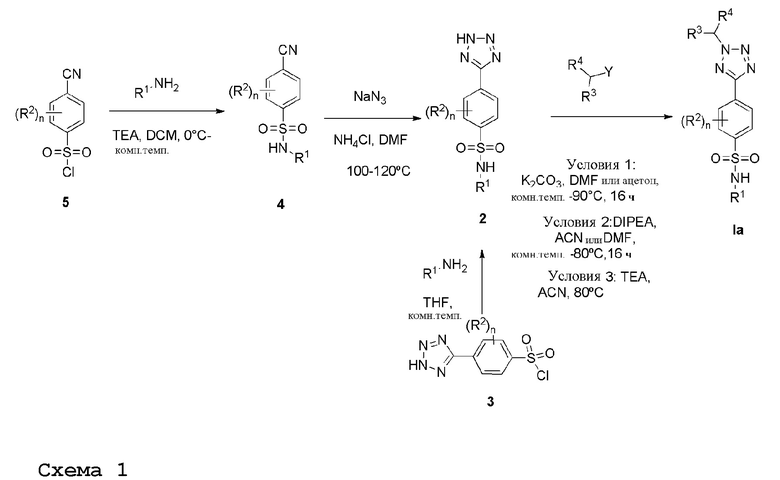
Получение соединений формулы (I), где R1 представляет метил, этил, С-связанный ацетамидо, метилацетат, 2-гидроксиэтил, 2-гидрокси-1-пропил, 1,3-дигидрокси-2-пропил или 1,2-дигидрокси-3-пропил; и R2 имеет значения, определенные в п.1.
Сульфонилхлорид 5 растворяют в подходящем растворителе, например, дихлорметане, и добавляют алкилирующий агент, имеющий формулу R1NH2, в присутствии TEA с получением соединений формулы 4. Реакцию проводят при подходящей температуре, например, от 0°С до комнатной температуры. Реакцию циклоприсоединения нитрила формулы 4 с азидом натрия и хлоридом аммония проводят при подходящей температуре, например, от 100°С до 120°С, с получением 5-замещенных 1Н-тетразолов 2.
Альтернативно, соединение формулы 2 можно получить взаимодействием сульфонилхлорида 3, где соединение 3 растворяют в подходящем растворителе, например, тетрагидрофуране, с последующим добавлением алкиламина, имеющего формулу R1NH2.
Затем соединение 2 растворяют в подходящем растворителе, например, ДМФА, ацетонитриле или ацетоне, и добавляют алкилирующий агент, имеющий формулу R3CH2Y (где Y представляет уходящую группу, такую как мезилат или галогенид, например, хлор или бром), в присутствии неорганического основания, например карбоната калия или органического основания, например N, N-диизопропилэтиламина или TEA, при подходящей температуре, например, от комнатной температуры до 90°С.
Когда R4 представляет метил, то условия, по существу соответствующие условиям 2, показанным на схеме 1, используются для стадии сочетания.
Соединения формулы Ia получают после очистки.
Соединения формулы Ib и Id, которые представляют производные тетразола (X=N) формулы (I), где R2 является другим, чем H (т.е. n равно 1 или 2), можно получить в соответствии со следующими общими процедурами, схематично показанными на схемах 2a и 2с.
Соединения формулы Ib, которые представляют производные тетразола (X=N) формулы (I), где R1 представляет 2-гидроксиэтил, и R2 не является H (например, как определено в п.1 формулы изобретения, когда n равно 1 или 2, более конкретно, метокси, фтор или метил) можно получить в соответствии со следующей общей процедурой, схематично показанной на схеме 2а.
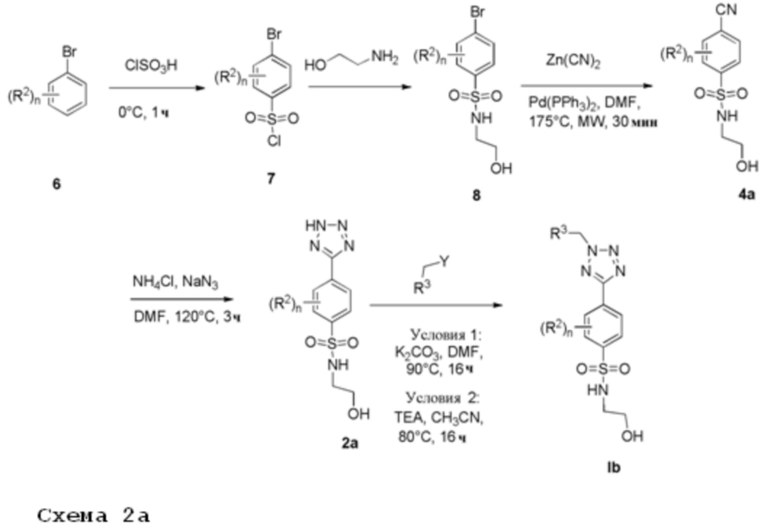
Соединение 6 подвергают взаимодействию с хлорсульфоновой кислотой при подходящей температуре, например, примерно 0°С, в течение от 1 ч до 3 ч. Затем соответствующий сульфонилхлорид 7 растворяют в подходящем растворителе, например, дихлорметане, и подвергают взаимодействию с алкиламином, имеющим формулу CH2(OH)CH2(NH2), в присутствии TEA с получением соединений формулы 8. Катализируемая палладием реакция цианирования арилгалогенида 8 с Zn(CN)2 при микроволновом облучении при подходящей температуре, например, 175°С в течение примерно 0,5 ч, дает нитрильное производное 4а. Циклоприсоединение нитрила 4а с азидом натрия и хлоридом аммония при подходящей температуре, например 120°С, дает 5-замещенный 1Н-тетразол 2а. Соединение 2а растворяют в подходящем растворителе, например, ДМФА или ACN, и подвергают взаимодействию с алкилирующим агентом, имеющим формулу R3CH2Y (где Y представляет собой уходящую группу, такую как мезилат или галогенид, например хлор или бром), в присутствии неорганического основания, например, карбоната калия, или органического основания, например, TEA, при температуре в диапазоне от 80°С до 90°C.
Соединения формулы Ib получают после очистки.
Соединения формулы Ic, которые представляют производные тетразола (X=N) формулы (I), где R1 представляет C-связанный ацетамидо, и где R2 является таким, как определено в п.1 формулы изобретения (в частности, OMe, F или Cl), и когда n равно 1, или где R2 отсутствует, т.е. n равно 0, можно получить в соответствии со следующей общей процедурой, схематично показанной на схеме 2b.
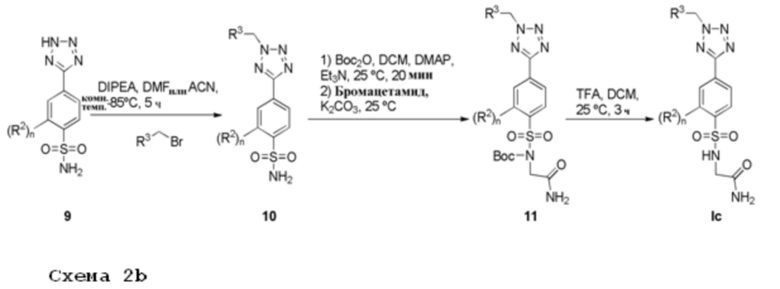
Соединение 9 растворяют в подходящем растворителе, например, ДМФА, и подвергают взаимодействию с алкилирующим агентом, имеющим формулу R3CH2Br, в присутствии органического основания, например DIPEA, при температуре в диапазоне от комнатной температуры до 80°С. Монозащита соединения 10 с использованием (Boc)2O, триэтиламина и каталитического количества 4-диметиламинопиридина в дихлорметане с последующим N-алкилированием с использованием 2-бромацетамида и карбоната калия, дает сульфонамид 11. Удаление защитной Boc-группы из соединения 11, например, трифторуксусной кислотой в дихлорметане при комнатной температуре дает соединения Ic.
Соединения формулы Ic получают после очистки.
Соединения формулы Id, которые представляют производные тетразола формулы (I), где R1 представляет цианометил, можно получить в соответствии со следующей общей процедурой, схематично показанной на схеме 2с.
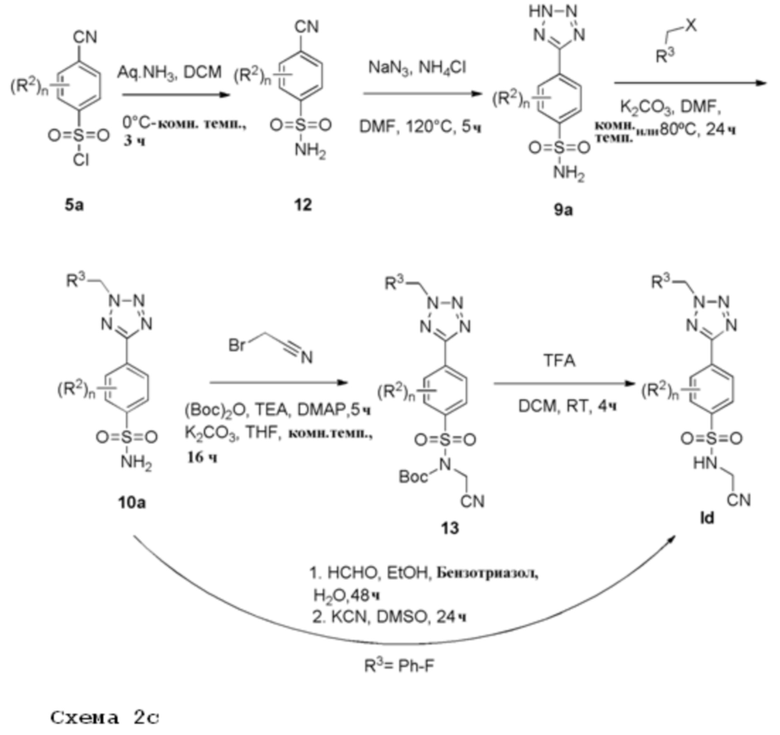
Арилсульфонилхлорид 5а растворяют в водном аммиаке и подходящем растворителе, например дихлорметане, при подходящей температуре, например от 0°С до комнатной температуры, с получением первичных сульфонамидов 12. Реакцию циклоприсоединения нитрила формулы 12 с азидом натрием и хлоридом аммония проводят при подходящей температуре, например 120°С, с образованием 5-замещенных 1Н-тетразолов 9а. Соединение 9а растворяют в подходящем растворителе, например, ДМФА, и подвергают взаимодействию с алкилирующим агентом, имеющим формулу R3CH2Y (где Y представляет уходящую группу, такую как мезилат или галогенид, например, хлор или бром), в присутствии неорганического основания, например, карбоната калия при температуре в диапазоне от комнатной температуры до 80°С. Монозащита соединения 10а с использованием (Boc)2O, триэтиламина и каталитического количества 4-диметиламинопиридина с последующим N-алкилированием с использованием алкилбромида и карбоната калия дает Boc-защищенный сульфонамид 13. Удаление защитной Boc-группы, например, трифторуксусной кислотой в дихлорметане при комнатной температуре дает соединения формулы Id. Продукты Id получают после очистки.
Альтернативно, сульфонамид 10а, бензотриазол и формальдегид в смеси этанол/вода перемешивают при комнатной температуре в течение примерно 48 ч, где аддукт бензотриазол-формальдегид растворяют в ДМСО и обрабатывают KCN в течение 24 ч, с получением соединения Id (когда n равно 0).
Соединения формулы Ie, которые представляют производные триазола (X=CH) формулы (I), где R1 и R2 имеют значения, определенные в п.1 фрмулы изобретения, исключая когда R1 представляет цианометил, можно получить в соответствии со следующей общей процедурой, схематично показанной на схеме 3.
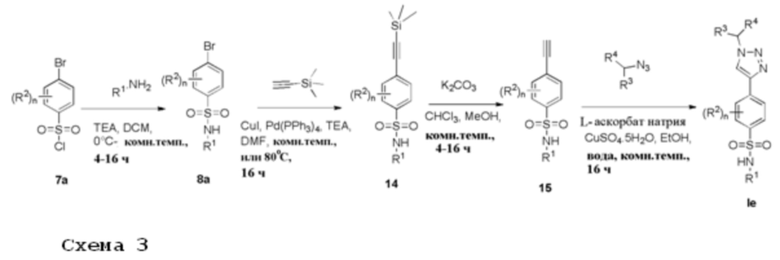
Арилсульфонилхлорид 7а растворяют в подходящем растворителе, например, дихлорметане, и подвергают взаимодействию с алкиламином, имеющим формулу R1NH2, в присутствии TEA при подходящей температуре, например, от 0°С до комнатной температуры, с получением соединений формулы 8а. Арилбромид формулы 9а сочетают в реакции типа Соногашира с (триметилсилил)ацетиленом, используя подходящую каталитическую систему, например тетракис(трифенилфосфин)палладий (0) и иодид меди (I), в подходящем растворителе, например ДМФА, в присутствии основания, например, триэтиламина, при температуре в диапазоне от комнатной температуры до 80°С, с получением соединения формулы 14. Снятие защиты с соединения 14 с получением соединения 15 может быть достигнуто с помощью основания, например, карбоната калия, в подходящем растворителе, например, смеси хлороформа/метанола, при комнатной температуре. 1,4-дизамещенные 1,2,3-триазолы формулы Ie получают 1,3-диполярным циклоприсоединением, катализируемым медью (I), между подходящими ароматическими азидами и терминальными ацетиленами формулы 15, где сульфата Cu (II) пентагидрат восстанавливается аскорбатом натрия в смеси этанола и воды при комнатной температуре. Соединения формулы Ie получают после очистки.
Соединения формулы (If), которые представляют производные триазола (X=CH) формулы (I), где R1 представляет цианометил, можно получить в соответствии со следующей общей процедурой, схематично показанной на схеме 4.
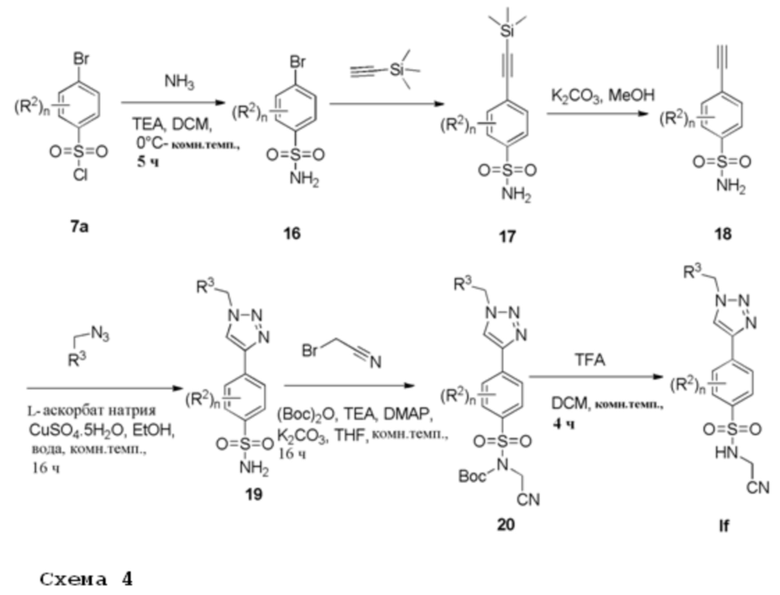
Арилсульфонилхлорид 7а растворяют в подходящем растворителе, например, дихлорметане, и подвергают взаимодействию с аммиаком в присутствии TEA при подходящей температуре, например, от 0°С до комнатной температуры, с получением соединений формулы 16. Арилбромид формулы 16 сочетают в реакции типа Соногашира с (триметилсилил)ацетиленом с использованием подходящей каталитической системы, например тетракис(трифенилфосфин)палладия (0) и иодида меди (I), в подходящем растворителе, например, ДМФА, в присутствии основания, например, триэтиламина, при 27°С, с получением соединения формулы 17. Снятие защиты с соединения формулы 17 с получением соединения формулы 18 может быть достигнуто с помощью основания, например карбоната калия, в подходящем растворителе, например, смеси хлороформа/метанола, при комнатной температуре. 1,4-дизамещенные 1,2,3-триазолы формулы 19 получают 1,3-диполярным циклоприсоединением, катализируемым медью (I), между соответствующими ароматическими азидами и терминальными ацетиленами формулы 18, где сульфат Cu (II) пентагидрат восстанавливается аскорбатом натрия в смеси этанола и воды при комнатной температуре. Монозащита соединения 19 с использованием (Boc)2O, триэтиламина и каталитического количества 4-диметиламинопиридина с последующим N-алкилированием с использованием алкилбромида и карбоната калия, дает сульфонамид 20. Удаление защитной Boc-группы, например, трифторуксусной кислотой в дихлорметане при комнатной температуре дает соединения If. Соединения формулы If получают после очистки.
Способы применения
В одном аспекте изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения в терапии.
В одном аспекте изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения в лечении микобактериальной инфекции. Микобактериальная инфекция представляет инфекцию, вызванную микобактериями.
Микобактерия может быть членом одной из следующих групп микобактерий: комплекс Mycobacterium tuberculosis (MTC), комплекс Mycobacterium avium (MAC), клад Mycobacterium gordonae, клад Mycobacterium kansasii, клад Mycobacterium chelonae, клад Mycobacterium fortucoitum, клад Mycobacterium parafortium или клад Mycobacterium vaccae. Микобактериями также могут представлять Mycobacterium ulcerans или Mycobacterium leprae.
В одном варианте осуществления микобактерия является членом комплекса Mycobacterium tuberculosis (MTC).
Члены комплекса Mycobacterium tuberculosis (MTC) включают Mycobacterium tuberculosis, Mycobacterium africanum, Mycobacterium bovis, Mycobacterium bovis BCG, Mycobacterium canetti, Mycobacterium caprae, Mycobacterium microti и Mycobacterium pinnipedii. Эти микобактерии являются возбудителями туберкулеза человека и животных. Mycobacterium tuberculosi является основным возбудителем туберкулеза человека.
В одном варианте осуществления инфекция представляет собой инфекцию, вызванную Mycobacterium tuberculosis. Другими словами, микобактериальная инфекция вызвана Mycobacterium tuberculosis.
В одном варианте осуществления Mycobacterium tuberculosis обладает множественной лекарственной устойчивостью.
Члены комплекса Mycobacterium avium (MAC) включают Mycobacterium avium, Mycobacterium avium paratuberculosis, Mycobacterium avium silaticum, Mycobacterium avium hominissuis, Mycobacterium columbiense и Mycobacterium indicus pranii.
Члены клада Mycobacterium gordonae включают Mycobacterium asiaticum и Mycobacterium gordonae.
Члены клада Mycobacterium kansasii включают Mycobacterium gastri и Mycobacterium kansasii.
Члены клада Mycobacterium chelonae включают Mycobacterium abscessus, Mycobacterium bolletii и Mycobacterium chelonae.
Члены клада Mycobacterium fortuitum включают Mycobacterium boenickei, Mycobacterium brisbanense, Mycobacterium cosmeticum, Mycobacterium fortuitum, Mycobacterium fortuitum subspecies acetamidolyticum, Mycobacterium houstonense, Mycobacterium mageritense, Mycobacterium neworleansense, Mycobacterium peregrinum, Mycobacterium porcinum, Mycobacterium senegalense и Mycobacterium septicum.
Члены клада Mycobacterium parafortuitum включают Mycobacterium austroafricanum, Mycobacterium diernhoferi, Mycobacterium frederiksbergense, Mycobacterium hodleri, Mycobacterium neoaurum и Mycobacterium parafortuitum.
Следовательно, микобактериальная инфекция может быть вызвана микобактериями, выбранными из следующего: Mycobacterium tuberculosis, Mycobacterium africanum, Mycobacterium bovis, Mycobacterium bovis BCG, Mycobacterium canetti, Mycobacterium caprae, Mycobacterium microti, Mycobacterium pinnipedii, Mycobacterium avium, Mycobacterium avium paratuberculosis, Mycobacterium avium silaticum, Mycobacterium avium hominissuis, Mycobacterium columbiense, Mycobacterium indicus pranii, Mycobacterium asiaticum, Mycobacterium gordonae, Mycobacterium gastri, Mycobacterium kansasii, Mycobacterium abscessus, Mycobacterium bolletii, Mycobacterium chelonae, включая Mycobacterium boenickei, Mycobacterium brisbanense, Mycobacterium cosmeticum, Mycobacterium fortuitum, Mycobacterium fortuitum subspecies acetamidolyticum, Mycobacterium houstonense, Mycobacterium mageritense, Mycobacterium neworleansense, Mycobacterium peregrinum, Mycobacterium porcinum, Mycobacterium senegalense, Mycobacterium septicum, Mycobacterium austroafricanum, Mycobacterium diernhoferi, Mycobacterium frederiksbergense, Mycobacterium hodleri, Mycobacterium neoaurum, Mycobacterium parafortuitum, Mycobacterium ulcerans и Mycobacterium leprae.
В еще одном аспекте изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения в лечении заболевания, вызванного микобактериями, где микобактерии выбраны из тех, которые описаны выше.
Заболевания, вызванные микобактериями, включают, не ограничиваясь этим, туберкулез (например, возбудитель Mycobacterium tuberculosis), проказу (например, возбудитель Mycobacterium leprae), болезнь Джонса (например, возбудитель Mycobacterium avium субвиды paratuberculosis), язву Бурулли или Бейрнсдейла (например, возбудитель Mycobacterium ulceran), болезнь Крона (например, возбудитель Mycobacterium avium субвиды paratuberculosis), заболевание легких или легочную инфекцию, пневмонию, бурсит, инфекцию в синовиальной сумке, инфекцию в сухожильном влагалище, локализованный абсцесс, лимфаденит, инфекции кожи и мягких тканей, синдром леди Уиндермир (например, возбудитель комплекс Mycobacterium avium (MAC)), болезнь легких МАС, диссеминированный комплекс Mycobacterium avium (DMAC), диссеминированный комплекс Mycobacterium avium intraceullulare (DMAIC), болезнь джакузи (например, возбудитель комплекс Mycobacterium avium), мастит MAC, пиомиозит MAC или гранулематозная болезнь.
В одном варианте осуществления заболевание представляет туберкулез. Таким образом, один аспект изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения в лечении туберкулеза.
В еще одном аспекте изобретение относится к способу лечения микобактериальной инфекции у млекопитающего, нуждающегося в этом, где указанное лечение включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Как описано здесь, микобактериальная инфекция является инфекцией, вызванной микобактериями. Микобактерии являются такими, как описано выше.
В одном варианте осуществления изобретение относится к способу лечения инфекции, вызванной Mycobacterium tuberculosis.
В еще одном аспекте изобретение относится к способу лечения заболевания, вызванного микобактериями, у млекопитающего, нуждающегося в этом, где указанное лечение включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В одном варианте осуществления заболевание представляет туберкулез. Следовательно, здесь также описан способ лечения туберкулеза у млекопитающего, нуждающегося в этом, где указанное лечение включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В одном варианте осуществления млекопитающее является человеком.
В еще одном аспекте изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли в производстве лекарственного средства для применения в лечении микобактериальной инфекции или в лечении заболевания, вызванного микобактериями.
Также здесь описывается применение соединения формулы (I) или его фармацевтически приемлемой соли в производстве лекарственного средства для применения в лечении туберкулеза.
В одном варианте осуществления лечение туберкулеза может быть направлено на лечение туберкулеза с множественной лекарственной устойчивостью, туберкулеза с широкой лекарственной устойчивостью или лекарственно-чувствительного туберкулеза. Кроме того, лечение может быть направлено на туберкулез легких и/или туберкулез, отличный от туберкулеза легких. Лечение также может быть направлено на лечение латентной формы туберкулеза.
В одном варианте осуществления соединение формулы (I) для применения в вышеописанных способах и способах лечения представляет 4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид, имеющий следующую структуру:
Фармацевтические композиции
Соединения формулы (I) или их фармацевтически приемлемые соли обычно, но не обязательно, формулируют в фармацевтические композиции перед введением пациенту. Следовательно, в еще одном аспекте обеспечивается фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент.
Фармацевтические композиции можно вводить любым подходящим путем, например, пероральным (включая трансбуккальный или сублингвальный), ингаляционным, интраназальным, местным (включая трансбуккальный, сублингвальный или трансдермальный), парентеральным (включая подкожный, внутримышечный, внутривенный или внутрикожный). В частности, фармацевтические композиции вводят пероральным путем введения.
Подходящие фармацевтически приемлемые эксципиенты включают следующие типы эксципиентов: носители, разбавители, наполнители, связующие вещества, разрыхлители, смазывающие вещества, скользящие вещества, гранулирующие агенты, покрывающие агенты, смачивающие агенты, растворители, сорастворители, суспендирующие агенты, эмульгаторы, подсластители, ароматизаторы, агенты, корригирующие вкус и запах, красящие агенты, антислеживающие агенты, увлажнители, хелатообразующие агенты, пластификаторы, повышающие вязкость агенты, антиоксиданты, консерванты, стабилизаторы, поверхностно-активные вещества и буферные агенты.
Подходящие способы формуляции соединений по изобретению известны специалистам в данной области, они описаны в Remington: The Science and Practice of Pharmacy, 21st Edition 2006 г.
Фармацевтические композиции могут быть представлены в разовых лекарственных формах, содержащих заранее определенное количество активного ингредиента на разовую дозу. Предпочтительными разовыми лекарственными формами являются композиции, содержащие суточную дозу или субдозу, или соответствующую их часть активного ингредиента. Следовательно, такие разовые дозы можно вводить более одного раза в день. Предпочтительными разовыми лекарственными формами являются композиции, содержащие суточную дозу или субдозу (для введения более одного раза в день), как указано выше, или соответствующую их часть активного ингредиента.
Когда соединение формулы (I) или его фармацевтически приемлемую соль используют для лечения туберкулеза, то их можно применять самостоятельно или в комбинации с дополнительным терапевтическим агентом, таким как дополнительный противомикобактериальный агент, в частности, дополнительный противотуберкулезный агент и/или противовирусный агент, включая противоретровирусные агенты.
Например, настоящее изобретение относится к комбинации (а) соединения формулы (I) или его фармацевтически приемлемой соли и (b) дополнительного противотуберкулезного агента. В одном варианте осуществления комбинация включает два, три, четыре, пять, шесть или семь дополнительных противотуберкулезных агентов. Например, при лечении туберкулеза с множественной лекарственной устойчивостью обычно пациентам вводят комбинации из четырех или более лекарственных препаратов. Например, при лечении лекарственно-чувствительного туберкулеза часто пациентам вводят комбинации из трех или четырех лекарственных препаратов.
Дополнительный противотуберкулезный агент является агентом, находящимся на стадии разработки, разрешенным или рекомендованным для лечения туберкулеза, и он может быть выбран из изониазида, рифампина, пиразинамида, этамбутола, моксифлоксацина, рифапентина, клофазимина, этионамида, протионамида, изоксила, тиоацетазона, диарилхинолина, такого как бедаквилин (TMC207) или TBAJ-587, нитроимидазооксазина PA-824 (претоманида), деламанида (OPC-67683), оксазолидинона, такого как линезолид, тедизолид, радезолид, сутезолид (PNU-100480), позизолид (AZD-5847) или TBI-223, аналога EMB SQ109, OPC-167832, GSK3036656A (также известного как GSK070), GSK2556286, GSK3211830, бензотиазинона, такого как BTZ043 или PBTZ169, азаиндола, такого как TBA-7371, динитробензамида или бета-лактама, такого как санфетринем, меропенем, фаропенем, эртапенем, тебипенем или комбинации бета-лактамов, такие как аугментин (амоксициллин-клавуланат).
В одном варианте осуществления дополнительный противотуберкулезный агент представляет собой агент, находящийся на стадии разработки, разрешенный или рекомендованный для лечения туберкулеза, и он может быть выбран из изониазида, рифампина, пиразинамида, этамбутола, моксифлоксацина, рифапентина, клофазимина, этионамида, протионамида, изоксила, тиоацетазона, бедаквилина (TMC207), нитроимидазооксазина PA-824, деламанида (OPC-67683), оксазолидинона, такого как линезолид, тедизолид, радезолид, сутезолид (PNU-100480) или позизолид (AZD-5847), аналога EMB OPC SQ109-167832, GSK3036656 (также известного как GSK070), GSK2556286, GSK3211830 и бензотиазинона или динитробензамида.
Комбинация по настоящему изобретению может дополнительно содержать противовирусный агент, включая противоретровирусные агенты.
Такие противоретровирусные агенты могут быть выбраны из зидовудина, диданозина, ламивудина, зальцитабина, абакавира, ставудина, адефовира, адефовира дипивоксила, фозивудина, тодоксила, эмтрицитабина, аловудина, амдоксовира, эльвуцитабина, невирапина, делавирдина, эфавиренза, ловирида, иммунокала, олтипраза, каправирина, лерсивирина, GSK2248761, TMC-278, TMC-125, этравирина, саквинавира, ритонавира, индинавира, нелфинавира, ампренавира, фосампренавира, бреканавира, дарунавира, атазанавира, типранавира, пилинавира, лазинавира, энфувиртида, Т-20, Т-1249, PRO-542, PRO-140, TNX-355, BMS-806, BMS-663068 и BMS-626529, 5-Helix, ралтегравира, элвитегравира, GSK1349572, GSK1265744, викривирока (Sch-C), Sch-D, TAK779, маравирока, TAK44, диданозина, тенофовира, лопинавира и дарунавира.
Комбинации могут быть удобно представлены для применения в форме фармацевтической композиции или состава. Следовательно, здесь также обеспечивается фармацевтическая композиция, содержащая (а) соединение формулы (I) или его фармацевтически приемлемую соль, как здесь описано, вместе с (b) дополнительным противотуберкулезным агентом и (с) необязательно противовирусным агентом, включая противоретровирусные агенты и (d) один или более фармацевтически приемлемых эксципиентов, как здесь описано.
Соединение формулы (I) или его фармацевтически приемлемую соль и дополнительный терапевтический агент можно вводить вместе или по отдельности, и при отдельном введении это может иметь место по отдельности или последовательно в любом порядке (одним и тем же или разными путями введения). Количество соединения по изобретению или его фармацевтически приемлемой соли и дополнительного терапевтически активного агента(ов) и относительные сроки введения будут выбраны для достижения желаемого комбинированного терапевтического эффекта.
Примеры
Теперь изобретение будет иллюстрировано с помощью следующих неограничивающих примеров. Несмотря на то, что ниже описаны конкретные варианты осуществления изобретения, специалист в данной области понимает, что могут быть сделаны различные изменения и модификации. Ссылки на получения, проводимые аналогичным образом или с помощью общего способа других получений, могут включать изменения в обычных параметрах, таких как время, температура, условия обработки, незначительные изменения в количестве реагентов и т.д.
В некоторых из следующих промежуточных продуктов и примеров исходные вещества идентифицируются посредством ссылки на номера других промежуточных продуктов или примеров. Это не означает, что фактическое вещество, полученное из какого-либо конкретного промежуточного соединения или примера, обязательно используется на последующей стадии, приведенной здесь в качестве примера, а используется в качестве условного средства для обозначения соответствующего названия соединения.
Сокращенные обозначения
В следующем списке приведены определения некоторых сокращений и символов, используемых в данном документе. Понятно, что данный список не является исчерпывающим, но значение таких сокращений и символов, которые здесь не определены ниже, будет хорошо очевидным для специалистов в данной области техники. При описании изобретения химические элементы идентифицируются в соответствии с Периодической системой химических элементов.
Аналитическое обоудование
1Н ЯМР спектры снимали на приборах Varian 400 МГц или Bruker Ultrashield DPX 400 МГц.
Химические сдвиги выражены в частях на миллион (ppm, δ единиц). Константы спин-спинового взаимодействия (J) выражены в герцах (Гц). Паттерны расщепления описывают видимые кратности и обозначаются как с (синглет), д (дублет), т (триплет), кв (квартет), дд (дублет дублета), дт (дублет триплета), м (мультиплет), br (шир.).
Все температуры указаны в градусах Цельсия.
МС и жидкостная хроматография: МС регистрировали на UPLC Waters с детектором PDA - Single Quad & Triple Quad с диапазоном анализа масс до M/Z=2000 или ВЭЖХ Agilent 1100 с системой Quadrupole 6120 (Agilent).
Данные ВЭЖХ/УВЭЖХ были получены на приборе Waters Alliance с детектором PDA или 2695 и Waters H-Class с детектором PDA или полупрепаративной ВЭЖХ Agilent 1200.
Промежуточные соединения
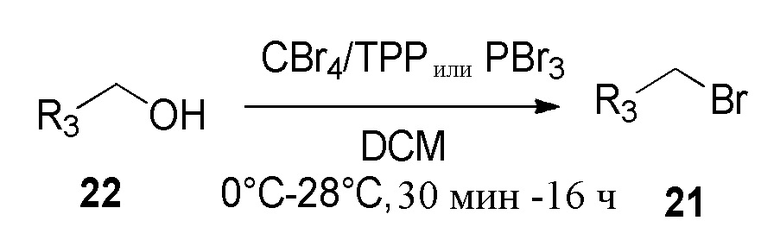
Синтез промежуточных соединений R3-Y
Y представляет собой уходящую группу, такую как мезилат или галогенид, например, атом хлора или брома.
Спиртовую функциональную группу формулы 22 преобразовали в соответствующий бромид 21 обработкой трибромидом фосфора (PBr3) в дихлорметане или трифенилфосфином (Ph3P) и тетрабромидом углерода (CBr4) в дихлорметане при температуре от 0°С до 28°C.
Промежуточное соединение 1: 4-(бромметил)пиримидин:
К перемешиваемому раствору пиримидин-4-илметанола (500 мг, 4,5454 ммоль, коммерческий источник: Manchestar) в дихлорметане (15 мл) добавляли тетрабромид углерода (2,26 г, 6,8181 ммоль) и трифенилфосфин (1,786 г, 6,8181 ммоль) при 0°C. Реакционную смесь перемешивали при 26°С в течение 2 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении при 35°С и неочищенное соединение очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 13% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-(бромметил)пиримидина (270 мг, 34%) в виде красной жидкости, которую охарактеризовывали 1H-ЯМР. Данное сырое вещество использовали без какой-либо очистки в следующей реакции. 1H ЯМР (400 МГц, CDCl3) δ 9,19 (д, J=1,4 Гц, 1H), 8,76 (д, J=5,2 Гц, 1H), 7,49 (дд, J=5,2, 1,4 Гц, 1H), 4,45 (с, 2H).
Промежуточное соединение 2: 2-(бромметил)-5-метилпиридин:
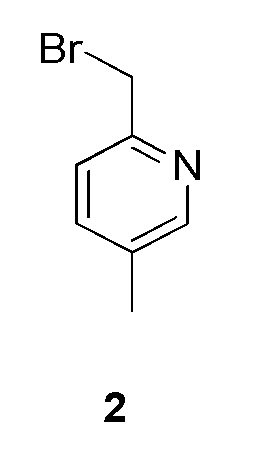
К раствору (5-метилпиридин-2-ил)метанола (150 мг, 0,0012 моль, коммерческий источник: Combi-Blocks) в дихлорметане (1,5 мл) добавляли трифенилфосфин (629 мг, 0,0024 моль) при 28°С. Реакционную смесь перемешивали в течение 15 мин при 0°С с последующим добавлением тетрабромида углерода (397 мг, 0,0012 моль). Реакционную смесь перемешивали при 28°С в течение 3 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении с получением 2-(бромметил)-5-метилпиридина (350 мг, неочищенное соединение) в виде бледно-желтого липкого твердого вещества. МС m/z (М, М+2) = 186,1, 188,1. Соединение использовали на следующей стадии без какой-либо дополнительной очистки.
Промежуточное соединение 3: 4-(бромметил)-1,1-дифторциклогексан:
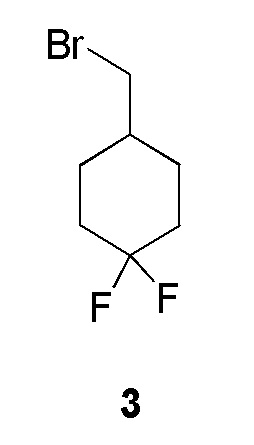
К раствору (4,4-дифторциклогексил)метанола (1 г, 0,0066 моль, коммерческий источник: J & W Pharma) в дихлорметане (5 мл) добавляли трифенилфосфин (3,4 г, 0,0133 моль) при 26°С с последующим добавлением по каплям раствора тетрабромида углерода (2,1 г, 0,0066 моль) в дихлорметане (5 мл) при 0°С. Реакционную смесь перемешивали при 26°С в течение 16 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении при температуре 26°С. Сырой продукт перемешивали с н-пентаном (100 мл) при 26°С в течение 10 мин. Слой н-пентана отделяли и концентрировали при пониженном давлении с получением 4-(бромметил)-1,1-дифторциклогексана (1,7 г) в виде бесцветной жидкости, которую охарактеризовывали 1H-ЯМР. Данное сырое вещество использовали без какой-либо очистки в следующей реакции. 1H ЯМР (400 МГц, CDCl3) δ 3,31 (д, J=6,4 Гц, 2H), 2,21-2,06 (м, 2H), 2,00-1,90 (м, 2H), 1,83-1,63 (м, 3H), 1,47- 1,32 (м, 2Н).
Промежуточное соединение 4: 2-(бромметил)-5-метоксипиридин гидробромид:
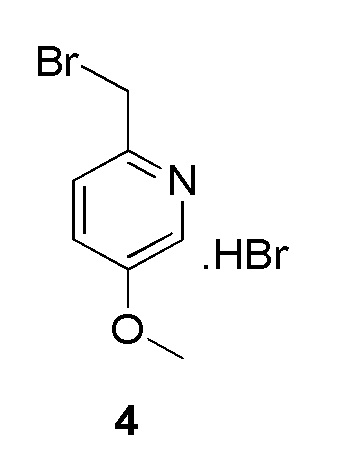
К раствору (5-метоксипиридин-2-ил)метанола (200 мг, 0,00143 моль, коммерческий источник: Combi-Blocks) в дихлорметане (2 мл) добавляли трибромид фосфора (0,4 мл) при 0°С. Реакционную смесь перемешивали при 28°С в течение 2 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении с получением 2-(бромметил)-5-метоксипиридина гидробромида (300 мг) в виде коричневого твердого вещества. МС m/z [М+Н]+ & [М+2Н]+ = 202,00 и 204,0. Соединение использовали на следующей стадии без какой-либо дополнительной очистки.
Промежуточное соединение 5: 2-(бромметил)-5-фторпиридин гидробромид:
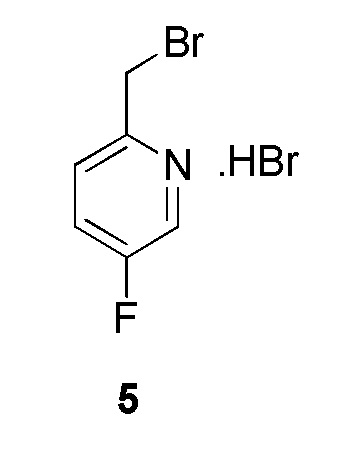
К перемешиваемому раствору (5-фторпиридин-2-ил)метанола (600 мг, 4,7244 ммоль, коммерческий источник: Combi-Blocks) в дихлорметане (15 мл) добавляли трибромид фосфора (1,5 мл) при 0°С. Реакционную смесь оставляли перемешиваться при 26°С в течение 3 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении с получением 2-(бромметил)-5-фторпиридина гидробромида (1,7 г) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,76-8,62 (м, 1Н), 8,12-7,94 (м, 1Н), 7,76-7,64 (м, 1Н), 4,66 (с, 2Н). МС m/z [М+Н]+ = 190,09. Соединение использовали на следующей стадии без какой-либо дополнительной очистки.
Реакционная схема синтеза промежуточного соединения 8
Этерификация соединения 23 с йодистым метилом (MeI) и карбонатом калия (K2CO3) в ДМФА при 26°С дает сложный эфир 24, который восстанавливают борогидридом натрия (NaBH4) в метаноле при температуре в диапазоне от 0°С до 26°С, с получением соответствующего спирта 22.
Промежуточное соединение 6: метил 5-хлорпиколинат:
К раствору 5-хлорпиколиновой кислоты (1 г, 6,34 ммоль, коммерческий источник: Combi-Blocks) в N, N-диметилформамиде (10 мл) добавляли карбонат калия (1,75 г, 12,6 ммоль) с последующим добавлением йодистого метила (0,8 мл, 12,6 ммоль) при 26°С. Реакционную смесь перемешивали в течение 12 ч при той же температуре. По окончании реакции реакционную смесь гасили ледяной водой (100 мл) и экстрагировали этилацетатом (200 мл). Органический слой промывали водой (2×100 мл) и насыщенным раствором соли (100 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением метил 5-хлорпиколината (1,2 г) в виде коричневой жидкости. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,77 (дд, J=2,4, 0,7 Гц, 1H), 8,14 (дд, J=8,4, 2,4 Гц, 1H), 8,07 (дд, J=8,4, 0,7 Гц, 1H), 3,89 (с, 3H). МС m/z [М+Н]+ = 172,25. Соединение использовали на следующей стадии без какой-либо дополнительной очистки.
Промежуточное соединение 7: (5-хлорпиридин-2-ил)метанол:
К раствору метил 5-хлорпиколината (1,2 г, 0,007 моль, промежуточное соединение 6) в метаноле (12 мл) добавляли порциями борогидрид натрия (0,53 г, 0,014 моль) при 0°С. Реакционную смесь перемешивали при 26°С в течение 13 ч. По окончании реакции реакционную смесь охлаждали до 0°С и гасили ледяной водой (5 мл). Реакционную смесь перемешивали в течение 30 мин при 26°С и экстрагировали этилацетатом (4×20 мл). Органический слой промывали водой (2×20 мл), высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением (5-хлорпиридин-2-ил)метанола (500 мг, 48,5%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,53 (д, J=2,3 Гц, 1H), 7,67 (дд, J=8,3, 2,4 Гц, 1H), 7,26-7,21 (м, 1H), 4,75 (с, 2H) 3,26 (с, 1Н). МС m/z [М+Н]+ = 144,1.
Промежуточное соединение 8: 2-(бромметил)-5-хлорпиридин:
К раствору (5-хлорпиридин-2-ил)метанола (500 мг, 0,003 моль, промежуточное соединение 7) в дихлорметане (4 мл) добавляли трифенилфосфин (1,57 г, 0,006 моль) при 26°С с последующим добавлением раствора тетрабромида углерода (1,15 г, 0,003 моль) в дихлорметане (1 мл) при 0°С. Реакционную смесь перемешивали при 26°С в течение 16 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 5% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении с получением 2-(бромметил)-5-хлорпиридина (200 мг, 30%) в виде бледно-желтой жидкости. 1H ЯМР (400 МГц, CDCl3) δ 8,53 (д, J=2,5 Гц, 1H), 7,67 (дд, J=8,3, 2,5 Гц, 1H), 7,40 (д, J=8,3 Гц, 1H), 4,52 (с, 2H). МС m/z [М] = 206,9.
Общая схема синтеза хлоридов промежуточных соединений 9, 10 и 11
Спиртовое производное формулы 22 превращают в соответствующие хлориды 25 с использованием тионилхлорида в дихлорметане при температуре от 0°С до 28°С.
Промежуточное соединение 9: 2-(хлорметил)-5-фторпиридин:
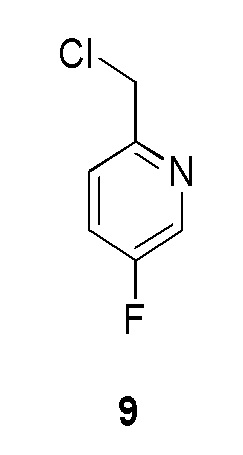
К раствору (5-фторпиридин-2-ил)метанола (10 г, 0,0787 моль, коммерческий источник: Combi-Blocks) в дихлорметане (200 мл) медленно добавляли тионилхлорид (18,72 г, 0,1574 моль, коммерческий источник: Avra) при 0°С. Реакционной смеси давали нагреться до 28°С и перемешивали в течение 2 ч. По окончании реакции реакционную смесь охлаждали до 0°С, гасили насыщенным раствором хлорида аммония (200 мл) и экстрагировали дихлорметаном (5-100 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 2-(хлорметил)-5-фторпиридина (7 г, 47%) в виде желтой жидкости, которую охарактеризовывали 1H-ЯМР. Данное сырое вещество использовали без какой-либо очистки в следующей реакции. 1H ЯМР (400 МГц, CDCl3) δ 8,43 (д, J=2,8 Гц, 1H), 7,53-7,40 (м, 2H), 4,67 (с, 2H).
Промежуточное соединение 9': 2-(хлорметил)-5-фторпиридин гидрохлорид:
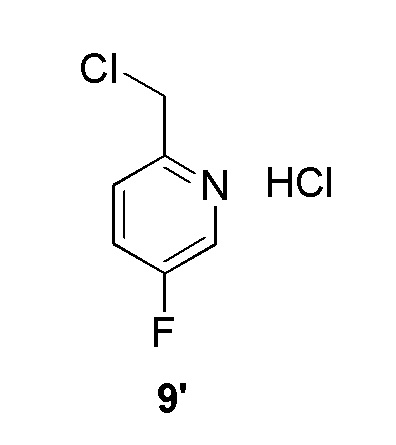
Тионилхлорид (0,419 мл, 5,74 ммоль, коммерческий источник: Aldrich) добавляли по каплям к перемешиваемому раствору (5-фторпиридин-2-ил)метанола (0,289 мл, 2,87 ммоль, коммерческий источник: Combi-Blocks) в дихлорметане (5,74 мл) в атмосфере азота при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 16 ч. Смесь концентрировали при пониженном давлении с получением 2-(хлорметил)-5-фторпиридина гидрохлорида (395 мг, 2,170 ммоль, 76%), который использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,68 (шир. с, 1H), 8,55 (д, J=3,0 Гц, 1H), 7,77 (дт, J=8,7, 3,0 Гц, 1H), 7,63 (дд, J=8,6, 4,5 Гц, 1H), 4,78 (с, 2H). МС m/z [М+Н]+ = 146,10.
Промежуточное соединение 10: 4-(хлорметил)-2-метилпиридин:
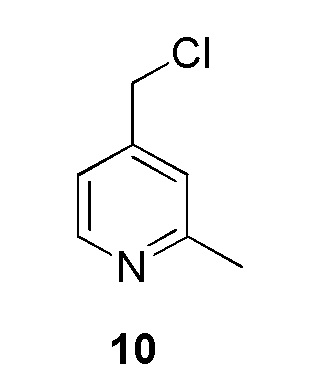
К раствору (2-метилпиридин-4-ил)метанола (500 мг, 4,06 ммоль, коммерческий источник: Combi-Blocks) в дихлорметане (20 мл) добавляли тионилхлорид (0,45 мл, 1,5 ммоль) при 0°С. Реакционной смеси давали нагреться до 27°С и перемешивали в течение 16 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Остаток нейтрализовали (рН 7) насыщенным раствором бикарбоната натрия (10 мл) и экстрагировали этилацетатом (2×50 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении с получением 4-(хлорметил)-2-метилпиридина (400 мг) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,49 (д, J=5,1 Гц, 1H), 7,18 (с, 1H), 7,11 (дд, J=5,2, 1,6 Гц, 1H), 4,50 (с, 2H), 2,57 (с, 3Н). МС m/z [М+Н]+ = 142,0.
Промежуточное соединение 11: 2-(хлорметил)-5-метилпиридин:
К раствору (5-метилпиридин-2-ил)метанола (500 мг, 4,06 ммоль, коммерческий источник: Pharma Blocks) в дихлорметане (20 мл) добавляли тионилхлорид (0,45 мл, 6,09 ммоль) при 0°С. Реакционную смесь перемешивали при 27°С в течение 16 ч. По окончании реакции реакционную смесь разбавляли водой (10 мл) и нейтрализовали насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали этилацетатом (2×50 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 2-(хлорметил)-5-метилпиридина (400 мг, 69%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,41 (с, 1H), 7,52 (д, J=7,5 Гц, 1H), 7,36 (д, J=7,9 Гц, 1H), 4,65 (с, 2H), 2,35 (с, 3Н). МС m/z [М+ Н]+ = 142,03.
Общая схема синтеза промежуточных соединений 12, 13, 14 и 15
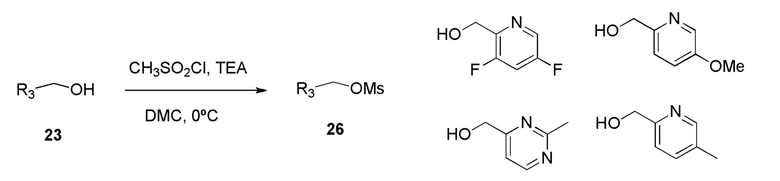
Ароматический спирт 23 формулы R3CH2OH превращают в соответствующий мезилат 26, используя метансульфонилхлорид и триэтиламин в дихлорметане (DCM) при температуре 0°С.
Промежуточное соединение 12: (3,5-дифторпиридин-2-ил)метилметансульфонат:
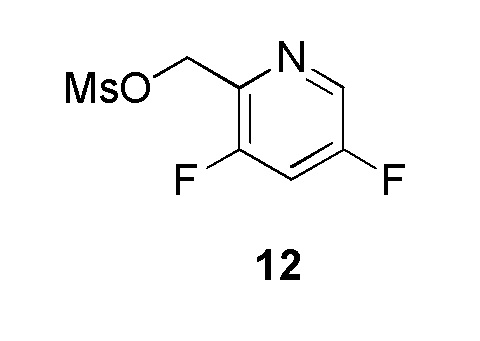
На ледяной бане в колбу загружали дихлорметан (1 мл), (3,5-дифторпиридин-2-ил)метанол (200 мг, 1,387 ммоль, коммерческий источник: Rennothech-China), триэтиламин (229 мкл, 1,654 ммоль) и в конце метансульфонилхлорид (118 мкл, 1,516 ммоль), который добавляли по каплям. Сырой продукт оставляли в тех же условиях на 1,5 ч. По окончании реакции смесь распределяли между водой и DCM и экстрагировали DCM (2×). Органические слои высушивали над MgSO4 (безводным) и фильтровали. Растворитель выпаривали в вакууме с получением (3,5-дифторпиридин-2-ил)метилметансульфоната (205 мг, 0,918 ммоль, 66,6%), который охарактеризовывали 1Н ЯМР. Данное неочищенное вещество использовали без какой-либо очистки в следующей реакции. 1H ЯМР (400 МГц, CDCl3) δ 8,40 (д, J=2,5 Гц, 1H), 7,32-7,27 (м, 1H), 5,40 (д, J=2,0 Гц, 2H) 3,11 (с, 3Н).
Промежуточное соединение 13: (2-метилпиримидин-4-ил)метилметансульфонат:
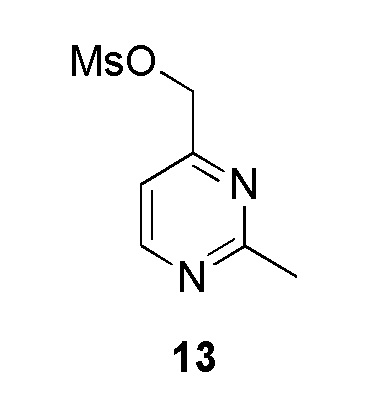
На ледяной бане в колбу загружали дихлорметан (1 мл), (2-метилпиримидин-4-ил)метанол (200 мг, 1,611 ммоль, коммерческий источник: Chembridge-USA), триэтиламин (268 мкл, 1,933 ммоль) и в конце метансульфонилхлорид (138 мкл, 1,772 ммоль), который добавляли по каплям. Неочищенное вещество оставляли в тех же условиях на 1,5 ч. По окончании реакции реакционную смесь распределяли между водой и DCM и экстрагировали DCM (2×). Органические слои высушивали над MgSO4 (безводным) и фильтровали. Растворитель выпаривали в вакууме с получением (2-метилпиримидин-4-ил)метилметансульфоната (245 мг, 1,212 ммоль, 75%), который охарактеризовывали 1Н ЯМР и ЖХМС. Данное сырое вещество использовали без какой-либо очистки в следующей реакции. 1H ЯМР (400 МГц, CDCl3) δ 8,71 (д, J=5,1 Гц, 1H), 7,31 (д, J=5,1 Гц, 1H), 5,25 (с, 2H), 2,74 (с, 3H). МС m/z [М+Н]+ = 203,21.
Промежуточное соединение 14: (5-метоксипиридин-2-ил)метилметансульфонат:
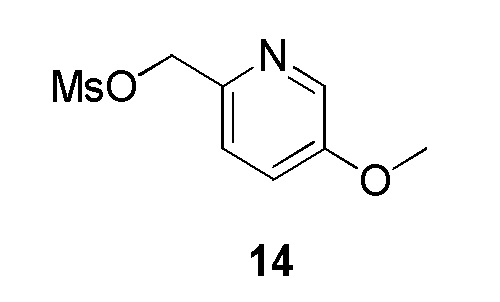
На ледяной бане в колбу загружали дихлорметан (1 мл), (5-метоксипиридин-2-ил)метанол (200 мг, 1,437 ммоль, коммерческий источник: ARK PHARM-USA AK), триэтиламин (239 мкл, 1,725 ммоль) и в конце метансульфонилхлорид (123 мкл, 1,581 ммоль), который добавляли по каплям. Неочищенное вещество оставляли в тех же условиях на 1,5 ч. По окончании реакции реакционную смесь распределяли между водой и DCM и экстрагировали DCM (2×). Органические слои высушивали над MgSO4 (безводным) и фильтровали. Растворитель выпаривали в вакууме с получением (5-метоксипиридин-2-ил)метилметансульфоната (312 мг, 1,437 ммоль), который охарактеризовывали ЖХМС. Данное сырое вещество использовали без какой-либо очистки в следующей реакции. МС m/z [М+Н]+ = 218,10.
Промежуточное соединение 15: (5-метилпиридин-2-ил)метилметансульфонат:
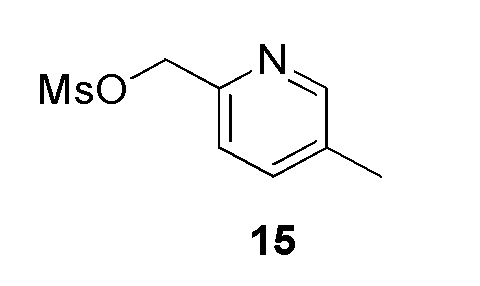
На ледяной бане в колбу загружали дихлорметан (1 мл), триэтиламин (270 мкл, 1,949 ммоль), (5-метилпиридин-2-ил)метанол (211 мкл, 1,624 ммоль, коммерческий источник: Asta Tech) и в конце метансульфонилхлорид (139 мкл, 1,786 ммоль), который добавляли по каплям. Неочищенное вещество оставляли в тех же условиях на 1,5 ч. По окончании реакции реакционную смесь распределяли между водой и DCM и экстрагировали DCM (2×). Органические слои высушивали над MgSO4 (безводным) и фильтровали. Растворитель выпаривали в вакууме с получением (5-метилпиридин-2-ил)метилметансульфоната (327 мг, 1,624 ммоль), который охарактеризовывали ЖХМС. Данное сырое вещество использовали без какой-либо очистки в следующей реакции. МС m/z [М+Н]+ = 202,10.
Промежуточное соединение 16: 4-циано-N-(2-гидроксиэтил)бензолсульфонамид:
К раствору 4-цианобензол-1-сульфонилхлорида (50 г, 0,2487 моль, коммерческий источник: Combi-Blocks), 2-аминоэтанола (18,2 г, 0,2985 моль, коммерческий источник: Combi-Blocks) в тетрагидрофуране (500 мл) добавляли триэтиламин (64 мл, 0,4974 моль, коммерческий источник: Alfa) при 0°С. Температуру повышали до 28°С и реакционную смесь перемешивали в течение 2 ч при той же температуре. По окончании реакции реакционную смесь растворяли в этилацетате (1000 мл) и промывали водой (3×300 мл). Объединенные органические слои высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 40% этилацетата в гексане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-циано-N-(2-гидроксиэтил)бензолсульфонамида (30 г, 53%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,07 (д, J=8,3 Гц, 2H), 7,94 (д, J=8,3 Гц, 2H), 7,93 (с, 1H), 4,69 (т, J=5,6 Гц), 1H), 3,39-3,32 (м, 2H), 2,86-2,79 (д, J=5,0 Гц, 2H). МС m/z [М-Н]- = 225,26.
Промежуточное соединение 17: N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил)бензолсульфонамид:
К раствору 4-циано-N-(2-гидроксиэтил)бензолсульфонамида (10 г, 0,0442 моль, промежуточное соединение 16) в N, N-диметилформамиде (1000 мл) добавляли азид натрия (28,7 г, 0,442 моль) и хлорид аммония (23,6 г, 0,442 моль) при 28°С. Реакционную смесь нагревали до 100°С и перемешивали в течение 3 ч при той же температуре. По окончании реакции реакционную смесь охлаждали до 0°С, гасили 1 Н HCl (500 мл) и перемешивали в течение 2 ч при 0°С. Осажденное твердое вещество отфильтровывали и высушивали в вакууме с получением N-(2-гидроксиэтил)-4-(2Н-тетразол-5-ил)бензолсульфонамида (6 г, 49%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,87 (шир. с, 1Н), 8,37-8,28 (м, 2Н), 8,02-7,97 (м, 2Н), 7,88-7,82 (м, 1Н), 3,38 (т, J=6,2 Гц, 2H), 2,90-2,80 (м, 2H). МС m/z [М+Н]+ = 270,1.
Промежуточное соединение 17’: N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил)бензолсульфонамид:
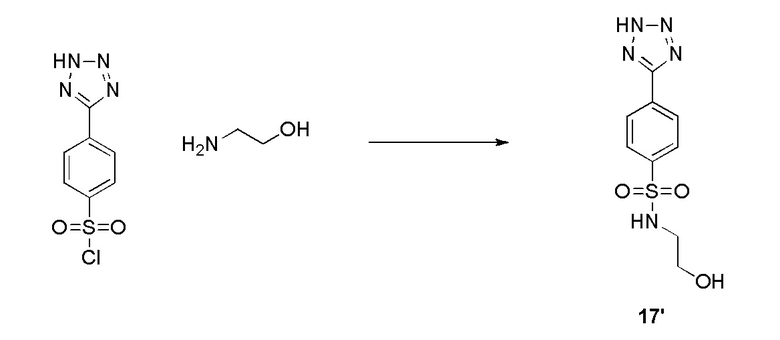
4-(2H-Тетразол-5-ил)бензолсульфонилхлорид (2,5 г, 10,22 ммоль, коммерческий источник: Achemblock-USA), растворенный в тетрагидрофуране (25,5 мл), добавляли по каплям к перемешиваемому раствору 2-аминоэтанола (1,248 г, 20,44 ммоль, коммерческий источник: Aldrich) в тетрагидрофуране (25,5 мл) при 0°С в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь гасили 1 Н HCl и экстрагировали DCM. Органический слой высушивали над Na2SO4 и концентрировали при пониженном давлении с получением N-(2-гидроксиэтил)-4-(1Н-тетразол-5-ил)бензолсульфонамида (2 г, 7,43 ммоль, 72,7%), который использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,26-8,20 (м, 2H), 8,02-7,97 (м, 2H), 7,80 (т, J=5,8 Гц, 1H), 4,63 (шир. с, 1H), 3,37 (т, J=6,2 Гц, 2H), 2,84 (кв, J=6,1 Гц, 2H). МС m/z [М+Н]+ = 270,18.
Промежуточное соединение 18: (R)-N-(2-гидроксипропил)-4- (2Н-тетразол-5-ил)бензолсульфонамид:
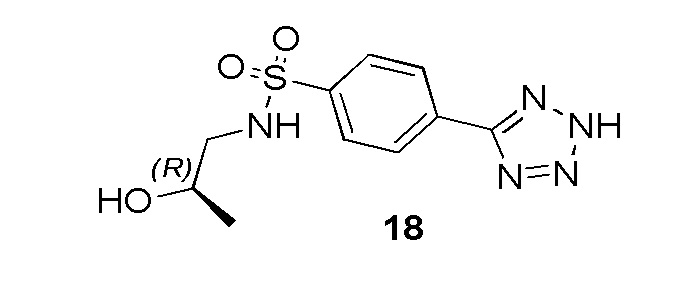
4-(2H-Тетразол-5-ил)бензолсульфонилхлорид (802 мг, 3,28 ммоль, коммерческий источник: Achemblock-USA), растворенный в тетрагидрофуране (0,7 мл), добавляли по каплям к перемешиваемому раствору (R)-1-аминопропан-2-ола (516 мкл, 6,56 ммоль, коммерческий источник: Aldrich) в тетрагидрофуране (0,7 мл) при комнатной температуре в атмосфере азота. Смесь перемешивали при комнатной температуре в течение ночи. После окончания реакции к реакционной смеси добавляли 5 Н HCl (7 мл) и экстрагировали 20 мл смеси DCM/MeOH (10%) (×4). Объединенные органические слои высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением белого твердого вещества, (R)-N-(2-гидроксипропил)-4-(2H-тетразол-5-ил)бензолсульфонамида (553 мг, 1,952 ммоль, 59,5%), который использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,24 (д, J=8,6 Гц, 2H), 8,01 (д, J=8,6 Гц, 2H), 7,79 (т, J=6,2 Гц, 1H), 3,62-3,57 (м, 1H), 2,76-2,66 (м, 2H), 1,00 (д, J=6,1 Гц, 3H). МС m/z [М+Н]+ = 284,17.
Промежуточное соединение 18’: (S)-N-(2-гидроксипропил)-4-(2H-тетразол-5-ил)бензолсульфонамид:
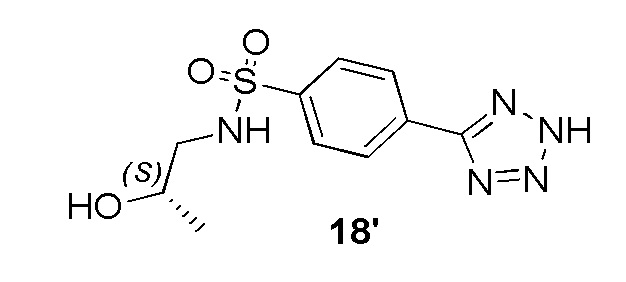
4-(2H-Тетразол-5-ил)бензолсульфонилхлорид (150 мг, 0,613 ммоль, коммерческий источник: Achemblock-USA), растворенный в тетрагидрофуране (1,2 мл), добавляли по каплям к перемешиваемому раствору (S)-1-аминопропан-2-ола (62,8 мкл, 0,797 ммоль, коммерческий источник: Aldrich) в тетрагидрофуране (1,2 мл) при комнатной температуре в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 1,5 ч. По окончании реакции к реакционной смеси добавляли 2 Н HCl и экстрагировали смесью DCM/ MeOH (10%). Объединенные органические слои высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением белого твердого вещества, (S)-N-(2-гидроксипропил)-4- (2H-тетразол-5-ил)бензолсульфонамида (110 мг, 0,388 ммоль, 63%), который использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,24 (д, J=8,6 Гц, 2H), 8,01 (д, J=8,3 Гц, 2H), 7,77 (т, J=6,2 Гц, 1H), 3,64-3,56 (м, 1H), 2,76-2,64 (м, 2H), 1,00 (д, J=6,1 Гц, 3H). МС m/z [М+Н]+ = 284,2.
Промежуточное соединение 19: N-(1,3-дигидроксипропан-2-ил)-4-(2Н-тетразол-5-ил)бензолсульфонамид:
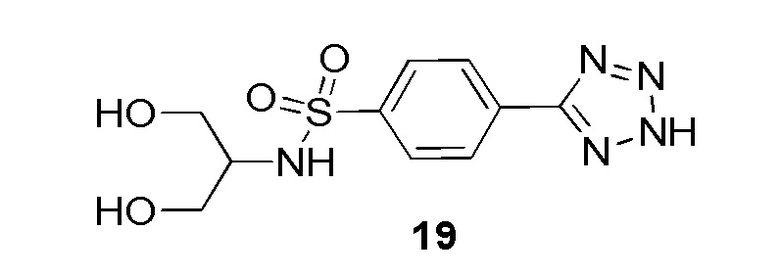
4-(2H-Тетразол-5-ил)бензолсульфонилхлорид (0,400 г, 1,635 ммоль, коммерческий источник: Achemblock-USA), растворенный в тетрагидрофуране (5,5 мл), добавляли по каплям к перемешиваемому раствору 2-аминопропана-1,3-диола (0,298 г, 3,27 ммоль, коммерческий источник: Aldrich) в тетрагидрофуране (5,5 мл) при 0°С в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь гасили 1 Н HCl и экстрагировали DCM. Органический слой высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением N-(1,3-дигидроксипропан-2-ил)-4-(2H-тетразол-5-ил)бензолсульфонамида (0,268 г, 0,716 ммоль, 43,8%). Продукт использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,27-8,16 (м, 2H), 8,08-8,02 (м, 2H), 8,02-7,98 (м, 1H), 7,82-7,77 (м, 1H), 7,68 (д J=7,8 Гц, 1Н), 3,44-3,23 (м, 5Н), 3,18-3,03 (м, 1Н).
Промежуточное соединение 20: N-(2,3-дигидроксипропил)-4- (2H-тетразол-5-ил)бензолсульфонамид:
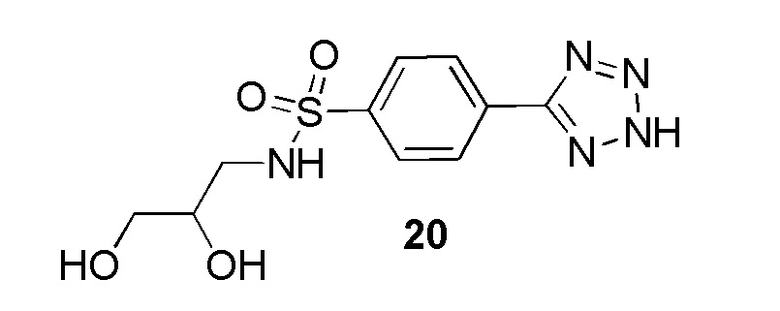
4-(2H-Тетразол-5-ил)бензолсульфонилхлорид (0,100 г, 0,409 ммоль, коммерческий источник: Achemblock-USA), растворенный в тетрагидрофуране (1,6 мл), добавляли по каплям к перемешиваемому раствору 3-аминопропан-1,2-диола (0,158 мл, 2,044 ммоль, коммерческий источник: Aldrich) в тетрагидрофуране (1,6 мл) при 0°С в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь гасили 1 Н HCl и экстрагировали DCM. Органический слой высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением N-(2,3-дигидроксипропил)-4-(2Н-тетразол-5-ил)бензолсульфонамида (60 мг, 0,200 ммоль, 49%). Продукт использовали на следующей стадии без дополнительной очистки. МС m/z [М+Н]+ = 300,21.
Промежуточное соединение 21: N-метил-4-(2H-тетразол-5-ил)бензолсульфонамид:
К раствору 4-циано-N-метилбензолсульфонамида (500 мг, 2,551 ммоль, коммерческий источник: Combi-Blocks) в N, N-диметилформамиде (15 мл) добавляли азид натрия (1,65 г, 25,51 ммоль) и хлорид аммония (1,36 г, 25,51 ммоль) при 27°С. Реакционную смесь нагревали до 120°С и перемешивали в течение 5 ч. По окончании реакции реакционную смесь охлаждали до 27°С, гасили 1 Н HCl (30 мл) и экстрагировали этилацетатом (3×60 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением N-метил-4-(2H-тетразол-5-ил)бензолсульфонамида (800 мг, неочищенное соединение) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,28-8,20 (м, 2H), 8,03-7,97 (м, 2H), 7,61 (кв, J=4,8 Гц, 1H), 2,47 (д, J=5,0 Гц, 3H). МС m/z [М+Н]+ = 240,08.
Промежуточное соединение 22: 4-циано-N-этилбензолсульфонамид:
К раствору 4-цианобензол-1-сульфонилхлорида (2 г, 9,92 ммоль, коммерческий источник: Aldrich) в дихлорметане (30 мл) добавляли этиламин гидрохлорид (890 мг, 10,91 ммоль, коммерческий источник: Avra) и триэтиламин (2,5 г, 24,75 ммоль) при 27°С. Реакционную смесь перемешивали при 27°С в течение 1 ч. По окончании реакции реакционную смесь выливали в воду (100 мл) и экстрагировали дихлорметаном (3×80 мл). Объединенный органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт промывали смесью дихлорметана (2 мл) и петролейного эфира (20 мл) с получением 4-циано-N-этилбензолсульфонамида (1,5 г, 69%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,03-7,95 (м, 2Н), 7,87-7,76 (м, 2Н), 4,43 (шир. с, 1Н), 3,12-3,03 (м, 2Н), 1,14 (т, J=7,2 Гц, 3H). МС m/z [М-Н]- = 209,05.
Промежуточное соединение 23: N-этил-4-(2H-тетразол-5-ил)бензолсульфонамид:
К раствору 4-циано-N-этилбензолсульфонамида (500 мг, 2,38 ммоль, промежуточное соединение 22) в N, N-диметилформамиде (10 мл) добавляли азид натрия (1,55 г, 23,809 ммоль) и хлорид аммония (1,27 г, 23,809 ммоль) при 27°С. Реакционную смесь нагревали до 120°С и перемешивали в течение 5 ч при той же температуре. По окончании реакции реакционную смесь охлаждали до 27°С, гасили 1 Н HCl (60 мл) и экстрагировали этилацетатом (3×50 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт промывали смесью дихлорметана (2 мл) и н-пентана (10 мл) с получением N-этил-4-(2H-тетразол-5-ил)бензолсульфонамида (700 мг, 97%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,33-8,18 (м, 2Н), 8,01 (д, J=8,5 Гц, 2Н), 7,74 (т, J=5,7 Гц, 1Н), 2,86-2,76 (м, 2H), 1,00 (т, J=7,2 Гц, 3H). МС m/z [М+Н]+ = 254,10.
Промежуточное соединение 24: 2-(4-цианофенилсульфонамидо) ацетамид:
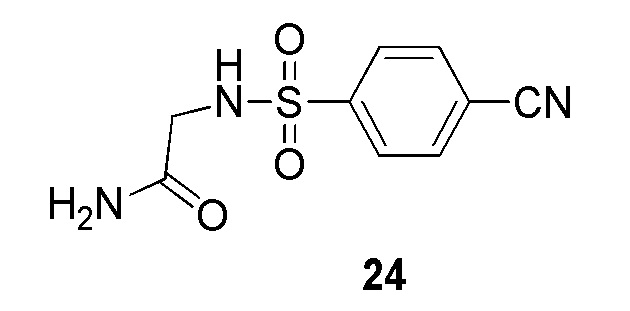
К раствору 4-цианобензол-1-сульфонилхлорида (5 г, 0,0248 моль, коммерческий источник: Combi-Blocks) в тетрагидрофуране (100 мл) добавляли триэтиламин (5 г, 0,0496 моль) при 28°С с последующим добавлением 2-аминоацетамида (1,84 г, 0,0248 моль, коммерческий источник: Combi-Blocks) при 0°С. Реакционную смесь оставляли нагреться до 28°С и перемешивали в течение 2 ч при той же температуре. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в этилацетате (100 мл) и промывали водой (3×50 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 2-(4-цианофенилсульфонамидо)ацетамида (4 г, 67%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,23 (с, 1H), 8,08 (д, J=8,5 Гц, 2H), 8,00-7,92 (м, 2H), 7,32 (с, 1H), 7,08 (с, 1H), 3,46 (с, 2H). МС m/z [М-Н]- = 238,2.
Промежуточное соединение 25: 2-(4-(2H-тетразол-5-ил)фенилсульфонамидо)ацетамид:
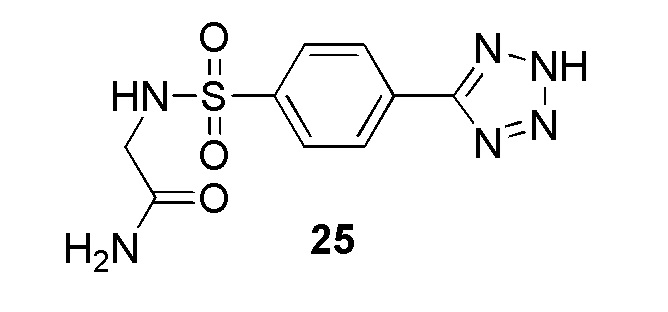
К раствору 2-(4-цианофенилсульфонамидо)ацетамида (4 г, 0,0167 моль, промежуточное соединение 24) в N, N-диметилформамиде (40 мл) добавляли азид натрия (10,8 г, 0,1673 моль) и хлорид аммония (8,9 г, 0,1673 моль) при 28°С. Реакционную смесь нагревали до 100°С и перемешивали в течение 3 ч. По окончании реакции реакционную смесь охлаждали до 28°С и медленно гасили 1 Н HCl (30 мл) при 0°С и перемешивали в течение 30 мин. Осажденное твердое вещество отфильтровывали и высушивали в вакууме с получением 2-(4-(2H-тетразол-5-ил)фенилсульфонамидо)ацетамида (3 г, 51%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,31-8,14 (м, 2Н), 8,13-7,99 (м, 3Н), 7,27 (с, 1Н), 7,06 (с, 1Н), 3,45 (д, J=6,1 Гц, 2Н). МС m/z [М+Н]+ = 283,15.
Промежуточное соединение 26: метил 2-(4-цианофенилсульфонамидо)ацетат:
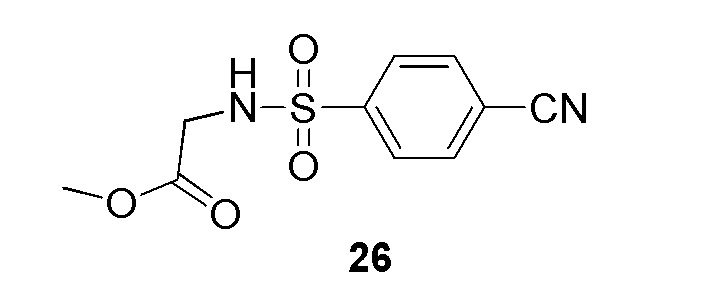
К раствору метил 2-аминоацетата гидрохлорида (310 мг, 0,0024 моль, коммерческий источник: Aldrich) в дихлорметане (5 мл) добавляли триэтиламин (0,64 мл, 0,0048 моль) при 28°С. Затем добавляли порциями 4-цианобензол-1-сульфонилхлорид (500 мг, 0,0024 моль, коммерческий источник: Combi-Blocks) при 0°С. Реакционную смесь перемешивали при 28°С в течение 16 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в воде (50 мл) и экстрагировали этилацетатом (3×50 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении с получением метил 2-(4-цианофенилсульфонамидо)ацетата (500 мг, 76%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,53 (т, J=6,1 Гц, 1H), 8,07 (д, J=8,5 Гц, 2H), 7,99-7,89 (м, 2H), 3,80 (д, J=6,1 Гц, 2H), 3,52 (с, 3H). МС m/z [М-Н]- = 253,05.
Промежуточное соединение 27: метил 2-(4-(2H-тетразол-5-ил)фенилсульфонамидо)ацетат:
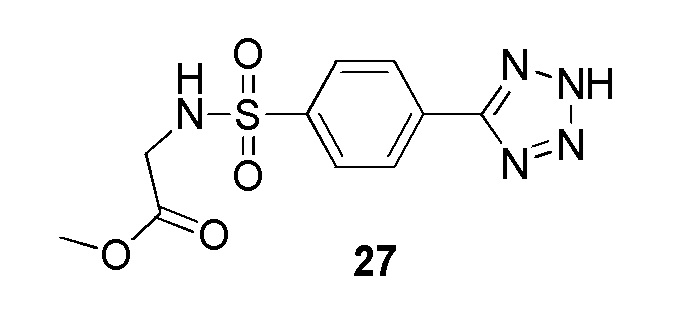
К раствору метил 2-(4-цианофенилсульфонамидо)ацетата (500 мг, 0,0019 моль, промежуточное соединение 26) в N, N-диметилформамиде (5 мл) добавляли азид натрия (1,2 г, 0,0196 моль) и хлорид аммония (1 г, 0,0196 моль) при 28°С. Реакционную смесь нагревали до 100°С и перемешивали в течение 3 ч при той же температуре. По окончании реакции реакционную смесь охлаждали до 0°С и гасили 1 Н HCl (2 мл) при 0°С. Осажденное твердое соединение отфильтровывали и высушивали в вакууме с получением метил 2-(4-(2H-тетразол-5-ил)фенилсульфонамидо)ацетата (400 мг, 57%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,40 (т, J=6,2 Гц, 1H), 8,26-8,19 (м, 2H), 8,00 (дд, J=8,7, 2,3 Гц, 2H), 3,79 (д, J=6,2 Гц, 2H), 3,52 (с, 3H). МС m/z [М-Н]- = 296,06.
Промежуточное соединение 28: 2-(4-(2-(4-фторбензил)-2Н-тетразол-5-ил)фенилсульфонамидо)уксусная кислота:
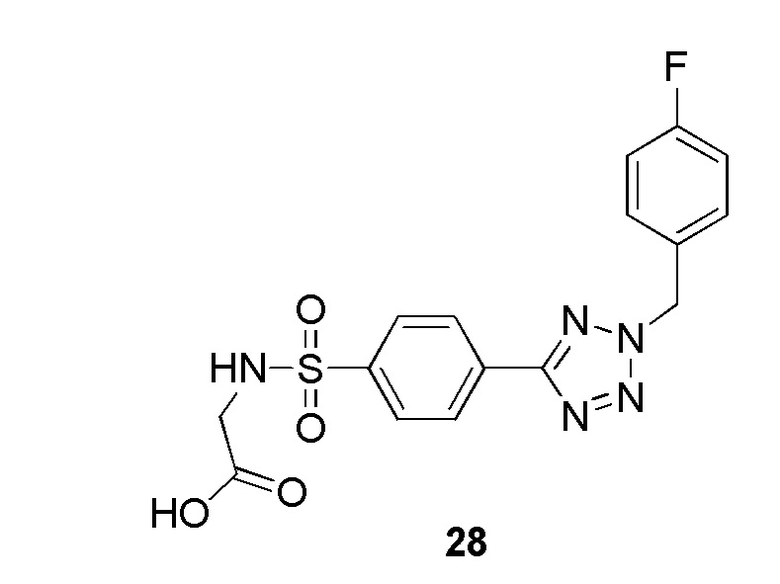
К раствору 2-(4-(2-(4-фторбензил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетата (380 мг, 0,0009 моль) в смеси тетрагидрофурана (3,8 мл) и метанола (3,8 мл) добавляли раствор гидроксида лития моногидрата (78 мг, 0,0013 моль) в воде (1,9 мл) при 28°С. Реакционную смесь перемешивали в течение 3 ч при той же температуре. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Остаток разбавляли водой (30 мл) и подкисляли (pH 2) 1 Н HCl (10 мл) при 0°С. Осажденное твердое соединение отфильтровывали и высушивали в вакууме с получением 2-(4-(2-(4-фторбензил)-2Н-тетразол-5-ил)фенилсульфонамидо)уксусной кислоты (220 мг, 59%) в виде белого твердого вещества. МС m/z [М+Н]+ = 392,14.
Промежуточное соединение 29: 2-(4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил)фенилсульфонамидо)уксусная кислота:
К раствору 2-(4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетата (416 мг, 0,001 моль) в смеси тетрагидрофурана (4,1 мл) и метанола (4,1 мл) добавляли раствор гидроксида лития моногидрата (89 мг, 0,0021 моль) в воде (2 мл) при 28°С. Реакционную смесь перемешивали при той же температуре в течение 2 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Остаток разбавляли водой (20 мл) и нейтрализовали (рН 7-8) 1 Н HCl (5 мл). Осажденное твердое вещество отфильтровывали и высушивали в вакууме с получением 2-(4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)фенилсульфонамидо)уксусной кислоты (245 мг, 65%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 12,84-12,56 (шир. с, 1H), 8,61 (д, J=5,7 Гц, 2H), 8,24 (д, J=8,3 Гц, 3H), 7,97 (д, J=8,5 Гц, 2H), 7,35 (д, J=5,9 Гц, 2H), 6,15 (с, 2H), 3,64 (д, J=5,0 Гц, 2H). МС m/z [М+Н]+ = 375,06.
Промежуточное соединение 30: 4-бром-2-метоксибензол-1-сульфонилхлорид:
К хлорсульфоновой кислоте (54 мл, коммерческий источник: Avra) добавляли по каплям 1-бром-3-метоксибензол (50 г, 0,568 моль, коммерческий источник: Avra), поддерживая температуру реакционной смеси при -5°С. Реакционную смесь перемешивали при 0°С в течение 3 ч. По окончании реакции реакционную смесь медленно выливали на лед (500 г) и экстрагировали этилацетатом (5×1 л). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 3% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-бром-2-метоксибензол-1-сульфонилхлорида (1,6 г, неочищенное соединение) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,82 (д, J=8,3 Гц, 1H), 7,28 (д, J=2,2 Гц, 1H), 7,24-7,26 (м, 1H), 4,07 (с, 3H).
Промежуточное соединение 31: 4-бром-N-(2-гидроксиэтил)-2-метоксибензолсульфонамид:
К раствору 4-бром-2-метоксибензол-1-сульфонилхлорида (1,6 г, 0,00565 моль, промежуточное соединение 30) в дихлорметане (20 мл) добавляли триэтиламин (1,1 г, 0,0113 моль) при 28°С и перемешивали в течение 10 мин. Затем добавляли 2-аминоэтанол (413 мг, 0,0067 моль, коммерческий источник: Alfa Aesar) при 28°С и перемешивали в течение 2 ч при той же температуре. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Остаток промывали н-пентаном (5×50 мл) и твердое соединение высушивали в вакууме с получением 4-бром-N-(2-гидроксиэтил)-2-метоксибензолсульфонамида (1,2 г, 55%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,65-7,60 (м, 1H), 7,44 (д, J=1,8 Гц, 1H), 7,30-7,26 (м, 1H), 7,24-7,17 (м, 1H), 4,67 (т, J=5,6 Гц, 1H), 3,92 (с, 3 H), 3,35-3,30 (м, 2 H), 2,86-2,76 (м, 2H). МС m/z [М+2Н]+ = 310,02.
Промежуточное соединение 32: 4-циано-N-(2-гидроксиэтил)-2-метоксибензолсульфонамид:
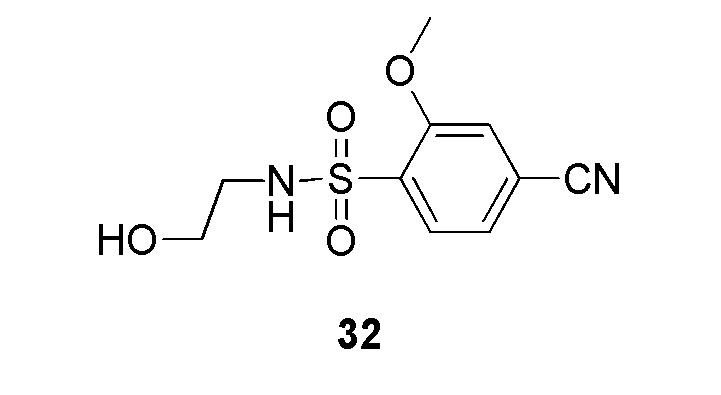
К продуваемому аргоном раствору 4-бром-N-(2-гидроксиэтил)-2-метоксибензолсульфонамида (1 г, 0,0032 моль, промежуточное соединение 31) и цианида цинка (2,4 г, 0,021 моль) в N, N-диметилформамиде (10 мл) добавляли тетракис(трифенилфосфин)палладий (0) (1,8 г, 0,0016 моль) при 28°С. Реакционную смесь перемешивали при 175°С в течение 30 мин в микроволновом реакторе. По окончании реакции реакционную смесь охлаждали до 28°С и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 60% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-циано-N-(2-гидроксиэтил)-2-метоксибензолсульфонамида (450 мг, 49%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,86 (д, J=8,1 Гц, 1H), 7,74 (д, J=1,3 Гц, 1H), 7,54 (дд, J=8, 1,4 Гц, 1H), 7,40 (т, J=5,8 Гц, 1H), 4,60 (т, J=5,6 Гц, 1H), 3,95 (с, 3H), 3,37-3,30 (м, 2H), 2,90-2,81 (м, 2H). МС m/z [М-Н]- = 255,14.
Промежуточное соединение 33: N-(2-гидроксиэтил)-2-метокси-4-(2H-тетразол-5-ил)бензолсульфонамид
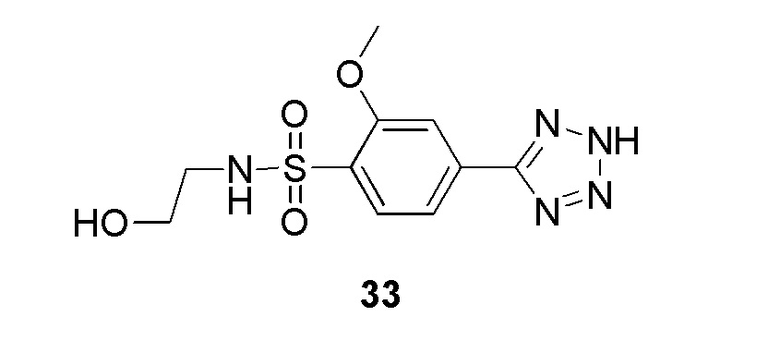
К раствору 4-циано-N-(2-гидроксиэтил)-2-метоксибензолсульфонамида (450 мг, 0,00175 моль, промежуточное соединение 32) в N, N-диметилформамиде (4,5 мл) добавляли азид натрия (1,1 г, 0,0175 моль) и хлорид аммония (0,93 г, 0,0175 моль) при 28°С. Реакционную смесь нагревали до 100°С и перемешивали при той же температуре в течение 3 ч. По окончании реакции реакционную смесь охлаждали до 0°С и гасили 1 М HCl (30 мл) при 0°С. Реакционную смесь концентрировали при пониженном давлении. Сырой продукт перемешивали со смесью 15% метанола в дихлорметане (10 мл) в течение 1 ч при 28°С. Фильтровали и фильтрат упаривали при пониженном давлении с получением N-(2-гидроксиэтил)-2-метокси-4-(2H-тетразол-5-ил)бензолсульфонамида (820 мг, неочищенное соединение) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,04-7,89 (м, 2Н), 7,40 (шир. с, 1Н), 7,32-7,22 (м, 1Н), 7,14 (шир. с, 1Н), 4,02 (с, 3Н), 3,45-3,37 (м, 2Н), 2,91-2,82 (м, 2Н). МС m/z [М+Н]+ = 300,19.
Реакционная схема синтеза промежуточных соединений 34, 35 и примера 51
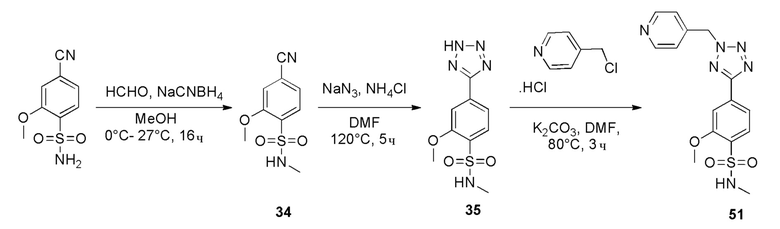
Промежуточное соединение 34: 4-циано-2-метокси-N-метилбензолсульфонамид:
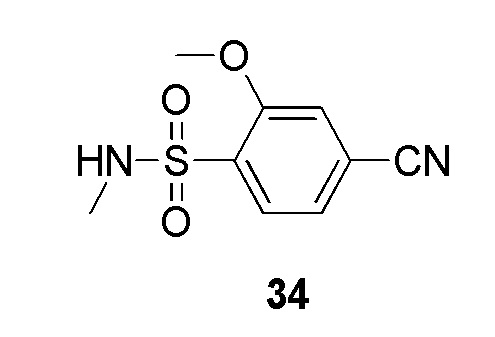
pH раствора 4-циано-2-метоксибензолсульфонамида (500 мг, 2,356 ммоль, коммерческий источник: Enamine) в метаноле (15 мл) доводили до 4 с помощью 1 Н HCl (0,2 мл). Затем добавляли формальдегид (30% водный раствор) (2,5 мл, коммерческий источник: Chemlabs) при 0°С с последующим добавлением цианоборогидрида натрия (296 мг, 4,71 ммоль) при 0°С. Температуру реакционной смеси медленно повышали до 27°С и перемешивали в течение 16 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Остаток подщелачивали раствором аммиака (30% водный раствор) и экстрагировали этилацетатом (3×50 мл). Органический слой промывали насыщенным раствором соли (2×50 мл), высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 50% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-циано-2-метокси-N-метилбензолсульфонамида (400 мг, 75%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,86 (д, J=8,1 Гц, 1H), 7,77 (д, J=1,1 Гц, 1H), 7,56 (дд, J=8,0, 1,2 Гц, 1H), 7,37 (м, 1H), 3,96 (с, 3H), 2,43 (д, J=5,0 Гц, 3H). МС m/z [М+Н]+ = 225,08.
Промежуточное соединение 35: 2-метокси-N-метил-4-(2H-тетразол-5-ил)бензолсульфонамид:
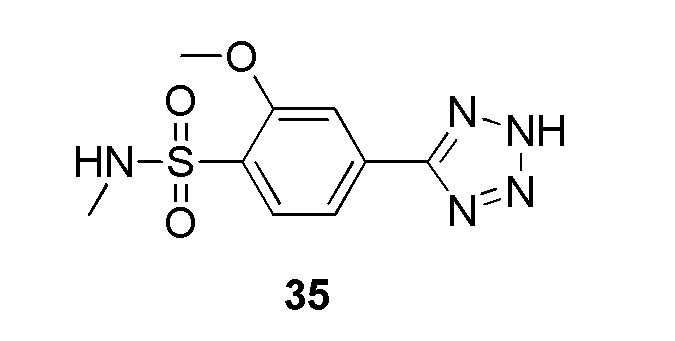
К раствору 4-циано-2-метокси-N-метилбензолсульфонамида (400 мг, 1,769 ммоль, промежуточное соединение 34) в N, N-диметилформамиде (10 мл) добавляли азид натрия (1,15 г, 17,69 ммоль) и хлорид аммония (950 мг, 17,76 ммоль) при 27°С. Реакционную смесь нагревали до 120°С и перемешивали при той же температуре в течение 5 ч. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь охлаждали до 27°С, гасили 1 Н HCl (30 мл) и экстрагировали этилацетатом (2×100 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Остаток перемешивали с дихлорметаном (10 мл) при 27°С в течение 20 мин. Твердое вещество отфильтровывали и высушивали в вакууме с получением 2-метокси-N-метил-4-(2H-тетразол-5-ил)бензолсульфонамида (300 мг, неочищенное соединение) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,93 (д, J=7,9 Гц, 1H), 7,84 (д, J=1,3 Гц, 1H), 7,77 (д, J=7,9 Гц, 1H), 7,25 (м, 1H), 4,01 (с, 3H), 2,44 (д, J=5,0 Гц, 3H). МС m/z [М-Н]- = 268,08.
Промежуточное соединение 36: 4-(2-(4-фторбензил)-2Н-тетразол-5-ил)-2-метоксибензолсульфонамид:
N-Этил-N-изопропилпропан-2-амин (356 мкл, 2,037 ммоль) добавляли к перемешиваемому раствору 2-метокси-4-(2H-тетразол-5-ил)бензолсульфонамида (260 мг, 1,019 ммоль, промежуточное соединение 51) и 1-(бромметил)-4-фторбензола (191 мкл, 1,528 ммоль, коммерческий источник: Aldrich) в N, N-диметилформамиде (ДМФА) (3395 мкл) в атмосфере азота. Раствор перемешивали при комнатной температуре в течение 5 ч. Реакционную смесь разбавляли водой и дважды экстрагировали DCM. Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением масла, которое промывали CH2Cl2, и белое твердое вещество отфильтровывали с получением 4-(2-(4-фторбензил)-2H-тетразол-5-ил)-2-метоксибензолсульфонамида (243 мг, 65,7%). 1H ЯМР (400 МГц, ДМСО-d6) δ 7,90 (д, J=7,8 Гц, 1H), 7,78-7,69 (м, 2H), 7,56-7,46 (м, 2Н), 7,31-7,19 (м, 4Н), 6,04 (с, 2Н), 4,01 (с, 3Н). МС m/z [М+Н]+ = 419,19.
Промежуточное соединение 37: трет-бутил (2-амино-2-оксоэтил) ((4-(2-(4-фторбензил)-2Н-тетразол-5-ил)-2-метоксифенил) сульфонил)карбамат:
К раствору 4-(2-(4-фторбензил)-2Н-тетразол-5-ил)-2-метоксибензолсульфонамида (240 мг, 0,660 ммоль, промежуточное соединение 36) в тетрагидрофуране (2,7 мл) добавляли ди-трет-бутилдикарбонат (0,455 мл, 1,981 ммоль), Et3N (0,110 мл, 0,793 ммоль) и DMAP (40,3 мг, 0,330 ммоль). Полученный белый раствор перемешивали при комнатной температуре в течение 20 мин. Практически сразу реакционная смесь становилась белой и более густой. Затем добавляли карбонат калия (183 мг, 1,321 ммоль) и 2-бромацетамид (193 мг, 1,400 ммоль). Реакционную смесь перемешивали при комнатной температуре. Через 90 мин реакционная смесь становилась желтой и не густела. Смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь гасили насыщенным раствором NaHCO3 и экстрагировали EtOAc (×2). Органический слой промывали насыщенным раствором NaCl, высушивали над безводным Na2SO4 и фильтровали. Растворитель выпаривали при пониженном давлении с получением трет-бутил (2-амино-2-оксоэтил)((4-(2-(4-фторбензил)-2Н-тетразол-5-ил)-2-метоксифенил)сульфонил)карбамата (240 мг, 0,461 ммоль, 69,8%). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,06 (д, J=8,1 Гц, 1H), 7,89-7,74 (м, 2H), 7,57-7,49 (м, 2H), 7,46 (шир. с, 1H), 7,31 -7,21 (м, 2Н), 7,09 (шир. с, 1Н), 6,05 (с, 2Н), 4,32 (с, 2Н), 4,01 (с, 3Н), 1,18 (с, 9Н). МС m/z [М+Н]+ = 521,25.
Промежуточное соединение 38: 4-(2-(4-цианобензил)-2Н-тетразол-5-ил)-2-метоксибензолсульфонамид:
N-этил-N-изопропилпропан-2-амин (233 мкл, 1,332 ммоль) добавляли к перемешиваемому раствору 2-метокси-4-(2H-тетразол-5-ил)бензолсульфонамида (170 мг, 0,666 ммоль, промежуточное соединение 51) и 4-(бромметил)бензонитрила (157 мг, 0,799 ммоль, коммерческий источник: Aldrich) в N, N-диметилформамиде (ДМФА) (2220 мкл) в атмосфере азота. Раствор перемешивали при комнатной температуре в течение 5 ч. Реакционную смесь разбавляли водой и дважды экстрагировали DCM. Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением масла, которое промывали CH2Cl2, с получением белого твердого вещества, которое отфильтровывали. Твердое вещество растирали с МеОН с получением белого твердого вещества, 4-(2-(4-цианобензил)-2Н-тетразол-5-ил)-2-метоксибензолсульфонамида (150 мг, 0,405 ммоль, 60,8%). 1H ЯМР (400 МГц, ДМСО-d6) δ 7,94-7,86 (м, 3H), 7,80-7,70 (м, 2H), 7,59 (д, J=8,1 Гц, 2H), 7,24 (с, 2H), 6,19 (с, 2Н), 4,01 (с, 3Н). МС m/z [М+Н]+ = 371,10.
Промежуточное соединение 39: трет-бутил (2-амино-2-оксоэтил) ((4-(2-(4-цианобензил)-2Н-тетразол-5-ил)-2-метоксифенил) сульфонил)карбамат:
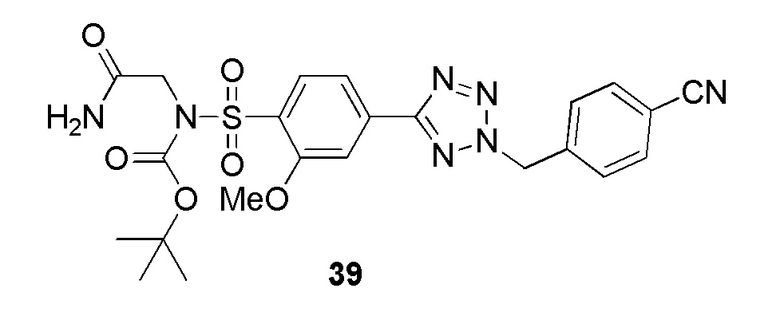
К раствору 4-(2-(4-цианобензил)-2Н-тетразол-5-ил)-2-метоксибензолсульфонамида (150 мг, 0,405 ммоль, промежуточное соединение 38) в тетрагидрофуране (7 мл) добавляли ди-трет-бутилдикарбонат (0,279 мл, 1,215 ммоль), Et3N (0,068 мл, 0,486 ммоль) и DMAP (24,74 мг, 0,202 ммоль). Полученный белый раствор перемешивали при комнатной температуре в течение 20 мин. Практически сразу реакционная смесь становилась белой и более густой. Затем добавляли карбонат калия (112 мг, 0,810 ммоль) и 2-бромацетамид (118 мг, 0,859 ммоль). Реакционную смесь перемешивали при комнатной температуре. Через 90 мин реакционная смесь становилась желтой и не густела. Смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь гасили насыщенным раствором NaHCO3 и экстрагировали EtOAc (×2). Органический слой промывали насыщенным раствором NaCl, высушивали над безводным Na2SO4 и фильтровали. Растворитель выпаривали при пониженном давлении с получением трет-бутил (2-амино-2-оксоэтил)((4-(2-(4-цианобензил)-2Н-тетразол-5-ил)-2-метоксифенил)сульфонил)карбамата (240 мг, 0,364 ммоль, 90%). Продукт использовали на следующей стадии без дополнительной очистки и охарактеризовывали на следующей стадии.
Промежуточное соединение 40: 2-метокси-4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил)бензолсульфонамид:
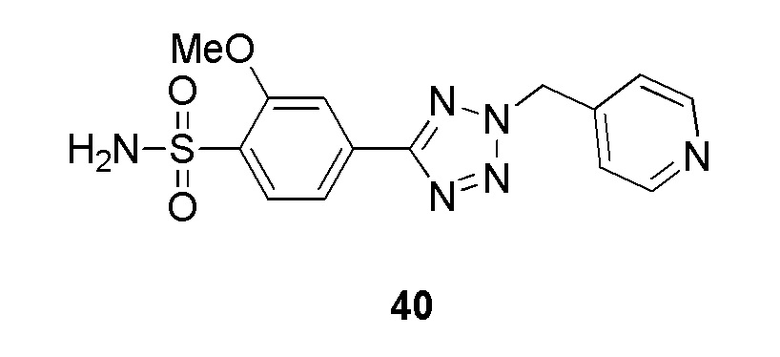
N-этил-N-изопропилпропан-2-амин (349 мкл, 1,998 ммоль) добавляли к перемешиваемому раствору 2-метокси-4-(2H-тетразол-5-ил)бензолсульфонамида (170 мг, 0,666 ммоль, промежуточное соединение 51) и 4-(бромметил)пиридина гидробромида (202 мг, 0,799 ммоль, коммерческий источник: Aldrich) в N, N-диметилформамиде (ДМФА) (2220 мкл) в атмосфере азота. Через 10 мин смесь становилась черной. Раствор перемешивали при комнатной температуре в течение 3 ч. Смесь перемешивали при комнатной температуре в течение выходных дней. Добавляли 4-(бромметил)пиридин гидробромид (238 мг, 0,940 ммоль, коммерческий источник: Aldrich) и N-этил-N-изопропилпропан-2-амин (411 мкл, 2,351 ммоль) и смесь перемешивали при комнатной температуре в течение 2 ч. Присутствовало некоторое количество исходного вещества, поэтому смесь нагревали при 50°C в течение 2 ч. Реакционную смесь разбавляли водой и дважды экстрагировали DCM. Неочищенное соединение очищали колоночной флэш-хроматографией (диоксид кремния; смесь EtOAc-циклогексан от 0/100 до 100/0) в течение 25 мин. Фракции собирали и концентрировали в вакууме с получением 2-метокси-4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил)бензолсульфонамида (150 мг, 0,433 ммоль, 65%) в виде белого твердого вещества. 1H ЯМР (ДМСО-d6) δ 8,65-8,56 (м, 1Н), 7,91 (д, J=8,6 Гц, 1Н), 7,80-7,71 (м, 2Н), 7,38-7,30 (м, 2Н), 7,25 (с, 2H), 6,15 (с, 2H), 4,01 (с, 3H). МС m/z [М+Н]+ = 347,10.
Промежуточное соединение 41: трет-бутил (2-амино-2-оксоэтил) ((2-метокси-4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил)фенил) сульфонил)карбамат:
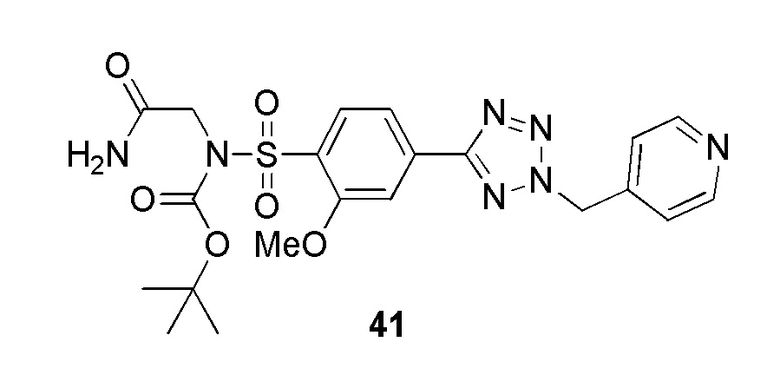
К раствору 2-метокси-4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил)бензолсульфонамида (150 мг, 0,433 ммоль, промежуточное соединение 40) в безводном тетрагидрофуране (10 мл) добавляли ди-трет-бутилдикарбонат (0,298 мл, 1,299 ммоль), Et3N (0,072 мл, 0,520 ммоль) и DMAP (26,5 мг, 0,217 ммоль). Полученный белый раствор перемешивали при комнатной температуре в течение 20 мин. Практически сразу реакционная смесь становилась белой и более густой. Затем добавляли карбонат калия (120 мг, 0,866 ммоль) и 2-бромацетамид (71,7 мг, 0,520 ммоль). Реакционную смесь перемешивали при комнатной температуре. Через 90 мин реакционная смесь становилась желтой и не густела. Смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь гасили насыщенным раствором NaHCO3 и экстрагировали EtOAc (×2). Органический слой промывали насыщенным раствором NaCl, высушивали над безводным Na2SO4 и фильтровали. Растворитель выпаривали при пониженном давлении с получением трет-бутил (2-амино-2-оксоэтил)((2-метокси-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)фенил)сульфонил)карбамата (35 мг, 0,070 ммоль, 16%). МС m/z [М+Н]+ = 504,20.
Промежуточное соединение 42: 4-цианобензолсульфонамид:
К раствору 4-цианобензол-1-сульфонилхлорида (10 г, 49,59 ммоль, коммерческий источник: Combi-Blocks) в дихлорметане (100 мл) добавляли водный раствор аммиака (25%) (50 мл) при 27°C и перемешивали в течение 3 ч при той же температуре. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Полученное твердое вещество перемешивали с 10% метанолом в дихлорметане (150 мл) при 27°C в течение 1 ч. Твердое вещество отфильтровывали и высушивали в вакууме с получением 4-цианобензолсульфонамида (8 г, неочищенное соединение) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,13-8,03 (м, 2Н), 7,99-7,92 (м, 2Н), 7,59-7,47 (шир. с, 2Н). МС m/z [М-Н]- = 180,97.
Промежуточное соединение 43: 4-(2Н-тетразол-5-ил) бензолсульфонамид:
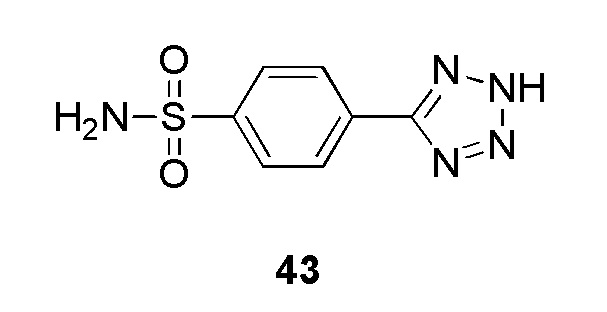
К раствору 4-цианобензолсульфонамида (3,8 г, 20,88 ммоль, промежуточное соединение 42 или коммерческий источник: Combi-Blocks) в N, N-диметилформамиде (50 мл) добавляли азид натрия (13,57 г, 208,73 ммоль) и хлорид аммония (11,17 г, 208,78 ммоль) при 27°C. Реакционную смесь нагревали до 120°C и перемешивали при той же температуре в течение 5 ч. По окончании реакции реакционную смесь охлаждали до 27°C, гасили 1 Н HCl (150 мл) и экстрагировали этилацетатом (3×100 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Остаток перемешивали с дихлорметаном (10 мл) при 27°C в течение 30 мин и фильтровали. Твердое вещество высушивали в вакууме с получением 4-(2H-тетразол-5-ил)бензолсульфонамида (3,2 г, 68%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,23 (д, J=8,5 Гц, 2H), 8,04 (д, J=8,5 Гц, 2H), 7,53 (с, 2H). МС m/z [М-Н]- = 224,03.
Промежуточное соединение 44: 4-(2-(4-фторбензил)-2Н-тетразол-5-ил)бензолсульфонамид:
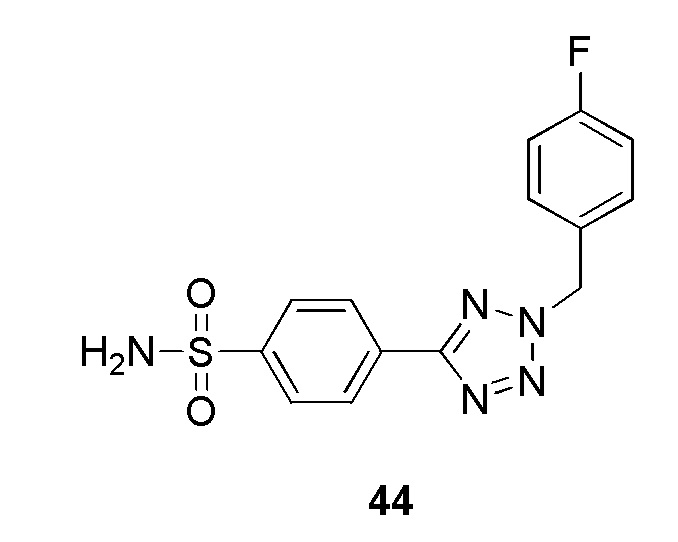
К раствору 4-(2H-тетразол-5-ил)бензолсульфонамида (1 г, 4,444 ммоль, промежуточное соединение 43 или коммерческий источник: Butt-Park) и карбоната калия (1,23 г, 8,9 ммоль) в N, N-диметилформамиде (20 мл) добавляли 4-фторбензилбромид (418 мг, 2,224 ммоль, коммерческий источник: Apollo Scientific) при 27°C. Полученную реакционную смесь перемешивали при 27°C в течение 2 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 60% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-(2-(4-фторбензил)-2Н-тетразол-5-ил)бензолсульфонамида (400 мг, 27%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,23 (д, J=8,5 Гц, 2H), 7,99 (д, J=8,5 Гц, 2H), 7,57-7,45 (м, 4H), 7,25 (с, 2H) 6,03 (с, 2Н). МС m/z [М+Н]+ = 333,92.
Промежуточное соединение 45: 4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил)бензолсульфонамид:
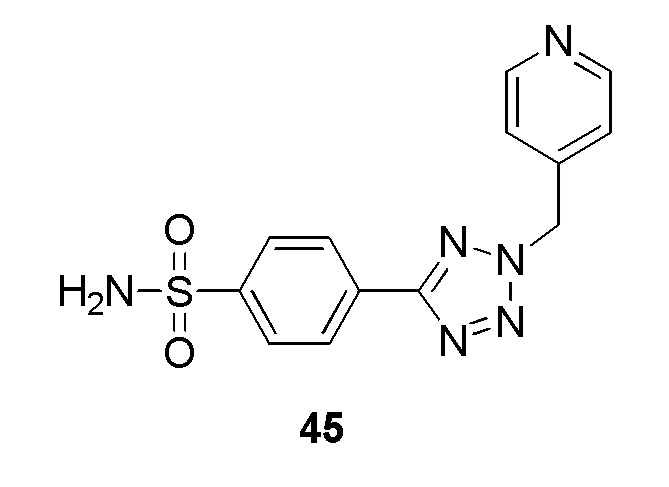
К раствору 4-(2H-тетразол-5-ил)бензолсульфонамида (1 г, 4,444 ммоль, промежуточное соединение 43 или коммерческий источник: Butt-Park) и карбоната калия (1,23 г, 8,9 ммоль) в N, N-диметилформамиде (20 мл) добавляли 4-(хлорметил)пиридин гидрохлорид (729 мг, 4,444 ммоль, коммерческий источник: Combi-Blocks) при 27°C и перемешивали при той же температуре в течение 16 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 10% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил)бензолсульфонамида (250 мг, 25%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,66-8,58 (м, 2Н), 8,31-8,22 (м, 2Н), 8,04-7,97 (м, 2Н), 7,56-7,49 (м, 2Н), 7,39-7,31 (м, 2Н), 6,14 (с, 2Н). МС m/z [М+Н]+ = 317,2.
Промежуточное соединение 46: трет-бутил (цианометил)((4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил)фенил)сульфонил)карбамат:
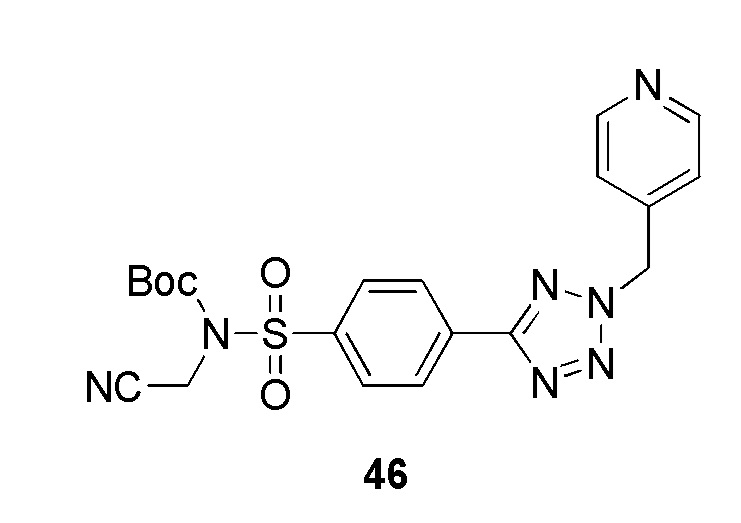
К раствору 4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил) бензолсульфонамида (250 мг, 0,79 ммоль, промежуточное соединение 45) и 4-(диметиламино)пиридина (48,2 мг, 0,395 ммоль) в тетрагидрофуране (10 мл) добавляли Boc-ангидрид (0,7 мл, 3,16 ммоль) и триэтиламин (0,15 мл, 1,106 ммоль) при 27°C. Реакционную смесь перемешивали при 27°C в течение 3 ч. Затем добавляли карбонат калия (218 мг, 1,58 ммоль) и бромацетонитрил (0,15 мл, 2,133 ммоль) при 27°C. Полученную реакционную смесь перемешивали в течение 16 ч при той же температуре. По окончании реакции реакционную смесь гасили насыщенным раствором бикарбоната натрия (100 мл) и экстрагировали этилацетатом (2×100 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 4% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением трет-бутил (цианометил)((4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил)фенил) сульфонил)карбамата (100 мг, неочищенное соединение) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,61 (д, J=5,9 Гц, 2H), 8,41-8,32 (м, 2H), 8,16 (д, J=8,3 Гц, 2H), 7,36 (д, J=5,9 Гц, 2H), 6,16 (с, 2H), 4,94 (с, 2H), 1,31 (с, 9H). МС m/z [М+Н]+ = 456,01.
Промежуточное соединение 47: 4-(2-((5-фторпиридин-2-ил) метил)-2Н-тетразол-5-ил)бензолсульфонамид:
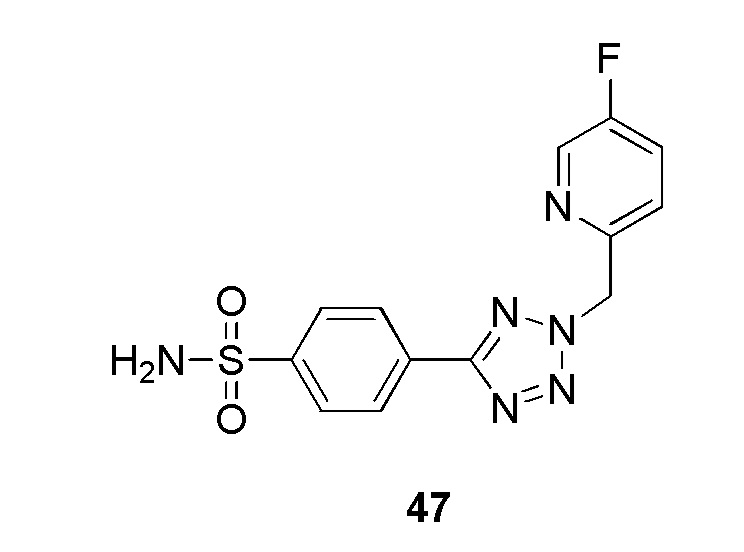
К раствору 4-(2H-тетразол-5-ил)бензолсульфонамида (1,3 г, 5,777 ммоль, промежуточное соединение 43 или коммерческий источник: Butt-Park) и карбоната калия (1,594 г, 11,555 ммоль) в N, N-диметилформамиде (25 мл) добавляли 2-(бромметил)-5-фторпиридин гидробромид (1,565 г, 5,7777 ммоль, промежуточное соединение 5) при 27°C и перемешивали при той же температуре в течение 24 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Неочищенное соединение очищали колоночной хроматографией (нейтральный оксид алюминия), используя смесь 6% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида (1,3 г, 53%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,55 (д, J=2,8 Гц, 1H), 8,24 (д, J=8,3 Гц, 2H), 7,99 (д, J=8,3 Гц, 2H), 7,84 (дт , J=8,7, 3,1 Гц, 1H), 7,66 (дд, J=8,8, 4,4 Гц, 1H), 7,52 (с, 2H), 6,17 (с, 2H). МС m/z [М+Н]+ = 335,08.
Промежуточное соединение 48: трет-бутил (цианометил)((4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)фенил)сульфонил) карбамат:
К перемешиваемому раствору 4-(2-((5-фторпиридин-2-ил)метил) -2Н-тетразол-5-ил)бензолсульфонамида (600 мг, 1,796 ммоль, промежуточное соединение 47) в тетрагидрофуране (25 мл) добавляли смесь 4-(диметиламино)пиридина (110 мг, 0,898 ммоль), Boc-ангидрида (1,17 г, 5,389 ммоль) и триэтиламина (218 мг, 2,155 ммоль). Реакционную смесь перемешивали при 27°C в течение 5 ч. Затем добавляли карбонат калия (496 мг, 3,592 ммоль) и бромацетонитрил (539 мг, 4,491 ммоль) при 26°C. Реакционную смесь перемешивали в течение 16 ч при той же температуре. По окончании реакции реакционную смесь гасили насыщенным раствором бикарбоната натрия (60 мл) и экстрагировали этилацетатом (3×60 мл). Объединенный органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (нейтральный оксид алюминия) и элюировали дихлорметаном. Чистые фракции собирали и концентрировали при пониженном давлении с получением трет-бутил (цианометил)((4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)фенил)сульфонил)карбамата (600 мг, 44%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,54 (д, J=2,6 Гц, 1H), 8,41-8,25 (м, 2H), 8,21-8,09 (м, 2H), 7,84 (дт, J=8,7, 2,9 Гц, 1H), 7,73-7,61 (м, 1H), 6,28-6,08 (м, 2H), 4,94 (с, 2H), 1,30 (с, 9H). МС m/z [М+Н]+ = 474,40.
Промежуточное соединение 49: 4-(2-(пиримидин-4-илметил)-2Н-тетразол-5-ил)бензолсульфонамид:
К раствору 4-(2H-тетразол-5-ил)бензолсульфонамида (350 мг, 1,5555 ммоль, промежуточное соединение 43 или коммерческий источник: Butt-Park) и карбоната калия (429 мг, 3,111 ммоль) в N, N-диметилформамиде (10 мл) добавляли 4-(бромметил)пиримидин (269 мг, 1,5555 ммоль, промежуточное соединение 1) при 26°C и перемешивали при той же температуре в течение 24 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырое вещество очищали колоночной хроматографией с использованием смеси 10% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении. Полученное соединение перемешивали со смесью дихлорметана (30 мл) и петролейного эфира (100 мл) при 26°C в течение 15 мин. Осажденное твердое соединение отфильтровывали и высушивали в вакууме с получением 4-(2-(пиримидин-4-илметил)-2Н-тетразол-5-ил) бензолсульфонамида (250 мг, 46%) в виде красного твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,14 (с, 1H), 8,88 (д, J=5,3 Гц, 1H), 8,26 (д, J=8,4 Гц, 2H), 8,00 (д, J=8,3 Гц), 2H), 7,60 (с, 1H), 7,52 (с, 2H), 6,28 (с, 2H). МС m/z [М+Н]+ = 318,40.
Промежуточное соединение 50: трет-бутил (цианометил)((4-(2- (пиримидин-4-илметил)-2Н-тетразол-5-ил)фенил)сульфонил) карбамат:
К перемешиваемому раствору 4-(2-(пиримидин-4-илметил)-2Н-тетразол-5-ил)бензолсульфонамида (250 мг, 0,7886 ммоль, промежуточное соединение 49) в тетрагидрофуране (15 мл) добавляли смесь 4-(диметиламино)пиридина (48 мг, 0,394 ммоль), Вос-ангидрида (516 мг, 2,365 ммоль) и триэтиламина (96 мг, 0,946 ммоль) при 26°C. Реакционную смесь перемешивали при 26°C в течение 5 ч. Затем добавляли карбонат калия (218 мг, 1,577 ммоль) и бромацетонитрил (236 мг, 1,9715 ммоль) при 26°C, и полученную реакционную смесь перемешивали в течение 16 ч при той же температуре. По окончании реакции реакционную смесь гасили насыщенным раствором бикарбоната натрия (40 мл) и экстрагировали этилацетатом (3×50 мл). Объединенный органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (нейтральный оксид алюминия), элюируя дихлорметаном. Чистые фракции собирали и концентрировали при пониженном давлении с получением трет-бутил (цианометил)((4-(2- (пиримидин-4-илметил)-2Н-тетразол-5-ил)фенил)сульфонил) карбамата (250 мг, 52%) в виде коричневой смолы. 1H ЯМР (400 МГц, CDCl3) δ 9,22 (д, J=1,2 Гц, 1H), 8,90-8,70 (м, 1H), 8,49-8,24 (м, 2H), 8,20-7,99 (м, 2H), 7,15 (дд, J=5,1, 1,2 Гц, 1H), 6,07-5,87 (м, 2H), 4,75 (с, 2H), 1,39 (с, 9H). МС m/z [М+Н]+ = 457,16.
Промежуточное соединение 51: 2-метокси-4-(2H-тетразол-5-ил) бензолсульфонамид:
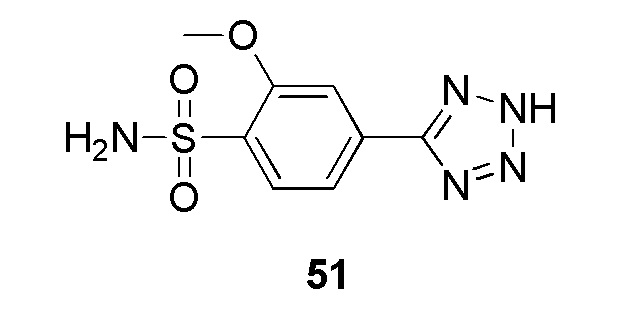
К раствору 4-циано-2-метоксибензолсульфонамида (1 г, 4,712 ммоль, коммерческий источник: Enamine) в N, N-диметилформамиде (20 мл) добавляли азид натрия (3,06 г, 47,07 ммоль) и хлорид аммония (2,52 г, 47,103 ммоль) 26°C. Реакционную смесь нагревали до 120°C и перемешивали в течение 4 ч при той же температуре. По окончании реакции реакционную смесь охлаждали до 26°C, гасили 1 Н HCl (40 мл) и экстрагировали этилацетатом (2×50 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Остаток перемешивали со смесью дихлорметана (10 мл) и диэтилового эфира (10 мл) при 27°C в течение 30 мин. Осажденное твердое вещество отфильтровывали и высушивали в вакууме с получением 2-метокси-4-(2H-тетразол-5-ил)бензолсульфонамида (890 мг, неочищенное соединение) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,94 (д, J=8,2 Гц, 1H), 7,82 (д, J=1,5 Гц, 1H), 7,74 (дд, J=8,1, 1,5 Гц, 1H), 7,28 (с, 2Н), 4,02 (с, 3Н).
Промежуточное соединение 52: 2-метокси-4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил)бензолсульфонамид:
К раствору 2-метокси-4-(2H-тетразол-5-ил)бензолсульфонамида (200 мг, 1,176 ммоль, промежуточное соединение 51) и карбоната калия (325 мг, 2,352 ммоль) в N, N-диметилформамиде (10 мл) добавляли 4-(хлорметил)пиридин гидрохлорид (193 мг, 1,177 ммоль, коммерческий источник: Combi-Blocks) при 26°C. Реакционную смесь нагревали до 80°C и перемешивали при той же температуре в течение 2 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 8% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 2-метокси-4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил)бензолсульфонамида (250 мг, неочищенное соединение) в виде коричневой смолы. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,65-8,58 (м, 2Н), 7,90-7,85 (м, 1Н), 7,85-7,69 (м, 2Н), 7,40-7,28 (м, 2Н), 7,22 (д J=4,4 Гц, 2H), 6,14 (с, 2H), 3,95 (с, 3H). МС m/z [М+Н]+ = 347,15.
Промежуточное соединение 53: трет-бутил (цианометил)((2-метокси-4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил)фенил) сульфонил)карбамат:
К раствору 2-метокси-4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил)бензолсульфонамида (250 мг, 0,725 ммоль, промежуточное соединение 52) и 4-(диметиламино)пиридина (44 мг, 0,36 ммоль) в тетрагидрофуране (10 мл) добавляли триэтиламин (0,14 мл, 1,01 ммоль) и Boc-ангидрид (0,5 мл, 2,176 ммоль) при 26°C и перемешивали в течение 3 ч. Затем добавляли карбонат калия (200 мг, 1,447 ммоль) и бромацетонитрил (0,14 мл, 2,009 ммоль) при 26°C. Полученную реакционную смесь перемешивали в течение 16 ч при той же температуре. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 5% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением трет-бутил (цианометил)((2-метокси-4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил)фенил)сульфонил)карбамата (300 мг, неочищенное соединение) в виде коричневой смолы. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,72-8,43 (м, 2H), 8,08 (д, J=8,2 Гц, 1H), 7,95-7,77 (м, 2H), 7,50-7,21 (м, 2H), 6,16 (с, 2H), 4,85 (с, 2H), 4,07 (с, 3H), 1,24 (с, 9H). МС m/z [М+Н]+ = 486,19.
Общая схема синтеза азидных промежуточных продуктов
Ароматический бромид 21 или спирт 22 превращают в соответствующий азид 27 с использованием азида натрия.
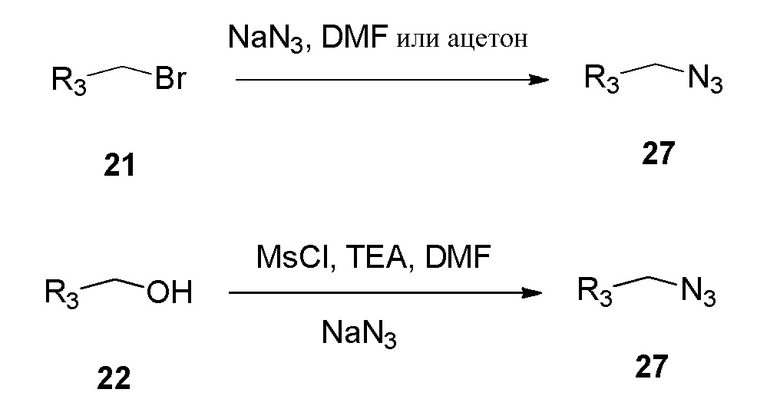
Промежуточное соединение 54: 1-(азидометил)-4-фторбензол:
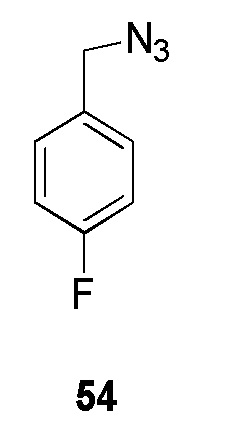
Раствор азида натрия (258 мг, 3,97 ммоль) в воде (8 мл) добавляли по каплям в течение 20 мин к раствору 1-(бромметил)-4-фторбензола (0,329 мл, 2,65 ммоль, коммерческий источник: Aldrich) в ацетоне (20 мл) при 0°C. Реакционной смеси давали нагреться до 25°C и перемешивали в течение 16 ч. Ацетон удаляли при пониженном давлении при 25°C и реакционную смесь экстрагировали гексаном. После этого объединенные органические слои объединяли и высушивали над безводным Na2SO4 и растворитель удаляли при пониженном давлении с получением 1-(азидометил)-4-фторбензола (320 мг, 2,117 ммоль, 80%) в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 7,22-7,39 (м, 2Н), 6,99-7,16 (м, 2Н), 4,33 (с, 2Н).
Промежуточное соединение 55: 4-(азидометил)пиридин:
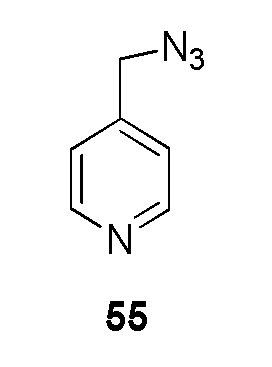
К раствору пиридин-4-илметанола (1 г, 9,163 ммоль, коммерческий источник: AK Scientific) и триэтиламина (1,3 мл, 9,337 ммоль) в N, N-диметилформамиде (10 мл) добавляли мезилхлорид (0,7 мл, 9,043 ммоль) при 0°C. Реакционной смеси давали нагреться до 26°C и перемешивали в течение 2 ч. Затем добавляли азид натрия (892 мг, 13,721 ммоль) при 26°C и перемешивали при той же температуре в течение 16 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 50% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-(азидометил)пиридина (450 мг, 36%) в виде желтой жидкости. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,69-8,53 (м, 2Н), 7,47-7,27 (м, 2Н), 4,58 (с, 2Н). МС m/z [М+Н]+ = 135,16.
Промежуточное соединение 56: 4-(азидометил)бензонитрил:
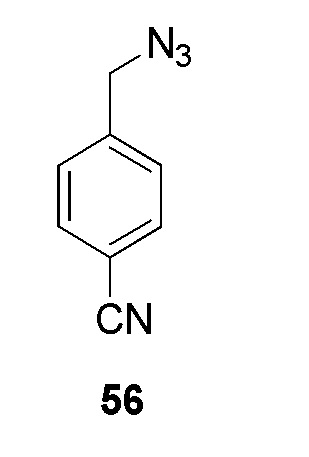
К раствору 4-(бромметил)бензонитрила (500 мг, 2,55 ммоль, коммерческий источник: Alfa Aesar) в N, N-диметилформамиде (10 мл) добавляли азид натрия (175 мг, 2,692 ммоль) при 26°C. Реакционную смесь перемешивали при той же температуре в течение 16 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 10% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-(азидометил)бензонитрила (360 мг, 89%) в виде желтой жидкости. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,91-7,81 (м, 2H), 7,58 (д, J=8,0 Гц, 2H), 4,61 (с, 2H). МС m/z[М] = 157,9.
Промежуточное соединение 57: 2-(азидометил)-5-фторпиридин:
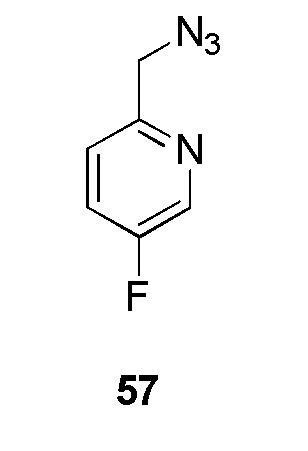
К перемешиваемому раствору (5-фторпиридин-2-ил)метанола (500 мг, 3,937 ммоль, коммерческий источник: Combi-Blocks) и триэтиламина (398 мг, 3,937 ммоль) в N, N-диметилформамиде (10 мл) добавляли мезилхлорид (451 мг, 3,937 ммоль) при 27°C. Реакционную смесь перемешивали при 27°C в течение 2 ч. Затем добавляли азид натрия (384 мг, 5,905 ммоль) при 27°C и реакционную смесь перемешивали в течение 12 ч при той же температуре. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырое соединение очищали колоночной хроматографией (силикагель 100-200 меш), используя смесь 5% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении с получением 2-(азидометил)-5-фторпиридина (150 мг, 25%) в виде коричневой жидкости. 1H ЯМР (400 МГц, CDCl3) δ 8,46 (д, J=2,6 Гц, 1H), 7,47-7,40 (м, 1H), 7,39-7,33 (м, 1H), 4,48 (с, 2H). МС m/z [М+Н]+ = 153,12.
Промежуточное соединение 58: 2-(азидометил)-5-метилпиридин:
К раствору (5-метилпиридин-2-ил)метанола (500 мг, 4,06 ммоль, коммерческий источник: Enamine) в N, N-диметилформамиде (10 мл) добавляли триэтиламин (0,6 мл, 4,317 ммоль) и мезилхлорид (0,3 мл, 3,876 ммоль) при 0°C. Реакционной смеси давали нагреться до 26°C и перемешивали в течение 3 ч. Затем добавляли азид натрия (396 мг, 6,091 ммоль) при 26°C и реакционную смесь перемешивали в течение 16 ч при той же температуре. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 40% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении с получением 2-(азидометил)-5-метилпиридина (300 мг, неочищенное соединение) в виде желтой жидкости. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,43 (д, J=2,2 Гц, 1H), 7,65 (дд, J=8,0, 2,2 Гц, 1H), 7,33 (д, J=7,9 Гц, 1H), 4,45 (с, 2Н), 2,30 (с, 3Н). МС m/z [М+Н]+ = 149,06.
Промежуточное соединение 59: 4-(азидометил)-1,1-дифторциклогексан:
К раствору 4-(бромметил)-1,1-дифторциклогексана (600 мг, 0,0028 моль, промежуточное соединение 3) в N, N-диметилформамиде (6 мл) добавляли азид натрия (549 мг, 0,0084 моль) при 26°C. Реакционную смесь нагревали до 100°C и перемешивали в течение 16 ч при той же температуре. По окончании реакции реакционную смесь использовали на следующей стадии без какой-либо дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 3,18 (т, J=6,6 Гц, 2H), 2,17-1,98 (м, 2H), 1,85-1,57 (м, 5H), 1,37-1,20 (м, 2H).
Промежуточное соединение 60: 4-бром-N-(2-гидроксиэтил) бензолсульфонамид:
К раствору 4-бромбензол-1-сульфонилхлорида (5 г, 19,568 ммоль, коммерческий источник: Alfa Aesar) в дихлорметане (100 мл) добавляли триэтиламин (8,15 мл, 58,941 ммоль) и 2-аминоэтанол (1,195 г, 19,59 ммоль, коммерческий источник: Avra) при 0°C. Температуру реакционной смеси повышали до 26°C и перемешивали в течение 3 ч при той же температуре. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 60% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-бром-N-(2-гидроксиэтил)бензолсульфонамида (3,5 г, 64%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,88-7,77 (м, 2H), 7,72 (д, J=8,8 Гц, 3H), 4,66 (с, 1H), 3,36 (кв, J=5,9 Гц, 2H), 2,80 (кв, J=6,1 Гц, 2H). МС m/z [М+Н]+ = 279,92.
Промежуточное соединение 61: N-(2-гидроксиэтил)-4- ((триметилсилил)этинил)бензолсульфонамид:
К раствору 4-бром-N-(2-гидроксиэтил)бензолсульфонамида (3 г, 10,714 ммоль, промежуточное соединение 60) в N, N-диметилформамиде (20 мл) добавляли триэтиламин (4,3 г, 42,856 ммоль) при 26°C. Реакционную смесь продували азотом в течение 10 мин, добавляли триметилсилилацетилен (1,578 г, 16,071 ммоль, коммерческий источник: Avra) и снова продували азотом в течение 15 мин. Затем добавляли иодид меди (I) (204 мг, 1,0714 ммоль) и тетракис(трифенилфосфин)палладий (0) (495 мг, 0,4285 ммоль) при 26°C. Полученную реакционную смесь перемешивали при той же температуре в течение 16 ч в герметичной пробирке. По окончании реакции реакционную смесь выливали в воду (100 мл) и экстрагировали этилацетатом (3×80 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 30% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении с получением N-(2-гидроксиэтил)-4-((триметилсилил)этинил)бензолсульфонамида (1,3 г, 36%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,80 (д, J=8,3 Гц, 2H), 7,59 (д, J=8,1 Гц, 2H), 4,86 (шир. с, 1H), 3,69 (кв, J=5,0 Гц, 2H), 3,11 (д, J=4,8 Гц, 2H), 1,70 (т, J=4,8 Гц, 1H), 0,27 (с, 9H). МС m/z [М+Н]+ = 298,03.
Промежуточное соединение 62: 4-этинил-N-(2-гидроксиэтил) бензолсульфонамид:
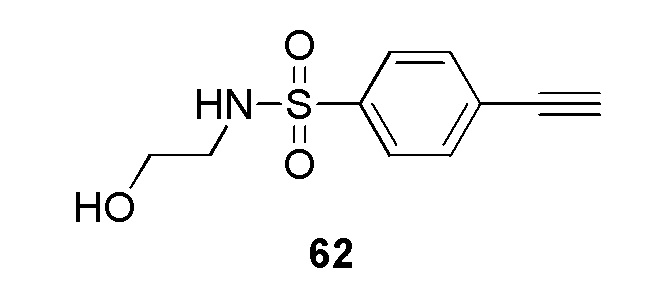
К раствору N-(2-гидроксиэтил)-4-((триметилсилил)этинил) бензолсульфонамида (1,3 г, 4,377 ммоль, промежуточное соединение 61) в смеси хлороформа (10 мл) и метанола (10 мл), добавляли карбонат калия (302 мг, 2,188 ммоль) при 26°C и перемешивали в течение 5 ч при той же температуре. По окончании реакции реакционную смесь упаривали при пониженном давлении. Остаток разводили 1 Н HCl (60 мл) и экстрагировали этилацетатом (3×60 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении с получением сырого 4-этинил-N-(2-гидроксиэтил)бензолсульфонамида (800 мг, 70%). 1H ЯМР (400 МГц, ДМСО-d6) δ 7,85-7,78 (м, 2H), 7,73 (с, 1H), 7,71-7,65 (м, 2H), 4,48 (с, 1H), 3,35 (т, J=6,4 Гц, 3H), 2,79 (кв, J=6,1 Гц, 2H). МС m/z [М-Н]- = 224,26.
Промежуточное соединение 63: 2-(4-бромфенилсульфонамидо) ацетамид:
К раствору 4-бромбензол-1-сульфонилхлорида (10 г, 0,0394 моль, коммерческий источник: Combi-Blocks) в тетрагидрофуране (150 мл) добавляли 2-аминоацетамид гидрохлорид (5,1 г, 0,047 моль, коммерческий источник: Alfa Aesar) при 28°C с последующим медленным добавлением триэтиламина (15 мл, 0,118 моль) при 0°C. Реакционную смесь перемешивали при 28°C в течение 2 ч. По окончании реакции реакционную смесь разбавляли этилацетатом (500 мл) и промывали водой (3×200 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 2-(4-бромфенилсульфонамидо)ацетамида (8 г, 63%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,94 (шир. с, 1H), 7,86-7,63 (м, 4H), 7,24 (шир. с, 1H), 7,04 (шир. с, 1H), 3,38 (д, J=4,2 Гц, 2H). МС m/z [М-Н]- = 291,02.
Промежуточное соединение 64: 2-(4-((триметилсилил)этинил) фенилсульфонамидо)ацетамид:
К продуваемому аргоном раствору 2-(4-бромфенилсульфонамидо)ацетамида (8 г, 0,0273 моль, промежуточное соединение 63) и триэтиламина (15,3 мл, 0,1092 моль) в N, N-диметилформамиде (40 мл) добавляли иодид меди (I) (519 мг, 0,0027 моль), этинилтриметилсилан (5,8 мл, 0,041 моль, коммерческий источник: Avra) и тетракис(трифенилфосфин)палладий (0) (1,26 г, 0,001 моль) при 28°C. Реакционную смесь перемешивали в течение 16 ч при той же температуре. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 80% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении с получением 2-(4-((триметилсилил)этинил)фенилсульфонамидо) ацетамида (4 г, 42%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,95 (т, J=6,2 Гц, 1H), 7,87-7,70 (м, 2H), 7,63 (дд, J=8,5, 1,9 Гц, 2H), 7,25 (шир. с, 1H), 7,04 (шир. с, 1H), 3,45-3,34 (м, 2H), 0,25 (с, 9H). МС m/z [М-Н]- = 309,17.
Промежуточное соединение 65: 2-(4-этинилфенилсульфонамидо) ацетамид:
К раствору 2-(4-((триметилсилил)этинил)фенилсульфонамидо)ацетамида (2 г, 0,0064 моль, промежуточное соединение 64) в смеси хлороформа (10 мл) и метанола (10 мл) добавляли карбонат калия (445 мг, 0,0032 моль) при 26°C. Реакционную смесь перемешивали в течение 16 ч при той же температуре. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в этилацетате (150 мл), промывали водой (2×50 мл) и насыщенным раствором соли (2×30 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении с получением 2-(4-этинилфенилсульфонамидо)ацетамида (1,2 г, 68%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,97 (с, 1H), 7,78 (д, J=8,5 Гц, 2H), 7,66 (д, J=8,5 Гц, 2H), 7,25 (с, 1H), 7,05 (с, 1Н), 4,45 (с, 1Н), 3,39 (с, 2Н). МС m/z [М+Н]+ = 239,06.
Промежуточное соединение 66: (R)-4-бром-N-(2-гидроксипропил)бензолсульфонамид
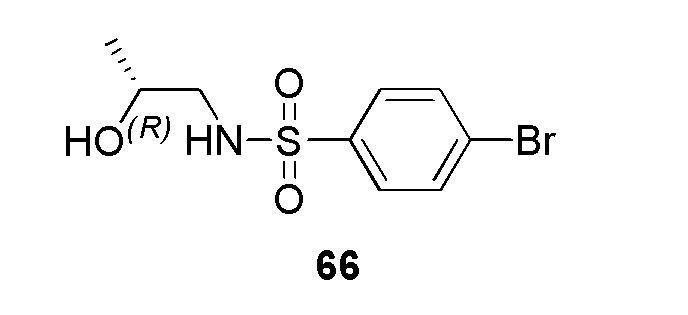
4-Бромбензолсульфонилхлорид (1023 мг, 4,00 ммоль, коммерческий источник: Aldrich), растворенный в тетрагидрофуране (1 мл), добавляли по каплям к перемешиваемому раствору (R)-1-аминопропан-2-ола (601 мг, 8,01 ммоль, коммерческий источник: Aldrich) в тетрагидрофуране (1 мл) при 0°C в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь гасили 1 Н HCl и экстрагировали DCM. Органический слой высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением (R)-4-бром-N-(2-гидроксипропил)бензолсульфонамида (1,3 мг, 4,00 ммоль, неочищенное соединение) в виде неочищенного твердого вещества, которое использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 7,76-7,70 (м, 2Н), 7,69-7,62 (м, 2Н), 5,31 (шир. с, 1Н), 3,96-3,84 (м, 1Н), 3,10-3,02 (м, 1H), 2,84-2,74 (м, 1H), 1,17 (д, J=6,3 Гц, 3H).
Промежуточное соединение 67: (R)-N-(2-гидроксипропил)-4- ((триметилсилил)этинил)бензолсульфонамид:
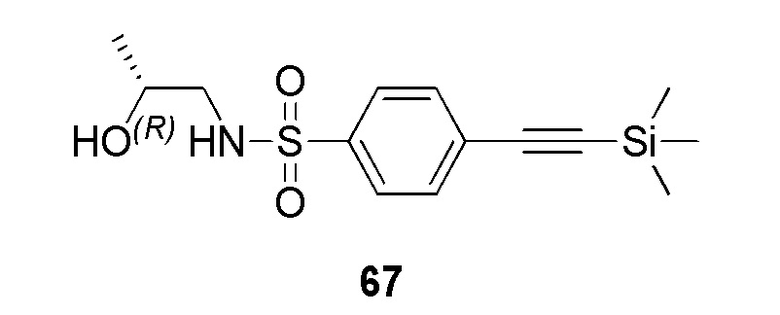
К перемешиваемому раствору (R)-4-бром-N-(2-гидроксипропил) бензолсульфонамида (700 мг, 2,380 ммоль, промежуточное соединение 66) и этинилтриметилсилана (0,501 мл, 3,57 ммоль) в триэтиламине (17 мл) добавляли иодид меди (I) (18,13 мг, 0,095 ммоль) и Pd(PPh3)4 (41,2 мг, 0,036 ммоль). Смесь нагревали до 80°C в течение 16 ч в атмосфере N2. Смесь фильтровали через целит и растворитель выпаривали с получением (R)-N-(2-гидроксипропил)-4-((триметилсилил)этинил)бензолсульфонамида (726 мг, 2,333 ммоль, неочищенное соединение) в виде оранжевого масла. МС m/z [М+Н]+ = 312,22.
Промежуточное соединение 68: (R)-4-этинил-N-(2-гидроксипропил)бензолсульфонамид:
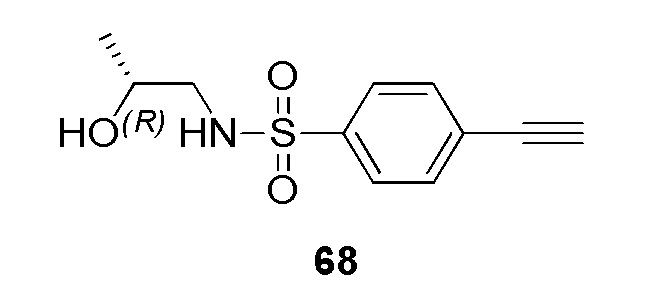
(R)-N-(2-гидроксипропил)-4-((триметилсилил)этинил)бензолсульфонамид (726 мг, 2,331 ммоль, промежуточное соединение 67) растворяли в метаноле (70 мл) и добавляли K2CO3 (1154 мг, 5,83 ммоль). Смесь перемешивали при комнатной температуре в течение 48 ч. Смесь концентрировали и распределяли между CH2Cl2 и H2O. Органический слой высушивали над MgSO4, фильтровали и упаривали досуха. Неочищенное соединение очищали колоночной флэш-хроматографией (диоксид кремния; смесь EtOAc-циклогексан от 0/100 до 50/50). Фракции собирали и концентрировали в вакууме с получением (R)-4-этинил-N-(2-гидроксипропил)бензолсульфонамида (260 мг, 1,087 ммоль, 46,6%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 7,86-7,80 (м, 2Н), 7,65-7,60 (м, 2Н), 5,18-5,13 (м, 1Н), 3,96-3,88 (м, 1Н), 3,27 (с, 1Н ), 3,11-3,05 (м, 1H), 2,84-2,75 (м, 1H), 2,08 (д, J=4,5 Гц, 1H), 1,17 (д, J=6,3 Гц, 2H). МС m/z [М+Н]+ = 240,2.
Промежуточное соединение 69: 4-бромбензолсульфонамид:
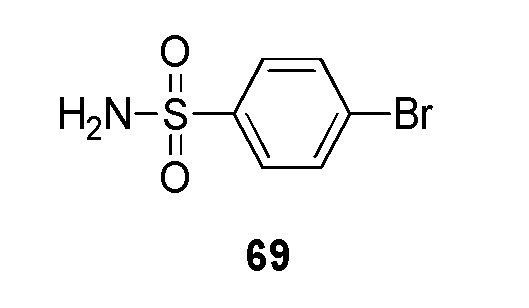
К раствору 4-бромбензол-1-сульфонилхлорида (5 г, 19,568 ммоль, коммерческий источник: Alfa Aesar) в дихлорметане (100 мл) добавляли раствор аммиака (35% водный раствор) (50 мл) при 27°C и перемешивали в течение 4 ч при 27°C. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 60% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-бромбензолсульфонамида (4 г, 86%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,83-7,71 (м, 4Н), 7,45 (с, 2Н). МС m/z [М-Н]- = 233,93.
Промежуточное соединение 70: 4-((триметилсилил)этинил) бензолсульфонамид:
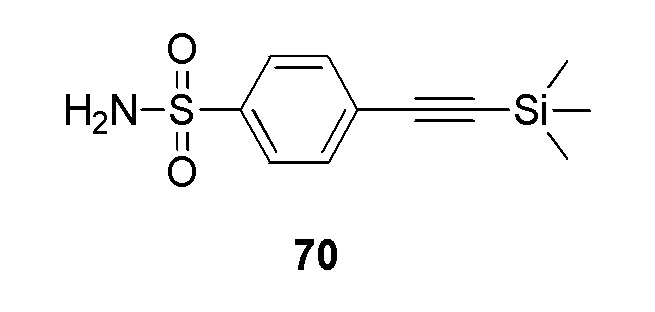
К продуваемому азотом раствору 4-бромбензолсульфонамида (2 г, 8,475 ммоль, промежуточное соединение 69), триэтиламина (4,71 мл, 33,851 ммоль) и триметилсилилацетилена (1,81 мл, 12,716 ммоль) в N, N-диметилформамиде (10 мл) добавляли иодид меди (I) (161 мг, 0,845 ммоль) и тетракис(трифенилфосфин)палладий (0) (392 мг, 0,339 ммоль) при 27°C и перемешивали при 27°C в течение 16 ч в герметичной пробирке. По окончании реакции реакционную смесь фильтровали через целит, и фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 60% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-((триметилсилил)этинил)бензолсульфонамида (1,8 г, 85,7%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,80 (д, J=8,5 Гц, 2H), 7,65 (д, J=8,3 Гц, 2H), 7,46 (с, 2H), 0,25 (с, 9H). МС m/z [М-Н]- = 252,00.
Промежуточное соединение 71: 4-этинилбензолсульфонамид:
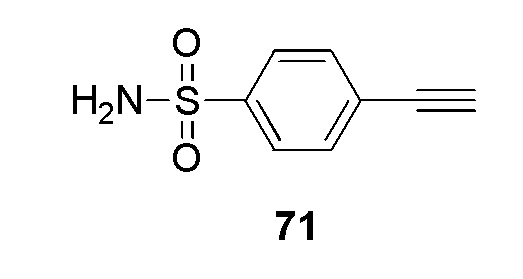
К раствору 4-((триметилсилил)этинил)бензолсульфонамида (1,8 г, 7,115 ммоль, промежуточное соединение 70) в смеси хлороформа (10 мл) и метанола (10 мл) добавляли карбонат калия (393 мг, 2,844 ммоль) при 27°C и перемешивали при 27°C в течение 16 ч. По окончании реакции реакционную смесь упаривали при пониженном давлении. Остаток разбавляли этилацетатом (200 мл) и промывали 1 Н HCl (150 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении с получением 4-этинилбензолсульфонамида (1,4 г, неочищенное соединение) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,82 (д, J=8,3 Гц, 2H), 7,68 (д, J=8,3 Гц, 2H), 7,44 (с, 2H), 4,42 (с, 1H). МС m/z [М-Н]- = 179,97.
Промежуточное соединение 72: 4-(1-(пиридин-4-илметил)-1H-1,2,3-триазол-4-ил)бензолсульфонамид:
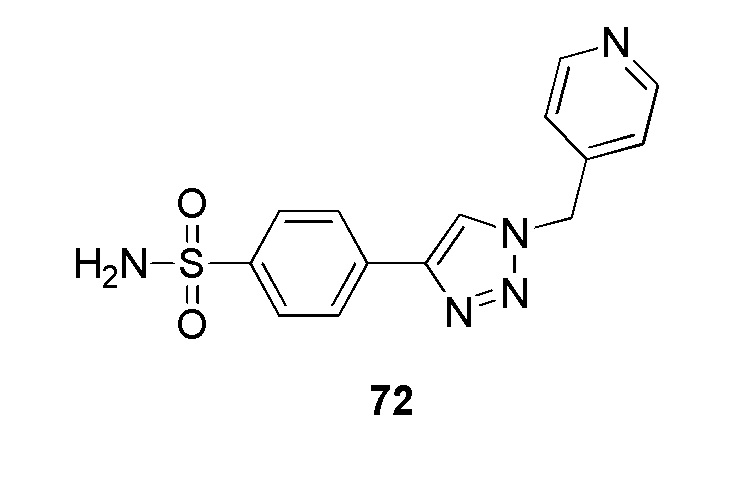
К раствору 4-этинилбензолсульфонамида (800 мг, 4,419 ммоль, промежуточное соединение 71) и 4-(азидометил)пиридина (583 мг, 4,417 ммоль, промежуточное соединение 55) в смеси этанола (10 мл) и воды (10 мл) добавляли сульфат меди пентагидрат (110 мг, 0,442 ммоль) и L-аскорбат натрия (262 мг, 1,332 ммоль) при 26°C и перемешивали при 26°C в течение 16 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 10% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-(1-(пиридин-4-илметил)-1H-1,2,3-триазол-4-ил)бензолсульфонамида (500 мг, неочищенное соединение) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,81 (с, 1Н), 8,65-8,55 (м, 2Н), 8,05 (д, J=8,4 Гц, 2Н), 7,89 (д, J=8,4 Гц, 2Н) 7,48-7,22 (м, 4H), 5,76 (д, J=6,7 Гц, 2H). МС m/z [М+Н]+ = 316,15.
Промежуточное соединение 73: трет-бутил (цианометил)((4-(1- (пиридин-4-илметил)-1H-1,2,3-триазол-4-ил)фенил)сульфонил) карбамат:
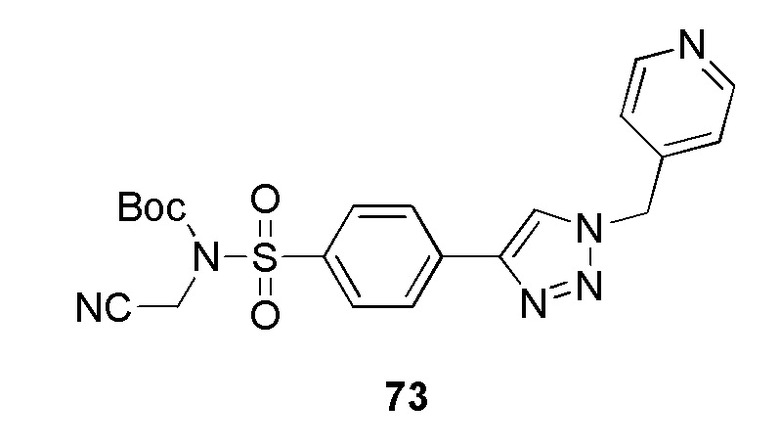
К раствору 4-(1-(пиридин-4-илметил)-1H-1,2,3-триазол-4-ил) бензолсульфонамида (500 мг, 1,587 ммоль, промежуточное соединение 72) и 4-(диметиламино)пиридина ( 97 мг, 0,794 ммоль) в тетрагидрофуране (20 мл) добавляли триэтиламин (0,3 мл, 2,158 ммоль) и ди-трет-бутилдикарбонат (1,09 мл, 4,747 ммоль) при 26°C и перемешивали при 26°C в течение 8 ч с последующим добавлением карбоната калия (438,7 мг, 3,174 ммоль) и бромацетонитрила (0,3 мл, 4,31 ммоль) при 26°C. Реакционную смесь перемешивали в течение 16 ч при той же температуре. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 10% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением трет-бутил (цианометил)((4-(1-(пиридин-4-илметил)-1H-1,2,3-триазол-4-ил)фенил)сульфонил)карбамата (500 мг, неочищенное соединение) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,91 (с, 1H), 8,60 (шир. с, 2H), 8,17 (д, J=8,7 Гц, 2H), 8,04 (д, J=8,7 Гц, 2H), 7,29 (д, J=4,9 Гц, 2H), 5,78 (с, 2H), 4,92 (с, 2H), 1,31 (с, 9H). МС m/z [М+Н]+ = 455,15.
Промежуточное соединение 74: 4-(1-((5-фторпиридин-2-ил) метил)-1Н-1,2,3-триазол-4-ил)бензолсульфонамид:
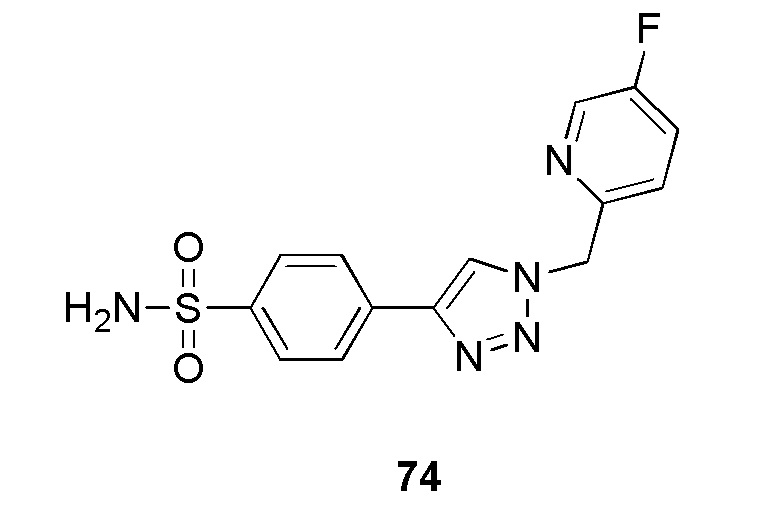
К раствору 4-этинилбензолсульфонамида (4 г, 22,099 ммоль, промежуточное соединение 71) и 2-(азидометил)-5-фторпиридина (3,36 г, 22,105 ммоль, промежуточное соединение 57) в смеси этанола (50 мл) и воды (50 мл) добавляли сульфат меди пентагидрат (550 мг, 2,209 ммоль) и L-аскорбат натрия (1,31 г, 6,616 ммоль) при 27°C и перемешивали в течение 16 ч при 27°C. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 4% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-(1-((5-фторпиридин-2-ил)метил)-1Н-1,2,3-триазол-4-ил)бензолсульфонамида (4,6 г, 63%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,77 (с, 1H), 8,59-8,54 (м, 1H), 8,05 (д, J=8,5 Гц, 2H), 7,88 (д, J=8,5 Гц, 2H) 7,84-7,76 (м, 1H), 7,58-7,49 (м, 1H), 7,36 (с, 2H), 5,79 (с, 2H). МС m/z [М+Н]+ = 334,37.
Промежуточное соединение 75: трет-бутил (цианометил)((4-(1- ((5-фторпиридин-2-ил)метил)-1Н-1,2,3-триазол-4-ил)фенил)сульфонил)карбамат:
К раствору 4-(1-((5-фторпиридин-2-ил)метил)-1Н-1,2,3-триазол-4-ил)бензолсульфонамида (2,6 г, 7,808 ммоль, промежуточное соединение 74) и 4-(диметиламино)пиридина (477 мг, 3,904 ммоль) в тетрагидрофуране (20 мл) добавляли ди-трет-бутилдикарбонат (5,4 мл, 23,505 ммоль) и триэтиламин (1,6 мл, 16 ммоль) добавляли при 27°C. Реакционную смесь перемешивали при 27°C в течение 5 ч с последующим добавлением карбоната калия (2,16 г, 15,629 ммоль) и бромацетонитрила (1,4 мл, 20,1 ммоль) при 27°C. Реакционную смесь перемешивали в течение 16 ч при той же температуре. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 2% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении. Полученное соединение промывали диэтиловым эфиром (2×20 мл) и высушивали в вакууме с получением трет-бутил (цианометил)((4-(1-((5-фторпиридин-2-ил) метил)-1H-1,2,3-триазол-4-ил)фенил)сульфонил)карбамата (2 г, 54%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,88 (с, 1H), 8,60-8,54 (м, 1H), 8,18 (д, J=8,5 Гц, 2H), 8,03 (д, J=8,5 Гц, 2H) 7,85-7,76 (м, 1H), 7,60-7,51 (м, 1H), 5,81 (с, 2H), 4,92 (с, 2H), 1,30 (с, 9H). МС m/z [М+Н]+ = 473,22.
Промежуточное соединение 76: (4-бром-2-метилбензолсульфонилхлорид):
К раствору 4-бром-2-метиланилина (5,0 г, 26,874 ммоль, коммерческий источник: Sigma Aldrich) в уксусной кислоте (12,5 мл) и концентрированной HCl (25 мл) медленно добавляли водный раствор NaNO2 (2,78 г, 40,31 ммоль, коммерческий источник: Finar) при 0°C и перемешивали в течение 30 мин. Эту реакционную смесь добавляли к приготовленному насыщенному раствору SO2 в уксусной кислоте (90 мл) и хлориде меди (II) (1,8 г, 13,43 ммоль, коммерческий источник: Alfa Aesar) при 0°C и перемешивали при комнатной температуре в течение 16 ч. По окончании реакции реакционную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Органический слой высушивали над безводным Na2SO4 и фильтровали. Фильтрат концентрировали при пониженном давлении с получением 4-бром-2-метилбензолсульфонилхлорида (5 г, неочищенное соединение) в виде коричневой смолы. 1H ЯМР (400 МГц, CDCl3) δ 7,94-7,91 (м, 1Н), 7,61-7,59 (м, 1Н), 7,54-7,51 (м, 1Н), 2,78 (с, 3Н). Соединение использовали без дополнительной очистки.
Промежуточное соединение 77: 4-бром-N-(2-гидроксиэтил)-2-метилбензолсульфонамид:
К раствору 4-бром-2-метилбензолсульфонилхлорида (5 г, 18,55 ммоль, промежуточное соединение 76) в тетрагидрофуране (100 мл) добавляли триэтиламин (7,2 мл, 51,94 ммоль, коммерческий источник: Finar) при 0°C с последующим добавлением 2-аминоэтанола (1,35 г, 22,26 ммоль, коммерческий источник: Avra). Реакционной смеси давали нагреться до 26°С и перемешивали в течение 3 ч при той же температуре. По окончании реакции реакционную смесь разбавляли этилацетатом (150 мл) и водой (50 мл). Водный слой экстрагировали этилацетатом (2×30 мл). Объединенные органические слои высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией, элюируя смесью 1,5% метанола в DCM. Чистые фракции концентрировали при пониженном давлении с получением 4-бром-N-(2-гидроксиэтил)-2-метилбензолсульфонамида (2,3 г, 40,8%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,76-7,72 (м, 2Н), 7,67-7,65 (м, 1Н), 7,60-7,58 (м, 1Н), 4,65-4,61 (м, 1Н), 3,35-3,31 (м, 2Н), 2,85-2,79 (м, 2Н), 2,6 (с, 3Н). МС m/z [М+Н]+ = 296,09.
Промежуточное соединение 78: 4-циано-N-(2-гидроксиэтил)-2-метилбензолсульфонамид:
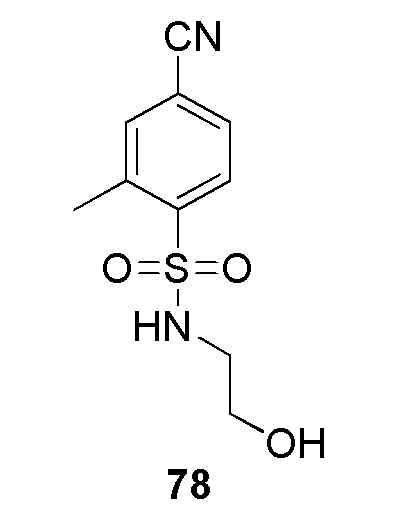
К раствору 4-бром-N-(2-гидроксиэтил)-2-метилбензолсульфонамида (2,2 г, 7,748 ммоль, промежуточное соединение 77) в N, N-диметилформамиде (22 мл) добавляли Zn(CN)2 (2,6 г, 22,43 ммоль, коммерческий источник: Sigma Aldrich) при 26°С. Реакционную смесь продували азотом в течение 10 мин, добавляли тетракис(трифенилфосфин)палладий (0) (0,863 г, 0,747 ммоль, коммерческий источник: Alfa Aesar) и снова продували азотом в течение 15 мин при 26°C. Полученную реакционную смесь перемешивали при 120°С в течение 40 мин в условиях микроволнового реактора. По окончании реакции реакционную смесь разбавляли этилацетатом (200 мл), фильтровали через слой целита и упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией, элюируя смесью 35% этилацетата в петролейном эфире. Чистые фракции концентрировали при пониженном давлении с получением 4-циано-N-(2-гидроксиэтил)-2-метилбензолсульфонамида (1,1 г, 42%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,95-7,91 (м, 3Н), 7,84-7,79 (м, 1Н), 4,65 (шир. С, 1Н), 3,33-3,27 (м, 2Н), 2,87-2,84 (м, 2Н), 2,65 (с, 3Н). МС m/z [М+Н]+ = 241,18.
Промежуточное соединение 79: N-(2-гидроксиэтил)-2-метил-4- (2H-тетразол-5-ил)бензолсульфонамид:
К раствору 4-циано-N-(2-гидроксиэтил)-2-метилбензолсульфонамида (1,0 г, 4,16 ммоль, промежуточное соединение 78) в тетрагидрофуране (10 мл) и воде (1 мл) добавляли азид натрия (1,02 г, 15,81 ммоль) и бромид цинка (1,78 г, 7,90 ммоль) при 26°С. Реакционную смесь нагревали до 85°С и перемешивали в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 0°С, разбавляли ледяной водой (20 мл) и подкисляли концентрированной HCl до pH 2. Осажденное твердое вещество отфильтровывали, промывали водой и высушивали с получением N-(2-гидроксиэтил)-2-метил-4-(2H-тетразол-5-ил)бензолсульфонамида (600 мг) в виде не совсем белого твердого вещества. МС m/z [М+Н]+ = 284,17. Вещество использовали на следующей стадии без какой-либо дополнительной очистки.
Промежуточное соединение 80: 4-бром-3-метилбензолсульфонилхлорид:
К раствору 4-бром-3-метиланилина (5,0 г, 26,87 ммоль, коммерческий источник: Combi-Blocks) в уксусной кислоте (12,0 мл) и концентрированной HCl (20 мл) медленно добавляли водный раствор NaNO2 (2,78 г, 40,31 ммоль, 20 мл воды, коммерческий источник: Avra) при 0°C и перемешивали в течение 30 мин. Эту реакционную смесь добавляли к приготовленному насыщенному раствору SO2 в уксусной кислоте (50 мл) и хлориде меди (II) (1,08 г, 8,06 ммоль, коммерческий источник: Combi-Blocks) при 10°C, и затем перемешивали при 27°C в течение 12 ч. По окончании реакции реакционную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (1 л). Объединенный органический раствор высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 4-бром-3-метилбензолсульфонилхлорида (5 г, неочищенное соединение) в виде коричневой смолы. 1H ЯМР (400 МГц, CDCl3) δ 7,92-7,88 (м, 1Н), 7,80-7,77 (м, 1Н), 7,73-7,69 (м, 1Н), 2,53 (с, 3Н).
Промежуточное соединение 81: 4-бром-N-(2-гидроксиэтил)-3-метилбензолсульфонамид:
К раствору 4-бром-3-метилбензолсульфонилхлорида (5 г, 18,65 ммоль, промежуточное соединение 80) в тетрагидрофуране (50 мл) добавляли триэтиламин (5,2 мл, 37,3 ммоль, коммерческий источник: RCP) с последующим добавлением 2-аминоэтанола (1,13 г, 18,65 ммоль, коммерческий источник: RCP) при 0°С. Реакционной смеси давали нагреться до 26°С и перемешивали при той же температуре в течение 5 ч. По окончании реакции реакционную смесь разбавляли этилацетатом (2 л) и промывали водой (1 л), насыщенным раствором соли (500 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 4-бром-N-(2-гидроксиэтил)-3-метилбензолсульфонамида (5 г, 91%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,80-7,77 (м, 1Н), 7,74-7,71 (м, 1Н), 7,61-7,58 (м, 1Н), 7,51-7,47 (м, 1Н), 4,62 (т , J=5,6 Гц, 1H), 3,33 (кв, J=6,0 Гц, 2H), 2,77 (кв, J=6,1 Гц, 2H), 2,56 (с, 3H). МС m/z [М+Н]+ = 294,08.
Промежуточное соединение 82: 4-циано-N-(2-гидроксиэтил)-3-метилбензолсульфонамид:
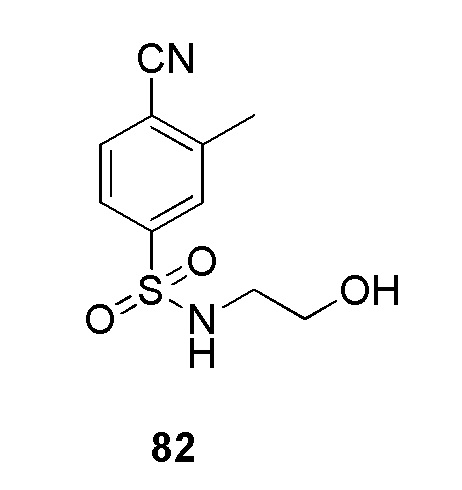
К раствору 4-бром-N-(2-гидроксиэтил)-3-метилбензолсульфонамида (4 г, 13,68 ммоль, промежуточное соединение 81) в N, N-диметилформамиде (40 мл) добавляли Zn(CN)2 (7,26 г, 41,08 ммоль, коммерческий источник: Sigma Aldrich) при 26°С. Реакционную смесь продували азотом в течение 10 мин, добавляли тетракис(трифенилфосфин)палладий (0) (1,6 г, 1,36 ммоль, коммерческий источник: Alfa Aesar) и снова продували азотом при 26°С в течение 15 мин. Полученную реакционную смесь перемешивали при 100°С в течение 12 ч. По окончании реакции реакционную смесь выливали в воду (1 л) и экстрагировали этилацетатом (2×700 мл). Объединенный органический раствор высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали с помощью системы флэш-хроматографии Reveleris®, элюируя смесью 20% этилацетата в петролейном эфире. Чистые фракции концентрировали при пониженном давлении с получением 4-циано-N-(2-гидроксиэтил)-3-метилбензолсульфонамида (2,5 г, 76%) в виде коричневой жидкости. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,04-8,0 (м, 1Н), 7,96 (с, 1Н), 7,90-7,86 (м, 2Н), 4,71-4,65 (м, 1Н), 3,37 (кв, J=6,0 Гц, 2H), 2,97 (т, J=6,1 Гц, 2H), 2,58 (с, 3H). МС m/z [М-Н]- = 239,19.
Промежуточное соединение 83: N-(2-гидроксиэтил)-3-метил-4- (2H-тетразол-5-ил)бензолсульфонамид:
К раствору 4-циано-N-(2-гидроксиэтил)-3-метилбензолсульфонамида (2,3 г, 9,58 ммоль, промежуточное соединение 82) в тетрагидрофуране (23 мл) и воде (3 мл) добавляли азид натрия (2,36 г, 36,4 ммоль, коммерческий источник: Avra) и бромид цинка (4,09 г, 18,2 ммоль, коммерческий источник: Combi-Blocks) при 26°С. Реакционную смесь нагревали до 85°С и перемешивали в течение 12 ч. По окончании реакции реакционную смесь охлаждали до 0°С и упаривали при пониженном давлении. Сырой продукт подкисляли концентрированной HCl (30 мл) до pH 2. Осажденное твердое вещество отфильтровывали, промывали водой и высушивали с получением N-(2-гидроксиэтил)-3-метил-4-(2H-тетразол-5-ил)бензолсульфонамида (1,5 г, неочищенное соединение) в виде не совсем белого твердого вещества. МС m/z [М-Н]- = 282,18.
Промежуточное соединение 84: 4-бром-2-фторбензол-1-сульфонилхлорид:
К раствору 4-бром-2-фторанилина (2,0 г, 10,58 ммоль, коммерческий источник: Combi-Blocks) в уксусной кислоте (5 мл) и водном растворе HCl (8 мл) медленно добавляли водный раствор NaNO2 (1,1 г, 15,87 ммоль, коммерческий источник: Avra) при 0°С и перемешивали в течение 45 мин. Эту реакционную смесь добавляли к приготовленному насыщенному раствору SO2 в уксусной кислоте (20 мл) и хлориде меди (II) (0,42 г, 3,17 ммоль, коммерческий источник: Combi-Blocks) при 0°С и перемешивали при комнатной температуре в течение 2 ч. По окончании реакции реакционную смесь экстрагировали этилацетатом (500 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 4-бром-2-фторбензол-1-сульфонилхлорида (2 г, неочищенное соединение) в виде смолы. 1H ЯМР (400 МГц, CDCl3) δ 7,92-7,88 (м, 1H), 7,58-7,54 (м, 2H).
Промежуточное соединение 85: 4-бром-2-фтор-N-(2-гидроксиэтил)бензолсульфонамид:
К раствору 4-бром-2-фторбензол-1-сульфонилхлорида (2 г, 7,35 ммоль, промежуточное соединение 84) в тетрагидрофуране (20 мл) добавляли триэтиламин (2 мл, 14,7 моль, коммерческий источник: RCP) с последующим добавлением 2-аминоэтанола (0,5 г, 7,35 ммоль, коммерческий источник: RCP) при 0°С. Реакционной смеси давали нагреться до 26°С и перемешивали при той же температуре в течение 5 ч. По окончании реакции реакционную смесь разбавляли этилацетатом (1 л). Органический слой промывали водой (500 мл), насыщенным раствором соли (200 мл), высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 4-бром-2-фтор-N-(2-гидроксиэтил)бензолсульфонамида (2 г, 91%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,89-7,85 (м, 1Н), 7,8-7,77 (м, 1Н), 7,72-7,65 (м, 1Н), 7,61-7,57 (м, 1Н), 4,62 (т, J=5,6 Гц, 1H), 3,33 (кв, J=6,0 Гц, 2H), 2,85 (кв, J=6,1 Гц, 2H). МС m/z [М+2Н]+ = 298,04.
Промежуточное соединение 86: 4-циано-2-фтор-N-(2-гидроксиэтил)бензолсульфонамид:
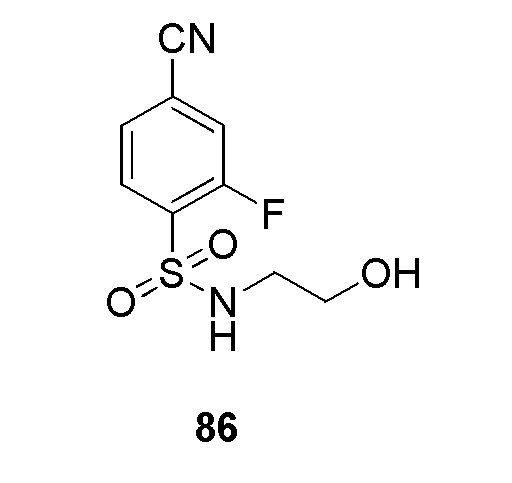
К раствору 4-бром-2-фтор-N-(2-гидроксиэтил) бензолсульфонамида (2 г, 6,71 ммоль, промежуточное соединение 85) в N, N-диметилформамиде (20 мл) добавляли Zn(CN)2 (3,56 г, 20,3 ммоль, коммерческий источник: Sigma Aldrich) при 26°С. Реакционную смесь продували азотом в течение 10 мин, добавляли тетракис(трифенилфосфин)палладий (0) (0,77 г, 0,67 ммоль, коммерческий источник: Alfa Aesar) и снова продували азотом при 26°С в течение 15 мин. Полученную реакционную смесь перемешивали при 100°С в течение 12 ч. По окончании реакции реакционную смесь выливали в воду (500 мл) и экстрагировали этилацетатом (2×500 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью системы флэш-хроматографии Reveleris®, элюируя смесью 15% этилацетата в петролейном эфире. Чистые фракции концентрировали при пониженном давлении с получением 4-циано-2-фтор-N-(2-гидроксиэтил)бензолсульфонамида (1,1 г, 68%) в виде коричневой жидкости. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,22 (с, 1H), 8,16-8,09 (м, 1H), 7,99-7,93 (м, 1H), 7,90-7,86 (м, 1H), 4,71-4,65 (м, 1H), 3,37 (кв, J=6,0 Гц, 2H), 2,97 (т, J=6,1 Гц, 2H). МС m/z [М+Н]+ = 243,12.
Промежуточное соединение 87: 2-фтор-N-(2-гидроксиэтил)-4- (2Н-тетразол-5-ил)бензолсульфонамид:
К раствору 4-циано-2-фтор-N-(2-гидроксиэтил) бензолсульфонамида (1,0 г, 4,098 ммоль, промежуточное соединение 86) в тетрагидрофуране (10 мл) и воде (1 мл) добавляли азид натрия (1,0 г, 15,57 ммоль) и бромид цинка (1,75 г, 7,78 ммоль) при 26°С. Реакционную смесь нагревали до 85°С и перемешивали в течение 12 ч. По окончании реакции реакционную смесь охлаждали до 0°С и подкисляли концентрированной HCl (30 мл) до pH 2. Осажденное твердое вещество отфильтровывали, промывали водой и высушивали с получением 2-фтор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил)бензолсульфонамида (1 г, 85%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,09-7,98 (м, 3H), 3,42-3,25 (м, J=6,4 Гц, 2H), 2,98 (кв, J=6,1 Гц, 2H). МС m/z [М+Н]+ = 288,13.
Промежуточное соединение 88: 4-(бензилтио)-3-хлорбензонитрил:
К раствору 3,4-дихлорбензонитрила (5 г, 29,25 ммоль, коммерческий источник: Alfa) в N, N-диметилформамиде (50 мл) добавляли карбонат калия (9,7 г, 70,19 ммоль) и фенилметантиол (4,35 г, 35,1 ммоль, коммерческий источник: Alfa) при 28°С. Полученную реакционную смесь перемешивали при 100°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 28°С и разбавляли этилацетатом (200 мл). Органический слой последовательно промывали ледяной водой (5×100 мл) и насыщенным раствором соли (50 мл). Органические слои высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 4-(бензилтио)-3-хлорбензонитрила (4 г, 28%, чистота=53%). МС m/z [М-Н]- = 258,42.
Промежуточное соединение 89: 2-хлор-4-цианобензол-1-сульфонилхлорид:
К раствору 4-(бензилтио)-3-хлорбензонитрила (4 г, 15,443 ммоль, промежуточное соединение 88) в уксусной кислоте (6 мл), воде (4 мл) и ацетонитриле (160 мл) добавляли 1,3-дихлор-5,5-диметилимидазолидин-2,4-дион (3,63 г, 18,531 ммоль, коммерческий источник: Aldrich) при 0°С. Реакционную смесь перемешивали при 28°С в течение 16 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении с получением сырого продукта, который разводили этилацетатом (200 мл) и водой (50 мл). Органический слой отделяли и промывали насыщенным раствором NaHCO3 (2×100 мл). Органический слой промывали насыщенным раствором NaCl (50 мл), высушивали над безводным Na2SO4 и фильтровали. Растворитель выпаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 10% этилацетата в гексане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 2-хлор-4-цианобензол-1-сульфонилхлорида (1,6 г, 24%, чистота 54%) в виде коричневого твердого вещества.
Промежуточное соединение 90: 2-хлор-4-циано-N-(2-гидроксиэтил)бензолсульфонамид:
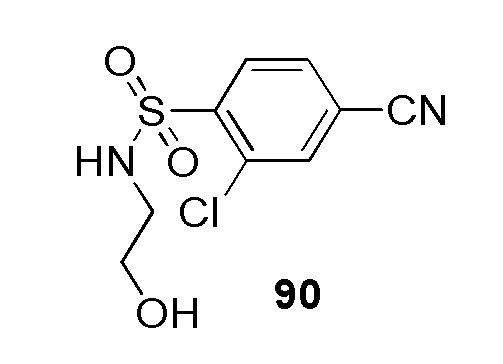
К раствору 2-хлор-4-цианобензол-1-сульфонилхлорида (1,6 г, 6,81 ммоль, промежуточное соединение 89) в тетрагидрофуране (32 мл) добавляли триэтиламин (2,3 мл, 17,026 ммоль, коммерческий источник: Finar) при 0°С с последующим добавлением 2-аминоэтанола (0,49 г, 8,17 ммоль, коммерческий источник: Avra). Реакционной смеси давали нагреться до 28°С и перемешивали в течение 3 ч при той же температуре. По окончании реакции реакционную смесь разбавляли этилацетатом (100 мл) и водой (50 мл). Органический слой отделяли и высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией, элюируя смесью 40% этилацетата в гексане. Чистые фракции концентрировали при пониженном давлении с получением 2-хлор-4-циано-N-(2-гидроксиэтил)бензолсульфонамида (1 г, 53,3%) в виде не совсем белого твердого вещества. МС m/z [М+Н]+ = 261,05.
Промежуточное соединение 91: 2-хлор-N-(2-гидроксиэтил)-4- (2Н-тетразол-5-ил)бензолсульфонамид:
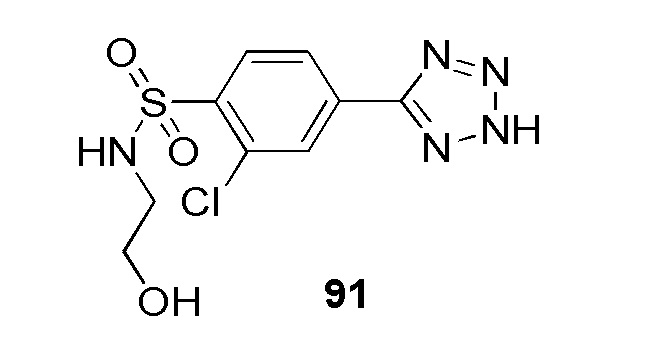
К раствору 2-хлор-4-циано-N-(2-гидроксиэтил) бензолсульфонамида (1,0 г, 3,8461 ммоль, промежуточное соединение 90) в тетрагидрофуране (10 мл) и воде (1 мл) добавляли азид натрия (0,95 г, 14,615 ммоль, коммерческий источник: Avra) и бромид цинка (1,6 г, 7,307 ммоль, коммерческий источник: Alfa Aesar) при 28°С. Реакционную смесь нагревали до 100°С и перемешивали в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 0°С и гасили ледяной водой (20 мл). рН раствора доводили, медленно добавляя концентрированную HCl (35 мл) до рН 2 при 0°С. Затем ТГФ удаляли при пониженном давлении и отфильтровывали осажденное твердое вещество, промывали водой и высушивали с получением 2-хлор-N-(2-гидроксиэтил)-4-(2Н-тетразол-5-ил)бензолсульфонамида (100 мг, 60%) в виде не совсем белого твердого вещества. МС m/z [М+Н]+ = 304,04.
Промежуточное соединение 92: 4-(бензилтио)-2-хлорбензонитрил:
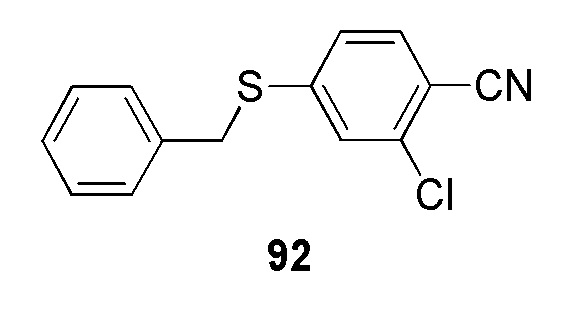
К раствору 2-хлор-4-фторбензонитрила (5 г, 32,26 ммоль, коммерческий источник: Combi-Blocks) в N, N-диметилформамиде (50 мл) добавляли карбонат калия (8,9 г, 64,52 ммоль) и фенилметантиол (4,8 г, 38,71 ммоль, коммерческий источник: Alfa) при 28°С. Полученную реакционную смесь перемешивали при 100°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 28°С и разбавляли этилацетатом (200 мл). Органический слой последовательно промывали ледяной водой (5×100 мл) и насыщенным раствором соли (100 мл). Органические слои объединяли и высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 18% этилацетата в гексане, с получением 4-(бензилтио)-2-хлорбензонитрила (3,5 г, 41,7%). МС m/z [М+2Н]+ = 261,13.
Промежуточное соединение 93: 3-хлор-4-цианобензол-1-сульфонилхлорид:
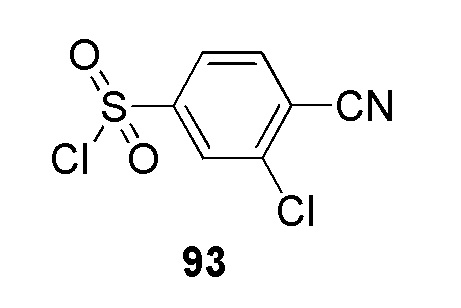
К раствору 4-(бензилтио)-2-хлорбензонитрила (3,5 г, 13,51 ммоль, промежуточное соединение 92) в ацетонитриле (140 мл) добавляли уксусную кислоту (5,25 мл), воду (3,5 мл) и 1,3-дихлор-5,5-диметилимидазолидин-2,4-дион (3,1 г, 16,21 ммоль, коммерческий источник: Aldrich) при 0°С. Реакционную смесь перемешивали при 28°С в течение 16 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении с получением сырого продукта, который разводили этилацетатом (200 мл). Органический слой отделяли и промывали насыщенным раствором NaHCO3 (3×50 мл). Органический слой промывали насыщенным раствором NaCl (50 мл), высушивали над безводным Na2SO4 и фильтровали. Растворитель выпаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 10% этилацетата в гексане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 3-хлор-4-цианобензол-1-сульфонилхлорида (1,6 г, 42,5%) в виде бледно-желтого твердого вещества.
Промежуточное соединение 94: 3-хлор-4-циано-N-(2-гидроксиэтил)бензолсульфонамид:
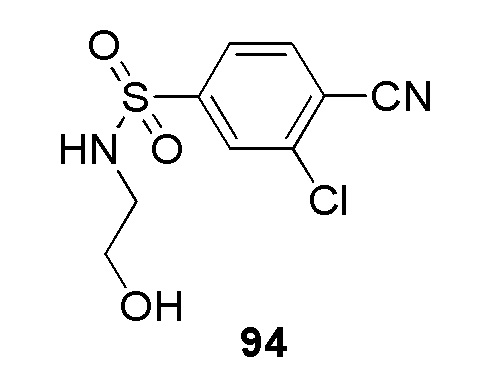
К раствору 3-хлор-4-цианобензол-1-сульфонилхлорида (1,5 г, 6,38 ммоль, промежуточное соединение 93) в тетрагидрофуране (30 мл) добавляли триэтиламин (1,8 мл, 12,77 ммоль, коммерческий источник: Finar) при 0°С с последующим добавлением 2-аминоэтанола (0,38 г, 6,38 ммоль, коммерческий источник: Avra). Реакционной смеси давали нагреться до 26°С и перемешивали в течение 1 ч при той же температуре. По окончании реакции реакционную смесь концентрировали при пониженном давлении с получением сырого продукта, который разводили этилацетатом (100 мл). Органический слой последовательно промывали водой (2×50 мл) и насыщенным раствором соли (50 мл). Органические слои объединяли и высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 50% этилацетата в гексане, с получением 3-хлор-4-циано-N-(2-гидроксиэтил)бензолсульфонамида (1 г, 59,6%) в виде не совсем белого твердого вещества. МС m/z [М-Н]- = 258,93.
Промежуточное соединение 95: 3-хлор-N-(2-гидроксиэтил)-4- (2H-тетразол-5-ил)бензолсульфонамид:
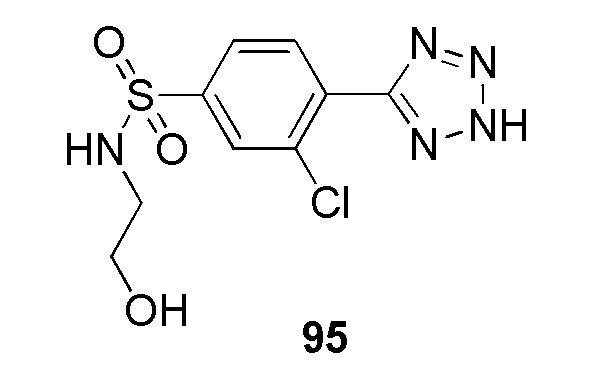
К раствору 3-хлор-4-циано-N-(2-гидроксиэтил) бензолсульфонамида (1,0 г, 3,846 ммоль, промежуточное соединение 94) в тетрагидрофуране (10 мл) и воде (1 мл) добавляли азид натрия (0,95 г, 14,61 ммоль) и бромид цинка (1,64 г, 7,31 ммоль) при 28°С. Реакционную смесь нагревали до 85°С и перемешивали в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 0°С и подкисляли концентрированной HCl (30 мл) до pH 2. Затем ТГФ выпаривали при пониженном давлении с получением 3-хлор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил)бензолсульфонамида (800 мг, 25%) в виде белого твердого вещества. Соединение использовали без дополнительной очистки.
Промежуточное соединение 96: 2-хлор-4-цианобензолсульфонамид:
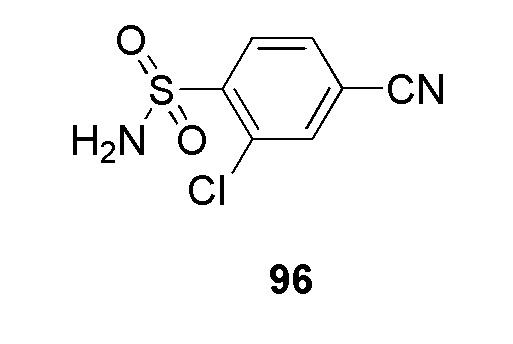
Перемешиваемый раствор 2-хлор-4-цианобензол-1-сульфонилхлорида (6,0 г, 25,416 ммоль, промежуточное соединение 89) в тетрагидрофуране (60 мл) охлаждали до 0°С и затем в течение 20 мин добавляли газообразный аммиак. После продувки реакционную смесь перемешивали при 0°С в течение 3 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении с получением сырого продукта, который разбавляли этилацетатом (500 мл) и водой (100 мл). Водный слой экстрагировали этилацетатом (3×30 мл). Органические слои объединяли и промывали насыщенным раствором NaCl (3×100 мл), высушивали над безводным Na2SO4 и фильтровали. Растворитель выпаривали при пониженном давлении с получением 2-хлор-4-цианобензолсульфонамида (5 г, 88,9%) в виде не совсем белого твердого вещества. МС m/z [М-Н]- = 215,16.
Промежуточное соединение 97: 2-хлор-4-(2H-тетразол-5-ил) бензолсульфонамид:
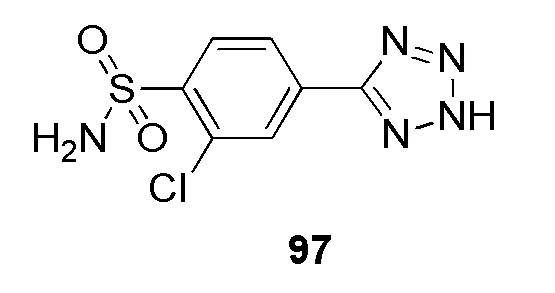
К раствору 2-хлор-4-цианобензолсульфонамида (5,0 г, 23,08 ммоль, промежуточное соединение 96) в тетрагидрофуране (50 мл) и воде (5 мл) добавляли азид натрия (5,7 г, 87,70 ммоль) и бромид цинка (9,8 г, 43,85 ммоль) при 26°С. Реакционную смесь нагревали до 85°С и перемешивали в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 0°С. Затем ее гасили холодной водой (50 мл) и подкисляли концентрированной HCl (30 мл) до pH 2. Затем ТГФ выпаривали при пониженном давлении, и полученное твердое вещество отфильтровывали с получением 2-хлор-4-(2H-тетразол-5-ил)бензолсульфонамида (5 г, 80,3%) в виде белого твердого вещества. Соединение использовали без дополнительной очистки. МС m/z [М+Н]+ = 260,14.
Промежуточное соединение 98: 2-хлор-4-(2-(4-фторбензил)-2Н-тетразол-5-ил)бензолсульфонамид:
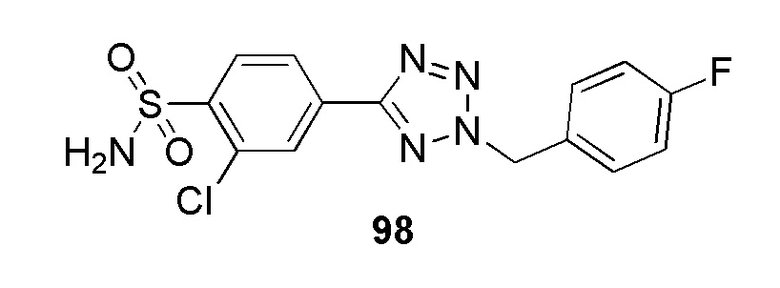
К раствору 2-хлор-4-(2H-тетразол-5-ил)бензолсульфонамида (5 г, 19,255 ммоль, промежуточное соединение 97) в ацетонитриле (100 мл) добавляли N, N-диизопропилэтиламин (6,7 мл, 38,51 ммоль, коммерческий источник: Finar) с последующим добавлением 2-(бромметил)-5-фторпиридина (0,169 г, 1,172 ммоль, коммерческий источник: Spectrochem) при 26°С. Реакционную смесь нагревали до 85°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и разбавляли этилацетатом (200 мл) и водой (50 мл). Смесь перемешивали в течение 15 мин и затем оба слоя разделяли. Водный слой экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным раствором соли (2×50 мл). Органический слой высушивали над безводным Na2SO4 и фильтровали. Фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 40% этилацетата в гексане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 2-хлор-4-(2-(4-фторбензил)-2Н-тетразол-5-ил)бензолсульфонамида (4,2 г) в виде не совсем белого твердого вещества. МС m/z [М+Н]+ = 368,15.
Промежуточное соединение 99: трет-бутил (2-амино-2-оксоэтил)((2-хлор-4-(2-(4-фторбензил)-2Н-тетразол-5-ил)фенил) сульфонил)карбамат:
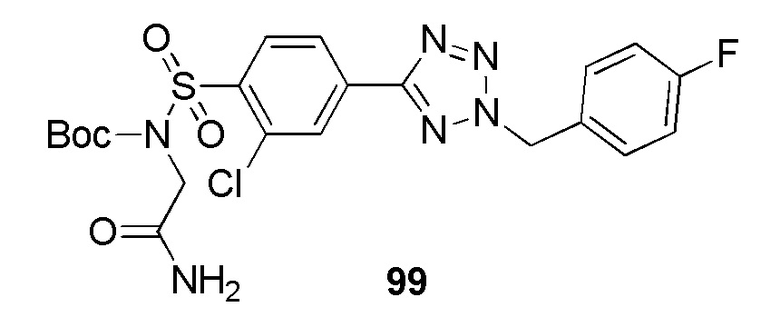
К раствору 2-хлор-4-(2-(4-фторбензил)-2Н-тетразол-5-ил) бензолсульфонамида (1 г, 2,719 ммоль, промежуточное соединение 98) в тетрагидрофуране (20 мл) добавляли 4-(диметиламино)пиридин (166 мг, 1,36 ммоль) и триэтиламин (0,57 мл, 4,08 ммоль) с последующим добавлением Boc-ангидрида (1,78 г, 8,157 ммоль) при 0°С. Реакционную смесь перемешивали при 26°С в течение 6 ч. Затем добавляли карбонат калия (0,75 г, 5,438 ммоль) и бромацетонитрил (0,56 г, 4,08 ммоль) при 26°С. Полученную реакционную смесь перемешивали при 26°С в течение 16 ч. По окончании реакции реакционную смесь гасили ледяной водой (50 мл) и экстрагировали этилацетатом (3×50 мл). Органический слой высушивали над безводным Na2SO4 и фильтровали. Фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 50% этилацетата в гексане. Чистые фракции собирали и концентрировали при пониженном давлении с получением трет-бутил (2-амино-2-оксоэтил)((2-хлор-4-(2-(4-фторбензил)-2Н-тетразол-5-ил)фенил) сульфонил)карбамата (440 мг, 27%) в виде не совсем белого твердого вещества. МС m/z [М+Н]+ = 525,40.
Промежуточное соединение 100: 4-амино-2-фторбензонитрил:
К раствору 4-бром-3-фторанилина (5,0 г, 26,46 ммоль, коммерческий источник: Matrix) в N, N-диметилформамиде (50 мл) добавляли Zn(CN)2 (9,3 г, 79,38 ммоль, коммерческий источник: Sigma Aldrich) при 26°С. Реакционную смесь продували азотом в течение 10 мин, добавляли тетракис(трифенилфосфин)палладий (0) (6,1 г, 5,29 ммоль, коммерческий источник: Alfa Aesar) и снова продували азотом в течение 10 мин при 26°С. Полученную реакционную смесь перемешивали при 130°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и разбавляли этилацетатом (2×500 мл). Органический слой промывали водой (2×100 мл), высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией с использованием диоксида кремния (100-200 меш), элюируя смесью 15% этилацетата в петролейном эфире. Чистые фракции концентрировали при пониженном давлении с получением 4-амино-2-фторбензонитрила (2,7 г, 72%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,38-7,31 (м, 1H), 6,47-6,38 (м, 2H), 4,26 (шир. с, 2H). МС m/z [М+Н]+ = 137,06.
Промежуточное соединение 101: 4-циано-3-фторбензол-1-сульфонилхлорид:
К раствору 4-амино-2-фторбензонитрила (2,0 г, 14,69 ммоль, промежуточное соединение 100) в уксусной кислоте (5 мл) и концентрированной HCl (10 мл) медленно добавляли водный раствор NaNO2 (1,52 г, 22,03 ммоль, коммерческий источник: Finar) при 0°С и перемешивали в течение 30 мин. Эту реакционную смесь добавляли к приготовленному насыщенному раствору SO2 в уксусной кислоте (36 мл) и хлориде меди (II) (0,987 г, 7,34 ммоль, коммерческий источник: Alfa Aesar) при -5°С и перемешивали при комнатной температуре в течение 16 ч. По окончании реакции реакционную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 4-циано-3-фторбензол-1-сульфонилхлорида (1,5 г, неочищенное соединение) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,01-7,91 (м, 1H), 7,63-7,56 (м, 1H), 7,39-7,31 (м, 1H).
Промежуточное соединение 102: 4-циано-3-фтор-N-(2-гидроксиэтил)бензолсульфонамид:
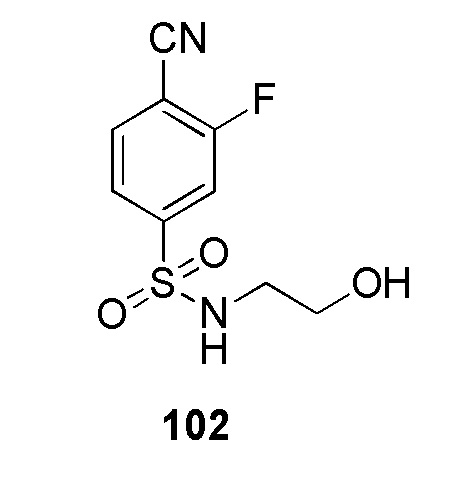
Триэтиламин (2,3 мл, 17,07 ммоль, коммерческий источник: Finar) добавляли к раствору 4-циано-3-фторбензол-1-сульфонилхлорида (1,5 г, 6,83 ммоль, промежуточное соединение 101) в тетрагидрофуране (30 мл) с последующим добавлением 2-аминоэтанола (500 мг, 8,14 ммоль, коммерческий источник: Avra) при 0°С. Реакционной смеси давали нагреться до 26°С и перемешивали в течение 3 ч при той же температуре. По окончании реакции реакционную смесь разбавляли этилацетатом (100 мл). Органический слой промывали водой (3×30 мл), высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией с использованием диоксида кремния (100-200 меш), элюируя смесью 2,5% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением 4-циано-3-фтор-N-(2-гидроксиэтил)бензолсульфонамида (700 мг, 42,8%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,21-8,16 (м, 1Н), 8,03 (шир. с, 1Н), 7,91-7,87 (м, 1Н), 7,83-7,73 (м, 1Н), 4,73-4,67 (м, 1Н), 3,41-3,35 (м, 2Н), 2,91-2,87 (м, 2Н). МС m/z [М-Н]- = 243,12.
Промежуточное соединение 103: 3-фтор-N-(2-гидроксиэтил)-4- (2H-тетразол-5-ил)бензолсульфонамид:
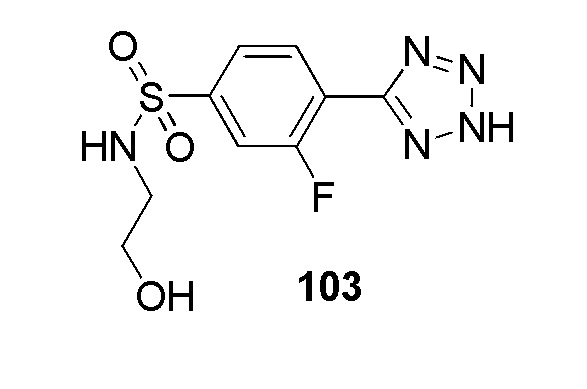
К раствору 4-циано-3-фтор-N-(2-гидроксиэтил)бензолсульфонамида (700 мг, 2,86 ммоль, промежуточное соединение 102) в тетрагидрофуране (7 мл) и воде (0,7 мл) добавляли азид натрия (708 мг, 10,89 ммоль) и бромид цинка (1,22 г, 5,44 ммоль, коммерческий источник: Alfa Aesar) при 26°С. Реакционную смесь нагревали до 85°С и перемешивали в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 0°С и подкисляли концентрированную HCl (40 мл) до pH 2. Осажденное твердое вещество отфильтровывали, промывали водой и высушивали с получением 3-фтор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил)бензолсульфонамида (500 мг, 58,9%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,32-8,25 (м, 1Н), 7,94-7,90 (м, 1Н), 7,88-7,82 (м, 2Н), 4,65 (шир. с, 1Н), 3,39 (т, J=6,1 Гц, 2H), 2,92-2,87 (м, 2H). МС m/z [М-Н]- = 286,15.
Промежуточное соединение 104: 4-бром-2,3-дифторбензолсульфонилхлорид:
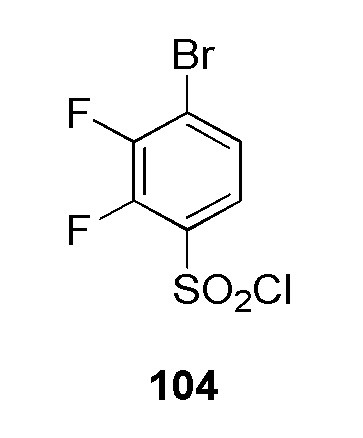
К раствору 4-бром-2,3-дифторанилина (5,0 г, 24,16 ммоль, коммерческий источник: Combi-Blocks) в уксусной кислоте (12,5 мл) и концентрированной HCl (25 мл) медленно добавляли водный раствор NaNO2 (2,5 г, 36,24 ммоль, коммерческий источник: Avra) при 0°С и перемешивали в течение 30 мин. Эту реакционную смесь добавляли к приготовленному насыщенному раствору SO2 в уксусной кислоте (90 мл) и хлориде меди (II) (1,62 г, 12,08 ммоль, коммерческий источник: Alfa Aesar) при -5°С, и затем перемешивали при комнатной температуре в течение 16 ч. По окончании реакции реакционную смесь разбавляли водой (200 мл), экстрагировали этилацетатом (3×100 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 4-бром-2,3-дифторбензолсульфонилхлорида (5 г, неочищенное соединение) в виде липкого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 6 7,68-7,65 (м, 1H), 7,61-7,57 (м, 1H).
Промежуточное соединение 105: 4-бром-2,3-дифтор-N-(2-гидроксиэтил)бензолсульфонамид:
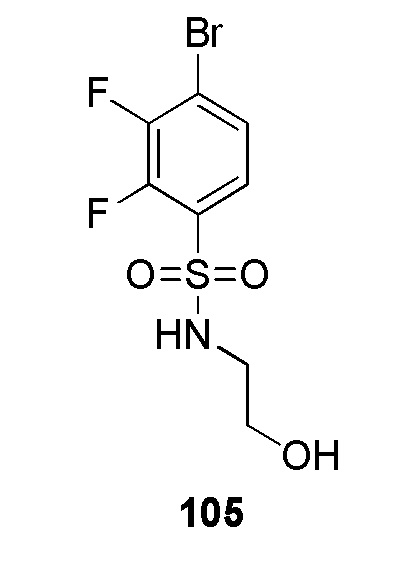
К раствору 4-бром-2,3-дифторбензолсульфонилхлорида (5 г, 17,24 ммоль, промежуточное соединение 104) в тетрагидрофуране (100 мл) добавляли триэтиламин (6,0 мл, 43,12 ммоль, коммерческий источник: Finar) с последующим добавлением 2-аминоэтанола (1,65 г, 20,69 ммоль, коммерческий источник: Avra) при 0°С. Реакционной смеси давали нагреться до 26°С и ее перемешивали в течение 3 ч при той же температуре. По окончании реакции реакционную смесь разбавляли этилацетатом (200 мл) и промывали водой (50 мл), насыщенным раствором соли (2×100 мл), высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией, элюируя смесью 50% этилацетата в петролейном эфире. Чистые фракции концентрировали при пониженном давлении с получением 4-бром-2,3-дифтор-N-(2-гидроксиэтил) бензолсульфонамида (2,8 г, 42%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,18-8,14 (м, 1H), 7,76-7,71 (м, 1H), 7,57-7,53 (м, 1H), 4,66-4,62 (м, 1H), 3,40-3,35 (м, 2Н), 2,98-2,94 (м, 2Н). МС m/z [М-Н]- = 316,14.
Промежуточное соединение 106: 4-циано-2,3-дифтор-N-(2-гидроксиэтил)бензолсульфонамид:
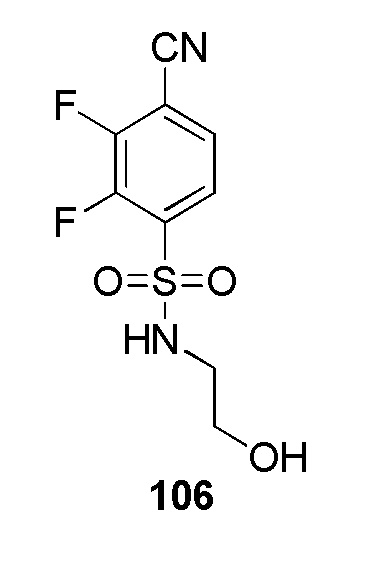
К раствору 4-бром-2,3-дифтор-N-(2-гидроксиэтил)бензолсульфонамида (2,8 г, 8,89 ммоль, промежуточное соединение 105) в N, N-диметилформамиде (28 мл) добавляли Zn(CN)2 (3,9 г, 33,78 ммоль, коммерческий источник: Sigma Aldrich) при 26°С. Реакционную смесь продували азотом в течение 10 мин, добавляли тетракис(трифенилфосфин)палладий (0) (2,0 г, 1,77 ммоль, коммерческий источник: Alfa Aesar) и снова продували азотом в течение 15 мин при 26°С. Полученную реакционную смесь перемешивали при 150°С в микроволновом реакторе в течение 30 мин. По окончании реакции реакционную смесь разбавляли этилацетатом (100 мл), фильтровали через слой целита и упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией, элюируя смесью 50% этилацетата в петролейном эфире. Чистые фракции концентрировали при пониженном давлении с получением 4-циано-2,3-дифтор-N-(2-гидроксиэтил)бензолсульфонамида (1,2 г, 41%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,42-8,38 (м, 1Н), 7,95-7,90 (м, 1Н), 7,76-7,72 (м, 1Н), 4,67-4,63 (м, 1Н), 3,41-3,36 (м, 2Н), 3,02-2,99 (м, 2Н). МС m/z [М-Н]- = 261,22.
Промежуточное соединение 107: 2,3-дифтор-N-(2-гидроксиэтил) -4-(2Н-тетразол-5-ил)бензолсульфонамид:
К раствору 4-циано-2,3-дифтор-N-(2-гидроксиэтил) бензолсульфонамида (1,2 г, 4,57 ммоль, промежуточное соединение 106) в тетрагидрофуране (12 мл) и воде (1,2 мл) добавляли азид натрия (1,13 г, 17,40 ммоль) и бромид цинка (1,95 г, 8,7 ммоль) при 26°С. Реакционную смесь нагревали до 90°С и перемешивали в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 0°С и подкисляли концентрированной HCl (10 мл) до pH 2. Осажденное твердое вещество отфильтровывали, промывали водой и высушивали с получением 2,3-дифтор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил)бензолсульфонамида (700 мг, 46,8%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,31-8,27 (м, 1Н), 8,07-8,01 (м, 1Н), 7,82-7,77 (м, 1Н), 3,42-3,38 (м, 2Н), 3,04-2,98 (м, 2Н). МС m/z [М-Н]- = 304,19.
Промежуточное соединение 108: 4-бром-2,6-дифторбензол-1-сульфонилхлорид:
К раствору 4-бром-2,6-дифторанилина (5,0 г, 24,03 ммоль, коммерческий источник: Combi-Blocks) в уксусной кислоте (12 мл) и концентрированной HCl (25 мл) медленно добавляли водный раствор NaNO2 (2,5 г, 36,05 ммоль, коммерческий источник: Avra) при 0°С и перемешивали в течение 45 мин при -5°С. Эту реакционную смесь добавляли к приготовленному насыщенному раствору SO2 в уксусной кислоте (50 мл) и хлориде меди (II) (0,96 г, 7,16 ммоль, коммерческий источник: Combi-Blocks) и перемешивали при комнатной температуре в течение 2 ч. По окончании реакции реакционную смесь экстрагировали этилацетатом (1 л). Органический слой промывали водой (500 мл) и насыщенным раствором соли (100 мл), высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 4-бром-2,6-дифторбензол-1-сульфонилхлорида (5 г, 71%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,38-7,30 (м, 2Н).
Промежуточное соединение 109: 4-бром-2,6-дифтор-N-(2-гидроксиэтил)бензолсульфонамид:
К раствору 4-бром-2,6-дифторбензол-1-сульфонилхлорида (5 г, 17,24 ммоль, промежуточное соединение 108) в тетрагидрофуране (50 мл) добавляли триэтиламин (4,8 мл, 34,38 ммоль, коммерческий источник: RCP) с последующим добавлением 2-аминоэтанола (1,05 г, 17,24 ммоль, коммерческий источник: RCP) при 0°С. Реакционной смеси давали нагреться до 28°С и перемешивали в течение 5 ч при той же температуре. По окончании реакции реакционную смесь разбавляли этилацетатом (700 мл). Органический слой промывали водой (500 мл) и насыщенным раствором соли (100 мл), высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью системы флэш-хроматографии Reveleris®, элюируя смесью 10-20% этилацетата в петролейном эфире. Чистые фракции концентрировали при пониженном давлении с получением 4-бром-2,6-дифтор-N-(2-гидроксиэтил)бензолсульфонамида (3 г, 55%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,31-8,27 (м, 1Н), 7,73-7,68 (м, 2Н), 4,71-4,66 (м, 1Н), 3,41-3,38 (м, 2Н), 3,03-2,97 (м, 2Н). МС m/z [М-Н]- = 314,13.
Промежуточное соединение 110: 4-циано-2,6-дифтор-N-(2-гидроксиэтил)бензолсульфонамид:
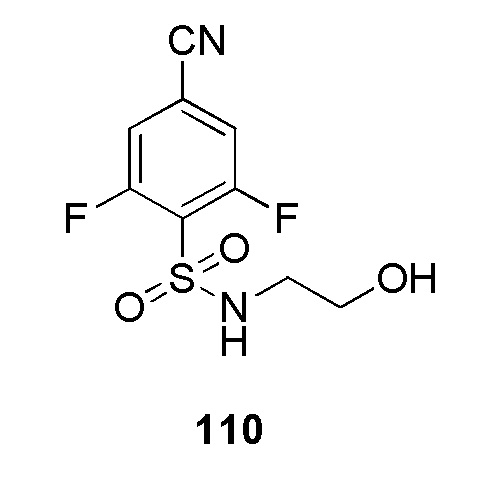
К раствору 4-бром-2,6-дифтор-N-(2-гидроксиэтил) бензолсульфонамида (3 г, 9,55 ммоль, промежуточное соединение 109) в N, N-диметилформамиде (30 мл) добавляли Zn(CN)2 (5,0 г, 28,66 ммоль, коммерческий источник: Sigma Aldrich) при 26°С. Реакционную смесь продували азотом в течение 10 мин, добавляли тетракис(трифенилфосфин)палладий (0) (1,1 г, 0,95 ммоль, коммерческий источник: Alfa Aesar) и снова продували азотом в течение 15 мин при 26°С. Полученную реакционную смесь перемешивали при 80°С в течение 18 ч. По окончании реакции реакционную смесь выливали в воду (500 мл) и экстрагировали этилацетатом (500 мл). Органический слой промывали водой (3×100 мл) и насыщенным раствором соли (50 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью системы флэш-хроматографии Reveleris®, элюируя смесью 20% этилацетата в петролейном эфире. Чистые фракции концентрировали при пониженном давлении с получением 4-циано-2,6-дифтор-N-(2-гидроксиэтил)бензолсульфонамида (1,5 г, 60%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,00-7,93 (м, 2Н), 7,59-7,52 (м, 1Н), 4,64 (т, J=5,4 Гц, 1Н), 3,39 (кв, J=5,9 Гц, 2H), 3,04 (кв, J=6,1 Гц, 2H). МС m/z [М-Н]- = 261,26.
Промежуточное соединение 111: 2,6-дифтор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил)бензолсульфонамид:
К раствору 4-циано-2,6-дифтор-N-(2-гидроксиэтил) бензолсульфонамида (1,5 г, 5,72 ммоль, промежуточное соединение 110) в тетрагидрофуране (15 мл) и воде (1,5 мл) добавляли азид натрия (1,4 г, 21,73 ммоль, коммерческий источник: Avra) и бромид цинка (2,4 г, 10,86 ммоль, коммерческий источник: Combi-Blocks) при 26°С. Реакционную смесь нагревали до 85°С и перемешивали в течение 12 ч. По окончании реакции реакционную смесь охлаждали до 0°С и подкисляли концентрированной HCl (30 мл) до pH 2. Осажденное твердое вещество отфильтровывали, промывали водой и высушивали с получением 2,6-дифтор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил)бензолсульфонамида (1 г, неочищенное соединение) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,75-7,67 (м, 2Н), 7,41-7,35 (м, 1Н), 4,75-4,68 (м, 1Н), 3,43-3,38 (м, 2Н), 3,06-2,98 (м, 2Н). МС m/z [М-Н]- = 304,22.
Промежуточное соединение 112: 4-(2-((тетрагидро-2Н-пиран-4-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамид:
К раствору 4-(2H-тетразол-5-ил)бензолсульфонамида (1 г, 4,439 ммоль, промежуточное соединение 43) в N, N-диметилформамиде (20 мл) добавляли карбонат калия (1,2 г, 8,69 ммоль, коммерческий источник: RCP) с последующим добавлением 4-(бромметил)тетрагидро-2H-пирана (874 мг, 4,88 ммоль, коммерческий источник: Sigma Aldrich) при 26°С. Реакционную смесь нагревали до 100°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией, элюируя смесью 3% метанола в DCM. Чистые фракции концентрировали при пониженном давлении с получением 4-(2- ((тетрагидро-2Н-пиран-4-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида (500 мг) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,30-8,19 (м, 2Н), 8,04-7,93 (м, 2Н), 7,48 (шир. с, 2Н), 4,76-4,66 (м, 2Н), 3,90-3,74 (м, 2Н), 3,26-3,16 (м, 1Н), 2,71-2,62 (м, 1Н), 2,34-2,25 (м, 1Н), 1,58-1,46 (м, 2Н), 1,41-1,30 (м, 2Н). МС m/z [М+Н]+ = 324,18. Соединение использовали без дополнительной очистки.
Промежуточное соединение 113: трет-бутил (цианометил)((4-(2 -((тетрагидро-2Н-пиран-4-ил)метил)-2Н-тетразол-5-ил)фенил) сульфонил)карбамат:
К раствору 4-(2-((тетрагидро-2Н-пиран-4-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида (500 мг, 1,548 ммоль, промежуточное соединение 112) в тетрагидрофуране (20 мл) добавляли DMAP (94,4 мг, 0,773 ммоль, коммерческий источник: Avra), триэтиламин (0,3 мл, 2,15 ммоль, коммерческий источник: RCP) с последующим добавлением Boc-ангидрида (0,71 мл, 3,09 ммоль, коммерческий источник: Avra) и перемешивали при 26°С в течение 5 ч. Затем добавляли карбонат калия (426,9 мг, 3,08 ммоль, коммерческий источник: RCP) с последующим добавлением бромацетонитрила (0,16 мл, 2,297 ммоль, коммерческий источник: Avra) и перемешивали при 26°С в течение 16 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией, элюируя смесью 40% этилацетата в петролейном эфире. Чистые фракции концентрировали при пониженном давлении с получением трет-бутил (цианометил)((4-(2-((тетрагидро-2Н-пиран-4-ил)метил)-2Н-тетразол-5-ил)фенил)сульфонил)карбамата (400 мг) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,38-8,32 (м, 2Н), 8,19-8,13 (м, 2Н), 4,94 (с, 2Н), 4,74-4,67 (м, 2Н), 3,91-3,79 (м, 2Н), 3,76-3,70 (м, 1Н), 3,34-3,29 (м, 2Н), 2,34-2,22 (м, 2Н), 2,04-1,94 (м, 1Н), 1,55 -1,45 (м, 1Н), 1,29-1,18 (м, 9Н). МС m/z [М+Н]+ = 463,30. Соединение использовали без дополнительной очистки.
Промежуточное соединение 114: этил 2-(4-цианофенилсульфонамидо)ацетат:
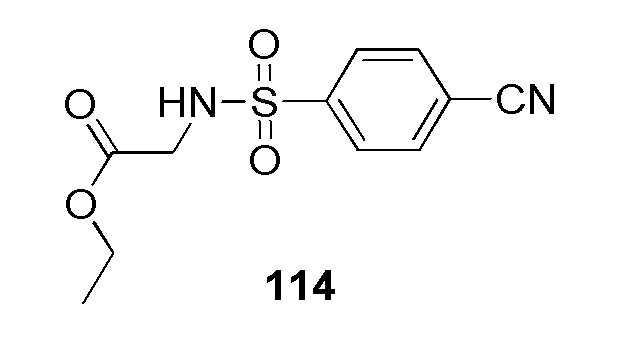
К раствору этил 2-аминоацетата гидрохлорида (8,27 г, 59,496 ммоль, коммерческий источник: Avra) в N, N-диметилформамиде (100 мл) добавляли триэтиламин (13,8 мл, 99,208 ммоль, коммерческий источник: RCP) при 27°С с последующим добавлением 4-цианобензол-1-сульфонилхлорида (10 г, 49,6 ммоль, коммерческий источник: Combi-Blocks) при 0°С. Реакционной смеси давали медленно нагреться до 27°С и перемешивали в течение 5 ч при той же температуре. По окончании реакции реакционную смесь выливали в ледяную воду (2 л). Осажденное твердое вещество отфильтровывали и высушивали в вакууме с получением этил 2-(4-цианофенилсульфонамидо)ацетата (8 г, неочищенное соединение) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,51 (шир. с, 1H), 8,07 (д, J=8,3 Гц, 2H), 7,96 (д, J=8,6 Гц, 2H), 3,97 (кв, J=7,0 Гц, 2H), 3,78 (с, 2H), 1,09 (т, J=7,1 Гц, 3H). МС m/z [М-Н]- = 267,07.
Промежуточное соединение 115: этил 2-(4-(2H-тетразол-5-ил) фенилсульфонамидо)ацетат:
К раствору этил 2-(4-цианофенилсульфонамидо)ацетата (20 г, 74,546 ммоль, промежуточное соединение 114) в N, N-диметилформамиде (250 мл) добавляли азид натрия (48,5 г, 746,04 ммоль, коммерческий источник: Spectrochem) и хлорид аммония (39,9 г, 745,79 ммоль, коммерческий источник: Chemlabs) при 27°С. Реакционную смесь нагревали до 80°С и перемешивали в течение 16 ч при той же температуре. По окончании реакции реакционную смесь охлаждали до 0°С и гасили 1 Н HCl (800 мл). Осажденное твердое вещество отфильтровывали и высушивали в вакууме с получением этил 2-(4-(2H-тетразол-5-ил)фенилсульфонамидо)ацетата (15 г, неочищенное соединение) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,40 (т, J=6,0 Гц, 1H), 8,27 (д, J=8,3 Гц, 2H), 8,01 (д, J=8,3 Гц, 2H), 3,98 (кв, J=7,0 Гц, 2H), 3,77 (д, J=6,1 Гц, 2H), 1,09 (т, J=7,0 Гц, 3H). МС m/z [М+Н]+ = 312,04.
Промежуточное соединение 116: 2-(хлорметил)пиразин:
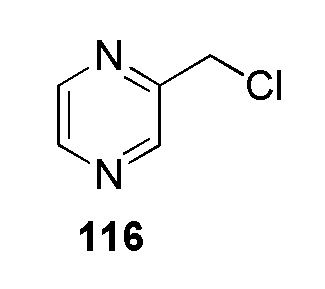
К раствору пиразин-2-илметанола (500 мг, 4,545 ммоль, коммерческий источник: Combi-Blocks) в дихлорметане (20 мл) добавляли тионилхлорид (0,4 мл, 5,514 ммоль, коммерческий источник: Avra) при 0°С. Реакционной смеси давали нагреться до 26°С и перемешивали в течение 16 ч при той же температуре. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Остаток нейтрализовали насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали этилацетатом (3×20 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 2-(хлорметил)пиразина (400 мг, неочищенное соединение) в виде коричневой жидкости. 1H ЯМР (400 МГц, CDCl3) δ 8,76 (с, 1H), 8,59-8,54 (м, 2H), 4,70 (с, 2H). МС m/z [М+Н]+ = 129,02. Соединение использовали без дополнительной очистки.
Промежуточное соединение 117: этил 2-(4-(2-(пиразин-2-илметил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетат:
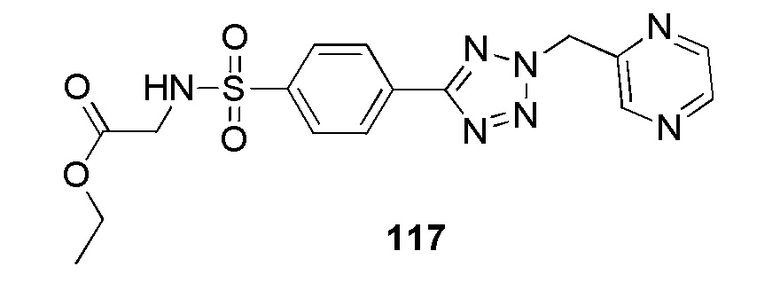
К раствору этил 2-(4-(2H-тетразол-5-ил)фенилсульфонамидо) ацетата (500 мг, 0,0016 моль, промежуточное соединение 115), 2-(хлорметил)пиразина (204 мг, 0,0016 моль, промежуточное соединение 116) в ацетонитриле (5 мл) добавляли N, N-диизопропилэтиламин (0,55 мл, 0,0032 моль, коммерческий источник: Finar) при 26°С. Реакционную смесь нагревали до 90°С и перемешивали в течение 8 ч при той же температуре. По окончании реакции реакционную смесь охлаждали до 26°С, растворяли в этилацетате (100 мл) и промывали водой (2×50 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении с получением этил 2-(4-(2-(пиразин-2-илметил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетата (400 мг, неочищенное соединение) в виде не совсем белого липкого твердого вещества. МС m/z [М+Н]+ = 403,21. Соединение использовали без дополнительной очистки.
Промежуточное соединение 118: 2-(4-(2-(пиразин-2-илметил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетамид:
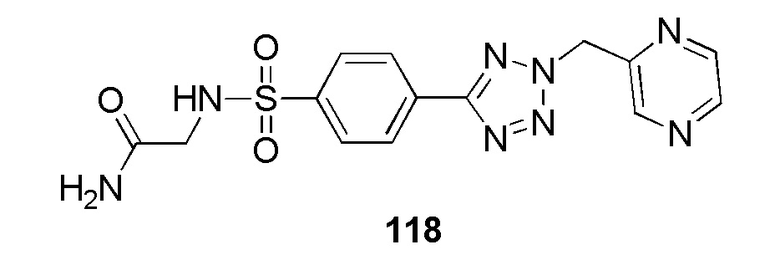
К раствору этил 2-(4-(2-(пиразин-2-илметил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетата (400 мг, 0,0009 моль, промежуточное соединение 117) в метаноле (8 мл) добавляли метанольный раствор аммиака (4 М раствор в МеОН) (4 мл, коммерческий источник: HYCHEM) при 26°С. Реакционную смесь нагревали до 60°С и перемешивали в течение 48 ч при той же температуре в герметичной пробирке. По окончании реакции реакционную смесь охлаждали до 26°С и концентрировали при пониженном давлении. Остаток растворяли в этилацетате (100 мл) и промывали водой (2×50 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 2-(4-(2-(пиразин-2-илметил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамида (200 мг, 41%) в виде не совсем белого твердого вещества. МС m/z [М+Н]+ = 375,05. Соединение использовали без дополнительной очистки.
Промежуточное соединение 119: 2-(хлорметил)-5-метилпиразин:
К раствору (5-метилпиразин-2-ил)метанола (500 мг, 4,028 ммоль, коммерческий источник: Combi-Blocks) в дихлорметане (20 мл) медленно добавляли тионилхлорид (0,35 мл, 4,825 ммоль, коммерческий источник: Avra) при 0°C. Реакционной смеси давали нагреться до 26°С и перемешивали в течение 16 ч при той же температуре. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Остаток нейтрализовали насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали этилацетатом (3×20 мл). Объединенный органический раствор высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 2-(хлорметил)-5-метилпиразина (500 мг, неочищенное соединение) в виде коричневой жидкости. 1H ЯМР (400 МГц, CDCl3) δ 8,60 (с, 1Н), 8,43 (с, 1Н), 4,67 (с, 2Н), 2,59 (с, 3Н). МС m/z [М+Н]+ = 143,04.
Промежуточное соединение 120: этил 2-(4-(2-((5-метилпиразин-2-ил)метил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетат:
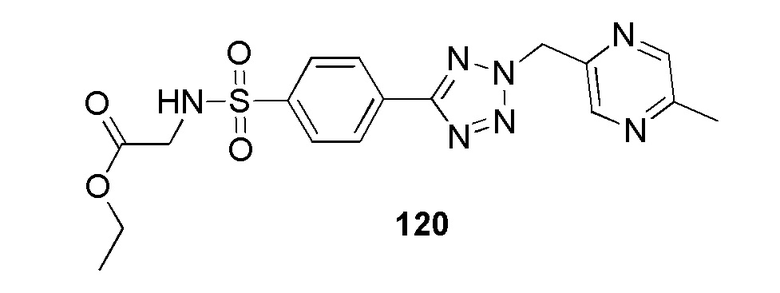
К раствору этил 2-(4-(2H-тетразол-5-ил)фенилсульфонамидо) ацетата (500 мг, 0,0016 моль, промежуточное соединение 115), 2- (хлорметил)-5-метилпиразина (228 мг, 0,0016 моль, промежуточное соединение 119) в ацетонитриле (5 мл) добавляли N, N-диизопропилэтиламин (0,55 мл, 0,0032 моль, коммерческий источник: Finar) при 26°С. Реакционную смесь нагревали до 80°С и перемешивали в течение 8 ч при той же температуре. По окончании реакции реакционную смесь охлаждали до 26°С, растворяли в этилацетате (100 мл) и промывали водой (2×30 мл). Органический раствор высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением этил 2-(4-(2-((5-метилпиразин-2-ил)метил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетата (400 мг, неочищенное соединение) в виде не совсем белого липкого твердого вещества. МС m/z [М+Н]+ = 418,23. Вещество использовали на следующей стадии без какой-либо дополнительной очистки.
Промежуточное соединение 121: 2-(4-(2-((5-метилпиразин-2-ил) метил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетамид:
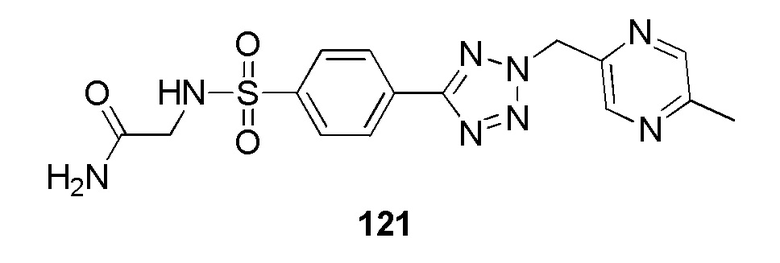
К раствору этил 2-(4-(2-((5-метилпиразин-2-ил)метил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетата (400 мг, 0,0009 моль, промежуточное соединение 120) в метаноле (8 мл) добавляли метанольный раствор аммиака (4 М раствор в МеОН) (4 мл, коммерческий источник: HYCHEM) при 26°С. Реакционную смесь нагревали до 60°С и перемешивали в течение 48 ч при той же температуре в герметичной пробирке. По окончании реакции реакционную смесь охлаждали до 26°С и концентрировали при пониженном давлении. Остаток растворяли в этилацетате (100 мл) и промывали водой (3×40 мл). Органический раствор высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении с получением 2-(4-(2-((5-метилпиразин-2-ил)метил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетамида (220 мг, неочищенное соединение) в виде не совсем белого твердого вещества. МС m/z [М+Н]+ = 389,17. Вещество использовали на следующей стадии без какой-либо дополнительной очистки.
Промежуточное соединение 122: 2-(хлорметил)-5-метоксипиразин:
К раствору (5-метоксипиразин-2-ил)метанола (200 мг, 0,0014 моль, коммерческий источник: Combi-Blocks) в дихлорметане (2 мл) медленно добавляли тионилхлорид (509 мг, 0,0042 моль, коммерческий источник: Avra) при 0°С. Реакционной смеси давали нагреться до 26°С и перемешивали в течение 1 ч при той же температуре. По окончании реакции реакционную смесь медленно выливали в насыщенный раствор бикарбоната натрия (50 мл) при 0°С и экстрагировали этилацетатом (3×30 мл). Органический раствор высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 2-(хлорметил)-5-метоксипиразина (250 мг, неочищенное соединение) в виде бледно-желтой жидкости. 1H ЯМР (400 МГц, CDCl3) δ 8,22-8,18 (2Н), 4,65 (с, 2Н), 3,98 (с, 3Н). МС m/z [М+Н]+ = 159,01.
Промежуточное соединение 123: 2-(4-(2-((5-метоксипиразин-2-ил)метил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетамид:
К раствору 2-(4-(2H-тетразол-5-ил)фенилсульфонамидо) ацетамида (400 мг, 0,0014 моль, промежуточное соединение 25), 2- (хлорметил)-5-метоксипиразина (246 мг, 0,0015 моль, промежуточное соединение 122) в ацетонитриле (4 мл) добавляли N, N-диизопропилэтиламин (0,48 мл, 0,0028 моль, коммерческий источник: Finar) при 26°С. Реакционную смесь нагревали до 80°С и перемешивали в течение 8 ч при той же температуре. По окончании реакции реакционную смесь охлаждали до 26°С, растворяли в этилацетате (100 мл) и промывали водой (2×50 мл). Органический раствор высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении с получением 2-((4-(2-((5-метоксипиразин-2-ил)метил)-2Н-тетразол-5-ил)фенил)сульфонамидо)ацетамида (250 мг, неочищенное соединение) в виде не совсем белого твердого вещества. МС m/z [М+Н]+ = 405,12. Вещество использовали на следующей стадии без какой-либо дополнительной очистки.
Промежуточное соединение 124: 4-(2-((6-метоксипиридин-3-ил) метил)-2Н-тетразол-5-ил)бензолсульфонамид:
К раствору 4-(2H-тетразол-5-ил)бензолсульфонамида (500 мг, 2,22 ммоль, промежуточное соединение 43) в ДМФА (20 мл) добавляли карбонат калия (613 мг, 4,44 ммоль, коммерческий источник: RCP) с последующим добавлением 5-(хлорметил)-2-метоксипиридина (420 мг, 2,66 ммоль, коммерческий источник: Enamine) при 26°С. Реакционную смесь нагревали до 80°С в течение 5 ч. По окончании реакции реакционную смесь охлаждали до 26°С и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией, элюируя смесью 3% метанола в DCM. Чистые фракции концентрировали при пониженном давлении с получением 4-(2-((6-метоксипиридин-3-ил)метил)-2Н-тетразол-5-ил) бензолсульфонамида (2) (500 мг, неочищенное соединение). МС m/z [М+Н]+ = 347,19. Соединение использовали без дополнительной очистки.
Промежуточное соединение 125: трет-бутил (цианометил)((4-(2 -((6-метоксипиридин-3-ил)метил)-2Н-тетразол-5-ил)фенил) сульфонил)карбамат:
К раствору 4-(2-((6-метоксипиридин-3-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида (500 мг, 1,445 ммоль, промежуточное соединение 124) в ТГФ (10 мл) добавляли DMAP (88 мг, 0,72 ммоль, коммерческий источник: Avra) и триэтиламин (0,3 мл, 2,158 ммоль, коммерческий источник: RCP) с последующим добавлением Boc-ангидрида (0,7 мл, 3,047 ммоль, коммерческий источник: Avra) и перемешивали при 26°С в течение 5 ч. Затем добавляли карбонат калия (400 мг, 2,89 ммоль, коммерческий источник: RCP) с последующим добавлением бромацетонитрила (0,15 мл, 2,15 ммоль, коммерческий источник: Avra) и перемешивали при 26°С в течение 16 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией, элюируя смесью 30% этилацетата в петролейном эфире. Чистые фракции концентрировали при пониженном давлении с получением трет-бутил (цианометил)((4-(2-((6-метоксипиридин-3-ил)метил)-2Н-тетразол-5-ил)фенил)сульфонил)карбамата (100 мг, неочищенное соединение) в виде смолы. МС m/z [М+Н]+ = 486,35. Соединение использовали без дополнительной очистки.
Промежуточное соединение 126: 4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамид:
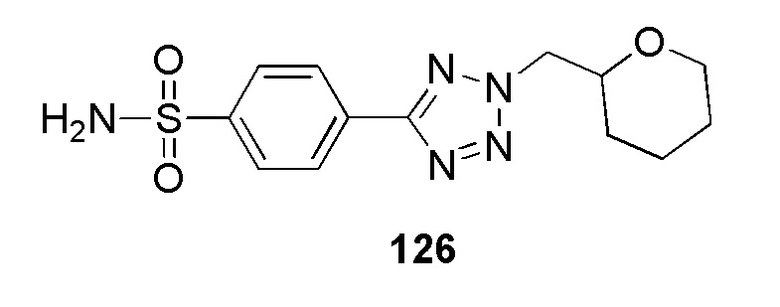
К раствору 4-(2H-тетразол-5-ил)бензолсульфонамида (2,0 г, 8,879 ммоль, промежуточное соединение 43) в ДМФА (50,0 мл) добавляли карбонат калия (2,45 г, 17,754 ммоль, коммерческий источник: RCP) с последующим добавлением 2-(бромметил) тетрагидро-2Н-пирана (1,91 г, 10,667 ммоль, коммерческий источник: Sigma Aldrich) при 26°С. Реакционную смесь нагревали до 80°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 4% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением 4-(2- ((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида (1,0 г, 34,8%). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,26-8,22 (м, 2H), 8,04-7,99 (м, 2H), 7,50 (шир. С, 2H), 4,80-4,76 (м, 2H), 3,95-3,89 (м, 1Н), 3,83-3,77 (м, 1Н), 1,87-1,79 (м, 1Н), 1,75-1,68 (м, 1Н), 1,57-1,50 (м, 1Н), 1,48-1,43 (м, 2Н), 1,39-1,24 (м, 2Н). МС m/z [М+Н]+ = 324,28.
Промежуточное соединение 127: трет-бутил (цианометил)((4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)фенил) сульфонил)карбамат:
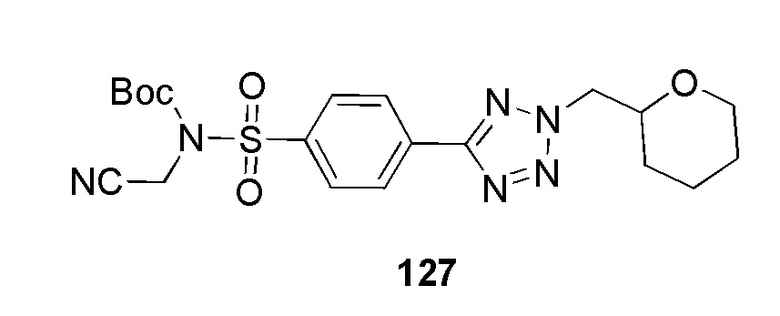
К раствору 4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида (1,0 г, 3,096 ммоль, промежуточное соединение 126) в ТГФ (20 мл) добавляли DMAP (189 мг, 1,547 ммоль, коммерческий источник: Avra) и триэтиламин (0,65 мл, 4,673 ммоль, коммерческий источник: RCP) с последующим добавлением Boc-ангидрида (1,42 мл, коммерческий источник: Avra) и перемешивали при 26°С в течение 6 ч. Затем добавляли карбонат калия (854 мг, 6,188 ммоль, коммерческий источник: RCP) с последующим добавлением бромацетонитрила (0,43 мл, 6,173 ммоль, коммерческий источник: Avra) и перемешивали при 26°С в течение 16 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией, элюируя смесью 30% этилацетата в петролейном эфире. Чистые фракции концентрировали при пониженном давлении с получением трет-бутил (цианометил)((4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)фенил)сульфонил)карбамата (1,0 г, неочищенное соединение) в виде смолы. Соединение использовали без дополнительной очистки.
Промежуточное соединение 128: 4-(2-((4,4-дифторциклогексил) метил)-2Н-тетразол-5-ил)-2-метоксибензолсульфонамид:
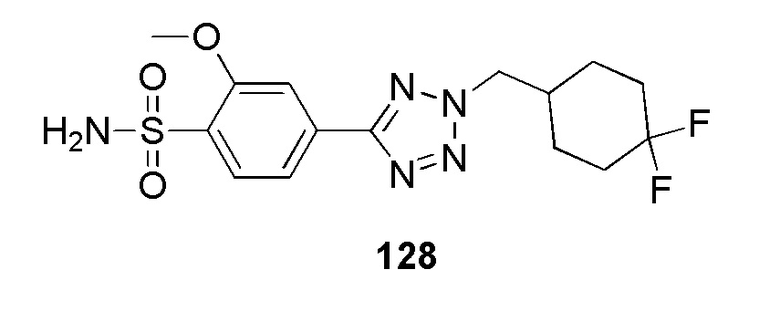
N, N-диизопропилэтиламин (164 мкл, 0,940 ммоль, коммерческий источник: Aldrich) добавляли к перемешиваемому раствору 2-метокси-4-(2H-тетразол-5-ил)бензолсульфонамида (120 мг, 0,470 ммоль, промежуточное соединение 51') и 4-(бромметил)-1,1-дифторциклогексана (70,8 мкл, 0,470 ммоль, коммерческий источник: Combi-Blocks) в N, N-диметилформамиде (ДМФА) (1,57 мл) в атмосфере азота. Смесь перемешивали при комнатной температуре в течение ночи и затем при 70°С в течение 24 ч. Добавляли 4-(бромметил)-1,1-дифторциклогексан (14,17 мкл, 0,094 ммоль, коммерческий источник: Combi-Blocks) и N, N-диизопропилэтиламин (82 мкл, 0,470 ммоль) и смесь перемешивали при 70°С в течение 6 ч. По окончании реакции реакционную смесь разбавляли водой и экстрагировали дихлорметаном (дважды). Объединенный органический раствор высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении с получением 4-(2-((4,4-дифторциклогексил)метил)-2Н-тетразол-5-ил)-2-метоксибензолсульфонамида (177 мг, неочищенное соединение), который использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение 129: трет-бутил (2-амино-2-оксоэтил)((4-(2-((4,4-дифторциклогексил)метил)-2Н-тетразол-5-ил) -2-метоксифенил)сульфонил)карбамат:
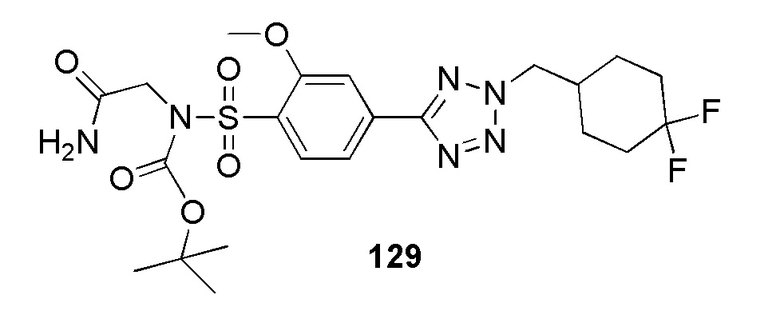
К раствору 4-(2-((4,4-дифторциклогексил)метил)-2Н-тетразол-5-ил)-2-метоксибензолсульфонамида (174 мг, 0,305 ммоль, промежуточное соединение 128) в безводном тетрагидрофуране (7 мл) добавляли ди-трет-бутилдикарбонат (0,21 мл, 0,916 ммоль), Et3N (0,051 мл, 0,366 ммоль) и DMAP (18,66 мг, 0,153 ммоль). Полученный раствор перемешивали при комнатной температуре в течение ночи. Добавляли карбонат калия (84 мг, 0,611 ммоль) и 2-бромацетамид (89 мг, 0,647 ммоль). Реакционную смесь перемешивали при комнатной температуре. Через 6 ч добавляли еще карбонат калия (42,2 мг, 0,305 ммоль) и 2-бромацетамид (42,1 мг, 0,305 ммоль) и смесь перемешивали в течение ночи. Затем реакционную смесь нагревали до 50°С и перемешивали при этой температуре в течение выходных дней. По окончании реакции реакционную смесь гасили насыщенным раствором NaHCO3 и экстрагировали этилацетатом (дважды). Органический слой промывали насыщенным раствором NaCl, высушивали над Na2SO4 и фильтровали. Фильтрат концентрировали при пониженном давлении с получением трет-бутил (2-амино-2-оксоэтил)((4-(2-((4,4-дифторциклогексил)метил)-2Н-тетразол-5-ил)-2-метоксифенил)сульфонил)карбамата (210 мг, неочищенное соединение), который использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение 130: 4-этоксибензилметансульфонат:
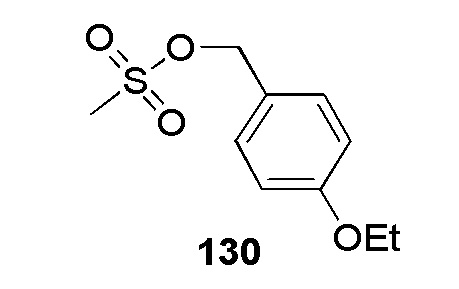
К раствору (4-этоксифенил)метанола (300 мг, 1,971 ммоль, коммерческий источник: Apollo SCI) и триэтиламина (328 мкл, 2,35 ммоль) в дихлорметане (15 мл) при 0°С добавляли по каплям метансульфонилхлорид (168 мкл, 2,168 ммоль). Смесь перемешивали при 0°С в течение 3 ч. По окончании реакции реакционную смесь разбавляли водой и DCM и экстрагировали DCM (2×20 мл). Объединенные органические слои высушивали над безводным MgSO4 и фильтровали. Фильтрат концентрировали при пониженном давлении с получением 4-этоксибензилметансульфоната (454 мг, 100%). Данный продукт использовали без какой-либо очистки в следующей реакции.
Промежуточное соединение 131: 3-(хлорметил)тетрагидро-2Н-пиран:
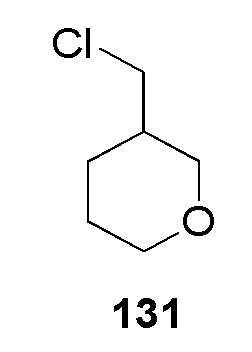
К раствору (тетрагидро-2H-пиран-3-ил)метанола (1 г, 8,621 ммоль, коммерческий источник: Frapps) в дихлорметане (20 мл) медленно добавляли тионилхлорид (1 мл, 13,785 ммоль, коммерческий источник: Avra) при 0°С и затем перемешивали при 26°С в течение 16 ч. По окончании реакции реакционную смесь выпаривали при пониженном давлении, разбавляли насыщенным раствором NaHCO3 (50 мл) и экстрагировали дихлорметаном (3×50 мл). Объединенный органический раствор высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 3-(хлорметил)тетрагидро-2Н-пирана (300 мг). 1H ЯМР (400 МГц, ДМСО-d6) δ 3,90-3,73 (м, 4H), 3,35-3,20 (м, 2H), 1,90-1,81 (м, 1H), 1,77-1,50 (м, 2H), 1,35-1,17 (м, 2Н).
Промежуточное соединение 132: 4-(хлорметил)пиридазин:
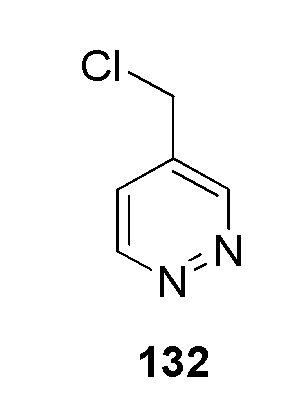
К раствору 4-метилпиридазина (1 г, 10,625 ммоль, коммерческий источник: Combi-Blocks) в ацетонитриле (10,0 мл) добавляли трихлоризоциануровую кислоту (1,2 г, 5,163 ммоль, коммерческий источник: Avra) при 26°С. Реакционную смесь перемешивали при 26°С в течение 3 ч. По окончании реакции реакционную смесь упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 80% этилацетата в петролейном эфире. Чистые фракции концентрировали при пониженном давлении с получением 4- (хлорметил)пиридазина (400 мг, неочищенное соединение). 1H ЯМР (400 МГц, CDCl3) δ 9,26-9,20 (м, 2Н), 7,55-7,51 (м, 1Н), 4,58 (с, 2Н). МС m/z [М+Н]+ = 129,1.
Промежуточное соединение 133: 3-(хлорметил)пиридазин:
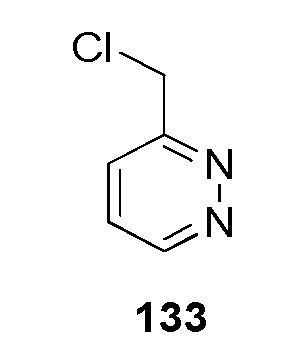
К раствору 3-метилпиридазина (300 мг, 3,18 ммоль, коммерческий источник: Alfa Aesar) в хлороформе (10,0 мл) добавляли трихлоризоциануровую кислоту (360 мг, 1,549 ммоль, коммерческий источник: Avra) при 26°С. Реакционную смесь перемешивали при 26°С в течение 3 ч. По окончании реакции реакционную смесь упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 70% этилацетата в петролейном эфире. Чистые фракции концентрировали при пониженном давлении с получением 3- (хлорметил)пиридазина (200 мг, неочищенное соединение). 1H ЯМР (400 МГц, ДМСО-d6) δ 9,18-9,14 (м, 1Н), 7,75-7,70 (м, 1Н), 7,58-7,51 (м, 1Н), 4,91 (с, 2Н). МС m/z [М+Н]+ = 129,20.
Промежуточное соединение 134: 2-(хлорметил)-5-метоксипиридин:
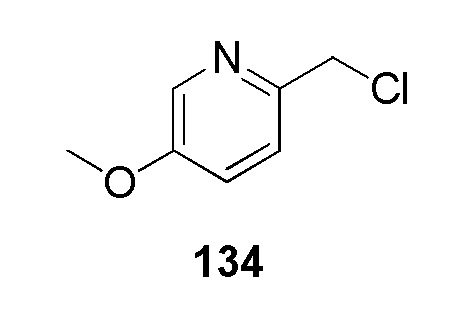
К раствору (5-метоксипиридин-2-ил)метанола (300 мг, 0,0021 моль, коммерческий источник: Combi-Blocks) в дихлорметане (3 мл) медленно добавляли тионилхлорид (0,509 мл, 0,0064 моль, коммерческий источник: Avra) при 0°С. Реакционной смеси давали нагреться до 26°С и перемешивали в течение 1 ч при той же температуре. По окончании реакции реакционную смесь медленно выливали в насыщенный раствор бикарбоната натрия (100 мл) при 0°С и экстрагировали этилацетатом (3×30 мл). Объединенный органический раствор высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 2-(хлорметил)-5-метоксипиразина (280 мг, 82%) в виде коричневой жидкости. 1H ЯМР (400 МГц, CDCl3) δ 8,29-8,26 (м, 1Н), 7,40-7,36 (м, 1Н), 7,23-7,20 (м, 1Н), 4,65-4,63 (м, 2Н), 3,87 (с, 3Н). МС m/z [М+Н]+ = 158,06.
Промежуточное соединение 135: 4-бром-2-фтор-5-метилбензол-1-сульфонилхлорид:
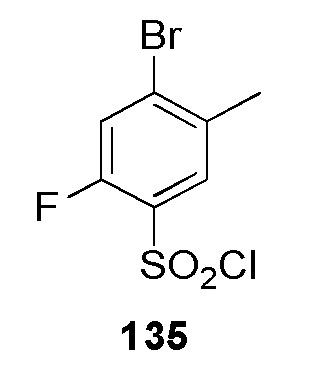
К 2-бром-4-фтор-1-метилбензолу (6,0 г, 31,9 ммоль, коммерческий источник: Combi-Blocks) добавляли по каплям хлорсульфоновую кислоту (6,5 г, 55 ммоль, коммерческий источник: Avra) при 0°С. Эту реакционную смесь перемешивали при комнатной температуре в течение 5 ч. По окончании реакции реакционную смесь гасили ледяной водой (60 мл) и экстрагировали этилацетатом (3×50 мл). Объединенный органический раствор промывали насыщенным раствором NaHCO3 (20 мл). Органическую фазу высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 4-бром-2-фтор-5-метилбензол-1-сульфонилхлорида (7 г, неочищенное соединение). Соединение использовали без дополнительной очистки.
Промежуточное соединение 136: 4-бром-2-фтор-N-(2-гидроксиэтил)-5-метилбензолсульфонамид:
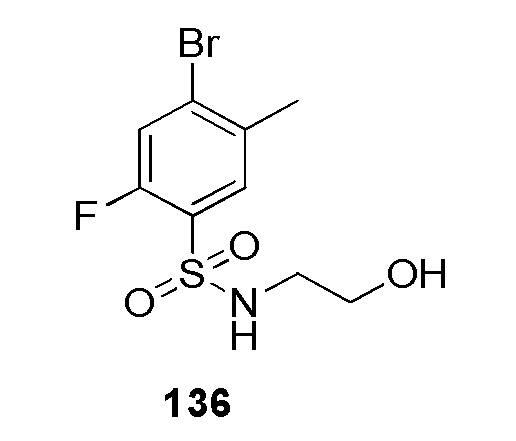
К раствору 4-бром-2-фтор-5-метилбензол-1-сульфонилхлорида (7 г, 24,34 ммоль, промежуточное соединение 135) в тетрагидрофуране (80 мл) добавляли 2-аминоэтанол (1,487 г, 24,34 ммоль, коммерческий источник: Avra) с последующим добавлением триэтиламина (6,7 мл, 48,69 ммоль, коммерческий источник: Avra) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 5 ч. По окончании реакции реакционную смесь выливали в ледяную воду (250 мл), и затем экстрагировали этилацетатом (2×500 мл) и промывали раствором бикарбоната натрия (200 мл). Органические слои высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией, элюируя смесью 50% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-бром-2-фтор-N-(2-гидроксиэтил)-5-метилбензолсульфонамида (5 г, 66,6%) в виде коричневой жидкости.
Промежуточное соединение 137: 4-циано-2-фтор-N-(2-гидроксиэтил)-5-метилбензолсульфонамид:
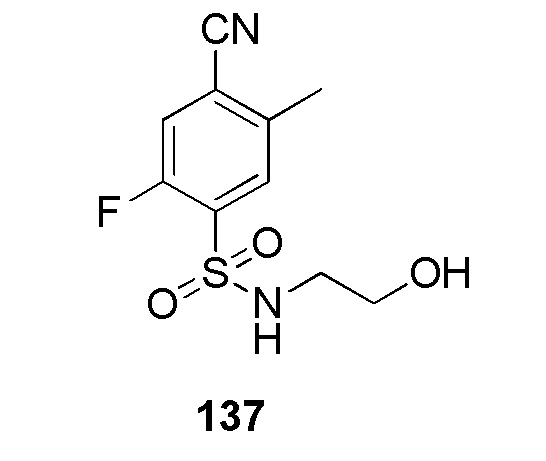
К раствору 4-бром-2-фтор-N-(2-гидроксиэтил)-5-метилбензолсульфонамида (4,5 г, 14,46 ммоль, промежуточное соединение 136) в N, N-диметилформамиде (45 мл) добавляли Zn(CN)2 (7,68 г, 43,4 ммоль коммерческий источник: Avra) в герметичной пробирке при комнатной температуре. Реакционную смесь продували азотом в течение 10 мин, добавляли тетракис(трифенилфосфин)палладий (0) (1,67 г, 1,44 ммоль, коммерческий источник: Alfa Aesar) и снова продували азотом при 26°С в течение 15 мин. Полученную реакционную смесь перемешивали при 100°С в течение ночи. По окончании реакции реакционную смесь разбавляли этилацетатом (200 мл) и промывали ледяной водой (2×500 мл). Органический раствор высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией, элюируя смесью 35% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-циано-2-фтор-N-(2-гидроксиэтил)-5-метилбензолсульфонамида (1,2 г, 32%, чистота=78%) в виде жидкости белого цвета. МС m/z [М-Н]- = 257,1. Соединение использовали без дополнительной очистки.
Промежуточное соединение 138: 2-фтор-N-(2-гидроксиэтил)-5-метил-4-(2H-тетразол-5-ил)бензолсульфонамид:
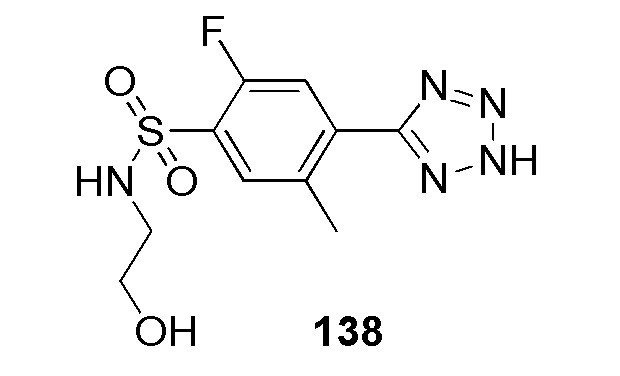
К раствору 4-циано-2-фтор-N-(2-гидроксиэтил)-5-метилбензолсульфонамида (1,5 г, 5,807 ммоль, промежуточное соединение 137) в тетрагидрофуране (10 мл) и воде (1 мл) добавляли бромид цинка (2,48 г, 11,034 ммоль, коммерческий источник: Spectrochem) при комнатной температуре с последующим добавлением азида натрия (1,623 г, 24,973 ммоль, коммерческий источник: Avra) при 0°С. Реакционную смесь нагревали до 85°С и перемешивали в течение ночи. По окончании реакции реакционную смесь охлаждали до 0°С и добавляли ледяную воду (100 мл) с последующим добавлением концентрированной HCl (10 мл) до pH 2. Затем ТГФ выпаривали при пониженном давлении и получали твердое вещество, которое фильтровали с получением 2-фтор-N-(2-гидроксиэтил)-5-метил-4-(2H-тетразол-5-ила)бензолсульфонамида (1 г, неочищенное соединение) в виде жидкости коричневого цвета. МС m/z [М+Н]+ = 302,1.
Синтез промежуточного соединения (2-(1-бромэтил)-5-фторпиридина)
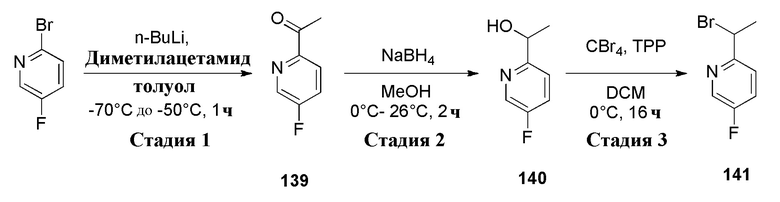
Промежуточное соединение 139: 1-(5-фторпиридин-2-ил)этанон:
К раствору 2-бром-5-фторпиридина (2 г, 0,0113 моль, коммерческий источник: Combi-Blocks) в сухом толуоле (20 мл) добавляли по каплям н-бутиллитий (2,5 М в н-гексане) (4,5 мл, 0,0113 ммоль, коммерческий источник: Hychem) при -70°С с последующим добавлением N, N-диметилацетамида (1,18 г, 0,0136 ммоль, коммерческий источник: Alfa) при -70°С. Реакционную смесь перемешивали при -50°С в течение 1 ч. По окончании реакции реакционную смесь гасили метанолом (20 мл) и перемешивали в течение 30 мин при 5-10°С, с последующим добавлением насыщенного раствора хлорида аммония (50 мл), перемешивали в течение 40 мин при 26°С и экстрагировали этилацетатом (3×70 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 1-(5-фторпиридин-2-ил)этанона (1,8 г, 85%) в виде бледно-желтой жидкости. 1H ЯМР (400 МГц, CDCl3) δ 8,50 (д, J=2,6 Гц, 1H), 8,16-8,07 (м, 1H), 7,55-7,46 (м, 1H), 2,70 (с, 3H). МС m/z [М+Н]+ = 140,16.
Промежуточное соединение 140: 1-(5-фторпиридин-2-ил)этанол:
К раствору 1-(5-фторпиридин-2-ил)этанона (1,8 г, 0,0129 моль, промежуточное соединение 139) в метаноле (18 мл) добавляли борогидрид натрия (0,95 г, 0,0258 моль, коммерческий источник: Aldrich) с избытком при 0°С. Реакционную смесь перемешивали при 26°С в течение 2 ч. По окончании реакции реакционную смесь гасили водой (30 мл) при 0°С и экстрагировали этилацетатом (3×30 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 1-(5-фторпиридин-2-ил)этанола (800 мг, 37%) в виде бледно-коричневой жидкости. 1H ЯМР (400 МГц, CDCl3) δ 8,40 (д, J=2,9 Гц, 1H), 7,45-7,38 (м, 1H), 7,33-7,27 (м, 1H), 4,94-4,86 (м, 1H), 3,76- 3,66 (м, 1Н), 1,54 (с, 3Н). МС m/z [М+Н]+ = 142,01.
Промежуточное соединение 141: 2-(1-бромэтил)-5-фторпиридин:
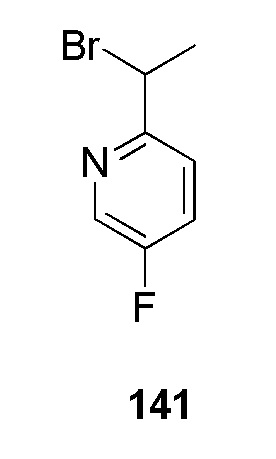
К раствору 1-(5-фторпиридин-2-ил)этанола (700 мг, 0,0049 моль, промежуточное соединение 140) в дихлорметане (3,5 мл) добавляли трифенилфосфин (2,5 г, 0,0098 моль, коммерческий источник: Aldrich) при 26°С. Реакционную смесь перемешивали в течение 15 мин при той же температуре. Затем добавляли по каплям раствор тетрабромида углерода (1,6 г, 0,0049 моль, коммерческий источник: Avra) в дихлорметане (3,5 мл) при 0°С. Реакционную смесь перемешивали в течение 16 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 10% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении с получением 2-(1-бромэтил)-5-фторпиридина (350 мг, 30%) в виде бледно-желтой жидкости. 1H ЯМР (400 МГц, CDCl3) δ 8,42 (д, J=2,6 Гц, 1H), 7,50-7,44 (м, 1H), 7,43-7,36 (м, 1H), 5,24 (кв, J=7,0 Гц, 1H) 2,07 (д, J=7,0 Гц, 3H). МС m/z [М+Н]+ & [М+2Н]+ = 204,10 и 206,11.
Синтез промежуточного соединения 1-(1-бромэтил)-4-фторбензола
Промежуточное соединение 142: 1-(1-бромэтил)-4-фторбензол:
К раствору 1-(4-фторфенил)этанола (500 мг, 0,0036 моль, коммерческий источник: Apollo) в дихлорметане (5 мл) при 0°С, медленно добавляли PBr3 (0,5 мл, коммерческий источник: TCI) и смесь перемешивали при 26°С в течение 16 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Добавляли раствор NaHCO3 (70 мл) и экстрагировали этилацетатом (3×30 мл). Органические слои объединяли и промывали насыщенным раствором соли (50 мл), высушивали над безводным Na2SO4, фильтровали и концентрировали с получением сырого 1-(1-бромэтил)-4-фторбензола (310 мг) в виде коричневой жидкости. Его использовали на следующей стадии без дополнительной очистки.
Синтез промежуточного соединения 1-(1-азидоэтил)-4-фторбензола
Промежуточное соединение 143: 1-(4-фторфенил)этилметансульфонат:
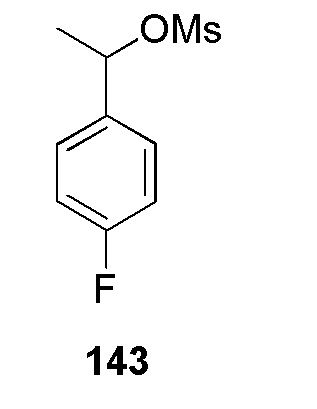
К раствору 1-(4-фторфенил)этанола (200 мг, 0,0014 моль, коммерческий источник: Aochem) в дихлорметане (4 мл) добавляли триэтиламин (0,4 мл, 0,0029 моль, коммерческий источник: Finar) и мезилхлорид (0,16 мл, 0,0021 моль, коммерческий источник: Avra) при 0°С. Реакционную смесь перемешивали при 26°С в течение 16 ч. По окончании реакции добавляли дихлорметан (60 мл) и воду (10 мл). Органический раствор высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением сырого 1-(4-фторфенил)этилметансульфоната (300 мг) в виде светло-желтой жидкости, которую использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение 144: 1-(1-азидоэтил)-4-фторбензол:
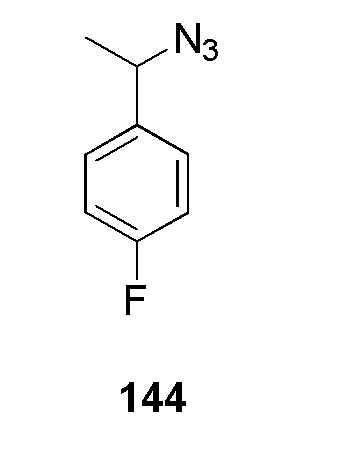
К раствору 1-(4-фторфенил)этилметансульфоната (220 мг, 0,0010 моль, промежуточное соединение 143) в ДМФА (2,2 мл) добавляли азид натрия (196 мг, 0,0030 моль, коммерческий источник: Avra) при 26°С и перемешивали в течение 16 ч. По окончании реакции добавляли 1 Н HCl (10 мл) при 0°С. Смесь экстрагировали этилацетатом, органические слои объединяли, промывали холодной водой (3×20 мл), высушивали над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением сырого 1-(1-азидоэтил)-4-фторбензола (290 мг) в виде коричневой липкой жидкости, которую использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение 145: этил 2-(4-(2-(1-(4-фторфенил) этил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетат:
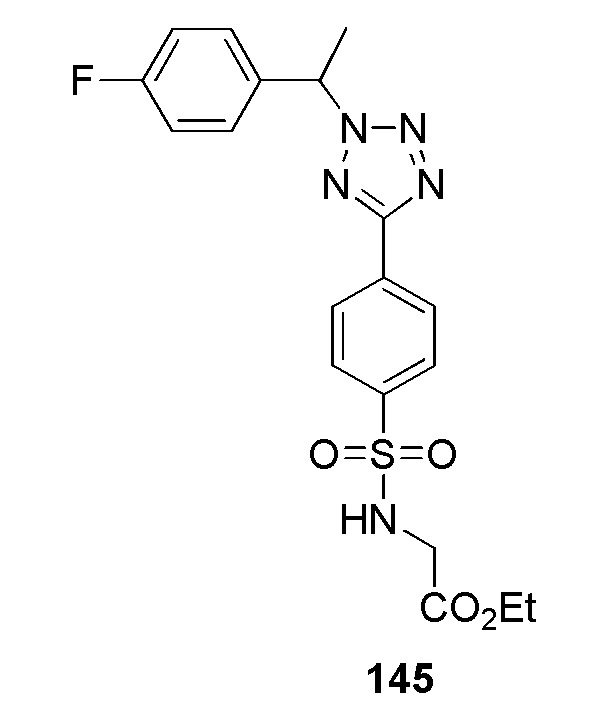
К раствору этил 2-(4-(2H-тетразол-5-ил)фенилсульфонамидо) ацетата (500 мг, 0,0016 моль, промежуточное соединение 115) в ацетонитриле (10 мл) добавляли N, N-диизопропилэтиламин (0,7 мл, 0,0040 моль, коммерческий источник: Finar) и 1-(1-бромэтил)-4-фторбензол (389 мг, 0,0019 моль, промежуточное соединение 142) при 26°С и смесь перемешивали при 85°С в течение 16 ч. По окончании реакции смесь охлаждали до 26°С и растворяли в этилацетате (100 мл) и воде (20 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,5% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением этил 2-(4-(2-(1-(4-фторфенил)этил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетата (300 мг, 41%) в виде не совсем белого твердого вещества. МС m/z [М+Н]+ = 434,26.
Промежуточное соединение 146: 2-(4-(2-(1-(4-фторфенил)этил) -2Н-тетразол-5-ил)фенилсульфонамидо)уксусная кислота:
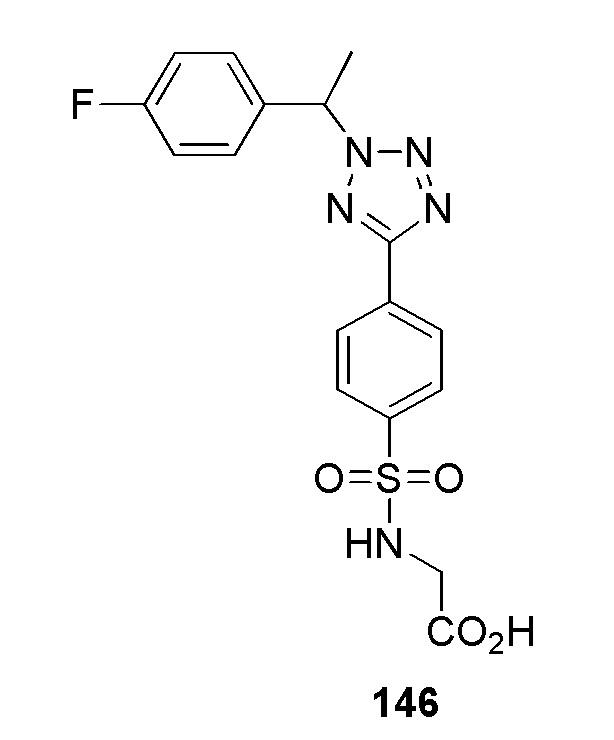
К раствору этил 2-(4-(2-(1-(4-фторфенил)этил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетата (300 мг, 0,00069 моль, промежуточное соединение 145) в ТГФ (3 мл) и метаноле (3 мл) добавляли LiOH⋅H2O (58 мг, 0,0014 моль, коммерческий источник: Fimar) при 26°С и перемешивали в течение 2 ч. По окончании реакции смесь концентрировали при пониженном давлении, растворяли в воде и промывали этилацетатом (2×20 мл). рН водного слоя подкисляли 2 Н HCl (20 мл) при 0°С. Образовывался осадок, который отфильтровывали и высушивали в вакууме с получением 2-(4-(2-(1-(4-фторфенил)этил)-2Н-тетразол-5-ил)фенилсульфонамидо)уксусной кислоты (140 мг, 50%) в виде белого твердого вещества. МС m/z [М+Н]+ = 405,05.
Промежуточное соединение 147: 4-(2-(1-(4-фторфенил)этил)-2Н-тетразол-5-ил)-2-метоксибензолсульфонамид:
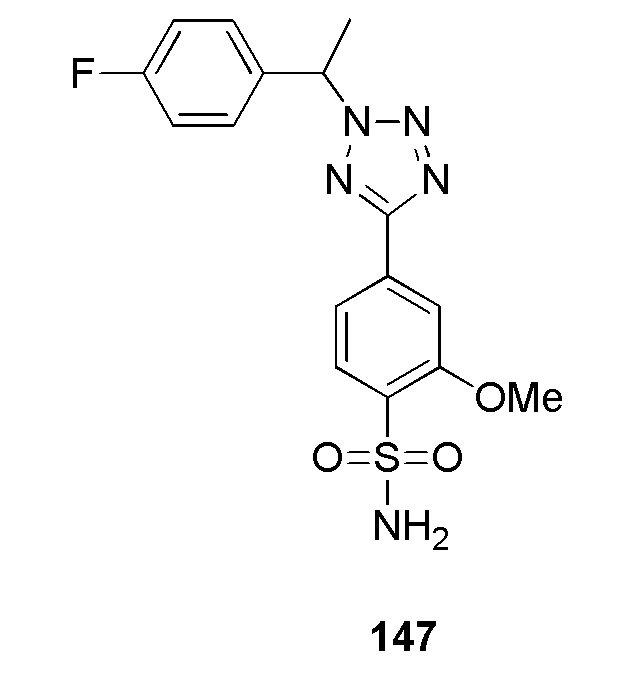
К раствору этил 2-(4-(2H-тетразол-5-ил)фенилсульфонамидо) ацетата (1,2 г, 0,0047 моль, промежуточное соединение 51) и 1-(1-бромэтил)-4-фторбензола (1,14 г, 0,0056 моль, промежуточное соединение 142) в ацетонитриле (24 мл) добавляли N, N-диизопропилэтиламин (2,0 мл, 0,0118 моль, коммерческий источник: Finar) при 26°С и смесь перемешивали при 85°С в течение 16 ч. По окончании реакции смесь охлаждали до 26°С и растворяли в этилацетате (100 мл) и воде (20 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-(2-(1-(4-фторфенил)этил)-2Н-тетразол-5-ил)-2-метоксибензолсульфонамида (500 мг, 27%) в виде не совсем белого твердого вещества. МС m/z [М+Н]+ = 378,52.
Промежуточное соединение 148: трет-бутил (2-амино-2-оксоэтил)((4-(2-(1-(4-фторфенил)этил)-2Н-тетразол-5-ил)-2-метоксифенил)сульфонил)карбамат:
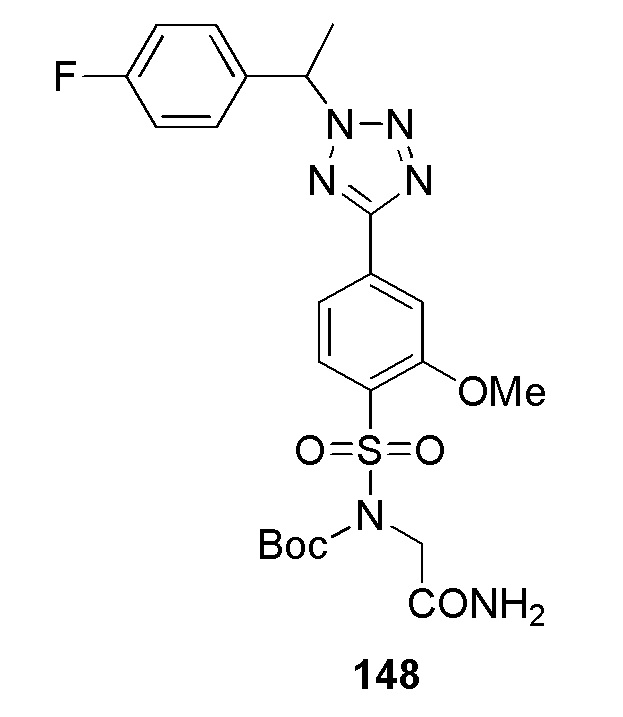
К раствору 4-(2-(1-(4-фторфенил)этил)-2Н-тетразол-5-ил)-2-метоксибензолсульфонамида (500 мг, 0,0013 моль, промежуточное соединение 147) в ТГФ (5 мл) добавляли N, N-диметиламинопиридин (80 мг, 0,00066 моль, коммерческий источник: Avra) и триэтиламин (0,27 мл, 0,0020 моль, коммерческий источник: Finar), с последующим добавлением (Boc)20 (868 мг, 0,0040 моль, коммерческий источник: Avra) при 0°С и смесь перемешивали при 26°С в течение 6 ч. Затем добавляли K2CO3 (366 мг, 0,0027 моль, коммерческий источник: Finar) и 2-бромацетамид (270 мг, 0,0020 моль, коммерческий источник: Alfa) и смесь перемешивали при 26°С в течение 16 ч. По окончании реакции добавляли этилацетат (100 мл) и воду (40 мл). Водный слой экстрагировали этилацетатом (2×30 мл). Объединенные органические слои высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 0,5% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением трет-бутил (2-амино-2-оксоэтил)((4-(2-(1-(4-фторфенил)этил)-2H-тетразол-5-ил)-2-метоксифенил)сульфонил)карбамата (300 мг, 41%) в виде не совсем белого твердого вещества. МС m/z [M-Boc+H]+ = 435,18.
Примеры
Пример 1: (4-(2-(4-фторбензил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид):
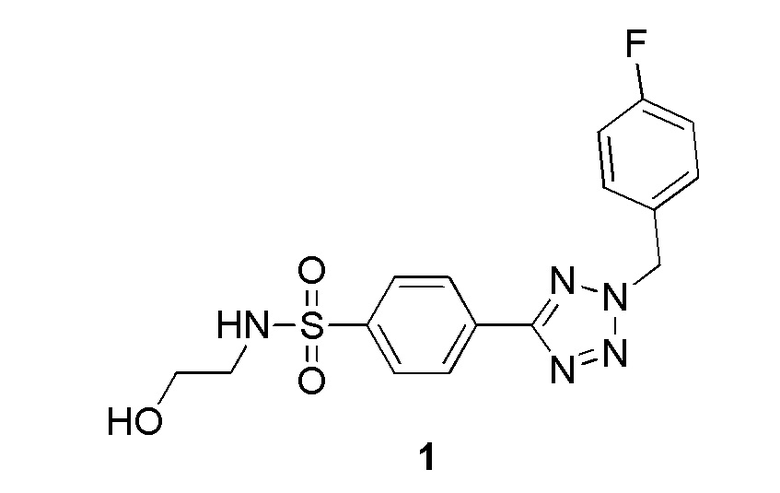
К раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (1,3 г, 4,832 ммоль, промежуточное соединение 17), карбоната калия (1,333 г, 9,665 ммоль) в N, N-диметилформамиде (15 мл) добавляли 1-(бромметил)-4-фторбензол (1,826 г, 9,665 ммоль, коммерческий источник: Aldrich) при 26°С. Реакционную смесь перемешивали при той же температуре в течение 5 ч. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь выливали в ледяную воду (200 мл) и осажденное твердое вещество отфильтровывали. Липкое твердое вещество растворяли в смеси 10% метанола в дихлорметане (50 мл) и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 2% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением (4-(2-(4-фторбензил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамида) (370 мг, 20%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,26-8,21 (м, 2H), 7,98-7,93 (м, 2H), 7,76-7,71 (м, 1H), 7,54-7,47 (м, 2H), 7,27-7,20 (м, 2H), 6,02 (с, 2H), 4,65 (т, J=5,6 Гц, 1H), 3,40-3,33 (м, 2H), 2,86-2,79 (м, 2H). МС m/z [М+Н]+ = 378,26.
Пример 2: N-(2-гидроксиэтил)-4-(2-(пиримидин-4-илметил)-2Н-тетразол-5-ил)бензолсульфонамид:
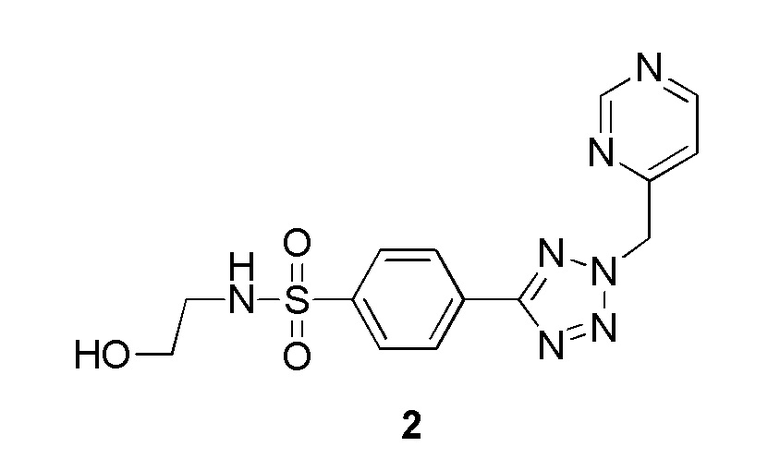
К раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (100 мг, 0,371 ммоль, промежуточное соединение 17), карбоната калия (103 мг, 0,746 ммоль) в N, N-диметилформамиде (10 мл) добавляли 4-(бромметил)пиримидин (64 мг, 0,37 ммоль, промежуточное соединение 1) при 26°С. Реакционную смесь перемешивали при 26°С в течение 16 ч. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 5% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении. Полученное соединение дополнительно очищали препаративной ВЭЖХ с получением N-(2-гидроксиэтил)-4-(2-(пиримидин-4-илметил)-2Н-тетразол-5-ил) бензолсульфонамида (22 мг, 16%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,13 (д, J=1,3 Гц, 1H), 8,87 (д, J=5,3 Гц, 1H), 8,27 (д, J=8,8 Гц, 2H), 7,97 (д, J=8,8 Гц, 2H), 7,56-7,62 (м, 1H), 6,27 (с, 2H), 4,67 (шир. с, 1H), 3,37 (т, J=6,4 Гц, 2H), 2,84 (т, J=6,3 Гц, 2H МС m/z [М+Н]+ = 362,12.
Условия препаративной ВЭЖХ:
Колонка: Kromasil PHENYL (250×25) мм, 10 мкм
Подвижная фаза: A-10 мМ бикарбонат аммония (водный раствор), B-ацетонитрил
Метод (время в мин/% B): A:B=68:32, изократический.
Скорость потока: 25 мл/мин
Температура: комнатная.
Пример 3: 4-(2-(4-фторбензил)-2Н-тетразол-5-ил)-N-метилбензолсульфонамид:
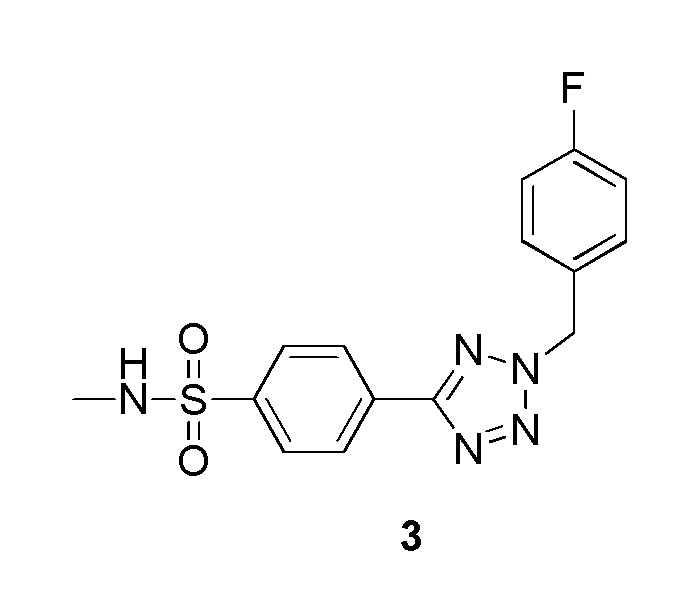
К раствору N-метил-4-(2H-тетразол-5-ил)бензолсульфонамида (296 мг, 1,238 ммоль, промежуточное соединение 21), карбоната калия (342 мг, 2,477 ммоль) в N, N-диметилформамиде (8 мл) добавляли 1-(бромметил)-4-фторбензол (233 мг, 1,2385 ммоль, коммерческий источник: Combi-Blocks) при 27°С. Полученную реакционную смесь перемешивали при 27°С в течение 2 ч. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь выливали в воду (60 мл). Осажденное твердое соединение отфильтровывали и высушивали в вакууме. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 3% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-(2-(4-фторбензил)-2Н-тетразол-5-ил)-N-метилбензолсульфонамида (104 мг, 24%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,28-8,22 (м, 2H), 7,97-7,91 (м, 2H), 7,60-7,54 (м, 1H), 7,54-7,48 (м, 2H), 7,28-7,20 (м, 2H), 6,02 (с, 2H), 2,44 (д, J=4,6 Гц, 3H). МС m/z [М+Н]+ = 348,14.
Пример 4: N-метил-4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил) бензолсульфонамид:
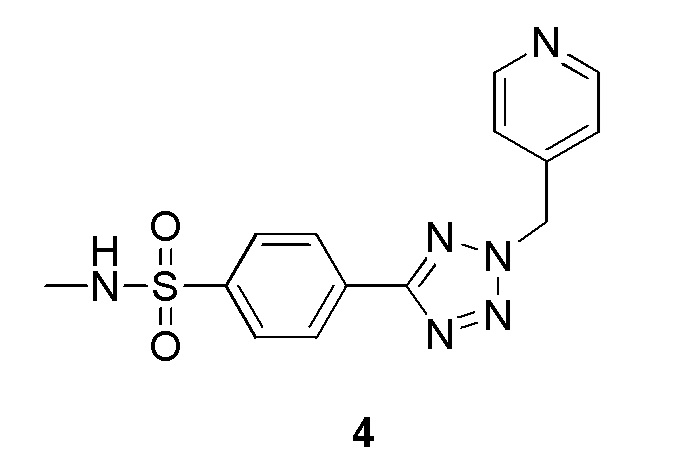
К раствору N-метил-4-(2H-тетразол-5-ил)бензолсульфонамида (296 мг, 1,238 ммоль, промежуточное соединение 21), карбоната калия (513 мг, 3,715 ммоль) в N, N-диметилформамиде (8 мл) добавляли 4-(хлорметил)пиридин гидрохлорид (203 мг, 1,238 ммоль, коммерческий источник: Alfa Aesar) при 27°С и реакционную смесь перемешивали в течение 2 ч. Полученную реакционную смесь нагревали до 80°С и перемешивали в течение 2 ч при той же температуре. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 3% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением N-метил-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамида (40 мг, 9%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,60 (д, J=5,9 Гц, 2H), 8,27 (д, J=8,3 Гц, 2H), 7,95 (д, J=8,3 Гц, 2H), 7,61-7,55 (м, 1H), 7,34 (д, J=5,5 Гц, 2H), 6,13 (с, 2H), 2,44 (д, J=5,0 Гц, 3H). МС m/z [М+Н]+ = 331,12.
Пример 5: N-этил-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил) бензолсульфонамид:
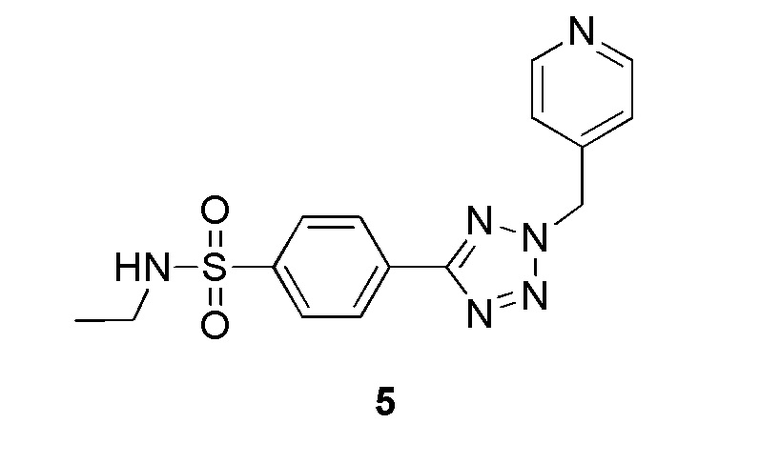
К раствору N-этил-4-(2H-тетразол-5-ил)бензолсульфонамида (350 мг, 1,383 ммоль, промежуточное соединение 23), карбоната калия (573 мг, 4,150 ммоль) в N, N-диметилформамиде (7 мл) добавляли 4-(хлорметил)пиридин гидрохлорид (227 мг, 1,383 ммоль, коммерческий источник: Alfa Aesar) при 27°С. Полученную реакционную смесь перемешивали при 27°С в течение 24 ч. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 3% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением N-этил-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамида (60 мг, 12%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,63-8,57 (м, 2Н), 8,29-8,23 (м, 2Н), 7,99-7,92 (м, 2Н), 7,69 (т, J=5,7 Гц, 1Н), 7,34 (д, J=5,9 Гц, 2H), 6,13 (с, 2H), 2,87-2,76 (м, 2H), 0,97 (т, J=7,2 Гц, 3H). МС m/z [М+Н]+ = 345,06.
Пример 6: N-(2-гидроксиэтил)-4-(2-(4-метоксибензил)-2Н-тетразол-5-ил)бензолсульфонамид:
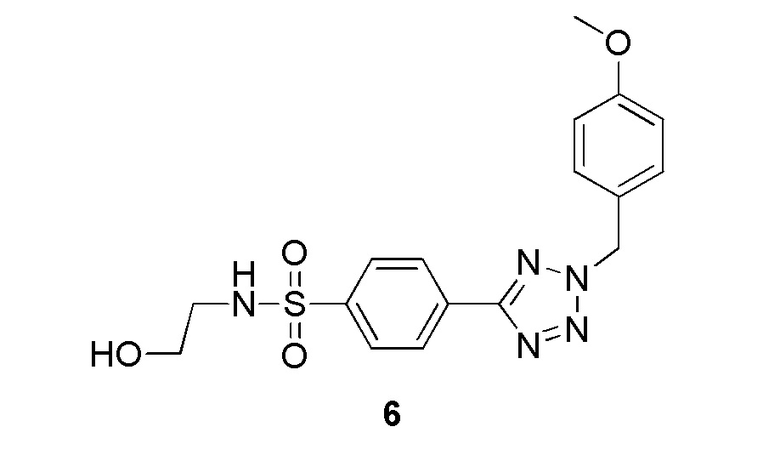
К раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (100 мг, 0,0003 моль, промежуточное соединение 17) в ацетонитриле (1 мл) добавляли N, N-диизопропилэтиламин (70 мг, 0,0006 моль) при 0°С и перемешивали в течение 10 мин при 0°С с последующим добавлением 1-(бромметил)-4-метоксибензола (89 мг, 0,0004 моль, коммерческий источник: Alfa Aesar) при 0°С. Затем реакционную смесь перемешивали при 28°С в течение 16 ч. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,5% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением N-(2-гидроксиэтил)-4-(2-(4-метоксибензил)-2Н-тетразол-5-ил)бензолсульфонамида (64 мг, 54%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,26-8,19 (м, 2H), 7,98-7,93 (м, 2H), 7,73 (т, J=5,7 Гц, 1H), 7,42-7,36 (м, 2H), 6,98-6,93 (м, 2H), 5,93 (с, 2H), 4,65 (т, J=5,6 Гц, 1H), 3,74 (с, 3H), 3,39-3,33 (м, 2H), 2,86-2,79 (м, 2H). МС m/z [М+Н]+ = 390,13.
Общая процедура алкилирования тетразолов
N, N-диизопропилэтиламин (1-4 экв) добавляли по каплям к перемешиваемому раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (1 экв) и алкилирующего агента (1 или 2 экв) в N, N-диметилформамиде (0,3 М) в атмосфере азота при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 1-16 ч. Для некоторых реакций требуется нагревание при 50°С и перемешивание в течение 16 ч, дополнительно дальнейшее нагревание до 70-80°С и перемешивание в течение еще 2 ч или в течение ночи. Смесь концентрировали при пониженном давлении. Сырое соединение очищали. Желаемые фракции собирали и концентрировали в вакууме с получением продуктов.
Пример 7: N-(2-гидроксиэтил)-4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамид:
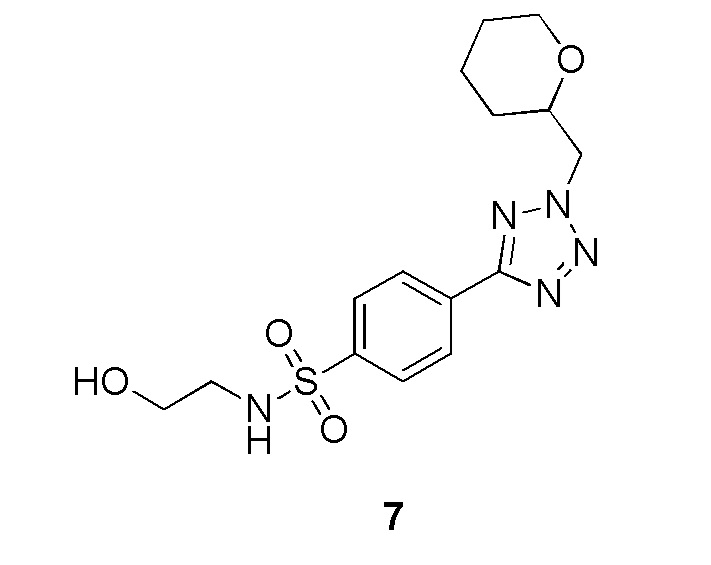
DIPEA (778 мкл, 4,46 ммоль) добавляли по каплям к перемешиваемому раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (300 мг, 1,114 ммоль, промежуточное соединение 17') и 2-(бромметил)тетрагидро-2Н-пирана (285 мкл, 2,228 ммоль, коммерческий источник: Aldrich) в N, N-диметилформамиде (ДМФА) (3714 мкл) при комнатной температуре в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 3 суток. Поскольку оставалось исходное вещество, то смесь перемешивали при 70°С в течение ночи. Смесь концентрировали при пониженном давлении. Неочищенное соединение очищали колоночной флэш-хроматографией (диоксид кремния; смесь EtOAc-циклогексан от 0/100 до 50/50). Желаемые фракции собирали и концентрировали в вакууме с получением продукта в виде рацемической смеси N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамида (49 мг, 0,133 ммоль, 28%). 1H ЯМР (400 МГц, ацетон-d6) δ 7,84-7,78 (м, 2H), 7,56-7,50 (м, 2H), 7,33 (т, J=5,9 Гц, 1H), 4,37-4,32 (м, 2H), 4,25 (т, J=5,6 Гц, 1H), 3,50-3,44 (м, 1H), 3,38-3,32 (м, 1H), 2,94-2,90 (м, 2H), 2,86-2,80 (м, 1H), 2,41-2,37 (м, 2Н), 1,40-1,25 (м, 2Н), 1,12-0,85 (м, 4Н). МС m/z [М-Н]- = 366,2.
Примеры 8-17 получали способами, аналогичными описанному для примера 7, с заменой алкилирующих реагентов и основных условий на условия, указанные в таблице 1. Метод, используемый для очистки, указан в виде сносок.
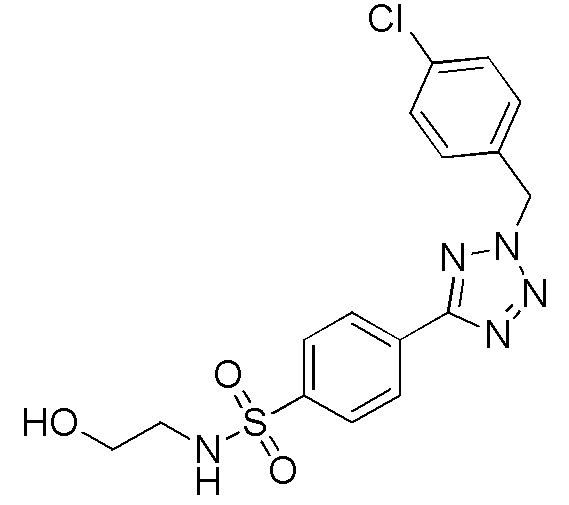
4-(2-(4-хлорбензил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид
(см. сноску а)
перемешивали при комнатной температуре в течение 16 ч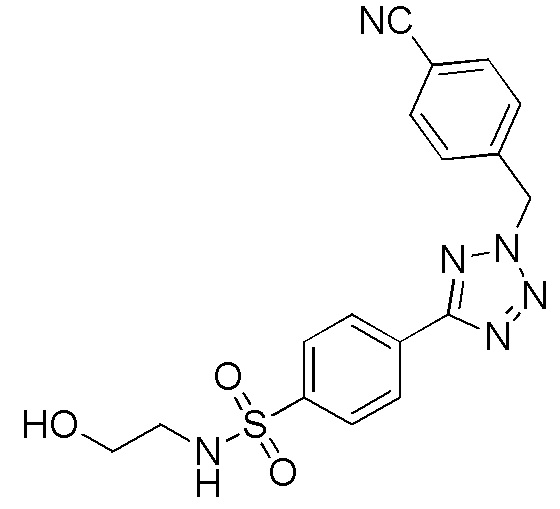
4-(2-(4-цианобензил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид
(см. сноску а)
(коммерческий источник: Aldrich);
перемешивали при комнатной температуре в течение 3 ч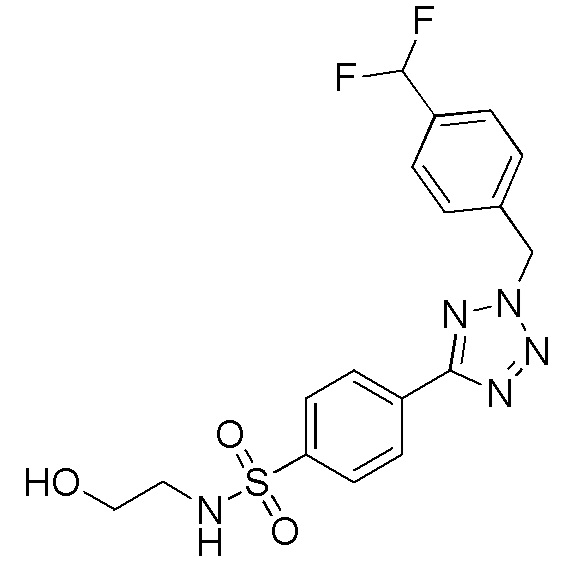
4-(2-(4-(дифторметил)бензил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид (см. сноску b)
бромид (1 экв, 99 мг, 0,446 ммоль)
(коммерческий источник: Apollo UK);
перемешивали при комнатной температуре в течение 16 ч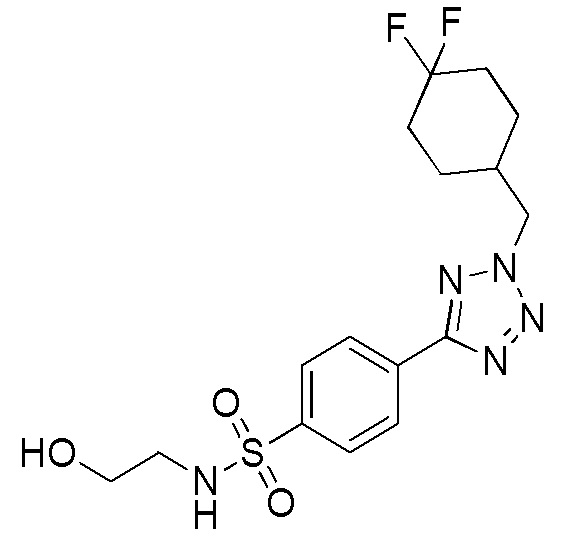
4-(2-((4,4-дифторциклогексил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид (см. сноску а)
50°С в течение 72 ч
(R)-N-(2-гидроксипропил)-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид (см. сноску с)
(R)-4-(2-((4,4-дифторциклогексил)метил)-2H-тетразол-5-ил)-N-(2-гидроксипропил) бензолсульфонамид
(см. сноску d)
(S)-4-(2-(4-фторбензил)-2Н-тетразол-5-ил)-N-(2-гидроксипропил)бензолсульфонамид (см. сноску е)
при комнатной температуре в течение 4 ч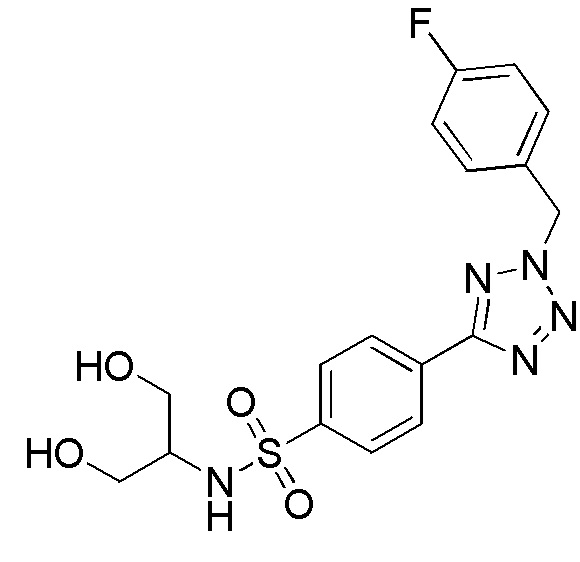
N-(1,3-дигидроксипропан-2-ил)-4-(2-(4-фторбензил)-2Н-тетразол-5-ил)бензолсульфонамид (см. сноску d)
при комнатной температуре в течение 3 ч
N-(1,3-дигидроксипропан-2-ил)-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид (см. сноску f)
50°С в течение 20 ч
N-(2,3-дигидроксипропил)-4-(2-(4-фторбензил)-2Н-тетразол-5-ил)бензолсульфонамид (см. сноску е)
1H ЯМР (400 МГц, ДМСО-d6) δ 8,26-8,20 (м, 2H), 7,99-7,93 (м, 2H), 7,70-7,64 (м, 1H), 7,53-7,48 (м, 2H), 7,27-7,21 (м, 2Н), 6,03 (с, 2Н), 4,75 (д, J=5,3 Гц, 1Н), 4,54-4,48 (м, 1Н), 3,47-3,40 (м, 1Н), 3,28-3,18 (м, 2Н), 2,93-2,86 (м, 1H), 2,68-2,58 (м, 1H). МС m/z [М+Н]+ = 408,09
b) Остаток очищали препаративной ВЭЖХ с использованием колонки X-Bridge (30×150 мм) в линейном градиенте 30-100% ACN/H2O (10 мМ NH4HCO3);
c) Остаток очищали флэш-хроматографией со смесью EtOAc/EtOH (3:1)-циклогексан от 0/100 до 50/50, затем EtOAc-циклогексан от 0/100 до 50/50;
d) Остаток очищали флэш-хроматографией со смесью EtOAc/EtOH (3:1)-циклогексан от 0/100 до 50/50;
е) Реакционную смесь гасили 1 Н HCl и экстрагировали этилацетатом. Органический слой высушивали над Na2SO4 и концентрировали при пониженном давлении;
f) Остаток очищали флэш-хроматографией со смесью EtOAc/EtOH (3:1)-циклогексан от 0/100 до 50/50, затем продукт растирали с ACN и, в конце очищали препаративной ВЭЖХ с использованием колонки X-Bridge (30×150 мм) в линейном градиенте 20-100% ACN/H2O (10 мМ NH4HCO3).
Общая процедура алкилирования тетразолов
N, N-диизопропилэтиламин (1-4 экв) добавляли к перемешиваемому раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (1 экв) в N, N-диметилформамиде (0,3 М). Затем добавляли алкилирующий агент (1 или 2 экв) и смесь перемешивали при комнатной температуре в течение, по меньшей мере, 16 ч. Смесь концентрировали при пониженном давлении. Неочищенное соединение очищали преимущественно колоночной флэш-хроматографией. Желаемые фракции собирали и концентрировали в вакууме с получением конечных продуктов.
Пример 18: 4-(2-((5-хлорпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид:
DIPEA (4 экв, 0,519 мл, 2,97 ммоль) добавляли к перемешиваемому раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (1 экв, 0,200 г, 0,743 ммоль, промежуточное соединение 17') в N, N-диметилформамиде (ДМФА) (2,476 мл). Затем добавляли 2-(бромметил)-5-хлорпиридин (2 экв, 0,307 г, 1,485 ммоль, коммерческий источник: Biogene Organics) и смесь перемешивали при комнатной температуре в течение 72 ч. Смесь концентрировали при пониженном давлении. Неочищенное соединение очищали колоночной флэш-хроматографией (диоксид кремния; смесь EtOAc-циклогексан от 0/100 до 100/0). Желаемые фракции собирали и концентрировали в вакууме с получением 4-(2-((5-хлорпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил) бензолсульфонамида (205 мг, 70%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,58 (д, J=2,3 Гц, 1H), 8,26-8,21 (м, 2H), 8,02 (дд, J=8,3, 2,5 Гц, 1H), 7,98-7,91 (м, 2H), 7,77 (т, J=5,9 Гц, 1H), 7,58 (д, J=8,3 Гц, 1H), 6,18 (с, 2H), 4,68 (т, J=5,6 Гц, 1H), 3,35 (кв, J=6,3 Гц, 2H), 2,82 (кв, J=6,2 Гц, 2H). МС m/z [М-Н]- = 393,05.
Примеры 19-28 получали способами, аналогичными описанному для примера 18, с заменой алкилирующих реагентов и основных условий на условия, указанные в таблице 2. Когда метод, использованный для очистки, отличался от метода для примера 18, то это указывалось.
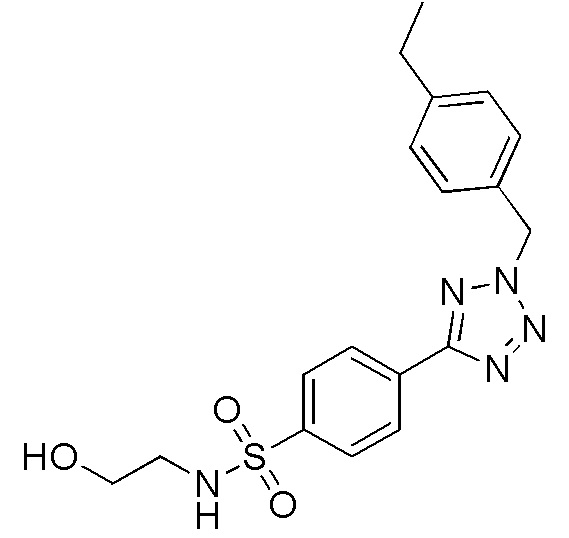
4-(2-(4-этилбензил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид (см. сноску a)
при комнатной температуре в течение 16 ч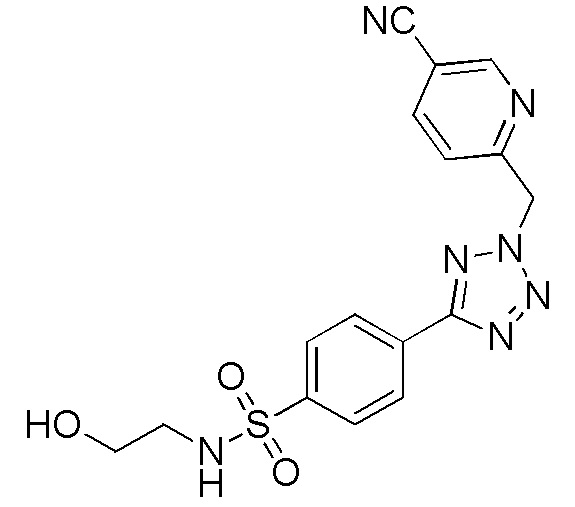
4-(2-((5-цианопиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид (см. сноски b, c)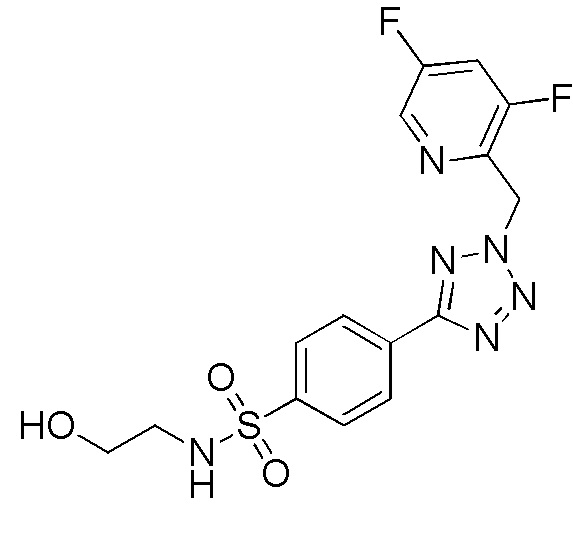
4-(2-((3,5-дифторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид (см. сноску d)
1H ЯМР (400 МГц, ДМСО-d6) δ 8,49 (д, J=2,5 Гц, 1H), 8,26-8,20 (м, 2H), 8,12-8,05 (м, 1H), 7,99-7,92 (м, 2H), 7,77 (т, J=5,9 Гц, 1H), 6,24 (м, 2H), 4,69 (т, J=5,6 Гц, 1H), 3,39-3,35 (м, 2H), 2,86-2,79 (м, 2H). МС m/z [М-Н]- = 395,1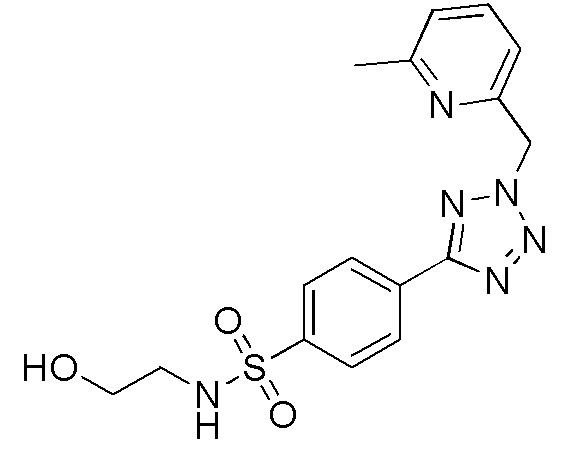
N-(2-гидроксиэтил)-4-(2-((6-метилпиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид
(см. сноски b, e)
1H ЯМР (400 МГц, ДМСО-d6) δ 8,27-8,21 (м, 2Н), 7,98-7,93 (м, 2Н), 7,78-7,71 (м, 2Н), 7,28-7,20 (м, 2Н), 6,07 (с), 2H), 4,67 (т, J=5,4 Гц, 1H), 3,39-3,32 (м, 2H), 2,82 (т, J=6,2 Гц, 2H), 2,40 (с, 3H). МС m/z [М+Н]+ = 375,3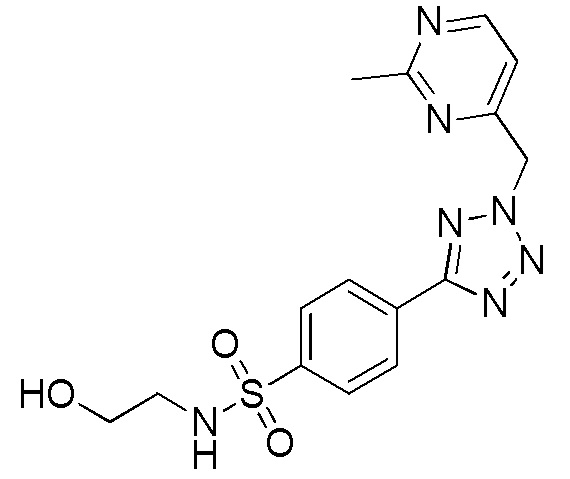
N-(2-гидроксиэтил)-4-(2-((2-метилпиримидин-4-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид (см. сноски f, g)
(R)-4-(2-(4-фторбензил)-2Н-тетразол-5-ил)-N-(2-гидроксипропил)бензолсульфонамид (см. сноску a)
1H ЯМР (400 МГц, ДМСО-d6) δ 8,26-8,19 (м, 2Н), 7,99-7,92 (м, 2Н), 7,73 (шир. с, 1Н), 7,53-7,48 (м, 2Н), 7,27-7,21 (м, 2H), 6,03 (с, 2H), 4,68 (д, J=4,8 Гц, 1H), 3,62-3,53 (м, 1H), 2,74-2,60 (м, 2H), 0,97 (д, J=6,1 Гц, 3Н). МС m/z [М-Н]- = 390,2
(R)-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксипропил)бензолсульфонамид (см. сноску a)
1H ЯМР (400 МГц, ДМСО-d6) δ 8,53 (д, J=3,0 Гц, 1H), 8,26-8,20 (м, 2H), 7,98-7,93 (м, 2H), 7,82 (дт, J=8,7, 3,0 Гц, 1H), 7,73 (т, J=5,7 Гц, 1H), 7,64 (дд, J=8,7, 4,4 Гц, 1H), 6,16 (с, 2H), 4,68 (д, J=4,8 Гц, 1H), 3,62-3,53 (м, 1H), 2,73-2,61 (м, 2H), 0,97 (д, J=6,1 Гц, 3H). МС m/z [М-Н]- = 391,1
(R)-N-(2-гидроксипропил)-4-(2-((5-метоксипиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид
ночи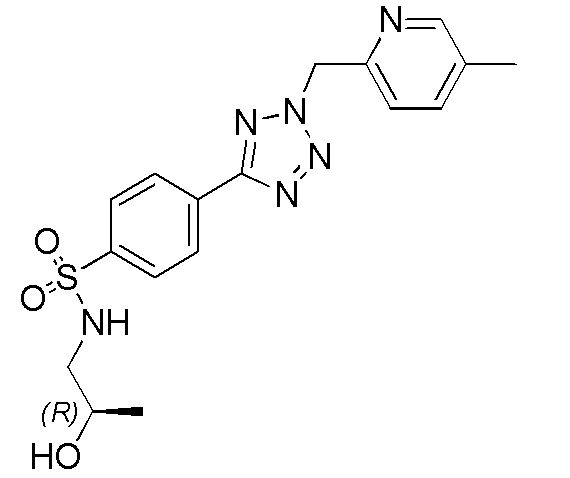
(R)-N-(2-гидроксипропил)-4-(2-((5-метилпиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид
50°C в течение 1 ч
1H ЯМР (400 МГц, ДМСО-d6) δ 8,37-8,33 (м, 1Н), 8,26-8,20 (м, 2Н), 7,98-7,92 (м, 2Н), 7,75-7,70 (м, 1Н), 7,70-7,65 (м, 1H), 7,42-7,36 (м, 1H), 6,08 (с, 2H), 4,67 (д, J=4,8 Гц, 1H), 3,62-3,53 (м, 1H), 2,75-2,60 (м, 2H ), 2,28 (с, 3H), 0,97 (д, J=6,1 Гц, 3H). МС m/z [М+Н]+ = 389,3 (R)-N-(2-гидроксипропил)-4-(2-(4-метоксибензил)-2H-тетразол-5-ил)бензолсульфонамид
(R)-N-(2-гидроксипропил)-4-(2-(4-метоксибензил)-2H-тетразол-5-ил)бензолсульфонамид
(см. сноску a)
1H ЯМР (400 МГц, ДМСО-d6) δ 8,22 (д, J=8,3 Гц, 2H), 7,95 (д, J=8,3 Гц, 2H), 7,73 (т, J=6,1 Гц, 1H), 7,39 (д , J=8,6 Гц, 2H), 6,95 (д, J=8,8 Гц, 2H), 5,93 (с, 2H), 4,67 (д, J=4,5 Гц, 1H), 3,73 (с, 3H), 3,62-3,53 (м, 1H), 2,74-2,61 (м, 2H), 0,97 (д, J=6,3 Гц, 3H). МС m/z [М-Н]- = 402,2
b) Реакционную смесь разбавляли водой и дважды экстрагировали DCM. Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении;
c) Остаток очищали флэш-хроматографией со смесью EtOAc-циклогексан от 0/100 до 100/0, затем EtOAc/EtOH (3:1)-циклогексан от 0/100 до 50/50. В конце остаток очищали препаративной ВЭЖХ с использованием колонки X-Bridge (19×150 мм) в линейном градиенте 20-100% ACN/H2O (10 мМ NH4HCO3);
d) Остаток очищали флэш-хроматографией со смесью EtOAc-циклогексан от 0/100 до 50/50 (дважды);
e) Остаток очищали флэш-хроматографией со смесью EtOAc-циклогексан от 0/100 до 30/70;
f) Остаток очищали флэш-хроматографией со смесью EtOAc-циклогексан от 0/100 до 100/0;
g)Остаток очищали препаративной ВЭЖХ на колонке X-Bridge (19×150 мм) в линейным градиенте 10-100% ACN/H2O (10 мМ NH4HCO3).
Пример 29 (получали через промежуточные соединения 9' и 17': 4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид:
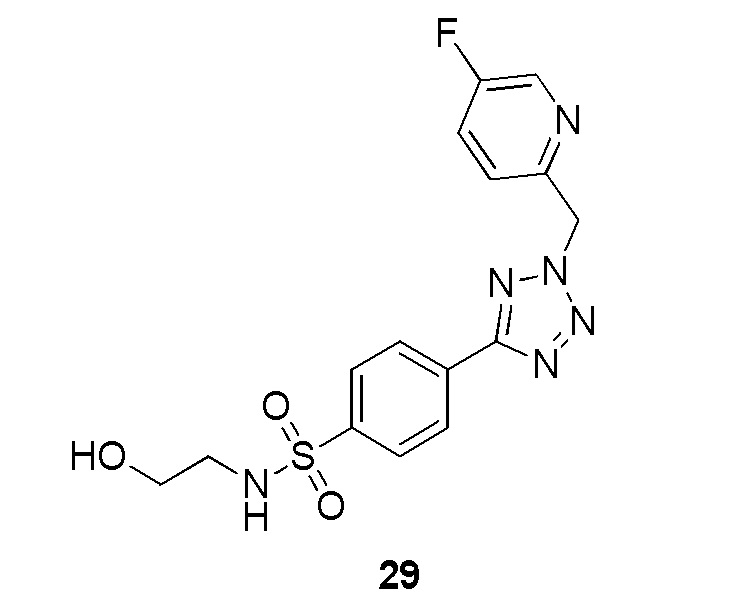
N, N-диизопропилэтиламин (1,507 мл, 8,63 ммоль, коммерческий источник: Aldrich) добавляли к перемешиваемому раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил)бензолсульфонамида (0,581 г, 2,158 ммоль, промежуточное соединение 17') в N, N-диметилформамиде (ДМФА) (7,19 мл). Затем добавляли 2-(хлорметил)-5-фторпиридин гидрохлорид (0,393 г, 2,158 ммоль, промежуточное соединение 9') и смесь перемешивали при комнатной температуре в течение 16 ч. Смесь концентрировали при пониженном давлении. Неочищенное соединение очищали колоночной флэш-хроматографией (силикагель; смесь EtOAc-циклогексан от 0/100 до 100/0). Желаемые фракции собирали и концентрировали в вакууме. Желтое масло осаждали Et2O и фильтровали с получением 4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил) бензолсульфонамида (178 мг, 0,447 ммоль, 20,7%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,53 (д, J=2,8 Гц, 1H), 8,24 (д, J=8,6 Гц, 2H), 7,98-7,92 (м, 2H), 7,82 (дт, J=8,7, 2,9 Гц, 1H), 7,78-7,74 (м, 1H), 7,64 (дд, J=8,6, 4,5 Гц, 1H), 6,16 (с, 2H), 4,68 (т, J=5,6 Гц, 1H), 3,35 (кв, J=6,2 Гц, 2H), 2,82 (кв, J=5,6 Гц, 2H). МС m/z [М-Н]- = 377,2.
Общая процедура алкилирования тетразолов
К раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (1 экв) И алкилирующего агента (1-1,6 экв) в ацетонитриле добавляли N, N-диизопропилэтиламин (2-4 экв) при 28°С. Реакционную смесь нагревали до 80°С и перемешивали в течение 16 ч при той же температуре. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь охлаждали до 28°С С, растворяли в этилацетате и промывали водой (3×). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырое соединение очищали. Желаемые фракции собирали и концентрировали в вакууме с получением продуктов.
Пример 29 (получали через промежуточные соединения 9 и 17): 4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид:
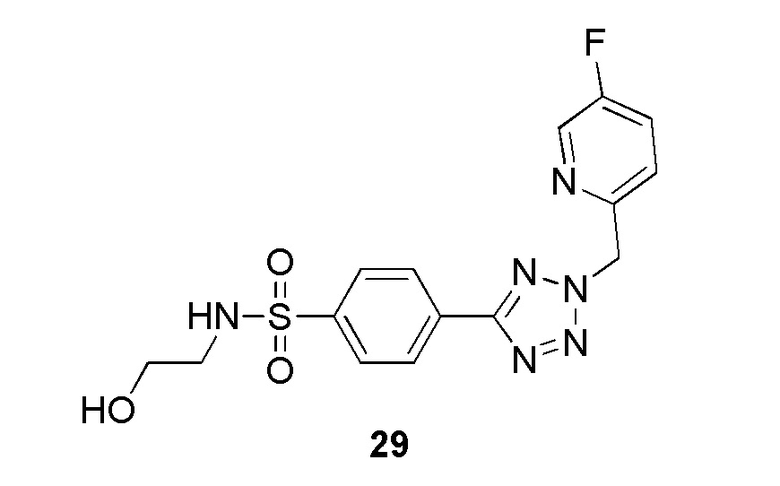
К раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (6 г, 0,0223 моль, промежуточное соединение 17), 2-(хлорметил)-5-фторпиридина (5 г, 0,0267 моль, промежуточное соединение 9) в ацетонитриле (90 мл) добавляли N, N-диизопропилэтиламин (11 мл, 0,0669 моль) при 28°С. Реакционную смесь нагревали до 80°С и перемешивали в течение 16 ч при той же температуре. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь охлаждали до 28°С, растворяли в этилацетате (800 мл) и промывали водой (3×200 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 2% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении. Полученное твердое соединение дополнительно очищали препаративной ВЭЖХ с получением после лиофилизации 4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамида (2,88 г, 34%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,53 (д, J=3,1 Гц, 1H), 8,26-8,21 (м, 2H), 7,98-7,93 (м, 2H), 7,85-7,78 (м, 1H), 7,74 (шир. с, 1H), 7,63 (дд, J=8,6, 4,4 Гц, 1H), 6,16 (с, 2H), 4,65 (шир. с, 1H), 3,36 (т, J=6,2 Гц, 2H), 2,83 (т, J=6,2 Гц, 2H). МС m/z [М+Н]+ = 379,05.
Условия препаративной ВЭЖХ:
Колонка: PURITAS C18 (250×30) мм, 10 мкм
Подвижная фаза: A-10 мМ бикарбонат аммония (водный раствор), B-ацетонитрил
Метод (время в мин/% В): 0/30, 1/30, 10/70, 10,5/100, 13/100, 13,5/30
Скорость потока: 30 мл/мин
Температура: комнатная.
Примеры 30-44 получали способами, аналогичными описанному для примера 29, с заменой алкилирующих реагентов и основных условий на условия, указанные в таблице 3. Метод, используемый для очистки, указан в виде сносок.
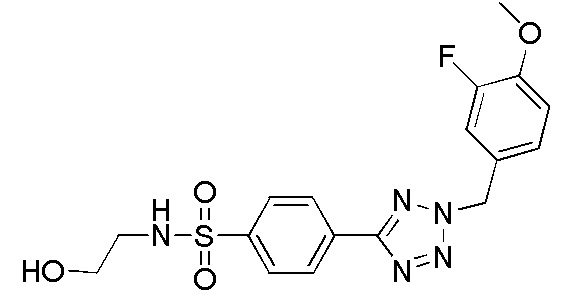
4-(2-(3-фтор-4-метоксибензил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид (см. сноску a)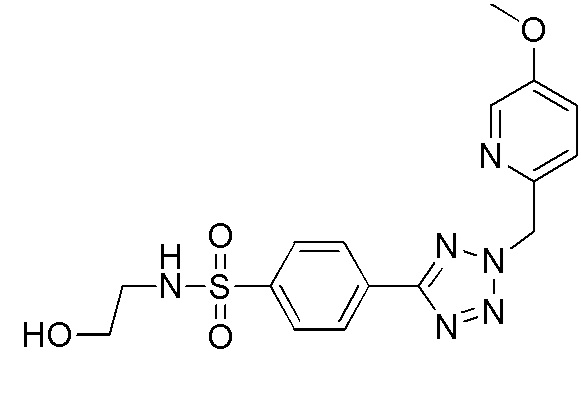
N-(2-гидроксиэтил)-4-(2-((5-метоксипиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид (см. сноску b)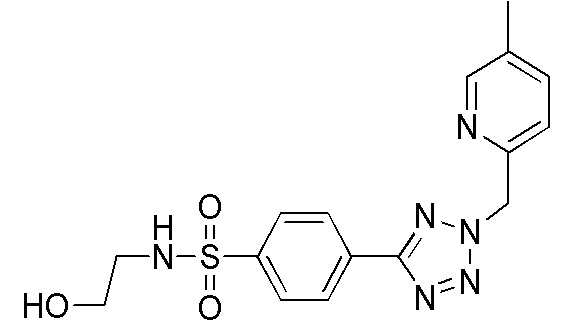
(N-(2-гидроксиэтил)-4-(2-((5-метилпиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид) (см. сноски с и d)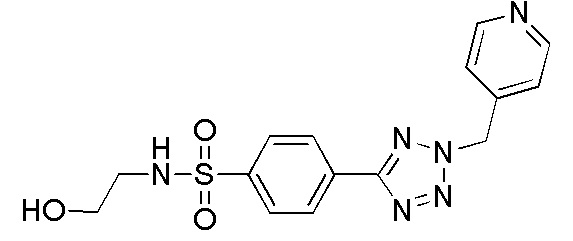
N-(2-гидроксиэтил)-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид (см. сноски e и f)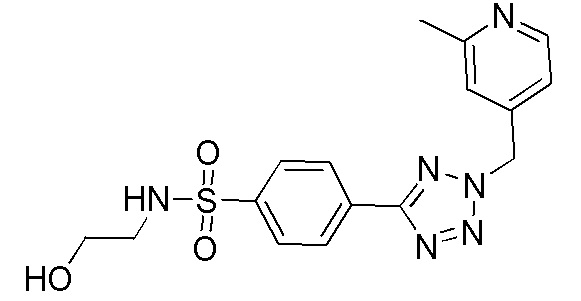
N-(2-гидроксиэтил)-4-(2-((2-метилпиридин-4-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид (см. сноску g)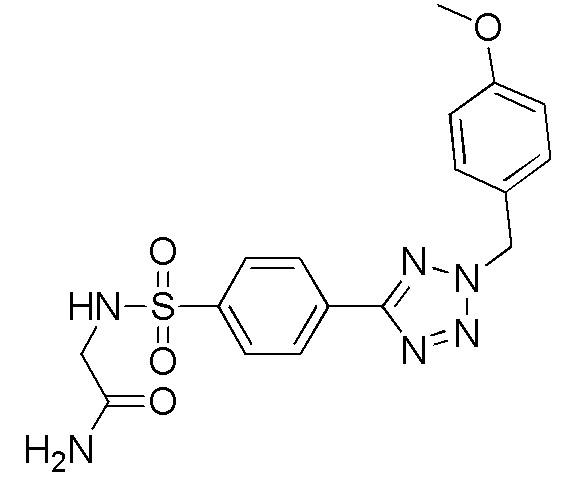
2-(4-(2-(4-метоксибензил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетамид (см. сноску h)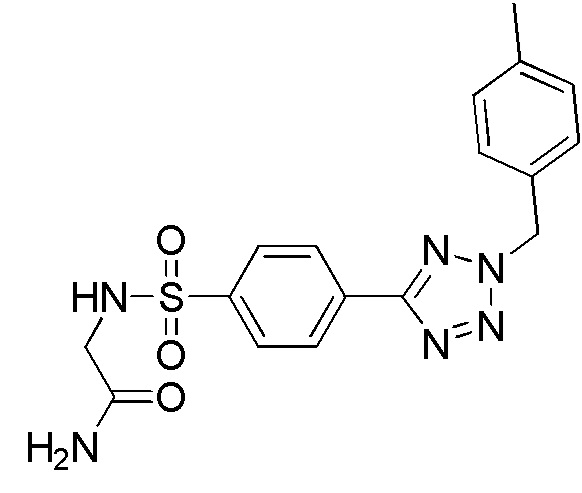
2-(4-(2-(4-метилбензил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетамид (см. сноску i)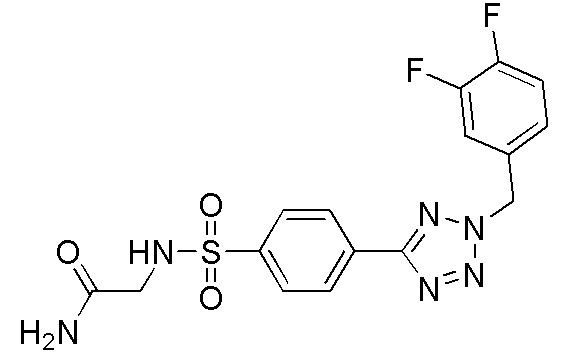
2-(4-(2-(3,4-дифторбензил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетамид (см. сноску j)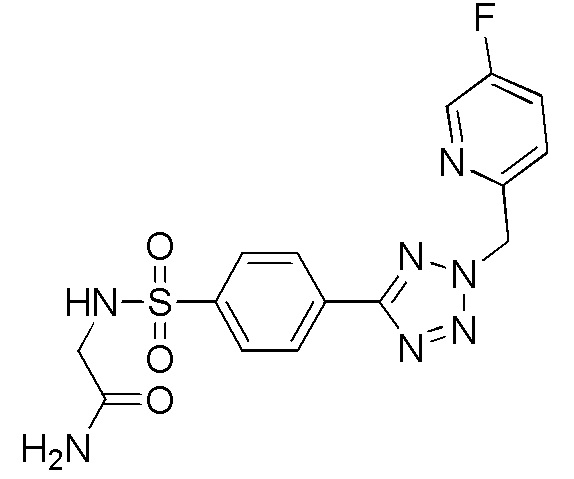
2-(4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид (см. сноску h)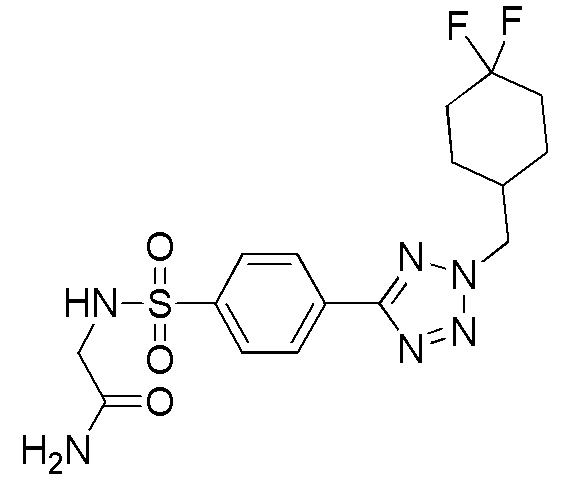
2-(4-(2-((4,4-дифторциклогексил)метил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид (см. сноску k)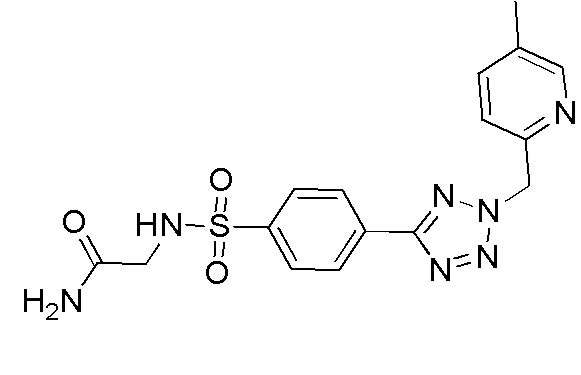
2-(4-(2-((5-метилпиридин-2-ил)метил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид (см. сноску l)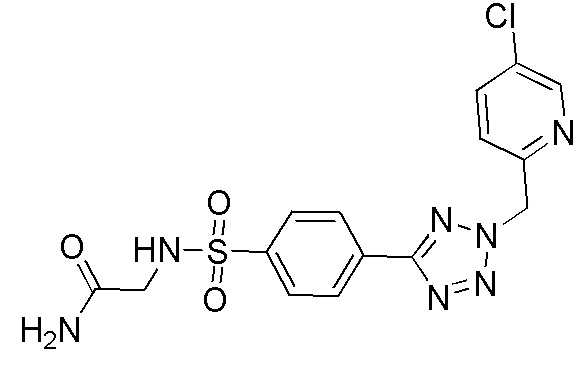
2-(4-(2-((5-хлорпиридин-2-ил)метил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид (см. сноску с)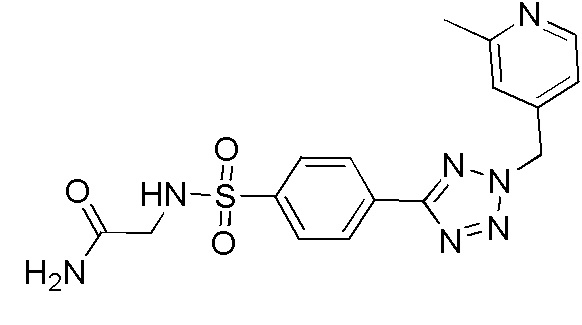
2-(4-(2-((2-метилпиридин-4-ил)метил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид (см. сноску m)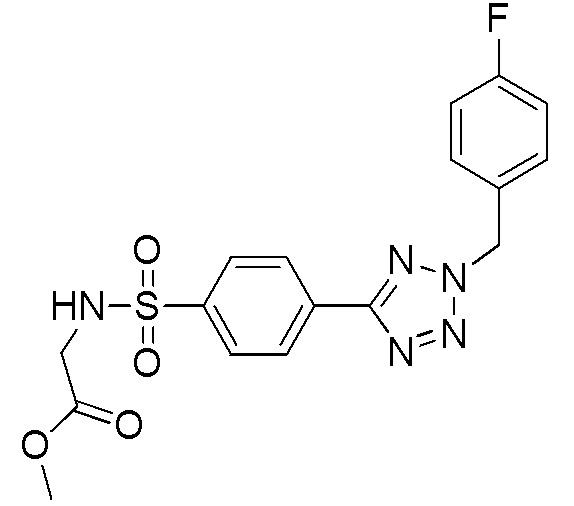
Метил 2-(4-(2-(4-фторбензил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетат (см. сноски с и n)
без обработки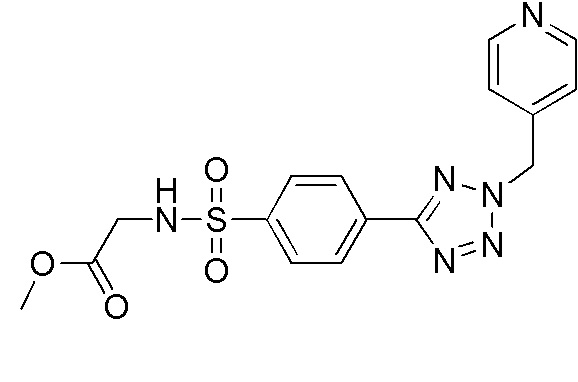
Метил 2-(4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетат (см. сноску о)
b) Остаток очищали препаративной ВЭЖХ на колонке YMC Triart C18 (25×150 мм, 10 мкм, скорость потока: 25 мл/мин). Подвижная фаза: A:H2O (10 мМ NH4HCO3) B:ACN; метод (время в мин/% В): 0/20, 8/55, 9/55, 9,2/98, 13/98, 13,1/20, 16/20;
c) Остаток очищали флэш-хроматографией со смесью DCM/MeOH (2%);
d) Остаток очищали препаративной ВЭЖХ на колонке Puritas C18 (30×550 мм, 10 мкм, скорость потока: 25 мл/мин). Подвижная фаза: A:H2O (10 мМ NH4HCO3) B:ACN; метод (время в мин/% В): 0/10, 10/60, 14/100, 14,3/100, 15/10, 18/10;
е) Остаток очищали флэш-хроматографией с использованием смеси 1,5% метанола в дихлорметане;
f) Остаток очищали препаративной ВЭЖХ на колонке Diasogel C18 (25×150 мм, скорость потока: 25 мл/мин). Подвижная фаза: A:H2O (10 мМ NH4HCO3) B:ACN; метод (время в мин/% В): 0/35, 1/40, 8/50, 12/50, 12,2/100;
g) Остаток очищали препаративной ВЭЖХ на колонке Kromasil C18 (25×150 мм, 10 мкм, скорость потока: 25 мл/мин). Подвижная фаза: A:H2O (10 мМ NH4HCO3) B:ACN; метод (время в мин/% В): 0/10, 1/10, 10/55, 10,5/ 100, 14/100, 14,5/10;
h) Остаток очищали препаративной ВЭЖХ на колонке X-SELECT C18 (19×150 мм, 5 мкм, скорость потока: 20 мл/мин). Подвижная фаза: A:H2O (10 мМ NH4HCO3) B:ACN; метод (время в мин/% В): 0/10, 10/60, 10,3/100, 12,7/100, 13/10, 15/10;
i) Остаток очищали препаративной ВЭЖХ на колонке Puritas C18 (30×250 мм, 10 мкМ, скорость потока: 30 мл/мин). Подвижная фаза: A:H2O (10 мМ NH4HCO3) B:ACN; метод (время в мин/% В): 0/35, 1/35, 8/75, 10,5/75, 11/100, 15,5/100, 16/35, 20/35;
j) Остаток очищали препаративной ВЭЖХ на колонке X-Terra RP C18 (19×150 мм, 5 мкм, скорость потока: 18 мл/мин). Подвижная фаза: A: H2O (10 мМ NH4HCO3) B:ACN; метод (время в мин/% В): 0/20, 1/20, 10/45, 11/45, 11,5/100, 14/100;
k) Остаток очищали препаративной ВЭЖХ на колонке X-Bridge C18 (19×150 мм, 5 мкм, скорость потока: 15 мл/мин). Подвижная фаза: A:H2O (10 мМ NH4HCO3) B:ACN; метод (время в мин/% В): 0/10, 8/45, 8,2/100, 12,5/100, 12,7/10, 15/10;
l) Остаток очищали препаративной ВЭЖХ на колонке Luna C18 (25×150 мм, 10 мкм, скорость потока: 25 мл/мин). Подвижная фаза: A:H2O (10 мМ NH4HCO3) B:ACN; метод (время в мин/% от В): 0/20, 8/50, 8,2/100, 9/100;
m) Остаток очищали препаративной ВЭЖХ на колонке YMC Triart C18 (25×150 мм, 10 мкм, скорость потока: 20 мл/мин). Подвижная фаза: A: H2O (10 мМ NH4HCO3) B:ACN; метод (время в мин/% от В): 0/20, 1/20, 10/50, 11,5/ 50, 12/100, 14/100, 14,5/100;
n) Остаток очищали препаративной ВЭЖХ на колонке Kromasil C18 (25×150 мм, 10 мкм, скорость потока: 25 мл/мин). Подвижная фаза: A:H2O (10 мМ NH4HCO3) B:ACN; метод (время в мин/% от В): 0/30, 1/30, 10/70, 10,5/ 100, 13/100, 13,5/30;
о) Остаток очищали флэш-хроматографией с использованием смеси 2,5% метанола в дихлорметане.
Пример 45: 2-(4-(2-(4-фторбензил)-2Н-тетразол-5-ил) фенилсульфонамидо)ацетамид:
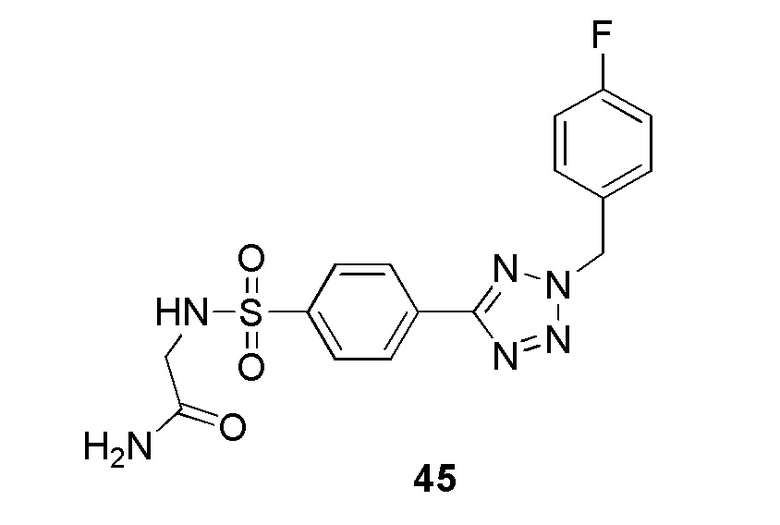
К раствору 2-(4-(2-(4-фторбензил)-2Н-тетразол-5-ил) фенилсульфонамидо)уксусной кислоты (220 мг, 0,00056 моль, промежуточное соединение 28), хлорида аммония (44 мг, 0,00084 моль) в N, N-диметилформамиде (2,2 мл) добавляли N, N-диизопропилэтиламин (0,24 мл, 0,0014 моль) и HATU (319 мг, 0,00084 моль) при 0°С. Реакционную смесь перемешивали при 28°С в течение 16 ч. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь охлаждали до 0°С и гасили ледяной водой (20 мл). Осажденное твердое соединение отфильтровывали и высушивали в вакууме. Сырой продукт очищали препаративной ВЭЖХ с получением 2-(4-(2-(4-фторбензил)-2Н-тетразол-5-ил) фенилсульфонамидо)ацетамида (62 мг, 28%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,24-8,19 (м, 2Н), 8,03 (шир. с, 1Н), 7,98-7,93 (м, 2Н), 7,53-7,47 (м, 2Н), 7,30-7,21 (м, 3Н), 7,06 (шир. с, 1Н), 6,03 (с, 2Н), 3,41 (с, 2Н). МС m/z [М+Н]+ = 391,05.
Условия препаративной ВЭЖХ:
Колонка: Kromasil C18 (150×25) мм, 10 мкм
Подвижная фаза: A-10 мМ бикарбонат аммония (водный раствор), B-ацетонитрил
Метод (время в мин/% В): 0/20, 1/20, 10/20, 10,2/100, 11/100, 11,2/20, 15/20
Скорость потока: 25 мл/мин
Температура: комнатная.
Пример 46: 2-(4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил) фенилсульфонамидо)ацетамид:
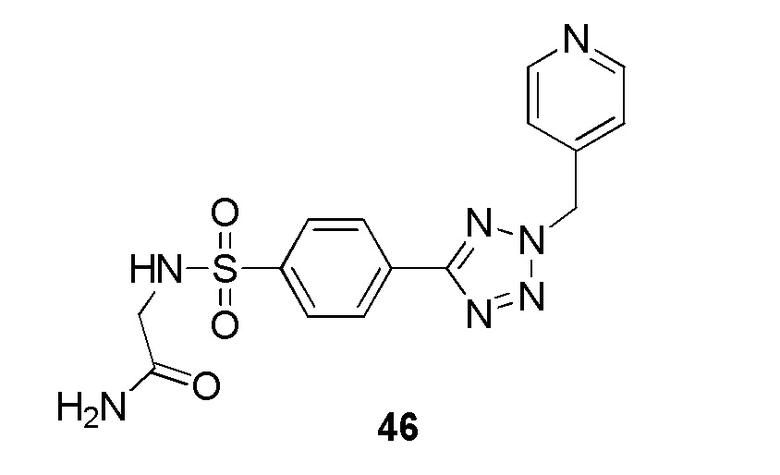
К раствору 2-(4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил) фенилсульфонамидо)уксусной кислоты (200 мг, 0,0005 моль, промежуточное соединение 29), хлорида аммония (42 мг, 0,0008) моль) в N,N-диметилформамиде (2 мл) добавляли N, N-диизопропилэтиламин (0,209 мл, 0,0012 моль) и HATU (304 мг, 0,0008 моль) при 0°С. Температуру реакционной смеси повышали до 28°С и перемешивали в течение 16 ч при той же температуре. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь охлаждали до 0°С, гасили ледяной водой (20 мл) и перемешивали в течение 15 мин. Осажденное твердое вещество отфильтровывали и высушивали в вакууме с получением 2-(4-(2- (пиридин-4-илметил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетамида (70 мг, 36%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,63-8,56 (м, 2Н), 8,28-8,20 (м, 2Н), 8,08-8,00 (м, 1Н), 8,00-7,92 (м, 2Н), 7,38-7,31 (м, 2Н), 7,28 (шир. с, 1Н), 7,06 (шир. с, 1Н), 6,14 (с, 2Н), 3,46-3,38 (м, 2Н). МС m/z [М+Н]+ = 374,06.
Пример 47: (4-(2-(4-фторбензил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)-2-метоксибензолсульфонамид):
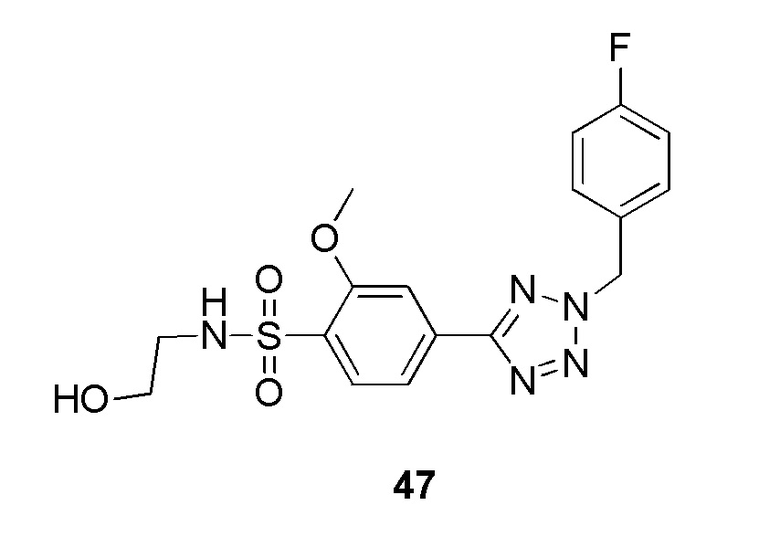
К раствору N-(2-гидроксиэтил)-2-метокси-4-(2H-тетразол-5-ил) бензолсульфонамида (200 мг, 0,0006 моль, промежуточное соединение 33), 1-(бромметил)-4-фторбензола (150 мг, 0,0008 моль, коммерческий источник: Combi-Blocks) в N, N-диметилформамиде (2 мл) добавляли карбонат калия (165 мг, 0,0012 моль) при 28°С. Реакционную смесь нагревали до 100°С и перемешивали в течение 16 ч при той же температуре. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь охлаждали до 28°С и концентрировали при пониженном давлении. Сырой продукт очищали препаративной ВЭЖХ с получением 4-(2-(4-фторбензил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)-2-метоксибензолсульфонамида (23 мг, 9%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,91-7,87 (м, 1H), 7,77-7,72 (м, 2H), 7,52-7,49 (м, 2H), 7,28-7,21 (м, 2H), 7,18 (шир. с, 1Н), 6,03 (с, 2Н), 4,60 (шир. с, 1Н), 3,99 (с, 3Н), 3,37-3,30 (м, 2Н), 2,87-2,81 (м, 2Н). МС m/z [М+Н]+ = 408,04.
Условия препаративной ВЭЖХ:
Колонка: YMC Triart C18 (150×25) мм, 10 мкм
Подвижная фаза: A-10 мМ бикарбонат аммония (водный раствор), B-ацетонитрил
Метод (время в мин/% В): 0/20, 10/60, 12/60, 12,3/100, 15,5/ 100, 15,8/20, 18/20
Скорость потока: 25 мл/мин
Температура: комнатная.
Пример 48: (N-(2-гидроксиэтил)-2-метокси-4-(2-(пиридин-4-илметил)-2Н-тетразол-5-ил)бензолсульфонамид):
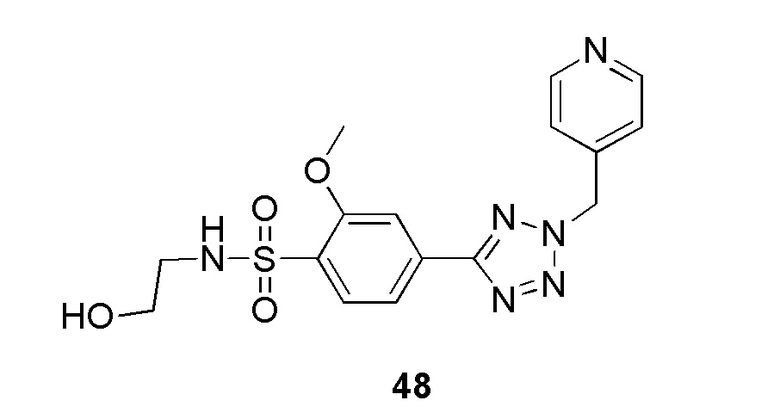
К раствору N-(2-гидроксиэтил)-2-метокси-4-(2H-тетразол-5-ил) бензолсульфонамида (200 мг, 0,0006 моль, промежуточное соединение 33) в ацетонитриле (4 мл) добавляли триэтиламин (121 мг, 0,0012 моль) при 28°С и перемешивали в течение 15 мин при 28°С. Затем добавляли 4-(хлорметил)пиридин гидрохлорид (131 мг, 0,0008 моль, коммерческий источник: Combi-Blocks) при 28°С. Реакционную смесь нагревали до 80°С и перемешивали в течение 16 ч при той же температуре. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь охлаждали до 28°С и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,5% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении. Полученное соединение очищали колоночной SCX с получением N-(2-гидроксиэтил)-2-метокси-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамида (21 мг, 8,6%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,62-8,58 (м, 2Н), 7,93-7,87 (м, 1Н), 7,80-7,74 (м, 2Н), 7,35-7,30 (м, 2Н), 7,23-7,17 (м, 1Н), 6,14 (с, 2Н), 4,63-4,58 (м, 1Н), 4,00 (с, 3Н), 3,37-3,30 (м, 2Н), 2,88-2,82 (м, 2Н). МС m/z [М+Н]+ = 391,23.
Пример 49: (4-(2-(4-цианобензил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)-2-метоксибензолсульфонамид):
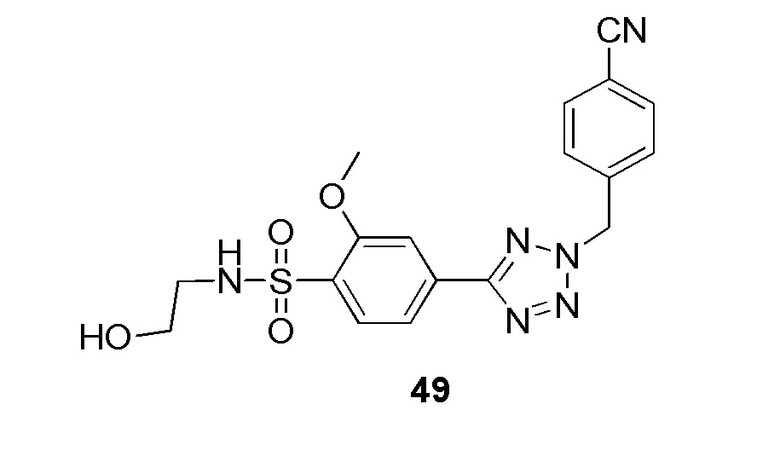
К раствору N-(2-гидроксиэтил)-2-метокси-4-(2H-тетразол-5-ил) бензолсульфонамида (200 мг, 0,0006 моль, промежуточное соединение 33) в ацетонитриле (2 мл) добавляли триэтиламин (0,16 мл, 0,0012 моль) при 28°С и перемешивали при 28°С в течение 5 мин. Затем добавляли 4-(бромметил)бензонитрил (156 мг, 0,0008 моль, коммерческий источник: AK Scientific) при 28°С. Реакционную смесь нагревали до 80°С и перемешивали в течение 16 ч при той же температуре. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь охлаждали до 28°С и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 2,5% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении (50 мг, неочищенное соединение). Реакцию повторяли два раза, каждый раз начиная с 200 мг промежуточного соединения 33, и полученные неочищенные продукты объединяли и очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 2,5% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-(2-(4-цианобензил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)-2-метоксибензолсульфонамида (28 мг, 11%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,92-7,86 (м, 3H), 7,78-7,73 (м, 2H), 7,60-7,55 (м, 2H), 7,21 (т, J=5,9 Гц, 1H) 6,18 (с, 2H), 4,61 (шир. с, 1H), 3,99 (с, 3H), 3,37-3,30 (м, 2H), 2,87-2,80 (м, 2H). МС m/z [М+Н]+ = 415,2.
Пример 50: (4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)-2-метоксибензолсульфонамид):
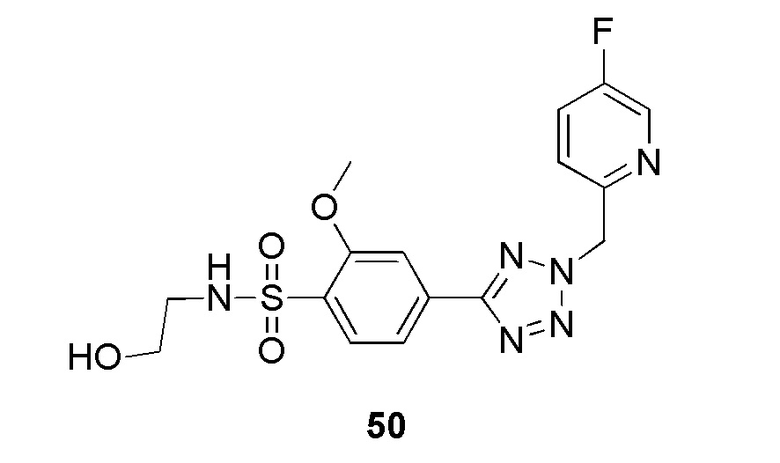
К раствору N-(2-гидроксиэтил)-2-метокси-4-(2H-тетразол-5-ил) бензолсульфонамида (220 мг, 0,0007 моль, промежуточное соединение 33), 2-(хлорметил)-5-фторпиридина (106 мг, 0,0007 моль, промежуточное соединение 9) в ацетонитриле (2,2 мл) добавляли N, N-диизопропилэтиламин (0,24 мл, 0,0014 моль) при 26°С. Реакционную смесь нагревали до 80°С и перемешивали в течение 16 ч при той же температуре. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь охлаждали до 26°С и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 2,1% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении. Полученное соединение очищали препаративной ТСХ, элюируя смесью 2% метанола в дихлорметане в качестве подвижной фазы. Чистую фракцию собирали и выделяли с получением 4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)-2-метоксибензолсульфонамида (28 мг, 9,6%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,53 (д, J=2,7 Гц, 1H), 7,91-7,87 (м, 1H), 7,84-7,78 (м, 1H), 7,75 (с, 1H), 7,76- 7-72 (м, 1Н), 7,66-7,61 (м, 1Н), 7,21-7,17 (м, 1Н), 6,16 (с, 2Н), 4,63-4,58 (м, 1Н), 3,99 (с, 3Н), 3,37 -3,30 (м, 2H), 2,84 (кв, J=6,4 Гц, 2H). МС m/z [М+Н]+ = 409,13.
Пример 51: (2-метокси-N-метил-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид):
К раствору 2-метокси-N-метил-4-(2H-тетразол-5-ил) бензолсульфонамида (300 мг, 1,114 ммоль, промежуточное соединение 35) в N, N-диметилформамиде (10 мл) добавляли карбонат калия (308 мг, 2,229 ммоль) и 4-(хлорметил)пиридин гидрохлорид (183 мг, 1,116 ммоль, коммерческий источник: Combi-Blocks) при 27°С. Реакционную смесь нагревали до 80°С и перемешивали в течение 3 ч при той же температуре. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь охлаждали до 27°С и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 3% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 2-метокси-N-метил-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамида (101 мг, 25%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,63-8,58 (м, 2Н), 7,92-7,87 (м, 1Н), 7,80-7,75 (м, 2Н), 7,35-7,30 (м, 2Н), 7,19-7,14 (м, 1H), 6,14 (с, 2H), 4,00 (с, 3H), 2,43 (д, J=4,8 Гц, 3H). МС m/z [М+Н]+ = 361,0.
Пример 52: (N-(цианометил)-4-(2-(4-фторбензил)-2Н-тетразол-5-ил)бензолсульфонамид):
К раствору 4-(2-(4-фторбензил)-2H-тетразол-5-ил) бензолсульфонамида (400 мг, 1,2 ммоль, промежуточное соединение 44) в этаноле (10 мл) добавляли бензотриазол (144 мг, 1,208 ммоль) и формальдегид (30% водный раствор) (0,44 мл, 5,867 ммоль) при 27°С и перемешивали при 27°С в течение 48 ч. Осажденную реакционную смесь отфильтровывали и твердое вещество промывали смесью этанол:диэтиловый эфир (1:1, 3×10 мл). Твердое вещество высушивали в вакууме и обрабатывали цианидом калия (400 мг, 6,142 ммоль) в воде (2 мл) в течение 24 ч при 27°С. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь фильтровали и промывали смесью этанол:диэтиловый эфир (1:1, 3×10 мл), фильтрат концентрировали. Остаток растворяли в воде (10 мл) и экстрагировали дихлорметаном (2×10 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали препаративной ВЭЖХ с получением N-(цианометил)-4-(2-(4-фторбензил)-2Н-тетразол-5-ил)бензолсульфонамида (39 мг, 8%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,75 (шир. с, 1H), 8,28-8,23 (м, 2H), 8,02-7,96 (м, 2H), 7,54-7,47 (м, 2H), 7,28-7,20 ( м, 2Н), 6,03 (с, 2Н), 4,13 (с, 2Н). МС m/z [М+Н]+ = 373,00.
Условия препаративной ВЭЖХ:
Колонка: KROMASIL phenyl (250×25) мм, 10 мкм
Подвижная фаза: A-10 M бикарбонат аммония (водный раствор), B-ацетонитрил
Метод (время в мин/% В): 0/20, 10/65, 10,2/100, 13/100, 13,2/100, 18/20
Скорость потока: 25 мл/мин
Температура: комнатная.
Общая процедура снятия Boc-защиты
К раствору Boc-производных тетразола (1 экв) в дихлорметане (0,2-0,04 М) добавляли 2,2,2-трифторуксусную кислоту (5 экв) при 0°С в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение 4-16 ч. Затем раствор разбавляли водой и дважды экстрагировали DCM. Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Неочищенное соединение очищали флэш-хроматографией со смесью EtOAc-циклогексан от 0/100 до 100/0. Альтернативно, когда реакционную смесь концентрировали при пониженном давлении, то неочищенное соединение очищали препаративной ВЭЖХ. Желаемые фракции собирали и концентрировали в вакууме с получением конечных продуктов.
Пример 53: 2-(4-(2-(4-фторбензил)-2Н-тетразол-5-ил)-2-метоксифенилсульфонамидо)ацетамид:
К раствору трет-бутил (2-амино-2-оксоэтил)((4-(2-(4-фторбензил)-2Н-тетразол-5-ил)-2-метоксифенил)сульфонил) карбамата (240 мг, 0,461 ммоль, промежуточное соединение 37) в дихлорметане (2,3 мл) добавляли 2,2,2-трифторуксусную кислоту (0,165 мл, 2,305 ммоль) при 0°С в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Раствор разбавляли водой и дважды экстрагировали DCM. Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Неочищенное соединение очищали колоночной флэш-хроматографией (силикагель; со смесью EtOAc-циклогексан от 0/100 до 100/0). Фракции собирали и концентрировали в вакууме с получением 2-(4-(2-(4-фторбензил)-2Н-тетразол-5-ил)-2-метоксифенилсульфонамидо)ацетамида (26 мг, 25%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,91-7,86 (м, 1Н), 7,78-7,70 (м, 2Н), 7,55-7,46 (м, 2Н), 7,40 (шир. с, 1Н), 7,29-7,22 (м, 2Н), 7,19 (шир. с, 1Н), 7,05 (шир. с, 1Н), 6,03 (с, 2Н), 3,98 (с, 3Н), 3,30 (с, 2Н). МС m/z [М+Н]+ = 421,2.
Примеры 54-61 получали способами, аналогичными описанному для примера 53, заменяя промежуточное соединение, растворитель и кислые условия на условия, указанные в таблице 4. Метод, используемый для очистки, указан в виде сносок.
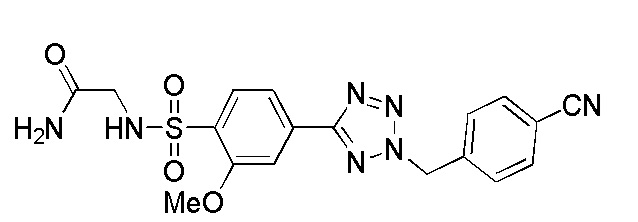
2-(4-(2-(4-цианобензил)2Н-тетразол-5-ил)-2-метоксифенилсульфонамидо)ацетамид (см. сноску a)
0,26 мл
1H ЯМР (400 МГц, ДМСО-d6) δ 7,96-7,85 (м, 3Н), 7,79-7,70 (м, 2Н), 7,59 (д, J=8,3 Гц, 2Н), 7,43 (т, J=5,9 Гц, 1H), 7,21 (шир. с, 1H), 7,07 (шир. с, 1H), 6,20 (с, 2H), 4,02-3,95 (м, 3H), 3,48 (д, J=5,8 Гц, 2H). МС m/z [М-Н]- = 426,11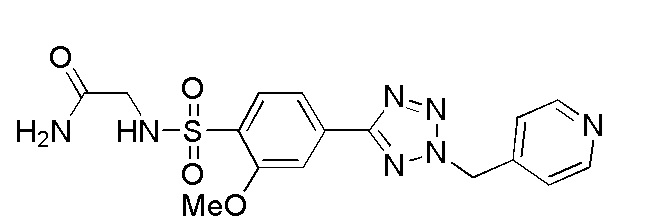
2-(2-метокси-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид (см. сноску a)
0,056 мл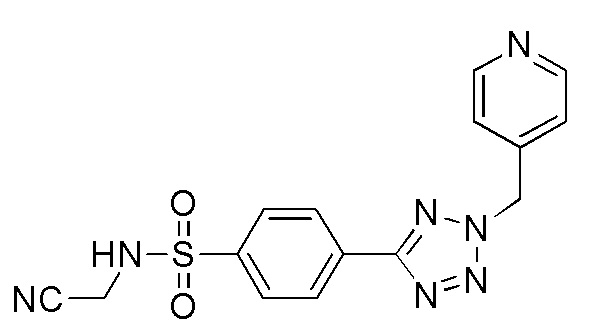
(N-(цианометил)-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид) (см. сноску b)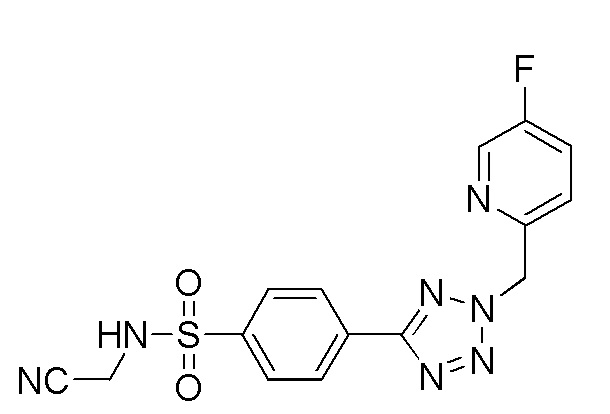
(N-(цианометил)-4-(2-((5-фторпиридин-2-ил)метил)-
2Н-тетразол-5-ил)бензолсульфонамид)
(см. сноску с)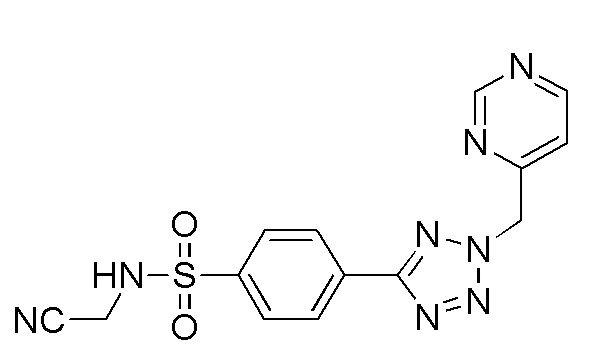
(N-(цианометил)-4-(2-(пиримидин-4-илметил)-2Н-тетразол-5-ил)бензолсульфонамид (см. сноску d)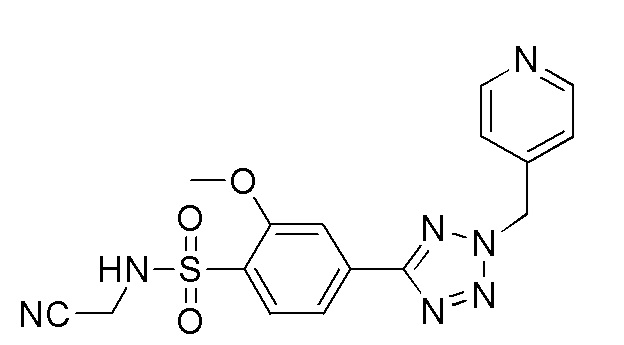
(N-(цианометил)-2-метокси-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид) (см. сноску е)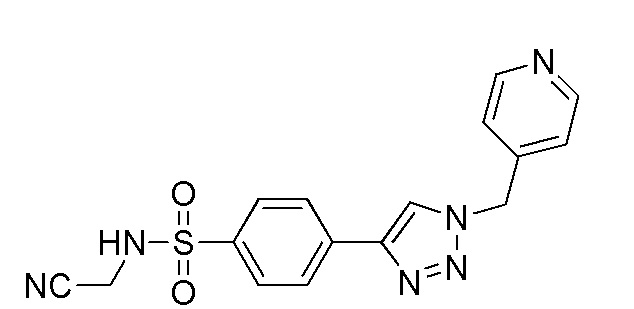
(N-(цианометил)-4-(1-(пиридин-4-илметил)-1Н-1,2,3-триазол-4-ил)бензолсульфонамид)
(см. сноску f)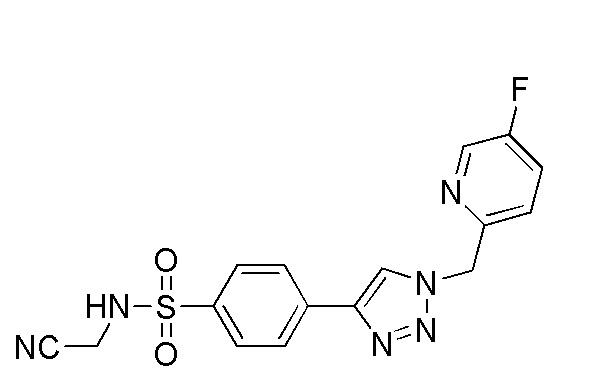
(N-(цианометил)-4-(1-((5-фторпиридин-2-ил)метил)-1Н-1,2,3-триазол-4-ил)бензолсульфонамид) (см. сноску g)
75)
Остаток очищали препаративной ВЭЖХ на колонке Kromasil C18 (25×150 мм, 10 мкм, скрость потока: 30 мл/мин). Подвижная фаза: A:H2O (10 мМ NH4HCO3) B: ACN; метод (время в мин/% В): 0/10, 1/10, 10/45, 10,5/ 100, 12,5/100, 13/10, 15/10;
Остаток очищали препаративной ВЭЖХ на колонке Diasogel C18 (25×150 мм, 8 мкм, скорость потока: 20 мл/мин). Подвижная фаза: A: H2O (10 мМ NH4HCO3) B: ACN; метод (время в мин/% В): 0/30, 1/30, 10/55, 11,5/55, 11,8/98, 14/98, 14,5/30, 17/30;
Остаток очищали препаративной ВЭЖХ на колонке Diasogel C18 (25×150 мм, 8 мкм, скорость потока: 20 мл/мин). Подвижная фаза: A: H2O (10 мМ NH4HCO3) B: ACN; метод (время в мин/% В): 0/35, 7,5/35, 8/100, 10/100, 10,2/35, 13/35;
Остаток очищали препаративной ВЭЖХ на колонке Phenomenex Luna C18 (21,2×250 мм, 5 мкм, скорость потока: 30 мл/мин). Подвижная фаза: A: H2O (10 мМ NH4HCO3) B: ACN+метанол; метод (время в мин/% В): 0/30, 1/20, 10/60, 11,5/100, 13/100, 13,5/30, 16/30;
Остаток очищали препаративной ВЭЖХ на колонке Luna C18 (21,2-250 мм, 5 мкм, скорость потока: 20 мл/мин). Подвижная фаза: A: H2O (10 мМ NH4HCO3) B: ACN; метод (время в мин/% В): 0/10, 10/60, 10,3/100, 12,7/100, 13/10, 15/10;
Остаток очищали препаративной ВЭЖХ на колонке ВЭЖХ Luna C18 (25×150 мм, 10 мкм, скорость потока: 25 мл/мин). Подвижная фаза: A: H2O (10 мМ NH4HCO3) B: ACN; метод (время в мин/% В): 0/30, 1/30, 7/60, 10/60, 10,2/100.
Общая процедура для 1,4-дизамещенного 1,2,3-триазола с клик циклоприсоединением
К раствору терминальных ацетиленов (1 экв) и ароматических азидов (1 экв) в смеси 1:1 этанола (0,2 М) и воды (0,2 М) добавляли сульфат меди пентагидрат (0,1 экв) И L-аскорбат натрия (0,3 экв) при комнатной температуре. Полученную реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырое соединение очищали. Желаемые фракции собирали и концентрировали в вакууме с получением продуктов.
Пример 62: (4-(1-((5-фторпиридин-2-ил)метил)-1Н-1,2,3-триазол-4-ил)-N-(2-гидроксиэтил)бензолсульфонамид):
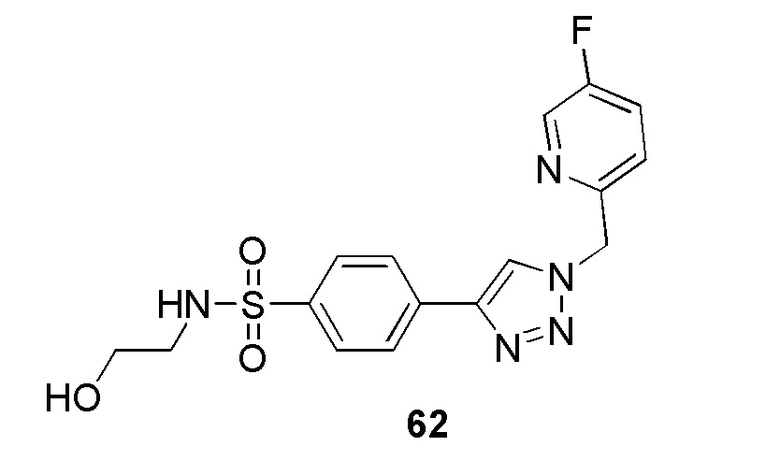
К раствору 4-этинил-N-(2-гидроксиэтил)бензолсульфонамида (200 мг, 0,8888 ммоль, промежуточное соединение 62) и 2- (азидометил)-5-фторпиридина (135 мг, 0,8888 ммоль, промежуточное соединение 57) в этаноле (5 мл) и воде (5 мл) добавляли сульфат меди пентагидрат (22 мг, 0,0888 ммоль) и L-аскорбат натрия (53 мг, 0,2666 ммоль) при 27°С. Полученную реакционную смесь перемешивали при 27°С в течение 16 ч. Ход реакции контролировали ТСХ. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Неочищенное соединение очищали колоночной хроматографией (силикагель 100-200 меш), используя смесь 2% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении. Полученное соединение очищали препаративной ВЭЖХ с получением 4-(1-((5-фторпиридин-2-ил)метил)-1Н-1,2,3-триазол-4-ил)-N-(2-гидроксиэтил)бензолсульфонамида (52 мг, 15%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,78 (с, 1H), 8,55 (д, J=3,1 Гц, 1H), 8,08-8,03 (м, 2H), 7,88-7,82 (м, 2H), 7,82- 7,75 (м, 1H), 7,60 (шир. с, 1H), 7,54-7,48 (м, 1H), 5,79 (с, 2H), 4,65 (шир. с, 1H), 3,36 (т, J=6,4 Гц, 2H) 2,81 (т, J=6,4 Гц, 2H). МС m/z [М+Н]+ = 378,40.
Условия препаративной ВЭЖХ:
Колонка: Puritas C18 (250×30) мм, 10 мкм
Подвижная фаза: A-10 мМ бикарбонат аммония (водный раствор), B-ацетонитрил
Метод (время в мин/% В) = 0/10, 10/60, 10,3/100, 12,7/ 100, 13/10, 15/10
Скорость потока: 25 мл/мин
Температура: комнатная.
Примеры 63-69 получали способами, аналогичными описанному для примера 62, с заменой промежуточных соединений на указанные в таблице 5. Метод, используемый для очистки, указан в виде сносок.
(N-(2-гидроксиэтил)-4-(1-(4-метоксибензил)-1Н-1,2,3-триазол-4-ил)бензолсульфонамид) (см. сноску a)
(4-(1-(4-фторбензил)-1Н-1,2,3-триазол-4-ил)-N-(2-гидроксиэтил)бензолсульфонамид) (см. сноску b)
(N-(2-гидроксиэтил)-4-(1-(пиридин-4-илметил)-1Н-1,2,3-триазол-4-ил)бензолсульфонамид) (см. сноску с)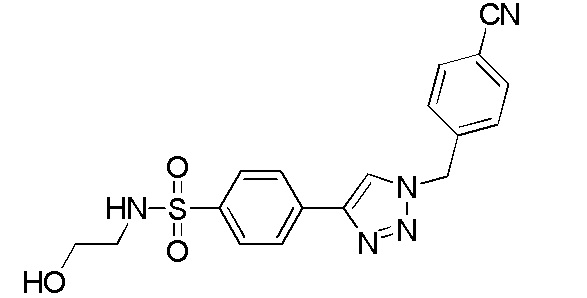
(4-(1-(4-цианобензил)-1Н-1,2,3-триазол-4-ил)-N- (2-гидроксиэтил)бензолсульфонамид) (см. сноску b)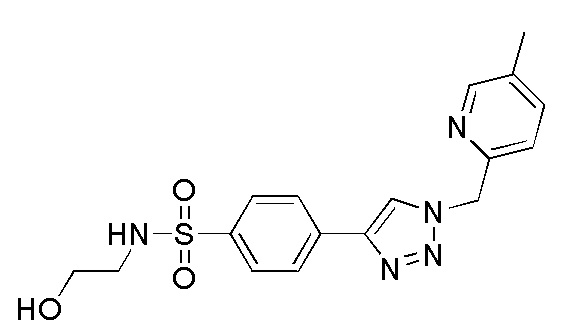
(N-(2-гидроксиэтил)-4-(1-((5-метилпиридин-2-ил)метил)-1Н-1,2,3-триазол-4-ил)бензолсульфонамид) (см. сноску b)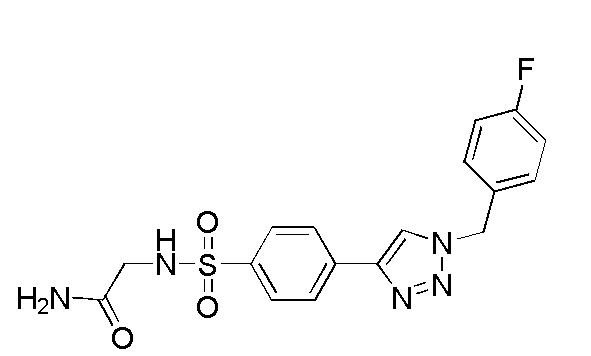
(2-(4-(1-(4-фторбензил)-1Н-1,2,3-триазол-4-ил)фенилсульфонамидо)ацетамид) (см. сноску d)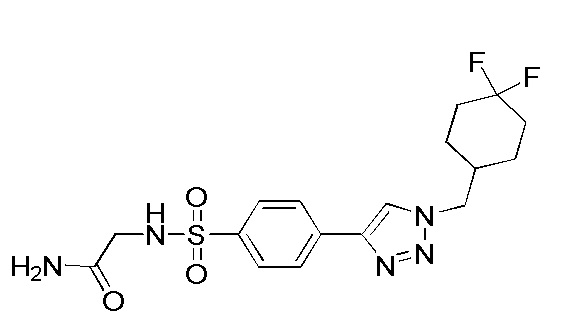
(2-(4-(1-((4,4-дифторциклогексил)метил)-1Н-1,2,3-триазол-4-ил)фенилсульфонамидо)ацетамид) (см. сноску е)
Остаток очищали флэш-хроматографией с использованием смеси 5% метанола в дихлорметане;
Остаток очищали флэш-хроматографией с использованием смеси 10% метанола в дихлорметане. В конце остаток очищали препаративной ВЭЖХ на колонке Kromasil Phenyl (25×150 мм, 10 мкм, скорость потока: 30 мл/мин). Подвижная фаза: A: H2O (10 мМ NH4HCO3) B: ACN; метод (время в мин/% В): 0/20, 1/20, 8,5/53,3, 9/100, 11,5/100, 12/20, 14/20;
Остаток очищали препаративной ВЭЖХ на колонке Kromasil C18 (25×150 мм, 10 мкм, скорость потока: 25 мл/мин). Подвижная фаза: A: H2O (10 мМ NH4HCO3) B: ACN; метод (время в мин/% В): 0/37, 7/37, 7,5/100, 9/100, 9,5/37;
Остаток очищали флэш-хроматографией с использованием смеси 5% метанола в дихлорметане. В конце остаток очищали препаративной ВЭЖХ на колонке Kromasil C18 (25×150 мм, 10 мкм, скорость потока: 25 мл/мин). Подвижная фаза: A: H2O (10 мМ NH4HCO3) B: ACN; метод (время в мин/% В): 0/10, 1/10, 10,5/ 100, 14/100, 14,5/10.
Пример 70: (R)-4-(1-(4-фторбензил)-1H-1,2,3-триазол-4-ил)-N-(2-гидроксипропил)бензолсульфонамид:
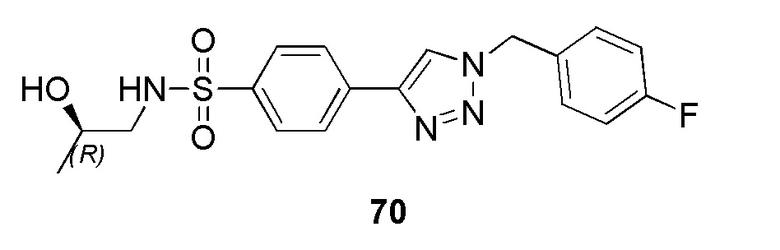
К раствору 1-(азидометил)-4-фторбензола (45 мг, 0,420 ммоль, промежуточное соединение 54) и (R)-4-этинил-N-(2-гидроксипропил) бензолсульфонамида (50 мг, 0,209 ммоль, промежуточное соединение 68) в смеси 1:1 этанол (1 мл)/вода (1 мл) добавляли CuSO4 (6 мг, 0,042 ммоль) и аскорбат натрия (24 мг, 0,126 ммоль), каждый одной порцией. Полученную суспензию перемешивали при комнатной температуре в течение 16 ч. Полученную смесь распределяли между водой (30 мл) и EtOAc (50 мл). Водный слой экстрагировали дополнительным количеством EtOAc (2×50 мл). Объединенные органические слои высушивали (MgSO4), фильтровали и концентрировали с получением (R)-4-(1-(4-фторбензил)-1H-1,2,3-триазол-4-ил)-N-(2-гидроксипропил)бензолсульфонамида (54 мг, 0,138 ммоль, 66,2%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,78 (с, 1H), 8,06-8,02 (м, 2H), 7,87-7,81 (м, 2H), 7,60 (т, J=6,2 Гц, 1H), 7,47-7,39 (м, 2H), 7,27-7,19 (м, 2H), 5,66 (с, 2H), 4,69 (д, J=4,5 Гц, 1H), 3,61-3,53 (м, 1H), 2,71-2,58 (м, 2H), 0,97 (д, J=6,1 Гц, 3H). МС m/z [М+Н]+ = 391,2.
Пример 71: 4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)-2-метилбензолсульфонамид:
К раствору N-(2-гидроксиэтил)-2-метил-4-(2H-тетразол-5-ил) бензолсульфонамида (250 мг, 0,882 ммоль, промежуточное соединение 79) в ацетонитриле (5 мл) добавляли N, N-диизопропилэтиламин (0,46 мл, 2,64 ммоль, коммерческий источник: Vinsa) с последующим добавлением 2-(хлорметил)-5-фторпиридина (192,6 мг, 1,32 ммоль, промежуточное соединение 9) при 26°С. Реакционную смесь нагревали до 85°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и разбавляли этилацетатом (50 мл) и промывали водой (20 мл). Органический слой отделяли, высушивали над безводным Na2SO4 и фильтровали. Фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,5% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением 4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)-2-метилбензолсульфонамида (89 мг, 25,4%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,53 (д, J=3,2 Гц, 1H), 8,07-7,97 (м, 3H), 7,84-7,75 (м, 2H), 7,66-7,63 (м, 1H), 6,16 (с, 2H), 4,63 (т, J=5,4 Гц, 1H), 3,33 (кв, J=6,4 Гц, 2H), 2,86 (кв, J=6,4 Гц, 2H), 2,66 (с, 3H). МС m/z [М+Н]+ = 393,06.
Пример 72: N-(2-гидроксиэтил)-2-метил-4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамид:
К раствору N-(2-гидроксиэтил)-2-метил-4-(2H-тетразол-5-ил) бензолсульфонамида (350 мг, 1,23 ммоль, промежуточное соединение 79) в ацетонитриле (7,0 мл) добавляли N, N-диизопропилэтиламин (1,07 мл, 6,17 ммоль, коммерческий источник: Vinsa) с последующим добавлением 2-(бромметил)тетрагидро-2Н-пирана (663 мг, 3,70 ммоль, коммерческий источник: Sigma Aldrich) при 26°С. Реакционную смесь нагревали до 85°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и разбавляли этилацетатом (100 мл) и промывали водой (20 мл). Органический слой отделяли, высушивали над безводным Na2SO4 и фильтровали. Фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,5% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением N-(2-гидроксиэтил)-2-метил-4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида, который подвергали хиральному разделению SFC.
Условия SFC:
Колонка/размеры: Chiralcel OJ-H (21×250 мм), 5 мкм
% CO2: 90,0%
% сорастворитель: 10,0% (100% МеОН)
Общий поток: 60,0 г/мин
Противодавление: 90,0 бар
УФ: 254 нм
Время стекового ввода: 4,3 мин
Загрузка/инжектирование: 3,7 мг.
Собранные фракции SFC концентрировали при пониженном давлении с получением изомера 1 (21 мг) в виде бледно-желтой смолы и еще одной фракции (25 мг).
Изомер 1: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,09-7,97 (м, 3Н), 7,79 (шир. с, 1Н), 4,79-4,72 (м, 2Н), 4,64 (шир. с, 1Н), 3,93- 3,88 (м, 1H), 3,83-3,78 (м, 1H), 3,38-3,33 (м, 2H), 3,29-3,27 (м, 1H), 2,89-2,85 (м, 2H), 2,67 (с, 3H), 1,85-1,68 (м, 2H), 1,56-1,31 (м, 4H). МС m/z [М-Н]- = 380,14. ее% = 98,30%.
Другая фракция (25 мг) была снова подвергнута хиральной SFC с получением изомера 2 (12 мг) в виде бледно-желтой смолы.
Условия SFC:
Колонка/размеры: Chiralcel OJ-H (21×250 мм), 5 мкм
% CO2: 80,0%
% сорастворитель: 20,0% (100% МеОН)
Общий поток: 60,0 г/мин
Противодавление: 90,0 бар
УФ: 254 нм
Время стекового ввода: 2,0 мин
Загрузка/инжектирование: 0,78 мг.
Изомер 2: 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,16-7,98 (м, 3Н), 7,82 (шир. с, 1Н), 4,82-4,76 (м, 2Н), 4,68 (т, J=5,5 Гц, 1Н), 3,95-3,88 (м, 1H), 3,83-3,77 (м, 1H), 3,37-3,34 (м, 2H), 3,28-3,23 (м, 1H), 2,86 (т, J=6,4 Гц, 2H), 2,67 (с, 3Н), 1,87-1,77 (м, 1Н), 1,75-1,69 (м, 1Н), 1,55-1,38 (м, 4Н), 1,28-1,23 (м, 1Н). МС m/z [М-Н]- = 380,05. ее% = 97,41%.
Абсолютную конфигурацию не определяли.
Пример 73: 4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)-3-метилбензолсульфонамид:
К раствору N-(2-гидроксиэтил)-3-метил-4-(2H-тетразол-5-ил) бензолсульфонамида (0,6 г, 2,11 ммоль, промежуточное соединение 83) в ацетонитриле (10 мл) добавляли N, N-диизопропилэтиламин (0,9 мл, 5,27 ммоль, коммерческий источник: Vinsa) с последующим добавлением 2-(хлорметил)-5-фторпиридина (0,43 г, 1,56 ммоль, промежуточное соединение 9) при 26°С. Реакционную смесь нагревали до 85°С в течение 16 ч. По окончании реакции реакционную смесь разбавляли этилацетатом (250 мл), промывали водой (2×100 мл) и насыщенным раствором соли (100 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали препаративной ВЭЖХ (условия препаративной ВЭЖХ: колонка XBridge C18 (250×19) мм, 5 мкм; подвижная фаза A: 10 мМ бикарбонат аммония (водный раствор), подвижная фаза B: ацетонитрил; метод (время в мин/% B): 0/10, 1/10, 10/30, 12,8/100, 17,8/100, 18/30, 21/30; скорость потока: 16 мл/мин, температура: комнатная). Чистые фракции собирали, лиофилизировали и растирали с пентаном с получением 4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)-3-метилбензолсульфонамида (145 мг, 34,7%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,56-8,53 (м, 1H), 8,14-8,10 (м, 1H), 7,86-7,75 (м, 3H), 7,66-7,60 (м, 1H), 7,0 (шир. с, 1H), 6,17 (с, 2H), 4,69-4,63 (м, 1H), 3,38 (кв, J=5,9 Гц, 2H), 2,84 (т, J=6,2 Гц, 2H), 2,62 (с, 3H). МС m/z [М+Н]+ = 391,17.
Пример 74: N-(2-гидроксиэтил)-3-метил-4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамид:
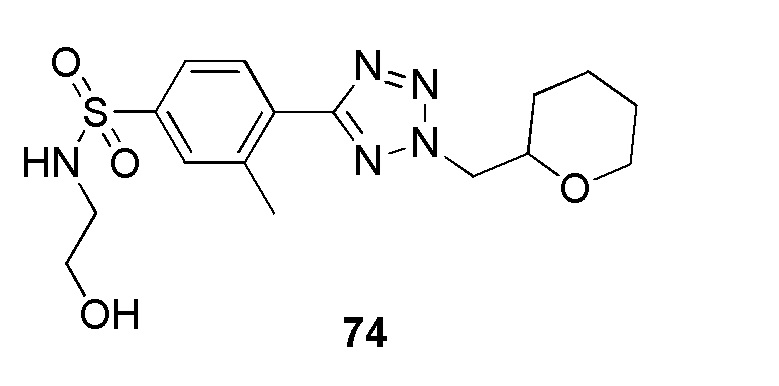
К раствору N-(2-гидроксиэтил)-3-метил-4-(2H-тетразол-5-ил) бензолсульфонамида (1,0 г, 3,32 ммоль, промежуточное соединение 83) в N, N-диметилформамиде (10,0 мл) добавляли карбонат калия (0,97 г, 7,04 ммоль, коммерческий источник: RCP) с последующим добавлением 2-(бромметил)тетрагидро-2Н-пирана (0,75 г, 4,22 ммоль, коммерческий источник: Sigma Aldrich) при 26°С. Реакционную смесь нагревали до 100°С в течение 12 ч. По окончании реакции реакционную смесь охлаждали до 26°С и гасили ледяной водой (100 мл). Смесь промывали водой (3×100 мл) и насыщенным раствором соли (50 мл). Органический экстракт высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией, элюируя смесью 2% метанола в DCM. Чистые фракции концентрировали при пониженном давлении с получением продукта, который затем очищали препаративной ВЭЖХ (колонка YMC Triart C18 (25×150 мм, 10 мкм, скорость потока: 25 мл/мин), подвижная фаза: A: H2O (10 мМ NH4HCO3) B: ACN; метод (время в мин/% B): 0/25, 1/25, 10/65, 105/100, 12/100, 12,5/25, 15/25; температура: комнатная). Чистые фракции собирали и лиофилизировали с получением N-(2-гидроксиэтил)-3-метил-4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида, который подвергали хиральному разделению SFC.
Условия SFC:
Колонка/ рзмеры: Lux Cellulose-2 (30×250 мм), 5 мкм
% CO2: 60,0%
% сорастворитель: 40,0% (100% МеОН)
Общий поток: 60,0 г/мин
Противодавление: 100,0 бар
УФ: 214 нм
Время стекового ввода: 3,5 мин.
Загрузка/инжектирование: 10,0 мг.
Собранные фракции SFC концентрировали при пониженном давлении с получением:
Изомер 1: (33 мг) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,14 (д, J=8,1 Гц, 1H), 7,81 (д, J=1,4 Гц, 1H), 7,81-7,76 (м, 1H), 7,73 (с, 1H) , 4,83-4,79 (м, 2H), 4,71 (т, J=5,5 Гц, 1H), 3,96-3,90 (м, 1H), 3,84-3,79 (м, 1H), 3,41-3,36 (м, 2H), 3,30 -3,27 (м, 1H), 2,84 (т, J=6,2 Гц, 2H), 2,66 (с, 3H), 1,86-1,79 (м, 1H), 1,76-1,70 (м, 1H), 1,56-1,35 (м, 4H). МС m/z [М-Н]- = 380,21. ее% = 99,80%.
Изомер 2: (33 мг) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,16-8,12 (м, 1H), 7,84-7,77 (м, 2H), 7,70-7,65 (м, 1H), 4,81-4,78 (м, 2H), 4,68-4,64 (м, 1Н), 3,97-3,90 (м, 1Н), 3,84-3,78 (м, 1Н), 3,41-3,35 (м, 2Н), 3,28-3,24 (м, 1Н), 2,88-2,81 (м, 2Н) 2,66 (с, 3Н), 1,85-1,71 (м, 2Н), 1,60-1,37 (м, 4Н). МС m/z [М-Н]- = 380,14. ее% = 94,23%.
Их абсолютную конфигурацию не определяли.
Пример 75: 2-фтор-4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид:
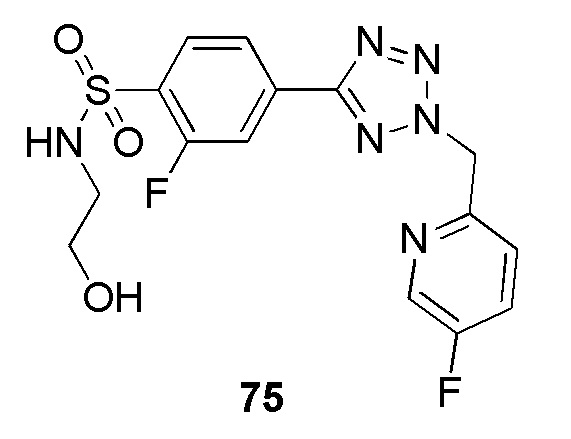
К раствору 2-фтор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (300 мг, 1,044 ммоль, промежуточное соединение 87) в ацетонитриле добавляли N, N-диизопропилэтиламин (0,54 мл, 3,13 ммоль, коммерческий источник: Vinsa) с последующим добавлением 2-(хлорметил)-5-фторпиридина (228 мг, 1,56 ммоль, промежуточное соединение 9) при 26°С. Реакционную смесь нагревали до 85°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и распределяли между этилацетатом (100 мл) и водой (20 мл). Органический слой отделяли, высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,3% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением 2-фтор-4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамида (145 мг, 34,7%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,53 (д, J=2,8 Гц, 1H), 8,06-7,97 (м, 4H), 7,85-7,80 (м, 1H), 7,69-7,62 (м, 1H), 6,18 (с, 2H), 4,65 (т, J=5,5 Гц, 1H), 3,41-3,36 (м, 2H), 2,99-2,94 (м, 2H). МС m/z [М+Н]+ = 397,06.
Пример 76: 2-фтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамид:
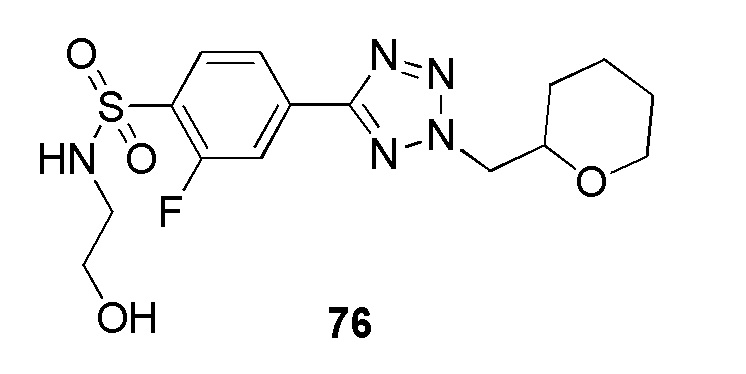
К раствору 2-фтор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (400 мг, 1,392 ммоль, промежуточное соединение 87) в ацетонитриле (8,0 мл) добавляли N, N-диизопропилэтиламин (1,21 мл, 6,96 ммоль, коммерческий источник: Vinsa) с последующим добавлением 2-(бромметил)тетрагидро-2Н-пирана (747 мг, 4,17 ммоль, коммерческий источник: Sigma Aldrich) при 26°С. Реакционную смесь нагревали до 85°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 25°С и разбавляли этилацетатом (100 мл) и промывали водой (20 мл). Органическую фазу отделяли, высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,5% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением 2-фтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида, который подвергали хиральному разделению SFC.
Условия SFC:
Колонка/размеры: Lux Cellulose-2 (30×250 мм), 5 мкм
% CO2: 65,0%
% сорастворитель: 35,0% (100% МеОН)
Общий поток: 60,0 г/мин
Противодавление: 90,0 бар
УФ: 254 нм
Время стекового ввода: 3,5 мин
Загрузка/инжектирование: 13,0 мг.
Собранные фракции SFC концентрировали при пониженном давлении с получением:
Изомер 1: (32 мг) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,07-7,97 (м, 4H), 4,83-4,78 (м, 2H), 4,65 (т, J=6,4 Гц, 1H), 3,95-3,88 (м, 1H), 3,83-3,77 (м, 1H), 3,39 (кв, J=6,1 Гц, 2H), 3,29-3,25 (м, 1H), 2,97 (т, J=6,4 Гц, 2H), 1,85-1,70 (м, 2H) 1,53-1,31 (м, 4H). МС m/z [М-Н]- = 384,24. ее% = 99,45%.
Изомер 2: (50 мг) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,08-7,96 (м, 4H), 4,84-4,78 (м, 2H), 4,66 (т, J=5,5 Гц, 1H), 3,96-3,89 (м, 1H), 3,84-3,78 (м, 1H), 3,42-3,36 (м, 2H), 3,29-3,25 (м, 1H), 2,97 (т, J=6,4 Гц, 2H), 1,88-1,67 (м, 2H), 1,56-1,33 (м, 4Н). МС m/z [М-Н]- = 384,17. ее% = 96,16%.
Их абсолютную конфигурацию не определяли.
Пример 77: 2-фтор-4-(2-(4-фторбензил)-2Н-тетразол-5-ил)-N- (2-гидроксиэтил)бензолсульфонамид:
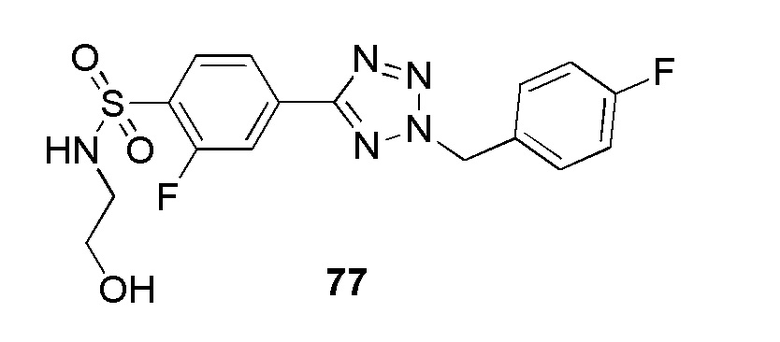
К раствору 2-фтор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (250 мг, 0,871 ммоль, промежуточное соединение 87) в ацетонитриле (5 мл) добавляли N, N-диизопропилэтиламин (0,3 мл, 1,742 ммоль, коммерческий источник: Finar) с последующим добавлением 1-(бромметил)-4-фторбензола (0,16 г, 0,871 ммоль, коммерческий источник: Alfa) при 26°С. Реакционную смесь нагревали до 85°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и распределяли между этилацетатом (80 мл) и водой (20 мл). Водный слой экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали насыщенным раствором соли (50 мл), высушивали над безводным Na2SO4 и фильтровали. Фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,5% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением 2-фтор-4-(2-(4-фторбензил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамида (137 мг, 38%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,02-7,97 (м, 4Н), 7,52-7,50 (м, 2Н), 7,27-7,23 (м, 2Н), 6,04 (с, 2Н), 4,66-4,64 (м, 1Н), 3,40-3,34 (м, 2Н), 2,98-2,94 (м, 2Н). МС m/z [М+Н]+ = 396,04.
Пример 78: 2-фтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2Н-пиран-4-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамид:
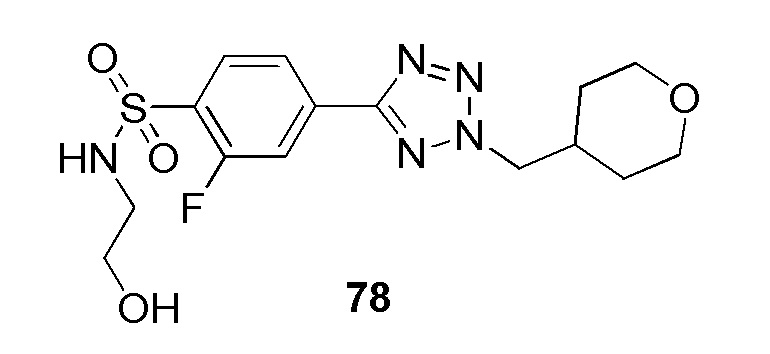
К раствору 2-фтор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (250 мг, 0,871 ммоль, промежуточное соединение 87) в ацетонитриле (5 мл) добавляли N, N-диизопропилэтиламин (0,3 мл, 1,742 ммоль, коммерческий источник: Finar) с последующим добавлением 4-(бромметил)тетрагидро-2Н-пирана (0,15 г, 0,871 ммоль, коммерческий источник: Apollo Scientific) при 26°С. Реакционную смесь нагревали до 85°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и распределяли между этилацетатом (100 мл) и водой (30 мл). Водный слой экстрагировали этилацетатом (2×40 мл). Объединенные органические слои высушивали над безводным Na2SO4 и фильтровали. Фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,5% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением 2-фтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-4-ил)метил)-2H-тетразол-5-ил)бензолсульфонамида (71 мг, 21%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,06-7,97 (м, 4H), 4,72-4,70 (м, 2H), 4,67-4,64 (м, 1H), 3,86-3,83 (м, 2H), 3,41-3,36 (м, 2Н), 3,00-2,94 (м, 2Н), 2,33-2,21 (м, 1Н), 1,52-1,45 (м, 2Н), 1,40-1,29 (м, 2Н). МС m/z [М+Н]+ = 386,10.
Пример 79: 4-(2-(циклогексилметил)-2Н-тетразол-5-ил)-2-фтор-N-(2-гидроксиэтил)бензолсульфонамид:
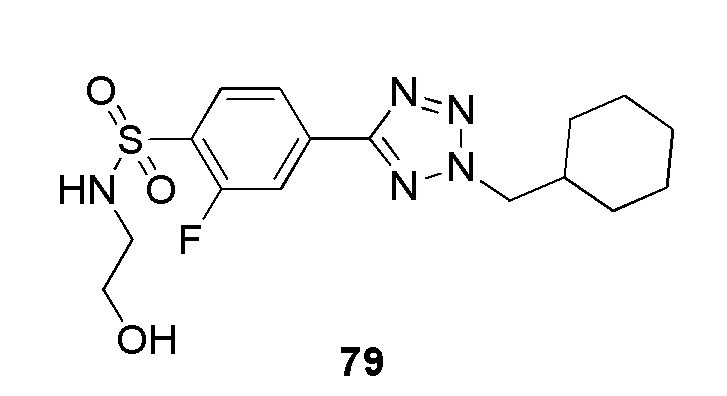
К раствору 2-фтор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (250 мг, 0,871 ммоль, промежуточное соединение 87) в ацетонитриле (5 мл) добавляли N, N-диизопропилэтиламин (0,3 мл, 1,742 ммоль, коммерческий источник: Finar) с последующим добавлением (бромметил)циклогексана (0,153 г, 0,871 ммоль, коммерческий источник: Apollo Scientific) при 26°С. Реакционную смесь нагревали до 85°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и распределяли между этилацетатом (100 мл) и водой (30 мл). Водный слой экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали насыщенным раствором соли (50 мл), высушивали над безводным Na2SO4 и фильтровали. Фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,5% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением 4-(2-(циклогексилметил)-2Н-тетразол-5-ил)-2-фтор-N-(2-гидроксиэтил)бензолсульфонамида (38 мг, 11%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,07-7,97 (м, 4Н), 4,68-4,62 (м, 3Н), 3,41-3,37 (м, 2Н), 3,01-2,94 (м, 2Н), 2,07-1,06 (м, 1Н), 1,72-1,54 (м, 5Н), 1,28-1,00 (м, 5Н). МС m/z [М+Н]+ = 384,10.
Пример 80: 2-хлор-4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид:
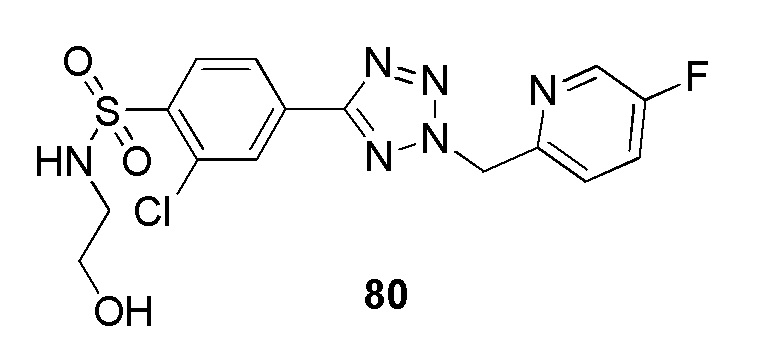
К раствору 2-хлор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (300 мг, 0,990 ммоль, промежуточное соединение 91) в ацетонитриле (6 мл) добавляли N, N-диизопропилэтиламин (0,34 мл, 1,98 ммоль, коммерческий источник: Finar) с последующим добавлением 2-(хлорметил)-5-фторпиридина (0,172 г, 1,188 ммоль, промежуточное соединение 9) при 26°С. Реакционную смесь нагревали до 85°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и ацетонитрил выпаривали. Сырой продукт разбавляли этилацетатом (100 мл) и промывали водой (3×50 мл), и затем насыщенным раствором соли (50 мл). Органический слой высушивали над безводным Na2SO4 и фильтровали. Фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,5% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением 2-хлор-4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамида (208 мг, 50,8%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,54-8,53 (м, 1Н), 8,20-8,18 (м, 1Н), 8,16-8,13 (м, 2Н), 7,97-7,94 (м, 1Н), 7,85-7,80 (м, 1Н), 7,67-7,64 (м, 1Н), 6,18 (с, 2Н), 4,66-4,63 (т, 1Н), 3,39-3,35 (м, 2Н), 2,96-2,92 (м, 2Н). МС m/z [М+Н]+ = 412,98.
Пример 81: 3-хлор-4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид:
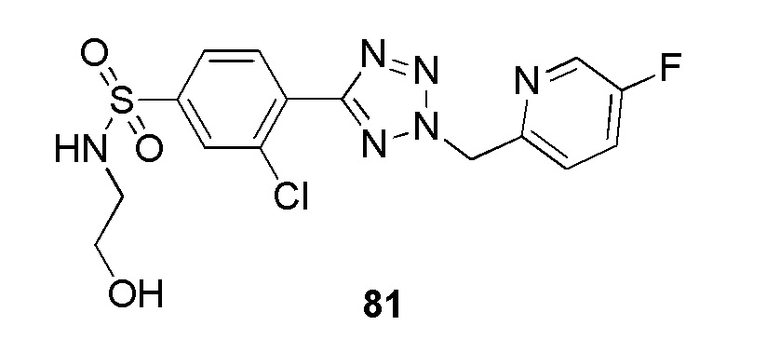
К раствору 3-хлор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (800 мг (неочищенное соединение, чистота 37%), 0,977 ммоль, промежуточное соединение 95) в ацетонитриле (5,9 мл), добавляли N, N-диизопропилэтиламин (0,34 мл, 1,95 ммоль, коммерческий источник: Finar) с последующим добавлением 2-(хлорметил)-5-фторпиридина (0,169 г, 1,172 ммоль, промежуточное соединение 9) при 26°С. Реакционную смесь нагревали до 85°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 28°С и разбавляли этилацетатом (80 мл). Сырой продукт промывали водой (2×40 мл) и затем насыщенным раствором соли (40 мл). Органический слой высушивали над безводным Na2SO4 и фильтровали. Фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении. Полученное твердое соединение дополнительно очищали препаративной ВЭЖХ с получением после лиофилизации 3-хлор-4-(2-((5-фторпиридин-2-ил) метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамида (22 мг, 5,4%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,55 (д, 1H), 8,16 (д, 1H), 8,02 (д, 1H), 7,92-7,81 (м, 3H), 7,66-7,63 (м, 1H), 6,20 (с, 2H), 4,71-4,68 (м, 1H), 3,41-3,37 (м, 2H), 2,90-2,87 (м, 2H). МС m/z [М+Н]+ = 412,98.
Условия препаративной ВЭЖХ:
Колонка: колонка Kromasil C18 (25×150 мм, 10 мкм)
Подвижная фаза: A: H2O (10 мМ NH4HCO3) B: ACN
Метод (время в мин/% В): 0/20, 1/20, 10/50, 10,5/100, 13/100, 13,1/20
Скорость потока: 25 мл/мин
Температура: комнатная.
Пример 82: 2-(2-хлор-4-(2-(4-фторбензил)-2Н-тетразол-5-ил) фенилсульфонамидо)ацетамид:
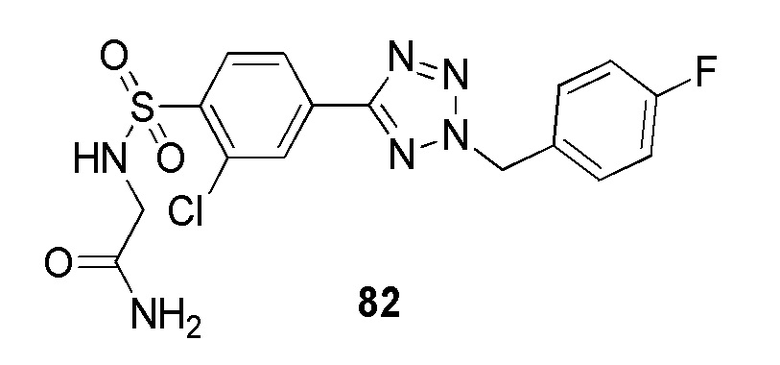
К раствору трет-бутил (2-амино-2-оксоэтил)((2-хлор-4-(2-(4-фторбензил)-2Н-тетразол-5-ил)фенил)сульфонил)карбамата (440 мг, 0,838 ммоль, промежуточное соединение 99) в 1,4-диоксане (4,4 мл) добавляли 4 М HCl в 1,4-диоксане (4,4 мл) при 26°С. Полученную реакционную смесь перемешивали при 26°С в течение 3 ч. После окончания реакции реакционную смесь концентрировали при пониженном давлении с получением сырого продукта, который растворяли в воде (30 мл). Затем раствор подщелачивали насыщенным раствором NaHCO3 (20 мл) и экстрагировали EtOAc (3×30 мл). Органические слои высушивали над безводным Na2SO4 и фильтровали. Растворитель выпаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 2% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 2-(2-хлор-4-(2-(4-фторбензил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетамида (140 мг, 37%) в виде не совсем белого твердого вещества. МС m/z [М-Н]- = 422,91.
Пример 83: 3-фтор-4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид:
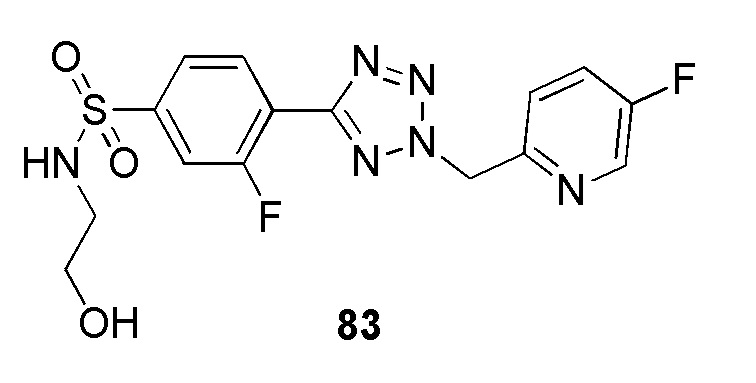
К раствору 3-фтор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (250 мг, 0,87 ммоль, промежуточное соединение 103) в ацетонитриле (5,0 мл) добавляли N, N-диизопропилэтиламин (0,53 мл, 3,04 ммоль, коммерческий источник: Vinsa) с последующим добавлением 2-(хлорметил)-5-фторпиридина (190 мг, 1,30 ммоль, промежуточное соединение 9) при 26°С. Реакционную смесь нагревали до 85°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и разбавляли этилацетатом (40 мл) и промывали водой (40 мл). Органическую фазу отделяли, высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,5% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 3-фтор-4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамида (160 мг, 45,7%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,55-8,53 (м, 1H), 8,32-8,28 (м, 1H), 7,91-7,80 (м, 4H), 7,67-7,62 (м, 1H), 6,20-6,18 (м, 2Н), 4,69-4,65 (м, 1Н), 3,38 (кв, J=6,0 Гц, 2Н), 2,91-2,84 (м, 2Н). МС m/z [М+Н]+ = 397,12.
Пример 84: 3-фтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамид:
К раствору 3-фтор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (200 мг, 0,696 ммоль, промежуточное соединение 103) в ацетонитриле (4,0 мл) добавляли N, N-диизопропилэтиламин (0,6 мл, 3,48 ммоль, коммерческий источник: Vinsa) с последующим добавлением 2-(бромметил)тетрагидро-2Н-пирана (373 мг, 2,08 ммоль, коммерческий источник: Sigma Aldrich) при 26°С. Реакционную смесь нагревали до 85°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и разбавляли этилацетатом (50 мл) и промывали водой (15 мл). Органические фазы объединяли, высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,5% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением 3-фтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида, который подвергали хиральному разделению SFC.
Условия SFC:
Колонка/размеры: Lux Cellulose-2 (4,6×250 мм), 5 мкм
% CO2: 60,0%
% сорастворитель: 40,0% (100% МеОН)
Общий поток: 60,0 г/мин
Противодавление: 100,0 бар.
Собранные фракции SFC концентрировали при пониженном давлении с получением:
Изомер 1: (22 мг) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,30 (т, J=7,4 Гц, 1H), 7,88 (шир. с, 1H), 7,81 (д, J=8,8 Гц, 2H), 4,84-4,78 (м, 2H), 4,71-4,67 (м, 1H), 3,94-3,89 (м, 1H), 3,83-3,78 (м, 1H), 3,41-3,36 (м, 2H), 3,28-3,24 (м, 1H), 2,89 (т J=6,1 Гц, 2Н), 1,86-1,80 (м, 1Н), 1,76-1,70 (м, 1Н), 1,53-1,29 (м, 4Н). МС m/z [М-Н]- = 384,17. ее% = 99,11%.
Изомер 2: (16 мг) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,29 (т, J=6,1 Гц, 1H), 7,96 (шир. с, 1H), 7,80 (д, J=8,8 Гц, 2H), 4,85-4,79 (м, 2H), 4,75-4,69 (м, 1H), 3,97-3,89 (м, 1H), 3,84-3,78 (м, 1H), 3,38 (т, J=6,1 Гц, 2H), 3,27-3,23 (м, 1H), 2,88 (т, J=6,1 Гц, 2H), 1,85-1,71 (м, 2H), 1,55-1,30 (м, 4H). МС m/z [М-Н]- = 384,17. ее=97,35%.
Их абсолютную конфигурацию не определяли.
Пример 85: 2,3-дифтор-4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид:
К раствору 2,3-дифтор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил)бензолсульфонамида (250 мг, 0,819 ммоль, промежуточное соединение 107) в ацетонитриле (5 мл) добавляли N, N-диизопропилэтиламин (0,42 мл, 2,488 ммоль, коммерческий источник: Vinsa) с последующим добавлением 2-(хлорметил)-5-фторпиридина (154 мг, 1,06 ммоль, промежуточное соединение 9) при 26°С. Реакционную смесь нагревали до 85°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и разбавляли этилацетатом (60 мл) и водой (10 мл). Органическую фазу промывали водой (2×30 мл), насыщенным раствором соли (2×30 мл), высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали препаративной ВЭЖХ. Чистые фракции лиофилизировали с получением 2,3-дифтор-4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N- (2-гидроксиэтил)бензолсульфонамида (196 мг, 57%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,52-8,49 (м, 1H), 8,22-8,18 (м, 1H), 8,06-8,00 (м, 1H), 7,84-7,79 (м, 1H), 7,76-7,72 (м, 1Н), 7,66-7,63 (м, 1Н), 6,19 (с, 2Н), 4,64-4,60 (м, 1Н), 3,39-3,34 (м, 2Н), 3,01-2,96 (м, 2Н). МС m/z [М-Н]- = 412,94.
Условия препаративной ВЭЖХ:
Колонка: YMC C8 (150×19) мм, 10 мкм
Подвижная фаза: A-10 мМ бикарбонат аммония (водный раствор), B-ацетонитрил
Метод (время в мин/% В): 0/10, 1/20, 8/55
Скорость потока: 30 мл/мин
Температура: комнатная.
Пример 86: 2,3-дифтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамид:
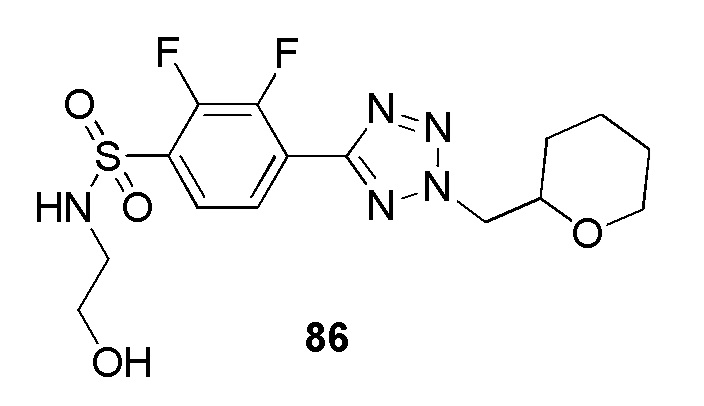
К раствору 2,3-дифтор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил)бензолсульфонамида (450 мг, 1,47 ммоль, промежуточное соединение 107) в ацетонитриле (9,0 мл) добавляли N, N-диизопропилэтиламин (1,28 мл, 7,37 ммоль, коммерческий источник: Vinsa) с последующим добавлением 2-(бромметил)тетрагидро-2Н-пирана (525 мг, 2,95 ммоль, коммерческий источник: Sigma Aldrich) при 26°С. Реакционную смесь нагревали до 85°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и разбавляли этилацетатом (100 мл) и промывали водой (20 мл). Органическую фазу отделяли, высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,5% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением 2,3-дифтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-2-ил) метил)-2H-тетразол-5-ил)бензолсульфонамида, который подвергали хиральному разделению SFC.
Условия SFC:
Колонка/размеры: Lux Cellulose-2 (30-250 мм), 5 мкм
% CO2: 60,0%
% cорастворитель: 40,0% (100% МеОН)
Общий поток: 90,0 г/мин
Противодавление: 90,0 бар
УФ: 254 нм
Время стекового ввода: 4,0 мин
Загрузка/инжектирование: 3,0 мг.
Собранные фракции SFC концентрировали при пониженном давлении с получением:
Изомер 1: (26 мг) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,23 (шир. с, 1H), 8,04 (т, J=6,7 Гц, 1H), 7,76 (т, J=6,7 Гц, 1H), 4,88-4,80 (м, 2H), 4,64 (шир. с, 1H), 3,94-3,89 (м, 1H), 3,82-3,77 (м, 1H), 3,42-3,35 (м, 2H), 3,03-2,94 (м, 2H), 1,87-1,71 (м, 2Н), 1,57-1,37 (м, 4Н), 1,25-1,20 (м, 1Н). МС m/z [М+Н]+ = 404,18. ее% = 99,48%.
Изомер 2: (22 мг) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,25 (шир. с, 1H), 8,05 (т, J=6,8 Гц, 1H), 7,76 (т, J=7,1 Гц, 1H), 4,88-4,79 (м, 2H), 4,65 (шир. с, 1H), 3,96-3,88 (м, 1H), 3,85-3,75 (м, 1H), 3,45-3,39 (м, 2H), 3,04-2,95 (м, 2H), 1,87-1,70 (м, 2Н), 1,56-1,32 (м, 4Н), 1,27-1,21 (м, 1Н). МС m/z [М+Н]+ = 404,18. ее% = 97,96%.
Их абсолютную конфигурацию не определяли.
Пример 87: 2,6-дифтор-4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид:
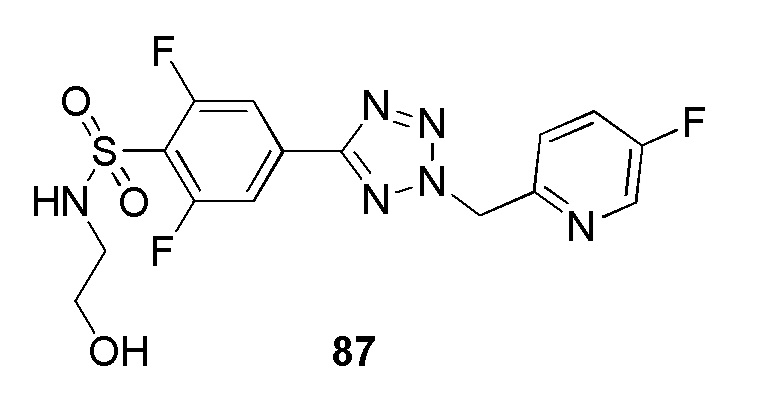
К раствору 2,6-дифтор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил)бензолсульфонамида (300 мг, 0,983 ммоль, промежуточное соединение 111) в ацетонитриле (6,0 мл) добавляли N, N-диизопропилэтиламин (0,51 мл, 2,95 ммоль, коммерческий источник: Vinsa) с последующим добавлением 2-(хлорметил)-5-фторпиридина (185 мг, 1,27 ммоль, промежуточное соединение 9) при 26°С. Реакционную смесь нагревали до 85°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и разбавляли этилацетатом (100 мл) и водой (20 мл). Органическую фазу отделяли, высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией, элюируя смесью 15% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении. Полученное твердое соединение дополнительно очищали препаративной ВЭЖХ (колонка Kromasil phenyl (25×150 мм, 10 мкм, скорость потока 25 мл/мин), подвижная фаза: A: H2O (10 мМ NH4HCO3), B: ACN; метод (время в мин/% В): 0/30, 1/30, 8/50, 10/50, 10,2/100, 12/100, 12,1/30, 14/30; температура: комнатная). Чистые фракции лиофилизировали с получением 2,6-дифтор-4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамида (45 мг, 10%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,52 (д, J=2,8 Гц, 1H), 7,85-7,81 (м, 3H), 7,69-7,63 (м, 2H), 6,18 (с, 2H), 4,61 (широкий с, 1H), 3,41-3,39 (м, 2H), 3,03 (т, J=6,3 Гц, 2H). МС m/z [М-Н]- = 413,01.
Пример 88: (R)-2,6-дифтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил) бензолсульфонамид :
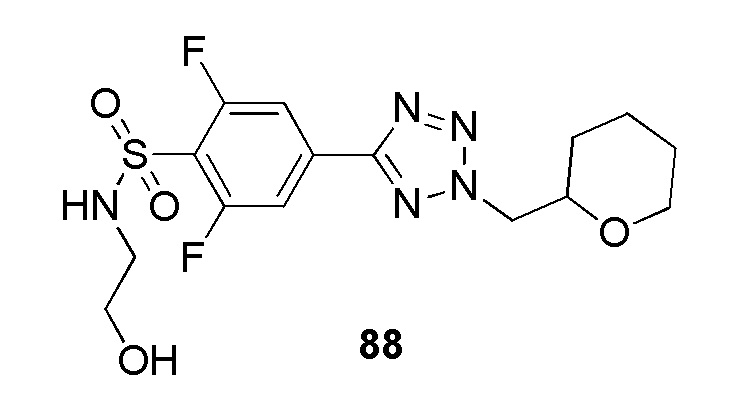
К раствору 2,6-дифтор-N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил)бензолсульфонамида (700 мг, 2,295 ммоль, промежуточное соединение 111) в ацетонитриле (14,0 мл) добавляли N, N-диизопропилэтиламин (2 мл, 11,475 ммоль, коммерческий источник: Vinsa) с последующим добавлением 2-(бромметил)тетрагидро-2Н-пирана (817 мг, 4,59 ммоль, коммерческий источник: Aldrich) при 26°С. Реакционную смесь нагревали до 85°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и разбавляли этилацетатом (50 мл) и водой (10 мл). Органическую фазу отделяли, высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией, элюируя смесью 1,5% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении. Полученное твердое соединение дополнительно очищали препаративной ВЭЖХ (хиральная колонка Lux Cellulose (30×250 мм, 5 мкм), % CO2: 60% сорастворителя: 40% (100% метанол); общий поток: 90 г/мин, противодавление: 90,0 бар, УФ: 254 нм, время стекового ввода: 4,2 мин, загрузка/инжектирование: 13 мг). Чистые фракции упаривали с получением одного известного энантиомера 2,6-дифтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида (5 мг) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,86-7,84 (м, 2Н), 4,82-4,79 (м, 2Н), 4,66 (т, J=5,5 Гц, 1Н), 3,82-3,77 (м, 1Н), 3,44-3,39 (м, 2Н), 3,10-2,99 (м, 2Н), 1,83 -1,24 (м, 8Н). МС m/z [М-Н]- = 402,05, хиральная чистота ее=99,4%.
Пример 89: N-(цианометил)-4-(2-((тетрагидро-2Н-пиран-4-ил) метил)-2Н-тетразол-5-ил)бензолсульфонамид:
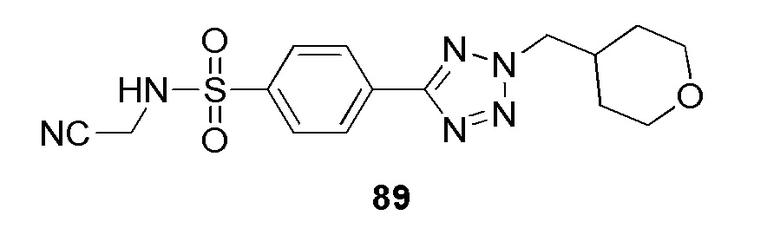
К раствору трет-бутил (цианометил)((4-(2-((тетрагидро-2Н-пиран-4-ил)метил)-2Н-тетразол-5-ил)фенил)сульфонил)карбамата (400 мг, 0,865 ммоль, промежуточное соединение 113) в DCM (10 мл), добавляли трифторуксусную кислоту (4,0 мл, коммерческий источник: Avra) при 0°С. Затем реакционной смеси давали достичь 26°С и перемешивали при 26°С в течение 4 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали препаративной ВЭЖХ.
Условия препаративной ВЭЖХ:
Колонка: YMC Trait C18 (150×25) мм, 10 мкм
Подвижная фаза A: 10 мМ бикарбонат аммония (водный раствор),
подвижная фаза B: ацетонитрил
Скорость потока: 25 мл/мин,
Метод (время в мин/% В): 0/10, 1/10, 10/60, 11/100
Температура: комнатная.
Чистые фракции лиофилизировали с получением N-(цианометил)-4-(2-((тетрагидро-2Н-пиран-4-ил)метил)-2Н-тетразол-5-ил) бензолсульфонамида (46 мг, 15%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,53 (шир. с, 1H), 8,29 (д, J=8,6 Гц, 2H), 8,02 (д, J=8,6 Гц, 2H), 4,71 (д, J=7,2 Гц, 2H), 4,15 (с, 2H), 3,88-3,78 (м, 2H), 3,35-3,30 (м, 1H), 3,28-3,21 (м, 1H), 2,35-2,23 (м, 1H), 1,52-1,46 (м, 2Н), 1,41-1,29 (м, 2Н). МС m/z [М+Н]+ = 363,10.
Пример 90. N-(цианометил)-4-(2-(пиразин-2-илметил)-2Н-тетразол-5-ил)бензолсульфонамид:
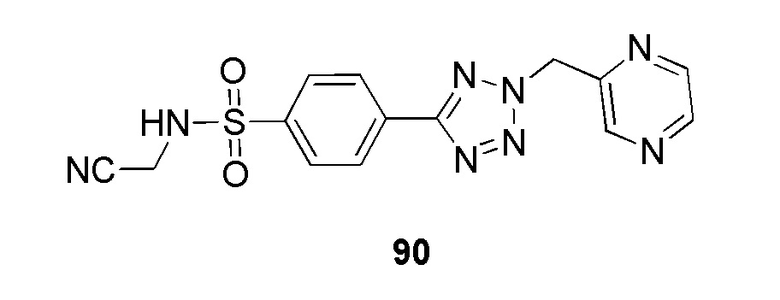
Раствор 2-(4-(2-(пиразин-2-илметил)-2H-тетразол-5-ил) фенилсульфонамидо)ацетамида (200 мг, 0,0005 моль, промежуточное соединение 118) в фосфорилхлориде (2 мл, коммерческий источник: Avra) нагревали до 100°С и перемешивали в течение 2 ч при той же температуре. После окончания реакции реакционную смесь охлаждали до 26°С, по каплям выливали в охлажденный насыщенный раствор бикарбоната натрия (0°С) (100 мл) и экстрагировали этилацетатом (4×30 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 2% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением N-(цианометил)-4-(2-(пиразин-2-илметил)-2Н-тетразол-5-ил)бензолсульфонамида (32 мг, 17%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,89 (д, J=1,5 Гц, 1H), 8,74-8,66 (м, 2H), 8,64-8,61 (м, 1H), 8,31-8,25 (м, 2H), 8,04-7,98 (м, 2Н), 6,28 (с, 2Н), 4,15 (с, 2Н). МС m/z [М-Н]- = 355,12.
Пример 91: N-(цианометил)-4-(2-((5-метилпиразин-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамид:
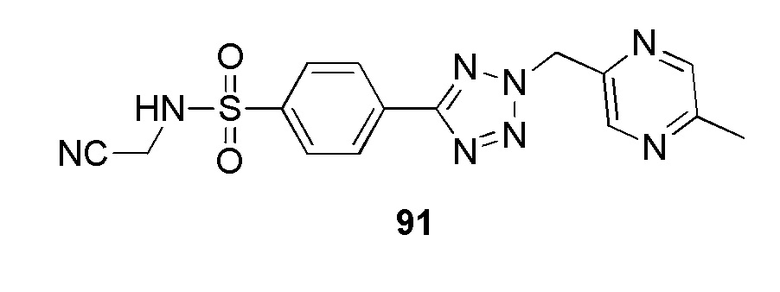
Раствор 2-(4-(2-((5-метилпиразин-2-ил)метил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетамида (220 мг, 0,0005 моль, промежуточное соединение 121) в фосфорилхлориде (2,2 мл, коммерческий источник: Avra) нагревали до 100°С и перемешивали в течение 2 ч при той же температуре. По окончании реакции реакционную смесь охлаждали до 26°С, по каплям выливали в охлажденный на льду водный насыщенный раствор бикарбоната натрия (0°С) (100 мл) и экстрагировали этилацетатом (4×30 мл). Объединенный органический раствор высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,5% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением N-(цианометил)-4-(2-((5-метилпиразин-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида (52 мг, 27,5%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,74 (с, 1H), 8,70 (с, 1H), 8,51 (с, 1H), 8,27 (д, J=6,6 Гц, 2H), 8,0 (д, J=6,6 Гц, 2H), 6,21 (с, 2H), 4,15 (с, 2H), 2,49 (с, 3H). МС m/z [М+Н]+ = 371,12.
Пример 92: N-(цианометил)-4-(2-((5-метоксипиразин-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамид:
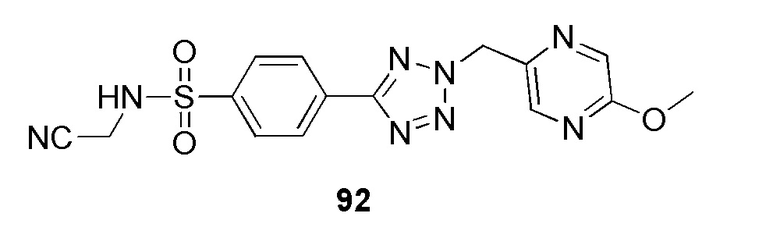
Раствор 2-(4-(2-((5-метоксипиразин-2-ил)метил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетамида (250 мг, 0,0006 моль, промежуточное соединение 123) в фосфорилхлориде (2,5 мл, коммерческий источник: Avra) нагревали до 100°С и перемешивали в течение 2 ч при той же температуре. По окончании реакции реакционную смесь охлаждали до 26°С и медленно выливали в насыщенный раствор бикарбоната натрия (100 мл) при 0°С. Полученную реакционную смесь экстрагировали этилацетатом (3×50 мл), высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,5% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением N-(цианометил)-4-(2-((5-метоксипиразин-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида (35 мг, 14%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,71 (шир. с, 1H), 8,49 (д, J=1,3 Гц, 1H), 8,31-8,24 (м, 3H), 8,04-7,98 (м, 2H), 6,13 (с, 2H), 4,15 (д, J=4,6 Гц, 2H), 3,93 (с, 3H). МС m/z [М+Н]+ = 387,06.
Пример 93: N-(цианометил)-4-(2-((6-метоксипиридин-3-ил) метил)-2Н-тетразол-5-ил)бензолсульфонамид:
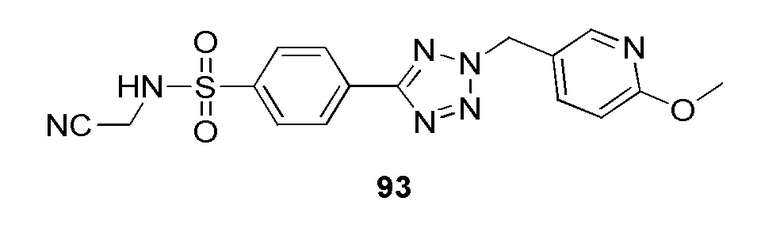
К раствору трет-бутил (цианометил)((4-(2-((тетрагидро-2Н-пиран-4-ил)метил)-2Н-тетразол-5-ил)фенил)сульфонил)карбамата (100 мг, 0,206 ммоль, промежуточное соединение 125) в DCM (10 мл) добавляли трифторуксусную кислоту (1,0 мл, коммерческий источник: Advent) и перемешивали при 26°С в течение 4 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали препаративной ВЭЖХ (условия препаративной ВЭЖХ: колонка: XBridge C18 (150×19) мм, 5 мкм; подвижная фаза A: 10 мМ бикарбонат аммония (водный раствор), подвижная фаза B: ацетонитрил, скорость потока: 16 мл/мин, метод (время в мин/% В): 0/10, 1/10, 13/50, 16,8/50, 17/100, 21,8/ 100, 22/10, 25/10; температура: комнатная). Чистые фракции собирали и лиофилизировали с получением N-(цианометил)-4-(2-((6-метоксипиридин-3-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида (15 мг, 19%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,46 (шир. с, 1H), 8,35-8,38 (м, 1H), 8,25-8,28 (м, 2H), 7,99-8,02 (м, 2H), 7,79-7,83 (м, 1Н), 6,85-6,88 (м, 1Н), 6,00 (с, 2Н), 4,14 (с, 2Н), 3,86 (с, 3Н). МС m/z [М-Н]- = 384,1.
Пример 94: N-(цианометил)-4-(2-((тетрагидро-2Н-пиран-2-ил) метил)-2Н-тетразол-5-ил)бензолсульфонамид:
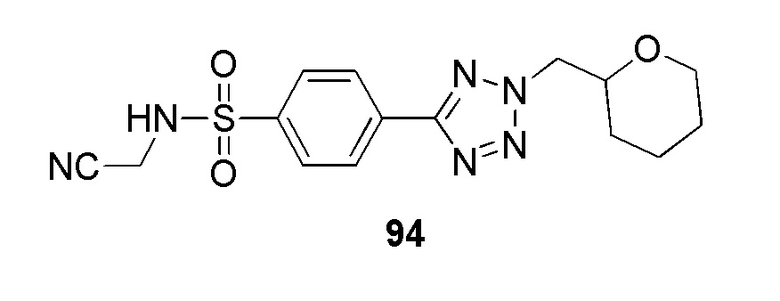
К раствору трет-бутил (цианометил)((4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)фенил)сульфонил)карбамата (1,0 г, 2,162 ммоль, промежуточное соединение 127) в DCM (20 мл) добавляли трифторуксусную кислоту (10,0 мл, коммерческий источник: Advent), и реакционную смесь перемешивали при 26°С в течение 4 ч. По окончании реакции реакционную смесь концентрировали при пониженном давлении с получением N-(цианометил)-4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида (1,1 г, неочищенное соединение). Две неочищенные партии (1,4 г и 300 мг) смешивали и высушивали в вакууме с получением объединенного неочищенного вещества (1,4 г), которое очищали препаративной ВЭЖХ (условия препаративной ВЭЖХ: колонка Kromasil C18 (150×25) мм, 10 мкм; подвижная фаза A: 10 мМ бикарбонат аммония (водный раствор), подвижная фаза B: ацетонитрил; метод (время в мин/% B): 0/30, 1/30, 10/70, 10,5/100, 12/100, 12,5/30, 15/30; скорость потока: 25 мл/мин, температура: комнатная). Чистые фракции собирали и лиофилизировали в течение 16 ч с получением N-(цианометил)-4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамида (250 мг), который подвергали препаративной SFC.
Условия SFC:
Колонка/размеры: Chiralpak AD-H (4,6×250 мм), 5 мкм
% CO2: 60,0%
% сорастворитель: 40,0% (100% МеОН)
Общий поток: 4,0 г/мин
Противодавление: 90,0 бар
УФ: 254 нм
Время стекового ввода: 10,0 мин
Загрузка/инжектирование: 8,5 мг.
Собранные фракции SFC концентрировали при пониженном давлении с получением изомера 1 (90 мг) в виде не совсем белого твердого вещества и изомера 2 (24 мг) в виде не совсем белого твердого вещества.
Изомер 1: 1Н ЯМР (400 МГц, ДМСО-d6) δ = ppm 8,76 (шир. с, 1Н), 8,32-8,25 (м, 2Н), 8,04-8,00 (м, 2Н), 4,85-4,76 (м, 2Н) , 4,1 (с, 2Н), 3,95-3,90 (м, 1Н), 3,83-3,77 (м, 1Н), 3,30-3,26 (м, 1Н), 1,87-1,71 (м, 2Н), 1,54-1,38 (м, 4H). МС m/z [М-Н]- = 361,11, хиральная чистота ее=99,82%.
Изомер 2: 1Н ЯМР (400 МГц, ДМСО-d6) δ = ppm 8,81 (шир. с, 1H), 8,25 (д, J=8,55 Гц, 2H), 7,98 (д, J=8,55 Гц, 2H), 4,83-4,77 (м, 2Н), 4,08 (с, 2Н), 3,94-3,88 (м, 1Н), 3,84-3,77 (м, 1Н), 3,30-3,25 (м, 1Н), 1,87-1,71 (м, 2Н), 1,57-1,36 (м, 4Н). МС m/z [М-Н]- = 361,03, хиральная чистота ее=99,82%.
Их абсолютную конфигурацию не определяли.
Пример 95: 2-((4-(2-((4,4-дифторциклогексил)метил)-2Н-тетразол-5-ил)-2-метоксифенил)сульфонамидо)ацетамид
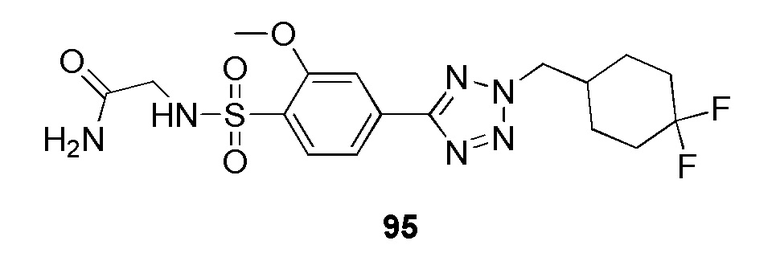
К раствору трет-бутил (2-амино-2-оксоэтил)((4-(2-((4,4-дифторциклогексил)метил)-2Н-тетразол-5-ил)-2-метоксифенил) сульфонил)карбамата (210 мг, 0,270 ммоль, промежуточное соединение 129) в дихлорметане (5 мл) по каплям добавляли 2,2,2-трифторуксусную кислоту (97 мкл, 1,350 ммоль) при 0°С в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли еще по каплям 2,2,2-трифторуксусную кислоту (97 мкл, 1,350 ммоль) при 0°С в атмосфере азота и смесь перемешивали при комнатной температуре в течение ночи. Сырой продукт разбавляли водой и дважды экстрагировали DCM. Органический слой высушивали над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали колоночной флэш-хроматографией (силикагель; смесь MeOH/CH2Cl2 от 0/100 до 20/80). Фракции собирали и концентрировали в вакууме с получением 2-((4-(2-((4,4-дифторциклогексил)метил)-2Н-тетразол-5-ил)-2-метоксифенил) сульфонамидо)ацетамида (20 мг, 16,7%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,90 (м, 1Н), 7,75 (м, 2Н), 7,42 (шир. с, 1Н), 7,21 (шир. с, 1Н), 7,08 (шир. с, 1Н), 4,75 (м, 2Н), 4,00 (с, 3Н), 3,48 (с, 2Н), 2,26-1,65 (м, 7Н), 1,34 (м, 2Н). МС m/z [М+Н]+ = 445,3.
Пример 96: 2-((4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)фенил)сульфонамидо)ацетамид:
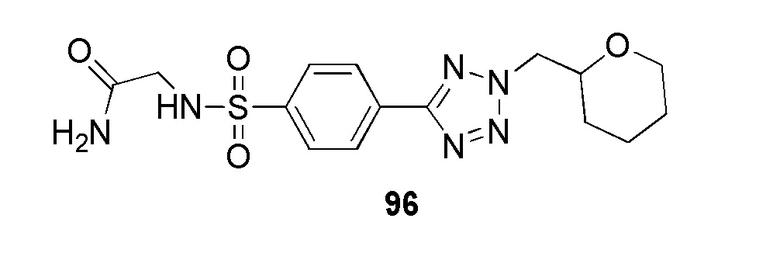
N-этил-N-изопропилпропан-2-амин (371 мкл, 2,126 ммоль, коммерческий источник: Aldrich) добавляли к перемешиваемому раствору 2-((4-(2H-тетразол-5-ил)фенил)сульфонамидо)ацетамида (200 мг, 0,709 ммоль, промежуточное соединение 25') в N, N-диметилформамиде (4 мл). Смесь нагревали при 70°С с последующим добавлением 2-(бромметил)тетрагидро-2H-пирана (91 мкл, 0,709 ммоль, коммерческий источник: Aldrich) и реакционную смесь перемешивали при 70°С в течение выходных дней. Реакционную смесь концентрировали при пониженном давлении, и сырой продукт очищали колоночной флэш-хроматографией (диоксид кремния; смесь MeOH/CH2Cl2 от 0/100 до 10/90). Фракции концентрировали в вакууме с получением 2-((4-(2-((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)фенил)сульфонамидо)ацетамида). Для удаления минимальных примесей твердое вещество растирали с Et2O и фильтровали. Затем его растворяли в DCM и промывали водой (дважды). Органический слой высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением 2-((4-(2- ((тетрагидро-2Н-пиран-2-ил)метил)-2Н-тетразол-5-ил)фенил) сульфонамидо)ацетамида (26 мг, 9,6%) в виде желтоватого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,24 (м, 2Н), 8,04 (м, 1Н), 7,98 (м, 2Н), 7,29 (шир. с, 1Н), 7,08 (шир. с, 1Н), 4,83-4,75 (м, 2Н), 3,95-3,89 (м, 1Н), 3,82-3,76 (м, 1Н), 3,43 (м, 2Н), 3,30-3,25 (м, 1Н), 1,83-1,72 (м, 2Н), 1,54-1,31 (м, 4H). МС m/z [М+Н]+ = 381,2.
Примеры 97-100 получали способами, аналогичными описанному для примера 18, заменяя алкилирующие реагенты и основные условия на условия, указанные в таблице 6. Метод, используемый для очистки, указан в виде сносок.
N-(2-гидроксиэтил)-4-(2-(4-(трифторметокси)бензил)-2Н-тетразол-5-ил)бензолсульфонамид (см. сноску a, затем b)
перемешивали при комнатной температуре в течение 16 ч, затем при
50°С в течение 2 ч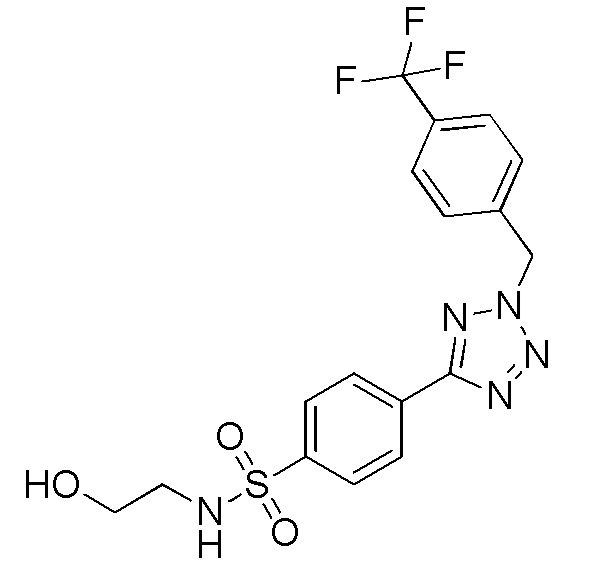
N-(2-гидроксиэтил)-4-(2-(4-(трифторметил)бензил)-2Н-тетразол-5-ил)бензолсульфонамид (см. сноску a)
перемешивали при комнатной температуре в течение ночи, затем при
50°С в течение 2 ч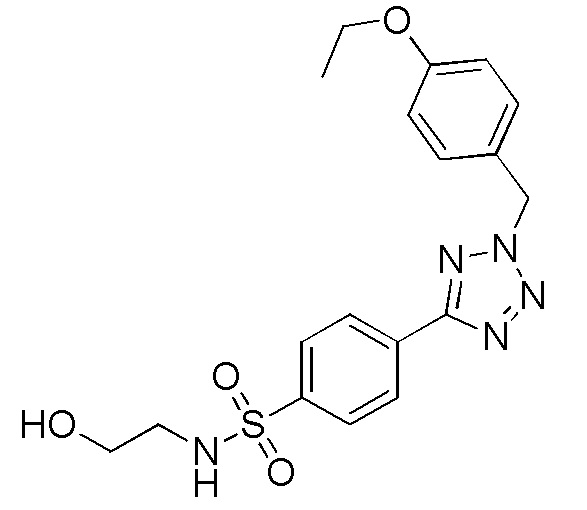
4-(2-(4-этоксибензил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид (см. сноски b, d)
перемешивали при комнатной температуре в течение ночи
Остаток очищали флэш-хроматографией со смесью EtOAc-циклогексан от 0/100 до 50/50;
Полученное твердое вещество промывали Et2O и фильтровали с получением конечного продукта;
В конце остаток очищали препаративной ВЭЖХ на колонке X-Bridge (19×150 мм) в линейном градиенте 50-100% ACN/H2O (10 мМ NH4HCO3).
Пример 100: 4-(2-(циклогексилметил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид:
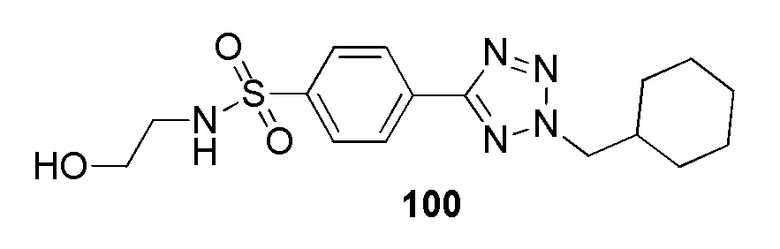
К раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (200 мг, 0,743 ммоль, промежуточное соединение 17) в N, N-диметилформамиде (ДМФА) (10,0 мл) добавляли карбонат калия (205 мг, 1,48 ммоль, коммерческий источник: RCP) с последующим добавлением (бромметил)циклогексана (158 мг, 0,89 ммоль, коммерческий источник: Sigma Aldrich) при 26°С. Реакционную смесь нагревали до 100°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 3% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-(2-(циклогексилметил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамида (50 мг, 18%). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,28-8,24 (м, 2Н), 8,00-7,96 (м, 2Н), 7,74 (шир. с, 1Н), 4,69-4,61 (м, 3Н), 3,37 (кв, J=6,4 Гц, 2H), 2,84 (т, J=6,4 Гц, 2H), 2,07-1,97 (м, 1H), 1,72-1,56 (м, 5H), 1,27-1,05 (м, 5H). МС m/z [М+Н]+ = 366,18.
Пример 101: N-(2-гидроксиэтил)-4-(2-((6-метилпиридин-3-ил) метил)-2Н-тетразол-5-ил)бензолсульфонамид:
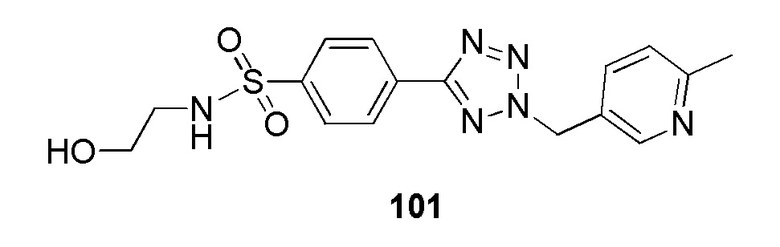
К раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (200 мг, 0,743 ммоль, промежуточное соединение 17) в N, N-диметилформамиде (ДМФА) (10,0 мл) добавляли карбонат калия (205 мг, 1,48 ммоль, коммерческий источник: RCP) с последующим добавлением 5-(хлорметил)-2-метилпиридина (126 мг, 0,89 ммоль, коммерческий источник: AstaTech) при 26°С. Реакционную смесь нагревали до 80°С в течение 4 ч. По окончании реакции реакционную смесь охлаждали до 26°С и упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 5% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением партии, которую очищали препаративной ВЭЖХ.
Условия препаративной ВЭЖХ:
Колонка: Kromasil C18 (150×25) мм, 10 мкм
Подвижная фаза: A-10 мМ бикарбонат аммония (водный раствор), B-ацетонитрил
Метод (время в мин/% В): 0/10, 1/10, 10/40, 10,5/100, 13/ 100, 13,5/10
Скорость потока: 25 мл/мин
Температура: комнатная.
Чистые фракции лиофилизовали с получением N-(2-гидроксиэтил)-4-(2-((6-метилпиридин-3-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида (100 мг, 36%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,59 (д, J=2,0 Гц, 1H), 8,25-8,23 (м, 2H), 7,97-7,94 (м, 2H), 7,75-7,72 (м, 1H), 7,29 (д, J=7,9 Гц, 1H), 6,05 (с, 2H), 4,66 (шир. с, 1H), 3,36 (т, J=6,4 Гц, 2H), 2,83 (т, J=6,3 Гц, 2H) 2,47 (с, 3Н). МС m/z [М+Н]+ = 375,12.
Пример 102: N-(2-гидроксиэтил)-4-(2-(пиридин-3-илметил)-2Н-тетразол-5-ил)бензолсульфонамид:
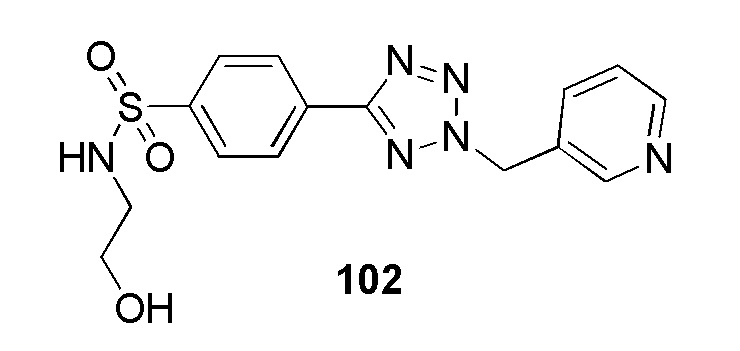
К раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (200 мг, 0,743 ммоль, промежуточное соединение 17) в ацетонитриле (10,0 мл) добавляли N, N-диизопропилэтиламин (191 мг, 1,487 ммоль, коммерческий источник: Avra) с последующим добавлением 3-(хлорметил)пиридина гидрохлорида (146 мг, 0,89 ммоль, коммерческий источник: Combi-Blocks) при 26°С. Реакционную смесь нагревали до 80°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 5% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением N-(2-гидроксиэтил)-4-(2-(пиридин-3-илметил)-2H-тетразол-5-ил)бензолсульфонамида (65 мг, 24%). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,73 (д, J=2,0 Гц, 1H), 8,59 (дд, J=1,6 Гц, 4,7 Гц, 1H), 8,27-8,23 (м, 2H), 7,99-7,96 (м, 2H), 7,88-7,86 (м, 1H), 7,78 (д, J=6,1 Гц, 1H), 7,49-7,42 (м, 4,8, 7,9 Гц, 1H), 6,12 (с, 2H), 4,69 (т, J=5,6 Гц, 1H), 3,40-3,34 (м, 2H), 2,83 (д, J=6,1 Гц, 1H). МС m/z [М+Н]+ = 361,12.
Пример 103: (N-(2-гидроксиэтил)-4-(2-((5-метоксипиридин-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамид):
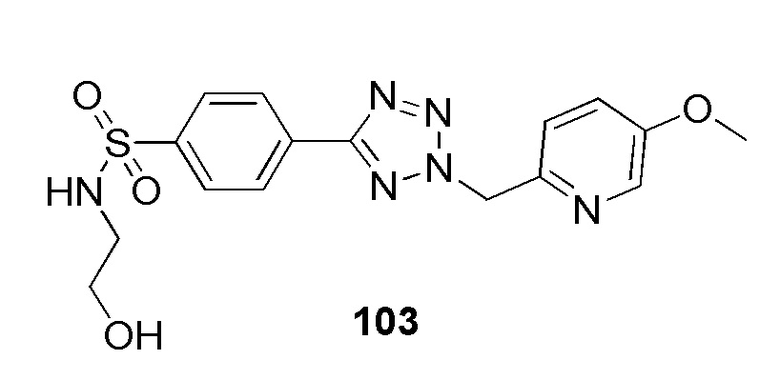
К раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (1) (200 мг, 0,743 ммоль, промежуточное соединение 17) в ацетонитриле (10,0 мл) добавляли карбонат калия (205 мг, 0,88 ммоль, коммерческий источник: RCP) с последующим добавлением 2-(хлорметил)-5-метоксипиридина (140 мг, 0,89 ммоль, коммерческий источник: Enamine) при 26°С. Реакционную смесь нагревали до 80°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 3% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением партии (100 мг, 63%), которую подвергали очистке препаративной ВЭЖХ.
Условия препаративной ВЭЖХ:
Подвижная фаза A: - 10 мМ бикарбонат аммония (водный раствор), подвижная фаза B: - ацетонитрил; колонка: XBridge C18 (150×19) мм, 5 мкм, скорость потока: - 16 мл/мин, метод (время в мин/% B): 0/10, 1/10, 10/55, 22/100, 22,2/35,5, 25/35,5. Растворимость: - ACN+ТГФ+вода; температура: комнатная.
Чистые фракции лиофилизировали с получением N-(2-гидроксиэтил)-4-(2-((5-метоксипиридин-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида (28 мг, 10%). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,36 (д, J=2,4 Гц, 1H), 8,26-8,23 (м, 2H), 7,98-7,96 (м, 2H), 7,83-7,79 (м, 2H), 6,86 (д, J=8,6 Гц, 1Н), 5,99 (с, 2Н), 4,65 (шир. с, 1Н), 3,85 (с, 3Н), 3,38-3,36 (м, 2Н), 2,86-2,83 (м, 2Н). МС m/z [М+Н]+ = 391,12.
Пример 104: N-(2-гидроксиэтил)-4-(2-((тетрагидро-2Н-пиран-3-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамид
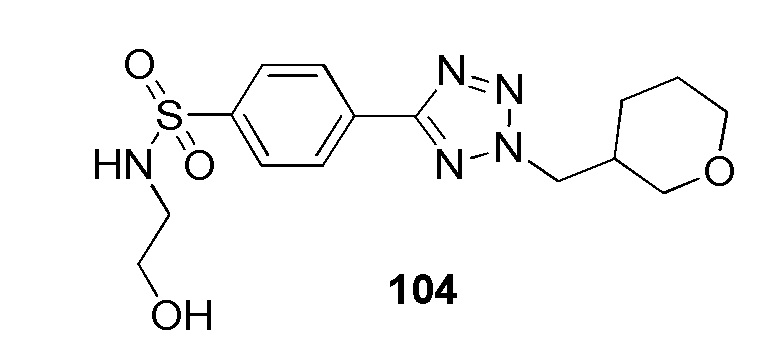
К раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (200 мг, 0,743 ммоль, промежуточное соединение 17) в N, N-диметилформамиде (10 мл) добавляли карбонат калия (205 мг, 1,487 ммоль, коммерческий источник: RCP) с последующим добавлением 3-(хлорметил)тетрагидро-2Н-пирана (120 мг, 0,89 ммоль, промежуточное соединение 131) при 26°С. Реакционную смесь нагревали до 100°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель, 100-200 меш), элюируя смесью 2% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением соединения с чистотой 80% по данным ЖХМС, которое затем очищали препаративной ВЭЖХ.
Условия препаративной ВЭЖХ:
Колонка: YMC-Triart C8 (150×25) мм, 10 мкм
Подвижная фаза A: 10 мМ бикарбонат аммония (водный раствор) B: ацетонитрил
Метод (время в мин/% В): 0/20, 1/20, 10/40, 10,5/100, 15/100, 15,5/20
Скорость потока: 25 мл/мин
Температура: комнатная.
Чистые фракции собирали и лиофилизировали с получением N- (2-гидроксиэтил)-4-(2-((тетрагидро-2Н-пиран-3-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида (28 мг, 24%). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,26 (д, J=8,3 Гц, 2H), 7,98 (д, J=8,3 Гц, 2H), 7,75 (шир. с, 1H), 4,80-4,63 (м, 3H), 3,77-3,66 (м, 2H), 3,43-3,35 (м, 3H), 3,26 (с, 1H), 2,90-2,80 (м, 2H), 2,26 (шир. с, 1H), 1,79-1,60 (м, 2H), 1,52-1,43 (м, 1H), 1,39-1,28 (м, 1H). МС m/z [М+Н]+ = 368,18.
Пример 105: N-(2-гидроксиэтил)-4-(2-((тетрагидро-2Н-пиран-4-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамид:
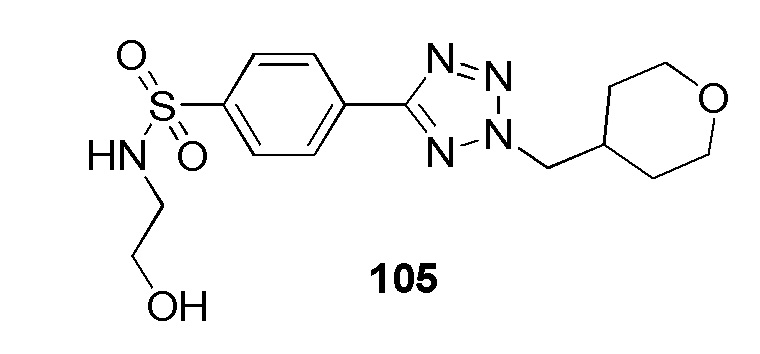
К раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (200 мг, 0,743 ммоль, промежуточное соединение 17) в N, N-диметилформамиде (10 мл) добавляли карбонат калия (205 мг, 1,485 ммоль, коммерческий источник: RCP) с последующим добавлением 4-(бромметил)тетрагидро-2Н-пирана (159 мг, 0,88 ммоль, коммерческий источник: Aldrich) при 26°С. Реакционную смесь нагревали до 80°С в течение 8 ч. По окончании реакции реакционную смесь охлаждали до 26°С С и упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель, 100-200 меш), элюируя смесью 5% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением N-(2-гидроксиэтил)-4-(2-((тетрагидро-2Н-пиран-4-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида (80 мг, 18%). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,28-8,24 (м, 2H), 7,98 (д, J=8,6 Гц, 2H), 7,79-7,72 (м, 1H), 4,73-4,65 (м, 3H), 3,88-3,80 (м, 2H), 3,38 (кв, J=6,1 Гц, 2H), 3,29-3,27 (м, 2H), 2,85 (кв, J=5,3 Гц, 2H), 2,35-2,23 (м, 1H) 1,52-1,46 (м, 2Н), 1,40-1,31 (м, 2Н). МС m/z [М+Н]+ = 368,12.
Пример 106: N-(2-гидроксиэтил)-4-(2-(пиридазин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид:
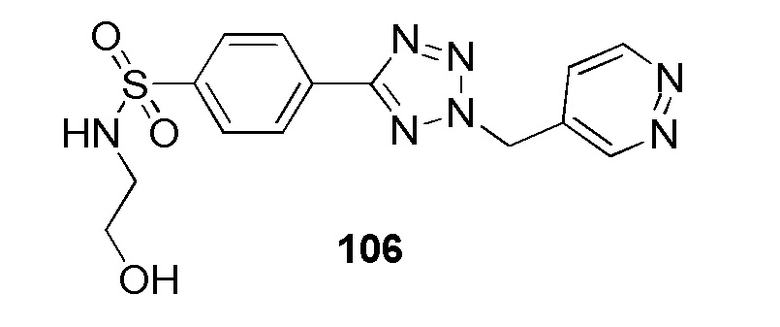
К раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (200 мг, 0,743 ммоль, промежуточное соединение 17) в N, N-диметилформамиде (10 мл) добавляли карбонат калия (205 мг, 1,485 ммоль, коммерческий источник: RCP) с последующим добавлением 4-(хлорметил)пиридазина (115 мг, 0,89 ммоль, промежуточное соединение 132) при 26°С. Реакционную смесь перемешивали при 26°С в течение 16 ч. По окончании реакции реакционную смесь упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 4% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением N-(2-гидроксиэтил)-4-(2-(пиридазин-4-илметил)-2Н-тетразол-5-ил) бензолсульфонамида (10 мг, 3,7%). 1H ЯМР (400 МГц, ДМСО-d6) δ 9,35-9,32 (м, 1H), 9,28-9,26 (м, 1H), 8,28-8,25 (м, 2H), 7,98 (д, J=8,6 Гц, 2H), 7,80-7,72 (м, 1H), 7,65-7,63 (м, 1H), 6,23-6,21 (м, 2H), 4,68-4,65 (м, 1H), 3,40-3,35 (м, 2H), 2,88-2,81 (м, 2H). МС m/z [М+Н]+ = 362,12.
Пример 107: N-(2-гидроксиэтил)-4-(2-(пиридазин-3-илметил)-2H-тетразол-5-ил)бензолсульфонамид:
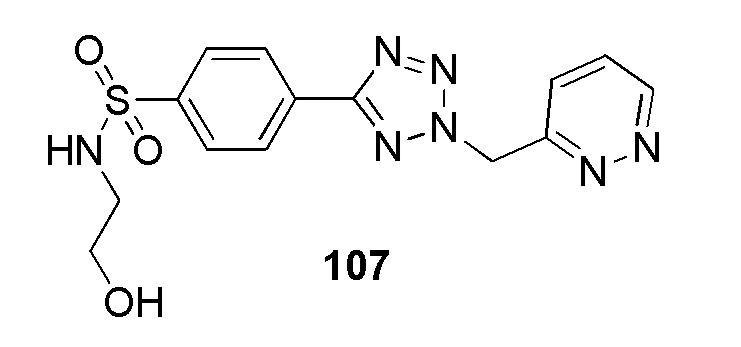
К раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (200 мг, 0,743 ммоль, промежуточное соединение 17) в N, N-диметилформамиде (10 мл) добавляли карбонат калия (205 мг, 1,485 ммоль, коммерческий источник: RCP) с последующим добавлением 3-(хлорметил)пиридазина (115 мг, 0,89 ммоль, промежуточное соединение 133) при 26°С. Реакционную смесь перемешивали при 26°С в течение 16 ч. По окончании реакции реакционную смесь упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 5% метанола в дихлорметане. Чистые фракции концентрировали при пониженном давлении с получением соединения с чистотой 77% по данным ЖХМС. Затем соединение очищали препаративной ВЭЖХ.
Условия препаративной ВЭЖХ:
Колонка: Kromasil C18 (150×25) мм, 10 мкм
Подвижная фаза: A - 10 мМ бикарбонат аммония (водный раствор), B-ацетонитрил
Метод (время в мин/% В): 0/40, 1/40, 10/75, 13/75, 13,2/ 100, 17/100, 17,2/40, 20/40
Скорость потока: 25 мл/мин,
Температура: комнатная.
Чистые фракции собирали и лиофилизировали с получением N- (2-гидроксиэтил)-4-(2-(пиридазин-3-илметил)-2Н-тетразол-5-ил) бензолсульфонамида (66 мг, 24,6%). 1H ЯМР (400 МГц, ДМСО-d6) δ 9,28-9,23 (м, 1Н), 8,29-8,24 (м, 2Н), 7,97 (д, J=8,3 Гц, 2Н), 7,85-7,73 (м, 3Н), 6,40 (м, 2H), 4,66 (д, J=5,4 Гц, 1H), 3,40-3,36 (м, 2H), 2,84 (д, J=6,3 Гц, 2H). МС m/z [М-Н]- = 360,12.
Пример 108: 4-(2-(4-цианобензил)-2Н-тетразол-5-ил)-N-метилбензолсульфонамид:
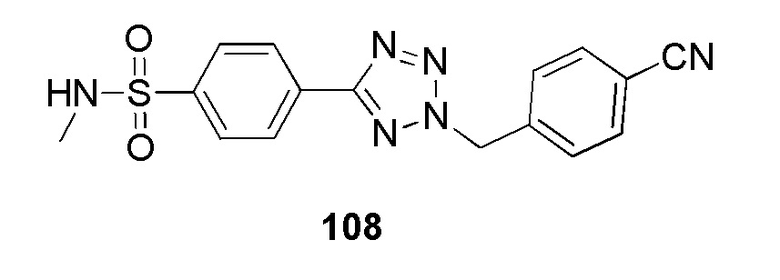
К раствору N-метил-4-(2H-тетразол-5-ил)бензолсульфонамида (500 мг, 2,092 ммоль, промежуточное соединение 21) в N, N-диметилформамиде (7 мл) добавляли карбонат калия (577 мг, 4,184 ммоль, коммерческий источник: Avra) с последующим добавлением 4- (бромметил)бензонитрила (410 мг, 2,092 ммоль, коммерческий источник: Alfa Aesar) при 26°С и перемешивали в течение 24 ч. По окончании реакции реакционную смесь упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 2% метанола в дихлорметане, и полученный продукт дополнительно очищали препаративной ВЭЖХ.
Условия препаративной ВЭЖХ:
Колонка: Xselect CSH Phenyl-Gexyl (150×19) мм, 5 мкм
Подвижная фаза: A-10 мМ бикарбонат аммония (водный раствор) B-ацетонитрил
Метод (время в мин/% В): 0/25, 10/55, 10,3/100, 12,7/100, 13/25, 15/25
Скорость потока: 20 мл/мин
Температура: комнатная.
Чистые фракции концентрировали при пониженном давлении с получением 4-(2-(4-цианобензил)-2Н-тетразол-5-ил)-N-метилбензолсульфонамида (170 мг, 22%). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,27 (д, J=8,6 Гц, 2H), 7,95 (д, J=8,6 Гц, 2H), 7,89 (д, J=8,3 Гц, 2H), 7,60 (д, J=8,3 Гц, 3H), 6,18 (с, 2H), 2,45 (с, 3H). МС m/z [М+Н]+ = 355,10.
Пример 109: N-(2-гидроксиэтил)-2-метокси-4-(2-((5-метоксипиридин-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамид:
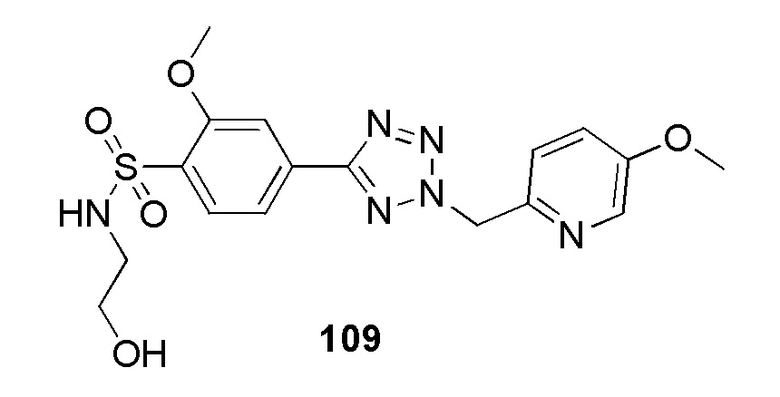
К раствору N-(2-гидроксиэтил)-2-метокси-4-(2H-тетразол-5-ил) бензолсульфонамида (380 мг, 0,0012 моль, промежуточное соединение 33) в ацетонитриле (7,6 мл) добавляли N, N-диизопропилэтиламин (0,79 мл, 0,0048 моль, коммерческий источник: Finar) с последующим добавлением 2-(хлорметил)-5-метоксипиридина (239 мг, 0,0015 моль, промежуточное соединение 134) при 26°С. Реакционную смесь нагревали до 80°С в течение 16 ч. По окончании реакции реакционную смесь охлаждали до 26°С и разбавляли этилацетатом (50 мл) и водой (10 мл). Водный слой экстрагировали этилацетатом (2×20 мл), высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией, элюируя смесью 2% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением N-(2-гидроксиэтил)-2-метокси-4-(2-((5-метоксипиридин-2-ил)метил)-2Н-тетразол-5-ил)бензолсульфонамида (7 мг) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,23 (д, J=2,9 Гц, 1H), 7,89 (д, J=7,9 Гц, 1H), 7,76-7,73 (м, 2H), 7,52-7,46 (м, 2H), 7,18 (т, J=5,9 Гц, 1H), 6,06 (с, 2H), 4,60 (т, J=5,9 Гц, 1H), 4,00 (с, 3H), 3,82 (с, 3H), 3,38-3,33 (м, 2Н), 2,88-2,83 (м, 2Н). МС m/z [М+Н]+ = 421,13.
Пример 110: 2-фтор-4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)-5-метилбензолсульфонамид:
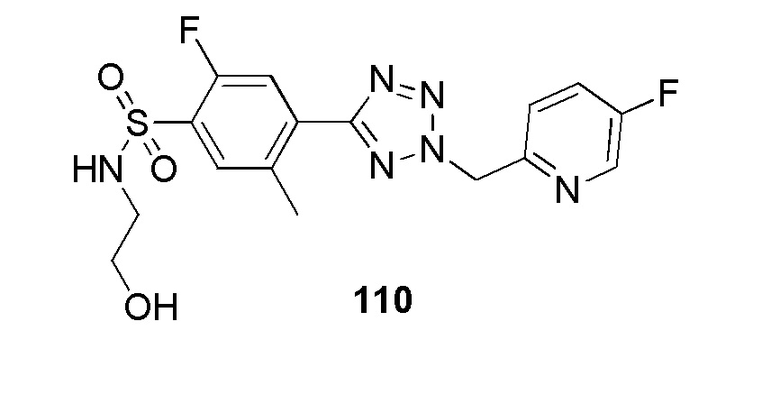
К раствору 2-фтор-N-(2-гидроксиэтил)-5-метил-4-(2H-тетразол-5-ил)бензолсульфонамида (1 г, 3,32 ммоль, промежуточное соединение 138) в ацетонитриле (10 мл) добавляли 2-(бромметил)-5-фторпиридин гидробромид (1,07 г, 3,98 ммоль, промежуточное соединение 5) и N, N-диизопропилэтиламин (1,71 мл, 9,96 ммоль, коммерческий источник: Avra) при комнатной температуре. Реакционную смесь перемешивали при 90°С в течение ночи. По окончании реакции реакционную смесь выливали в ледяную воду (200 мл) и экстрагировали этилацетатом (2×500 мл). Объединенные органические слои высушивали над безводным Na2SO4, фильтровали и фильтрат упаривали при пониженном давлении. Сырой продукт очищали колоночной хроматографией, элюируя смесью 30% этилацетата в петролейном эфире. Чистые фракции собирали и концентрировали при пониженном давлении, и затем очищали препаративной ВЭЖХ (условия препаративной ВЭЖХ: колонка XBridge C18 (150×19) мм, 5 мкм; подвижная фаза A: 10 мМ бикарбонат аммония (водный раствор), подвижная фаза B: ацетонитрил; метод (время в мин/% В): 0/10, 15/10; скорость потока: 16 мл/мин, температура: комнатная). Чистые фракции собирали и концентрировали при пониженном давлении с получением 2-фтор-4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)-5-метилбензолсульфонамида (157 мг, 11,4%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,55-8,54 (м, 1H), 8,01-7,88 (м, 2H), 7,85-7,80 (м, 2H), 7,66-7,63 (м, 1H), 6,18 (с, 2H), 4,65 (шир.с, 1H), 3,38 (т, J=6,5 Гц, 2H), 2,96 (т, J=6,3 Гц, 2H), 2,58 (с, 3H). МС m/z [М+Н]+ = 411,10.
Пример 111: (4-(2-(1-(5-фторпиридин-2-ил)этил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид:
К раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (400 мг, 0,0014 моль, промежуточное соединение 17), 2-(1-бромэтил)-5-фторпиридина (301 мг, 0,0014 моль, промежуточное соединение 141) в ацетонитриле (4 мл) добавляли N, N-диизопропилэтиламин (0,46 мл, 0,0028 моль, коммерческий источник: Finar) при 26°С. Реакционную смесь нагревали до 80°С и перемешивали в течение 16 ч при той же температуре. После окончания реакции реакционную смесь растворяли в этилацетате (100 мл) и промывали водой (2×30 мл). Органический слой концентрировали при пониженном давлении. Сырой продукт очищали препаративной ВЭЖХ и чистую фракцию высушивали при лиофилизации с получением 4-(2-(1-(5-фторпиридин-2-ил)этил) -2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамида (200 мг, 36%) в виде не совсем белого твердого вещества.
Условия препаративной ВЭЖХ:
Колонка: YMC Triart C8 (150×25) мм, 10 мкм
Подвижная фаза: H2O (10 мМ NH4HCO3) B: ACN
Метод (время в мин/% В): 0/30, 1/30, 10/65, 11/65, 11,5/ 100, 13,5/100, 14/30, 16/30
Скорость потока: 20 мл/мин
Температура: комнатная.
Полученное рацемическое соединение подвергали хиральной препаративной SFC для разделения изомеров.
Условия препаративной SFC:
Колонка: Chiralpak AD-H (30×250 мм), 5 мкм
% CO2: 70%:% сорастворитель: 30% (100% этанол)
Поток: 70 г/мм, противодавление: 100 бар, УФ: 253 нм, время стекового ввода: 3,5 мин
Загрузка: 4 мг, растворимость: MeOH, число инжектирований: 55
Модель прибора: марка/модель: SFC-80
Две чистые фракции высушивали при лиофилизации с получением:
(S)-4-(2-(1-(5-фторпиридин-2-ил)этил)-2Н-тетразол-5-ил)-N- (2-гидроксиэтил)бензолсульфонамид:
(78 мг, 14%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,54 (д, J=2,9 Гц, 1H), 8,24 (д, J=8,6 Гц, 2H), 7,96 (д, J=8,6 Гц, 2H), 7,85-7,75 (м, 1H), 7,74 (шир. с, 1H), 7,66-7,58 (м, 1H), 6,48 (кв, J=6,9 Гц, 1H), 4,66 (т, J=5,5 Гц, 1H), 3,36 (кв , J=6,1 Гц, 2H), 2,83 (кв, J=5,8 Гц, 2H), 2,06 (д, J=7,0 Гц, 3H). МС m/z [М+Н]+ = 393,14, хиральная чистота: ее% = 99,86%. Абсолютную конфигурацию определяли ab initio колебательным круговым дихроизмом (VCD).
(R)-4-(2-(1-(5-фторпиридин-2-ил)этил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид:
(69 мг, 13%) в виде бледно-желтой смолы. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,54 (д, J=2,9 Гц, 1H), 8,24 (д, J=8,6 Гц, 2H), 7,96 (д, J=8,3 Гц, 2H), 7,85-7,77 (м, 1H), 7,74 (т, J=5,8 Гц, 1H), 7,65-7,58 (м, 1H), 6,48 (кв, J=6,9 Гц, 1H), 4,66 (т, J=5,6 Гц, 1H), 3,36 (кв, J=6,1 Гц, 2H), 2,83 (кв, J=6,1 Гц, 2H), 2,06 (д, J=7,0 Гц, 3H). МС m/z [М+Н]+ = 393,14, хиральная чистота: ее% = 97,5%. Абсолютную конфигурацию определяли ab initio колебательным круговым дихроизмом (VCD).
Пример 112: 4-(2-(1-(5-фторпиридин-2-ил)этил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид:
К раствору N-(2-гидроксиэтил)-4-(2H-тетразол-5-ил) бензолсульфонамида (300 мг, 0,0011 моль, промежуточное соединение 17) и N, N-диизопропилэтиламина (0,38 мл, 0,0022 моль, коммерческий источник: Finar) в ацетонитриле (6 мл) добавляли 1- (1-бромэтил)-4-фторбензол (271 мг, 0,0013 моль, промежуточное соединение 142) при 26°С. Реакционную смесь нагревали до 85°С и перемешивали в течение 16 ч при той же температуре. По окончании реакции реакционную смесь охлаждали до 26°С, растворяли в этилацетате (10 мл) и промывали водой (30 мл) и насыщенным раствором соли (40 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,5% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением (4-(2-(1-(5-фторпиридин-2-ил) этил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамида (110 мг, 26%) в виде не совсем белого твердого вещества.
Полученное рацемическое соединение подвергали хиральной препаративной SFC для разделения изомеров.
Условия препаративной SFC:
Колонка: Chiralpak IG (30×250 мм), 5 мкм
% CO2: 55%:% сорастворитель: 45% (100% метанол)
Поток: 90 г/мм, противодавление: 90 бар, УФ: 254 нм, время стекового ввода: 5,5 мин
Загрузка: 8,6 мг, растворимость: MeOH, число инжектирований: 20
Модель прибора: марка/модель: SFC-200-002.
Две чистые фракции высушивали при лиофилизации с получением:
Изомер 1: (43 мг, 10%) в виде коричневого липкого твердого вещества и в виде одного неизвестного энантиомера. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,24 (д, J=8,1 Гц, 2H), 7,96 (д, J=8,3 Гц, 2H), 7,52-7,49 (м, 2H), 7,26-7-21 ( м, 2H), 6,44-6,39 (м, 1H), 3,38-3,35 (м, 2H), 2,85-2,82 (м, 2H), 2,02 (д, J=7 Гц, 3H). МС m/z [М+Н]+ = 392,05, хиральная чистота: ее% = 99,53%.
Изомер 2: (49 мг, 11%) в виде коричневого липкого твердого вещества и в виде одного неизвестного энантиомера. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,25 (д, J=8,3 Гц, 2H), 7,97 (д, J=8,3 Гц, 2H), 7,76-7,73 (м, 1H), 7,52-7,49 (м, 2H), 7,26-7-21 (м, 2H), 6,44-6,39 (м, 1H), 4,67-4,64 (м, 1H), 3,39-3,34 (м, 2H), 2,86-2,81 (м, 2H), 2,03-2,01 (м, 3Н). МС m/z [М+Н]+ = 392,02, хиральная чистота: ее% = 99,80%.
Пример 113: 4-(1-(1-(4-фторфенил)этил)-1Н-1,2,3-триазол-4-ил)-N-(2-гидроксиэтил)бензолсульфонамид:
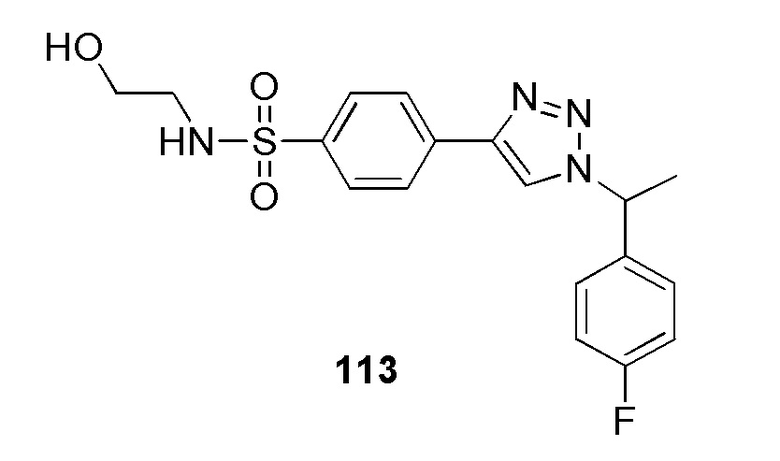
К раствору 4-этинил-N-(2-гидроксиэтил)бензолсульфонамида (310 мг, 0,0014 моль, промежуточное соединение 62) и 1-(1-азидоэтил)-4-фторбензола (227 мг, 0,0014 моль, промежуточное соединение 144) в этаноле (6,2 мл) и воде (6,2 мл) добавляли L-аскорбат натрия (81,8 мг, 0,00041 моль, коммерческий источник: Aldrich) и CuSO4⋅5H2O (34,3 мг, 0,00014 моль, коммерческий источник: Finar) и реакционную смесь перемешивали в течение 16 ч при 26°С. По окончании реакции этанол удаляли в вакууме. Реакционную смесь растворяли в этилацетате (100 мл), промывали насыщенным раствором соли (2×30 мл), высушивали над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,8% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 4-(1-(1-(4-фторфенил)этил)-1Н-1,2,3-триазол-4-ил)-N- (2-гидроксиэтил)бензолсульфонамида (170 мг, 32%).
Полученное рацемическое соединение подвергали хиральной препаративной SFC для разделения изомеров.
Условия препаративной SFC:
Колонка: (R, R) Whelk-01 (30×250 мм), 5 мкм
% CO2: 70%:% сорастворитель: 30% (100% изопропанол)
Поток: 90 г/мм, противодавление: 90 бар, УФ: 261 нм, время стекового ввода: 6,4 мин
Загрузка: 6 мг, растворимость: MeOH, число инжектирований: 30
Модель прибора: марка/модель: SFC-200-003.
Две чистые фракции высушивали при лиофилизации с получением:
Изомер 1: (68 мг, 12%) в виде не совсем белого твердого вещества и в виде одного неизвестного энантиомера. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,88 (с, 1H), 8,05 (д, J=8,1 Гц, 2H), 7,85 (д, J=8,3 Гц, 2H), 7,59 (шир. с, 1H), 7,45-7,42 (м, 2H), 7,24-7-20 (м, 2H), 6,07-6,01 (м, 1H), 4,66-4,64 (м, 1H), 3,39-3,34 (м, 2H), 2,83-2,80 (м, 2H), 1,93 (д, J=7,2 Гц, 3H). МС m/z [М+Н]+ = 391,04, хиральная чистота: ее% = 99,38%.
Изомер 2: (43 мг, 8%) в виде не совсем белого твердого вещества и в виде одного неизвестного энантиомера. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,89 (с, 1H), 8,05 (д, J=8,6 Гц, 2H), 7,85 (д, J=8,3 Гц, 2H), 7,61-7,58 (м, 1H) 7,45-7,42 (м, 2Н), 7,24-7-20 (м, 2Н), 6,07-6,01 (м, 1Н), 4,67-4,64 (м, 1Н), 3,38-3,34 (м, 2Н), 2,84-2,79 (м, 2H), 1,93 (д, J=7 Гц, 3H). МС m/z [М+Н]+ = 391,10, хиральная чистота: ее% = 97,59%.
Пример 114: 2-(4-(2-(1-(4-фторфенил)этил)-2Н-тетразол-5-ил) фенилсульфонамидо)ацетамид:
К раствору 2-(4-(2-(1-(4-фторфенил)этил)-2Н-тетразол-5-ил) фенилсульфонамидо)уксусной кислоты (140 мг, 0,00035 моль, промежуточное соединение 146) в ДМФА (1,4 мл) добавляли N, N-диизопропилэтиламин (0,12 мл, 0,00069 моль, коммерческий источник: Finar), NH4Cl (27 мг, 0,00052 моль, коммерческий источник: Finar) и HATU (144 мг, 0,00038 моль, коммерческий источник: Aldrich) и смесь перемешивали при 26°С в течение 16 ч. После окончания реакции реакционную смесь растворяли в этилацетате (100 мл) и промывали холодной водой (4×50 мл) и насыщенным раствором соли (50 мл). Органический слой высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 2% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 2-(4-(2-(1-(4-фторфенил)этил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетамида (130 мг, 92%).
Полученное рацемическое соединение подвергали хиральной препаративной SFC для разделения изомеров.
Условия препаративной SFC:
Колонка: Chiralpak AD-H (30×250 мм), 5 мкм
% CO2: 70%:% сорастворитель: 30% (100% метанол)
Поток: 70 г/мм, противодавление: 90 бар, УФ: 254 нм, время стекового ввода: 11,0 мин
Загрузка: 4,5 мг, растворимость: MeOH+MeCN, число инжектирований: 30
Модель прибора: марка/модель: SFC-80.
Две чистые фракции высушивали при лиофилизации с получением:
Изомер 1: (23 мг, 16%) в виде не совсем белого твердого вещества и одного неизвестного энантиомера. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,23 (д, J=8,3 Гц, 2H), 8,01 (шир. с, 1H), 7,97 (д, J=8,3 Гц, 2H), 7,52-7,49 (м, 2H), 7,26-7,21 (м, 3H), 7,04 (шир. с, 1H), 6,44-6,39 (м, 1H), 3,43 (с, 2H), 2,02 (д, J=6,8 Гц, 3H). МС m/z [М+Н]+ = 405,05, хиральная чистота: ее% = 99,34%.
Изомер 2: (30 мг, 21%) в виде не совсем белого твердого вещества и в виде одного неизвестного энантиомера. Хиральная чистота: ее% = 90,09%. Его снова очищали хиральной SFC (колонка: Chiralpak AD-H (30×250 мм), 5 мкм; % CO2: 70%:% сорастворитель: 30% (100% метанол); поток: 70 г/мм, противодавление: 90 бар, УФ: 254 нм, время стекового ввода: 12,0 мин; загрузка: 2,3 мг; растворимость: MeOH+MeCN; количество инжектирований: 10; модель прибора: марка/модель: SFC-80). Чистое соединение высушивали при лиофилизации с получением 2-(4-(2-(1-(4-фторфенил)этил)-2Н-тетразол-5-ил)фенилсульфонамидо)ацетамида (14 мг, 10%) в виде не совсем белого твердого вещества и одного неизвестного энантиомера. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,22 (д, J=8,3 Гц, 2H), 7,96 (д, J=8,3 Гц, 2H), 7,52-7,49 (м, 2H), 7,26-7,21 (м, 3H), 7,03 (шир. с, 1H), 6,44-6,39 (м, 1H), 3,43 (с, 2H), 2,02 (д, J=7 Гц, 3H). МС m/z [М+Н]+ = 405,05, хиральная чистота: ее% = 99,26%.
Пример 115: 2-(4-(2-(1-(4-фторфенил)этил)-2Н-тетразол-5-ил)-2-метоксифенилсульфонамидо)ацетамид:
К раствору трет-бутил (2-амино-2-оксоэтил)((4-(2-(1-(4-фторфенил)этил)-2Н-тетразол-5-ил)-2-метоксифенил)сульфонил) карбамата (300 мг, 0,00056 моль, промежуточное соединение 148) в 1,4-диоксане (3 мл) медленно добавляли 4 М HCl (1,5 мл, коммерческий источник: Hychem) при 0°С и смесь перемешивали при 26°C в течение 5 ч. По окончании реакции реакционную смесь концентрировали и растворяли в этилацетате (100 мл) и насыщенном растворе NaHCO3. Органический слой отделяли, и водный слой экстрагировали этилацетатом (2×30 мл). Объединенные органические слои концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией (силикагель 100-200 меш), элюируя смесью 1,5% метанола в дихлорметане. Чистые фракции собирали и концентрировали при пониженном давлении с получением 2-(4-(2-(1-(4-фторфенил)этил)-2H-тетразол-5-ил)-2-метоксифенилсульфонамидо)ацетамида (220 мг, 90%).
Полученное рацемическое соединение подвергали хиральной препаративной SFC для разделения изомеров.
Условия препаративной SFC:
Колонка: Chiralpak OJ-H (21×250 мм), 5 мкм
% CO2: 90%:% сорастворитель: 10% (100% этанол)
Поток: 70 г/мм, противодавление: 90 бар, УФ: 214 нм, время стекового ввода: 6,5 мин
Загрузка: 5,1 мг, растворимость: MeOH+MeCN, число инжектирований: 40
Модель прибора: марка/модель: SFC-80.
Две чистые фракции высушивали при лиофилизации с получением:
Изомер 1: (58 мг, 24%) в виде не совсем белого твердого вещества и в виде одного неизвестного энантиомера. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,90-7,88 (м, 1H), 7,74-7,73 (м, 2H), 7,51-7,48 (м, 2H), 7,38 (шир. с, 1H), 7,26-7,21 (м, 2Н), 7,18 (шир. с, 1Н), 7,04 (шир. с, 1Н), 6,44-6,39 (м, 1Н), 4,00 (с, 3Н), 3,47 (с, 2Н), 2,02 (д, J=7 Гц, 3H). МС m/z [М+Н]+ = 435,05, хиральная чистота: ее% = 99,61%.
Изомер 2: (58 мг, 24%) в виде белого твердого вещества и в виде одного неизвестного энантиомера. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,90-7,88 (м, 1H), 7,74-7,73 (м, 2H), 7,51-7,48 (м, 2H), 7,38 (шир. с, 1H), 7,26-7,21 (м, 2Н), 7,18 (шир. с, 1Н), 7,04 (шир. с, 1Н), 6,44-6,39 (м, 1Н), 4,00 (с, 3Н), 3,47 (с, 2Н), 2,02 (д, J=7 Гц, 3H). МС m/z [М+Н]+ = 435,11, хиральная чистота: ее% = 98,69%.
Биологическая активность
Анализ ингибирования Mycobacterium tuberculosis H37Rv (цельноклеточный анализ)
Определение минимальной ингибирующей концентрации (MIC) в отношении M. tuberculosis H37Rv для каждого тестируемого соединения проводили в микротитрационных 96-луночных плоскодонных полистироловых планшетах в конечном объеме 200 мкл. Десять двукратных разведений препарата в чистом ДМСО, начиная с 80 мкМ, готовили в ряду 1-10. Изониазид (INH) (Sigma Aldrich) использовали в качестве положительного контроля как дозозависимый контроль, где двукратные разведения начинали с 4 мкг/мл в ряду 11. В G-12 и H-12 рифампицин разливали в концентрации 1 мкг/мл в качестве контроля с отсутствием роста. От A12 до F12 разливали ДМСО в качестве контроля роста.
Инокулят стандартизировали примерно до 1×107 КОЕ/мл и разводили 1 к 200 бульоном Мидлбрук 7H9, с добавлением ADC (Difco). Этот инокулят (200 мкл и 104 КОЕ/лунку) вносили во все лунки планшета.
Все планшеты помещали в герметичную коробку для предотвращения высыхания периферических лунок и инкубировали при 37°С без встряхивания в течение шести суток.
Раствор резазурина готовили растворением одной таблетки резазурина (таблетки резазурина для анализа молока; Ref 330884Y' VWR International Ltd) в 30 мл стерильного PBS (забуференный фосфатом физиологический раствор). Из этого раствора 25 мкл добавляли в каждую лунку.
Измеряли флуоресценцию (Spectramax M5 Molecular Devices, Exitation).
Результаты анализа ингибирования Mycobacterium tuberculosis H37Rv (цельноклеточный анализ)
Все примеры тестировали по существу в соответствии с описанным выше цельноклеточным анализом.
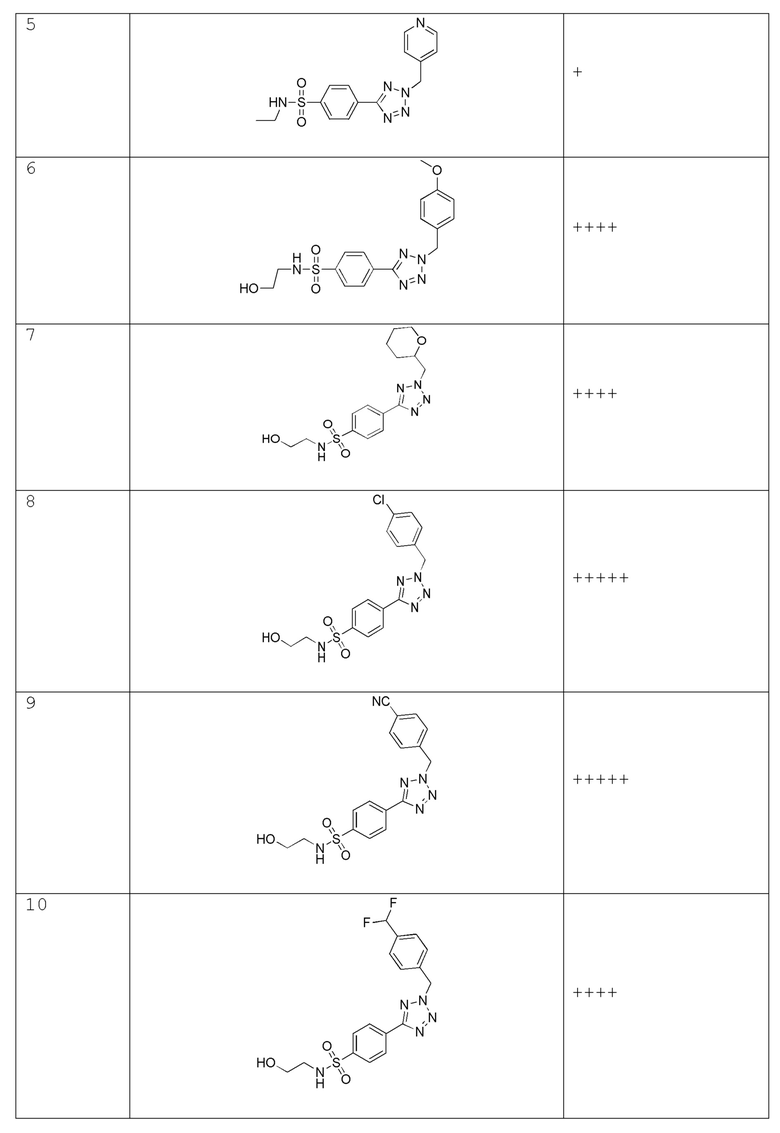
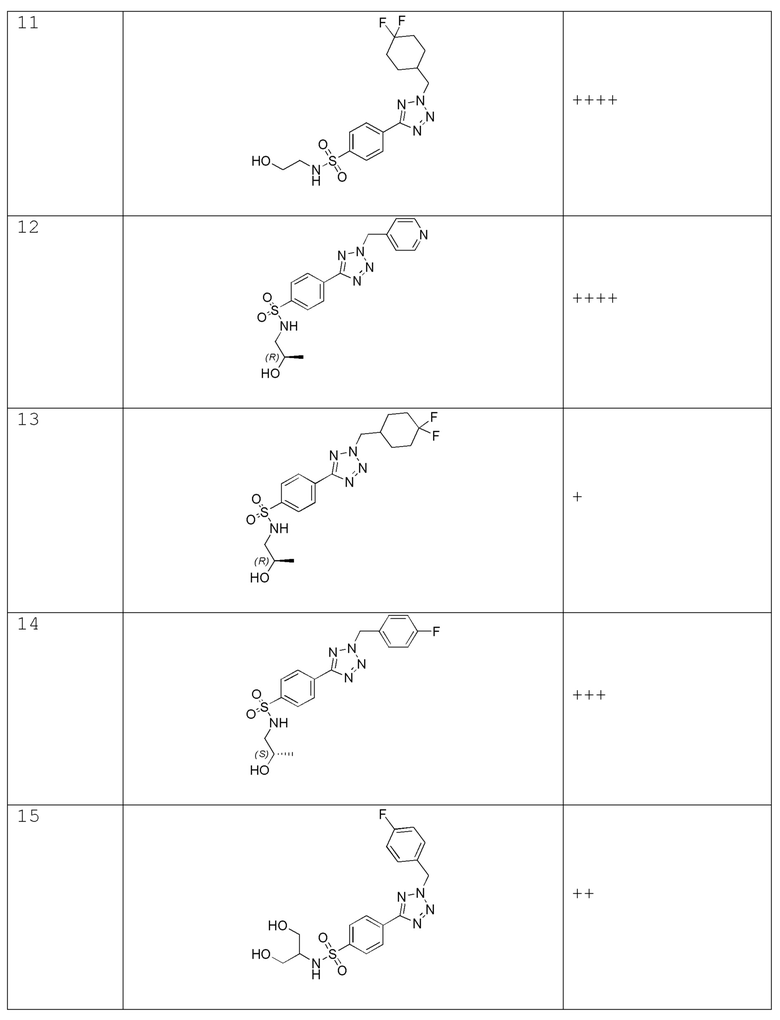
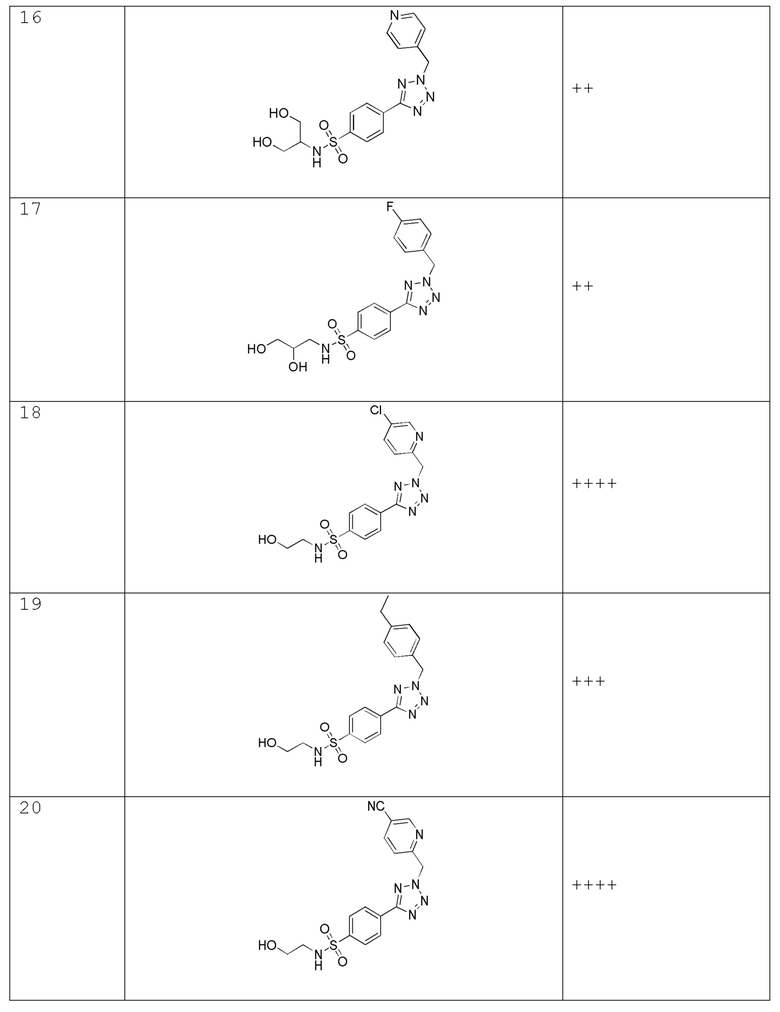
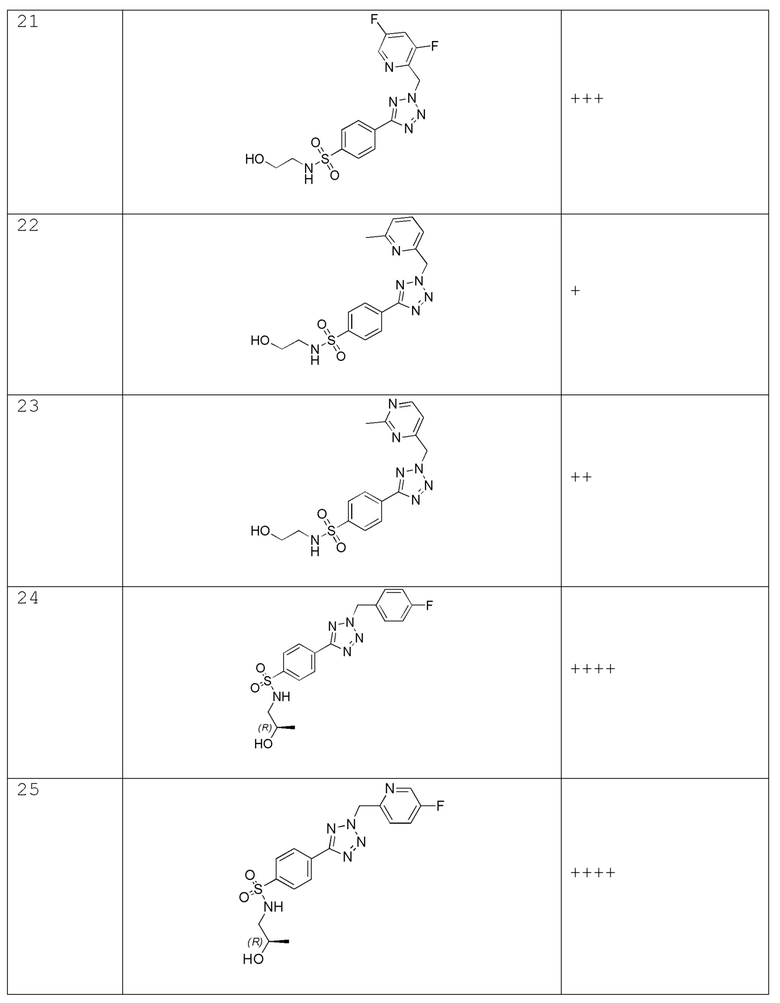
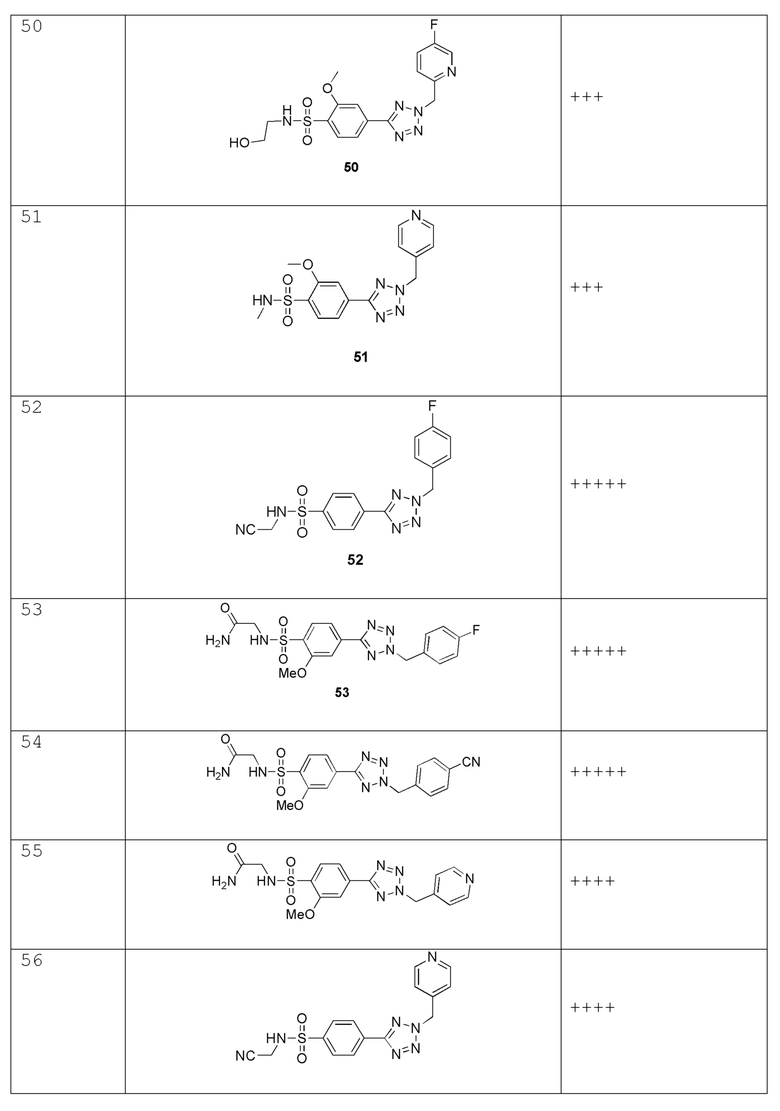
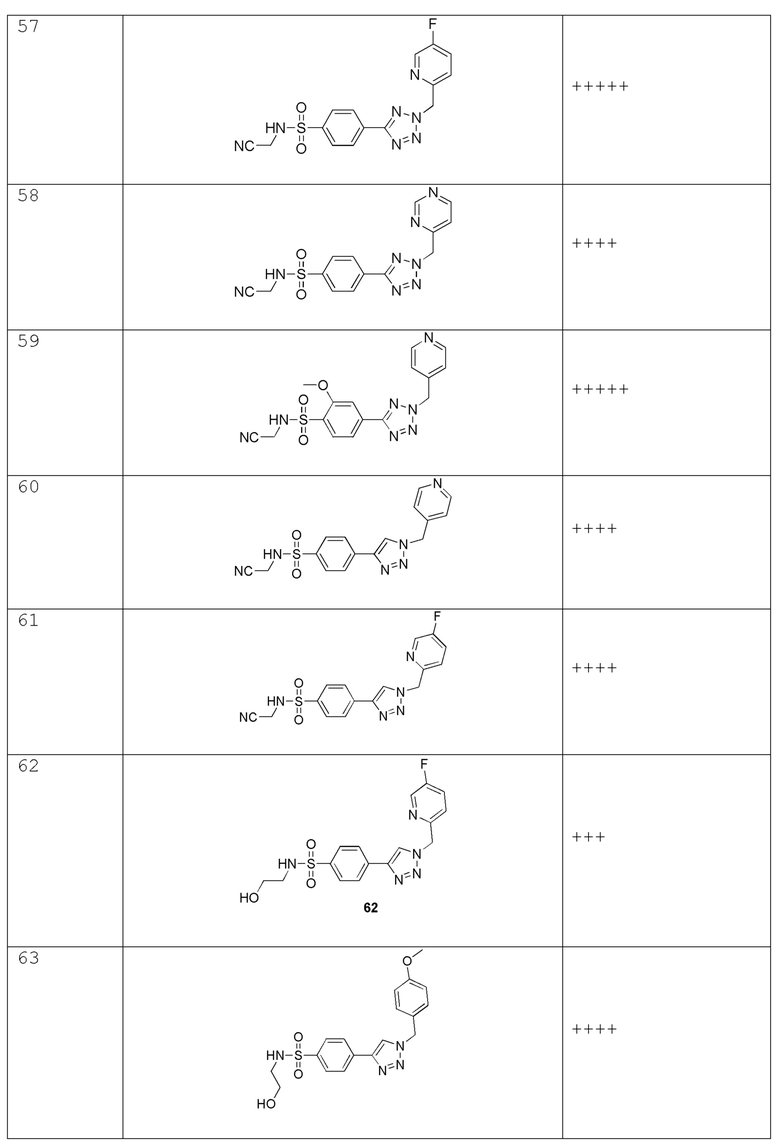
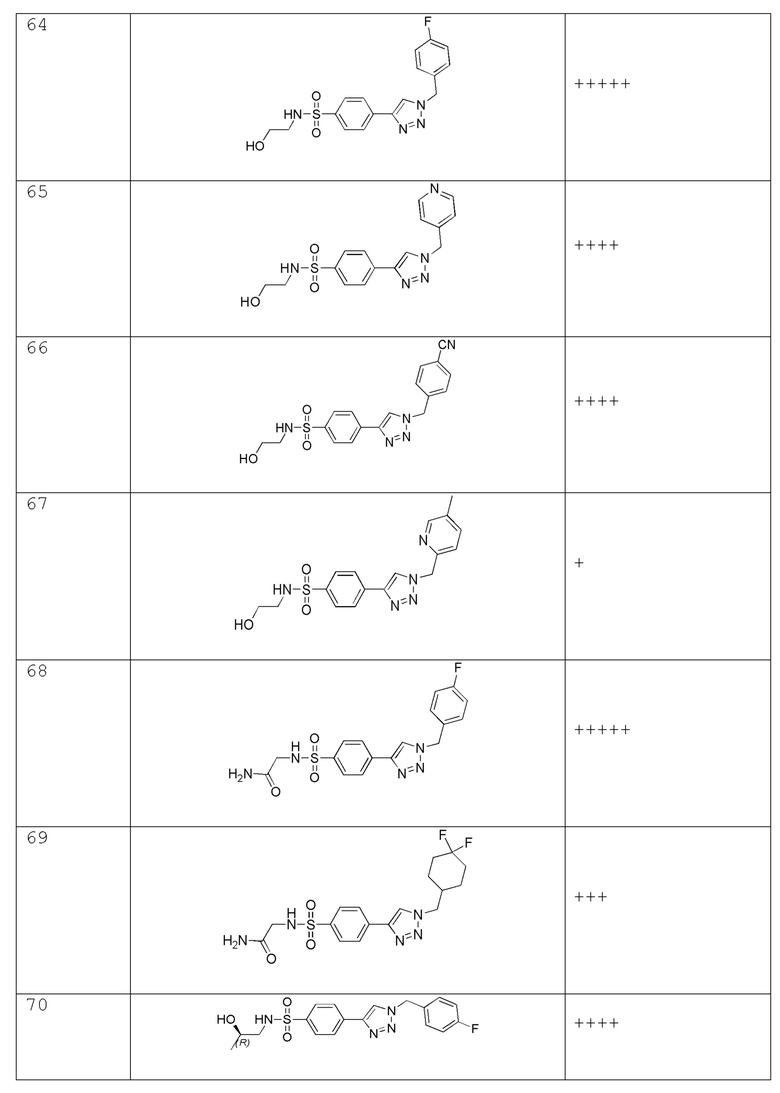
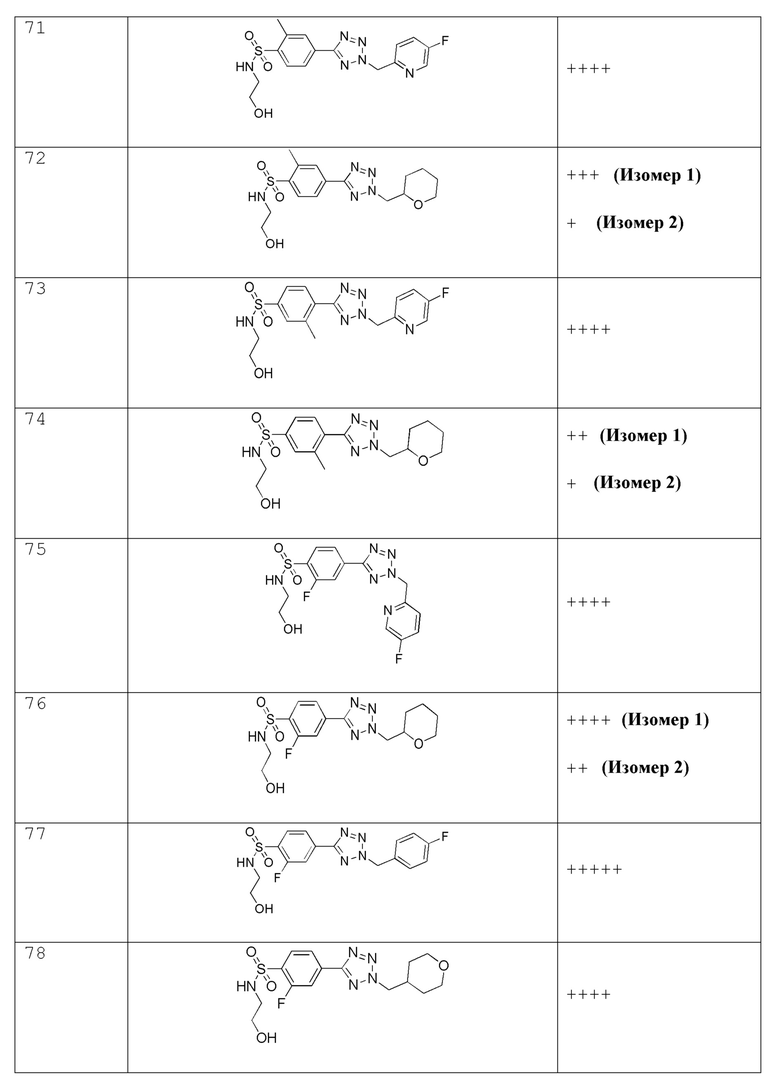
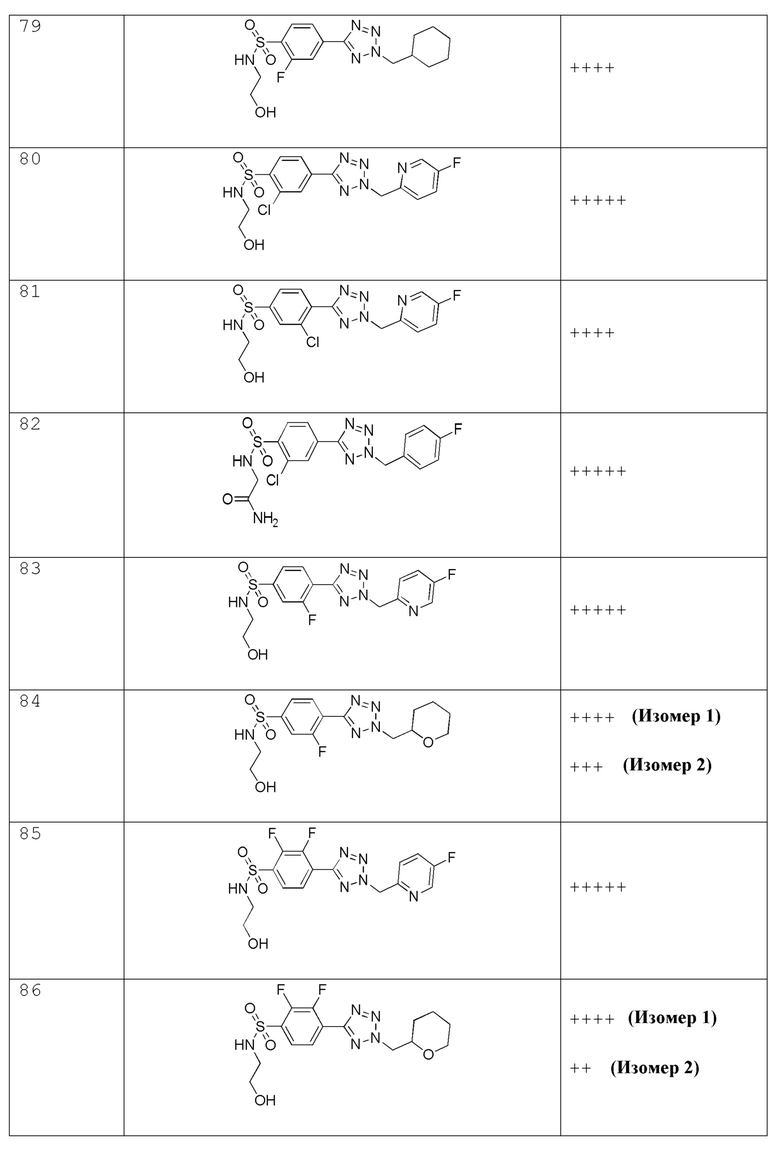
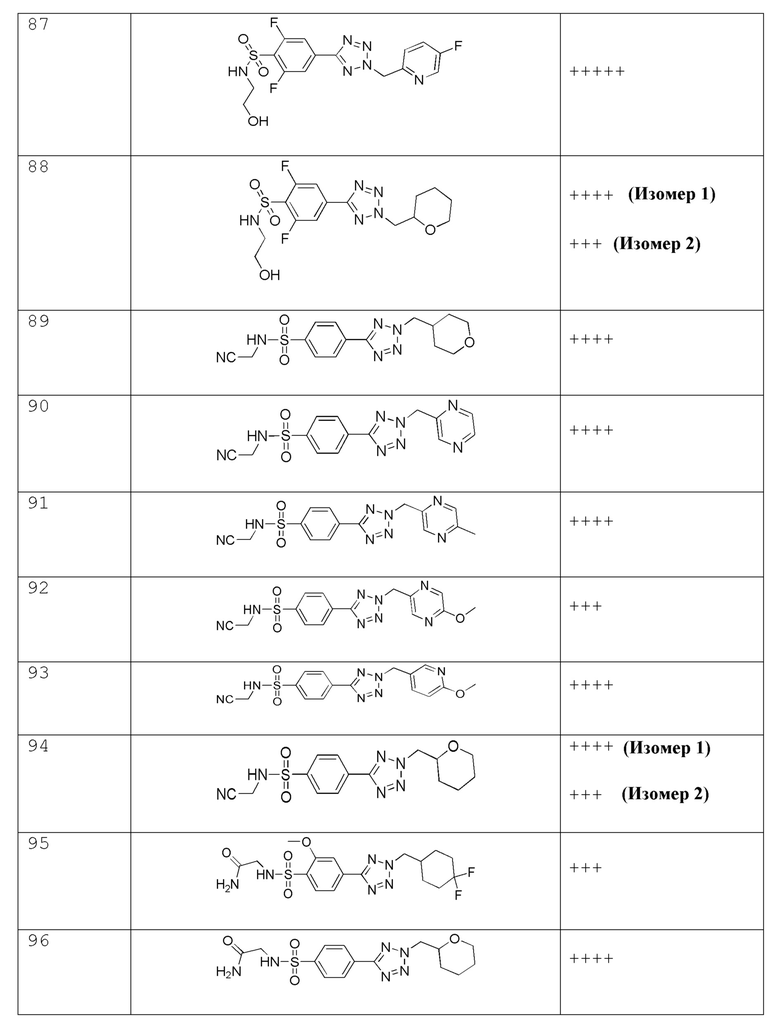
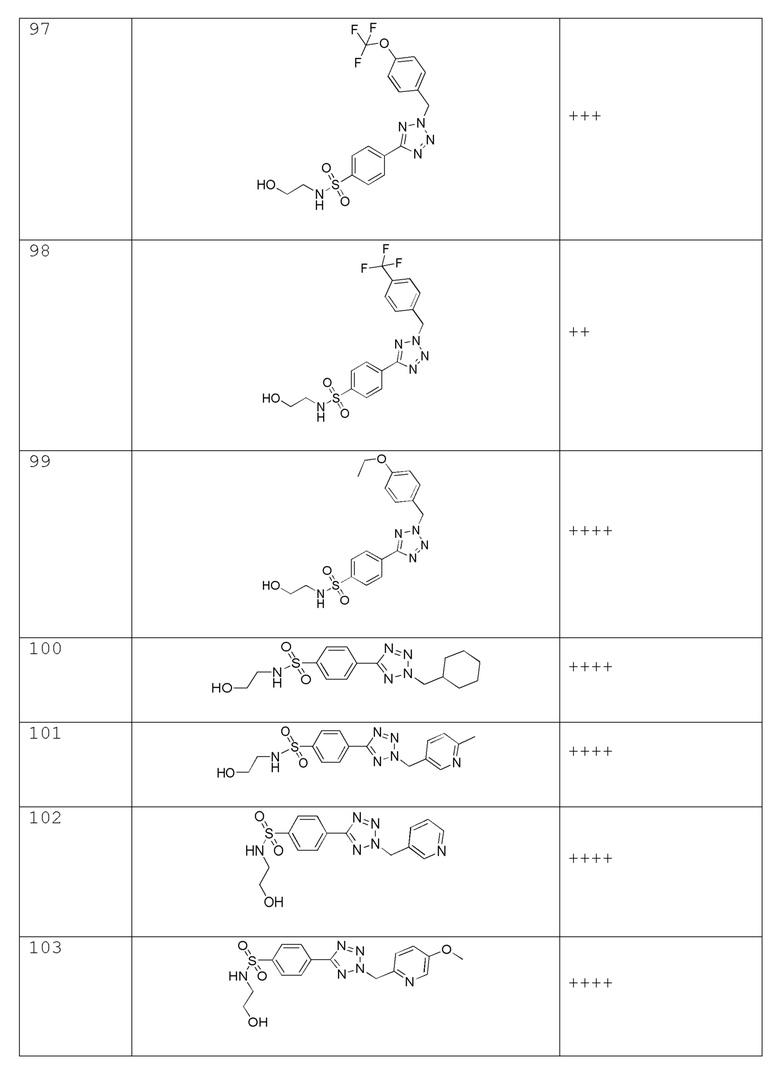
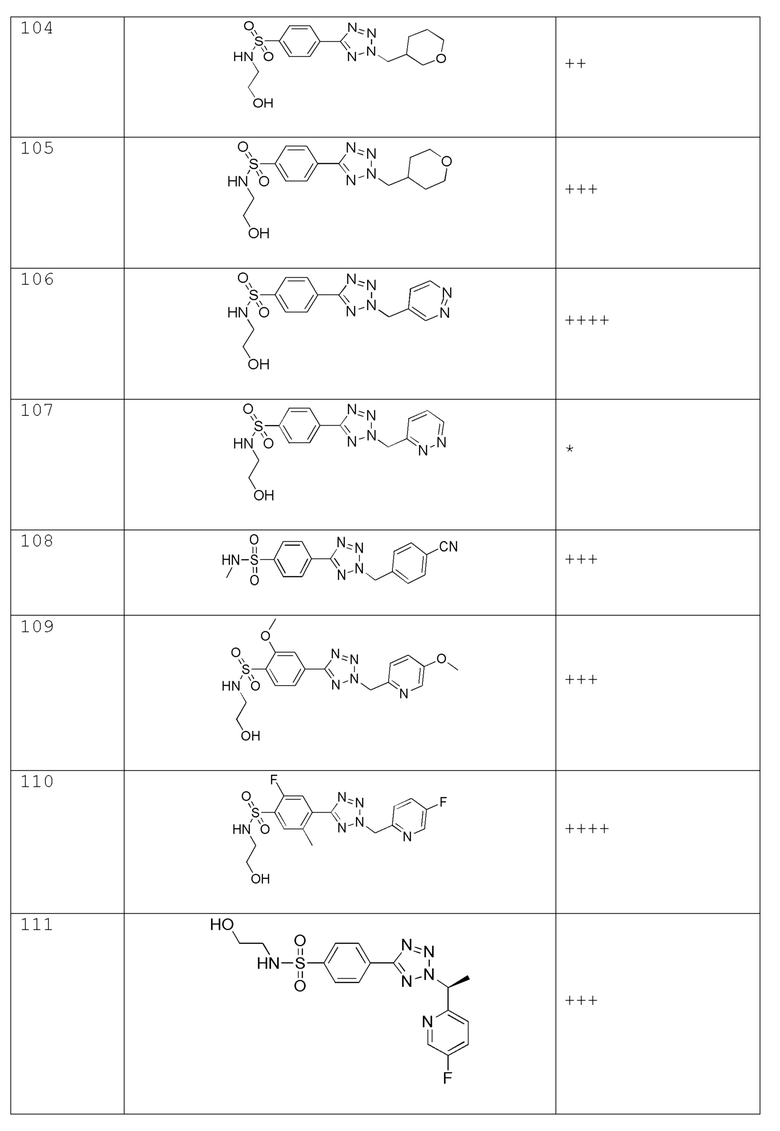
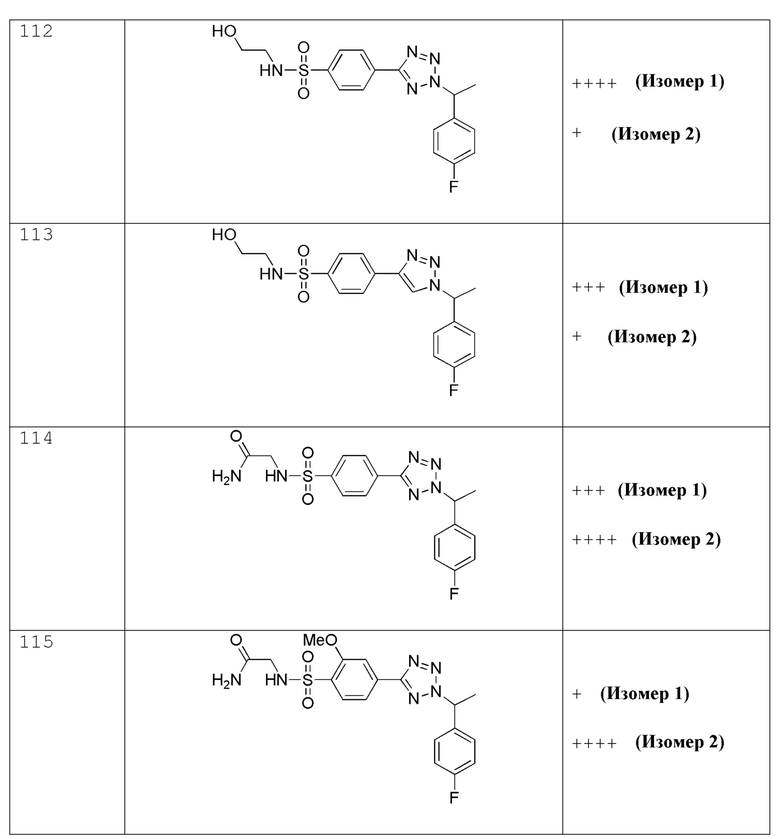
<0,1 мкМ = +++++
≥0,1 мкМ до <1 мкМ = ++++
≥1 мкМ до <5 мкМ = +++
≥5 мкМ до <10 мкМ = ++
≥10 мкМ до ≤25 мкМ = +
В частности, было установлено, что значение MIC примера 25 составляет 0,6 мкМ (что определено двукратным тестированием соединения).
Было установлено, что пример 29 имеет значение MIC в диапазоне от 0,07 до 0,16 мкМ (чир определено двенадцатикратным тестированием соединения).
Было установлено, что пример 53 имеет значение MIC 0,04 мкМ (что определено шестикратным тестированием соединения).
Было установлено, что пример 75 имеет значение MIC 0,16 мкМ (что определено двукратным тестированием соединения).
Было установлено, что пример 87 имеет значение MIC ≤0,04 мкМ (что определено четырехкратным тестированием соединения, давая ≤0,16 мкМ и один раз ≤0,04 мкМ)
Было установлено, что пример 111 (S-изомер) имеет значение MIC в диапазоне от 0,6 до 1,875 мкМ (что определено трехкратным тестированием соединения).
Было установлено, что пример 115 изомер 2 имеет значение MIC в диапазоне от 0,45 до 1,25 мкМ (что определено двукратным тестированием соединения).
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ MKK4 ДЛЯ СТИМУЛЯЦИИ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ УМЕНЬШЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2788000C2 |
| ПРОИЗВОДНЫЕ ПИРИДООКСАЗИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ TNAP | 2016 |
|
RU2715704C2 |
| АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1996 |
|
RU2198878C2 |
| БЕНЗОКСЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2557658C2 |
| Новые 3-индол замещенные производные, фармацевтические композиции и способы применения | 2016 |
|
RU2672252C1 |
| КОМПОЗИЦИИ ТЕТРАГИДРОХИНОЛИНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ БРОМОДОМЕНА ВЕТ | 2014 |
|
RU2727169C2 |
| МОДУЛЯТОРЫ КАЛЬПАИНА И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2773288C2 |
| ПЕРИ-ЗАМЕЩЕННЫЕ АРИЛСУЛЬФОНАМИДНЫЕ БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ПРЕДНАЗНАЧЕННЫЕ ДЛЯ ЛЕЧЕНИЯ ОККЛЮЗИОННОГО ЗАБОЛЕВАНИЯ АРТЕРИЙ | 2005 |
|
RU2407736C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2011 |
|
RU2652638C2 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
Изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, где Х является СН или N; n равно 0, 1 или 2; R1 представляет метил, этил, цианометил, С-связанный ацетамидо, метилацетат, 2-гидроксиэтил, 2-гидрокси-1-пропил, 1,3-дигидрокси-2-пропил или 1,2-дигидрокси-3-пропил; R2 независимо выбран из атома галогена, C1-2 алкила или C1-2 алкокси; R3 представляет фенил, пиридил, пиримидинил, пиразинил или пиридазинил, где каждая из этих групп может быть необязательно замещена одним или двумя заместителями, выбранными из атома галогена, циано, C1-2 алкила, необязательно замещенного не более чем тремя атомами фтора, и C1-2 алкокси, необязательно замещенного не более чем тремя атомами фтора, где заместители могут быть одинаковыми или различными; или R3 представляет циклогексил, который может быть необязательно замещен одним или двумя атомами фтора или хлора, где каждый заместитель может быть присоединен к одному и тому же атому углерода и каждый заместитель может быть одинаковым или различным; или R3 представляет тетрагидропиран; и R4 является Н или метилом. Изобретение также относится к фармацевтической композиции на основе соединения формулы (I). Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине для лечения микобактериальных инфекций или в лечении заболеваний, вызванных микобактериями, таких как туберкулез. 5 н. и 14 з.п. ф-лы, 6 табл., 115 пр.
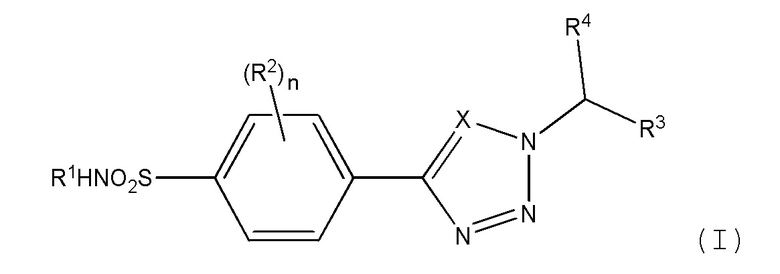
1. Соединение формулы (I) или его фармацевтически приемлемая соль
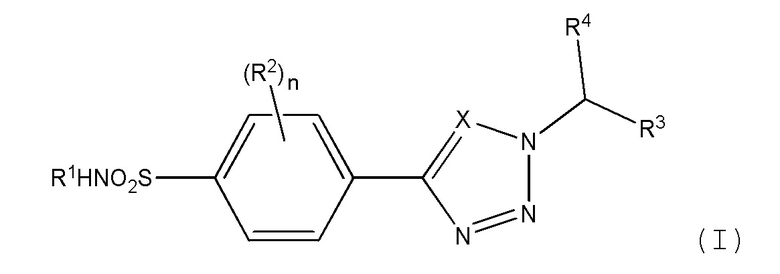
где:
Х является СН или N;
n равно 0, 1 или 2;
R1 представляет метил, этил, цианометил, С-связанный ацетамидо, метилацетат, 2-гидроксиэтил, 2-гидрокси-1-пропил, 1,3-дигидрокси-2-пропил или 1,2-дигидрокси-3-пропил;
R2 независимо выбран из атома галогена, C1-2 алкила или C1-2 алкокси;
R3 представляет фенил, пиридил, пиримидинил, пиразинил или пиридазинил, где каждая из этих групп может быть необязательно замещена одним или двумя заместителями, выбранными из атома галогена, циано, C1-2 алкила, необязательно замещенного не более чем тремя атомами фтора, и C1-2 алкокси, необязательно замещенного не более чем тремя атомами фтора, где заместители могут быть одинаковыми или различными; или
R3 представляет циклогексил, который может быть необязательно замещен одним или двумя атомами фтора или хлора, где каждый заместитель может быть присоединен к одному и тому же атому углерода и каждый заместитель может быть одинаковым или различным; или
R3 представляет тетрагидропиран; и
R4 является Н или метилом.
2. Соединение или его фармацевтически приемлемая соль по п.1, где X представляет N.
3. Соединение или его фармацевтически приемлемая соль по п.1 или 2, где R4 представляет Н.
4. Соединение или его фармацевтически приемлемая соль по любому из предшествующих пунктов, где n равно 0.
5. Соединение или его фармацевтически приемлемая соль по любому из предшествующих пунктов, где R1 представляет метил, цианометил, С-связанный ацетамидо, 2-гидроксиэтил, (R)-2-гидрокси-1-пропил или (S)-2-гидрокси-1-пропил.
6. Соединение или его фармацевтически приемлемая соль по любому из предшествующих пунктов, где R3 представляет фенил, пиридил или пиримидинил, где каждая из этих групп может быть необязательно замещена одним заместителем, выбранным из атома фтора, хлора, циано, метила, дифторметила (-CHF2), трифторметила, метокси и трифторметокси (-OCF3).
7. Соединение или его фармацевтически приемлемая соль по любому из пп.1-4, где R3 представляет фенил или пиридил, где каждая из этих групп может быть необязательно замещена одним заместителем, выбранным из атома галогена, циано, C1-2 алкила, необязательно замещенного не более чем тремя атомами фтора, и C1-2 алкокси, необязательно замещенного не более чем тремя атомами фтора.
8. Соединение или его фармацевтически приемлемая соль по любому из предшествующих пунктов, где R3 представляет пиридил, необязательно замещенный атомом фтора или хлора.
9. Соединение или его фармацевтически приемлемая соль по любому из предшествующих пунктов, где R3 представляет 2-пиридил, необязательно замещенный атомом фтора.
10. Соединение или его фармацевтически приемлемая соль по любому из предшествующих пунктов, включающее:
(4-(2-(4-фторбензил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид);
N-(2-гидроксиэтил)-4-(2-(пиримидин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
4-(2-(4-фторбензил)-2H-тетразол-5-ил)-N-метилбензолсульфонамид;
N-метил-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
N-этил-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-(4-метоксибензил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
4-(2-(4-хлорбензол)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(2-(4-цианобензол)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(2-(4-(дифторметил)бензил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(2-((4,4-дифторциклогексил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
(R)-N-(2-гидроксипропил)-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид
(R)-4-(2-((4,4-дифторциклогексил)метил)-2H-тетразол-5-ил)-N-(2-гидроксипропил)бензолсульфонамид;
(S)-4-(2-(4-фторбензил)-2H-тетразол-5-ил)-N-(2-гидроксипропил)бензолсульфонамид;
N-(1,3-дигидроксипропан-2-ил)-4-(2-(4-фторбензил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(1,3-дигидроксипропан-2-ил)-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2,3-дигидроксипропил)-4-(2-(4-фторбензил)-2H-тетразол-5-ил)бензолсульфонамид;
4-(2-((5-хлорпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(2-(4-этилбензил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(2-((5-цианопиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(2-((3,5-дифторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-((6-метилпиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-((2-метилпиримидин-4-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
(R)-4-(2-(4-фторбензил)-2H-тетразол-5-ил)-N-(2-гидроксипропил)бензолсульфонамид;
(R)-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксипропил)бензолсульфонамид;
(R)-N-(2-гидроксипропил)-4-(2-((5-метоксипиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
(R)-N-(2-гидроксипропил)-4-(2-((5-метилпиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
(R)-N-(2-гидроксипропил)-4-(2-(4-метоксибензил)-2H-тетразол-5-ил)бензолсульфонамид;
4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(2-(3-фтор-4-метоксибензил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-((5-метоксипиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
(N-(2-гидроксиэтил)-4-(2-((5-метилпиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид);
N-(2-гидроксиэтил)-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-((2-метилпиридин-4-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
2-(4-(2-(4-метоксибензил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
2-(4-(2-(4-метилбензил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
2-(4-(2-(3,4-дифторбензил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
2-(4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
2-(4-(2-((4,4-дифторциклогексил)метил)-2H-тетразол-5-ил) фенилсульфонамидо)ацетамид;
2-(4-(2-((5-метилпиридин-2-ил)метил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
2-(4-(2-((5-хлорпиридин-2-ил)метил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
2-(4-(2-((2-метилпиридин-4-ил)метил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
Метил 2-(4-(2-(4-фторбензил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетат;
Метил 2-(4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетат;
2-(4-(2-(4-фторбензил)-2H-тетразол-5-ил) фенилсульфонамидо)ацетамид;
2-(4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил) фенилсульфонамидо)ацетамид;
(4-(2-(4-фторбензил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)-2-метоксибензолсульфонамид);
(N-(2-гидроксиэтил)-2-метокси-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид);
(4-(2-(4-цианобензил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)-2-метоксибензолсульфонамид);
(4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)-2-метоксибензолсульфонамид);
(2-метокси-N-метил-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид);
(N-(цианометил)-4-(2-(4-фторбензил)-2H-тетразол-5-ил)бензолсульфонамид);
2-(4-(2-(4-фторбензил)-2H-тетразол-5-ил)-2-метоксифенилсульфонамидо)ацетамид;
2-(4-(2-(4-цианобензил)-2H-тетразол-5-ил)-2- метоксифенилсульфонамидо)ацетамид;
2-(2-метокси-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид;
(N-(цианометил)-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид);
(N-(цианометил)-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид);
(N-(цианометил)-4-(2-(пиримидин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
(N-(цианометил)-2-метокси-4-(2-(пиридин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид);
(N-(цианометил)-4-(1-(пиридин-4-илметил)-1H-1,2,3-триазол-4-ил)бензолсульфонамид);
(N-(цианометил)-4-(1-((5-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензолсульфонамид);
(4-(1-((5-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-N-(2-гидроксиэтил)бензолсульфонамидо);
(N-(2-гидроксиэтил)-4-(1-(4-метоксибензил)-1H-1,2,3-триазол-4-ил)бензолсульфонамид);
(4-(1-(4-фторбензил)-1H-1,2,3-триазол-4-ил)-N-(2-гидроксиэтил)бензолсульфонамид);
(N-(2-гидроксиэтил)-4-(1-(пиридин-4-илметил)-1H-1,2,3-триазол-4-ил)бензолсульфонамид);
(4-(1-(4-цианобензил)-1H-1,2,3-триазол-4-ил)-N-(2-гидроксиэтил)бензолсульфонамид);
(N-(2-гидроксиэтил)-4-(1-((5-метилпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензолсульфонамид);
(2-(4-(1-(4-фторбензил)-1H-1,2,3-триазол-4-ил)фенилсульфонамидо)ацетамид);
(2-(4-(1-((4,4-дифторциклогексил)метил)-1H-1,2,3-триазол-4-ил)фенилсульфонамидо)ацетамид);
(R)-4-(1-(4-фторбензил)-1H-1,2,3-триазол-4-ил)-N-(2-гидроксипропил)бензолсульфонамид;
4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)-2-метилбензолсульфонамид;
N-(2-гидроксиэтил)-2-метил-4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)-3-метилбензолсульфонамид;
N-(2-гидроксиэтил)-3-метил-4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
2-фтор-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
2-фтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
2-фтор-4-(2-(4-фторбензил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
2-фтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-4-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
4-(2-(циклогексилметил)-2H-тетразол-5-ил)-2-фтор-N-(2-гидроксиэтил)бензолсульфонамид;
2-хлор-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
3-хлор-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
2-(2-хлор-4-(2-(4-фторбензил)-2H-тетразол-5-ил) фенилсульфонамидо)ацетамид;
3-фтор-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
3-фтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
2,3-дифтор-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
2,3-дифтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
2,6-дифтор-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
(R)-2,6-дифтор-N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(цианометил)-4-(2-((тетрагидро-2H-пиран-4-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(цианометил)-4-(2-(пиразин-2-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(цианометил)-4-(2-((5-метилпиразин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(цианометил)-4-(2-((5-метоксипиразин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(цианометил)-4-(2-((6-метоксипиридин-3-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(цианометил)-4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
2-((4-(2-((4,4-дифторциклогексил)метил)-2H-тетразол-5-ил)-2-метоксифенил)сульфонамидо)ацетамид;
2-((4-(2-((тетрагидро-2H-пиран-2-ил)метил)-2H-тетразол-5-ил)фенил)сульфонамидо)ацетамид;
N-(2-гидроксиэтил)-4-(2-(4-(трифторметокси)бензил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-(4-(трифторметил)бензил)-2H-тетразол-5-ил)бензолсульфонамид;
4-(2-(4-этоксибензил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(2-(циклогексилметил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-((6-метилпиридин-3-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-(пиридин-3-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
(N-(2-гидроксиэтил)-4-(2-((5-метоксипиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид);
N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-3-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-((тетрагидро-2H-пиран-4-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-(пиридазин-4-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
N-(2-гидроксиэтил)-4-(2-(пиридазин-3-илметил)-2H-тетразол-5-ил)бензолсульфонамид;
4-(2-(4-цианобензил)-2H-тетразол-5-ил)-N-метилбензолсульфонамид;
N-(2-гидроксиэтил)-2-метокси-4-(2-((5-метоксипиридин-2-ил)метил)-2H-тетразол-5-ил)бензолсульфонамид;
2-фтор-4-(2-((5-фторпиридин-2-ил)метил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)-5-метилбензолсульфонамид;
(4-(2-(1-(5-фторпиридин-2-ил)этил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(2-(1-(5-фторпиридин-2-ил)этил)-2H-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
4-(1-(1-(4-фторфенил)этил)-1H-1,2,3-триазол-4-ил)-N-(2-гидроксиэтил)бензолсульфонамид;
2-(4-(2-(1-(4-фторфенил)этил)-2H-тетразол-5-ил)фенилсульфонамидо)ацетамид; и
2-(4-(2-(1-(4-фторфенил)этил)-2H-тетразол-5-ил)-2-метоксифенилсульфонамидо)ацетамид.
11. Соединение или его фармацевтически приемлемая соль по п.10, где соединение представляет 4-(2-((5-фторпиридин-2-ил)метил)-2Н-тетразол-5-ил)-N-(2-гидроксиэтил)бензолсульфонамид, имеющий следующую структуру:
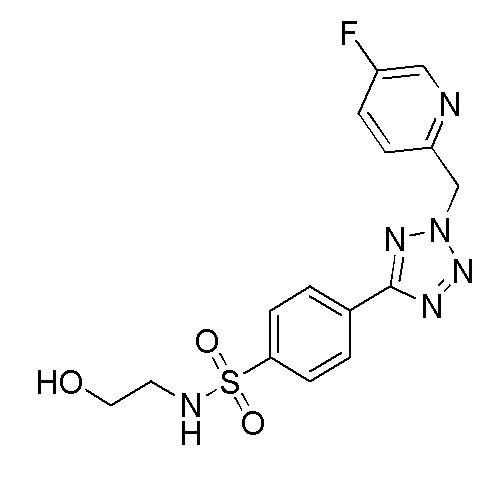 .
.
12. Соединение или его фармацевтически приемлемая соль по любому из пп.1-11 для применения в лечении туберкулеза.
13. Соединение или его фармацевтически приемлемая соль по любому из пп.1-11 для применения в лечении микобактериальной инфекции, где микобактериальная инфекция представляет собой инфекцию Mycobacterium tuberculosis.
14. Соединение или его фармацевтически приемлемая соль по любому из пп.1-11 для применения в лечении заболевания, вызванного инфекцией Mycobacterium tuberculosis.
15. Соединение или его фармацевтически приемлемая соль для применения по п.14, где заболевание представляет туберкулез.
16. Способ лечения микобактериальной инфекции, вызванной Mycobacterium tuberculosis, у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по любому из пп.1-11.
17. Способ лечения заболевания, вызванного инфекцией Mycobacterium tuberculosis, у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по любому из пп.1-11.
18. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-11 в производстве лекарственного средства для применения в лечении микобактериальной инфекции или заболевания, вызванного инфекцией Mycobacterium tuberculosis.
19. Фармацевтическая композиция для лечения микобактериальной инфекции или заболевания, вызванного Mycobacterium tuberculosis, содержащая (а) терапевтически эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп.1-11; и (b) фармацевтически приемлемый эксципиент.
| WO 2008124703 A2, 16.10.2008 | |||
| WO 2017066964 A1, 27.04.2017 | |||
| RANGAPPA S | |||
| KERI et al., Chemical Biology & Drug Design., vol | |||
| Пюпитр для работы на пишущих машинах | 1922 |
|
SU86A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Устройство анодов катодных ламп | 1923 |
|
SU410A1 |
| ПРОТИВОИНФЕКЦИОННЫЕ СОЕДИНЕНИЯ | 2011 |
|
RU2576662C2 |
| КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ГЛОМЕРУЛОПАТИИ, НОВЫЕ СОЕДИНЕНИЯ, В НЕЕ ВХОДЯЩИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ МАТРИКСНЫХ МЕТАЛЛОПРОТЕИНАЗ И КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ КОЛЛАГЕНАЗ ТИПА IV | 1998 |
|
RU2207849C2 |

