Настоящее изобретение относится к способам диагностики или прогноза болезни Альцгеймера. Оно относится также к наборам для диагностики или прогноза болезни Альцгеймера.
Болезнь Альцгеймера представляет собой нейродегенеративное заболевание, которое поражает значительную часть населения пожилого возраста. Эта болезнь характеризуется, в клиническом плане, потерей познавательных функций и, в невропатологическом плане, наличием в головном мозге внутриклеточных нейрофибриллярных отложений и внеклеточных отложений β-амилоидного пептида (Аβ), образующего амилоидные бляшки. Амилоидные бляшки в большинстве своем образованы пептидами Аβ из 40 или 42 аминокислот, которые генерируются в результате протеолитического процесса из белка-предшественника β-амилоидного пептида (АРР). Внеклеточные отложения Аβ являются специфическими для болезни Альцгеймера. Они представляют собой раннюю и неизменную характеристику всех форм болезни Альцгеймера, включая семейные формы.
Семейные формы болезни проявляются относительно рано (в возрасте от 40 до 60 лет). Они, по-видимому, возникают, по меньшей мере частично, вследствие мутаций в гене АРР и в генах презенилина-1 (PS1) и презенилина-2 (PS2). Мутации в этих трех генах индуцируют изменения в протеолизе АРР, приводящие к сверхпродуцированию Аβ и к раннему появлению патологии и симптомов, которые подобны таковым спорадических форм болезни Альцгеймера.
Связь между холестерином и болезнью Альцгеймера также была установлена в ходе недавних эпидемиологических исследований, на основании результатов биохимических исследований и клеточной биологии (см. обзор Hartmann T., TINS, 24, 45-48 (2001)). Повышенное содержание холестерина у взрослого, а также повышенное артериальное кровяное давление значительно повышают риск болезни Альцгеймера (Kivipelto и др., Br. Med. J., 322, 1447 (2001)).
Напротив, очень уменьшенный риск отмечается у людей, подвергаемых лечению с помощью гипохолестеринемических агентов типа статинов (Wolozin и др., Arch. Neurol., 57, 1439 (2000); Jick и др., Lancet, 356, 1627 (2000)).
Недавно была установлена имеющаяся, по-видимому, молекулярная связь. In vitro и in vivo, повышенное содержание холестерина увеличивает продуцирование пептида А-β и ускоряет появление амилоидных бляшек (Sparks и др., Exp. Neurol., 126, 88-94 (1994); Refolo и др., Neurobiol. Dis., 7, 321-331 (2000); Puglielli и др., Nat. Cell Biol., 3, 905 (2001); Shie и др., Neuroreport, 13, 455 (2002)), тогда как ингибиторы пути синтеза холестерина их уменьшают (Simons и др., PNAS USA, 95, 6460-6464 (1998); Fassbender и др., PNAS USA, 98, 5856 (2001); Refolo и др., Neurobiol. Dis., 8, 890-899 (2001)).
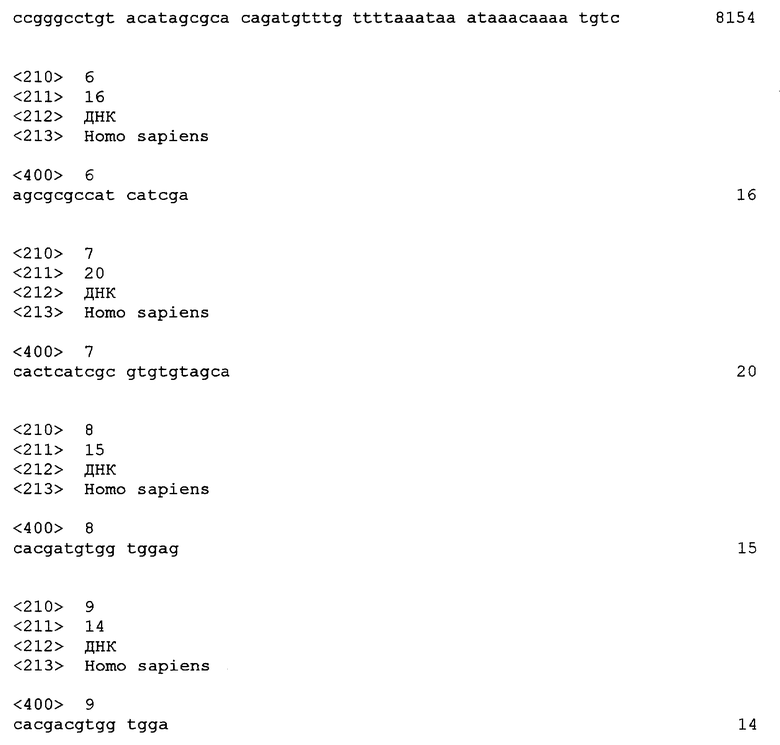
Несмотря на значительные подвижки в этой области, медицинский персонал все еще сталкивается с отсутствием молекул, действительно эффективных против болезни Альцгеймера. Одна из причин этого отсутствия заключается в трудности нахождения мишеней, позволяющих осуществлять эффективный скрининг молекул, способных оказывать терапевтическое действие против этой болезни.
С другой стороны, раннее обнаружение этой болезни является определяющим, чтобы проводить лечение до того, как появляются первые симптомы, в высокой степени вызывающие потерю трудоспособности у больных.
Кроме того, вследствие разнообразных форм этой болезни необходимо целенаправленно проводить лечение в зависимости, с одной стороны, от особенностей пациента и, с другой стороны, от молекулы, которая может быть использована для лечения. Этот фармакогеномный или фармакогенетический подход приобретает все более и более важное значение.
Белок АВСА2 представляет собой белок, относящийся к многочисленному семейству переносчиков холестерина типа АВС (АТФ-связывающий кластер). Этот переносчик специфически экспрессируется в головном мозге.
Клонирование гена АВСА2 описано в 2000 г. Zhao и др. (Biochem. J., 350, 865-872). Последовательность, кроме того, составила объект заявки РСТ WO-01/14414, поданной фирмой ACTIVEPASS PHARMACEUTICALS. В этой заявке сформулированы различные гипотезы в отношении роли АВСА2, но ни один результат не получен в пользу их поддержания.
Уже идентифицированы различные полиморфизмы гена АВСА2. Их последовательности, в частности, доступны из базы данных dbSNP NCBI. Среди различных полиморфизмов, перечисленных в этой базе данных, описан полиморфизм rs908832.
Этот полиморфизм характеризуется содержащимся в значительном количестве у людей индоевропейской расы аллелем, который представляет собой гуанин, локализованный в положении 348 последовательности SEQ ID №2, и содержащимся в незначительном количестве у людей индоевропейской расы аллелем, который представляет собой аденин, локализованный в положении 348 последовательности SEQ ID №1.
Альтернативно, этот мононуклеотидный полиморфизм представлен последовательностями SEQ ID №3 и SEQ ID №4, частично комплементарными соответственно последовательностям SEQ ID №1 и SEQ ID №2. Этот полиморфизм в таком случае характеризуется содержащимся в значительном количестве у людей индоевропейской расы аллелем, который представляет собой цитозин, локализованный в положении 201 последовательности SEQ ID №4, и содержащимся в незначительном количестве у людей индоевропейской расы аллелем, который представляет собой тимидин, локализованный в положении 201 последовательности SEQ ID №3.
Этот полиморфизм является несмысловым, то есть он не изменяет выраженной последовательности белка. Два аллеля полиморфизма составляют часть кодонов, которые кодируют аспарагиновую кислоту. Этот кодирующий полиморфизм локализован на экзоне 14 транскрипта, имеющего последовательность SEQ ID №5, в положении 2185.
Заявитель неожиданно обнаружил, что лица, обладающие содержащимся в незначительном количестве аллелем полиморфизма rs908832 гена АВСА2, имеют повышенный риск преждевременного развития болезни Альцгеймера.
Это открытие особенно важно, так как оно впервые дает, по данным заявителя, доказательство связи между белком АВСА2 и, в частности, полиморфизмом гена, кодирующим этот белок, и патологическим состоянием, таким как болезнь Альцгеймера.
Оно позволяет подтверждать и/или прогнозировать тяжесть поражения у пациентов, в случае которых болезнь Альцгеймера уже была диагностирована, а также вероятную эффективность предусматриваемых лечений, или еще прогнозировать риск появления болезни у субъектов, не обладающих симптомами болезни Альцгеймера, включая индивиды, относящиеся к пациентам, диагностика которых была подтверждена.
Оно позволяет, кроме того, подтверждать преобладающую функциональную роль АВСА2 в физиопатологии болезни Альцгеймера, оправдывающую осуществление тестов скрининга молекул, способных оказывать стимулирующее или ингибирующее воздействие на активность АВСА2, с целью терапевтического применения против болезни Альцгеймера.
Первым объектом настоящего изобретения, таким образом, является способ диагностики или прогноза болезни Альцгеймера у пациента. Вышеуказанный способ включает по меньшей мере одну стадию обнаружения аллелей полиморфизма гена АВСА2. Преимущественно, таким полиморфизмом является полиморфизм, принимающий активное участие в болезни Альцгеймера. Он предпочтительно представляет собой полиморфизм rs908832, но он может быть любым другим полиморфизмом без генетической связи с полиморфизмом rs908832.
Соответствующими лицами, например, могут быть:
- люди, которые не имеют симптомов болезни Альцгеймера;
- люди, у которых уже обнаружен риск развития болезни Альцгеймера, но которые еще не имеют симптомов болезни; и
- люди, которые предварительно диагностированы как пораженные болезнью Альцгеймера и для которых желательно подтверждение диагностики.
Стадию или стадии обнаружения, одновременные или нет, наличия или отсутствия содержащегося в незначительном количестве аллеля полиморфизма rs908832 осуществляют прямо или непрямо с помощью любого соответствующего способа, исходя из биологических образцов.
Настоящее изобретение, следовательно, также относится к способу скрининга биологических образцов, взятых на анализ у лиц, в частности, лиц, не имеющих симптомов болезни Альцгеймера, для выявления лиц, которые в высокой степени способны подвергаться развитию болезни Альцгеймера. Такой скрининг включает поиск, проводимый одновременно или нет, в вышеуказанных биологических образцах наличия содержащегося в незначительном количестве аллеля полиморфизма rs908832.
Вышеуказанные образцы, содержащие идентифицируемые ДНК или белки, могут быть различных происхождений. Речь может идти, например, об образцах крови, спермы, волос (с их корнями) или о любом другом образце, включающем ядросодержащие клетки. Предпочтительно, анализируемые биологические образцы представляют собой образцы крови. В этом случае идентифицируемую ДНК выделяют из лейкоцитов.
В смысле настоящего изобретения, под термином «биологический образец» понимают образцы, прямо происходящие от лица, для которого желательна диагностика или прогноз без другого превращения, или образцы, подвергаемые одной или нескольким стадиям получения, чтобы сохранить из них только одну фракцию, пригодную для стадий обнаружения, как, например, сырой клеточный экстракт.
Способы, позволяющие обнаруживать или идентифицировать наличие или отсутствие ДНК, несущей содержащийся в незначительном количестве аллель полиморфизма rs908832, могут быть комбинациями способов полимеразной цепной реакции (“PCR”), гибридизации, саузерн-блоттинга, рестрикции нуклеазами, анализа полиморфизма длин рестрикционных фрагментов (“RFLP”) и/или прямого секвенирования продуктов PCR. Все эти способы известны специалисту в данной области.
Как правило, все способы идентификации ДНК, несущей содержащийся в незначительном количестве аллель полиморфизма rs908832, которые могут быть использованы в рамках настоящего изобретения, включают предварительную стадию сбора биологического образца или биологических образцов, содержащих идентифицируемую ДНК, и стадию экстракции геномной ДНК стандартными способами, хорошо известными специалисту в данной области, например, согласно способу Smith и др. (The Lancet, 339, 1375-1377 (1992)).
Такой способ индентификации может включать:
а) экстракцию ДНК вышеуказанного лица;
b) амплификацию вышеуказанной выделенной ДНК с помощью праймеров, способных амплифицировать последовательности, соответствующие каждому из аллелей полиморфизма rs908832 гена АВСА2; и
с) обнаружение по меньшей мере одного из аллелей полиморфизма rs908832 гена АВСА2 в амплифицированной ДНК.
Согласно одному предпочтительному способу осуществления используют способ PCR. Таким образом, стадия b) преимущественно включает стадию полимеразной цепной реакции. Этот способ состоит с самого начала в синтезе олигонуклеотидов, комплементарных последовательности областей, которые ограничивают амплифицируемый сегмент ДНК (праймеры). Эти олигонуклетиды служат праймерами ДНК-полимеразы. Затем, осуществляют стадии тепловой денатурации (92-95°С) для разделения двух нитей ДНК, гибридизации с двумя специфическими праймерами за счет снижения температуры (50-55°С) и удлинения праймеров с помощью ДНК-полимеразы при температуре 70-72°.
Такие праймеры, позволяющие осуществлять амплификацию каждого из аллелей полиморфизма rs908832 гена АВСА2, и, в частности, содержащегося в незначительном количестве аллеля, а также их комплементарные последовательности составляют другие объекты настоящей заявки. Преимущественно, они имеют соответствующие последовательности, такие, что число оснований между их соответствующими сайтами гибридизации в молекуле ДНК составляет от 25 пар оснований до 2500 пар оснований и, предпочтительно, от 100 пар оснований до 500 пар оснований.
Преимущественно, такие праймеры находятся между примерно 15-30 смежными нуклеотидами последовательности SEQ ID №3 или SEQ ID №4. Предпочтительно речь идет о паре праймеров, имеющих последовательности SEQ ID №6 и SEQ ID №7.
Согласно предпочтительному варианту осуществления, объектом настоящей заявки является способ, отличающийся тем, что амплифицированную ДНК, несущую содержащийся в незначительном количестве аллель полиморфизма rs908832, отличают от амплифицированной ДНК, не несущей этого аллеля, путем специфической гибридизации двух зондов, каждый из которых является специфическим для одной из двух форм полиморфизма. Эти зонды, также как их комплементарные последовательности, составляют другие объекты настоящей заявки.
Эти зонды преимущественно состоят из соответственно примерно 12-17 смежных нуклеотидов последовательности SEQ ID №3 или SEQ ID №4. Предпочтительно, они имеют соответственно последовательности SEQ ID №8 и SEQ ID №9.
Амплифицированную ДНК, несущую содержащийся в незначительном количестве аллель полиморфизма rs908832, также можно отличать от амплифицированной ДНК, не несущей этого аллеля, с помощью способа, в случае которого используют активность 5'-нуклеазы ДНК-полимеразы I (TaqMan).
Согласно предпочтительному способу осуществления, объектом настоящей заявки является способ, отличающийся тем, что амплифицированную ДНК, несущую полиморфизм rs908832, отличают от амплифицированной ДНК, не несущей этого полиморфизма, путем анализа полиморфизма длин рестрикционных фрагментов (RFLP). Предпочтительно, рестрикционные фрагменты получают путем расщепления амплифицированной ДНК с помощью фермента рестрикции до миграции в геле агарозы, переноса на мембрану методом саузерн-блоттинга и гибридизации.
Для достижения сразу гораздо большей чувствительности и наилучшей специфичности, также можно осуществлять две последовательные PCR, используя две разные пары праймеров (“внутригенная PCR”): первую пару внешних праймеров, которая позволяет получать фрагмент амплифицированной ДНК, как в классической PCR, и вторую пару внутренних праймеров для амплификации фрагмента ДНК, полученного путем первой PCR.
Для обнаружения генетического отпечатка желательной области, фрагменты ДНК, полученные путем PCR, также могут быть разделены путем электрофореза по их размеру и визуализированы благодаря ВЕТ (этидиумбромид) и ультрафиолетовым лучам.
Особенно представляющая интерес возможность в настоящем случае заключается в осуществлении PCR при использовании первого праймера, имеющего на своем 3'-конце мутированную нуклеотидную последовательность, и второго праймера, имеющего на своем 3'-конце исходную нуклеотидную последовательность. Различие температуры денатурации в этих двух случаях и, следовательно, эффективности амплификации позволяет отличать мутантную ДНК от исходной ДНК.
Согласно другой альтернативе, амплифицированные фрагменты ДНК прямо идентифицируют путем переноса через точечные отверстия в матрице (метод дот-блоттинга), который состоит в помещении образца фрагментов ДНК, полученных путем PCR, на найлоновый фильтр, в осуществлении денатурации фрагментов ДНК, их гибридизации со специфическим радиоактивным зондом и промывке для удаления избытка незафиксированного радиоактивного продукта с последующим снятием авторадиограммы. Обнаружение также может быть осуществлено другими способами благодаря использованию специфических зондов, включающих маркер, другой, чем радиоактивный, например, краситель или еще флуоресцентный маркер.
Согласно другой альтернативе, также можно обнаруживать наличие или отсутствие мутаций ДНК, включающих полиморфизм rs908832, путем прямого секвенирования всей или части амплифицированных фрагментов ДНК. Такой способ состоит в обнаружении нуклеотидной последовательности на уровне полиморфизма rs908832. Секвенирование может быть осуществлено любым способом, известным специалисту в данной области, например, методом Sanger или еще методом Maxam и Gilbert.
Согласно следующей альтернативе, обнаружение наличия или отсутствия ДНК, несущей полиморфизм rs908832, может быть осуществлено, используя метод переноса по Саузерну (саузерн-блоттинг), который состоит в осуществлении электрофореза фрагментов ДНК, полученных после воздействия одного или нескольких ферментов рестрикции. Гель затем денатурируют и осуществляют перенос на найлоновую мембрану. Эта мембрана предназначена для осуществления гибридизации со специфическим зондом. После промывки для удаления избытка незафиксированного радиоактивного продукта на мембрану наносят пленку. Таким образом могут быть выявлены одна или несколько полос, соответствующих фрагментам ДНК, распознаваемым зондом.
Также можно использовать способ RFLP, ассоциированный с методом саузерн-блоттинга и/или PCR, для обнаружения наличия или отсутствия полиморфизма rs908832. RFLP позволяет сравнивать ДНК различных индивидов и исследовать, проявились ли точечные мутации, указывая на наличие или отсутствие сайтов рестрикции. Две ДНК с идентичной последовательностью, обработанные ферментами рестрикции, дают идентичные фрагменты и получаемые с этими фрагментами саузерн-блоты, следовательно, будут идентичными. Напротив, если сайт рестрикции исчез или же появился вследствие мутации, фрагменты не будут иметь идентичные размеры, что будет видимым на авторадиограммах. И это точно также, если новый сайт рестрикции появился вследствие мутации.
Еще можно обнаруживать полиморфизм rs908832 гена АВСА2 путем определения нуклеотида в положении 2185 транскрипта, кодирующего белок АВСА2.
Все способы, описанные выше, хорошо известны специалисту в данной области. Может быть использован любой другой известный способ, также приспособленный для обнаружения наличия или отсутствия полиморфизма rs908832. На этом основании можно назвать, например, методы “лигазной цепной реакции”, “амплификации с замещением цепи”, “базирующейся на транскрипции амплификации” и т.д. Предпочтительно, вышеуказанные стадии обнаружения осуществляют путем PCR, RFLP, саузерн-блоттинга и/или путем комбинации этих способов согласно методам, описанным в литературе (см., например: Gough и др., Nature, 347, 773 (1990); Kagimoto и др., J. Biol. Chem., 265, 17209 (1990); Wolf и др., The Lancet, 336, 1452 (1990); Hayashi и др., Nucleic Acids Res., 19, 4797 (1991); Daly и др., Pharmacogenetics, 1, 33 (1991); Spurr и др., Methods Enzymol., 206, 149 (1991); Armstrong и др., The Lancet, 339, 1017 (1992); Kurth и др., Am. J. of Med. Genet., 48, 166 (1993); McCann и др., J. Neurol. Sci., 153, 50 (1997); Stroombergen и др., Hum and Exper. Toxicol., 18, 141 (1999)).
Все эти способы обнаружения особенно пригодны, так как они представляют собой основу, позволяющую обнаруживать, может ли быть то или иное лицо поражено болезнью Альцгеймера или же оно обладает повышенным риском развития болезни Альцгеймера.
Таким образом, другой объект настоящего изобретения относится к способу анализа биологических образцов, взятых у лица, который состоит в:
а) определении генотипа гена АВСА2 вышеуказанного субъекта; и
b) конвертировании полученных в п. а) данных для прогнозирования риска развития болезни Альцгеймера у вышеуказанного субъекта и эффективности терапевтических обработок, предусматриваемых для этого заболевания.
Согласно другому аспекту, настоящее изобретение относится также к наборам или комплектам для диагностики или прогноза болезни Альцгеймера у лица.
Такие наборы могут находиться в форме упаковки с отделениями для помещения различных емкостей, таких как, например, ампулы или пробирки. Каждая из этих емкостей включает различные компоненты, необходимые для осуществления обнаружения наличия или отсутствия ДНК, несущей полиморфизм rs908832.
Вышеуказанные компоненты, позволяющие осуществлять реакцию или реакции обнаружения, выбирают среди вышеуказанных. Речь может идти, например, о:
- паре праймеров, гибридизирующихся с определенной областью гена АВСА2, и, в случае необходимости, средствах, необходимых для осуществления реакции амплификации; или
- олигонуклеотидных зондах, в случае необходимости, иммобилизированных на носителе и включающих обнаруживаемый маркер, и, в случае необходимости, реагентах, необходимых для осуществления реакции гибридизации.
Другим объектом настоящего изобретения является способ лечения болезни Альцгеймера, отличающийся тем, что он включает:
- по меньшей мере одну стадию обнаружения наличия содержащегося в незначительном количестве аллеля полиморфизма rs908832 у лица; и
- введение соединения или смеси соединений, известных своей активностью против болезни Альцгеймера, лицу, имеющему содержащийся в незначительном количестве аллель полиморфизма rs908832.
Предпочтительно, болезнью является болезнь Альцгеймера раннего типа.
Следующим объектом настоящего изобретения является способ селекции соединения, предназначенного для введения индивиду с заболеванием, ассоциированным с содержащимся в незначительном количестве аллелем полиморфизма rs908832 гена АВСА2, отличающийся тем, что он включает:
а. по меньшей мере одну стадию обнаружения наличия содержащегося в незначительном количестве аллеля полиморфизма rs908832 гена АВСА2 в биологическом образце вышеуказанного индивида, и
b. селекцию соответствующего соединения, если имеется вышеуказанный аллель.
Другим объектом настоящего изобретения является способ селекции соединения, предназначенного для введения индивиду с болезнью Альцгеймера, отличающийся тем, что он включает:
а. по меньшей мере одну стадию обнаружения наличия содержащегося в незначительном количестве аллеля полиморфизма rs908832 гена АВСА2 в биологическом образце вышеуказанного индивида, и
b. селекцию соответствующего соединения, если имеется вышеуказанный аллель.
Настоящая заявка, кроме того, относится к применению соединения или смеси соединений, известных своей активностью против болезни Альцгеймера, для получения лекарственного средства в целях лечения пациента, пораженного болезнью Альцгеймера, у которого до лечения обнаружено наличие содержащегося в незначительном количестве аллеля полиморфизма rs908832.
Вышеуказанными соединениями, известными своей активностью против болезни Альцгеймера, могут быть, например, ингибиторы ацетилхолинэстеразы (AchEIs, такие как донепецил (арисепт), галантамин (реминил) или экселон (ривастигмин)), антагонисты рецептора канала NMDA (такие как мемантин (эбикса)), ингибиторы продуцирования амилоидного пептида, такие как BMS 299897, или будущие агенты-модуляторы активности АВСА2.
Соединения ассоциации могут быть введены перорально, парентерально, трансдермально или ректально, либо одновременно, либо раздельно, либо распределенно во времени.
Соединения используют для приготовления лекарственных средств в виде фармацевтических композиций, содержащих ассоциацию одного или нескольких соединений, таких как указанные выше, с фармацевтически приемлемым наполнителем.
В качестве твердых композиций для перорального введения могут быть использованы таблетки, пилюли, порошки (желатиновые капсулы, крахмальные облатки) или гранулы. В этих композициях действующие начала смешаны, в токе аргона, с одним или несколькими инертными разбавителями, такими как крахмал, целлюлоза, сахароза, лактоза или диоксид кремния. Эти композиции также могут включать вещества, другие, чем разбавители, например, одну или несколько смазок, таких как стеарат магния или тальк, краситель, защитное покрытие (драже) или лак.
В качестве жидких композиций для перорального введения можно использовать фармацевтически приемлемые растворы, суспензии, эмульсии, сиропы и эликсиры, содержащие инертные разбавители, такие как вода, этанол, глицерин, растительные масла или парафиновое масло. Эти композиции могут включать вещества, другие, чем разбавители, например, смачиватели, подсластители, загустители, ароматизаторы или стабилизаторы.
Стерильными композициями для парентерального введения могут быть, предпочтительно, водные или неводные растворы, суспензии или эмульсии. В качестве растворителя или наполнителя можно использовать воду, пропиленгликоль, полиэтиленгликоль, растительные масла, в частности, оливковое масло, сложные органические эфиры для инъекций, например, этилолеат, или другие подходящие органические растворители. Эти композиции также могут содержать добавки, в частности, смачиватели, изотонизирующие средства, эмульгаторы, диспергаторы и стабилизаторы. Стерилизацию можно осуществлять несколькими способами, например путем асептизирующей фильтрации, за счет включения в композицию стерилизующих агентов, путем облучения или путем нагревания. Они также могут быть получены в виде твердых стерильных композиций, которые могут быть растворены в момент употребления в стерильной воде или любой другой стерильной инъецируемой среде.
Композициями для ректального введения являются суппозитории или ректальные капсулы, которые содержат, кроме активного продукта, эксципиенты, такие как масло какао, полусинтетические глицериды или полиэтиленгликоли.
Фармацевтические композиции, включающие ассоциацию, такую как указанная выше, обычно содержат 0,1-500 мг соединения.
Дозы зависят от искомого эффекта, продолжительности лечения и используемого пути введения; они обычно составляют 0,1-500 мг соединения в сутки перорально для взрослого.
Как правило, врач определяет соответствующую дозировку в зависимости от возраста, массы и всех других факторов, присущих подвергаемому лечению пациенту.
Настоящее изобретение относится, кроме того, к трансгенному животному, в геном которого встроена по меньшей мере одна последовательность экзогенной ДНК, несущая содержащийся в незначительном количестве аллель полиморфизма rs908832, так, что функция гена АВСА2 является модифицированной.
Под трансгенным животным понимают любое животное, исключая человека, имеющее искусственно модифицированный геном. Модификация генома может происходить в результате альтерации или изменения одного или нескольких генов путем инсерции (“knock-in”) или делеции (“knock-out”) (инактивация/модификация генов путем гомологичной рекомбинации) или за счет сверхпродуцирования человеческого гена под контролем специфических промоторов нейронных (таких как ThyI, PDGF или прион) или глиальных (как GFAP) клеточных типов в случае мыши. Эта модификация может быть вызвана воздействием классических модифицирующих или мутагенных факторов или по меньшей мере может быть осуществлена путем стабильной инсерции полигенного экспрессирующего кластера, позволяющего осуществлять экспрессию гибридного гена. В частности, можно, например, поступать согласно способам, идентичным или аналогичным таковым, описанным в Международных заявках WO 01/02552 или еще WO 01/13176.
Модификация генома может также происходить в результате инсерции гена (генов) или замены гена (генов) в его (их) исходной или мутированной форме.
Модификации преимущественно осуществляют в репродуктивных исходных клетках.
В настоящее время, модификация генома при использовании технологии инсерции (“knock-in”) и делеции (“knock-out”) в случае мыши в качестве модели ограничена тем, что доступны единственные исходные клетки (так называемые клетки “ES”), обладающие способностью колонизировать зародышевую линию, происходящую из эмбриона мыши. Когда клеточные линии ES будут установлены для других видов, эти технологии могут быть легко применены специалистом к этим другим видам для генерирования моделей КО и/или KI. Кроме того, подходы, основанные на использовании олигонуклеотидов (ДНК, РНК или гибридов), индивидуальных или связанных с ферментами, модифицирующими ДНК/РНК, могут быть использованы для введения определенной модификации/мутации в предопределенный локус в геноме. Точно также облучения или химические мутагены, которые индуцируют алеаторные модификации в геноме, могут быть использованы, если они комбинированы с эффективным набором биологических маркеров (фенотип) и с процессом позиционного клонирования в большом объеме.
Наиболее прямым подходом к модификации генома лабораторных животных (мыши, крысы, коровы, свиньи, овцы...), однако, является алеаторная интеграция трансгенов путем микроинъекции линеаризированной ДНК в один или два пронуклеуса оплодотворенных ооцитов на стадии одной клетки (предпочтительно, для избежания генерации химерных животных, хотя инъекция на стадии двух или более клеток также может быть осуществлена). Как правило, трансген состоит из двух частей: элементы регуляции, которые осуществляют пространственно-временной контроль в отношении экспрессии РНК, кодируемой ДНК, и расположенная рядом ДНК (кДНК или геномный фрагмент). Эти два элемента (элемент регуляции и ДНК, кодирующая желательный белок) могут быть гомологичны или же гетерологичны геному-мишени. Представляющих интерес трансгенных животных, как правило, выбирают среди млекопитающих, исключая человека. Речь может идти, например, о мышиных, а именно о мышах, крысах и морских свинках, кроликах, кошках, собаках, овцах или еще быках. Предпочтительно речь идет о мышах, крысах или кроликах, получаемых классическими способами трансгенеза. Другими объектами настоящего изобретения являются исходные клеточные линии и клеточные линии, образующиеся путем дифференцировки из этих исходных клеточных линий, в геном которых встроена по меньшей мере одна последовательность геномной экзогенной ДНК, которая несет содержащийся в незначительном количестве аллель полиморфизма rs908832.
Вкратце, получение трансгенных животных, исходных клеточных линий и клеточных линий, образующихся путем дифференцировки, согласно изобретению заключается в способе осуществления генерации трансгенных животных за счет инсерции путем гомологичной рекомбинации геномной экзогенной ДНК, кодирующей белок АВСА2, в соответствующий ген животного (инсерцию трансгена целенаправленно осуществляют непосредственно после промотора гена животного так, чтобы обеспечить функциональный профиль экспрессии трансгена и предотвратить экспрессию эндогенного гена животного: “элиминируют”) или же за счет инсерции специфической мутации согласно настоящему изобретению, соответствующей изоформе человеческого гена АВСА2, в эндогенный ген животного (модификацию осуществляют путем гомологичной рекомбинации в исходных клетках: “встраивают”) или же путем сверхпродуцирования гена АВСА2, несущего содержащийся в незначительном количестве аллель полиморфизма rs908832.
В случае мышей эти животные могут быть преимущественно скрещены с трансгенными мышами, несущими человеческий ген АРР с мутациями типа Альцгеймера и которые обеспечивают амилоидные бляшки. Таким образом полученные “двойные” трансгенные животные репродуцируют генотип, наблюдаемый у пациентов, пораженных болезнью Альцгеймера или обладающих риском этой болезни. Контроль генотипа полиморфизма rs908832 таким образом полученного новорожденного животного может быть осуществлен при использовании уже описанных выше способов, в частности, при использовании реакции амплификации (PCR) и/или саузерн-блоттинга.
Такие трансгенные животные особенно интересны, так как они представляют собой предпочтительную модель для понимания болезни Альцгеймера, которая очень точно воспроизводит характеристики болезни Альцгеймера. По сравнению с известными моделями, эта модель позволяет, в частности, выявить соединения, особенно применимые для лечения болезни Альцгеймера, особенно такой, как описанная у человека. Этими соединениями могут быть химические молекулы, пептидные или белковые молекулы, антитела, химерные молекулы, а также антисмысловые ДНК или рибозимы.
Таким образом, выявленные соединения могут быть использованы в качестве лекарственного средства, индивидуально или в сочетании с фармацевтически приемлемым наполнителем, чтобы получить фармацевтическую композицию. Речь может идти, в частности, о стерильных изотонических солевых растворах (как растворы мононатрийфосфата, динатрийфосфата, хлорида натрия, хлорида калия, хлорида кальция или хлорида магния, и т.д., или растворы смесей таких солей) или о сухих композициях, особенно лиофилизированных, которые путем добавления, в зависимости от случая, стерильной воды или физиологической сыворотки позволяют получать инъецируемые растворы. Введения могут быть осуществлены стереотаксическим, локальным, оральным, парентеральным, внутриносовым, внутривенным, внутримышечным, подкожным, внутриглазным, трансдермальным и т.д. путем.
Выявление вышеописанных соединений базируется на введении в контакт, в частности, путем введения, такого как, например, инъекция, животной модели согласно изобретению с соединением или смесью соединений, предположительно обладающим (обладающих) активностью, и определении затем эффекта или эффектов соединений, особенно на церебральном уровне модели, в отношении различных биохимических и/или гистологических изменений.
Кроме факта возможности тестирования in vivo терапевтических соединений, позволяющих предупреждать, уменьшать интенсивность или излечивать болезнь Альцгеймера, эти трансгенные животные также позволяют располагать животной моделью болезни Альцгеймера, пригодной для скрининга относящихся к окружающей среде факторов, которые индуцируют или ускоряют патогенез, или еще для исследования состояния во время развития болезни и изучения различных биологических механизмов, которые принимают активное участие, например, с целью изучения новых лекарственных средств или определения эффективных количеств лекарственных средств и токсичности. Таким образом, другой объект настоящего изобретения относится к применению трансгенного животного, исходных клеточных линий или образующихся путем дифференцировки клеточных линий, таких как указанные выше, для тестирования активности соединений или способов, предназначенных для предупреждения и/или лечения болезни Альцгеймера. Следующий объект изобретения относится к клетке, извлеченной из трансгенных животных, таких как указанные выше, а также к ее применению для выявления соединений, предназначенных для лечения болезни Альцгеймера.
Выявление вышеуказанных соединений базируется на введении в контакт клеточных экстрактов из животной модели согласно изобретению с соединением или смесью соединений, предположительно обладающим (обладающих) активностью, и определении затем эффекта или эффектов соединений на уровне целых клеток, в гомогенатах клеток или в субклеточной фракции по различным параметрам, таким как, например, гибель клетки.
Кроме вышеуказанных положений, настоящее изобретение включает также другие характеристики и преимущества, относящиеся к примерам и фигурам, которые следуют и которые должны рассматриваться как поясняющие изобретение, без ограничения ими его объема охраны.
ПРИМЕРЫ
Пример 1
Исследование генотипа полиморфизма rs908832 гена АВСА2 в образце, взятом на анализ у пациентов
Была использована коллекция ДНК, происходящих от пациентов индоевропейской расы, пораженных болезнью Альцгеймера, и контрольных индивидов, после их согласия.
Частоту встречаемости полиморфизмов определяли по генотипажу образца 47 индивидов из набора, выпускаемого институтом Coriell (США).
Десять полиморфизмов (в том числе полиморфизм rs908832) были отобраны на основании их локализации в гене и их частоты встречаемости для генотипирования в случае пациентов, пораженных болезнью Альцгеймера, и контрольных индивидов.
Генотипы были получены методом анализа 5'-нуклеазы (способ различения аллелей; фирма Applied Biosystems, Foster City, США), затем осуществляли статистический анализ типа Chi2, тестируя ассоциацию каждого из этих маркеров с болезнью Альцгеймера (тест на гетерогенность). Для анализа группы различали в зависимости от возраста, в котором происходит появление болезни ((ранний (до или в 65 лет) или поздний (после 65 лет)), и ее происхождения (спорадическое (первый известный случай) или семейное (другие, уже известные в том же самом семействе случаи)).
Результаты, полученные для полиморфизма rs908832, являются следующими:
- частота встречаемости содержащегося в незначительном количестве аллеля (Т) у пациентов (440 генотипов): 7,4
- частота встречаемости содержащегося в незначительном количестве аллеля (Т) у контрольных индивидов (519 генотипов): 3,4.
Пропорции Hardy Weinberg были подтверждены у пациентов и контрольных индивидов: не наблюдают значительного отклонения, что подтверждает, что нет ошибки генотипирования.
Результаты тестов, осуществленных в случае пациентов, которые обладают ранней формой заболевания (до или в 65 лет):
- 136 спорадических случаев в противоположность 272 контрольным;
- тест на гетерогенность: chi2 (1dd1) = 22,69 р = 2.10-6;
- аллельный тест (частота встречаемости (Т) в противоположность частоте встречаемости (С)): chi2 (1dd1)=21,27 р=4.10-6;
- 104 семейных случая в противоположность 272 контрольным;
- тест на гетерогенность: chi2 (2dd1)=7,80 р=0,02;
- аллельный тест (частота встречаемости (Т) в противоположность частоте встречаемости (С)): chi2 (1dd1)=7,27 р=7.10-3.
Нечетное отношение оценивали путем логической регрессии у 240 пациентов с ранней формой заболевания (выравнивание в отношении пола и состояния АРОЕ-ε4). Результат также является показательным: OR=3,97; IC=[2,23-7,09].
Полученные значения остаются значительными (порог фиксирован при 0,05) даже после корректировки для множественных тестов по методу Bonferroni.
Эти результаты указывают, что ген АВСА2 представляет собой важный ген в этиологии болезни Альцгеймера.
В отношении риска этот результат величины того же порядка, как и риск, придаваемый аполипротеином Е4, который представляет собой наиболее сильный, известный на сегодняшний день фактор риска для болезни Альцгеймера.
Пример 2
Получение трансгенных мышей
1. Генерация трансгенной мыши, экспрессирующей человеческий белок АВСА2
Мутагенез человеческого АВСА2 осуществляли за счет использования системы мутагенеза in vitro, как SculptorTM (Amersham, Франция). Кодирующую область АВСА2 субклонировали в векторе клонирования типа Bluescriprt (Stratagéne) и мутации вводили согласно протоколу, выпускаемому изготовителем, путем использования олигонуклеотидов, содержащих желательную мутацию. Мутированные последовательности подтверждали путем анализа последовательности.
2. Генерация и идентификация трансгенных мышей АВСА2
Для конструкции трансгена, геномную ДНК, кодирующую АВСА2 и имеющую содержащийся в незнечительном количестве аллель полиморфизма rs908832 человека, субклонировали в полилинкере специфического трансгенного экспрессирующего вектора некоторых тканей/типов клеток, как THYI (Lüthi и др., J. Neuroscience, 17, 4688-99), PDGF или прион для нейронных типов или GFAP для астроцитарных типов. Набор для получения плазмиды (Quiagen) использовали для получения суперспирализованной ДНК. Для микроинъекции, последовательности вектора удаляли путем рестрикции с помощью определенного фермента рестрикции, оставляющего интактной всю совокупность трансгена и отделяющего его от непригодных последовательностей вектора клонирования. Затем фрагмент, содержащий полигенный экспрессирующий кластер, очищали путем электрофореза на геле агарозы.
Аликвоты, предназначенные для микроинъекции, подвергали диализу против буфера ТЕ (10 мМ Трис, рН 7,4; 0,1 мМ ЭДТУ) на плавающем фильтре (Millipore; тип мембраны: VS; 0,025 мкм), затем отфильтровывали (Spin-X; Costar; полиацетатная мембрана; 0,22 мкм). ДНК для микроинъекции разводили до конечной концентрации 1-2 нг/мкл. Очищенный фрагмент инъецировали в один из двух пронуклеусов оплодотворенных ооцитов мыши. Выжившие эмбрионы немедленно трансплантировали в яйцевод приемных матерей (“несущие в себе псевдозародыш”). Наличие трансгена у новорожденных определяли либо путем PCR, либо путем саузерн-блоттинга, используя специфические зонды/последовательности. За счет совокупности этих анализов могут быть исключены все основные реаранжировки или делеции трансгена у исходных мышей и их потомства.
3. Генерация трансгенных животных, включающих человеческий белок АВСА2 в их геноме
Используют технологию гомологичной рекомбинации в исходных клетках для введения гена АВСА2, имеющего содержащийся в незначительном количестве аллель полиморфизма rs908832 человека, в желательное предопределенное положение в гене мыши. Праймеры и образцы, описанные в настоящем изобретении, могут быть легко использованы для скрининга изогенных геномных библиотек мыши (библиотеки лямбда, ВАС, YAC...), чтобы выделить и клонировать соответствующий ген мыши. Вышеуказанный ген мыши охарактеризовывали по его геномной организации и его последовательности, используя стандартные способы, известные специалисту в данной области (картография сайтов рестрикции, секвенирования, биоаналитические инструменты), чтобы определить точное место, куда человеческая ДНК должна быть встроена для получения желательного профиля экспрессии человеческой ДНК, полностью прерывая экспрессию мышиного гена. Как только идентифицировано точное положение, конструируют стандартный вектор для скрининга для исходных клеток (то есть вектор, имеющий маркеры для селекции желательных “явлений”, причем все вышеуказанные маркеры хорошо известны специалисту в данной области). Кластер для селекции (ген устойчивости к антибиотику) быстро фланкируют 3'- и 5'-концами фрагментов геномной ДНК мыши (2-6 т.н.), идентичными 3'- и 5'-удлиняющим сегментам последовательности мышиного гена АВСА2, расположенного непосредственно после селекционированного сайта инсерции.
На основе знания мышиного гена генерировали вектор положительного контроля для скрининга рекомбинантных исходных клеток (вектор репродуцирует локус мышиного гена, благоприятствуя интеграции) с целью оптимизации и осуществления процесса скрининга в большом объеме.
Согласно стандартным методикам ДНК очищали для опытов по скринингу. Вектор для скрининга вводили в исходные клетки, используя стандартизированные способы электропорации, после чего исходные клеточные клоны подвергали процессу последовательного скрининга (антибиотики) для благоприятствования исходным клеточным клонам, несущим желательную рекомбинацию. Вообще, скрининг занимает около двух недель. Резистентные исходные клеточные клоны подвергали скринингу путем PCR и/или саузерн-блоттинга.
Исходные клеточные клоны, обладающие желательной рекомбинацией без другой модификации, обнаруживаемой в их геноме, продуцировали для получения достаточного количества клеток. Затем вышеуказанные клетки инъецировали в эмбрионы в возрасте трех с половиной суток, полученные от самки, оплодотворенной естественным путем. Выжившие бластоциты (включающие исходные клетки) имплантировали в самку-реципиента, в которой происходит развитие бластоцитов до их предела и которая производит на свет новорожденных мышат, состоящих из клеток, происходящих от бластоцитов-хозяев и исходных клеточных клонов. Этот тип животного называют “химерным животным”.
Этих химер (предпочтительно самцов, так как большинство исходных клеточных линий получено самцами) спаривали с мышами дикого типа, чтобы получить гетерозиготных животных для модификации. Отбор путем спаривания гетерозиготных животных между собой позволяет генерировать гомозиготных животных.
4. Генерация трансгенных животных, включающих содержащийся в незначительном количестве аллель полиморфизма rs908832 человеческого гена АВСА2 в их соответствующем гене
Таким же образом, как описано выше, мышиный ген выделяли и охарактеризовывали. Для этого типа модификации существенным является сравнение человеческого гена и мышиного гена для идентификации точного положения, где находящийся у человека сайт мутации должен быть введен в мышиный ген. На этой стадии существенным является биоанализ. С этой целью получали вектор для скрининга и связывали таким же образом, как описано выше. После успешной гомологичной рекомбинации ничего другого, чем желательный сайт мутации, не заменяли в кодирующей последовательности мышиного гена. Некоторые маркеры селекции могут оставаться в интроне, но они обычно не оказывают никакого влияния на экспрессию гена. Если необходимо, они могут быть удалены путем использования второй генерации векторов для скрининга, включающих элементы распознавания рекомбиназы. При сборе конструкции стадии идентичны таковым процесса, описанного выше.
Как правило, эти трансгенные мыши АВСА2 благоприятно скрещивают с трансгенными мышами, экспрессирующими ген АРР, несущий мутации семейных форм болезни Альцгеймера, чтобы способствовать отложению амилоидных бляшек.
5. Нейрогистопатология
Получение церебральной ткани
Мыши должны быть подвергнуты глубокой анестезии (пентобарбитал: 60 мг/мл/кг, интраперитонеально; кетамин: 40 мг/мл/кг интраперитонеально), затем перфузии транскардиально с помощью физиологической сыворотки, затем параформальдегида (4% в забуференном фосфатом физиологическом растворе). Головной мозг мышей затем извлекали, потом постфиксировали в том же самом растворе для фиксации в течение 24 часов при температуре 4°С. После фиксации головной мозг мышей разделяли на левую и правую половины, после чего, согласно классическому протоколу, наносили защитное покрытие при использовании парафина.
Левые половины головного мозга с нанесенным защитным покрытием из парафина трансгенных и нетрансгенных мышей, а также блоки ткани посмертного человеческого головного мозга (фронтальная кора головного мозга) субъектов, пораженных болезнью Альцгеймера, и контрольного субъекта разрезали до толщины 6 мкм (серийные срезы) путем использования микротома (LEICA RM 2155, Франция). Соответствующие блоки ткани правых половин головного мозга трансгенных и нетрансгенных мышей разрезали до толщины 25 мкм.
Иммунореактивность против амилоидного пептида Аβ обнаруживается, как обычно описывается, специалистом.






Изобретение относится к области молекулярной биологии и может быть использовано в медицине. Предложен способ прогноза болезни Альцгеймера, который предусматривает тестирование ДНК исследуемого индивида с целью определения полиморфизма rs 908832 в гене АВСА2. При этом основанием для заключения о предрасположенности индивида к развитию болезни Альцгеймера служит выявление по меньшей мере одного редко встречающегося аллеля, характеризующегося наличием аденина в данном полиморфном сайте гена АВСА2. 6 з.п. ф-лы.
1. Способ прогноза болезни Альцгеймера у лица, отличающийся тем, что он включает:
a) отбор ДНК-содержащего биологического образца;
b) экстракцию ДНК из вышеуказанного образца;
c) амплификацию вышеуказанной выделенной ДНК с помощью праймеров, ограничивающих последовательность, включающую сайт полиморфизма rs908832 гена АВСА2;
d) анализ полиморфизма rs908832 гена АВСА2 в амплифицированной ДНК и
e) заключение о предрасположенности к болезни Альцгеймера в случае выявления, по меньшей мере, одного редко встречающегося аллеля, характеризующегося тем, что он содержит аденин, локализованный в положении 348 последовательности SEQ ID NO:1.
2. Способ прогноза болезни Альцгеймера по п.1, отличающийся тем, что вышеуказанными биологическими образцами являются образцы крови, спермы или волос.
3. Способ прогноза болезни Альцгеймера по п.1, отличающийся тем, что вышеуказанные стадии анализа полиморфизма rs908832 осуществляют способами полимеразной цепной реакции (PCR), гибридизации, переноса на мембрану по методу Саузерна, рестрикции с помощью нуклеаз, анализа полиморфизма длин рестрикционных фрагментов (RFLP) или прямого секвенирования, или их комбинацией.
4. Способ по п.1, отличающийся тем, что стадия амплификации с) включает полимеразную цепную реакцию (PCR).
5. Способ по п.1, отличающийся тем, что амплифицированную ДНК, несущую редко встречающийся аллель полиморфизма rs908832, отличают от амплифицированной ДНК, не несущей этого аллеля, с помощью способа с использованием активности 5'-нуклеазы ДНК-полимеразы I (TaqMan).
6. Способ по п.1, отличающийся тем, что амплифицированную ДНК, несущую редко встречающийся аллель полиморфизма rs908832, отличают от амплифицированной ДНК, не несущей этого аллеля, путем анализа полиморфизма длин рестрикционных фрагментов (RFLP).
7. Способ по п.6, отличающийся тем, что рестрикционные фрагменты получают путем рестрикции амплифицированной ДНК с помощью фермента рестрикции до миграции в геле агарозы, переноса на мембрану по методу Саузерна и гибридизации.
| DATABASE SNP, Ac.no RS 908832, 06.09.2000 | |||
| DATABASE GenBank, Ac.no AF 327671, 01.03.2001 | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

