Изобретение относится к химико-фармацевтической промышленности и касается создания антитромботического средства, влияющего на регуляцию функциональной активности тромбоцитов.
Повышение функциональной активности тромбоцитов ведет к увеличению тромбогенного потенциала крови и может быть причиной тромбоэмболии сосудов головного мозга, легких, почек (Духанин А.С., Губаева Ф.Р. Фармакологическая регуляция активности тромбоцитов. Экспериментальная и клиническая фармакология, 1998, №4, с.66-71).
Показано участие тромбоцитарного звена в патогенезе заболеваний, сопровождающихся диссеминированным внутрисосудистым свертыванием крови и атеросклерозом (Панченко Е.П. Концепция атеротромбоза - основа патогенеза сердечно-сосудистых заболеваний. Основные направления антитромботической терапии. Русский медицинский. журнал, 2005, том 13, №7, с.433-438; Ишемическая болезнь сердца, Лупанов В.П. Применение ацетилсалициловой кислоты с целью вторичной профилактики коронарной болезни сердца. Русский медицинский журнал, 2005, том 13, №15, с.1053-1056, диабетической ангиопатией, Балаболкин М.И. Диабетология, М., 2000, с.672.
Исследованиями установлено, что на мембране тромбоцитов имеются три подтипа пуринорецепторов: P2X1, P2Y1 и P2Y12. P2Y1 - рецепторы играют ключевую роль в тромбообразовании и представляют собой мишень для создания препаратов для терапии ряда заболеваний, сопровождающихся увеличением тромбогенного потенцииала крови, Kunapuli S.P., Multiple P2 receptor subtypes on platelets: A new interpritation of their function. Trends Pharmacol. Sci., 1998, V.19, p 391-394.
В настоящее время в клинической практике отсутствуют препараты, действующие на P2Y1-рецепторы, однако однако используются блокаторы Р2У12-рецепторов, например тиклопидин, Регистр лекарственных средств, России, РЛС, Энциклопедия лекарств. Выпуск 15, Издательство: РЛС-2007, 2006, с.1488.
Тиклопидин имеет ряд побочных эффектов, а именно тромбо-цитопеническую пурпуру, лейкоцитоз, агранулоцитоз, Quinn M.J., Fitzgerald D.J. Ticlopidine and clopidogrel, Circulation, 1999, V.100, p.1667-1672; Matsuda N, Yoshihara A, Nishikata R, Matsuda T, Endo K, Yamamoto T.A, Case of ticlopidine-associated thrombotic thrombocytopenic purpura: special emphasis on diffusion weighted MRI, Rinsho Shinkeigaku, 2006, V.46, №10, Р.693-698, а это требует обязательного лабораторного контроля, поскольку указанные выше побочные эффекты представляют потенциальную угрозу для жизни больного.
Кроме того, возможны желудочно-кишечные кровотечения, кожная сыпь, диарея, диспептические расстройства, тошнота, гастрит, нарушения функции печени, Yim H.B., Lieu P.K., Choo P.W. Ticlopidine induced cholestatic jaundice, Singapure Med. J., 1997, V.38, p.132-133; Skurnik Y.D. et al. Ticlopidine-induced cholestatic hepatitis, Ann. Pharmacother., 2003, V.37, p.371-375.
Слабым местом препарата является также его фармакокинетика, а именно достаточно позднее развитие основного антитромбоцитарного действия, что связано с биотрансформацией исходного вещества в печени с участием цитохрома С450 до активных метаболитов, Yoneda К, Iwamura R, Kishi H, Mizukami Y, Mogami К, Kobayashi S., Identification of the active metabolite of ticlopidine from rat in vitro me-tabolites, Br. J. Pharmacol, 2004, V.142, №3, p.551-557.
Задачей изобретения является получение антитромботического средства, влияющего на регуляцию функциональной активности тромбоцитов, лишенных недостатков используемых в настоящее время препаратов.
Поставленная задача достигается тем, что антитромботическое средство, влияющее на регуляцию функциональной активности тромбоцитов, содержит антагонист пуриновых P2Y1 рецепторов в виде дигидрохлорида 2-(4-метоксифенил)-(9-морфолиноэтил)имидазо[1,2-а]бензимидазола формулы I:
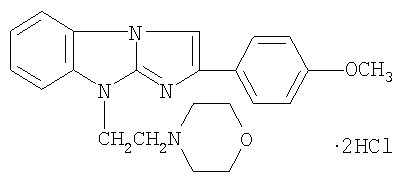
обладающий антитромботической активностью.
Вышеупомянутый антагонист пуриновых P2Y1 - рецепторов является представителем ряда соединений 2,9-дизамещенных имида-зо[1,2-а]бензимидазола, у которого была выявлена способность ингибировать ФДЭ ЦАМФ и снижать возбудимость предсердий, В.А.Анисимова, А.А.Спасов, В.А.Косолапов, А.Ф.Кучерявенко, О.В.Островский, Н.П.Ларионов, Р.Е. Либинзон. Фармакологическая активность, 2-метоксифенилзамещенных-9-диалкиламиноэтил-имидазо[1,2-а]-бензимидазолов, Химико - фарм. журнал, 2005, т.39, №9, с.26-32.
Синтез вышеупомянутого антагониста пуриновых P2Y1 рецепторов заключается во взаимодействии 1-морфолиноэтил-2-аминобензимидазола с 4-метоксифенацилбромидом и дальнейшей термической циклизации образующегося при этом бромида 1-морфолиноэтил-2-амино-3-(4-метоксифенацил)бензимидазолия в трициклическое основание I, которое переводят обычными способами в дигидрохлорид I
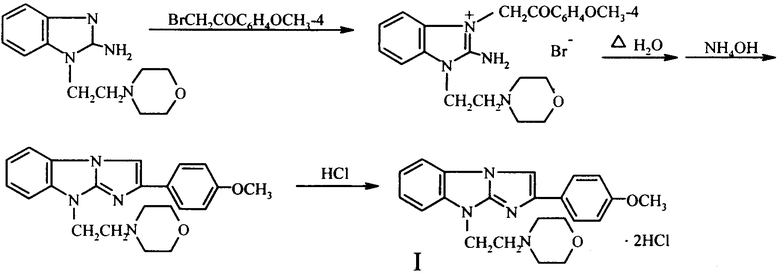
МЕТОДИКА СИНТЕЗА СОЕДИНЕНИЯ I:
Дигидрохлорид-2-(4-метоксифенил)-9-морфолино-этилимидазо[1,2-а]-бензимидазола (I). В раствор 2,46 г (10 ммоль) 1-морфоли-ноэтил-2-аминобензимидазола в 140 мл ацетона, приготовленный при нагревании до кипения, вносят 2,3 г (~10 ммоль) 4-метоксифенацилбромида, полученного бромированием 4-метоксиа-цетофенона бромом в ЕtOН или 2-РrOН, Хим.-фарм. журнал, 2005, т.39. №9, с.27.
Смесь тщательно перемешивают, полученный раствор нагревают до кипения и реакционную массу оставляют стоять при комнатной температуре на 3-4 часа или кипятят при перемешивании 1 час. Выпавший осадок бромида 1-морфолиноэтил-2-амино-3-(4-метокси-
менацил)бензимидазолия отфильтровывают, промывают ацетоном, сушат на воздухе и вводят в реакцию циклизации без дополнительной очистки. Выход 4,5-4,6 г (94,3-96,8%). Т.пл. 199-200°С (разложение).
Найдено, %: С 55,5; Н 5,8; Вг 16,5; N 12,0.
[C22H27N4O3]+Br-.
Вычислено, %: С 55,6; Н 5,7; Вг 16,8; N 11,8.
ИК-спектр, λmах, см-1 1660 (C=N), 1680 (C=O), 3205,3340 (NH2).
Кипятят 4,76 г (10 ммоль) полученной четвертичной соли в 120 мл воды до полного протекания реакции (2-3 часа, контроль - ТСХ). Раствор охлаждают, подщелачивают 22%-ным водным раствором аммиака и выделившееся трициклическое основание - 2-(4-метоксифенил)-9-морфолиноэтил)имидазо [1,2-а] бензимидазол экстрагируют хлороформом. Экстракт упаривают до небольшого объема (-15 мл) и пропускают через слой Аl2О3, элюируя продукт циклизации хлороформом. В остатке после испарения последнего из элюата получают белые кристаллы с т.пл 110°С. Их далее используют для получения соли, но в случае необходимости перекристаллизовывают из СН3СN. Выход 92,8-94,5%.
Найдено, %: С 70,0; Н 6,4; N 15,0.
C22H24N4O2.
Вычислено, %: С 70,2; Н 6,4; N14,9.
ИК-спектр, λmах, см-1:1510, 1605 (С=С), 1615 (C=N).
Раствор трициклического основания в ацетоне подкисляют конц. НСl или насыщенным раствором НСl в 2-РrОН до рН 2-3. Выпавшие белоснежные или слегка розоватые кристаллы дигидрохлорида I через час отфильтровывают, промывают ацетоном, перекристаллизовывают из 90%-ного водного этанола и сушат при 105-110°С. Т.пл. 260-261°С (разложение).
Найдено, %: С 58,9; Н 6,0; С1 15,5; N 12,7.
C22H24N4O2·2 НС1.
Вычислено, %: С 58,8; Н 5,8; С1 15,8; N12,6.
ИК-спектр, λmах, см-1: 1660 (C=N), 2360-2650 (широкая полоса N+TH).
Спектр 1H-ЯMP(HMCO-d6 - CCl4), δ, м.д.: 3,46 (ушир.с, 4Н, N(CH2)2), 3,72 (т, 2Н, CH2N), 3,85 (с, 3Н, ОСН3), 3,94 (т, 4Н, (СН2)2O), 5,23 (т, 2Н, NCH2), 7,02 (д, 2Н, 2′,6′-Н), 7,40-7,57 (дт, 2Н, 6,7-Н), 7,95 (м, 3Н, 3′,5′-Н, 5-Н), 8,13 (д, 1Н, 8-Н), 8,44 (с, 1Н, 3-Н), H+ в обмене.
Синтез соединения I можно проводить без выделения продукта взаимодействия 1-морфолиноэтил-2-аминобензимидазола с 4-метокси-фенацилбромидом. Для этого после протекания реакции кватернизации ацетон из реакционной массы упаривают досуха, к остатку приливают воду, кипятят смесь до полной циклизации бромида в производное имидазо[1,2-а]бензимидазола и дальше выделение искомого соединения I проводят, как описано выше.
ФАРМАКОЛОГИЧЕСКИЕ СВОЙСТВА СОЕДИНЕНИЯ I.
А. Р2Y1-Антагонистическая активность соединения I на тромбоцитах in vitro.
Влияние соединения I на пуриновые рецепторы тромбоцитов изучали с использованием метода малоуглового светорассеяния, Сакаев М.Р., Миндукшев И.В., Лесиовская Е.Е. и др. Оценка эффективности действия пуриновых нуклеотидов на Р2-рецепторы тромбоцитов методом малоуглового светорассеяния, Экспериментальная и клиническая фармакология, 2000, №.3, С.65-69.
Эксперименты проводили на лазерном анализаторе малоуглового светорассеяния «Лайт-скан» («Люмекс ЛТД», Россия) в солевой среде, содержащей 140 мМ NaCl; 10 мМ трис-HCl буфера; 5 мМ ЭДТА (рН 7, 8), с разбавлением плазмы, обогащенной тромбоцитами (концентрация тромбоцитов не превышала 107 клеток/мл). Для экспериментов взята плазма 6 кроликов породы «Шиншилла» массой 4-4,5 кг. В качестве индукторов активации тромбоцитов использовали динатриевую соль аденозин-5-дифосфорной кислоты (АДФ) («Реанал», Венгрия) в концентрации 70 нМ и селективный агонист P2Y1-рецепторов 2-метилтио-АТФ («Sigma», США) в концентрации 3 нМ. В качестве препарата сравнения использовали тиклопидин (Тиклид, "Sanofi-syntelabo", Франция).
Статистическую обработку результатов экспериментов производили в пакете прикладных программ «Statistika 6.0» с использованием парного критерия Стьюдента при предварительной проверке выборки на нормальность распределения.
Соединение I в концентрации 10-6 М достоверно подавляло АДФ-индуцированную активацию тромбоцитов в безкальциевой среде на -29,4±2,6%, в то время как тиклопидин не продемонстрировал ингибирующей активности.
При использовании в качестве индуктора активации тромбоцитов селективного P2Y1-агониста 2-метилтио-АТФ соединение I в концентрациях 10-4-10-5 М выражено подавляло активацию тромбоцитов (табл.1) в отличие от тиклопидина, показавшего статистически не значимый эффект.
Б. Антитромботические свойства соединения I.
Антитромботическую активность соединения на модели артериального тромбоза, индуцированного поверхностной аппликацией 50% раствора хлорида железа (III), Kurz K.D., Main B.W., Sandusky G.E. Rat model of arterial thrombosis induced by ferric chloride. Thromb. Res., 1990, V.15, P.269-280, оценивали по времени, необходимом для достижения 50%-ного уровня кровотока, времени полной окклюзии сонной артерии, а также по средней объемной и средней линейной скорости артериального кровотока.
Эффективность соединений на модели артериального тромбоза, индуцированного постоянным электрическим током (12В, 50 мА), uglielmi G. et al. Electrothrombosis of saccular aneurysms via endovascular approach. Part 1: Electrochemical basis, technique, and experimental results. J. Neurosurg., 1991, V.75, P.1-7), оценивали по интервалу времени от момента начала стимуляции до полной окклюзии каротидной артерии.
Моделирование генерализованного тромбоза (DiMinno G., Silver M.J.Mouse antithrombotic assay: a simple method for the evaluation of antithrombotic agents in vivo. Potentiation of antithrombotic activity by ethyl alcohol, J. Pharmacol. Exp.Ther., 1983, V.225, P.57-60) проводили введением смеси растворов коллагена и адреналина (0,5 мг/кг и 0,06 мг/кг, соответственно) в хвостовую вену мышей. Критерием эффективности исследуемых соединений служило количество выживших животных по сравнению с контрольной группой и наличие тромбов в сосудах легких.
Исследуемое соединение I и препарат сравнения тиклопидин (Тиклид, "Sanofi-syntelabo", Франция) вводили перорально в дозе, эквимоляльной 6 мг/кг тиклопидина, за 2 часа до моделирования тромбоза. Воспроизведение моделей артериальных тромбозов проводили под нембуталовым (35 мг/кг) наркозом с регистрацией уровня кровотока с помощью ультразвукового доплерографа «Минимакс-Доплер-К» («СП Минимакс», С.-Петербург, Россия).
Антитромботическую активность изучали на 40 половозрелых нелинейных белых крысах-самцах массой 330-440 г. Для моделирования генерализованного тромбоза использовали 35 белых нелинейных мышей-самцов массой 20-26 г.
Статистическую обработку результатов экспериментов производили в пакете прикладных программ «Statistika 6.0» с использованием критерия Манна-Уитни, ANOVA (Newman-Keuls test) и точного критерия Фишера.
Установлено, что в группе крыс, получивших соединение I, за время действия нембуталового наркоза (2,5 часа) не происходит не только полной окклюзии сосуда, но и снижения уровня кровотока до 50% по сравнению с исходными показателями (табл.2). В группе крыс, получивших тиклопидин, время достижения 50%-ного уровня кровотока составляет 29 мин, и к 42 минуте после начала воздействия повреждающего фактора наблюдается полная окклюзия (табл.2). Таким образом, соединение I проявляет выраженные антитромботические свойства и превосходит тиклопидин.
На модели артериального тромбоза, индуцированного электрическим током, соединение I, введенное перорально однократно в дозе, эквимоляльной 6 мг/кг тиклопидина, достоверно увеличивает время кровотока, превосходя при этом тиклопидин в 2,4 раза (табл.3).
При изучении генерализованного коллаген-адреналинового тромбоза соединение I и тиклопидин, введенные перорально за 2 часа до моделирования тромбоза, в разной степени уменьшали гибель животных. Выживаемость в контроле составляла 6,3%. Соединение I достоверно уменьшало гибель мышей по сравнению с контролем, при этом выживаемость составила 70,0%. В экспериментальной группе, получивших тиклопидин, выживаемость достоверно не отличалась от контрольной группы и составила 30,0%.
Таким образом, выживаемость животных в группе, получавших соединение I, в 2,3 раза превышала таковую в группе, получавших тиклопидин.
ОСТРАЯ ТОКСИЧНОСТЬ СОЕДИНЕНИЯ I.
Исследование острой токсичности проводили на белых нелинейных мышах-самцах массой 20-22 грамма при внутрибрюшинном введении. Гибель животных регистрировали в течение двух недель. Руководство по экспериментальному (доклиническому) изучению новых фармакологических веществ. Под общей редакцией члена-корреспондента РАМН, профессора Р.У.Хабриева. - 2-изд., перераб. и доп. - М.: ОАО Издательство «Медицина», 2005, с.170-204. Величину токсикологического показателя - LD50 рассчитывали по методу Личфилда-Вилкоксона. По результатам изучения острой токсичности показатель
ЛД50 при внутрибрюшинном введении для соединения I составил 282,8 мг/кг, таким образом, оно является умеренно токсичным.
# -данные достоверны по отношению к группе тиклопидина (р≤0,01)
| название | год | авторы | номер документа |
|---|---|---|---|
| ДИГИДРОХЛОРИД 1-(3-МОРФОЛИНОПРОПИЛ)-2-ФЕНИЛИМИДАЗО[1,2-a]-БЕНЗИМИДАЗОЛА, ПРОЯВЛЯЮЩИЙ СВОЙСТВА АНТАГОНИСТА ПУРИНОВЫХ P2Y-РЕЦЕПТОРОВ, АНТИАГРЕГАНТНУЮ И АНТИТРОМБОТИЧЕСКУЮ АКТИВНОСТЬ | 2008 |
|
RU2391345C2 |
| СРЕДСТВО, ПРОЯВЛЯЮЩЕЕ АНТИАГРЕГАНТНУЮ И АНТИТРОМБОГЕННУЮ АКТИВНОСТИ | 2010 |
|
RU2453312C1 |
| СРЕДСТВО, ОБЛАДАЮЩЕЕ АНТИТРОМБОГЕННОЙ АКТИВНОСТЬЮ | 2010 |
|
RU2440814C1 |
| СРЕДСТВО, ИНГИБИРУЮЩЕЕ Na+/H+-ОБМЕН, И ДИГИДРОХЛОРИД 2-(3,4-МЕТИЛЕНДИОКСИФЕНИЛ)-9-МОРФОЛИНОЭТИЛИМИДАЗО[1,2-a]БЕНЗИМИДАЗОЛА | 2013 |
|
RU2518740C1 |
| СРЕДСТВО, ОБЛАДАЮЩЕЕ КАРДИОПРОТЕКТОРНЫМ ДЕЙСТВИЕМ, И ГАЛОГЕНИДЫ 1,3-ДИЗАМЕЩЕННЫХ 2-АМИНОБЕНЗИМИДАЗОЛИЯ | 2013 |
|
RU2526902C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ АНТИТРОМБОТИЧЕСКОГО ДЕЙСТВИЯ В ТВЕРДОЙ ЛЕКАРСТВЕННОЙ ФОРМЕ В ВИДЕ ТАБЛЕТОК | 2019 |
|
RU2696869C1 |
| СРЕДСТВА, ПРОЯВЛЯЮЩИЕ СВОЙСТВА АНТАГОНИСТОВ СЕРОТОНИНОВЫХ 5-HT-РЕЦЕПТОРОВ | 2011 |
|
RU2465901C2 |
| СРЕДСТВО, ПРОЯВЛЯЮЩЕЕ СВОЙСТВА АНТАГОНИСТА СЕРОТОНИНОВЫХ 5-HT-РЕЦЕПТОРОВ | 2010 |
|
RU2438669C1 |
| ГИДРОГАЛОГЕНИДЫ 11-ФЕНОКСИЭТИЛ- И 11-БЕНЗИЛЗАМЕЩЁННЫХ 2,3,4,5-ТЕТРАГИДРО[1,3]ДИАЗЕПИНО[1,2-а]БЕНЗИМИДАЗОЛА, ОБЛАДАЮЩИЕ АНТИАГРЕГАНТНОЙ АКТИВНОСТЬЮ | 2015 |
|
RU2582618C1 |
| ДИГИДРОБРОМИД 2-(3,4-ДИГИДРОКСИФЕНИЛ)-9-ДИЭТИЛАМИНОЭТИЛИМИДАЗО[1,2-a] БЕНЗИМИДАЗОЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2008 |
|
RU2391979C2 |
Изобретение относится к области химико-фармацевтической промышленности и касается лекарственного средства, обладающего антитромботической активностью, содержащего антагонист пуриновых Р2У1-рецепторов, а именно дигидрохлорид 2-(4-метоксифенил)-(9-морфолиноэтил)имидазо[1,2-а]бензимидазола формулы I. 3 табл.
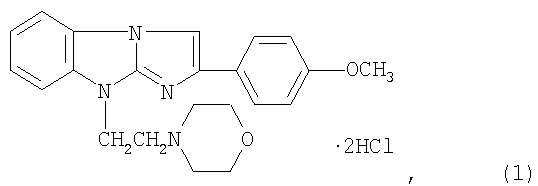
Антитромботическое средство, влияющее на регуляцию функциональной активности тромбоцитов, содержащее антагонист пуриновых P2Y1 рецепторов в виде дигидрохлорида 2-(4-метоксифенил)-(9-морфолиноэтил)имидазо[1,2-а]бензимидазола формулы I:
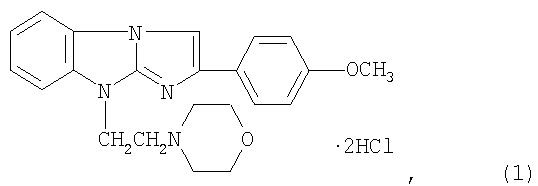
обладающего антитромботической активностью.
| RU 2005136383 А, 10.06.2007 | |||
| N-ГУАНИДИНОАЛКИЛАМИДЫ, ИХ ПОЛУЧЕНИЕ, ИХ ПРИМЕНЕНИЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ | 2000 |
|
RU2253651C2 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |

