Изобретение относится к новым производным дикетопиперазина, обладающим сильным и избирательным антагонистическим действием на рецептор окситоцина, способам их получения, фармацевтическим композициям, содержащим эти соединения, и их применению в медицине.
Гормон окситоцин является сильнодействующим средством для сокращения матки и используется для стимуляции или усиления родов. Также плотность маточных рецепторов окситоцина значительно увеличивается более чем в 100 раз во время беременности и принимает максимальное значение во время родов (преждевременных и своевременных).
Преждевременные роды/рождение (между 24 и 37 неделями) являются причиной около 60% смертности/заболеваемости среди новорожденных и таким образом соединение, которое ингибирует сокращения матки под действием окситоцина, например антагонисты окситоцина, должно быть полезным для предотвращения или управления преждевременными родами.
Международная патентная заявка WO 99/47549 описывает производные дикетопиперазина, в том числе производные 3-бензил-2,5-дикетопиперазина, в качестве ингибиторов фруктозы 1,6-бифосфата (FBPase).
Международная патентная заявка WO 03/053443 описывает класс производных дикетопиперазина, который демонстрирует, в частности, значительный уровень активности, как антагонисты избирательного действия на рецептор окситоцина. Предпочтительный класс соединений, описанный в патентной заявке, представляют формулой (А)
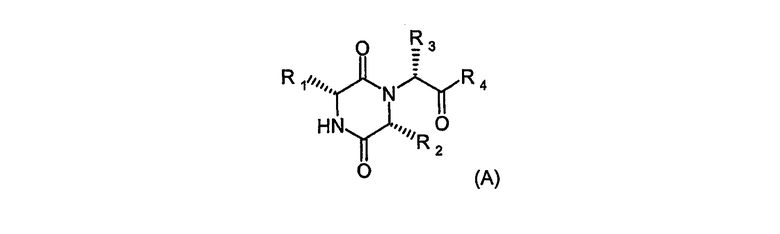
Данные соединения включают такие, в которых inter alia R1 означает 2-инданил, R2 означает С3-4алкил, R3 означает 5- или 6-членную гетероарильную группу, связанную с остальной частью молекулы через атом углерода в кольце, R4 представляет группу NR5R6, в которой R5 и R6, каждый, представляет алкил, например метил, или R5 и R6 вместе с атомом азота, к которому они присоединены, образуют 3-7-членное насыщенное гетероциклическое кольцо, этот гетероцикл может содержать дополнительный гетероатом, выбранный из кислорода.
Международная патентная заявка WO 2005/000840 описывает производные дикетопиперазина формулы (В)
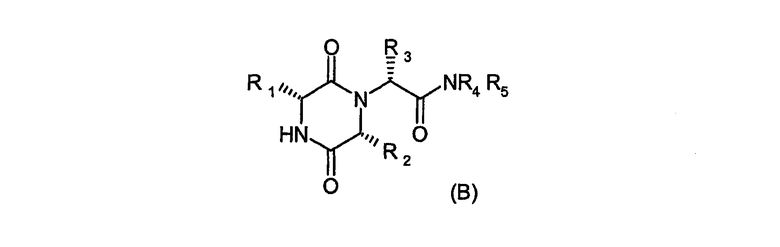
в которых R1 означает 2-инданил, R2 означает 1-метилпропил, R3 означает 2-метил-1,3-оксазол-4-ил, и R4 и R5 совместно с атомом азота, к которому они присоединены, представляют морфолиногруппу.
В настоящее время авторами патентной заявки обнаружена группа антагонистов избирательного действия рецептора окситоцина, которые демонстрируют особенно выгодный фармакокинетический профиль.
Настоящее изобретение таким образом относится к, по крайней мере одному, химическому соединению, выбранному из соединений формулы (I)
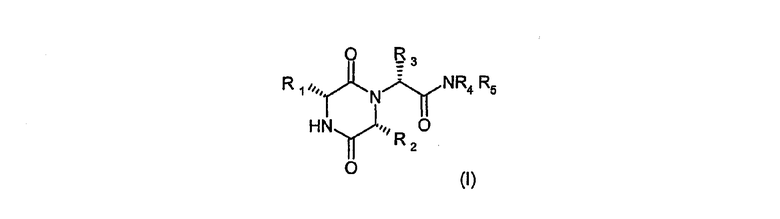
в котором R1 означает 2-инданил, R2 означает 1-метилпропил, R3 является группой, выбранной из 2,6-диметил-3-пиридила или 4,6-диметил-3-пиридила, R4 обозначает метил и R5 обозначает водород, или метил, или R4 и R5 совместно с атомом азота, к которому они присоединены, представляют морфолиногруппу, и его фармацевтически приемлемым производным.
Следует понимать, что соединения формулы (I) обладают абсолютной стереохимией, показанной у асимметричных атомов углерода, несущих группы R1, R2 и R3, т.е. стереохимия в этих положениях является всегда (R). Тем не менее, также следует понимать, что хотя такие соединения в основном свободны от (S)-эпимера при каждом R1, R2 и R3, каждый эпимер может присутствовать в небольших количествах, например может присутствовать 1% или меньше (S)-эпимера.
Также следует понимать, что группа R2 содержит асимметричный атом углерода и, что изобретение включает как их (R)-эпимеры, так и (S)-эпимеры.
В одном варианте осуществления настоящего изобретения R2 означает (1S)-1-метилпропил. В другом варианте осуществления настоящего изобретения R2 означает (1R)-1-метилпропил.
Одним вариантом осуществления настоящего изобретения являются соединения, получение которых, в частности, описывается в примерах 1-3 и 6. Другим вариантом осуществления настоящего изобретения являются соединения, получение которых, в частности, описывается в примерах 1-3. Еще одним вариантом осуществления настоящего изобретения являются соединения, получение которых, в частности, описывается в примерах 3 и 6. Кроме того, еще одним вариантом осуществления настоящего изобретения являются соединения, получение которых, в частности, описывается в примере 3. Другим вариантом осуществления настоящего изобретения являются соединения, получение которых, в частности, описывается в примере 1.
В одном аспекте химические соединения настоящего изобретения могут быть, по крайней мере, одним химическим соединением, выбранным из
(2R)-2-{(3R,6R)-3-(2,3-дигидро-1H-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-2-(2,6-диметил-3-пиридинил)-N-метилэтанамида;
(2R)-2-{(3R,6R)-3-(2,3-дигидро-1H-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-2-(2,6-диметил-3-пиридинил)-N,N-диметилэтанамида;
(3R,6R)-3-(2,3-дигидро-1H-инден-2-ил)-1-[(1R)-1-(2,6-диметил-3-пиридинил)-2-(4-морфолинил)-2-оксоэтил]-6-[(1S)-1-метилпропил]-2,5-пиперазиндиона;
(2R)-2-{(3R,6R)-3-(2,3-дигидро-1H-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-2-(4,6-диметил-3-пиридинил)-N,N-диметилэтанамида;
(2R)-2-{(3R,6R)-3-(2,3-дигидро-1H-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-2-(4,6-диметил-3-пиридинил)-N-метилэтанамида;
(3R,6R)-3-(2,3-дигидро-1H-инден-2-ил)-1-[(1R)-1-(4,6-диметил-3-пиридинил)-2-(4-морфолинил)-2-оксоэтил]-6-[(1S)-1-метилпропил]-2,5-пиперазиндиона, и их фармацевтически приемлемые производные.
Используемый в описании термин «фармацевтически приемлемый» означает соединение, которое можно использовать в фармацевтических целях. Соли и сольваты соединений настоящего изобретения, которые пригодны для использования в медицине, это те соединения, в которых противоион или ассоциированный растворитель являются фармацевтически приемлемыми. Однако соли и сольваты, имеющие нефармацевтически приемлемые противоионы или ассоциированные растворители, входят в объем настоящего изобретения, например, для использования в качестве промежуточных продуктов при получении других соединений настоящего изобретения и их фармацевтически приемлемых солей и сольватов.
Используемый в описании термин «фармацевтически приемлемая производная» подразумевает любую фармацевтически приемлемую соль, сольват или пролекарство, например сложный эфир соединения настоящего изобретения, который при назначении реципиенту способен обеспечить (прямо или косвенно) соединение настоящего изобретения, или активный метаболит, или их остаток. Такие производные признаны специалистами в этой области без необходимости экспериментирования. Тем не менее, ссылка делается на изучение Burger's Medical Chemistry and Drug Discovery, 5th Edition, Vol.1: Principles and Practice, которое включается в данную патентную заявку путем ссылки на углубленное изучение таких производных. В одном аспекте фармацевтически приемлемыми производными являются соли, сольваты, сложные эфиры, карбаматы и фосфаты сложных эфиров. В другом аспекте фармацевтически приемлемыми производными являются соли, сольваты и сложные эфиры. В следующем аспекте фармацевтически приемлемыми производными являются соли и сольваты. В другом аспекте фармацевтически приемлемыми производными являются физиологически приемлемые соли.
Допустимые физиологически приемлемые соли соединений настоящего изобретения включают кислотно-аддитивные соли, образованные с физиологически приемлемыми неорганическими кислотами или органическими кислотами. Примеры таких кислот включают соляную кислоту, бромистоводородную кислоту, азотную кислоту, фосфорную кислоту, серную кислоту, сульфоновые кислоты, например метансульфоновую, этансульфоновую, бензолсульфоновую и п-толуолсульфоновую, лимонную кислоту, виннокаменную кислоту, молочную кислоту, пировиноградную кислоту, уксусную кислоту, янтарную кислоту, фумаровую кислоту и малеиновую кислоту.
Настоящее изобретение также касается сольватов соединений формулы (I), например гидратов, или сольватов с фармацевтически приемлемыми растворителями. Неограничивающие примеры растворителей включают спирты, например этанол, изопропанол, ацетон, простые эфиры, сложные эфиры, например этилацетат.
Соединения настоящего изобретения можно также использовать в комбинации с другими терапевтическими агентами. Таким образом, настоящее изобретение относится, с другой стороны, к комбинации, содержащей соединение настоящего изобретения, или его фармацевтически приемлемой производной, вместе с другим терапевтическим агентом.
При использовании соединения настоящего изобретения или его фармацевтически приемлемой производной в комбинации со вторым терапевтическим агентом, активным против такого же состояния при заболевании, доза каждого соединения может отличаться от дозы, когда используется только соединение. Соответствующие дозы легко подобрать специалистам в данной области. Следует понимать, что количество соединения настоящего изобретения, требуемое для лечения, будет варьироваться в зависимости от природы, возраста и состояния пациента и будет оставаться на усмотрении лечащего врача или ветеринара. Соединения настоящего изобретения можно использовать в комбинации с лекарственными средствами, снижающими родовую деятельность (токолитическими) или профилактическими средствами. Неограничивающие примеры включают бета-агонисты, такие как тербуталин и ритодрин, блокаторы кальциевых каналов, например нифедепин, нестероидные противовоспалительные лекарственные средства, такие как индометацин, соли магния, такие как сульфат магния, другие антагонисты окситоцина, такие как атосибан, и агонисты прогестерона и рецептуры. Кроме того, соединения настоящего изобретения можно использовать в комбинации с антенатальными стероидами, включающими, но без ограничения, бетаметазон и дексаметазон, пренатальные витамины, особенно фолатные добавки, антибиотиками, включающими, но без ограничения, ампициллин, амоксициллин/клавуланат, метронидазол, клиндамицин, и анксиолитиками (транквилизаторами).
Комбинации, на которые ссылаются выше, могут быть удобно представлены для использования в виде фармацевтической рецептуры и, таким образом, фармацевтические рецептуры, содержащие комбинацию, как определено выше, вместе с фармацевтически приемлемым носителем или наполнителем являются следующим аспектом настоящего изобретения. Индивидуальные компоненты таких комбинаций можно вводить либо последовательно, либо одновременно в раздельных или комбинированных фармацевтических рецептурах с помощью любого удобного способа.
При последовательном введении либо соединение настоящего изобретения, либо второй терапевтический агент можно применять первым. При одновременном введении комбинацию можно применять либо в одной и той же, либо в другой фармацевтической композиции.
При комбинировании в одной и той же рецептуре следует понимать, что два соединения должны быть стабильными и совместимыми друг с другом и другими компонентами рецептуры. При составлении рецептуры раздельно они могут быть представлены в любой из подходящих рецептур, известных для таких соединений в этой области.
Соединения формулы (I) обладают большим сродством с рецепторами окситоцина в матке крыс и людей, и это может определять использование традиционных процедур. Например сродство рецепторов окситоцина в матке крыс может определяться с помощью процедуры Pettibone et al., Drug Development Research 30, 129-142 (1993). Соединения настоящего изобретения также демонстрируют большое сродство с рекомбинированным рецептором окситоцина человека в СНО клетках, и это может быть обычно продемонстрировано при использовании процедуры, описанной Wyatt et al., Bioorganic & Medical Letters, 2001 (11), p.1301-1305.
Соединения настоящего изобретения демонстрируют благоприятный фармакокинетический профиль, включающий хорошую биоактивность, взаимосвязанную с хорошей растворимостью в воде. В одном аспекте соединения настоящего изобретения показывают высокую активность и низкий собственный клиренс. В другом аспекте соединения настоящего изобретения показывают низкий собственный клиренс.
Вследствие этого соединения настоящего изобретения успешно применяются при лечении или предотвращении заболеваний и/или состояний, вызванных действием окситоцина. Примеры таких заболеваний и/или состояний включают преждевременные роды, дисменорею, эндометриоз и доброкачественную гиперплазию простаты.
Соединения также могут успешно использоваться для задерживания родов перед рекомендуемым кесаревым сечением или переводе пациента в высокоспециализированный медицинский центр, лечения сексуальной дисфункции (у мужчин и женщин), в частности при преждевременной эякуляции, ожирении, расстройствах пищевого поведения, острой сердечной недостаточности, артериальной гипертензии, циррозе печени, почечной или офтальмологической гипертензии, обсессивно-компульсивного расстройства и психоневрологических расстройств. Соединения настоящего изобретения можно также успешно использовать для улучшения коэффициентов фертильности у животных, например в животноводстве.
В силу вышесказанного настоящее изобретение относится к по меньшей мере одному химическому соединению, выбранному из соединения формулы (I) и/или его фармацевтически приемлемого производного для применения в терапии, в частности для применения в терапии и ветеринарии, и, в частности, применения в качестве лекарственного средства для противодействия влиянию окситоцина на рецептор окситоцина.
Изобретение также относится к применению, по меньшей мере, одного химического соединения, выбранного из группы соединения формулы (I) и/или его фармацевтически приемлемых производных для получения лекарственного средства для противодействия влиянию окситоцина на рецептор окситоцина.
В соответствии со следующим аспектом изобретение также обусловливает способ противодействия влиянию окситоцина на рецептор окситоцина, заключающийся во введении пациенту, нуждающемуся в таком лечении, антагонистического количества, по меньшей мере, одного химического соединения, выбранного из группы соединения формулы (I) и/или его фармацевтически приемлемых производных.
Специалистам ясно, что ссылка в данном описании на лечение распространяется на профилактику, а также на лечение установленных заболеваний или синдромов.
В дальнейшем следует понимать, что количество соединения настоящего изобретения, требующееся для лечения, будет варьироваться в зависимости от характера заболевания, способа введения, возраста и состояния пациента и будет установлено, в конце концов, по усмотрению лечащего врача или ветеринара. В целом, однако, дозы, использованные для лечения взрослых пациентов, будут находиться в диапазоне от 2 до 1000 мг в день, в зависимости от способа введения.
Таким образом, при парентеральном применении дневная доза будет обычно составлять от 2 до 50 мг, предпочтительно от 5 до 25 мг в день. При оральном применении дневная доза обычно будет находиться в диапазоне от 10 до 1000 мг, например 50 до 500 мг в день.
Желаемое количество лекарственного средства может быть обычно представлено как одноразовая доза или как дробные дозы, назначенные с соответствующими интервалами, например, как две, три, четыре или больше субдоз в день.
Хотя возможно, чтобы при использовании в терапевтических целях соединение настоящего изобретения могло быть введено в виде сырого химического продукта, в то же время предпочтительно представить активный ингредиент как фармацевтическую композицию.
Таким образом, изобретение в дальнейшем относится к фармацевтической композиции, содержащей соединение формулы (I) и/или его фармацевтически приемлемые производные вместе с одним или несколькими их фармацевтически приемлемыми носителями и, возможно, другими терапевтическими и/или профилактическими ингредиентами. Носитель(и) должен быть «приемлемым» в смысле совместимости с другими ингредиентами рецептуры и неопасными для их реципиентов.
Эти композиции настоящего изобретения включают такие композиции, которые имеют формы, специально составленные для орального, буккального, парентерального, ингаляционного или инсуффляции (вдувания), имплантационного, вагинального или ректального применения.
Таблетки и капсулы для орального применения могут содержать традиционные наполнители, такие как связующие вещества, например сироп, гуммиарабик, желатин, сорбит, трагакант, клейкое вещество крахмала или поливинилпирролидона; наполнители, например лактоза, сахар, микрокристаллическая целлюлоза, маисовый крахмал, фосфат кальция или сорбит; лубриканты, например стеарат магния, стеариновая кислота, тальк, полиэтиленгликоль или кремнезем; агенты, дезинтегранты, например картофельный крахмал или крахмалгликолят натрия, или смачивающие агенты, такие как лаурилсульфат натрия. Таблетки могут быть покрыты пленкой в соответствии со способами, известными в данной области. Оральные жидкие препараты могут быть в форме, например, водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров либо могут быть представлены как сухой продукт, который разводят водой или другим подходящим растворителем перед использованием. Такие жидкие препараты могут содержать традиционные добавки, такие как суспендирующие агенты, например сироп сорбита, сироп глюкозы/сахара, желатин, гидроксиэтилцеллюлоза, карбоксиметилцеллюлоза, гель стеарата алюминия или гидрогенизированные пищевые жиры; эмульгирующие агенты, например лецитин, моноолеат сорбита или гуммиарабик; безводные растворители (которые могут включать пищевые масла), например миндальное масло, фракционированное кокосовое масло, масляные сложные эфиры, пропиленгликоль или этиловый спирт; солюбилизаторы, такие как поверхностно-активные вещества, например полисорбаты или другие агенты, такие как циклодекстрины; и консерванты, например метил- или пропил п-гидроксибензоаты или аскорбиновая кислота. Композиции могут также иметь форму суппозиториев, например содержащих обычные суппозиторные основы, такие как кокосовое масло или другие глицериды.
Для буккального введения композиция может иметь форму таблеток или лепешек, полученных традиционным способом.
Композиция в соответствии с настоящим изобретением может иметь форму, удобную для парентерального введения посредством инъекции или непрерывной инфузии. Композиции для инъекции можно приготовить в виде одноразовой дозы в ампулах или в мультидозовых контейнерах с добавленным консервантом. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных растворителях, и могут содержать композиционные агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно активный ингредиент может быть в порошкообразном виде, который можно разводить подходящим растворителем, например стерильной, апирогенной водой перед использованием.
Композиции в соответствии с настоящим изобретением могут содержать от 0,1 до 99% активного ингредиента, традиционно от 1 до 50% для таблеток и капсул и 3-50% для жидких лекарственных средств.
Эффективный фармакокинетический профиль соединений настоящего изобретения легко продемонстрировать, используя обычные процедуры для измерения фармакокинетических свойств биологически активных соединений.
Соединения настоящего изобретения и их фармацевтически приемлемые производные можно получить с помощью способов, описанных ниже, вышеупомянутые способы формируют следующий аспект настоящего изобретения. В следующем описании группы, как определено выше, состоят из соединений настоящего изобретение, если нет другого утверждения.
Соединения формулы (I) могут быть получены реакцией карбоновых кислот (II), в которых R1, R2 и R3 имеют значения, определенные в формуле (I), и хиральность R3 является либо R, либо S или их смесь
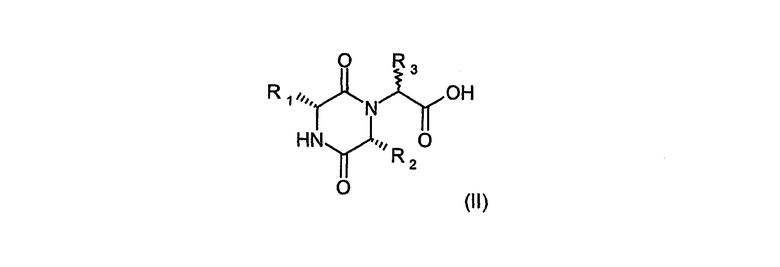
или их активированной производной с амином HNR4R5, в котором R4 и R5 имеют значения, определенные в формуле (I), в стандартных условиях получения амидов из карбоновой кислоты или ее активированной производной и амина.
Следует помнить, что смесь диастереомеров соединений формулы (I), полученных путем вышеуказанной реакции, можно разделить, используя стандартные методики по разделению продуктов, хорошо известные в данной области, например колоночную хроматографию.
Таким образом, амид формулы (I) можно получить с помощью обработки карбоновой кислоты формулы (I) активирующим агентом, таким как ВОР (гексафторфосфат бензотриазол-1-илокси-трис(диметиламино)фосфония), TBTU (тетрафторборат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония), BOP-Cl (хлорангидрид бис(2-оксо-3-оксазолидинил)фосфиновой кислоты), оксалилхлорид или 1,1'-карбонилдиимидазол в апротонном растворителе, таком как дихлорметан, необязательно, в присутствии четвертичного амина, такого как триэтиламин, и последующей реакцией продукта, таким образом, образованным, например, активированной производной соединения формулы (II), с амином HNR4R5.
Альтернативно амид формулы (I) можно получить взаимодействием смешанного ангидрида, полученного из карбоновой кислоты (II), в котором R1, R2 и R3 имеют значения, определенные в формуле (I) с амином HNR4R5 в апротонном растворителе, таком как тетрагидрофуран. Удобно провести реакцию при низких температурах, например, от 25 до -90°С, удобно при приблизительно -78°С.
Смешанный ангидрид удобно получить путем взаимодействия карбоновой кислоты (II) с подходящим хлорангидридом, например пивалоилхлоридом, в апротонном растворителе, таком как этилацетат в присутствии четвертичного органического основания, такого как триалкиламин, например триэтиламин, и при низких температурах, например от 25 до -90°С, удобно при приблизительно -78°С.
Соединения формулы (I) можно также получить путем взаимодействия соединения формулы (III)

в которых R1, R2 и R3 имеют значения, определенные в формуле (I), и R6 обозначает 2-гидроксифенил, с 1,1'-карбонилдиимидазолом или 1,1'-тиокарбонилдиимидазолом в соответствующем растворителе, таком как дихлорметан и последующей реакцией продуктов, образованных таким образом, с амином HNR4R5.
Соединения формулы (II) можно также получить из соединения формулы (III), в котором R6 обозначает 2-гидроксифенил, с помощью реакции с 1,1'-карбонилдиимидазолом или 1,1'-тиокарбонилдиимидазолом в соответствующем растворителе, таком как дихлорметан, и последующей реакцией продуктов, образованных таким образом, с водным раствором ацетона.
Соединения формулы (III), в которых R6 обозначает 2-гидроксифенил, можно получить из соответствующих соединений формулы (III), в которых R6 обозначает 2-бензилоксифенилгруппу путем гидрогенолиза, используя водород и палладиевый катализатор.
Альтернативно соединения формулы (III), в которых R6 обозначает 2-гидроксифенил, можно получить из соединений формулы (IV)
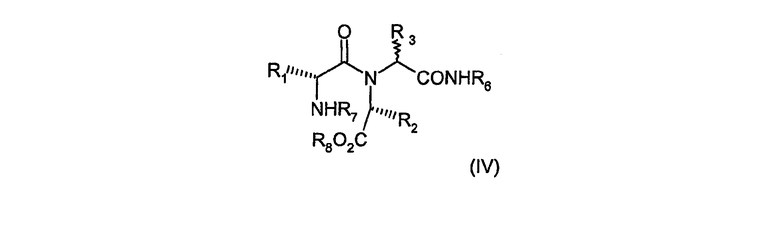
в которых R1, R2 и R3 имеют значения, определенные в формуле (I), R6 обозначает 2-бензилоксифенил, R7 обозначает бензилоксикарбонил и R8 обозначает C1-6алкил, путем реакции с водородом в присутствии катализатора палладия-на-угле и уксусной кислоты. Эта реакция успешно выполняется в растворителе, таком как этанол, трифторэтанол или их смеси.
Соединения формулы (IV) можно получить путем взаимодействия гидрохлорида сложного аминоэфира (V)
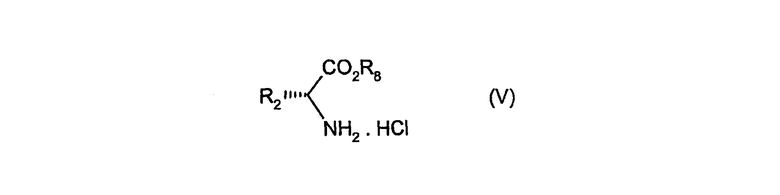
в котором R1 имеет значение, определенное в формуле (I), и R8 обозначает C1-6алкил, с альдегидом R3CHO (VI), в котором R3 имеет значение, определенное в формуле (I), в присутствии триэтиламина и в растворителе, таком как трифторэтанол, и затем взаимодействием полученного в результате продукта с соединением формулы (VII)
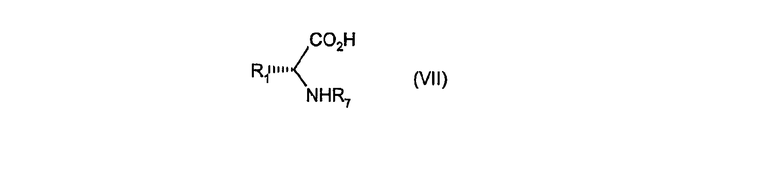
в котором R1 имеет значение, определенное в формуле (I), и R7 обозначает трет-бутилоксикарбонил или бензилоксикарбонил, и изоцианидом CNR6 (VIII), в котором R6 обозначает 2-бензилоксифенильную группу, в растворителе, таком как трифторэтанол.
Соединения формулы (III), в которых R6 обозначает 2-бензилоксифенильную группу, можно получить из соединения формулы (IV), в котором R1, R2 и R3 имеют значения, определенные в формуле (I), и R6 обозначает 2-бензилоксифенил, и R7 обозначает трет-бутилоксикарбонил путем реакции с хлористым водородом в диоксане, с последующим взаимодействием с триэтиламином в растворителе, таком как дихлорметан.
Соединение формулы (IV), в котором R7 обозначает трет-бутилоксикарбонил, можно получить с помощью способа, описанного выше в тексте патентной заявки, используя соединение формулы (VII), в котором R7 обозначает трет-бутилоксикарбонил.
Заместитель R2 представляет собой 1-метилпропиловую группу, и соединение формулы (I), в котором R2 обозначает 1-метилпропильную группу, имеющую (S)- или (R)-конфигурацию, можно получить, начиная реакцию с гидрохлорид сложного аминоэфира (V), в котором R2 группа обладает (S)- или (R)-конфигурацией.
Гидрохлорид сложного аминоэфира (V), в котором R1 имеет значение, определенное в формуле (I) и R8 обозначает С1-6алкил, можно получить из соответствующих коммерчески доступных аминокислот, D-аллоизолейцин или D-изолейцин, с помощью способа, описанного Schmidt, U.; Kroner, M.; Griesser, H. Synthesis (1989), (11), 832-5.
Альдегиды R3CHO (VI), в которых R3 имеет значение, определенное в формуле (I), являются либо коммерчески доступными, либо их можно получить с помощью способов, описанных в литературе (Comins, Daniel L.; Weglarz, Michael A.; J. Org. Chem.; 53, 19, 1988; 4437-4442).
Производная аминокислоты (VII), в которой R1 имеет значение, определенное в формуле (I) и R7 обозначает трет-бутилоксикарбонил, является коммерчески доступной; производную аминокислоты (VII), в которой R1 имеет значение, определенное в формуле (I) и R7 обозначает бензилоксикарбонил, можно получить из соответствующей коммерчески доступной аминокислоты (R)-R1CH(NH2)CO2H (IX), в которой R1 имеет значение, определенное в формуле (I), путем взаимодействия с N-(бензилоксикарбонилокси)сукцинамидом и триэтиламином в растворителе, таком как диоксан в воде.
Изоцианид CNR6 (VIII) можно получить в соответствии с методами, описанными в литературе (Obrecht, Roland; Herrmann, Rudolf; Ugi, Ivar, Synthesis, 1985, 4, 400-402).
Кислотно-аддитивные соли соединения формулы (I) можно получить с помощью традиционных способов, например, путем обработки раствора соединения в подходящем растворителе, таком как дихлорметан или ацетон, подходящим раствором соответствующей неорганической или органической кислоты.
Следующие неограничивающие примеры иллюстрируют варианты осуществления настоящего изобретения.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Номенклатура
Все промежуточные соединения и примеры названы с использованием ACD Name Pro 6.02 в ISIS Draw.
Аббревиатура
CV: Объем колонки. Один объем колонки определяется как объем, заполненный сорбентом, в насадочной колонке. Его можно приблизительно вычислить из массы и плотности определенного используемого сорбента (1CV=масса, деленная на плотность).
Основные методы очистки и анализа
Аналитическую жидкостную хроматографию HPLC провели на Supelcosil LCABZ+PLUS колонке (3,3 см × 4,6 мм ID), выполняя элюирование 0,1% HCO2H и 0,01 М ацетатом аммония в воде (растворитель А), и 0,05% HCO2H и 5% воды в ацетонитриле (растворитель В), используя либо градиентное элюирование 1, 0-0,7 мин 0%В, 0,7-4,2 мин 0-100%В, 4,2-5,3 мин 100%В, 5,3-5,5 мин 0%В, либо градиентное элюирование 2, 0-0,7 мин 0%В, 0,7-4,2 мин 0-100%В, 4,2-4,6 мин 100%В, 4,6-4,8 мин 0%В при скорости потока 3 мл/мин. Время удерживания (Rt) приводится в минутах. Масс-спектр (MS) зарегистрировали на Waters ZQ 2000 масс-спектрометре, используя электроспрей положительный [ES+, чтобы получить MH+и M(NH4)+молекулярные ионы] или электроспрей отрицательный [ES-, чтобы получить M-H- молекулярный ион] типы. 1H ЯМР спектры зарегистрировали, используя спектрометр Bruker DPX 400 МГц, используя тетраметилсилан в качестве внешнего стандарта.
Очистка, использующая силикагелевые картриджи, касается хроматографии, выполненной с использованием Combiflash® CompanionTM с Redisep® картриджами, поставляемыми Presearch. Гидрофобные фритты относятся к фильтрационным трубкам, поставленным Whatman. SPE (экстракция твердой фазы) касается использования картриджей, поставляемых International Sorbent Technology Ltd. TLC (тонкослойная хроматография) касается использования пластин для тонкослойной хроматографии, поставляемых Merck, покрытых силикагелем 60 F254.
Промежуточное соединение 1
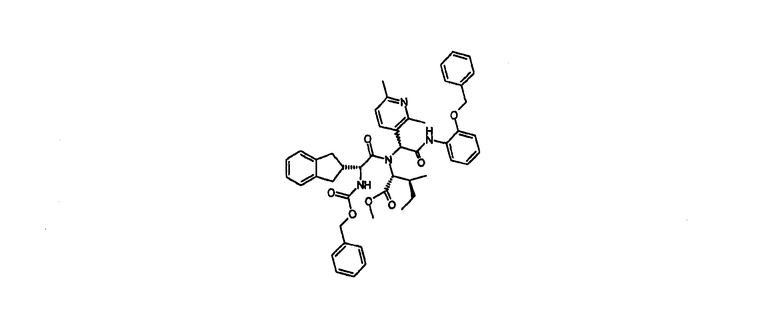
Метил N-[(2R)-2-(2,3-дигидро-1Н-инден-2-ил)-2-({[(фенилметил)окси]карбонил}амино)ацетил]-N-[1-(2,6-диметил-3-пиридинил)-2-оксо-2-({2-[(фенилметил)окси]фенил}амино)этил]-D-аллоизолейцинат
2,6-Диметилпиридин-3-карбоксальдегид (Aurora Feinchemie GmbH) (2,00 г, 16,1 ммол) и (D)-аллоизолейцин метиловый эфир гидрохлорид (2,93 г, 16,1 ммол) в метаноле (50 мл) и 2,2,2-трифторэтанол (50 мл) обрабатывали триэтиламином (2,24 мл, 16,1 ммол) и смесь перемешивали в атмосфере азота при комнатной температуре в течение 20 час.
(2R)-2,3-дигидро-1Н-инден-2-ил-({[(фенилметил)окси]карбонил}амино)уксусная кислота (5,24 г, 16,1 ммол) и 2-бензилоксифенилизонитрил (3,37 г, 16,1 ммол) добавляли и смесь перемешивали в атмосфере азота при комнатной температуре в течение 4 дней. Смесь концентрировали при пониженном давлении, затем разделили между этилацетатом (150 мл) и водой (150 мл) плюс насыщенным водным бикарбонатом натрия (6 мл). Водную фазу экстрагировали обратно этилацетатом (50 мл) и объединенные органические экстракты успешно промывали полунасыщенными водными растворами бикарбоната натрия, хлорида аммония и хлорида натрия (100 мл каждого), сушили над обезвоженным сульфатом магния и выпаривали под вакуумом, чтобы получить неочищенный продукт (12,01 г). Этот продукт очищали на Redisep силикагелевой колонке (330 г), элюировали 20-50% этилацетатом в циклогексане с получением 7,46 г указанного в заголовке соединения в виде пары диастереомеров.
HPLC Rt=3,88 и 3,96 мин (градиент 1); m/z [M+H]+=797
Промежуточное соединение 2
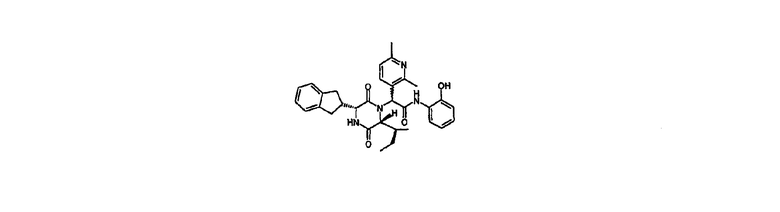
2-{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-2-(2,6-диметил-3-пиридинил)-N-(2-гидроксифенил)ацетамид
Неочищенный метил N-[(2R)-2-(2,3-дигидро-1Н-инден-2-ил)-2-({[(фенилметил)окси]карбонил}амино)ацетил]-N-[1-(2,6-диметил-3-пиридинил)-2-оксо-2-({2-[(фенилметил)окси]фенил}амино)этил]-D-аллоизолейцинат (промежуточное соединение 1) (7,46 г) растворили в этаноле (150 мл) и уксусной кислоте (10 мл), смесь гидрировали при 1 атмосфере Н2 над 10% палладия-на-угле (Degussa тип) (1,8 г смоченного водой 1:1 масс./масс.) в течение 18 час. Реакционную смесь упаривали под вакуумом и остаток разделяли между этилацетатом и водой с насыщенным водным бикарбонатом натрия, добавленным пока водная фаза была основной (рН 8). Водную фазу экстрагировали этилацетатом и объединенные органические экстракты промывали насыщенным водным бикарбонатом натрия и водой в соотношении 3:1 (100 мл), затем насыщенным соляным раствором перед сушкой над обезвоженным сульфатом магния и выпариванием под вакуумом. Неочищенный продукт очищали на Redisep силикагелевой колонке (120 г), элюировали 0-10% метанолом в этилацетате с получением указанного в заголовке соединения в виде пары диастереомеров (2,94 г).
HPLC Rt=2,75 и 2,81 мин (градиент 2); m/z [M+H]+=541
Промежуточное соединение 3
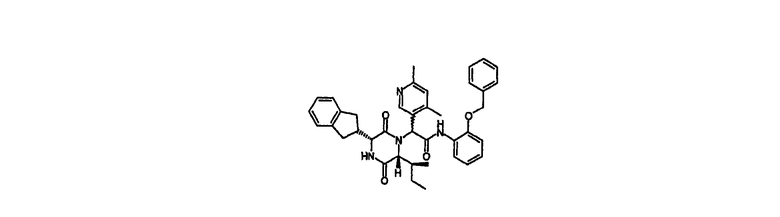
2-{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-2-(4,6-диметил-3-пиридинил)-N-{2-[(фенилметил)окси]фенил}ацетамид
4,6-Диметил-3-пиридинкарбальдегид1 (2,52 г) и метил D-аллоизолейцин гидрохлорид (3,4 г) растворили в 2,2,2-трифторэтаноле (50 мл). К этой смеси добавляли триэтиламин (2,61 мл) и реакционную смесь оставляли выстаиваться в течение 18 час. (2R)-2,3-дигидро-1Н-инден-2-ил({[(1,1-диметилэтил)окси]карбонил}амино)уксусную кислоту (5,44 г) и 2-[(фенилметил)окси]фенилизоцианид (4,18 г) с метанолом (10 мл) добавляли к реакционной смеси и раствор перемешивали при комнатной температуре в течение 3 дней. Растворитель удаляли in vacuo и остаток разделяли между дихлорметаном и водой. Органическую фазу пропускали через гидрофобный фритт и упаривали в вакууме. Осадок растворяли в 4N хлористом водороде в диоксане (50 мл) и реакционную смесь оставляли выстаиваться в течение 4 час. Растворитель удаляли in vacuo и осадок растворяли в дихлорметане (200 мл). К нему добавляли триэтиламин (20 мл) и реакционную смесь оставляли выстаиваться в течение 20 час. Реакционную смесь разделяли между дихлорметаном и водой. Органическую фазу пропускали через гидрофобный фритт и упаривали in vacuo. Осадок направляли в 4×90 г Biotage колонки и элюировали циклогексаном/этилацетатом (1:1, 1:2 об./об.) и этилацетатом. Требуемые фракции объединяли и выпаривали in vacuo с получением 2-{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-2-(4,6-диметил-3-пиридинил)-N-{2-[(фенилметил)окси]фенил}ацетамида (5,55 г, 47%) в виде желто-коричневой пены.
HPLC Rt=3,43, 3,45 мин (градиент 1); m/z [M+H]+=631
Ссылка:
1 Comins, Daniel L.; Weglarz, Michael A.; J. Org. Chem.; 53; 19; 1988; 4437-4442.
Промежуточное соединение 4
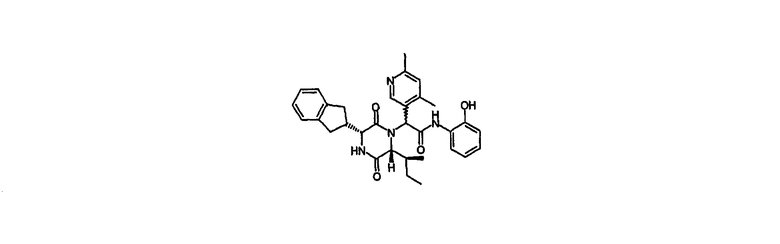
2-{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-2-(4,6-диметил-3-пиридинил)-N-(2-гидроксифенил)ацетамид
2-{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-2-(4,6-диметил-3-пиридинил)-N-{2-[(фенилметил)окси]фенил}ацетамид (промежуточное соединение 3) (3,30 г) растворяли в этаноле (75 мл) и гидрировали над палладием-на-угле (смоченным 10% Pd, 0,50 г) в течение 20 час. Катализатор удаляли фильтрованием и промывали дихлорметаном. Объединенные фильтрат и продукты промывания выпаривали in vacuo. Осадок направляли в 90 г Biotage колонку и элюировали этилацетатом. Требуемые фракции объединяли и упаривали in vacuo с получением 2-{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-2-(4,6-диметил-3-пиридинил)-N-(2-гидроксифенил)ацетамида (2,43 г, 87%) в виде бледно-желтого твердого вещества.
HPLC Rt=2,86 мин (градиент 1); m/z [M+H]+=541.
Промежуточное соединение 5
Гидрохлорид {(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}(2,6-диметил-3-пиридинил)уксусной кислоты
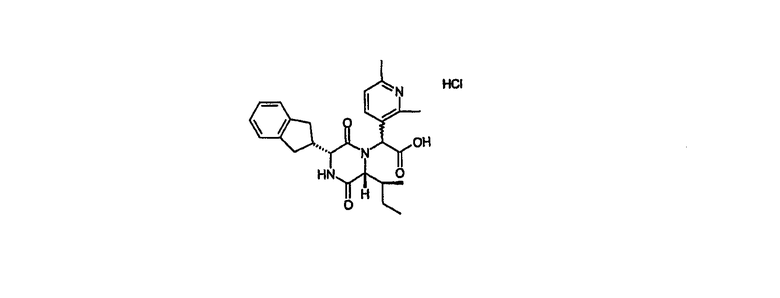
2-{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-2-(4,6-диметил-3-пиридинил)-N-(2-гидроксифенил)ацетамид (24,25 г, 45 ммол) (промежуточное соединение 2) и 1,1'-карбонилдиимидазол (11,7 г, 72 ммол) растворяли в сухом дихлорметане (200 мл) и оставляли выстаиваться в атмосфере азота в течение 20 час. Растворитель удаляли in vacuo и осадок растворяли в ацетоне (200 мл) и 2N соляной кислоте (20 мл). После перемешивания в течение 20 час растворитель удаляли in vacuo и осадок растворяли в метаноле (50 мл). Раствор наносили на аминопропиловый картридж (2×70 г) и элюировали метанолом (250 мл), а затем 10% уксусной кислоты в метаноле (250 мл). Требуемые фракции объединяли и выпаривали in vacuo. Осадок обрабатывали 2N соляной кислотой и полученный в результате раствор выпаривали in vacuo с получением указанного в заголовке соединения гидрохлорид {(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}(2,6-диметил-3-пиридинил)уксусной кислоты в виде желто-коричневого твердого вещества (12,21 г, 56%).
HPLC Rt=2,48 мин (градиент 2); m/z [M+H]+=450.
Промежуточное соединение 6
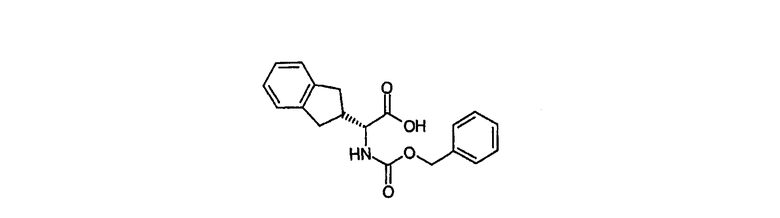
(2R)-2,3-дигидро-1Н-инден-2-ил({[(фенилметил)окси]карбонил}амино)уксусная кислота
(2R)-амино(2,3-дигидро-1Н-инден-2-ил)уксусную кислоту (1,91 г, 10 ммол) суспендировали в диоксане (10 мл) и воде (10 мл). К этой смеси добавляли триэтиламин (1,7 мл) и N-(бензилоксикарбонилокси)сукцинимид (2,54 г), реакционную смесь быстро перемешивали при комнатной температуре в течение 2 дней. Реакционную смесь выливали в воду (50 мл) и экстрагировали хлороформом (100 мл). Органическую фазу промывали 1N соляной кислотой (50 мл) и водой (50 мл). Далее высушивали над сульфатом магния, растворитель удаляли in vacuo с получением указанного в заголовке соединения (3,06 г, 94%):
1Н ЯМР (CDCl3) δ 7,40-7,29 (м, 5H), 7,21-7,11 (м, 4H), 5,28 (д, 1H, J=8,6 Гц), 5,11 (с, 2H), 4,57 (м, 1H), 3,14-2,79 (м, 5H); LCMS m/z 326 (MH+), Rt 3,35 мин (градиент 2).
Пример 1
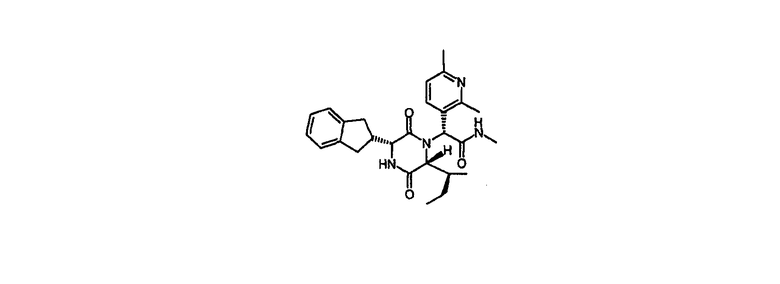
(2R)-2-{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-2-(2,6-диметил-3-пиридинил)-N-метилэтанамид
2-{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-2-(4,6-диметил-3-пиридинил)-N-(2-гидроксифенил)ацетамид (промежуточное соединение 2) (0,400 г, 0,74 ммол) и 1,1'-карбонилдиимидазол (0,192 г, 1,18 ммол) в сухом дихлорметане (10 мл) перемешивали при комнатной температуре в атмосфере N2 в течение 7 час. Смесь обрабатывали 2М раствором метиламина в тетрагидрофуране (1,849 мл, 3,70 ммол) и оставляли выстаиваться на ночь при комнатной температуре. Растворители удаляли продуванием в атмосфере N2 и осадок очищали на Redicep силикагелевой колонке (35 г), элюировали 0-10% метанолом в этилацетате с последующей очисткой на Kromasil KR100-10-C18 колонке обратного чередования фаз, элюированной водным ацетонитрилом (20-45% MeCN), содержащим 0,1% муравьиной кислоты. Это дает указанное в заголовке соединение в виде белого лиофилизата (30%) после сублимационной сушки из 1,4-диоксана.
HPLC Rt=2,44 мин (градиент 1); m/z [M+H]+=463
1Н ЯМР (CDCl3) δ 7,63 (д, 1H), 7,25-7,15 (м, 4H), 7,05 (д, 1H), 6,79 (д, 1H), 5,96 (кв., 1H), 5,35 (с, 1H), 4,07 (дд, 1H), 3,88 (д, 1H), 3,19-2,88 (м, 4H), 2,85 (д, 3H), 2,81-2,73 (м, 1H), 2,56 (с, 3H), 2,55 (с, 3H), 1,82-1,67 (м, 2H), 1,20-1,08 (м, 1H), 0,99 (д, 3H), 0,90 (т, 3H).
Аналогично приготавливали из промежуточного соединения 2 и диметиламина (2,0М в тетрагидрофуране):
Пример 2
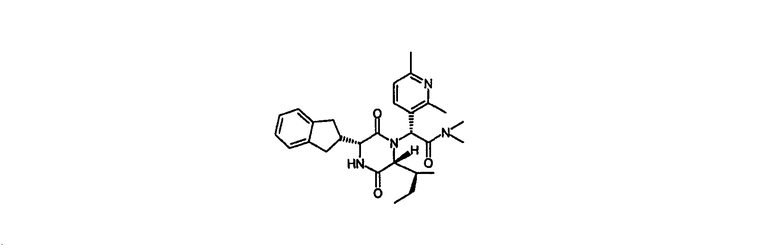
(2R)-2-{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-2-(2,6-диметил-3-пиридинил)-N,N-диметилэтанамид
в виде белого лиофилизата (33%) после сублимационной сушки из 1,4-диоксана.
HPLC Rt=2,69 мин (градиент 1); m/z [M+H]+=477
1Н ЯМР (CDCl3) δ 7,49 (д, 1H), 7,27-7,15 (м, 4H), 7,08 (д, 1H), 6,66 (с, 1H), 6,30 (д, 1H), 4,10 (дд, 1H), 4,05 (д, 1H), 3,22-3,08 (м, 3H), 2,99-2,84 (м, 4H), 2,80-2,70 (м, 4H), 2,63 (с, 3H), 2,58 (с, 3H), 1,65-1,53 (м, 1H), 0,97-0,78 (м, 2H), 0,71 (т, 3H), 0,46 (д, 3H).
Аналогично приготавливали из промежуточного соединения 2 и морфолина (3,7 ммол):
Пример 3 (Способ А)
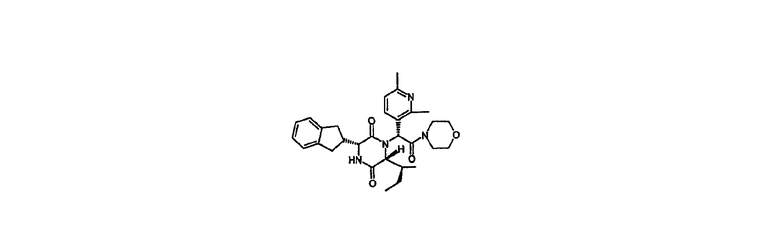
(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-1-[(1R)-1-(2,6-диметил-3-пиридинил)-2-(4-морфолинил)-2-оксоэтил]-6-[(1S)-1-метилпропил]-2,5-пиперазиндион
в виде белого лиофилизата (88 мг, 23%) после сублимационной сушки из 1,4-диоксана.
HPLC Rt=2,70 мин (градиент 2); m/z [M+H]+=519
1Н ЯМР (CDCl3) δ 7,49 (д, 1H), 7,27-7,15 (м, 4H), 7,10 (д, 1H), 6,68 (с, 1H), 6,40 (д, 1H), 4,10 (дд, 1H), 4,01 (д, 1H), 3,74-3,52 (м, 5H), 3,28-3,07 (м, 5Н), 2,97-2,84 (м, 2H), 2,79-2,71 (м, 1H), 2,62 (с, 3H), 2,59 (с, 3H), 1,65-1,53 (м, 1H), 0,98-0,80 (м, 2H), 0,70 (т, 3H), 0,45 (д, 3H).
Пример 3 (Способ B)

(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-1-[(1R)-1-(2,6-диметил-3-пиридинил)-2-(4-морфолинил)-2-оксоэтил]-6-[(1S)-1-метилпропил]-2,5-пиперазиндион
Суспензию гидрохлорида {(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}(2,6-диметил-3-пиридинил)уксусной кислоты (5,0 г, 10,3 ммол) (промежуточное соединение 5) в сухом дихлорметане (50 мл) обрабатывали 1,1-карбонилдиимидазолом (2,6 г, 16 ммол) и реакционную смесь перемешивали в атмосфере азота в течение 18 час. Морфолин (4,8 мл, 55 ммол) добавляли и полученный в результате раствор оставляли выстаиваться в атмосфере азота в течение 18 час. Растворитель удаляли in vacuo и осадок разделяли между этилацетатом и водой. Органическую фазу промывали солевым раствором и сушили над обезвоженным сульфатом магния. Растворитель удаляли in vacuo и осадок растворяли в дихлорметане. Полученную смесь наносили на основный картридж из оксида алюминия (240 г) и элюировали, используя градиент 0-7,5% метанола в диэтиловом эфире (9CV), 7,5-10% метанола в диэтиловом эфире (1CV) и 10% метанола в диэтиловом эфире (1СV). Требуемые фракции объединяли и выпаривали in vacuo с получением (3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-1-[(1R)-1-(2,6-диметил-3-пиридинил)-2-(4-морфолинил)-2-оксоэтил]-6-[(1S)-1-метилпропил]-2,5-пиперазиндиона в виде белого твердого вещества (2,4 г, 45%).
HPLC Rt=2,72 мин (градиент 2); m/z [M+H]+=519.
Пример 4
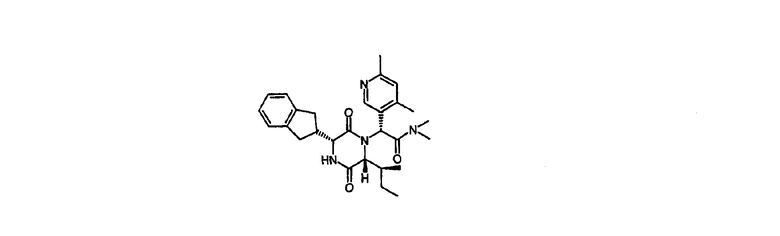
(2R)-2-{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-2-(4,6-диметил-3-пиридинил)-N,N-диметилэтанамид
2-{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-2-(4,6-диметил-3-пиридинил)-N-(2-гидроксифенил)ацетамид (промежуточное соединение 4) (0,700 г) и 1,1'-карбонилдиимидазол (0,324 г) растворяли в сухом дихлорметане (20 мл) и оставляли выстаиваться в течение 20 час. К одной половине данного раствора (10 мл) добавляли 2,0 М раствор диметиламина в тетрагидрофуране (5 мл) и реакционную смесь оставляли выстаиваться в течение 3 дней. Реакционную смесь разделяли между дихлорметаном и насыщенным водным раствором бикарбоната натрия. Органическую фазу пропускали через гидрофобный фритт и выпаривали в вакууме. Осадок наносили на силикагелевый картридж (10 г) и элюировали этилацетатом, а затем 5% метанолом в этилацетате. Требуемые фракции упаривали in vacuo и осадок очищали, используя Mass Directed AutoPrep. Это дает (2R)-2-{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-2-(4,6-диметил-3-пиридинил)-N,N-диметилэтанамид (0,10 г, 32%) в виде белой пены.
HPLC Rt=2,82 мин (градиент 1); m/z [M+H]+=477
1Н ЯМР (CDCl3) δ 8,32 (с, 1H), 7,26-7,15 (м, 4H), 7,08 (с, 1H), 6,71 (с, 1H), 6,16 (д, 1H), 4,17 (д, 1H), 4,10 (дд, 1H), 3,22-3,06 (м, 3H), 2,98 (с, 3H), 2,91 (м, 1H), 2,74 (дд, 1H), 2,67 (с, 3H), 2,57 (с, 3H), 2,39 (с, 3H), 1,56 (м, 1H), 0,93 (м, 1H), 0,85 (м, 1H), 0,68 (т, 3H), 0,45 (д, 3H).
Аналогично приготавливали из промежуточного соединения 4 и метиламина:
Пример 5
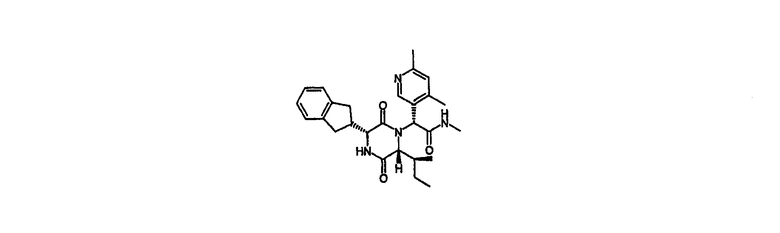
(2R)-2-{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-2-(4,6-диметил-3-пиридинил)-N-метилэтанамид
HPLC Rt=2,60 мин (градиент 1); m/z [M+H]+=463
1Н ЯМР (CDCl3) δ 8,48 (с, 1H), 7,25-7,14 (м, 4H), 7,04 (с, 1H), 6,72 (д, 1H), 6,07 (кв., 1H), 5,45 (с, 1H), 4,07 (дд, 1H), 3,90 (д, 1H), 3,17-3,04 (м, 3H), 2,92 (м, 1H), 2,86 (д, 3H), 2,76 (дд, 1H), 2,53 (с, 3H), 2,33 (с, 3H), 1,70 (м, 2H), 1,12 (м, 1H), 0,94 (д, 3H), 0,87 (т, 3H).
Аналогично приготавливали из промежуточного соединения 4 и морфолина:
Пример 6
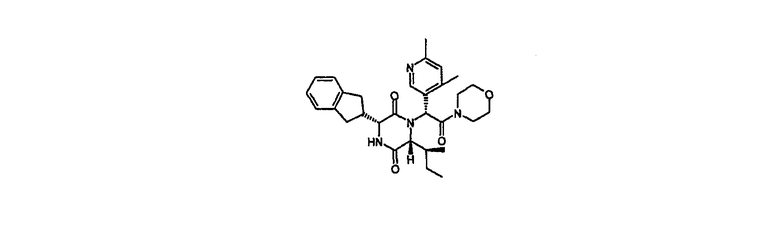
(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-1-[(1R)-1-(4,6-диметил-3-пиридинил)-2-(4-морфолинил)-2-оксоэтил]-6-[(1S)-1-метилпропил]-2,5-пиперазиндион
HPLC Rt=2,94 мин (градиент 2); m/z [M+H]+=519
1Н ЯМР (CDCl3) δ 8,34 (с, 1H), 7,25-7,15 (м, 4H), 7,09 (с, 1H), 6,71 (с, 1H), 6,21 (д, 1H), 4,14-4,07 (м, 2H), 3,73-3,47 (м, 5H), 3,23-3,05 (м, 5H), 2,95-2,83 (м, 2H), 2,74 (дд, 1H), 2,58 (с, 3H), 2,38 (с, 3Н), 1,56 (м, 1H), 0,94 (м, 1H), 0,86 (м, 1H), 0,68 (т, 3H), 0,44 (д, 3H).
Биологическая активность
Примеры 1-6 настоящего изобретения протестировали во всех биологических испытаниях, описанных ниже. Результаты по каждому соединению представлены в таблице ниже. Для сравнения в таблицу также включено соединение Х.
Биологическое испытание 1
Определение антагонистического сродства у рецепторов окситоцина-1 человека с использованием FLIPR
Культура клеток
Адгезивные клетки яичника китайского хомячка (CHO), стабильно экспрессирующие рекомбинантный рецептор окситоцина-1 человека (hOT), поддерживали в культуре в среде DMEM:F12 (Sigma, cat. No D6421), добавляли 10% теплую инактивированную околоплодную сыворотку теленка (Gibco/Invitrogen, cat. No. 01000-147), 2 мМ L-глутамина (Gibco/Invitrogen, cat. No. 25030-024) и 0,2 мг/мл G418 (Gibco/Invitrogen, cat. No. 10131-027). Клетки выращивали в виде монослоев в атмосфере воздух:СО2 в соотношении 95%:5% при температуре 37°С и пересевали каждые 3-4 дня, используя TrypLETM Express (Gibco/Invitrogen, cat. No. 12604-013).
Измерение [Ca 2+ ] с использованием FLIPR TM
CHO-hOT клетки высевали в черные обрамленные 384-луночные планшеты (Nunc) с прозрачной основой при плотности 10000 клеток на лунку в культуральной среде, как описано выше, и выдерживали в течение ночи (95%:5%, воздух:СО2, при 37°С). После удаления культуральной среды клетки инкубировали в течение 1 час при 37°С в Tyrode среде (NaCl, 145 мМ; KCl, 2,5 мМ; HEPES, 10 мМ; глюкоза 10 мМ; MgCl2, 1,2 мМ; CaCl2, 1,5 мМ), содержащей пробенацид (0,7 мг/мл), индикатор цитоплазматического кальция, Fluo-4 (4 мкМ; Teflabs, USA) и гасящий агент Brilliant Black (Бриллиантовый черный) (250 мкМ; Molecular Devices, UK). Затем клетки инкубировали в течение дополнительных 30 мин при 37°С либо только с буфером, либо с буфером, содержащим ОТ антагонист, до помещения клеток в FLIPRTM (Molecular Devices, UK) для монитора клеточной флуоресценции (λex=488 нм, λEM=540 нм) до или после добавления субмаксимальной концентрации окситоцина (ЕС80).
Анализ данных
Функциональные реакции с применением FLIPR анализировали, используя Activity Base Version 5.0.10.
Биологическое испытание 2
Анализ связывания окситоцина
Препараты
Мембраны готовили из СНО клеток, экспрессирующих рекомбинантные рецепторы окситоцина человека. Мембранный препарат замораживали в аликвотах при -70°С до использования.
Протокол анализа связывания
Мембраны (~50 мкг) инкубировали в 200 мкл аналитического буфера (50 мМ Tris, 10 мМ MgCl2 и 0,1% бычий сывороточный альбумин, рН 7,5), содержащего ~2,4 нМ [3H]-окситоцина при отсутствии (полное связывание) или в присутствии (без специфического связывания) 1 мкМ немаркированного окситоцина и повышенных концентраций соединений примеров 1-6 или сравнительных соединений. Инкубацию выполняли при комнатной температуре в течение 60 мин. Реакции останавливали 3 мл ледяного буфера и отфильтровывали через Whatman GF/C фильтровальную бумагу, предварительно смоченную в 0,3% полиэтиленимина. Фильтры промывали 4 раза 3 мл буфера, используя клеточный коллектор Брандела. Фильтры включали 3 мл Ready Safe сцинтилляционной жидкости (Beckman).
Специфическое связывание составило приблизительно 90% от общего связывания.
Анализ данных
IC50 значения определили из экспериментов конкурентного связывания, используя анализ нелинейной регрессии (GraphPad), и конвертировали до Ki, используя метод Cheng и Prusoff, 1974. Данные представлены в виде средних значений.
Биологическое испытание 3
Определение in vitro собственного клиренса в микросомах
NADP регенерационный буфер для использования в инкубациях был свежеприготовлен в день проведения анализа. Он содержал 7,8 мг глюкозо-6-фосфат (мононатриевая соль), 1,7 мг NADP и 6 единиц глюкозо-6-фосфат дегидрогеназы на 1 мл 2% бикарбоната натрия. Микросомы (человека, женские; макак-крабоедов, самок; собак, сук; крыс, самок), приготовленные в рН 7,4 фосфатного буфера, и содержали 0,625 мг белка/мл.
Если не указано особо, все последующие шаги осуществляли с помощью Tecan Genesis 150/8 RSP. 1,25 мМ маточного раствора соединений приготавливали в смеси ацетонитрил/вода (1:1). 25 мкл 1,25 мМ маточного раствора добавляли к 600 мкл смеси ацетонитрил/вода (1:1) с получением 50 мкМ раствора. Для каждого образца 50 мкМ растворов (10 мкл) добавляли к микросомам (790 мкл) в микропланшет (Porvair, 96 лунок, квадратный).
400 мкл микросомального раствора, содержащего соединение, перенесли в микропланшет (Porvair, 96 лунок, круглый) и предварительно промывали при 37°С в течение 5 мин перед тем, как инициировать инкубации. Все инкубации инициировали добавлением 100 мкл NADP регенерационной системы к предварительно промытым микросомам. Смеси инкубировали при 37°С в Techne обогревающем блоке. Следующие 0, 3, 6, 12 и 30 мин инкубации брали 20 мкл аликвоты и добавляли к 100 мкл ацетонитрила, содержащего внутренний стандарт.
Для определения скорости метаболизма инкубации выполняли при концентрации соединения 0,5 мкМ и концентрации белка 0,5 мг/мл. Концентрация растворителя при инкубации составила 0,5%.
Концентрации тестируемых соединений определяли с помощью LC/MC/MC; результаты представили как соотношение анализируемое вещество:площадь пика внутреннего стандарта.
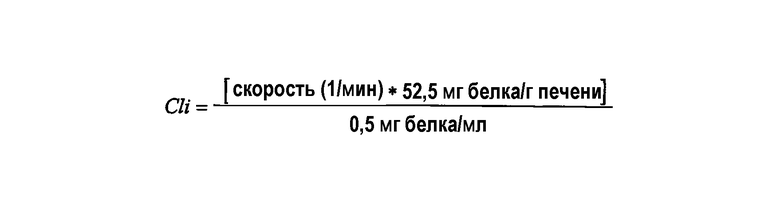
Скорость исчезновения вычисляли с помощью подбора единичного экспоненциального спада по кривой зависимости концентрация - время, используя Excel, и собственный клиренс вычисляли, используя следующую формулу:
Результаты
Примеры 1-6 настоящего изобретения и также сравнительное соединение Х=(2R)-2-[(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-изобутил-2,5-диоксопиперазин-1-ил]-N,N-диметил-2-(6-метилпиридин -3-ил)этанамид (пример 209 в WO 03/053443) протестировали в вышеописанных анализах.
Сравнительное соединение Х при тестировании в биологических испытаниях 1 и 2 показало активность, сходную с той, что продемонстрировали соединения 1-6 настоящего изобретения, фактически каждое из этих соединений продемонстрировало fpKi между 8,1 и 9,2 (Биологическое испытание 1) и pKi между 8,8 и 10,5 (Биологическое испытание 2).
Однако соединения настоящего изобретения демонстрировали поразительное улучшение in vitro собственного клиренса в микросомах (биологическое испытание 3) при сопоставлении со сравнительным соединением Х.
Ключ к таблице:
+соответствует 1-8 мл/мин/мг;
++соответствует 9-15 мл/мин/мг;
+++соответствует 16-20 мл/мин/мг;
++++соответствует 21-30 мл/мин/мг;
+++++соответствует>31 мл/мин/мг.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ДИКЕТОПИПЕРАЗИНЫ КАК АНТАГОНИСТЫ ОКСИТОЦИНА | 2002 |
|
RU2303032C2 |
| ЗАМЕЩЕННЫЕ ДИКЕТОПИПЕРАЗИНЫ И ИХ ИСПОЛЬЗОВАНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ ОКСИТОЦИНА | 2004 |
|
RU2343152C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| ПРОИЗВОДНОЕ ПИПЕРИДИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2730508C2 |
| ПРОИЗВОДНЫЕ БОРОНОВОЙ КИСЛОТЫ | 2018 |
|
RU2793315C2 |
| СП0СОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ ДИГИДРОИНДЕНАМИДА, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИИ, СОДЕРЖАЩИЕ ДАННЫЕ СОЕДИНЕНИЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗЫ | 2009 |
|
RU2528408C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛОВ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2689777C1 |
| КОМБИНИРОВАННОЕ ЛЕЧЕНИЕ АГОНИСТОМ ТОЛЛ-ПОДОБНОГО РЕЦЕПТОРА (TLR7) И ИНГИБИТОРОМ СБОРКИ КАПСИДА ВИРУСА ГЕПАТИТА В | 2016 |
|
RU2718917C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| ПИРИДАЗИНОНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PARP | 2008 |
|
RU2490265C2 |
Описываются пиперазиндионы общей формулы (I), в которой R1 означает 2-инданил, R2 означает 1-метилпропил, R3 и R4 совместно с атомами азота, к которому они присоединены, представляют морфолиногруппу и его фармацевтически приемлемые соли. Соединения обладают антагонистическим действием на рецептор окситоцина. Описывается также фармацевтическая композиция на основе соединения формулы (I). 2 н. и 4 з.п. ф-лы, 1 табл.
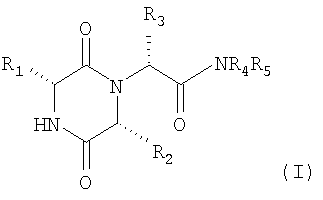
1. Пиперазиндионы общей формулы (I)
в котором R1 означает 2-инданил, R2 означает 1-метилпропил, R3 означает 2,6-диметил-3-пиридил, R4 и R5 совместно с атомом азота, к которому они присоединены, представляют морфолиногруппу, и его фармацевтически приемлемая соль присоединения кислоты, в которой кислота выбрана из соляной кислоты, бромистоводородной кислоты, азотной кислоты, фосфорной кислоты, серной кислоты, метансульфоновой кислоты, этансульфоновой кислоты, бензолсульфоновой и п-толуолсульфоновой кислоты, лимонной кислоты, виннокаменной кислоты, молочной кислоты, пировиноградной кислоты, уксусной кислоты, янтарной кислоты, фумаровой кислоты и малеиновой кислоты.
2. Соединение по п.1, представляющее собой (3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-1-[(1R)-1-(2,6-диметил-3-пиридинил)-2-(4-морфолинил)-2-оксоэтил]-6-[(1S)-1-метилпропил]-2,5-пиперазиндион бензолсульфонат.
3. Соединение по п.1, представляющее собой (3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-1-[(1R)-1-(2,6-диметил-3-пиридинил)-2-(4-морфолинил)-2-оксоэтил]-6-[(1S)-1-метилпропил]-2,5-пиперазиндион.
4. Фармацевтическая композиция, обладающая антагонистическим действием на рецептор окситоцина, включающая соединение по п.1 в эффективном количестве и фармацевтически приемлемый носитель.
5. Фармацевтическая композиция по п.4, включающая соединение по п.2 и фармацевтически приемлемый носитель.
6. Фармацевтическая композиция по п.4, включающая соединение по п.3 и фармацевтически приемлемый носитель.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| RU 95108387 A1, 10.12.1996. | |||

