Перекрестные ссылки на родственные заявки
Эта заявка имеет приоритет предварительной заявки США №60/56,821, поданной 16 апреля 2004 года, которая включена сюда в виде ссылки.
ПРАВИТЕЛЬСТВЕННЫЕ ПРАВА
Изобретение было сделано при поддержке правительства. Номера правительственных грантов EY l 1254 и HL 16411 национального института здоровья. Правительство имеет определенные права на изобретение.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение относится к способу модулирования васкуляризации. В частности, изобретение относится к способу модулирования васкуляризации для стимулирования или ингибирования неоваскуляризации у животных, посредством модулирования сигнального пути PAR-2.
УРОВЕНЬ ТЕХНИКИ
Ангиогенез - образование новых сосудов из предсуществующей сосудистой сети, играет критическую роль для нормального развития, регенерации тканей при ранениях и для восстановления тканей после ишемии. Неоваскуляризация при увеличении опухоли и при ишемической ретинопатии является примером ангиогенеза, лежащего в основе прогрессирования болезни (патологическая неоваскуляризация). Улучшенное понимание основных механизмов, которые регулируют патологический и физиологический ангиогенез, помогут разработать про- и анти- ангиогенезисную терапию. Тканевой фактор (TF) является инициатором протеаз системы свертывания крови, который генерирует протеолитические фрагменты, обладающие мощным регуляторным влиянием на ангиогенез. TF действует как внеклеточный ко-рецептор, который активирует и представляет протеазы свертывания крови для активации протеаз-активируемых рецепторов (PARs), посредством связанных G-белков. PARs активируются посредством уникального механизма, включающего внеклеточный протеолиз рецептора. In vitro, PAR-2 активируется комплексом TF-VIIa, также как и фактором Ха. Фактор Ха может также расщеплять PAR-I - первый обнаруженный рецептор тромбина. Фактор Ха наиболее эффективен в тройном комплексе TF-VIIa-Ха.
На основании данных in vitro, предполагают, что TF служит ко-рецептором при активации PAR, однако недостаточно ясно определена роль цитоплазматического домена TF при активации PAR in vivo. TF, экспрессируемый опухолевыми клетками, способствует развитию опухоли. Была предположена зависимость активации фактора роста эндотелиальных клеток (VEGF) от цитоплазматического домена TF, хотя полностью это не было подтверждено, которая способствует патологическому ангиогенезу. Кроме того, TF находится рядом с эндотелием при злокачественном раке груди, и было показано, что прямое ингибирование TF приводит к остановке роста опухоли и ангиогенеза. PAR-1 и PAR-2 также вовлечены в ангиогенез, но остаются редкими данные, полученные in vivo, связывающие ангиогенез с активацией PAR посредством индуцированной TF коагуляции.
Хорошо известно, что развитие опухоли связано с неоваскуляризацией. Например, ингибиторы ангиогенеза, например ингибиторы сигнального пути VEGF, как было показано, замедляют или останавливают рост опухоли. В настоящее время продолжают делать попытки найти новые физиологические пути, участвующие в неоваскуляризации опухоли, с тем чтобы найти новые мишени для ингибирования известных путей неоваскуляризации.
Возрастная дегенерация желтого пятна (ARMD) и диабетическая ретинопатия (ДР) являются основными причинами потери зрения в индустриально развитых странах, что является причиной патологической ретинальной неоваскуляризации. Поскольку сетчатка состоит из хорошо выраженных слоев нервных клеток, глиальных клеток и сосудов, относительно небольшие изменения, вызванные, например, васкулярной пролиферацией или отеком, могут привести к значительной потере зрительной функции. Наследственные заболевания, связанные с дегенерацией сетчатки, такие как пигментная дегенерация сетчатки (ПДС), также связаны с сосудистыми нарушениями, такими как сужение артериол и сосудистая атрофия. Ретролетальная фиброплазия (РФ) - заболевание, связанное с патологией сетчатки у недоношенных детей. РФ заключается в ненормальном росте кровяных сосудов в сетчатке, который начинается в течение первых нескольких дней жизни и может прогрессировать очень быстро (например, в течение нескольких недель), что приводит к потере зрения. Когда ребенок рождается недоношенным, нормальный рост сосудов может остановиться и может начаться патологический рост сосудов, который со временем может привести к образованию фиброзной ткани в сетчатке, что, в свою очередь, приводит к отслоению сетчатки, что является причиной слепоты. Несмотря на значительный прогресс в определении факторов, которые способствуют или ингибируют ангиогенез, в настоящее время не предложено ни одного способа лечения глазных неоваскулярных заболеваний.
Сейчас имеется потребность в разработке способов лечения заболеваний, включающих патологическую неоваскуляризацию, к которым относится опухолевый рост, и ишемические ретинопатии. Настоящее изобретение удовлетворяет этой потребности.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение обеспечивает способ модулирования васкуляризации в тканях млекопитающих. Способ включает управление сигнальным путем PAR, таким как сигнальный путь PAR-I или PAR-2 в ткани. Сигнальным путем можно управлять, регулируя фосфорилирование цитоплазматического домена тканевого фактора в ткани. Сигнальным путем PAR можно управлять введением ингибитора сигнального пути PAR в ткань.
Способ изобретения полезен для лечения заболеваний, характеризующихся патологической неоваскуляризацией, в особенности у человека.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фигуре 1 изображен усиленный опухолевый рост и ангиогенез у мышей TFΔCT. (а) Объем опухоли сингенной фиброкарциномы Т241 и финальный вес на 14 день были определены для мышей дикого типа (ДТ) и TFΔCT мышей (n=5, *t-критерий, p<0,05). Усиленное распространение опухоли было подтверждено в другой опухолевой модели карциномы легкого Льюиса (данные не приведены). Правая панель: плотность кровеносных сосудов в Т241 опухолях по окрашиванию CD31. (b) Ex vivo ангиогенез. Верхняя панель: изображение кусочков аорты дикого типа и TFΔCT, полученных с помощью светового микроскопа на 3 день после помещения в матригель (20х кратное увеличение). Нижняя панель: конфокальная флуоресцентная микроскопия после окрашивания, как указано (10х кратное увеличение). (с) Количество отростков из аорт дикого типа и TFΔCT на третий день (среднее ± стандартная ошибка, n=74; *t-критерий, p<0,05). (d) Экспрессия TF: полуколичественный ПЦР для TF и β-актина на четвертый день в аортах дикого типа и TFΔCT.
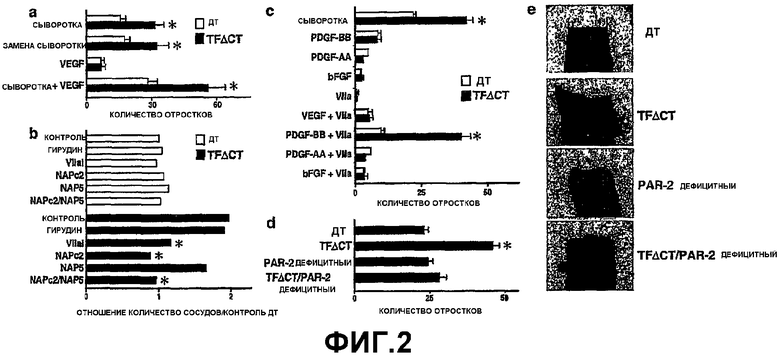
На Фигуре 2 изображен синергизм TF-VIIa и PDGF-BB при ангиогенезе. (а) Проростки в аорте дикого типа (ДТ) и TFΔCT на третий день при указанных условиях (среднее±стандартная ошибка, n=10-15, *t-критерий, p<0,05). (b) Аорты дикого типа и TFΔCT инкубировали в сыворотке с добавлением ингибиторов протеаз, как указано в течение трех дней (*значительно отличается от TFΔCT контроля; t-критерий, p<0,05). (с) Проростки на четвертый день в аортах дикого типа и TFΔCT в среде, содержащей EGM, как указано (среднее ± стандартная ошибка, n=5-19, *значительно отличается от дикого типа, t-критерий p<0,05). (d) Для усиленного образования отростков в аортах TFΔCT необходима экспрессия PAR-2: образование отростков на 4 день в аорте дикого типа, TFΔCT, PAR-2 дефицитной и TFΔCT/ PAR-2 дефицитной (среднее±стандартная ошибка, n=21-37, * значительно отличается от дикого типа, t-критерий, p<0,05). (e) Образцы кусочков аорты соответствующих генотипов (20х кратное увеличение).
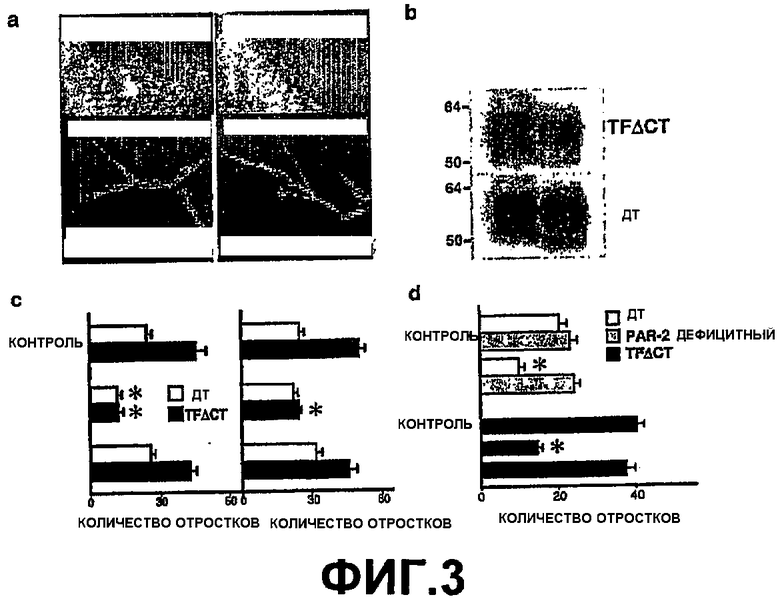
На Фигуре 3 показано, что цитоплазматический домен тканевого фактора супрессирует PAR-2-зависимый ангиогенез. (а) Верхняя панель: Флуоресцентная микроскопия на третий день кусочков TFΔCT аорты, одновременно трансдуцированных зеленым флуоресцентным белком (GFP) и TF(1-263) человека или TF(1-243) человека при высоких дозах вируса (увеличение 20х). Нижняя панель: Конфокальная флуоресцентная микроскопия проростков TFΔCT аорты, трансдуцированных TF(1-263) человека (левая) или TF(1-243) человека и GFP (правая), и затем окрашенные, как указано (увеличение 10х). (b) TF человека был осажден с помощью антител из экстрактов трансдуцированных (высокая доза) кусочков аорты, полученных с помощью детергентов на 4 день и определенных с помощью вестерн-блоттинга с помощью поликлональных антител к TF. (c) Отростки аорт дикого типа (ДТ) и TFΔCT трансдуцированных TF (1-263) человека или TF(1-243) человека при высоких (левая панель) или низких (правая панель) дозах вируса (среднее ± стандартная ошибка, n>13; *значительное отличие от контроля, t-критерий, p<0,05). (d) Для супрессии образования отростков, вызванных TF (1-263) человека, необходима экспрессия PAR-2 и внеклеточная активность TF: дикий тип, TFΔCT и PAR-2 дефицитные аорты были трансдуцированы большими дозами TF(1-263) человека и затем проинкубированы в сыворотке с (+ анти-TF) или без антител к внеклеточному домену TF человека. Количество отростков было определено на четвертый день (среднее±стандартная ошибка, n>17; * значительное отличие от контроля, t-критерий, p<0,05).
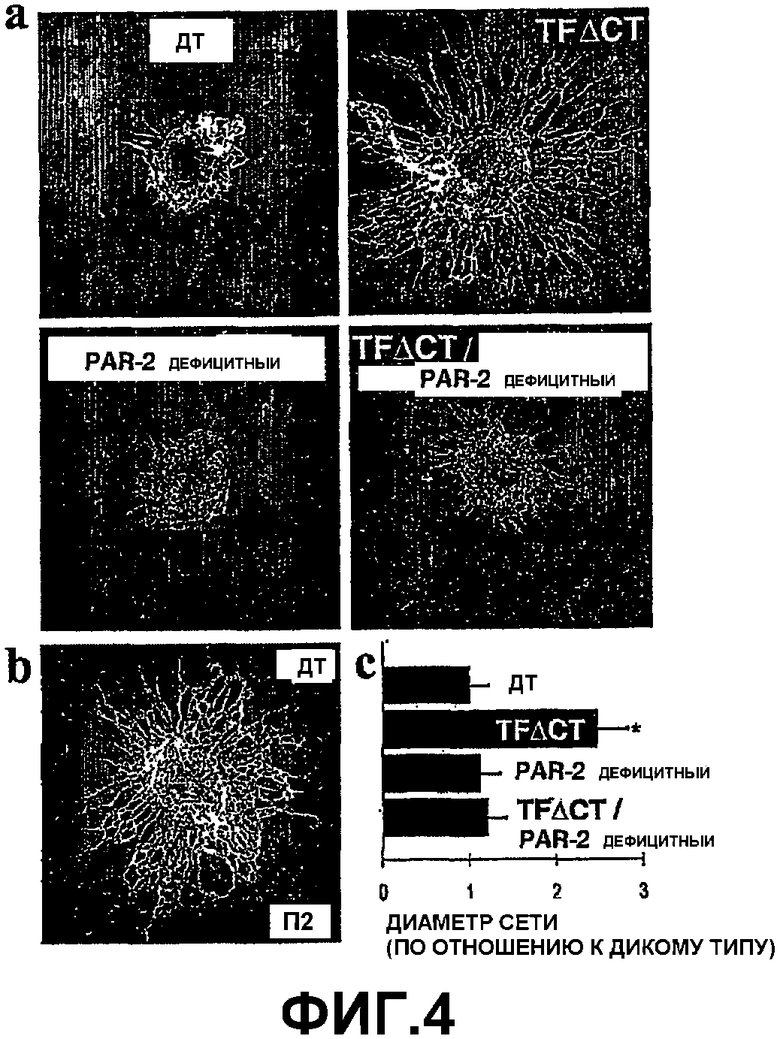
На Фигуре 4 показано ускоренное развитие ангиогенеза у мышей TFΔCT, (а) Показана сетчатка в день П0 из мышей дикого типа (ДТ), TFΔCT, PAR-2 дефицитных и TFΔCT/ PAR-2 дефицитных. (b) В качестве сравнения показана сетчатка дикого типа в день П2. Картинки были получены с помощью монтажа четырех отдельных картинок, полученных при 20х увеличении. (с) Количественная оценка среднего диаметра капиллярной сетки сетчатки в день П0 из указанных генотипов. Планки погрешностей показывают стандартную ошибку измерения.
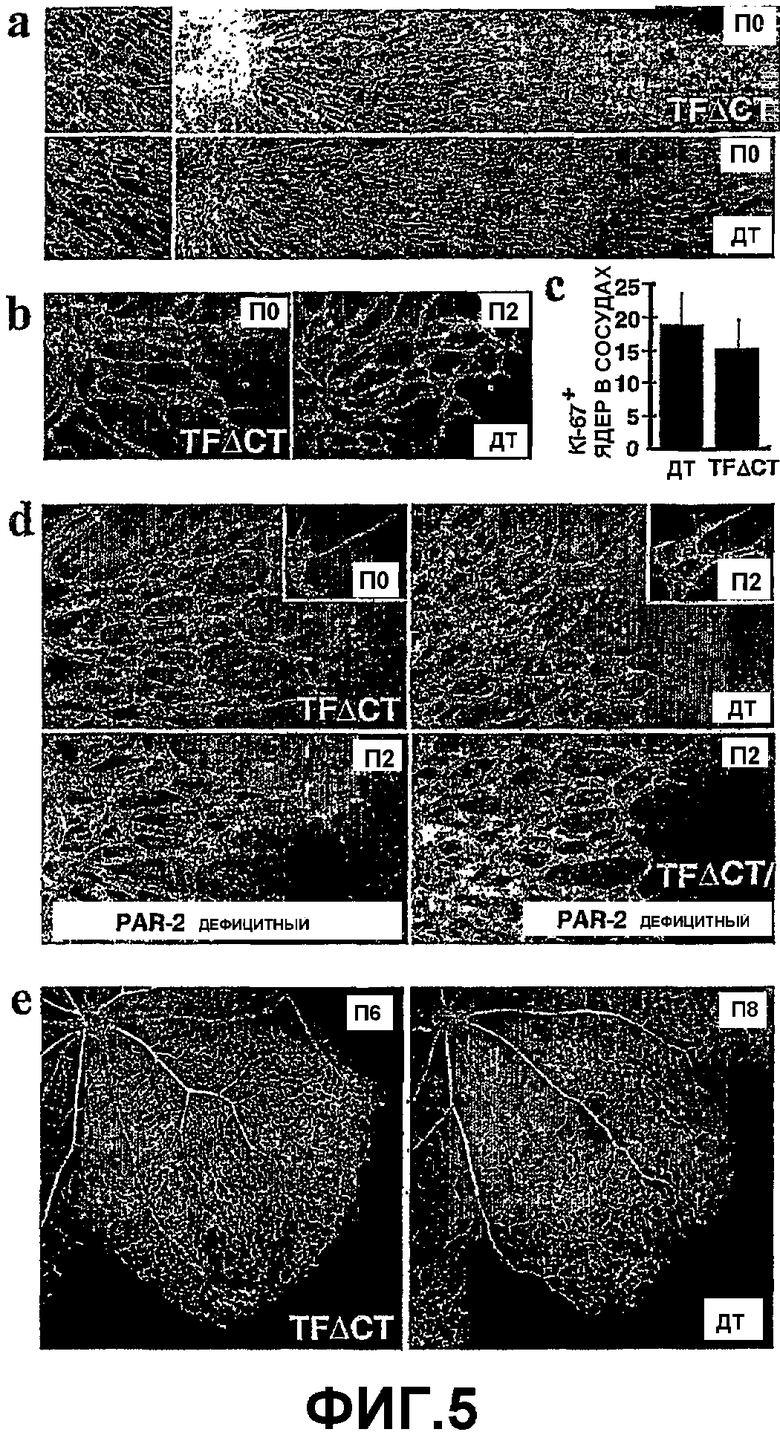
На Фигуре 5 показана нормальная морфология астроцитов и набор перицитов у мышей TFΔCT. (а) Окрашивание GFAP показало сходный рисунок астроцитов в сетчатке на день П0 дикого типа (ДТ) и TFΔCT (монтаж изображений, полученных при 20х увеличении). На левой панели изображена при большом увеличении (40х увеличение) развивающаяся кровеносная сеть (красная). (b) Окрашивание Ki-67 ядер, принадлежащих сосудам, не показало отличий в пролиферативной активности клеток сосудов между сетчатками TFΔCT на день П0 и П2 дикого типа на день П2 (20х увеличение). (с) Количественная оценка Ki-67+ ядер (планки погрешностей показывают стандартное среднее отклонение). (d) Набор перицитов (SMA) был сходен в П0 TFΔCT, и П2 для дикого типа, дефицитном по PAR-2 или дефицитном по TFΔCT/ PAR-2 (20х увеличение, вставки взяты при увеличении 60х). (е) Реконструированная архитектура поверхностной кровеносной сети была также сходна в сетчатках П6 TFΔCT и П8 для дикого типа (монтаж изображений, полученных при 10х увеличении). Во всех случаях, сосуды были окрашены изолектином из griffonia simplicifolia.
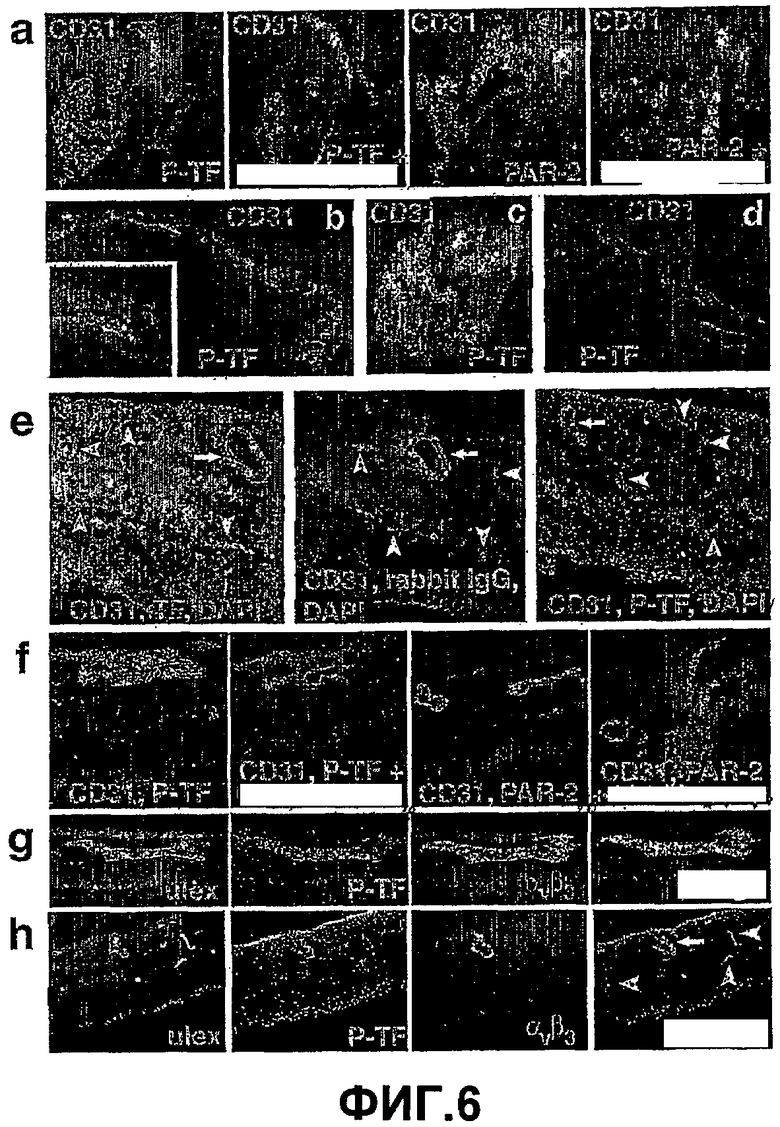
На Фигуре 6 показано фосфорилирование TF и экспрессия PAR-2 в областях неоваскуляризации в глазу. (а) Образец радужной оболочки #1, окрашенный специфическим антителом (Р-TF) к фосфорилированному Ser258 цитоплазматического домена TF или поликлональным антителом к PAR-2 (подтверждение блокировки расщепления PAR-2). Для демонстрирования специфичности было проведено также окрашивание в присутствии пептидного иммуногена (увеличение 20х). (b, c) На дополнительных независимых фрагментах радужной оболочки, полученных из пациентов с диабетом, было показано фосфорилирование TF в патологических сосудах, (b) монтаж изображений с 10х увеличением, вставка с 40х увеличением, (с) с 20х увеличением. (d) Фрагмент радужной оболочки, принадлежащий пациенту с глаукомой, на котором показано отсутствие фосфорилирования TF. (e-h) фрагмент, взятый у пациентов с диагнозом пролиферативная диабетическая ретинопатия (10х увеличение). (е) Окрашивание поликлональными антителами к внеклеточному домену TF, на фигуре 6 видна обширная экспрессия TF в сетчатке. Патологические (стрелка), но не нормальные (острие стрелки) сосуды были окрашены для выявления фосфорилированного TF (увеличение 40х). (f) Окрашивание PAR-2 наблюдалось как в самих патологических новых сосудах в сетчатке, так и около них (увеличение 40х). (g, h) Фосфорилированный TF находится в патологических новых сосудах в сетчатке, что было подтверждено окрашиванием также с антителом LM609, специфичным к интегрину αvβ3 ((g) 40х увеличение, (h) 20х увеличение).
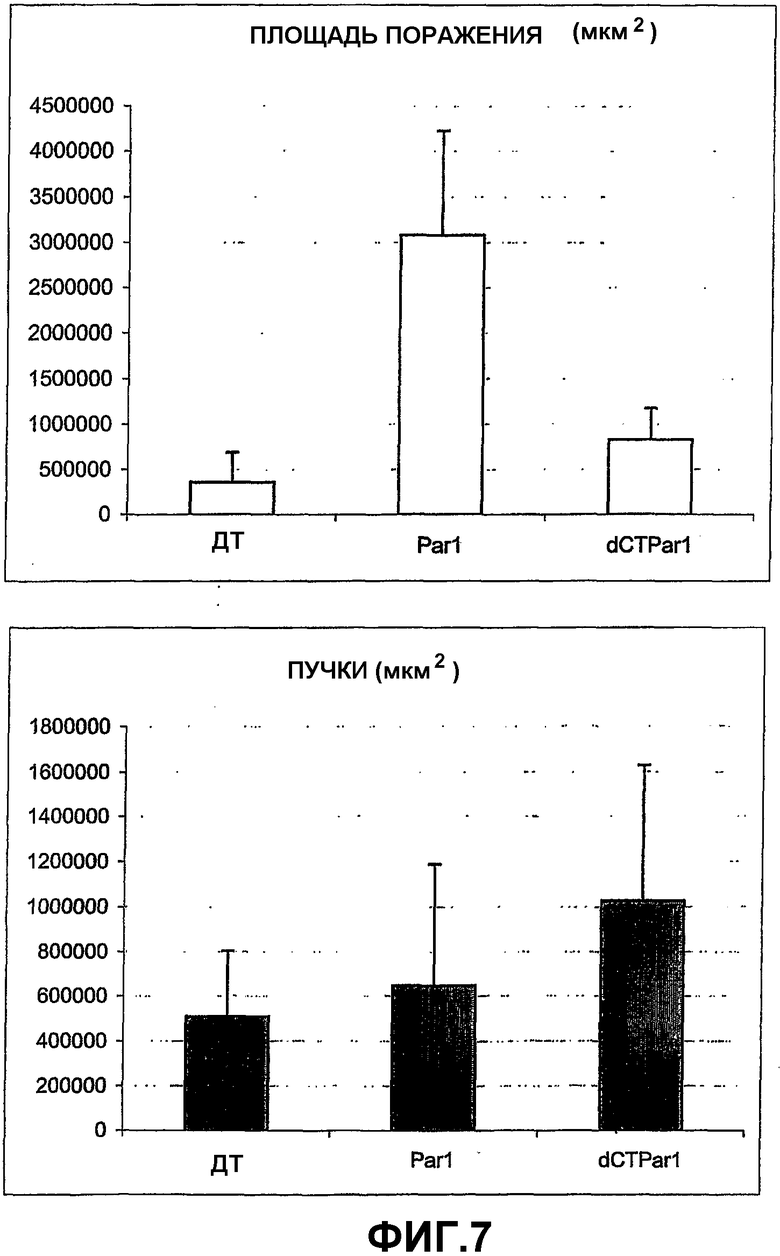
На фигуре 7 графически показана степень разрушения сосудов (верхняя панель) и образование пучков новых сосудов (нижняя панель) у неонатальных мышей, подвергнутых гипероксии (дп = мыши дикого типа; Par-1 = мыши, дефицитные по PAR-I; dCTParl = мыши, дефицитные по PAR-I и имеющие мутацию TFΔCT).
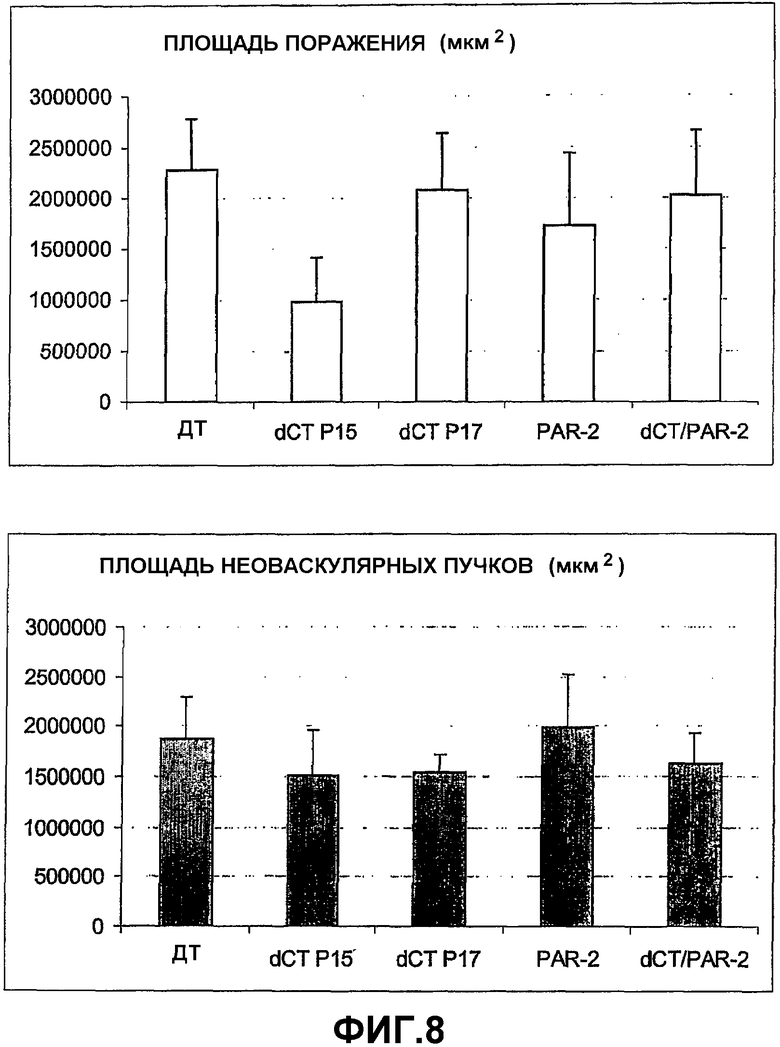
На фигуре 8 графически показана степень разрушения сосудов (верхняя панель) и образования пучков новых сосудов (нижняя панель) у неонатальных мышей, подвергнутых гипероксии (ДТ = мыши дикого типа; dCT = мыши, имеющие мутацию TFΔCT; Par-2 = мыши, дефицитные по PAR-2; dCT/PPAR-2 = мыши, дефицитные по PAR-2, и имеющие мутацию TFΔCT; П15 = постнатальный день 15; П17 = постнатальный день 17).
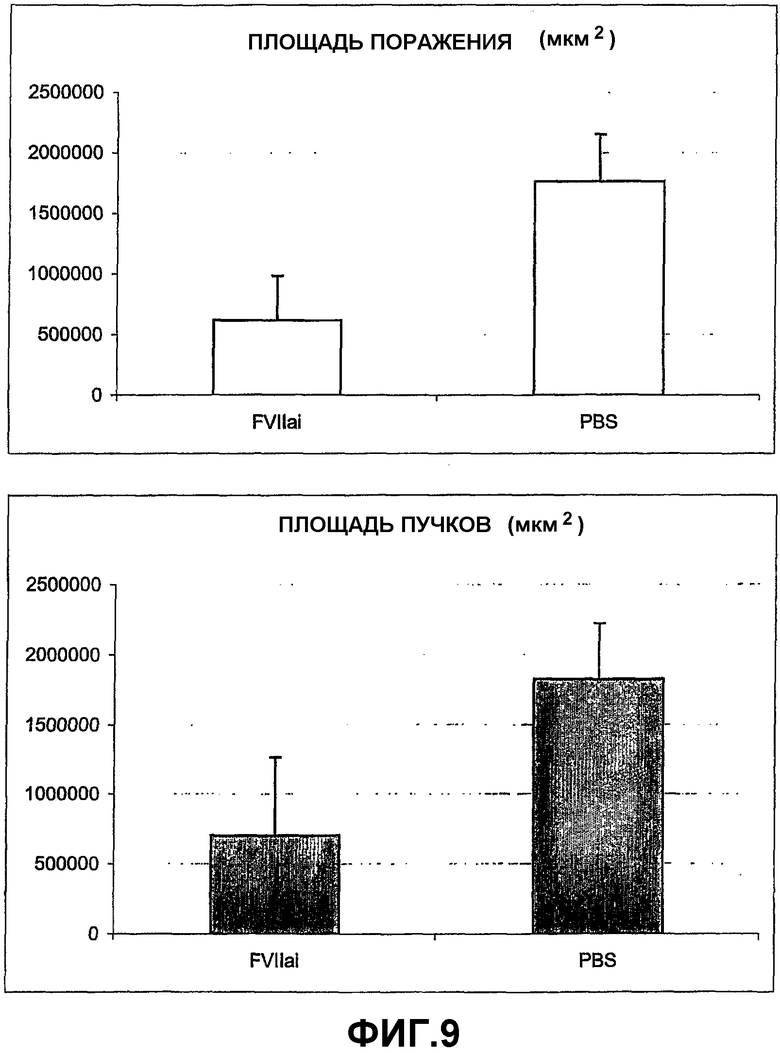
На фигуре 9 графически показана степень разрушения сосудов (верхняя панель) и образование пучка новых сосудов (нижняя панель) у неонатальных мышей дикого типа, подвергнутых гипероксии, которым был введен ингибитор сигнального пути PAR по отношению к контрольному веществу (FVIIai = мыши, которым был введен активный центр мутантного фактора VII; PBS = контрольные мыши, обработанные только фосфатным буфером).
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНОГО СПОСОБА ОСУЩЕСТВЛЕНИЯ
Как используется здесь и в приложенной формуле изобретения, термин терапевтически эффективное количество относится к ингибитору сигнального пути PAR, например сигнального пути PAR-2 или PAR-1, и включают ингибиторы TF-VIIa, β-рецепторы PDGF и ингибиторы фосфорилирования цитоплазматического домена тканевого фактора, означает количество ингибитора, которое при введении животному, страдающему от патологической неоваскуляризации, уменьшает или полностью останавливает патологическую неоваскуляризацию. Введение может быть одноразовое или многоразовое в течение определенного периода времени или неопределенного. Терапевтически эффективное количество может быть легко определено квалифицированным медицинским работником.
Сигнальный путь PAR может влиять на неоваскуляризацию в тканях млекопитающих. Фосфорилирование цитоплазматического домена тканевого фактора (например, фосфорилирование Ser258 цитоплазматического хвоста TF) стимулирует экспрессию PAR в тканях, где такое фосфорилирование происходит, что приводит к патологической неоваскуляризации. В соответствии с этим управление сигнальным путем PAR, например фосфорилированием цитоплазматического домена тканевого фактора, может быть использовано для модулирования васкуляризации (например, для усиления или ингибирования ангиогенеза).
Модулирование неоваскуляризации посредством сигнального пути PAR включает ряд факторов и пересекается с другими сигнальными путями, среди которых сигнальный путь комплекса TF-VIIa, сигнальный путь фактора Ха, сигнальный путь β рецептора фактора роста тромбоцитов (PDGF). Способ модулирования васкуляризации в тканях млекопитающих включает контроль PAR сигнального пути в тканях, предпочтительно контроль PAR-2 сигнального пути.
Лечение патологической неоваскуляризации у животных заключается во введении животным, страдающим от патологической неоваскуляризации, терапевтически эффективных количеств ингибитора PAR сигнального пути. Предпочтительно, чтобы млекопитающим был человек. Примеры предпочтительных ингибиторов PAR сигнального пути включают ингибиторы TF-VIIa, ингибиторы β рецептора PDGF и ингибиторы фосфорилирования цитоплазматического домена тканевого фактора, но ими не ограничиваются.
Один предпочтительный способ настоящего изобретения включает введение животным, страдающим от патологической неоваскуляризации, терапевтически эффективных количеств ингибитора TF-VIIa сигнального пути. Примеры предпочтительных ингибиторов TF-VIIa сигнального пути включают активный центр ингибированного VIIa (VIIai)), антикоагулянтный пептид с2 нематод (NAPc2), антитела, специфичные к фактору Vila, и антитела, специфичные к комплексу TF-VIIa, и им подобные, но этим не ограничиваются.
Другой предпочтительный вариант способа настоящего изобретения включает введение животным, страдающим от патологической неоваскуляризации, терапевтически эффективных количеств ингибитора сигнального пути β рецептора PDGF. Примеры таких ингибиторов включают антитела, специфичные к PDGF-BB, и тому подобное, но этим не ограничиваются.
Еще одним предпочтительным вариантом способа настоящего изобретения является введение животным, страдающим от патологической неоваскуляризации, терапевтически эффективных количеств ингибитора фосфорилирования цитоплазматического домена тканевого фактора.
Примеры заболеваний, при которых происходит патологическая неоваскуляризация, которые можно лечить способом настоящего изобретения, включают развитие опухоли при раковых заболеваниях (например, при раке груди, легкого и тому подобное) и ишемические ретинопатические заболевания, такие как диабетическая ретинопатия, возрастная дегенерация желтого пятна, ретинопатия у недоношенных детей и тому подобное, но ими не ограничиваются.
ПРИМЕР 1
Линии мышей и реагенты. Линия мышей TFΔCT, у которых в цитоплазматическом домене TF отсутствуют 18 концевых карбоксильных остатков, и мыши, дефицитные по PAR-2 (любезно предоставленные P. Andrade-Gordon из компании Johnson & Johnson Pharmaceutical Research & Development), были обратно скрещены с получением>90% гомогенности с C57/BL6 генетическим фоном. Мыши с двойным нокаутом TFΔCT/ PAR-2 были получены с помощью интербридинга после пяти генераций обратного скрещивания. Источники реагентов были следующие: Матригель (Beckton & Dickinson), ростовая среда для эндотелиальных клеток (EGM, Clonetics), DMEM (GIBCO), факторы роста (R&D Systems), TOPRO и изолектин из griffonia simplicifolia (Molecular Probes), антитела к CD31 (Santa Cruz) и к SMA и GFAP (SIGMA), Ki-67 (NOVO Laboratories). Козьи антитела и моноклональные антитела к TF, VIIai, гирудину, VIIa были описаны ранее Riewald, M. и Ruf W. Proc. Natl. Acad. Sci USA 98, 7742-7747 (2001). NAPc2 и NAP5 были любезно предоставлены G. Vlasuk (Corvas International). Аденовирусные конструкции TF(1-263) человека и TF(1-243) человека были описаны Dorfleutner A. и Ruf W. Blood 102, 3998-4005 (2003). Активируемый протеиназами рецептор 2 индуцируется воспалительными медиаторами в эндотелиальных клетках человека, и серологичные векторы Ad5, коэкпрессирующие GFP были получены сходным образом.
Рост опухоли. Все тесты на животных были одобрены институтом по защите животных и исполнительным комитетом Scripps Research Institute. 4×105 клеток фибросаркомы T241 были введены подкожно мышам дикого типа и TFΔCT мышам в возрасте 7-9 недель и >97% C57BL/6. На 14 день были определены объемы опухоли и конечные веса, после чего опухоли поместили в OCT. 10-мкм срезы из замороженной ткани были зафиксированы в ацетоне, окрашены на присутствие CD31, и с помощью флуоресцентной микроскопии была определена плотность сосудов/микроскопическое поле в 6-8 секциях двух опухолей, каждая из которых из мышей дикого типа и TFΔCT.
Ангиогенный анализ. Был использован адаптированный ex vivo ангиогенный анализ для крысиной модели отростков аорты, описанной Masson et al. Biol. Proced. 4, 24-31 (2002) и Nicosia et al. Lab Invest. 63,115-122 (1990). Грудные аорты мышей в возрасте 8-11 недель дикого типа, TFΔCT, PAR-2 дефицитных и TFΔCT/PAR-2 дефицитных любого пола были внесены в матригель (Matrigel) и окружены EGM, содержащей 5% сыворотки, факторы роста или ингибиторы в следующих концентрациях: VEGF, bFGF и PDGF: 20 нг/мл; гирудин: 500 нМ, VIIai: 100 нМ, NAPc2: 200 нМ, NAP5: 1 мкМ, VIla: 50 нМ. В большинстве случаев, количество отростков аорты определяли на день 3 и 4, не зная генотип. РНК из кольца аорты была выделена с помощью экстракции с тризолем (Invitrogen) по стандартной методике, расщеплена ДНКазой I, и затем проводили RT-PCR для определения β-актина и TF. Кусочки аорты были трансдуцированы аденовирусом, содержащим конструкцию полноразмерного TF (1-263) человека или обрезанного TF (1-243) человека в среде, не содержащей DMEM, в течение от 20 до 24 часов до внесения в тест на образование отростков. В тесте использовали две различные дозы вируса, которые назывались высокая доза (1,1×1010 вирусных частиц/мл) и низкая (5×109 частиц/мл). Для конфокальной микроскопии кусочки аорты с минимумом окружающего матригеля фиксировали в 4% параформальдегиде и метаноле, инкубировали с первичными и вторичными антителами (24 часа с каждым) и затем помещали в фиксирующую среду на предметном стекле (Vector laboratories). С другой стороны, срезы аорт, помещенные в OCT и полученные из замороженного состояния, были зафиксированы в ацетоне и окрашены, как описано выше.
Для оценки неонатального ангиогенеза были получены полные срезы сетчатки, и количественную оценку ангиогенеза проводили как описано Dorrell et al. Invest. Ophthalmol. Vis. Sd. 43, 3500-3510 (2002). Для соответствующего генотипа использовали следующее количество сетчаток и различные пометы: дикий тип (20 сетчаток от 6 пометов), TFΔCT (24 сетчатки от 5 пометов), PAR-2 дефицитные (10 сетчаток от 3 пометов) и TFΔCT/ PAR-2 дефицитные (16 сетчаток от 4 пометов). Препарированные сетчатки фиксировали в 4% параформальдегиде и затем в метаноле, инкубировали с первичным антителом или с флуоресцентным коньюгатом изолектина из griffonia simplicifolia в течение ночи, затем инкубировали со вторичным антителом и получали срезы. Снимки сетчатки были получены с тем же увеличением, разрешением и с теми же условиями съемки. Изображения были смонтированы как изображения одной сетчатки, и диаметр васкуляризации оценивали с использованием программы LaserPix (BioRad) (6 измерений диаметров в 1, 2, 3, 4, 5 и 6 часов + два случайных измерения диаметров). Общее количество васкулярных ядер Ki-67+ были определены фокусированием в плоскости сосуда. Фокусирование в этой плоскости позволяло исключить из подсчета пролиферирующие нейрональные клетки.
Анализ фрагментов глаза. Все тесты с использованием тканей человека были проведены в соответствии с одобренным протоколом анализа тканей человека и с разрешения информированного пациента. Образцы радужной оболочки, которые должны были взять по плану для клинического исследования, были немедленно погружены в 20% сахарозу при 4°С до получения замороженных срезов. Фрагменты сетчатки с неоваскуляризацией были получены из глазного банка в Сан Диего. Эти фрагменты глаз брались в течение 25 лет у пациентов с клиническим диагнозом диабетическая ретинопатия. После приготовления срезов сетчатку фиксировали в 4% PFA в течение ночи при 4°С, затем ее помещали в 20% сахарозу, после чего получали замороженные срезы. Замороженные срезы обрабатывали первичными антителами и затем окрашивали вторичными антителами, коньюгированными либо с alexa 488, alexa 568 или alexa 633 (Molecular Probes) для конфокальной микроскопии. Использовали мышиные антитела к CD31 (Biocare Medical, 1:50) и к интегрину αvβ3 (LM609, 1:500), агглютинин 1 из Ulex Europaeus, конъюгированный с родамином (Vector Laboratories, 1:1000), антитела кролика к внеклеточному домену TF (R4563, 25 мкг/мл), к фосфорилированному Ser258 цитоплазматического домена TF (R6936, 25 мкг/мл) и к PAR-2 (R6797, 25 мкг/мл). Пептиды, используемые как иммуногены для R6936 и R6797, были добавлены в концентрации примерно 50 мкг/мл для контроля специфичности.
Удаление цитоплазматического домена TF приводило к усилению ангиогенеза. Для оценки роли цитоплазматического домена TF в опухолевом ангиогенезе, авторы изучили рост сингенных опухолей в мышах, у которых в TF был удален цитоплазматический домен (TFΔCT) по сравнению с потомством дикого типа. (Фиг.1, панель (а)). Увеличение опухоли и конечный вес опухоли были примерно в два раза больше в TFΔCT мышах по сравнению с мышами дикого типа. Однако конечная плотность сосудов в опухолях из мышей дикого типа и TFΔCT была одинаковой (Фиг.1, панель (а)), это совпадает с тем, что увеличение опухоли следует после увеличения снабжения кровью и что раковые клетки формируют сходную неоваскулатуру в этих мышах. Однако эти данные не исключают возможность того, что TF, экспрессируемый стромальными клетками хозяина, может быть причиной усиленного ангиогенеза в TFΔCT мышах. Для прямого анализа регуляторной роли цитоплазматического домена TF в клетках сосудов при определенных условиях, авторы использовали анализ с использованием артериального кольца, который проводился в присутствии аутогенной мышиной сыворотки для стимулирования ангиогенеза. Образовавшихся микрососудов в TFΔCT мышиной аорте было в два раза больше по сравнению с аортой дикого типа (Фиг.1, панели (b, c)). Клетки в отростках были главным образом эндотелиальными, что было показано положительным окрашиванием на CD31 и негативным окрашиванием актина в клетках гладких мышц (SMA) (Фиг.1, панель (b)). Сходный уровень экспрессии TF наблюдался в отростках аорты TFΔCT и дикого типа (Фиг.1, панель (d)), что указывало на то, что отсутствие цитоплазматического хвоста TF, а не нерегулируемая экспрессия TF, является причиной усиленного образованием отростков эндотелиальными клетками в TFΔCT мышах.
Поскольку сигнал от цитоплазматического домена TF вовлечен в регуляцию экспрессии VEGF в опухолевых клетках, авторы протестировали возможность аорты дикого типа проявлять уменьшенное образование отростков, вызванное относительной недостаточностью VEGF. Сыворотка, содержащая VEGF, все равно приводила к различиям в образовании отростков аорты TFΔCT по отношению к аорте дикого типа. Однако у аорты, стимулированной VEGF в отсутствии сыворотки, формировались очень ограниченные отростки, что показывало, что сыворотка необходима для усиления ангиогенеза для TFΔCT аорт (Фиг.2, панель (а)). Образование отростков от аорты дикого типа в присутствии сыворотки TFΔCT мыши (замена сыворотки) не было повышенным, что указывает на то, что TF, экспрессируемый TFΔCT клетками сосудов, а не сывороточный фактор или повышенный уровень циркулирующего TF, способствует про-ангиогенному фенотипу (Фиг.2, панель (а).
Сигналы от TF-VIIa усиливают ангиогенез в TFΔACT аортах. Фенотип отростков TFΔCT, зависящий от сыворотки, предполагает, что генетическое удаление цитоплазматического хвоста TF может демаскировать коагуляционный фактор с про-ангиогенной активностью. Был исследован ингибиторный эффект заблокированных протеаз свертывания крови на модели образования отростков у кольца аорты (Фиг.2, панель (b)). Ингибирование тромбина с помощью гирудина, а также инактивация Ха антикоагулянтным пептидом 5 нематоды (NAP) не оказало никакого эффекта на образование отростков, что исключает влияние протеаз, участвующих в последующих этапах свертывания крови. Активный центр ингибированного VIIa (VIIai), который является высоко аффинным конкурентным антагонистом, блокирующим образование TF-VIIa комплекса, а также нематодный ингибитор NAPc2, который ингибирует TF-VIIa за счет образования экранированного TF- VIIa-Xa комплекса, изменили фенотип отростков TFΔCT, но не оказали влияние на отростки аорты дикого типа. Эти результаты показывают, что цитоплазматический домен TF негативно регулирует сигналы от TF-VIIa протеазы.
Для непосредственного анализа роли фактора VIIa ("VIIa") в ангиогенезе, сыворотка была заменена на VIIa в модели с кольцом аорты. TF-VIIa неэффективно индуцирует образование отростков у аорт как дикого типа, так и TFΔCT (Фиг.2, панель (с)). Поскольку образование эндотелиальными клетками отростков обычно зависит от сигналов ростовых факторов, авторы также исследовали образование отростков TFΔCT аортой в присутствии определенных про-ангиогенных факторов, например, VEGF, фактора роста тромбоцитов (PDGF) АА, PDGF-BB, или основного фактора роста фибробластов (bFGF). Ни один из этих факторов не обеспечил значительного усиления образования отростков, что совпадает с предыдущими данными, и про-ангиогенный фенотип TFΔCT не появлялся в присутствии любого одного из этих факторов. Однако комбинация TF-VIIa с PDGF-BB селективно восстанавливала про-ангиогенный фенотип TFΔCT, наблюдаемый в присутствии сыворотки (Фиг.2, панель (с)). Не было получено никаких доказательств аддитивного эффекта PDGF-BB и VIIa на образование отростков аортой дикого типа, как описано для миграции фибробластов. PDGF-AA является селективным агонистом α рецептора PDGF, но не может активировать β рецептор PDGF. Поскольку VIIa действительно усиливает ангиогенезис в присутствии PDGF-BB, но не PDGF-AA, сигналы TF-VIIa, по-видимому, действуют синергично с сигналами β рецептора PDGF, когда отсутствует негативный регуляторный контроль цитоплазматического домена TF.
Взаимное влияние сигнала TF-VIIa и PAR-2 регулирует ангиогенез. TF-VIIa зависимая активация PAR-2 усиливает ангиогенез у мышей, у которых отсутствует цитоплазматический домен в TF, при одновременном действии PDGF-BB. Образование отростков от кольца аорты в TFΔCT/ PAR-2 дефицитных двойных трансгенных мышах возвращалось к уровню дикого типа (Фиг.2, панель (d)), показывая, что потеря цитоплазматического домена TF приводит к PAR-2 зависимому усилению ангиогенеза. Отсутствие фенотипа в PAR-2 дефицитных аортах также указывает на то, что цитоплазматический домен TF высокоэффективен для подавления про-ангиогенного эффекта, вызванного PAR-2, что, в свою очередь, также подтверждается открытием, что ингибиторы TF (VIIai и NAPc2) не уменьшают образование отростков у аорты дикого типа (Фиг.2, панель (b)).
С целью исключить, что фенотип TFΔCT мыши не относится к сигналам цитоплазматического домена TF, либо полноразмерный TF(1-263) человека, или, в качестве контроля, TF(1-243) человека без цитоплазматического домена был реконструирован с помощью аденовирусной трансдукции в аортах дикого типа и TFΔCT. Ко-экспрессия зеленого флуоресцентного белка (GFP) и окрашивание антителами, специфичными к TF человека, показали, что миграция эндотелиальных клеток отростков в окружающий матригель была подавлена TF(1-263) человека, но не TF(1-243) человека (Фиг.3, панель (а)). Ко-локализация TF человека с CD31 также определяла эндотелиальные клетки как мишени для аденовирусной трансдукции. Уровень экспрессии TF человека определяли в экстрактах из кольца аорты с помощью вестерн-блоттинга, подтверждая одинаковый уровень экспрессии обоих форм TF (Фиг.3, панель (b)). При высокой дозе вируса, TF(1-263) человека подавляла образование отростков, как в аорте дикого типа, так и в TFΔCT (Фиг.3, панель (c), левая), тогда как при более низкой дозе вируса, образование отростков TFΔCT селективно возвращалось к уровню дикого типа (Фиг.3, панель (с), правая). Во всех случаях, обрезанный TF(1-243) человека не оказывал никакого влияния, что доказывает, что супрессия зависит от цитоплазматического домена TF (Фиг.3, панель (с)). Эти данные подтверждают идею о том, что при введении достаточной дозы, цитоплазматический домен TF может восстановить негативное регуляторное управление сигнального пути PAR-2 в ангиогенезе.
С целью еще большего понимания механизма того, как цитоплазматический домен TF подавляет PAR-2 сигналы в ангиогенезе, была исследована инверсия проангиогенного фенотипа TFΔCT аорт, для определения необходимости сигналов и сборки внеклеточных протеаз с введенным TF человека. Блокада внеклеточного домена TF(1-263) специфическими моноклональными антителами человека предотвращала реверсию фенотипа с усиленным образованием отростков у мышей TFΔCT (Фиг.3, панель (d)). Участие PAR-2 позволило задуматься над возможностью использования открытия, что высокий уровень экспрессии TF(1-263) человека подавляет образование отростков аорты дикого типа. Такие же дозы вируса не уменьшили образование отростков у PAR-2 дефицитной аорты, что доказывает, что для супрессирующей функции цитоплазматического домена TF необходима экспрессия PAR-2 (Фиг.3(d)). Суммируя все вышесказанное, можно сказать, что эти данные показывают, что отрицательное регуляторное управление ангиогенеза с помощью цитоплазматического домена TF, происходит специфично при участи PAR-2 сигнального пути.
Цитоплазматический домен TF регулирует физиологический ангиогенез. Для дальнейшего изучения роли цитоплазматического домена TF in vivo был исследован физиологический ангиогенез в неонатальной сетчатке, в результате которого формируется кровеносная сеть, происходящая из оптического диска стереотипным способом. Диаметр поверхностной кровеносной сети у неонатальных мышей TFΔCT был в два раза больше, чем у мышей дикого типа, показывая, что цитоплазматический хвост TF негативно регулирует in vivo ангиогенез во время постнатального развития (Фиг.4, панель (а)). Степень васкуляризации сетчатки TFΔCT у новорожденных была сравнима с двухдневной сетчаткой (П2) дикого типа (Фиг.4. панель (b)). В соответствии с данными по анализу кольца аорты, васкуляризация сетчатки из неонатальных PAR-2 дефицитных мышей, а также TFΔCT/ PAR-2 двойных дефицитных трансгенных мышей, была в соответствии с возрастом (Фиг.4, панель (а)). Оценка, по меньшей мере, десяти сетчаток, полученных из, по меньшей мере, трех различных беременных мышей каждого генотипа, подтвердила состояние наблюдаемого фенотипа TFΔCT мышей и его изменение при одновременной делеции PAR-2 (Фиг.4, панель (с)).
Нахождение TF в клетках сосудов в TFΔCT сетчатках было трудно определить из-за заметной экспрессии TF астроцитами, установленными клетками ЦНС, которые экспрессируют TF, а также из-за возможной экспрессии TF нижележащими нервными волокнами. Глиальный фибриллярный кислый белок (GFAP), окрашивающий астроциты, показал, что астроциты сходным образом протягиваются к периферии сетчатки новорожденных мышей дикого типа и TFΔCT, без заметного изменения рисунка окрашивания (Фиг.5 панель (а)). Таким образом, формирование сосудов прямо следует за усиленной развивающейся миграцией астроцитов в TFΔCT сетчатках. Апоптоз кровеносных сосудов не часто наблюдается у мышей дикого типа на этой стадии развития, и защита от апоптоза не является вероятной причиной усиленного ангиогенеза у мышей TFΔCT. Усиленное развитие сосудов может быть результатом повышенной скорости клеточной пролиферации, но количество пролиферирующих клеток сосудов на основании окрашивания Ki-67 было примерно одинаковым в сетчатках новорожденных TFΔCT мышей и дикого типа на П2 (Фиг.5, панель (а), (b)). Сеть TFΔCT сетчаток на П0 кажется более обширной по сравнению с сетчатками дикого типа на П2 (Фиг.4, панель (а), (b)). Это отражает повышенную миграцию эндотелиальных клеток, совпадающую с направляющей функцией PAR-2 для расположения МАР киназного пути на лидирующем кончике мигрирующей клетки. TF экпрессируется в ангиогенных эндотелиальных клетках, связанных со злокачественных раком груди. Исследования in vitro показали прямое влияние PDGF-BB на главную миграцию эндотелиальных клеток и на образование узлов/сосудов посредством активации рецептора-β PDGF, который можно обнаружить на эндотелиальных клетках капилляров in vivo.
PDGF-BB сигнальный путь также важен для привлечения и распространения популяций париетальных клеток/перицитов, которые стабилизируют и регулируют изменение архитектуры кровеносных сосудов. Более того, полное удаление гена TF приводит к неправильной перестройке сосудов эмбриональной кровеносной сети в желточном мешке, что связано с пониженным количеством привлеченных перицитов. Тесная ассоциация между эндотелиальными и париетальными клетками во время ангиогенеза позволяет сделать предположение о различии между аутокринным эффектом PDGF-BB на эндотелиальные клетки и вторичным, паракринным эффектом на используемые париетальные клетки. Используя окрашивание SMA в качестве маркера, специфичного к перицитам, сходный рисунок окрашивания наблюдался в кровеносной сети сетчатки из новорожденных мышей TFΔCT и П2 дикого типа (Фиг.5, панель (d)). Окрашивание перицитов в каждом случае доходило до кончиков отростков (Фиг.5, панель (d), вставка). Кровеносная сеть П2 PAR-2 дефицитных или TFΔCT/PAR-2 дефицитных мышей была неотличима от П2 для мышей дикого типа, за исключением возможности, что неправильное развитие сосудов в PAR-2 дефицитных мышах было незаметным в начальные периоды времени. Перициты играют важную роль в перестройке и развитии сосудистой сети сетчатки. Одинаково увеличенная поверхностная кровеносная сеть сетчаток П6 TFΔCT и П8 дикого типа также показала сравнимую плотность капиллярной сети, распределение артерий и вен и рисунок окрашивания SMA (Фиг.5, панель (е)). Эти сходства на поздних стадиях васкуляризации сетчатки говорят против измененной функции перицитов. Усиленное развитие сосудов в TFΔCT сетчатках продолжается, по меньшей мере, до дня П6, во время которого наблюдалось прорастание несозревших эндотелиальных клеток в более глубокие слои сетчатки. В совокупности, эти данные согласуются с фенотипом усиленной миграции эндотелиальных клеток при развитии поверхностной сосудистой сетки, но не с анормальным использованием перицитов в TFΔCT мышах.
Фосфорилирование цитоплазматического домена TF при неоваскулярных заболеваниях глаз. Для того чтобы проверить, происходит ли фосфорилирование TF и в других случаях патологического ангиогенеза, были проанализированы образцы неоваскуляризированной радужной оболочки, взятые у пациентов с диабетом.
Как правило, цитоплазматический домен TF в эндотелиальных клетках не фосфорилирован. Фосфорилирование может привести к негативным регуляторным эффектам цитоплазматического домена TF и, таким образом, способствовать патологическому ангиогенезу. В самом деле, окрашивание антителами, которые специфично узнают фосфорилированный Ser258 цитоплазматического домена TF, показало, что фосфорилирование цитоплазматического домена TF в образцах, взятых у шести различных пациентов, происходит только в местах неоваскуляризации (Фиг.6, панели, (а, b, c)). Фосфорилирование в этих патологических сосудах ко-локализовано с экспрессией PAR-2 (Фиг.6, панель (а)), подтверждая роль неконтролируемой работы PAR-2 во время патологической неоваскуляризации. Важно заметить, что фосфорилированный TF и окрашивание PAR-2 не наблюдались в контрольном образце радужной оболочки, взятой у пациента с глаукомой, у которого в истории болезни не было диабета или патологической неоваскуляризации (Фиг.6, панель (d)).
Фосфорилирование TF и активация PAR-2 также наблюдались специфично в новых сосудах в образцах сетчатки, взятых у пациентов с диабетической ретинопатией. Окрашивание антителом к внеклеточному домену TF показало повышенную экспрессию TF в глиальных и нейрональных типах клеток, зрелых сосудах (Фиг.6, панель (е), белая стрелка) и в местах неоваскуляризации (Фиг.6, панель (е), белая стрелка). Однако фосфорилирование TF наблюдалось только в увеличенных патологических сосудах (Фиг.6, панель (е)). Окрашивание фосфорилированного TF в сосудах было невозможно из-за конкуренции с антигенным пептидом (Фиг.6, панель (а, f)). Неспецифическое точечное окрашивание в результате частичного конкурирования с иммуногеном иногда наблюдалось на внутренних и наружных ограничивающих мембранах, областях, известных за их неспецифическое окрашивание различными антителами в образцах сетчатки.
Фосфорилирование TF не наблюдалось в нормальных, зрелых сосудах сетчатки, что подтверждает специфическую роль фосфорилирования TF для патологической неоваскуляризации. В серийных срезах хорошо выраженная экспрессия PAR-2 специфично наблюдалась только в тех сосудах, где наблюдалось фосфорилирование TF (Фиг.6, панель (f)). Для подтверждения того, что фосфорилирование TF было специфичным для новых сосудов, авторы окрасили в сетчатке, полученной из диабетического больного, хорошо известный маркер сосудистой пролиферации αvβ3 интегрин (Фиг.6, панель (g, h)). Фосфорилирование TF точно совпадало с αvβ3 положительными сосудами, тогда как нормальные микрососуды в сетчатке не окрашивались ни тем, ни другим (Фиг.6, панели (g, h)).
ПРИМЕР 2
Этот пример демонстрирует роль р53 в передаче сигнала тканевого фактора. Сетчатка мышей TFΔCT и двойных мутантов TFΔCT/p53 была исследована в постнатальный день 0 (П0) и в постнатальный день 6 (П6). Фенотип сетчатки неонатальных мышей (TFΔCT), содержащих делецию в цитоплазматическом хвосте тканевого фактора, который проявляет повышенную васкуляризацию сетчатки во время развития, был противоположным, чем у TFΔCT/p53 двойных мутантных мышей, что указывает на то, что р53, который первоначально был открыт как белок, супрессирующий опухоль, взаимодействует с сигналами TF.
ПРИМЕР 3
Этот пример демонстрирует влияние гипероксии на TFΔCT, TFΔCT/PAR-2 и TFΔCT/PAR-1 мышей. Роль цитоплазматического хвоста тканевого фактора и активируемых протеазами рецепторов (PAR) 1 и 2 в патологическом ангиогенезе была исследована на мышиной модели ретинопатии, индуцированной кислородом (OIR). Неонатальные мыши дикого типа (ДТ), TFΔCT, PAR-2 дефицитные и PAR-1 дефицитные (любезно предоставленные Johnson & Johnson Pharmaceutical Research & Development), а также TFΔCT/PAR-2 дефицитные и TFΔCT/PAR-1 дефицитные двойные мутанты были подвергнуты гипероксии (75% кислорода), начиная с дня П7 в течение 5 дней. Поскольку в TFΔCT наблюдалась повышенная скорость васкуляризации в сетчатке, мыши были подвергнуты гипероксии на день П5, когда васкуляризация сетчатки сравнима с васкуляризацией дикого типа на день П7. На день П12 и П17 (сразу после и 5 дней спустя после снятия гипероксии, соответственно), сетчатка была препарирована, зафиксирована в 4% PFA и проинкубирована с флуоресцентным коньюгатом изолектина из Griffonia simplicifolia. Были получены картинки сетчатки с использованием конфокальной микроскопии и количественно были оценены разрушения и образования пучков новых сосудов.
Сразу после воздействия гипероксии, степень разрушения сосудов была примерно одинаковой у всех мышей. На фигуре 7 показаны графические сравнения TFΔCT мышей и TFΔCT/PAR-1 дефицитных двойных мутантов с мышами дикого типа. На верхней панели на фигуре 7 показаны сетчатки мышей дикого типа и двойных мутантов, которые имели сходный уровень разрушения сосудов, тогда как в PAR-1 дефицитных мышах, реваскуляризация разрушенных областей была значительно замедленна по сравнению с мышами дикого типа. В TFΔCT/PAR-1 дефицитных двойных мутантах эта задержка реваскуляризации, как видно из образования пучков новых сосудов (Фигура 7, нижняя панель), была частично меньше.
На фигуре 8 графически сравниваются TFΔCT мыши и TFΔCT/PAR-2 дефицитные двойные мутанты с мышами дикого типа. На верхней панели фигуры 8 показано, что на день П17, в TFΔCT мышах наблюдается значительно меньшее разрушение сосудов сетчатки по сравнению с мышами дикого типа, что говорит о том, что потеря цитоплазматического хвоста TF приводит к повышенной реваскуляризации разрушенных областей. TFΔCT/PAR-2 дефицитные двойные мутанты возвращались к TFΔCT фенотипу, демонстрируя, что PAR-2 сигналы регулируются цитоплазматическим хвостом тканевого фактора в патологическом ангиогенезе. Не наблюдалось значительных изменений в степени разрушения в PAR-2 нокаутах. В противоположность изменениям, наблюдаемым при реваскуляризации разрушенных областей, не наблюдалось значительных отличий в образовании пучков новых сосудов у любой трансгенной мыши, по сравнению с мышами дикого типа на день П17 (Фигура 8, нижняя панель).
ПРИМЕР 4
Этот пример демонстрирует влияние инъекции ингибитора активации PAR (активный центр ингибированного фактора VII (FVIIai), полученный по методу, описанному Dickinson и Ruf, J. Biological Chem., 1997; 272:19875-19879)) на мышах в модели OIR. Для дальнейшего изучения роли активации TF на мышиной модели OIR, рекомбинантный активный центр мутированного ингибитора фактора VII (FVIIai), который обладает высокой аффинностью к TF по сравнению с природным фактором VII, был введен в стекловидное тело сразу после прекращения гипероксии. В противоположные контрольные глаза мышей, которым был введен FVIIai, был введен PBS в качестве контроля. Сетчатку анализировали на четвертый день после инъекции. Инъекция FVIIai усилила реваскуляризацию разрушенных областей (Фигура 9, верхняя панель), тогда как образование пучков новых сосудов было меньше (Фигура 9, нижняя панель).
ОБСУЖДЕНИЕ
Ангиогенез является важным компонентом патологии, наблюдаемой при раке, неоваскулярных заболеваниях глаз и артритах, где преобладает активация коагуляции. Вообще-то коагуляция может опосредованно способствовать ангиогенезу многими путями, которые включают образование промежуточного обогащенного фибриллами внеклеточного матрикса, освобождение про- и анти- ангиогенных факторов из активированных тромбоцитов, и активацию тромбина через PAR-I эндотелиальных клеток. Приведенные данные показывают нам новый и неожиданный взгляд на то, как активация коагуляции регулирует ангиогенез, показывая нам, что активация PAR-2
жестко контролируется цитоплазматическим доменом TF. Генетическая делеция цитоплазматического домена TF приводит к усиленному патологическому и физиологическому ангиогенезу. Таким образом, утрата негативного регуляторного контроля цитоплазматическим доменом TF является новым путем, посредством которого проангиогенная активация PAR-2 может быть включена.
Тогда как PAR-I конститутивно экпрессируется в эндотелиальных клетках, PAR-2 специфически активируется при стимуляции цитокинов воспаления, которые также индуцируют TF. Однако экспрессия TF синергично усиливается одновременной активацией VEGF в эндотелиальных клетках. Экспрессия TF и PAR-2 и функционирование TF-PAR-2 сигнального пути, таким образом, зависит от доступности как ангиогенных факторов роста, так и цитокинов воспаления. Продукция цитокинов воспаления активированными моноцитами/макрофагами признается важной для ангиогенеза и дополнительного роста сосудов. Этот адаптивный процесс сходен с процессом при заживлении ран, который обычно связан с активностью врожденного иммунитета для удаления патогенов из поврежденных тканей. Усиленный ангиогенез при заживлении ран при одновременном воспалении может быть физиологической функцией TF-PAR-2 сигнального пути и, таким образом, объясняет эволюционную консервативность структуры цитоплазматического домена и регуляторных элементов у позвоночных.
Пути физиологического ответа зачастую приводят к патологии, при отсутствии негативного регуляторного контроля. В эндотелиальных клетках цитоплазматический домен TF является мишенью для посттрансляционной модификации, заключающейся в фосфорилировании серина, через PKCα-зависимый путь. TF первоначально не фосфорилирован, и присоединение пальмитиновой кислоты подавляет индуцированное агонистами фосфорилирование. Кроме того, активация PAR-2, но не PAR-1 приводит к фосфорилированию цитоплазматического домена TF в эндотелиальных клетках. Таким образом, отсутствие присоединения пальмитиновой кислоты совместно с активацией PAR-2 определяет степень фосфорилирования цитоплазматического домена TF. Эта концепция подтверждается данными, полученными in vivo, на тканях глаза диабетических больных; удивительная одновременная локализация активированной PAR-2 с фосфорилированным TF наблюдалась только в новых сосудах. Таким образом, фосфорилирование TF является возможным механизмом, который выключает негативную регуляторную функцию и способствует патологическому PAR-2 - зависимому ангиогенезу.
TF-PAR-2 активация взаимно усиливается только PDGF-BB, но не VEGF, bFGF или PDGF-AA в TFΔCT аортах. PDGF-BB может быть легко получен, либо при высвобождении активированными тромбоцитами при условии местной коагуляции или синтезом из отростков эндотелиальных клеток. Тогда как направленная на VEGF анти-ангиогенная терапия бывает достаточно эффективной при некоторых заболеваниях, дополнительные выгоды можно получить из комбинационной терапии молекулами, для которых мишенями являются альтернативные и кооперативные пути. Например, ингибирование рецепторов PDGF обладает большим синергичным эффектом в комбинации с подходом, направленным на VEGF. Поскольку сигнальный путь PDGF-BB является критическим для стабилизации привлечения перицитов и архитектуры зрелых сосудов, общая блокада PDGF рецепторов, очевидно, является лимитирующей. В самом деле, пониженная плотность перицитов в сосудах в результате специфичного для эндотелия уменьшения PDGF-BB является причиной микроваскулярной ангиопатии у мышей.
Многочисленные вариации и модификации способов осуществления, описанные выше, могут быть эффективными и находятся в духе и в границах элементов новизны изобретения. Например, ишемию можно лечить системным или местным введением пациентам, которым необходимо такое лечение, терапевтически эффективного количества TF, имеющего фосфорилированный цитоплазматический домен. Никакие ограничения по отношению к особенностям проиллюстрированных здесь способов осуществления не имеются в виду и не подразумеваются.

Изобретение относится к медицине, в частности к онкологии, и касается модулирования процесса васкуляризации. Для этого вводят эффективное количество ингибитора сигнального пути TF-VIIa. В качестве такого ингибитора используют активный центр ингибированного фактора VIIa (VIIai) или антитело, специфичное к фактору VIIa. Способ обеспечивает ингибирование патологической васкуляризации, в том числе в тканях злокачественных опухолей. 3 з.п. ф-лы, 9 ил.
1. Способ ингибирования васкуляризации в тканях млекопитающих, который включает ингибирование сигнального пути рецептора, активируемого протеазами (PAR), в указанных тканях путем введения млекопитающему, страдающему от патологической неоваскуляризации, эффективного количества ингибитора сигнального путем TF-VIIa, выбранного из группы, состоящей из активного центра ингибированного фактора VIIa (VIIai) и антитела, специфичного к фактору VIIa.
2. Способ по п.1, где заболеванием является рак, включающий прогрессирование опухоли, или ишемическое ретинопатическое заболевание.
3. Способ по п.1, где млекопитающим является человек.
4. Способ по п.1, где ингибитор сигнального пути TF-VIIa представляет собой антитело, специфичное к фактору VIIa.
| HEMBROUGH T.A | |||
| et al | |||
| "Tissue Factor/Factor VIIa inhibitors block angiogenesis and tumor growth through a nonhemostatic mechanism" | |||
| Cancer Recaerch, 2003, June 1; 63:2997-3000/ | |||
| RU 99118499 A, 20.09.2001 | |||
| RU 2002102602 A, 10.10.2003 | |||
| US 5506134 A, 09.04.1996 | |||
| JAMES N.J | |||
| et al | |||
| "Inhibition of tissue factor activity reduces the density of cellular |

