ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к низкомолекулярным ингибиторам обогащенной лейциновыми повторами киназы 2 (LRRK2). Данное изобретение также относится к способам ингибирования у млекопитающих, в том числе людей, LRRK2 путем введения низкомолекулярных ингибиторов LRRK2. Настоящее изобретение также относится к лечению болезни Паркинсона (PD) и других нейродегенеративных и/или неврологических расстройств у млекопитающих, в том числе людей, ингибиторами LRRK2. Более конкретно, данное изобретение относится к новым соединениям имидазо[4,5-с]хинолинов и имидазо[4,5-с][1,5]нафтиридинов, полезным для лечения нейродегенеративных и/или неврологических расстройств, таких как PD, болезнь Альцгеймера (AD) и другие LRRK2-ассоциированные расстройства.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
LRRK2 представляет собой белок, имеющий молекулярную массу 286 килодальтон (кДа), из семейства содержащих ROCO-домены белков со сложной мультидоменной структурой. Белковые мотивы, которые были определены для LRRK2, включают армадиллоподобный (ARM) домен, анкириноподобный (ANK) домен, домен с лейцин-обогащенными повторами (LRR), Ras (ренин-ангиотензиновая система)-подобный комплексный (ROC) домен, С-концевой ROC (COR) домен, киназный домен и С-концевой WD40 домен. ROC домен связывается с гуанозинтрифосфатом (ГТФ), a COR домен может представлять собой регулятор ГТФазной активности ROC домена. Киназный домен имеет структурную гомологию с киназами киназы митоген-активируемой протеин(МАР)киназы (MAPKKK), и показано, что он фосфорилирует ряд клеточных белков in vitro, однако эндогенный субстрат еще предстоит определить. LRRK2 обнаружена в различных участках головного мозга, а также в ряде периферических тканей, включая сердце, легкое, селезенку и почку.
По всей вероятности, LRRK2 участвует в сложных многочисленных клеточных процессах вследствие своего мультидоменного строения, при этом каждый из доменов ассоциирован с предполагаемыми белок-белковыми взаимодействиями, гуанозинтрифосфатазной (ГТФаза) активностью и киназной активностью. Например, LRRK2 ассоциирована с ингибированием NFAT (ядерный фактор активации Т-клеток) в иммунной системе и связана с миграцией везикул, пресинаптическим гомеостазом, с передачей сигнала, опосредуемой мишенью рапамицина у млекопитающих (mTOR), передачей сигнала через рецепторную тирозинкиназу МЕТ при папиллярном раке почки и раке щитовидной железы, динамикой цитоскелета, с путем митоген-активируемой протеинкиназы (MAPK), с опосредуемым фактором некроза опухоли-α (TNF-α) путем, с сигнальным путем Wnt и аутофагией. Благодаря недавно проведенному полногеномному поиску ассоциаций (GWA) высказано предположение о причастности LRRK2 к патогенезу различных заболеваний у человека, таких как PD, воспалительное заболевание кишечника (болезнь Крона), рак и лепра (Lewis Р.А. and Manzoni С. Science Signaling, 2012, 5(207), ре2).
Болезнь Паркинсона (PD) представляет собой относительно распространенное возрастное нейродегенеративное расстройство, возникающее вследствие прогрессирующей потери дофамин-продуцирующих нейронов и которое поражает до 4% населения в возрасте старше 80 лет. PD характеризуется моторными симптомами, такими как тремор в состоянии покоя, ригидность, акинезия и постуральная неустойчивость, а также немоторными симптомами, такими как нарушение когнитивной способности, сна и обоняния. Благодаря GWA поиску высказано предположение о связи LRRK2 с PD, и многие пациенты с точечными мутациями в гене LRRK2 имеют симптомы, которые неотличимы от симтомов, характерных для идиопатической PD. Свыше 20 мутаций в гене LRRK2 ассоциированы с аутосомно-доминантной формой паркинсонизма, а миссенс-мутации R1441C, R1441G, R1441H, Y1699C, G2019S, I2020T и N1437H считаются патогенными. Показано, что мутация R1441G в LRRK2 усиливает высвобождение провоспалительных цитокинов (с более высокими уровнями TNF-α, IL-1β, IL-12 и более низкими уровнями IL-10) в микроглиальных клетках из трансгенных мышей, и это, таким образом, может выражаться в непосредственном токсичном воздействии на нейроны (Gillardon, F. et al. Neuroscience, 2012, 208, 41-48). В мышиной модели нейровоспаления наблюдали индуцирование LRRK2 в микроглии, и ингибирование киназной активности LRRK2 низкомолекулярными ингибиторами LRRK2 (LRRK2-IN-1 или сунитинибом), или нокаут гена LRRK2 приводил к ослаблению секреции TNF-α и индуцированию синтазы оксида азота (iNOS) (Moehle, М. et al. J. Neurosci., 2012, 32(5), 1602-1611). Самая распространенная из мутаций в LRRK2, G2019S, имеется более чем у 85% пациентов с PD, несущих мутации в LRRK2. Эта мутация, которая находится в киназном домене LRRK2, приводит к повышению киназной активности LRRK2. В головном мозге человека самая высокая степень экспрессии LRRK2 наблюдается в тех же участках головного мозга, которые затрагиваются при PD, и LRRK2 обнаруживают в тельцах Леви, что является важнейшим признаком PD. Недавние исследования указывают на то, что сильнодействующий, селективный, проникающий в головной мозг киназный ингибитор LRRK2 можно было бы использовать для терапевтического лечения PD.
Деменция является результатом целого ряда различных патологических процессов. Самыми распространенными патологическими процессами, приводящими к деменции, являются AD (болезнь Альцгеймера), церебральная амилоидная ангиопатия (СМ) и прион-опосредуемые заболевания (см. например, Haan et al., Clin. Neurol. Neurosurg., 1990, 92(4): 305-310; Glenner et al., J. Neurol. Sci., 1989, 94: 1-28). AD представляет собой прогрессирующее нейродегенеративное расстройство, характеризующееся ослаблением памяти и когнитивной дисфункцией. AD поражает почти половину всех людей в возрасте старше 85 лет, самой быстро увеличивающейся по численности части населения Соединенных Штатов Америки. В связи с этим ожидается, что к 2050 году в Соединенных Штатах Америки число пациентов с AD возрастет от примерно 4 миллионов до примерно 14 миллионов. Мутации в LRRK2 ассоциированы с AD-подобной патологией, и это позволяет высказать предположение, что возможно имеется частичное «перекрывание» нейродегенеративных путей как в случае AD, так и в случае PD (Zimprach, A. et al. Neuron, 2004, 44, 601-607). Помимо этого, с увеличением уровня заболеваемости AD в некоторой популяции ассоциирован вариант LRRK2 с мутацией R1628P (в COR домене), что возможно является следствием усиления апоптоза и гибели клеток (Zhao, Y. et al.; Neurobiology of Aging, 2011, 32, 1990-1993).
Сообщалось об увеличении уровня заболеваемости некоторыми не связанными с кожей видами рака, такими как рак почки, рак молочной железы, рак легкого и рак предстательной железы, а также острый миелогенный лейкоз (AML), у пациентов с болезнью Паркинсона, имеющих мутацию G2019S в LRRK2 (Saunders-Pullman, R. et al.; Movement Disorders, 2010, 25(15), 2536-2541). Поскольку мутация G2019S ассоциируется с повышением киназной активности LRRK2, то ингибирование этой активности может быть полезным в лечении рака, такого как рак почки, рак молочной железы, рак легкого, рак предстательной железы и рак крови.
Воспалительное заболевание кишечника (IBD) или болезнь Крона (CD) представляет собой сложное заболевание, и считается, что оно является следствием несоответствующего иммунного ответа на микрофлору кишечного тракта. Благодаря недавно проведенному GWA поиску ген LRRK2 идентифицирован как основной ген восприимчивости к болезни Крона, в частности, это касается М2397Т полиморфизма в WD40 домене (Liu, Z. et al. Nat. Immunol., 2011, 12, 1063-1070). В недавно проведенном исследовании было обнаружено, что дефицитные по LRRK2 мыши более восприимчивы к индуцированному декстрансульфатом натрия колиту, чем их сородичи дикого типа, что указывает на возможное участие LRRK2 в патогенезе IBD (Liu Z. and Lenardo М., Cell Research, 2012, 1-3).
Уже описаны и неселективные, и селективные низкомолекулярные соединения с LRRK2-ингибируюшей активностью, такие как стауроспорин, сунитиниб, LRRK2-IN-1, CZC-25146, ТАЕ684 и соединения из WO 2011/141756, WO 2012/028629 и WO 2012/058193. Желательно разработать соединения, которые являются мощными и селективными ингибиторами LRRK2 с благоприятным фармакокинетическим профилем и способностью преодолевать гематоэнцефалический барьер. Соответственно, настоящее изобретение относится к новым соединениям имидазо[4,5-с]хинолинов и имидазо[4,5-с][1,5]нафтиридинов с LRRK2-ингибирующей активностью и к применению этих соединений в лечении заболеваний, ассоциированных с LRRK2, таких как нейродегенеративные заболевания, включая PD.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Первым воплощением первого аспекта настоящего изобретения является соединение формулы (I)
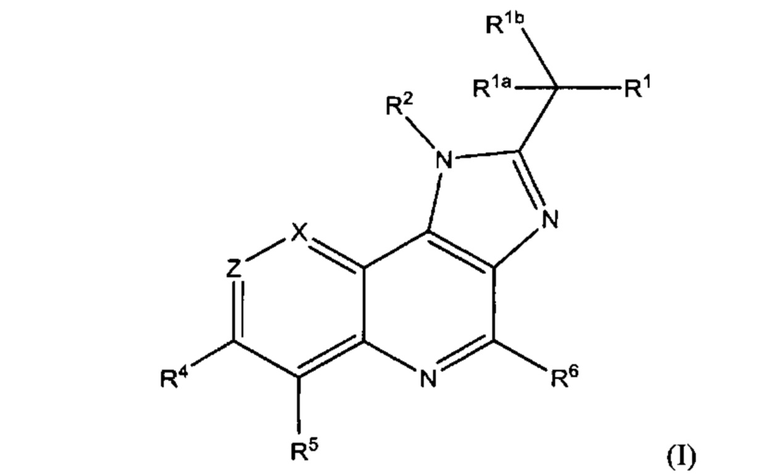
или его фармацевтически приемлемая соль; где X представляет собой CR7 или N; Z представляет собой CR3 или N; R1 выбран из группы, состоящей из водорода, циано и 5-10-членного гетероарила, который содержит 1-5 гетероатомов, независимо выбранных из N, О и S; при этом 5-10-членный гетероарил возможно замещен 1-3 группами R8; каждый из R1a и R1b независимо представляет собой водород, галоген, гидрокси или C1-С3алкил; или R1a и R1b вместе с атомом углерода, к которому они присоединены, представляют собой С3-С6циклоалкил; R2 представляет собой C1-С6алкил, С3-С7циклоалкил или 4-7-членный гетероциклоалкил, который содержит 1-3 гетероатома, независимо выбранных из NR, О и S; при этом каждый из С3-С7циклоалкила и 4-7-членного гетероциклоалкила возможно замещен 1-3 группами R9; и при этом С1-С6алкил возможно замещен 1-3 группами R10; R представляет собой водород, C1-С6алкил или отсутствует; каждый из R3, R4, R5, R6 и R7 независимо выбран из группы, состоящей из водорода, дейтеро, амино, галогена, гидрокси, циано, C1-С6алкила, С3-С6циклоалкила и C1-С6алкокси; при этом каждый из C1-С6алкила, С3-С6циклоалкила и C1-С6алкокси возможно замещен 1-3 группами галоген или C1-С3алкокси; R8 в каждом случае независимо выбран из группы, состоящей из галогена, -C(O)NH2, -C(O)NH(C1-С3алкил), -C(O)N(C1-С3алкил)2, C1-C6алкила, C1-С6алкокси и С3-С6циклоалкила; при этом каждый из C1-С6алкила, C1-С6алкокси и С3-С6циклоалкила возможно замещен 1-3 группами галоген, циано, гидрокси или C1-С3алкокси; R9 в каждом случае независимо выбран из группы, состоящей из галогена, гидрокси, C1-С6алкила, C1-С6алкокси и C1-С6алкоксиC1-С6алкила, при этом C1-С6алкил, C1-С6алкокси и C1-C6алкоксиC1-C6алкил возможно замещены одной-тремя группами галоген или циано; и R10 в каждом случае независимо выбран из группы, состоящей из галогена, C1-С6алкокси, C1-С6тиоалкокси, амино, C1-С6алкиламино и ди(С1-С6алкил)амино.
Вторым воплощением первого аспекта настоящего изобретения является соединение по первому воплощению первого аспекта или его фармацевтически приемлемая соль, где X представляет собой CR7; Z представляет собой CR3; R3 представляет собой водород, бром, хлор, фтор, метокси или циано; и каждый из R4, R5, R6 и R7 представляет собой водород или дейтеро.
Третьим воплощением первого аспекта настоящего изобретения является соединение по второму воплощению первого аспекта или его фармацевтически приемлемая соль, где R1 представляет собой 5-10-членный гетероарил, который содержит 1-4 гетероатома, независимо выбранных из N, О и S; при этом 5-10-членный гетероарил возможно замещен 1-2 группами R8; каждый из R1a и R1b представляет собой водород; и R8 в каждом случае независимо выбран из группы, состоящей из галогена, C1-С3алкила, C1-С3алкокси и С3-С6циклоалкила; при этом C1-С3алкил возможно замещен 1-3 атомами фтора, группами гидрокси или С1-С3алкокси.
Четвертым воплощением первого аспекта настоящего изобретения является соединение по третьему воплощению первого аспекта или его фармацевтически приемлемая соль, где R1 представляет собой 5-10-членный гетероарил, выбранный из группы, состоящей из оксазолила, изоксазолила, оксадиазолила, тиазолила, пиразолила, триазолила, тетразолила, пиридинила, бензоксазолила, бензоизоксазолила, бензопиразолила, бензотриазолила, имидазотиазолила и имидазотиадиазолила; каждый из которых возможно замещен группой R8; и R8 выбран из группы, состоящей из метила, трифторметила, изопропила, 2-гидроксиизопропила, метокси, метоксиметила, циклопропила и хлора.
Пятым воплощением первого аспекта настоящего изобретения является соединение по четвертому воплощению первого аспекта или его фармацевтически приемлемая соль, где R1 выбран из группы, состоящей из
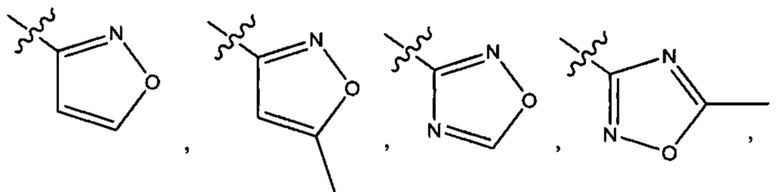
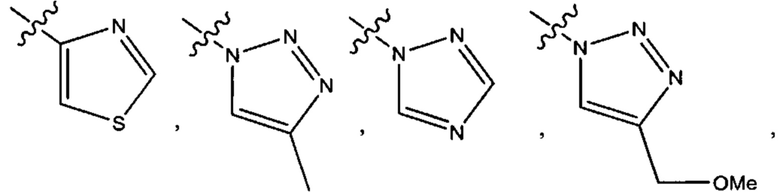
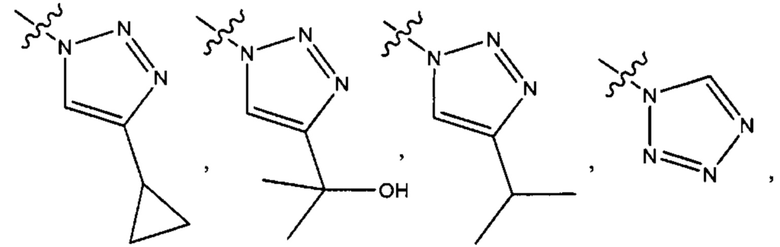
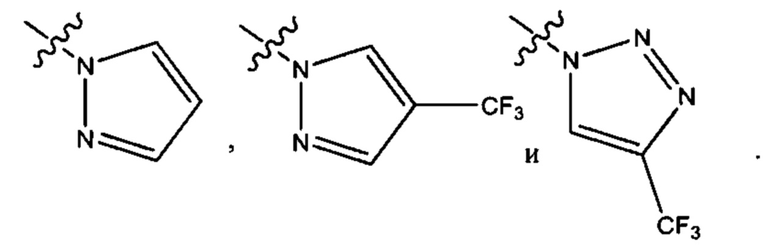
Шестым воплощением первого аспекта настоящего изобретения является соединение по четвертому воплощению первого аспекта или его фармацевтически приемлемая соль, где R1 выбран из группы, состоящей из
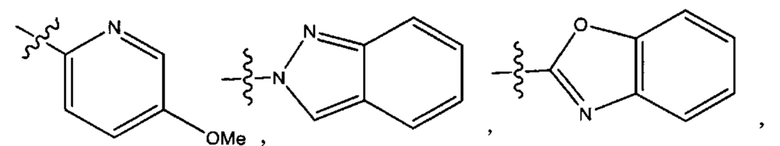
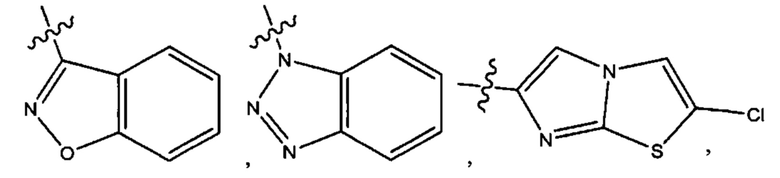
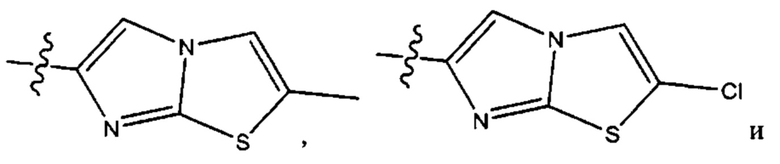
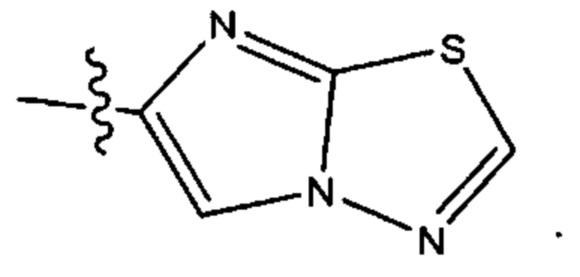
Седьмым воплощением первого аспекта настоящего изобретения является соединение по четвертому воплощению первого аспекта или его фармацевтически приемлемая соль, где R2 представляет собой тетрагидропиранил, циклопентил или циклогексил; каждый из которых возможно замещен 1-2 группами R9; и R9 в каждом случае представляет собой независимо метил, этил, цианометил, гидрокси или фтор.
Восьмым воплощением первого аспекта настоящего изобретения является соединение по седьмому воплощению первого аспекта или его фармацевтически приемлемая соль, где R2 выбран из группы, состоящей из
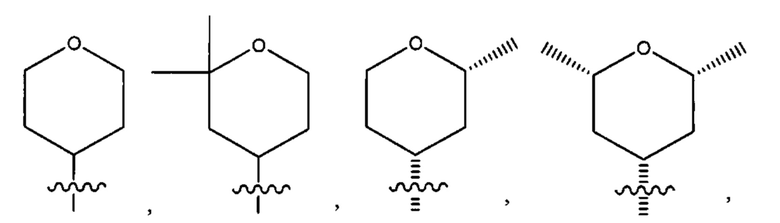
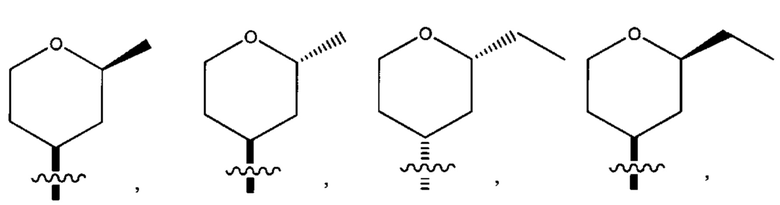
Девятым воплощением первого аспекта настоящего изобретения является соединение по восьмому воплощению первого аспекта или его фармацевтически приемлемая соль, где R2 представляет собой
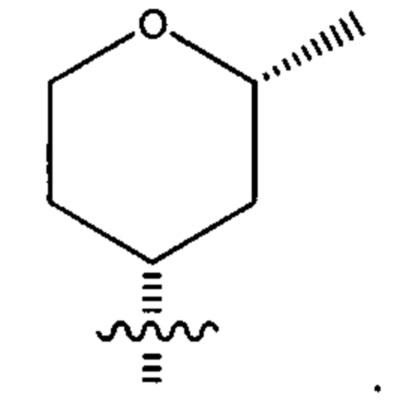
Десятым воплощением первого аспекта настоящего изобретения является соединение по седьмому воплощению первого аспекта или его фармацевтически приемлемая соль, где R2 выбран из группы, состоящей из
Одиннадцатым воплощением первого аспекта настоящего изобретения является соединение по п. 1 или его фармацевтически приемлемая соль, где X представляет собой N; Z представляет собой CR3; R1 представляет собой 5-10-членный гетероарил, выбранный из группы, состоящей из оксазолила, изоксазолила, оксадиазолила, тиазолила, пиразолила, триазолила, тетразолила, пиридинила, бензоксазолила, бензоизоксазолила, бензопиразолила, бензотриазолила, имидазотиазолила и имидазотиадиазолила; каждый из которых возможно замещен группой R8; каждый из R1a и R1b представляет собой водород; и R8 представляет собой метил, трифторметил, изопропил, 2-гидроксиизопропил, метокси, метоксиметил, циклопропил или хлор.
Двенадцатым воплощением первого аспекта настоящего изобретения является соединение по одиннадцатому воплощению первого аспекта или его фармацевтически приемлемая соль, где R2 представляет собой
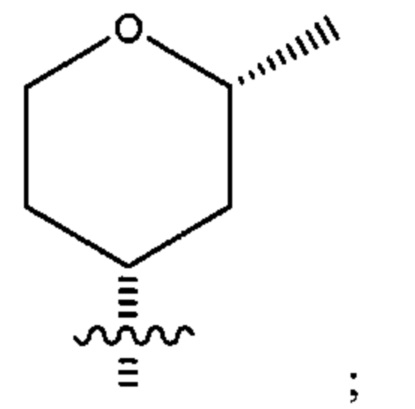
и каждый из R3, R4, R5 и R6 представляет собой водород или дейтеро.
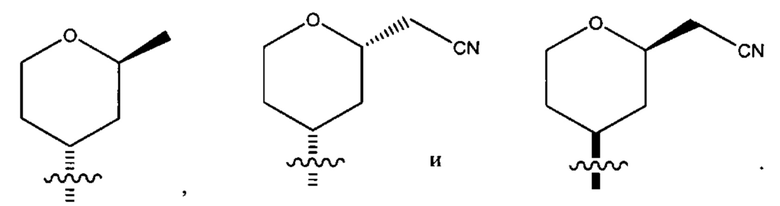
Тринадцатым воплощением первого аспекта настоящего изобретения является соединение по первому воплощению первого аспекта или его фармацевтически приемлемая соль, где X представляет собой CR7; Z представляет собой CR3; R1 представляет собой водород или циано; каждый из R1a и R1b представляет собой водород;
R2 представляет собой тетрагидропиранил или циклопентил; каждый из которых возможно замещен 1-2 группами R9; и R9 в каждом случае представляет собой независимо метил, цианометил или фтор.
Четырнадцатым воплощением первого аспекта настоящего изобретения является соединение по тринадцатому воплощению первого аспекта или его фармацевтически приемлемая соль, где R2 представляет собой
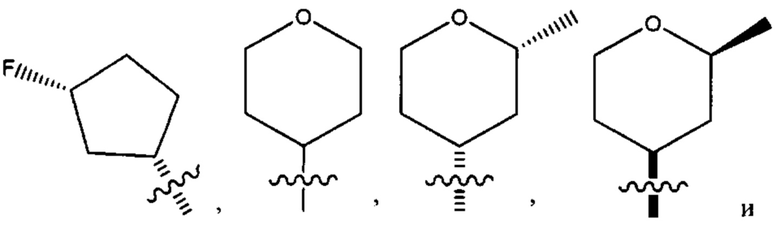
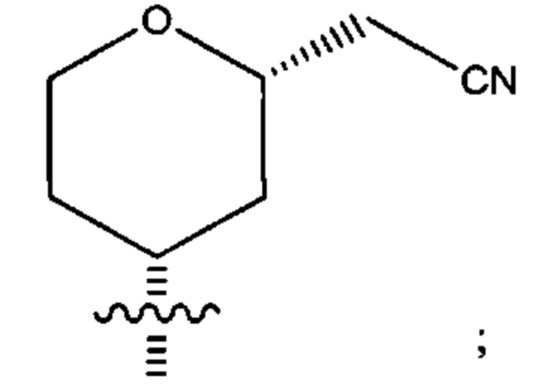
R3 представляет собой водород, бром, хлор, метокси или циано; и каждый из R4, R5, R6 и R7 представляет собой водород или дейтеро.
Пятнадцатым воплощением первого аспекта настоящего изобретения является соединение или его фармацевтически приемлемая соль, которые описаны ниже в примерах 1-92.
Шестнадцатым воплощением первого аспекта настоящего изобретения является соединение по первому воплощению первого аспекта, выбранное из группы, состоящей из
8-хлор-2-[(5-метоксипиридин-2-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
2-[(5-метил-1,2-оксазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолин-8-карбонитрила;
8-хлор-2-[(5-метил-1,2-оксазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
8-хлор-2-[(5-метил-1,2,4-оксадиазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
8-хлор-2-(имидазо[2,1-b][1,3,4]тиадиазол-6-илметил)-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
{8-хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолин-2-ил}ацетонитрила;
8-хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)(4-2Н)-1H-имидазо[4,5-с]хинолина и
8-хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-[(4-метил-1H-1,2,3-триазол-1-ил)метил]-1H-имидазо[4,5-с]хинолина;
или его фармацевтически приемлемая соль.
Семнадцатым воплощением первого аспекта настоящего изобретения является соединение по первому воплощению первого аспекта или его фармацевтически приемлемая соль, где X представляет собой CR7; Z представляет собой CR3; каждый из R1a, R1b, R4, R5, R6 и R7 представляет собой водород; и R3 представляет собой хлор или циано.
Восемнадцатым воплощением первого аспекта настоящего изобретения является соединение по семнадцатому воплощению первого аспекта или его фармацевтически приемлемая соль, где R2 представляет собой 1-метилпирролидинил или 2-метилтетрагидропиранил.
Девятнадцатым воплощением первого аспекта настоящего изобретения является соединение по восемнадцатому воплощению первого аспекта или его фармацевтически приемлемая соль, где R1 выбран из группы, состоящей из изоксазолила, пиразолила, триазолила, оксадиазолила, тиадиазолила, пиримидинила и пиразинила; каждый из которых возможно замещен группой R8; и R8 представляет собой метил или метокси.
Двадцатым воплощением первого аспекта настоящего изобретения является соединение по девятнадцатому воплощению первого аспекта или его фармацевтически приемлемая соль, где R1 выбран из группы, состоящей из метилизоксазолила, метоксипиразолила, метилтриазолила, метилоксадиазолила, метилтиадиазолила, метилпиримидинила и метилпиразинила; R2 представляет собой (2R,4R)-2-метилтетрагидро-2H-пиран-4-ил; и R3 представляет собой хлор.
Двадцать первым воплощением первого аспекта настоящего изобретения является соединение по девятнадцатому воплощению первого аспекта или его фармацевтически приемлемая соль, где R1 выбран из группы, состоящей из метилизоксазолила, метоксипиразолила, метилтриазолила, метилоксадиазолила, метилтиадиазолила, метилпиримидинила и метилпиразинила; R2 представляет собой 1-метилпирролидинил; и R3 представляет собой циано.
Двадцать вторым воплощением первого аспекта настоящего изобретения является соединение по девятнадцатому воплощению первого аспекта, выбранное из группы, состоящей из
8-хлор-2-[(5-метил-1,2-оксазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1Н-имидазо[4,5-с]хинолина;
8-хлор-2-[(5-метил-1,2,4-оксадиазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1Н-имидазо[4,5-с]хинолина;
8-хлор-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-2-[(4-метил-1Н-1,2,3-триазол-1-ил)метил]-1Н-имидазо[4,5-с]хинолина;
8-хлор-2-[(6-метилпиримидин-4-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1Н-имидазо[4,5-с]хинолина;
8-хлор-2-[(5-метилпиразин-2-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1Н-имидазо[4,5-с]хинолина;
8-хлор-2-[(4-метокси-1Н-пиразол-1-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1Н-имидазо[4,5-с]хинолина и
8-хлор-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-2-[(5-метил-1,3,4-тиадиазол-2-ил)метил]-1Н-имидазо [4,5-с]хинолина;
или его фармацевтически приемлемая соль.
Двадцать третьим воплощением первого аспекта настоящего изобретения является соединение по первому воплощению первого аспекта или его фармацевтически приемлемая соль, где, помимо определений всех переменных, которые приведены в данном описании, R1a и R1b вместе с атомом углерода, к которому они присоединены, также могут представлять собой С(О).
Первым воплощением второго аспекта настоящего изобретения является фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по любому из воплощений от первого до двадцать третьего воплощения первого аспекта или его фармацевтически приемлемой соли вместе с фармацевтически приемлемым носителем.
Первым воплощением третьего аспекта настоящего изобретения является способ лечения болезни Крона или болезни Паркинсона у пациента, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по любому из воплощений от первого до двадцать третьего воплощения первого аспекта изобретения.
Другим воплощением настоящего изобретения является соединение или его фармацевтически приемлемая соль по любому из воплощений от первого до двадцать третьего воплощения первого аспекта настоящего изобретения для применения в лечении болезни Крона или болезни Паркинсона.
Другим воплощением настоящего изобретения является способ ингибирования LRRK2 у пациента, включающий введение LRRK2-ингибирующего количества соединения или его фармацевтически приемлемой соли по любому из воплощений от первого до двадцать третьего воплощения первого аспекта.
Другим воплощением настоящего изобретения является способ лечения нейродегенеративного заболевания у пациента, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по любому из воплощений от первого до двадцать третьего воплощения первого аспекта.
Соответственно, данное изобретение также относится к способам лечения пациента (предпочтительно человека) от заболеваний, в которые вовлечена киназа LRRK2, таких как болезнь Паркинсона, путем введения терапевтически эффективного количества соединения по любому из воплощений формулы (I) или его фармацевтически приемлемой соли и фармацевтически приемлемого носителя.
Данное изобретение также относится к способам ингибирования киназной активности LRRK2 путем введения терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли и фармацевтически приемлемого носителя млекопитающему или пациенту, нуждающемуся в этом. Данное изобретение также относится к способам лечения расстройств, отвечающих на ингибирование киназной активности LRRK2, таких как неврологические расстройства (в частности, болезнь Паркинсона), определенные раковые заболевания и определенные иммунологические расстройства (такие как болезнь Крона и лепра), путем введения терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли и фармацевтически приемлемого носителя млекопитающему или пациенту, нуждающемуся в этом.
Данное изобретение также относится к способам лечения состояний или заболеваний центральной нервной системы и неврологических расстройств, в которые вовлечена киназа LRRK2, в частности болезни Паркинсона (но в том числе также и других неврологических заболеваний, которые могут включать мигрень; эпилепсию; болезнь Альцгеймера; травму головного мозга; инсульт); нарушений мозгового кровообращения (включая церебральный артериосклероз, церебральную амилоидную ангиопатию, наследственную церебральную геморрагию и обусловленную гипоксией ишемию головного мозга); когнитивных расстройств (включая амнезию, сенильную деменцию, ВИЧ-ассоциированную деменцию, болезнь Альцгеймера, болезнь Гентингтона, деменцию с тельцами Леви, сосудистую деменцию, связанную с приемом лекарственных средств деменцию, позднюю дискинезию, миоклонические судороги, дистонию, делирий, болезнь Пика, болезнь Крейцфельдта-Якоба, ВИЧ-ассоциированное заболевание, синдром Жиль де ля Туретта, эпилепсию, мышечные спазмы и расстройства, ассоциированные с мышечной спастичностью или слабостью, в том числе треморы, и умеренную когнитивную недостаточность); умственной неполноценности (включая спастичность, синдром Дауна и синдром ломкой Х-хромосомы); расстройств сна (включая гиперсомнию, расстройство циркадного ритма сна, бессонницу, парасомнию и недосыпание) и психических расстройств, таких как тревога (включая острое стрессовое расстройство, генерализованное тревожное расстройство, социальное тревожное расстройство, паническое расстройство, посттравматическое стрессовое расстройство, агорафобию и обсессивно-компульсивное расстройство); симулятивных расстройств (включая истерическое сумеречное помрачение сознания); расстройств контроля импульсивного поведения (включая компульсивное влечение к азартным играм и интермиттирующее эксплозивное расстройство); расстройств настроения (включая биполярное расстройство I типа, биполярное расстройство II типа, манию, смешанное аффективное состояние, глубокую депрессию, хроническую депрессию, сезонную депрессию, психотическую депрессию, сезонную депрессию, предменструальный синдром (PMS), предменструальное дисфорическое расстройство (PDD) и послеродовую депрессию); психомоторных расстройств; психотических расстройств (включая шизофрению, шизоаффективное расстройство, шизофреноформное и бредовое расстройство); лекарственной зависимости (включая наркотическую зависимость, алкоголизм, амфетаминовую зависимость, кокаинизм, никотиновую зависимость и абстинентный наркотический синдром); расстройств приема пищи (включая анорексию, булимию, компульсивное переедание, гиперфагию, ожирение, компульсивные расстройства приема пищи и пагофагию); расстройств, связанных с нарушением половой функции; недержания мочи; вызванных нейрональным повреждением расстройств (включая нарушение зрения, ретинопатию или макулярную дегенерацию глаза, шум в ушах, нарушение и потерю слуха и отек головного мозга) и психических расстройств у детей (включая синдром дефицита внимания, синдром дефицита внимания с гиперактивностью, кондуктивное расстройство и аутизм), у млекопитающего, предпочтительно человека, включающим введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Соединения по настоящему изобретению формулы (I) могут быть, в частности, пригодны для лечения таких заболеваний или расстройств, как деменция с тельцами Леви, лобно-височная деменция, кортикобазальная деменция, прогрессирующий супрануклеарный паралич, лепра, воспалительное заболевание кишечника, синдром раздраженного кишечника, болезнь Альцгеймера, таупатий, альфа-синуклеопатия, болезнь Паркинсона, болезнь Паркинсона с деменцией, синдром риска болезни Паркинсона, вариант болезни Альцгеймера с тельцами Леви, комбинация болезни Паркинсона и болезни Альцгеймера, множественная системная атрофия, стриатонигральная дегенерация, оливопонтоцеребеллярная атрофия, синдром Шая-Дрейджера, язвенный колит, ювенильный паркинсонизм, болезнь Стила-Ричардсона-Олыневского, болезнь LyticoBodig или комплекс паркинсонизм-деменция-ALS (амиотрофический боковой склероз) с о. Гуам, дегенерация коры больших полушарий и базальных ганглиев, прогрессирующая паллидарная атрофия, комплекс паркинсонизм-деменция, стриопаллидарный синдром, наследственный ювенильный паркинсонизм с дистоническими проявлениями, аутосомнодоминантная болезнь с тельцами Леви, болезнь Гентингтона, болезнь Вильсона, наследственная недостаточность церулоплазмина, болезнь Галлервордена-Шпатца, оливопонтоцеребеллярная и спиноцеребеллярная дегенерации, болезнь Мачадо-Джозефа, наследственный комплекс «амиотрофия-деменция-паркинсонизм», комплекс «расторможенность-деменция-паркинсонизм-амиотрофия», болезнь Герстманна-Штраусслера-Шейнкера, наследственный прогрессирующий субкортикальный глиоз, синдром Lubag (сцепленый с Х-хромосомой паркинсонизм с дистоническими проявлениями), наследственная кальцификация базальных ганглиев, митохондрииальные цитопатии с некрозом полосатого тела, цероидный липофусциноз, наследственный паркинсонизм с периферической нейропатией, паркинсонизм-пирамидальный синдром, нейроакантоцитоз и наследственный гемохроматоз.
В пересмотренной версии четвертого издания Руководства по диагностике и статистике психических расстройств (DSM-IV-TR) (2000, Американская ассоциация психиатров, Вашингтон, D.C.) приведен диагностический инструмент для идентификации многих расстройств, изложенных в данном описании. Специалисту в данной области будет известно, что имеются альтернативные номенклатуры, нозологии и системы классификации для расстройств, изложенных в данном описании, включая расстройства, которые описаны в DMS-IV-TR, и что терминология и системы классификации дорабатываются в соответствии с научным прогрессом в медицине.
Предпочтительные способы предназначены для лечения неврологического расстройства, наиболее предпочтительно болезни Паркинсона (но также и других неврологических расстройств, таких как мигрень; эпилепсия; болезнь Альцгеймера; болезнь Ниманна-Пика типа С; травма головного мозга; инсульт; нарушение мозгового кровообращения; когнитивное расстройство; расстройство сна) или психического расстройства (такого как тревога; симулятивное расстройство; расстройство контроля импульсивного поведения; расстройство настроения; психомоторное расстройство; психотическое расстройство; лекарственная зависимость; расстройство приема пищи; и психическое расстройство у детей) у млекопитающего, предпочтительно человека, включающие введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Помимо этого, соединения формулы (I) и их фармацевтически приемлемые соли также могут быть применены в способах лечения других расстройств, ассоциированных с LRRK2, таких как болезнь Крона, лепра и определенные раковые заболевания, такие как рак почки, рак молочной железы, рак легкого, рак предстательной железы, рак легкого и рак крови.
Кроме того, согласно данному изобретению предложены композиции, содержащие фармацевтически эффективное количество одного или более соединений, описанных в данной заявке, и фармацевтически приемлемый наполнитель, носитель или эксципиент.
Настоящее изобретение также относится к применению комбинации LRRK2-ингибирующего соединения формулы (I) и одного или более чем одного дополнительного фармацевтически активного агента.
Другие признаки и преимущества данного изобретения будут очевидны на основании данного описания и прилагаемой формулы изобретения, которые описывают данное изобретение.
Определения
Термин «алкил» относится к насыщенному углеводородному заместителю с линейной или разветвленной цепью (т.е. заместителю, получаемому из углеводорода путем удаления атома водорода); в одном из воплощений, состоящему из одного-шести атомов углерода (т.е. C1-С6алкилу); в другом воплощении, состоящему из одного-трех атомов углерода (т.е. C1-С3алкилу). Примеры таких заместителей включают метил, этил, пропил (в том числе н-пропил и изопропил), бутил (в том числе н-бутил, изобутил, втор-бутил и трет-бутил), пентил, изоамил, гексил и тому подобное.
Термин «алкокси» относится к насыщенному углеводородному заместителю с линейной или разветвленной цепью (т.е. заместителю, получаемому из углеводорода путем удаления атома водорода), который в свою очередь присоединен к атому кислорода; в одном из воплощений, состоящему из одного-шести атомов углерода (т.е. C1-С6алкокси); в другом воплощении, состоящему из одного-трех атомов углерода (т.е. С1-С3алкокси). Примеры таких заместителей включают метокси, этокси, пропокси (в том числе н-пропокси и изопропокси), бутокси (в том числе н-бутокси, изобутокси, втор-бутокси и трет-бутокси), пентокси и тому подобное.
Термин «циклоалкил» относится к карбоциклическому заместителю, получаемому путем удаления атома водорода из насыщенной карбоциклической молекулы и имеющему конкретное число атомов углерода. В одном из воплощений циклоалкильный заместитель имеет три-семь атомов углерода (т.е. С3-С7циклоалкил). Примеры циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. В другом воплощении циклоалкильный заместитель имеет три-шесть атомов углерода (т.е. С3-С6циклоалкил). Термин «циклоалкил» включает в себя моно-, би- и трициклические насыщенные карбоциклы, а также мостиковые и конденсированные кольцевые карбоциклы, а также спиро-конденсированные кольцевые системы.
В некоторых случаях число атомов в циклическом заместителе, содержащем один или более гетероатомов (т.е. гетероариле или гетероциклоалкиле) указывается префиксом «(x-y)-членный», где x представляет собой минимальное, а y представляет собой максимальное число атомов, образующих циклическую группировку заместителя. Термин «гетероциклоалкил» относится к заместителю, образуемому в результате удаления атома водорода из насыщенных или частично насыщенных кольцевых структур, содержащих конкретное число атомов в кольце, при этом по меньшей мере один из атомов в кольце представляет собой гетероатом (т.е. атом кислорода, азота или серы), причем остальные атомы в кольце независимо выбраны из группы, состоящей из атомов углерода, кислорода, азота и серы. Если гетероциклоалкильный заместитель в свою очередь замещен группой или заместителем, то такие группа или заместитель могут быть связаны с гетероатомом азота или могут быть связаны с атомом углерода в кольце, при необходимости. Использованный в данном описании термин «гетероциклоалкил», как он использован в данном описании, относится к моноциклической кольцевой системе, содержащей гетероатомы NR, О или S, как указано. Так, например, «четырех-семи-членный гетероциклоалкил» относится к гетероциклоалкилу, содержащему от 4 до 7 атомов, включая один или более гетероатомов, в циклической группировке гетероциклоалкила. Число гетероатомов, присутствующих в заданном гетероцикле, может быть таким, как указано. Если гетероциклоалкильная группа имеет азот-содержащую группировку NR и является насыщенной, то следует понимать, что R представляет собой водород или C1-С6алкил. Если гетероциклоалкильная группа имеет азот-содержащую группировку NR и эта группировка NR присоединена к соседнему атому в кольце двойной связью, то следует понимать, что R отсутствует.
Примеры состоящих из одного кольца гетероциклоалкилов включают тетрагидропиранил, азетидинил, оксетанил, тиетанил, дигидрофуранил, тетрагидрофуранил, дигидротиофенил, тетрагидротиофенил, пирролинил, пирролидинил, имидазолинил, имидазолидинил, пиразолинил, пиразолидинил, тиазолинил, изотиазолинил, тиазолидинил, изотиазолидинил, дигидропиранил, пиперидинил, морфолинил, пиперазинил, азепинил, оксепинил, тиепинил и диазепинил.
Термин «водород» относится к заместителю, представляющему собой атом водорода, и может быть изображен как -Н. Термин «дейтеро» относится к заместителю, представляющему собой атом дейтерия, и может быть изображен как -D.
Термин «гидрокси» или «гидроксил» относится к группе -ОН. Соединения, несущие атом углерода, к которому присоединен один или более чем один заместитель гидрокси, включают, например, спирты, енолы и фенол.
Термин «галогено» или «галоген» относится к атому фтора (который может быть изображен как -F), хлора (который может быть изображен как -Cl), брома (который может быть изображен как -Br) или йода (который может быть изображен как -I).
Термин «гетероарил» относится к ароматическим кольцевым структурам, содержащим конкретное число атомов в кольце, в которых по меньшей мере один из атомов в кольце представляет собой гетероатом (т.е. атом кислорода, азота или серы), при этом остальные атомы в кольце независимо выбраны из группы, состоящей из атомов углерода, кислорода, азота и серы. Пяти-шести-членный гетероарил представляет собой ароматическую кольцевую систему, которая имеет пять или шесть атомов в кольце, при этом по меньшей мере одним из атомов в кольце является N, О или S. Аналогичным образом пяти-десяти-членный гетероарил представляет собой ароматическую кольцевую систему, которая имеет от пяти до десяти атомов в кольце, при этом по меньшей мере один из атомов в кольце представляет собой N, О или S. Гетероарил может представлять собой одиночное кольцо или 2 конденсированных друг с другом кольца. Примеры гетероарильных заместителей включают 6-членные кольцевые заместители, такие как пиридил, пиразил, пиримидинил и пиридазинил; 5-членные кольцевые заместители, такие как триазолил, имидазолил, фуранил, тиофенил, пиразолил, оксазолил, изоксазолил, тиазолил, 1,2,3-, 1,2,4-, 1,2,5- или 1,3,4-оксадиазолил и изотиазолил; 6/5-членные конденсированные кольцевые заместители, такие как бензотиофуранил, изобензотиофуранил, бензизоксазолил, бензоксазолил, пуринил и антранилил; и 6/6-членные конденсированные кольца, такие как хинолинил, изохинолинил, циннолинил, хиназолинил и 1,4-бензоксазинил. В случае группы, имеющей гетероарильный заместитель, атом в кольце гетероарильного заместителя, который участвует в образовании связи с этой группой, может представлять собой по меньшей мере один гетероатом или может представлять собой атом углерода в кольце, причем этот атом углерода в кольце может находиться в том же самом кольце, что и по меньшей мере один гетероатом, или при этом атом углерода в кольце может находиться в другом кольце, отличном от того кольца, в котором находится по меньшей мере один гетероатом. Аналогичным образом, если гетероарильный заместитель, в свою очередь, замещен группой или заместителем, то такие группа или заместитель могут участвовать в образовании связи по меньшей мере с одним гетероатомом или могут участвовать в образовании связи с атомом углерода в кольце, при этом атом углерода в кольце может находиться в том же самом кольце, что и по меньшей мере один гетероатом, или при этом атом углерода в кольце может находиться в другом кольце, отличном от того кольца, в котором находится по меньшей мере один гетероатом. Термин «гетероарил» также включает в себя пиридил-N-оксиды и группы, содержащие N-оксид пиридинового кольца.
Примеры состоящих из 2-х конденсированных колец гетероарилов включают, индолизинил, пиранопирролил, 4H-хинолизинил, пуринил, нафтиридинил, пиридопиридинил (включая пиридо[3,4-b]-пиридинил, пиридо[3,2-b]-пиридинил или пиридо[4,3-b]-пиридинил) и птеридинил, индолил, изоиндолил, индоленинил, изоиндазолил, бензазинил, фталазинил, хиноксалинил, хиназолинил, бензодиазинил, бензопиранил, бензотиопиранил, бензоксазолил, индоксазинил, антранилил, бензодиоксолил, бензодиоксанил, бензоксадиазолил, бензофуранил, изобензофуранил, бензотиенил, изобензотиенил, бензотиазолил, бензотиадиазолил, бензимидазолил, бензотриазолил, бензоксазинил, бензизоксазинил, пирролопиридинил, пиразолопиридинил и имидазотиазолил.
Другие примеры конденсированных кольцевых гетероарилов включают бензо-конденсированные гетероарилы, такие как индолил, изоиндолил, индоленинил, изоиндазолил, бензазинил (включая хинолинил или изохинолинил), фталазинил, хиноксалинил, хиназолинил, бензодиазинил (включая циннолинил или хиназолинил).
Упомянутые группы, происходящие из перечисленных выше групп, могут быть C-присоединенными или N-присоединенными, если такое возможно. Например, группа, происходящая из пиррола, может представлять собой пиррол-1-ил (N-присоединенный) или пиррол-3-ил (C-присоединенный). Кроме того, группа, происходящая из имидазола, может представлять собой имидазол-1-ил (N-присоединенный) или имидазол-2-ил (C-присоединенный).
Если написано, что заместители «независимо выбраны» из группы, то каждый вариант заместителя выбран независимо от другого. Вследствие этого каждый заместитель может быть идентичен другому(им) заместителю(ям) или отличаться от него(них).
Использованные в данной заявке термины «формула I» », «Формула I», «формула (I)» или «Формула (I)» могут рассматриваться как «соединение(ия) по изобретению». Кроме того, такие термины сформулированы с целью включения всех форм соединения формулы I, в том числе гидратов, сольватов, изомеров, кристаллических и некристаллических форм, изоморфов, полиморфов и их метаболитов. Например, соединения по изобретению или их фармацевтически приемлемые соли могут существовать в несольватированной и в сольватированной формах. Когда растворитель или вода связаны прочно, комплекс будет иметь хорошо определяемую стехиометрию независимо от влажности. Однако, когда растворитель или вода связаны слабо, как в канальных сольватах и гигроскопичных соединениях, содержание воды/растворителя будет зависеть от влажности или условий сушки. В таких случаях нормой будет отсутствие стехиометрии.
Соединения по изобретению могут существовать в виде клатратов или других комплексов. В объем данного изобретения включены такие комплексы, как клатраты, комплексы включения лекарственное средство-носитель, где лекарственное средство и носитель присутствуют в стехиометрических или нестехиометрических количествах. Также включены комплексы на основе соединений по изобретению, содержащие два или более органических и/или неорганических компонентов, которые могут находиться в стехиометрических или нестехиометрических количествах. Полученные комплексы могут быть ионизированными, частично ионизированными или неионизированными. В качестве обзора по таким комплексам см. Haleblian в J. Pharm. Sci., 64 (8), 1269-1288 (август 1975 г.).
Соединения по изобретению могут иметь асимметрические атомы углерода. Углерод-углеродные связи соединений по изобретению могут быть изображены в данном описании с использованием сплошной линии  сплошного клина
сплошного клина  или штрихованного клина
или штрихованного клина 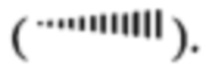 Применение сплошной линии для изображения связей с асимметрическими атомами углерода означает указание на включение всех стереоизомеров (например, конкретных энантиомеров, рацемических смесей и т.д.) относительно этого атома углерода. Использование либо сплошного, либо штрихованного клина для изображения связей с асимметрическими атомами углерода означает указание на присутствие только показанного стереоизомера. Возможно, что соединения формулы (I) могут содержать более одного асимметрического атома углерода. Использование в этих соединениях сплошной линии для изображения связей с асимметрическими атомами углерода означает указание на то, что должны быть включены все возможные стереоизомеры. Например, если не указано иное, то подразумевается, что соединения формулы (I) могут существовать в виде энантиомеров и диастереомеров или в виде рацематов и их смесей. Использование сплошной линии для изображения связей с одним или более асимметрическими атомами углерода в соединении формулы (I) и использование сплошного или штрихованного клина для изображения связей с другими асимметричными атомами углерода в том же соединении означает указание на присутствие смеси диастереомеров.
Применение сплошной линии для изображения связей с асимметрическими атомами углерода означает указание на включение всех стереоизомеров (например, конкретных энантиомеров, рацемических смесей и т.д.) относительно этого атома углерода. Использование либо сплошного, либо штрихованного клина для изображения связей с асимметрическими атомами углерода означает указание на присутствие только показанного стереоизомера. Возможно, что соединения формулы (I) могут содержать более одного асимметрического атома углерода. Использование в этих соединениях сплошной линии для изображения связей с асимметрическими атомами углерода означает указание на то, что должны быть включены все возможные стереоизомеры. Например, если не указано иное, то подразумевается, что соединения формулы (I) могут существовать в виде энантиомеров и диастереомеров или в виде рацематов и их смесей. Использование сплошной линии для изображения связей с одним или более асимметрическими атомами углерода в соединении формулы (I) и использование сплошного или штрихованного клина для изображения связей с другими асимметричными атомами углерода в том же соединении означает указание на присутствие смеси диастереомеров.
Стереоизомеры соединений формулы (I) включают цис- и транс-изомеры, оптические изомеры, такие как R- и S-энантиомеры, диастереомеры, геометрические изомеры, поворотные изомеры, конформационные изомеры и таутомеры соединений по изобретению, включая соединения, демонстрирующие более одного типа изомерии, и их смеси (такие как рацематы и диастереомерные пары). Также включены соли присоединения кислоты и соли присоединения основания, в которых противоион является оптически активным, например, D-лактат или L-лизин, или рацемическим, например, DL-тартрат или DL-аргинин.
Когда происходит кристаллизация какого-либо рацемата, возможно образование кристаллов двух разных типов. Первый тип относится к рацемическому соединению (истинному рацемату), упомянутому выше, при этом образуется одна гомогенная форма кристалла, содержащая оба энантиомера в эквимолярных количествах. Второй тип представляет собой рацемическую смесь или конгломерат, при этом образуются две формы кристалла в эквимолярных количествах, каждая из которых содержит единственный энантиомер.
Настоящее изобретение содержит таутомерные формы соединений по изобретению. Если взаимопревращение структурных изомеров протекает через низкий энергетический барьер, то может иметь место таутомерная изомерия («таутомерия»). Она может принимать форму протонной таутомерии в соединениях по изобретению, содержащих, например, группу имино, кето или оксима, или так называемой валентной таутомерии в соединениях, которые содержат ароматическую группировку. Из этого следует, что одно соединение может демонстрировать изомерию более чем одного типа. Различные соотношения таутомеров в твердой и жидкой форме зависят от наличия различных заместителей в молекуле, а также конкретного метода кристаллизации, использованного для выделения соединения.
Соединения по данному изобретению могут быть использованы в форме солей, происходящих из неорганических или органических кислот. В зависимости от конкретного соединения соль соединения может иметь преимущества благодаря наличию у этой соли одного или более физических свойств, таких как повышенная фармацевтическая стабильность при изменении температур и влажности или желаемая растворимость в воде или масле. В некоторых случаях соль соединения также может быть использована в качестве вспомогательного средства при выделении, очистке и/или отделении соединения.
Когда соль предназначена для введения пациенту (в отличие, например, от ее использования в условиях in vitro), то такая соль предпочтительно является фармацевтически приемлемой. Термин «фармацевтически приемлемая соль» относится к соли, получаемой путем объединения соединения формулы (I) с кислотой, анион которой обычно принято считать подходящим для потребления человеком, или с основанием, катион которого обычно принято считать подходящим для потребления человеком. Фармацевтически приемлемые соли особенно полезны в качестве продуктов, получаемых способами по настоящему изобретению, благодаря их более высокой растворимости в воде по сравнению с исходным соединением. Что касается использования в медицине, то соли соединений по данному изобретению являются нетоксичными «фармацевтически приемлемыми солями». Соли, охватываемые термином «фармацевтически приемлемые соли», относятся к нетоксичным солям соединений по данному изобретению, которые обычно получают путем приведения во взаимодействие свободного основания с подходящей органической или неорганической кислотой.
Подходящие фармацевтически приемлемые соли присоединения кислоты соединений по настоящему изобретению, когда возможно, включают соли, происходящие из неорганических кислот, таких как соляная, бромистоводородная, фтористоводородная, борная, фторборная, фосфорная, метафосфорная, азотная, угольная, сульфоновая и серная кислоты, и органических кислот, таких как уксусная, бензолсульфоновая, бензойная, лимонная, этансульфоновая, фумаровая, глюконовая, гликолевая, изотионовая, молочная, лактобионовая, малеиновая, яблочная, метансульфоновая, трифторметансульфоновая, янтарная, толуолсульфоновая, винная и трифторуксусная кислоты. Подходящие органические кислоты обычно включают кислоты классов алифатических, циклоалифатических, ароматических, аралифатических, гетероциклических, карбоновых и сульфоновых органических кислот.
Конкретные примеры подходящих органических кислот включают ацетат, трифторацетат, формиат, пропионат, сукцинат, гликолят, глюконат, диглюконат, лактат, малат, винную кислоту, цитрат, аскорбат, глюкуронат, малеат, фумарат, пируват, аспартат, глутамат, бензоат, антраниловую кислоту, стеарат, салицилат, n-гидроксибензоат, фенилацетат, манделат, эмбонат (памоат), метансульфонат, этансульфонат, бензолсульфонат, пантотенат, толуолсульфонат, 2-гидроксиэтансульфонат, суфанилат, циклогексиламиносульфонат, β-гидроксибутират, галакторат, галактуронат, адипат, альгинат, бутират, камфорат, камфорсульфонат, циклопентанпропионат, додецилсульфат, гликогептаноат, глицерофосфат, гептаноат, гексаноат, никотинат, 2-нафталинсульфонат, оксалат, пальмоат, пектинат, 3-фенилпропионат, пикрат, пивалат, тиоцианат и ундеканоат.
Кроме того, если соединения по изобретению несут кислотную группировку, то их подходящие фармацевтически приемлемые соли могут включать соли щелочных металлов, т.е. соли натрия или калия; соли щелочноземельных металлов, например, соли кальция или магния; и соли, образованные с участием подходящих органических лигандов, например, соли четвертичного аммония. В другом воплощении основные соли получают из оснований, которые образуют нетоксичные соли, в том числе соли алюминия, аргинина, бензатина, холина, диэтиламина, диоламина, глицина, лизина, меглумина, оламина, трометамина и цинка.
Органические соли могут быть получены на основе солей вторичных, третичных или четвертичных аминов, таких как трометамин, диэтиламин, N,N'-бензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, этилендиамин, меглумин (N-метилглюкамин) и прокаин. Основные азот-содержащие группы могут быть кватернизированы с использованием таких агентов, как (низший (С1-С6)алкил)-галогениды (например, метил-, этил-, пропил- и бутил-хлориды, -бромиды и -иодиды), диалкилсульфаты (т.е. диметил-, диэтил-, дибутил- и диамил-сульфаты), длинноцепочечные галогениды (т.е. децил-, лаурил-, миристил- и стеарил-хлориды, -бромиды и -иодиды), арилалкилгалогениды (т.е. бензил- и фенетил-бромиды) и другие.
В одном из воплощений также могут быть образованы гемисоли кислот и оснований, например гемисульфатные и гемикальциевые соли.
Кроме того, в объем настоящего изобретения включены так называемые «пролекарства» соединения по изобретению. Так, некоторые производные соединения по изобретению, которые сами по себе могут обладать незначительной или вообще не обладать фармакологической активностью, при введении внутрь организма или нанесении на тело могут превращаться в соединение по изобретению, обладающее желаемой активностью, например, в результате гидролитического расщепления. Такие производные называются «пролекарствами». Дополнительную информацию по применению пролекарств можно найти в "Pro-drugs as Novel Delivery Systems", Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) и "Bioreversible Carriers in Drug Design", Pergamon Press, 1987 (ed. E.B. Roche, American Pharmaceutical Association). Пролекарства по изобретению могут быть получены, например, путем замены соответствующих функциональных групп, присутствующих в соединениях любой из формул (I), определенными группировками, известными специалистам в данной области техники в качестве «прогруппировок», как описано, например, в "Design of Prodrugs" под ред. Н. Bundgaard (Elsevier, 1985).
Настоящее изобретение также включает меченные изотопами соединения, которые идентичны соединениям, описанным формулой I, если не считать, что один или более атомов заменены атомом, имеющим атомную массу или массовое число, отличную(ое) от атомной массы или массового числа, обычно встречающихся в природе. Примеры изотопов, которые могут быть инкорпорированы в соединения по настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как 2Н, 3Н, 13С, 11С, 14С, 15N, 18O, 17O, 32Р, 35S, 18F и 36Cl, соответственно. Соединения по настоящему изобретению, их пролекарства и фармацевтически приемлемые соли указанных соединений или указанных пролекарств, которые содержат вышеупомянутые изотопы и/или другие изотопы других атомов, включены в объем данного изобретения. Некоторые меченные изотопом соединения по настоящему изобретению, например те, в которые инкорпорированы такие радиоактивные изотопы, как 3Н и 14С, полезны в анализах распределения лекарственного средства и/или субстрата в тканях. Изотопы тритий, т.е. 3Н, и углерода-14, т.е. 14С, особенно предпочтительны ввиду простоты их получения и детектирования. Кроме того, замена на более тяжелые изотопы, такие как дейтерий, т.е. 2Н, может давать некоторые терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличенный in vivo период полувыведения или снижение дозировки, и поэтому может быть предпочтительной в некоторых обстоятельствах. В общем случае, меченные изотопом соединения формулы (I) по данному изобретению и их пролекарства могут быть получены путем осуществления методик, описанных ниже на схемах и/или в разделах Примеры и Подготовительные примеры, с заменой не меченного изотопом реагента легко доступным меченным изотопом реагентом.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Обычно соединение по изобретению вводят в количестве, эффективном для лечения состояния, которое описано в данной заявке. Соединения по изобретению вводят любым подходящим путем в форме фармацевтической композиции, адаптированной для такого пути, и в дозе, эффективной для предполагаемого лечения. Терапевтически эффективные дозы соединений, необходимых для лечения прогрессирования медицинского расстройства, легко определяются специалистом обычной квалификации в данной области техники с использованием доклинических и клинических подходов, известных в области медицины.
Термин «лечение», как он использован в данном описании и если не указано иное, означает реверсирование, облегчение, торможение развития или предупреждение расстройства или состояния, к которому применяется такой термин, или одного или более симптомов такого расстройства или состояния. Термин «лечение», как он использован в данном описании и если не указано иное, также относится к акту лечения как «лечения», которое определено непосредственно выше. Термин «лечение» также включает в себя адъювантное и неоадъювантное лечение субъекта.
Соединения по изобретению можно вводить перорально. Пероральное введение может включать проглатывание, при котором соединение поступает в желудочно-кишечный тракт, или можно применить трансбуккальное или сублингвальное введение, при котором соединение поступает в кровоток непосредственно из полости рта.
В другом воплощении соединения по изобретению также могут быть введены непосредственно в кровоток, в мышцу или во внутренний орган. Подходящие средства для парентерального введения включают внутривенные, интраартериальные, внутрибрюшинные, интратекальные, интравентрикулярные, интрауретральные, интрастернальные, интракраниальные, внутримышечные и подкожные средства. Подходящие устройства для парентерального введения включают игольные (в том числе микроигольные) инжекторы, безыгольные инжекторы и средства для осуществления инфузии.
В другом воплощении соединения по изобретению также могут быть введены местно в кожу или слизистую оболочку, т.е. дермально или трансдермально. В другом воплощении соединения по изобретению также могут быть введены интраназально или посредством ингаляции. В другом воплощении соединения по изобретению также могут быть введены ректально или вагинально. В другом воплощении соединения по изобретению также могут быть введены непосредственно в глаз или ухо.
Режим введения соединений и/или композиций, содержащих данные соединения, основывается на ряде факторов, включая тип, возраст, массу, пол и состояние здоровья пациента; тяжесть состояния; путь введения и активность конкретного используемого соединения. Такой режим введения может варьировать в широких пределах. Для лечения указанных выше состояний полезны уровни дозировок порядка от примерно 0,01 мг до примерно 100 мг на килограмм массы тела в сутки. В одном из воплощений общая суточная доза соединения по изобретению (вводимого в однократной или разделенных дозах) обычно составляет от примерно 0,01 до примерно 100 мг/кг. В другом воплощении общая суточная доза соединения по изобретению составляет от примерно 0,1 до примерно 50 мг/кг, а в другом воплощении от примерно 0,5 до примерно 30 мг/кг (т.е. один мг соединения по изобретению на один кг массы тела). В одном из воплощений доза составляет от 0,01 до 10 мг/кг/сутки. В другом воплощении доза составляет от 0,1 до 1,0 мг/кг/сутки. Композиции в дозированной форме могут содержать такие количества или их дольные единицы для составления суточной дозы. Во многих случаях введение соединения будет повторено несколько раз в сутки (обычно не более 4 раз). Для увеличения общей суточной дозы, при желании, обычно можно использовать многократные введения доз в сутки.
В случае перорального введения композиции могут быть представлены в форме таблеток, содержащих от примерно 0,01 мг до примерно 500 мг активного ингредиента или, в другом воплощении, от примерно 1 мг до примерно 100 мг активного ингредиента. В случае внутривенного введения дозы могут находиться в диапазоне от примерно 0,1 до примерно 10 мг/кг/минута в процессе осуществляемой с постоянной скоростью инфузии.
Подходящие субъекты по настоящему изобретению включают млекопитающих. Млекопитающие по настоящему изобретению включают, но не ограничиваются этим, собак, кошек, крупный рогатый скот, коз, лошадей, овец, свиней, грызунов, зайцеобразных, приматов и тому подобных и охватывают млекопитающих в утробе. В одном из воплощений подходящими субъектами являются люди. Люди могут быть любого пола и любого возраста.
В другом воплощении изобретение включает применение одного или более соединений по изобретению для приготовления лекарственного средства для лечения состояний, перечисленных в данном описании.
Для лечения упомянутых выше состояний соединение по изобретению можно вводить в виде соединения в чистом виде. Альтернативно, для применений в медицине подходят фармацевтически приемлемые соли вследствие их более высокой растворимости в воде по сравнению с исходным соединением.
В другом воплощении настоящее изобретение включает фармацевтические композиции. Такие фармацевтические композиции содержат соединение по изобретению, представленное вместе с фармацевтически приемлемым носителем. Носитель может представлять собой твердое вещество, жидкость или и то и другое, и вместе с соединением может быть использован для приготовления композиции с единичной дозой, например, таблетки, которая может содержать активные соединения в количестве от 0,05% до 95% по массе. Соединение по изобретению может быть соединено с подходящими полимерами, используемыми в качестве носителей для направленной доставки лекарственного средства. Также могут присутствовать другие фармакологически активные вещества.
Соединения по настоящему изобретению можно вводить любым подходящим путем, предпочтительно в форме фармацевтической композиции, адаптированной к такому пути, и в дозе, эффективной для предполагаемого лечения. Активные соединения и композиции, например, можно вводить перорально, ректально, парентерально или местно.
Твердая лекарственная форма для перорального введения может быть представлена, например, в дискретных единицах, таких как твердые или мягкие капсулы, пилюли, облатки, пастилки или таблетки, каждая из которых содержит предварительно заданное количество по меньшей мере одного соединения по настоящему изобретению. В другом воплощении пероральное введение может быть осуществлено с использованием формы порошка или гранулы. В другом воплощении пероральная лекарственная форма является сублингвальной, такой как, например, пастилка. В таких твердых лекарственных формах соединения по настоящему изобретению, как правило, скомбинированы с одним или более адъювантами. Такие капсулы или таблетки могут содержать композицию с контролируемым высвобождением. В случае капсул, таблеток и пилюль лекарственные формы также могут содержать буферные агенты или могут быть приготовлены с энтеросолюбильными покрытиями.
В другом воплощении пероральное введение может быть осуществлено с использованием жидкой лекарственной формы. Жидкие лекарственные формы для перорального введения включают, например, фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, содержащие инертные разбавители, обычно используемые в данной области техники (например, воду). Такие композиции также могут содержать адъюванты, такие как увлажняющие, эмульгирующие, суспендирующие, ароматизирующие агенты (например, подсластитель) и/или отдушки.
В другом воплощении настоящее изобретение включает парентеральную лекарственную форму. «Парентеральное введение» включает, например, подкожные инъекции, внутривенные инъекции, внутрибрюшинные инъекции, внутримышечные инъекции, интрастернальные инъекции и инфузию. Препараты для инъекций (например, стерильные инъекционные водные или масляные суспензии) могут быть приготовлены согласно известным в данной области техники методам с использованием подходящих диспергирующих, увлажняющих и/или суспендирующих агентов.
В другом воплощении настоящее изобретение включает лекарственную форму для местного применения. «Местное введение» включает, например, трансдермальное введение, в частности, посредством трансдермальных пластырей или устройств для ионофореза, интраокулярное введение или интраназальное либо ингаляционное введение. Композиции для местного введения также включают, например, гели, спреи, мази и кремы для местного применения. Композиция для местного применения может включать соединение, которое усиливает всасывание или проникновение активного ингредиента через кожу или другие пораженные участки. Если соединения по данному изобретению вводят с использованием устройства для трансдермального введения, то такое введение будет осуществлено с использованием пластыря либо резервуарного и мембранного (из пористого материала) типа, либо твердоматричного варианта. Типичные для этой цели композиции включают гели, гидрогели, лосьоны, растворы, кремы, мази, присыпки, повязки, пены, пленки, накожные пластыри, облатки, имплантаты, губки, волокна, перевязочные материалы и микроэмульсии. Также можно использовать липосомы. Типичные носители включают спирт, воду, минеральное масло, жидкий вазелин, белый вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. Могут быть включены усилители проницаемости; см., например, Finnin and Morgan, J. Pharm. Sci., 88 (10), 955-958 (October 1999).
Композиции, подходящие для местного введения в глаз, включают, например, глазные капли, при этом соединение по данному изобретению растворено или суспендировано в подходящем носителе. Типичная композиция, подходящая для введения в глаз или ухо, может быть представлена в форме капель микронизированной суспензии или раствора в изотоническом, с подведенным значением pH, стерильном физиологическом растворе. Другие композиции, подходящие для введения в глаз и ухо, включают мази, биоразлагаемые (например, рассасывающиеся гелевые губки, коллаген) и не являющиеся биоразлагаемыми (например, силиконовые) имплантаты, облатки, линзы и дисперсные или везикулярные системы, такие как ниосомы или липосомы. Такой полимер, как поперечно-сшитая полиакриловая кислота, поливиниловый спирт, гиалуроновая кислота, целлюлозный полимер, например гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза или метилцеллюлоза, или гетерополисахаридный полимер, например геллановая камедь, могут быть инкорпорированы вместе с консервантом, таким как бензалкония хлорид. Такие композиции также можно доставлять ионофорезом.
В случае интраназального введения или введения посредством ингаляции активные соединения по изобретению подходящим образом доставляют в форме раствора или суспензии из контейнера с распылительной помпой, который сдавливается или приводится в действие пациентом, или в (лекарственной) форме с аэрозольным распылением из находящегося под давлением контейнера или небулайзера, с использованием подходящего пропеллента. Композиции, подходящие для интраназального введения, обычно вводят в форме сухого порошка (либо композиции самой по себе, либо в виде смеси, например в виде сухой смеси с лактозой, либо в виде многокомпонентной частицы, например, в смеси с фосфолипидами, такими как фосфатидилхолин) из ингалятора сухого порошка, или в виде аэрозольного спрея из находящегося под давлением контейнера, насоса, распылителя, атомайзера (предпочтительно атомайзера, в котором используется электрогидродинамический принцип работы для создания тонкодисперсного тумана) или небулайзера, с использованием или без использования подходящего пропеллента, такого как 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3-гептафторпропан. Порошок для интраназального применения может содержать биоадгезивный агент, например, хитозан или циклодекстрин.
В другом воплощении настоящее изобретение включает ректальную лекарственную форму. Такая ректальная лекарственная форма может иметь форму, например, суппозитория. Традиционной основой для суппозиториев является масло какао, но при необходимости можно использовать различные альтернативные вещества.
Также можно использовать другие вещества-носители и способы введения, известные в области фармацевтики. Фармацевтические композиции по изобретению могут быть приготовлены любым из хорошо известных методов фармацевтики, таких как эффективные технологии приготовления композиций и методики введения. Приведенные выше соображения относительно эффективных технологий приготовления композиций и методик введения хорошо известны в данной области техники и описаны в стандартных руководствах. Технология приготовления лекарственных средств обсуждается, например, в Hoover, John Е., Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pennsylvania, 1975; Liberman et al., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980; и Kibbe et al., Eds., Handbook of Pharmaceutical Excipients (3rd Ed.), American Pharmaceutical Association, Washington, 1999.
Соединения по настоящему изобретению можно использовать, по отдельности или в комбинации с другими терапевтическими агентами, для лечения различных заболеваний или болезненных состояний. Соединение(ия) по настоящему изобретению и другой(ие) терапевтический(ие) агент(ы) можно вводить одновременно (либо в одной и той же лекарственной форме, либо в отдельных лекарственных формах) или последовательно.
Два или более соединений можно вводить совместно, одновременно или последовательно. Кроме того, одновременное введение может быть осуществлено путем смешивания соединений перед введением или посредством введения соединений в один и тот же момент времени, но в разные анатомические места либо с использованием разных путей введения.
Фразы «осуществляемое совместно введение», «совместное введение», «одновременное введение» и «вводимые одновременно» означают, что данные соединения вводят в комбинации.
Настоящее изобретение включает применение комбинации LRRK2-ингибирующего соединения, которое представлено формулой (I), и одного или более чем одного дополнительного фармацевтически активного агента. Если вводят комбинацию активных агентов, то их можно вводить последовательно или совместно в отдельных лекарственных формах или объединенных в разовой лекарственной форме. Соответственно, настоящее изобретение также включает фармацевтические композиции, содержащие некоторое количество: (а) первого агента, содержащего соединение формулы (I) или фармацевтически приемлемую соль соединения; (б) второго фармацевтически активного агента; и (в) фармацевтически приемлемого носителя, наполнителя или разбавителя.
Для применения вместе с соединениями формулы (I) могут быть выбраны различные фармацевтически активные агенты в зависимости от подлежащего лечению заболевания, расстройства или состояния. Например, фармацевтическая композиция для применения в лечении болезни Паркинсона может содержать соединение формулы (I) или его фармацевтически приемлемую соль вместе с другим агентом, таким как дофамин (леводопа, либо сама по себе, либо вместе с ингибитором DOPA(L-3,4-дигидроксифенилаланин)-декарбоксилазы), ингибитор моноаминоксидазы (МАО), ингибитор катехол-О-метилтрансферазы (СОМТ) или антихолинергический агент либо любая их комбинация. В частности, предпочтительные агенты для объединения с соединениями формулы (I) для применения в лечении болезни Паркинсона включают леводопу, карбидопу, толкапон, энтакапон, селегилин, бензтропин и тригексифенидил или любую их комбинацию. Фармацевтически активные агенты, которые можно использовать в комбинации с соединениями формулы (I) и композициями на их основе, включают, без ограничения:
(1) леводопу (или ее метиловый или этиловый эфир), по отдельности или в комбинации с ингибитором DOPA-декарбоксилазы (например, с карбидопа (СИНЕМЕТ, КАРБИЛЕВ (carbilev), ПАРКОПА), бенсеразидом (МАДОПАРОМ), α-метилдопа, монофторметилдопа, дифторметилдопа, брокрезином или м-гидроксибензилгидразином);
(2) антихолинергические средства, такие как амитриптилин (ЭЛАВИЛ, ЭНДЕП), бутриптилин, бензтропина мезилат (КОГЕНТИН), тригексифенидил (АРТАН), дифенгидрамин (БЕНАДРИЛ), орфенадрил (НОРФЛЕКС), гиосциамин, атропин (АТРОПЕН), скополамин (ТРАНСДЕРМ-СКОП), скополамина метилбромид (ПАРМИН (parmine)), дицикловерин (БЕНТИЛ, БИКЛОМИН, ДИБЕНТ, ДИЛОМИН), толтеродин (ДЕТРОЛ), оксибутинин (ДИТРОПАН, ЛИРИНЕЛ XL, ОКСИТРОЛ), пентиената бромид, пропантелин (ПРОБАНТИН), циклизин, имипрамина гидрохлорид (ТОФРАНИЛ), имипрамина малеат (СУРМОНТИЛ), лофепрамин, дезипрамин (НОРПРАМИН), доксепин (СИНЕКВАН, ЗОНАЛОН), тримипрамин (СУРМОНТИЛ) и гликопирролат (РОБИНУЛ);
(3) ингибиторы катехол-О-метилтрансферазы (СОМТ), такие как нитекапон, толкапон (ТАСМАР), энтакапон (КОМТАН) и трополон;
(4) ингибиторы моноаминоксидазы (МАО), такие как селегилин (ЭМСАМ), селегилина гидрохлорид (L-депренил, ЭЛДЕПРИЛ, ЗЕЛАПАР), диметилселегилен, брофаромин, фенелзин (НАРДИЛ), транилципромин (ПАРНАТ), моклобемид (АУРОРИКС, МАНЕРИКС), бефлоксатон, сафинамид, изокарбоксазид (МАРПЛАН), ниаламид (НИАМИД), разагилин (АЗИЛЕКТ), ипрониазид (МАРСИЛИД, ИПРОЗИД, ИПРОНИД), ипроклозид, толоксатон (ГУМОРИЛ, ПЕРЕНУМ), бифемелан, дезоксипеганин, гармин (также известный как телепатин или банастерин), гармалин, линезолид (ЗИВОКС, ЗИВОКСИД) и паргилин (ЭУДАТИН, СУПИРДИЛ);
(5) ингибиторы ацетилхолинэстеразы, такие как донепезила гидрохлорид (ARICEPT®, МЕМАК (МЕМАС)), физостигмина салицилат (ANTILIRIUM®), физостигмина сульфат (ЭЗЕРИН), ганстигмин, ривастигмин (EXELON®), ладостигил, NP-0361, галантамина гидробромид (RAZADYNE®, REMINYL®, NIVALIN®), такрин (COGNEX®), толсерин, мемоквин, гуперзин A (HUP-A; NeuroHitech), фенсерин, биснорцимсерин (также известный как BNC) и INM-176;
(6) амилоид-β (Аβ) (или его фрагменты), такие как Аβ1-15, конъюгированный с универсальным эпитопом, связывающимся с HLA DR (молекулами главного комплекса гистосовместимости II класса) (PADRE®), АСС-001 (Elan/Wyeth) и аффитоп;
(7) антитела к амилоиду-β (или его фрагментам), такие как понезумаб, соланезумаб, бапинейзумаб (также известный как ААВ-001), ААВ-002 (Wyeth/Elan), гантенерумаб, внутривенный Ig (GAMMAGARD®), LY2062430 (гуманизированное m266; Lilly) и таковые, описанные в публикациях международных заявок №№ WO 04/032868, WO 05/025616, WO 06/036291, WO 06/069081, WO 06/118959, в публикациях заявок на патент США №№ US 2003/0073655, US 2004/0192898, US 2005/0048049, US 2005/0019328, в европейских патентных публикациях №№ ЕР 0994728 и 1257584 и в патенте США №5750349;
(8) снижающие содержание амилоида или ингибирующие амилоид агенты (в том числе агенты, которые снижают образование, накопление и фибриллизацию амилоида), такие как эпродизат, целекоксиб, ловастатин, анапсос, колостринин, пиоглитазон, клиохинол (также известный как РВТ1), РВТ2 (Prana BioTechnology), флурбипрофен (ANSAID®, FROBEN®) и его R-энантиомер таренфлурбил (FLURIZAN®), нитрофлурбипрофен, фенопрофен (FENOPRON, NALFON®), ибупрофен (ADVIL®, MOTRIN®, NUROFEN®), ибупрофена лизинат, меклофенамовая кислота, меклофенамат натрия (MECLOMEN®), индометацин (INDOCIN®), диклофенак натрия (VOLTAREN®), диклофенак калия, сулиндак (CLINORIL®), сулиндака сульфид, дифлунизал (DOLOBID®), напроксен (NAPROSYN®), напроксен натрия (ANAPROX®, ALEVE®), разрушающий инсулин фермент (также известный как инсулизин), экстракт гингко билоба EGb-761 (ROKAN®, TEBONIN®), трамипрозат (CEREBRIL®, ALZHEMED®), (KIACTA®), неприлизин (также известный как нейтральная эндопептидаза (NEP)), сцилло-инозитол (также известный как сциллитол), аторвастатин (LIPITOR®), симвастатин (ZOCOR®), ибутаморена мезилат, ингибиторы ВАСЕ (фермент, расщепляющий бета-сайт предшественника амилоидного белка), такие как LY450139 (Lilly), BMS-782450, GSK-188909; модуляторы и ингибиторы гамма-секретазы, такие как ELND-007, BMS-708163 (Avagacestat) и DSP8658 (Dainippon); и ингибиторы RAGE (рецептор для конечных продуктов усиленного гликозилирования), такие как ТТР488 (Transtech) и ТТР4000 (Transtech) и таковые, описанные в патенте США №7285293, включая PTI-777;
(9) агонисты альфа-адренергических рецепторов и агенты, блокирующие бета-адренергические рецепторы (бета-блокаторы); антихолинергические средства; противосудорожные средства; антипсихотические средства; блокаторы кальциевых каналов; ингибиторы катехол-О-метилтрансферазы (СОМТ); стимуляторы центральной нервной системы; кортикостероиды; агонисты и антагонисты дофаминовых рецепторов; ингибиторы обратного захвата дофамина; агонисты рецепторов гамма-аминомасляной кислоты (GABA); иммуносупрессоры; интерфероны; агонисты мускариновых рецепторов; нейропротекторные лекарственные средства; агонисты никотиновых рецепторов; ингибиторы обратного захвата норэпинефрина (норадреналина); хинолины; и трофические факторы;
(10) антагонисты гистаминовых рецеторов 3 типа (Н3), такие как PF-3654746 и таковые, описанные в публикациях заявок на патент США №№US 2005-0043354, US 2005-0267095, US 2005-0256135, US 2008-0096955, US 2007-1079175 и US 2008-0176925; публикациях международных патентных заявок №№ WO 2006/136924, WO 2007/063385, WO 2007/069053, WO 2007/088450, WO 2007/099423, WO 2007/105053, WO 2007/138431 и WO 2007/088462; и патенте США №7115600;
(11) антагонисты N-метил-D-аспартатных (NMDA) рецепторов, такие как мемантин (НАМЕНДА, АКСУРА, АБИКСА), амантадин (СИММЕТРЕЛ), акампросат (КАМПРАЛ), безонпродил, кетамин (КЕТАЛАР), делуцемин, дексанабинол, дексефароксан, декстрометорфан, декстрорфан, траксопродил, СР-283097, гимантан, идантадол, ипеноксазон, L-701252 (Merck), ланцицемин, леворфанол (ДРОМОРАН), метадон (ДОЛОФИН), нерамексан, перзинфотел (perzinfotel), фенциклидин, тианептин (СТАБЛОН), дизоцилпин (также известный как MK-801), ибогаин, воакангин, тилетамин, рилузол (РИЛУТЕК), аптиганель (ЦЕРЕСТАТ), гавестинел и ремацимид;
(12) ингибиторы фосфодиэстераз (PDE), в том числе (а) ингибиторы PDE1; (б) ингибиторы PDE2; (в) ингибиторы PDE3; (г) ингибиторы PDE4; (д) ингибиторы PDE5; (е) ингибиторы PDE9 (например, PF-04447943, BAY 73-6691 (Bayer AG) и таковые, описанные в публикациях заявок на патент США №№ US 2003/0195205, US 2004/0220186, US 2006/0111372, US 2006/0106035 и USSN (United States Serial Number) 12/118062 (поданной 9 мая 2008 года)); и (ж) ингибиторы PDE10, такие как 2-({4-[1-метил-4-(пиридин-4-ил)-1H-пиразол-3-ил]фенокси}метил)хинолин (PF-2545920);
(13) антагонисты рецепторов серотонина (5-гидрокситриптамина) 1А (5-HT1A), такие как спиперон, лево-пиндолол, лекозотан;
(14) агонисты рецепторов серотонина (5-гидрокситриптамина) 2С (5-НТ2с), такие как вабикасерин и зикронапин; агонисты/антагонисты рецепторов серотонина (5-гидрокситриптамина) 4 (5-НТ4), такие как PRX-03140 (Epix) и PF-04995274;
(15) антагонисты рецепторов серотонина (5-гидрокситриптамина) 3С (5-НТ3с), такие как ондансетрон (зофран);
(16) антагонисты рецепторов серотонина (5-гидрокситриптамина) 6 (5-НТ6), такие как миансерин (ТОРВОЛ, БОЛЬВИДОН, НОРВАЛ), метиотепин (также известный как метитепин), ритансерин, SB-271046, SB-742457 (GlaxoSmithKline), Lu АЕ58054 (Lundbeck A/S), SAM-760 и PRX-07034 (Epix);
(17) ингибиторы обратного захвата серотонина (5-НТ), такие как алапроклат, циталопрам (ЦЕЛЕКСА, ЦИПРАМИЛ), эсциталопрам (ЛЕКСАПРО, ЦИПРАЛЕКС), кломипрамин (АНАФРАНИЛ), дулоксетин (СИМБАЛТА), фемоксетин (МАЛЕКСИЛ), фенфлурамин (ПОНДИМИН), норфенфлурамин, флуоксетин (ПРОЗАК), флувоксамин (ЛУВОКС), индалпин, милнаципран (ИКСЕЛ), пароксетин (ПАКСИЛ, СЕРОКСАТ), сертралин (ЗОЛОФТ, ЛУСТРАЛ), тразодон (ДЕЗИРЕЛ, МОЛИПАКСИН), венлафаксин (ЭФФЕКСОР), зимелидин (НОРМУД, ЗЕЛМИД), бицифадин, десвенлафаксин (ПРИСТИК), бразофензин, вилазодон, карипразин и тезофензин;
(18) ингибиторы переносчика-1 глицина, такие как палифлутин (paliflutine), ORG-25935 и ORG-26041; и модуляторы метаботропных глутаматных рецепторов (mGluR), такие как AFQ-059 и амантидин;
(19) модуляторы глутаматных рецепторов АМРА(альфа-амино-3-гидрокси-5-метил-4-изоксазол-пропионовая кислота)-типа, такие как перампанел, мибампатор (mibampator), селурампанел, GSK-729327 и N-{(3S,4S)-4-[4-(5-цианотиофен-2-ил)фенокси]тетрагидро-фуран-3-ил}пропан-2-сульфонамид;
(20) ингибиторы цитохрома Р450, такие как ритонавир;
(21) направленные на лечение таупатии средства, такие как давунетид; и тому подобное.
Настоящее изобретение также включает наборы, которые подходят для использования в осуществлении способов лечения, описанных выше. В одном из воплощений набор включает в себя первую лекарственную форму, содержащую одно или более соединений по настоящему изобретению, и контейнер для приготовления доз, в количествах, достаточных для осуществления способов по настоящему изобретению.
В другом воплощении набор по настоящему изобретению содержит одно или более соединений по изобретению.
Общие схемы синтеза
Соединения формулы (I) могут быть получены методами, описанными ниже, в сочетании с методами синтеза, известными в области органической химии, или с применением модификаций и преобразований, которые известны специалистам обычной квалификации в данной области техники. Исходные вещества, используемые в данном изобретении, имеются в продаже или могут быть получены традиционными методами, известными в данной области техники (как например, методы, изложенные в стандартных справочниках, таких как Compendium of Organic Synthetic Methods, Vol. I-XII (опубликованный Wiley-Interscience)). Предпочтительные способы включают, но не ограничиваются этим, способы, описанные ниже.
При выполнении любой из приведенных далее последовательностей синтеза может оказаться необходимым и/или желательным защитить чувствительные или реакционноспособные группы в любой из рассматриваемых молекул. Этого можно достичь путем использования традиционных защитных групп, например, описанных в T.W. Greene, Protective Groups in Organic Chemistry, John Wiley & Sons, 1981; T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Chemistry, John Wiley & Sons, 1991, и T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Chemistry, John Wiley & Sons, 1999, которые тем самым включены посредством ссылки.
Соединения формулы (I) или их фармацевтически приемлемые соли могут быть получены в соответствии с реакционными схемами, рассмотренными в данном описании ниже. Если не указано иное, то заместители на схемах являются такими, как определено выше. Выделение и очистку продуктов осуществляют, используя стандартные методики, известные химику средней квалификации.
Специалисту в данной области техники будет известно, что во многих случаях указанные на схемах 1-4 соединения могут образовываться в виде смеси диастереомеров и/или энантиомеров; они могут быть разделены на различных стадиях схем синтеза с использованием традиционных методов или сочетания таких методов, как например, но не ограничиваясь этим, кристаллизация, нормально-фазовая хроматография, обращенно-фазовая хроматография и хиральная хроматография, с получением отдельных энантиомеров по изобретению.
Специалисту в данной области техники будет понятно, что различные символы, надстрочные и подстрочные индексы, используемые на схемах, в способах и примерах, применяются для удобства представления и/или для отражения порядка, в котором они введены на схемах, и подразумевается, что они не обязательно должны соответствовать символам, надстрочным или подстрочным индексам в прилагаемой формуле изобретения. Схемы являются репрезентативными примерами способов, полезных для синтеза соединений по настоящему изобретению. Они не ограничивают каким-либо образом объема данного изобретения.
Реакции для получения соединений по изобретению могут быть выполнены в подходящих растворителях, которые могут быть легко выбраны специалистом в области органического синтеза. Подходящие растворители по существу не должны вступать в реакцию с исходными веществами (реактивами), промежуточными соединениями или продуктами при температурах проведения реакций, например, при температурах, которые могут изменяться в диапазоне от температуры замерзания растворителя до температуры кипения растворителя. Заданную реакцию можно проводить в одном растворителе или в смеси, содержащей более одного растворителя. В зависимости от конкретной стадии реакции подходящие растворители для конкретной стадии реакции могут быть выбраны специалистом в данной области.
Ход реакций можно отслеживать в соответствии с любым подходящим способом, известным в данной области техники. Например, мониторинг образования продуктов может быть выполнен спектроскопическими методами, такими как спектроскопия ядерного магнитного резонанса (например, 1Н или 13С), инфракрасная спектроскопия, спектрофотометрия (например, в ультрафиолетовой (УФ) и видимой областях), масс-спектрометрия, или хроматографическими методами, такими как высокоэффективная жидкостная хроматография (HPLC) или тонкослойная хроматография (TLC).
Соединения формулы (I) и их промежуточные соединения могут быть получены в соответствии со следующими далее реакционными схемами и прилагаемым обсуждением. Если не указано иное, то R1, R1a, R1b, R2, R3, R4, R5, R6, X и Z на реакционных схемах и в обсуждениях, которые следуют далее, являются такими, как определено в таком же месте выше. Как правило, соединения по данному изобретению могут быть получены способами, которые включают способы, аналогичные известным в области химии, в частности, с учетом описания, содержащегося в данной заявке. Некоторые способы получения соединений по данному изобретению и их промежуточных соединений приведены в качестве других признаков данного изобретения, и они проиллюстрированы следующими далее реакционными схемами. Другие способы могут быть описаны в экспериментальном разделе. Схемы и примеры, приведенные в данном описании (включая соответствующее описание) даются только для иллюстрации и не предназначены для ограничения объема настоящего изобретения.
Реакционная схема 1
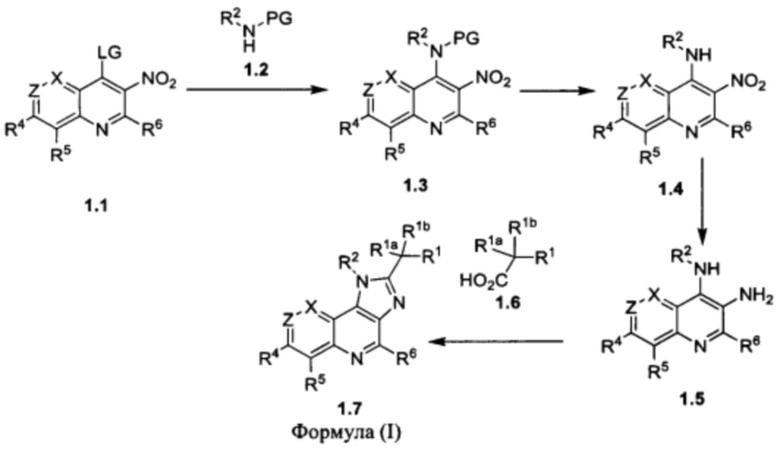
На реакционной схеме 1 показано получение соединений формулы (I). Приведенные на схеме 1 соединения 1.1 и 1.2 либо имеются в продаже, либо могут быть получены методами, изложенными в данном описании, или другими методами, хорошо известными специалистам в данной области техники. Группа в соединении формулы 1.1, обозначенная как LG, представляет собой соответствующую уходящую группу, такую как галогенид (например, хлор или бром) или трифлат, которая подходит для осуществления реакции нуклеофильного замещения при взаимодействии с амином формулы 1.2. Группа в амин-содержащем соединении формулы 1.2, обозначенная как PG, представляет собой соответствующую защитную группу амина, такую как кислотолабильная защитная группа, выбранная из 2,4-диметоксибензила (DMB), 4-метоксибензила (РМВ) и трет-бутоксикарбонила (Boc). Соединения формул 1.1 и 1.2 могут быть приведены во взаимодействие, например, в присутствии соответствующего основания, такого как 1 N,N-диизопропилэтиламин (основание Хюнига) или триэтиламин, в подходящем растворителе, таком как ацетонитрил или N,N-диметилформамид (DMF), с получением соединения формулы 1.3. Обычно эту реакцию проводят при повышенной температуре, как например, от 50 до 100°C, в течение периода времени от 1 до 48 часов. Удаление защитной группы, такой как кислотолабильная защитная группа (PG), из соединения формулы 1.3 обычно может быть осуществлено путем обработки соединения 1.3 соответствующей кислотой, такой как уксусная кислота, трифторуксусная кислота или соляная кислота, с получением соединения формулы 1.4. Кроме того, следует понимать, что в некоторых случаях соединение формулы 1.1 может быть приведено во взаимодействие с незащищенным амином формулы R2-NH2 с получением непосредственно соединения формулы 1.4. После восстановления нитрогруппы в соединении формулы 1.4 с использованием условий, соответствующих присутствующей функциональности, получают соединение формулы 1.5. Например, нитрогруппу в соединении формулы 1.4 можно восстановить до соответствующего амина формулы 1.5 путем обработки соединения 1.4 цинковой пылью и гидроксидом аммония в метаноле или, альтернативно, путем гидрирования соединения 1.4 с использованием соответствующего катализатора, такого как оксид платины(IV), в соответствующем растворителе, таком как метанол, ацетонитрил или их смесь. Затем в результате сочетания диамин-содержащего соединения 1.5 с карбоновой кислотой формулы 1.6 образуется желаемое соединение формулы (I), также указанное как 1.7. Реакция сочетания с участием диамина формулы 1.5 и карбоновой кислоты формулы 1.6 может быть осуществлена в соответствующем растворителе, таком как N,N-диметилформамид, в присутствии соответствующего основания, такого как диизопропилэтиламин, и реагента сочетания, такого как 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфирана.
Реакционная схема 2
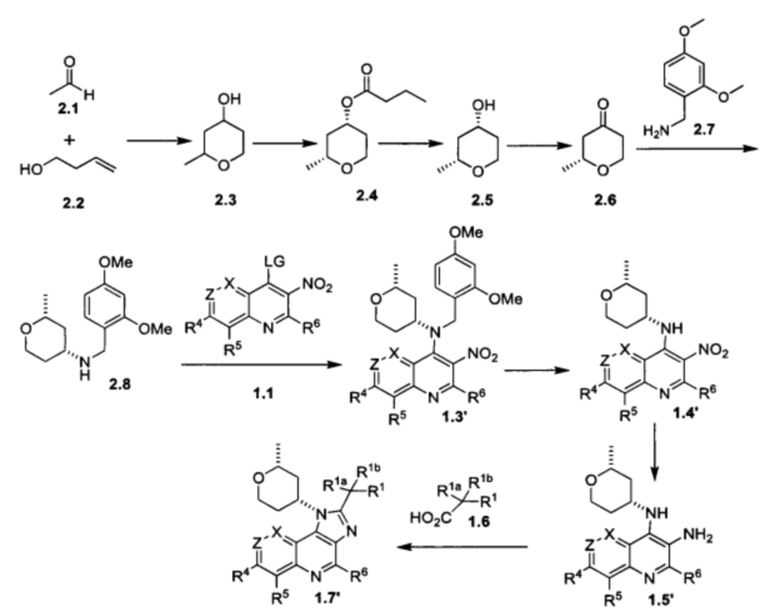
На реакционной схеме 2 показано получение соединения формулы 1.7', представляющего собой соединение формулы (I), в котором R2 обозначает показанную хиральную 2-метилтетрагидропиран-4-ильную группировку. В результате осуществления взаимодействия соединения 2.1 с соединением 2.2 в соответствии с опубликованной методикой реакции Принса образовывался пиран 2.3. После выполнения основанного на применении ферментов метода хирального разделения с целью получения отдельных энантиомеров получали соединение формулы 2.5 после гидролиза выделенного сложного эфира 2.4. В результате окисления соединения 2.5 получали кетон 2.6, который приводили во взаимодействие с соединением формулы 2.7, используя химические реакции восстановительного аминирования, с получением защищенного амина формулы 2.8. Защищенный амин формулы 2.8 можно привести во взаимодействие с соединением формулы 1.1 способом, аналогичным описанному ранее на схеме 1, с получением соединения формулы 1.3'. Затем могут быть получены соединения формул 1.4', 1.5' и 1.7' способом, аналогичным способам, описанным на схеме 1 для соединений формул 1.4, 1.5 и 1.7, соответственно.
Реакционная схема 3
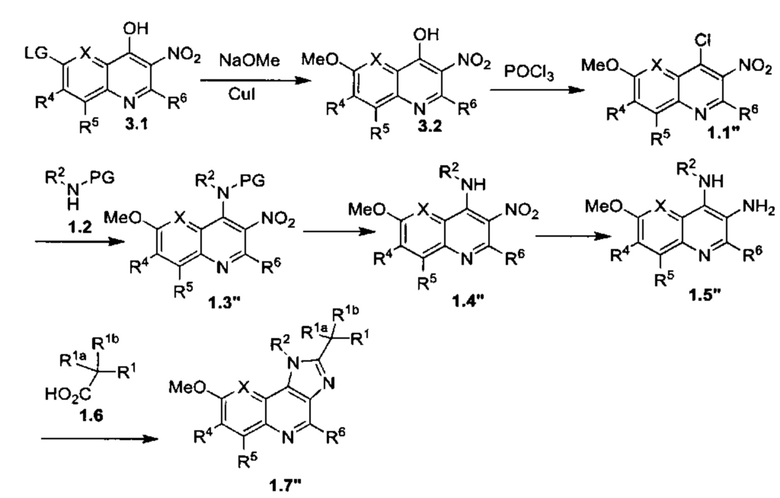
На реакционной схеме 3 показано, как можно на начальной стадии синтеза модифицировать функциональную группу в положении R3 соединения формулы (I) (т.е. когда Z представляет собой CR3). Выполненная в начале синтеза модификация соединения, например, имеющегося в продаже соединения 3.1 (где LG представляет собой бром), позволяет специалисту в данной области техники вводить такие группы, как метокси, которые являются достаточно устойчивыми при осуществлении полной схемы синтеза способом, аналогичным описанному для схемы 1. Соединение формулы 3.1 можно привести во взаимодействие с метилатом натрия в присутствии иодида меди с получением метокси-содержащего соединения формулы 3.2. Затем соединение формулы 3.2 можно обработать оксихлоридом фосфора, чтобы преобразовать группу гидрокси, присутствующую в соединении формулы 3.1, в соответствующий хлорид формулы 1.1''. Далее соединение формулы 1.1'' можно привести во взаимодействие с амином формулы 1.2 с получением соединения 1.3'' способом, описанным ранее для схемы 1. После этого соединение формулы 1.3'' можно обработать далее для получения соединений формул 1.4'', 1.5'' и 1.7'' способом, аналогичным соответствующим стадиям, описанным ранее для схемы 1.
Реакционная схема 4
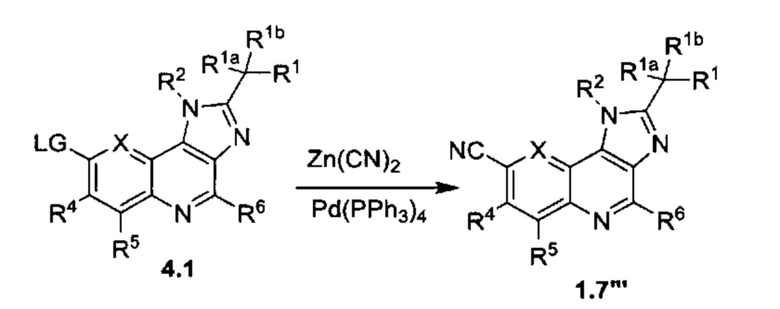
На реакционной схеме 4 показана последняя стадия преобразования соединения формулы 4.1 в соединение 1.7''', способа, который может быть применен для получения некоторых соединений формулы (I), где Z представляет собой CR3, и в которых присутствующая функциональная группа R3 не согласуется с полной схемой пути синтеза, приведенного на схеме 1. Например, нитрильная группа (-CN), присутствующая в положении R3 в соединении формулы 1.7''', не смогла бы сохраниться при осуществлении стадии восстановления, необходимой для преобразования соединения 1.4 в соединение 1.5, как описано на схеме 1 (восстановление нитрогруппы до соответствующего амина). Соединением формулы 4.1 на схеме 4 является соединение, в котором LG представляет собой подходящую уходящую группу, такую как галогенид (например, бром). Соединение формулы 4.1 может быть приведено во взаимодействие с цианидом цинка в присутствии соответствующего катализатора, такого как тетракис(трифенилфосфин)палладий, в соответствующем растворителе, таком как N,N-диметилформамид. Обычно эту реакцию проводят в диапазоне температур от приблизительно температуры окружающей среды до 100°C в течение периода времени от 1 до 48 часов с получением соединения формулы 1.7'''.
Способы, описанные в общих чертах на схемах 1-4, не следует истолковывать в ограничительном смысле. Специалисту в данной области техники следует понимать, что для получения соединений формулы (I) можно изменять порядок выполнения некоторых стадий и условия реакции. Специалистом в области органического синтеза может быть сделан выбор относительно того, который подход является наилучшим для применения. Более конкретные примеры способов, использованных для получения соединений формулы (I), приведены ниже в разделе Примеры, и аналогичным образом специалисту в данной области техники также не следует истолковывать эти способы в ограничительном смысле.
Экспериментальные методики
Далее показан синтез различных соединений по настоящему изобретению. Дополнительные соединения, включенные в объем данного изобретения, могут быть получены с использованием способов, проиллюстрированных в этих примерах, либо по отдельности, либо в сочетании с методами, общеизвестными в данной области техники.
Как правило, эксперименты проводили в атмосфере инертного газа (азота или аргона), в особенности, в случаях использования чувствительных к кислороду или влаге реагентов или промежуточных соединений. Имеющиеся в продаже растворители и реагенты обычно использовали без дополнительной очистки. Там, где это целесообразно, использовали безводные растворители, обычно продукты AcroSeal® от Acros Organics или продукты DriSolv® от EMD Chemicals. В других случаях, имеющиеся в продаже растворители пропускали через колонки, упакованные  молекулярными ситами, пока не были достигнуты следующие стандарты контроля качества (QC), касающиеся содержания воды: а) меньше 100 млн-1 для дихлорметана, толуола, N,N-диметилформамида и тетрагидрофурана; б) меньше 180 млн-1 для метанола, этанола, 1,4-диоксана и диизопропиламина. В случае очень чувствительных реакций растворители дополнительно обрабатывали металлическим натрием, гидридом кальция или молекулярными ситами и подвергали дистилляции непосредственно перед применением. Обычно продукты сушили под вакуумом, после чего передавали на следующие стадии реакции или подвергали биологическому тестированию. Масс-спектрометрические данные регистрируют на оборудовании либо для жидкостной хроматографии/масс-спектрометрии (LCMS), с химической ионизацией при атмосферном давлении (APCI), либо газовой хроматографии/масс-спектрометрии (GCMS). Химические сдвиги для данных ядерного магнитного резонанса (ЯМР) выражают в числе частей на миллион (млн-1, δ) по отношению к остаточным пикам от применяемых дейтерированных растворителей. В некоторых примерах проводили хиральные разделения, чтобы разделить энантиомеры некоторых соединений по изобретению (в некоторых примерах разделенные энантиомеры обозначены как ENT-1 и ENT-2, согласно порядку их элюирования). В некоторых примерах измеряли оптическое вращение энантиомера, используя поляриметр. Согласно обнаруженным для него данным по вращению (или данным о его удельном вращении), энантиомер с правосторонним вращением обозначали как (+)-энантиомер, а энантиомер с левосторонним вращением обозначали как (-)-энантиомер. На принадлежность к рацемическим соединениям указывает присутствие знака (+/-), расположенного рядом со структурой; в этих случаях, показанная стереохимия указывает на относительную (а не абсолютную) конфигурацию заместителей данного соединения.
молекулярными ситами, пока не были достигнуты следующие стандарты контроля качества (QC), касающиеся содержания воды: а) меньше 100 млн-1 для дихлорметана, толуола, N,N-диметилформамида и тетрагидрофурана; б) меньше 180 млн-1 для метанола, этанола, 1,4-диоксана и диизопропиламина. В случае очень чувствительных реакций растворители дополнительно обрабатывали металлическим натрием, гидридом кальция или молекулярными ситами и подвергали дистилляции непосредственно перед применением. Обычно продукты сушили под вакуумом, после чего передавали на следующие стадии реакции или подвергали биологическому тестированию. Масс-спектрометрические данные регистрируют на оборудовании либо для жидкостной хроматографии/масс-спектрометрии (LCMS), с химической ионизацией при атмосферном давлении (APCI), либо газовой хроматографии/масс-спектрометрии (GCMS). Химические сдвиги для данных ядерного магнитного резонанса (ЯМР) выражают в числе частей на миллион (млн-1, δ) по отношению к остаточным пикам от применяемых дейтерированных растворителей. В некоторых примерах проводили хиральные разделения, чтобы разделить энантиомеры некоторых соединений по изобретению (в некоторых примерах разделенные энантиомеры обозначены как ENT-1 и ENT-2, согласно порядку их элюирования). В некоторых примерах измеряли оптическое вращение энантиомера, используя поляриметр. Согласно обнаруженным для него данным по вращению (или данным о его удельном вращении), энантиомер с правосторонним вращением обозначали как (+)-энантиомер, а энантиомер с левосторонним вращением обозначали как (-)-энантиомер. На принадлежность к рацемическим соединениям указывает присутствие знака (+/-), расположенного рядом со структурой; в этих случаях, показанная стереохимия указывает на относительную (а не абсолютную) конфигурацию заместителей данного соединения.
Реакции, протекающие с образованием детектируемых промежуточных соединений, как правило, сопровождались выполнением LCMS анализа и проводились до завершения превращения перед добавлением последующих реагентов. Что касается процедур синтеза с отсылкой к методикам из других примеров или способов, то реакционные условия (продолжительность реакции и температура) могут варьировать. В общем случае ход реакций отслеживали по тонкослойной хроматографии или масс-спектрометрии и при необходимости реакционные смеси подвергали дополнительной обработке. Процедуры очистки могут варьировать от эксперимента к эксперименту: в общем случае растворители и соотношения растворителей, использованных для элюентов/градиентов, выбирали с целью получения соответствующих значений фактора удерживания (Rf) или времени удерживания.
Подготовительный пример Р1
цис-N-(2,4-Диметоксибензил)-2-метилтетрагидро-2Н-пиран-4-амин (Р1)
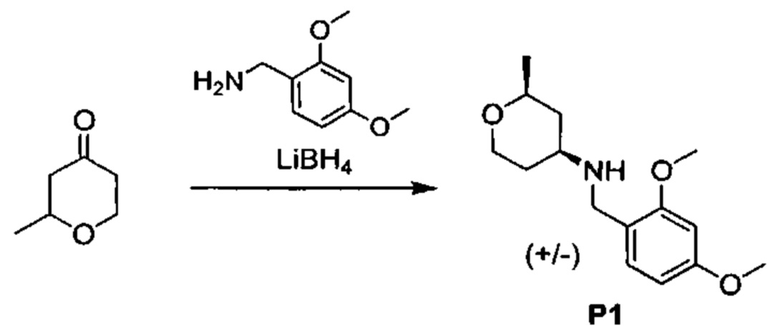
К раствору 2-метилтетрагидро-4H-пиран-4-она (500 мг; 4,4 ммоль) в метаноле (10 мл) добавляли 1-(2,4-диметоксифенил)метанамин (1,97 мл; 13,1 ммоль). После перемешивания в течение 1 часа при комнатной температуре реакционную смесь охлаждали до -78°C и по каплям добавляли раствор боргидрида лития (98%-ного; 85 мг; 3,8 ммоль) в тетрагидрофуране (1,5 мл). Реакционную смесь оставляли медленно нагреваться до комнатной температуры в течение ночи, после чего ее охлаждали до -20°C и гасили путем осторожного добавления насыщенного водного раствора бикарбоната натрия. Добавляли этилацетат (25 мл) и воду в количестве достаточном для солюбилизации осадка, и водный слой экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 0% до 15% (10:1 метанол/концентрированный раствор гидроксида аммония) в этилацетате) получали продукт в виде бесцветного масла. Выход: 936 мг; 3,53 ммоль; 80%. 1Н ЯМР (400 МГц, CDCl3) δ 7.13 (d, J=8,0 Гц, 1Н), 6.46 (d, половина квартета АВ, J=2,2 Гц, 1H), 6.44 (dd, половина АВХ системы, J=8,1; 2,3 Гц, 1Н), 4.00 (ddd, J=11,6; 4,6; 1,6 Гц, 1Н), 3.82 (s, 3Н), 3.81 (s, 3Н), 3.76 (s, 2Н), 3.37-3.46 (m, 2Н), 2.63-2.72 (m, 1Н), 1.85-1.92 (m, 1H), 1.78-1.85 (m, 1Н), 1.37 (dddd, J=13; 12; 11; 4,6 Гц, 1Н), 1.20 (d, J=6,2 Гц, 3Н), 1.10 (ddd, J=12, 11, 11 Гц, 1H).
Альтернативное получение Р1
цис-N-(2,4-Диметоксибензил)-2-метилтетрагидро-2Н-пиран-4-амин (Р1)
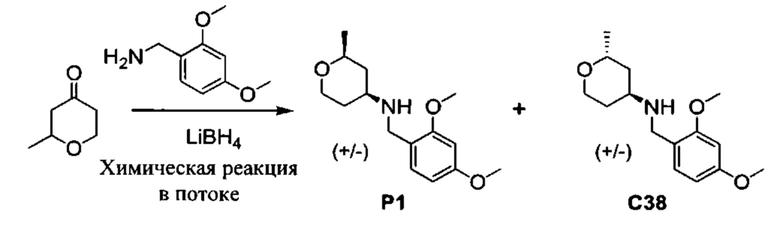
Используя шприцевой насос, к раствору 1-(2,4-диметоксифенил)метанамина (9,21 мл; 61,3 ммоль) в метаноле (137 мл) добавляли 2-метилтетрагидро-4H-пиран-4-он (7,00 г; 61,3 ммоль) в течение 3,5 часа (2 мл/час). По завершении добавления реакционную смесь оставляли перемешиваться при комнатной температуре в течение 1 часа. Затем этот раствор приводили во взаимодействие с боргидридом лития (0,48 М раствором в тетрагидрофуране; 153,2 мл; 73,5 ммоль), используя проточный реактор (реактор емкостью 25 мл, состоящий из стеклянного чипа емкостью 1 мл с двумя подающими каналами и подводящими шлангами из перфторалкокси-сополимера (объем 24 мл); температура: -78°C; концентрация в реакционной смеси: 0,2 М; продолжительность обработки: 10 минут; скорость потока: 1,25 мл/минута в обоих потоках). Собранную реакционную смесь разбавляли насыщенным водным раствором бикарбоната натрия и экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. 1Н ЯМР-анализ на этом этапе показал, что соотношение цис : транс изомеров составляло 10,7:1. После хроматографии на силикагеле (градиент: от 0% до 20% метанола в этилацетате) получали цис-продукт Р1. Выход: 11,59 г; 43,68 ммоль; 71%. 1Н ЯМР (400 МГц, CDCl3) δ 7.16 (d, J=8,0 Гц, 1Н), 6.41-6.48 (m, 2H), 4.00 (ddd, J=11,7; 4,7; 1,8 Гц, 1H), 3.82 (s, 3H), 3.80 (s, 3H), 3.78 (s, 2H), 3.36-3.46 (m, 2H), 2.70 (tt, J=11,2; 4,1 Гц, 1H), 1.87-1.94 (m, 1H), 1.79-1.87 (m, 1H), 1.35-1.47 (m, 1H), 1.20 (d, J=6,2 Гц, 3H), 1.08-1.19 (m, 1H).
Также выделяли транс-изомер C38. Выход: 1,24 г; 4,67 ммоль; 7,6%. 1Н ЯМР (400 МГц, CDCl3) δ 7.14 (d, J=8,2 Гц, 1Н), 6.42-6.48 (m, 2Н), 3.84-3.94 (m, 2Н), 3.82 (s, 3Н), 3.81 (s, 3Н), 3.69-3.77 (m, 3Н), 2.97-3.02 (m, 1Н), 1.72-1.82 (m, 1Н), 1.44-1.66 (m, 3Н), 1.14 (d, J=6,2 Гц, 3Н).
Подготовительный пример Р2
(2R,4R)-N-(2,4-Диметоксибензил)-2-метилтетрагидро-2Н-пиран-4-амин (Р2)
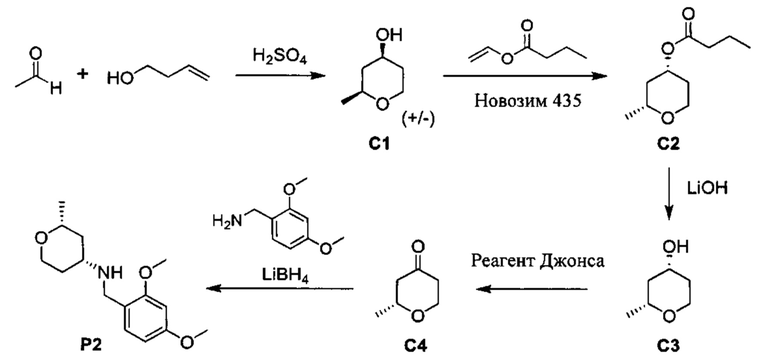
Стадия 1. Синтез цис-2-метилтетрагидро-2Н-пиран-4-ола (С1)
Бут-3-ен-1-ол (39,0 мл; 453 ммоль) и ацетальдегид (25,5 мл; 454 ммоль) объединяли в водном растворе серной кислоты (20% (масс./масс.), 565 г) и перемешивали при 80°C в течение 5 суток. Реакционную смесь охлаждали до комнатной температуры и экстрагировали диэтиловым эфиром, а затем дихлорметаном; объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 0% до 25% этилацетата в гептане) получали продукт в виде бесцветного масла. Выход: 11,2 г; 96,4 ммоль; 21%. 1Н ЯМР (400 МГц, CDCl3) δ 3.99 (ddd, J=11,8; 4,9; 1,7 Гц, 1Н), 3.71-3.80 (m, 1Н), 3.35-3.46 (m, 2Н), 1.82-1.98 (m, 3Н), 1.48 (dddd, J=12,5; 12,4; 11,1; 4,9 Гц, 1Н), 1.21 (d, J=6,2 Гц, 3Н), 1.14-1.24 (m, 1H).
Стадия 2. Синтез (2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил бутаноата (С2)
Этенилбутаноат (78,6 мл; 620 ммоль) и новозим 435 (иммобилизованную липазу В из Candida antarctica, 25 г) добавляли к раствору соединения С1 (150 г; 1,29 моль) в тетрагидрофуране (1,3 л). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, после чего ее фильтровали через набивку диатомовой земли, которую затем дважды промывали дихлорметаном. Объединенные фильтраты концентрировали в вакууме и очищали посредством хроматографии на силикагеле (градиент: от 0% до 10% этилацетата в гептане), получая продукт в виде масла. Выход: 51,5 г; 276 ммоль, 45%. Абсолютные конфигурации соединения С2 и последующих промежуточных соединений подтверждали посредством рентгеноструктурного анализа, выполненного для соединения С14 (см. пример 2). 1Н ЯМР (400 МГц, CDCl3) δ 4.82-4.92 (m, 1Н), 3.99 (ddd, J=11,9; 4,9; 1,7 Гц, 1H), 3.42-3.52 (m, 2H), 2.25 (t, J=7,4 Гц, 2H), 1.92-2.00 (m, 1H), 1.84-1.91 (m, 1H), 1.52-1.69 (m, 3H), 1.28 (ddd, J=12, 11, 11 Гц, 1H), 1.20 (d, J=6,2 Гц, 3Н), 0.94 (t, J=7,4 Гц, 3Н).
Стадия 3. Синтез (2R,4R)-2-метилтетрагидро-2Н-пиран-4-ола (С3)
Раствор соединения С2 (51,5 г; 276 ммоль) в метаноле и тетрагидрофуране (1:1, 700 мл) обрабатывали раствором гидроксида лития (19.9 г; 831 ммоль) в воде (120 мл) и реакционную смесь перемешивали в течение ночи при комнатной температуре. После удаления органических растворителей посредством концентрирования при пониженном давлении водный остаток экстрагировали 4 раза дихлорметаном; объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме, получая продукт в виде бесцветного масла. Выход: 27,3 г; 235 ммоль; 85%. 1Н ЯМР (400 МГц, CDCl3) δ 3.99 (ddd, J=11,8; 4,8; 1,7 Гц, 1H), 3.71-3.80 (m, 1H), 3.35-3.47 (m, 2H), 1.82-1.98 (m, 3H), 1.48 (dddd, J=12,5; 12,4; 11,1; 4,8 Гц, 1H), 1.21 (d, J=6,2 Гц, 3Н), 1.14-1.24 (m, 1H).
Стадия 4. Синтез (2R)-2-метилтетрагидро-4Н-пиран-4-она (C4)
Раствор соединения С3 (27.3 г; 235 ммоль) в ацетоне (980 мл) охлаждали в ледяной бане и по каплям обрабатывали реагентом Джонса (2,5 М; 103 мл; 258 ммоль). Реакционную смесь перемешивали в течение 10 минут при 0°C, затем нагревали до комнатной температуры, перемешивали в течение еще 30 минут и охлаждали до 0°C. Добавляли 2-пропанол (18 мл; 240 ммоль) и перемешивание продолжали в течение 30 минут. После окончания концентрирования смеси в вакууме остаток распределяли между водой и дихлорметаном; водный слой экстрагировали 3 раза дихлорметаном и объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая продукт в виде светло-желтого масла. Выход: 23 г; 200 ммоль; 85%. 1Н ЯМР (400 МГц, CDCl3) δ 4.25 (ddd, J=11,5; 7,4; 1,3 Гц, 1Н), 3.70 (dqd, J=12,2; 6,1; 2,7 Гц, 1H), 3.64 (ddd, J=12,2; 11,6; 2,8 Гц, 1H), 2.55 (dddd, J=14,6; 12,4; 7,4; 1,0 Гц, 1H), 2.37 (ddd, J=14,4; 2,3; 2,3 Гц, 1H), 2.21-2.31 (m, 2H), 1.29 (d, J=6,2 Гц, 3Н).
Стадия 5. Синтез (2R,4R)-N-(2,4-диметоксибензил)-2-метилтетрагидро-2H-пиран-4-амина (Р2)
1-(2,4-Диметоксифенил)метанамин (20,3 мл; 135 ммоль) добавляли к раствору соединения С4 (10,3 г; 90,2 ммоль в метаноле (200 мл) и реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Затем ее охлаждали до -78°С; по каплям добавляли раствор боргидрида лития (2M в тетрагидрофуране; 45,1 мл; 90,2 ммоль) и перемешивание продолжали при -78°С в течение 2 часов. После медленного нагревания до комнатной температуры в течение ночи реакционную смесь гасили путем осторожного добавления насыщенного водного раствора бикарбоната натрия. Добавляли этилацетат (250 мл) и воду в количестве, достаточном для солюбилизации осадка, и водный слой экстрагировали этилацетатом; объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 0% до 5% метанола в дихлорметане) получали продукт в виде бесцветного масла (10,4 г). После аналогичной очистки смешанных фракций получали дополнительное количество продукта (3,7 г). Объединенный выход: 14,1 г; 53,1 ммоль; 59%. 1Н ЯМР (400 МГц, CDCl3) δ 7.13 (d, J=8,0 Гц, 1Н), 6.42-6.47 (m, 2H), 3.99 (ddd, J=11,6; 4,6; 1,5 Гц, 1H), 3.82 (s, 3H), 3.80 (s, 3H), 3.76 (s, 2H), 3.36-3.45 (m, 2H), 2.63-2.73 (m, 1H), 1.85-1.92 (m, 1H), 1.78-1.85 (m, 1H), 1.38 (dddd, J=13; 12; 11; 4,7 Гц, 1H), 1.20 (d, J=6,2 Гц, 3Н), 1.10 (ddd, J=11, 11, 11 Гц, 1H).
Альтернативное получение P2
(2R,4R)-N-(2,4-Диметоксибензил)-2-метилтетрагидро-2Н-пиран-4-амин (P2)
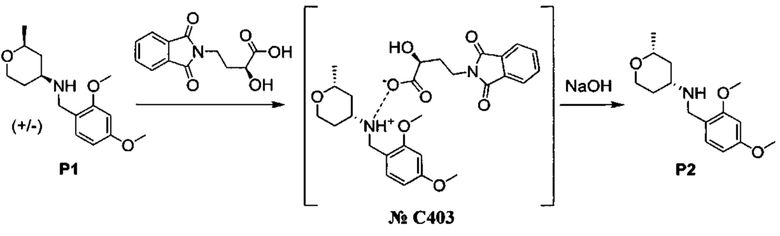
Раствор соединения P1 (200 мг; 0,754 ммоль) в ацетонитриле (0,05 М) добавляли к суспензии (+)-(2S)-4-(1,3-диоксо-1,3-дигидро-2H-изоиндол-2-ил)-2-гидроксибутановой кислоты (93,9 мг; 0,377 ммоль) в ацетонитриле (0,15 М). Реакционную смесь нагревали до 75°С для осуществления полного растворения, затем оставляли охлаждаться до комнатной температуры и перемешиваться в течение еще 18 часов. Полученное твердое вещество (С39) собирали фильтрованием, промывали ацетонитрилом и растворяли в дихлорметане. Этот раствор промывали три раза 1 М водным раствором гидроксида натрия и один раз насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая продукт в виде бесцветного масла. Показанную абсолютную конфигурацию устанавливали посредством хиральной HPLC в сравнении с известным образцом соединения Р2. Энантиомерный избыток этой партии соединения Р2 составлял 77,5% по результатам определения посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AS от Chiral Technologies, 5 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: этанол, содержащий 0,2% гидроксида аммония; градиент: от 5% до 60% В). В этой системе Р2 представлял собой элюирующийся вторым энантиомер. Выход: 68 мг; 0,26 ммоль; 69%. 1Н ЯМР (400 МГц, CDCl3) δ 7.13 (d, J=8,0 Гц, 1Н), 6.46 (d, половина квартета АВ, J=2,3 Гц, 1Н), 6.44 (dd, половина АВХ системы, J=8,1; 2,4 Гц, 1Н), 4.00 (ddd, J=11,7; 4,7; 1,8 Гц, 1Н), 3.82 (s, 3Н), 3.81 (s, 3Н), 3.76 (s, 2Н), 3.37-3.46 (m, 2Н), 2.63-2.72 (m, 1Н), 1.85-1.92 (m, 1H), 1.78-1.85 (m, 1Н), 1.38 (dddd, J=12,7; 12,5; 11,3; 4,7 Гц, 1Н), 1.20 (d, J=6,2 Гц, 3Н), 1.10 (ddd, J=12,3; 11,3; 11,1 Гц, 1Н).
Подготовительный пример Р3
цис-3-Фторциклопентанамин, гидрохлоридная соль (Р3)
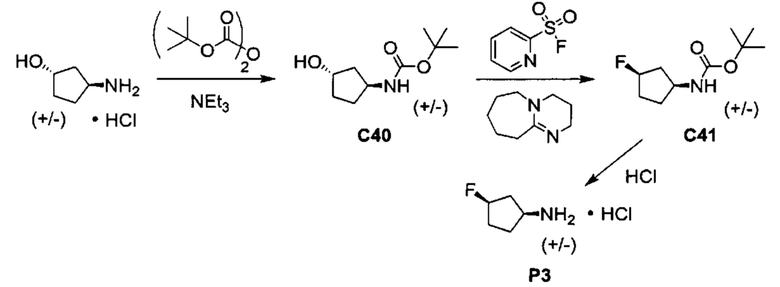
Стадия 1. Синтез трет-бутил-(транс-3-гидроксициклопентил)карбамата (С40)
транс-3-Аминоциклопентанол, гидрохлоридную соль (9,7 г; 70 ммоль) смешивали с дихлорметаном (120 мл), после чего добавляли триэтиламин (21,6 мл; 155 ммоль), затем ди-трет-бутилдикарбонат (16,9 г; 77,4 ммоль). После окончания перемешивания при комнатной температуре в течение ночи в реакционную смесь добавляли воду и полученную смесь экстрагировали дихлорметаном. Органический слой промывали водой, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая светло-желтое масло, которое отвердевало после добавления гептана. Это вещество собирали фильтрованием, промывали гептаном и кристаллизовали из смеси дихлорметан/гептан, получая продукт в виде белого твердого вещества. Выход: 11,86 г; 58,93 ммоль; 84%. 1Н ЯМР (400 МГц, CDCl3) δ 4.36-4.54 (m, 2Н), 4.10-4.25 (br m, 1Н), 2.16-2.28 (m, 1Н), 1.97-2.09 (m, 2Н), 1.55-1.71 (m, 2Н), 1.45 (s, 9Н), 1.36-1.48 (m, 2Н).
Стадия 2. Синтез трет-бутил-(цис-3-фторциклопентил)карбамата (С41)
1,8-Диазабицикло[5.4.0]ундец-7-ен (7,43 мл; 49,7 ммоль) добавляли к смеси соединения С40 (5,00 г; 24,8 ммоль), толуола (25 мл) и пиридин-2-сульфонилфторида (PyFluor; 4,40 г; 27,3 ммоль). Через 16 часов выдерживания при комнатной температуре реакционную смесь разбавляли насыщенным водным раствором бикарбоната натрия (50 мл) и экстрагировали гептаном (3×100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 0% до 30% этилацетата в гептане) получали продукт в виде твердого вещества. Выход: 3,78 г; 18,6 ммоль; 75%. 1Н ЯМР (400 МГц, CDCl3) δ (5,20-5.26 (m) и 5.07-5.13 (m), JHF=54 Гц, суммарно 1Н), 4.75-4.89 (br m, 1Н), 4.10-4.24 (br m, 1Н), 1.99-2.21 (m, 3Н), 1.66-1.95 (m, 3Н), 1.45 (s, 9Н).
Стадия 3. Синтез цис-3-фторциклопентанамина, гидрохлоридной соли (Р3)
Хлористый водород (4 М раствор в 1,4-диоксане; 46,2 мл; 185 ммоль) добавляли при 0°C к раствору соединения С41 (3,76 г; 18,5 ммоль) в тетрагидрофуране (54 мл) и реакционную смесь оставляли медленно нагреваться до комнатной температуры в течение ночи. Растворители удаляли в вакууме и остаток перекристаллизовывали из смеси 2-пропанол/гептан, получая продукт в виде белого твердого вещества. Выход: 2,45 г; 17,6 ммоль; 95%. 1Н ЯМР (400 МГц, D2O) δ (5,31-5.35 (m) и 5.18-5.22 (m), JHF=53 Гц, суммарно 1H), 3.76-3.84 (m, 1Н), 2.00-2.40 (m, 4Н), 1.75-1.98 (m, 2Н).
Подготовительный пример Р4
Бензил-[(1R, 3S)-3-фторциклопентил]карбамат (Р4)
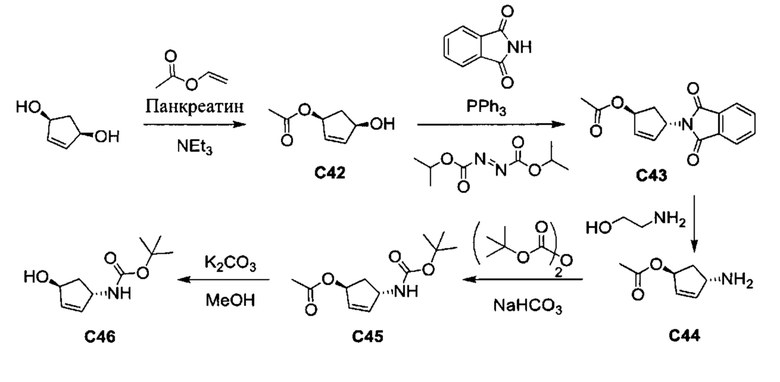
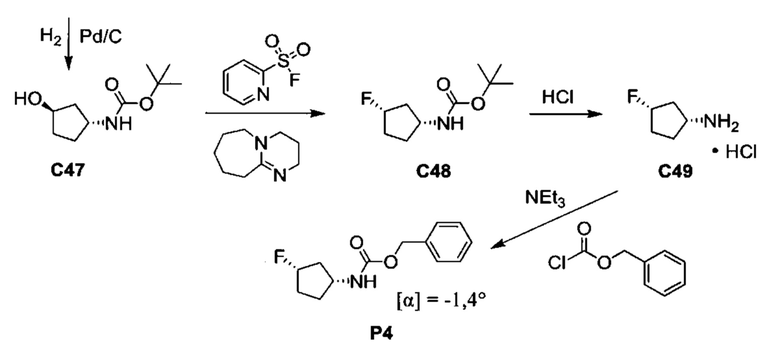
Стадия 1. Синтез (1S,4R)-4-гидроксициклопент-2-ен-1-илацетата (С42)
Используя метод S. Specklin и др. (Tetrahedron Lett., 2014, 55, 6987-6991), к перемешиваемому раствору цис-циклопент-4-ен-1,3-диола (3,04 г; 30,4 ммоль), винилацетата (19,6 мл; 213 ммоль) и триэтиламина (29,6 мл; 212 ммоль) в тетрагидрофуране (76 мл) добавляли панкреатин (Sigma, из поджелудочной железы свиньи, 4 × согласно USP (Фармакопея США); 15,2 г). Полученную суспензию перемешивали в течение 22 часов при комнатной температуре, после чего ее фильтровали через набивку диатомовой земли. После промывания фильтрующей набивки этилацетатом (50 мл) объединенные фильтраты концентрировали в вакууме и очищали посредством хроматографии на силикагеле (градиент: от 20% до 33% этилацетата в циклогексане), получая продукт в виде желтого твердого вещества. Выход: 2,28 г; 16,0 ммоль; 53%. 1Н ЯМР (400 МГц, CDCl3) δ 6.12 (ddd, J=5,5; 1,9; 1,3 Гц, 1Н), 5.99 (ddd, J=5,5; 2,1; 1,2 Гц, 1H), 5.48-5.53 (m, 1H), 4.70-4.75 (m, 1H), 2.76-2.86 (m, 1H), 2.06 (s, 3H), 1.66 (ddd, J=14,6; 3,9; 3,7 Гц, 1H).
Стадия 2. Синтез (1S,4S)-4-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)циклопент-2-ен-1-илацетата (С43)
Диизопропилазодикарбоксилат (94%-ный; 2,73 мл; 13,0 ммоль) медленно добавляли к смеси соединения С42 (1,68 г; 11,8 ммоль), тетрагидрофурана (50 мл), 1Н-изоиндол-1,3(2H)-диона (1,92 г; 13,0 ммоль) и трифенилфосфина (98,5%-ного; 3,47 г; 13,0 ммоль). После окончания перемешивания реакционной смеси при комнатной температуре в течение 18 часов ее пропускали через короткую набивку силикагеля (100 г), через которую затем дополнительно пропускали этилацетат. Фракции, содержащие продукт, объединяли, концентрировали в вакууме и подвергали хроматографии на силикагеле (градиент: от 0% до 40% этилацетата в гептане), получая продукт в виде белого твердого
вещества (4,96 г). По данным 1Н ЯМР это вещество содержало в виде примесей существенное количество вещества, происходящего из диизопропилазодикарбоксилата; часть переносили на следующую стадию без дополнительной очистки. GCMS m/z 211,0 [М - АсОН]+. 1Н ЯМР (400 МГц, CDCl3), только пики продукта: δ 7.81-7.84 (m, 2Н), 7.70-7.73 (m, 2Н), 6.16 (ddd, J=5,7; 2,3; 2,2 Гц, 1Н), 6.01-6.06 (m, 1Н), 5.98 (ddd, J=5,7; 2,2; 1,0 Гц, 1Н), 5.52-5.58 (m, 1Н), 2.57 (ddd, J=14,4; 7,2; 4,7 Гц, 1Н), 2.27 (ddd, J=14,5; 8,5; 2,9 Гц, 1Н), 2.07 (s, 3Н).
Стадия 3. Синтез (1S,4S)-4-аминоциклопент-2-ен-1-илацетата (С44)
2-Аминоэтанол (2,13 мл; 35,3 ммоль) добавляли к раствору соединения С43 (с предыдущей стадии; 2,40 г; не более 6,29 ммоль) в этилацетате (20 мл) и реакционную смесь нагревали при температуре дефлегмации в течение 18 часов. 2-Аминоэтанол (1,0 мл; 17 ммоль) добавляли еще раз и нагревание продолжали в течение еще 4 часов. После удаления растворителя при пониженном давлении остаток очищали с использованием хроматографии на силикагеле (градиент: от 0% до 10% (2 М аммиак в метаноле) в дихлорметане), получая продукт в виде бесцветного масла (1,25 г). Это вещество переносили непосредственно на следующую стадию.
Стадия 4. Синтез (1S,4S)-4-[(трет-бутоксикарбонил)амино]циклопент-2-ен-1-илацетата (С45)
К раствору соединения С44 (с предыдущей стадии; ≤6,29 ммоль) в дихлорметане (30 мл) добавляли бикарбонат натрия (3,72 г; 44,3 ммоль) и ди-трет-бутилдикарбонат (3,86 г; 17,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, после чего ее концентрировали в вакууме и использовали непосредственно на следующей стадии.
Стадия 5. Синтез трет-бутил-[(1S,4S)-4-гидроксициклопент-2-ен-1-ил]карбамата (С46)
Карбонат калия (2,44 г; 17,7 ммоль) добавляли к раствору соединения С45 (с предыдущей стадии; не более 6,29 ммоль) в метаноле (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, после чего ее разбавляли водой (50 мл) и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 0% до 60% этилацетата в гептане) получали продукт в виде белого твердого вещества. Выход: 783 мг; 3,93 ммоль; 62% за 4 стадии. GCMS m/z 143,0 [М - 2-метилпроп-1-ен]+. 1Н ЯМР (400 МГц, CDCl3) δ 5.96-6.00 (m, 1H), 5.92-5.96 (m, 1H), 4.85-5.01 (m, 2H), 2.19 (ddd, J=14,4; 7,4; 3,1 Гц, 1H), 1.95 (ddd, J=14,4; 7,0; 4,3 Гц, 1H), 1.45 (s, 9H).
Стадия 6. Синтез трет-бутил-[(1R,3R)-3-гидроксициклопентил]карбамата (C47)
Смесь соединения С46 (315 мг; 1,58 ммоль) и 10%-ного палладия на угле (150 мг) в метаноле (20 мл) гидрировали при давлении 60 ф/кв. дюйм (414 кПа) в течение 4 часов. Катализатор удаляли посредством фильтрования, фильтрат концентрировали в вакууме и объединяли с неочищенным продуктом аналогичной реакции, проведенной с использованием соединения С46 (151 мг; 0,758 ммоль). После хроматографии на силикагеле (градиент: от 0% до 60% этилацетата в гептане) получали продукт в виде белого твердого вещества. Объединенный выход: 286 мг; 1,42 ммоль, 61%. GCMS m/z 145,0 [М - 2-метилпроп-1-ен]+. 1Н ЯМР (400 МГц, CDCl3) δ 4.49 (br s, 1Н), 4.36-4.42 (m, 1H), 4.09-4.24 (br m, 1H), 2.15-2.26 (m, 1H), 1.95-2.08 (m, 2H), 1.8-2.0 (v br s, 1H), 1.55-1.69 (m, 2H), 1.44 (s, 9H), 1.33-1.45 (m, 1H).
Стадия 7. Синтез трет-бутил-[(1R,3S)-3-фторциклопентил]карбамата (C48)
Пиридин-2-сульфонилфторид (252 мг; 1,56 ммоль) добавляли к смеси соединения С47 (286 мг; 1,42 ммоль) в толуоле (1,4 мл). Затем добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (0,425 мл; 2,84 ммоль) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли насыщенный водный раствор бикарбоната натрия (10 мл) и полученную смесь экстрагировали диэтиловым эфиром (3×10 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и очищали посредством хроматографии на силикагеле (градиент: от 0% до 30% этилацетата в гептане), получая продукт в виде белого твердого вещества. Выход: 181 мг; 0,890 ммоль; 63%. 1Н ЯМР (400 МГц, CDCl3) δ (5.20-5.25 (m) и 5.07-5.12 (m), JHF=54 Гц, суммарно 1H), 4.76-4.88 (br m, 1Н), 4.10-4.23 (br m, 1H), 1.99-2.20 (m, 3H), 1.66-1.94 (m, 3H), 1.45 (s, 9H).
Стадия 8. Синтез (1R,3S)-3-фторциклопентанамина, гидрохлоридной соли (С49)
Раствор хлористого водорода в 1,4-диоксане (4 М; 2,2 мл; 8,8 ммоль) добавляли к соединению С48 (181 мг; 0,890 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. После концентрирования в вакууме получали продукт в виде белого твердого вещества. Выход: 121 мг; 0,867 ммоль; 97%. 1Н ЯМР (400 МГц, CD3OD) δ (5.25-5.29 (m) и 5.11-5.16 (m), JHF=53 Гц, суммарно 1Н), 3.67-3.76 (m, 1Н), 2.35 (dddd, J=36,0; 15,6; 8,6; 4,7 Гц, 1Н), 1.79-2.27 (m, 5Н).
Стадия 9. Синтез бензил-[(1R,3S)-3-фторциклопентил]карбамата (Р4)
Триэтиламин (2,6 ммоль) и бензилхлорформиат (0,136 мл; 0,953 ммоль) добавляли к суспензии соединения С49 (121 мг; 0,867 ммоль) в дихлорметане (5 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем ее концентрировали в вакууме и очищали посредством хроматографии на силикагеле (градиент: от 0% до 40% этилацетата в гептане), получая продукт в виде белого твердого вещества. Выход: 159 мг; 0,670 ммоль; 77%. Удельное вращение: [α] -1,4° (с 1,52; дихлорметан). GCMS m/z 237,0 [М+]. 1Н ЯМР (400 МГц, CDCl3) δ 7.29-7.40 (m, 5Н), 5.10 (s, 2Н), 5.00-5.27 (m, 2Н), 4.20-4.31 (br m, 1Н), 2.00-2.20 (m, 3H), 1.69-1.98 (m, 3H).
Альтернативное получение P4
Бензил-[(1R,3S)-3-фторциклопентил]карбамат (P4)
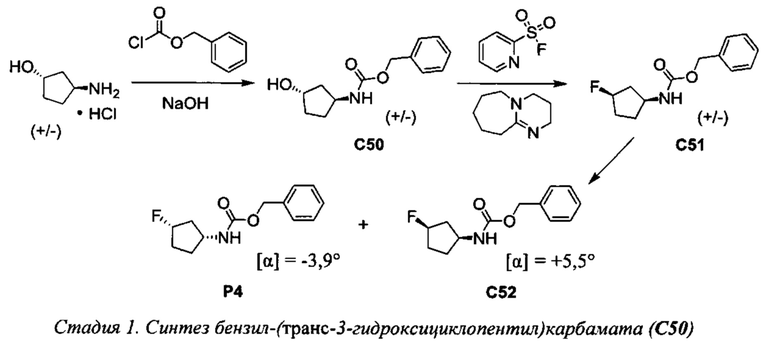
Смесь гидрохлоридной соли транс-3-аминоциклопентанола (2,30 г; 16,7 ммоль) в воде (15 мл) охлаждали до 0°С. Добавляли поочередно водный раствор гидроксида натрия (3 М; 12,3 мл; 36,9 ммоль) и бензилхлорформиат (2,62 мл; 18,4 ммоль). По завершении добавления реакционную смесь перемешивали при 0°С в течение 3 часов, после чего ее разбавляли водой и экстрагировали дихлорметаном (3×30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток перекристаллизовывали из смеси дихлорметан/гептан, получая продукт в виде белого твердого вещества (2,88 г). Маточные жидкости концентрировали и перекристаллизовывали из смеси дихлорметан/гептан, получая дополнительный продукт (286 мг). Объединенный выход: 3,17 г; 13,5 ммоль; 81%. 1Н ЯМР (400 МГц, CDCl3) δ 7.29-7.40 (m, 5Н), 5.10 (br s, 2Н), 4.60-4.77 (br s, 1H), 4.38-4.46 (m, 1H), 4.19-4.33 (m, 1H), 2.18-2.32 (m, 1H), 1.98-2.13 (m, 2H), 1.57-1.74 (m, 2H), 1.38-1.49 (m, 1H), 1.38 (d, J=3,5 Гц, 1H).
Стадия 2. Синтез бензил-(цис-3-фторциклопентил)карбамата (C51)
Пиридин-2-сульфонилфторид (2,17 г; 13,5 ммоль), затем 1,8-диазабицикло[5.4.0]ундец-7-ен (3,67 мл; 24,5 ммоль) добавляли к раствору соединения С50 (2,88 г; 12,2 ммоль) в толуоле (20 мл). Реакционную смесь перемешивали в течение 64 часов, после чего добавляли насыщенный водный раствор бикарбоната натрия (20 мл). Полученную смесь экстрагировали этилацетатом (3×20 мл); объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 0% до 40% этилацетата в гептане) получали продукт в виде твердого вещества. Выход: 2,23 г; 9,40 моль; 77%. 1Н ЯМР (400 МГц, CDCl3) δ 7.29-7.41 (m, 5Н), 5.10 (br s, 2H), 5.00-5.27 (m, 2H), 4.20-4.31 (br m, 1H), 2.00-2.20 (m, 3H), 1.69-1.98 (m, 3H).
Стадия 3. Выделение бензил-[(1R,3S)-3-фторциклопентил]карбамата (P4) и бензил-[(1S,3R)-3-фторциклопентил]карбамата (C52)
Входящие в состав соединения С51 энантиомеры (1,60 г) разделяли, используя сверхкритическую жидкостную хроматографию (колонка: Lux Amylose-2, 5 мкм, от Phenomenex; подвижная фаза: смесь 9:1 двуокись углерода/(этанол, содержащий 0,2% гидроксида аммония)). Энантиомером, элюирующимся первым, было соединение Р4, а энантиомером, элюирующимся вторым, было соединение С52. Показанные абсолютные конфигурации присваивали энантиомерам путем сравнения соответствующих им характеристик вращения с образцом соединения Р4, синтезированного в подготовительном примере Р4.
Для Р4 - выход: 612 мг; 38% для этого разделения. Удельное вращение: [α] -3,9° (с 0,455; дихлорметан). LCMS m/z 238,5 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 7.30-7.39 (m, 5Н), 5.10 (s, 2Н), 5.01-5.27 (m, 2Н), 4.20-4.31 (br m, 1Н), 2.00-2.21 (m, 3H), 1.69-1.98 (m, 3H).
Для C52 - выход: 647 мг; 40% для этого разделения. Удельное вращение: [α] +5,5° (с 0,445; дихлорметан). LCMS m/z 238,5 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 7.29-7.39 (m, 5Н), 5.10 (s, 2Н), 5.01-5.27 (m, 2Н), 4.20-4.31 (br m, 1H), 2.01-2.20 (m, 3H), 1.69-1.98 (m, 3H).
Пример 1
8-Метокси-2-[(5-метил-1,2-оксазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1H-имидазо[4,5-с]хинолин (1)
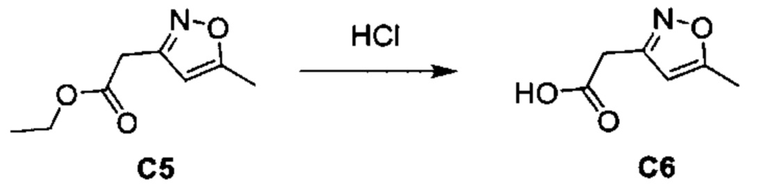
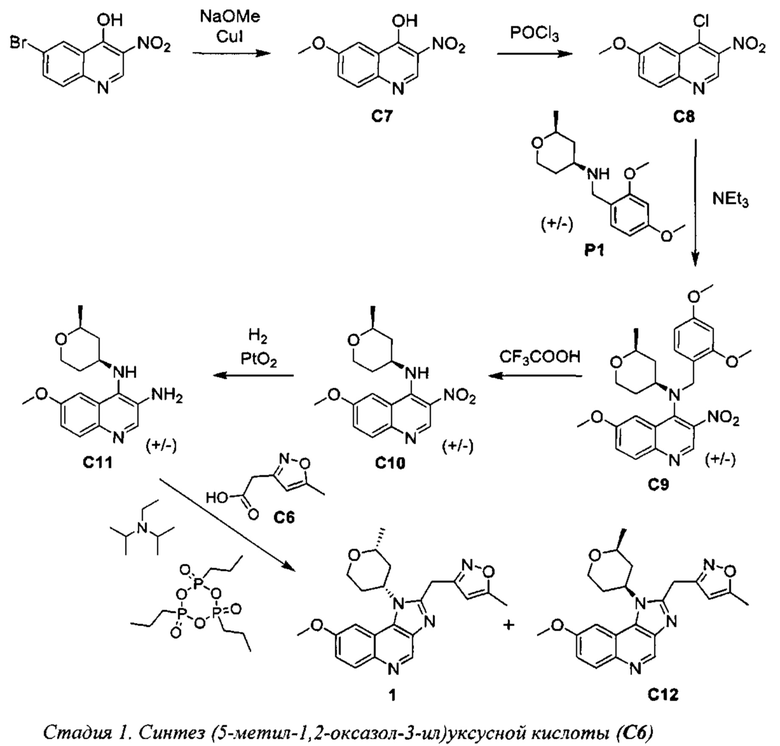
Смесь соединения С5 (которое может быть получено в соответствии с J. Gainer et al., J. Chem. Soc., Perkin Trans., 1 (1972-1999) 1976, 9, 994-997; 400 мг; 2,36 ммоль) и концентрированной соляной кислоты (5 мл) нагревали при 50°С в течение ночи. Реакционную смесь концентрировали с получением продукта. Выход: 300 мг; 2,1 ммоль, 89%. 1Н ЯМР (400 МГц, DMSO(диметилсульфоксид)-d6) δ 6.18 (br s, 1Н), 3.62 (s, 2Н), 2.37 (d, J=0,6 Гц, 3Н).
Стадия 2. Синтез 6-метокси-3-нитрохинолин-4-ола (С7)
Смесь металлического натрия (1,3 г; 56 ммоль) в метаноле (50 мл) перемешивали при комнатной температуре в течение 30 минут, после чего вносили N,N-диметилформамид (50 мл). Добавляли иодид меди(I) (4,25 г; 22,3 ммоль) и 6-бром-3-нитрохинолин-4-ол (5,00 г; 18,6 ммоль) и реакционную смесь нагревали при 100°С в течение 3 суток. Затем ее охлаждали и фильтровали; фильтрат концентрировали в вакууме и остаток разбавляли водой (200 мл). После подведения рН до 5-6 путем добавления концентрированной соляной кислоты смесь фильтровали еще раз и осадок на фильтре промывали водой (40 мл), получая продукт в виде коричневого твердого вещества. Выход: 2,8 г; 13 ммоль; 70%. 1Н ЯМР (400 МГц, DMSO-d6) δ 9.12 (br s, 1H), 7.68 (br d, J=8,5 Гц, 1H), 7.65 (d, J=2,3 Гц, 1H), 7.42 (dd, J=8,8; 2,8 Гц, 1H), 3.87 (s, 3H).
Стадия 3. Синтез 4-хлор-6-метокси-3-нитрохинолина (C8)
Оксихлорид фосфора (11,7 г; 76,3 ммоль) по каплям добавляли к раствору соединения С7 (5,8 г; 26 ммоль) в N,N-диметилформамиде (50 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов, после чего ее выливали в ледяную воду (100 мл). Полученную смесь фильтровали и осадок на фильтре промывали водой (300 мл), получая продукт в виде коричневого твердого вещества. Выход: 4,5 г; 19 ммоль; 73%.
Стадия 4. Синтез N-(2,4-диметоксибензил)-6-метокси-N-(цис-2-метилтетрагидро-2Н-пиран-4-ил)-3-нитрохинолин-4-амина (С9)
Этот эксперимент проводили в трех партиях. К смеси соединения С8 (1,5 г; 6,3 ммоль) и соединения Р1 (2,18 г; 8,22 ммоль) в N,N-диметилформамиде (15 мл) добавляли триэтиламин (1,3 г; 13 ммоль) и смесь нагревали при 80°С в течение ночи. Эти три реакционные смеси объединяли, разбавляли водой (300 мл) и экстрагировали дихлорметаном (3×150 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (3×100 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме; после очистки посредством хроматографии на силикагеле (элюент: смесь 5:1 петролейный эфир/этилацетат) получали продукт в виде желтого масла. Выход: 4,8 г; 10 ммоль; 53%. 1Н ЯМР (400 МГц, CDCl3) δ 8.94 (s, 1Н), 7.97 (d, J=9,2 Гц, 1H), 7.51 (d, J=2,9 Гц, 1H), 7.42 (dd, J=9,1; 2,8 Гц, 1H), 6.91 (d, J=8,3 Гц, 1H), 6.24 (dd, половина ABX системы, J=8,3; 2,4 Гц, 1H), 6.21 (d, половина квартета AB, J=2,3 Гц, 1H), 4.32 (квартет AB, JAB=14,8 Гц, ΔνAB=8,0 Гц, 2H), 3.98-4.05 (m, 1H), 3.88 (s, 3H), 3.73-3.84 (m, 1H), 3.70 (s, 3H), 3.48 (s, 3H), 3.38-3.47 (m, 2H), 1.82-2.00 (m, 3H), 1.51-1.62 (m, 1H), 1.18 (d, J=6,2 Гц, 3H).
Стадия 5. Синтез 6-метокси-N-(цис-2-метилтетрагидро-2Н-пиран-4-ил)-3-нитрохинолин-4-амина (С10)
Раствор соединения С9 (4,8 г; 10 ммоль) в трифторуксусной кислоте (30 мл) перемешивали при комнатной температуре в течение 30 минут, после чего ее разбавляли дихлорметаном (200 мл). Добавляли насыщенный водный раствор бикарбоната натрия (200 мл) и водный слой экстрагировали дихлорметаном (3×100 мл); объединенные органические слои промывали насыщенным водным раствором хлорида натрия (3×100 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток промывали этилацетатом (30 мл), получая продукт в виде желтого твердого вещества. Выход: 2,5 г; 7,9 ммоль, 79%. 1H ЯМР (400 МГц, CDCl3) δ 9.26 (s, 1Н), 8.87 (br d, J=8,9 Гц, 1H), 7.97 (d, J=10,0 Гц, 1H), 7.42-7.48 (m, 2H), 4.23-4.35 (m, 1H), 4.11 (br dd, J=12; 5 Гц, 1H), 3.93 (s, 3H), 3.45-3.55 (m, 2H), 2.09-2.19 (m, 2H), 1.7-1.84 (m, 1H), 1.48 (ddd, J=12; 12; 11 Гц, 1H), 1.26 (d, J=6,3 Гц, 3H).
Стадия 6. Синтез 6-метокси-N4-(цис-2-метилтетрагидро-2Н-пиран-4-ил)хинолин-3,4-диамина (С11)
К раствору соединения С10 (2,5 г; 7,9 ммоль) в смеси метанола (25 мл) и ацетонитрила (100 мл) добавляли оксид платины(IV) (500 мг; 2,2 ммоль). Реакционную смесь дегазировали и продували водородом три раза, затем перемешивали при комнатной температуре в течение 3 часов с использованием баллона, содержащего водород. Реакционную смесь фильтровали и фильтрат концентрировали, получая продукт в виде черного твердого вещества, которое использовали без дополнительной очистки. Выход: 2,0 г; 7,0 ммоль; 89%. LCMS m/z 287,9 [М+Н]+.
Стадия 7. Синтез 8-метокси-2-[(5-метил-1,2-оксазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1Н-имидазо[4,5-с]хинолина (1) и 8-метокси-2-[(5-метил-1,2-оксазол-3-ил)метил]-1-[(2S,4S)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина (С12)
К раствору соединения С11 (350 мг; 1,22 ммоль) и соединения С6 (200 мг; 1,4 ммоль) в N,N-диметилформамиде (15 мл) добавляли N,N-диизопропилэтиламин (346 мг; 2,68 ммоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 2,3 г; 3,6 ммоль) и реакционную смесь нагревали при 120°С в течение 5 часов. Затем ее разбавляли водой (80 мл) и экстрагировали этилацетатом (3×50 мл); объединенные органические слои промывали насыщенным водным раствором хлорида натрия (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После очистки посредством обращенно-фазовой HPLC (колонка: Durashell С18, 5 мкм, от Agela; подвижная фаза А: 0,225% муравьиной кислоты в воде; подвижная фаза В: ацетонитрил; градиент от 18% до 38% В) получали рацемический продукт в виде белого твердого вещества, которое затем разделяли на составляющие его энантиомеры, используя сверхкритическую жидкостную хроматографию (колонка: Chiralpak AD-3, 3 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: метанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В). Соединением, элюирующимся первым, было соединение 1, выделенное в виде белого твердого вещества. Выход: 9,2 мг; 23 мкмоль; 2%. LCMS m/z 393,0 [М+Н]+. Время удерживания: 5,51 минуты (аналитическая колонка: Chiralpak AD-3, 4,6×50 мм, 3 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: метанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В; скорость потока: 1,5 мл/минута). 1Н ЯМР (400 МГц, DMSO-d6) δ 9.01 (s, 1Н), 8.07 (d, J=9,2 Гц, 1H), 7.85-7.94 (br m, 1H), 7.35 (br d, J=9 Гц, 1H), 6.24 (s, 1H), 5.04-5.20 (br m, 1H), 4.60 (br s, 2H), 4.12-4.23 (br m, 1H), 3.97 (s, 3H), 3.54-3.72 (br m, 2H), 2.6-2.72 (br m, 1H, предположительно; частично закрыт пиком от растворителя), 2.39 (s, 3Н), 2.24-2.35 (br m, 1H), 1.93-2.05 (br m, 1Н), 1.78-1.90 (br m, 1H), 1.21 (d, J=5,9 Гц, 3Н).
Энантиомером, элюирующимся вторым, было соединение С12, также полученное в виде белого твердого вещества. Выход: 11,3 мг; 28,8 мкмоль; 2,4%. LCMS m/z 393,0 [М+Н]+. Время удерживания: 6,6 минуты (условия анализа идентичны условиям для соединения 1) 1Н ЯМР (400 МГц, DMSO-d6) δ 9.01 (s, 1Н), 8.07 (d, J=9,2 Гц, 1H), 7.85-7.94 (br m, 1H), 7.35 (dd, J=9,3; 2,5 Гц, 1H), 6.24 (s, 1H), 5.05-5.19 (br m, 1H), 4.59 (br s, 2H), 4.12-4.23 (br m, 1H), 3.97 (s, 3H), 3.55-3.72 (br m, 2H), 2.57-2.72 (br m, 1H), 2.39 (s, 3H), 2.22-2.36 (br m, 1H), 1.93-2.06 (br m, 1H), 1.78-1.91 (br m, 1H), 1.21 (d, J=6,0 Гц, 3Н). Абсолютные конфигурации соединений 1 и С12 присваивали с учетом их относительной биологической активности (см. приведенную ниже Таблицу 3, рентгеноструктурный анализ кристалла для соединения С14, и обсуждение в примере 5, стадия 3).
Пример 2
8-Хлор-2-[(5-метоксипиридин-2-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1H-имидазо[4,5-с]хинолин (2)
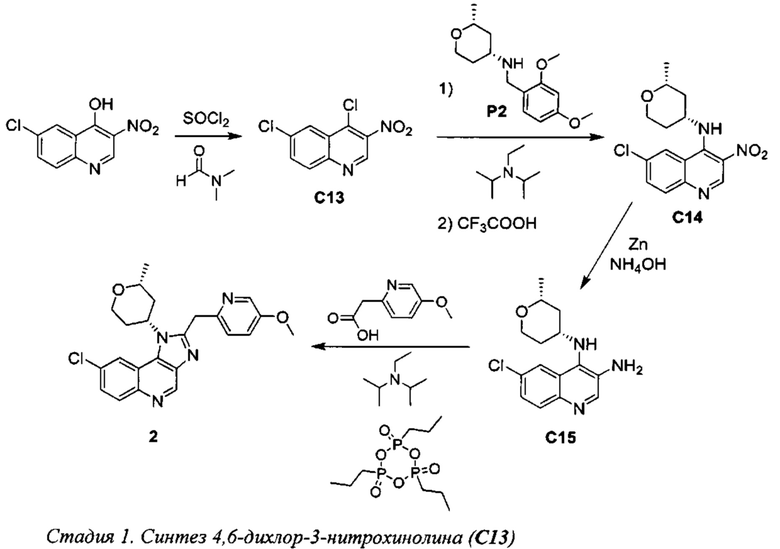
К суспензии 6-хлор-3-нитрохинолин-4-ола (15,38 г; 68,48 ммоль) в дихлорметане (140 мл) добавляли N,N-диметилформамид (3,1 мл; 40 ммоль) и тионилхлорид (97%-ный; 6,9 мл; 93 ммоль) и реакционную смесь нагревали при температуре дефлегмации. Через 5 часов ее охлаждали до комнатной температуры, разбавляли дополнительным количеством дихлорметана (25 мл) и выливали в насыщенный водный раствор бикарбоната натрия (250 мл). Водный слой экстрагировали дихлорметаном (100 мл), затем пропускали через набивку диатомовой земли, которую промывали дихлорметаном (50 мл). Объединенные органические слои и органический фильтрат сушили над сульфатом магния, фильтровали и концентрировали в вакууме, получая продукт в виде бледно-коричневого твердого вещества. Выход: 16,8 г; количественный. 1Н ЯМР (400 МГц, CDCl3) δ 9.25 (s, 1Н), 8.42 (d, J=2,2 Гц, 1Н), 8.17 (d, J=8,9 Гц, 1H), 7.89 (dd, J=9,0; 2,2 Гц, 1Н).
Стадия 2. Синтез 6-хлор-N-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-3-нитрохинолин-4-амина (С14)
Соединение С13 (12,2 г; 50,2 ммоль) добавляли к раствору соединения Р2 (13,3 г; 50,1 ммоль) и N,N-диизопропилэтиламина (13,1 мл; 75,2 ммоль) в ацетонитриле (250 мл) и реакционную смесь нагревали до 55°С в течение ночи. После концентрирования в вакууме остаток распределяли между водным раствором бикарбоната натрия (100 мл) и дихлорметаном (150 мл). Водный слой экстрагировали дихлорметаном (2×50 мл) и объединенные органические слои обрабатывали трифторуксусной кислотой (25 мл). (Осторожно: экзотермический эффект!). Через 20 минут добавляли порциями насыщенный водный раствор карбоната натрия (150 мл) и смесь оставляли перемешиваться в течение 10 минут. Водный слой дважды экстрагировали дихлорметаном и объединенные органические слои концентрировали в вакууме, получая красноватое твердое вещество (17,3 г); его растирали с диэтиловым эфиром (230 мл), получая желтое твердое вещество (14,0 г). Часть этого твердого вещества (10 г) подвергали очистке посредством сверхкритической жидкостной хроматографии (колонка: Lux Amylose-2, 5 мкм; подвижная фаза: смесь 65:35 двуокись углерода/метанол), получая продукт в виде кристаллического вещества. Указанную абсолютную конфигурацию определяли посредством рентгеноструктурного анализа монокристалла, используя это вещество: см. ниже. Выход: 7,1 г; 22 ммоль; 62% (выход скорректирован для вещества, которое не подвергали очистке). 1Н ЯМР (400 МГц, CDCl3) δ 9.36 (s, 1Н), 9.11 (br d, J=9 Гц, 1H), 8.12 (d, J=2,0 Гц, 1H), 7.98 (d, J=8,9 Гц, 1H), 7.73 (dd, J=8,9; 2,2 Гц, 1H), 4.21-4.33 (m, 1H), 4.08-4.15 (m, 1H), 3.50-3.60 (m, 2H), 2.11-2.22 (m, 2H), 1.77 (dddd, J=12; 12; 12; 5 Гц, 1H), 1.49 (ddd, J=12; 12; 11 Гц, 1H), 1.28 (d, J=6,2 Гц, 3H).
Рентгеноструктурный анализ монокристалла соединения С14
Рентгеноструктурный анализ монокристалла
Сбор данных проводили на дифрактометре APEX от Bruker при комнатной температуре. Сбор данных заключался в получении сканограмм по углам омега и фи.
Структуру разрешали прямыми методами, используя пакет программ SHELX в пространственной группе P212121. Впоследствии структуру уточняли, используя метод наименьших квадратов в полноматричном приближении. Определяли все атомы, не являющиеся атомами водорода, и их координаты уточняли с использованием анизотропных параметров смещений.
Атом водорода, расположенный при атоме азота, был найден на основании разностной карты Фурье и уточнен исходя из ограничений по расстояниям. Остальные атомы водорода помещали в рассчитанные положения и для них допускали колебания относительно их атомов-носителей. Окончательное уточнение заключалось в использовании изотропных параметров смещений для всех атомов водорода.
Анализ абсолютной структуры с применением методов максимального правдоподобия (Hooft, 2008) проводили, используя программу PLATON (Spek, 2010). Результаты указывают на то, что абсолютная структура идентифицирована корректно. С помощью этого метода вычислено, что вероятность того, что данная структура является корректной, составляет 100,0. Как сообщается, параметр Хуфта составляет 0,017 с ESD 0,09.
Окончательное значение R-фактора составляло 4,8%. Окончательная разностная карта Фурье не выявила никаких пропусков или смещения электронной плотности.
Информация о соответствующем кристалле, сборе данных и уточнении структуры суммирована в Таблице А. Координаты атомов, длины связей, углы связей, торсионные углы и параметры смещений приведены в Таблицах В-Е.
Программное обеспечение и ссылки
SHELXTL, версия 5.1, Bruker AXS, 1997.
PLATON, A.L. Spek J. Appl. Cryst., 2003, 36, 7-13.
MERCURY, C.F. Macrae, P.R. Edington, P. McCabe, E. Pidcock, G.P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Cryst., 2006, 39, 453-457.
OLEX2, O.V. Dolomanov, L.J. Bourhis, R.J. Gildea, J.A.K. Howard and H. Puschmann, J. Appl. Cryst., 2009, 42, 339-341.
R.W.W. Hooft, L.H. Straver and A.L. Spek, J. Appl. Cryst., 2008, 41, 96-103.
H.D. Flack, Acta Cryst., 1983, A39, 867-881.


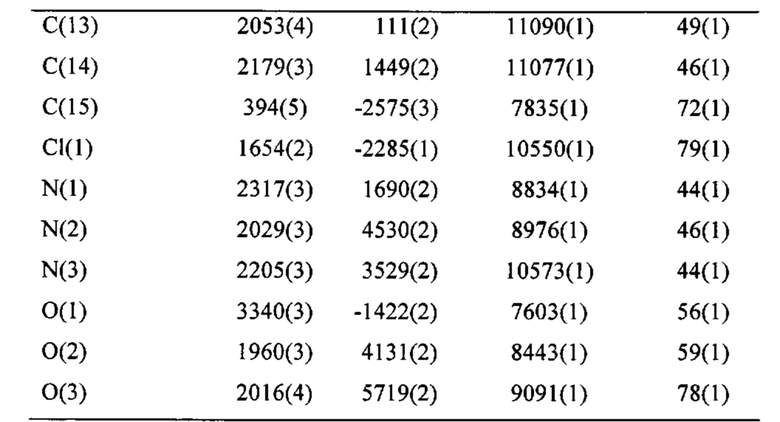



Для создания эквивалентных атомов использовали преобразования симметрии.


Стадия 3. Синтез 6-хлор-N4-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]хинолин-3,4-диамина (С15)
Цинковую пыль (97,5%-ную; 12,3 г; 183 ммоль) добавляли в виде одной порции к суспензии соединения С14 (7,40 г; 23,0 ммоль) в метаноле (100 мл) и концентрированном растворе гидроксида аммония (100 мл). Через 1 час реакционную смесь фильтровали через диатомовую землю; фильтрующую набивку промывали дихлорметаном (70 мл). Фильтрат разбавляли водой и водный слой экстрагировали дихлорметаном (2×60 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и очищали посредством хроматографии на силикагеле (градиент: от 40% до 100% этилацетата в гептане), получая продукт. Выход: 3,68 г; 12,6 ммоль; 55%. 1Н ЯМР (400 МГц, CDCl3) δ 8.48 (s, 1Н), 7.91 (d, J=8,9 Гц, 1H), 7.74 (d, J=2,2 Гц, 1H), 7.40 (dd, J=8,9; 2,2 Гц, 1H), 4.02 (br dd, J=12; 5 Гц, 1H), 3.88 (br s, 2H), 3.29-3.56 (m, 4H), 1.82-1.96 (m, 2H), 1.56 (dddd, J=12; 12; 12; 5 Гц, 1H), 1.21-1.31 (m, 1H), 1.21 (d, J=6,2 Гц, 3H).
Стадия 4. Синтез 8-хлор-2-[(5-метоксипиридин-2-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1Н-имидазо[4,5-с]хинолина (2)
К смеси соединения С15 (400 мг; 1,37 ммоль) и (5-метоксипиридин-2-ил)уксусной кислоты (229 мг; 1,37 ммоль) в N,N-диметилформамиде (3 мл) добавляли N,N-диизопропилэтиламин (532 мг; 4,12 ммоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (1,31 г; 4,12 ммоль; в виде 50%-ного раствора в этилацетате). Реакционную смесь нагревали при 100°С в течение ночи, после чего ее охлаждали до комнатной температуры, объединяли с двумя мелкомасштабными реакционными смесями, полученными после проведения аналогичных реакций с соединением С15 (в целом 40 мг; 0,14 ммоль), и разбавляли водой (100 мл). Полученную смесь экстрагировали дихлорметаном (2×200 мл) и объединенные органические слои концентрировали в вакууме. После хроматографии на силикагеле (элюент: 2% метанола в этилацетате), затем обращенно-фазовой HPLC (колонка: Diamonsil (2) С18, 5 мкм от DIKMA; подвижная фаза А: 0,225% муравьиной кислоты в воде; подвижная фаза В: ацетонитрил; градиент: от 22% до 42% В), получали продукт в виде желтого твердого вещества. Выход: 147 мг; 0,348 ммоль; 23%. LCMS m/z 423,0 [М+Н]+. 1Н ЯМР (400 МГц, CD3OD) δ 9.16 (s, 1Н), 8.70-8.82 (br m, 1H), 8.17-8.22 (m, 2H), 7.75 (dd, J=8,8; 2,1 Гц, 1H), 7.35-7.43 (m, 2H), 5.23-5.42 (br m, 1H), 4.69 (s, 2H), 4.18-4.26 (m, 1H), 3.86 (s, 3H), 3.61-3.76 (br m, 2H), 2.56-2.69 (br m, 1H), 2.24-2.41 (br m, 1H), 1.75-1.91 (br m, 1H), 1.61-1.75 (br m, 1H), 1.28 (d, J=6,2 Гц, 3Н).
Пример 3
2-[(5-Метил-1,2-оксазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1H-имидазо[4,5-с]хинолин-8-карбонитрил (3)
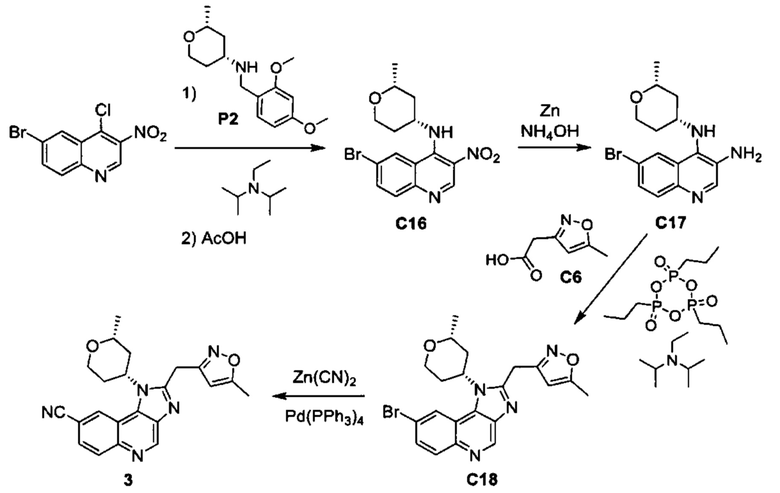
Стадия 1. Синтез 6-бром-N-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-3-нитрохинолин-4-амина (С16)
6-Бром-4-хлор-3-нитрохинолин (1,93 г; 6,71 ммоль) добавляли к раствору соединения Р2 (2,35 г; 8,86 ммоль) и N,N-диизопропилэтиламина (3,4 мл; 20 ммоль) в ацетонитриле (39 мл) и реакционную смесь нагревали до 45°С в течение 18 часов. Затем добавляли уксусную кислоту (1,8 мл; 24 ммоль) и перемешивание продолжали в течение 5 часов при 100°С, после чего реакционную смесь оставляли охлаждаться до комнатной температуры и перемешиваться в течение 18 часов. После удаления растворителя в вакууме остаток переносили в дихлорметан и промывали насыщенным водным раствором бикарбоната натрия. Органический слой загружали в колонку с силикагелем и элюировали (градиент: от 0% до 5% метанола в дихлорметане), получая продукт в виде коричневого масла. Выход: 1,40 г; 3,82 ммоль; 57%. LCMS m/z 366,0; 368,2 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.37 (s, 1Н), 9.13 (br d, J=9 Гц, 1H), 8.30 (br d, J=2,0 Гц, 1H), 7.91 (br d, половина квартета AB, J=8,8 Гц, 1H), 7.86 (dd, половина ABX системы, J=8,9; 2,0 Гц, 1H), 4.21-4.32 (m, 1H), 4.12 (ddd, J=12,1; 4,7; 1,7 Гц, 1H), 3.52-3.60 (m, 2H), 2.11-2.21 (m, 2H), 1.78 (dddd, J=12; 12; 11; 5 Гц, 1H), 1.49 (ddd, J=13; 11; 11 Гц, 1H), 1.28 (d, J=6,2 Гц, 3H).
Стадия 2. Синтез 6-бром-N4-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]хинолин-3,4-диамина (С17)
Цинк (97,5%-ный; 2,33 г; 34,7 ммоль) добавляли в виде одной порции при 0°С к суспензии соединения С16 (1,40 г; 3,82 ммоль) в метаноле (6 мл) и концентрированном растворе гидроксида аммония (6 мл) и реакционную смесь перемешивали при 0°С в течение 30 минут. Затем ее оставляли нагреваться до комнатной температуры и перемешиваться в течение 45 минут, после чего ее фильтровали через диатомовую землю. Осадок на фильтре промывали дихлорметаном и объединенные фильтраты разбавляли водой. Водный слой экстрагировали дихлорметаном, объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 0% до 3% метанола в дихлорметане) получали продукт в виде рыжевато-коричневой пены. Выход: 836 мг; 2,49 ммоль; 65%. LCMS m/z 336,1; 338,2 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 8.49 (s, 1Н), 7.92 (d, J=2,1 Гц, 1H), 7.84 (d, J=8,8 Гц, 1H), 7.53 (dd, J=8,9; 2,1 Гц, 1H), 4.03 (ddd, J=11,8; 4,7; 1,7 Гц, 1H), 3.88 (br s, 2H), 3.33-3.56 (m, 4H), 1.82-1.96 (m, 2H), 1.50-1.62 (m, 1H), 1.26 (ddd, J=12; 11; 11 Гц, 1H), 1.21 (d, J=6,2 Гц, 3H).
Стадия 3. Синтез 8-бром-2-[(5-метил-1,2-оксазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1H-имидазо[4,5-с]хинолина (С18)
Смесь соединения С17 (836 мг; 2,49 ммоль), соединения С6 (281 мг; 1,99 ммоль), 2,4,6-триоксида 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ного раствора в этилацетате; 1,9 мл; 3,2 ммоль) и N,N-диизопропилэтиламина (0,87 мл; 5,0 ммоль) в этилацетате (10 мл) перемешивали при 50°С в течение ночи. Добавляли уксусную кислоту (1 эквивалент) и нагревание продолжали при 115°С в течение 5 часов, после чего реакционную смесь оставляли охлаждаться до комнатной температуры и перемешиваться в течение 18 часов. После удаления летучих веществ в вакууме остаток переносили в дихлорметан и промывали насыщенным водным раствором бикарбоната натрия. Органический слой загружали в колонку с силикагелем и элюировали (градиент: от 0% до 5% метанола в дихлорметане), получая продукт в виде рыжевато-коричневого твердого вещества. Выход: 507 мг; 1,15 ммоль; 58%. LCMS m/z 441,2; 443,3 [М+Н]+.
Стадия 4. Синтез 2-[(5-метил-1,2-оксазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1H-имидазо[4,5-с]хинолин-8-карбонитрила (3)
Тетракис(трифенилфосфин)палладий(0) (262 мг; 0,227 ммоль) добавляли к смеси соединения С18 (500 мг; 1,13 ммоль) и цианида цинка (99%-ного; 644 мг; 5,43 ммоль) в N,N-диметилформамиде (5 мл) и реакционную колбу подвергали трем циклам вакуумирования, после чего заполняли азотом. Затем реакционную смесь нагревали при 100°С в течение 20 часов, после чего ее распределяли между водой и этилацетатом и фильтровали через диатомовую землю. Осадок на фильтре промывали этилацетатом и водный слой из объединенных фильтратов дважды экстрагировали этилацетатом. Объединенные органические слои промывали 5 раз водой, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент от 0% до 3% метанола в метиленхлориде) получали смесь продукта и соединения С18 (324 мг; приблизительно 1:1), ввиду чего эту смесь повторно обрабатывали в данных реакционных условиях. К смеси цианида цинка (99%-ного; 422 мг; 3,56 ммоль) и вещества, содержащего соединения С18 и 3 (324 мг), в N,N-диметилформамиде (2 мл) добавляли тетракис(трифенилфосфин)палладий(0) (172 мг; 0,149 ммоль) и реакционную колбу подвергали трем циклам вакуумирования, после чего заполняли азотом. Затем реакционную смесь нагревали при 100°С в течение 2 часов, распределяли между водой и этилацетатом и фильтровали через диатомовую землю. Осадок на фильтре промывали этилацетатом и водный слой из объединенных фильтратов дважды экстрагировали этилацетатом. Объединенные органические слои промывали 5 раз водой, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент от 0% до 5% метанола в дихлорметане) получали масло, которое растирали с диэтиловым эфиром, получая рыжевато-коричневое твердое вещество. Его перекристаллизовывали из смеси этилацетат/гептан, получая продукт в виде беловатого твердого вещества. Выход: 97 мг; 0,25 ммоль; 22%. LCMS m/z 388,2 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.41 (s, 1Н), 8.9-9.1 (br m, 1H), 8.38 (d, J=8,7 Гц, 1H), 7.86 (dd, J=8,6; 1,5 Гц, 1H), 6.02 (br s, 1H), 5.15-5.28 (br m, 1H), 4.53 (s, 2H), 4.32 (br dd, J=12; 5 Гц, 1H), 3.66-3.79 (br m, 2H), 2.53-2.69 (br m, 1H), 2.41 (s, 3H), 2.23-2.4 (br m, 1H), 1.66-1.96 (br m, 2H), 1.36 (d, J=6,2 Гц, 3H).
Пример 4
8-Хлор-2-[(5-метил-1,2-оксазол-3-ил)метил-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолин (4)
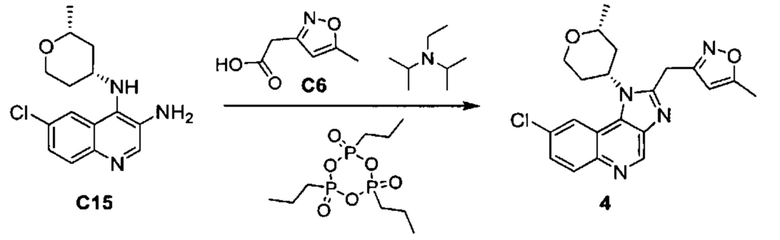
Смесь соединения С15 (400 мг; 1,4 ммоль), (5-метил-1,2-оксазол-3-ил)уксусной кислоты (155 мг; 1,10 ммоль), 2,4,6-триоксида 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ного раствора в этилацетате; 1,0 мл; 1,7 ммоль) и N,N-диизопропилэтиламина (0,48 мл; 2,8 ммоль) в этилацетате (10 мл) перемешивали при 50°С в течение ночи. Реакционную смесь концентрировали в вакууме для удаления большей части этилацетата, затем разбавляли уксусной кислотой и нагревали до 115°С. Когда по данным LCMS анализа делалось заключение о завершении реакции, реакционную смесь концентрировали при пониженном давлении, переносили в дихлорметан и промывали насыщенным водным раствором бикарбоната натрия. Водный слой один раз экстрагировали дихлорметаном, объединенные органические слои адсорбировали на силикагеле и хроматографировали (элюент:этилацетат). Продукт (405 мг) смешивали с диэтиловым эфиром и оставляли перемешиваться в течение 2 суток, после чего твердое вещество собирали фильтрованием и промывали смесью 3:1 гептан/диэтиловый эфир, получая продукт (239 мг) в виде твердого вещества. С применением дифракции рентгеновских лучей на порошке было показано, что это вещество является кристаллическим. Объединенные фильтраты концентрировали в вакууме, смешивали с диэтиловым эфиром (4 мл) и перемешивали в течение 2 часов, после чего добавляли гептан (1 мл). Через 2 часа снова добавляли гептан (2 мл) и перемешивание продолжали в течение ночи. Добавляли дополнительное количество гептана (1 мл) и после перемешивания в течение еще одной ночи имеющееся твердое вещество выделяли посредством фильтрования и промывали гептаном, получая дополнительное количество продукта (99 мг). Общий выход: 338 мг; 0,852 ммоль; 77%. LCMS m/z 397,3; 399,3 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3), характерные пики: δ 9.29 (s, 1Н), 8.58-8.73 (br m, 1Н), 8.23 (d, J=9,0 Гц, 1H), 7.64 (dd, J=8,8; 2,0 Гц, 1H), 6.00 (br s, 1H), 5.09-5.25 (br m, 1H), 4.51 (s, 2H), 4.30 (br dd, J=12; 5 Гц, 1H), 3.65-3.79 (br m, 2H), 2.59-2.77 (br m, 1H), 2.40 (s, 3H), 2.32-2.47 (m, 1H), 1.73-1.88 (br m, 1H), 1.35 (d, J=6,2 Гц, 3Н).
Пример 5
8-Хлор-2-[(5-метил-1,2,4-оксадиазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1H-имидазо[4,5-с]хинолин (5)
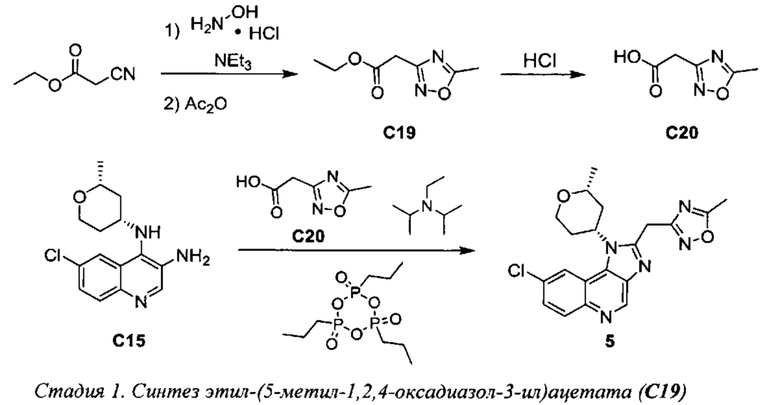
Этот эксперимент проводили в двух идентичных партиях. При 0°С к смеси гидроксиламина гидрохлорида (39,3 г; 566 ммоль) в этаноле (1,2 л) добавляли триэтиламин (86 г; 850 ммоль). Затем эту смесь перемешивали в течение 10 минут по каплям добавляли этилцианоацетат (32 г; 280 ммоль) и реакционную смесь оставляли нагреваться до комнатной температуры и перемешиваться в течение ночи. Вносили дополнительное количество триэтиламина (86 г; 850 ммоль), после этого уксусный ангидрид (89,5 г; 877 ммоль) и перемешивание продолжали в течение 2 часов при комнатной температуре. Затем реакционную смесь перемешивали в течение ночи при 90°С. На этом этапе две партии объединяли и концентрировали в вакууме. Остаток распределяли между этилацетатом (1 л) и соляной кислотой (1 М, 500 мл) и водный слой экстрагировали этилацетатом (2×100 мл); объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия (1 л) до достижения рН равного 7, затем сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. После хроматографии на силикагеле (градиент: от 0% до 20% этилацетата в петролейном эфире) получали продукт в виде бесцветного масла. Выход: 20,0 г; 118 ммоль; 21%. 1Н ЯМР (400 МГц, CDCl3) δ 4.20 (q, J=7,1 Гц, 2Н), 3.76 (s, 2Н), 2.58 (s, 3Н), 1.26 (t, J=7,2 Гц, 3Н).
Стадия 2. Синтез (5-метил-1,2,4-оксадиазол-3-ил)уксусной кислоты (С20)
Смесь соединения С19 (4,30 г; 25,3 ммоль) и соляной кислоты (2 М; 50 мл; 100 ммоль) перемешивали в течение 2 суток при комнатной температуре, затем нагревали до 50°С в течение 2 суток. Добавляли концентрированную соляную кислоту (2 мл) и нагревание продолжали при 50°С в течение 66 часов, после чего реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме до объема приблизительно 10 мл. Эту смесь экстрагировали восемь раз дихлорметаном и объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая продукт в виде белого твердого вещества. Выход: 2,85 г; 20,1 ммоль; 79%. 1Н ЯМР (400 МГц, CDCl3) δ 9.43 (br s, 1Н), 3.86 (s, 2H), 2.62 (s, 3H).
Стадия 3. Синтез 8-хлор-2-[(5-метил-1,2,4-оксадиазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1H-имидазо[4,5-с]хинолина (5)
Смесь соединения С15 (770 мг; 2,64 ммоль), соединения С20 (300 мг; 2,11 ммоль), 2,4,6-триоксида 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ного раствора в этилацетате; 2,0 мл; 3,4 ммоль) и N,N-диизопропилэтиламина (0,92 мл; 5,3 ммоль) в этилацетате (10 мл) нагревали при 60°С в течение 2 часов, затем при температуре дефлегмации в течение 2 часов. Вносили дополнительное количество 2,4,6-триоксида 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ного раствора в этилацетате; 2,0 мл; 3,4 ммоль) и нагревание при температуре дефлегмации продолжали в течение ночи. Реакционную смесь концентрировали в вакууме для удаления большей части этилацетата, затем разбавляли уксусной кислотой и нагревали до 100°С в течение ночи. После удаления растворителей при пониженном давлении остаток переносили в дихлорметан и промывали насыщенным водным раствором бикарбоната натрия; водный слой экстрагировали дихлорметаном и объединенные органические слои адсорбировали на диатомовой земле и хроматографировали с использованием силикагеля (градиент: от 0% до 5% метанола в дихлорметане). Продукт (739 мг) смешивали с диэтиловым эфиром (7 мл) и перемешивали в течение 2 суток. Полученное твердое вещество собирали фильтрованием и промывали смесью 3:1 гептан/диэтиловый эфир, получая продукт в виде беловатого твердого вещества (329 мг). Объединенные фильтраты концентрировали в вакууме, растворяли в диэтиловом эфире (3 мл) и перемешивали в течение 2 суток. После фильтрования и промывки осадка на фильтре смесью 3:1 гептан/диэтиловый эфир получали дополнительное количество продукта в виде беловатого твердого вещества. Объединенный выход: 390 мг; 0,98 ммоль; 46%. LCMS m/z 398,2; 400,2 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.29 (s, 1Н), 8.56-8.78 (br m, 1Н), 8.24 (d, J=8,8 Гц, 1H), 7.65 (dd, J=8,9; 1,9 Гц, 1H), 4.94-5.17 (br m, 1H), 4.60 (s, 2H), 4.28-4.39 (m, 1H), 3.63-3.81 (br m, 2H), 2.67-2.88 (br m, 1H), 2.60 (s, 3H), 2.38-2.6 (br m, 1H), 1.80-2.08 (br m, 2H), 1.38 (d, J=6,2 Гц, 3H).
По сравнению с соединением примера 5 энантиомер соединения примера 5 (пример 51) оказался значительно менее эффективным (см. Таблицу 3 с данными по биологической активности). Абсолютные конфигурации разделенных энантиомеров, описанных в данной заявке, присваивали на основании их относительной биологической активности, соответствующей этим двум соединениям.
Пример 6
8-Бром-1-[(1S,3R)-3-фторциклопентил]-2-метил-1Н-имидазо[4,5-с]хинолин (6)
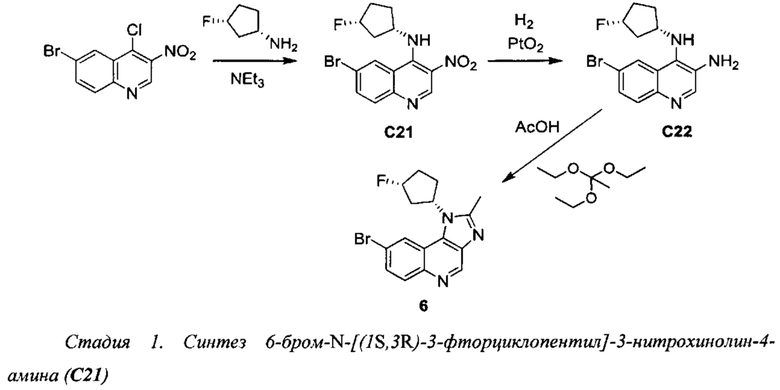
Триэтиламин (364 мг; 3,60 ммоль) добавляли к смеси 6-бром-4-хлор-3-нитрохинолина (515 мг; 1,79 ммоль) и (1S,3R)-3-фторциклопентанамина (250 мг; 2,4 ммоль) в тетрагидрофуране (10 мл) и реакционную смесь нагревали при 45°С в течение 2 часов. Затем ее разбавляли водой (50 мл) и экстрагировали этилацетатом (3×20 мл); объединенные органические слои промывали насыщенным водным раствором хлорида натрия (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая продукт в виде желтого твердого вещества. Выход: 439 мг; 1,24 ммоль; 69%. LCMS m/z 355,6 [М+Н]+.
Стадия 2. Синтез 6-бром-N4-[(1S,3R)-3-фторциклопентил]хинолин-3,4-диамина (С22)
К смеси соединения С21 (500 мг; 1,4 ммоль) в метаноле (50 мл) и ацетонитриле (10 мл) добавляли оксид платины(IV) (50 мг; 0,22 ммоль). Суспензию дегазировали и трижды продували водородом, после чего реакционную смесь перемешивали при комнатной температуре в течение 1,5 часа в атмосфере водорода с использованием баллона. После фильтрования реакционной смеси осадок на фильтре промывали ацетонитрилом (3×10 мл) и объединенные фильтраты концентрировали в вакууме, получая продукт в виде желтого масла. Выход: 400 мг; 1,2 ммоль; 86%. LCMS m/z 323,8 [М+Н]+.
Стадия 3. Синтез 8-бром-1-[(1S,3R)-3-фторциклопентил]-2-метил-1H-имидазо[4,5-с]хинолина (6)
Раствор соединения С22 (90 мг; 0,28 ммоль) и уксусной кислоты (каталитическое количество) в 1,1,1-триэтоксиэтане (5 мл) перемешивали при 110°С в течение ночи, после чего реакционную смесь концентрировали в вакууме. После очистки посредством обращенно-фазовой HPLC (колонка: Triart С18 серии YMC-Actus, 5 мкм; подвижная фаза А: 0,225% муравьиной кислоты в воде; подвижная фаза В: ацетонитрил; градиент: от 25% до 45% В) получали продукт в виде желтого твердого вещества. Выход: 30,2 мг; 86,7 мкмоль; 31%. LCMS m/z 350,0 [М+Н]+. 1Н ЯМР (400 МГц, DMSO-d6) δ 9.22 (s, 1Н), 8.69-8.73 (m, 1H), 8.11 (d, J=9,0 Гц, 1H), 7.86 (dd, J=8,9; 1,9 Гц, 1H), 5.40-5.53 (m, 1.5H), 5.31-5.38 (m, 0.5H), 2.8-2.96 (m, 1H), 2.78 (s, 3H), 2.01-2.5 (m, 5H).
Пример 7
1-[(2R,4R)-2-Метилтетрагидро-2Н-пиран-4-ил]-2-(1,3-тиазол-4-илметил)-1Н-имидазо[4,5-с][1,5]нафтиридин (7)
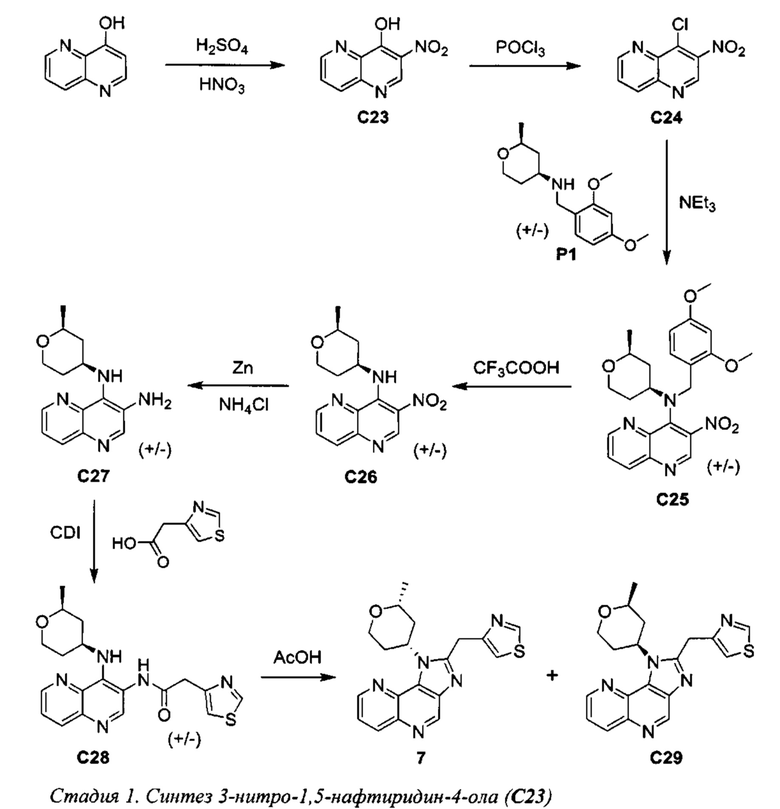
Концентрированную азотную кислоту (1,5 мл) добавляли к раствору 1,5-нафтиридин-4-ола (600 мг; 4,1 ммоль) в концентрированной серной кислоте (4,5 мл) и реакционную смесь перемешивали при 90°С в течение ночи. Затем ее выливали в воду, охлаждали в ледяной бане и рН подводили до 6-7, добавляя водный раствор гидроксида аммония. Полученную смесь перемешивали в ледяной бане в течение 10 минут, затем фильтровали; собранное твердое вещество промывали водой, получая продукт в виде желтого твердого вещества. Выход: 0,60 г; 3,1 ммоль; 76%. 1Н ЯМР (400 МГц, DMSO-d6) δ 8.96 (s, 1Н), 8.55-8.60 (m, 1H), 7.98 (br d, J=8,2 Гц, 1H), 7.54 (dd, J=8,3; 4,3 Гц, 1H).
Стадия 2. Синтез 4-хлор-3-нитро-1,5-нафтиридина (С24)
Оксихлорид фосфора (624 мг; 4,08 ммоль) по каплям добавляли к раствору соединения С23 (0,60 г; 3,1 ммоль) в N,N-диметилформамиде (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, после чего ее выливали в ледяную воду (80 мл). Полученную смесь фильтровали и осадок на фильтре промывали водой (30 мл), получая продукт в виде желтого твердого вещества. Выход: 0,36 г; 1,7 ммоль; 55%. 1Н ЯМР (400 МГц, DMSO-d6) δ 9.50 (s, 1Н), 9.28 (dd, J=4,1; 1,5 Гц, 1H), 8.65 (dd, J=8,5; 1,5 Гц, 1Н), 8.09 (dd, J=8,5; 4,1 Гц, 1Н).
Стадия 3. Синтез N-(2,4-диметоксибензил)-N-(цис-2-метилтетрагидро-2Н-пиран-4-ил)-3-нитро-1,5-нафтиридин-4-амина (С25)
Триэтиламин (580 мг; 5,7 ммоль) добавляли к смеси соединения С24 (600 мг; 2,9 ммоль) и соединения Р1 (761 мг; 2,87 ммоль) в N,N-диметилформамиде (10 мл). Реакционную смесь нагревали при 50°С в течение 1 часа, после чего ее разбавляли водой (50 мл) и экстрагировали этилацетатом (3×30 мл). После промывания объединенных органических слоев насыщенным водным раствором хлорида натрия (100 мл) их сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 0% до 40% этилацетата в петролейном эфире) получали продукт в виде желтого твердого вещества. Выход: 1,0 г; 2,3 ммоль; 80%. 1Н ЯМР (400 МГц, CDCl3) δ 8.96 (s, 1Н), 8.90 (dd, J=4,1; 1,7 Гц, 1Н), 8.29 (dd, J=8,5; 1,7 Гц, 1Н), 7.65 (dd, J=8,5; 4,1 Гц, 1Н), 6.89 (d, J=9,0 Гц, 1Н), 6.16-6.20 (m, 2Н), 4.76-4.86 (m, 1Н), 4.56 (квартет АВ, JAB=16,1 Гц, ΔνAB=18,6 Гц, 2Н), 4.07-4.14 (m, 1Н), 3.69 (s, 3Н), 3.47-3.55 (m, 2Н), 3.46 (s, 3Н), 2.25-2.34 (m, 2Н), 2.04-2.16 (m, 1Н), 1.76-1.88 (m, 1Н), 1.27 (d, J=6,3 Гц, 3Н).
Стадия 4. Синтез N-(цис-2-метилтетрагидро-2Н-пиран-4-ил)-3-нитро-1,5-нафтиридин-4-амина (С26)
Смесь соединения С25 (1,0 г; 2,3 ммоль) и трифторуксусной кислоты (20 мл) перемешивали при комнатной температуре в течение 30 минут, после чего ее концентрировали в вакууме. После подведения рН оставшегося раствора до 7-8 посредством добавления насыщенного водного раствора бикарбоната натрия (100 мл) его экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая продукт в виде желтого твердого вещества. Выход: 0,60 г; 2,1 ммоль; 91%. 1Н ЯМР (400 МГц, CDCl3), характерные пики: δ 9.41 (br s, 1Н), 8.83 (dd, J=4,1; 1,6 Гц, 1H), 8.29 (br dd, J=8,4; 1,6 Гц, 1H), 7.69 (dd, J=8,5; 4,1 Гц, 1H), 4.11 (br dd, J=12; 4 Гц, 1H), 3.59-3.69 (m, 2H), 2.15-2.30 (m, 2H), 1.61-1.74 (m, 1H), 1.35-1.45 (m, 1H), 1.28 (d, J=6,3 Гц, 3Н).
Стадия 5. Синтез N4-(цис-2-метилтетрагидро-2Н-пиран-4-ил)-1,5-нафтиридин-3,4-диамина (С27)
К суспензии соединения С26 (600 мг; 2,1 ммоль) в тетрагидрофуране (10 мл) и воде (5 мл) добавляли цинковую пыль (677 мг; 10,4 ммоль) и хлорид аммония (551 мг; 10,3 ммоль). Затем реакционную смесь перемешивали при 60°С в течение 40 минут, после чего ее разбавляли водой (50 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая продукт в виде желтого твердого вещества. Выход: 0,40 г; 1,5 ммоль; 71%. LCMS m/z 259,0 [М+Н]+.
Стадия 6. Синтез N-{4-[(цис-2-метилтетрагидро-2H-пиран-4-ил)амино]-1,5-нафтиридин-3-ил}-2-(1,3-тиазол-4-ил)ацетамида (С28)
1,1'-Карбонилдиимидазол (CDI; 250 мг; 1,54 ммоль) добавляли к смеси соединения С27 (200 мг; 0,77 ммоль) и 1,3-тиазол-4-илуксусной кислоты (138 мг; 0,964 ммоль) в N,N-диметилформамиде (3 мл). Реакционную смесь нагревали при 50°С в течение ночи, после чего ее разбавляли водой (30 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая продукт, который переносили непосредственно на следующую стадию. LCMS m/z 384,2 [М+Н]+.
Стадия 7. Синтез 1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-2-(1,3-тиазол-4-илметил)-1Н-имидазо[4,5-с][1,5]нафтиридина (7) и 1-[(2S,4S)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с][1,5]нафтиридина (С29)
Соединение С28 (с предыдущей стадии; 295 мг; не более 0,77 ммоль) и уксусную кислоту (2 мл) объединяли в герметично закрытой пробирке и нагревали в микроволновом реакторе при 155°С в течение 20 минут. Реакционную смесь концентрировали в вакууме и очищали обращенно-фазовой HPLC (колонка: Triart С18 серии YMC-Actus, 5 мкм; подвижная фаза А: 0,225% муравьиной кислоты в воде; подвижная фаза В: ацетонитрил; градиент: от 23% до 43% В), получая рацемическую смесь продуктов в виде желтого твердого вещества. Выход: 25 мг; 68 мкмоль; 9% за 2 стадии. Входящие в состав энантиомеры разделяли посредством сверхкритической жидкостной хроматографии (колонка: Chiralcel OD-3, 3 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: метанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В).
Соединение примера 7, элюирующийся вторым энантиомер, выделяли в виде желтого твердого вещества. Выход: 9,0 мг; 25 мкмоль; 3% за две стадии. Время удерживания: 6,37 минуты (аналитическая колонка: Chiralcel OD-3, 4,6×150 мм, 3 мкм; градиент такой же, который указан выше; скорость потока: 1,5 мл/минута). LCMS m/z 366,0 [М+Н]+. 1Н ЯМР (400 МГц, DMSO-d6) δ 9.26 (s, 1Н), 9.05 (d, J=1,9 Гц, 1H), 9.02 (dd, J=4,3; 1,6 Гц, 1H), 8.53 (dd, J=8,5; 1,7 Гц, 1H), 7.74 (dd, J=8,5; 4,3 Гц, 1H), 7.65 (br s, 1H), 4.86-5.05 (br m, 1H), 4.76 (s, 2H), 3.96-4.05 (m, 1H), 3.44-3.60 (m, 2H), 3.13-3.3 (br m, 1H), 2.85-3.07 (br m, 1H), 1.31-1.55 (br m, 2H), 1.13 (d, J=6,2 Гц, 3H).
Энантиомер C29 элюировался первым, и его также выделяли в виде желтого твердого вещества. Выход: 6,5 мг; 18 мкмоль; 2% за две стадии. Время удерживания: 6,16 минуты с использованием идентичной системы аналитической HPLC. LCMS m/z 366,0 [М+Н]+. 1Н ЯМР (400 МГц, DMSO-d6) δ 9.26 (s, 1Н), 9.05 (d, J=2,0 Гц, 1H), 9.02 (dd, J=4,1; 1,6 Гц, 1H), 8.53 (dd, J=8,4; 1,6 Гц, 1H), 7.74 (dd, J=8,4; 4,3 Гц, 1H), 7.65 (br s, 1H), 4.86-5.05 (br m, 1H), 4.76 (s, 2H), 3.97-4.04 (m, 1H), 3.44-3.60 (m, 2H), 3.14-3.28 (br m, 1H), 2.86-3.08 (br m, 1H), 1.31-1.56 (br m, 2H), 1.13 (d, J=6,2 Гц, 3H).
Пример 8
8-Хлор-2-(имидазо[2,1-b][1,3,4]тиадиазол-6-илметил)-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1Н-имидазо[4,5-с]хинолин (8)
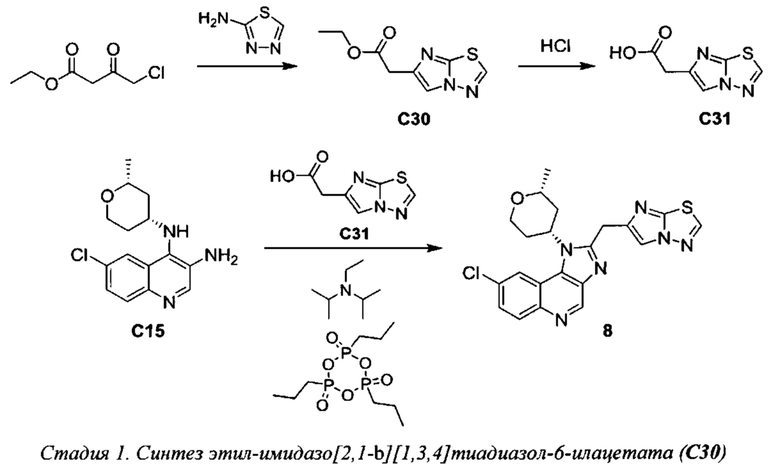
Раствор 1,3,4-тиадиазол-2-амина (3,0 г; 30 ммоль) и этил-4-хлор-3-оксобутаноата (7,4 г; 45 ммоль) в безводном этаноле (50 мл) нагревали при температуре дефлегмации в течение 24 часов, после чего реакционную смесь концентрировали в вакууме. Остаток растворяли о 10%-ной соляной кислоте и промывали хлороформом (3×50 мл); затем водный слой нейтрализовали бикарбонатом натрия и экстрагировали хлороформом (3×50 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлорида натрия (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая продукт в виде желтого масла. Выход: 1,0 г; 4,7 ммоль; 16%. 1H ЯМР (400 МГц, CDCl3) δ 8.51 (s, 1H), 7.80 (t, J=0,7 Гц, 1Н), 4.21 (q, J=7,2 Гц, 2Н), 3.77 (d, J=0,6 Гц, 2Н), 1.29 (t, J=7,2 Гц, 3Н).
Стадия 2. Синтез имидазо[2,1-b][1,3,4]тиадиазол-6-илуксусной кислоты (С31)
Раствор соединения С30 (1,0 г; 4,7 ммоль) в соляной кислоте (5 мл) нагревали при температуре дефлегмации в течение ночи. Затем реакционную смесь концентрировали в вакууме и остаток промывали дихлорметаном (10 мл), получая продукт в виде коричневого твердого вещества. Выход: 1 г; количественный. LCMS m/z 184,0 [M+H]+. 1Н ЯМР (400 МГц, DMSO-d6) δ 9.40 (s, 1Н), 8.28 (br s, 1H), 3.79 (br s, 2H).
Стадия 3. Синтез 8-хлор-2-(имидазо[2,1-b][1,3,4]тиадиазол-6-илметил)-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1H-имидазо[4,5-с]хинолина (8)
К смеси соединений С15 (850 мг; 2,91 ммоль) и С31 (640 мг; 3,5 ммоль) в N,N-диметилформамиде (20 мл) добавляли N,N-диизопропилэтиламин (828 мг; 6,41 ммоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 5,5 г; 8,6 ммоль). Реакционную смесь нагревали при 100°С в течение ночи, после чего ее разбавляли водой (50 мл) и экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (150 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (элюент: смесь 10:1 дихлорметан/метанол) получали продукт в виде желтого твердого вещества. Выход: 372 мг; 0,848 ммоль; 29%. LCMS m/z 438,9 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.29 (s, 1Н), 8.60-8.75 (br m, 1Н), 8.53 (s, 1H), 8.22 (d, J=8,8 Гц, 1H), 7.79 (s, 1H), 7.63 (dd, J=8,7; 1,9 Гц, 1H), 5.29-5.42 (m, 1H), 4.58 (br s, 2H), 4.30 (br dd, J=12; 5 Гц, 1H), 3.65-3.80 (br m, 2H), 2.61-2.82 (br m, 1H), 2.34-2.54 (br m, 1H), 1.71-1.97 (br m, 2H), 1.35 (d, J=6,2 Гц, 3Н).
Пример 9
{8-Хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолин-2-ил}ацетонитрил (9)
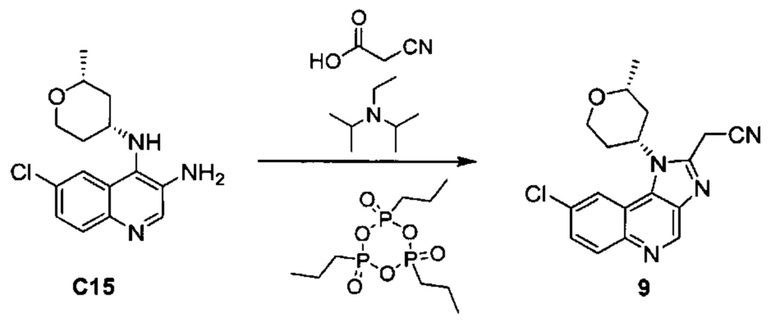
Смесь соединения С15 (280 мг; 0,96 ммоль), цианоуксусной кислоты (65,3 мг; 0,768 ммоль), 2,4,6-триоксида 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (635 мг; 2,00 ммоль; в виде 50%-ного раствора в этилацетате) и N,N-диизопропилэтиламина (0,34 мл; 2,0 ммоль) в этилацетате (8 мл) перемешивали в течение 1 часа, затем обрабатывали дополнительным количеством 2,4,6-триоксида 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ного раствора в этилацетате; 1,0 мл; 1,7 ммоль) и нагревали при температуре дефлегмации в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли дополнительным количеством этилацетата и промывали насыщенным водным раствором бикарбоната натрия. Водный слой экстрагировали этилацетатом и объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 50% до 100% этилацетата в гептане) получали продукт в виде белого твердого вещества. Выход: 159 мг; 0,466 ммоль; 61%. LCMS m/z 341,2; 343,2 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.30 (s, 1Н), 8.5-8.8 (v br m, 1H), 8.26 (d, J=9,0 Гц, 1H), 7.69 (dd, J=8,8; 2,0 Гц, 1H), 4.8-5.1 (v br m, 1H), 4.35-4.43 (m, 1H), 4.29 (s, 2H), 3.73-3.86 (m, 2H), 2.35-2.95 (v br m, 2H), 2.05-2.29 (br m, 2H), 1.41 (d, J=6,0 Гц, 3Н).
Пример 10
8-Хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)(4-2Н)-1H-имидазо[4,5-с]хинолин (10)
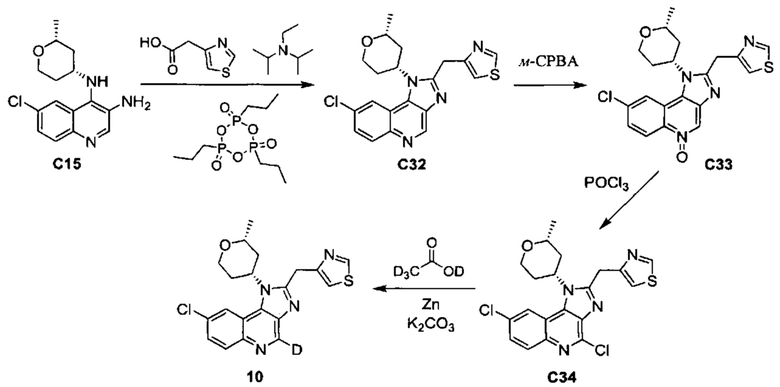
Стадия 1. Синтез 8-хлор-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с]хинолина (С32)
Смесь соединения С15 (889 мг; 3,05 ммоль), 1,3-тиазол-4-илуксусной кислоты (438 мг; 2,44 ммоль), 2,4,6-триоксида 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ного раствора в этилацетате; 2,3 мл; 3,9 ммоль) и N,N-диизопропилэтиламина (1,1 мл; 6,3 ммоль) в этилацетате (14 мл) перемешивали в течение 1 часа и 45 минут при комнатной температуре, затем нагревали при 50°С в течение 1 часа. Добавляли уксусную кислоту (30 мл) и реакционную смесь перемешивали при 115°С в течение 66 часов. Растворители удаляли в вакууме; остаток разбавляли насыщенным водным раствором бикарбоната натрия и трижды экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. После хроматографии на силикагеле (градиент: от 0% до 5% метанола в дихлорметане) вещество, полученное из очищенных фракций, растворяли в этилацетате, обрабатывали активированным углем и фильтровали. Фильтрат концентрировали в вакууме и очищали посредством хроматографии на силикагеле (элюент: диэтиловый эфир), получая продукт в виде белой пены. Выход: 584 мг; 1,46 ммоль; 60%. LCMS m/z 399,2; 401,0 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3), характерные пики: δ 9.29 (s, 1Н), 8.80-8.83 (m, 1Н), 8.58-8.71 (br m, 1Н), 8.22 (d, J=8,9 Гц, 1Н), 7.63 (dd, J=9,0; 2,0 Гц, 1Н), 7.24 (br s, 1Н), 5.20-5.34 (m, 1Н), 4.72 (s, 2H), 4.29 (br dd, J=12; 5 Гц, 1H), 3.60-3.76 (br m, 2H), 2.60-2.80 (br m, 1H), 2.33-2.51 (br m, 1H), 1.7-1.87 (br m, 1H), 1.34 (d, J=6,0 Гц, 3H).
Стадия 2. Синтез 8-хлор-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-2-(1,3-тиазол-4-илметил)-1Н-имидазо[4,5-с]хинолин-5-оксида (С33)
3-Хлорпероксибензойную кислоту (м-СРВА; 547 мг; 3,17 ммоль) добавляли к раствору соединения С32 (972 мг; 2,44 ммоль) в дихлорметане (12 мл). После перемешивания при комнатной температуре в течение ночи реакционную смесь обрабатывали насыщенным водным раствором бикарбоната натрия (30 мл) и перемешивали в течение еще 20 минут. Водный слой трижды экстрагировали дихлорметаном и объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 0% до 5% метанола в дихлорметане) получали продукт в виде желтого твердого вещества. Выход: 1,0 г; 2,4 ммоль; 98%. LCMS m/z 415,3; 417,2 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3), характерные пики: δ 9.05 (d, J=9,4 Гц, 1H), 9.03 (s, 1H), 8.81 (d, J=1,8 Гц, 1Н), 8.63-8.76 (br m, 1Н), 7.70 (dd, J=9,4; 1,8 Гц, 1H), 5.23-5.36 (m, 1H), 4.68 (s, 2H), 4.30 (dd, J=12,1; 5,1 Гц, 1H), 3.61-3.80 (m, 2H), 2.53-2.71 (br m, 1H), 2.25-2.42 (br m, 1H), 1.78-1.93 (br m, 1H), 1.65-1.78 (br m, 1H), 1.34 (d, J=6,2 Гц, 3Н).
Стадия 3. Синтез 4,8-дихлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с]хинолина (С34)
Оксихлорид фосфора (98%-ный; 0,17 мл; 1,8 ммоль) добавляли к раствору соединения С33 (300 мг; 0,72 ммоль) в хлороформе (4 мл) и реакционную смесь нагревали до 70°С в течение 1,5 часа. После охлаждения до комнатной температуры ее выливали в перемешиваемую смесь воды и дихлорметана и оставляли перемешиваться в течение 20 минут. Смесь подщелачивали путем добавления насыщенного водного раствора бикарбоната натрия; водный слой один раз экстрагировали дихлорметаном и объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 50% до 100% этилацетата в гептане) получали продукт в виде белой пены. Выход: 290 мг; 0,669 ммоль; 93%. LCMS m/z 433,2; 435,2; 437,1 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 8.78 (d, J=1,8 Гц, 1Н), 8.56-8.67 (br m, 1Н), 8.07 (d, J=8,9 Гц, 1H), 7.59 (dd, J=9,0; 2,0 Гц, 1H), 7.23-7.29 (br m, 1H), 5.23-5.35 (m, 1H), 4.75 (s, 2H), 4.26 (dd, J=11,9; 4,9 Гц, 1H), 3.57-3.72 (m, 2H), 2.53-2.74 (br m, 1H), 2.26-2.46 (br m, 1H), 1.69-1.83 (br m, 1H), 1.55-1.69 (br m, 1H), 1.31 (d, J=6,2 Гц, 3Н).
Стадия 4. Синтез 8-хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)(4-2Н)-1Н-имидазо[4,5-с]хинолина (10)
Соединение С34 (45 мг; 0,10 ммоль) объединяли с цинковой пылью (98%-ной; 55,5 мг; 0,832 ммоль) в (2Н4)уксусной кислоте (0,5 мл) и нагревали при 100°С в течение 15 минут. Реакционную смесь охлаждали до комнатной температуры, обрабатывали 1 М водным раствором гидроксида натрия и экстрагировали дихлорметаном. Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток смешивали с уксусной кислотой (2 мл) и нагревали до 100°С в течение 10 минут; после удаления растворителя при пониженном давлении остаток растворяли в дихлорметане и промывали насыщенным водным раствором бикарбоната натрия. Водный слой один раз экстрагировали дихлорметаном и объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (элюент: этилацетат, затем градиент от 0% до 5% метанола в дихлорметане) получали продукт в виде белого твердого вещества. Выход: 10,1 мг; 25,3 мкмоль; 25%. По данным 1Н ЯМР анализа для этого вещества продемонстрировало включение приблизительно 85% дейтерия. LCMS m/z 400,3; 402,2 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3), характерные пики: δ 9.29 (пик остаточных протонов (residual protio peak)), 8.81 (d, J=1,8 Гц, 1H), 8.59-8.70 (br m, 1H), 8.22 (d, J=9,0 Гц, 1H), 7.63 (dd, J=9,0; 2,1 Гц, 1H), 7.22-7.25 (m, 1H), 5.20-5.33 (m, 1H), 4.73 (s, 2H), 4.29 (br dd, J=12; 5 Гц, 1H), 3.61-3.75 (br m, 2H), 2.61-2.79 (br m, 1H), 2.33-2.52 (br m, 1H), 1.70-1.85 (br m, 1H), 1.34 (d, J=6,2 Гц, 3Н).
Пример 11
8-Хлор-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-2-[(4-метил-1H-1,2,3-триазол-1-ил)метил]-1H-имидазо[4,5-с]хинолин (11)
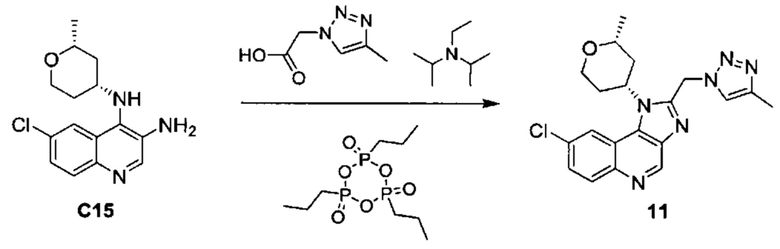
N,N-Диизопропилэтиламин (828 мг; 6,41 ммоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 5,5 г; 8,7 ммоль) добавляли к смеси соединения С15 (850 мг; 2,91 ммоль) и (4-метил-1H-1,2,3-триазол-1-ил)уксусной кислоты (493 мг; 3,49 ммоль) в N,N-диметилформамиде (20 мл). Реакционную смесь нагревали при 100°С в течение ночи, после чего ее разбавляли водой (50 мл) и экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (150 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После очистки посредством обращенно-фазовой HPLC (колонка: Triart С18 серии YMC-Actus, 5 мкм; подвижная фаза А: вода, содержащая 0,225% муравьиной кислоты; подвижная фаза В: ацетонитрил; элюент: 42% В) получали продукт в виде желтого твердого вещества. Выход: 340 мг; 0,86 ммоль; 30%. LCMS m/z 396,9 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.31 (s, 1Н), 8.58-8.72 (br m, 1H), 8.23 (d, J=8,9 Гц, 1H), 7.67 (dd, J=8,9; 2,0 Гц, 1H), 7.47 (br s, 1H), 5.99 (s, 2H), 5.30-5.42 (m, 1H), 4.29 (br dd, J=12; 5 Гц, 1H), 3.68-3.81 (m, 2H), 2.56-2.74 (br m, 1H), 2.32 (s, 3H), 2.3-2.46 (br m, 1H), 1.43-1.90 (2 br m, 2H, предположительно; частично закрыт пиком от воды), 1.34 (d, J=6,0 Гц, 3Н).
Альтернативный синтез соединения примера 11
8-Хлор-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-2-[(4-метил-1H-1,2,3-триазол-1-ил)метил]-1H-имидазо[4,5-с]хинолин (11)
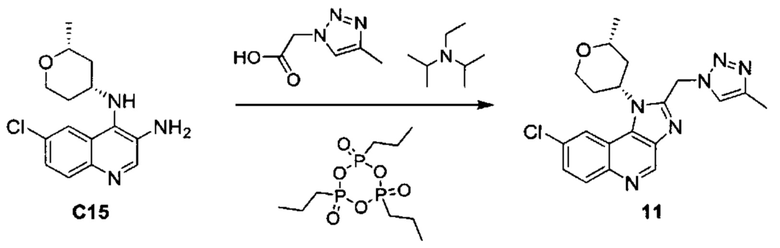
Смесь соединения С15 (500 мг; 1,71 ммоль) и (4-метил-1H-1,2,3-триазол-1-ил)уксусной кислоты (247 мг; 1,75 ммоль) трижды продували азотом и затем смешивали с толуолом (5,7 мл). Добавляли N,N-диизопропилэтиламин (0,30 мл; 1,72 ммоль), затем 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 1,48 мл; 2,49 ммоль). Реакционную смесь нагревали до 70°С в течение 70 минут, после чего LCMS анализ показал полное расходование исходного вещества и соотношение промежуточного амида и соединения 11, равное приблизительно 2:1. Затем реакционную смесь нагревали при 110°С в течение 3 часов, после чего ее охлаждали, разбавляли этилацетатом и промывали насыщенным водным раствором бикарбоната натрия. Органический слой сушили над сульфатом магния, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 0% до 10% метанола в дихлорметане) получали продукт в виде твердого вещества. Выход: 585 мг; 1,47 ммоль; 86%. LCMS m/z 397,4 (обнаружен пик изотопа хлора) [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.30 (s, 1Н), 8.55-8.73 (br m, 1H), 8.23 (d, J=9,0 Гц, 1H), 7.66 (dd, J=8,8; 2,2 Гц, 1H), 7.43-7.50 (br m, 1H), 5.99 (s, 2H), 5.29-5.42 (m, 1H), 4.29 (br dd, J=12,1; 4,7 Гц, 1H), 3.65-3.81 (m, 2H), 2.54-2.75 (br m, 1H), 2.31 (s, 3H), 2.24-2.47 (br m, 1H), 1.43-1.75 (br m, 2H), 1.34 (d, J=6,1 Гц, 3Н).
Пример 93
8-Хлор-1-(цис-3-фторциклопентил)-2-[(4-метил-1H-1,2,3-триазол-1-ил)метил]-1H-имидазо[4,5-с]хинолин, ENT-1 (93)
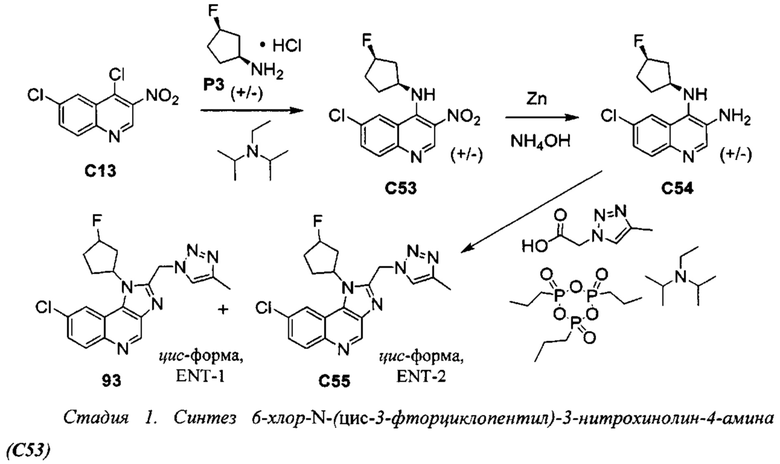
N,N-Диизопропилэтиламин (8,33 мл; 47,8 ммоль) добавляли к суспензии соединения С13 (3,32 г; 13,7 ммоль) и соединения Р3 (2,00 г; 14,3 ммоль) в ацетонитриле (80 мл). Реакционную смесь перемешивали при комнатной температуре в течение 5 минут и затем нагревали до 55°С в течение 6 часов, после чего ее охлаждали до комнатной температуры. После добавления водного раствора бикарбоната натрия смесь экстрагировали дихлорметаном и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 0% до 40% этилацетата в гептане) получали продукт в виде твердого вещества. Выход: 3,78 г; 12,2 ммоль; 89%. LCMS m/z 310,3 (обнаружен пик изотопа хлора) [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.79 (br d, 1Н), 9.35 (s, 1H), 8.23 (d, J=2,3 Гц, 1H), 7.95 (d, J=9,0 Гц, 1H), 7.71 (dd, J=9,0; 2,2 Гц, 1H), (5.38-5.43 (m) и 5.25-5.30 (m), суммарно 1H), 4.71-4.80 (m, 1H), 2.43-2.54 (m, 1H), 2.27-2.43 (m, 3H), 2.15-2.27 (m, 1H), 1.87-2.08 (m, 1H).
Стадия 2. Синтез 6-хлор-N4-(цис-3-фторциклопентил)хинолин-3,4-диамина (C54)
Цинк (8,66 г; 132 ммоль) добавляли в виде одной порции к смеси соединения С53 (4,00 г; 12,9 ммоль) в метаноле (64 мл) и концентрированном растворе гидроксида аммония (64 мл). Через 2 часа выдерживания при комнатной температуре реакционную смесь фильтровали через диатомовую землю и фильтрующую набивку промывали дихлорметаном и метанолом. Объединенные фильтраты концентрировали в вакууме; остаток разбавляли водой и экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. После хроматографии на силикагеле (градиент: от 0% до 60% этилацетата в гептане, затем 100% этилацетата) и последующего растирания с диэтиловым эфиром получали продукт в виде твердого вещества. Выход: 1,68 г; 6,01 ммоль; 47%. LCMS m/z 280,4 (обнаружен пик изотопа хлора) [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 8.47 (s, 1Н), 7.89 (d, J=9,0 Гц, 1H), 7.85 (d, J=2,2 Гц, 1H), 7.39 (dd, J=8,9; 2,2 Гц, 1H), [5.36-5.41 (m) и 5.23-5.28 (m), JHF=54 Гц, суммарно 1H], 4.16-4.26 (m, 1H), 3.81-3.92 (m, 3H), 1.78-2.34 (m, 6H).
Стадия 3. Синтез 8-хлор-1-(цис-3-фторциклопентил)-2-[(4-метил-1Н-1,2,3-триазол-1-ил)метил]-1H-имидазо[4,5-с]хинолина, ENT-1 (93), и 8-хлор-1-(цис-3-фторциклопентил)-2-[(4-метил-1H-1,2,3-триазол-1-ил)метил]-1H-имидазо[4,5-с]хинолина, ENT-2 (С55)
К смеси соединения С54 (150 мг; 0,536 ммоль) и (4-метил-1H-1,2,3-триазол-1-ил)уксусной кислоты (75,7 мг; 0,536 ммоль) в этилацетате (3,2 мл) добавляли N,N-диизопропилэтиламин (0,280 мл; 1,61 ммоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 0,958 мл; 1,61 ммоль). Реакционную смесь нагревали при 80°С в течение ночи, после чего ее разбавляли этилацетатом и промывали водой. Водный слой один раз экстрагировали этилацетатом, объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 0% до 5% метанола в дихлорметане) с последующим растираниес с гептаном, содержащим небольшое количество этилацетата, получали смесь соединений 93 и С55 в виде беловатого твердого вещества. Выход рацемического продукта: 148 мг; 0,384 ммоль; 72%. Входящие в состав энантиомеры разделяли, используя сверхкритическую жидкостную хроматографию (колонка: Lux Amylose-1, 5 мкм, от Phenomenex; подвижная фаза: смесь 7:3 двуокись углерода/(1:1 ацетонитрил/метанол)). Элюирующийся первым энантиомер растирали с диэтиловым эфиром, получая соединение 93, в виде белого твердого вещества. Выход: 52 мг; 0,135 ммоль; 35% для этого разделения. LCMS m/z 385,4 (обнаружен пик изотопа хлора) [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.31 (s, 1Н), 8.49-8.53 (m, 1H), 8.22 (d, J=9,0 Гц, 1H), 7.67 (dd, J=9,0; 2,2 Гц, 1H), 7.47 (br s, 1H), 5.99 (квартет AB, JAB=15,6 Гц, ΔνAB=11,0 Гц, 2H), [5.43-5.56 (m) и 5.32-5.38 (m), суммарно 2H], 2.42-2.78 (m, 4Н), 2.33 (d, J=0,6 Гц, 3Н), 1.98-2.18 (m, 1H), 1.88-1.98 (m, 1H).
Энантиомером, элюирующимся вторым, было соединение С55, которое также выделяли в виде белого твердого вещества после растирания с диэтиловым эфиром. Выход: 58 мг; 0,151 ммоль; 39% для этого разделения. LCMS m/z 385,4 (обнаружен пик изотопа хлора) [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.31 (s, 1H), 8.49-8.53 (m, 1H), 8.22 (d, J=9,0 Гц, 1H), 7.67 (dd, J=9,0; 2,2 Гц, 1H), 7.47 (br s, 1H), 5.99 (квартет AB, JAB=15,6 Гц, ΔνAB=11,0 Гц, 2H), [5.43-5.56 (m) и 5.32-5.38 (m), суммарно 2H], 2.42-2.77 (m, 4H), 2.33 (d, J=0,6 Гц, 3Н), 1.98-2.18 (m, 1H), 1.88-1.98 (m, 1H).
Пример 94
1-[цис-3-Фторциклопентил]-2-[(4-метил-1H-пиразол-1-ил)метил]-1H-имидазо[4,5-с]хинолин-8-карбонитрил, ENT-1 (94)
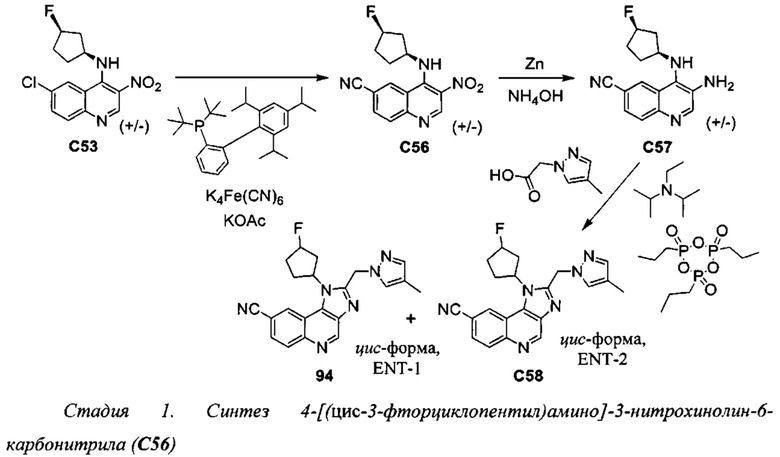
Реакционный сосуд, содержащий смесь соединения С53 (6,00 г; 19,4 ммоль), тригидрата ферроцианида(II) калия (4,09 г; 9,68 ммоль), метансульфоната [(2-ди-трет-бутилфосфино-2',4',6'-триизопропил-1,1'-бифенил)-2-(2'-амино-1,1'-бифенил))палладия(II) (предкатализатора tBuXPhos-Pd-G3; 769 мг; 0,968 ммоль) и ди-трет-бутил[2',4',6'-три(пропан-2-ил)бифенил-2-ил]фосфана (411 мг; 0,968 ммоль), вакуумировали и заполняли азотом. Такой цикл вакуумирования повторяли дважды и затем добавляли 1,4-диоксан (предварительно дегазированный путем барботирования через него азота в течение 2 часов при энергичном перемешивании; 39 мл) и водный раствор ацетата калия (0,0625 М; полученный с использованием дегазированной деионизованной воды; 38,7 мл; 2,42 ммоль). Реакционную смесь помещали в предварительно нагретую до 100°С масляную баню и перемешивали при 100°С в течение 2 часов, после чего ее удаляли из масляной бани, охлаждали до комнатной температуры и распределяли между этилацетатом и насыщенным водным раствором бикарбоната натрия. Водный слой экстрагировали этилацетатом (3×100 мл) и дихлорметаном (100 мл) и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток растирали с дихлорметаном и гептаном и полученное твердое вещество перекристаллизовывали из смеси дихлорметан/гептан, получая продукт в виде твердого вещества. Выход: 4,70 г; 15,6 ммоль; 80%. 1Н ЯМР (400 МГц, CDCl3) δ 9.98-10.09 (br m, 1Н), 9.46 (s, 1H), 8.61 (d, J=1,8 Гц, 1H), 8.09 (d, половина квартета AB, J=8,6 Гц, 1H), 7.92 (dd, половина ABX системы, J=8,8; 1,8 Гц, 1H), [5.42-5.46 (m) и 5.29-5.33 (m), суммарно 1H], 4.71-4.80 (m, 1H), 2.48-2.59 (m, 1H), 2.29-2.46 (m, 3H), 2.19-2.29 (m, 1H), 1.92-2.13 (m, 1H).
Стадия 2. Синтез 3-амино-4-[(цис-3-фторциклопентил)амино]хинолин-6-карбонитрила (С57)
Цинк (4,46 г; 66,4 ммоль) добавляли в виде одной порции к смеси соединения С56 (2,00 г; 6,63 ммоль) в метаноле (33 мл) и концентрированного раствора гидроксида аммония (33 мл). Через 1 час реакционную смесь фильтровали через набивку диатомовой земли, фильтрующую набивку промывали дихлорметаном и небольшим количеством метанола и объединенные фильтраты разбавляли смесью 1:1 воды и насыщенного водного раствора хлорида натрия. Водный слой экстрагировали дихлорметаном и объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После растирания остатка с диэтиловым эфиром в течение 30 минут получали продукт в виде желтого твердого вещества. Выход: 1,49 г; 5,51 ммоль; 83%. LCMS m/z 271,4 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 8.58 (s, 1H), 8.28 (d, J=1,6 Гц, 1Н), 8.02 (d, J=8,6 Гц, 1Н), 7.60 (dd, J=8,7; 1,7 Гц, 1H), [5.39-5.44 (m) и 5.26-5.30 (m), JHF=53 Гц, суммарно 1Н], 4.23-4.33 (m, 1Н), 3.98-4.07 (m, 1Н), 3.91 (br s, 2Н), 2.20-2.36 (m, 1Н), 2.04-2.18 (m, 2Н), 1.81-2.03 (m, 3Н).
Стадия 3. Синтез 1-[цис-3-фторциклопентил]-2-[(4-метил-1H-пиразол-1-ил)метил]-1Н-имидазо[4,5-с]хинолин-8-карбонитрила, ENT-1 (94), и 1-[цис-3-фторциклопентил]-2-[(4-метил-1H-пиразол-1-ил)метил]-1Н-имидазо[4,5-с]хинолин-8-карбонитрила, ENT-2 (С58)
N,N-Диизопропилэтиламин (0,374 мл; 2,15 ммоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 1,28 мл; 2,15 ммоль) добавляли к смеси соединения С57 (200 мг; 0,740 ммоль) и (4-метил-1H-пиразол-1-ил)уксусной кислоты (100 мг; 0,714 ммоль) в этилацетате (4,4 мл) и реакционную смесь нагревали при 80°С в течение ночи. Затем ее распределяли между этилацетатом и водой. Водный слой экстрагировали этилацетатом и объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 0% до 5% метанола в дихлорметане), затем растирания с диэтиловым эфиром получали смесь соединений 94 и С58 в виде беловатого твердого вещества. Выход рацемического продукта: 203 мг; 0,542 ммоль; 76%. Его разделяли на составляющие его энантиомеры, используя сверхкритическую жидкостную хроматографию (колонка: Chiralpak AD-H, 5 мкм, от Chiral Technologies; подвижная фаза: смесь 4:1 двуокись углерода/(этанол, содержащий 0,2% гидроксида аммония)). Энантиомером, элюирующимся первым, было соединение 94, выделенное в виде белого твердого вещества. Выход: 78 мг; 0,21 ммоль; 39% для этого разделения. LCMS m/z 375,5 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.43 (s, 1Н), 8.94-9.00 (m, 1H), 8.36 (d, J=8,6 Гц, 1H), 7.86 (dd, J=8,6; 1,6 Гц, 1H), 7.37 (s, 1H), 7.28 (s, 1H), 5.75 (s, 2H), 5.53-5.65 (m, 1H), [5.47-5.53 (m) и 5.34-5.40 (m), JHF=54 Гц, суммарно 1H], 2.43-2.70 (m, 4H), 2.04 (s, 3H), 1.92-2.14 (m, 1H), 1.82-1.92 (m, 1H).
Элюирующимся вторым соединением, также полученным в виде белого твердого вещества, было соединение С58. Выход: 91 мг; 0,24 ммоль; 44% для этого разделения. LCMS m/z 375,5 [М+Н]+. 1H ЯМР (400 МГц, CDCl3) δ 9.43 (s, 1Н), 8.95-9.00 (m, 1H), 8.36 (d, J=8,6 Гц, 1H), 7.86 (dd, J=8,7; 1,7 Гц, 1H), 7.37 (s, 1H), 7.28 (s, 1H), 5.75 (s, 2H), 5.52-5.65 (m, 1H), [5.48-5.53 (m) и 5.34-5.40 (m), JHF=54 Гц, суммарно 1H], 2.43-2.70 (m, 4H), 2.04 (s, 3Н), 1.92-2.14 (m, 1H), 1.82-1.92 (m, 1H).
Пример 95
2-[(3-Метил-1,2-оксазол-5-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолин-8-карбонитрил (95)
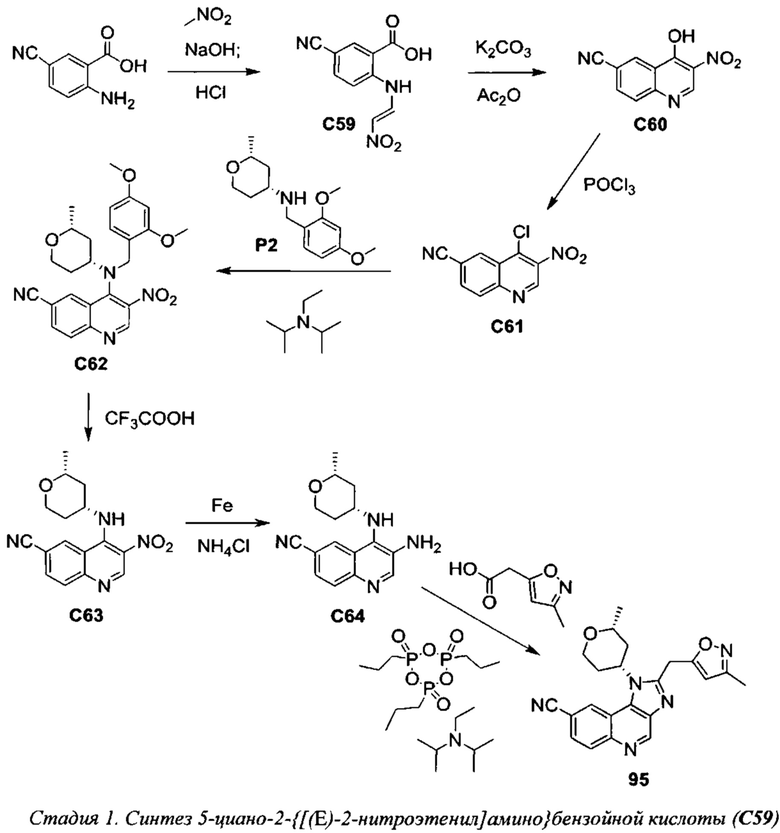
Этот эксперимент проводили в двух идентичных партиях. {Осторожно: эту реакцию не следует проводить в масштабе свыше 1 грамма ввиду наличия высокоэнергетических реагентов и промежуточных соединений. Необходимо соблюдать надлежащие меры безопасности и применять взрывозащитный экран}. Нитрометан (4,71 г; 77,2 ммоль) добавляли по каплям к раствору гидроксида натрия (3,95 г; 98,8 ммоль) в воде (25 мл) и полученный раствор оставляли нагреваться до 45°С в течение 5 минут, после чего его охлаждали в водяной бане и обрабатывали концентрированной соляной кислотой (12 М, 10 мл) пока рН раствора не будет соответствовать кислотной среде. Затем его добавляли к суспензии 2-амино-5-цианобензойной кислоты (5,0 г; 31 ммоль) в воде (50 мл), ацетоне (10 мл) и концентрированной соляной кислоте (12 М, 50 мл) при 25°С и реакционную смесь оставляли перемешиваться при 25°С в течение 15 часов. На этом этапе две партии объединяли и полученную суспензию фильтровали; собранное твердое вещество промывали водой, получая продукт в виде желтого твердого вещества. По данным 1Н ЯМР анализа продукт предположительно существовал в виде смеси ротамеров. Выход: 13,8 г; 59,2 ммоль; 95%. 1Н ЯМР (400 МГц, DMSO-d6) δ [13.15 (s) и 13.12 (s), суммарно 1Н], 8.37 (d, J=2,0 Гц, 1Н), 8.07-8.15 (m, 2Н), 7.92 (d, половина квартета АВ, J=9,0 Гц, 1Н), 6.86 (d, J=6,0 Гц, 1Н).
Стадия 2. Синтез 4-гидрокси-3-нитрохинолин-6-карбонитрила (С60)
Карбонат калия (39,1 г; 283 ммоль) добавляли к суспензии соединения С59 (22,0 г; 94,4 ммоль) в уксусном ангидриде (200 мл). После нагревания реакционной смеси до 90°С в течение 2 часов ее фильтровали и собранное вещество промывали трет-бутил-метиловым эфиром (100 мл) и водой (400 мл), получая продукт в виде коричневого твердого вещества. Выход: 17,0 г; 79,0 ммоль; 84%. LCMS m/z 215,9 [М+Н]+. 1Н ЯМР (400 МГц, DMSO-d6) δ 9.14 (s, 1Н), 8.55 (dd, J=2,0; 0,5 Гц, 1Н), 7.98 (dd, J=8,5; 2,0 Гц, 1H), 7.77 (dd, J=8,5; 0,5 Гц, 1Н).
Стадия 3. Синтез 4-хлор-3-нитрохинолин-6-карбонитрила (С61)
Превращение соединения С60 в продукт осуществляли, используя способ, описанный для синтеза соединения С8 из соединения С7 в примере 1. Продукт выделяли в виде коричневого твердого вещества. Выход: 9,1 г; 39 ммоль; 86%. 1Н ЯМР (400 МГц, DMSO-d6) δ 9.26 (s, 1Н), 8.59 (d, J=1,8 Гц, 1H), 8.16 (dd, J=8,7; 1,9 Гц, 1H), 7.93 (d, J=8,8 Гц, 1H).
Стадия 4. Синтез 4-{(2,4-диметоксибензил)[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]амино}-3-нитрохинолин-6-карбонитрила (С62)
К раствору соединения С61 (8,81 г; 37,7 ммоль) в ацетонитриле (80 мл) добавляли соединение Р2 (11,0 г; 41,5 ммоль), затем N,N-диизопропилэтиламин (5,85 г; 45,3 ммоль). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре, после чего ее концентрировали в вакууме и очищали посредством хроматографии на силикагеле (элюент: 4:1 петролейный эфир/этилацетат), получая продукт в виде вязкого оранжевого масла, которое медленно отвердевало. Выход: 15,0 г; 32,4 ммоль; 86%. LCMS m/z 313,0 [М-(2,4-диметоксибензил)+Н]+. 1Н ЯМР (400 МГц, DMSO-d6) δ 9.18 (s, 1Н), 8.55 (br dd, J=1,3; 1 Гц, 1H), 8.15 (d, J=1,0 Гц, 2H), 6.88 (d, J=8,0 Гц, 1H), 6.24-6.30 (m, 2H), 4.33 (br квартет AB, JAB=14,5 Гц, ΔνAB=12 Гц, 2H), 3.76-3.92 (m, 2H), 3.62 (s, 3H), 3.42 (s, 3H), 3.3-3.4 (m, 2H, предположительно; в значительной мере закрыт пиком от воды), 1.83-2.00 (m, 2Н), 1.70-1.83 (m, 1Н), 1.42-1.54 (m, 1Н), 1.09 (d, J=6,0 Гц, 3Н).
Стадия 5. Синтез 4-{[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]амино}-3-нитрохинолин-6-карбонитрила (С63)
Смесь соединения С62 (15,0 г; 32,4 ммоль) и трифторуксусной кислоты (18,5 г; 162 ммоль) в дихлорметане (150 мл) перемешивали при комнатной температуре в течение 30 минут, после чего ее концентрировали до объема 20 мл и обрабатывали насыщенным водным раствором бикарбоната натрия (200 мл). Водный слой экстрагировали дихлорметаном (3×150 мл) и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая продукт в виде желтого твердого вещества. Выход: 5,68 г; 18,2 ммоль; 56%. LCMS m/z 313,0 [М+Н]+. 1Н ЯМР (400 МГц, DMSO-d6) δ 9.06-9.09 (m, 2Н), 8.30 (br d, J=9,0 Гц, 1Н), 8.14 (dd, половина ABX системы, J=8,7; 1,6 Гц, 1H), 8.01 (d, половина квартета AB, J=8,8 Гц, 1H), 3.87-3.93 (m, 1H), 3.69-3.82 (m, 1H), 3.3-3.5 (m, 2H, предположительно; в значительной мере закрыт пиком от воды), 1.87-2.03 (m, 2Н), 1.60-1.72 (m, 1Н), 1.36-1.47 (m, 1Н), 1.11 (d, J=6,0 Гц, 3Н).
Стадия 6. Синтез 3-амино-4-{[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]амино}хинолин-6-карбонитрила (С64)
Этанол (60 мл) и воду (15 мл) добавляли к смеси соединения С63 (5,68 г; 18,2 ммоль), железа (10,2 г; 183 ммоль) и хлорида аммония (9,73 г; 182 ммоль). Реакционную смесь нагревали до 80°С в течение 1 часа, после чего ее разбавляли этанолом (100 мл) и фильтровали. Фильтрат концентрировали в вакууме и полученное твердое вещество распределяли между насыщенным водным раствором бикарбоната натрия (100 мл) и дихлорметаном (300 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая продукт в виде коричневого твердого вещества. Выход: 4,73 г; 16,8 ммоль; 92%. LCMS m/z 282,9 [М+Н]+. 1Н ЯМР (400 МГц, CD3OD) δ 8.55 (d, J=1,2 Гц, 1Н), 8.51 (s, 1Н), 7.90 (d, J=8,8 Гц, 1H), 7.60 (dd, J=8,5; 1,8 Гц, 1H), 3.92-4.00 (m, 1Н), 3.58-3.69 (m, 1Н), 3.39-3.50 (m, 2Н), 1.78-1.94 (m, 2Н), 1.56-1.69 (m, 1Н), 1.29-1.40 (m, 1Н), 1.17 (d, J=6,0 Гц, 3Н).
Стадия 7. Синтез 2-[(3-метил-1,2-оксазол-5-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1H-имидазо[4,5-с]хинолин-8-карбонитрила (95)
2,4,6-Триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 1,8 г; 2,8 ммоль) и N,N-диизопропилэтиламин (439 мг; 3,40 ммоль) добавляли к смеси соединения С64 (320 мг; 1,13 ммоль) и (3-метил-1,2-оксазол-5-ил)уксусной кислоты (192 мг; 1,36 ммоль) в этилацетате (5 мл) при комнатной температуре (18°С). После нагревания реакционной смеси при 80°С в течение 2,5 суток ее охлаждали до комнатной температуры (18°С) и распределяли между насыщенным водным раствором хлорида натрия (40 мл) и этилацетатом (6×40 мл). Объединенные органические слои концентрировали в вакууме и очищали посредством хроматографии на силикагеле (градиент: от 0% до 8% метанола в дихлорметане), получая коричневое смолообразное вещество, которое растирали со смесью петролейного эфира и этилацетата (2:1, 30 мл). Полученное твердое вещество промывали смесью петролейного эфира и этилацетата (1:1, 10 мл) и затем петролейным эфиром (10 мл), получая продукт в виде коричневатого твердого вещества. Выход: 160 мг; 0,413 ммоль; 37%. LCMS m/z 388,0 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.40 (s, 1Н), 8.80-9.15 (br m, 1H), 8.39 (d, J=8,5 Гц, 1H), 7.88 (br d, J=8,5 Гц, 1H), 6.10 (s, 1H), 4.99-5.25 (br m, 1H), 4.63 (s, 2H), 4.35 (br dd, J=12; 5 Гц, 1H), 3.65-3.83 (m, 2H), 2.51-2.78 (br m, 1H), 2.22-2.48 (br m, 1H), 2.29 (s, 3H), 1.75-2.19 (br m, 2H), 1.38 (d, J=6,0 Гц, 3Н).
Пример 96
2-[(5-Метоксипиридин-2-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1H-имидазо[4,5-с]хинолин-8-карбонитрил, формиатная соль (96)
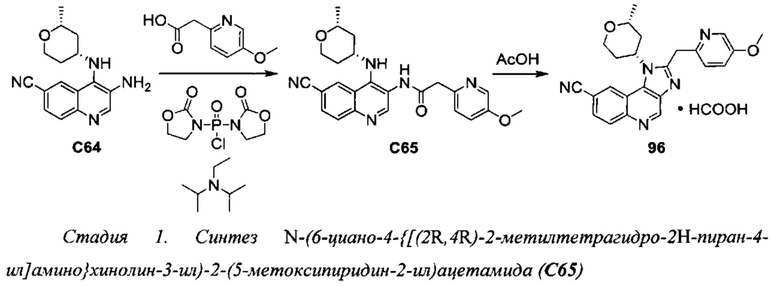
Раствор соединения С64 в N,N-диметилацетамиде (0,1 М; 1,0 мл; 100 мкмоль) добавляли к (5-метоксипиридин-2-ил)уксусной кислоте (25 мг; 150 мкмоль). Добавляли N,N-диизопропилэтиламин (50 мкл, 300 мкмоль), затем бис(2-оксо-1,3-оксазолидин-3-ил)фосфинхлорид (ВОР-Cl; 38,1 мг; 150 мкмоль), флакон с реакционной смесью закрывали и встряхивали при 30°С в течение 16 часов. После удаления растворителя с использованием концентратора Speedvac® остаток промывали и экстрагировали этилацетатом (3×1,5 мл). Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме, получая продукт, который переносили непосредственно на следующую стадию.
Стадия 2. Синтез 2-[(5-метоксипиридин-2-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1H-имидазо[4,5-с]хинолин-8-карбонитрила, формиатной соли (96)
Уксусную кислоту (1 мл) добавляли к соединению С65 (с предыдущей стадии, не более 100 мкмоль) и флакон с реакционной смесью закрывали и встряхивали при 80°С в течение 16 часов. После очистки посредством обращенно-фазовой HPLC (колонка: Durashell С18, 5 мкм, от Agela; подвижная фаза А: 0,225% муравьиной кислоты в воде; подвижная фаза В: ацетонитрил; градиент: от 20% до 50% В) получали продукт. Выход: 4,0 мг; 8,7 мкмоль; 9% за 2 стадии. LCMS m/z 414 [М+Н]+. Время удерживания: 2,44 минуты при проведении аналитической HPLC (колонка: XBridge С18, 2,1×50 мм, 5 мкм, от Waters; подвижная фаза А: 0,0375% трифторуксусной кислоты в воде; подвижная фаза В: 0,01875% трифторуксусной кислоты в ацетонитриле; градиент: от 1% до 5% В в течение 0,6 минуты; от 5% до 100% В в течение 3,4 минуты; скорость потока: 0,8 мл/минута).
Пример 97
1-[(1R,3S)-3-Фторциклопентил]-2-[(5-метил-1,2-оксазол-3-ил)метил]-1H-имидазо[4,5-с]хинолин-8-карбонитрил (97)
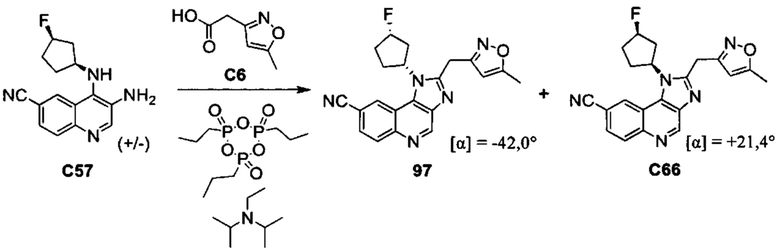
N,N-Диизопропилэтиламин (0,387 мл; 2,22 ммоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 1,32 мл; 2,22 ммоль) добавляли к смеси соединения С57 (200 мг; 0,740 ммоль) и соединения С6 (104 мг; 0,737 ммоль) в этилацетате (4,4 мл) и реакционную смесь нагревали при 80°С в течение ночи. Затем ее разбавляли дополнительным количеством этилацетата и промывали водой. Водный слой один раз экстрагировали этилацетатом и объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (элюент: этилацетат), затем растирания с диэтиловым эфиром получали смесь соединений 97 и С66 в виде беловатого твердого вещества. Выход рацемического продукта: 141 мг; 0,376 ммоль; 51%. Этот продукт разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Lux Amylose-1, 5 мкм, от Phenomenex; подвижная фаза: смесь 4:1 двуокись углерода/этанол). Энантиомером, элюирующимся первым, было соединение 97, полученное в виде белого твердого вещества. Выход: 63,4 мг; 0,169 ммоль; 45% для этого разделения. LCMS m/z 376,2 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.40 (s, 1Н), 8.92-8.97 (m, 1H), 8.35 (d, J=8,6 Гц, 1H), 7.85 (dd, J=8,6; 1,6 Гц, 1H), 6.00 (s, 1H), [5.48-5.54 (m) и 5.32-5.44 (m), суммарно 2H], 4.53 (s, 2H), 2.46-2.76 (m, 4H), 2.40 (s, 3H), 1.92-2.15 (m, 2H). Для образца соединения 97, синтезированного и выделенного таким же путем, получали значение удельного вращения [α] -42,0° (с 0,105; дихлорметан).
Рентгеноструктурный анализ (см. ниже) выполняли для образца соединения 97, которое кристаллизовали из смеси гептан/этилацетат; в результате этого была подтверждена цис-конфигурация при атомах азота и фтора на циклопентановом кольце. Указанная абсолютная стереохимическая конфигурация соединения 97 убедительно вытекает из описанного ниже альтернативного синтеза соединения примера 97; абсолютная конфигурация реагента С49 будет идентична конфигурации его предшественника Р4, которая предсказана на основе его ферментативного синтеза в подготовительном примере Р4.
Элюирующимся вторым энантиомером, также выделенным в виде белого твердого вещества, было соединение С66, 1-[(1S,3R)-3-фторциклопентил]-2-[(5-метил-1,2-оксазол-3-ил)метил]-1H-имидазо[4,5-с]хинолин-8-карбонитрил. Выход: 65,3 мг; 0,174 ммоль; 46% для этого разделения. LCMS m/z 376,2 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.40 (s, 1Н), 8.92-8.97 (m, 1H), 8.35 (d, J=8,8 Гц, 1H), 7.85 (dd, J=8,6; 1,6 Гц, 1H), 6.00 (s, 1H), [5.48-5.54 (m) и 5.32-5.44 (m), суммарно 2H], 4.53 (s, 2H), 2.45-2.76 (m, 4H), 2.40 (s, 3H), 1.92-2.15 (m, 2H). Для образца соединения C66, синтезированного и выделенного таким же путем, получали значение удельного вращения [α] +21,4° (с 0,180; дихлорметан).
Рентгеноструктурный анализ монокристалла соединения 97
Рентгеноструктурный анализ монокристалла
Сбор данных проводили на дифрактометре APEX от Bruker при -150°С. Сбор данных заключался в получении сканограмм по углам омега и пси.
Структуру разрешали прямыми методами, используя пакет программ SHELX в пространственной группе триклинического класса Р1 в виде двух молекул на одну асимметрическую ячейку. Впоследствии структуру уточняли, используя метод наименьших квадратов в полноматричном приближении. Определяли все атомы, не являющиеся атомами водорода, и их координаты уточняли с использованием анизотропных параметров смещений.
Остальные атомы водорода помещали в рассчитанные положения и для них допускали колебания относительно их атомов-носителей. Окончательное уточнение заключалось в использовании изотропных параметров смещений для всех атомов водорода.
Анализ абсолютной структуры с применением методов максимального правдоподобия (Hooft, 2008) проводили, используя программу PLATON (Spek, 2010). В этом случае оказалось невозможным определить абсолютную структуру с использованием данного анализа по причине слабой интенсивности фриделевых пар.
Окончательное значение R-фактора составляло 7,5%. Окончательная разностная карта Фурье не показала никакой пропущенной или неправильно размещенной электронной плотности.
Информация о соответствующем кристалле, сборе данных и уточнении структуры суммирована в Таблице F. Координаты атомов, длины связей, углы связей, торсионные углы и параметры смещений приведены в Таблицах G, Н и J.
Программное обеспечение и ссылки
SHELXTL, версия 5.1, Bruker AXS, 1997.
PLATON, A.L. Spek J. Appl. Cryst., 2003, 36, 7-13.
MERCURY, C.F. Macrae, P.R. Edington, P. McCabe, E. Pidcock, G.P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Cryst., 2006, 39, 453-457.
OLEX2, O.V. Dolomanov, L.J. Bourhis, R.J. Gildea, J.A.K. Howard and H. Puschmann, J. Appl. Cryst., 2009, 42, 339-341.
R.W.W. Hooft, L.H. Straver and A.L. Spek, J. Appl. Cryst., 2008, 41, 96-103.
H.D. Flack, Acta Cryst., 1983, A39, 867-881.


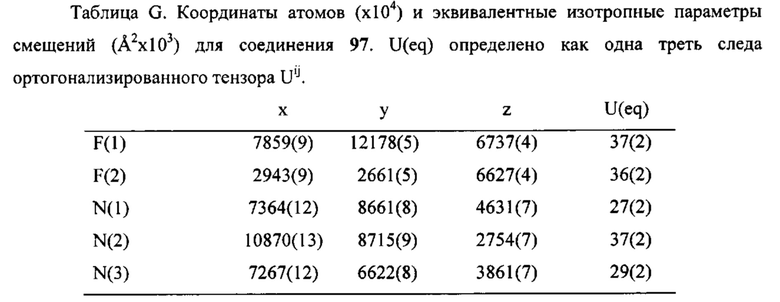






Для создания эквивалентных атомов использовали преобразования симметрии.


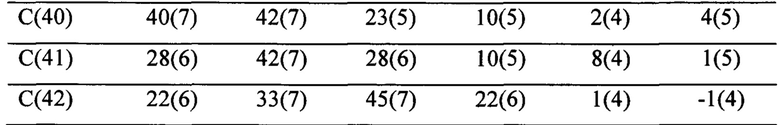
Альтернативный синтез соединения примера 97
1-[(1R,3S)-3-Фторциклопентил]-2-[(5-метил-1,2-оксазол-3-ил)метил]-1H-имидазо[4,5-с]хинолин-8-карбонитрил (97)
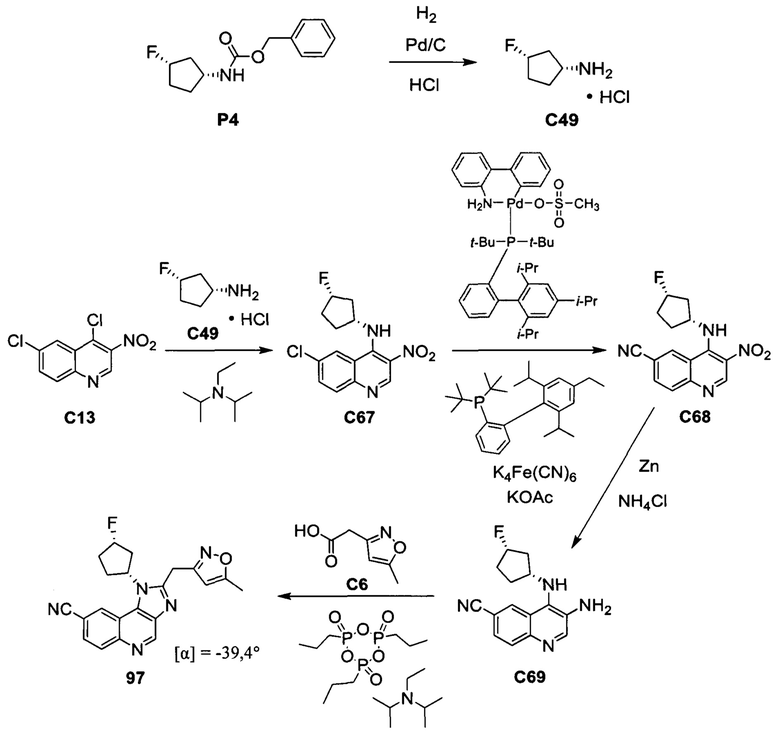
Стадия 1. Синтез (1R,3S)-3-фторциклопентанамина, гидрохлоридной соли (С49)
Соединение Р4 (см. Альтернативное получение соединения Р4, выше; 250 мг; 1,05 ммоль) растворяли в растворе хлористого водорода в метаноле (1,25 М; 12,6 мл; 15,8 ммоль). Добавляли палладий на угле (10%-ный; 250 мг) и в реакционном сосуде трижды создавали давление до 100 ф/кв. дюйм (689,5 кПа) с использованием азота, затем трижды создавали давление до 40 ф/кв. дюйм (275,8 кПа) с использованием водорода. Далее реакционную смесь гидрировали при комнатной температуре и давлении 40 ф/кв. дюйм (275,8 кПа) в течение ночи, после чего ее трижды продували азотом и объединяли с реакционной смесью из аналогичной реакции, проведенной с использованием соединения Р4 (270 мг; 1,14 ммоль). После фильтрования смеси через фильтр из полиэтилена фильтрат концентрировали в вакууме, один раз азеотропно отгоняли с толуолом и дважды промывали гептаном, получая продукт в виде белого твердого вещества. Выход: 315 мг; предположительно количественный. 1Н ЯМР (400 МГц, CD3OD) δ [5.24-5.29 (m) и 5.11-5.16 (m), JHF=53 Гц, суммарно 1Н], 3.67-3.77 (br m, 1H), 2.35 (dddd, J=35,9; 15,6; 8,6; 4,7 Гц, 1Н), 1.79-2.27 (m, 5H).
Стадия 2. Синтез 6-хлор-N-[(1R,3S)-3-фторциклопентил]-3-нитрохинолин-4-амина (С67)
Взаимодействие соединения С13 с соединением С49 осуществляли, используя способ, описанный для синтеза соединения С53 из соединения С13 в примере 93. В этом случае очищенное вещество после проведения хроматографии на силикагеле кристаллизовали из смеси дихлорметан/гептан, получая продукт в виде твердого вещества. Выход: 685 мг; 2,21 ммоль; 89%. 1Н ЯМР (400 МГц, CDCl3) δ 9.80 (br d, J=1 Гц, 1H), 9.36 (s, 1H), 8.24 (d, J=2,3 Гц, 1H), 7.96 (d, J=9,0 Гц, 1H), 7.71 (dd, J=8,9; 2,2 Гц, 1H), [5.38-5.43 (m) и 5.25-5.30 (m), JHF=53 Гц, суммарно 1H], 4.71-4.81 (m, 1H), 2.43-2.54 (m, 1H), 2.28-2.43 (m, 3Н), 2.16-2.27 (m, 1H), 1.88-2.08 (m, 1H).
Стадия 3. Синтез 4-{[(1R,3S)-3-фторциклопентил]амино}-3-нитрохинолин-6-карбонитрила (С68)
Превращение соединения С67 в продукт осуществляли, используя способ, описанный для синтеза соединения С56 из соединения С53 в примере 94. В этом случае очистку проводили, используя хроматографию на силикагеле (градиент: от 0% до 60% этилацетата в гептане, затем 100% этилацетата), получая продукт в виде твердого вещества. Выход: 332 мг; 1,11 ммоль; 50%. 1Н ЯМР (400 МГц, CDCl3) δ 10.04 (br d, J=7 Гц, 1H), 9.46 (s, 1H), 8.61 (d, J=1,8 Гц, 1H), 8.09 (d, половина квартета АВ, J=8,8 Гц, 1H), 7.92 (dd, половина АВХ системы, J=8.7, 1,7 Гц, 1H), [5.42-5.46 (m) и 5.29-5.33 (m), суммарно 1H], 4.71-4.80 (m, 1H), 2.48-2.59 (m, 1H), 2.29-2.46 (m, 3Н), 2.19-2.29 (m, 1H), 1.92-2.13 (m, 1H).
Стадия 4. Синтез 3-амино-4-{[(1R,3S)-3-фторциклопентил]амино}хинолин-6-карбонитрила (С69)
Цинк (97,5%-ный; 0,739 г; 11,0 ммоль) добавляли в виде одной порции к смеси соединения С68 (331 мг; 1,10 ммоль) в метаноле (5,5 мл) и концентрированном растворе гидроксида аммония (5,5 мл). Через 1 час выдерживания при комнатной температуре реакционную смесь фильтровали через диатомовую землю и фильтрующую набивку промывали метанолом. Объединенные фильтраты концентрировали в вакууме и остаток очищали посредством хроматографии на силикагеле (градиент: от 0% до 10% метанола в этилацетате). Полученное вещество растирали с диэтиловым эфиром и промывали гептаном, получая продукт. Выход: 114 мг; 0,422 ммоль; 38%. 1Н ЯМР (400 МГц, CDCl3) δ 8.57 (s, 1Н), 8.28 (d, J=1,6 Гц, 1H), 8.02 (d, J=8,6 Гц, 1H), 7.60 (dd, J=8,6; 1,8 Гц, 1Н), [5.39-5.43 (m) и 5.26-5.30 (m), JHF=53,5 Гц, суммарно 1H], 4.23-4.33 (m, 1H), 3.99-4.07 (m, 1H), 3.91 (br s, 2H), 2.20-2.35 (m, 1H), 2.04-2.17 (m, 2H), 1.82-2.03 (m, 3H).
Стадия 5. Синтез 1-[(1R,3S)-3-фторциклопентил]-2-[(5-метил-1,2-оксазол-3-ил)метил]-1Н-имидазо[4,5-с]хинолин-8-карбонитрила (97)
N,N-Диизопропилэтиламин (39,1 мкл; 0,224 ммоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 0,191 мл; 0,321 ммоль) добавляли к смеси соединения С69 (60 мг; 0,22 ммоль) и соединения С6 (31,3 мг; 0,222 ммоль) в толуоле (2,2 мл). Реакционную смесь нагревали при 70°С в течение 1 часа и затем при 110°С в течение 3 часов, после чего ее охлаждали до комнатной температуры и непосредственно подвергали двум хроматографическим очисткам на силикагеле (градиент: от 0% до 20% метанола в этилацетате). Полученное вещество растирали с этилацетатом и диэтиловым эфиром, получая продукт в виде твердого вещества с окраской от беловатой до светло-желтой. Выход: 41,2 мг; 0,110 ммоль; 50%. Удельное вращение: [α] -39,4° (с 0,120; дихлорметан). 1H ЯМР (400 МГц, CDCl3) δ 9.41 (s, 1H), 8.92-8.97 (m, 1H), 8.36 (d, J=8,8 Гц, 1H), 7.85 (dd, J=8,7; 1,7 Гц, 1H), 6.00 (br s, 1H), 5.32-5.54 (m, 2H), 4.53 (s, 2H), 2.46-2.76 (m, 4H), 2.41 (br s, 3H), 1.92-2.15(m, 2H).
Пример 98
1-[(2R,4R)-2-Метилтетрагидро-2Н-пиран-4-ил]-2-(пиразин-2-илметил)-1Н-имидазо[4,5-c] хинолин-8-карбонитрил, формиатная соль (98)
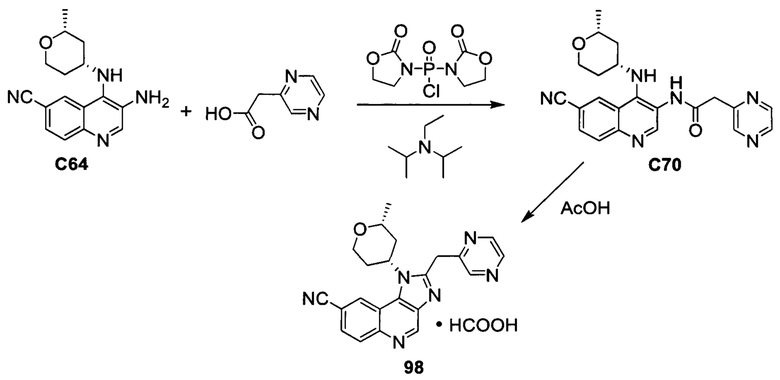
Стадия 1. Синтез N-(6-циано-4-{[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]амино}хинолин-3-ил)-2-(пиразин-2-ил)ацетамида (С70)
Соединение С64 приводили во взаимодействие с пиразин-2-илуксусной кислотой, используя способ, описанный в примере 96 для синтеза соединения С65 из соединения С64. Продукт переносили непосредственно на следующую стадию.
Стадия 2. Синтез 1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-2-(пиразин-2-илметил)-1}1-имидазо[4,5-с]хинолин-8-карбонитрила, формиатной соли (98)
Превращение соединения С70 в продукт осуществляли, используя способ, описанный для синтеза соединения 96 из соединения С65 в примере 96. После очистки посредством обращенно-фазовой HPLC (колонка: Durashell С18, 5 мкм, от Agela; подвижная фаза А: водный раствор аммиака, рН 10; подвижная фаза В: ацетонитрил; градиент: от 18% до 48% В) получали продукт. Выход: 3,0 мг; 7,0 мкмоль; 7%. LCMS m/z 385 [М+Н]+. Время удерживания: 2,30 минуты в случае аналитической HPLC (колонка: XBridge С18, 2,1×50 мм; 5 мкм, от Waters; подвижная фаза А: 0,05% гидроксида аммония в воде; подвижная фаза В: ацетонитрил; градиент: 5% В в течение 0,5 минуты; от 5% до 100% В в течение 2,9 минуты; 100% В в течение 0,8 минуты; скорость потока: 0,8 мл/минута).
Пример 99
8-Хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-{[4-(трифторметил)-1H-1,2,3-триазол-1-ил]метил}-1H-имидазо[4,5-с]хинолин, формиатная соль (99)
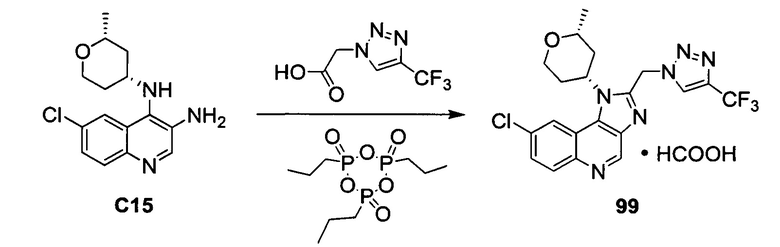
Готовили смесь соединения С15 (29 мг; 100 мкмоль), [4-(трифторметил)-1Н-1,2,3-триазол-1-ил]уксусной кислоты (см. М.D. Andrews et al., US 2015/0218172 A1, Aug 6, 2015) (23 мг; 120 мкмоль) и 2,4,6-триоксида 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ного раствора в этилацетате; 1,0 мл; 1,7 ммоль) во флаконе, который затем закрывали и встряхивали при 120°С в течение 16 часов. После удаления растворителя с использованием концентратора Speedvac® остаток очищали посредством обращенно-фазовой HPLC (колонка: Durashell С18, 5 мкм, от Agela; подвижная фаза А: 0,225% муравьиной кислоты в воде; подвижная фаза В: ацетонитрил; градиент: от 17% до 57% В), получая продукт. Выход: 10,2 мг; 20,5 мкмоль; 20%. LCMS m/z 451 [М+Н]+. Время удерживания: 2,90 минуты в случае аналитической HPLC (колонка: XBridge C18, 2,1×50 мм; 5 мкм, от Waters; подвижная фаза А: 0,0375% трифторуксусной кислоты в воде; подвижная фаза В: 0,01875% трифторуксусной кислоты в ацетонитриле; градиент: от 1% до 5% В в течение 0,6 минуты; от 5% до 100% В в течение 3,4 минуты; скорость потока: 0,8 мл/минута).
Пример 100
8-Хлор-2-[(5-метилпиридин-2-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолин (100)
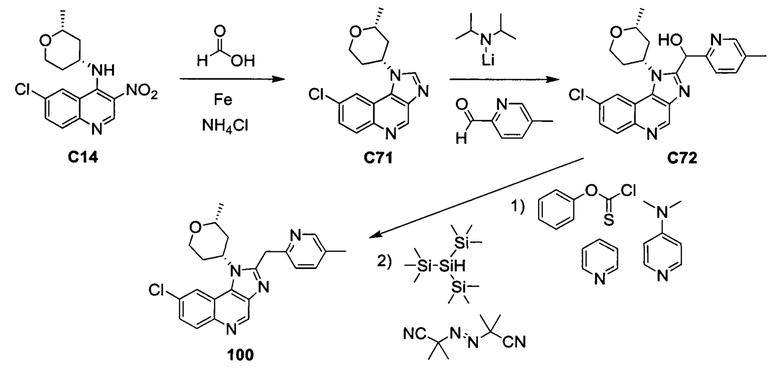
Стадия 1. Синтез 8-хлор-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1Н-имидазо[4,5-с]хинолина (С71)
Муравьиную кислоту (310 мл) добавляли к смеси порошка железа (34,7 г; 621 ммоль), хлорида аммония (33,2 г; 621 ммоль) и соединения С14 (20 г; 62,2 ммоль) в 2-пропаноле (310 мл) при комнатной температуре (14°С). Реакционную смесь нагревали при 80°С в течение 16 часов, после чего ее разбавляли этанолом (300 мл) и фильтровали. Собранные твердые вещества промывали 2-пропанолом (200 мл) и дихлорметаном (100 мл) и объединенные фильтраты концентрировали в вакууме, затем упаривали совместно с этанолом (200 мл). Остаток разбавляли дихлорметаном (300 мл), подщелачивали путем добавления насыщенного водного раствора бикарбоната натрия (500 мл) и затем фильтровали через диатомовую землю; фильтрующую набивку промывали дихлорметаном (300 мл). Водный слой объединенных фильтратов экстрагировали дихлорметаном (4×100 мл) и объединенные органические слои промывали насыщенным водным раствором хлорида натрия (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. После хроматографии на силикагеле (градиент: от 0% до 5% метанола в дихлорметане) получали твердое вещество, которое промывали смесью петролейного эфира и этилацетата (3:1, 100 мл) и петролейным эфиром (50 мл), получая продукт в виде бежевого твердого вещества. Выход: 10,05 г; 33,3 ммоль; 54%. LCMS m/z 301;8 (обнаружен пик изотопа хлора) [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.35 (s, 1Н), 8.25 (d, J=9,0 Гц, 1H), 8.19 (s, 1H), 8.09 (d, J=2,3 Гц, 1Н), 7.66 (dd, J=8,8; 2,3 Гц, 1H), 5.02 (tt, J=12,0; 3,8 Гц, 1H), 4.30 (ddd, J=11,9; 4,6; 1,6 Гц, 1Н), 3.77-3.89 (m, 2H), 2.33-2.46 (m, 2H), 2.09-2.22 (m, 1H), 1.83-1.95 (m, 1H), 1.38 (d, J=6,3 Гц, 3Н).
Стадия 2. Синтез {8-хлор-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1Н-имидазо[4,5-с]хинолин-2-ил}(5-метилпиридин-2-ил)метанола (С72)
Раствор диизопропиламида лития в смеси гептан/тетрагидрофуран/этилбензол (2 М; 3,0 мл; 6,0 ммоль) добавляли к раствору соединения С71 (1,64 г; 5,43 ммоль) в тетрагидрофуране (28 мл) при -78°С и реакционную смесь оставляли перемешиваться при -78°С в течение 15 минут. Раствор 5-метилпиридин-2-карбальдегида (29 мг; 0,24 ммоль) в тетрагидрофуране (0,4 мл) охлаждали до -78°С и обрабатывали порцией С71-содержащей реакционной смеси (0,9 мл; приблизительно 0,15 ммоль); перемешивание продолжали при -78°С в течение 15 минут, после чего охлаждающую баню удаляли и реакционную смесь оставляли нагреваться до комнатной температуры. Затем ее распределяли между водой (1,5 мл) и этилацетатом (2,4 мл) при перемешивании вихревым способом. Органический слой пропускали через картридж для твердофазной экстракции (6 мл), в который был загружен сульфат натрия (приблизительно 1 г); такую методику экстракции повторяли дважды и объединенные элюаты концентрировали в вакууме и использовали непосредственно на следующей стадии.
Стадия 3. Синтез 8-хлор-2-[(5-метилпиридин-2-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1H-имидазо[4,5-с]хинолина (100)
К соединению С72 (с предыдущей стадии; не более 0,15 ммоль) добавляли пиридин (45 мкл; 0,56 ммоль), затем раствор 4-(диметиламино)пиридина (2,5 мг; 20 мкмоль) в 1,2-дихлорэтане (0,3 мл). Реакционный сосуд вакуумировали и заполняли азотом; этот цикл вакуумирования повторяли дважды и затем добавляли раствор O-фенил-карбонохлоридотиоата (52 мг; 0,30 ммоль) в 1,2-дихлорэтане (0,3 мл). После встряхивания реакционной смеси при комнатной температуре в течение 2 часов ее распределяли между водой (1,5 мл) и этилацетатом (2,4 мл) при перемешивании вихревым способом. Органический слой пропускали через картридж для твердофазной экстракции (6 мл), в который был загружен сульфат натрия (приблизительно 1 г); такую методику экстракции повторяли дважды и объединенные элюаты концентрировали в вакууме и использовали непосредственно на следующей стадии. Полученное вещество обрабатывали раствором 2,2'-азо-бис-изобутиронитрила (2 мг; 10 мкмоль) в толуоле (0,6 мл) и 1,1,1,3,3,3-гексаметил-2-(триметилсилил)трисиланом (40 мкл; 0,13 ммоль) и реакционную смесь встряхивали при 110°С в течение 20 часов. Затем ее распределяли между водой (1,5 мл) и этилацетатом (2,4 мл) при перемешивании вихревым способом и органический слой пропускали через картридж для твердофазной экстракции (6 мл), в который был загружен сульфат натрия (приблизительно 1 г); такую методику экстракции повторяли дважды и объединенные элюаты концентрировали в вакууме и очищали, используя обращенно-фазовую HPLC (колонка: XBridge С18, 5 мкм, от Waters; подвижная фаза А: 0,05% гидроксида аммония в воде; подвижная фаза В: 0,05% гидроксида аммония в ацетонитриле; градиент: от 5% до 100% В). Выход: 4,7 мг; 12 мкмоль; 8% за 2 стадии. LCMS m/z 407,4 (обнаружен пик изотопа хлора) [М+Н]+. Время удерживания: 1,89 минуты в случае аналитической HPLC (колонка: Atlantis dC18; 4,6×50 мм; 5 мкм, от Waters; подвижная фаза А: 0,05% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,05% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 20% до 95% В, линейный в течение 4,0 минуты; скорость потока: 2 мл/минута).
Пример 101
1-(цис-3-Фторциклопентил)-2-[(4-метил-1H-1,2,3-триазол-1-ил)метил]-1Н-имидазо[4,5-c]хинолин-8-карбонитрил, ENT-2 (101)
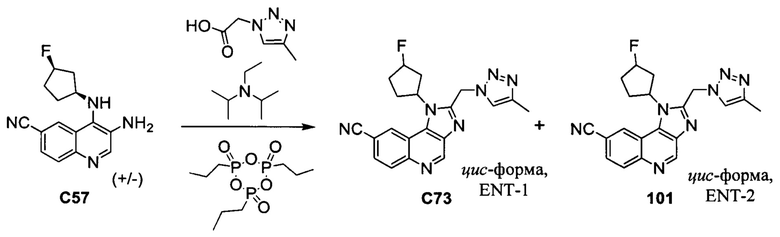
Взаимодействие соединения С57 с (4-метил-1H-1,2,3-триазол-1-ил)уксусной кислотой проводили с использованием способа, описанного для синтеза соединения 97 из соединения С57 и соединения С6 в примере 97, получая рацемическую смесь соединений С73 и 101 в виде беловатого твердого вещества. Выход рацемического продукта: 54,0 мг; 0,144 ммоль; 40%. LCMS m/z 376,4 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.43 (s, 1H), 8.93-8.99 (m, 1H), 8.37 (d, J=8,6 Гц, 1H), 7.89 (dd, J=8,6; 1,6 Гц, 1H), 7.48 (br s, 1H), 6.01 (квартет AB, JAB=15,4 Гц, ΔνAB=11,7 Гц, 2Н), [5.49-5.63 (m) и 5.36-5.42 (m), суммарно 2H], 2.46-2.75 (m, 4H), 2.33 (br s, 3H), 1.92-2.19 (m, 2Н).
Входящие в состав энантиомеры разделяли, используя сверхкритическую жидкостную хроматографию (колонка: Lux Cellulose-2, 5 мкм, от Phenomenex; подвижная фаза: смесь 1:1 двуокись углерода/(метанол, содержащий 0,2% гидроксида аммония)). Энантиомером, элюирующимся первым, выделенным в виде белого твердого вещества, было соединение С73, 1-(цис-3-фторциклопентил)-2-[(4-метил-1H-1,2,3-триазол-1-ил)метил]-1H-имидазо[4,5-с]хинолин-8-карбонитрил, ENT-1. Выход: 8,4 мг; 22 мкмоль; 16% для этого разделения. LCMS m/z 376,1 [М+Н]+. Время удерживания: 8,32 минуты в случае аналитической HPLC (колонка: Lux Cellulose-2, 4,6×100 мм, 5 мкм, от Phenomenex; подвижная фаза: смесь 1:1 двуокись углерода/(метанол, содержащий 0,2% гидроксида аммония); скорость потока: 1,5 мл/минута). Энантиомером, элюирующимся вторым, было соединение 101, также полученное в виде белого твердого вещества. Выход: 6,6 мг; 18 мкмоль; 12% для этого разделения. LCMS m/z 376,0 [М+Н]+. Время удерживания: 9,93 минуты (условия аналитической HPLC идентичны условиям, описанным выше для соединения С73).
Пример 102
8-Хлор-2-[(6-метилпиримидин-4-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-1-пиран-4-ил]-1H-имидазо[4,5-с]хинолин (102)
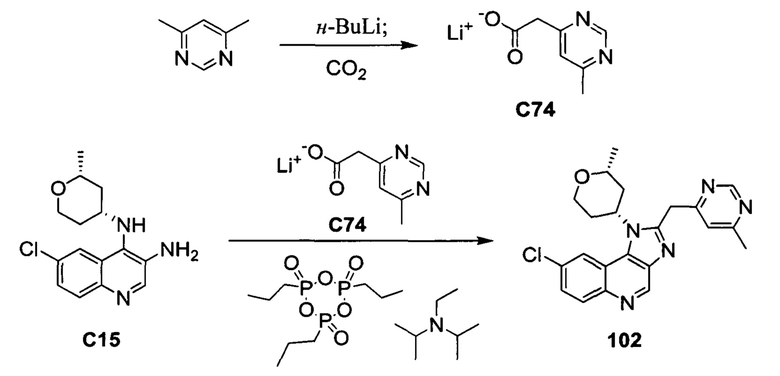
Стадия 1. Синтез (6-метилпиримидин-4-ил)ацетата лития (С74)
н-Бутиллитий (2,5 М в гексанах; 5,00 мл; 12,5 ммоль) медленно по каплям добавляли к раствору (-78°С) 4,6-диметилпиримидина (1,08 г; 9,99 ммоль) в тетрагидрофуране (20 мл). После окончания перемешивания в течение 20 минут при -78°С в реакционную смесь добавляли твердую двуокись углерода (в виде сухого льда; 5,0 г), реакционную смесь нагревали до комнатной температуры (15°С) и перемешивали в течение 1 часа. Затем добавляли воду (3,0 мл) и полученную смесь концентрировали в вакууме, получая продукт в виде белого твердого вещества. Выход: 1,53 г; 9,68 ммоль; 97%. 1Н ЯМР (400 МГц, D2O) δ 8.78 (s, 1H), 7.28 (s, 1H), [3.60 (s) и 3.59 (br s), суммарно 2H], 2.43 (s, 3H).
Стадия 2. Синтез 8-хлор-2-[(6-метилпиримидин-4-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1Н-имидазо[4,5-с]хинолина (102)
2,4,6-Триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 795 мг; 1,25 ммоль) и N,N-диизопропилэтиламин (194 мг; 1,50 ммоль) добавляли к смеси соединения С15 (146 мг; 0,500 ммоль) и соединения С74 (87,5 мг; 0,553 ммоль) в этилацетате (2 мл) при комнатной температуре (15°С). Реакционную смесь нагревали при 80°С в течение 16 часов, после чего ее объединяли с реакционной смесью из аналогичной реакции, проведенной с использованием соединения С15 (100 мг; 0,343 ммоль). Смесь распределяли между водой (40 мл) и этилацетатом (40 мл) и водный слой экстрагировали этилацетатом (6×40 мл). Объединенные органические слои концентрировали в вакууме и очищали посредством обращенно-фазовой HPLC (колонка: Durashell, 5 мкм, от Agela; подвижная фаза А: 0,05% гидроксида аммония в воде; подвижная фаза В: ацетонитрил; градиент: от 26% до 56% В), получая продукт в виде желтого твердого вещества. Выход: 195 мг; 0,478 ммоль; 57%. После хроматографии на силикагеле (градиент: от 0% до 10% метанола в дихлорметане), затем растирания с диэтиловым эфиром получали дополнительно очищенный образец в виде светло-желтого твердого вещества. По данным дифракции рентгеновских лучей на порошке этот образец был кристаллическим. LCMS m/z 408,4 (обнаружен пик изотопа хлора) [М+Н]+. 1Н ЯМР (400 МГц, DMSO-d6), характерные пики: δ 9.19 (s, 1Н), 8.94 (s, 1H), 8.56-8.75 (br m, 1H), 8.20 (d, J=9,0 Гц, 1H), 7.75 (dd, J=9,0; 2,0 Гц, 1H), 7.46 (br s, 1H), 5.10-5.34 (br m, 1H), 4.72 (s, 2H), 4.06-4.22 (br m, 1H), 3.48-3.77 (br m, 2H), 2.46 (s, 3H), 2.10-2.28 (br m, 1H), 1.93-2.09 (br m, 1H), 1.76-1.93 (br m, 1H), 1.21 (d,J=5,9 Гц, 3H).
Пример 103
8-Хлор-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-2-[(4-метил-2Н-1,2,3-триазол-2-ил)метил]-1Н-имидазо[4,5-с]хинолин (103)
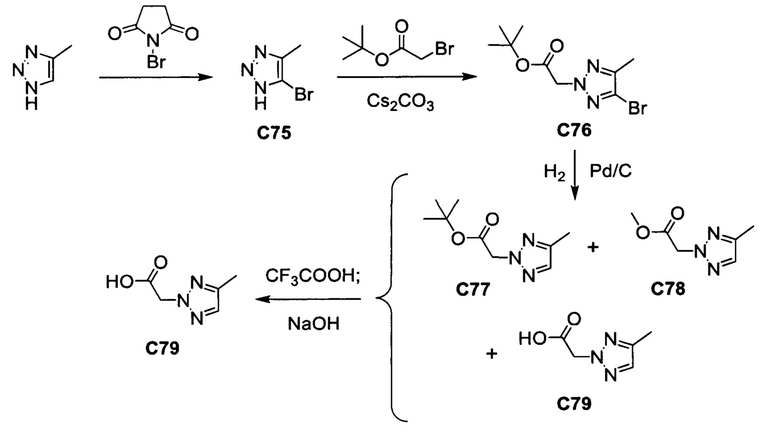
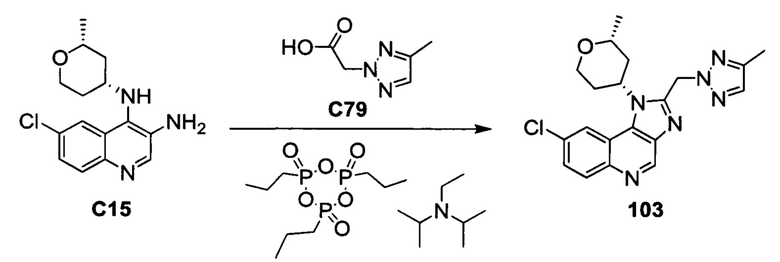
Стадия 1. Синтез 4-бром-5-метил-1H-1,2,3-триазола (С75)
N-Бромсукцинимид (5,89 г; 33,1 ммоль) добавляли к раствору 4-метил-1H-1,2,3-триазола (2,50 г; 30,1 ммоль) в хлороформе (30 мл) и реакционную смесь перемешивали в течение 16 часов при комнатной температуре (15°С). Затем ее разбавляли дихлорметаном (100 мл), промывали водой (2×100 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая продукт в виде белого твердого вещества (4,9 г), которое использовали непосредственно на следующей стадии.
Стадия 2. Синтез трет-бутил-(4-бром-5-метил-2H-1,2,3-триазол-2-ил)ацетата (С76)
трет-Бутил-бромацетат (8,8 г; 45 ммоль) добавляли в виде одной порции к смеси соединения С75 (с предыдущей стадии; 4,9 г; не более 30,1 ммоль) и карбоната цезия (17,6 г; 54,0 ммоль) в N,N-диметилформамиде (80 мл). Реакционную смесь перемешивали при комнатной температуре (20°С) в течение 16 часов, после чего ее разбавляли водой (100 мл) и экстрагировали этилацетатом (2×80 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (2×100 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 0% до 15% этилацетата в петролейном эфире) получали продукт в виде бесцветного масла. Выход: 4,00 г; 14,5 ммоль; 48% за 2 стадии.
Стадия 3. Синтез трет-бутил-(4-метил-2H-1,2,3-триазол-2-ил)ацетата (С77), метил-(4-метил-2H-1,2,3-триазол-2-ил)ацетата (С78) и (4-метил-2H-1,2,3-триазол-2-ил)уксусной кислоты (С79)
Смесь соединения С76 (3,50 г; 12,7 ммоль) и палладия на угле (10%-ного; 500 мг) в метаноле (35 мл) перемешивали в атмосфере водорода (40 ф/кв. дюйм (275,8 кПа)) в течение 4 часов при комнатной температуре (17°С). Реакционную смесь фильтровали и фильтрат концентрировали в вакууме, получая желтое масло (3,00 г). На основании данных 1Н ЯМР продукт идентифицировали как смесь соединения С77 (трет-бутилового эфира), соединения С78 (метилового эфира) и соединения С79 (карбоновой кислоты); это вещество переносили непосредственно на следующую стадию для гидролиза сложного эфира. Пики 1Н ЯМР (400 МГц, CD3OD) δ [7.50 (s) и 7.49 (s), суммарно 1Н], [5.23 (s), 5.17 (s) и 5.10 (s), суммарно 2Н], 3.75 (s, от метилового эфира), 2.30 (s, 3Н), 1.46 (s, от трет-бутилового эфира).
Стадия 4. Синтез (4-метил-2Н-1,2,3-триазол-2-ил)уксусной кислоты (С79)
Смесь соединений С77, С78 и С79 (с предыдущей стадии; 3,00 г; не более 12,7 ммоль) в трифторуксусной кислоте (3 мл) перемешивали в течение 2 часов при комнатной температуре (17°С). После удаления растворителя в вакууме остаток растворяли в тетрагидрофуране (10 мл) и обрабатывали водным раствором гидроксида натрия (2 М, 10 мл). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре (17°С), концентрировали в вакууме и распределяли между водой (50 мл) и дихлорметаном (20 мл). Водный слой экстрагировали дихлорметаном (2×20 мл) и затем подкисляли 1 М водным раствором соляной кислоты до рН 1. Этот кислотный водный слой экстрагировали этилацетатом (3×40 мл) и объединенные этилацетатные слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая продукт в виде желтого твердого вещества. Выход: 1,9 г; 13 ммоль; 100% за 2 стадии. 1H ЯМР (400 МГц, CDCl3) δ 7.46 (s, 1Н), 5.25 (s, 2Н), 2.34 (s, 3Н).
Стадия 5. Синтез 8-хлор-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-2-[(4-метил-2Н-1,2,3-триазол-2-ил)метил]-1Н-имидазо[4,5-с]хинолина (103)
Взаимодействие соединения С15 с соединением С79 осуществляли, используя способ, описанный для синтеза соединения 95 из соединения С64 и (3-метил-1,2-оксазол-5-ил)уксусной кислоты в примере 95. Очистку проводили посредством обращенно-фазовой HPLC (колонка: Durashell С18, 5 мкм, от Agela; подвижная фаза А: 0,05% гидроксида аммония в воде; подвижная фаза В: ацетонитрил; градиент: от 35% до 55% В), получая продукт в виде бледно-желтого смолообразного вещества. Выход: 95 мг; 0,24 ммоль; 48%. LCMS m/z 397,0 (обнаружен пик изотопа хлора) [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.33 (s, 1Н), 8.54-8.70 (br m, 1Н), 8.23 (d, J=9,0 Гц, 1Н), 7.65 (dd, J=8,9; 2,1 Гц, 1Н), 7.44 (br s, 1Н), 6.02 (s, 2Н), 5.15-5.30 (m, 1Н), 4.29 (dd, J=12; 5 Гц, 1Н), 3.58-3.78 (m, 2Н), 2.55-2.81 (br m, 1Н), 2.31 (s, 3Н), 2.3-2.52 (br m, 1Н), 1.62-1.78 (br m, 1Н), 1.44-1.62 (br m, 1Н), 1.34 (d, J=6,0 Гц, 3Н).
Пример 104
8-Хлор-2-[(5-метилпиразин-2-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1Н-мидазо[4,5-с]хинолин (104)
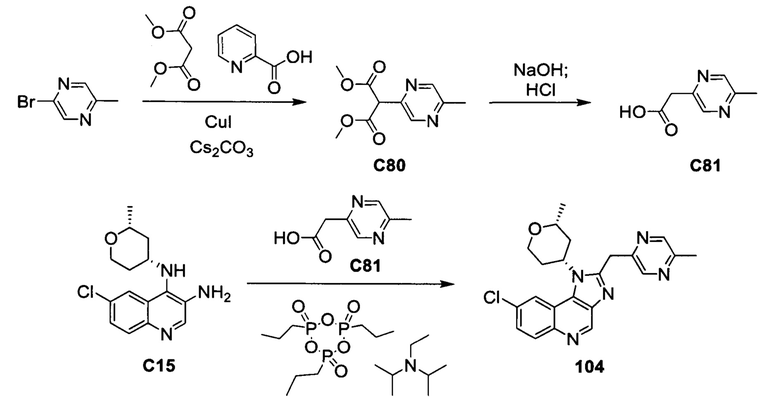
Стадия 1. Синтез диметил-(5-метилпиразин-2-ил)пропандиоата (С80)
К раствору 2-бром-5-метилпиразина (5,0 г; 28,9 ммоль) в 1,4-диоксане (150 мл) добавляли диметилпропандиоат (11,5 г; 87,0 ммоль), пиридин-2-карбоновую кислоту (712 мг; 5,78 ммоль), иодид меди(I) (2,20 г; 11,6 ммоль) и карбонат цезия (28,2 г; 86,6 ммоль). Реакционную смесь перемешивали при 95°С в течение 16 часов, после чего ее охлаждали до температуры окружающей среды и объединяли с аналогичной реакционной смесью, проведенной с использованием 2-бром-5-метилпиразина (100 мг; 0,578 ммоль). Объединенный продукт разбавляли этилацетатом (150 мл), промывали насыщенным водным раствором хлорида натрия (150 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 1% до 67% этилацетата в петролейном эфире) получали продукт в виде желтого твердого вещества. Выход: 5,1 г; 23 ммоль; 78%. LCMS m/z 224,9 [М+Н]+. 1H ЯМР (400 МГц, CDCl3) δ 8.62 (d, J=1,5 Гц, 1Н), 8.42-8.44 (m, 1H), 4.94 (s, 1H), 3.80 (s, 6H), 2.58 (s, 3H).
Стадия 2. Синтез (5-метилпиразин-2-ил)уксусной кислоты (С81)
Водный раствор гидроксида натрия (2 M; 44,6 мл; 89,2 ммоль) добавляли к раствору соединения С80 (5,00 г; 22,3 ммоль) в тетрагидрофуране (15 мл) при 10°С. После окончания перемешивания реакционной смесь в течение 16 часов ее объединяли с аналогичной реакционной смесью, проведенной с использованием соединения С80 (100 мг; 0,45 ммоль) и промывали 4-метилпентан-2-оном. Затем рН водного слоя подводили до 3 путем добавления 6 М водного раствора соляной кислоты с поддержанием при этом температуры смеси от 20°С до 25°С. После концентрирования смеси досуха остаток экстрагировали 4-метилпентан-2-оном (2×150 мл) и два объединенных органических слоя сушили над сульфатом магния, фильтровали и концентрировали в вакууме. После перекристаллизации из смеси дихлорметан/трет-бутил-метиловый эфир (1:20, 50 мл) получали продукт в виде желтого твердого вещества. Выход: 1,80 г; 11,8 ммоль; 52%. LCMS m/z 153,0 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 8.33 (s, 1H), 8.20 (s, 1H), 3.62 (s, 2H), 2.45 (s, 3H).
Стадия 3. Синтез 8-хлор-2-[(5-метилпиразин-2-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1H-имидазо[4,5-с]хинолинаа (104)
2,4,6-Триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 4,30 г; 6,76 ммоль) и N,N-диизопропилэтиламин (1,05 г; 8,12 ммоль) добавляли к смеси соединения С15 (788 мг; 2,70 ммоль) и соединения С81 (452 мг; 2,97 ммоль) в этилацетате (11 мл) при комнатной температуре (15°С). Реакционную смесь нагревали при 80°С в течение 44 часов, после чего ее охлаждали до комнатной температуры и объединяли с аналогичной реакционной смесью, проведенной с использованием соединения С15 (87,5 мг; 0,300 ммоль). Смесь распределяли между водой (40 мл) и дихлорметаном (100 мл) и водный слой экстрагировали дихлорметаном (6×100 мл). Объединенные органические слои концентрировали в вакууме и очищали с использованием хроматографии на силикагеле (градиент: от 0% до 10% метанола в дихлорметане), затем обращенно-фазовой HPLC (колонка: Durashell С18, 5 мкм, от Agela; подвижная фаза А: 0,05% гидроксида аммония в воде; подвижная фаза В: ацетонитрил; градиент: от 35% о 65% В). Продукт получали в виде бледно-желтого смолообразного вещества. Выход: 490 мг; 1,20 ммоль; 40%. LCMS m/z 408,0 (обнаружен пик изотопа хлора) [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.26 (s, 1H), 8.6-8.70 (br m, 1H), 8.58 (s, 1H), 8.38 (s, 1H), 8.21 (d, J=8,8 Гц, 1H), 7.62 (dd, J=8,9; 2,1 Гц, 1H), 5.18-5.35 (br m, 1H), 4.65 (s, 2H), 4.30 (br dd, J=11,8; 5,0 Гц, 1H), 3.58-3.80 (br m, 2H), 2.61-2.82 (br m, 1H), 2.55 (s, 3H), 2.34-2.54 (br m, 1H), 1.58-1.91 (brm,2H), 1.34 (d, J=6,3 Гц, 3Н).
Возможное улучшение для стадии 3 (синтез соединения 104), продемострированное с использованием рацемата соединения С15
2,4,6-Триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 436 мг; 0,685 ммоль) добавляли к раствору рацемата соединения С15 (100 мг; 0,343 ммоль), соединения С81 (52,1 мг; 0,342 ммоль) и N,N-диизопропилэтиламина (66 мкл; 0,38 ммоль) в этилацетате (3 мл). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 1,5 часа, после чего LCMS анализ показал полное превращение в нециклизованный амид (LCMS m/z 426,4 [М+Н]+). Реакционную смесь концентрировали в вакууме для удаления этилацетата и полученное масло растворяли в толуоле (5 мл) и нагревали до 105°С в течение 1 часа и 40 минут. Реакционную смесь распределяли между этилацетатом и насыщенным водным раствором бикарбоната натрия и органический слой промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 10% до 20% метанола в этилацетате) получали масло, которое растворяли в минимальном количестве этилацетата и обрабатывали гептаном. После концентрирования в вакууме получали рацемат соединения 104 в виде почти бесцветного твердого вещества. Выход: 78 мг; 0,19 ммоль; 55%. LCMS m/z 408,3 (обнаружен пик изотопа хлора) [М+Н]+. 1Н ЯМР (600 МГц, DMSO-d6), характерные пики: δ 9.16 (br s, 1Н), 8.59-8.71 (m, 2H), 8.46 (s, 1H), 8.19 (d, J=8,8 Гц, 1H), 7.74 (br d, J=8,8 Гц, 1H), 5.20-5.35 (br m, 1H), 4.76 (s, 2H), 4.10-4.20 (br m, 1H), 3.54-3.76 (br m, 2H), 2.48 (s, 3H), 2.12-2.28 (br m, 1H), 1.92-2.07 (br m, 1H), 1.78-1.92 (br m, 1H), 1.22 (d, J=5,9 Гц, 3Н).
Пример 105
8-Хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-{[5-(трифторметил)пиразин-2-ил]метил}-1Н-имидазо[4,5-с]хинолин (105)
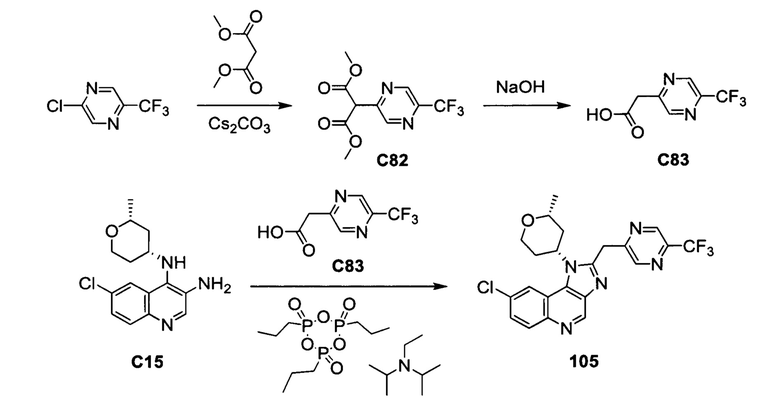
Стадия 1. Синтез диметил-[5-(трифторметил)пиразин-2-ил]пропандиоата (С82)
Смесь 2-хлор-5-(трифторметил)пиразина (6,10 г; 33,4 ммоль), диметил-пропандиоата (4,64 г; 35,1 ммоль) и карбоната цезия (12,0 г; 36,8 ммоль) в N,N-диметилформамиде (40 мл) перемешивали при 15°С в течение 16 часов. Затем реакционную смесь распределяли между этилацетатом (200 мл) и насыщенным водным раствором хлорида натрия (150 мл) и органический слой промывали насыщенным водным раствором хлорида натрия (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После очистки посредством хроматографии на силикагеле (градиент: от 0% до 30% этилацетата в петролейном эфире) получали продукт в виде желтого масла (6,1 г). По данным 1Н ЯМР было установлено, что продукт содержал диметилпропандиоат. Выход, скорректированный с учетом примеси диметилпропандиоата: 4,30 г; 15,5 ммоль; 46%. 1Н ЯМР (400 МГц, CDCl3), только пики продуктов: δ 8.91 (s, 2Н), 5.08 (s, 1Н), 3.83 (s, 6H).
Стадия 2. Синтез [5-(трифторметил)пиразин-2-ил]уксусной кислоты (С83)
К раствору соединения С82 (2,78 г с предыдущей стадии; скорректировано с учетом примеси диметилпропандиоата: 1,96 г; 7,05 ммоль) в тетрагидрофуране (15 мл) добавляли водный раствор гидроксида натрия (2 М; 20 мл; 40 ммоль) в виде одной порции и реакционную смесь перемешивали при 45°С в течение 16 часов. После охлаждения до 20°С реакционную смесь промывали трет-бутил-метиловым эфиром (2×30 мл). Затем водный слой подкисляли до рН 3 путем добавления 6 М водного раствора соляной кислоты и экстрагировали этилацетатом (2×40 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (2×20 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая продукт в виде желтого масла. Выход: 1,0 г; 4,9 ммоль; 70%. 1Н ЯМР (400 МГц, CDCl3) δ 8.93 (br s, 1Н), 8.75 (br s, lH), 4.07 (s, 2Н).
Стадия 3. Синтез 8-хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-{[5-(трифторметил)пиразин-2-ил]метил}-1Н-имидазо[4,5-с]хинолина (105)
N,N-Диизопропилэтиламин (111 мг; 0,859 ммоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 545 мг; 0,856 ммоль) добавляли к раствору соединения С15 (100 мг; 0,343 ммоль) и соединения С83 (70,6 мг; 0,343 ммоль) в этилацетате (2 мл) при комнатной температуре (19°С). Реакционную смесь перемешивали при 80°С в течение 40 часов, после чего ее промывали последовательно водой (3×50 мл) и насыщенным водным раствором хлорида натрия (100 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После обращенно-фазовой HPLC (колонка: Durashell, 5 мкм, от Agela; подвижная фаза А: 0,05% гидроксида аммония в воде; подвижная фаза В: ацетонитрил; градиент: от 44% до 74% В) получали продукт в виде коричневого твердого вещества. Выход: 125 мг; 0,271 ммоль 479%. LCMS m/z 462,0 (обнаружен пик изотопа хлора) [М+Н]+. 1Н ЯМР (400 МГц, CD3OD) δ 9.09 (br s, 1Н), 8.98 (br s, 1Н), 8.96 (br s, 1Н), 8.75-8.90 (br m, 1Н), 8.19 (d, J=9,0 Гц, 1Н), 7.74 (dd, J=8,9; 2,1 Гц, 1Н), 5.25-5.45 (br m, 1Н), 4.93-4.98 (m, 2Н), 4.28 (br dd, J=12,0; 5,3 Гц, 1Н), 3.69-3.86 (m, 2H), 2.62-2.83 (br m, 1H), 2.32-2.52 (br m, 1H), 1.93-2.22 (br m, 2H), 1.34 (d, J=6,0 Гц, 3Н).
Пример 106
1-[(2R,4R)-2-Метилтетрагидро-2Н-пиран-4-ил]-2-[(4-метил-1Н-1,2,3-триазол-1-ил)метил]-1H-имидазо[4,5-с]хинолин-8-карбонитрил (106)
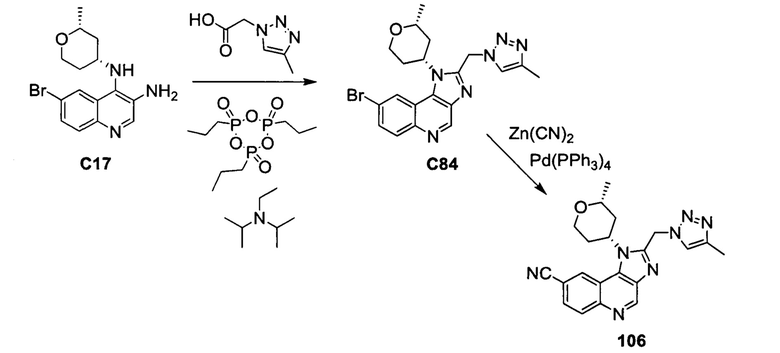
Стадия 1. Синтез 8-бром-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-2-[(4-метил-1H-1,2,3-триазол-1-ил)метил]-1Н-имидазо[4,5-с]хинолина (С84)
N,N-Диизопропилэтиламин (169 мг; 1,31 ммоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 1,2 г; 1,9 ммоль) добавляли к смеси соединения С17 (200 мг; 0,595 ммоль) и (4-метил-1H-1,2,3-триазол-1-ил)уксусной кислоты (101 мг; 0,716 ммоль) в N,N-диметилформамиде (10 мл) и реакционную смесь нагревали при 100°С в течение ночи. Затем ее разбавляли водой (30 мл) и экстрагировали дихлорметаном (3×20 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После обращенно-фазовой HPLC (колонка: Triart С18 серии YMC-Actus, 5 мкм; подвижная фаза А: вода, содержащая 0,225% муравьиной кислоты; подвижная фаза В: ацетонитрил; градиент: от 31% до 51% В) получали продукт в виде желтого твердого вещества. Выход: 18,9 мг; 42,8 мкмоль; 7%. LCMS m/z 442,8 (обнаружен пик изотопа брома) [М+Н]+. 1Н ЯМР (400 МГц, DMSO-d6), характерные пики: δ 9.24 (s, 1H), 8.70-8.89 (m, 1H), 8.13 (d, J=8,5 Гц, 1H), 7.97 (s, 1H), 7.88 (br dd, J=9, 2 Гц, 1H), 6.22 (s, 2H), 5.21-5.40 (br m, 1H), 4.11-4.23 (m, 1H), 3.54-3.78 (m, 2H), 2.25 (s, 3Н), 2.05-2.24 (br m, 1H), 1.69-2.04 (br m, 2H), 1.23 (d, J=6,0 Гц, 3Н).
Стадия 2. Синтез 1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-2-[(4-метил-1H-1,2,3-триазол-1-ил)метил]-1H-имидазо[4,5-с]хинолин-8-карбонитрила (106)
Тетракис(трифенилфосфин)палладий(0) (52,4 мг; 45,3 мкмоль) и цианид цинка (426 мг; 3,63 ммоль) добавляли к раствору соединения С84 (200 мг; 0,453 ммоль) в N,N-диметилформамиде (15 мл) и реакционный сосуд вакуумировали и заполняли азотом. Такой цикл вакуумирования повторяли дважды и затем реакционную смесь нагревали при 140°С в течение ночи. После фильтрования реакционной смеси фильтрат разбавляли водой (50 мл) и экстрагировали этилацетатом (3×50 мл); объединенные органические слои промывали насыщенным водным раствором хлорида натрия (50 мл), сушили над сульфатом натрия и концентрировали в вакууме. После очистки посредством обращенно-фазовой HPLC (колонка: Gemini С18 от Phenomenex, 8 мкм; подвижная фаза А: водный аммиак, рН 10; подвижная фаза В: ацетонитрил; градиент: от 21% до 41% В) получали продукт в виде белого твердого вещества. Выход: 43,6 мг; 0,113 ммоль; 25%. LCMS m/z 387,9 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.43 (s, 1Н), 8.91-9.10 (br m, 1H), 8.39 (d, J=8,8 Гц, 1H), 7.90 (dd, J=9; 1 Гц, 1H), 7.45-7.51 (br s, 1H), 6.01 (s, 2H), 5.34-5.48 (br m, 1H), 4.31 (br dd, J=12; 5 Гц, 1H), 3.68-3.83 (m, 2H), 2.50-2.67 (br m, 1H), 2.33 (s, 3Н), 2.21-2.38 (br m, 1H), 1.48-1.82 (br m, 2H, предположительно; частично закрыт пиком от воды), 1.35 (d, J=6,0 Гц, 3Н).
Пример 107
8-Хлор-l-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-[(1-метил-1Н-1,2,3-триазол-4-ил)метил]-1H-имидазо[4,5-с]хинолин (107)
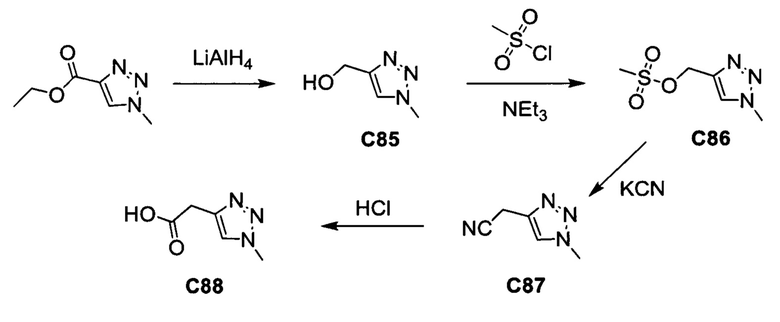
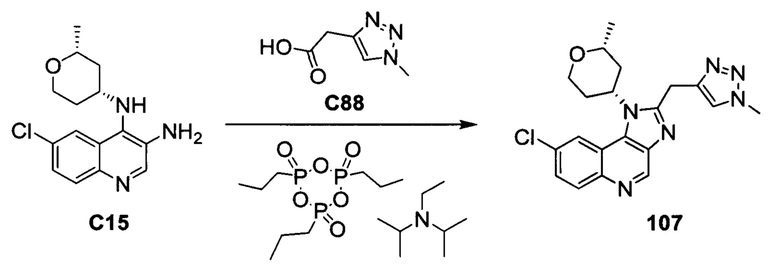
Стадия 1. Синтез (1-метил-1H-1,2,3-триазол-4-ил)метанола (С85)
Алюмогидрид лития (685 мг; 18,0 ммоль) добавляли к суспензии этил-1-метил-1Н-1,2,3-триазол-4-карбоксилата (1,40 г; 9,02 ммоль) в тетрагидрофуране (20 мл) при 0°С и реакционную смесь перемешивали при 0°С в течение 1 часа. Затем по каплям добавляли воду при 0°С до тех пор, пока не прекращалось выделение газа, после чего добавляли сульфат натрия и смесь перемешивали в течение 10 минут. Затем смесь фильтровали и фильтрат концентрировали в вакууме, получая продукт в виде желтого масла. Выход: 700 мг; 6,19 ммоль; 69%. 1Н ЯМР (400 МГц, DMSO-d6) δ 7.90 (s, 1H), 5.15 (t, J=5,5 Гц, 1Н), 4.49 (d, J=5,5 Гц, 2Н), 4.01 (s, 3Н).
Стадия 2. Синтез (1-метил-1Н-1,2,3-триазол-4-ил)метил-метансульфоната (С86)
Метансульфонилхлорид (851 мг; 7,43 ммоль) добавляли при 0°С к раствору соединения С85 (700 мг; 6,19 ммоль) и триэтиламина (1,00 г; 9,88 ммоль) в дихлорметане (20 мл). Реакционную смесь перемешивали при 0°С в течение 2 часов, после чего добавляли воду (100 мл) и смесь экстрагировали дихлорметаном (2×100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая продукт в виде желтого масла, которое использовали непосредственно на следующей стадии. Выход: 800 мг; 4,18 ммоль; 68%.
Стадия 3. Синтез (1-метил-1H-1,2,3-триазол-4-ил)ацетонитрила (С87)
К раствору соединения С86 (800 мг; 4,18 ммоль) в ацетонитриле (20 мл) добавляли цианид калия (1,50 г; 23,0 ммоль). Реакционную смесь перемешивали при 60°С в течение ночи, после чего ее обрабатывали водой (150 мл) и экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (80 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая продукт в виде коричневого твердого вещества. Выход: 200 мг; 1,64 ммоль; 39%. 1Н ЯМР (400 МГц, CDCl3) δ 7.61 (s, 1H), 4.13 (s, 3Н), 3.89 (br s, 2Н).
Стадия 4. Синтез (1-метил-1H-1,2,3-триазол-4-ил)уксусной кислоты (С88)
Раствор соединения С87 (200 мг; 1,64 ммоль) в концентрированной соляной кислоте (4 мл) перемешивали при 60°С в течение 2 часов. После охлаждения реакционной смеси до комнатной температуры ее разбавляли водой (10 мл) и промывали трет-бутил-метиловым эфиром (2×20 мл). Затем водный слой концентрировали досуха, получая продукт в виде коричневого твердого вещества. Выход: 200 мг; 1,42 ммоль; 87%. LCMS m/z 142,0 [М+Н]+. 1Н ЯМР (400 МГц, DMSO-d6) δ 7.94 (s, 1H), 4.01 (s, 3Н), 3.66 (s, 2H).
Стадия 5. Синтез 8-хлор-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-2-[(1-метил-1H-1,2,3-триазол-4-ил)метил]-1H-имидазо[4,5-с]хинолина (107)
N,N-Диизопропилэтиламин (133 мг; 1,03 ммоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (327 мг; 1,03 ммоль) добавляли к смеси соединения C15 (100 г; 0,343 ммоль) и соединения С88 (100 мг; 0,709 ммоль) в N,N-диметилформамиде (2 мл). Реакционную смесь нагревали при 100°С в течение ночи, после чего ее охлаждали до комнатной температуры, разбавляли насыщенным водным раствором хлорида натрия (30 мл) и экстрагировали дихлорметаном (2×30 мл). Объединенные органические слои концентрировали в вакууме и очищали с использованием обращенно-фазовой HPLC (колонка: Gemini С18, 8 мкм, от Phenomenex; подвижная фаза А: водный раствор аммиака, рН 10; подвижная фаза В: ацетонитрил; градиент: от 25% до 45% В), получая продукт в виде белого твердого вещества. Выход: 30,6 мг; 77,1 мкмоль; 22%. LCMS m/z 396,9 (обнаружен пик изотопа хлора) [М+Н]+. 1Н ЯМР (400 МГц, DMSO-d6) δ 9.18 (s, 1H), 8.57-8.71 (br m, 1H), 8.19 (d, J=8,8 Гц, 1H), 8.03 (br s, 1H), 7.74 (dd, J=9,0; 2,0 Гц, 1H), 5.22-5.39 (br m, 1H), 4.62 (s, 2H), 4.11-4.21 (br m, 1H), 4.02 (s, 3Н), 3.55-3.76 (br m, 2H), 2.36-2.5 (br m, 1H, предположительно; частично закрыт пиком от растворителя), 2.09-2.25 (br m, 1H), 1.73-2.04 (br m, 2H), 1.22 (d, J=6,0 Гц, 3Н).
Пример 108
2-[(5-Метилпиразин-2-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолин-8-карбонитрил (108)
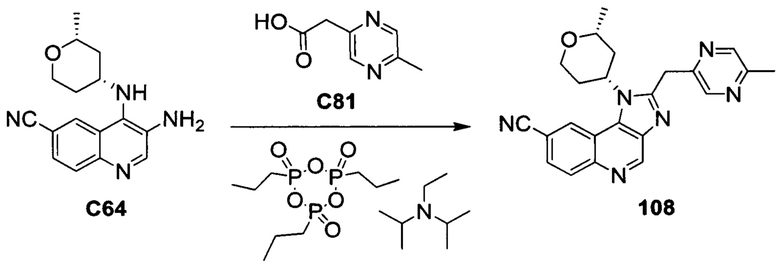
N,N-Диизопропилэтиламин (150 мг; 1,16 ммоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 0,493 мл; 0,828 ммоль) добавляли к смеси соединения С64 (148 мг; 0,524 ммоль) и соединения С81 (80 мг; 0,53 ммоль) в N,N-диметилформамиде (2 мл) и реакционную смесь перемешивали при 110°С в течение 15 часов. Затем ее выливали в воду (10 мл) и экстрагировали дихлорметаном (3×20 мл). Объединенные органические слои промывали водой (2×20 мл), сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении и очищали с использованием обращенно-фазовой HPLC (колонка: Durashell, 5 мкм, от Agela; подвижная фаза А: 0,225% муравьиной кислоты в воде; подвижная фаза В: ацетонитрил; градиент: от 25% до 55% В), получая продукт в виде светло-желтого твердого вещества. Выход: 41,1 мг; 0,103 ммоль; 20%. LCMS m/z 399,1 [М+Н]+. 1Н ЯМР (400 МГц, CD3OD) δ 9.23 (s, 1Н), 9.07-9.20 (br m, 1H), 8.64 (s, 1H), 8.47 (s, 1H), 8.32 (d, J=9,0 Гц, 1Н), 7.97 (br d, J=8,5 Гц, 1H), 5.35-5.54 (br m, 1H), 4.81 (s, 2H), 4.22-4.33 (m, 1H), 3.68-3.86 (br m, 2H), 2.57-2.75 (br m, 1H), 2.55 (s, 3H), 2.24-2.44 (br m, 1H), 1.84-2.21 (br m, 2H), 1.33 (d, J=6,0 Гц, 3Н).
Пример 109
1-(цис-3-Фторциклопентил)-2-[(5-метилпиразин-2-ил)метил]-1Н-имидазо[4,5-с]хинолин-8-карбонитрил, ENT-1 (109)
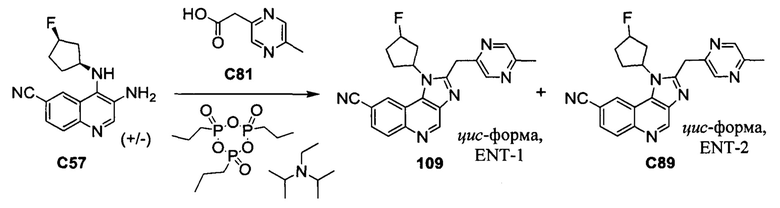
N,N-Диизопропилэтиламин (1,29 мл; 7,41 ммоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 3,53 г; 5,55 ммоль) добавляли к смеси соединения С57 (500 мг; 1,85 ммоль) и соединения С81 (296 мг; 1,94 ммоль) в N,N-диметилформамиде (9,2 мл). Реакционную смесь нагревали до 110°С в течение ночи, после чего ее охлаждали до комнатной температуры и распределяли между водой и этилацетатом. Водный слой трижды экстрагировали этилацетатом и объединенные органические слои промывали водой (3×20 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 0% до 10% метанола в этилацетате) получали смесь соединений 109 и С89 в виде твердого вещества. Выход рацемического продукта: 444 мг; 1,15 ммоль; 62%. Его объединяли с продуктом аналогичной реакции (14 мг) и разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AS-H, 5 мкм, от Chiral Technologies; подвижная фаза: смесь 4:1 двуокись углерода/(этанол, содержащий 0,2% гидроксида аммония)). Энантиомером, элюирующимся первым, было соединение 109, полученное в виде твердого вещества. Выход: 164 мг; 36% для этого разделения. LCMS m/z 387,5 [М+Н]+. 1H ЯМР (400 МГц, CDCl3) δ 9.39 (s, 1H), 8.90-8.95 (m, 1H), 8.61 (s, 1H), 8.38 (s, 1H), 8.35 (d, J=8,6 Гц, 1Н), 7.85 (br d, J=8,6 Гц, 1H), 5.35-5.58 (m, 2H), 4.69 (s, 2H), 2.61-2.81 (m, 3Н), 2.57 (s, 3H), 2.46-2.61 (m, 1H), 1.90-2.18 (m, 2H).
Энантиомером, элюирующимся вторым, также выделенным в виде твердого вещества, было соединение С89, 1-(цис-3-фторциклопентил)-2-[(5-метилпиразин-2-ил)метил]-1H-имидазо[4,5-с]хинолин-8-карбонитрил, ENT-2. Выход: 179 мг; 39% для этого разделения. LCMS m/z 387,5 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.39 (s, 1H), 8.90-8.95 (m, 1H), 8.60 (br s, 1H), 8.38 (br s, 1H), 8.35 (d, J=9,0 Гц, 1Н), 7.85 (dd, J=8,6; 1,2 Гц, 1H), 5.35-5.58 (m, 2H), 4.68 (s, 2H), 2.61-2.80 (m, 3Н), 2.57 (s, 3Н), 2.46-2.61 (m, 1H), 1.90-2.17(m, 2H).
Пример 110
8-Хлор-2-[(4-метокси-1Н-пиразол-1-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1Н-имидазо[4,5-с]хинолин (110)
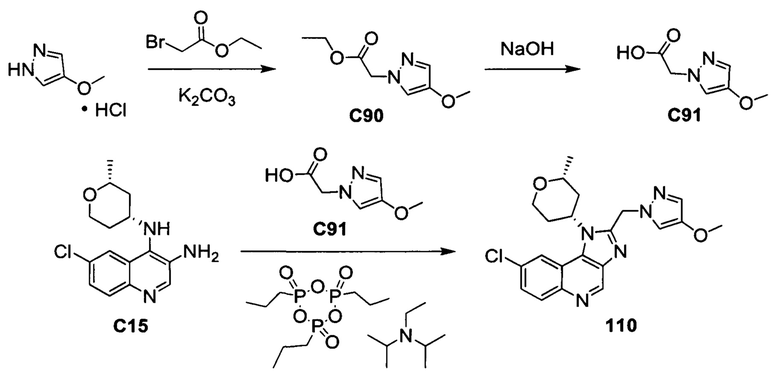
Стадия 1. Синтез этил-(4-метокси-1H-пиразол-1-ил)ацетата (С90)
Этилбромацетат (2,59 г; 15,5 ммоль) добавляли в виде одной порции к смеси 4-метокси-1H-пиразола, гидрохлоридной соли (1,90 г; 14,1 ммоль) и карбоната калия (4,10 г; 29,7 ммоль) в N,N-диметилформамиде (20 мл) и реакционную смесь перемешивали при комнатной температуре (20°С) в течение 60 часов. Затем ее разбавляли водой (100 мл) и экстрагировали этилацетатом (3×80 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (2×150 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 0% до 30% этилацетата в петролейном эфире) получали продукт в виде бесцветного масла. Выход: 1,90 г; 10,3 ммоль; 73%. 1Н ЯМР (400 МГц, CDCl3) δ 7.30 (s, 1H), 7.15 (s, 1H), 4.80 (s, 2H), 4.24 (q, J=7,2 Гц, 2Н), 3.76 (s, 3Н), 1.29 (t, J=7,2 Гц, 3Н).
Стадия 2. Синтез (4-метокси-1H-пиразол-1-ил)уксусной кислоты (С91)
Водный раствор гидроксида натрия (2 М; 10,3 мл; 20,6 ммоль) добавляли в виде одной порции к раствору соединения С90 (1,90 г; 10,3 ммоль) в тетрагидрофуране (10 мл) при комнатной температуре (17°С) и реакционную смесь перемешивали при комнатной температуре (17°С) в течение 3 часов. После удаления тетрагидрофурана в вакууме остаток растворяли в воде (20 мл) и промывали дихлорметаном (2×20 мл). Водную фазу подкисляли до рН 1, используя 1 М раствор соляной кислоты, и затем экстрагировали этилацетатом (3×30 мл). Объединенные этилацетатные слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая продукт в виде белого твердого вещества. Выход: 1,5 г; 9,6 ммоль; 93%. 1Н ЯМР (400 МГц, CDCl3) δ 7.35 (s, 1H), 7.15 (s, 1H), 4.87 (s, 2H), 3.77 (s, 3Н).
Стадия 3. Синтез 8-хлор-2-[(4-метокси-1Н-пиразол-1-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина (110)
2,4,6-Триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 436 мг; 0,685 ммоль) и N,N-диизопропилэтиламин (106 мг; 0,820 ммоль) добавляли к смеси соединения С15 (80 мг; 0,27 ммоль) и соединения С91 (42,8 мг; 0,274 ммоль) в этилацетате (2 мл). Реакционную смесь нагревали при 85°С в течение 16 часов, после чего ее распределяли между этилацетатом (10 мл) и водой (30 мл). Органический слой последовательно промывали водой (2×30 мл) и насыщенным водным раствором хлорида натрия (50 мл), сушили, фильтровали и концентрировали в вакууме. После обращенно-фазовой HPLC (колонка: XBridge C18 OBD, 5 мкм, от Waters; подвижная фаза А: вода, содержащая 0,05% гидроксида аммония; подвижная фаза В: ацетонитрил; градиент: от 5% до 95% В) получали продукт в виде белого твердого вещества. Выход: 64,6 мг; 0,157 ммоль; 58%. LCMS m/z 412,0 (обнаружен пик изотопа хлора) [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.32 (s, 1H), 8.57-8.70 (br m, 1H), 8.23 (d, J=8,5 Гц, 1H), 7.66 (dd, J=9,0; 2,0 Гц, 1H), 7.29 (s, 1H), 7.14 (s, 1H), 5.70 (s, 2H), 5.27-5.41 (m, 1H), 4.28 (br dd, J=12,0; 5,0 Гц, 1H), 3.67 (s, 3Н), 3.63-3.77 (m, 2H), 2.53-2.74 (br m, 1H), 2.26-2.47 (br m, 1H), 1.56-1.7 (br m, 1H, предположительно; частично закрыт пиком от воды), 1.40-1.56 (br m, 1H), 1.33 (d, J=6,0 Гц, 3Н).
Пример 111
1-(2,2-Дифторциклогексил)-2-[(5-метил-1,2-оксазол-3-ил)метил]-1H-имидазо[4,5-с] хинолин-8-карбонитрил, ENT-1 (111)
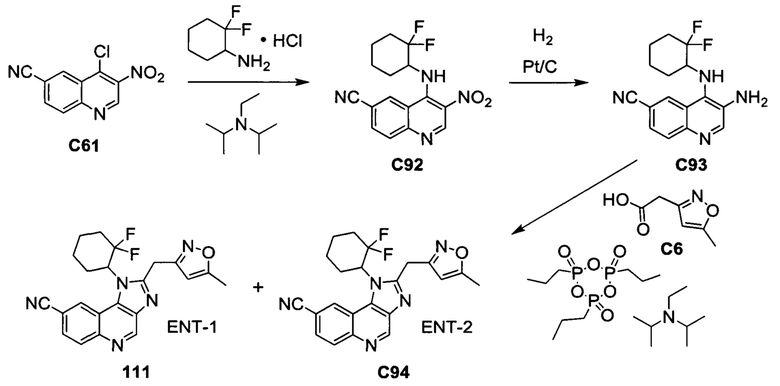
Стадия 1. Синтез 4-[(2,2-дифторциклогексил)амино]-3-нитрохинолин-6-карбонитрила (С92)
Эту реакцию проводили в двух идентичных партиях. 2,2-Дифторциклогексанамин, гидрохлоридную соль (410 мг; 2,39 ммоль), и N,N-диизопропилэтиламин (900 мг; 6,96 ммоль) добавляли к смеси соединения С61 (620 мг; 2,6 ммоль) в ацето нитриле (10 мл) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Эти две партии, концентрировали в вакууме и очищали с использованием хроматографии на силикагеле (градиент: от 0% до 30% этилацетата в петролейном эфире), получая продукт в виде желтого твердого вещества. Выход: 790 мг; 2,38 ммоль; 50%. LCMS m/z 332;7 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.49 (s, 1H), 9.05 (br d, J=9,8 Гц, 1H), 8.43 (br s, 1H), 8.15 (d, J=8,5 Гц, 1H), 7.96 (dd, J=8,8; 1,8 Гц, 1H), 4.10-4.24 (m, 1H), 2.22-2.42 (m, 2H), 1.43-2.01 (m, 6H, предположительно; частично закрыт пиком от воды).
Стадия 2. Синтез 3-амино-4-[(2,2-дифторциклогексил)амино]хинолин-6-карбонитрила (С93)
Платину на угле (5%-ную, 81 мг) добавляли в виде одной порции к смеси соединения С92 (690 мг; 2,08 ммоль) в тетрагидрофуране (50 мл). Реакционную смесь трижды продували азотом и затем трижды продували водородом, после чего ее гидрировали в течение 2 часов при комнатной температуре (приблизительно 20°С) при давлении водорода 40 ф/кв. дюйм (275,8 кПа). После выдерживания реакционной смеси при комнатной температуре в течение 16 часов ее фильтровали через диатомовую землю; фильтрующую набивку последовательно промывали тетрагидрофураном (150 мл) и этилацетатом (50 мл) и объединенные фильтраты концентрировали в вакууме, получая продукт в виде оранжевого твердого вещества. Выход: 650 мг; количественный. LCMS m/z 302,7 [M+H]+.
Стадия 3. Синтез 1-(2,2-дифторциклогексил)-2-[(5-метил-1,2-оксазол-3-ил)метил]-1H-имидазо[4,5-с]хинолин-8-карбонитрила, ENT-1 (111), и 1-(2,2-дифторциклогексил)-2-[(5-метил-1,2-оксазол-3-ил)метил]-1H-имидазо[4,5-с]хинолин-8-карбонитрила, ENT-2 (C94)
N,N-Диизопропилэтиламин (80 мг; 0,62 ммоль) добавляли к смеси соединения С93 (100 мг; 0,33 ммоль) и соединения С6 (68 мг; 0,48 ммоль) в толуоле (1 мл). Затем добавляли 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 411 мг; 0,646 ммоль) и реакционную смесь нагревали при 70°С в течение 45 минут и затем при 105°С в течение 2,5 суток. После охлаждения до комнатной температуры ее объединяли с аналогичной реакционной смесью, проведенной с использованием соединения С93 (20 мг; 66 мкмоль), и полученную смесь переносили в этилацетат (40 мл) и промывали насыщенным водным раствором бикарбоната натрия (20 мл). Водный слой экстрагировали этилацетатом (2×30 мл) и объединенные органические слои промывали насыщенным водным раствором хлорида натрия (30 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. После очистки с использованием обращенно-фазовой HPLC (колонка: Durashell, 5 мкм, от Agela; подвижная фаза А: 0,225% муравьиной кислоты в воде; подвижная фаза В: ацетонитрил; градиент: от 35% до 65% В) получали рацемическую смесь соединений 111 и C94 в виде желтого твердого вещества. На основании анализа 1Н ЯМР-спектра сделано предположение, что это вещество представляло собой смесь ротамеров. Выход рацемического продукта: 40 мг; 98 мкмоль; 25%. LCMS m/z 407,8 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ [9.40 (s) и 9.40 (s), суммарно 1Н], [8.94 (br s) и 8.51 (br s), суммарно 1Н], [8.39 (d, J=8,8 Гц) и 8.33 (d, J=8,5 Гц), суммарно 1Н], [7.87 (dd, J=8,7; 1,6 Гц) и 7.82 (dd, J=8,7; 1,6 Гц), суммарно 1Н], [6.11-6.13 (m) и 6.04-6.06 (m), суммарно 1Н], 5.18-5.42 (m, 1Н), [4.62 (квартет АВ, JAB=16,7 Гц, ΔνAB=21,8 Гц) и 4.51 (квартет АВ, JAB=15,8 Гц, ΔνAB=10.7 Гц), суммарно 2Н], 2.47-2.88 (m, 2H), [2.43 (d, J=1,0 Гц) и 2.40 (d, J=0,8 Гц), суммарно 3Н], 2.03-2.25 (m, 4H), 1.78-1.98 (m, 2H). Рацемический продукт (34,3 мг) разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AD-H от Chiral Technologies, 5 мкм; подвижная фаза: смесь 95:5 двуокись углерода/(метанол, содержащий 0,2% гидроксида аммония)). Энантиомером, элюирующимся первым, было соединение 111. Выход: 5,6 мг; 16% для этого разделения. LCMS m/z 408,4 [М+Н]+. Время удерживания: 3,66 минуты в случае аналитической HPLC (колонка: AD-H, 4,6×100 мм, 5 мкм, от Chiral Technologies; подвижная фаза: смесь 90:10 двуокись углерода/(метанол, содержащий 0,2% гидроксида аммония); скорость потока: 1,5 мл/минута).
Энантиомером, элюирующимся вторым, было соединение С94. Выход: 4,3 мг; 12% для этого разделения. LCMS m/z 408,1 [М+Н]+. Время удерживания 4,63 минуты (условия аналитической HPLC идентичны использованым выше для соединения 111).
Пример 112
2-[(5-Метил-1,2,4-оксадиазол-3-ил)метил]-1-[(3R)-1-метилпирролидин-3-ил]-1H-имидазо[4,5-с]хинолин-8-карбонитрил (112)
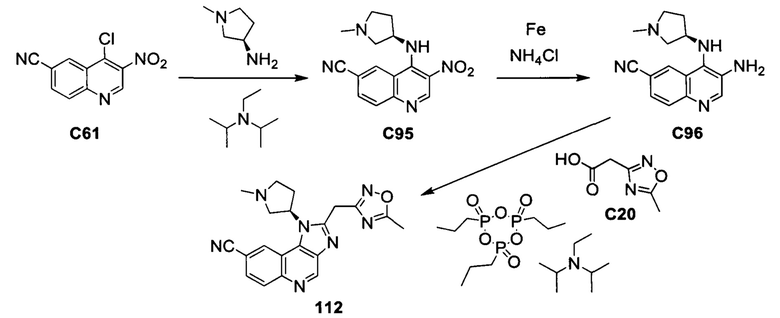
Стадия 1. Синтез 4-{[(3R)-1-метилпирролидин-3-ил]амино}-3-нитрохинолин-6-карбонитрила (С95)
К раствору (20°С) соединения С61 (210 мг; 0,899 ммоль) и (3R)-1-метилпирролидин-3-амин (77,9 мг; 0,778 ммоль) в ацетонитриле (3 мл) добавляли N,N-диизопропилэтиламин (251 мг; 1,94 ммоль). Реакционную смесь перемешивали при 20°С в течение 2 часов, после чего ее концентрировали в вакууме. После очистки остатка посредством хроматографии на силикагеле (градиент: от 0% до 1% метанола в дихлорметане) получали продукт в виде желтого твердого вещества. Выход: 210 мг; 0,706 ммоль; 91%. LCMS m/z 297,9 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 10.04-10.15 (br m, 1H), 9.45 (s, 1H), 8.55 (d, J=1,5 Гц, 1H), 8.07 (d, половина квартета АВ, J=8,5 Гц, 1H), 7.92 (dd, половина ABX системы, J=8,5; 1,8 Гц, 1H), 4.65-4.74 (m, 1H), 3.02-3.10 (m, 1H), 2.84-2.90 (m, 1H), 2.80 (dd, половина ABX системы, J=9,9; 5,6 Гц, 1H), 2.61-2.71 (m, 1H) 2.46 (s, 3Н), 2.41-2.50 (m, 1H), 2.06-2.16 (m, 1H).
Стадия 2. Синтез 3-амино-4-{[(3R)-1-метилпирролидин-3-ил]амино}хинолин-6-карбонитрила (С96)
К раствору соединения С95 (100 мг; 0,336 ммоль) в смеси этанола (1 мл) и воды (0,25 мл) добавляли хлорид аммония (36 мг; 0,673 ммоль) и порошок железа (75,1 мг; 1,34 ммоль) и реакционную смесь перемешивали при 80°С в течение 1 часа. Затем ее фильтровали и осадок на фильтре промывали метанолом (30 мл). Органический слой из объединенных фильтратов концентрировали в вакууме и очищали посредством хроматографии на силикагеле (градиент: от 0% до 15% метанола в дихлорметане), получая продукт в виде желтого твердого вещества. Выход: 112 мг; предположительно количественный. 1Н ЯМР (400 МГц, DMSO-d6), характерные пики: δ 8.65-8.71 (br s, 1H), 8.58 (s, 1H), 7.89 (d, J=8,5 Гц, 1H), 7.62 (dd, J=8,5; 2,0 Гц, 1H), 5.56-5.70 (br s, 1H), 5.43 (d, J=10,5 Гц, 1H), 4.32-4.46 (br m, 1H), 2.81 (s, 3Н), 1.84-1.95 (m, 1H).
Стадия 3. Синтез 2-[(5-метил-1,2,4-оксадиазол-3-ил)метил]-1-[(3R)-1-метилпирролидин-3-ил]-1Н-имидазо[4,5-с]хинолин-8-карбонитрила (112)
N,N-Диизопропилэтиламин (25,4 мг; 0,196 ммоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 238 мг; 0,374 ммоль) добавляли к раствору соединений С96 (50 мг; 0,19 ммоль) и С20 (27,1 мг; 0,191 ммоль) в толуоле (1 мл) и реакционную смесь перемешивали при 70°С в течение 1 часа. LCMS на этом этапе показала превращение в промежуточный амид (LCMS m/z 392,2 [М+Н]+, и тогда реакционную смесь перемешивали при 105°С в течение 16 часов, после чего ее концентрировали в вакууме и очищали обращенно-фазовой HPLC (колонка: Durashell, 5 мкм, от Agela; подвижная фаза А: 0,05% гидроксида аммония в воде; подвижная фаза В: ацетонитрил; градиент: от 27% до 47% В), получая продукт в виде желтого твердого вещества. Выход: 13,0 мг; 34,8 мкмоль; 18%. LCMS m/z 374,1 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 10.00-10.26 (br s, 1H), 9.39 (s, 1H), 8.32 (d, J=8,6 Гц, 1Н), 7.84 (dd, J=8,7; 1,6 Гц, 1H), 5.50-5.62 (m, 1H), 4.72 (br квартет AB, JAB=16,3 Гц, ΔνAB=20,5 Гц, 2Н), 3.38-3.48 (m, 2H), 2.86 (dd, J=11,0; 10,8 Гц, 1H), 2.60 (s, 3Н), 2.57 (s, 3Н), 2.42-2.63 (m, 2H), 2.32-2.42 (br m, 1H).
Пример 113
1-[(3R)-1-Метилпирролидин-3-ил]-2-(пиразин-2-илметил)-1Н-имидазо[4,5-с]хинолин-8-карбонитрил (113)
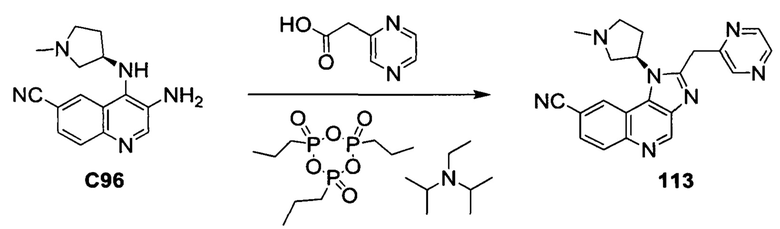
N,N-Диизопропилэтиламин (25,4 мг; 0,196 ммоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 238 мг; 0,374 ммоль) добавляли к раствору соединения С96 (50 мг; 0,19 ммоль) и пиразин-2-илуксусной кислоты (26,4 мг; 0,191 ммоль) в толуоле (1 мл). Реакционную смесь перемешивали при 70°С в течение 1 часа и затем при 105°С в течение 16 часов. После удаления растворителя в вакууме получали остаток, который очищали с использованием обращенно-фазовой HPLC (колонка: Durashell, 5 мкм, от Agela; подвижная фаза А: 0,05% гидроксида аммония в воде; подвижная фаза В: ацетонитрил; градиент: от 25% до 55% В), получая продукт в виде желтого твердого вещества. Выход: 10,3 мг; 30,6 мкмоль; 16%. LCMS m/z 370,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 10.18-10.32 (br s, 1H), 9.38 (s, 1H), 8.72 (d, J=1,3 Гц, 1H), 8.52-8.54 (m, 2H), 8.32 (d, J=8,5 Гц, 1H), 7.83 (dd, J=8,6; 1,6 Гц, 1H), 5.64-5.74 (m, 1H), 4.78 (br s, 2H), 3.40-3.46 (m, 1H), 3.38 (dd, J=11,0; 4,3 Гц, 1H), 2.79 (dd, J=11,0; 10,8 Гц, 1H), 2.56 (s, 3H), 2.53-2.61 (m, 1H), 2.41-2.52 (m, 1H), 2.15-2.27 (br m, 1H).
Пример 114
2-[(5-Метил-1,2,4-оксадиазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-8-(трифторметил)-1H-имидазо[4,5-с]хинолин (114)
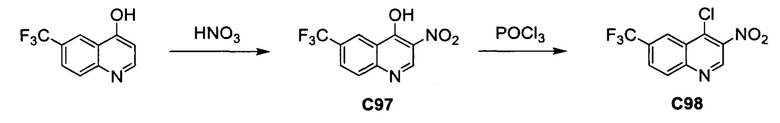
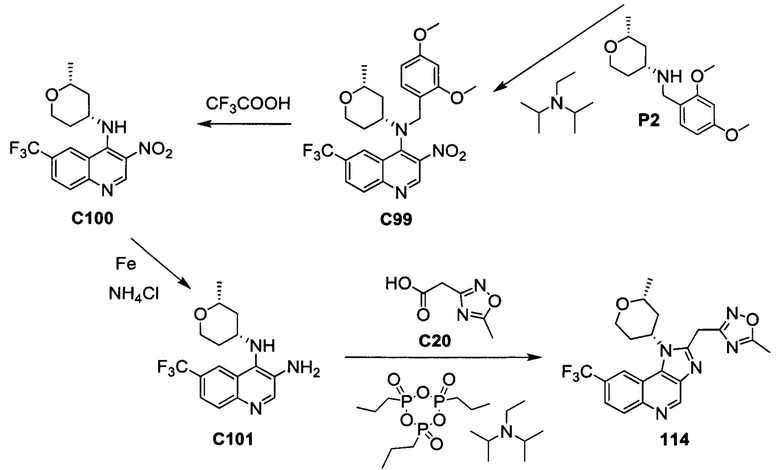
Стадия 1. Синтез 2-нитро-6-(трифторметил)хинолин-4-ола (С97)
Раствор 6-(трифторметил)хинолин-4-ола (2,00 г; 9,38 ммоль) в концентрированной азотной кислоте (10 мл) перемешивали в течение 14 часов при 50°С, после чего его выливали в воду (50 мл). Полученное твердое вещество выделяли фильтрованием, получая продукт в виде бледно-желтого твердого вещества. Выход: 1,80 г; 6,97 ммоль; 74%. 1Н ЯМР (400 МГц, DMSO-d6) δ 9.29 (s, 1H), 8.46 (s, 1H), 8.11 (d, J=9,0 Гц, 1Н), 7.92 (d, J=8,5 Гц, 1Н).
Стадия 2. Синтез 4-хлор-3-нитро-6-(трифторметил)хинолина (С98)
Оксихлорид фосфора (3,25 мл; 34,9 ммоль) добавляли при 15°С к раствору соединения С97 (3,00 г; 11,6 ммоль) в N,N-диметилформамиде (10 мл) и реакционную смесь перемешивали в течение 2 часов при 15°С. Затем ее выливали в воду (80 мл). После сбора осадка посредством фильтрования получали продукт в виде твердого вещества (2,40 г). По данным 1Н ЯМР анализа это вещество оказалось неочищенным, и его переносили непосредственно на следующую стадию. 1Н ЯМР (400 МГц, DMSO-d6), только пики продуктов: δ 9.22 (s, 1H), 8.40 (br s, 1H), 8.03 (br d, J=8,5 Гц, 1H), 7.92-7.97 (m, 1H).
Стадия 3. Синтез N-(2,4-диметоксибензил)-N-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-3-нитро-6-(трифторметил)хинолин-4-амина (С99)
N,N-Диизопропилэтиламин (3,36 г; 26,0 ммоль) и соединение Р2 (2,43 г; 9,16 ммоль) медленно добавляли при 15°С к раствору соединения С98 (с предыдущей стадии; 2,40 г; не более 8,68 ммоль) в ацетонитриле (30 мл) и реакционную смесь перемешивали в течение 30 минут при 80°С. Добавляли воду (100 мл) и полученную смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические слои концентрировали в вакууме и остаток очищали посредством хроматографии на силикагеле (градиент: от 9% до 25% этилацетата в петролейном эфире), получая продукт в виде желтого твердого вещества. Выход: 3,40 г; 6,73 ммоль; 58% за 2 стадии. 1Н ЯМР (400 МГц, CDCl3) δ 9.11 (s, 1H), 8.60 (br s, 1H), 8.15 (d, J=9,0 Гц, 1H), 7.92 (dd, J=8,8; 1,8 Гц, 1H), 6.84 (d, J=8,0 Гц, 1H), 6.22 (dd, J=8,3; 2,3 Гц, 1H), 6.16 (d, J=2,0 Гц, 1H), 4.33-4.44 (m, 2H), 4.02-4.10 (m, 1H), 3.77-3.87 (m, 1H), 3.68 (s, 3H), 3.50 (s, 3H), 3.36-3.46 (m, 2H), 1.95-2.10 (m, 3H), 1.67-1.78 (m, 1H), 1.23(d, J=6,0 Гц, 3H).
Стадия 4. Синтез N-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-3-нитро-6-(трифторметил)хинолин-4-амина (С100)
Трифторуксусную кислоту (7,67 г; 67,3 ммоль) добавляли при 15°С к раствору соединения С99 (3,40 г; 6,73 ммоль) в дихлорметане (30 мл) и реакционную смесь перемешивали в течение 30 минут при 15°С. Растворители удаляли в вакууме и остаток разбавляли водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои концентрировали в вакууме, получая продукт (2,50 г) в виде бледно-желтого твердого вещества, часть которого использовали непосредственно на следующей стадии. LCMS m/z 355,8 [М+Н]+.
Стадия 5. Синтез N4-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-6-(трифторметил)хинолин-3,4-диамина (С101)
Порошок железа (314 мг; 5,62 ммоль) и хлорид аммония (301 мг; 5,63 ммоль) добавляли к раствору соединения С100 (с предыдущей стадии; 200 мг; не более 0,54 ммоль) в этаноле (5 мл) и воде (1 мл) и реакционную смесь перемешивали в течение 1 часа при 80°С. Затем ее фильтровали через диатомовую землю и фильтрат концентрировали в вакууме. После хроматографии на силикагеле (градиент: от 9% до 33% этилацетата в петролейном эфире) получали продукт в виде бледно-серого твердого вещества. Выход: 140 мг; 0,430 ммоль; 80% за 2 стадии. LCMS m/z 325,9 [М+Н]+.
Стадия 6. Синтез 2-[(5-метил-1,2,4-оксадиазол-3-ил}метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-8-(трифторметил)-1Н-имидазо[4,5-с]хинолина (114)
К раствору соединения С20 (60,0 мг; 0,422 ммоль) в N,N-диметилформамиде (2 мл) добавляли соединение С101 (137 мг; 0,421 ммоль), N,N-диизопропилэтиламин (161 мг; 1,25 ммоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 0,39 мл; 0,655 ммоль). Реакционную смесь перемешивали в течение 2 часов при 110°С, после чего ее разбавляли водой (80 мл) и экстрагировали этилацетатом (3×80 мл). Объединенные органические слои концентрировали в вакууме и очищали обращенно-фазовой HPLC (колонка: Durashell, 5 мкм, от Agela; подвижная фаза А: 0,05% гидроксида аммония в воде; подвижная фаза В: ацетонитрил; градиент: от 40% до 70% В), получая продукт в виде бледно-серого твердого вещества. Выход: 16,8 мг; 38,9 мкмоль; 9%. LCMS m/z 432,0 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.41 (s, 1H), 8.94-9.11 (br m, 1H), 8.41 (d, J=8,8 Гц, 1H), 7.90 (dd, J=8,8; 1,8 Гц, 1H), 4.99-5.19 (br m, 1H), 4.62 (s, 2H), 4.33 (br dd, J=12; 5 Гц, 1H), 3.64-3.79 (m, 2H), 2.67-2.87 (br m, 1H), 2.61 (s, 3Н), 2.38-2.63 (br m, 1H), 1.80-2.09 (br m, 2H), 1.35 (d, J=6,0 Гц, 3Н).
Пример 115
8-Хлор-2-[(3-метил-1,2-оксазол-5-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолин (115)
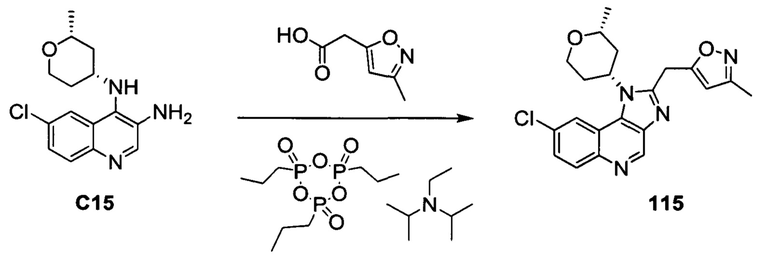
N,N-Диизопропилэтиламин (71,6 мкл; 0,411 ммоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 0,245 мл; 0,412 ммоль) добавляли к смеси соединения С15 (40,0 мг; 0,137 ммоль) и (3-метил-1,2-оксазол-5-ил)уксусной кислоты (19,3 мг; 0,137 ммоль) в этилацетате (0,8 мл) и реакционную смесь нагревали при 80°С в течение ночи. Затем ее распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом и водный слой дважды экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. После хроматографии на силикагеле (градиент: от 0% до 10% метанола в дихлорметане), затем растирания с диэтиловым эфиром получали продукт в виде желтого твердого вещества. Выход: 33,2 мг; 83,6 мкмоль; 61%. LCMS m/z 397,3 [M+H]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.28 (s, 1H), 8.55-8.75 (br m, 1H), 8.24 (d, J=8,6 Гц, 1H), 7.66 (dd, J=9,0; 2,0 Гц, 1H), 6.07 (s, 1H), 4.90-5.13 (br m, 1H), 4.61 (s, 2H), 4.34 (br dd, J=11,7; 4,3 Гц, 1H), 3.64-3.82 (m, 2H), 2.62-2.88 (br m, 1H), 2.36-2.59 (br m, 1H), 2.28 (s, 3Н), 1.71-2.02 (br m, 2H), 1.37 (d, J=5,9 Гц, 3Н).
Пример 116
8-Хлор-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-2-[(5-метил-1,3,4-тиадиазол-2-ил)метил]-1Н-имидазо[4,5-с]хинолин (116)
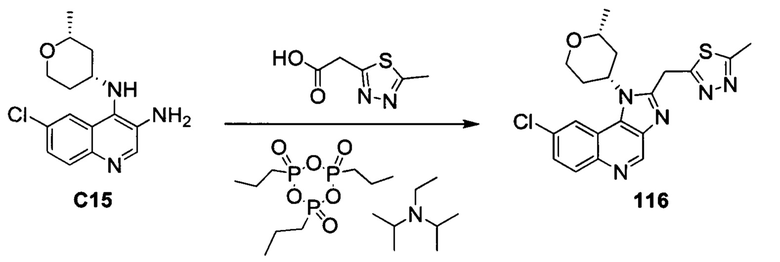
N,N-Диизопропилэтиламин (52 мг; 0,40 ммоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 480 мг; 0,75 ммоль) добавляли к раствору соединения С15 (102 мг; 0,350 ммоль) и (5-метил-1,3,4-тиадиазол-2-ил)уксусной кислоты (60 мг; 0,38 ммоль) в толуоле (3 мл). Реакционную смесь нагревали до 70°С в течение 2 часов и затем при 105°С в течение 18 часов. Добавляли насыщенный водный раствор бикарбоната натрия (10 мл) и полученную смесь экстрагировали этилацетатом (6×10 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После очистки посредством обращенно-фазовой HPLC (колонка: Durashell, 5 мкм, от Agela; подвижная фаза А: 0,225 муравьиной кислоты в воде; подвижная фаза В: ацетонитрил; градиент: от 34% до 54% В) получали продукт в виде красного твердого вещества. Выход: 38 мг; 92 мкмоль; 26%. LCMS m/z 414,0 (обнаружен пик изотопа хлора) [М+Н]+. 1Н ЯМР (400 МГц, CDCl3) δ 9.28 (s, 1H), 8.56-8.76 (br m, 1H), 8.23 (d, J=9,0 Гц, 1Н), 7.65 (dd, J=8,9; 2,1 Гц, 1Н), 5.23-5.37 (m, 1H), 4.94 (s, 2H), 4.31 (br dd, J=12; 5 Гц, 1H), 3.68-3.82 (m, 2H), 2.76 (s, 3Н), 2.57-2.80 (br m, 1H), 2.31-2.52 (br m, 1H), 1.58-1.9 (br m, 2H, предположительно; частично закрыт пиком от воды), 1.36 (d, J=6,0 Гц, 3Н).
Способ А
Превращение бициклических гетероароматических соединений, содержащих вицинальные атом хлора и нитрогруппу, в 1,2-дизамещенные имидазо[4,5-с]-конденсированные трициклические соединения M1
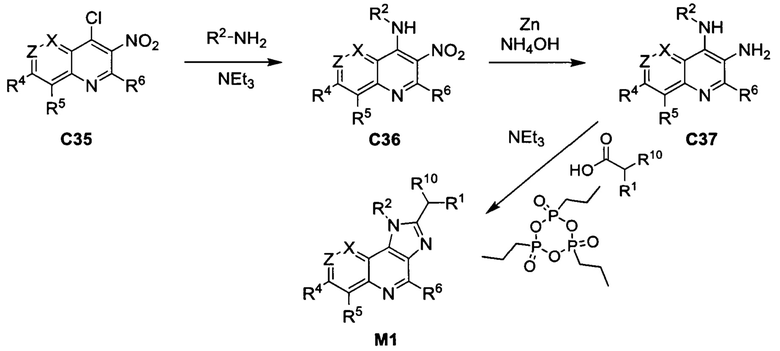
Стадия 1. Синтез бициклического гетероароматического соединения С36, содержащего вициналъные аминогруппу и нитрогруппу
Бициклическое гетероароматическое исходное вещество С35, содержащее вицинальные атом хлора и нитрогруппу (1 ммоль), объединяли во флаконе с амином R2-NH2 (1,2 ммоль) и N,N-диметилформамидом (4 мл). Добавляли триэтиламин (300 мкл; 2 ммоль), флакон герметично закрывали и реакционную смесь встряхивали при 30°С в течение 16 часов. Растворитель удаляли с использованием концентратора Speedvac®, получая продукт.
Стадия 2. Синтез бициклического гетероароматического соединения С37, содержащего две вицинальные аминогруппы
Соединение С36 с предыдущей стадии смешивали с метанолом (2 мл) и водным раствором гидроксида аммония (2 мл). Во флакон добавляли активированную цинковую пыль (650 мг; 10 ммоль), затем его герметично закрывали и встряхивали при 30°С в течение 1 часа. Реакционную смесь фильтровали и фильтрат концентрировали с использованием концентратора Speedvac®. К остатку добавляли воду (10 мл) и смесь экстрагировали этилацетатом (3×10 мл); объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали, получая продукт.
Стадия 3. Синтез 1,2-дизамещенного имидазо[4,5-с]-конденсированного трициклического соединения M1
Раствор соединения С37 в 1,4-диоксане (0,125 М; 800 мкл; 100 мкмоль) добавляли к карбоновой кислоте (R1)(R10)CHCOOH (100 мкмоль). Добавляли триэтиламин (45 мкл; 320 мкмоль) и 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ный раствор в этилацетате; 80 мкл; 130 мкмоль), флакон герметично закрывали и реакционную смесь встряхивали при 130°С в течение 16 часов. После концентрирования с использованием Speedvac® продукт очищали, применяя одну из следующих систем для обращенно-фазовой HPLC: 1) колонка: Gemini C18, 8 мкм, от Phenomenex; градиент: ацетонитрил в водном растворе гидроксида аммония (рН 10); 2) колонка: Diamonsil(2) C18, 5 мкм, от DIKMA; градиент: ацетонитрил в воде, содержащей 0,225% муравьиной кислоты; 3) колонка: Triart C18 серии YMC-Actus, 5 мкм; градиент: ацетонитрил в водном растворе гидроксида аммония (рН 10).
Способ В
Превращение бициклических гетероароматических соединений, содержащих вициналъные атом хлора и нитрогруппу, в 1,2-дизамещенные имидазо[4,5-с]-конденсированные трициклические соединения M1
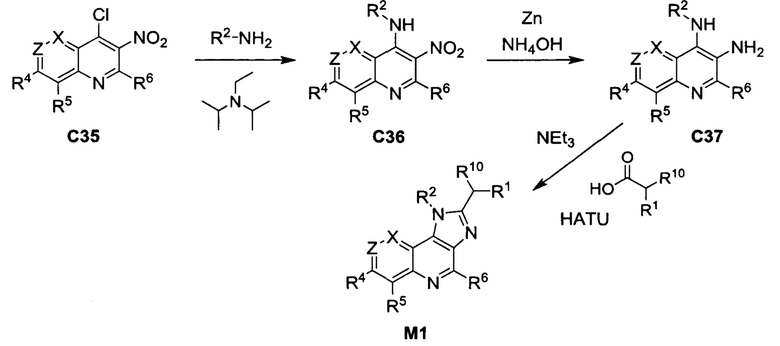
Стадия 1. Синтез бициклического гетероароматического соединения С36, содержащего вициналъные аминогруппу и нитрогруппу
Стадия 1. Синтез бициклического гетероароматического соединения С36, содержащего вициналъные аминогруппу и нитрогруппу
Соединение С35 (0,15 ммоль) объединяли с амином R2NH2 (0,18 ммоль) и N,N-диизопропилэтиламином (0,10 мл; 0,6 ммоль) в ацетонитриле (0,5 мл) и флакон с реакционной смесью встряхивали при 45°С в течение 2 часов. Затем реакционную смесь распределяли между водой (1,5 мл) и этилацетатом (2,4 мл) при перемешивании вихревым способом. Органический слой пропускали через картридж для твердофазной экстракции (6 мл), в который был загружен сульфат натрия (приблизительно 1 г); такой способ экстракции повторяли дважды и растворитель удаляли в вакууме, получая продукт.
Стадия 2. Синтез бициклического гетероароматического соединения С37, содержащего две вицинальные аминогруппы
Соединение С36 (с предыдущей стадии; приблизительно 0,15 ммоль) обрабатывали метанолом (0,3 мл) и водным раствором гидроксида аммония (0,3 мл). Добавляли цинковую пыль (приблизительно 100 мг; 1,5 ммоль) и реакционную смесь встряхивали при комнатной температуре в течение 1 часа, затем фильтровали через диатомовую землю. Фильтрующую набивку промывали этилацетатом (2×2,5 мл) и объединенные фильтраты концентрировали в вакууме. Остаток распределяли между водой (1,5 мл) и этилацетатом (2,4 мл) при перемешивании вихревым способом. Органический слой пропускали через картридж для твердофазной экстракции (6 мл), в который был загружен сульфат натрия (приблизительно 1 г); такой способ экстракции повторяли дважды и растворитель удаляли при пониженном давлении, получая продукт.
Стадия 3. Синтез 1,2-дизамещенного имидазо[4,5-с]-конденсированного трициклического соединения M1
Соединение С37 (с предыдущей стадии; приблизительно 0,15 ммоль) растворяли в 1-метилпирролидин-2-оне (0,4 мл) и добавляли к карбоновой кислоте (R1)(R10)CHCOOH (0,19 ммоль). Добавляли триэтиламин (23 мкл; 0,16 ммоль) и раствор гексафторфосфата O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU; 71 мг; 0,19 ммоль) в 1-метилпирролидин-2-оне (0,3 мл). (Применяли дополнительный эквивалент триэтиламина в случае использования карбоновой кислоты в виде гидрохлоридной соли.) Реакционную смесь встряхивали при 100°С в течение 20 часов, затем распределяли между водой (1,5 мл) и этилацетатом (2,4 мл) при перемешивании вихревым способом. Органический слой пропускали через картридж для твердофазной экстракции (6 мл), в который был загружен сульфат натрия (приблизительно 1 г); такой способ экстракции повторяли дважды и растворитель удаляли при пониженном давлении, получая продукт. Очистку осуществляли посредством градиентного элюирования, применяя одну из следующих систем для обращенно-фазовой HPLC: 1) колонка: Sunfire C18, 5 мкм, от Waters; подвижная фаза А: 0,05% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,05% трифторуксусной кислоты в ацетонитриле (об./об.); или 2) колонка: XBridge C18, 5 мкм, от Waters; подвижная фаза А: 0,03% гидроксида аммония в воде (об./об.); подвижная фаза В: 0,03% гидроксида аммония в ацетонитриле (об./об.).
В Таблице 1, ниже, приведены способ получения, структура и физико-химические данные для соединений из примеров 12-92 и 117-145.
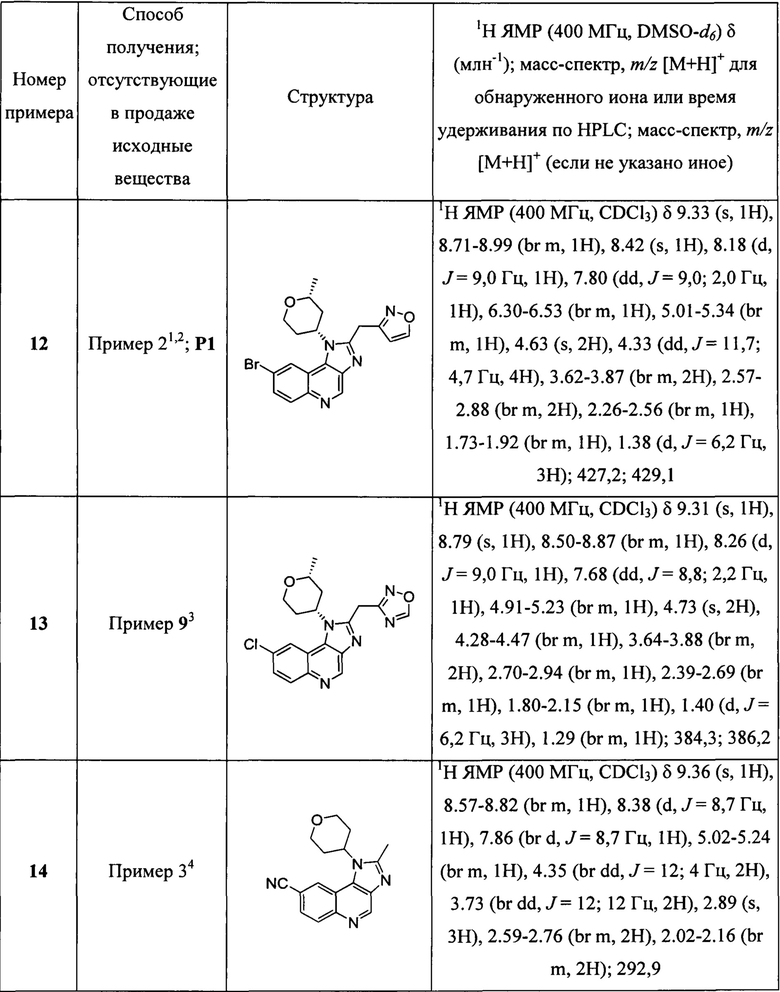
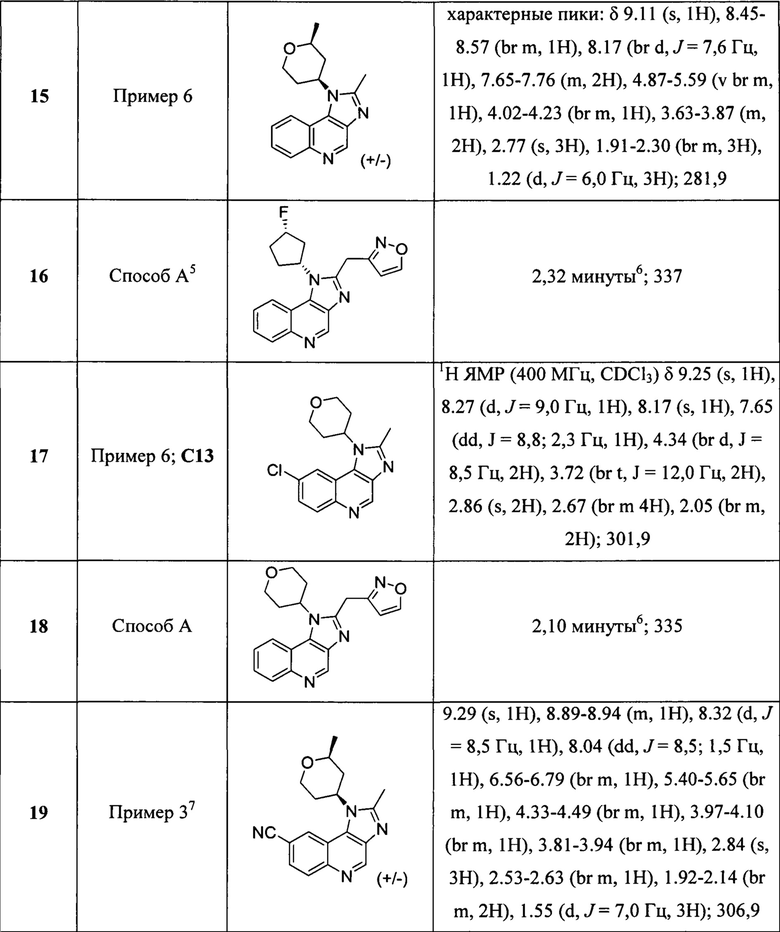
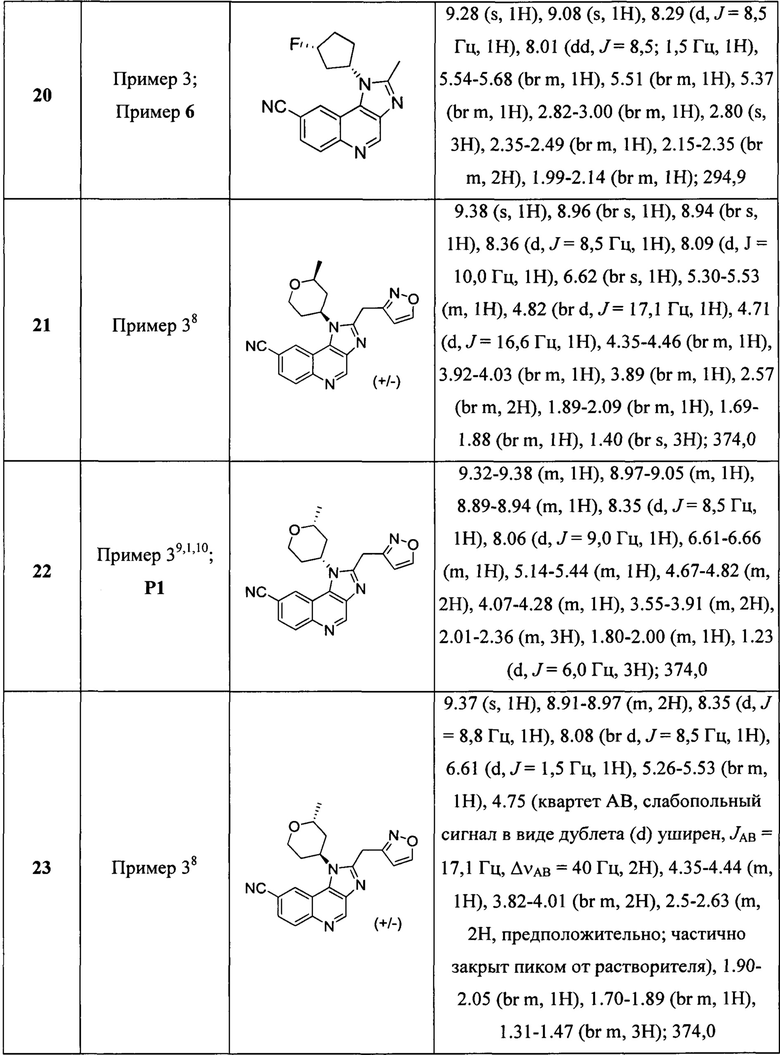
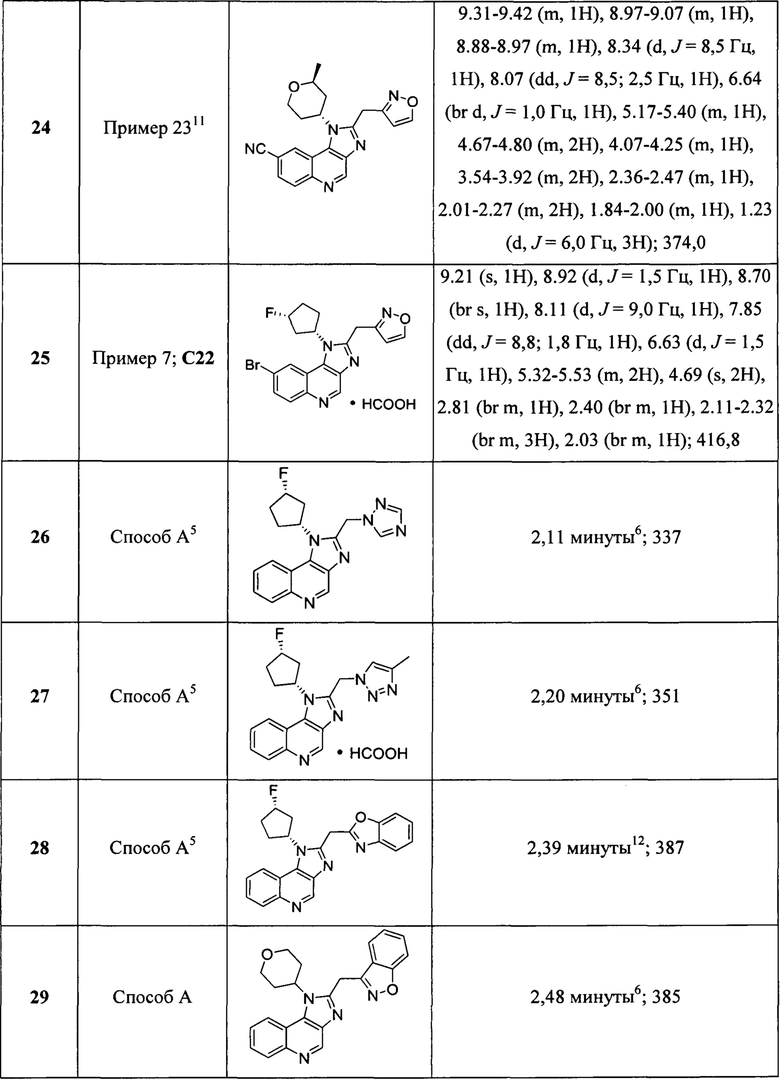
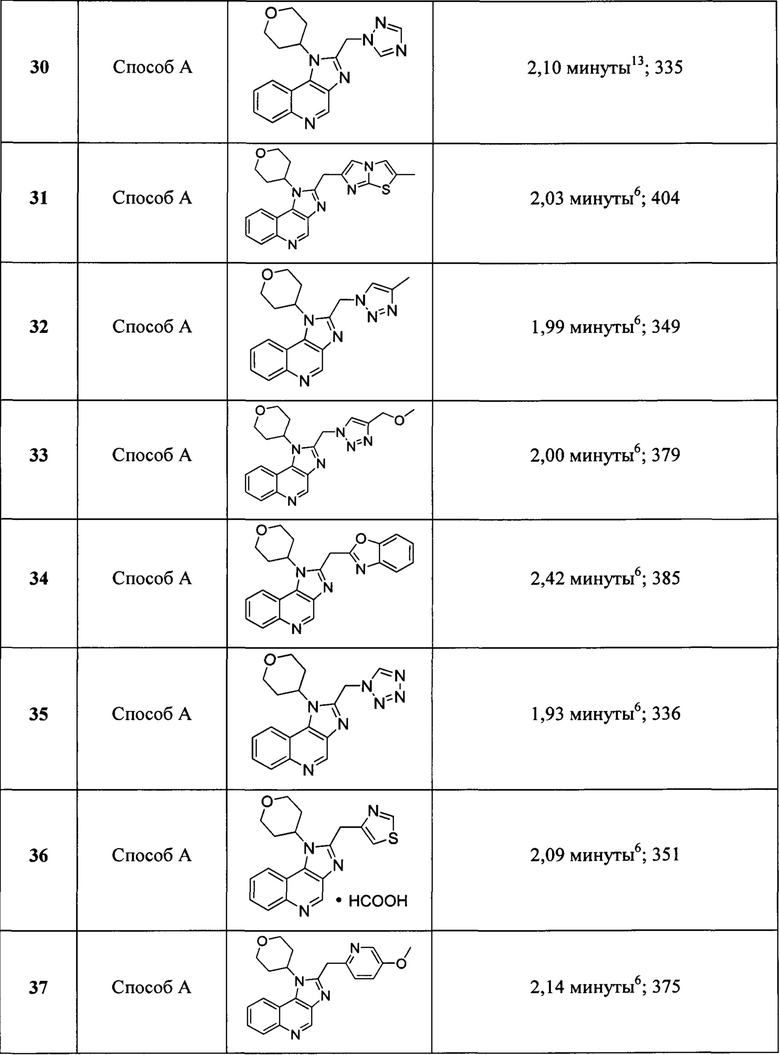
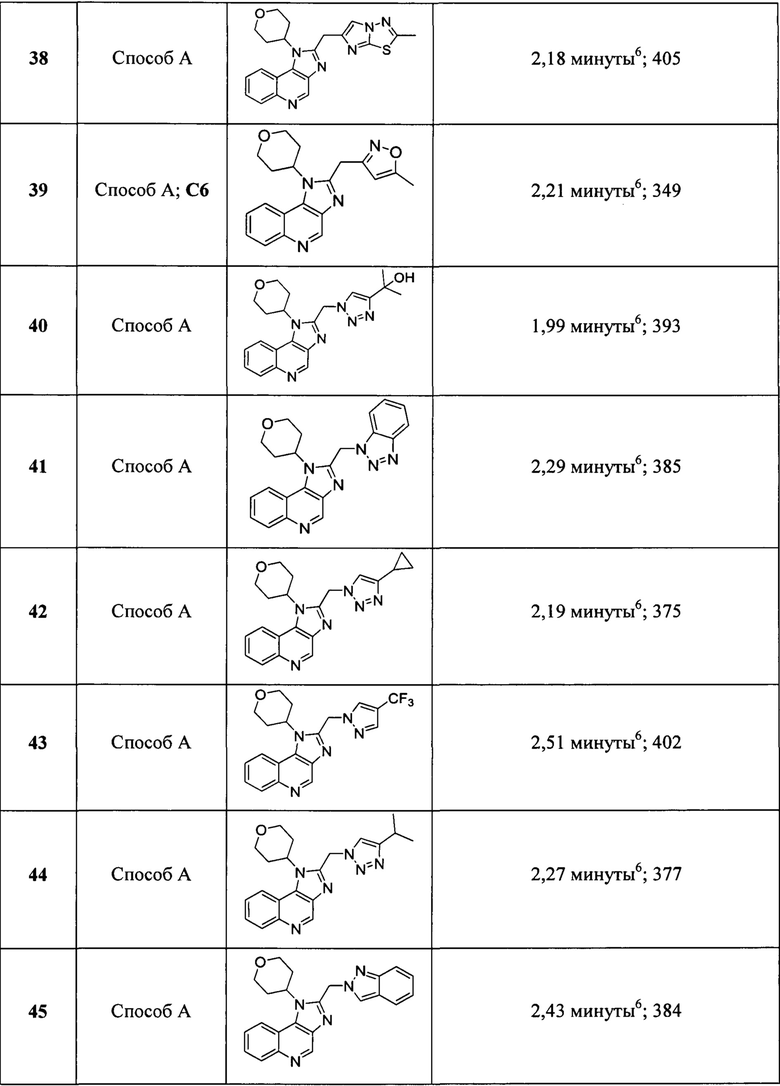
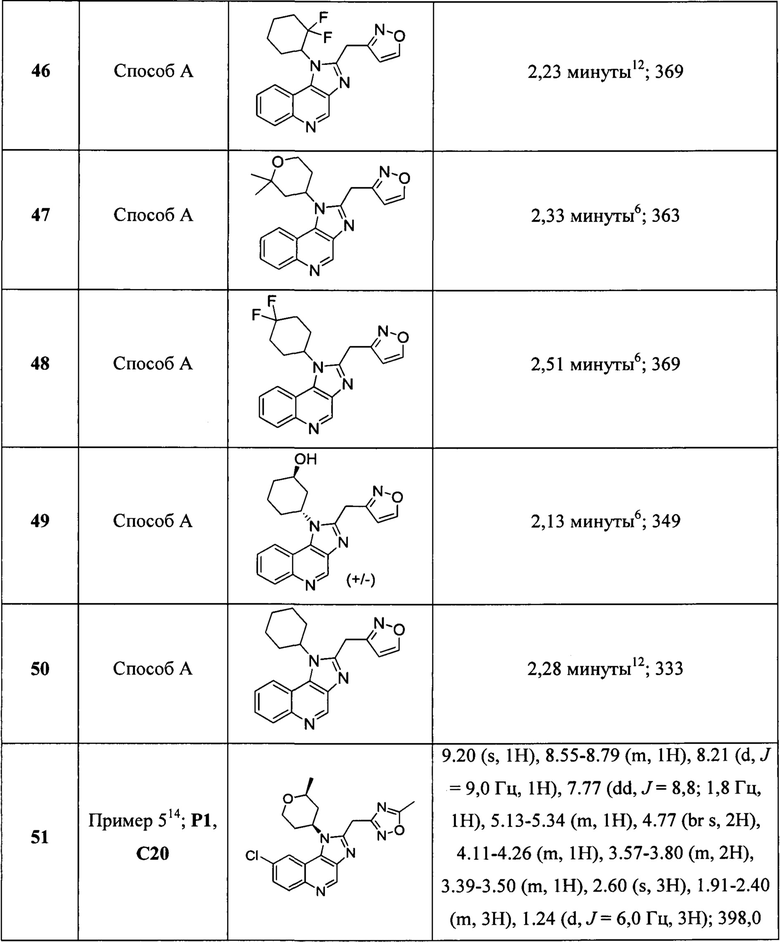
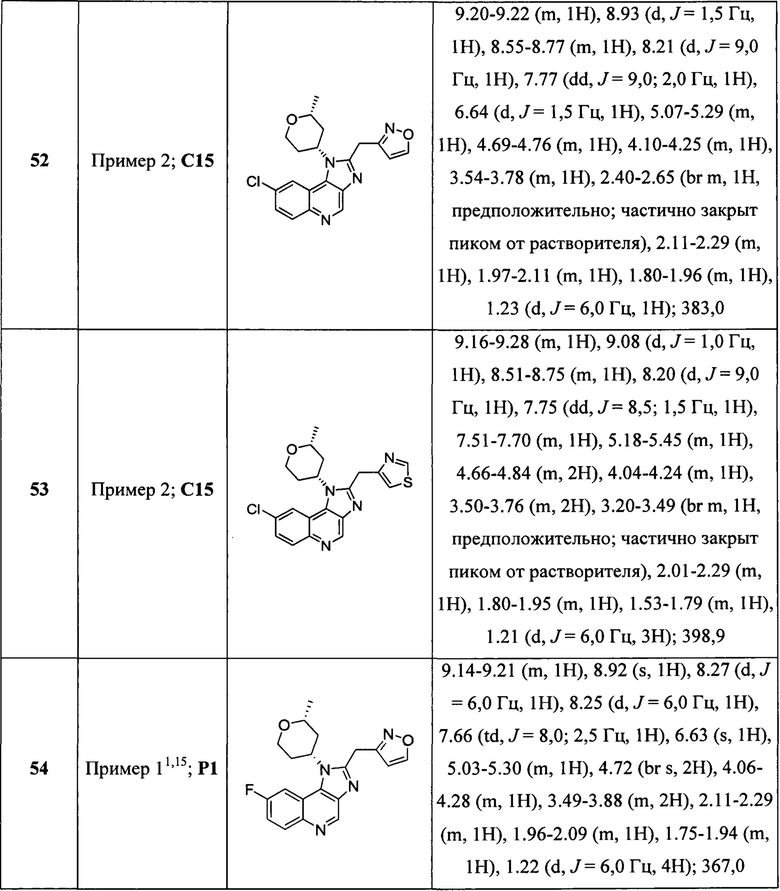
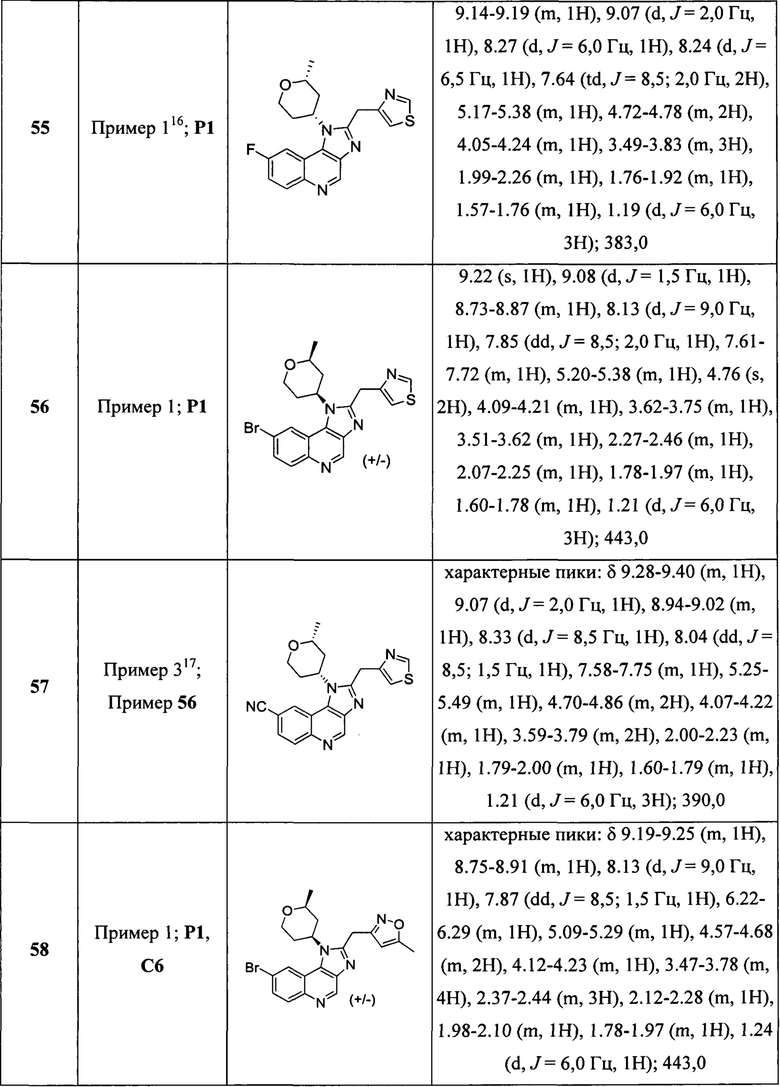
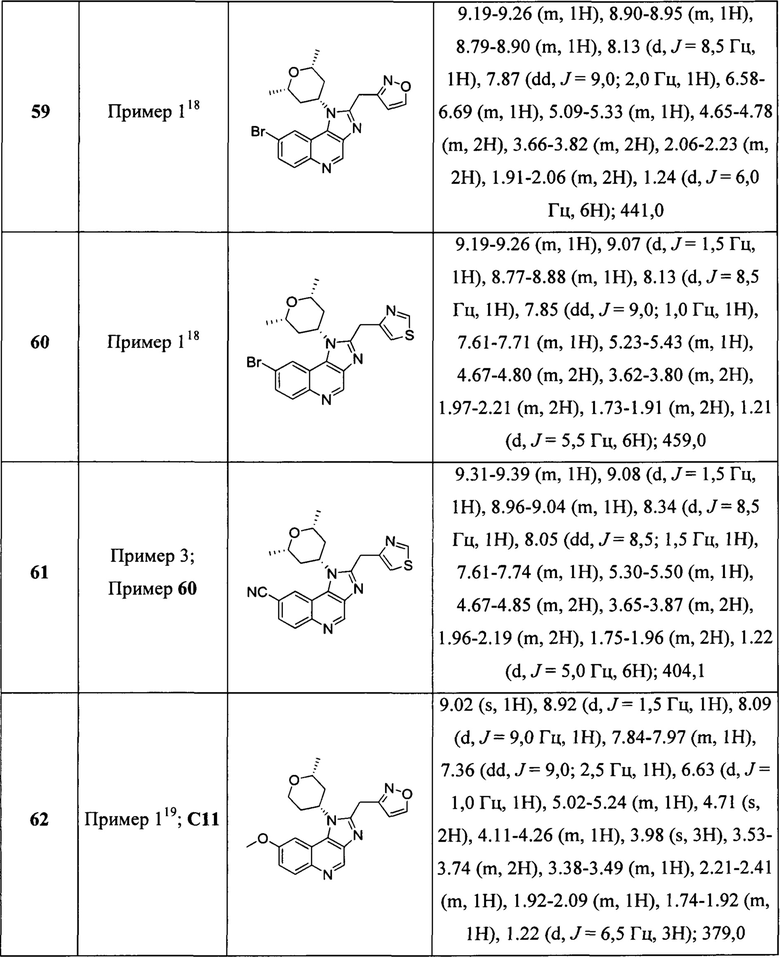
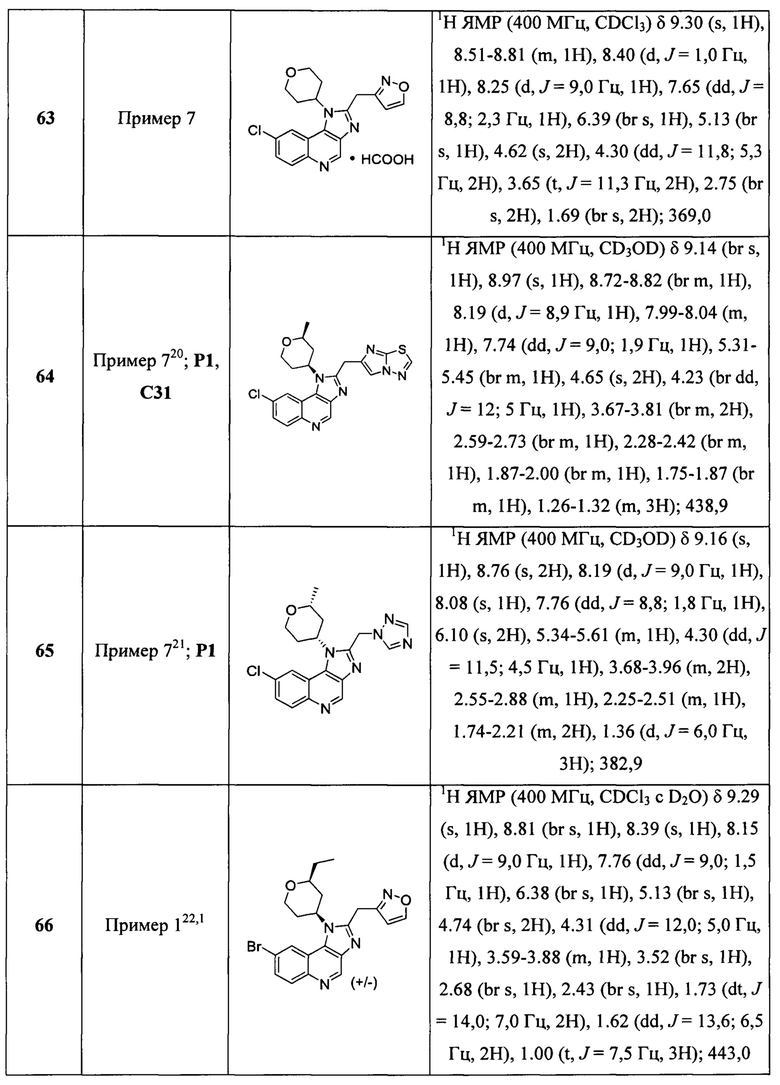
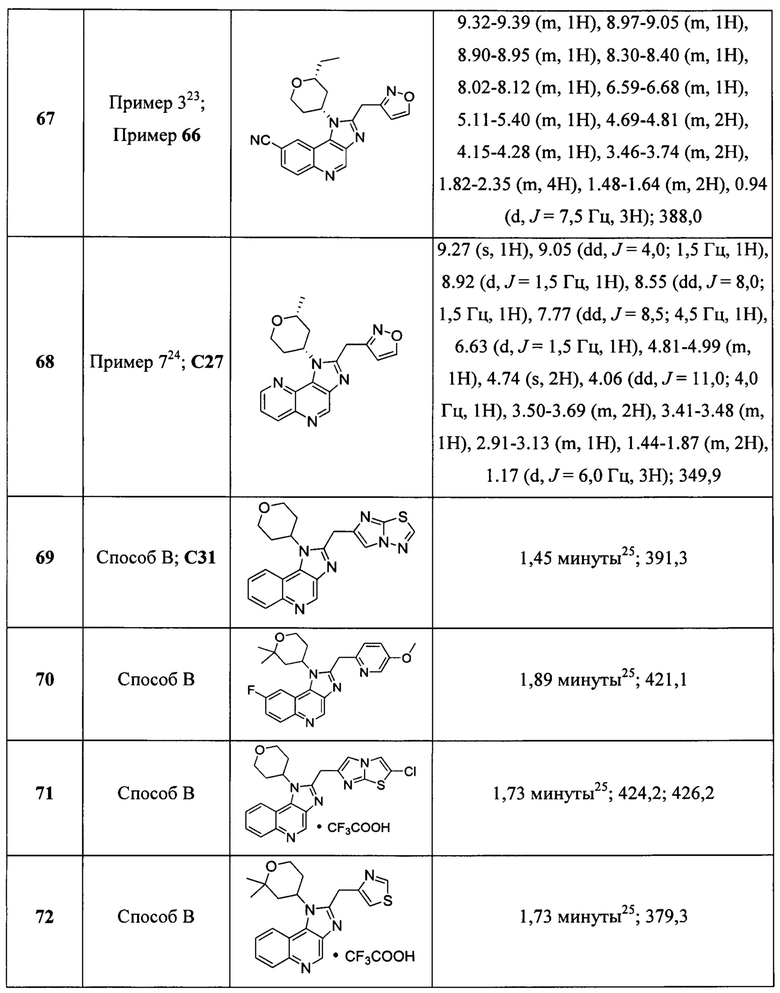
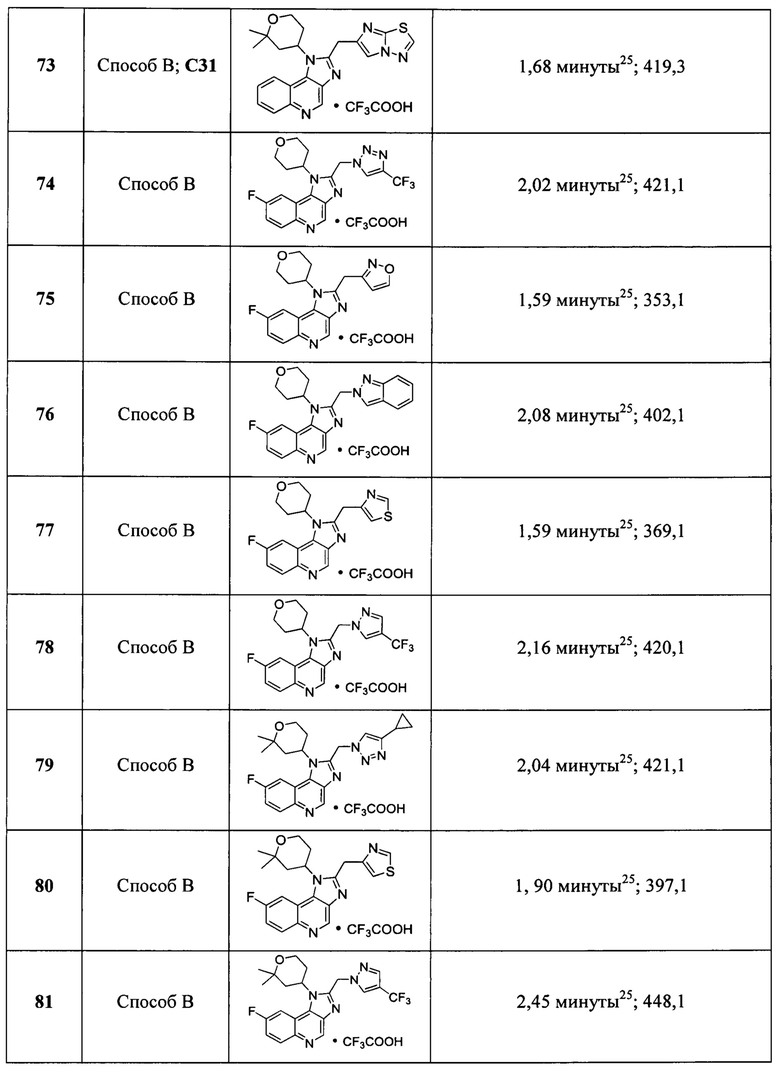
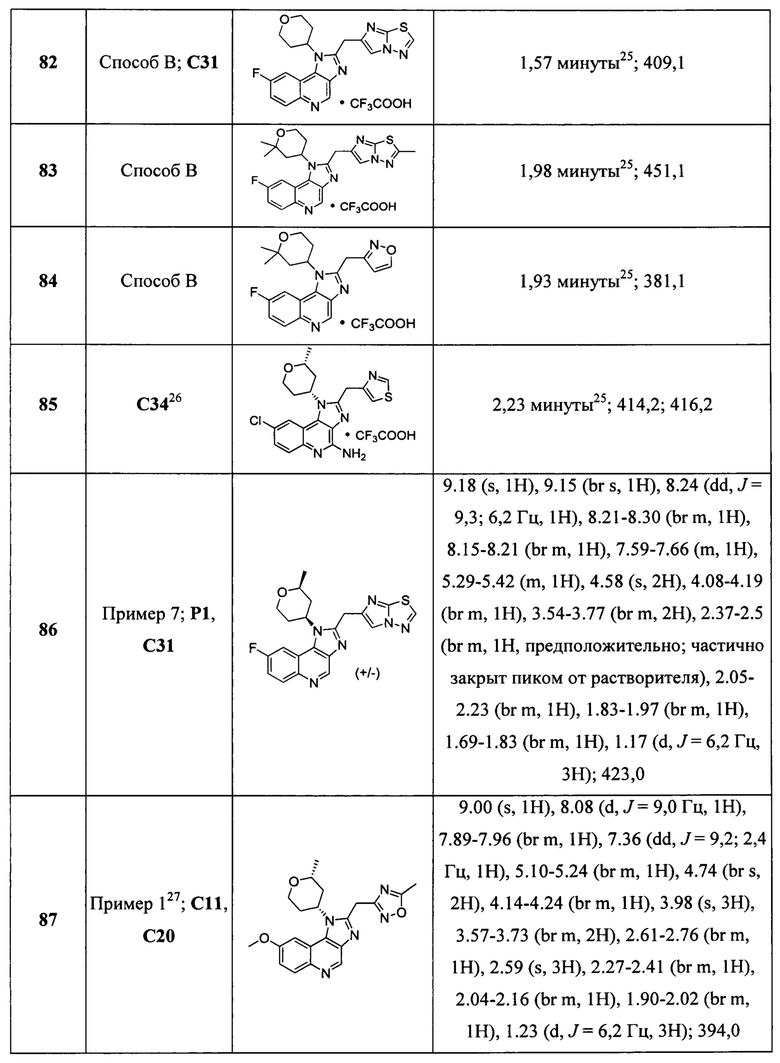
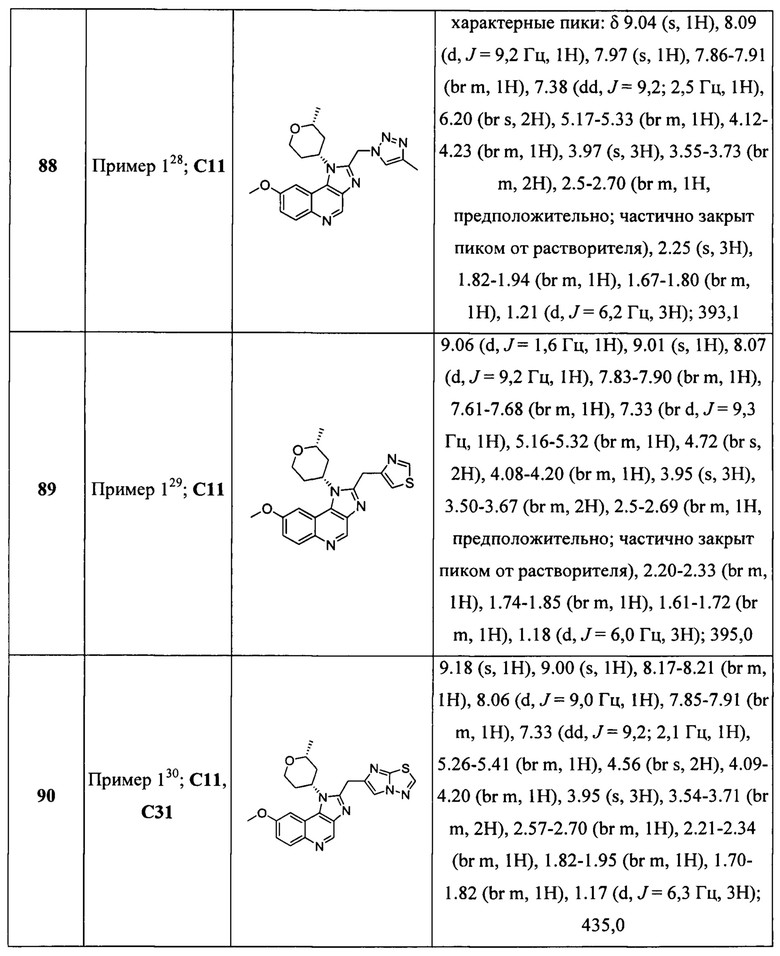
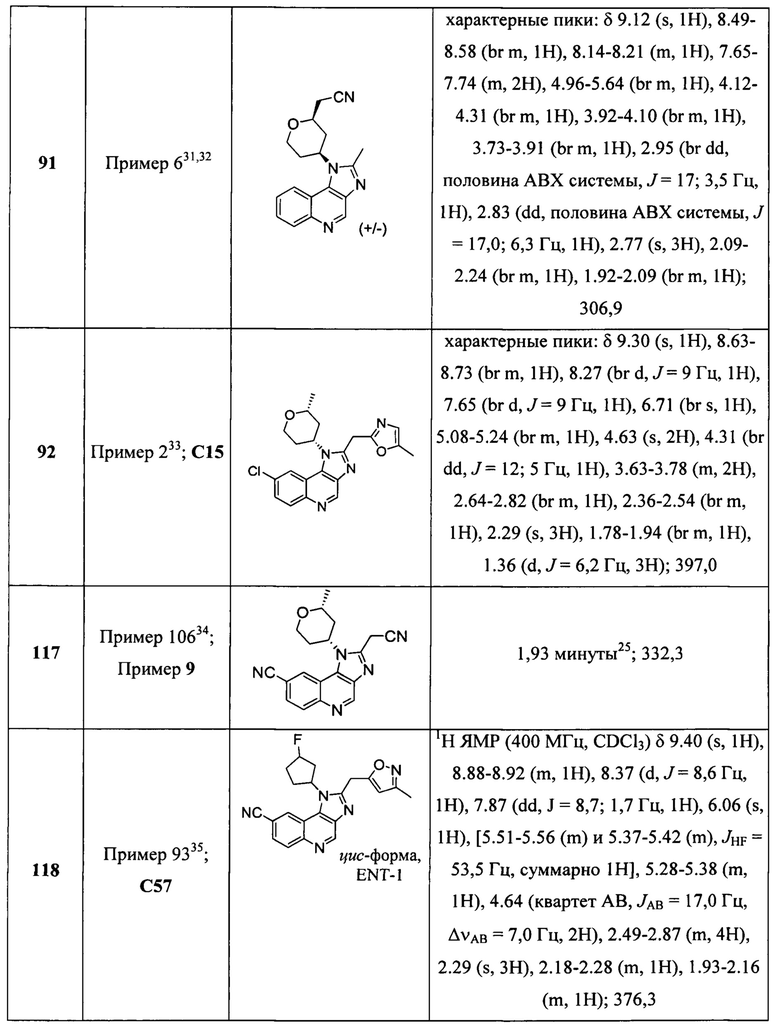
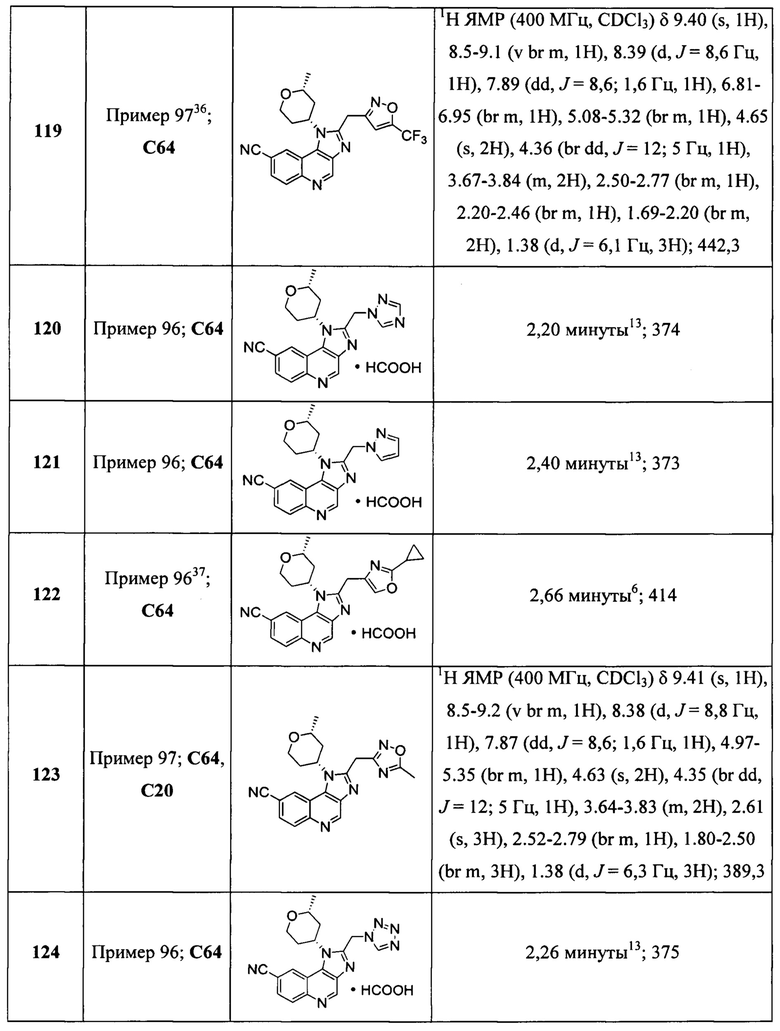
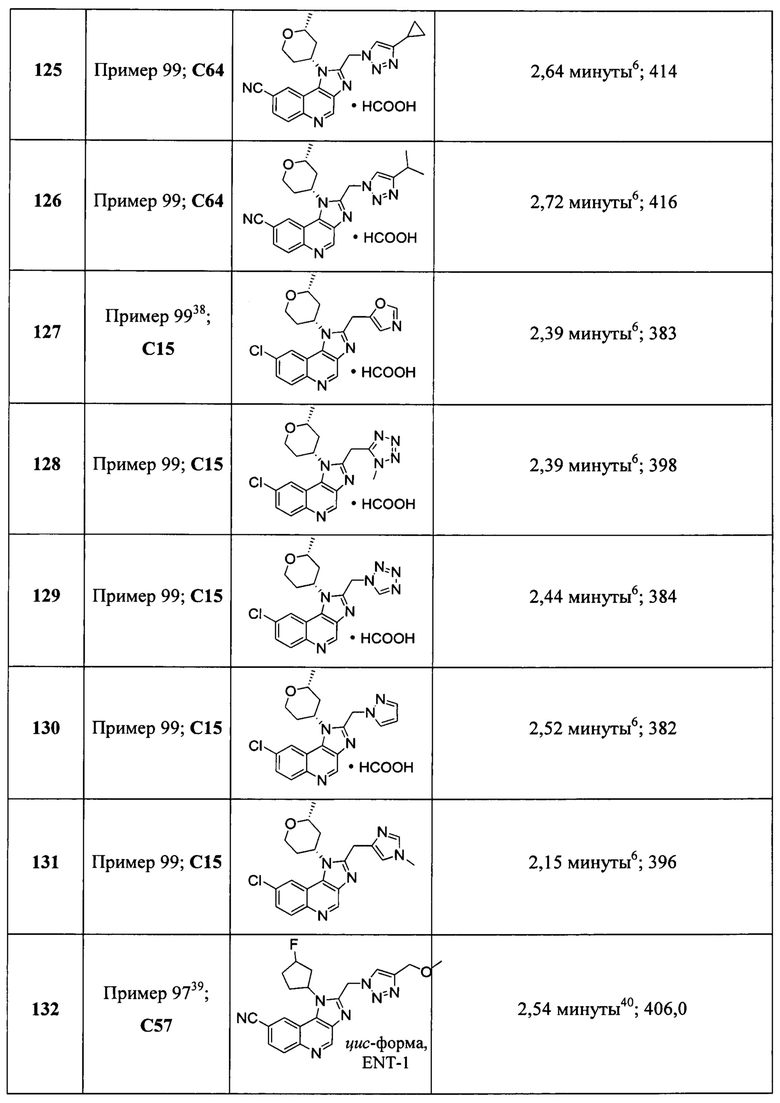
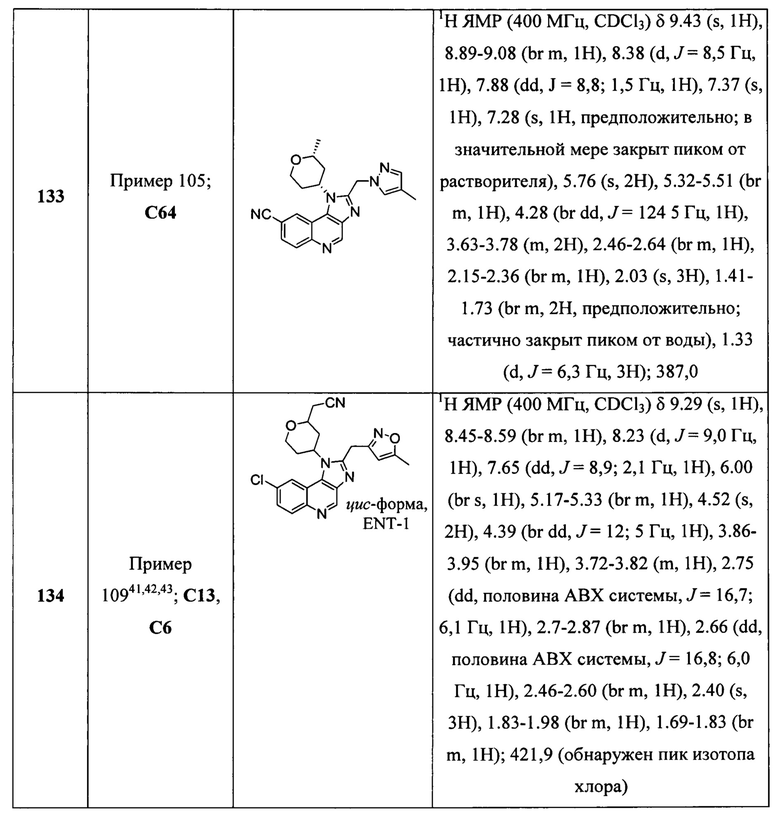
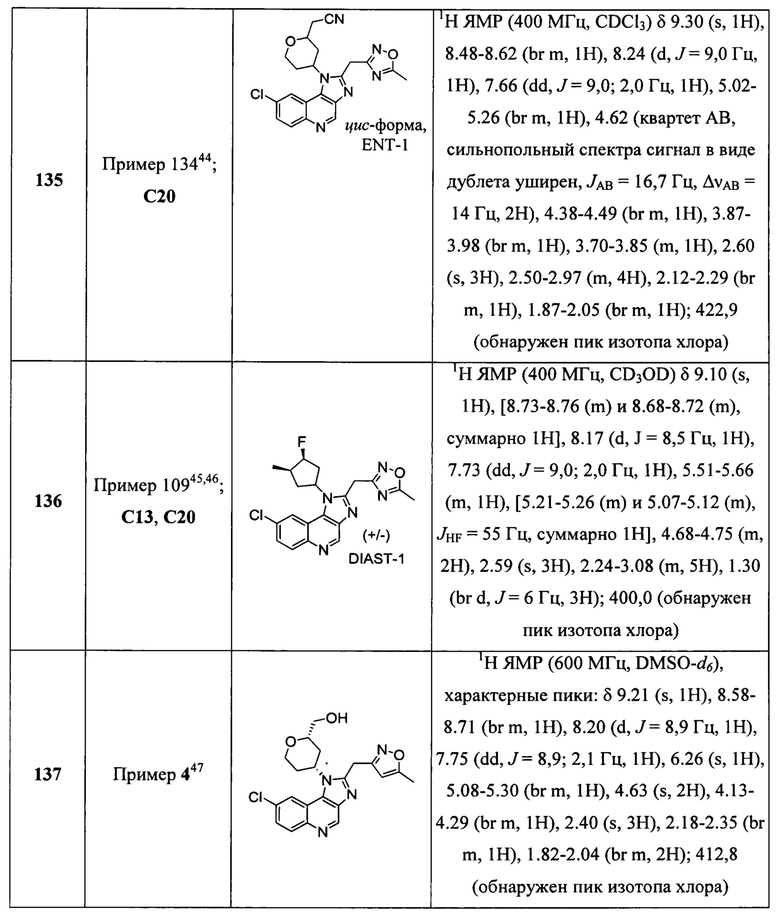
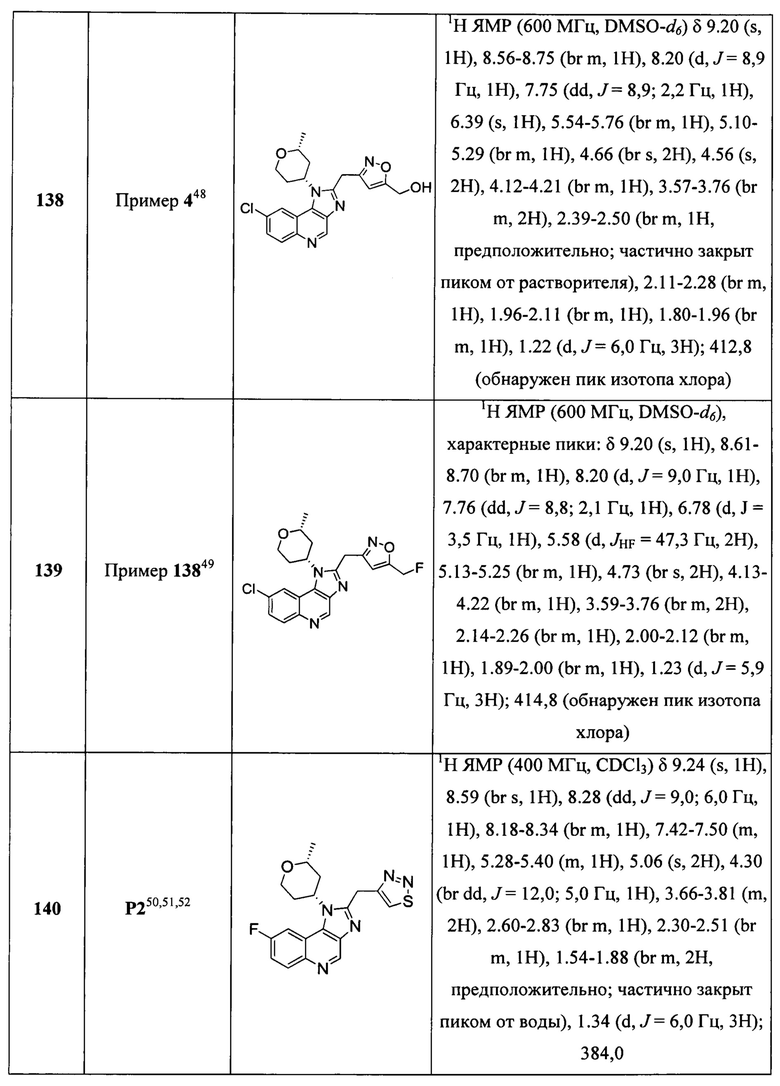
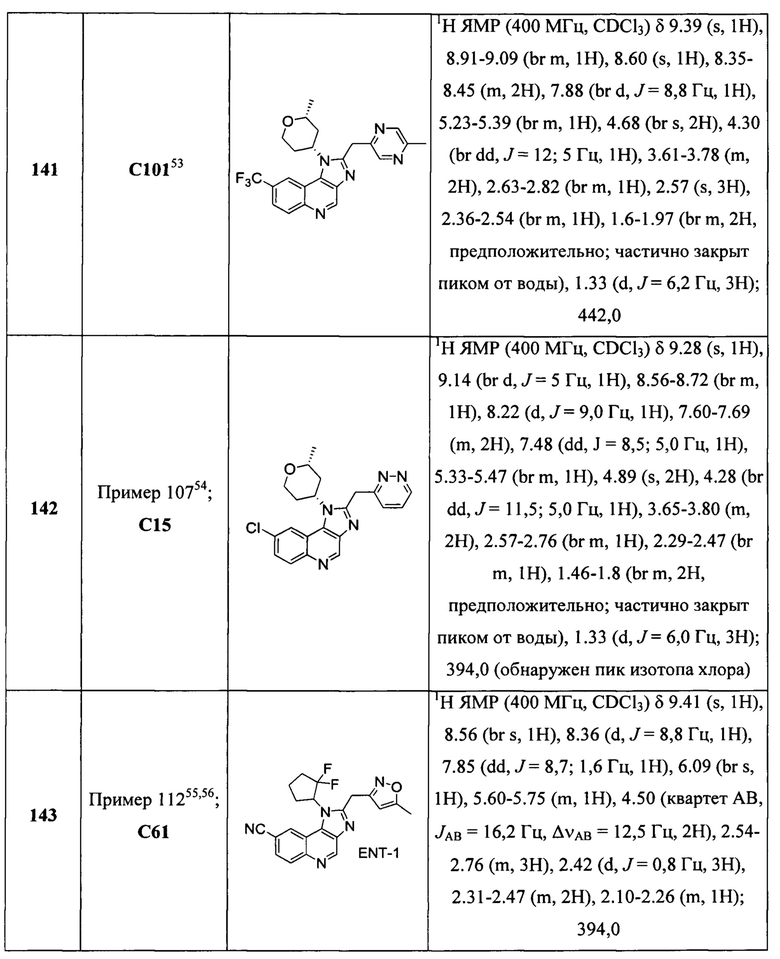
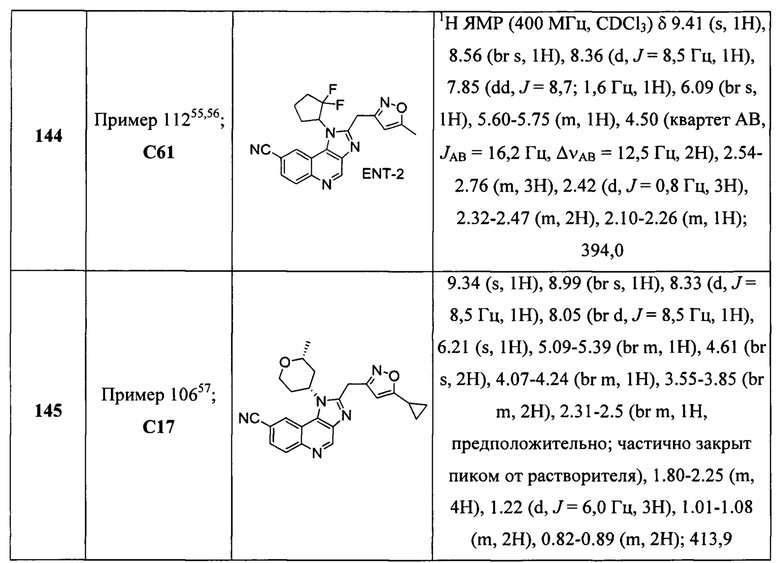
1. В этом случае 2,4-диметоксибензильную защитную группу удаляли с использованием нитрата аммония-церия(IV).
2. Рацемический продукт разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Lux Cellulose-1, 5 мкм; элюент: смесь 4:1 двуокись углерода/метанол). Элюирующимся вторым соединением было соединение примера 12. Энантиомер соединения примера 12, 8-бром-1-[(2S,4S)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолин, представлял собой элюирующийся первым энантиомер и демонстрировал следующие биологические данные: LRRK2, формат 1, IC50=510 нМ для WT (дикий тип); LRRK2, формат 1, IC50=226 нМ для формы с мутацией G2019S.
3. Соединение примера 9 приводили во взаимодействие с гидроксиламином и N,N-диизопропилэтиламином в этаноле; полученный 2-{8-хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолин-2-ил}-N'-гидроксиэтанимидамид подвергали реакции циклизации с использованием триметилортоформиата и и-толуолсульфоновой кислоты, получая соединение примера 13.
4. Необходимый 8-бром-2-метил-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолин получали, используя общий способ из примера 6.
5. После взаимодействия трет-бутил-[(1R,3R)-3-гидроксициклопентил]карбамата с трифторидом (диэтиламино)серы с последующей обработкой хлористым водородом в этилацетате получали (1R,3S)-3-фторциклопентанамин.
6. Условия для аналитической HPLC. Колонка: XBridge С18, 2,1×50 мм, 5 мкм, от Waters; подвижная фаза А: 0,0375% трифторуксусной кислоты в воде; подвижная фаза В: 0,01875% трифторуксусной кислоты в ацетонитриле; градиент: от 1% до 5% В в течение 0,6 минуты; от 5% до 100% В в течение 3,4 минуты; скорость потока: 0,8 мл/минута.
7. Необходимый 8-бром-2-метил-1-(2-метилтетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолин получали, используя общий способ из примера 6.
8. 8-Бром-1-(2-метилтетрагидро-2H-пиран-4-ил)-2-(1,2-оксазол-3-илметил)-1Н-имидазо[4,5-с]хинолин синтезировали, используя способ из примера 7. Конечный продукт получали в виде смеси соединений примеров 21 и 23, которые разделяли посредством обращенно-фазовой HPLC (колонка: Triart С18 серии YMC-Actus, 5 мкм; подвижная фаза А: 0,225% муравьиной кислоты в воде; подвижная фаза В: ацетонитрил; градиент: от 29% до 49% В).
9. Необходимый 8-бром-1-(цис-2-метилтетрагидро-2H-пиран-4-ил)-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолин получали, используя общий способ из примера 1.
10. Рацемический продукт разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AD-3, 3 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: метанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В). При проведении аналитической HPLC (колонка: Chiralpak AD-3, 4,6×50 мм, 3 мкм; градиентная система такая же) соединение примера 22 демонстрировало время удерживания 1,18 минуты. Энантиомер соединения примера 22, 1-[(2S,4S)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолин-8-карбонитрил, имел время удерживания 1,37 минуты в тех же условиях. Энантиомер соединения примера 22, LCMS m/z 374,0 [М+Н]+, демонстрировал следующие биологические данные: LRRK2, формат 1, IC50=534 нМ для WT; LRRK2, формат 1, IC50=258 нМ для формы с мутацией G2019S.
11. Соединение примера 23 разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AD-3, 3 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: метанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В). При проведении аналитической HPLC (колонка: Chiralpak AD-3, 4,6×50 мм, 3 мкм; градиентная система такая же) соединение примера 24 демонстрировало время удерживания 1,37 минуты. Энантиомер соединения примера 24, 1-[(2R,4S)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолин-8-карбонитрил, имел время удерживания 1,51 минут в тех же условиях. Энантиомер соединения примера 24, LCMS m/z 374,1 [М+Н]+, демонстрировал следующие биологические данные: LRRK2, формат 1, IC50=267 нМ для WT, LRRK2, формат 1, IC50=134 нМ для формы с мутацией G2019S.
12. Условия для аналитической HPLC. Колонка: XBridge С18, 2,1×50 мм, 5 мкм, от Waters; подвижная фаза А: 0,0375% трифторуксусной кислоты в воде; подвижная фаза В: 0,01875% трифторуксусной кислоты в ацетонитриле; градиент: от 10% до 100% В в течение 4,0 минуты; скорость потока: 0,8 мл/минута.
13. Условия для аналитической HPLC. Колонка: XBridge С18, 2,1×50 мм, 5 мкм, от Waters; подвижная фаза А: 0,05% гидроксида аммония в воде; подвижная фаза В: ацетонитрил; градиент: 5% В в течение 0,5 минуты; от 5% до 100% В в течение 2,9 минуты; 100% В в течение 0,8 минуты; скорость потока: 0,8 мл/минута.
14. Соединение этого примера получали в виде рацемата; энантиомеры разделяли посредством сверхкритической жидкостной хроматографии. Соединение примера 51 представляло собой элюирующийся вторым энантиомер; время удерживания 6,21 минуты (аналитическая колонка: Chiralpak AD-3, 4,6×150 мм, 3 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: этанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В; скорость потока: 1,5 мл/минута). Энантиомер из примера 51 (соединения примера 5) демонстрировал время удерживания 5,65 минуты в этой аналитической системе.
15. Рацемический продукт разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AD-H, 5 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: метанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В). При проведении аналитической HPLC (колонка: Chiralpak AD-H, 4,6×250 мм, 5 мкм; градиентная система такая же) соединение примера 54 демонстрировало время удерживания 6,28 минуты. Энантиомер соединения примера 54, 8-фтор-1-[(2S,4S)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолин, имел время удерживания 6,66 минуты в тех же условиях. Энантиомер соединения примера 54, LCMS m/z 366,9 [М+Н]+, демонстрировал следующие биологические данные: LRRK2, формат 1, IC50=332 нМ для WT; LRRK2, формат 1, IC50=236 нМ для формы с мутацией G2019S.
16. Рацемический продукт разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Chiralcel OD-H, 5 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: метанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В). При проведении аналитической HPLC (колонка: Chiralpak AS-H, 4,6×250 мм, 5 мкм; подвижная фаза: 10% этанола (содержащего 0,05% диэтиламина) в двуокиси углерода) соединение примера 55 демонстрировало время удерживания 5,85 минуты. Энантиомер соединения примера 55, 8-фтор-1-[(2S,4S)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с]хинолин, LCMS m/z 383,0 [М+Н]+, имел время удерживания 6,02 минуты в тех же условиях. Энантиомер соединения примера 55, LCMS m/z 366,9 [М+Н]+, демонстрировал следующие биологические данные: LRRK2, формат 1, IC50=725 нМ для WT; LRRK2, формат 1, IC50=380 нМ для формы с мутацией G2019S.
17. Рацемический продукт разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Chiralcel OD-3, 3 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: метанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В). При проведении аналитической HPLC (колонка: Chiralcel OD-3, 4,6×150 мм, 3 мкм; градиентная система такая же; скорость потока: 1,5 мл/минута) соединение примера 57 демонстрировало время удерживания 8,22 минуты. Энантиомер соединения примера 57, 1-[(2S,4S)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с]хинолин-8-карбонитрил, имел время удерживания 7,29 минуты в тех же условиях. Энантиомер соединения примера 57, LCMS m/z 390,0 [М+Н]+, демонстрировал следующие биологические данные: LRRK2, формат 1, IC50=382 нМ для WT; LRRK2, формат 1, IC50=196 нМ для формы с мутацией G2019S.
18. После гидрирования 2,6-диметил-4H-пиран-4-она над палладием на угле получали цис-2,6-диметилтетрагидро-4H-пиран-4-он, который преобразовывали в необходимый (2R,4r,6S)-N-(2,4-диметоксибензил)-2,6-диметилтетрагидро-2H-пиран-4-амин, используя способ, описанный для синтеза соединения Р1 в подготовительном примере P1.
19. Рацемический продукт разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AD-3, 3 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: метанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В). При проведении аналитической HPLC (колонка: Chiralpak AD-3, 4,6×150 мм, 3 мкм; градиентная система такая же) соединение примера 62 демонстрировало время удерживания 4,19 минуты. Энантиомер соединения примера 62, 8-метокси-1-[(2S,4S)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолин, имел время удерживания 5,07 минуты в тех же условиях. Энантиомер соединения примера 62, LCMS m/z 379,0 [М+Н]+, демонстрировал следующие биологические данные: LRRK2, формат 1, IC50=1713 нМ для WT; LRRK2, формат 1, IC50=508 нМ для формы с мутацией G2019S.
20. Соединение этого примера получали в виде рацемата; энантиомеры разделяли посредством сверхкритической жидкостной хроматографии. Соединение примера 64 представляло собой элюирующийся вторым энантиомер; время удерживания 8,87 минуты (аналитическая колонка: Chiralpak AD-H, 4,6×250 мм, 5 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: метанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В). Энантиомер из примера 64 (соединения примера 8) демонстрировал время удерживания 6,98 минуты в этой аналитической системе.
21. Соединение этого примера получали в виде рацемата; энантиомеры разделяли посредством сверхкритической жидкостной хроматографии. Соединение примера 65 представляло собой элюирующийся вторым энантиомер; время удерживания 8,73 минуты (аналитическая колонка: Chiralpak AD-H, 4,6×250 мм, 5 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: метанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В). Энантиомер соединения примера 65, 8-хлор-1-[(2S,4S)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1H-1,2,4-триазол-1-илметил)-1H-имидазо[4,5-с]хинолин, имел время удерживания 7,97 минуты в тех же условиях. Энантиомер соединения примера 65, LCMS m/z 382,9 [М+Н]+, демонстрировал следующие биологические данные: LRRK2, формат 1, IC50=687 нМ для WT; LRRK2, формат 1, IC50=241 нМ для формы с мутацией G2019S.
22. Необходимый цис-N-(2,4-диметоксибензил)-2-этилтетрагидро-2H-пиран-4-амин получали из пропаналя и бут-3-ен-1-ола по аналогии с синтезами соединений Р1 и Р2 за исключением того, что вместо реагента Джонса использовали хлорхромат пиридиния.
23. Рацемический продукт разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AD-3, 3 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: метанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В). При проведении аналитической HPLC (колонка: Chiralpak AD-3, 4,6×150 мм, 3 мкм; градиентная система такая же) соединение примера 67 демонстрировало время удерживания 1,17 минуты. Энантиомер соединения примера 67, 1-[(2S,4S)-2-этилтетрагидро-2H-пиран-4-ил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолин-8-карбонитрил, имел время удерживания 1,38 минуты в тех же условиях. Энантиомер соединения примера 67, LCMS m/z 388,0 [М+Н]+, демонстрировал следующие биологические данные: LRRK2, формат 1, IC50=699 нМ для WT; LRRK2, формат 1, IC50=403 нМ для формы с мутацией G2019S.
24. Рацемический продукт разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AD-3, 3 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: метанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В). При проведении аналитической HPLC (колонка: Chiralpak AD-3, 4,6×150 мм, 3 мкм; градиентная система такая же) соединение примера 68 демонстрировало время удерживания 5,76 минуты. Энантиомер соединения примера 68, 1-[(2S,4S)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с][1,5]нафтиридин, имел время удерживания 6,14 минуты в тех же условиях. Энантиомер соединения примера 68, LCMS m/z 349,9 [М+Н]+, демонстрировал следующие биологические данные: LRRK2, формат 1, IC50=853 нМ для WT; LRRK2, формат 1, IC50=632 нМ для формы с мутацией G2019S.
25. Условия для аналитической HPLC. Колонка: Atlantis dC18, 4,6×50 мм, 5 мкм, от Waters; подвижная фаза А: 0,05% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,05% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 5,0% до 95% В, линейный в течение 4,0 минуты; скорость потока: 2 мл/минута.
26. Соединение С34 объединяли с раствором аммиака в метаноле (7 М) и нагревали в микроволновом реакторе при 160°С, получая соединение примера 85.
27. Рацемический продукт разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AD-H, 5 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: этанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В). При проведении аналитической HPLC (колонка: Chiralpak AD-H, 4,6×250 мм, 5 мкм; градиентная система такая же) соединение примера 87 демонстрировало время удерживания 6,39 минуты. Энантиомер соединения примера 87, 8-метокси-2-[(5-метил-1,2,4-оксадиазол-3-ил)метил]-1-[(2S,4S)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолин, имел время удерживания 7,57 минуты в тех же условиях. Энантиомер соединения примера 87, LCMS m/z 394,1 [М+Н]+, демонстрировал следующие биологические данные: LRRK2, формат 1, IC50=2853 нМ для WT; LRRK2, формат 1, IC50=929 нМ для формы с мутацией G2019S.
28. Рацемический продукт разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AD-H, 5 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: метанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В). При проведении аналитической HPLC (колонка: Chiralpak AD-H, 4,6×250 мм, 5 мкм; градиентная система такая же) соединение примера 88 демонстрировало время удерживания 6,96 минуты. Энантиомер соединения примера 88, 8-метокси-1-[(2S,4S)-2-метилтетрагидро-2H-пиран-4-ил]-2-[(4-метил-1H-1,2,3-триазол-1-ил)метил]-1H-имидазо[4,5-с]хинолин, имел время удерживания 7,78 минуты в тех же условиях. Энантиомер соединения примера 88, LCMS m/z 393,1 [М+Н]+, демонстрировал следующие биологические данные: LRRK2, формат 1, IC50=1055 нМ для WT; LRRK2, формат 1, IC50=372 нМ для формы с мутацией G2019S.
29. Рацемический продукт разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AD-H, 5 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: метанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В). При проведении аналитической HPLC (колонка: Chiralpak AD-H, 4,6×250 мм, 5 мкм; градиентная система такая же) соединение примера 89 демонстрировало время удерживания 7,54 минуты. Энантиомер соединения примера 89, 8-метокси-1-[(2S,4S)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с]хинолин, имел время удерживания 8,17 минуты в тех же условиях. Энантиомер соединения примера 89, LCMS m/z 395,0 [М+Н]+, демонстрировал следующие биологические данные: LRRK2, формат 1, IC50=1218 нМ для WT; LRRK2, формат 1, IC50=743 нМ для формы с мутацией G2019S.
30. Рацемический продукт разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AD-H, 5 мкм; подвижная фаза А: двуокись углерода; подвижная фаза В: этанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В). При проведении аналитической HPLC (колонка: Chiralpak AD-H, 4,6×250 мм, 5 мкм; градиентная система такая же) соединение примера 90 демонстрировало время удерживания 8,60 минуты. Энантиомер соединения примера 90, 2-(имидазо[2,1-b][1,3,4]тиадиазол-6-илметил)-8-метокси-1-[(2S,4S)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолин, имел время удерживания 9,48 минуты в тех же условиях. Энантиомер соединения примера 90, LCMS m/z 435,1 [М+Н]+, демонстрировал следующие биологические данные: LRRK2, формат 1, IC50=623 нМ для WT; LRRK2, формат 1, IC50=245 нМ для формы с мутацией G2019S.
31. Реагент цис-2-[(бензилокси)метил]-N-(2,4-диметоксибензил)тетрагидро-2H-пиран-4-амин получали из (бензилокси)ацетальдегида и бут-3-ен-1-ола по аналогии с примечанием 22.
32. С промежуточного 1-{цис-2-[(бензилокси)метил]тетрагидро-2H-пиран-4-ил}-2-метил-1H-имидазо[4,5-с]хинолина удаляли защитную группу, используя трихлорид бора, и полученный спирт преобразовывали в 4-метилбензолсульфонатное производное. После замещения с использованием цианида тетраэтиламмония получали соединение примера 91.
33. Необходимую (5-метил-1,3-оксазол-2-ил)уксусную кислоту получали, используя метод из A.S.K. Hashmi et al., Org. Lett., 2004, 6, 4391-4394.
34. В этом случае для реакции с цианидом цинка применяли трис(дибензилиденацетон)дипалладий(0) и дициклогексил(2',6'-диметоксибифенил-2-ил)фосфан, а не тетракис(трифенилфосфин)палладий(0), и реакцию проводили с использованием облучения микроволнами.
35. Рацемический продукт разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Lux Cellulose-1, 5 мкм, от Phenomenex; элюент: смесь 4:1 двуокись углерода/(этанол, содержащий 0,2% гидроксида аммония)). Соединением, элюирующимся первым, было соединение примера 118. Энантиомер соединения примера 118, 1-(цис-3-фторциклопентил]-2-[(3-метил-1,2-оксазол-5-ил)метил]-1H-имидазо[4,5-с]хинолин-8-карбонитрил, ENT-2, представлял собой элюирующийся вторым энантиомер и демонстрировал следующие биологические данные: LRRK2, формат 2, IC50=22,4 нМ для WT; LRRK2, формат 2, IC50=21,6 нМ для формы с мутацией G2019S.
36. В результате взаимодействия этил-5-(трифторметил)-1,2-оксазол-3-карбоксилата с боргидридом натрия с последующим превращением первичного спирта в соответствующий мезилат и замещением с использованием цианида калия получали [5-(трифторметил)-1,2-оксазол-3-ил]ацетонитрил. Затем после гидролиза данного нитрила с использованием концентрированной соляной кислоты получали необходимую [5-(трифторметил)-1,2-оксазол-3-ил]уксусную кислоту.
37. Необходимую (2-циклопропил-1,3-оксазол-4-ил)уксусную кислоту можно получить, используя метод, описанный в М.D. Andrews et al., РСТ Int. Appl., 2012137089, Oct 11, 2012.
38. В результате взаимодействия 5-(хлорметил)-1,3-оксазола с цианидом натрия при последующем гидролизе нитрила с использованием водного раствора гидроксида натрия получали 1,3-оксазол-5-илуксусную кислоту.
39. Рацемический продукт разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AD-H, 5 мкм, от Chiral Technologies; подвижная фаза: смесь 1:1 двуокись углерода/(метанол, содержащий 0,2% гидроксида аммония)). Соединением, элюирующимся первым, было соединение примера 132. Энантиомер соединения примера 132, 1-(цис-3-фторциклопентил)-2-{[4-(метоксиметил)-1Н-1,2,3-триазол-1-ил]метил}-1H-имидазо[4,5-с]хинолин-8-карбонитрил, ENT-2, представлял собой элюирующийся вторым энантиомер и демонстрировал следующие биологические данные: LRRK2, формат 2, IC50=26,8 нМ для WT; LRRK2, формат 2, IC50=34,5 нМ для формы с мутацией G2019S.
40. Условия для аналитической HPLC. Колонка: Chiralpak AD-H, 4,6×100 мм, 5 мкм, от Chiral Technologies; подвижная фаза: смесь 1:1 двуокись углерода/(метанол, содержащий 0,2% гидроксида аммония); скорость потока: 3,0 мл/минута.
41. В результате взаимодействия бут-3-ен-1-ола и (бензилокси)ацетальдегида в присутствии серной кислоты получали 2-[(бензилокси)метил]тетрагидро-2H-пиран-4-ол, который окисляли с использованием хлорхромата пиридиния, получая 2-[(бензилокси)метил]тетрагидро-4H-пиран-4-он. По окончании последующего восстановительного аминирования с использованием 1-(2,4-диметоксифенил)метанамина и боргидрида лития получали цис-2-[(бензилокси)метил]-N-(2,4-диметоксибензил)тетрагидро-2H-пиран-4-амин. Его приводили во взаимодействие с соединением С13 и триэтиламином и с продукта удаляли защитную группу с использованием трифторуксусной кислоты, получая N-{цис-2-[(бензилокси)метил]тетрагидро-2H-пиран-4-ил}-6-хлор-3-нитрохинолин-4-амин; после гидрирования нитрогруппы над оксидом платины(IV) получали N4-{цис-2-[(бензилокси)метил]тетрагидро-2H-пиран-4-ил}-6-хлорхинолин-3,4-диамин.
42. 1-{(2R,4S)-2-[(Бензилокси)метил]тетрагидро-2H-пиран-4-ил}-8-хлор-2-[(5-метил-1,2-оксазол-3-ил)метил]-1H-имидазо[4,5-с]хинолин (продукт, полученный после взаимодействия соединения С6 и N4-{цис-2-[(бензилокси)метил]тетрагидро-2H-пиран-4-ил}-6-хлорхинолин-3,4-диамина, описанного в примечании 41) приводили во взаимодействие с трихлоридом бора. Полученный первичный спирт преобразовывали в соответствующее мезилатное производное и проводили замещение с использованием цианида калия в присутствии каталитического количества цианида тетраэтиламмония, получая рацемат примера 134.
43. Рацемат примера 134 разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AD-3, 4,6×150 мм, 3 мкм, от Chiral Technologies; подвижная фаза А: двуокись углерода; подвижная фаза В: метанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В). Соединением, элюирующимся первым, было соединение примера 134. Энантиомер соединения примера 134, [цис-4-{8-хлор-2-[(5-метил-1,2-оксазол-3-ил)метил]-1H-имидазо[4,5-с]хинолин-1-ил}тетрагидро-2H-пиран-2-ил]ацетонитрил, ENT-2, представлял собой элюирующийся вторым энантиомер и демонстрировал следующие биологические данные: LRRK2, формат 1, IC50=353 нМ для WT; LRRK2, формат 1, IC50=327 нМ для формы с мутацией G2019S.
44. Рацемат примера 135 разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AD-3, 4,6×150 мм, 3 мкм, от Chiral Technologies; подвижная фаза А: двуокись углерода; подвижная фаза В: метанол, содержащий 0,05% диэтиламина; градиент: от 5% до 40% В). Соединением, элюирующимся первым, было соединение примера 135. Энантиомер соединения примера 135, [цис-4-{8-хлор-2-[(5-метил-1,2,4-оксадиазол-3-ил)метил]-1H-имидазо[4,5-с]хинолин-1-ил}тетрагидро-2H-пиран-2-ил]ацетонитрил, ENT-2, представлял собой элюирующийся вторым энантиомер и демонстрировал следующие биологические данные: LRRK2, формат 1, IC50=1450 нМ для WT; LRRK2, формат 1, IC50=1220 нМ для формы с мутацией G2019S.
45. В результате взаимодействия трет-бутил-циклопент-3-ен-1-илкарбамата с 3-хлорпероксибензойной кислотой с последующим раскрытием эпоксидного кольца при использовании метилмагнийбромида в присутствии иодида меди(I) получали трет-бутил-[отн-(3R,4R)-3-гидрокси-4-метилциклопентил]карбамат. Превращение вторичного спирта в соответствующий фторид осуществляли с использованием трифторида (диэтиламино)серы; после удаления защитной группы с использованием хлористого водорода получали необходимый отн-(3S,4R)-3-фтор-4-метилциклопентанамин. Его приводили во взаимодействие с соединением С13 в присутствии триэтиламина и нитрогруппу продукта гидрировали над оксидом платины(IV), получая 6-хлор-N4-[отн-(3S,4R)-3-фтор-4-метилциклопентил]хинолин-3,4-диамин.
46. Смесь диастереомерных продуктов разделяли на составляющие его рацемические изомеры посредством обращенно-фазовой HPLC (колонка: Kromasil Eternity XT С18, 10 мкм; подвижная фаза А: 0,225% муравьиной кислоты в воде; подвижная фаза В: ацетонитрил; градиент: от 26% до 46% В). Соединением, элюирующимся первым, было соединение примера 136. Диастереомер соединения примера 136, 8-хлор-1-[отн-(3S,4R)-3-фтор-4-метилциклопентил]-2-[(5-метил-1,2,4-оксадиазол-3-ил)метил]-1H-имидазо[4,5-с]хинолин, DIAST-2, представлял собой элюирующееся вторым соединение и демонстрировал следующие биологические данные: LRRK2, формат 1, IC50=156 нМ для WT; LRRK2, формат 1, IC50=105 нМ для формы с мутацией G2019S; LRRK2, формат 2, IC50=63,2 нМ для WT; LRRK2, формат 2, IC50=69,2 нМ для формы с мутацией G2019S.
47. MCYP-RXN буфер (545,0 мг; Codex®) обрабатывали деионизованной водой (19,2 мл) и в него загружали раствор реагента MCYP0016 (41,38 мг; Codex® MicroCyp®), растворенного в буфере на основе фосфата калия (0,1 М; 4,0 мл) при рН 8,0. Смесь обрабатывали раствором соединения примера 4 (5,72 мг), растворенного в диметилсульфоксиде (0,6 мл) и буфере на основе фосфата калия (0,1 М; 0,6 мл) при рН 8,0. Реакционную смесь встряхивали при 30°С в течение 12 часов. После извлечения посредством обращенно-фазовой HPLC (колонка: Gemini NX С18, 5 мкм, от Phenomenex; подвижная фаза А: вода, содержащая 0,1% муравьиной кислоты; подвижная фаза В: ацетонитрил, содержащий 0,1% муравьиной кислоты; градиент: от 5% до 90% В) получали соединение примера 137.
48. Соединение примера 4 инкубировали с Codex® MicroCyp® MCYP0030 при 30°С, используя общую методику, описанную в примечании 47. После извлечения посредством обращенно-фазовой HPLC (колонка: Gemini NX С18, 5 мкм, от Phenomenex; подвижная фаза А: вода, содержащая 0,1% муравьиной кислоты; подвижная фаза В: ацетонитрил, содержащий 0,1% муравьиной кислоты; градиент: от 5% до 90% В) получали соединение примера 138.
49. Соединение примера 138 приводили во взаимодействие с трифторидом (диэтиламино)серы, получая соединение примера 139.
50. Необходимый 6-фтор-N4-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]хинолин-3,4-диамин синтезировали из 6-фтор-3-нитрохинолин-4-ола, используя общий способ, описанный в примере 1 для синтеза соединения С11 из соединения С7, за исключением того, что вместо соединения Р1 использовали соединение Р2 и гидрирование проводили над платиной на угле, а не над оксидом платины(IV).
51. В результате взаимодействия 1,2,3-тиадиазол-4-илметанола с метансульфонилхлоридом, затем замещения с использованием цианида калия и гидролиза в концентрированной соляной кислоте получали необходимую 1,2,3-тиадиазол-4-илуксусную кислоту.
52. В этом случае заключительную реакцию сочетания и циклизации проводили за две стадии: взаимодействие 6-фтор-N4-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]хинолин-3,4-диамина (примечание 50) с 1,2,3-тиадиазол-4-илуксусной кислотой (примечание 51) осуществляли в присутствии 2,4,6-триоксида 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана и триэтиламина при 50°С и выделяли промежуточный N-(6-фтор-4-{[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]амино}хинолин-3-ил)-2-(1,2,3-тиадиазол-4-ил)ацетамид. В результате последующего взаимодействия в присутствии 2,4,6-триоксида 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана и N,N-диизопропилэтиламина при 110°С получали соединение примера 140.
53. Заключительную реакцию сочетания и циклизации проводили за две стадии, как описано для соединения примера 140 в примечании 52.
54. После взаимодействия метилпиридазин-3-илацетата с гидроксидом лития получали пиридазин-3-илацетат лития.
55. 3-Амино-4-[(2,2-дифторциклопентил)амино]хинолин-6-карбонитрил синтезировали из соединения С61, используя способ, описанный для получения соединения С54 из соединения С13 в примере 93.
56. Рацемический продукт разделяли на составляющие его энантиомеры посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AS, 5 мкм, от Chiral Technologies; элюент: 4:1 двуокись углерода/2-пропанол, содержащий 0,1% гидроксида аммония)). Соединением, элюирующимся первым, было соединение примера 143, а соединение примера 144 представляло собой элюирующийся вторым энантиомер.
57. Превращение (5-циклопропил-1,2-оксазол-3-ил)метанола в необходимую (5-циклопропил-1,2-оксазол-3-ил)уксусную кислоту осуществляли, используя способ, описанный в примечании 51.
В приведенной ниже Таблице 2 приведены структура и масс-спектральные данные для соединений примеров 146-250.
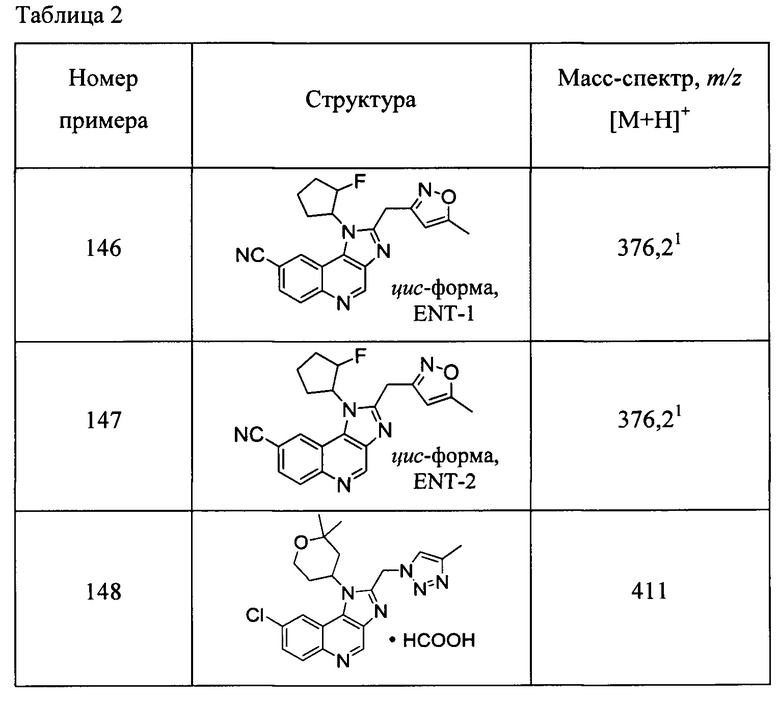
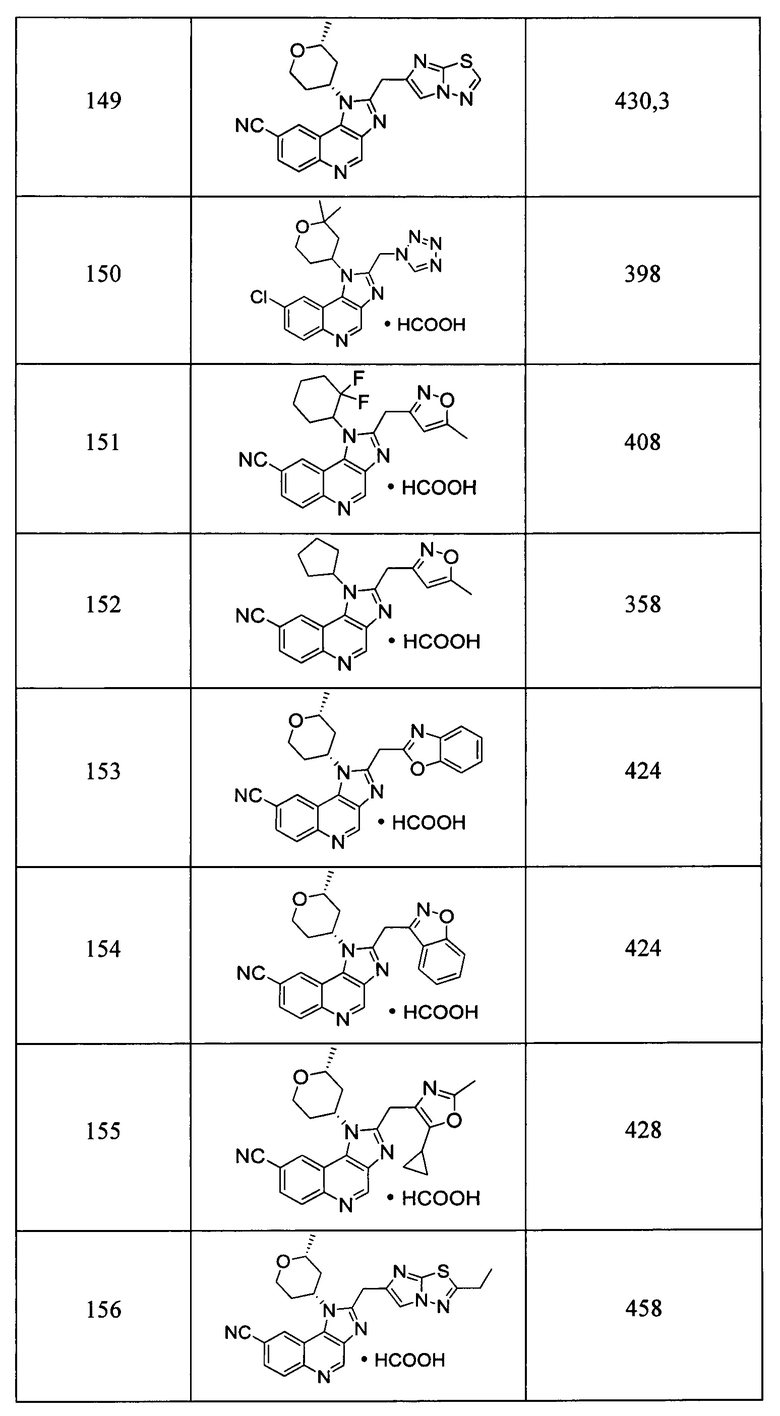
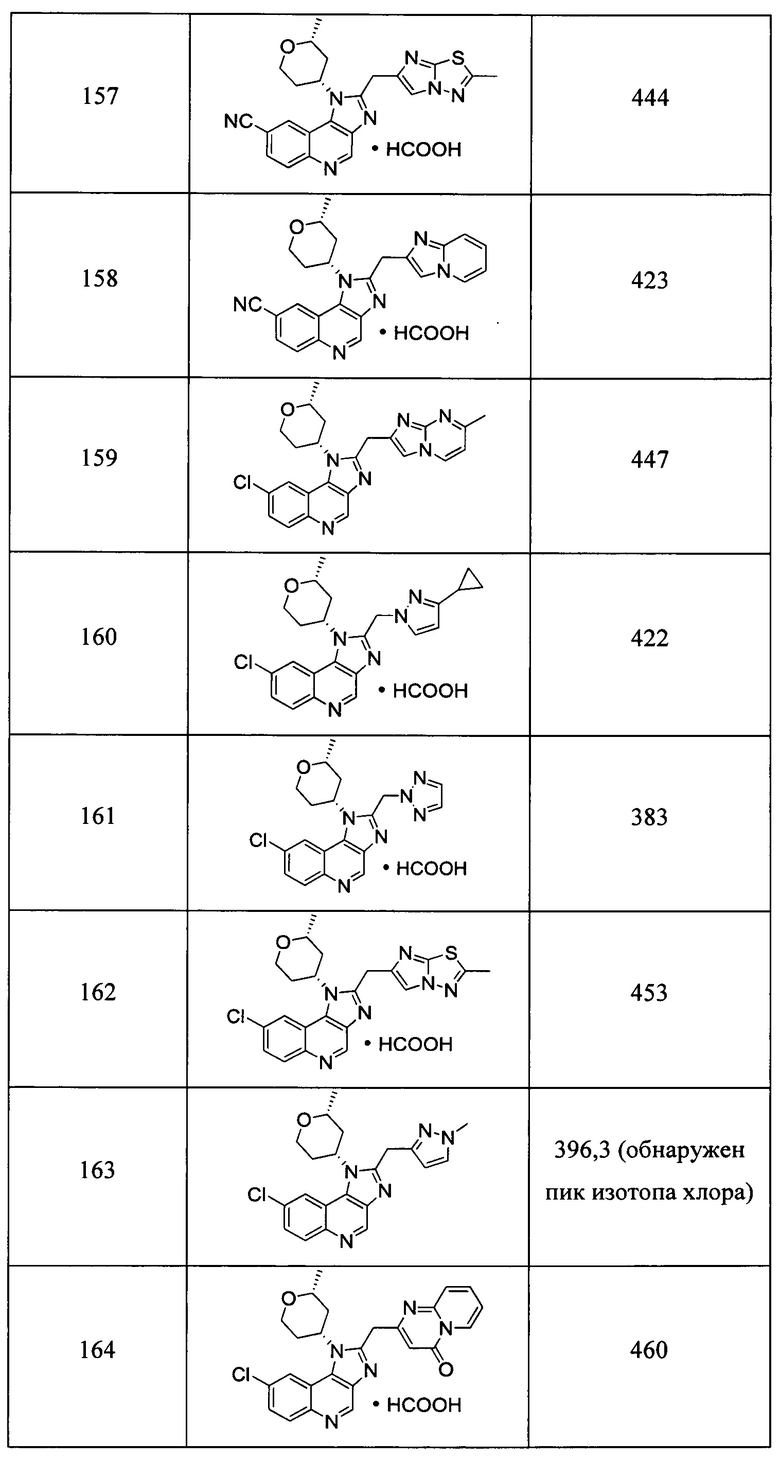
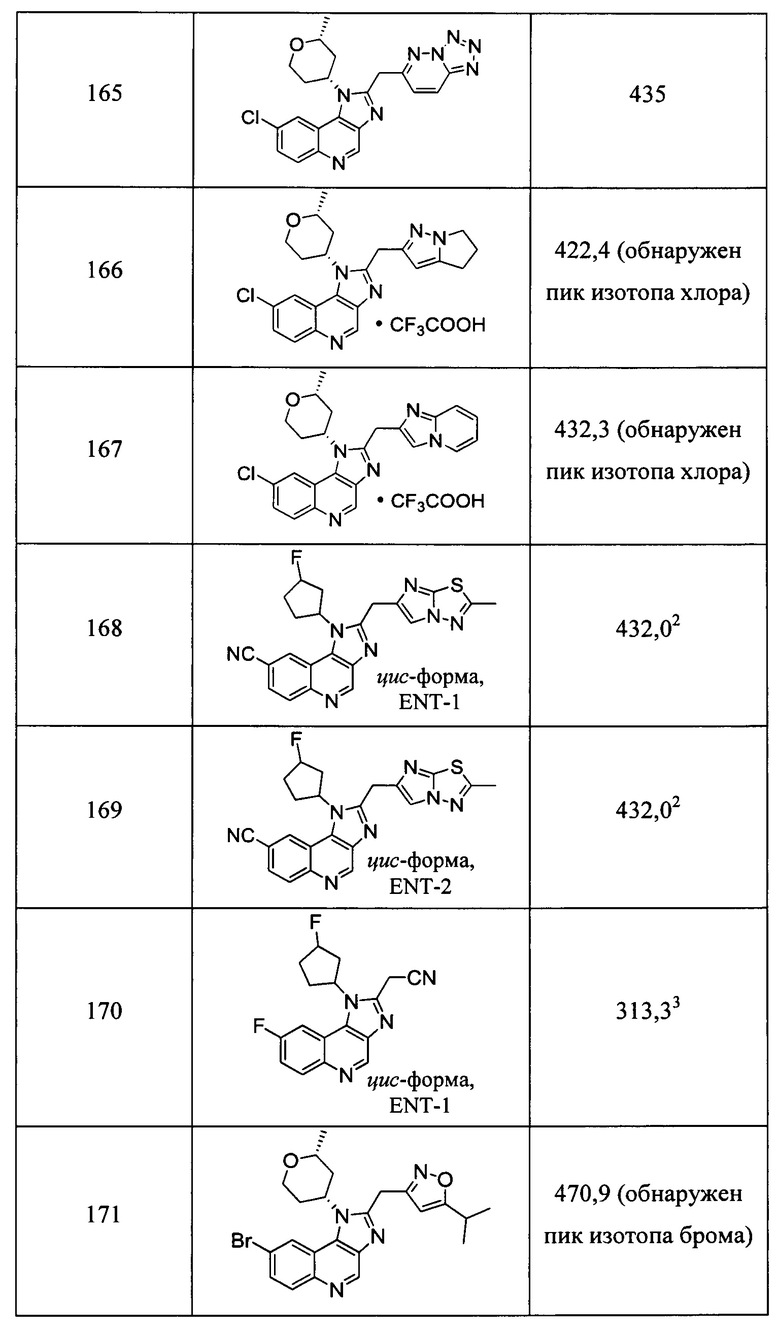
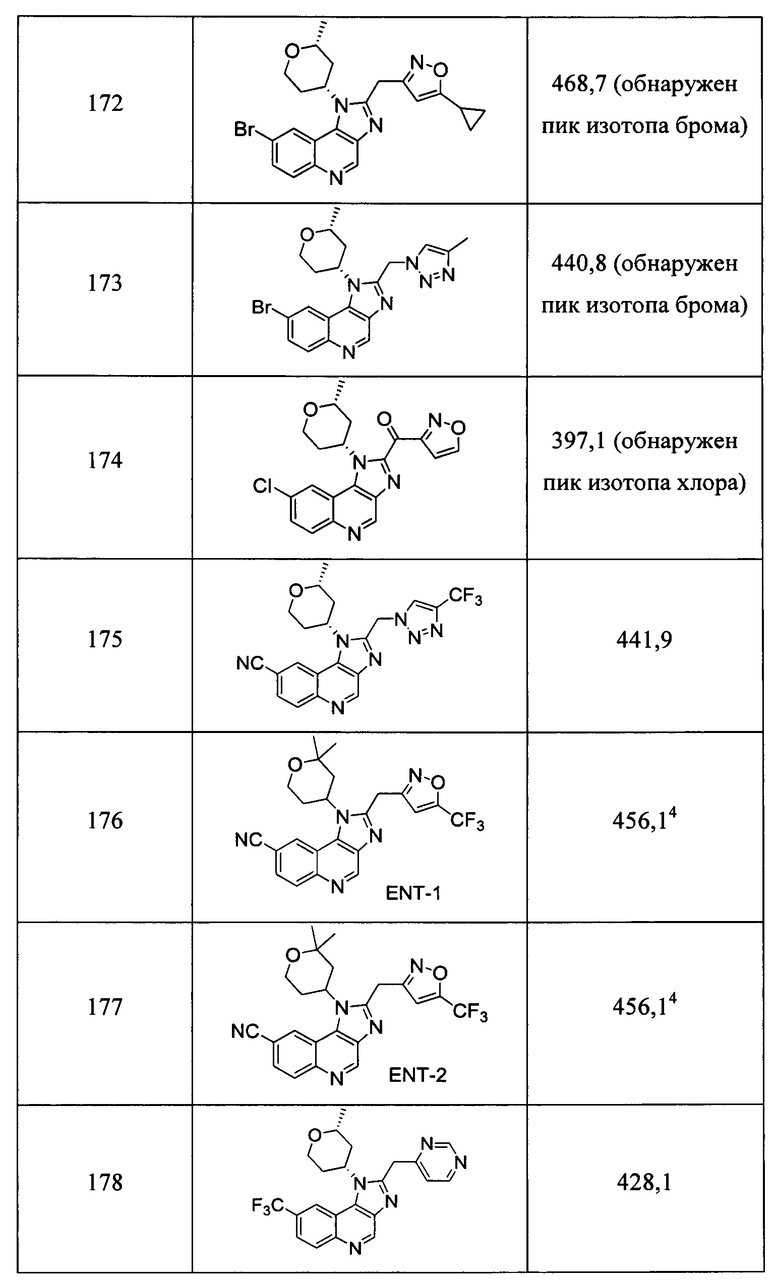
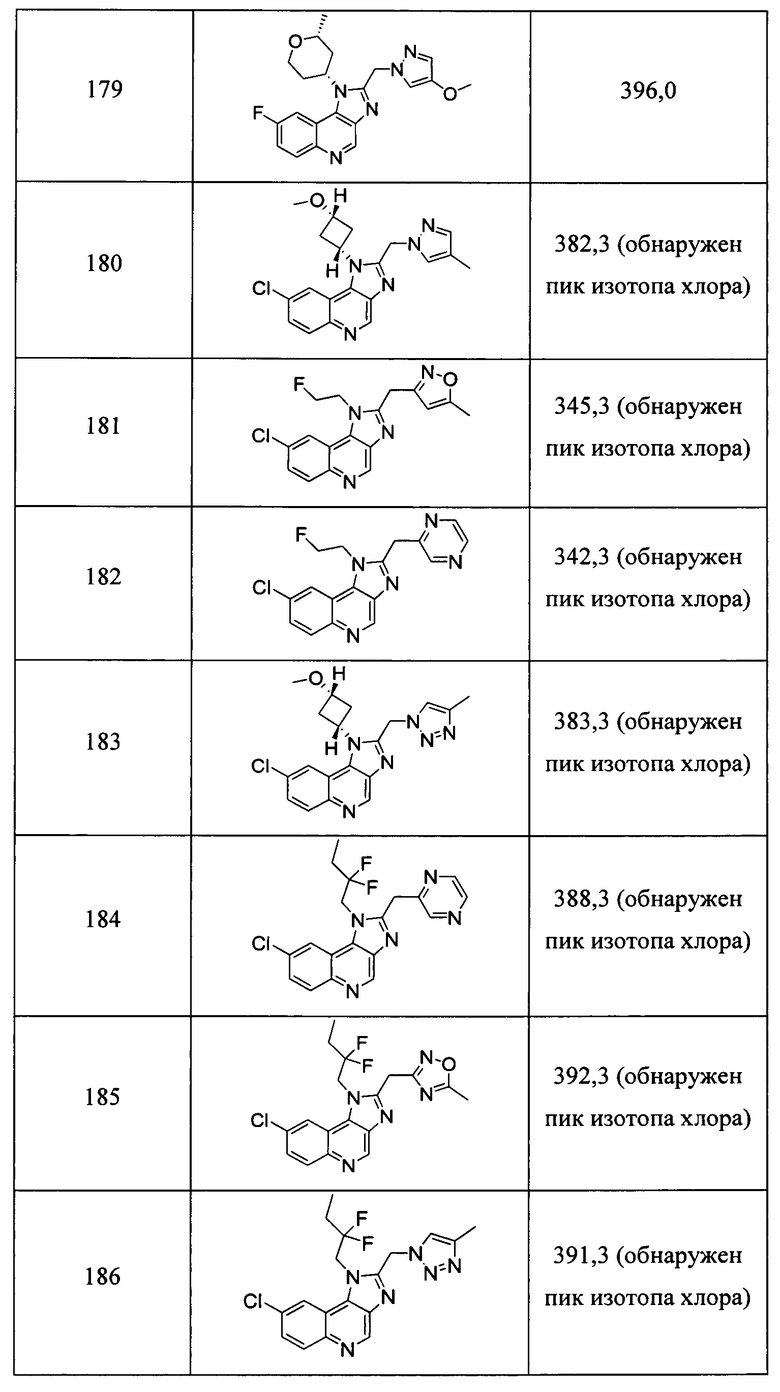
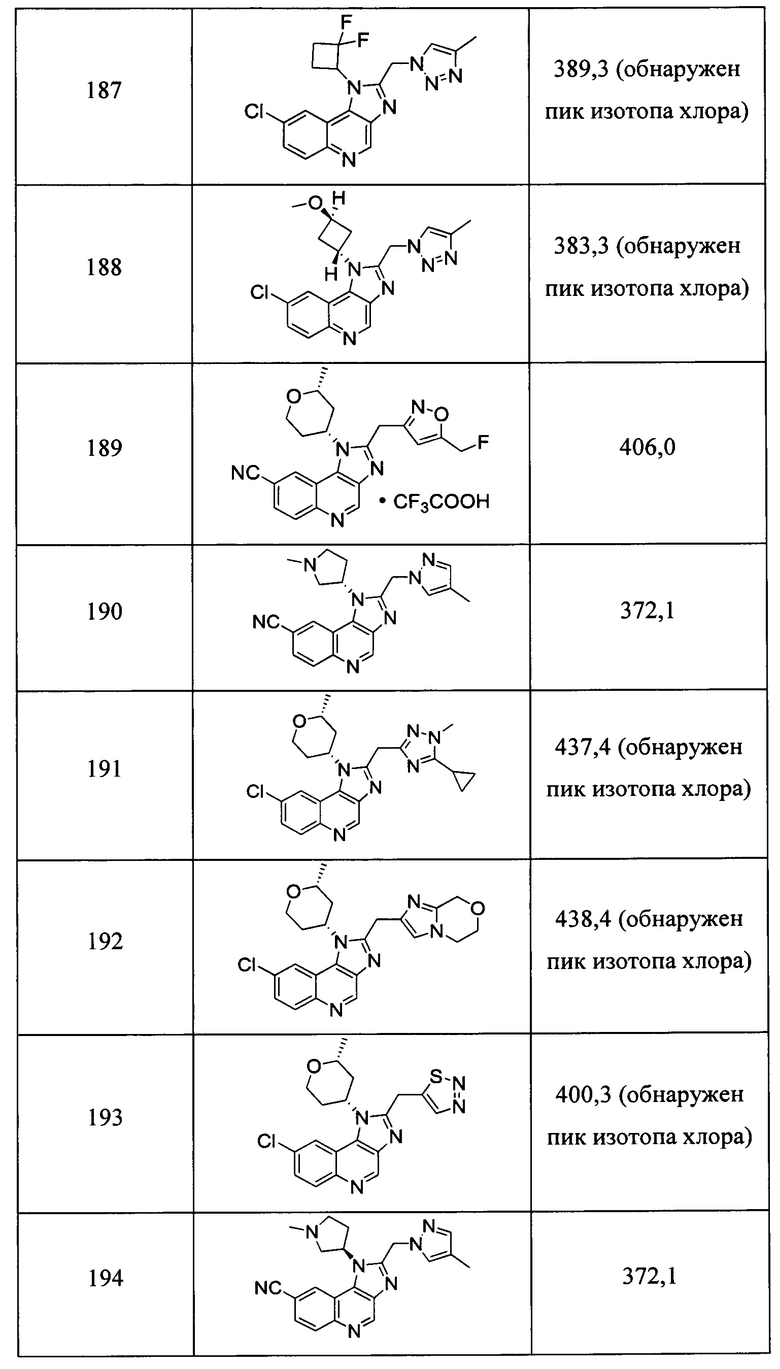
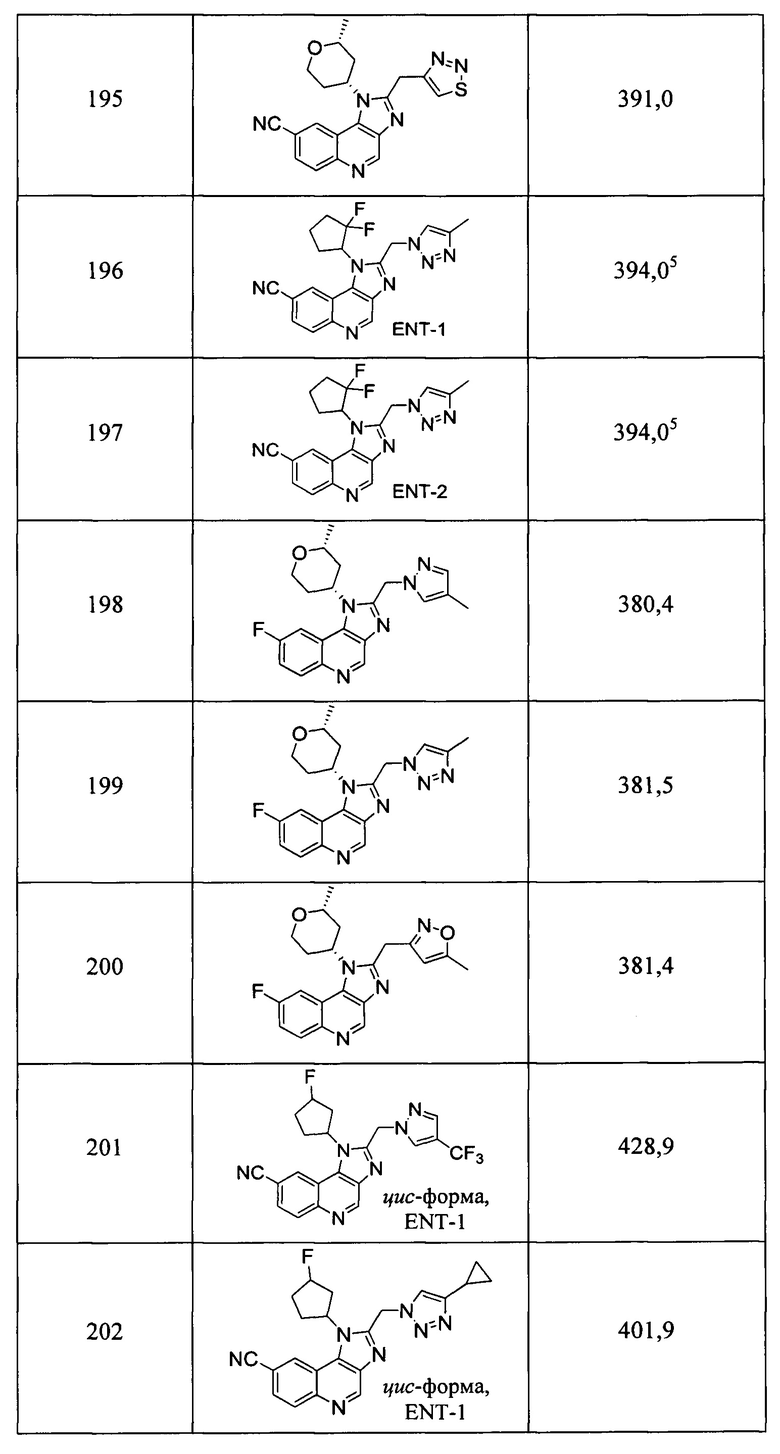
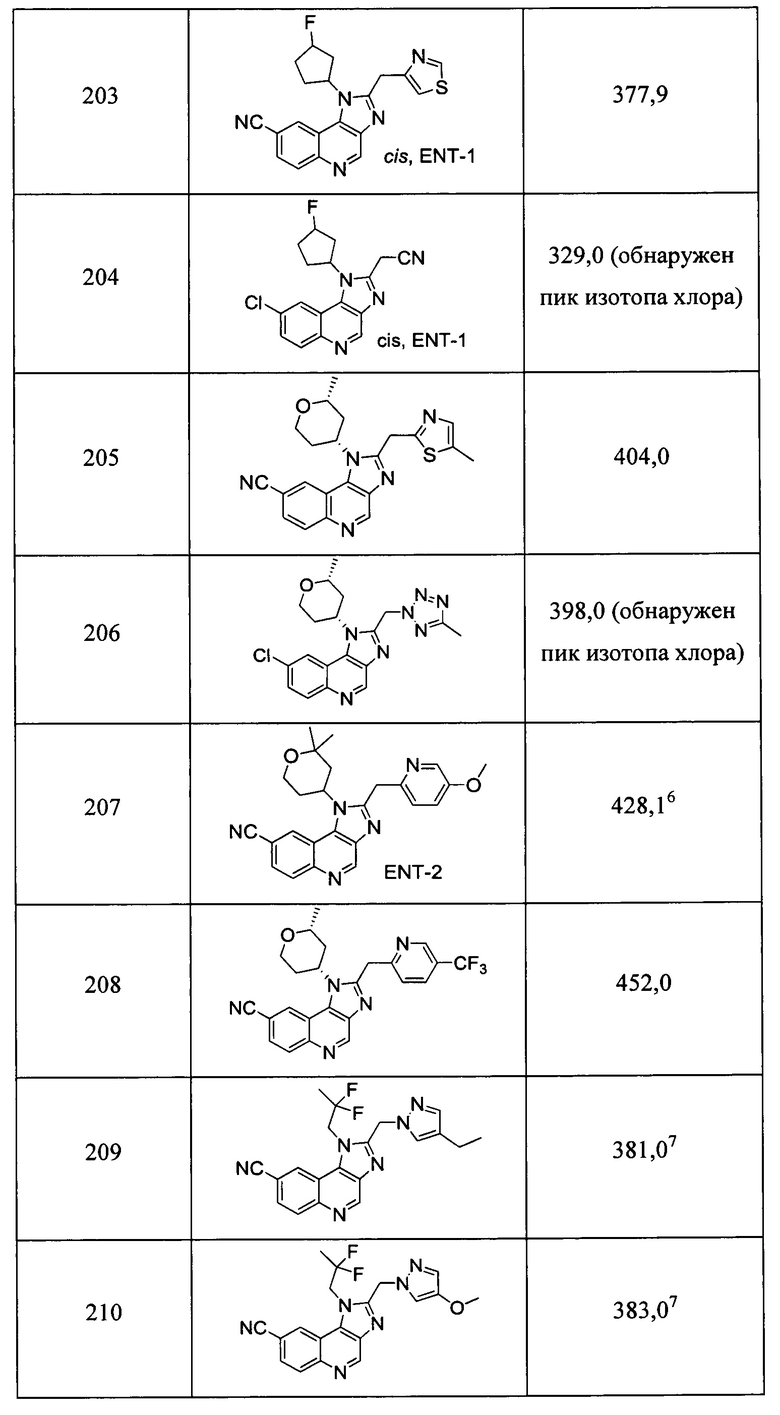
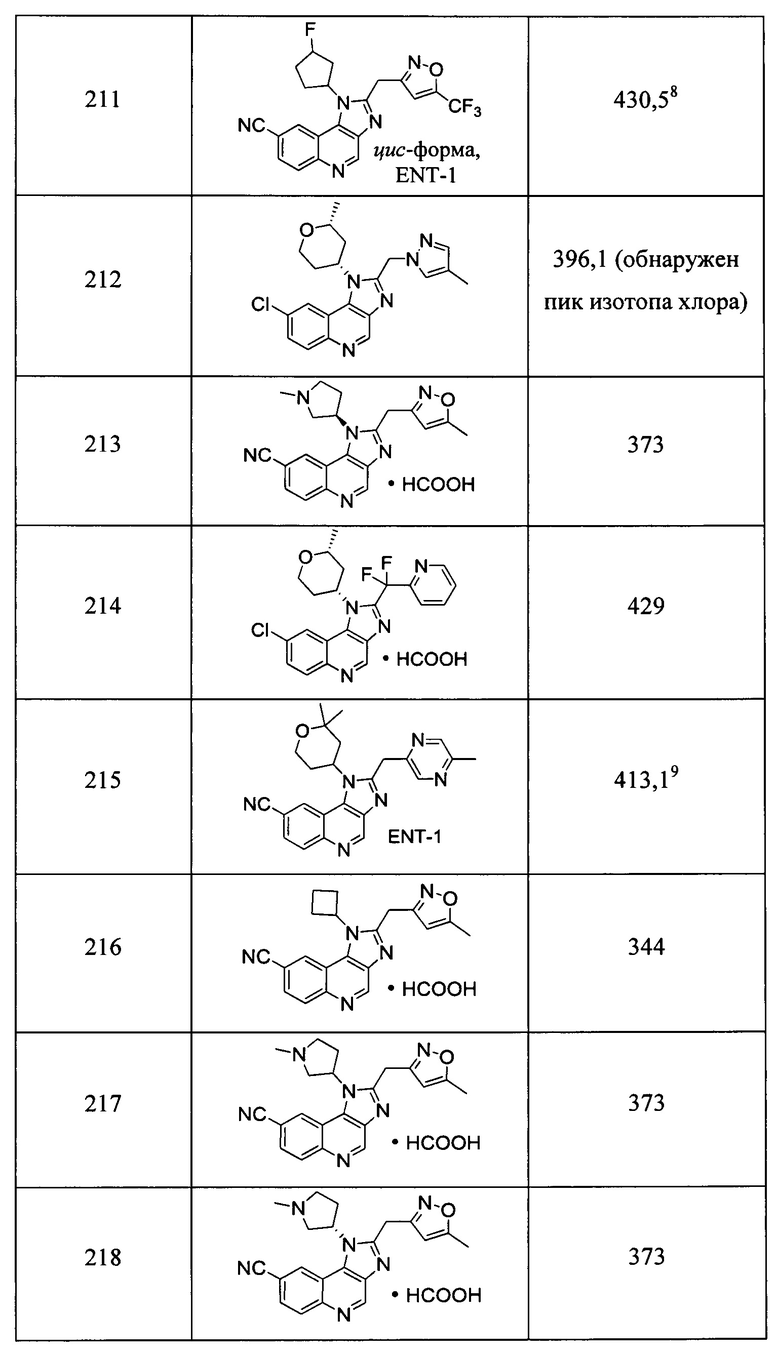
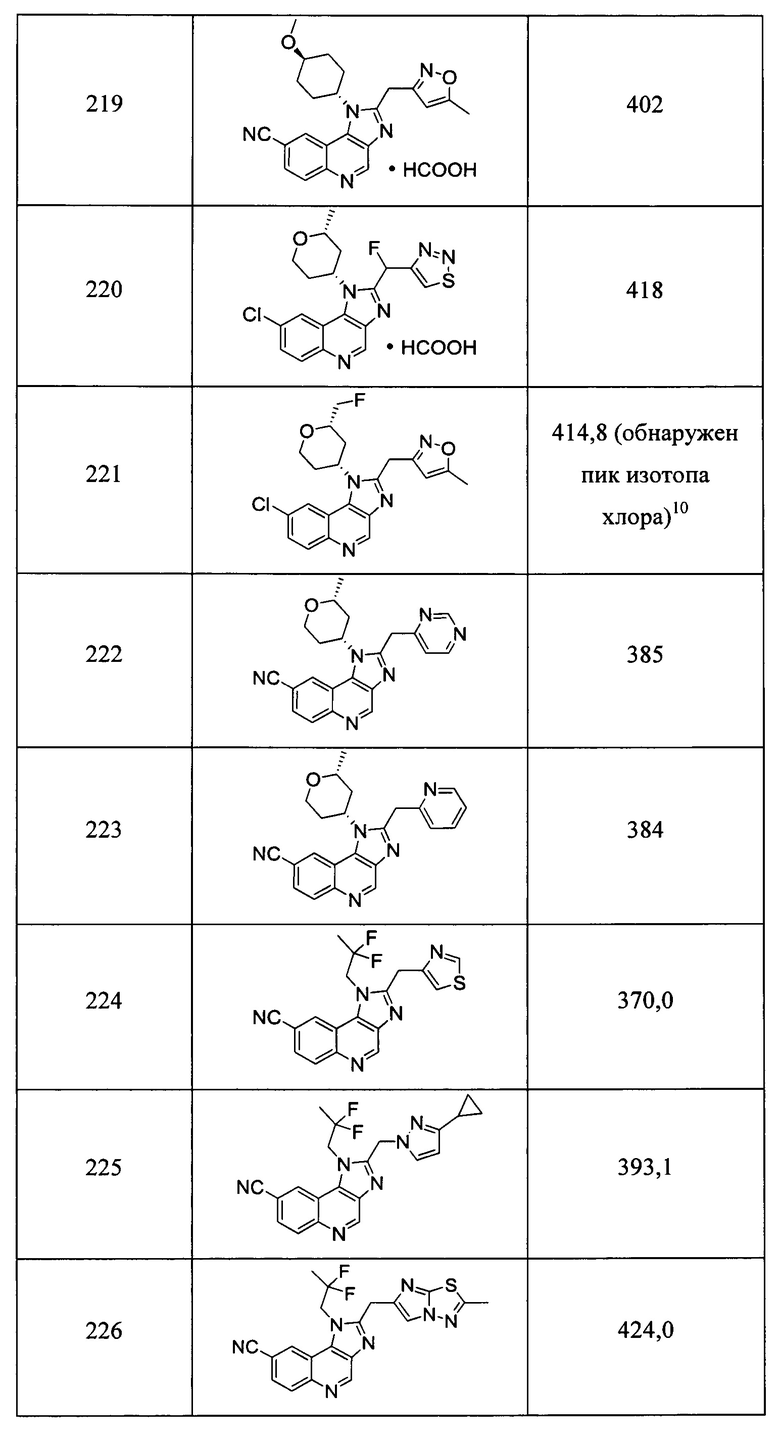
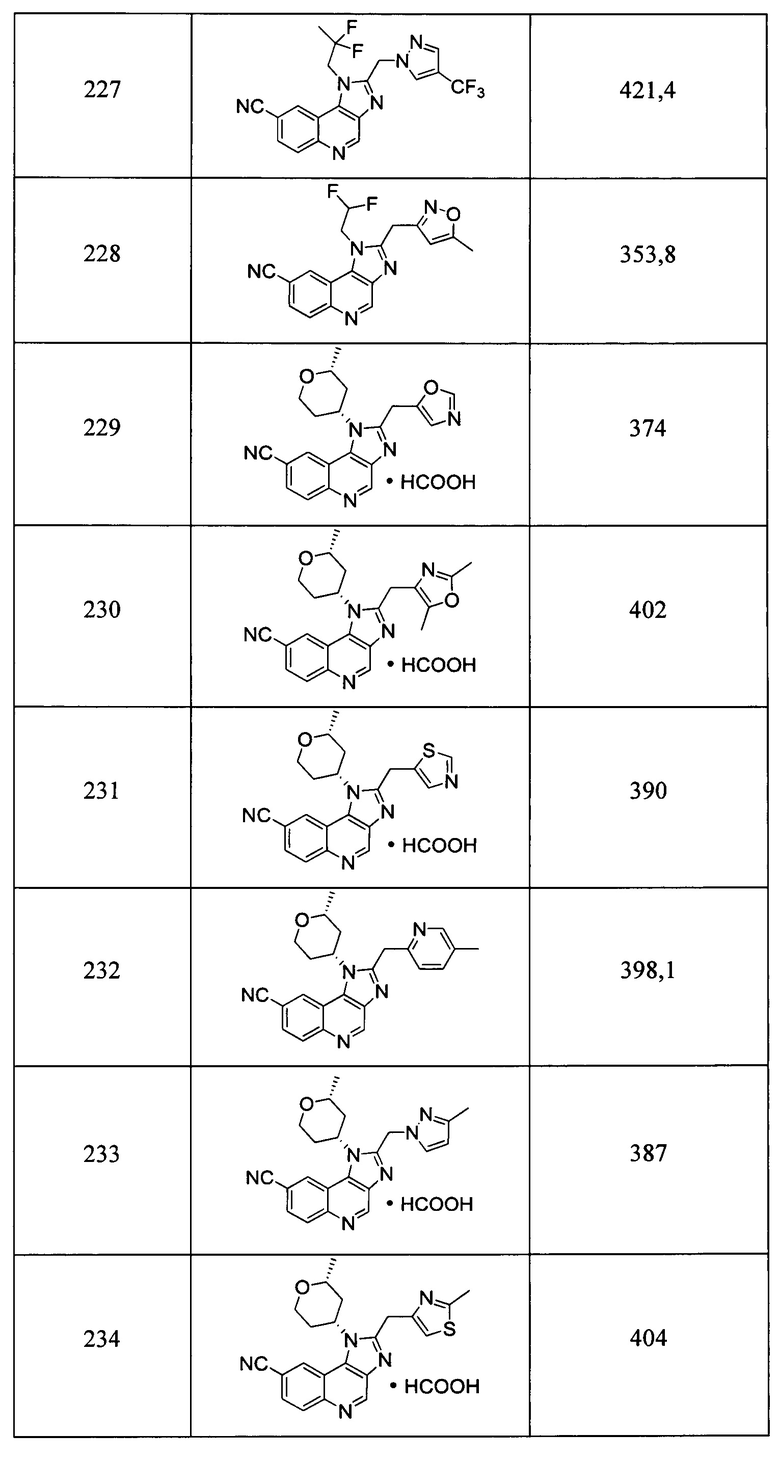
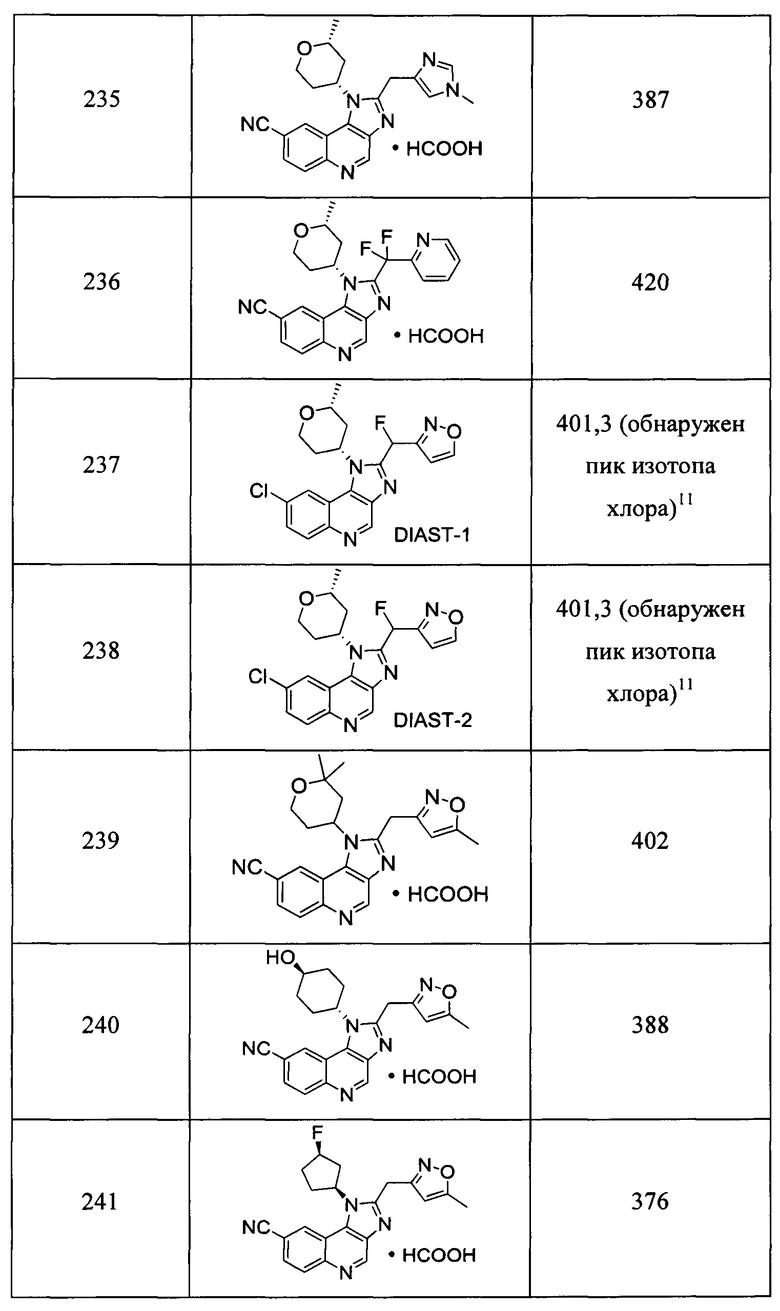
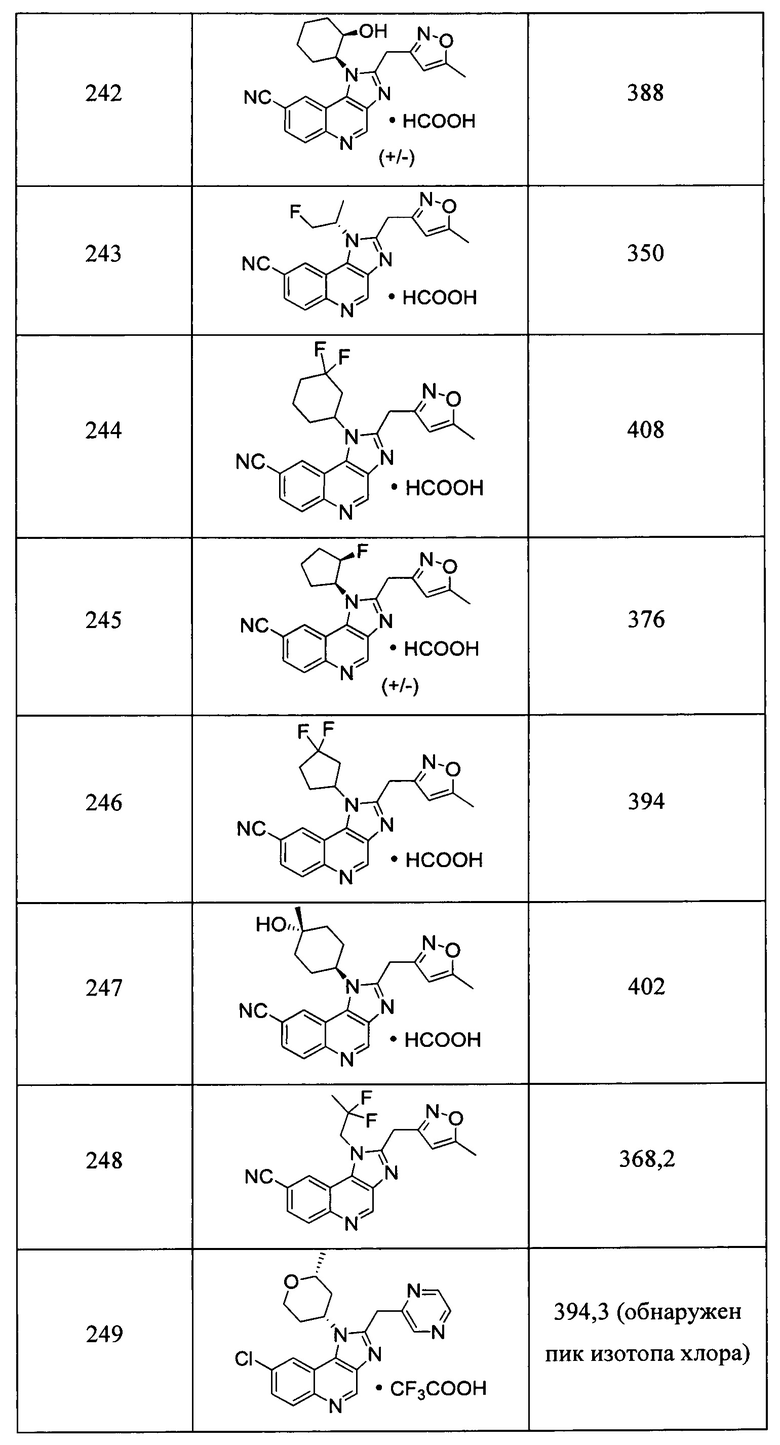
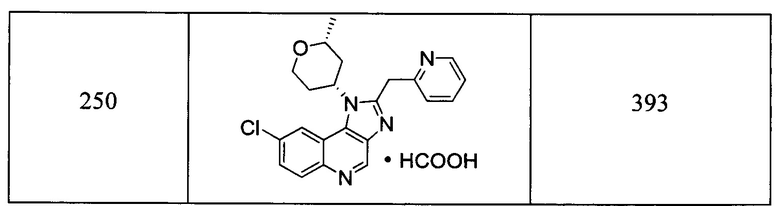
1. Соединения примеров 146 и 147 синтезировали в виде рацемической смеси и затем разделяли на индивидуальные энантиомеры, используя сверхкритическую жидкостную хроматографию (колонка: Lux Amylose-1, 5 мкм, от Phenomenex; подвижная фаза: смесь 85:15 двуокись углерода/этанол). Соединение примера 146 представляло собой элюирующийся первым энантиомер, за которым следовало соединение примера 147.
2. Соединения примеров 168 и 169 синтезировали в виде рацемической смеси и затем разделяли на индивидуальные энантиомеры, используя сверхкритическую жидкостную хроматографию (колонка: Chiralcel OD-H, 5 мкм, от Phenomenex; подвижная фаза: смесь 85:15 двуокись углерода/(метанол, содержащий 0,05% гидроксида аммония)). Соединение примера 168 представляло собой элюирующийся первым энантиомер, соединение примера 169.
3. Соединение примера 170 выделяли из соответствующей рацемической смеси посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AD-H, 5 мкм, от Chiral Technologies; подвижная фаза: смесь 85:15 двуокись углерода/(метанол, содержащий 0,2% гидроксида аммония)). Соединение примера 170 представляло собой элюирующийся первым энантиомер.
4. Соединения примеров 176 и 177 синтезировали в виде рацемической смеси и затем разделяли на индивидуальные энантиомеры, используя сверхкритическую жидкостную хроматографию (колонка: Chiralpak AS, 5 мкм, от Chiral Technologies; подвижная фаза: смесь 85:15 двуокись углерода/(2-пропанол, содержащий 0,1% гидроксида аммония)). Соединение примера 176 представляло собой элюирующийся первым энантиомер, соединение примера 177.
5. Соединения примеров 196 и 197 синтезировали в виде рацемической смеси. Для разделения и очистки потребовались две хроматографические стадии: после сверхкритической жидкостной хроматографии (колонка: Lux Cellulose-2, 10 мкм, от Phenomenex; подвижная фаза: смесь 3:2 двуокись углерода/(метанол, содержащий 0,1% гидроксида аммония)) получали соединение примера 196 в качестве элюирующегося первым энантиомера и соединение примера 197 в качестве элюирующегося вторым энантиомера. Дополнительную очистку проводили, используя обращено-фазовую HPLC (колонка: XBridge С18 OBD, 5 мкм, от Waters; подвижная фаза А: вода, содержащая 0,05% гидроксида аммония; подвижная фаза В: ацетонитрил; градиент: от 25% до 55% В).
6. Соединение примера 207 выделяли из соответствующей рацемической смеси посредством сверхкритической жидкостной хроматографии (колонка: Chiralpak AD, 5 мкм, от Chiral Technologies; подвижная фаза: смесь 3:1 двуокись углерода/(этанол, содержащий 0,1% гидроксида аммония)). Соединение примера 207 представляло собой элюирующийся вторым энантиомер.
7. В результате взаимодействия соединения С61 с 2,2-дифторпропан-1-амином и 2 N,N-диизопропилэтиламином получали 4-[(2,2-дифторпропил)амино]-3-нитрохинолин-6-карбонитрил, который восстанавливали с использованием железа в присутствии соляной кислоты, получая необходимый промежуточный 3-амино-4-[(2,2-дифторпропил)амино]хинолин-6-карбонитрил.
8. Соединение примера 211 выделяли из соответствующей рацемической смеси посредством сверхкритической жидкостной хроматографии (колонка: Lux Amylose-1, 5 мкм, от Phenomenex; подвижная фаза: смесь 85:15 двуокись углерода/(метанол, содержащий 0,2% гидрооксида аммония). Соединение примера 211 представляло собой элюирующийся первым энантиомер.
9. Соединение примера 215 выделяли из соответствующей рацемической смеси посредством сверхкритической жидкостной хроматографии. При проведении аналитической HPLC (колонка: Lux Cellulose-2, 3 мкм, от Phenomenex; подвижная фаза: смесь 3:2 двуокись углерода/(2-пропанол, содержащий 0,05% диэтиламина); скорость потока: 2,5 мл/минута) соединение примера 215 представляло собой элюирующийся первым энантиомер.
10. Соединение примера 221 синтезировали из соединения примера 137 посредством фторирования с использованием трифторида (диэтиламино)серы.
11. Соединения примеров 237 и 238 синтезировали в виде диастереомерной смеси и затем разделяли на индивидуальные диастереомеры, используя сверхкритическую жидкостную хроматографию (колонка: Chiralcel OJ-H, 5 мкм, от Phenomenex; подвижная фаза: смесь 9:1 двуокись углерода/(метанол, содержащий 0,2% гидроксида аммония)). Соединение примера 237 представляло собой элюирующийся первым диастереомер, за которым следовало соединение примера 238.
Биологические анализы
Анализ LRRK2, формат 1
Киназную активность LRRK2 измеряли с использованием технологии Lantha Screen от Invitrogen. Слитую с GST (глутатион-S-трансфераза) укороченную LRRK2 от Invitrogen (№ по кат. PV4874) инкубировали с меченным флуоресцеином пептидным субстратом на основе белков эзрин/радиксин/моэзинового (ERM) семейства, также известным как LRRKtide (Invitrogen, № по кат. PR8976A), в присутствии дающего дозозависимый ответ соединения. По завершении инкубации анализ останавливали и проводили детектирование, используя меченное тербием антитело к фосфо-ERM (Invitrogen, № по кат. PR8975A). Анализ проводили согласно следующему протоколу: 3 мкл рабочего раствора субстрата (233 нМ LRRKtide, 117 мкМ АТФ), приготовленного в буфере для анализа (50 мМ HEPES (N-2-гидроксиэтил-пиперазин-N-2-этансульфоновая кислота), рН 7,5; 3 мМ MgCl2, с добавленными свежеприготовленными 2 мМ дитиотреитом (DTT) и 0,01%-ным Брием 35), добавляли в 384-луночный планшет от Greiner с лунками малого объема. Образцы для кривой зависимости ответа от дозы соединения по 11 точкам готовили путем разведения соединения в максимальной концентрации 3,16 мМ в 100%-ном DMSO и выполнения серийных полулогарифмических разведений в DMSO. Аликвоты (3,5 мкл) образцов в 100%-ном DMSO для кривой доза-ответ смешивали с 46,5 мкл воды, затем 1 мкл этой смеси добавляли к 3 мкл содержащей субстрат смеси в 384-луночном планшете. Киназную реакцию инициировали, используя 3 мкл рабочего раствора фермента LRRK2 в концентрации 4 мкг/мл. Использовали следующие конечные концентрации в реакционной смеси: 100 нМ LRRKtide, 50 мкМ АТФ, 1,7 мкг/мл для фермента LRRK2 и образец для кривой зависимости ответа от дозы соединения в максимальной дозе 32 мкМ. Реакцию проводили при комнатной температуре в течение двух часов и затем останавливали, добавляя 7 мкл буфера для детекции (20 мМ трис, рН 7,6; 0,01% NP-40 (нонидет Р-40); 0,02% NaN3, 6 мМ EDTA (этилендиаминтетерауксусная кислота) с 2 нМ раствором меченного тербием антитела к фосфо-ERM). После инкубирования в течение 1 часа при комнатной температуре планшет прочитывали на приборе Envision с длиной волны возбуждения 340 нм и регистрировали излучение как на 520 нм, так и на 495 нм. Для анализа данных использовали соотношение излучения при 520 нм и 495 нм.
Ингибирование мутантной G2019S LRRK2 (Invitrogen, № по кат. PV4881) измеряли точно таким же способом. Все конечные концентрации субстрата, АТФ и фермента были такими же. Однако, поскольку фермент с мутацией является более активным, продолжительность реакции уменьшали до 90 минут, чтобы гарантировать измерение ингибирования в стационарном состоянии до того, как могло произойти исчерпание субстрата.
Анализ LRRK2, формат 2
Киназную активность LRRK2 измеряли с использованием технологии Lantha Screen от Invitrogen. Слитую с GST укороченную LRRK2 от Invitrogen (№ по кат. PV4874) инкубировали с меченным флуоресцеином пептидным субстратом на основе белков эзрин/радиксин/моэзинового (ERM) семейства, также известным как LRRKtide (Invitrogen, № по кат. PR8976A), в присутствии дающего дозозависимый ответ соединения. По завершении инкубации анализ останавливали и проводили детектирование, используя меченное тербием антитело к фосфо-ERM (Invitrogen, № по кат. PR8975A). Анализ проводили согласно следующему протоколу: образцы для кривой зависимости ответа от дозы соединения по 11 точкам готовили путем разведения соединения в максимальной концентрации 0,3 мМ в 100%-ном DMSO и выполнения серийных полулогарифмических разведений в DMSO, 100х конечная концентрация в анализе. С использованием акустического диспенсера (acoustic dispensing) Echo переносили по 60 нл растворов соединения в 384-луночный используемый в анализе планшет Corning с лунками малого объема. В лунки используемого в анализе планшета, содержащие по 60 нл раствора соединения, добавляли по 3 мкл рабочего раствора субстрата (200 нМ LRRKtide, 2000 мМ АТФ), приготовленного в буфере для анализа (50 мМ HEPES, рН 7,5; 3 мМ MgCl2, с добавленными свежеприготовленными 2 мМ DTT и 0,01%-ным Брием 35). Киназную реакцию инициировали, используя 3 мкл рабочего раствора фермента LRRK2 в концентрации 4 мг/мл. Использовали следующие конечные концентрации в реакционной смеси: 100 нМ LRRKtide, 1000 мМ АТФ, 2 мг/мл для фермента LRRK2 и образец для кривой зависимости ответа от дозы соединения в максимальной концентрации 3 мМ. Реакцию проводили при комнатной температуре в течение 30 минут и затем останавливали, добавляя 6 мл буфера для детекции (20 мМ трис, рН 7,6; 0,01% NP-40; 0,02% NaN3, 6 мМ EDTA с 2 нМ раствором меченного тербием антитела к фосфо-ERM). После инкубирования в течение 1 часа при комнатной температуре планшет прочитывали на приборе Envision с длиной волны возбуждения 340 нм и регистрировали излучение как на 520 нм, так и на 495 нм. Для анализа данных использовали соотношение излучения при 520 нм и 495 нм. Ингибирование имеющей мутацию G2019S LRRK2 (Invitrogen, № по кат. PV4881) измеряли точно таким же способом. Все конечные концентрации субстрата, АТФ и фермента были такими же.
В приведенных ниже Таблицах 3 и 4 представлены данные по IC50 в отношении LRRK2 для соединений по изобретению.
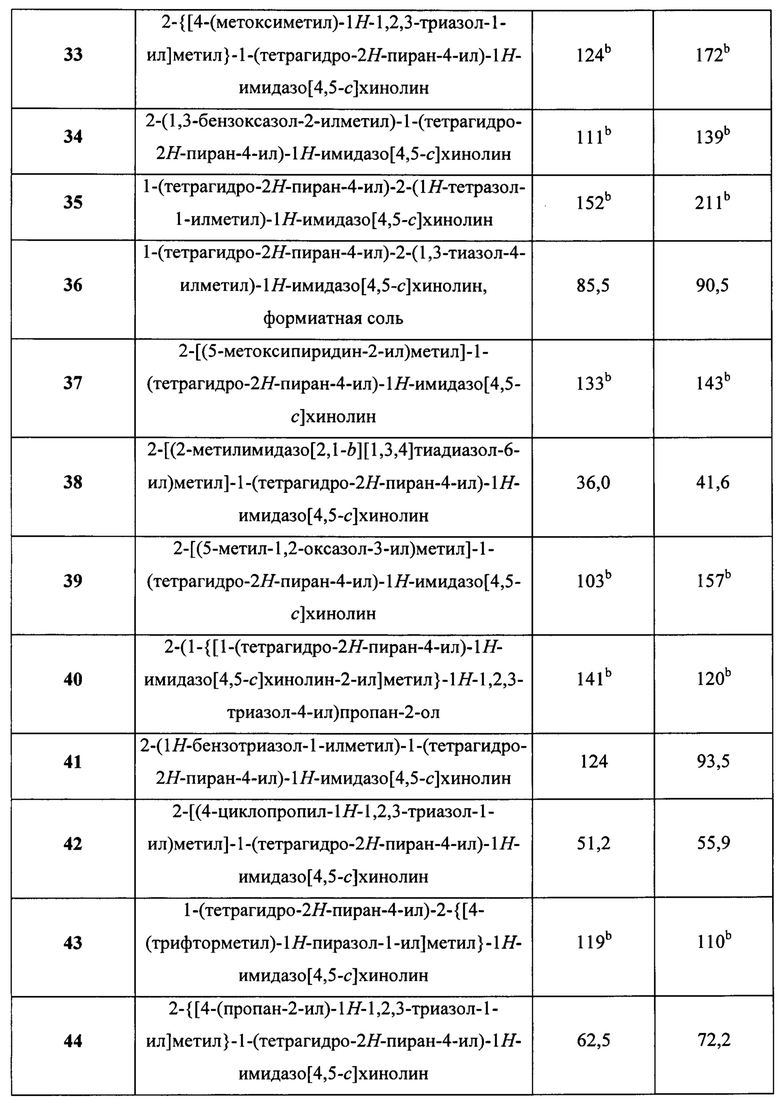
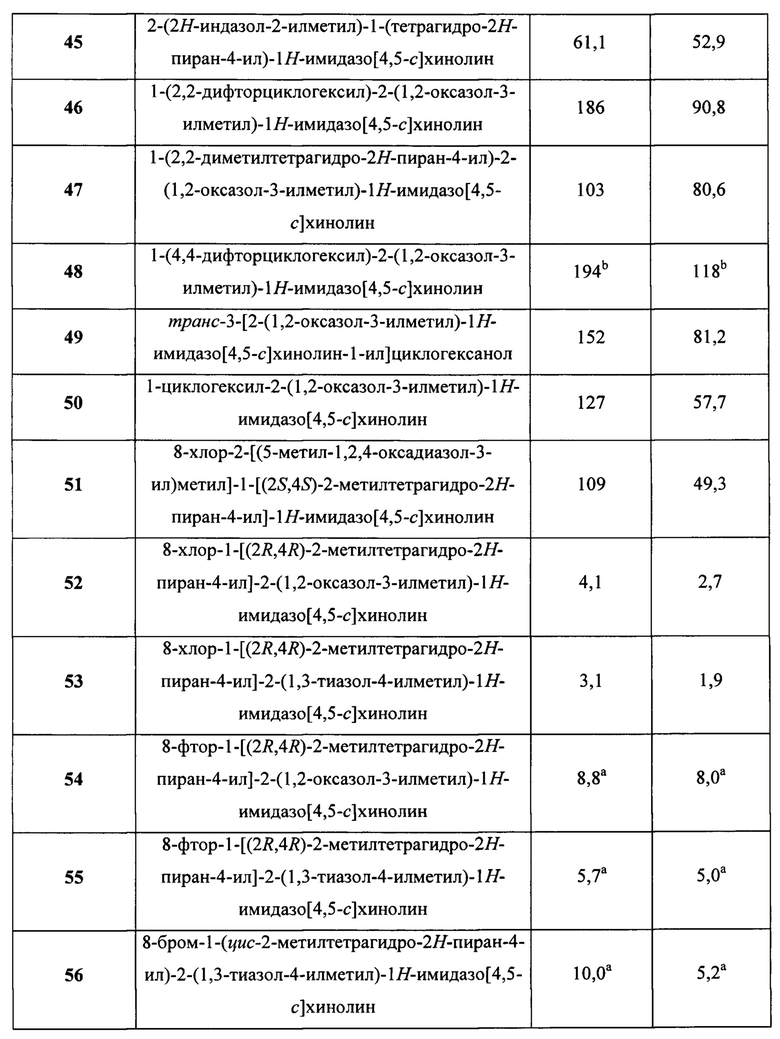
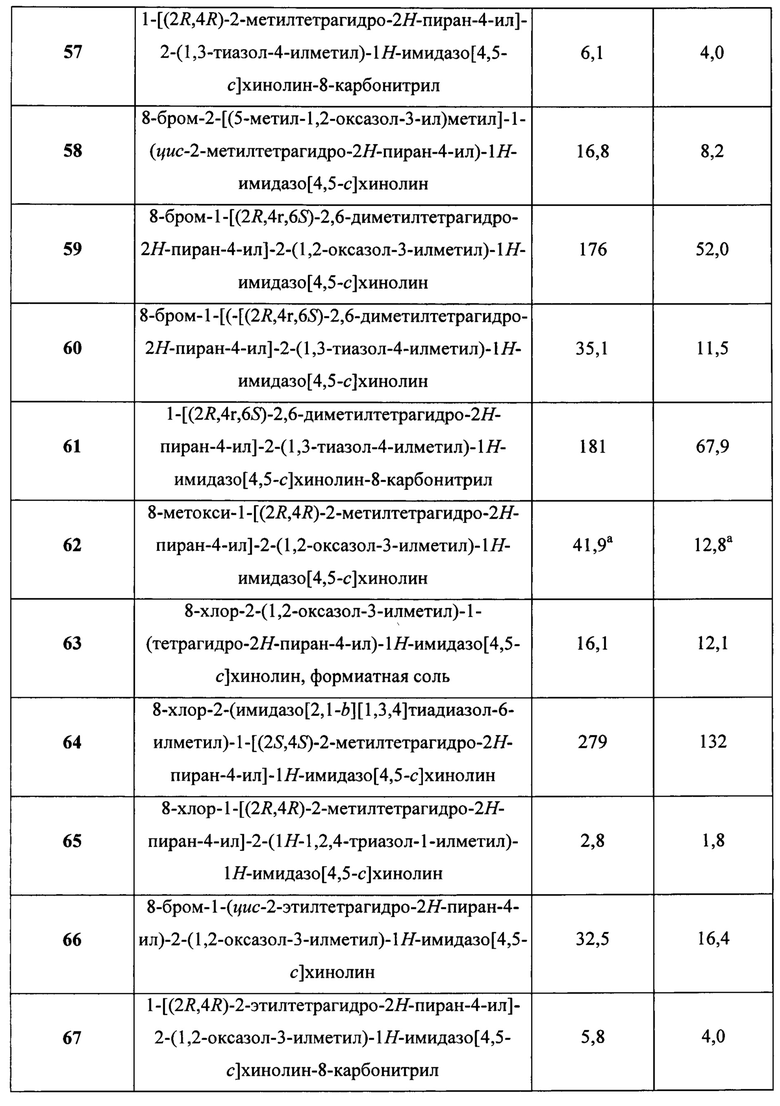
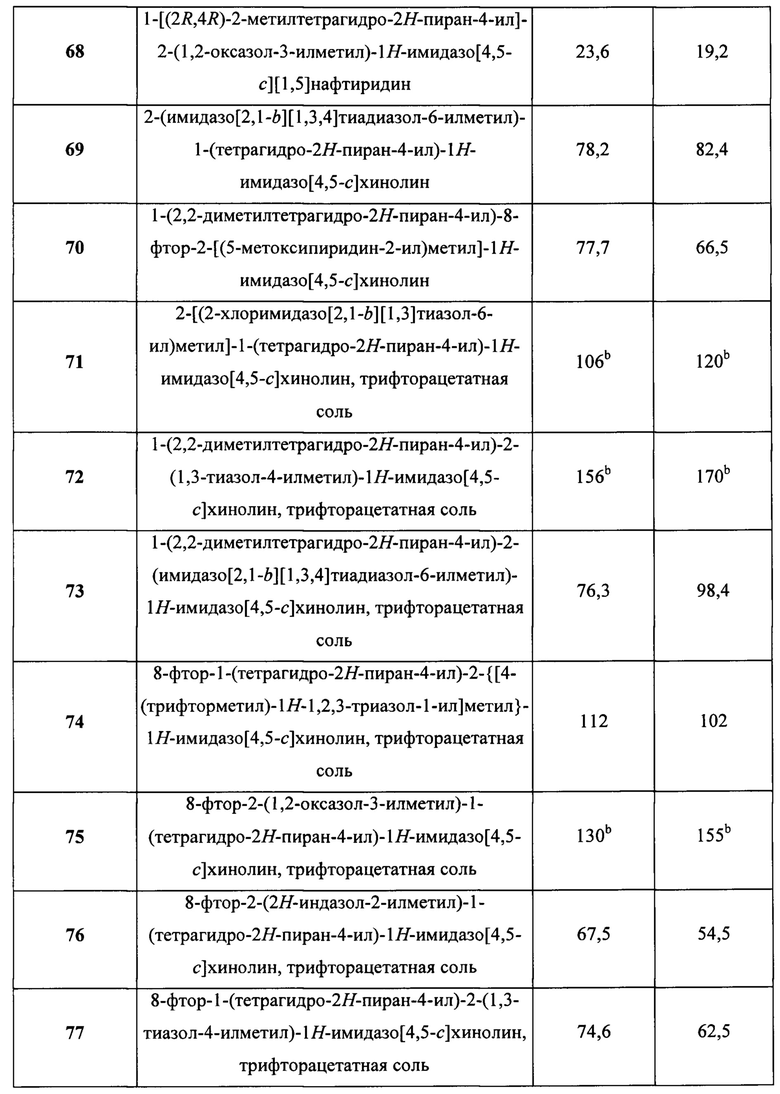
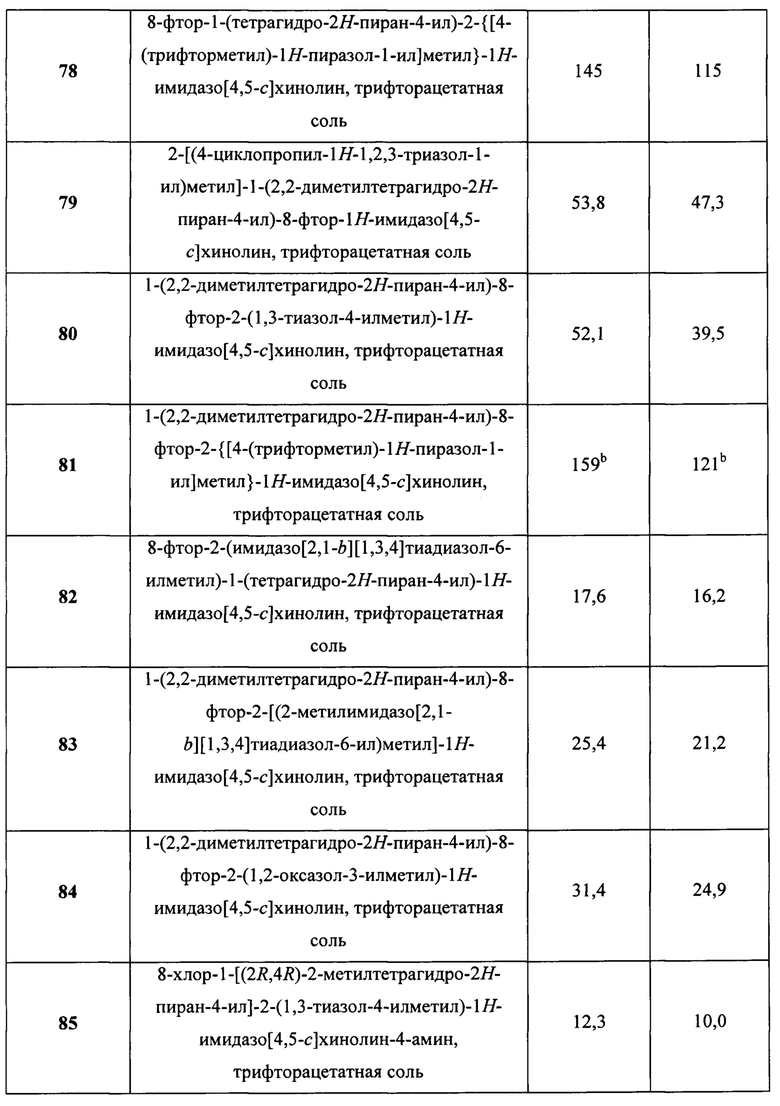
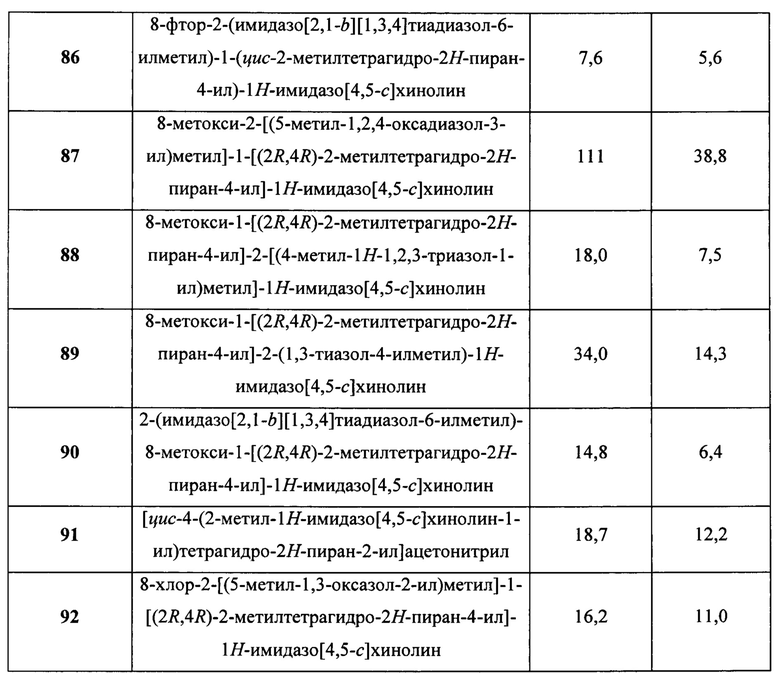
* Среднее геометрическое из 2-4 определений, если не указано иное.
a. Значение IC50 представляет собой среднее геометрическое не менее чем из 5 определений.
b. Значение IC50 относится к однократному определению.
c. Это значение определяли для трифторацетатной соли соединения из примера.
Соединения из примеров, представленные в Таблице 4, могут быть получены с использованием способов, проиллюстрированных в отношении синтезов соединений примеров 1-92, либо по отдельности, либо в сочетании с методами, общеизвестными в данной области техники.

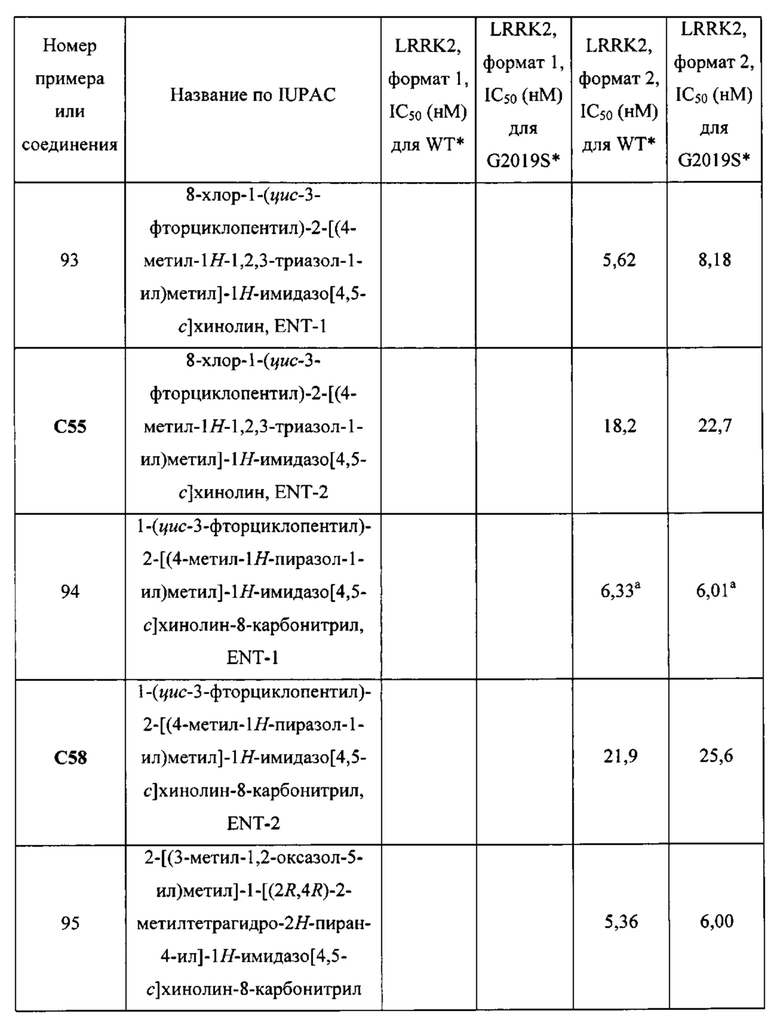
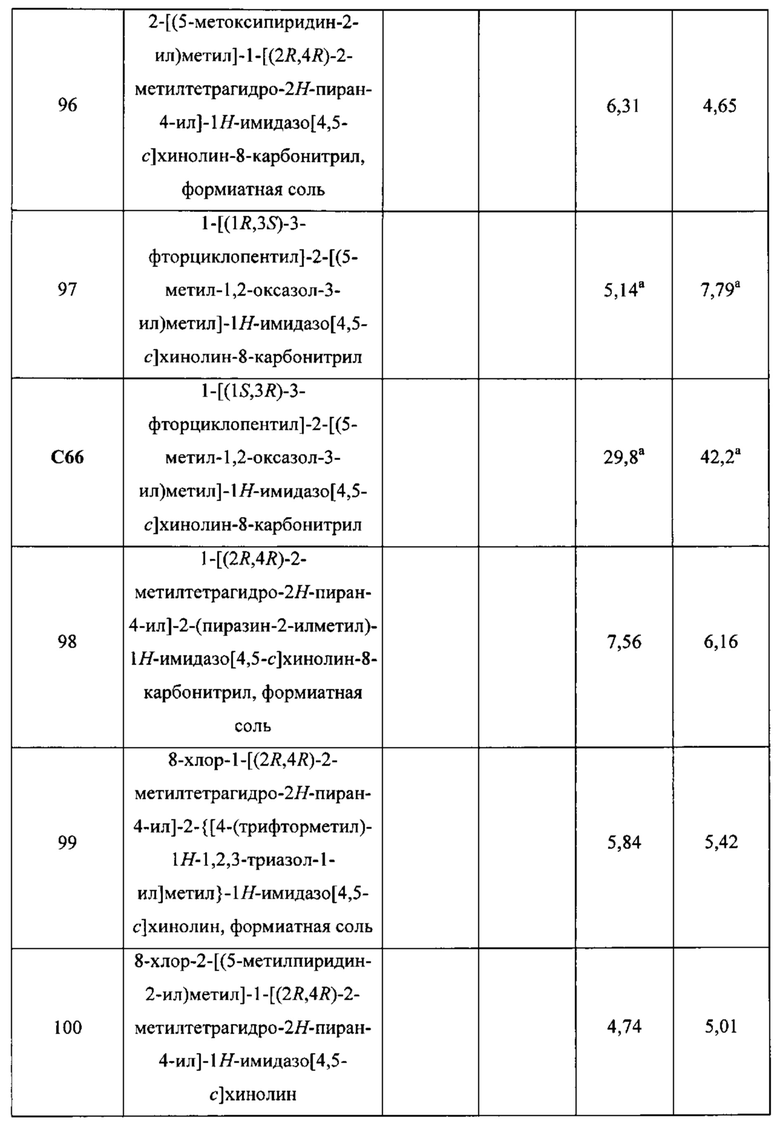
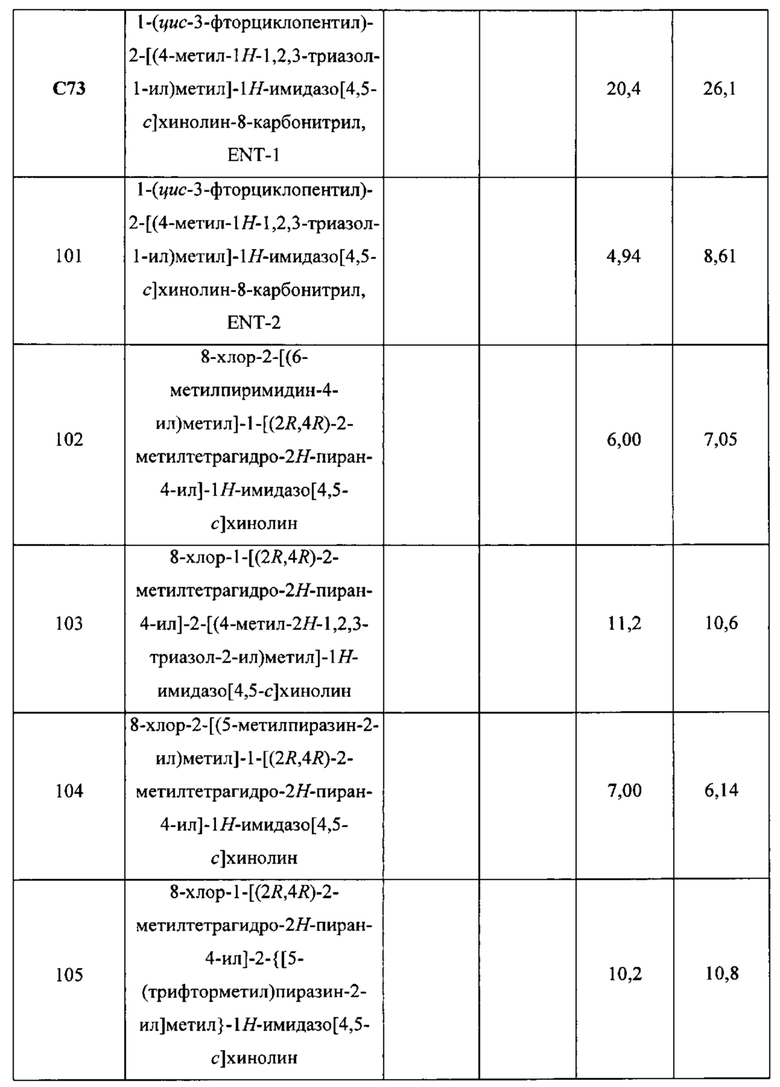
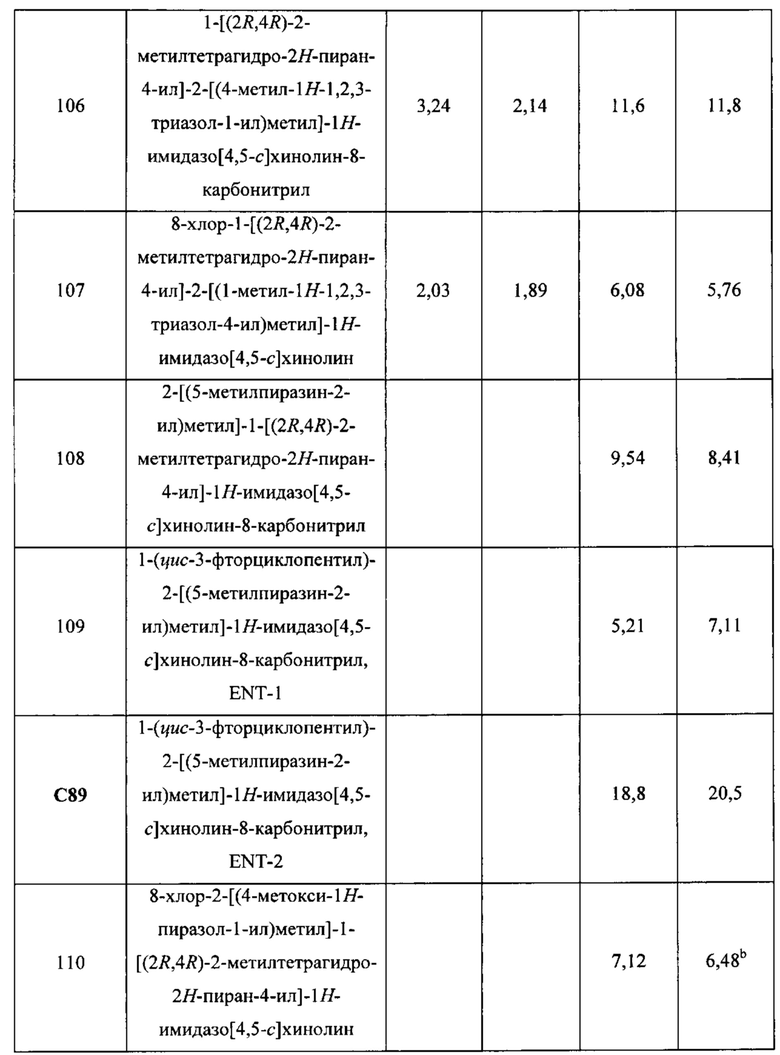
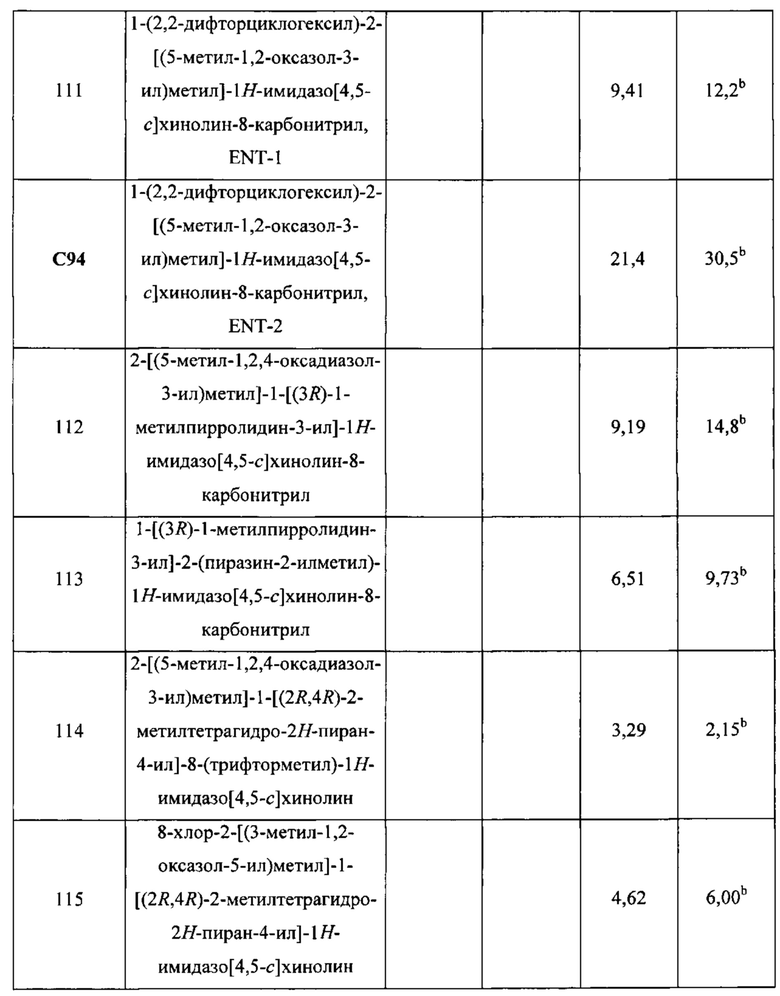
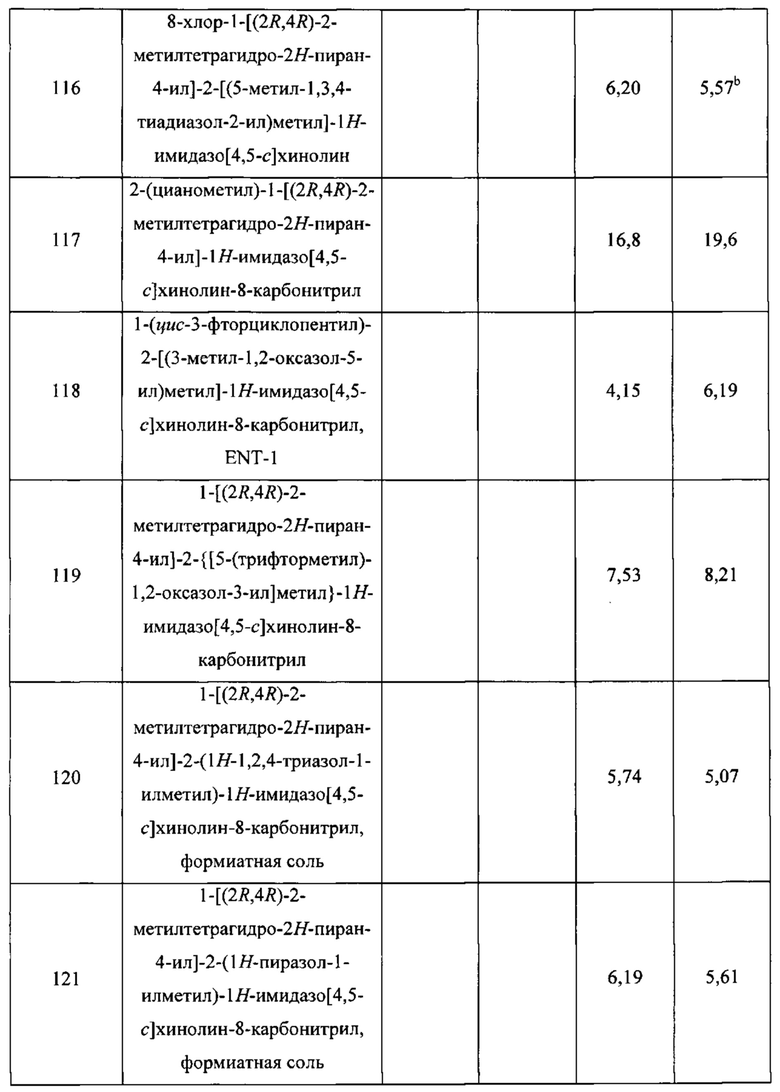
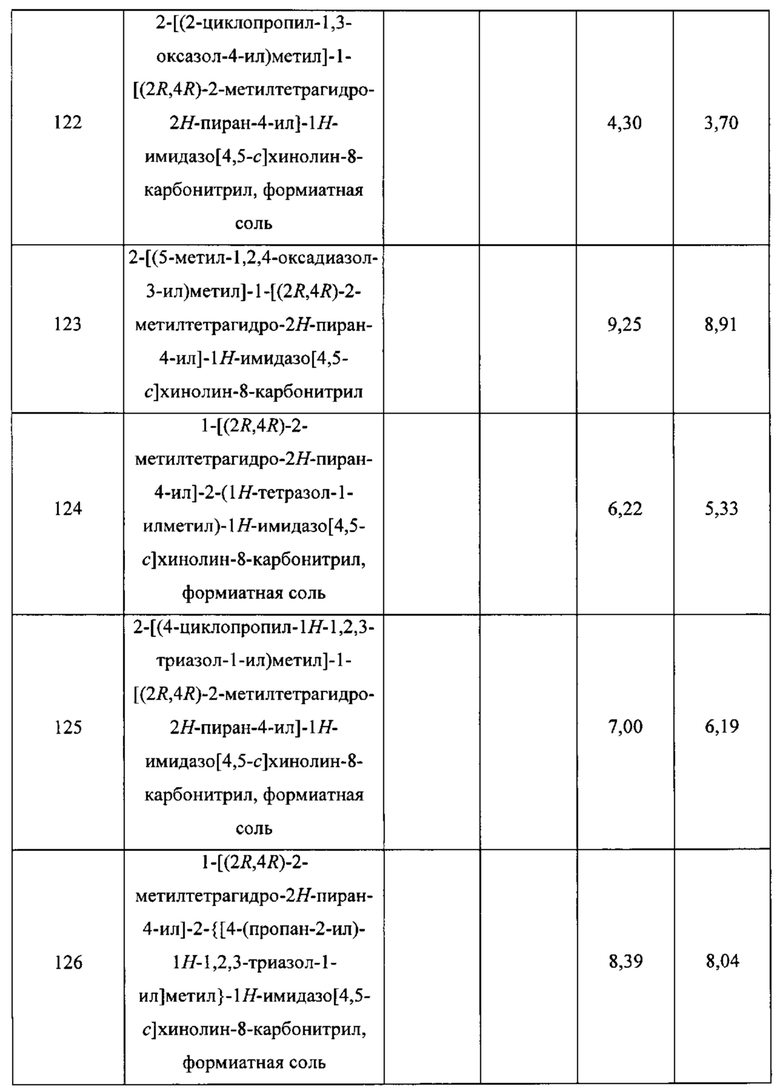
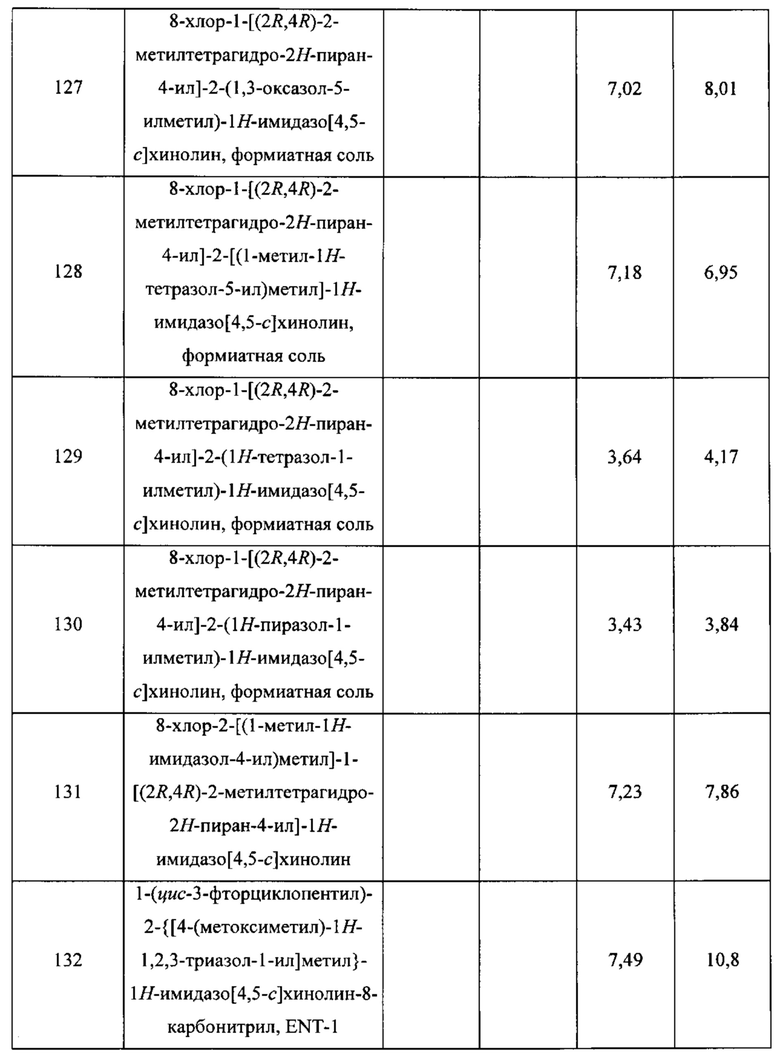
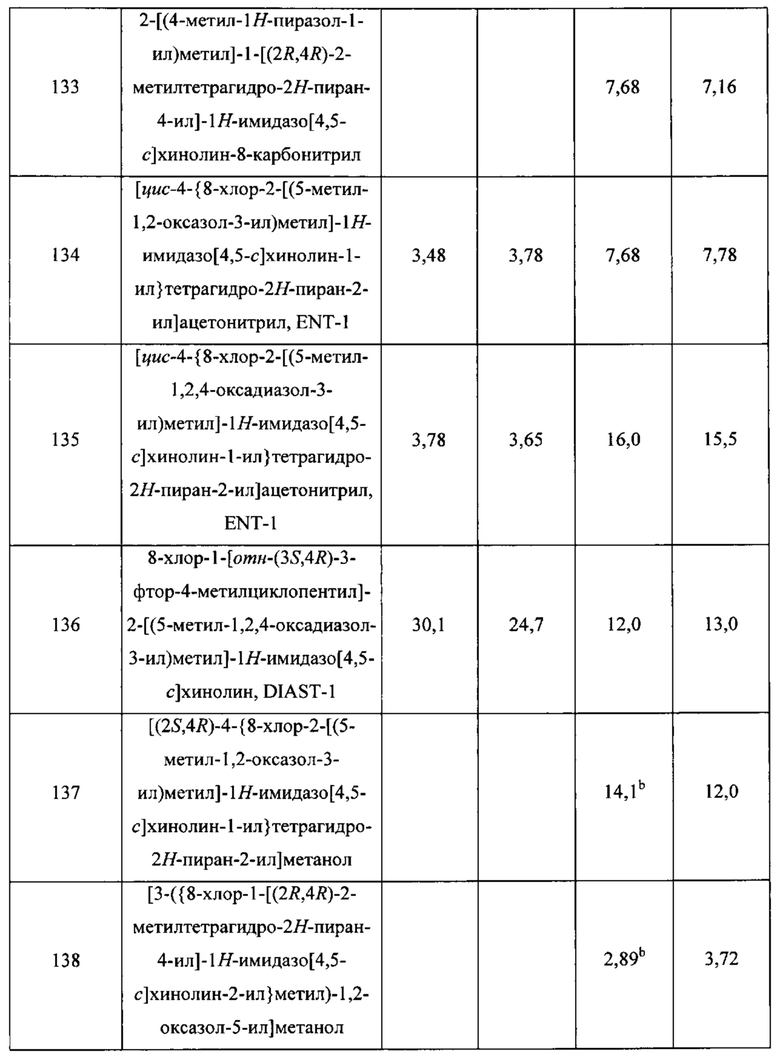
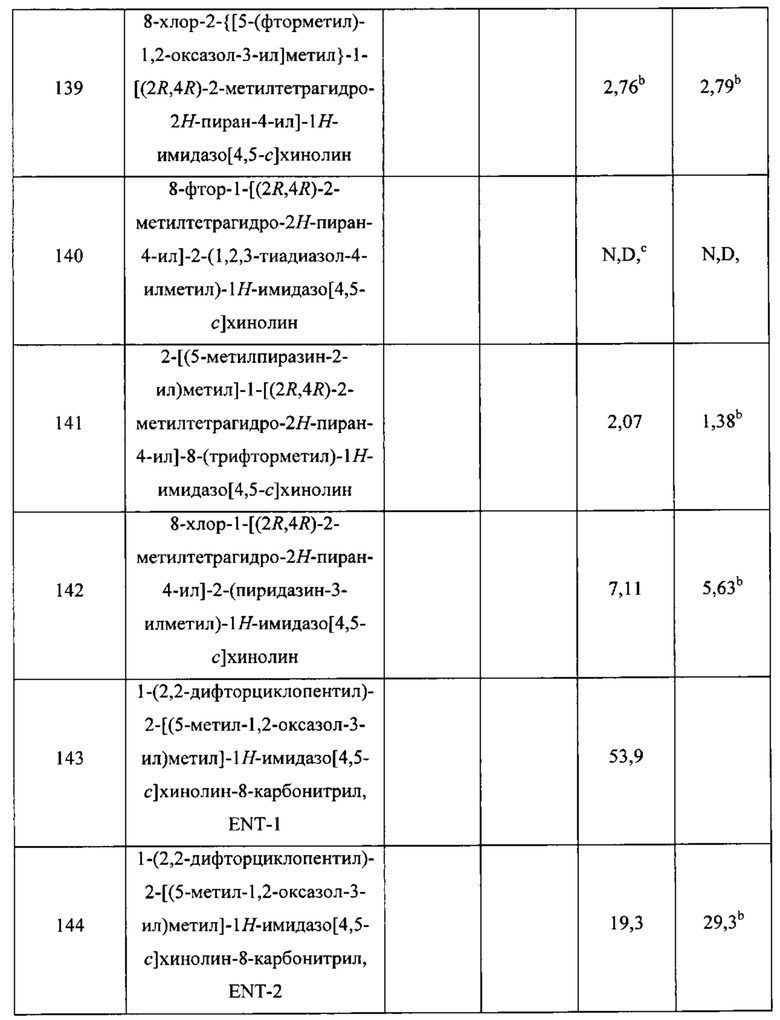
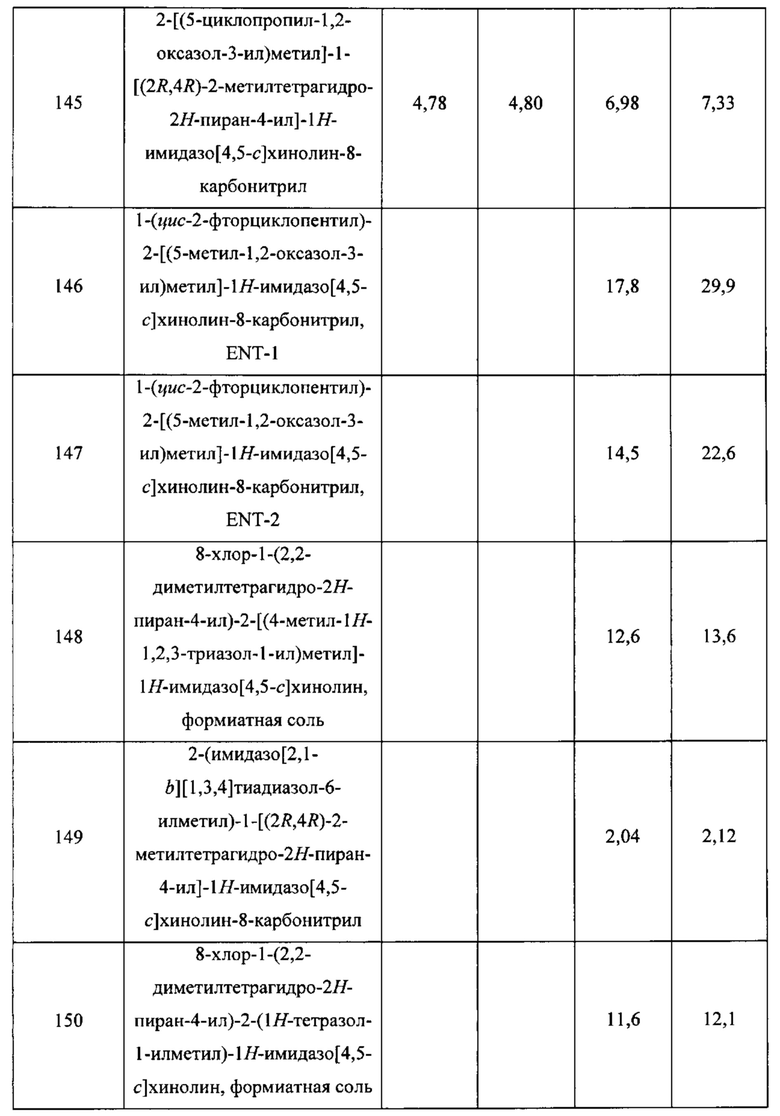
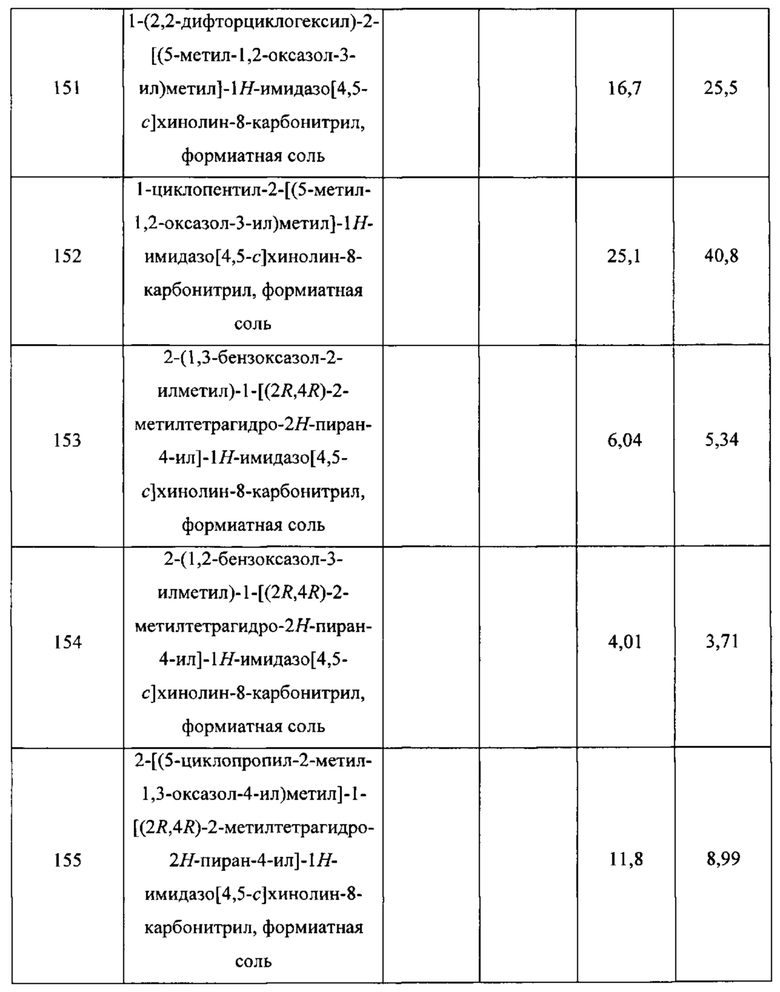
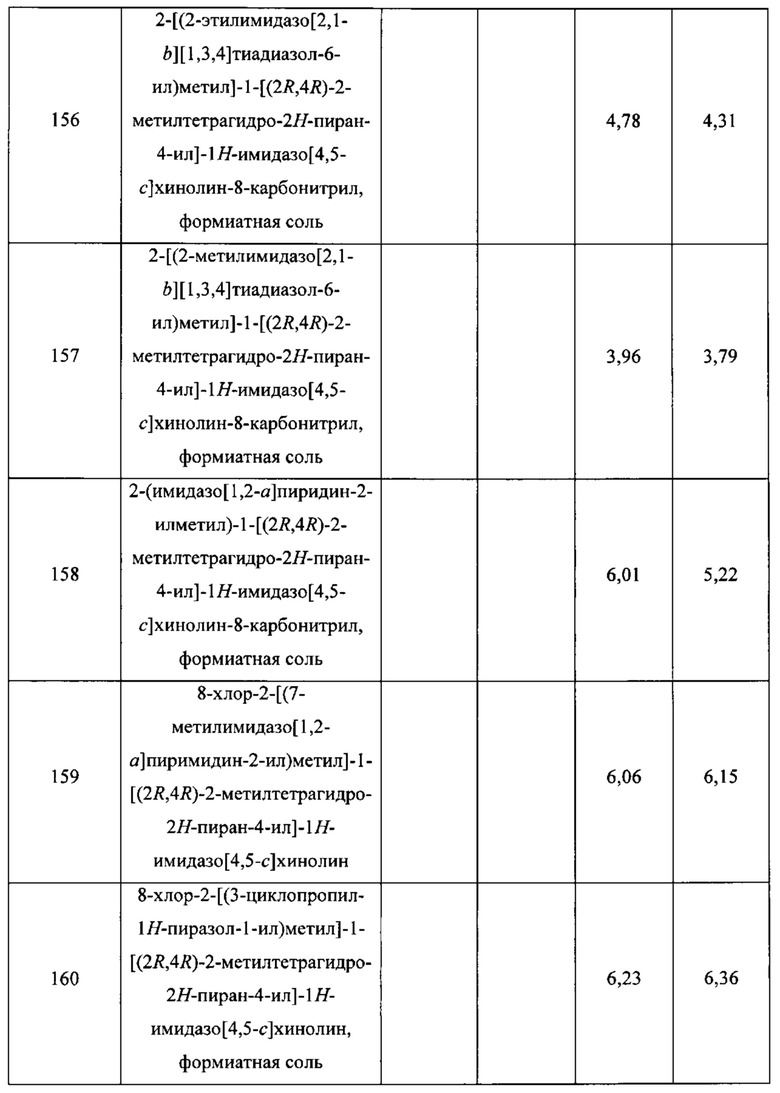
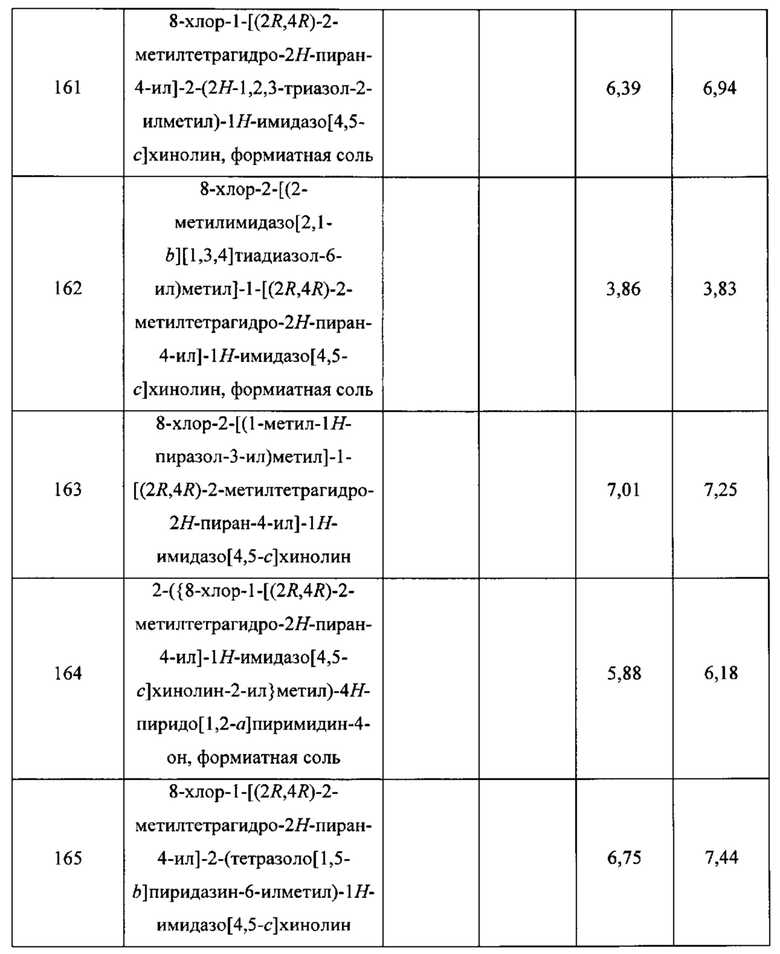
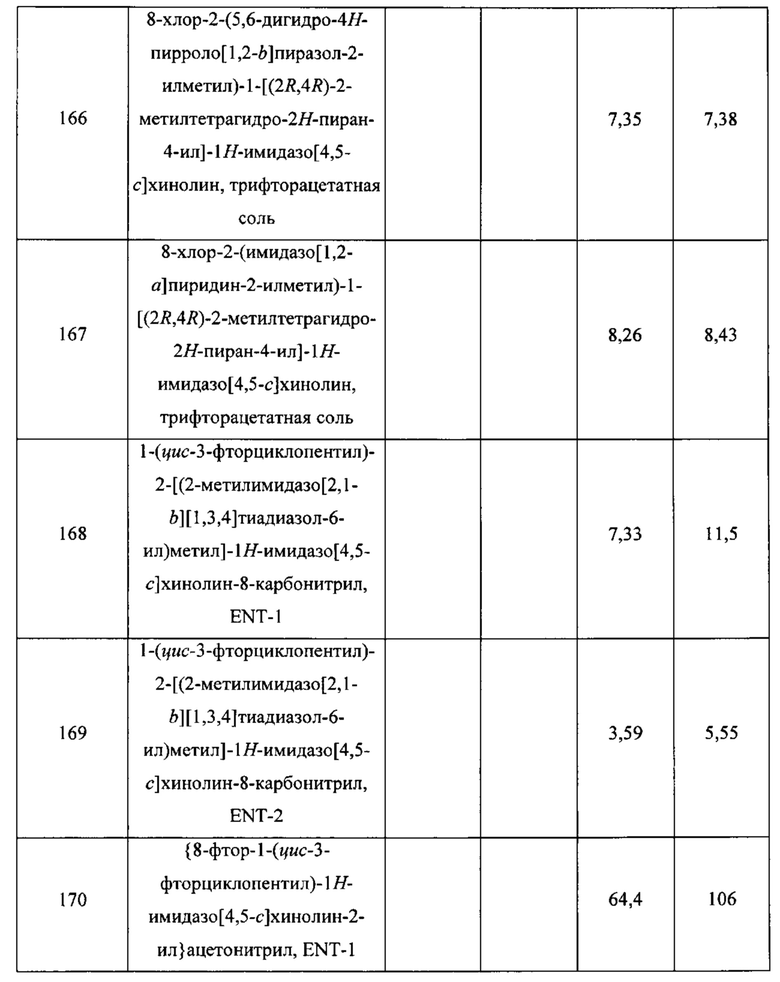
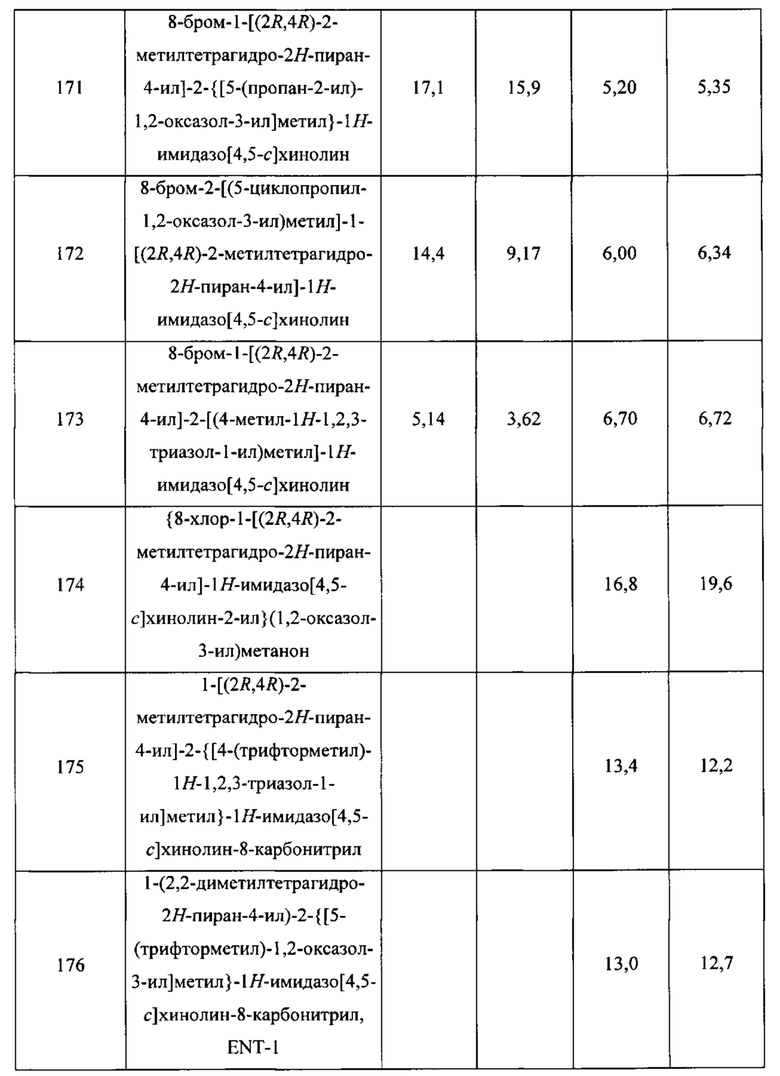
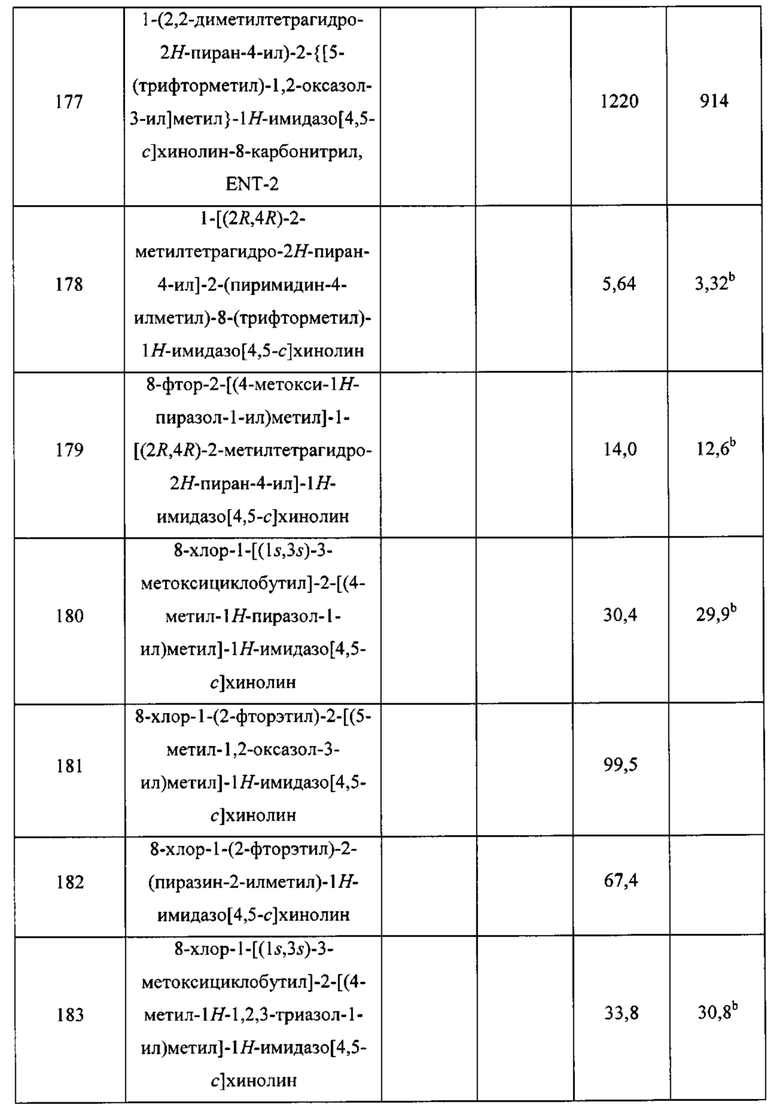
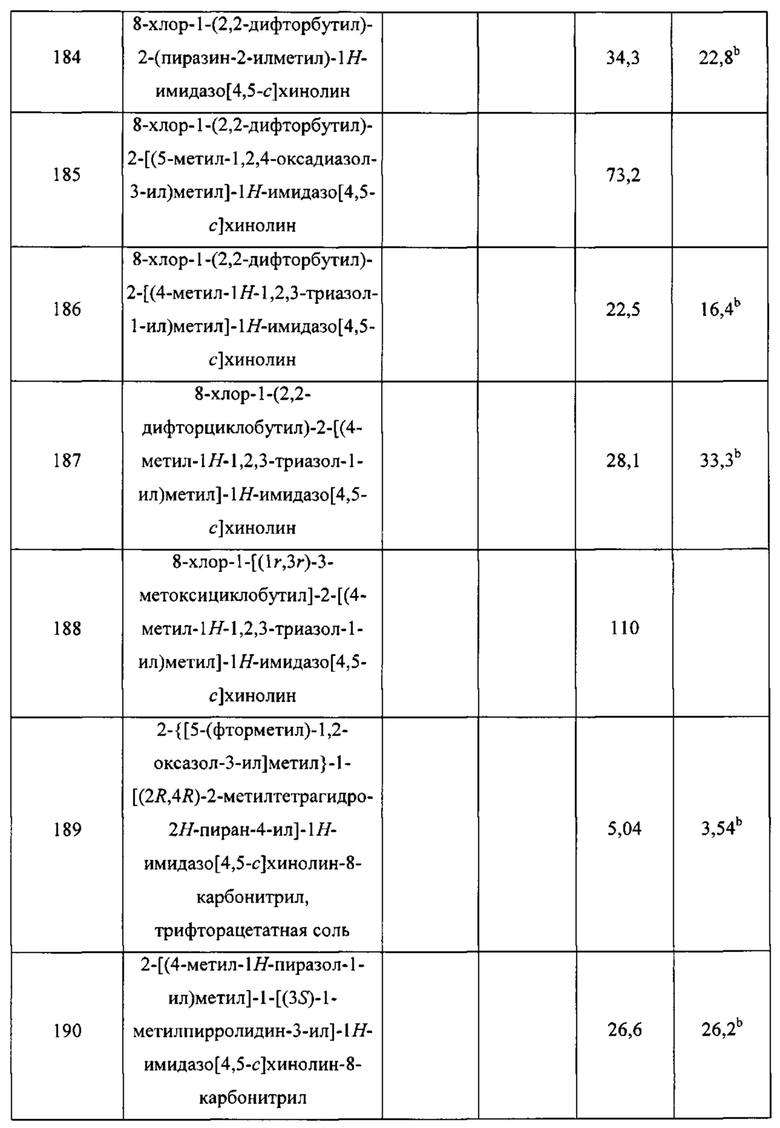
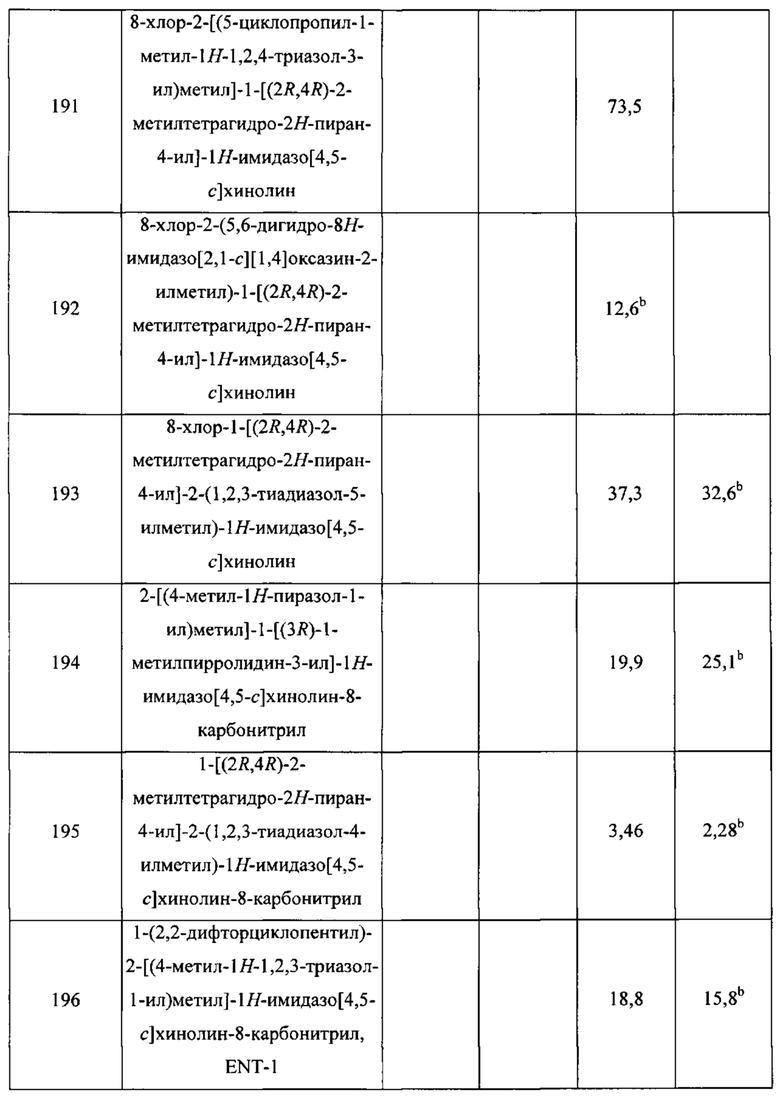
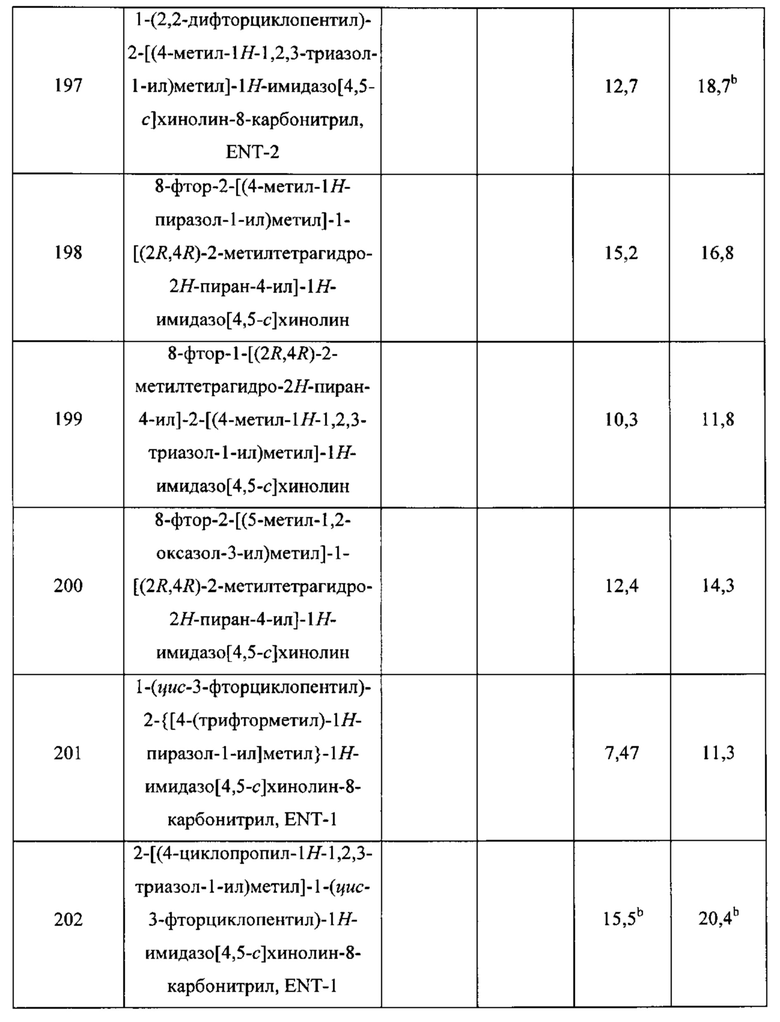
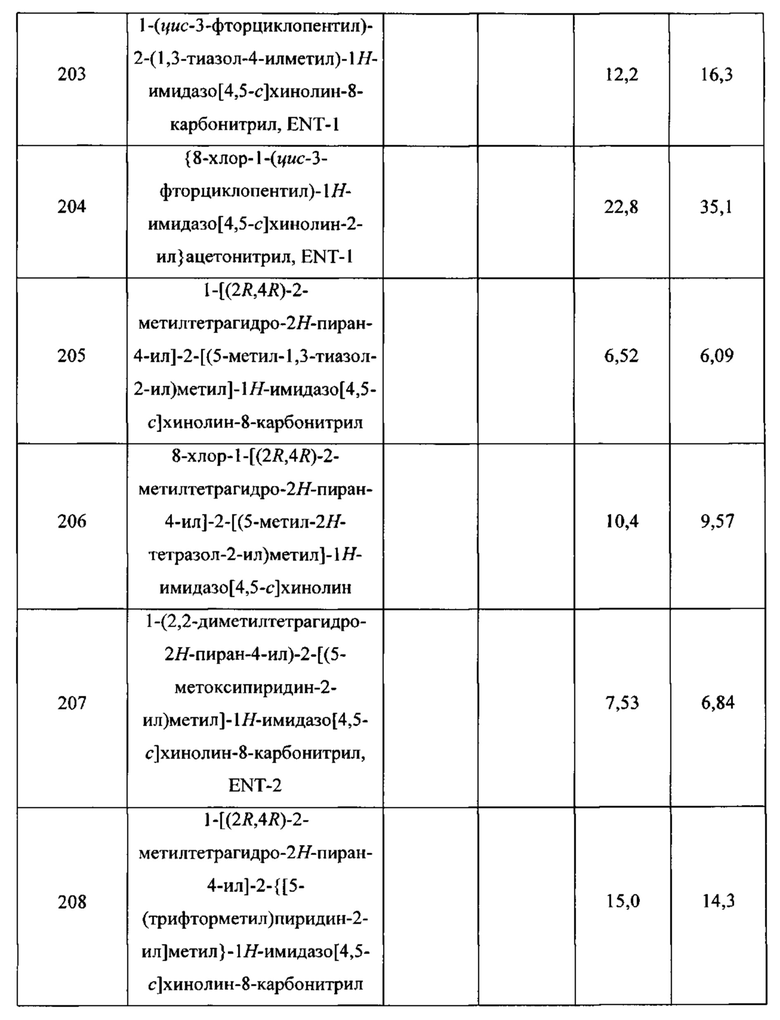
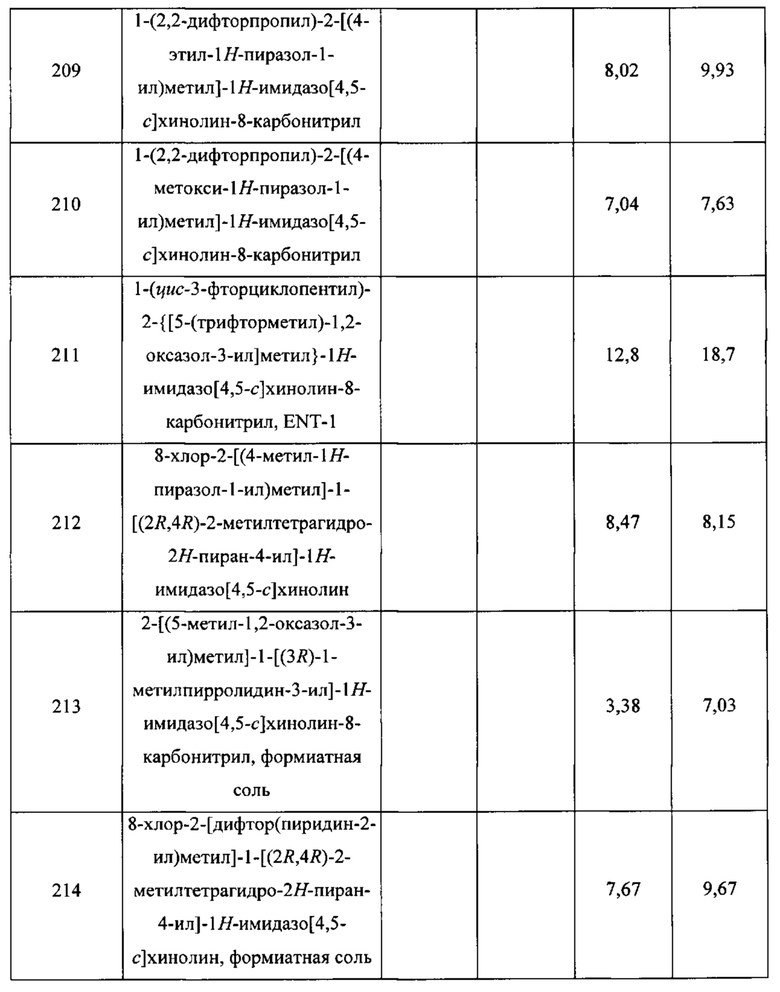
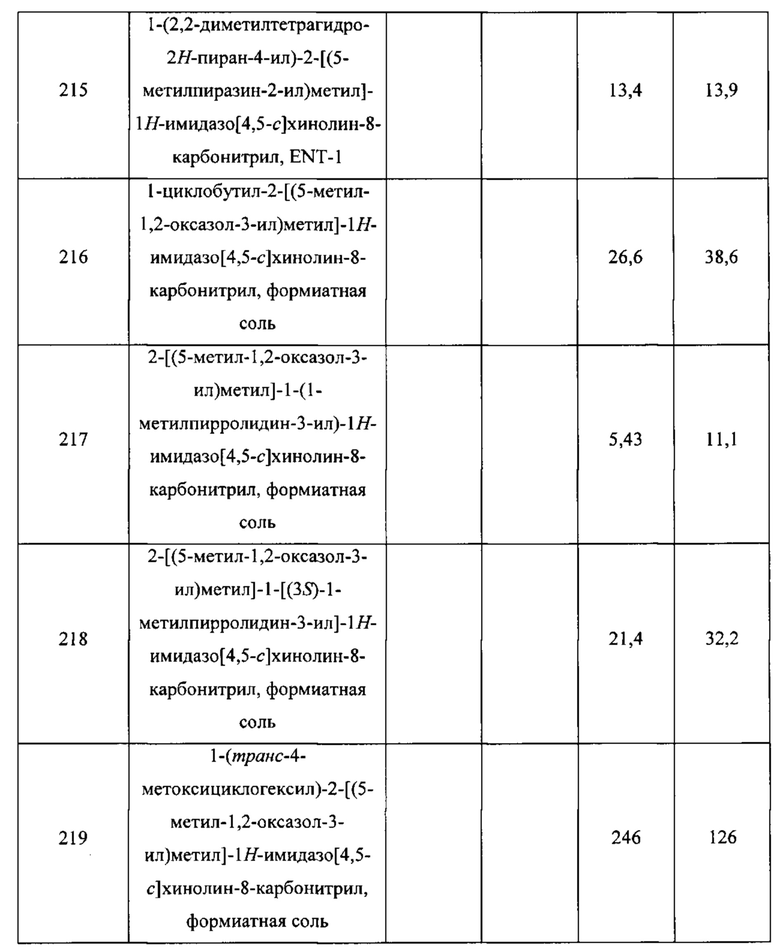
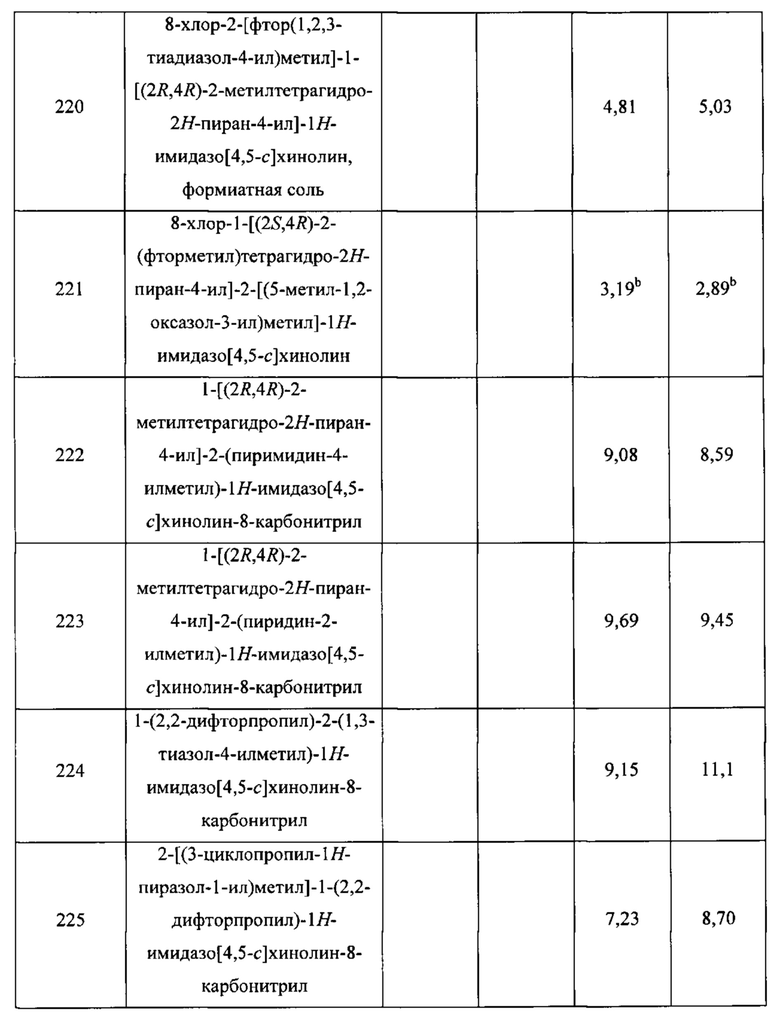
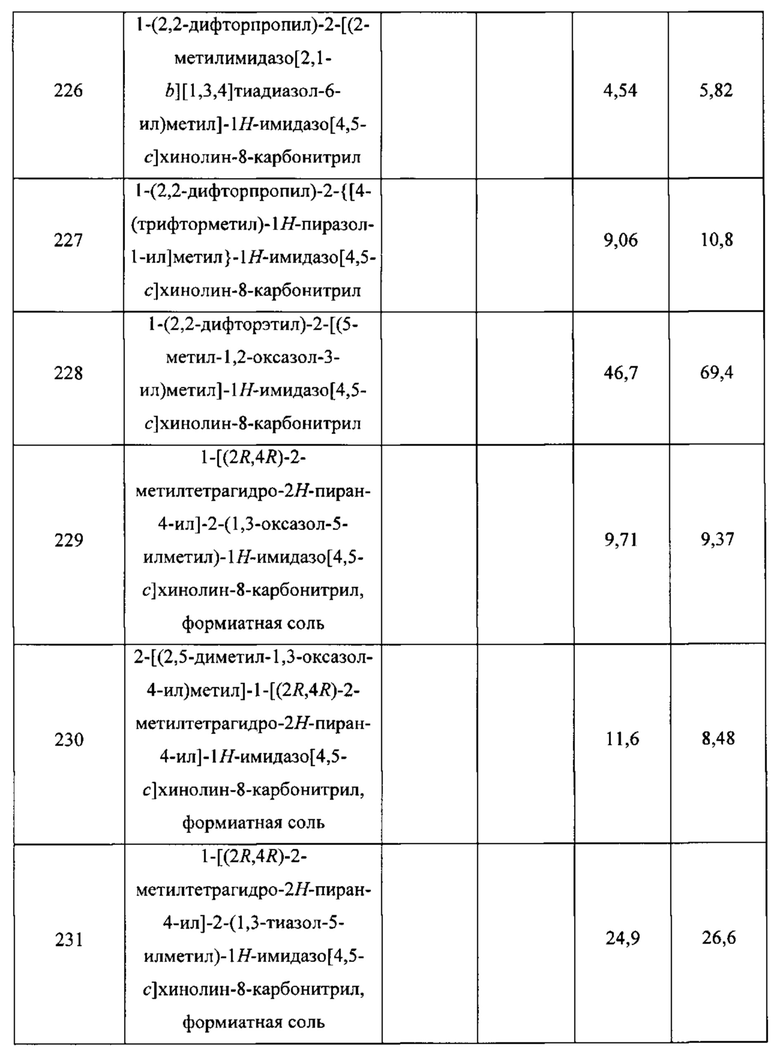
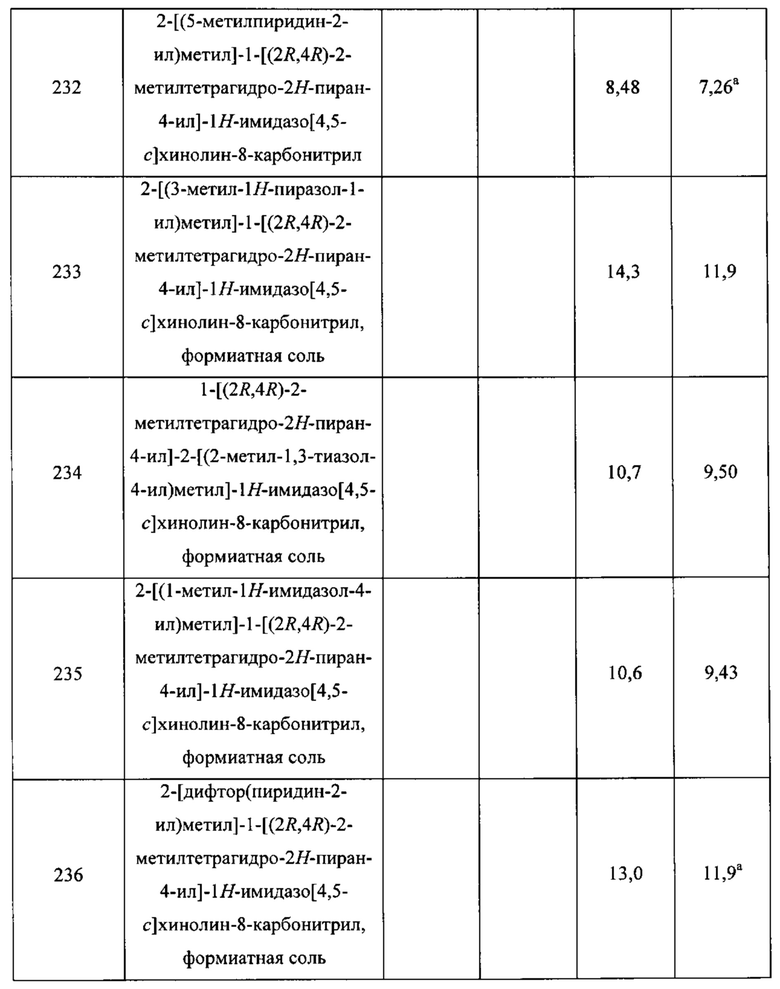
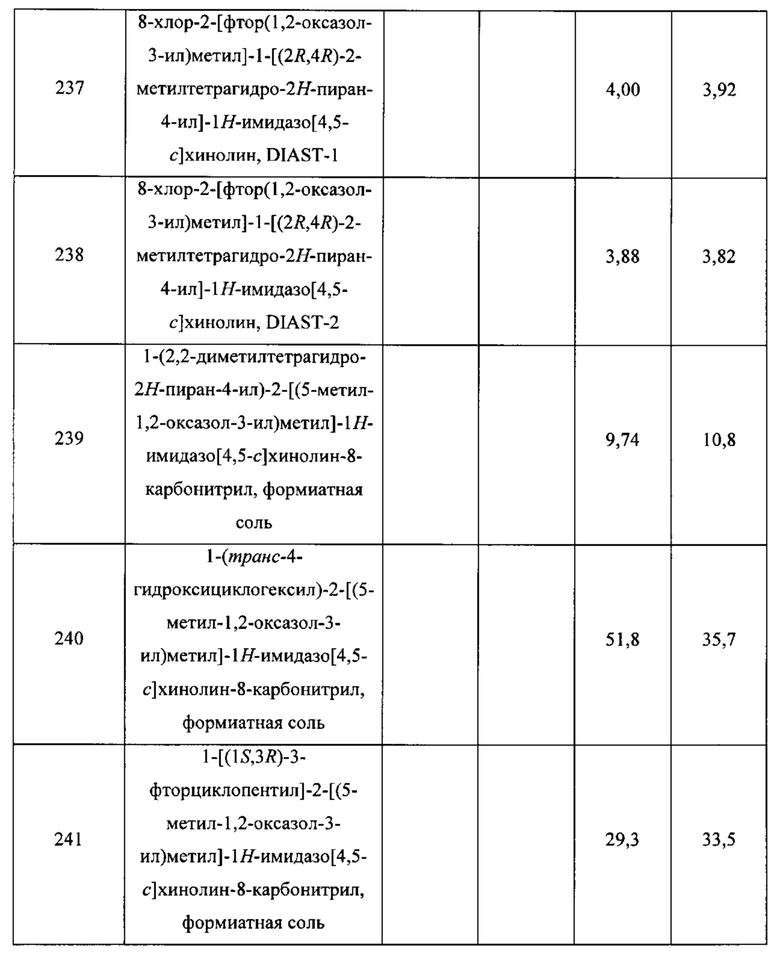
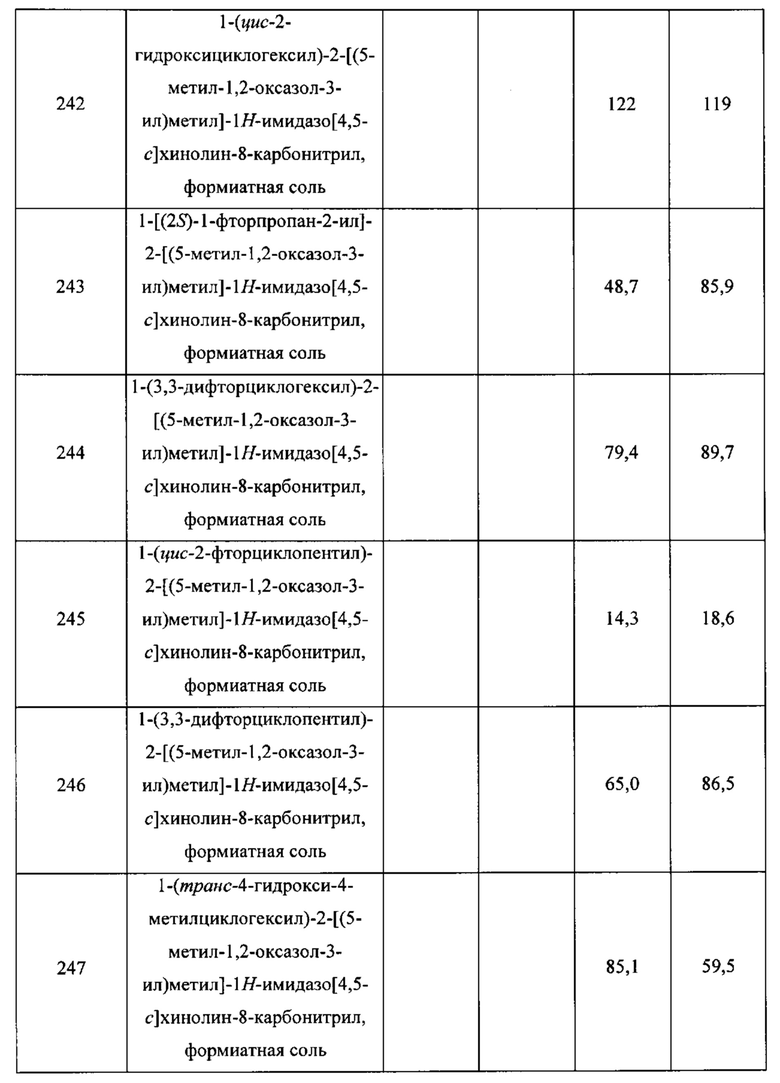
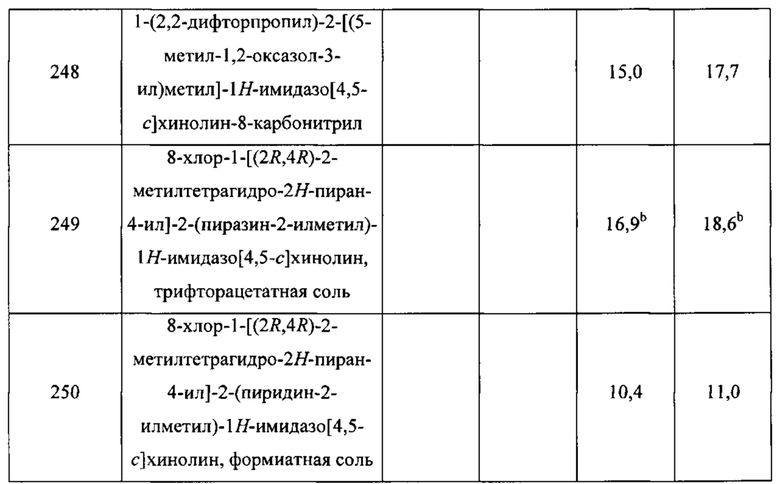
* Среднее геометрическое из 2-4 определений, если не указано иное.
a. Значение IC50 представляет собой среднее геометрическое не менее чем из 5 определений.
b. Значение IC50 относится к однократному определению.
c. Не определяли.
В Таблице 5, ниже, приведены данные по селективности в отношении киназ для соединений примеров 3, 4, 5 и 22. Соединения тестировали, используя имеющийся в продаже набор для анализа селективности в отношении киназ, поставляемый CarnaBio USA, Inc. 209 West Central St., Suite 307, Natick, MA 01760 USA. Соединения примеров 3, 4, 5 и 22 тестировали в данном анализе в концентрации 1 мкМ с использованием АТФ в концентрации 1 мМ. В Таблице 5А, ниже, приведены данные по селективности в отношении киназ из последующего анализа для соединений примеров 4, 11, 5, 104, 102 и 116.

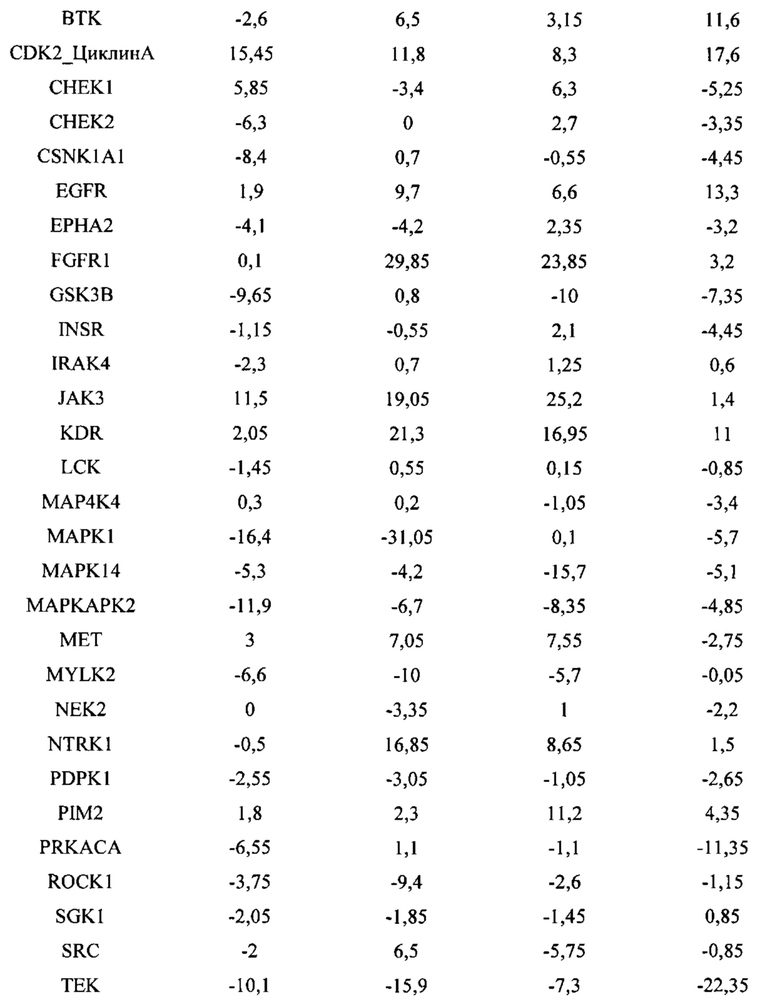
При этом ABL означает тирозинкиназу Абельсона; AKT1 означает представитель 1 семейства протеинкиназ В; AURKA означает аврора-киназу А; BTK означает тирозинкиназу Брутона; CDK означает циклинзависимую киназу; CHEK означает киназу контрольной точки (клеточного цикла); CSNK1A1 означает казеиновую киназу 1, субъединицу альфа 1; EGFR означает рецептор эпидермального фактора роста; ЕРНА2 означает рецептор эфрина А2; FGFR означает рецептор фактора роста фибробластов; GSK3B означает киназу-3В гликогенсинтазы; INSR означает инсулиновый рецептор; IRAK4 означает киназу, ассоциированную с рецептором интерлейкина-4; JAK3 означает янус-киназу 3; KDR означает рецептор, содержащий домен вставки киназы; LCK означает протеин-тирозинкиназу лимфоцитов; MAP4K4 означает митоген-активируемую киназу киназы 4 протеинкиназы 4; MAPKAPK2 означает протеинкиназу 2, активируемую митоген-активируемой протеинкиназой; MYLK означает киназу легких цепей миозина; NEK2 означает центросомальную киназу человека; NTRK1 означает нейротрофную рецепторную тирозинкиназу 1; PDPK1 означает 3-фосфоинозитид-зависимую протеинкиназу 1; PIM означает сайт интеграции провируса вируса Молони; PRKACA означает каталитическую альфа-субъединицу протеинкиназы A; ROCK1 означает Rho-ассоциированную протеинкиназу 1; SGK1 означает сыворотка- и глюкокортикоид-индуцибельную киназу 1; SRC означает киназу из семейства протоонкогенных тирозинкиназ; TEK означает эндотелиальную рецепторную тирозинкиназу.
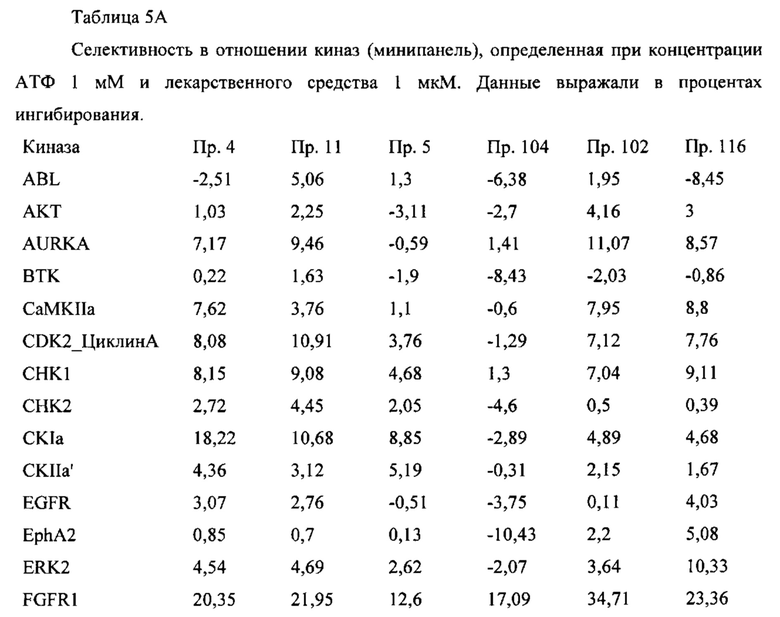
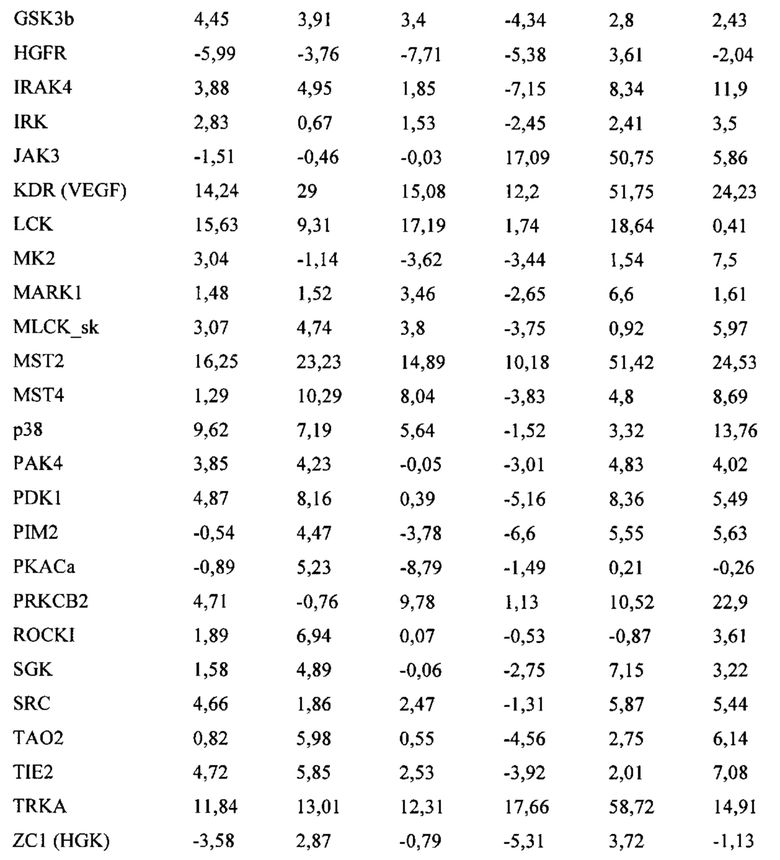
При этом CaMKIIa означает альфа-субъединицу кальций/кальмодулин-зависимой протеинкиназы типа II; CKIa означает казеиновую киназу I, изоформу альфа; ERK2 означает внеклеточную сигнал-регулируемую киназу 2; HGFR означает рецептор фактора роста гепатоцитов; IRK означает киназу рецептора инсулина; VEGF означает сосудистый эндотелиальный фактор роста; MK2 означает протеинкиназу 2, активированную митоген-активированной протеинкиназой; MLCK_sk означает киназу легких цепей миозина скелетных мышц; MST означает ste20-подобную киназу млекопитающих; PAK означает р21-активируемую киназу; PDK1 означает фосфоинозитид-зависимую киназу 1; PKACa означает каталитическую альфа-субъединицу протеинкиназы A; PRKCB2 означает протеинкиназу С, тип В2; TAO2 означает киназу 2 из подсемейства ТАО ,TIE2 означает тирозинкиназу с гомологичными иммуноглобулин-подобному и эпидермальному фактору роста доменами; TRKA означает тирозинкиназу A; ZC1 означает киназу 1 из подсемейства ZC киназ, HGK означает киназу, подобную киназе клеток-предшественников гепатоцитов/подобную киназе зародышевого центра.
| название | год | авторы | номер документа |
|---|---|---|---|
| Новые производные имидазо[4,5-с]хинолина в качестве ингибиторов LRRK2 | 2018 |
|
RU2773516C2 |
| СОЕДИНЕНИЯ ТИЕНОПИРИМИДИНА | 2013 |
|
RU2637925C2 |
| N-{2-[4-АМИНО-2-(ЭТОКСИМЕТИЛ)-1Н-ИМИДАЗО-[4,5-с]-ХИНОЛИН-1-ИЛ]-1,1-ДИМЕТИЛЭТИЛ}-МЕТАНСУЛЬФОНАМИД И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2004 |
|
RU2374246C2 |
| ПРОИЗВОДНЫЕ 6-ФЕНИЛ-1Н-ИМИДАЗО[4,5,-с] ПИРИДИН-4-КАРБОНИТРИЛА В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА К И S | 2007 |
|
RU2400482C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗО[4,5-С]ПИРИДИНА В КАЧЕСТВЕ АГОНИСТОВ TOLL-ПОДОБНОГО РЕЦЕПТОРА | 2020 |
|
RU2785124C1 |
| ИМИДАЗОПИРАЗИНОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE1 | 2016 |
|
RU2712219C2 |
| ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА ИЛИ ТЕТРАГИДРОПИРАНА, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ИЛИ ГИДРАТЫ, ИЛИ СОЛЬВАТЫ ЭТИХ СОЕДИНЕНИЙ, ИЛИ ИХ СОЛЕЙ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ И ФУНГИЦИДНОЕ СРЕДСТВО | 1992 |
|
RU2084439C1 |
| ПРОИЗВОДНЫЕ 6-ФЕНИЛ-1Н-ИМИДАЗО[4,5-c]ПИРИДИН-4-КАРБОНИТРИЛА В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА | 2008 |
|
RU2485119C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |
| АНАЛОГИ СОЕДИНЕНИЙ 4Н-ПИРАЗОЛО[1,5-А]БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ PARP | 2015 |
|
RU2672722C2 |
Изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы (I) или его фармацевтически приемлемой соли, где Х представляет собой CR7 или N; Z представляет собой CR3; R1 выбран из группы, состоящей из циано и 5-9-членного гетероарила, который содержит 1-5 гетероатомов, независимо выбранных из N, О и S; где 5-9-членный гетероарил возможно замещен 1-2 группами R8; каждый из R1a и R1b независимо представляет собой водород или галоген; или R1a и R1b вместе с атомом углерода, к которому они присоединены, представляют собой С(O); R2 представляет собой C1-С6алкил, С3-С7циклоалкил или 5-6-членный гетероциклоалкил, который содержит 1-3 гетероатома, независимо выбранных из NR и О; где каждый из С3-С7циклоалкила и 5-6-членного гетероциклоалкила возможно замещен 1-2 группами R9; и где C1-С6алкил возможно замещен 1-2 группами R10; R представляет собой C1-С6алкил; R3 выбран из группы, состоящей из водорода, дейтеро, амино, галогена, циано, C1-С6алкила и C1-С6алкокси; где каждый из C1-С6алкила и C1-С6алкокси возможно замещен 1-3 группами галоген; R4, R5 и R7 независимо представляют собой водород или дейтеро; R6 представляет собой водород, дейтеро, галоген или амино; R8 в каждом случае независимо выбран из группы, состоящей из галогена, C1-С6алкила, C1-С6алкокси и С3-С6циклоалкила; где C1-С6алкил возможно замещен 1-3 группами галоген, гидрокси или C1-С3алкокси; R9 в каждом случае независимо выбран из группы, состоящей из галогена, гидрокси, C1-С6алкила и C1-С6алкокси, где C1-С6алкил и C1-С6алкокси возможно замещены одной-тремя группами галоген или циано; и R10 в каждом случае представляет собой галоген. Также изобретение относится к конкретным соединениям, фармацевтической композиции на основе соединения формулы (I) и способу лечения болезни Крона, болезни Паркинсона, деменции с тельцами Леви, лепры, болезни Альцгеймера и таупатии. Технический результат: получены новые гетероциклические соединения, обладающие активностью ингибирования обогащенной лейциновыми повторами киназы 2 (LRRK2). 4 н. и 22 з.п. ф-лы, 15 табл., 250 пр.
1. Соединение формулы (I)
или его фармацевтически приемлемая соль; где
Х представляет собой CR7 или N;
Z представляет собой CR3;
R1 выбран из группы, состоящей из циано и 5-9-членного гетероарила, который содержит 1-5 гетероатомов, независимо выбранных из N, О и S; где 5-9-членный гетероарил возможно замещен 1-2 группами R8;
каждый из R1a и R1b независимо представляет собой водород или галоген; или
R1a и R1b вместе с атомом углерода, к которому они присоединены, представляют собой С(O);
R2 представляет собой C1-С6алкил, С3-С7циклоалкил или 5-6-членный гетероциклоалкил, который содержит 1-3 гетероатома, независимо выбранных из NR и О; где каждый из С3-С7циклоалкила и 5-6-членного гетероциклоалкила возможно замещен 1-2 группами R9; и где C1-С6алкил возможно замещен 1-2 группами R10;
R представляет собой C1-С6алкил;
R3 выбран из группы, состоящей из водорода, дейтеро, амино, галогена, циано, C1-С6алкила и C1-С6алкокси; где каждый из C1-С6алкила и C1-С6алкокси возможно замещен 1-3 группами галоген;
R4, R5 и R7 независимо представляют собой водород или дейтеро;
R6 представляет собой водород, дейтеро, галоген или амино;
R8 в каждом случае независимо выбран из группы, состоящей из галогена, C1-С6алкила, C1-С6алкокси и С3-С6циклоалкила; где C1-С6алкил возможно замещен 1-3 группами галоген, гидрокси или C1-С3алкокси;
R9 в каждом случае независимо выбран из группы, состоящей из галогена, гидрокси, C1-С6алкила и C1-С6алкокси, где C1-С6алкил и C1-С6алкокси возможно замещены одной-тремя группами галоген или циано; и
R10 в каждом случае представляет собой галоген.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где
Х представляет собой CR7;
Z представляет собой CR3;
R3 представляет собой водород, бром, хлор, фтор, метокси или циано; и
каждый из R4, R5, R6 и R7 представляет собой водород или дейтеро.
3. Соединение по п. 2 или его фармацевтически приемлемая соль, где
R1 представляет собой 5-9-членный гетероарил, который содержит 1-4 гетероатома, независимо выбранных из N, О и S; где 5-9-членный гетероарил возможно замещен 1-2 группами R8;
каждый из R1a и R1b представляет собой водород; и
R8 в каждом случае независимо выбран из группы, состоящей из галогена, C1-С3алкила, C1-С3алкокси и С3-С6циклоалкила; где C1-С3алкил возможно замещен 1-3 фторами, группами гидрокси или C1-С3алкокси.
4. Соединение по п. 3 или его фармацевтически приемлемая соль, где
R1 представляет собой 5-9-членный гетероарил, выбранный из группы, состоящей из оксазолила, изоксазолила, оксадиазолила, тиазолила, пиразолила, триазолила, тетразолила, пиридинила, бензоксазолила, бензоизоксазолила, бензопиразолила, бензотриазолила, имидазотиазолила и имидазотиадиазолила; каждый из которых возможно замещен группой R8; и R8 выбран из группы, состоящей из метила, трифторметила, изопропила, 2-гидроксиизопропила, метокси, метоксиметила, циклопропила и хлора.
5. Соединение по п. 4 или его фармацевтически приемлемая соль, где R1 выбран из группы, состоящей из
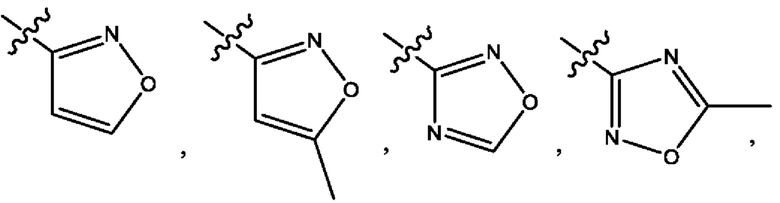
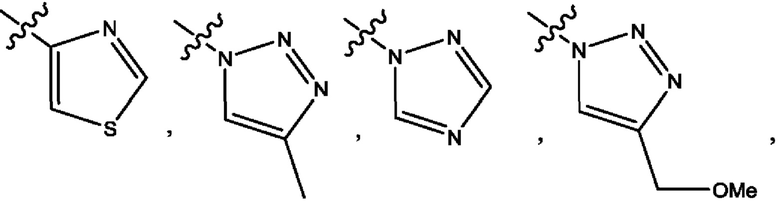
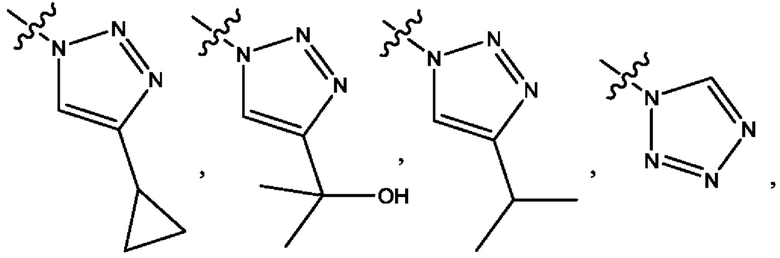
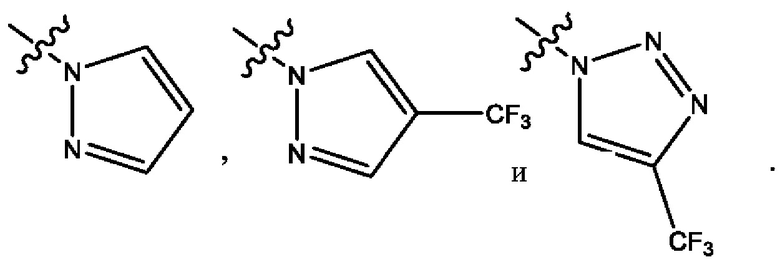
6. Соединение по п. 4 или его фармацевтически приемлемая соль, где R1 выбран из группы, состоящей из
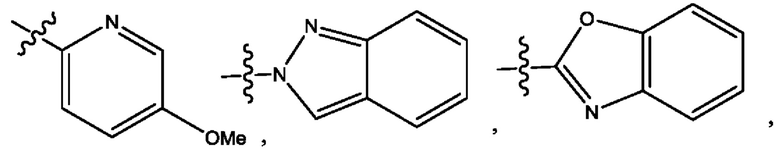
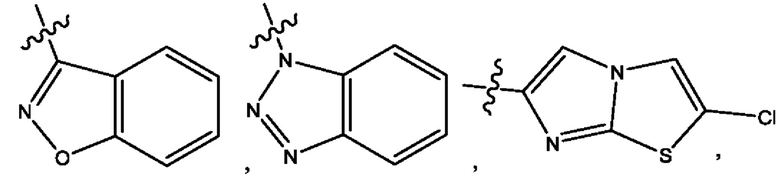
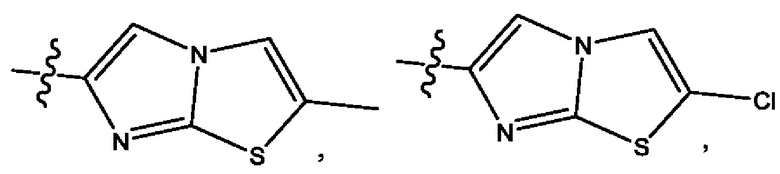 и
и
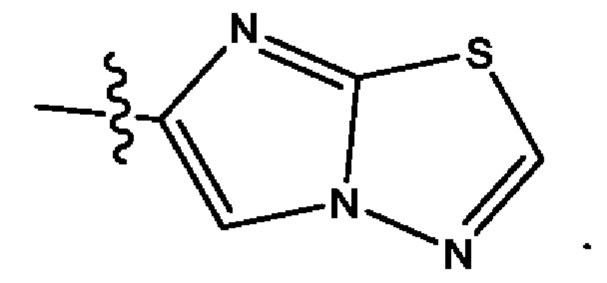
7. Соединение по п. 4 или его фармацевтически приемлемая соль, где R2 представляет собой тетрагидропиранил, циклопентил или циклогексил; каждый из которых возможно замещен 1-2 группами R9; и
R9 в каждом случае независимо представляет собой метил, этил, цианометил, гидрокси или фтор.
8. Соединение по п. 7 или его фармацевтически приемлемая соль, где
R2 выбран из группы, состоящей из
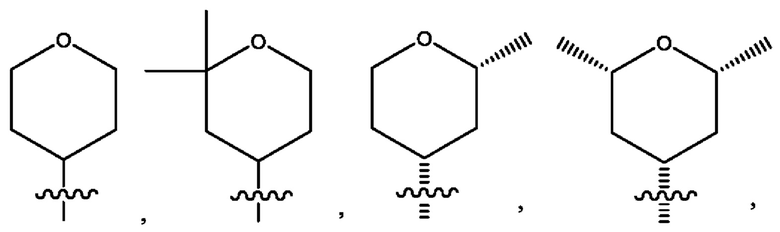
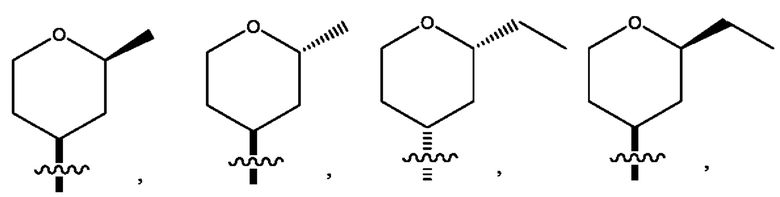
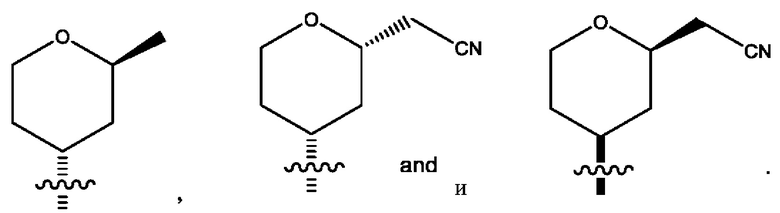
9. Соединение по п. 8 или его фармацевтически приемлемая соль, где R2 представляет собой
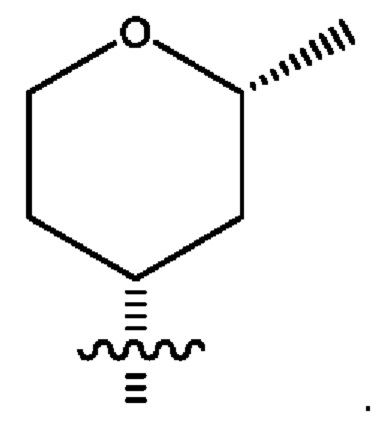
10. Соединение по п. 7 или его фармацевтически приемлемая соль, где R2 выбран из группы, состоящей из
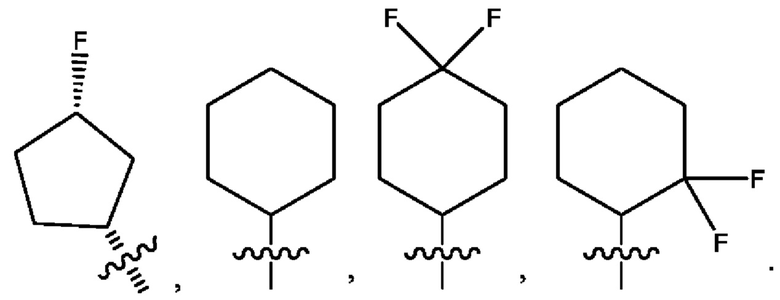 и
и
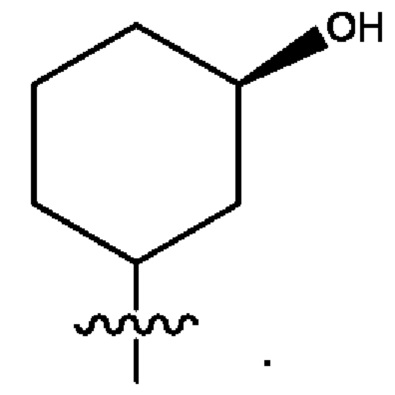
11. Соединение по п. 1 или его фармацевтически приемлемая соль, где
Х представляет собой N;
Z представляет собой CR3;
R1 представляет собой 5-9-членный гетероарил, выбранный из группы, состоящей из оксазолила, изоксазолила, оксадиазолила, тиазолила, пиразолила, триазолила, тетразолила, пиридинила, бензоксазолила, бензоизоксазолила, бензопиразолила, бензотриазолила, имидазотиазолила и имидазотиадиазолила; каждый из которых возможно замещен группой R8;
каждый из R1a и R1b представляет собой водород; и
R8 представляет собой метил, трифторметил, изопропил, 2-гидроксиизопропил, метокси, метоксиметил, циклопропил или хлор.
12. Соединение по п. 11 или его фармацевтически приемлемая соль, где
R2 представляет собой
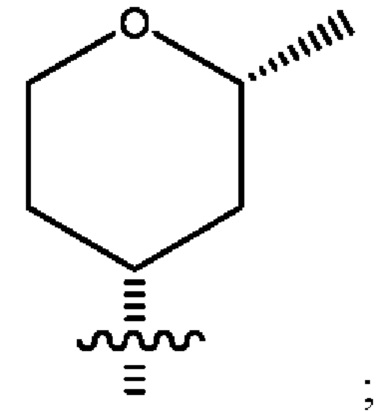 и
и
каждый из R3, R4, R5 и R6 представляет собой водород или дейтеро.
13. Соединение по п. 1 или его фармацевтически приемлемая соль, где
Х представляет собой CR7;
Z представляет собой CR3;
R1 представляет собой циано;
каждый из R1a и R1b представляет собой водород;
R2 представляет собой тетрагидропиранил или циклопентил; каждый из которых возможно замещен 1-2 группами R9; и
R9 в каждом случае независимо представляет собой метил, цианометил или фтор.
14. Соединение по п. 13 или его фармацевтически приемлемая соль; где
R2 представляет собой
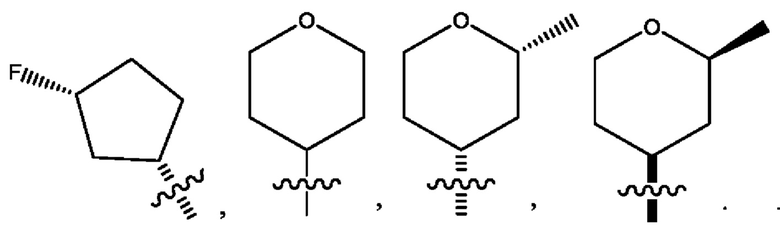 и
и
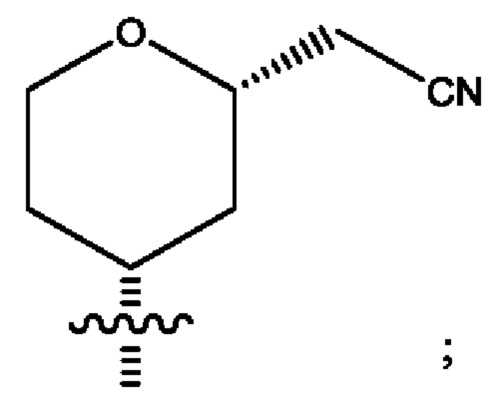
R3 представляет собой водород, бром, хлор, метокси или циано; и
каждый из R4, R5, R6 и R7 представляет собой водород или дейтеро.
15. Соединение, выбранное из группы, состоящей из
8-метокси-2-[(5-метил-1,2-оксазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
8-метокси-2-[(5-метил-1,2-оксазол-3-ил)метил]-1-[(2S,4S)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
8-хлор-2-[(5-метоксипиридин-2-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
2-[(5-метил-1,2-оксазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолин-8-карбонитрила;
8-хлор-2-[(5-метил-1,2-оксазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
8-хлор-2-[(5-метил-1,2,4-оксадиазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
8-бром-1-[(1S,3R)-3-фторциклопентил]-2-метил-1H-имидазо[4,5-с]хинолина;
1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с][1,5]нафтиридина;
1-[(2S,4S)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с][1,5]нафтиридина;
8-хлор-2-(имидазо[2,1-b][1,3,4]тиадиазол-6-илметил)-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
{8-хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолин-2-ил}ацетонитрила;
8-хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)(4-2H)-1H-имидазо[4,5-c]хинолина;
8-хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-[(4-метил-1Н-1,2,3-триазол-1-ил)метил]-1H-имидазо[4,5-с]хинолина;
8-бром-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолина;
8-хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,2,4-оксадиазол-3-илметил)-1H-имидазо[4,5-с]хинолина;
2-метил-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолин-8-карбонитрила;
2-метил-1-(цис-2-метилтетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
1-[(1R,3S)-3-фторциклопентил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолина;
8-хлор-2-метил-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
2-(1,2-оксазол-3-илметил)-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
2-метил-1-(цис-2-метилтетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолин-8-карбонитрила;
1-[(1S,3R)-3-фторциклопентил]-2-метил-1H-имидазо[4,5-с]хинолин-8-карбонитрила;
1-(цис-2-метилтетрагидро-2H-пиран-4-ил)-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолин-8-карбонитрила;
1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолин-8-карбонитрила;
1-(транс-2-метилтетрагидро-2H-пиран-4-ил)-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолин-8-карбонитрила;
1-[(2S,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолин-8-карбонитрила;
8-бром-1-[(1S,3R)-3-фторциклопентил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолина;
1-[(1R,3S)-3-фторциклопентил]-2-(1H-1,2,4-триазол-1-илметил)-1H-имидазо[4,5-с]хинолина;
1-[(1R,3S)-3-фторциклопентил]-2-[(4-метил-1H-1,2,3-триазол-1-ил)метил]-1H-имидазо[4,5-с]хинолина;
2-(1,3-бензоксазол-2-илметил)-1-[(1R,3S)-3-фторциклопентил]-1H-имидазо[4,5-с]хинолина;
2-(1,2-бензоксазол-3-илметил)-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
1-(тетрагидро-2H-пиран-4-ил)-2-(1H-1,2,4-триазол-1-илметил)-1H-имидазо[4,5-с]хинолина;
2-[(2-метилимидазо[2,1-b][1,3]тиазол-6-ил)метил]-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
2-[(4-метил-1Н-1,2,3-триазол-1-ил)метил]-1-(тетрагидро-2H-пиран-4-ил)-1Н-имидазо[4,5-с]хинолина;
2-{[4-(метоксиметил)-1H-1,2,3-триазол-1-ил]метил}-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
2-(1,3-бензоксазол-2-илметил)-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
1-(тетрагидро-2H-пиран-4-ил)-2-(1H-тетразол-1-илметил)-1H-имидазо[4,5-с]хинолина;
1-(тетрагидро-2H-пиран-4-ил)-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с]хинолина;
2-[(5-метоксипиридин-2-ил)метил]-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
2-[(2-метилимидазо[2,1-b][1,3,4]тиадиазол-6-ил)метил]-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
2-[(5-метил-1,2-оксазол-3-ил)метил]-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
2-(1-{[1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолин-2-ил]метил}-1H-1,2,3-триазол-4-ил)пропан-2-ола;
2-(1H-бензотриазол-1-илметил)-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
2-[(4-циклопропил-1H-1,2,3-триазол-1-ил)метил]-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
1-(тетрагидро-2H-пиран-4-ил)-2-{[4-(трифторметил)-1H-пиразол-1-ил]метил}-1H-имидазо[4,5-с]хинолина;
2-{[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]метил}-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
2-(2H-индазол-2-илметил)-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
1-(2,2-дифторциклогексил)-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолина;
1-(2,2-диметилтетрагидро-2H-пиран-4-ил)-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолина;
1-(4,4-дифторциклогексил)-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолина;
транс-3-[2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолин-1-ил]циклогексанола;
1-циклогексил-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолина;
8-хлор-2-[(5-метил-1,2,4-оксадиазол-3-ил)метил]-1-[(2S,4S)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
8-хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолина;
8-хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с]хинолина;
8-фтор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолина;
8-фтор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с]хинолина;
8-бром-1-(цис-2-метилтетрагидро-2H-пиран-4-ил)-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с]хинолина;
1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с]хинолин-8-карбонитрила;
8-бром-2-[(5-метил-1,2-оксазол-3-ил)метил]-1-(цис-2-метилтетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
8-бром-1-[(2R,4r,6S)-2,6-диметилтетрагидро-2H-пиран-4-ил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолина;
8-бром-1-[(2R,4r,6S)-2,6-диметилтетрагидро-2H-пиран-4-ил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолина;
8-бром-1-[(-[(2R,4r,6S)-2,6-диметилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с]хинолина;
1-[(2R,4r,6S)-2,6-диметилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с]хинолин-8-карбонитрила;
8-метокси-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолина;
8-хлор-2-(1,2-оксазол-3-илметил)-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
8-хлор-2-(имидазо[2,1-b][1,3,4]тиадиазол-6-илметил)-1-[(2S,4S)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
8-хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1H-1,2,4-триазол-1-илметил)-1H-имидазо[4,5-с]хинолина;
8-бром-1-(цис-2-этилтетрагидро-2H-пиран-4-ил)-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолина;
1-[(2R,4R)-2-этилтетрагидро-2H-пиран-4-ил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолин-8-карбонитрила;
1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с][1,5]нафтиридина;
2-(имидазо[2,1-b][1,3,4]тиадиазол-6-илметил)-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
1-(2,2-диметилтетрагидро-2H-пиран-4-ил)-8-фтор-2-[(5-метоксипиридин-2-ил)метил]-1H-имидазо[4,5-с]хинолина;
2-[(2-хлоримидазо[2,1-b][1,3]тиазол-6-ил)метил]-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
1-(2,2-диметилтетрагидро-2H-пиран-4-ил)-2-(1,3-тиазол-4-илметил)-1Н-имидазо[4,5-с]хинолина;
1-(2,2-диметилтетрагидро-2H-пиран-4-ил)-2-(имидазо[2,1-b][1,3,4]тиадиазол-6-илметил)-1H-имидазо[4,5-с]хинолина;
8-фтор-1-(тетрагидро-2H-пиран-4-ил)-2-{[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]метил}-1H-имидазо[4,5-с]хинолина;
8-фтор-2-(1,2-оксазол-3-илметил)-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
8-фтор-2-(2H-индазол-2-илметил)-1-(тетрагидро-2H-пиран-4-ил)-1Н-имидазо[4,5-с]хинолина;
8-фтор-1-(тетрагидро-2H-пиран-4-ил)-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с]хинолина;
8-фтор-1-(тетрагидро-2H-пиран-4-ил)-2-{[4-(трифторметил)-1H-пиразол-1-ил]метил}-1H-имидазо[4,5-с]хинолина;
2-[(4-циклопропил-1H-1,2,3-триазол-1-ил)метил]-1-(2,2-диметилтетрагидро-2H-пиран-4-ил)-8-фтор-1H-имидазо[4,5-с]хинолина;
1-(2,2-диметилтетрагидро-2H-пиран-4-ил)-8-фтор-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с]хинолина;
1-(2,2-диметилтетрагидро-2H-пиран-4-ил)-8-фтор-2-{[4-(трифторметил)-1H-пиразол-1-ил]метил}-1H-имидазо[4,5-с]хинолина;
8-фтор-2-(имидазо[2,1-b][1,3,4]тиадиазол-6-илметил)-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
1-(2,2-диметилтетрагидро-2H-пиран-4-ил)-8-фтор-2-[(2-метилимидазо[2,1-b][1,3,4]тиадиазол-6-ил)метил]-1H-имидазо[4,5-с]хинолина;
1-(2,2-диметилтетрагидро-2H-пиран-4-ил)-8-фтор-2-(1,2-оксазол-3-илметил)-1H-имидазо[4,5-с]хинолина;
8-хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с]хинолин-4-амина;
8-фтор-2-(имидазо[2,1-b][1,3,4]тиадиазол-6-илметил)-1-(цис-2-метилтетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолина;
8-метокси-2-[(5-метил-1,2,4-оксадиазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
8-метокси-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-[(4-метил-1H-1,2,3-триазол-1-ил)метил]-1H-имидазо[4,5-с]хинолина;
8-метокси-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)-1H-имидазо[4,5-с]хинолина;
2-(имидазо[2,1-b][1,3,4]тиадиазол-6-илметил)-8-метокси-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
[цис-4-(2-метил-1H-имидазо[4,5-с]хинолин-1-ил)тетрагидро-2H-пиран-2-ил]ацетонитрила и
8-хлор-2-[(5-метил-1,3-оксазол-2-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
или его фармацевтически приемлемая соль.
16. Соединение по п. 1, выбранное из группы, состоящей из
8-хлор-2-[(5-метоксипиридин-2-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
2-[(5-метил-1,2-оксазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолин-8-карбонитрила;
8-хлор-2-[(5-метил-1,2-оксазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
8-хлор-2-[(5-метил-1,2,4-оксадиазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
8-хлор-2-(имидазо[2,1-b][1,3,4]тиадиазол-6-илметил)-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолина;
{8-хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-1H-имидазо[4,5-с]хинолин-2-ил}ацетонитрила;
8-хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-(1,3-тиазол-4-илметил)(4-2H)-1H-имидазо[4,5-c]хинолина и
8-хлор-1-[(2R,4R)-2-метилтетрагидро-2H-пиран-4-ил]-2-[(4-метил-1Н-1,2,3-триазол-1-ил)метил]-1H-имидазо[4,5-с]хинолина;
или его фармацевтически приемлемая соль.
17. Соединение по п. 1 или его фармацевтически приемлемая соль, где
Х представляет собой CR7;
Z представляет собой CR3;
каждый из R1a, R1b, R4, R5, R6 и R7 представляет собой водород; и
R3 представляет собой хлор или циано.
18. Соединение по п. 17 или его фармацевтически приемлемая соль, где
R2 представляет собой 1-метилпирролидинил или 2-метилтетрагидропиранил.
19. Соединение по п. 18 или его фармацевтически приемлемая соль, где
R1 выбран из группы, состоящей из изоксазолила, пиразолила, триазолила, оксадиазолила, тиадиазолила, пиримидинила и пиразинила; каждый из которых возможно замещен группой R8; и R8 представляет собой метил или метокси.
20. Соединение по п. 19 или его фармацевтически приемлемая соль, где
R1 выбран из группы, состоящей из метилизоксазолила, метоксипиразолила, метилтриазолила, метилоксадиазолила, метилтиадиазолила, метилпиримидинила и метилпиразинила;
R2 представляет собой (2R,4R)-2-метилтетрагидро-2H-пиран-4-ил; и
R3 представляет собой хлор.
21. Соединение по п. 19 или его фармацевтически приемлемая соль, где
R1 выбран из группы, состоящей из метилизоксазолила, метоксипиразолила, метилтриазолила, метилоксадиазолила, метилтиадиазолила, метилпиримидинила и метилпиразинила;
R2 представляет собой 1-метилпирролидинил; и
R3 представляет собой циано.
22. Соединение по п. 19, выбранное из группы, состоящей из
8-хлор-2-[(5-метил-1,2-оксазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1Н-имидазо[4,5-с]хинолина;
8-хлор-2-[(5-метил-1,2,4-оксадиазол-3-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1Н-имидазо[4,5-с]хинолина;
8-хлор-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-2-[(4-метил-1Н-1,2,3-триазол-1-ил)метил]-1Н-имидазо[4,5-с]хинолина;
8-хлор-2-[(6-метилпиримидин-4-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1Н-имидазо[4,5-с]хинолина;
8-хлор-2-[(5-метилпиразин-2-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1Н-имидазо[4,5-с]хинолина;
8-хлор-2-[(4-метокси-1Н-пиразол-1-ил)метил]-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-1Н-имидазо[4,5-с]хинолина и
8-хлор-1-[(2R,4R)-2-метилтетрагидро-2Н-пиран-4-ил]-2-[(5-метил-1,3,4-тиадиазол-2-ил)метил]-1Н-имидазо[4,5-с]хинолина;
или его фармацевтически приемлемая соль.
23. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения, обладающего активностью ингибирования обогащенной лейциновыми повторами киназы 2 (LRRK2), по любому из пп. 1-22 или его фармацевтически приемлемой соли вместе с фармацевтически приемлемым носителем.
24. Способ лечения заболевания или расстройства, выбранного из группы, состоящей из болезни Крона, болезни Паркинсона, деменции с тельцами Леви, лепры, болезни Альцгеймера и таупатии у пациента, включающий введение пациенту, нуждающемуся в лечении, терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по любому из пп. 1-22 или фармацевтической композиции по п. 23.
25. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-22 для использования в лечении заболевания или расстройства, выбранного из группы, состоящей из болезни Крона, болезни Паркинсона, деменции с тельцами Леви, лепры, болезни Альцгеймера и таупатии.
26. Фармацевтическая композиция по п. 23 для использования в лечении заболевания или расстройства, выбранного из группы, состоящей из болезни Крона, болезни Паркинсона, деменции с тельцами Леви, лепры, болезни Альцгеймера и таупатии.
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| US 4689338 А, 25.08.1987 | |||
| US 5389640 А1, 14.02.1995 | |||
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| 0 |
|
RU2221798C | |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| ИМИДАЗОХИНОЛИНОВЫЕ СОЕДИНЕНИЯ | 2005 |
|
RU2415857C2 |

