Область техники
Данное изобретение относится к соединениям и способам ингибирования ферментов. В частности, изобретение относится к терапевтическим способам, основанным на ингибировании ферментов.
Уровень техники
У эукариот деградация белка преимущественно опосредована метаболическим путем убиквитина, в котором белки, которые становятся мишенью для разрушения, лигируются с полипептидом из 76 аминокислот убиквитином. Ставшие мишенью, убиквитинированные белки затем служат субстратами для 26S протеасомы, мультикаталитической протеазы, которая расщепляет белки на короткие пептиды посредством действия ее трех основных протеолитических активностей. Обладая основной функцией во внутриклеточном метаболизме белка, опосредованная протеасомой деградация также играет ключевую роль во многих процессах, таких как презентация главного комплекса гистосовместимости (MHC) класса I, апоптоз, деление клетки и активация NF-κB.
20S протеасома представляет собой 700 кДа комплекс мультикаталитических протеаз цилиндрической формы, содержащий 28 субъединиц, организованных в четыре кольца, который играет важную роль при регуляции роста клеток, презентации главного комплекса гистосовместимости (MHC) класса I, апоптозе, процессинге антигенов, активации NF-κB и трансдукции провоспалительных сигналов. В дрожжах и других эукариотах 7 различных субъединиц α формируют внешние кольца и 7 различных субъединиц β составляют внутренние кольца. Субъединицы α служат в качестве участков связывания для 19S (PA700) и 11S (PA28) регуляторных комплексов, а также в качестве физического барьера для внутренней протеолитической полости, образованной двумя кольцами из субъединиц β. Таким образом, in vivo, протеасома, как полагают, существует в виде частицы 26S («26S протеасома»). Эксперименты in vivo показали, что можно легко найти зависимость между ингибированием формы 20S протеасомы и ингибированием 26S протеасомы. Расщепление N-концевой пропоследовательности субъединицы β во время образования частицы высвобождает N-концевые остатки треонина, которые служат каталитическими нуклеофилами. Субъединицы, ответственные за каталитическую активность в протеасоме, таким образом, содержат N-концевой нуклеофильный остаток, и данные субъединицы принадлежат семейству гидролаз с N-концевой нуклеофильной группой (Ntn) (где N-концевой нуклеофильный остаток представляет собой, например, Cys, Ser, Thr и другие нуклеофильные группы). Данное семейство включает, например, пенициллин-G-ацилазу (PGA), пенициллин-V-ацилазу (PVA), глутамин-ФРПФ-амидотрансферазу (GAT) и бактериальную гликозиласпарагиназу. В дополнение к распространенным экспрессируемым субъединицам β, высшие позвоночные животные также имеют три индуцируемые γ-интерфероном субъединицы β (LMP7, LMP2 и MECL1), которые замещают их нормальные соответствующие части, X, Y и Z соответственно, таким образом изменяя каталитическую активность протеасомы. При помощи различных пептидных субстратов три основные протеолитические активности были определены для 20S протеасомы эукариот: химотрипсино-подобная активность (CT-L), при которой происходит расщепление после больших гидрофобных остатков; трипсино-подобная активность (T-L), при которой происходит расщепление после основных остатков, и петидилглутамил-пептидгидролазная активность (PGPH), при которой происходит расщепление после кислотных остатков. Протеасоме также приписывают две дополнительные менее изученные активности: активность BrAAP, при которой происходит расщепление после аминокислот с разветвленной цепью, и активность SNAAP, при которой происходит расщепление после небольших нейтральных аминокислот. В основные протеолитические активности протеасомы, по-видимому, вносят вклад различные каталитические участки, поскольку ингибиторы, точечные мутации в субъединицах β и замена индуцируемых γ-интерфероном субъединиц β изменяют данные активности в различной степени.
Существует несколько примеров небольших молекул, которые использовали для ингибирования активности протеасомы; однако данным соединениям, в основном, недостает специфичности, устойчивости или активности, необходимой для исследования и использования роли протеасомы на клеточном и молекулярном уровне. Поэтому синтез низкомолекулярного ингибитора(ов) с повышенной специфичностью к месту связывания, с улучшенной устойчивостью и растворимостью и с увеличенной активностью необходим для того, чтобы осуществить исследование роли протеасомы на клеточном и молекулярном уровне.
Сущность изобретения
Изобретение относится к классам молекул, известным как α',β'-эпоксиды пептидов и α',β'-азиридины пептидов. Подразумевается, что исходные молекулы эффективно, необратимо и селективно связываются с гидролазами, содержащими N-концевые нуклеофильные группы (Ntn), и могут специфически ингибировать определенную активность ферментов, обладающих множественной каталитической активностью.
Ранее предполагали, что протеасома только удаляет денатурированные белки и белки с неправильной укладкой, а теперь обнаружили, что протеасома образуют протеолитический аппарат, который регулирует уровни разнообразных внутриклеточных белков посредством их деградации зависимым от сигналов способом. Следовательно, существует большой интерес к обнаружению реагентов, которые могут специфически изменять активности протеасомы и других гидролаз Ntn и, таким образом, использоваться в качестве зондов для изучения роли данных ферментов в биологических процессах. Здесь описаны, синтезированы и исследованы соединения, которые действуют на гидролазы Ntn. Описаны и заявлены эпоксиды пептидов и азиридины пептидов, которые могут сильно, селективно и необратимо ингибировать определенные активности протеасомы.
В отличие от некоторых других ингибиторов на основе пептидов описанные здесь эпоксиды пептидов и азиридины пептидов, как ожидают, существенно не ингибируют непротеасомные протеазы, такие как трипсин, химотрипсин, катепсин В, папаин и кальпаин, при концентрациях вплоть до 50 мкМ. При более высоких концентрациях можно наблюдать ингибирование, но следовало бы ожидать, что оно будет конкурентным и не необратимым, если ингибитор просто конкурирует с субстратом. Также ожидают, что новые эпоксиды пептидов и азиридины пептидов ингибируют активацию NF-κB и стабилизируют уровни p53 в культуре клеток. Кроме того, данные соединения, как ожидается, обладают противовоспалительной активностью. Таким образом, данные соединения могут представлять собой уникальные молекулярные зонды, которые обладают многоцелевым назначением для исследования функции фермента Ntn при нормальных биологических и патологических процессах.
В одном аспекте изобретение относится к ингибиторам, включающим трехчленное кольцо, содержащее гетероатом. Такие ингибиторы могут ингибировать каталитическую активность ферментов гидролаз с N-концевой нуклеофильной группой (например, 20S протеасома или 26S протеасома), если указанные ингибиторы присутствуют в концентрациях ниже приблизительно 50 мкМ. Относительно 20S протеасомы, определенные ингибиторы гидролазы ингибируют химотрипсино-подобную активность 20S протеасомы, когда ингибитор присутствует в концентрациях ниже приблизительно 5 мкМ, и не ингибируют трипсино-подобную активность или активность PGPH 20S протеасомы, когда присутствует в концентрациях ниже приблизительно 5 мкМ. Ингибитор гидролазы может представлять собой, например, α',β'-эпоксикетон или α',β'-азиридинкетон, и пептид может представлять собой тетрапептид. Пептид может включать разветвленные или линейные боковые цепи, такие как водород, C1-6-алкил, C1-6-гидроксиалкил, C1-6-алкоксиалкил, арил, C1-6-аралкил, C1-6-алкиламид, C1-6-алкиламин, C1-6-карбоновая кислота, C1-6-эфир карбоновой кислоты, C1-6-алкилтиол или C1-6-алкилтиоэфир, например изобутил, 1-нафтил, фенилметил и 2-фенилэтил. α'-Углерод α',β'-эпоксикетона или α',β'-азиридинкетона может представлять собой хиральный атом углерода, такой как углерод в конфигурации (R) или β, как они определены здесь.
В другом аспекте изобретение относится к фармацевтическим композициям, включающим фармацевтически приемлемый носитель и фармацевтически эффективное количество ингибитора гидролазы, который среди прочего облегчает симптомы нейродегенеративного заболевания (такого как болезнь Альцгеймера), заболевания, вызывающего мышечное истощение, рака, хронических инфекционных заболеваний, лихорадки, бездействия мускулатуры, денервации, поражения нерва, голодания и связанных с иммунной системой состояний.
В другом аспекте изобретение относится к противовоспалительным композициям.
В другом аспекте изобретение относится к следующим способам: ингибирования или уменьшения инфицирования ВИЧ у пациента; влияния на уровень экспрессии вирусного гена у пациента; изменения множества антигенных пептидов, продуцируемых протеасомой в организме; определения, регулируются ли протеолитической активностью определенной гидролазы Ntn клеточный процесс, процесс развития или физиологический процесс или продуцирование в организме; лечения болезни Альцгеймера у пациента; снижения скорости деградации мышечных белков в клетке; снижения скорости внутриклеточной деградации белка в клетке; снижения скорости деградации белка p53 в клетке; ингибирования роста связанных с p53 раковых образований у пациента; ингибирование презентации антигена в клетке; подавления иммунной системы пациента; ингибирования деградации IκB-α в организме; уменьшения содержания NF-κB в клетке, мускулатуре, органе или у пациента; воздействия на циклин-зависимые циклы эукариотических клеток; лечения пролиферативных заболеваний у пациента; воздействия на зависимое от протеасомы регулирование онкогенных белков в клетке; лечения роста опухоли у пациента; лечения связанного с p53 апоптоза у пациента; и скрининга белков, процессируемых гидролазами с N-концевой нуклеофильной группой в клетке. Каждый из данных способов включает введение или контактирование с эффективным количеством композиции, содержащей описанные здесь ингибиторы гидролазы, пациенту, в клетку, в ткань, орган или организм.
Другие характеристики и преимущества изобретения станут очевидны из следующего ниже подробного описания и из формулы изобретения.
Подробное описание изобретения
Изобретение относится к композициям, применимым в качестве ингибиторов фермента. Данные композиции, главным образом, применимы для ингибирования ферментов, содержащих нуклеофильную группу на N-конце. Например, активность ферментов или субъединиц ферментов, содержащих N-концевые аминокислоты с нуклеофильными группами в их боковых цепях, такие как треонин, серин или цистеин, можно успешно ингибировать описанными здесь ингибиторами фермента. Активности ферментов или субъединиц ферментов, содержащих неаминокислотные нуклеофильные группы на их N-концах, такие как, например, защитные группы или углеводороды, можно также успешно ингибировать описанными здесь ингибиторами фермента.
Несмотря на то, что это не связано с какой-либо определенной теорией процесса, полагают, что такая N-концевая нуклеофильная группа Ntn образует ковалентные аддукты с эпоксидной функциональной группой описанных здесь ингибиторов фермента. Например, в субъединице β5/Pre2 20S протеасомы N-концевой треонин, как полагают, необратимо образует морфолиновый или пиперазиновый аддукт при реакции с эпоксидом или азиридином пептида, таких как описано ниже. Такое образование аддукта повлекло бы за собой расщепление с размыканием цикла эпоксида или азиридина.
В вариантах осуществления, включающих такие группы, связанные с α'-углеродами, стереохимическая конфигурация α'-углерода (углерод, который образует часть эпоксидного или азиридинового кольца) может представлять собой (R) или (S). Изобретение частично базируется на структурно-функциональной информации, описанной здесь, которая предполагает следующее предпочтительное стереохимическое взаиморасположение. Следует отметить, что предпочтительное соединение может содержать большое количество стереоцентров, обозначенных как взаиморасположенные сверху-снизу (или β-α, где β, как изображено здесь, находится выше плоскости страницы) или как (R)-(S) (то есть не требуется, чтобы каждый стереоцентр в соединении соответствовал установленному предпочтению). В некоторых предпочтительных вариантах осуществления стереохимия α'-углерода представляет собой (R), то есть атом X находится в расположении β или выше плоскости молекулы.
Что касается стереохимии, правила Кана-Ингольда-Прелога для определения абсолютной стереохимии представляют собой следующее. Данные правила описаны, например, в Organic Chemistry, Fox and Whitesell; Jones and Bartlett Publishers, Boston, MA (1994); Section 5-6, pp 177-178, раздел которой таким образом включен сюда путем ссылки. Пептиды могут содержать повторяющиеся структуры основной цепи с боковыми цепями, расходящимися от звена основной цепи. Вообще, каждое звено основной цепи содержит боковую цепь, связанную с ней, хотя в некоторых случаях боковая цепь представляет собой атом водорода. В других вариантах осуществления не каждое звено основной цепи содержит связанную боковую цепь. Пептиды, используемые для получения эпоксидов пептида или азиридинов пептида, имеют два или более звеньев основной цепи. В некоторых вариантах осуществления, применимых для ингибирования химотрипсино-подобной активности (CT-L) протеасомы, присутствует от двух до восьми звеньев основной цепи, и в некоторых вариантах осуществления для ингибирования CT-L присутствует от двух до шести звеньев основной цепи.
Боковые цепи, отходящие от звеньев основной цепи, могут включать природные алифатические или ароматические боковые цепи аминокислот, такие как водород (глицин), метил (аланин), изопропил (валин), втор-бутил (изолейцин), изобутил (лейцин), фенилметил (фенилаланин), и боковую цепь, представляющую собой аминокислоту пролин. Боковые цепи могут также представлять собой разветвленные или линейные алифатические или ароматические группы, такие как этил-, н-пропил-, н-бутил-, трет-бутил- и арилзамещенные производные, такие как 1-фенилэтил, 2-фенилэтил, (1-нафтил)метил, (2-нафтил)метил, 1-(1-нафтил)этил, 1-(2-нафтил)этил, 2-(1-нафтил)этил, 2-(2-нафтил)этил и подобные соединения. Арильные группы могут дополнительно замещаться разветвленными или линейными C1-6-алкильными группами, или замещенными алкильными группами, ацетилом и тому подобным или дополнительно арильными группами, или замещенными арильными группами, такими как бензоил и тому подобное. Гетероарильные группы также можно использовать в качестве заместителей боковых цепей. Гетероарильные группы включают азот-, кислород- и серосодержащие арильные группы, такие как тиенил, бензотиенил, нафтотиенил, тиантренил, фурил, пиранил, изобензофуранил, хроменил, пирролил, имидазолил, пиразолил, пиридил, пиразинил, индолил, пуринил, хинолил и тому подобное.
В некоторых вариантах осуществления полярные или заряженные остатки можно вводить в эпоксиды пептидов или азиридины пептидов. Например, можно вводить встречающиеся в природе аминокислоты, такие как содержащие гидроксигруппу (Thr, Tyr, Ser) или серосодержащие (Met, Cys), а также и заменимые аминокислоты, например таурин, карнитин, цитруллин, цистин, орнитин, норлейцин и другие. Также можно вводить не встречающиеся в природе заместители боковых цепей с заряженными или полярными группами, такими как, например, цепи C1-6-алкил или C6-12-арильные группы с одной или более гидрокси-, алкокси- с короткой цепью, сульфидной, тио-, карбоксильной, эфирной, фосфо-, амидо- и аминогруппами, или такими заместителями, замещенными одним или более атомами галогенов. В некоторых предпочтительных вариантах осуществления существует, по меньшей мере, одна арильная группа, присутствующая в боковой цепи пептидной части.
В некоторых вариантах осуществления звенья основной цепи представляют собой амидные звенья [-NH-CHR-C(=O)-], в которых R представляет собой боковую цепь. Такое обозначение не исключает встречающуюся в природе аминокислоту пролин или другие не встречающиеся в природе циклические вторичные аминокислоты, которые известны специалистам в данной области.
В других вариантах осуществления звенья основной цепи представляют собой N-алкилированные амидные звенья (например, N-метил и тому подобное), олефиновые аналоги (в которых одна или более амидных связей заменены на олефиновые связи), аналоги тетразола (в которых кольцо тетразола придает цис-конфигурацию основной цепи) или комбинации таких связей в основной цепи. В еще одних вариантах осуществления α-углерод аминокислоты модифицирован введением α-алкильного заместителя, например аминоизомасляная кислота. В некоторых дополнительных вариантах осуществления боковые цепи локально модифицированы, например, ΔE- или ΔZ-дегидромодификацией, в которой присутствует двойная связь между α и β атомами боковой цепи, или, например, ΔE- или циклопропильной ΔZ-модификацией, в которой присутствует циклопропильная группа между α и β атомами боковой цепи. В еще одних дополнительных вариантах осуществления, использующих группы аминокислот, можно использовать D-аминокислоты. Дополнительные осуществления могут включать в себя циклизацию боковой цепи к основной цепи, образование дисульфидной связи, образование лактама, азосвязь и другие модификации, обсуждаемые в книге "Peptides and Mimics, Design of Conformationally Constrained" by Hruby and Boteju, in "Molecular Biology and Biotechnology: A Comprehensive Desk Reference", ed. Robert A. Meyers, VCH Publishers (1995), pp. 658-664, которая, таким образом, включена сюда в качестве ссылки.
Один аспект изобретения относится к соединениям, имеющим структуру формулы (I), или их фармацевтически приемлемой соли.
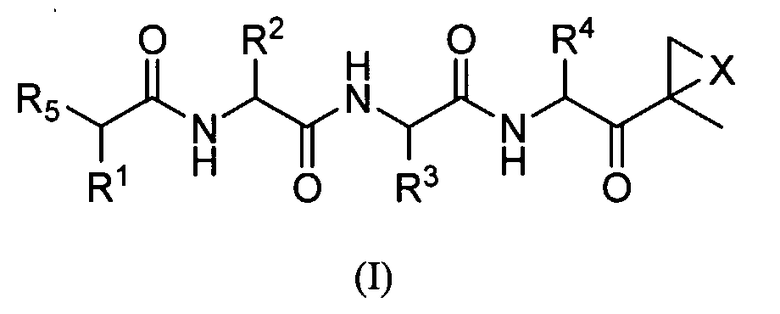
где каждый A независимо выбран из C=O, C=S и SO2, предпочтительно C=O;
каждый B независимо выбран из C=O, C=S и SO2, предпочтительно C=O;
D отсутствует или представляет собой C1-8-алкил;
G выбран из О, NH и N-C1-6-алкила;
K отсутствует или выбран из C=O, C=S и SO2, предпочтительно K отсутствует или представляет собой C=O;
L отсутствует или выбран из C=O, C=S и SO2, предпочтительно L отсутствует или представляет собой C=O;
M отсутствует или представляет собой C1-8-алкил;
Q отсутствует или выбран из О, NH и N-C1-6-алкила, предпочтительно Q отсутствует, представляет собой О или NH, наиболее предпочтительно Q отсутствует;
X выбран из О, S, NH и N-C1-6-алкила, предпочтительно представляет собой О;
каждый V независимо отсутствует или выбран из О, S, NH и N-C1-6-алкила, предпочтительно V отсутствует или представляет собой О;
W отсутствует или независимо выбран из О, S, NH и N-C1-6-алкила, предпочтительно представляет собой О;
Y отсутствует или выбран из О, NH, N-C1-6-алкила, S, SO, SO2, CHOR10 и CHCO2R10;
каждый Z независимо выбран из О, S, NH и N-C1-6-алкила, предпочтительно представляет собой О;
R1, R2, R3 и R4 каждый независимо выбран из C1-6-алкила, C1-6-гидроксиалкила, C1-6-алкоксиалкила, арила, C1-6-аралкила и R14DVKOC1-3-алкила-, где, по меньшей мере, один из R1 и R3 представляет собой R14DVKOC1-3-алкил-;
R5 представляет собой N(R6)LQR7;
R6 выбран из водорода, OH и C1-6-алкила, предпочтительно представляет собой C1-6-алкил;
R7 представляет собой дополнительную цепь аминокислот, водород, защитную группу, арил или гетероарил, любой из которых необязательно замещен галогеном, карбонилом, нитро, гидрокси, арилом, C1-5-алкилом; или R7 выбран из C1-6-алкила, C1-6-алкенила, C1-6-алкинила, C1-6-аралкила, C1-6-гетероаралкила, R8ZA-C1-8-алкила-, R11Z-C1-8-алкила-, (R8O)(R9O)P(=O)O-C1-8-алкил-ZAZ-C1-8-алкила-, (R8O)(R9O)P(=O)O-C1-8-алкил-Z-C1-8-алкила-, R8ZA-C1-8-алкил-ZAZ-C1-8-алкила-, гетероциклилMZAZ-C1-8-алкила-, (R8O)(R9O)P(=O)O-C1-8-алкила-, (R10)2N-C1-8-алкила-, (R10)3N+-C1-8-алкила-, гетероциклилM-, карбоциклилM-, R11SO2C1-8-алкила- и R11SO2NH; или
R6 и R7, взятые вместе, представляют собой C1-6-алкил-Y-C1-6-алкил, C1-6-алкил-ZA-C1-6-алкил, A-C1-6-алкил-ZA-C1-6-алкил, A-C1-6-алкил-A или C1-6-алкил-A, предпочтительно C1-2-алкил-Y-C1-2-алкил, C1-2-алкил-ZA-C1-2-алкил, А-C1-2-алкил-ZA-C1-2-алкил, A-C1-3-алкил-A или C1-4-алкил-A, с образованием цикла, предпочтительно R6 представляют собой водород и R7 представляют собой C1-6-алкил;
R8 и R9 независимо выбраны из водорода, катиона металла, C1-6-алкила, C1-6-алкенила, C1-6-алкинила, арила, гетероарила, C1-6-аралкила и C1-6-гетероаралкила, предпочтительно из водорода, катиона металла и C1-6-алкила или R8 и R9, взятые вместе, представляют собой C1-6-алкил, с образованием цикла;
каждый R10 независимо выбран из водорода и C1-6-алкила, предпочтительно из C1-6-алкила;
каждый R11 независимо выбран из водорода, OR10, C1-6-алкила, C1-6-алкенила, C1-6-алкинила, карбоциклила, гетероциклила, арила, гетероарила, C1-6-аралкила и C1-6-гетероаралкила;
R14 выбран из водорода, (R15O)(R16O)P(=O)W-, R15GB-, гетероциклила-, (R17)2N-, (R17)3N+-, R17SO2GBG- и R15GBC1-8-алкила-, где группа C1-8-алкил необязательно замещена OH, C1-8-алкилW (необязательно замещенным галогеном, предпочтительно фтором), арилом, гетероарилом, карбоциклилом, гетероциклилом и C1-6-аралкилом, предпочтительно, по меньшей мере, наличие одного R14, который является отличным от водорода;
R15 и R16 независимо выбраны из водорода, катиона металла, C1-6-алкила, C1-6-алкенила, C1-6-алкинила, арила, гетероарила, C1-6-аралкила и C1-6-гетероаралкила, предпочтительно из водорода, катиона металла и C1-6-алкила, или R15 и R16, взятые вместе, представляют собой C1-6-алкил, с образованием цикла; и
каждый R17 независимо выбран из водорода, OR10, C1-6-алкила, C1-6-алкенила, C1-6-алкинила, карбоциклила, гетероциклила, арила, гетероарила, C1-6-аралкила и C1-6-гетероаралкила;
при условии, что R6 представляет собой H, L представляет собой C=O и Q отсутствует, R7 не является водородом, C1-6-алкилом или замещенным или незамещенным арилом или гетероарилом и
D, G, V, K и W выбраны так, что не существует связей О-О, N-O, S-N или S-O.
Подходящие N-концевые защитные группы, известные в области синтеза пептидов, включают трет-бутоксикарбонил (Boc), бензоил (Bz), флуорен-9-илметоксикарбонил (Fmoc), трифенилметил (тритил) и трихлорэтоксикарбонил (Troc) и тому подобное. Использование различных N-защитных групп, например бензилоксикарбонильной группы или трет-бутоксикарбонильной группы (Boc), различных конденсирующих реагентов, например, дициклогексилкарбодиимид (DCC), 1,3-диизопропилкарбодиимид (DIC), 1-(3-диметиламинопропил)-3-этилкарбодиимид (EDC), N-гидроксиазабензотриазол (HATU), карбонилдиимидазол или моногидрат 1-оксибензотриазола (HOBT) и различных условий расщепления: например, трифторуксусная кислота (TFA), HCl в диоксане, гидрирование Pd-C в органических растворителях (таких как метанол или этилацетат), трис-трифторацетат бора и бромциан, и реакции в растворе с выделением и очисткой промежуточных продуктов хорошо известны в области пептидного синтеза и в равной степени применимы для получения рассматриваемых соединений.
В некоторых вариантах осуществления R1, R2, R3 и R4 каждый независимо выбран из C1-6-алкила, C1-6-гидроксиалкила, C1-6-алкоксиалкила, арила, C1-6-аралкила и R14DVKOC1-3-алкила-, где, по меньшей мере, один из R1 и R3 представляет собой R14DVKOC1-3-алкил-. В предпочтительных вариантах осуществления один из R1 и R3 представляет собой C1-6-аралкил и другой представляет собой R14DVKOC1-3-алкил- и R2 и R4 независимо представляет собой C1-6-алкил. В наиболее предпочтительном осуществлении один из R1 и R3 представляет собой 2-фенилэтил или фенилметил и другой представляет собой R14DVKOCH2- и R14DVKO(CH3)СН-, и оба R2 и R4 представляют собой изобутил.
В некоторых вариантах осуществления каждый R11 независимо выбран из водорода, C1-6-алкила, C1-6-алкенила, C1-6-алкинила, карбоциклила, гетероциклила, арила, гетероарила, C1-6-аралкила и C1-6-гетероаралкила.
В некоторых вариантах осуществления каждый R17 независимо выбран из водорода, C1-6-алкила, C1-6-алкенила, C1-6-алкинила, карбоциклила, гетероциклила, арила, гетероарила, C1-6-аралкила и C1-6-гетероаралкила.
В некоторых вариантах осуществления L и Q отсутствуют и R7 выбран из водорода, дополнительной цепи аминокислот, C1-6-ацила, защитной группы, арила, гетероарила, C1-6-алкила, C1-6-алкенила, C1-6-алкинила, C1-6-аралкила и C1-6-гетероаралкила. В некоторых таких вариантах осуществления R6 представляет собой C1-6-алкил и R7 выбран из бутила, аллила, пропаргила, фенилметила, 2-пиридила, 3-пиридила и 4-пиридила.
В других вариантах осуществления L представляет собой SO2, Q отсутствуют и R7 выбран из C1-6-алкила и арила. В некоторых таких вариантах осуществления R7 выбран из метила и фенила.
В некоторых вариантах осуществления L представляет собой C=O и R7 выбран из
C1-6-алкила, C1-6-алкенила, C1-6-алкинила, арила, C1-6-аралкила, гетероарила и C1-6-гетероаралкила, R8ZA-C1-8-алкила-, R11Z-C1-8-алкила-, (R8O)(R9O)P(=O)O-C1-8-алкила-, (R8O)(R9O)P(=O)O-C1-8-алкил-ZAZ-C1-8-алкила-, (R8O)(R9O)P(=O)O-C1-8-алкил-Z-C1-8-алкила-, R8ZA-C1-8-алкил-ZAZ-C1-8-алкила-, гетероциклилMZAZ-C1-8-алкила-, (R10)2N-C1-8-алкила-, (R10)3N+-C1-8-алкила-, гетероциклилM-, карбоциклилM-, R11SO2C1-8-алкила- и R11SO2NH-. В некоторых вариантах осуществления L представляет собой C=O, Q отсутствует и R7 представляет собой H.
В некоторых вариантах осуществления R6 представляет собой C1-6-алкил, R7 представляет собой C1-6-алкил, Q отсутствует и L представляет собой C=O. В некоторых таких вариантах осуществления R7 представляет собой этил, изопропил, 2,2,2-трифторэтил или 2-(метилсульфонил)этил.
В других вариантах осуществления L представляет собой C=O, Q отсутствует и R7 представляет собой C1-6-аралкил. В некоторых таких вариантах осуществления R7 выбран из 2-фенилэтила, фенилметила, (4-метоксифенил)метила, (4-хлорфенил)метила и (4-фторфенил)метила.
В других вариантах осуществления L представляет собой C=O, Q отсутствует, R6 представляет собой C1-6-алкил и R7 представляет собой арил. В некоторых таких вариантах осуществления R7 представляет собой замещенный или незамещенный фенил.
В некоторых вариантах осуществления L представляет собой C=O, Q отсутствует или представляет собой О и R7 представляет собой (CH2)nкарбоциклил. В некоторых таких вариантах осуществления R7 представляет собой циклопропил или циклогексил.
В некоторых вариантах осуществления L и A представляют собой C=O, Q отсутствует, Z представляют собой О и R7 выбран из R8ZA-C1-8-алкила-, R11Z-C1-8-алкила-, R8ZA-C1-8-алкил-ZAZ-C1-8-алкила-, (R8O)(R9O)P(=O)O-C1-8-алкил-ZAZ-C1-8-алкила-, (R8O)(R9O)P(=O)O-C1-8-алкил-Z-C1-8-алкила- и гетероциклилMZAZ-C1-8-алкила-. В некоторых таких вариантах осуществления R7 представляет собой гетероциклилMZAZ-C1-8-алкил-, где гетероциклил представляет собой замещенный или незамещенный оксодиоксоленил или N(R12)(R13), где R12 и R13, взятые вместе, представляют собой C1-6-алкил-Y-C1-6-алкил, предпочтительно C1-3-алкил-Y-C1-3-алкил, с образованием цикла.
В некоторых предпочтительных вариантах осуществления L представляет собой C=O, Q отсутствует и R7 выбран из (R8O)(R9O)P(=O)O-C1-8-алкила-, (R10)2N-C1-8-алкила, (R10)3N+(СН2)n- и гетероциклил-M-. В некоторых таких вариантах осуществления R7 представляет собой -C1-8-алкилN(R10)2 или -C1-8-алкилN+(R10)3, где R10 представляет собой C1-6-алкил. В некоторых других таких вариантах осуществления R7 представляет собой гетероциклилM-, где гетероциклил выбран из морфолиновой группы, пиперидиновой группы, пиперазиновой группы и пирролидиновой группы.
В некоторых вариантах осуществления L представляет собой C=O, R6 представляет собой C1-6-алкил, Q выбран из О и NH и R7 выбран из C1-6-алкила, циклоалкил-M, C1-6-аралкила и C1-6-гетероаралкила. В других вариантах осуществления L представляет собой C=O, R6 представляет собой C1-6-алкил, Q выбран из О и NH и R7 представляет собой C1-6-алкил, где C1-6-алкил выбран их метила, этила и изопропила. В дополнительных вариантах осуществления L представляет собой C=O, R6 представляет собой C1-6-алкил, Q выбран из О и NH и R7 представляет собой C1-6-аралкил, где аралкил представляет собой фенилметил. В других вариантах осуществления L представляет собой C=O, R6 представляет собой C1-6-алкил, Q выбран из О и NH и R7 представляет собой C1-6-гетероалкил, где гетероалкил представляет собой (4-пиридил)метил.
В некоторых вариантах осуществления L отсутствует или представляет собой C=O и R6 и R7, взятые вместе, представляют собой C1-6-алкил-Y-C1-6-алкил, C1-6-алкил-ZA-C1-6-алкил или C1-6-алкил-А, с образованием цикла. В некоторых предпочтительных вариантах осуществления L представляет собой C=O, Q и Y отсутствуют и R6 и R7, взятые вместе, представляют собой C1-3-алкил-Y-C1-3-алкил. В другом предпочтительном осуществлении L и Q отсутствуют и R6 и R7, взятые вместе, представляют собой C1-3-алкил-Y-C1-3-алкил. В другом предпочтительном осуществлении L представляет собой C=O, Q отсутствует, Y выбран из NH и N-C1-6-алкила и R6 и R7, взятые вместе, представляют собой C1-3-алкил-Y-C1-3-алкил. В другом предпочтительном осуществлении L представляет собой C=O, Y отсутствует и R6 и R7, взятые вместе, представляют собой C1-3-алкил-Y-C1-3-алкил. В другом предпочтительном осуществлении L и A представляют собой C=O и R6 и R7, взятые вместе, представляют собой C1-2-алкил-ZA-C1-2-алкил. В другом предпочтительном осуществлении L и A представляют собой C=O и R6 и R7, взятые вместе, представляют собой C2-3-алкил-A.
В некоторых вариантах осуществления R14 представляет собой (R15O)(R16O)P(=O)W-. В некоторых таких вариантах осуществления D, V, K и W отсутствуют. В других таких вариантах осуществления V и K отсутствуют, D представляет собой C1-8-алкил и W представляет собой O. В еще одних таких вариантах осуществления D представляет собой C1-8-алкил, K представляет собой C=O и V и W представляют собой O.
В некоторых вариантах осуществления R14 представляет собой R15GB-. В предпочтительных вариантах осуществления B представляет собой C=O, G представляет собой О, D представляет собой C1-8-алкил, V представляет собой О и K представляет собой C=O.
В некоторых вариантах осуществления R14 представляет собой гетероциклил-. В предпочтительных таких вариантах осуществления D представляет собой C1-8-алкил. В некоторых таких вариантах осуществления V представляет собой О, K представляет собой C=O и гетероциклил представляет собой оксодиоксоленил. В других таких вариантах осуществления V отсутствует, K отсутствует или представляет собой C=O и гетероциклил представляет собой N(R18)(R19), где R18 и R19, взятые вместе, представляют собой J-T-J, J-WB-J или B-J-T-J, T отсутствует или выбран из О, NR17, S, SO, SO2, CHOR17, CHCO2R15, C=O, CF2 и CHF и J отсутствует или представляет собой C1-3-алкил.
В некоторых вариантах осуществления R14 представляет собой (R17)2N- или (R17)3N+- и предпочтительно V отсутствует. В предпочтительных таких вариантах осуществления D представляет собой C1-8-алкил и K отсутствует или представляет C=O. В некоторых вариантах осуществления, где V отсутствует и R14 представляет собой (R17)2N-, D отсутствует, K отсутствует или представляет собой C=O, предпочтительно K представляет собой C=O.
В некоторых вариантах осуществления R14 представляет собой R17SO2GBG-. В предпочтительных таких вариантах осуществления B представляет собой C=O, D, V и K отсутствуют и G представляет собой NH или NC1-6-алкил.
В некоторых вариантах осуществления R14 представляет собой R15GBC1-8-алкил. В предпочтительных вариантах осуществления B представляет собой C=O, G представляет собой О и группа C1-8-алкил необязательно замещена OH, C1-8-алкилом (необязательно замещенным галогеном, предпочтительно фтором), C1-8-алкилW, арилом, гетероарилом, карбоциклилом, гетероциклилом и C1-6-аралкилом. В некоторых таких вариантах осуществления группа C1-8-алкил представляет собой незамещенный, моно- или дизамещенный C1-алкил.
В некоторых вариантах осуществления соединения формулы I имеет следующую стереохимическую конфигурацию:
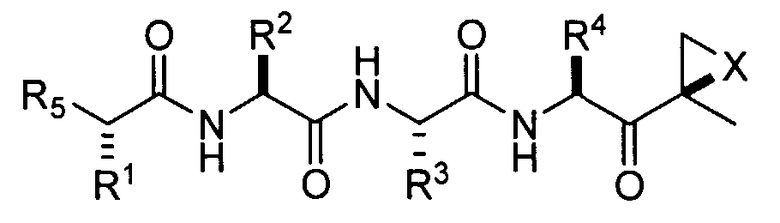
В предпочтительных вариантах осуществления ингибитор имеет структуру формулы II или ее фармацевтически приемлемой соли
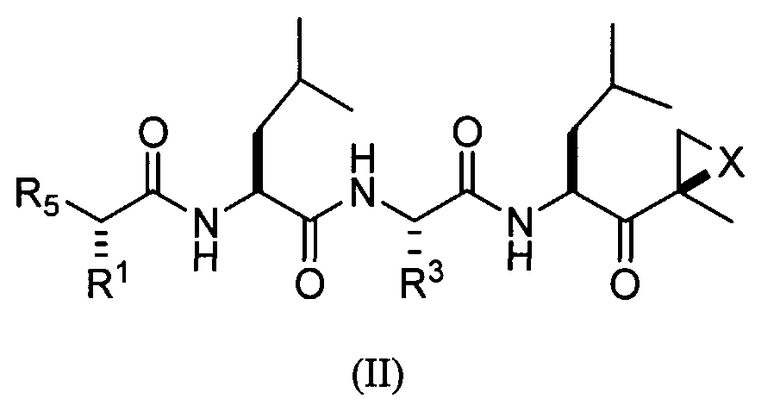
где каждый A независимо выбран из C=O, C=S и SO2, предпочтительно C=O;
каждый B независимо выбран из C=O, C=S и SO2, предпочтительно C=O;
D отсутствует или представляет собой C1-8-алкил;
G выбран из О, NH и N-C1-6-алкила;
K отсутствует или выбран из C=O, C=S и SO2, предпочтительно K отсутствует или представляет собой C=O;
L отсутствует или выбран из C=O, C=S и SO2, предпочтительно L отсутствует или представляет собой C=O;
M отсутствует или представляет собой C1-8-алкил;
Q отсутствует или выбран из О, NH и N-C1-6-алкила, предпочтительно Q отсутствует, представляет собой О или NH, наиболее предпочтительно Q отсутствует или представляет собой О;
X выбран из О, S, NH и N-C1-6-алкила, предпочтительно представляет собой О;
каждый V независимо отсутствует или выбран из О, S, NH и N-C1-6-алкила, предпочтительно V отсутствует или представляет собой О;
W отсутствует или независимо выбран из О, S, NH и N-C1-6-алкила, предпочтительно представляет собой О;
Y отсутствует или выбран из О, NH, N-C1-6-алкила, S, SO, SO2, CHOR10 и CHCO2R10;
каждый Z независимо выбран из О, S, NH и N-C1-6-алкила, предпочтительно представляет собой О;
R1 и R3 каждый независимо выбран из C1-6-алкила, C1-6-гидроксиалкила, C1-6-алкоксиалкила, арила, C1-6-аралкила и R14DVKOC1-3-алкила-, где, по меньшей мере, один из R1 и R3 представляет собой R14DVKOC1-3-алкил-;
R5 представляет собой N(R6)LQR7;
R6 выбран из водорода, OH и C1-6-алкила, предпочтительно представляет собой C1-6-алкил;
R7 представляет собой дополнительную цепь аминокислот, водород, защитную группу, арил или гетероарил, любой из которых необязательно замещен галогеном, карбонилом, нитро, гидрокси, арилом, C1-5-алкилом, или R7 выбран из C1-6-алкила, C1-6-алкенила, C1-6-алкинила, C1-6-аралкила, C1-6-гетероаралкила, R8ZA-C1-8-алкила-, R11Z-C1-8-алкила-, (R8O)(R9O)P(=O)O-C1-8-алкил-ZAZ-C1-8-алкила-, (R8O)(R9O)P(=O)O-C1-8-алкил-Z-C1-8-алкила-, R8ZA-C1-8-алкил-ZAZ-C1-8-алкила-, гетероциклилMZAZ-C1-8-алкила-, (R8O)(R9O)P(=O)O-C1-8-алкила-, (R10)2N-C1-8-алкила-, (R10)3N+-C1-8-алкила-, гетероциклилM-, карбоциклилM-, R11SO2C1-8-алкила- и R11SO2NH или
R6 и R7, взятые вместе, представляют собой C1-6-алкил-Y-C1-6-алкил, C1-6-алкил-ZA-C1-6-алкил, A-C1-6-алкил-ZA-C1-6-алкил, A-C1-6-алкил-A или C1-6-алкил-A, предпочтительно C1-2-алкил-Y-C1-2-алкил, C1-2-алкил-ZA-C1-2-алкил, А-C1-2-алкил-ZA-C1-2-алкил, A-C1-3-алкил-A или C1-4-алкил-A, с образованием цикла;
R8 и R9 независимо выбраны из водорода, катиона металла, C1-6-алкила, C1-6-алкенила, C1-6-алкинила, арила, гетероарила, C1-6-аралкила и C1-6-гетероаралкила, предпочтительно из водорода, катиона металла и C1-6-алкила или R8 и R9, взятые вместе, представляют собой C1-6-алкил, с образованием цикла;
каждый R10 независимо выбран из водорода и C1-6-алкила, предпочтительно из C1-6-алкила и
каждый R11 независимо выбран из водорода, OR10, C1-6-алкила, C1-6-алкенила, C1-6-алкинила, карбоциклила, гетероциклила, арила, гетероарила, C1-6-аралкила и C1-6-гетероаралкила;
R14 выбран из водорода, (R15O)(R16O)P(=O)W-, R15GB-, гетероциклила-, (R17)2N-, (R17)3N+-, R17SO2GBG- и R15GBC1-8-алкила, где группа C1-8-алкил необязательно замещена OH, C1-8-алкилW (необязательно замещенным галогеном, предпочтительно фтором), арилом, гетероарилом, карбоциклилом, гетероциклилом и C1-6-аралкилом, предпочтительно, по меньшей мере, наличие одного R14, который является отличным от водорода;
R15 и R16 независимо выбраны из водорода, катиона металла, C1-6-алкила, C1-6-алкенила, C1-6-алкинила, арила, гетероарила, C1-6-аралкила и C1-6-гетероаралкила, предпочтительно из водорода, катиона металла и C1-6-алкила, или R15 и R16, взятые вместе, представляют собой C1-6-алкил, с образованием цикла;
каждый R17 независимо выбран из водорода, OR10, C1-6-алкила, C1-6-алкенила, C1-6-алкинила, карбоциклила, гетероциклила, арила, гетероарила, C1-6-аралкила и C1-6-гетероаралкила;
при условии, что R6 представляет собой H, L представляет собой C=O и Q отсутствует, R7 не является водородом, C1-6-алкилом или замещенным или незамещенным арилом или гетероарилом, и
D, G, V, K и W выбраны так, что не существует связей О-О, N-O, S-N или S-O.
В некоторых вариантах осуществления R1 и R3 каждый независимо выбран из C1-6-алкила, C1-6-гидроксиалкила, C1-6-алкоксиалкила, арила, C1-6-аралкила и R14DVKOC1-3-алкила-, где, по меньшей мере, один из R1 и R3 представляет собой R14DVKOC1-3-алкил. В предпочтительных вариантах осуществления один из R1 и R3 представляет собой C1-6-аралкил и другой представляет собой R14DVKOC1-3-алкил-. В наиболее предпочтительном осуществлении один из R1 и R3 представляет собой 2-фенилэтил или фенилметил и другой представляет собой R14DVKOCH2 или R14DVKO(CH3)СН-.
В некоторых вариантах осуществления каждый R11 независимо выбран из водорода, C1-6-алкила, C1-6-алкенила, C1-6-алкинила, карбоциклила, гетероциклила, арила, гетероарила, C1-6-аралкила и C1-6-гетероаралкила.
В некоторых вариантах осуществления каждый R17 независимо выбран из водорода, C1-6-алкила, C1-6-алкенила, C1-6-алкинила, карбоциклила, гетероциклила, арила, гетероарила, C1-6-аралкила и C1-6-гетероаралкила.
В некоторых вариантах осуществления L и Q отсутствуют и R7 выбран из водорода, дополнительной цепи аминокислот, C1-6-ацила, защитной группы, арила, гетероарила, C1-6-алкила, C1-6-алкенила, C1-6-алкинила, C1-6-аралкила и C1-6-гетероаралкила. В некоторых таких вариантах осуществления R6 представляет собой C1-6-алкил и R7 выбран из бутила, аллила, пропаргила, фенилметила, 2-пиридила, 3-пиридила и 4-пиридила.
В других вариантах осуществления L представляет собой SO2, Q отсутствуют и R7 выбран из C1-6-алкила и арила. В некоторых таких вариантах осуществления R7 выбран из метила и фенила.
В некоторых вариантах осуществления L представляет собой C=O и R7 выбран из C1-6-алкила, C1-6-алкенила, C1-6-алкинила, арила, C1-6-аралкила, гетероарила и C1-6-гетероаралкила, R8ZA-C1-8-алкила-, R11Z-C1-8-алкила-, (R8O)(R9O)P(=O)O-C1-8-алкила, (R8O)(R9O)P(=O)O-C1-8-алкил-ZAZ-C1-8-алкила-, (R8O)(R9O)P(=O)O-C1-8-алкил-Z-C1-8-алкила-, R8ZA-C1-8-алкил-ZAZ-C1-8-алкила-, гетероциклилMZAZ-C1-8-алкила-, (R10)2N-C1-8-алкила-, (R10)3N+-C1-8-алкила-, гетероциклилM-, карбоциклилM-, R11SO2C1-8-алкила- и R11SO2NH-. В некоторых вариантах осуществления L представляет собой C=O, Q отсутствует и R7 представляет собой H.
В некоторых вариантах осуществления R6 представляет собой C1-6-алкил, R7 представляет собой C1-6-алкил, Q отсутствует и L представляет собой C=O. В некоторых таких вариантах осуществления R7 представляет собой этил, изопропил, 2,2,2-трифторэтил или 2-(метилсульфонил)этил.
В других вариантах осуществления L представляет собой C=O, Q отсутствует и R7 представляет собой C1-6-аралкил. В некоторых таких вариантах осуществления R7 выбран из 2-фенилэтила, фенилметила, (4-метоксифенил)метила, (4-хлорфенил)метила и (4-фторфенил)метила.
В других вариантах осуществления L представляет собой C=O, Q отсутствует, R6 представляет собой C1-6-алкил и R7 представляет собой арил. В некоторых таких вариантах осуществления R7 представляет собой замещенный или незамещенный фенил.
В некоторых вариантах осуществления L представляет собой C=O, Q отсутствует или представляет собой О и R7 представляет собой (CH2)nкарбоциклил. В некоторых таких вариантах осуществления R7 представляет собой циклопропил или циклогексил.
В некоторых вариантах осуществления L и A представляют собой C=O, Q отсутствует, Z представляют собой О и R7 выбран из R8ZA-C1-8-алкила-, R11Z-C1-8-алкила-, R8ZA-C1-8-алкил-ZAZ-C1-8-алкила-, (R8O)(R9O)P(=O)O-C1-8-алкил-ZAZ-C1-8-алкила-, (R8O)(R9O)P(=O)O-C1-8-алкил-Z-C1-8-алкила- и гетероциклилMZAZ-C1-8-алкила. В некоторых таких вариантах осуществления R7 представляет собой гетероциклилMZAZ-C1-8-алкил-, где гетероциклил представляет собой замещенный или незамещенный оксодиоксоленил или N(R12)(R13), где R12 и R13, взятые вместе, представляют собой C1-6-алкил-Y-C1-6-алкил, предпочтительно C1-3-алкил-Y-C1-3-алкил, с образованием цикла.
В некоторых предпочтительных вариантах осуществления L представляет собой C=O, Q отсутствует и R7 выбран из (R8O)(R9O)P(=O)O-C1-8-алкила, (R10)2N-C1-8-алкила-, (R10)3N+(СН2)n- и гетероциклилM-. В некоторых таких вариантах осуществления R7 представляет собой -C1-8-алкилN(R10)2 или -C1-8-алкилN+(R10)3, где R10 представляет собой C1-6-алкил. В некоторых других таких вариантах осуществления R7 представляет собой гетероциклилM-, где гетероциклил выбран из морфолиновой группы, пиперидиновой группы, пиперазиновой группы и пирролидиновой группы.
В некоторых вариантах осуществления L представляет собой C=O, R6 представляет собой C1-6-алкил, Q выбран из О и NH и R7 выбран из C1-6-алкила, циклоалкилM, C1-6-аралкила и C1-6-гетероаралкила. В других вариантах осуществления L представляет собой C=O, R6 представляет собой C1-6-алкил, Q выбран из О и NH и R7 представляет собой C1-6-алкил, где C1-6-алкил выбран их метила, этила и изопропила. В дополнительных вариантах осуществления L представляет собой C=O, R6 представляет собой C1-6-алкил, Q выбран из О и NH и R7 представляет собой C1-6-аралкил, где аралкил представляет собой фенилметил. В других вариантах осуществления L представляет собой C=O, R6 представляет собой C1-6-алкил, Q выбран из О и NH и R7 представляет собой C1-6-гетероалкил, где гетероалкил представляет собой (4-пиридил)метил.
В некоторых вариантах осуществления L отсутствует или представляет собой C=O и R6 и R7, взятые вместе, представляют собой C1-6-алкил-Y-C1-6-алкил, C1-6-алкил-ZA-C1-6-алкил или C1-6-алкил-А, с образованием цикла. В некоторых предпочтительных вариантах осуществления L представляет собой C=O, Q и Y отсутствуют и R6 и R7, взятые вместе, представляют собой C1-3-алкил-Y-C1-3-алкил. В другом предпочтительном осуществлении L и Q отсутствуют и R6 и R7, взятые вместе, представляют собой C1-3-алкил-Y-C1-3-алкил. В другом предпочтительном осуществлении L представляет собой C=O, Q отсутствует, Y выбран из NH и N-C1-6-алкила и R6 и R7, взятые вместе, представляют собой C1-3-алкил-Y-C1-3-алкил. В другом предпочтительном осуществлении L представляет собой C=O, Y отсутствует и R6 и R7, взятые вместе, представляют собой C1-3-алкил-Y-C1-3-алкил. В другом предпочтительном осуществлении L и A представляют собой C=O и R6 и R7, взятые вместе, представляют собой C1-2-алкил-ZA-C1-2-алкил. В другом предпочтительном осуществлении L и A представляют собой C=O и R6 и R7, взятые вместе, представляют собой C2-3-алкил-A.
В некоторых вариантах осуществления R14 представляет собой (R15O)(R16O)P(=O)W-. В некоторых таких вариантах осуществления D, V, K и W отсутствуют. В других таких вариантах осуществления V и K отсутствуют, D представляет собой C1-8-алкил и W представляет собой O. В еще одних таких вариантах осуществления D представляет собой C1-8-алкил, K представляет собой C=O и V и W представляют собой O.
В некоторых вариантах осуществления R14 представляет собой R15GB-. В предпочтительных вариантах осуществления B представляет собой C=O, G представляет собой О, D представляет собой C1-8-алкил, V представляет собой О и K представляет собой C=O.
В некоторых вариантах осуществления R14 представляет собой гетероциклил-. В предпочтительных таких вариантах осуществления D представляет собой C1-8-алкил. В некоторых таких вариантах осуществления V представляет собой О, K представляет собой C=O и гетероциклил представляет собой оксодиоксоленил. В других таких вариантах осуществления V отсутствует, K отсутствует или представляет собой C=O и гетероциклил представляет собой N(R18)(R19), где R18 и R19, взятые вместе, представляют собой J-T-J, J-WB-J или B-J-T-J, T отсутствует или выбран из О, NR17, S, SO, SO2, CHOR17, CHCO2R15, C=O, CF2 и CHF и J отсутствует или представляет собой C1-3-алкил.
В некоторых вариантах осуществления R14 представляет собой (R17)2N- или (R17)3N+- и предпочтительно V отсутствует. В предпочтительных таких вариантах осуществления D представляет собой C1-8-алкил и K отсутствует или представляет C=O. В некоторых вариантах осуществления, где V отсутствует и R14 представляет собой (R17)2N-, D отсутствует, K отсутствует или представляет собой C=O, предпочтительно K представляет собой C=O.
В некоторых вариантах осуществления R14 представляет собой R17SO2GBG-. В предпочтительных таких вариантах осуществления B представляет собой C=O, D, V и K отсутствуют и G представляет собой NH или NC1-6-алкил.
В некоторых вариантах осуществления R14 представляет собой R15GBC1-8-алкил. В предпочтительных вариантах осуществления B представляет собой C=O, G представляет собой О и группа C1-8-алкил необязательно замещена OH, C1-8-алкилом (необязательно замещенным галогеном, предпочтительно фтором), C1-8-алкилW, арилом, гетероарилом, карбоциклилом, гетероциклилом и C1-6-аралкилом. В некоторых таких вариантах осуществления группа C1-8-алкил представляет собой незамещенный, моно- или дизамещенный C1-алкил.
Другой аспект изобретения относится к соединениям, имеющим структуру формулы (III) или формулы (IV), или их фармацевтически приемлемой соли
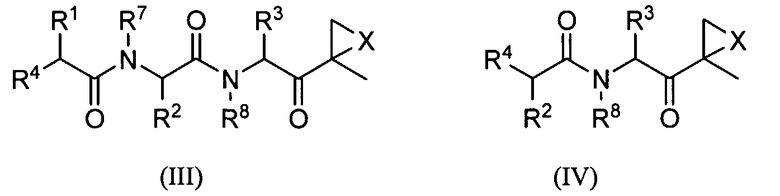
где каждый Ar независимо представляет собой ароматическую или гетероароматическую группу, необязательно замещенную от 1 до 4 заместителями;
L отсутствует или выбран из C=O, C=S и SO2, предпочтительно представляет собой SO2 или C=O;
X выбран из О, S, NH и N-C1-6-алкила, предпочтительно представляет собой О;
Y отсутствует или выбран из C=O и SO2;
Z отсутствует или представляет собой C1-6-алкил;
R1, R2 и R3 каждый независимо выбран из C1-6-алкила, C1-6-гидроксиалкила, C1-6-алкоксиалкила, арила и C1-6-аралкила, любой из которых необязательно замещен одним или более амидным, аминным заместителями, карбоновой кислотой (или ее солью), сложноэфирным (включая C1-6-алкиловый эфир, C1-5-алкиловый эфир и ариловый эфир), тиольным или тиоэфирным заместителями;
R4 представляет собой N(R5)L-Z-R6;
R5 выбран из водорода, OH и C1-6-аралкил-Y и C1-6-алкил-Y, предпочтительно представляет собой водород;
R6 выбран из водорода, OR7, C1-6-алкенила, Ar-Y-, карбоциклила и гетероциклила; и
R7 и R8 независимо выбраны из водорода, C1-6-алкила и C1-6-аралкила, предпочтительно представляют собой водород.
В некоторых вариантах осуществления L выбран из C=O, C=S и SO2, предпочтительно представляет собой SO2 или C=O.
В некоторых вариантах осуществления R5 выбран из водорода, OH, C1-6-аралкила и C1-6-алкила, предпочтительно представляет собой водород.
В некоторых вариантах осуществления R6 выбран из водорода, C1-6-алкенила, Ar-Y-, карбоциклила и гетероциклила.
В некоторых вариантах осуществления X представляет собой О и R1, R2 и R3 каждый независимо выбран из C1-6-алкила, C1-6-гидроксиалкила и C1-6-аралкила. В предпочтительных таких вариантах осуществления R1 и R3 независимо представляют собой C1-6-алкил и R2 представляет собой C1-6-аралкил. В более предпочтительных таких вариантах осуществления R1 и R3 оба представляют изобутил и R2 представляет собой фенилметил.
В некоторых вариантах осуществления R5 представляет собой водород, L представляет собой C=O или SO2, R6 представляет собой Ar-Y- и каждый Ar независимо выбран из фенила, индолила, бензофурила, нафтила, хинолинила, хинолонила, тиенила, пиридила, пиразила и тому подобного. В некоторых таких вариантах осуществления Ar можно замещать Ar-Q-, где Q выбран из прямой связи, -О- и C1-6-алкила. В некоторых других таких вариантах осуществления, где Z представляет собой C1-6-алкил, Z можно замещать предпочтительно Ar, например фенилом.
В некоторых вариантах осуществления R5 представляет собой водород, Z отсутствует, L представляет собой C=O или SO2 и R6 выбран из Ar-Y или гетероциклила. В некоторых предпочтительных таких вариантах осуществления гетероциклил выбран из хромонильной, хроманильной, морфолиновой и пиперидиновой группы. В некоторых других таких предпочтительных вариантах осуществления Ar выбран из фенила, индолила, бензофурила, нафтила, хинолинила, хинолонила, тиенила, пиридила, пиразила и тому подобного.
В некоторых вариантах осуществления R5 представляет собой водород, L представляет собой C=O или SO2, Z отсутствует и R6 представляет собой C1-6-алкенил, где C1-6-алкенил представляет собой замещенную винильную группу, где заместитель предпочтительно представляет арильную или гетероарильную группу, более предпочтительно, фенильную группу, необязательно замещенную 1-4 заместителями.
В некоторых вариантах осуществления R7 и R8 независимо выбраны из водорода и C1-6-алкила. В некоторых таких предпочтительных вариантах осуществления R7 и R8 независимо выбраны из водорода и метила. В более предпочтительных вариантах осуществления как R7, так и R8 представляют собой водород.
В некоторых вариантах осуществления соединения формулы (III) или формулы (IV) имеет следующую стереохимическую конфигурацию
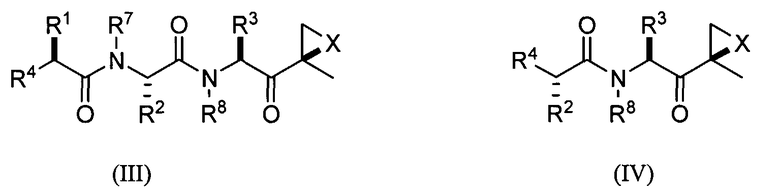
В предпочтительных вариантах осуществления ингибитор имеет структуру формулы (V) или формулы (VI) или его фармацевтически приемлемой соли
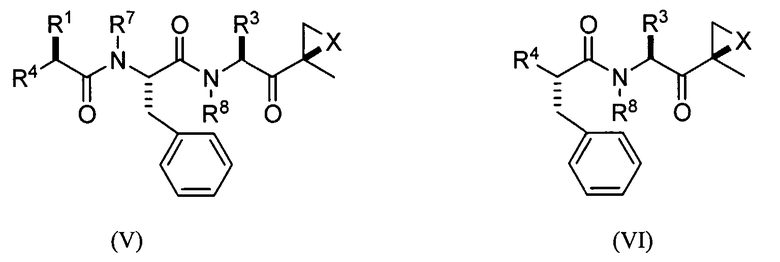
где каждый Ar независимо представляет собой ароматическую или гетероароматическую группу, необязательно замещенную 1-4 заместителями;
L отсутствует или выбран из C=O, C=S и SO2, предпочтительно представляет собой SO2 или C=O;
X выбран из О, S, NH и N-C1-6-алкила, предпочтительно представляет собой О;
Y отсутствует или выбран из C=O и SO2;
Z отсутствует или представляет собой C1-6-алкил;
R1 и R3 каждый независимо выбран из C1-6-алкила, C1-6-гидроксиалкила, C1-6-алкоксиалкила, арила и C1-6-аралкила, любой из которых необязательно замещен одним или более амидным, аминным заместителями, карбоновой кислотой (или ее солью), сложноэфирным (включая C1-6-алкиловый эфир и ариловый эфир), тиольным или тиоэфирным заместителями;
R4 представляет собой N(R5)L-Z-R6;
R5 выбран из водорода, OH и C1-6-аралкил-Y и C1-6-алкил-Y, предпочтительно представляет собой водород;
R6 выбран из водорода, OR7, C1-6-алкенила, Ar-Y-, карбоциклила и гетероциклила; и
R7 и R8 независимо выбраны из водорода, C1-6-алкила и C1-6-аралкила, предпочтительно представляют собой водород.
В некоторых вариантах осуществления L выбран из C=O, C=S и SO2, предпочтительно представляет собой SO2 или C=O.
В некоторых вариантах осуществления R5 выбран из водорода, OH, C1-6-аралкила и C1-6-алкила, предпочтительно представляет собой водород.
В некоторых вариантах осуществления R6 выбран из водорода, C1-6-алкенила, Ar-Y-, карбоциклила и гетероциклила.
В некоторых вариантах осуществления X представляет собой О и R1 и R3 каждый независимо выбран из C1-6-алкила, C1-6-гидроксиалкила и C1-6-аралкила. В таких предпочтительных вариантах осуществления R1 и R3 независимо представляют собой C1-6-алкил. В более предпочтительных таких вариантах осуществления R1 и R3 представляют собой изобутил.
В некоторых вариантах осуществления R5 представляет собой водород, L представляет собой C=O или SO2 и R6 представляет собой Ar-Y, каждый Ar выбран из фенила, индолила, бензофуранила, нафтила, хинолинила, хинолонила, тиенила, пиридила, пиразила и тому подобного. В некоторых таких вариантах осуществления Ar можно замещать Ar-Q-, где Q выбран из прямой связи, -О- и C1-6-алкила. В некоторых других таких вариантах осуществления, где Z представляют собой C1-6-алкил, Z можно замещать, например, предпочтительно Ar, более предпочтительно фенилом.
В некоторых вариантах осуществления R5 представляет собой водород, Z отсутствует, L представляет собой C=O или SO2 и R6 выбран из Ar-Y или гетероциклила. В некоторых предпочтительных таких вариантах осуществления гетероциклил выбран из хромонильной, хроманильной, морфолиновой и пиперидиновой группы. В некоторых других таких предпочтительных вариантах осуществления Ar выбран из фенила, индолила, бензофуранила, нафтила, хинолинила, хинолонила, тиенила, пиридила, пиразила и тому подобного.
В некоторых вариантах осуществления R5 представляет собой водород, L представляет собой C=O или SO2, Z отсутствует и R6 представляет собой C1-6-алкенил, где C1-6-алкенил представляет собой замещенную винильную группу, где заместитель предпочтительно представляет собой арильную или гетероарильную группу, более предпочтительно заместитель представляет собой фенильную группу, необязательно замещенную от одного до четырех заместителей.
В некоторых вариантах осуществления R7 и R8 независимо выбраны из водорода и C1-6-алкила. В некоторых таких предпочтительных вариантах осуществления R7 и R8 независимо выбраны из водорода и метила. В более предпочтительных таких вариантах осуществления как R7, так и R8 представляют собой водород.
В некоторых вариантах осуществления -L-Z-R6 выбран из
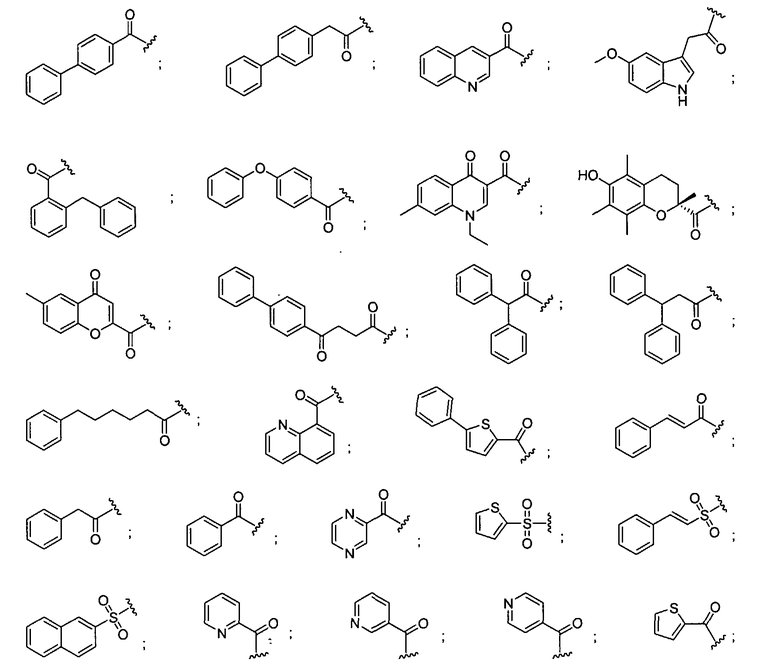
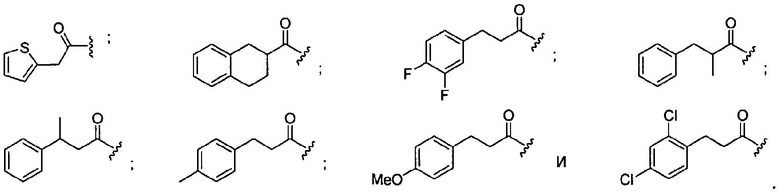
Один аспект изобретения относится к медицинскому устройству, содержащему описанную здесь композицию, которая содержит ингибитор, имеющий структуру по любой из формул с I по VI. В одном варианте осуществления композиция содержится в медицинском устройстве. В некоторых вариантах осуществления медицинское устройство представляет собой гель, содержащий полимерный матрикс или керамический матрикс и ингибитор. Указанный полимер может быть либо природного происхождения, либо синтетическим. В другом осуществлении указанный гель используется в качестве депо препарата, адгезива, шовного материала, барьера или изолирующего вещества.
Другой аспект изобретения относится к медицинскому устройству, содержащему субстрат, обладающему поверхностью, на которой размещен ингибитор, имеющий структуру любой из формул с I по VI. В одном варианте осуществления ингибитор непосредственно помещен в медицинское устройство. В другом варианте осуществления покрытие размещено таким образом, что покрытие содержит полимерный матрикс или керамический матрикс с распределенным или растворенным в нем ингибитором, имеющим структуру любой из формул с I по VI.
В одном варианте осуществления медицинское устройство представляет собой коронарный, сосудистый, периферийный или желчный стент. Более подробно, стент по настоящему изобретению представляет собой расширяющийся стент. При нанесении на поверхность матрикса, содержащего ингибитор, имеющий структуру по любой из формул с I по VI, матрикс является гибким для адаптации к сжатому и расширенному состоянию такого расширяющегося стента. В другом варианте осуществления данного изобретения стент содержит, по меньшей мере, участок, который является вставным или имплантируемым в организм пациента, где участок обладает поверхностью, которая адаптирована к воздействию на ткань организма, и где, по меньшей мере, участок поверхности имеет покрытие с ингибитором, имеющим структуру по любой из формул с I по VI, или представляет собой покрытие, несущее матрикс, содержащий ингибитор, имеющий структуру по любой из формул с I по VI, распределено или растворено в нем. Пример подходящего стента описан в патенте США № 4733665, который полностью включен сюда путем ссылки.
В другом варианте осуществления медицинское устройство по настоящему изобретению представляет собой хирургический инструмент, такой как сосудистый протез, внутрипросветное устройство, хирургический изолирующий материал или сосудистую подложку. Более подробно, медицинское устройство по настоящему изобретению представляет собой катетер, имплантируемый проход для сосудистого доступа, центральный венозный катетер, артериальный катетер, сосудистый трансплантат, внутриаортальную балонную контрпульсацию, шовный материал, желудочковый вспомогательный насос, содержащую лекарство перегородку, пластырь, сосудистую манжету, экстра/периваскулярную подложку, фильтр для крови или фильтр, адаптированный для развертывания в кровеносном сосуде, имеющие покрытие с ингибитором, имеющим структуру любой из формул с I по VI, либо непосредственно, либо посредством матрикса, содержащего ингибитор, имеющий структуру по любой из формул с I по VI.
В некоторых вариантах осуществления внутрипросветное устройство имеет покрытие с ингибитором, имеющим структуру любой из формул от I до VI или представляет собой покрытие, содержащее биологически лишенный иммуногенных свойств матрикс и распределенный в полимере ингибитор, имеющий структуру любой из формул с I по VI, причем указанное устройство имеет внутреннюю поверхность и внешнюю поверхность, имеющую покрытие, по меньшей мере, на части внутренней поверхности внешней поверхности или на обеих частях.
В некоторых вариантах осуществления медицинское устройство можно использовать для предотвращения рестеноза после ангиопластики. Медицинское устройство можно также применять для лечения различных заболеваний и состояний, обеспечивая локализованное введение ингибитора, имеющего структуру по любой из формул с I по VI. Такие заболевания и состояния включают рестеноз, воспаление, ревматоидный артрит, повреждение ткани вследствие воспаления, гиперпролиферативные заболевания, тяжелый или подагрический псориаз, заболевание, вызывающее мышечное истощение, хронические инфекционные заболевания, аномальную иммунную реакцию, состояния, вызванные разрывом легко повреждаемых атеросклеротических бляшек, нарушения, связанные с ишемическими состояниями, и вирусную инфекцию и пролиферацию. Примеры заболеваний и состояний, которые поддаются лечению, включающему использование покрытых лекарственным средством медицинских устройств по настоящему изобретению, включают атеросклероз, острый коронарный синдром, болезнь Альцгеймера, рак, лихорадку, мышечное бездействие (атрофию), денервацию, окклюзии сосудов, удар, инфицирование ВИЧ, поражение нерва, почечную недостаточность, связанную с ацидозом, и печеночную недостаточность. См., например, Goldberg, патент США № 5340736.
Термин «Cx-y-алкил» обозначает замещенные или незамещенные насыщенные углеводородные группы, включающие алкильные группы с линейной цепью и алкильные группы с разветвленной цепью, которые содержат от x до y атомов углерода в цепи, включая галогеналкильные группы, такие как трифторметил и 2,2,2- трифторэтил и т.д. C0-алкил обозначает водород, где группа находится в концевом положении, если связь внутренняя. Термины «С2-y-алкенил» и «С2-y-алкинил» обозначают замещенные или незамещенные ненасыщенные алифатические группы, аналогичные по длине и возможному замещению описанным выше алкилам, но которые содержат, по меньшей мере, одну двойную или тройную связь соответственно.
Термин «алкокси» обозначает алкильную группу, содержащую присоединенный к ней кислород. Типичные представители алкоксигрупп включают метокси-, этокси-, пропокси-, трет-бутоксигруппу и тому подобное. «Простой эфир» представляет собой два углеводорода, ковалентно связанных с кислородом. Соответственно, заместитель алкила, который участвует в образовании такого алкилового эфира, представляет собой или подобен алкоксигруппе.
Термин «C1-6-алкоксиалкил» обозначает C1-6-алкильную группу, замещенную алкоксигруппой, тем самым образующей простой эфир.
Используемый здесь термин «C1-6-аралкил» обозначает C1-6-алкильную группу, замещенную арильной группой.
Термины «амин» и «амино-» приняты в данной области и обозначают как незамещенные, так и замещенные амины и их соли, например, фрагменты которых можно представить общими формулами:
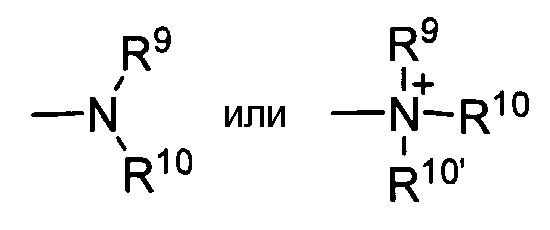
где R9, R10 и R10' каждый независимо представляет собой водород, алкил, алкенил, -(CH2)m-R8 или R9 и R10, взятые вместе с атомом N, к которому они присоединены, составляют гетероцикл, содержащий от 4 до 8 атомов в структуре кольца; R8 представляет собой арил, циклоалкил, циклоалкенил, гетероциклил или полициклил и m представляет собой ноль или целое число от 1 до 8. В предпочтительных вариантах осуществления только один из R9 или R10 может представлять собой карбонил, например, R9, R10 и азот, взятые вместе, не образуют имид. В еще более предпочтительных вариантах осуществления R9 и R10 (и необязательно R10') каждый независимо представляет собой водород, алкил, алкенил, -(CH2)m-R8. В некоторых вариантах осуществления аминогруппа является основной, обозначая, что протонированная форма имеет pKa≥7,00.
Термины «амид» и «амидо-» приняты в данной области в качестве аминозамещенных карбонилов и включают в себя фрагмент, который можно представить общей формулой:
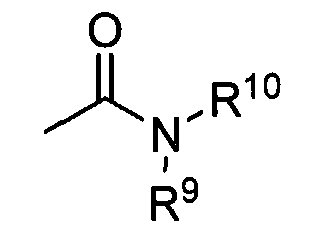
где R9, R10 представляют собой заместители, как описано выше. Предпочтительные варианты осуществления амида не включают имиды, которые могут быть нестабильными.
Используемый здесь термин «арил» включает 5-, 6- и 7-членные замещенные или незамещенные однокольцевые ароматические группы, в которых каждый атом в кольце представляет собой углерод. Термин «арил» также включает полициклические кольцевые системы, в которых два или более атомов углерода, общие для двух смежных колец, где, по меньшей мере, одно из колец является ароматическим, например, другие циклические кольца могут представлять собой циклоалкилы, циклоалкенилы, циклоалкинилы, арилы, гетероарилы и/или гетероциклилы. Арильные группы включают бензол, нафталин, фенантрен, фенол, анилин и тому подобное.
Используемый здесь термин «карбоцикл» и «карбоциклил» обозначает неароматическое замещенное или незамещенное кольцо, в котором каждый атом в кольце представляет собой углерод. Термин «карбоцикл» и «карбоциклил» также включает полициклические кольцевые системы, содержащие два или более циклических кольца, в которых два или более атомов углерода являются общими для двух смежных колец, где, по меньшей мере, одно из колец является карбоциклическим, например, другие циклические кольца могут представлять собой циклоалкилы, циклоалкенилы, циклоалкинилы, арилы, гетероарилы и/или гетероциклилы.
Термин «карбонил» известен в данной области и включает такие фрагменты, которые можно представить общей формулой:
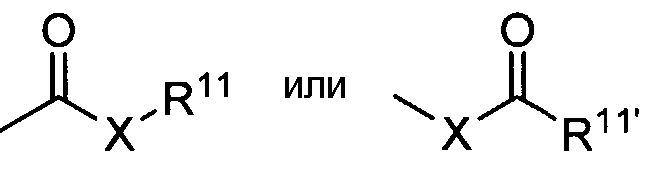
где X является связью или представляет собой кислород или серу и R11 представляет собой водород, алкил, алкенил, -(CH2)m-R8 или фармацевтически приемлемую соль, R11' представляет собой водород, алкил, алкенил, -(CH2)m-R8, где m и R8 являются такими же, как описано выше.
В тех случаях, когда X представляет собой кислород и R11 или R11' не является водородом, формула представляет собой «сложный эфир». В тех случаях, когда X представляет собой кислород и R11 является водородом, формула представляет собой «карбоновую кислоту».
Используемый здесь термин «фермент» может представлять собой любую частично или полностью белковую молекулу, которая осуществляет химическую реакцию каталитическим способом. Такие ферменты могут являться нативными ферментами, ферментами слияния, проферментами, апоферментами, денатурированными ферментами, фарнезилированными ферментами, убиквитинированными ферментами, ферментами, ацилированными жирными кислотами, геранил-геранилированными ферментами, GPI-связанными ферментами, связанными с липидами ферментами, пренилированными ферментами, встречающимися в природе или искусственно получаемыми мутантными ферментами, ферментами с модификациями боковых цепей или основной цепи, ферментами, содержащими лидерные последовательности, и ферментами, находящимися в комплексе с небелковым веществом, такими как протеогликаны и протеолипосомы. Ферменты можно получать любыми способами, включая природную экспрессию, экспрессию с промотором, клонирование, различные методы синтеза пептидов в растворе и на твердой фазе и подобные способы, известные специалистам в данной области.
Используемый здесь термин «C1-6-гетероаралкил» обозначает C1-6-алкильную группу, замещенную гетероарильной группой.
Термин «гетероарил» включает замещенные или незамещенные ароматические 5-7-членные кольцевые структуры, более предпочтительно 5-6-членные циклы, чьи кольцевые структуры включают в себя от одного до четырех гетероатомов. Термин «гетероарил» также включает полициклические кольцевые системы, содержащие два или более циклических кольца, в которых два или более атомов углерода являются общими для двух смежных колец, где, по меньшей мере, одно из колец является гетероароматическим, например, другие циклические кольца могут представлять собой циклоалкилы, циклоалкенилы, циклоалкинилы, арилы, гетероарилы и/или гетероциклилы. Гетероарильные группы включают, например, пиррол, фуран, тиофен, имидазол, оксазол, тиазол, триазол, пиразол, пиридин, пиразин, пиридазин, пиримидин и тому подобное.
Используемый здесь термин «гетероатом» обозначает атом любого элемента, отличного от углерода или водорода. Предпочтительные гетероатомы представляют собой азот, кислород, фосфор и серу.
Термин «гетероциклил» или «гетероциклическая группа» обозначает замещенные или незамещенные неароматические 3-10-членные кольцевые структуры, более предпочтительно 3-7-членные циклы, чьи кольцевые структуры включают от одного до четырех гетероатомов. Термин «гетероциклил» или «гетероциклическая группа» также включает полициклические кольцевые системы, содержащие два или более циклических кольца, в которых два или более атомов углерода являются общими для двух смежных колец, где, по меньшей мере, одно из колец является гетероциклическим, например, другие циклические кольца могут представлять собой циклоалкилы, циклоалкенилы, циклоалкинилы, арилы, гетероарилы и/или гетероциклилы. Гетероциклические группы включают, например, пиперидин, пиперазин, пирролидин, морфолин, лактоны, лактамы и тому подобное.
Термин «C1-6-гидроксиалкил» обозначает C1-6-алкильную группу, замещенную гидроксигруппой.
Используемый здесь термин «ингибитор» предназначен для описания соединения, которое блокирует или снижает активность фермента (например, ингибирование протеолитического расщепления стандартного флуорогенного пептидного субстрата, такого как suc-LLVY-AMC, Box-LLR-AMC и Z-LLE-AMC, ингибирование различных каталитических активностей 20S протеасомы). Ингибитор может действовать по типу конкурентного, бесконкурентного или неконкурентного ингибирования. Ингибитор может связываться обратимо или необратимо, и поэтому термин включает соединения, которые являются суицидными субстратами фермента. Ингибитор может модифицировать один или более участков в активном центре или около активного центра фермента, или он может вызывать конформационное изменение где-то в другом участке фермента.
Используемый здесь термин «пептид» включает не только стандартную амидную связь со стандартными α-заместителями, но и широко используемые пептидомиметики, другие модифицированные связи, не встречающиеся в природе боковые цепи и модификации боковых цепей, как подробно описано ниже.
Термины «полициклил» или «полициклический» обозначает два или более кольца (например, циклоалкилы, циклоалкенилы, циклоалкинилы, арилы, гетероарилы и/или гетероциклилы), в которых два или более атомов углерода являются общими для двух соседних колец, например кольца представляют собой конденсированные кольца. Каждое из колец полицикла может быть замещенным или незамещенным.
Термин «предотвращение» известен в данной области и при использовании в отношении состояния, такого как местный рецидив (например, боль), заболевания, такого как рак, симптомокомплекса, такого как сердечная недостаточность или любого другого медицинского состояния, хорошо понятен в данной области и включает введение композиции, которая уменьшает частоту возникновения или задерживает наступление симптомов медицинского состояния у пациента относительно пациента, который не принимает композицию. Таким образом, предотвращение рака включает, например, сокращение количества обнаруживаемых раковых опухолей в популяции пациентов, получающих профилактическое лечение, относительно не получающей лечения контрольной популяции, и/или задержку появления обнаруживаемых раковых опухолей у получающей лечение популяции по сравнению с не получающей лечения контрольной популяцией, и/или задержку появления обнаруживаемых раковых опухолей у получающей лечение популяции по сравнению с не получающей лечения контрольной популяцией, например, по статистически и/или клинически существенному количеству. Предотвращение развития инфекции включает, например, уменьшение количества диагностирования инфекции получающей лечение популяции по сравнению с не получающей лечения контрольной популяцией и/или задержку появления симптомов инфицирования у получающей лечение популяции по сравнению с не получающей лечения контрольной популяцией. Предотвращение возникновения боли включает, например, уменьшение силы, или альтернативно задержку, ощущения боли, испытываемого пациентами получающей лечение популяции по сравнению с не получающей лечения контрольной популяцией.
Термин «пролекарство» включает соединения, которые при физиологических условиях преобразуются в терапевтически активные средства. Стандартный способ получения пролекарства должен включать выбранные фрагменты, которые гидролизируются при физиологических условиях, чтобы позволить обнаружить необходимую молекулу. В других вариантах осуществления пролекарство преобразуют посредством ферментативной активности организма-хозяина животного.
Термин «профилактическое или терапевтическое» лечение известен в данной области и включает введение в организм-хозяин одного или более необходимых веществ. Если вещество вводят до клинического проявления нежелательного состояния (например, заболевания или другого нежелательного состояния организма-хозяина животного), тогда лечение является профилактическим (то есть оно защищает организм-хозяин от развития нежелательного состояния), тогда как, если композицию вводят после проявления нежелательного состояния, лечение является терапевтическим (то есть оно предназначено для уменьшения, облегчения или стабилизации существующего нежелательного состояния или его побочных эффектов).
Используемый здесь термин «протеасома» включает иммуно- и конститутивные протеасомы.
Термин «замещенный» обозначает фрагменты, имеющие заместители, заменяющие водород на один или более атомов углерода в основной цепи. Понятно, что термины «замещение» и «замещен» включают неявное условие, что такое замещение находится в соответствии с разрешенной валентностью замещаемого атома и заместителя и что замещение приводит к стабильному соединению, которое, например, не подвергается спонтанно преобразованию, такому как перегруппировка, циклизация, элиминирование и так далее. Используемый здесь термин «замещенный», как подразумевают, включает все допустимые заместители органических соединений. В широком аспекте допустимые заместители включают нециклические и циклические, разветвленные и линейные, карбоциклические и гетероциклические, ароматические и неароматические заместители органических соединений. Допустимые заместители могут представлять собой один или более и те же самые или различные заместители для соответствующих органических соединений. В целях данного изобретения геретоатомы, такие как азот, могут иметь водородные заместители и/или любые допустимые заместители описанных здесь органических соединений, которые удовлетворяют валентности гетероатомов. Заместители могут включать, например, галогеновые, гидроксильные, карбонильные (такие как карбоксил, алкоксикарбонил, формил или ацил), тиокарбонильные (такие как сложный тиоэфир, тиоацетат или тиоформиат), алкоксильные, фосфорильные, фосфатные, фосфонатные, фосфинатные, амино-, амидо-, амидиновые, иминовые, циано-, нитро-, азидо-, сульфгидрильные, алкилтио-, сульфатные, сульфонатные, сульфамоильные, сульфонамидо-, сульфонильные, гетероциклильные, аралкильные, или ароматические или гетероароматические фрагменты. Специалистам в данной области будет понятно, что сами группы, замещаемые на углеводородной цепи, если целесообразно, можно замещать.
«Терапевтически эффективное количество» соединения в отношении используемого способа лечения обозначает количество соединения(ий) в препарате, которое при введении в качестве части необходимой схемы дозировки (млекопитающему, предпочтительно человеку) смягчает симптом, облегчает состояние или замедляет возникновение болезненных состояний согласно клинически приемлемым стандартам для нарушений или состояний, которые необходимо вылечить, или для косметической цели, например, при разумном соотношении выгоды/риска, применимым при любом медицинском лечении.
Термин «тиоэфир» обозначает алкильную группу, как описано выше, содержащую присоединенный к ней сернистый фрагмент. В предпочтительных вариантах осуществления «тиоэфир» представлен -S-алкилом. Типичные представители тиоэфирных групп включают метилтио-, этилтиогруппу и тому подобное.
Используемый здесь термин «лечить» или «лечение» включает изменение, уменьшение или купирование симптомов, клинических признаков и лежащего в основе патологии состояния путем улучшения или стабилизации состояния пациента.
Селективность в отношении 20S протеасомы
Описанные здесь ингибиторы фермента полезны, в частности, потому, что они ингибируют активность 20S протеасомы. Дополнительно, в отличие от других ингибиторов 20S протеасомы, описанные здесь соединения высокоселективны в отношении 20S протеасомы по сравнению с другими протеазными ферментами. То есть соединения по изобретению проявляют селективность в отношении 20S протеасомы по сравнению с другими протеазами, такими как катепсины, кальпаины, папаины, химотрипсин, трипсин, трипептидилпепсидаза II. Селективность ингибиторов фермента в отношении 20S протеасомы такова, что при концентрациях ниже приблизительно 50 мкМ ингибиторы фермента проявляют ингибирование каталитической активности 20S протеасомы, не проявляя в то же самое время ингибирования каталитической активности других протеаз, таких как катепсины, кальпаины, папаины, химотрипсин, трипсин, трипептидилпепсидаза II. В предпочтительных вариантах осуществления ингибиторы фермента проявляют ингибирование каталитической активности 20S протеасомы при концентрациях ниже приблизительно 10 мкМ, не проявляя в то же самое время ингибирования каталитической активности других протеаз при таких концентрациях. В еще более предпочтительных вариантах осуществления ингибиторы фермента проявляют ингибирование каталитической активности 20S протеасомы при концентрациях ниже приблизительно 1 мкМ, не проявляя в то же самое время ингибирования каталитической активности других протеаз при таких концентрациях. Исследования ферментативной кинетики описаны в заявке на патент США с серийным номером 09/569748, пример 2, и Stein et al., Biochem. (1996), 35, 3899-3908.
Селективность в отношении химотрипсино-подобной активности
Определенные варианты осуществления описанных здесь ингибирующих фермент соединений далее применимы, поскольку они могут эффективно и селективно ингибировать химотрипсино-подобную активность 20S протеасомы по сравнению с трипсин-подобной и PGPH-активностью. Химотрипсино-подобная активность 20S протеасомы характеризуется расщеплением петидов в непосредственной близости от больших гидрофобных остатков. В частности, химотрипсино-подобную активность Ntn-гидролаз можно определить по расщеплению стандартного субстрата. Примеры таких субстратов известны в данной области. Например, можно использовать производное лейцилвалинилтирозина. Исследования ферментативной кинетики описаны в заявке на патент США с серийным номером 09/569748, пример 2, и Stein et al., Biochem. (1996), 35, 3899-3908.
Применение ингибиторов фермента
Биологические последствия ингибирования протеасомы обширны. На клеточном уровне сообщали о накоплении полиубиквитинированных белков, о морфологических изменениях клетки и об апоптозе при обработке клеток различными ингибиторами протеасомы. Ингибирование протеасомы также предлагают в качестве возможной противоопухолевой терапевтической стратегии. Тот факт, что эпоксомицин был первоначально обнаружен при проведении скрининга на противоопухолевую активность, позволяет оценивать протеасому как мишень для противоопухолевой химиотерапии. Соответственно, данные соединения применимы для лечения рака. Ингибирование протеасомы также ассоциируется с активацией ингибирования NF-κВ и стабилизацией уровней p53. Таким образом, соединения по изобретению можно также использовать для того, чтобы ингибировать активацию NF-κВ и стабилизировать уровни p53 в культуре клеток. Так как NF-κВ представляет собой ключевой регулятор воспаления, он является привлекательной мишенью для противовоспалительного терапевтического воздействия. Таким образом, соединения по изобретению можно применять для лечения состояний, связанных с хроническим воспалением, включая в качестве неограничивающих примеров COPD, псориаз, бронхит, эмфизему и кистозный фиброз.
Описанные соединения можно использовать для лечения состояний, опосредованных напрямую протеолитической функцией протеасомы, таких как мышечное истощение, или косвенно опосредованных через белки, которые процессируются протеасомой, такие как NF-κВ. Протеасома участвует в быстром удалении и посттрансляционном процессинге белков (например, ферментов), вовлеченных в клеточную регуляцию (например, клеточного цикла, транскрипции генов и метаболических путей), межклеточных контактах и иммунной реакции (например, презентация антигена). Определенные примеры, обсуждаемые ниже, включают в себя β-амилоидный белок и регуляторные белки, такие как циклины, TGF-β и фактор транскрипции NF-κВ.
Другое осуществление изобретения представляет собой применение описанных здесь соединений для лечения нейродегенеративных заболеваний и состояний, включающих в качестве неограничивающих примеров удар, ишемическое поражение нервной системы, нервную травму (например, сотрясение головного мозга, повреждение спинного мозга и травматическое повреждение нервной системы), рассеянный склероз и другие иммуно-опосредованные невропатии (например, синдром Гийена-Барре и его варианты, острую моторную аксональную невропатию, острую воспалительную демиелинизирующую полиневропатию и синдром Фишера), комплексную деменцию при ВИЧ/СПИД, axonomy, диабетическую невропатию, болезнь Паркинсона, болезнь Хантингтона, рассеянный склероз, бактериальный, паразитический, грибковый и вирусный менингит, энцефалит, сосудистую деменцию, мультиинфарктную деменцию, деменцию телец Леви, деменцию лобной доли, такую как болезнь Пика, подкорковую деменцию (такую как паралич Хантингтона или прогрессирующий надъядерный паралич), синдром очаговой кортикальной атрофии (такой как первичная афазия), обменно-токсическую деменцию (такую как хронический гипотиреоз или B12-недостаточность) и деменцию, вызванную инфекцией (такой как сифилис или хронический менингит).
Болезнь Альцгеймера характеризуется внеклеточными отложениями β-амилоидного белка (β-AP) в виде старческих бляшек в мозговых сосудах. β-AP представляет собой фрагмент из от 39 до 42 аминокислот, получающихся из предшественника амилоидного белка (АРР). Известны, по меньшей мере, три изоформы АРР (695, 751 и 770 аминокислот). Альтернативный сплайсинг мРНК приводит к возникновению изоформ; нормальный процессинг затрагивает часть последовательности β-AP, таким образом предотвращая появление β-AP. Полагают, что процессинг аномального белка протеасомой вносит свой вклад в избыток β-AP в мозге при болезни Альцгеймера. АРР-процессирующий фермент у крыс содержит приблизительно десять различных субъединиц (22 кДа - 32 кДа). Субъдиница 25 кДа содержит N-концевую последовательность X-Gln-Asn-Pro-Met-X-Thr-Gly-Thr-Ser, которая идентична β-субъединице человеческого макропаина (Kojima, S. et al., Fed. Eur. Biochem. Soc., (1992) 304: 57-60). АРР-процессирующий фермент расщепляет связь Gln15-Lys16; в присутствии иона кальция фермент также расщепляет связь Met-1-Asp1 и связи Asp1-Ala2 для удаления внеклеточного домена β-AP.
Один вариант осуществления поэтому представляет собой способ лечения болезни Альцгеймера, охватывающий введение пациенту эффективного количества соединения (например, фармацевтической композиции), описанного здесь. Такое лечение включает уменьшение скорости образования бляшек β-AP, уменьшение скорости образования β-AP и уменьшение клинических признаков болезни Альцгеймера.
Другие осуществления изобретения относятся к кахексии и заболеваниям, вызывающим мышечное истощение. Протеасома участвует в деградации множества белков в созревающих ретикулоцитах и растущих фибробластах. В клетках, лишенных инсулина или сыворотки, скорость протеолиза почти удваивается. Ингибирование протеасомы уменьшает протеолиз, таким образом уменьшая и потерю мышечного белка, и азотную нагрузку на почки или печень. Ингибиторы по изобретению применимы для лечения состояний, таких как рак, хронические инфекционные заболевания, лихорадка, мышечное бездействие (атрофия), денервация, поражение нерва, голодание, почечная недостаточность, связанная с ацидозом, диабет и печеночная недостаточность. См., например, Goldberg, патент США № 5340736. Варианты осуществления изобретения поэтому охватывают способы уменьшения скорости деградации мышечного белка в клетке; уменьшения скорости внутриклеточной деградации белка; уменьшения скорости деградации белка p53 в клетке и ингибирования роста связанных с p53 раковых образований. Каждый из данных способов включает контакт клетки (in vivo или in vitro, например, мускулатуры пациента) с эффективным количеством описанного здесь соединения (например, фармацевтической композиции).
Фиброз представляет собой чрезмерное и постоянное образование ткани шрама, являющееся результатом гиперпролиферативного роста фибробластов, и он связан с активацией пути передачи сигналов TGF-β. Фиброз включает обширное отложение внеклеточного матрикса и может иметь место в пределах фактически любой ткани или среди нескольких различных тканей. Обычно уровень внутриклеточного сигнального белка (Smad), который активирует транскрипцию необходимых генов при стимуляции TGF-β, регулируется активностью протеасом (Xu et al., 2000). Однако увеличенную деградацию сигнальных компонентов TGF-β наблюдали при раках и других гиперпролиферативных состояниях. Таким образом, некоторые осуществления изобретения относятся к способу лечения гиперпролиферативных состояний, таких как диабетическая ретинопатия, дегенерация желтого пятна, диабетическая нефропатия, гломерулосклероз, IgA-нефропатия, цирроз, атрезия желчных протоков, застойная сердечная недостаточность, склеродермия, вызванный радиацией фиброз и легочный фиброз (идиопатический легочный фиброз, коллагеновая сосудистая болезнь, саркоидоз, интерстиальные легочные заболевания и приобретенные легочные нарушения). Лечение жертв ожогов часто затруднено фиброзом, таким образом, дополнительный вариант осуществления изобретения представляет собой местное и систематическое введение ингибиторов для лечения ожогов. Ушивание раны после хирургической операции часто связано с обезображивающими рубцами, которые можно предотвратить ингибированием фиброза. Таким образом, в некоторых вариантах осуществления изобретение относится к способу предупреждения возникновения или уменьшения рубцевания.
Другим белком, процессируемым протеасомой, является NF-κВ, член белкового семейства Rel. Семейство Rel транскрипционных белков-активаторов можно подразделить на две группы. Для первой группы необходим протеолитический процессинг, и она включает p50 (NF-κВ 1, 105 кДа) и p52 (NF-κ2, 100 кДа). Для второй группы не требуется протеолитический процессинг, и она включает в себя p65 (RelA, Rel (c-Rel) и RelB). Как гомо-, так и гетеродимеры могут образовываться членами Rel-семества; NF-κВ, например, представляет собой гетеродимер p50-p65. После фосфорилирования и убиквитинирования IκВ и p105 два белка деградируются и процессируются для получения активного NF-κВ, который перемещается из цитоплазмы в ядро. Убиквитинированный p105 также процессируется очищенными протеасомами (Palombella et al., Cell (1994) 78: 773-785). Активный NF-κВ образует стереоспецифический энхансерный комплекс с другими транскрипционными активаторами и, например, HMG I (Y), индуцирующим селективную экспрессию определенного гена.
NF-κВ регулирует гены, вовлеченные в иммунную и воспалительную реакцию и митотические явления. Например, NF-κВ необходим для экспрессии гена легкой цепи κ иммуноглобулина, гена α-цепи рецептора IL-2, гена главного комплекса гистосовместимости класса I большого количество генов цитокинов, кодирующих, например, IL-2, IL-6, гранулоцитарный колониестимулирующий фактор и IFN-β (Palombella et al., Cell (1994) 78: 773-785). Некоторые осуществления изобретения охватывают способы влияния на уровень экспрессии IL-2, MHC-I, IL-6, TNFα, IFN-β или любых других ранее упоминавшихся белков, причем каждый способ включает введение пациенту эффективного количества описанного здесь соединения. Комплексы, включающие p50, являются быстрыми медиаторами острого воспаления и иммунной реакции (Thanos, D. and Maniatis, T., Cell (1995) 80: 529-532).
NF-κВ также участвует в экспрессии генов адгезии клеток, которые кодируют E-селектин, P-селектин, ICAM и VCAM-1 (Collins, T., Lab. Invest. (1993) 68:499-508). Один вариант осуществления изобретения представляет собой способ ингибирования адгезии клеток (например, адгезии клеток, опосредованной E-слектином, P-селектином, ICAM или VCAM-1), охватывающий взаимодействие клетки с (или введение пациенту) эффективным количеством описанного здесь соединения (или фармацевтической композиции).
Ишемические состояния и реперфузионные повреждения приводят к гипоксии, условию, при котором существует недостаточность поступающего в ткани организма кислорода. Данное состояние вызывает увеличенную деградацию Iκ-Bα, тем самым приводя к активации NF-κB (Koong et al., 1994). Было продемонстрировано, что серьезность повреждения, приводящего к гипоксии, можно уменьшить при введении ингибитора протеасомы (Gao et al., 2000; Bao et al., 2001; Pye et al., 2003). Поэтому некоторые варианты осуществления изобретения относятся к способу лечения ишемических состояний или реперфузионных повреждений, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества описанного здесь соединения. Примерами таких состояний или поражений в качестве неограничивающих примеров являются острый коронарный синдром (легко повреждаемые атеросклеротические бляшки), окклюзионное поражение артерии (сердечная, церебральная, периферическая артериальная и сосудистая окклюзии), атеросклероз (склероз коронарных сосудов, болезнь коронарных артерий), инфаркты, сердечная недостаточность, панкреатит, гипертрофия миокарда, стеноз и рестеноз.
NF-κB также специфически связывается с ВИЧ-энхансером/промотором. При сравнении с Nef mac239 регуляторный белок ВИЧ Nef pbj14 отличается двумя аминокислотами в области, которая контролирует связывание протеинкиназы. Полагают, что протеинкиназа передает сигнал о фосфорилировании IκB, вызывая деградацию IκB через путь убиквитин-протеасома. После деградации NF-κB высвобождается в ядро, таким образом увеличивая транскрипцию ВИЧ (Cohen, J., Science, (1995) 267: 960). Два варианта осуществления изобретения представляют собой способ ингибирования или уменьшения инфицирования ВИЧ у пациента и способ снижения уровня экспрессии вирусного гена, причем каждый способ включает введение пациенту эффективного количества описанного здесь соединения.
Избыточное продуцирование индуцируемых липополисахаридами (LPS) цитокинов, таких как TNFα, как полагают, является центральным процессом, связанным с септическим шоком. Кроме того, общепринято, что первая стадия в активации клеток LPS представляет собой связывание LPS с определенными мембранными рецепторами. α- и β-Субъединицы комплекса 20S протеасомы идентифицировали в качестве связывающих LPS белков, предполагая, что индуцируемая LPS трансдукция может представлять собой важную терапевтическую мишень для лечения и предотвращения развития сепсиса (Qureshi, N. et al., J. Immun. (2003) 171: 1515-1525). В некоторых вариантах осуществления поэтому соединения по изобретению можно использовать для ингибирования TNFα для того, чтобы предотвратить развитие и/или лечить септический шок.
При внутриклеточном протеолизе образуются небольшие пептиды для представления T-лимфоцитам, чтобы вызвать опосредованный MHC класса I иммунный ответ. Иммунная система защищает от аутологичных клеток, которые заражены вирусом или претерпели онкогенную трансформацию. Один вариант осуществления представляет собой способ ингибирования презентации антигена в клетке, охватывающий воздействие на клетку описанным здесь соединением. Соединение по изобретению можно использовать для лечения связанных с иммунным ответом состояний, таких как аллергия, астма, отторжение органа/ткани (реакция «трансплантат против хозяина») и аутоиммунных заболеваний, включающих в качестве неограничивающих примеров волчанку, ревматоидный артрит, псориаз, рассеянный склероз воспалительные заболевания кишечника (такие как язвенный колит и болезнь Крона). Таким образом, дополнительное осуществление представляет собой способ подавления иммунной системы пациента (например, ингибирование отторжения трансплантата, аллергий, аутоиммунных заболеваний и астмы), включающий введение пациенту эффективного количества описанного здесь соединения.
Другое дополнительное осуществление представляет собой способ изменения разнообразия антигенных пептидов, продуцируемых протеасомой или другими Ntn с мультикаталитической активностью. Например, если активность PGPH 20S протеасомы селективно ингибировать, то другой ряд антигенных пептидов будет продуцироваться протеасомой и представляться молекулам MHC на поверхности клеток, которые продуцировались и представлялись бы либо без ингибирования какого-либо фермента, либо, например, при селективном ингибировании химотрипсино-подобной активности протеасомы.
Некоторые ингибиторы протеасомы блокируют как деградацию, так и процессинг убиквитинированного NF-κB in vitro и in vivo. Ингибиторы протеасомы также блокируют деградацию IκB-α и активацию NF-κB (Palombella, et al. Cell (1994) 78: 773-785; Traenckner, et al., EMBO J. (1994) 13: 5433-5441). Один вариант осуществления изобретения представляет собой способ ингибирования деградации IκB-α, охватывающий воздействие на клетку описанным здесь соединением. Дополнительное осуществление представляет собой способ уменьшения клеточного содержания NF-κB в клетке, мускулатуре, органе или в организме пациента, включающий воздействие на клетку, мускулатуру, орган или организм пациента описанным здесь соединением.
Другие эукариотические факторы транскрипции, для которых необходим протеолитический процессинг, включают основной фактор транскрипции TFIIA, вспомогательный белок вируса простого герпеса VP16 (фактор клетки-хозяина), белок индуцируемого вирусом регуляторного фактора 2 IFN и белок 1, связывающий мембраносвязанный регуляторный элемент стерола.
Другие варианты осуществления изобретения представляют собой способы воздействия на циклинзависимые циклы эукариотических клеток, охватывающие воздействие на клетку (in vitro или in vivo) описанным здесь соединением. Циклины представляют собой белки, вовлеченные в контролирование клеточного цикла. Протеасома участвует в деградации циклинов. Примеры циклинов включают в себя митотические циклины, циклины G1 и циклин B. Деградация циклинов позволяет клетке выйти из одной стадии клеточного цикла (например, митоза) и войти в другую (например, деление). Полагают, что все циклины ассоциированы с протеинкиназой p34.sup.cdc2 или родственными киназами. Сигнал позиционирования для протеолиза локализован в аминокислотах 42-RAALGNISEN-50 (бокс деструкции). Существует доказательство, что циклин превращается в форму, легко поддающуюся действию убиквитинлигазы, или что специфическая для циклина лигаза активируется во время митоза (Ciechanover, A., Cell, (1994) 79:13-21). Ингибирование протеасомы ингибирует деградацию циклина и, следовательно, ингибирует пролиферацию клеток, например, связанные с циклином раковые образования (Kumatori et al., Proc. Natl. Acad. Sci. USA (1990) 87: 7071-7075). Один вариант осуществления изобретения представляет собой способ лечения пролиферативных заболеваний пациента (например, рак, псориаз или рестеноз), включающий введение пациенту эффективного количества описанного здесь соединения. Изобретение также включает способ лечения связанного с циклином воспаления у пациента, включающий введение пациенту терапевтически эффективного количества описанного здесь соединения.
Дополнительные варианты осуществления изобретения представляют собой способы воздействия на зависимую от протеасомы регуляцию онкогенных белков и способы лечения или ингибирования роста раковых образований, причем каждый способ включает воздействие на клетку (in vivo, например, на пациента, или in vitro) описанным здесь соединением. Белки E6, полученные из HPV-16 и HPV-18, стимулируют АТФ- и убиквитин-зависимую конъюгацию и деградацию p53 в неочищенных лизатах ретикулоцитов. Рецессивный онкоген p53, как было показано, накапливается при не позволяющей развиваться температуре в клеточной линии с мутированным термостабильным E1. Увеличенные уровни p53 могут приводить к апоптозу. Примеры прото-онкогенных белков, деградируемых системой убиквитина, включают в себя c-Mos, c-Fos и c-Jun. Один вариант осуществления представляет собой способ лечения связанных с p53 апоптозов, включающий введение пациенту терапевтически эффективного количества описанного здесь соединения.
В другом варианте осуществления описанные соединения применимы для лечения паразитических инфекций, таких как инфекции, вызванные простейшими паразитами. Протеасома данных паразитов, как полагают, вовлечена прежде всего в дифференцировку клетки и репликационные активности (Paugam et al., Trends Parasitol. 2003, 19(2): 55-59). Кроме того, виды энтамебы, как было показано, теряют способность к инкапсулированию при воздействии ингибиторов протеасом (Gonzales, et al., Arch. Med. Res. 1997, 28, Spec No: 139-140). В некоторых таких вариантах осуществления описанные соединения применимы для лечения паразитических инфекций у людей, вызванных простейшими паразитами, выбранными из видов Plasmodium (включающих P. falciparum, P. vivax, P. Malariae и P. ovale, которые вызывают малярию), видов Trypanosoma (включающих T. cruzi, которая вызывает болезнь Шагаса, и T. Brucei, которая вызывает африканскую сонную болезнь), видов Leishmania (включающих L. amazonensis, L. donovani, L. infantum, L. Mexicana и так далее), Pneumocystis carinii (простейшее, которое, как известно, вызывает пневмонию у пациентов со СПИД и другими иммуносупрессивными заболеваниями), Toxoplasma gondii, Entamoeba histolytica, Entamoeba invadens и Giardia lamblia. В некоторых вариантах осуществления описанные соединения применимы для лечения паразитических инфекций у животных и крупного рогатого скота, вызванных простейшими паразитами, выбранными из Plasmodium hermani, видов Cryptosporidium, Echinococcus granulosus, Eimeria tenella, Sarcocystis neurona и Neurospora crassa. Другие соединения, применимые в качестве ингибиторов протеасом при лечении вызванных паразитами заболеваний, описаны в WO 98/10779, которая полностью включена сюда.
В некоторых вариантах осуществления описанные соединения необратимо ингибируют активность протеасомы в паразитах. Такое необратимое ингибирование, как было показано, вызывает прекращение работы фермента без восстановления в красных клетках крови и белых клетках крови. В некоторых таких вариантах осуществления продолжительный период полужизни клеток крови может обеспечить пролонгированную защиту в качестве терапии против повторного воздействия паразитов. В некоторых вариантах осуществления продолжительный период полужизни клеток крови может обеспечить пролонгированную защиту в качестве химиопрофилактики будущей инфекции.
Также было продемонстрировано, что ингибиторы, которые связываются с 20S протеасомой, стимулируют образование кости в органных культурах кости. Кроме того, когда такие ингибиторы систематически вводили мышам, некоторые ингибиторы протеасомы привели к увеличению объема кости и скорости образования кости более чем на 70% (Garrett, I.R. et al. , J. Clin. Invest. (2003) 111: 1771-1782), предполагая, следовательно, что убиквитин-протеасомный аппарат регулирует дифференцировку остеобластов и образование кости. Поэтому описанные соединения можно применять для лечения и/или предотвращения развития заболеваний, связанных с потерей костной массы, такой как остеопороз.
Костная ткань является превосходным источником факторов, которые обладают способностью стимулировать костные клетки. Таким образом, экстракты бычьей костной ткани содержат не только структурные белки, которые ответственны за поддержание структурной целостности кости, но также и за биологически активные факторы роста кости, которые могут стимулировать пролиферацию клеток кости. Среди таких последних факторов находится недавно описанное семейство, названное костные морфогенетические белки (BMP). Все данные факторы роста воздействуют на другие типы клеток, также как и на клетки кости, охватывая Hardy, M.H., et al., Trans Genet (1992) 8: 55-61, который описывает доказательство, что костные морфогенетические белки (BMP) по разному экспрессированы в волосяных фолликулах в процессе развития. Harris, S.E., et al., J Bone Miner Res (1994) 9: 855-863, описывают влияние TGFβ на экспрессию BMP-2 и другие вещества в костных клетках. Экспрессия BMP-2 в зрелых фолликулах также имеет место при созревании и после периода пролиферации клетки (Hardy, et al. (1992, выше). Таким образом, соединения по изобретению можно также использовать для стимуляции роста волосяных фолликул.
В заключение, описанные соединения также применимы в качестве диагностических средств (например, в диагностических наборах или для использования в клинических лабораториях) для скрининга белков (например, ферментов, транскрипционных факторов), процессируемых гидролазами Ntn, включающих в себя протеасому. Описанные соединения также применимы в качестве реагентов для исследований специфического связывания субъединицы X/MB 1 или α-цепи и ингибирования связанной с ней протеолитической активности. Например, можно определять активность (и специфические ингибиторы) других субъединиц протеасомы.
Большинство клеточных белков подвергаются протеолитическому процессингу во время созревания или активации. Описанные здесь ингибиторы фермента можно использовать для того, чтобы определить, регулируются ли клеточный процесс, процесс развития или физиологический процесс или продукция протеолитической активностью определенной гидролазой Ntn. Один такой способ включает получение организма, препарата интактных клеток или клеточного экстракта; воздействие на организм, препарат клеток или клеточный экстракт описанным здесь соединением; воздействие на подвергнутый воздействию соединением организм, препарат клеток или клеточный экстракт сигналом и мониторинг процесса или продукции. Высокая селективность описанных здесь соединений позволяет осуществлять быстрое и точное удаление или вовлечение Ntn (например, 20S протеасомы) в конкретный клеточный процесс, процесс развития или физиологический процесс.
Введение
Полученные, как описано здесь, соединения можно вводить в различных видах в зависимости от расстройства, которое необходимо вылечить, и возраста, состояния и веса тела пациента, как это хорошо известно в данной области. Например, в тех случаях, когда соединения вводят перорально, их можно приготовить в виде таблеток, капсул, гранул, порошков или сиропов, или для парентерального введения их можно приготовить в виде инъекций (внутривенных, внутримышечных или подкожных), препаратов для капельного вливания или суппозиториев. Для введения через мембрану слизистой глаза их можно приготовить в виде глазных капель или глазной мази. Данные лекарственные формы можно приготовить общепринятыми способами, и, если необходимо, активный компонент можно смешать с любой обычной добавкой или наполнителем, таким как связующее средство, разрыхлитель, смазывающее вещество, модификатор лекарственных веществ, солюбилизирующее средство, суспензирующее средство, эмульгирующее средство, средство покрытия, циклодекстрин и/или буфер. Хотя дозировка будет меняться в зависимости от симптомов, возраста и веса тела пациента, природы и серьезности нарушения, которое необходимо вылечить или предотвратить его развитие, способа введения и формы лекарственного средства, в общем случае для взрослого человека рекомендована ежедневная дозировка от 0,01 до 2000 мг соединения, и ее можно вводить в виде однократной дозы или в виде разделенных доз. Количество активного компонента, который можно объединить с веществом носителя, чтобы получить лекарственную форму для однократной дозы, в общем случае будет представлять собой такое количество соединения, которое оказывает терапевтический эффект.
Точное время введения и/или количество соединения, которое приведет к самым эффективным результатам в отношении эффективности лечения данного пациента, будет зависеть от активности, фармакокинетики и биодоступности определенного соединения, физиологического состояния пациента (включая сюда возраст, пол, тип заболевания и стадию, общее физическое состояние, восприимчивость данной дозировки и тип лечения), способа введения и так далее. Тем не менее упомянутые выше рекомендации можно использовать в качестве основы для точного подбора лечения, например, определяя оптимальное время и/или количества для введения, которая не потребует большего, чем обычное экспериментирование, состоящее из контроля над пациентом и подбора дозировки и/или выбора времени.
Используемая здесь фраза «фармацевтически приемлемый» обозначает такие лиганды, вещества, композиции и/или лекарственные формы, которые, с медицинской точки зрения, подходят для использования при контактировании с тканями людей и животных без чрезмерной токсичности, раздражения, аллергического ответа или другой проблемы или осложнения, соразмерного с разумным соотношением выгода/риск.
Используемая здесь фраза «фармацевтически приемлемый носитель» обозначает фармацевтически приемлемое вещество, композицию или носитель, такой как жидкий или твердый заполнитель, разбавитель, наполнитель, растворитель или инкаспулирующее вещество. Каждый носитель должен быть «приемлемым» в том смысле, что должен быть совместимым с другими ингредиентами препарата и не должен быть вредным для пациента. Некоторые примеры веществ, которые могут служить фармацевтически приемлемыми носителями, включают (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал, картофельный крахмал и замещенный и незамещенный β-циклодекстрин; (3) целлюлозу и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы; (4) порошкообразный трагакант; (5) солод; (6) желатин; (7) тальк; (8) наполнители, такие как масло какао и воск для суппозиториев; (9) масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоли; (11) полиолы типа глицерина, сорбита, маннита и полиэтиленгликоля; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) буферирующие средства, такие как гидроксид магния и гидроксид алюмия; (15) альгиновую кислоту; (16) апирогенную воду; (17) изотонический раствор; (18) раствор Рингера; (19) этиловый спирт; (20) фосфатно-буферный раствор и (21) другие нетоксичные совместимые вещества, используемые в фармацевтических препаратах. В некоторых вариантах осуществления фармацевтические композиции по настоящему изобретению являются непирогенными, то есть не вызывают значительного повышения температуры при введении пациенту.
Термин «фармацевтически приемлемая соль» обозначает относительно нетоксичные аддитивные соли неорганических и органических кислот ингибитора(ов). Такие соли можно получить in situ во время конечного выделения или очистки ингибитора(ов), по отдельности подвергая очищенный ингибитор(ы) в виде его свободного основания реакции с подходящей органической или неорганической кислотой и выделяя таким образом образовавшуюся соль. Типичные примеры соли включают гидробромид, гидрохлорид, сульфат, бисульфат, фосфат, нитрат, ацетат, валерат, олеат, пальмитат, стеарат, лаурат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептонат, лактобионат, лаурилсульфанатные соли, соли аминокислот и тому подобное. (См., например, Berge et al. (1977) "Pharmaceutical Salts", J. Pharm. Sci. 66: 1-19).
В других случаях ингибиторы, применимые в способах по настоящему изобретению, могут содержать одну или более кислотных функциональных групп и, таким образом, способных к образованию фармацевтически приемлемых солей с фармацевтически приемлемыми основаниями. Термин «фармацевтически приемлемые соли» в этих случаях обозначает относительно нетоксичные аддитивные соли неорганических и органических оснований ингибитора(ов). Такие соли можно аналогичным способом получить in situ во время конечного выделения или очистки ингибитора(ов), по отдельности подвергая реакции очищенный ингибитор(ы) в его свободной кислотной форме с подходящим органическим или неорганическим основанием, таким как гидроксид, карбонат или бикарбонат фармацевтически приемлемого катиона металла, с аммиаком или с фармацевтически приемлемым органическим первичным, вторичным или третичным амином. Типичные примеры оснований или щелочноземельных солей включают литий, натрий, калий, кальций, магний и соли алюминия и тому подобное. Типичные примеры органических аминов, применимых для образования аддитивных солей оснований, включают этиламин, диэтиламин, этилендиамин, этаноламин, диэтаноламин, пиперазин и тому подобное (см., например, Berge et al., выше).
Смачивающие средства, эмульгаторы и смазывающие вещества, такие как лаурилсульфат натрия и стеарат магния, а также и красящие вещества, разделительные средства, средства покрытия, подслащивающие, вкусовые и ароматизирующие добавки, консерванты и антиоксиданты также могут присутствовать в композициях.
Примеры фармацевтически приемлемых антиоксидантов включают (1) растворимые в воде антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфит натрия, сульфит натрия и тому подобное; (2) растворимые в масле антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол (ВНА), бутилированный гидрокситолуол (ВНТ), лецитин, пропилгаллат, альфа-токоферол и тому подобное, и (3) хелатирующие металл агенты, такие как лимонная кислота, этилендиаминтетрауксусная кислота (EDTA), сорбитол, винная кислота, фосфорная кислота и тому подобное.
Препараты, подходящие для перорального введения, могут находиться в виде капсул, саше, пилюль, таблеток, леденцов (использующих ароматизированную основу, обычно сахарозу и гуммиарабик или трагакант), порошков, гранул или в виде раствора или суспензии в водной или неводной жидкости, или в виде жидкой эмульсии типа масло-в-воде или вода-в-масле, или в виде эликсира или сиропа, или в виде пастилок (использующих инертный матрикс, такой как желатин и глицерин или сахарозу и гуммиарабик) и/или в виде жидкости для полоскания рта и тому подобного, причем каждый содержит предварительно определенное количество ингибитора(ов) в качестве активного ингредиента. Композицию можно также вводить в виде болюса, электуария или пасты.
В твердых лекарственных формах для перорального введения (капсулы, таблетки, пилюли, драже, порошки, гранулы и тому подобное) активный ингредиент смешан с одним или несколькими фармацевтически приемлемыми носителями, такими как цитрат натрия или дикальцийфосфат и/или любым из следующего ниже: (1) заполнители или наполнители, такие как крахмалы, циклодекстрины, лактоза, сахароза, глюкоза, маннит и/или кремниевая кислота; (2) связующие вещества, такие как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпироллидон, сахароза и/или гуммиарабик; (3) увлажняющие средства, такие как глицерин; (4) дезинтегрирующие средства, такие как агар-агар, карбонат кальция, картофельный крахмал или крахмал из тапиоки, альгиновая кислота, некоторые силикаты и карбонат натрия; (5) растворы ингибирующих средств, такие как парафин; (6) ускорители впитывания, такие как соединения четвертичного аммония; (7) смачивающие средства, такие как, например, ацетиловый спирт и моностеарат глицерина; (8) абсорбенты, такие как каолиновая и бентонитовая глина; (9) смазывающие вещества, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси, и (10) окрашивающие средства. В случае капсул, таблеток и пилюль фармацевтические композиции могут также содержать буферизующие средства. Твердые композиции подобного типа можно также использовать в качестве наполнителей в мягких и полностью заполненных желатиновых капсулах, используя такие наполнители, как лактоза или молочный сахар, так же, как и высокомолекулярные полиэтиленгликоли и тому подобное.
Таблетку можно изготовить прессованием или литьем под давлением, необязательно с одним или более дополнительными ингредиентами. Прессованные таблетки можно изготовить, используя связующее вещество (например, желатин или гидроксипропилметилцеллюлозу), смазывающее вещество, инертный разжижитель, консервант, разрыхлитель (например, натрия крахмала гликолят или перекрестносвязанную карбоксиметилцеллюлозу натрия), поверхностно-активное или диспергирующее вещество. Отлитые под давлением таблетки можно изготовить, отливая в подходящем устройстве смесь порошкового ингибитора(ов), увлажненного инертным жидким разжижителем.
Таблетки и другие твердые лекарственные формы, такие как драже, капсулы, пилюли и гранулы, можно необязательно изготовить с насечкой или с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, известные в области фармацевтических препаратов. Их также можно изготовить так, чтобы обеспечить медленное или контролируемое высвобождение активного компонента, используя в них, например, гидроксипропилметилцеллюлозу в различных пропорциях для того, чтобы обеспечить необходимый профиль высвобождения, другие полимерные матриксы, липосомы и/или микросферы. Их можно стерилизовать, например, фильтрацией через удерживающий бактерии фильтр или добавляя стерилизующие средства в виде стерильных твердых композиций, которые можно растворить в стерильной воде, или некоторой другой стерильной инъецируемой среде непосредственно перед использованием. Такие композиции могут также необязательно содержать опалесцирующие компоненты и могут принадлежать к композициям, которые высвобождают только активный компонент(ы) или предпочтительно только в определенной части желудочно-кишечного тракта, необязательно, пролонгированным способом. Примеры встроенных композиций, которые можно использовать, включают в себя полимерные вещества и воски. Активный компонент можно также инкапсулировать в форму микрокапсулы, если это целесообразно, с одним или более из описанных выше наполнителей.
Жидкие лекарственные формы для перорального введения включают в себя фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активному ингредиенту жидкие лекарственные формы могут содержать инертные разжижители, обычно используемые в данной области, такие как, например, вода или другие растворители, солюбилизирующие средства и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (в частности, хлопковое, арахисовое, кукурузное, масло из жмыха зародышей, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и сложный эфир жирной кислоты и сорбитана и их смеси.
Помимо инертных разжижителей композиции для перорального введения могут также включить в себя адъюванты, такие как смачивающие средства, эмульгирующие и суспендирующие средства, подслащивающие, придающие вкус, окрашивающие, отдушивающие и консервирующие средства.
Суспензии в дополнение к активному ингибитору(ам) содержат суспендирующие средства, такие как, например, этоксилированный изостеариловый спирт, полиоксиэтиленовые эфиры сорбита и сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант и их смеси.
Препараты для ректального или вагинального введения могут представлять собой суппозиторий, который можно изготовить, смешивая один или более ингибитор(ов) с одним или более подходящими нераздражающими наполнителями или носителями, содержащими, например, масло какао, полиэтиленгликоль, воск для суппозиториев или салицилат, который является твердым при комнатной температуре, но жидким при температуре тела и который поэтому будет таять в прямой кишке или вагинальной полости и высвобождать активный агент.
Препараты, которые подходят для вагинального введения, также включают пессарии, тампоны, кремы, гели, пасты, пенки или препараты в виде спрея, содержащие такие носители, которые, как известно в данной области, являются подходящими.
Лекарственные формы для местного или трансдермального введения ингибитора(ов) включают в себя порошки, спреи, мази, пасты, кремы, лосьоны, гели, растворы, пластыри и ингаляторы. Активный компонент можно смешивать в стерильных условиях с фармацевтически приемлемым носителем и с любыми консервантами, буферами или пропеллентами, которые могут потребоваться.
Мази, пасты, кремы и гели в дополнение к ингибитору(ам) могут содержать наполнители, такие как животные и растительные жиры, масла, воски, парафины, крахмал, трагакант, производные целлюлозы, полиэтиленгликоли, силиконы, бентониты, кремниевая кислота, тальк и оксид цинка или их смеси.
Порошки и спреи в дополнение к ингибитору(ам) могут содержать наполнители, такие как лактоза, тальк, кремниевая кислота, гидроксид алюминия, силикаты кальция и порошок на основе полиамидов или смеси данных веществ. Спреи могут дополнительно содержать общепринятые пропелленты, такие как хлорфторуглеводороды, и летучие незамещенные углеводороды, такие как бутан и пропан.
Ингибитор(ы) альтернативно вводят аэрозолем. Это осуществляется посредством приготовления водного аэрозоля, липосомального препарата или твердых частиц, содержащих композицию. Можно использовать неводную суспензию (например, фторуглеродный пропеллент). Ультразвуковые распылители предпочтительны, поскольку они минимизируют воздействие на средство сдвига слоев, который может привести к деградации соединения.
Обычно водный аэрозоль получают, приготавливая водный раствор или суспензию агента вместе с общепринятыми фармацевтически приемлемыми носителями и стабилизаторами. Носители и стабилизаторы изменяются в зависимости от требований для определенной композиции, но типично включают неионогенные поверхностно-активные вещества (твин, плюроник, эфир сорбитана, лецитин, кремофоры), фармацевтически приемлемые сорастворители, такие как полиэтиленгликоли, нетоксичные белки, такие как альбумин сыворотки, олеиновая кислота, аминокислоты, такие как глицин, буферы, соли, сахара или сахарные спирты. Аэрозоли обычно приготавливают из изотонических растворов.
Трансдермальные пластыри имеют дополнительное преимущество обеспечения контролируемой доставки ингибитора(ов) в организм. Такие лекарственные формы можно приготовить, растворяя или диспергируя агент в соответствующей среде. Ускорители абсорбции можно также использовать, чтобы увеличить поступление ингибитора(ов) на кожу. Скорость такого потока можно контролировать либо при помощи контролирующей скорость мембраны, либо диспергируя ингибитор(ы) в полимерном матриксе или геле.
Фармацевтические композиции по данному изобретению, подходящие для парентерального введения, содержат один или более ингибитор(ов) в комбинации с одним или более фармацевтически приемлемыми стерильными водными или неводными растворами, дисперсиями, суспензиями или эмульсиями или стерильными порошками, которые можно превратить в стерильные инъецируемые растворы или дисперсии непосредственно перед использованием, которые могут содержать антиоксиданты, буферы, бактериостаты, растворы, которые придают препарату изотоничность крови предполагаемого получателя, или суспендирующие или загущающие средства.
Примеры подходящих водных и неводных носителей, которые можно использовать в фармацевтических композициях по изобретению, включают в себя воду, этанол, полиолы (такие как глицерин, пропиленгликоль, полиэтиленгликоль и тому подобное) и подходящие их смеси, растительные масла, такие как оливковое масло, и инъецируемые органические сложные эфиры, такие как этилолеат. Соответствующую текучесть можно поддерживать, например, при помощи веществ покрытия, таких как лецитин, сохраняя необходимый размер частиц в случае дисперсий и используя поверхностно-активные вещества.
Данные композиции могут также содержать адъюванты, такие как консерванты, смачивающие средства, эмульгирующие средства и диспергирующие средства. Предотвращение действия микроорганизмов можно обеспечить добавлением различных противомикробных и противогрибковых агентов, например, парабенов, хлорбутанола, фенола сорбиновой кислоты и тому подобного. Также желательно добавлять в композиции поддерживающие тоничность средства, такие как сахара, хлорид натрия и тому подобное. Кроме того, пролонгированное всасывание инъецируемой фармацевтической формы можно вызвать добавлением агентов, которые задерживают поглощение, такие как моностеарат алюминия и желатин.
В некоторых случаях, чтобы продлить действие препарата, необходимо замедлить всасывание препарата при подкожной или внутримышечной инъекции. Например, замедленное всасывание парентерально вводимой формы препарата осуществляют, растворяя или суспендируя лекарственное средство в масляном носителе.
Инъецируемые лекарственные формы пролонгированного действия получают, образуя микроинкапсулированные матриксы с ингибитором(ами) в биодеградируемых полимерах, таких как полилактид-полигликолид. В зависимости от соотношения лекарственного средства и полимера и природы определенного используемого полимера можно контролировать скорость высвобождения лекарственного средства. Примеры других биодеградируемых полимеров включают в себя поли(ортоэфиры) и поли(ангидриды). Инъецируемые препараты пролонгированного действия также получают, заключая лекарственное средство в липосомы или микроэмульсии, которые являются совместимыми с тканью организма.
Препараты лекарственных средств можно вводить перорально, парентерально, местно или ректально. Им, конечно, придают формы, подходящие для каждого способа введения. Например, их вводят в виде таблеток или капсул, инъекцией, ингаляцией, в виде глазного лосьона, мази, суппозитория, вливания; местно в виде лосьона или мази; ректально суппозиторием. Предпочтительно пероральное введение.
Используемые здесь фразы «парентеральное введение» и «вводимый парентерально» обозначают способы введения, отличные от энтерального и местного введения, обычно инъекцией, и без ограничения включают внутривенную, внутримышечную, внутриартериальную, подоболочечную, внутрикапсульную, внутриорбитальную, внутрисердечную, внутрикожную, внутрибрюшинную, транстрахеальную, подкожную, под кутикулу, внутрисуставную, подкапсульную, подпаутинную, внутриспинную и надчревную инъекцию и инфузию.
Используемые здесь фразы «системное введение», «вводимый системно», «периферическое введение» и «вводимый периферически» обозначают введение лиганда, лекарственного средства или другого вещества способом, за исключением непосредственного введения в центральную нервную систему, такое, что препарат вводят в систему пациента и, таким образом, он подвергается метаболизму и другим подобным процессам, например, подкожное введение.
Данные ингибиторы можно вводить человеку и другим животным для лечения любым подходящим способом введения, включая сюда перорально, назально как, например, посредством спрея, ректально, интравагинально, парентерально, интрацистернально и местно, как посредством порошков, мазей или капель, включая сюда буккально и подъязычно.
Независимо от выбранного способа введения ингибитор(ы), которые можно использовать в подходящем гидратированном виде и/или в фармацевтических композициях по настоящему изобретения, получают в фармацевтически приемлемых лекарственных формах общепринятыми способами, известными специалистам в данной области.
Фактические уровни дозировки активных ингредиентов в фармацевтических композиция по данному изобретению можно изменять, с тем чтобы получить количество активного ингредиента, которое эффективно для достижения необходимого терапевтического ответа для определенного пациента, композиции и способа введения, не являясь токсичным для пациента.
Концентрация описанного соединения в фармацевтически приемлемой смеси будет меняться в зависимости от нескольких факторов, включающих дозировку вводимого соединения, фармакокинетические характеристика соединения(ий) и способ введения. В общем случае композиции по данному изобретению можно предоставить в водном растворе, содержащем приблизительно 0,1-10% мас./об. описанного здесь соединения среди других веществ, для парентерального введения. Типичные диапазоны доз находятся в диапазоне от приблизительно 0,01 до приблизительно 50 мг/кг веса тела в день, при введении в 1-4 разделенных дозах. Каждая разделенная доза может содержать те же самые или различные соединения по изобретению. Дозировка будет представлять собой эффективное количество в зависимости от нескольких факторов, включающих общее здоровье пациента и препарат и способ введения выбранного соединения(ий).
Другой аспект изобретения относится к комбинационной терапии, где одно или более различные терапевтические средства вводят с ингибитором протеасомы. Такое комбинационное лечение можно достичь посредством одновременного, последовательного или отдельного дозирования индивидуальных компонентов лечения.
В некоторых вариантах осуществления соединение по изобретению вводят совместно с одним или несколькими другими ингибитором(ами) протеасомы.
В некоторых вариантах осуществления соединение по изобретению вводят совместно с химиотерапевтическим препаратом. Подходящий химиотерапевтический препарат может включать природные продукты, такие как алкалоиды барвинка (то есть винбластин, винкристин и винорелбин), паклитаксел, эпидиподофиллотоксины (то есть этопозид, тенипозид), антибиотики (дактиномицин (актиномицин D) даунорубицин, доксорубицин и идарубицин), антрациклины, митоксантрон, блеомицины, пликамицин (митрамицин) и митомицин, ферменты (L-аспарагиназа, которая систематически метаболизирует L-аспарагин и удаляет клетки, которые не обладают способностью синтезировать свой собственный аспарагин); антитромбоцитарные средства; антипролиферативные/антимитотические алкилирующие средства, такие как азотистый иприт (мехлорэтамин, циклофосфамид и аналоги, мелфалан, хлорамбуцил), этиленимины и метилмеламины (гексаметилмеламин и тиотепа), алкилированные сульфонаты (бусульфан), нитрозомочевина (кармустин (BCNU) и аналоги, стрептозоцин), триазен-дакарбазинин (DTIC); антипролиферативные/антимитотические антиметаболиты, такие как аналоги фолиевой кислоты (метотрексат), аналоги пиримидина (фторурацил, флоксуридин и цитарабин), аналоги пурина и родственные ингибиторы (меркаптопурин, тиогуанин, пентостатин и 2-хлордезоксиаденозин); ингибиторы ароматазы (анастрозол, экземестан и летрозол); и платиновые координационные комплексы (цисплатин, карбоплатин), прокарбазин, гидроксимочевина, митотан, аминоглутетимид; гормоны (то есть эстроген) и гормональные агонисты, такие как агонисты лютропин-высвобождающего гормона (LHRH) (гозерелин, леупролид и трипторелин). Другие химиотерапевтические средства могут включать мехлоретамин, камптотецин, ифосфамид, тамоксифен, ралоксифен, гемцитабин, навельбин или любой аналог или производный вариант из упомянутого выше.
В некоторых вариантах осуществления соединение по изобретению вводят совместно со стероидом. Подходящие стероиды могут включать в качестве неограничивающих примеров 21-ацетоксипрегненолон, алклометазон, алгестон, амцинонид, беклометазон, бетаметазон, будезонид, хлорпреднизон, клобетазол, клокортолон, клопреднол, кортикостерон, кортизон, кортивазол, дефлазокорт, дезонид, дезоксиметазон, дексаметазон, дифлоразон, дифлукортолон, дифупреднат, эноксолон, флуазакорт, флуклоронид, флуметазон, флунизолид, флуоцинолонацетонид, флуоцинонид, флуокортинбутил, флуокортолон, флуорометолон, флуперолонацетат, флупреднидена ацетат, флупреднизолон, флурандренолид, флутиказона пропионат, формокортал, галцинонид, галобетазола пропионат, галометазон, гидрокортизон, лотепреднол этабонат, мазипредон, медризон, мепреднизон, метилпреднизолон, мометазона фуроат, параметазон, предникарбат, преднизолон, преднизолона 25-диэтиламиноацетат, преднизолона фосфат натрия, преднизон, преднивал, преднилиден, римексолон, тиксокортол, триамцинолон, триамцинолонацетонид, триамцинолонбенетонид, триамцинолонгексацетонид и их соли и/или производные.
В некоторых вариантах осуществления соединение по изобретению вводят совместно с иммунотерапевтическим средством. Подходящие иммунотерапевтические средства могут включать в качестве неограничивающих примеров циклоспорин, талидомид и моноклональные антитела. Моноклональные антитела могут быть либо простыми, либо конъюгированными, такими как ритуксимаб, тозитумомаб, алемтузумаб, епратузумаб, ибритутомаб тиуксетан, гемтузумаб озогамицин, бевацизумаб, цетуксимаб, эрлотиниб и трастузумаб.
ПРИМЕРЫ
Схема 1: синтез по примеру 1
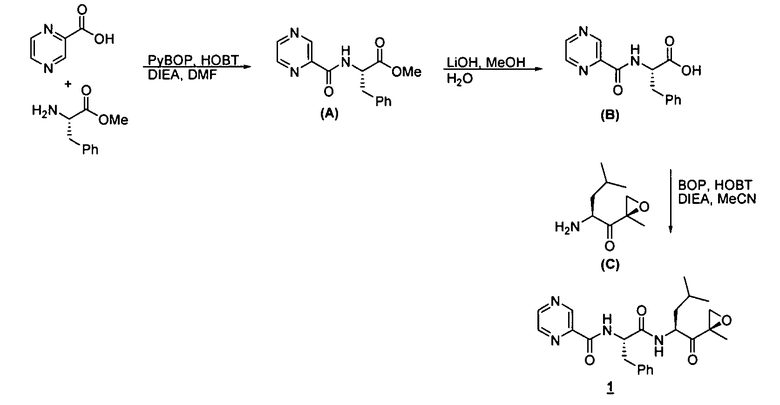
Синтез (A)
К раствору гидрохлорида метилового эфира фенилаланина (2,31 ммоль, 0,5 г) в 25 мл DMF добавляли пиразинкарбоновую кислоту (2,31 ммоль, 0,28 г), DIEA (9,24 ммоль, 1,19 г, 1,61 мл) и HOBT (3,70 ммоль, 0,50 г) и раствор охлаждали до 0ºC. К охлажденному раствору несколькими порциями добавляли PyBOP (3,70 ммоль, 1,93 г) и смесь убирали с бани со льдом и давали ей нагреться до комнатной температуры при перемешивании в атмосфере аргона в течение 6 часов. Смесь разбавляли насыщенным солевым раствором (50 мл) и экстрагировали EtOAc (5×15 мл). Органические слои объединяли, промывали H2O (2×15 мл) и насыщенным солевым раствором (2×15 мл) и высушивали над MgSO4. MgSO4 удаляли фильтрацией и летучие вещества удаляли при пониженном давлении. Неочищенное вещество очищали флэш-хроматографией, чтобы получить (A) (0,64 г).
Синтез (B)
К взвеси (A) (1,03 ммоль, 0,298 г) в 15 мл MeOH, охлажденной до 0ºC, добавляли LiOH (10,31 ммоль, 0,247 г), растворенного в 5 мл H2O. Через 4 часа при 0ºC реакцию гасили 20 мл насыщенного NH4Cl и pH реакционной смеси доводили до 2 с использованием 1 н. HCl. Летучие вещества удаляли при пониженном давлении и твердые вещества собирали фильтрацией, чтобы получить (B) (0,200 г).
Синтез соединения 1
К перемешиваемому раствору (C) [см. Bioorg. Med. Chem. Letter 1999, 9, 2283-88] (0,08 ммоль) в MeCN (2 мл) добавляли (B) (0,08 ммоль, 0,021 г), DIEA (0,320 ммоль, 0,055 мл) и HOBT (0,128 ммоль, 0,017 г). Смесь охлаждали до 0ºC на бане со льдом и несколькими порциями добавляли BOP (0,128 ммоль, 0,057 г). Смесь перемешивали при 5ºC в атмосфере аргона в течение ночи. Затем реакционную смесь разбавляли H2O (15 мл) и эстрагировали EtOAc (3×5 мл). Органические слои объединяли, промывали водой (2×5 мл), насыщенным NaHCO3 (2×5 мл) и насыщенным солевым раствором (2×5 мл) и высушивали над безводным MgSO4. MgSO4 удаляли фильтрацией и летучие вещества удаляли при пониженном давлении. Неочищенное вещество очищали флэш-хроматографией, чтобы получить 1 (0,017 г).
Схема 2: синтез по примеру 2
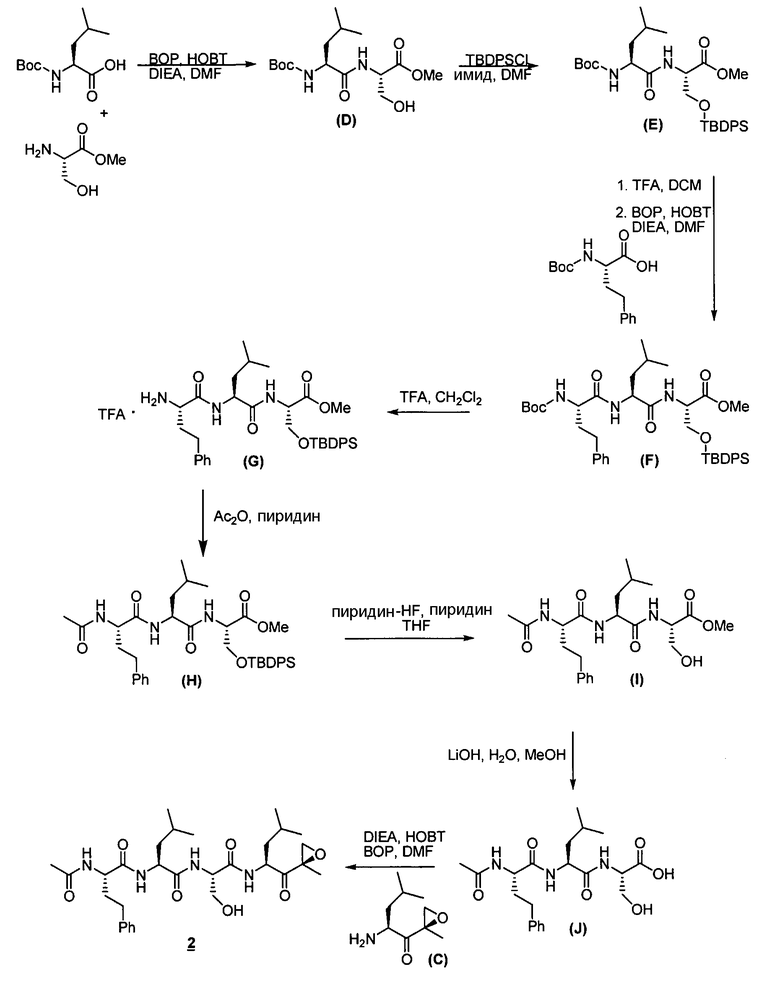
Синтез (D)
К раствору NBOC лейцина (40,0 ммоль, 9,25 г) и метилового эфира серина (40,0 ммоль, 6,22 г) в 400 мл DMF добавляли HOBT (64,0 ммоль, 8,65 г) и DIEA (160,0 ммоль, 20,68 г, 28 мл). Смесь охлаждали до 0ºC на бане со льдом и несколькими порциями в течение более пяти минут добавляли BOP (64,0 ммоль, 28,30 г). Смесь помещали в атмосферу аргона и перемешивали в течение ночи. Реакционную смесь разбавляли насыщенным солевым раствором (1000 мл) и экстрагировали EtOAc (6×200 мл). Органические слои объединяли, промывали водой (10×100 мл) и солевым раствором (3×200 мл) и высушивали над MgSO4. MgSO4 удаляли фильтрацией и летучие вещества удаляли при пониженном давлении, чтобы получить (D) (13,65 г).
Синтез (E)
К раствору имидазола (99,3 ммоль, 6,76 г) в DMF (330 мл) добавляли TBDPSCl (49,64 ммоль, 13,65 г) и смесь перемешивали в течение 15 минут в атмосфере аргона. Добавляли соединение (D) (33,10 ммоль, 11,0 г) и смесь перемешивали в течение ночи. Смесь разбавляли насыщенным солевым раствором (750 мл) и экстрагировали EtOAc (6×200 мл). Органические слои объединяли, промывали H2O (3×300 мл) и насыщенным солевым раствором (2×200 мл) и высушивали над MgSO4. MgSO4 удаляли фильтрацией, и летучие вещества удаляли при пониженном давлении. Неочищенное вещество очищали флэш-хроматографией, получая (E) (13,78 г).
Синтез (F)
К 50 мл охлажденного до 0ºC раствора 80% TFA/DCM добавляли (E) (12,26 ммоль, 7,0 г). Раствор перемешивали и позволяли ему нагреться до комнатной температуры в течение 2 ч. Летучие вещества удаляли при пониженном давлении. К маслу добавляли BocNHhPhe (12,26 ммоль, 3,42 г), 125 мл DMF, HOBT (19,62 ммоль, 2,65 г) и DIEA (49,04 ммоль, 6,34 г, 8,5 мл). Смесь охлаждали до 0ºC на бане со льдом и несколькими порциями в течение более пяти минут добавляли BOP (19,62 ммоль, 8,67 г). Реакционную смесь помещали под аргон и позволяли ей нагреться до комнатной температуры в течение ночи. Реакционную смесь разбавляли DCM в количестве 100 мл и выливали в 600 мл H2O. Органические слои разделяли и промывали H2O (1×100 мл), насыщенным NaHCO3 (1×100 мл) и насыщенным солевым раствором (1×100 мл) и высушивали над MgSO4. MgSO4 удаляли фильтрацией и летучие вещества удаляли при пониженном давлении, чтобы получить (F) (8,14 г).
Синтез (G)
К раствору (F) (1,6 ммоль, 1,0 г) в DCM (10 мл) при 0ºC на бане со льдом медленно добавляли раствор 20% TFA/DCM (20 мл). Раствор медленно нагревали до комнатной температуры и перемешивали в течение ночи. Летучие вещества удаляли в вакууме и остаток разводили с DCM и упаривали (3×). Полученное масло помещали в высокий вакуум на 2 ч, чтобы получить (G).
Синтез (H)
К раствору промежуточного продукта (G) (1,6 ммоль) в пиридине (10 мл) при 0ºC на бане со льдом медленно добавляли уксусный ангидрид (10 мл). Полученный раствор нагревали до комнатной температуры и перемешивали в течение ночи. Летучие вещества удаляли в вакууме и остаток разводили DCM и упаривали. Полученное твердое вещество растворяли в горячем EtOH (20 мл) и медленно выливали на лед (200 мл). Твердые вещества собирали фильтрацией и высушивали на воздухе, получая (H) (0,735 г, 1,3 ммоль).
Синтез (I)
К раствору фторгидрата пиридиния (109 ммоль, 2,2 г) в THF (10 мл) и пиридина (5,7 мл) медленно добавляли (H) (0,36 ммоль, 0,20 г). Полученный раствор перемешивали в течение 2,5 ч и затем гасили медленным добавлением насыщенного NaHCO3 (10 мл). Смесь экстрагировали EtOAc (3x10 мл) и органические слои объединяли, промывали H2O (3×10 мл) и насыщенным солевым раствором (10 мл) и высушивали над MgSO4. MgSO4 удаляли фильтрацией и летучие вещества удаляли при пониженном давлении, чтобы получить (I) (0,22 ммоль, 0,095 г).
Синтез (J)
К гетерогенному раствору (I) (0,12 ммоль, 0,050 г) в MeOH (1,5 мл) и H2O (0,5 мл) при 0ºC по каплям добавляли раствор LiOH (1,0 ммоль, 0,024 г) в H2O (0,25 мл). Полученный раствор медленно нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь разводили насыщенным NH4Cl (10 мл) и выливали на лед (10 мл) и pH раствора доводили до величины 2 1 н. HCl. Полученный раствор экстрагировали EtOAc (3×15 мл) и органические слои объединяли, промывали H2O (1×10 мл) и насыщенным солевым раствором (1×10 мл) и высушивали над MgSO4, MgSO4 удаляли фильтрацией и летучие вещества удаляли при пониженном давлении, чтобы получить полутвердое вещество. Полутвердое вещество помещали в высокий вакуум на 2 часа, чтобы получить (J) (0,023 г, 0,05 ммоль).
Синтез соединения 2
К перемешиваемому раствору (C) (см. Bioorg. Med. Chem. Letter 1999, 9, 2283-88] (15 мг, 0,19 ммоль) в DMF (2 мл) добавляли (J) (0,05 ммоль, 0,02 г), DIEA (0,02 ммоль, 0,033 мл) и HOBT (0,02 ммоль, 0,010 г). Смесь охлаждали до 0ºC на бане со льдом и добавляли несколькими порциями BOP (0,07 ммоль, 0,035 г). Смесь перемешивали при 5ºC в атмосфере аргона в течение ночи. Реакционную смесь разбавляли насыщенным солевым раствором (15 мл) и экстрагировали EtOAc. Органический слой промывали водой, насыщенным NaHCO3 и насыщенным солевым раствором и высушивали над безводным MgSO4. MgSO4 удаляли фильтрацией и летучие вещества удаляли при пониженном давлении. Неочищенный материал очищали флэш-хроматографией с использованием от 20 до 40% смеси EtOAc/гексан в качестве элюента, чтобы получить 2 (3,6 мг). IC50 CT-L 20S <50 нМ, IC50 CT-L на клетках <500 нМ.
Схема 3: синтез по примеру 3
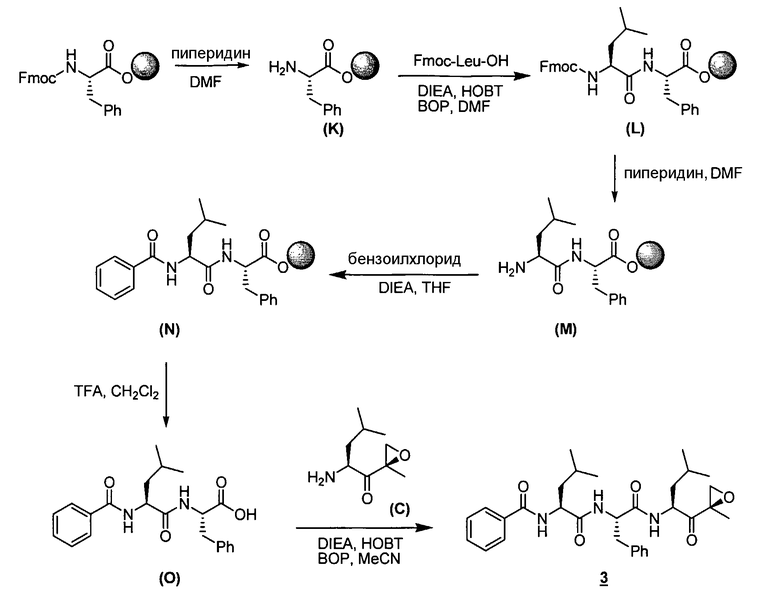
Синтез (K)
К смоле Fmoc-Phe-Wang (4,0 ммоль, 5,0 г) добавляли смесь 20% пиперидин/DMF (50 мл). Гетерогенную смесь встряхивали в течение 20 минут, фильтровали и смолу промывали DMF (100 мл), MeOH (100 мл) и DCM (100 мл) и высушивали на воздухе. Смолу подвергали описанным выше условиям реакции второй раз, чтобы получить (K).
Синтез (L)
К смеси (K) (4,0 ммоль) и DMF (40 мл) добавляли по порядку Fmoc-Leu-OH (7,9 ммоль, 2,82 г), DIEA (13,3 ммоль, 2,23 мл) и HOBT (8,10 ммоль, 1,24 г). К описанной выше смеси медленно добавляли BOP (7,98 ммоль, 3,53 г) и разводили в DMF (40 мл). Реакционную смесь встряхивали в течение ночи. Реакционную смесь фильтровали и смолу промывали DMF (150 мл), MeOH (150 мл), DCM (150 мл) и высушивали на воздухе, чтобы получить (L).
Синтез (M)
К (L) (4,0 ммоль) добавляли смесь 20% пиперидин/DMF (50 мл). Гетерогенную смесь встряхивали в течение 20 минут. Раствор фильтровали и смолу промывали DMF (100 мл), MeOH (100 мл) и DCM (100 мл) и высушивали на воздухе. Смолу подвергали описанным выше условиям реакции второй раз, чтобы получить (M).
Синтез (N)
К (M) (0,10 ммоль, 0,130 г) добавляли THF (2 мл), DIEA (0,40 ммоль, 0,08 мл) и бензоилхлорид (0,34 ммоль, 0,04 мл). Полученную смесь встряхивали в течение 30 минут. Реакционную смесь фильтровали и смолу промывали DMF (20 мл), водой (20 мл), MeOH (20 мл) и DCM (20 мл) и высушивали на воздухе, чтобы получить (N).
Синтез (О)
К (N) (0,10 ммоль) добавляли 50% TFA/DCM (2 мл) и смесь встряхивали в течение 20 минут (смола становилась пурпурной). Смесь фильтровали и смолу промывали DCM (10 мл). Летучие вещества удаляли при пониженном давлении и полученное масло разбавляли DCM (10 мл) и упаривали в целом три раза, чтобы получить (О).
Синтез соединения 3
К перемешиваемому раствору (C) (см. Bioorg. Med. Chem. Letter 1999, 9, 2283-88] (19 мг, 0,11 ммоль) в MeCN (2 мл) добавляли (О) (0,1 ммоль), DIEA (2,9 ммоль, 0,5 мл), HOBT (0,2 ммоль, 0,032 г) и BOP (0,23 ммоль, 0,103 г). Смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь разбавляли насыщенным солевым раствором (15 мл) и экстрагировали EtOAc. Органический слой промывали водой, насыщенным NaHCO3 и насыщенным солевым раствором и высушивали над безводным MgSO4. MgSO4 удаляли фильтрацией и летучие вещества удаляли при пониженном давлении. Неочищенное вещество очищали флэш-хроматографией с использованием от 20 до 40% смесей EtOAc/гексан в качестве элюента, чтобы получить 3 (13,8 мг). IC50 CT-L 20S <50 нМ, IC50 CT-L на клетках <50 нМ.
Схема 4: синтез по примеру 4
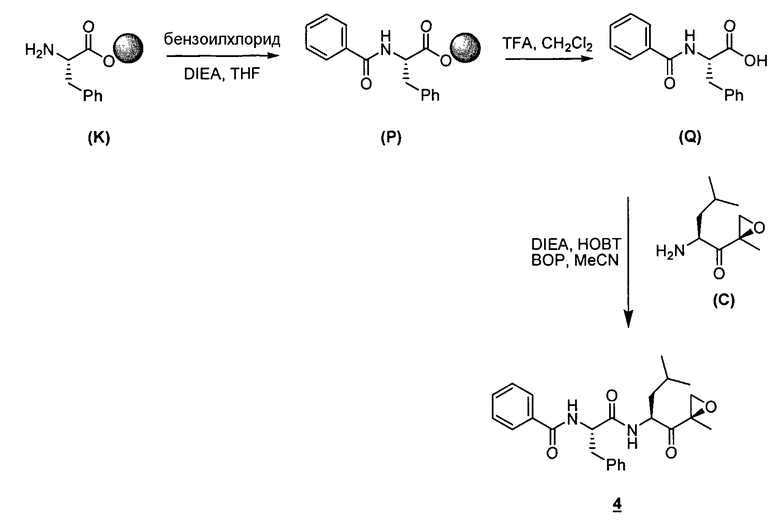
Синтез (P)
К промежуточному продукту (K) (0,10 ммоль, 0,130 г) добавляли THF (2 мл), DIEA (0,40 ммоль, 0,08 мл) и бензоилхлорид (0,34 ммоль, 0,04 мл). Полученную смесь встряхивали в течение 30 минут. Реакционную смесь фильтровали и смолу промывали DMF (20 мл), водой (20 мл), MeOH (20 мл) и DCM (20 мл) и высушивали на воздухе, чтобы получить (P).
Синтез (Q)
К (P) добавляли 50% TFA/DCM (2 мл) и встряхивали в течение 20 минут (смола становилась пурпурной). Реакционную смесь фильтровали и смолу промывали DCM (10 мл). Летучие вещества удаляли при пониженном давлении и полученное масло разбавляли DCM (10 мл) и упаривали в целом три раза, чтобы получить (Q).
Синтез соединения 4
К перемешиваемому раствору (C) [см. Bioorg. Med. Chem. Letter 1999, 9, 2283-88] (19 мг, 0,11 ммоль) в MeCN (2 мл) добавляли (Q) (0,1 ммоль), DIEA (2,9 ммоль, 0,5 мл), HOBT (0,2 ммоль, 0,032 г) и BOP (0,23 ммоль, 0,103 г). Затем смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли насыщенным солевым раствором (15 мл) и экстрагировали EtOAc. Органический слой промывали водой, насыщенным NaHCO3 и насыщенным солевым раствором и высушивали над безводным MgSO4. MgSO4 удаляли фильтрацией и летучие вещества удаляли при пониженном давлении. Неочищенное вещество очищали методом флэш-хроматографии с использованием от 20 до 40% смесей EtOAc/гексан в качестве элюента, чтобы получить 4 (12,7 мг).
Схема 5: синтез по примеру 5
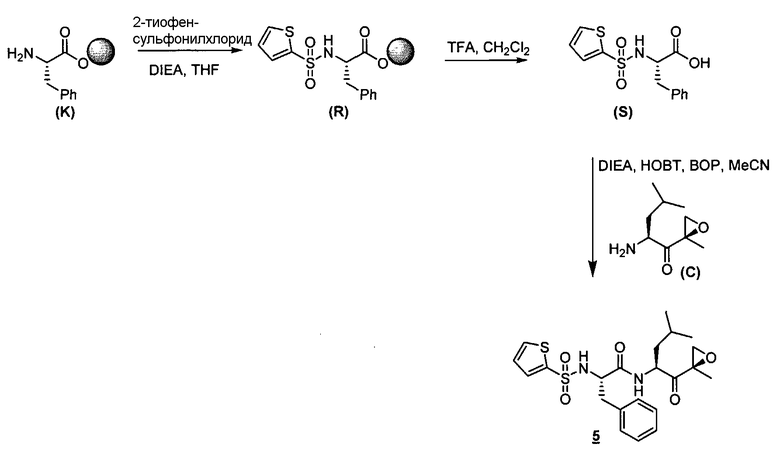
Синтез (R)
К (K) (0,10 ммоль, 0,130 г) добавляли THF (2 мл), DIEA (0,40 ммоль, 0,08 мл) и 2-тиофенсульфонилхлорид (0,34 ммоль, 0,064 г). Полученную смесь встряхивали в течение 30 минут. Затем реакционную смесь фильтровали и смолу промывали DMF (20 мл), водой (20 мл), MeOH (20 мл) и DCM (20 мл) и высушивали на воздухе, чтобы получить (R).
Синтез (S)
К (R) (0,10 ммоль) добавляли 50% TFA/DCM (2 мл) и встряхивали в течение 20 минут (смола становилась пурпурной). Затем реакционную смесь фильтровали и смолу промывали DCM (10 мл). Летучие вещества удаляли при пониженном давлении и полученное масло разбавляли DCM (10 мл) и упаривали всего три раза, чтобы получить (S).
Синтез соединения 5
К перемешиваемому раствору (C) [см. Bioorg. Med. Chem. Letter 1999, 9, 2283-88] (0,11 ммоль, 0,019 г) в MeCN (2 мл) добавляли (S) (0,1 ммоль), DIEA (2,9 ммоль, 0,5 мл), HOBT (0,2 ммоль, 0,032 г) и BOP (0,23 ммоль, 0,103 г) и смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь разбавляли насыщенным солевым раствором (15 мл) и экстрагировали EtOAc. Органический слой промывали водой, насыщенным NaHCO3 и насыщенным солевым раствором и высушивали над безводным MgSO4. MgSO4 удаляли фильтрацией и летучие вещества удаляли при пониженном давлении. Неочищенное вещество очищали флэш-хроматографией с использованием от 20 до 40% смеси EtOAc/гексан в качестве элюента, чтобы получить 5 (13,1 мг).
Схема 6: синтез по примеру 6
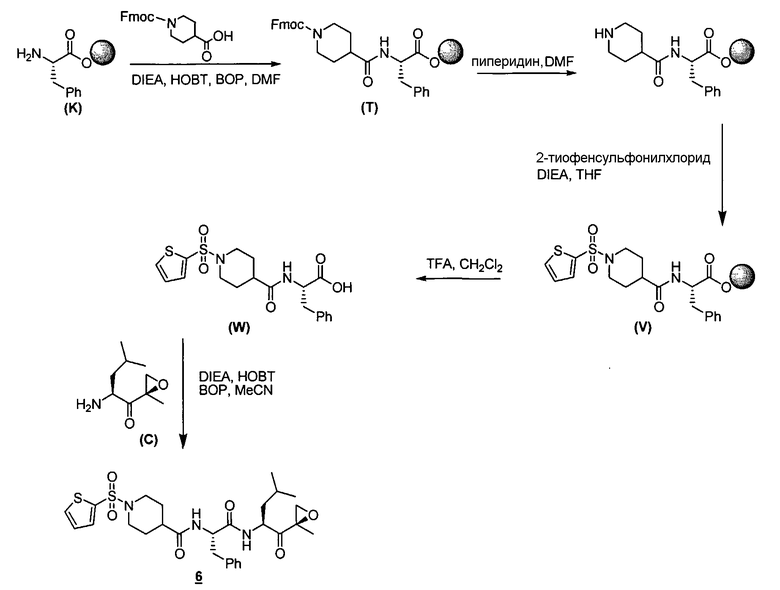
Синтез (T)
К (K) (0,8 ммоль, 1,0 г) добавляли DMF (20 мл), Fmoc- изонипекотиновую кислоту (2,8 ммоль, 0,320 g), DIEA (4,8 ммоль, 0,445 мл), HOBT (0,9 ммоль, 0,140 г) и BOP (1,0 ммоль, 0,450 г) и реакционную смесь встряхивали в течение ночи. Реакционную смесь фильтровали и смолу промывали DMF (150 мл), MeOH (150 мл) и DCM (150 мл) и высушивали на воздухе, чтобы получить (T).
Синтез (U)
К (T) (0,8 ммоль, 1,0 г) добавляли смесь 20% пиперидин/DMF (30 мл) и гетерогенную смесь встряхивали в течение 20 минут. Смесь фильтровали и смолу промывали DMF (150 мл), MeOH (150 мл) и DCM (150 мл) и высушивали на воздухе. Смолу подвергали описанным выше условиям реакции второй раз, чтобы получить (U).
Синтез (V)
К (U) (0,10 ммоль, 0,130 г) добавляли THF (2 мл), DIEA (0,40 ммоль, 0,08 мл) и 2-тиофенсульфонилхлорид (0,34 ммоль, 0,064 г) и полученную смесь встряхивали в течение 30 минут. Реакционную смесь фильтровали и смолу промывали DMF (20 мл), водой (20 мл), MeOH (20 мл) и DCM (20 мл) и высушивали на воздухе, чтобы получить (V).
Синтез (W)
К (V) (0,10 ммоль) добавляли 50% TFA/DCM (2 мл) и смесь встряхивали в течение 20 минут (смола становилась пурпурной). Затем реакционную смесь фильтровали и смолу промывали DCM (10 мл). Летучие вещества удаляли при пониженном давлении, полученное масло разбавляли DCM (10 мл) и упаривали в общей сложности три раза, чтобы получить (W).
Синтез соединения 6
К перемешиваемому раствору (C) [см. Bioorg. Med. Chem. Letter 1999, 9, 2283-88] (19 мг, 0,11 ммоль) в MeCN (2 мл) добавляли (W) (0,1 ммоль), DIEA (2,9 ммоль, 0,5 мл), HOBT (0,2 ммоль, 0,032 г) и BOP (0,23 ммоль, 0,103 г) и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли насыщенным солевым раствором (15 мл) и экстрагировали EtOAc. Органический слой промывали водой, насыщенным NaHCO3 и насыщенным солевым раствором и высушивали над безводным MgSO4. MgSO4 удаляли фильтрацией и летучие вещества удаляли при пониженном давлении. Неочищенное вещество очищали флэш-хроматографией с использованием от 20 до 40% смеси EtOAc/гексан в качестве элюента для получения 6 (7,4 мг).
Схема 7: синтез по примеру 7
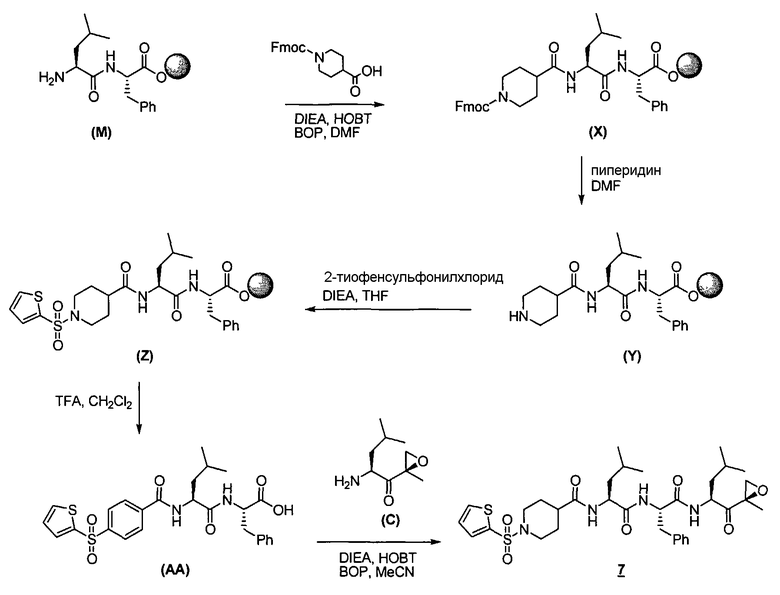
Синтез (X)
К (M) (0,8 ммоль, 1,0 г) добавляли DMF (20 мл), Fmoc-изонипекотиновую кислоту (2,8 ммоль, 0,320 г), DIEA (4,8 ммоль, 0,445 мл), HOBT (0,9 ммоль, 0,140 г) и BOP (1,0 ммоль, 0,450 г) и реакционную смесь встряхивали в течение ночи. Реакционную смесь фильтровали и смолу промывали DMF (150 мл), MeOH (150 мл) и DCM (150 мл) и высушивали на воздухе, чтобы получить (X).
Синтез (Y)
К (X) (0,8 ммоль, 1,0 г) добавляли 20% пиперидин/DMF (30 мл) и полученную гетерогенную смесь встряхивали в течение 20 минут. Раствор фильтровали и смолу промывали DMF (150 мл), MeOH (150 мл) и DCM (150 мл) и высушивали на воздухе. Смолу подвергали описанным выше условиям реакции второй раз, чтобы получить (Y).
Синтез (Z)
К (Y) (0,10 ммоль, 0,130 г) добавляли THF (2 мл), DIEA (0,40 ммоль, 0,08 мл) и 2-тиофенсульфонилхлорид (0,34 ммоль, 0,064 г) и полученную смесь встряхивали в течение 30 минут. Реакционную смесь фильтровали и смолу промывали DMF (20 мл), водой (20 мл), MeOH (20 мл) и DCM (20 мл) и высушивали на воздухе, чтобы получить (Z).
Синтез (AA)
К (Z) (0,10 ммоль) добавляли 50% TFA/DCM (2 мл) и смесь встряхивали в течение 20 минут (смола становилась пурпурной). Реакционную смесь фильтровали и смолу промывали DCM (10 мл). Летучие вещества удаляли при пониженном давлении, полученное масло разбавляли DCM (10 мл) и упаривали в общей сложности три раза, чтобы получить (AA).
Синтез соединения 7
К перемешиваемому раствору (C) [см. Bioorg. Med. Chem. Letter 1999, 9, 2283-88] (0,11 ммоль, 0,019 г) в MeCN (2 мл) добавляли (AA) (0,1 ммоль), DIEA (2,9 ммоль, 0,5 мл), HOBT (0,2 ммоль, 0,032 г) и BOP (0,23 ммоль, 0,103 г) и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли насыщенным солевым раствором (15 мл) и экстрагировали EtOAc. Органический слой промывали водой, насыщенным NaHCO3 и насыщенным солевым раствором и высушивали над безводным MgSO4. MgSO4 удаляли фильтрацией и летучие вещества удаляли при пониженном давлении. Неочищенное вещество очищали флэш-хроматографией с использованием от 20 до 40% смеси EtOAc/гексан в качестве элюента, чтобы получить 7 (18,2 мг). IC50 CT-L 20S <500 нМ, IC50 CT-L на клетках <500 нМ.
Схема 8: синтез по примеру 8
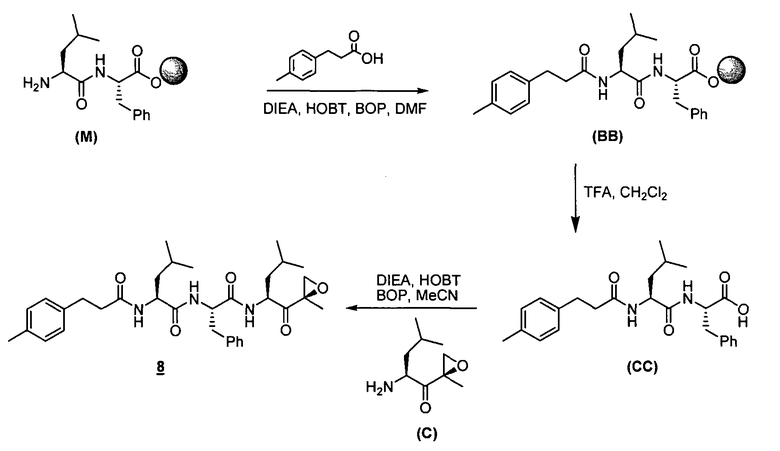
Синтез (BB)
К (M) (0,12 ммоль, 0,100 г) добавляли DMF (2 мл), 3-(п-толил)пропионовую кислоту (0,09 ммоль, 0,015 г), DIEA (0,17 ммоль, 0,029 мл), HOBT (0,11 ммоль, 0,016 г) и BOP (0,11 ммоль, 0,051 г) и реакционную смесь встряхивали в течение ночи. Реакционную смесь фильтровали и смолу промывали DMF (15 мл), MeOH (15 мл) и DCM (15 мл) и высушивали на воздухе, чтобы получить (BB).
Синтез (CC)
К (BB) (0,12 ммоль) добавляли 50% TFA/DCM (2 мл) и смесь встряхивали в течение 20 минут (смола становилась пурпурной). Реакционную смесь фильтровали и смолу промывали DCM (10 мл). Летучие вещества удаляли при пониженном давлении, полученное масло разбавляли DCM (10 мл) и упаривали в общей сложности три раза, чтобы получить (CC).
Синтез соединения 8
К перемешиваемому раствору (C) [см. Bioorg. Med. Chem. Letter 1999, 9, 2283-88] (0,11 ммоль, 0,019 г) в MeCN (2 мл) добавляли (CC) (0,1 ммоль), DIEA (2,9 ммоль, 0,5 мл), HOBT (0,2 ммоль, 0,032 г) и BOP (0,23 ммоль, 0,103 г) и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли насыщенным солевым раствором (15 мл) и экстрагировали EtOAc. Органический слой промывали водой, насыщенным NaHCO3, насыщенным солевым раствором и высушивали над безводным MgSO4. MgSO4 удаляли фильтрацией и летучие вещества удаляли при пониженном давлении. Неочищенное вещество очищали флэш-хроматографией с использованием от 20 до 40% смеси EtOAc/гексан в качестве элюента, чтобы получить 8 (3,9 мг). IC50 CT-L 20S <50 нМ, IC50 CT-L на клетках <50 нМ.
Схема 9: синтез по примеру 9
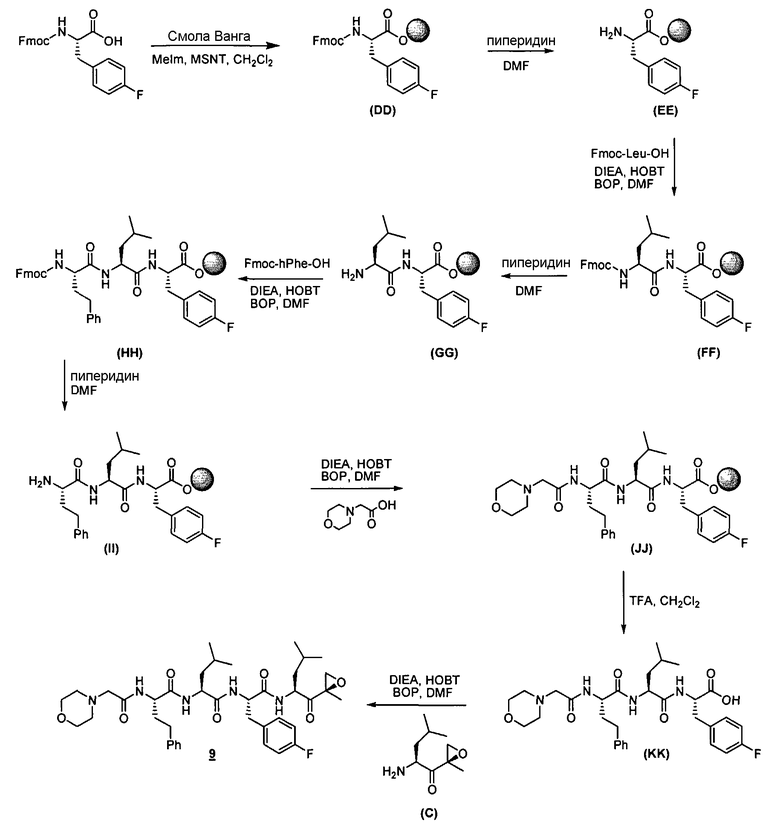
Синтез (DD)
К раствору Fmoc-Phe(4-F)-OH (2,4 ммоль, 1,0 г) в DCM (20 мл) добавляли MeIm (6,7 ммоль, 0,370 мл). Когда раствор становился гомогенным, добавляли MSNT (2,9 ммоль, 0,870 г). Когда MSNT растворялся, реакционную смесь добавляли к смоле Ванга (0,8 ммоль, 1,0 г) и полученный раствор встряхивали в течение 45 минут. Смолу фильтровали и промывали DMF (50 мл), MeOH (50 мл) и DCM (50 мл). Полученную смолу высушивали на воздухе, чтобы получить (DD).
Синтез (EE)
К (DD) (0,40 ммоль, 0,5 г) добавляли 20% пиперидин/DMF (10 мл) и полученный гетерогенный раствор встряхивали в течение 20 минут. Смесь фильтровали, смолу промывали DMF (20 мл), MeOH (20 мл) и DCM (20 мл) и высушивали на воздухе. Смолу подвергали описанным выше условиям реакции второй раз, чтобы получить (EE).
Синтез (FF)
К (EE) (0,40 ммоль) добавляли DMF (20 мл), Fmoc-Leu-OH (0,40 ммоль, 0,143 г), DIEA (1,6 ммоль, 0,12 мл), HOBT (0,64 ммоль, 0,062 мг) и BOP (0,64 ммоль, 0,178 г) и реакционную смесь встряхивали в течение ночи. Реакционную смесь фильтровали и смолу промывали DMF (40 мл), MeOH (40 мл) и DCM (40 мл) и высушивали на воздухе, чтобы получить (FF).
Синтез (GG)
К (FF) (0,08 ммоль, 0,10 г) добавляли 20% пиперидин/DMF (2 мл) и полученный гетерогенный раствор встряхивали в течение 20 минут. Раствор фильтровали, смолу промывали DMF (10 мл), MeOH (10 мл) и DCM (10 мл) и высушивали на воздухе. Смолу подвергали описанным выше условиям реакции второй раз, чтобы получить (GG).
Синтез (HH)
К (GG) (0,08 ммоль) добавляли DMF (20 мл), Fmoc-hPhe-OH (0,40 ммоль, 0,143 г), DIEA (1,6 ммоль, 0,12 мл), HOBT (0,64 ммоль, 0,062 мг) и BOP (0,64 ммоль, 0,178 г) и реакционную смесь встряхивали в течение ночи. Реакционную смесь фильтровали и смолу промывали DMF (40 мл), MeOH (40 мл) и DCM (40 мл) и высушивали на воздухе, чтобы получить (HH).
Синтез (II)
К (HH) (0,08 ммоль) добавляли 20% пиперидин/DMF (2 мл) и полученный гетерогенный раствор встряхивали в течение 20 минут. Раствор фильтровали и смолу промывали DMF (10 мл), MeOH (10 мл) и DCM (10 мл) и высушивали на воздухе. Смолу подвергали описанным выше условиям реакции второй раз, чтобы получить (II).
Синтез (JJ)
К (II) (0,08 ммоль) добавляли DMF (2 мл), 4-морфолиноуксусную кислоту (0,10 ммоль, 0,015 г), DIEA (0,17 ммоль, 0,029 мл), HOBT (0,11 ммоль, 0,016 г) и BOP (0,11 ммоль, 0,051 г) и реакционную смесь встряхивали в течение ночи. Реакционную смесь фильтровали и смолу промывали DMF (15 мл), MeOH (15 мл) и DCM (15 мл) и высушивали на воздухе, чтобы получить (JJ).
Синтез (KK)
К (JJ) (0,08 ммоль) добавляли 50% TFA/DCM (2 мл) и смесь встряхивали в течение 20 минут (смола становилась пурпурной). Реакционную смесь фильтровали и смолу промывали DCM (10 мл). Летучие вещества удаляли при пониженном давлении, полученное масло разбавляли DCM (10 мл) и упаривали в общей сложности три раза, чтобы получить (KK).
Синтез соединения 9
К перемешиваемому раствору (C) [см. Bioorg. Med. Chem. Letter 1999, 9, 2283-88] (0,11 ммоль, 0,019 г) в MeCN (2 мл) добавляли (KK) (0,1 ммоль), DIEA (2,9 ммоль, 0,5 мл), HOBT (0,2 ммоль, 0,032 г) и BOP (0,23 ммоль, 0,103 г) и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли насыщенным солевым раствором (15 мл) и экстрагировали EtOAc. Органический слой промывали водой, насыщенным NaHCO3 и насыщенным солевым раствором и высушивали над безводным MgSO4. MgSO4 удаляли фильтрацией и летучие вещества удаляли при пониженном давлении. Неочищенное вещество очищали флэш-хроматографией с использованием от 20 до 40% смеси EtOAc/гексан в качестве элюента, чтобы получить 9 (7,7 мг). IC50 CT-L 20S <50 нМ, IC50 CT-L на клетках <50 нМ.
Схема 10: синтез по примеру 10
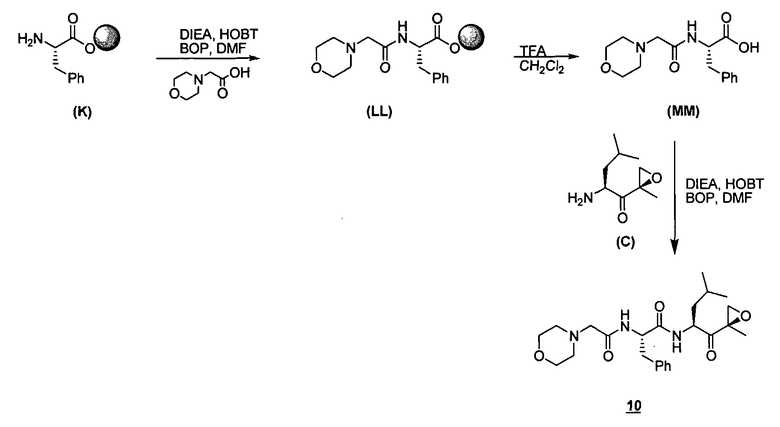
Синтез (LL)
К (K) (0,12 ммоль, 0,100 г) добавляли DMF (2 мл), 4-морфолиноуксусную кислоту (0,12 ммоль, 0,018 г), DIEA (0,17 ммоль, 0,029 мл), HOBT (0,11 ммоль, 0,016 г) и BOP (0,11 ммоль, 0,051 г) и реакционную смесь встряхивали в течение ночи. Реакционную смесь фильтровали, смолу промывали DMF (15 мл), MeOH (15 мл) и DCM (15 мл) и высушивали на воздухе, чтобы получить (LL).
Синтез (MM)
К (LL) (0,12 ммоль) добавляли 50% TFA/DCM (2 мл) и смесь встряхивали в течение 20 минут (смола становилась пурпурной). Реакционную смесь фильтровали и смолу промывали DCM (10 мл). Летучие вещества удаляли при пониженном давлении, полученное масло разбавляли DCM (10 мл) и упаривали в общей сложности три раза, чтобы получить (MM).
Синтез соединения 10
К перемешиваемому раствору (C) [см. Bioorg. Med. Chem. Letter 1999, 9, 2283-88] (0,11 ммоль, 0,019 г) в MeCN (2 мл) добавляли (MM) (0,1 ммоль), DIEA (2,9 ммоль, 0,5 мл), HOBT (0,2 ммоль, 0,032 г) и BOP (0,23 ммоль, 0,103 г) и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли насыщенным солевым раствором (15 мл) и экстрагировали EtOAc. Органический слой промывали водой, насыщенным NaHCO3 и насыщенным солевым раствором и высушивали над безводным MgSO4. MgSO4 удаляли фильтрацией и летучие вещества удаляли при пониженном давлении. Неочищенное вещество очищали флэш-хроматографией с использованием от 20 до 40% смеси EtOAc/гексан в качестве элюента, чтобы получить 10 (14 мг).
Схема 11: синтез по примеру 11
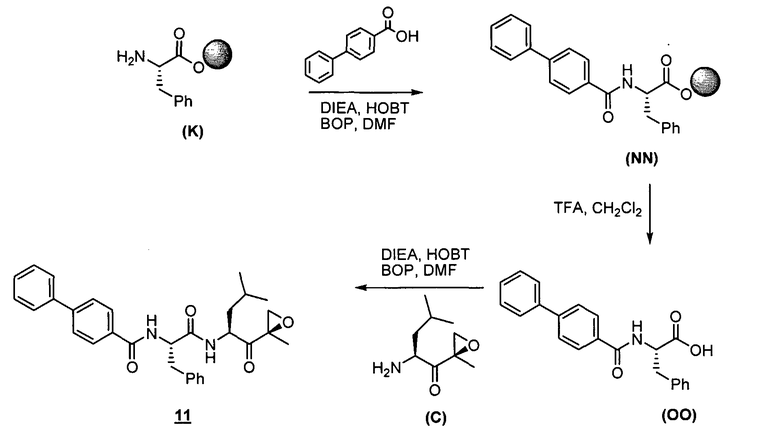
Синтез (NN)
К (K) (0,12 ммоль, 0,100 г) добавляли DMF (2 мл), бифенил-4-карбоновую кислоту (0,12 ммоль, 0,025 г), DIEA (0,17 ммоль, 0,029 мл), HOBT (0,11 ммоль, 0,016 г) и BOP (0,11 ммоль, 0,051 г) и реакционную смесь встряхивали в течение ночи. Реакционную смесь фильтровали и смолу промывали DMF (15 мл), MeOH (15 мл) и DCM (15 мл) и высушивали на воздухе, чтобы получить (NN).
Синтез (ОО)
К (NN) (0,12 ммоль) добавляли 50% TFA/DCM (2 мл) и встряхивали в течение 20 минут (смола становилась пурпурной). Реакционную смесь фильтровали и смолу промывали DCM (10 мл). Летучие вещества удаляли при пониженном давлении, полученное масло разбавляли DCM (10 мл) и упаривали в общей сложности три раза, чтобы получить (ОО).
Синтез соединения 11
К перемешиваемому раствору (C) [см. Bioorg. Med. Chem. Letter 1999, 9, 2283-88] (0,11 ммоль, 0,019 г) в MeCN (2 мл) добавляли (ОО) (0,1 ммоль), DIEA (2,9 ммоль, 0,5 мл), HOBT (0,2 ммоль, 0,032 г) и BOP (0,23 ммоль, 0,103 г) и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли насыщенным солевым раствором (15 мл) и экстрагировали EtOAc. Органический слой промывали водой, насыщенным NaHCO3, насыщенным солевым раствором и высушивали над безводным MgSO4. MgSO4 удаляли фильтрацией и летучие вещества удаляли при пониженном давлении. Неочищенное вещество очищали флэш-хроматографией с использованием от 20 до 40% смеси EtOAc/гексан в качестве элюента, чтобы получить 11 (7,4 мг).
Схема 12: синтез по примерам 12, 13 и 14
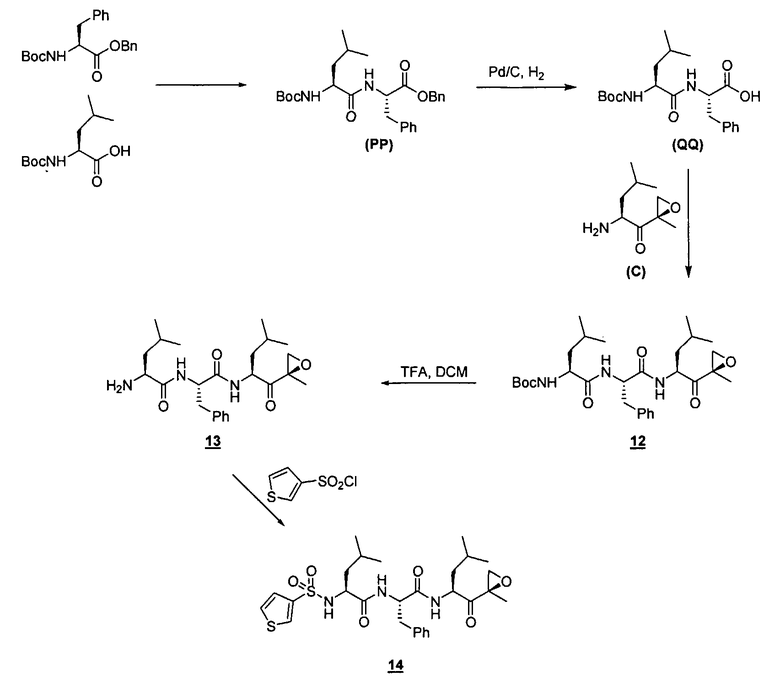
Синтез (PP)
К раствору Nboc лейцина (85,67 ммоль, 19,81 г, 1,0 экв.) и бензилового эфира фенилаланина (85,67 ммоль, 25,0 г, 1,0 экв.) в 900 мл MeCN добавляли DIEA (342,68 ммоль, 44,29 г, 60 мл, 4,0 экв.) и смесь охлаждали до 0ºC на бане со льдом. К смеси добавляли HOBT (137,08 ммоль, 18,52 г, 1,6 экв.), после чего следовал PyBOP (137,08 ммоль, 71,33 г, 1,6 экв.), который добавляли несколькими порциями в течение пяти минут. Реакционную смесь помещали в атмосферу аргона и перемешивали в течение ночи. Летучие вещества удаляли при пониженном давлении и оставшееся вещество переносили к 500 мл EtOAc и промывали насыщенным NaHCO3, H2O и насыщенным солевым раствором и высушивали над безводным MgSO4. MgSO4 удаляли фильтрацией и летучие вещества удаляли при пониженном давлении, чтобы получить (PP) (37,3 г).
Синтез (QQ)
Соединение (PP) (4,27 ммоль, 2,0 г) растворяли в MeOH/EtOAc (1:1, 40 мл) и добавляли Pd-C (5%, 800 мг). Смесь перемешивали при комнатной температуре при давлении 1 атмосфера водорода в течение 2 часов и затем фильтровали через целит и концентрировали, чтобы получить (QQ).
Синтез соединения 12
К перемешиваемому раствору (C) [см. Bioorg. Med. Chem. Letter 1999, 9, 2283-88] (6,0 ммоль, 1,8 г, 1,4 экв.) в DMF (50 мл) добавляли (QQ) (4,27 ммоль, 2,0 г, 1 экв.), DIEA (0,02 моль, 3,5 мл, 4 экв.) и затем HOBT (32 ммоль, 4,3 г, 1,6 экв.). Смесь охлаждали до 0ºC на бане со льдом и несколькими порциями добавляли PyBOP (32 ммоль, 16,6 г, 1,6 экв.). Смесь перемешивали при 5ºC в атмосфере азота в течение ночи. Реакционную смесь разбавляли насыщенным NaCl и экстрагировали EtOAc. Органический слой промывали водой и насыщенным солевым раствором и высушивали над безводным MgSO4, фильтровали и концентрировали до масла, которое очищали флэш-хроматографией, чтобы получить 12 (0,50 г).
Синтез соединения 13
Соединение 12 (0,046 ммоль, 24,5 мг) смешивали с TFA/DCM (80%) и перемешивали при комнатной температуре в течение одного часа, во время которого смесь концентрировалась, и ее помещали в высокий вакуум на 2 часа, получая 13.
Синтез соединения 14
К раствору DCM (10 мл) и 13 добавляли 3-тиофенсульфонилхлорид (0,055 ммоль, 0,012 г, 1,2 экв.) и TEA (0,184 ммоль, 0,026 мл, 4,0 экв.). Смесь перемешивали при комнатной температуре в течение 2 ч и затем концентрировали до сухого вещества. Остаток помещали в EtOAc, промывали водой и насыщенным солевым раствором и высушивали над безводным MgSO4, фильтровали и концентрировали до масла, которое очищали препаративной ВЭЖХ, чтобы получить 14 (1 мг). IC50 CT-L 20S <50 нМ, IC50 CT-L на клетках <50 нМ.
Схема 13: синтез по примеру 15
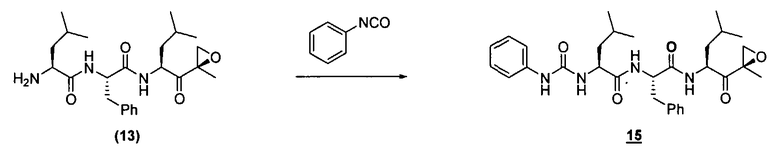
Синтез соединения 15
К раствору DCM (2 мл) и 13 (0,092 ммоль) добавляли фенилизоцианат (0,14 ммоль, 0,015 мл, 1,5 экв.) и DIEA (0,276 ммоль, 0,050 мл, 3 экв.) и смесь перемешивали при комнатной температуре в течение ночи. Неочищенную смесь затем концентрировали до сухого вещества и остаток помещали в EtOAc. Органические слои промывали водой и насыщенным солевым раствором и высушивали над безводным MgSO4, фильтровали и концентрировали до масла, которое очищали препаративной ВЭЖХ, чтобы получить 15 (2,8 мг).
Схема 14: синтез по примеру 16
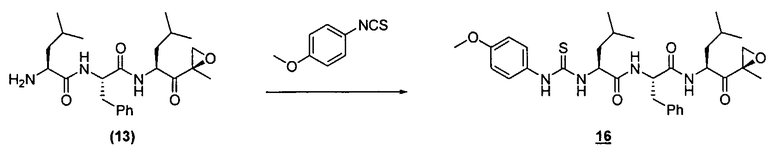
Синтез соединения 16
К раствору DCM (2 мл) и 13 (0,07 ммоль) добавляли 4-метоксифенилизотиоцианат (0,1 ммоль, 0,015 мл, 1,5 экв.) и DIEA (0,21 ммоль, 0,040 мл, 3 экв.) и смесь перемешивали при комнатной температуре в течение ночи. Неочищенное вещество концентрировали до сухого вещества и остаток помещали в EtOAc. Органический слой промывали водой и насыщенным солевым раствором и высушивали над безводным MgSO4, фильтровали и концентрировали до масла, которое очищали флэш-хроматографией, чтобы получить 16 (2 мг). IC50 CT-L 20S <50 нМ, IC50 CT-L на клетках <50 нМ.
Схема 15: синтез по примеру 17
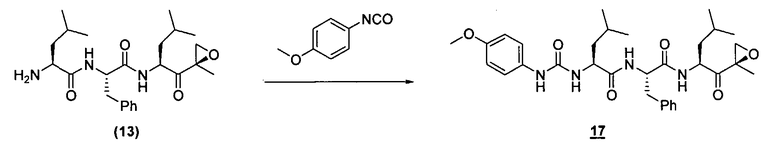
Синтез соединения 17
К раствору DCM (2 мл) и 13 (0,092 ммоль) добавляли 4-метоксифенилизотиоцианат (0,14 ммоль, 0,018 мл, 1,5 экв.) и DIEA (0,276 ммоль, 0,050 мл, 3 экв.) и смесь перемешивали при комнатной температуре в течение ночи. Неочищенное вещество концентрировали до сухого вещества и остаток помещали в EtOAc. Органический слой промывали водой и насыщенным солевым раствором, высушивали над безводным MgSO4, фильтровали и концентрировали до масла, которое очищали флэш-хроматографией, чтобы получить 17 (2,5 мг). IC50 CT-L 20S <50 нМ, IC50 CT-L на клетках <50 нМ.
Схема 16: синтез по примеру 18
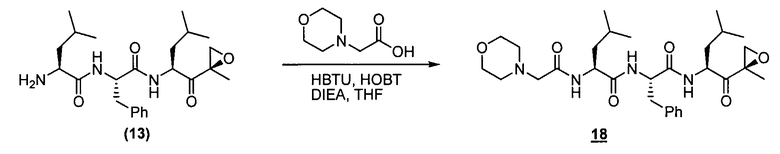
Синтез соединения 18
К раствору THF (15 мл) и 13 (1,4 ммоль) при 0ºC добавляли 4-морфолиноуксусную кислоту (1,79 ммоль, 0,260 г), HOBT (1,92 ммоль, 0,260 г), HBTU (1,72 ммоль, 0,656 г) и DIEA (11,45 ммоль, 2,0 мл), смесь перемешивали и давали ей нагреться до комнатной температуры в течение ночи. Реакционный раствор разбавляли насыщенным NaHCO3 (15 мл) и отделяли слои. Водный слой экстрагировали EtOAc (3×5 мл), органические слои объединяли, промывали H2O (1×15 мл) и насыщенным солевым раствором (1×15 мл) и высушивали над MgSO4. MgSO4 удаляли фильтрацией, и летучие вещества удаляли при пониженном давлении. Неочищенное вещество очищали флэш-хроматографией, чтобы получить 18 (0,423 г). IC50 CT-L 20S <50 нМ, IC50 CT-L на клетках <50 нМ.
Схема 17: синтез по примеру 19
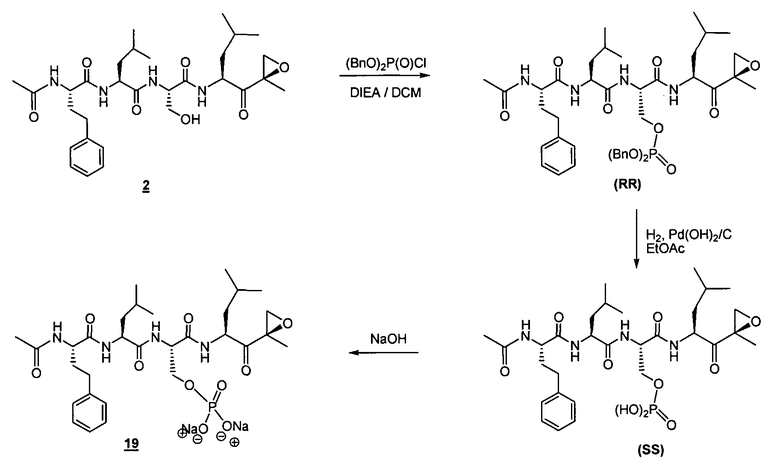
Синтез (RR)
К раствору 2 (1 ммоль) и DIEA (1,5 ммоль) в сухом CH2Cl2 (100 мл) при 0ºC добавляли по каплям раствор дибензилфосфорилхлорида (1 ммоль) в дихлорметане (10 мл). Реакционную смесь полностью перемешивали при комнатной температуре, промывали холодным 1 н. HCl и затем насыщенным солевым раствором, высушивали над MgSO4, фильтровали и выпаривали, чтобы получить (RR).
Синтез (SS)
Раствор (RR) (1 ммоль) и жидкого 20% Pd(OH)2/C (100 мг; катализатор Перлмана) в EtOAc (10 мл) подвергали полному гидрированию при атмосферном давлении и комнатной температуре. Реакционную смесь фильтровали через целит и выпаривали, чтобы получить необходимое соединение (SS). Данное вещество, таким образом выделенное, можно использовать как есть либо на следующей стадии, либо для тестирования в любом исследовании.
Синтез соединения 19
Соединение (SS) (1 ммоль) разбавляли MeOH (5 мл) и обрабатывали стандартизованным раствором 1,00 н. NaOH (2,00 ммоль) в H2O (2 мл). Раствор перемешивали в течение 2 часов и лиофилизировали, чтобы получить 19.
Схема 18: синтез по примеру 20
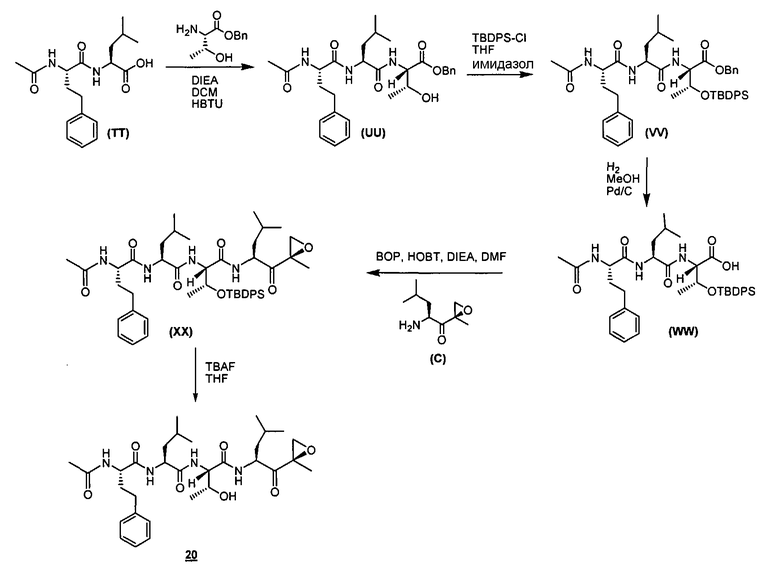
Синтез (UU)
К раствору (TT) (1 ммоль) [полученому стандартными способами синтеза пептидов в растворах], бензилового раствора треонина (1 ммоль) и DIEA (2,5 ммоль) в сухом CH2Cl2 (100 мл) добавляли HBTU (2,5 ммоль). Реакционную смесь полностью перемешивали при комнатной температуре, промывали холодным 1 н. HCl и водой, высушивали над MgSO4, фильтровали и выпаривали, чтобы получить (UU).
Синтез (VV)
К соединению (UU) (1 ммоль) в сухом CH2Cl2 (100 мл) добавляли TBDPSCl (1 ммоль) и имидазол (1,2 ммоль). Реакционную смесь полностью перемешивали при комнатной температуре, промывали холодным 1 н. HCl и водой, высушивали над MgSO4, фильтровали и выпаривали, чтобы получить (VV).
Синтез (WW)
Раствор (VV) (1 ммоль) и 10% Pd/C (100 мг) в MeOH (10 мл) подвергали полному гидрированию при атмосферном давлении и комнатной температуре. Реакционную смесь фильтровали через целит и выпаривали, чтобы получить (WW).
Синтез (XX)
К раствору (WW) (1 ммоль) и E (1 ммоль) [см. Bioorg. Med. Chem. Letter 1999, 9, 2283-88] в DMF (10 мл) добавляли HOBT (1,60 ммоль) и DIEA (4 ммоль). Смесь охлаждали до 0ºC и в течение нескольких минут добавляли BOP (1,60 ммоль). Полученный раствор нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь разбавляли насыщенным солевым раствором и экстрагировали EtOAc. Объединенный органический слой экстрагировали H2O и насыщенным солевым раствором, высушивали над MgSO4 и выпаривали, чтобы получить (XX).
Синтез соединения 20
К раствору (XX) (1 ммоль) в THF (100 мл) при комнатной температуре медленно добавляли TBAF (1 ммоль) и полученную смесь полностью перемешивали. Выпариванием и хроматографией полученного осадка получали 20.
Схема 19: синтез по примеру 21
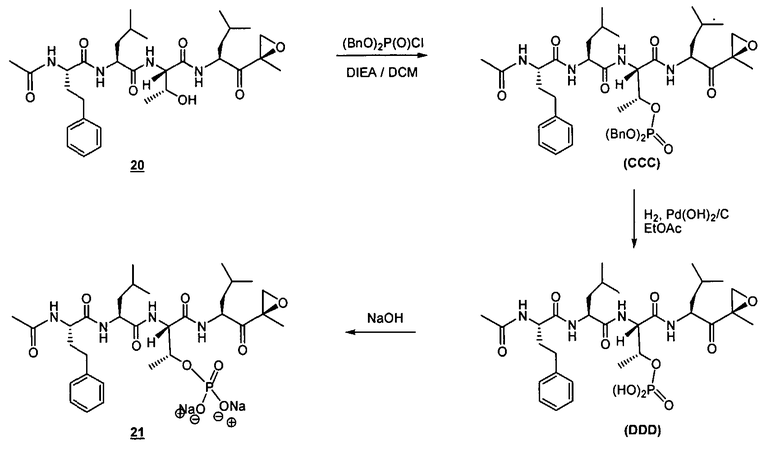
Синтез (CCC)
Целевое соединение получают по существу после процедуры превращения 2 в (RR), за исключением того, что 20 используют вместо 2.
Синтез (DDD)
Целевое соединение получают по существу после процедуры превращения (RR) в (SS), за исключением того, что (CCC) используют вместо (RR).
Синтез соединения 21
Целевое соединение получают по существу после процедуры превращения (SS) в 19, за исключением того, что (DDD) используют вместо (SS).
Схема 20: синтез по примеру 22
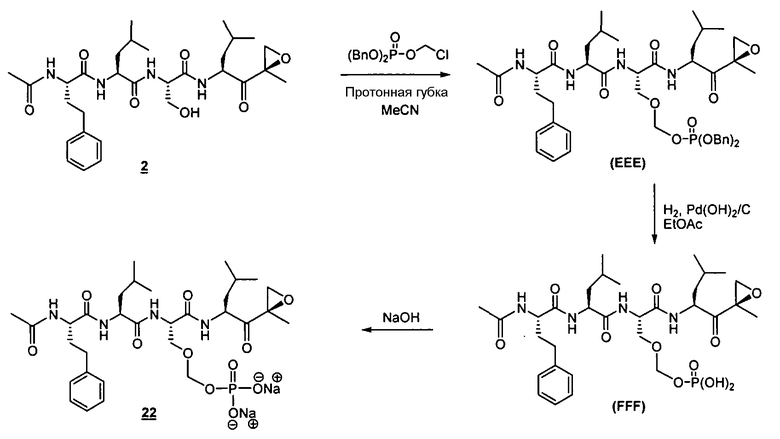
Синтез (EEE)
[См. J. Med. Chem. 1999, 42(16), 3094-3100.] К раствору дибензилхлорметилфосфата (1 ммоль) [патент США 6204257] в безводном ацетонитриле в аргоне добавляли соединение 2 (1 ммоль) и 1,2,2,6,6-пентаметилпиперидин (1,2 ммоль), после чего следовало полное перемешивание при 70ºC. Затем растворитель удаляли в вакууме, чтобы получить (EEE).
Синтез (FFF)
Целевое соединение получают по существу после процедуры превращения (RR) в (SS), за исключением того, что (EEE) используют вместо (RR).
Синтез соединения 22
Целевое соединение получают по существу после процедуры превращения (SS) в 19, за исключением того, что (FFF) используют вместо (SS).
Схема 21: синтез по примеру 23
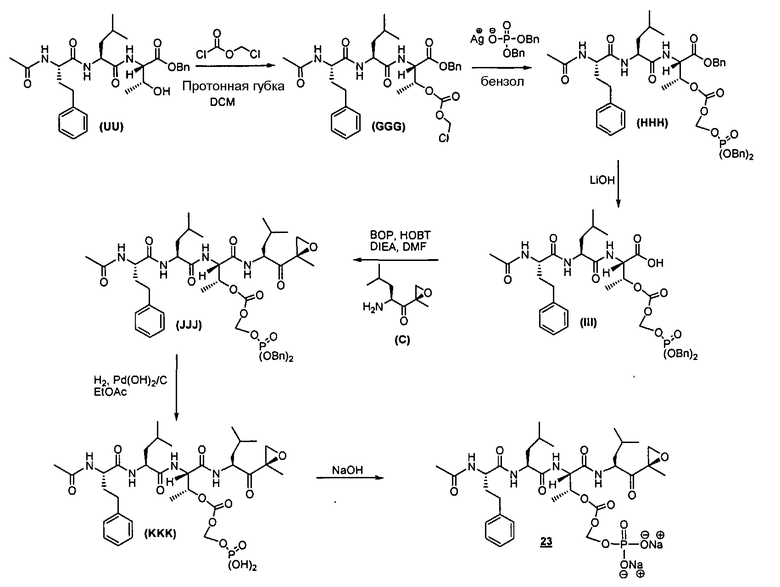
Синтез (GGG)
(См. Pharmaceutical Research 1993, 10(9), 1350-55.) К раствору (UU) (1 ммоль) и протонной губке (1 ммоль) в сухом CH2Cl2 (10 мл) при 0ºC добавляли по каплям раствор хлорметилового эфира хлормуравьиной кислоты (1 ммоль) в дихлорметане (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, промывали холодным 1 н. HCl и водой, высушивали над MgSO4, фильтровали и выпаривали, чтобы получить (GGG).
Синтез (HHH)
Раствор (GGG) (1 ммоль) и дибензилфосфата серебра (1,5 ммоль) в сухом бензоле (10 мл) кипятили с обратным холодильником в течение ночи. Реакционную смесь охлаждали, фильтровали, промывали насыщенным раствором Na2CO3, высушивали над MgSO4 и выпаривали, чтобы получить (HHH).
Синтез (III)
К раствору (HHH) (1 ммоль) в 3:1 MeOH/H2O (20 мл) при 0ºC добавляли по каплям раствор LiOH (5 ммоль) в H2O (2,5 мл). Реакционную смесь помещали в аргон и содержали при 5ºC в течение ночи. Реакцию гасили при 0ºC добавлением насыщенного NH4Cl (50 мл) и разбавляли холодной водой (150 мл). Значение pH доводили до 2 при 0ºC холодным 6 н. HCl и полученные твердые вещества собирали вакуумной фильтрацией, чтобы получить (III).
Синтез (JJJ)
Целевое соединение получают по существу после процедуры превращения (WW) в (XX), за исключением того, что (III) используют вместо (WW).
Синтез (KKK)
Целевое соединение получают по существу после процедуры превращения (RR) в (SS), за исключением того, что (JJJ) используют вместо (RR).
Синтез соединения 23
Целевое соединение получают по существу после процедуры превращения (SS) в 19, за исключением того, что (KKK) используют вместо (SS).
Схема 22: синтез по примеру 24
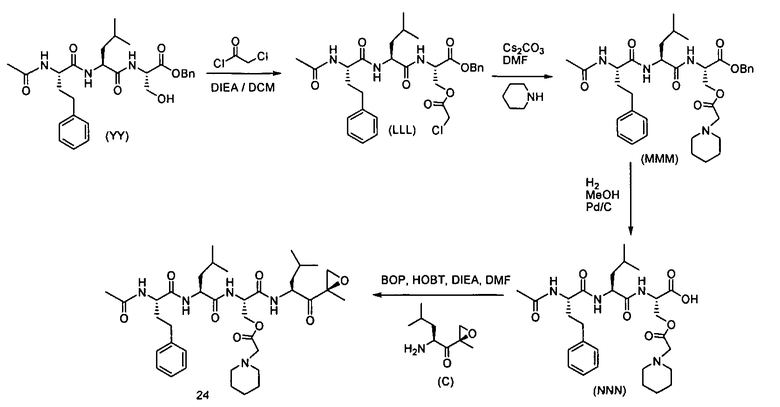
Синтез (LLL)
Целевое соединение (LLL) получают по существу после процедуры превращения 2 в (RR), заменяя соединение 2 на соединение (YY) [полученное стандартными способами синтеза пептидов в растворах] и хлорангидрид дибензилфосфорной кислоты на хлорангидрид хлоруксусной кислоты.
Синтез (MMM)
К раствору (LLL) (1 ммоль) в DMF (10 мл) при комнатной температуре в аргоне добавляли Cs2CO3 (1 ммоль), после чего добавляли пиперидин (1 ммоль). Перемешивание продолжали до тех пор, пока реакция не оканчивалась. Затем смесь вливали в насыщенный солевой раствор и промывали EtOAc. EtOAc высушивали над MgSO4, фильтровали и выпаривали при пониженном давлении, чтобы получить соединение (MMM).
Синтез (NNN)
Целевое соединение получают по существу после процедуры превращения (VV) в (WW), за исключением того, что (MMM) используют вместо (VV).
Синтез соединения 24
Целевое соединение получают по существу после процедуры превращения (WW) в (XX), за исключением того, что (NNN) используют вместо (WW).
Схема 23: синтез по примеру 25
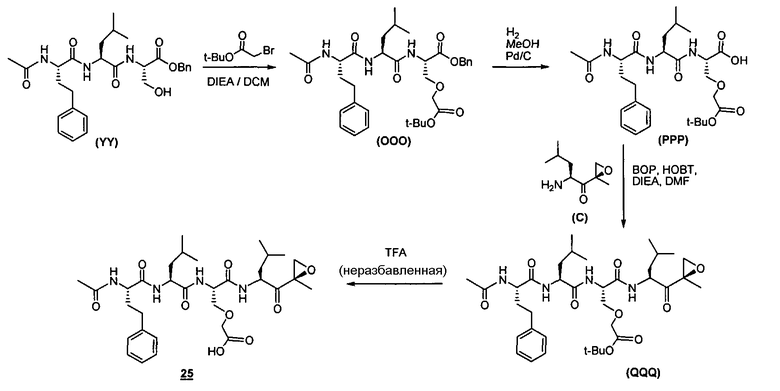
Синтез (ООО)
Целевое соединение получают по существу после процедуры превращения 2 в (RR), за исключением того, что (YY) используют вместо 2 и трет-бутилбромацетат используют вместо хлорангидрида дибензилфосфорной кислоты.
Синтез (PPP)
Целевое соединение получают по существу после процедуры превращения (VV) в (WW), за исключением того, что (ООО) используют вместо (VV).
Синтез (QQQ)
Целевое соединение получают по существу после процедуры превращения (WW) в (XX), за исключением того, что (PPP) используют вместо (WW).
Синтез соединения 25
Обработка (QQQ) (1 ммоль) с неразбавленной TFA (10 мл) при 0ºC сопровождалась выпариванием TFA при пониженном давлении, см. Bioorg. Med. Chem. Letter 1999, 9, 2283-88], чтобы получить 25.
Схема 24: синтез по примеру 26
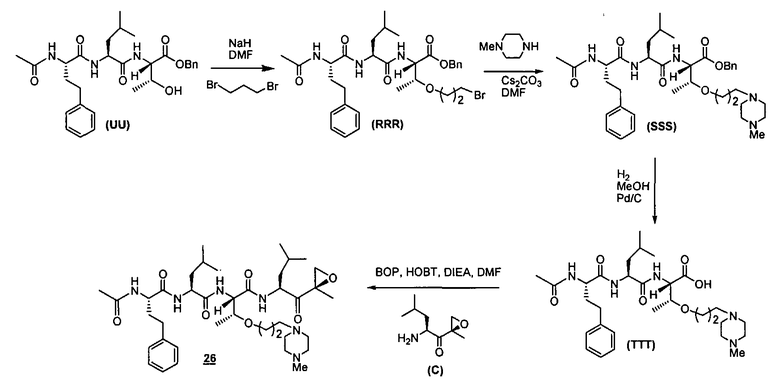
Синтез (RRR)
К раствору (UU) (1 ммоль) в безводном DMF (10 мл) при комнатной температуре осторожно добавляли NaH (1 ммоль). После перемешивания в течение 0,5 ч смесь обрабатывали 1,3-дибромпропаном (1,2 ммоль) и реакционную смесь перемешивали до окончания реакции. Раствор затем осторожно гасили добавлением насыщенного NH4Cl (1 мл), после чего вливали смесь в холодный насыщенный солевой раствор (10 мл). Полученную смесь промывали CH2Cl2, высушивали над MgSO4, фильтровали и растворитель выпаривали при пониженном давлении, чтобы получить (RRR).
Синтез (SSS)
К раствору (RRR) (1 ммоль) в безводном DMF (10 мл) при комнатной температуре в аргоне добавляли Cs2CO3 (1 ммоль), после чего добавляли N-метилпиперазин (1 ммоль). Перемешивание продолжали до тех пор, пока реакция не оканчивалась. Затем смесь вливали в насыщенный солевой раствор и промывали EtOAc. EtOAc высушивали над MgSO4, фильтровали и выпаривали при пониженном давлении, чтобы получить соединение (SSS).
Синтез (TTT)
Целевое соединение получают по существу после процедуры превращения (VV) в (WW), за исключением того, что (SSS) используют вместо (VV).
Синтез соединения 26
Целевое соединение получают по существу после процедуры превращения (WW) в (XX), за исключением того, что (TTT) используют вместо (WW).
Схема 25: синтез по примеру 27
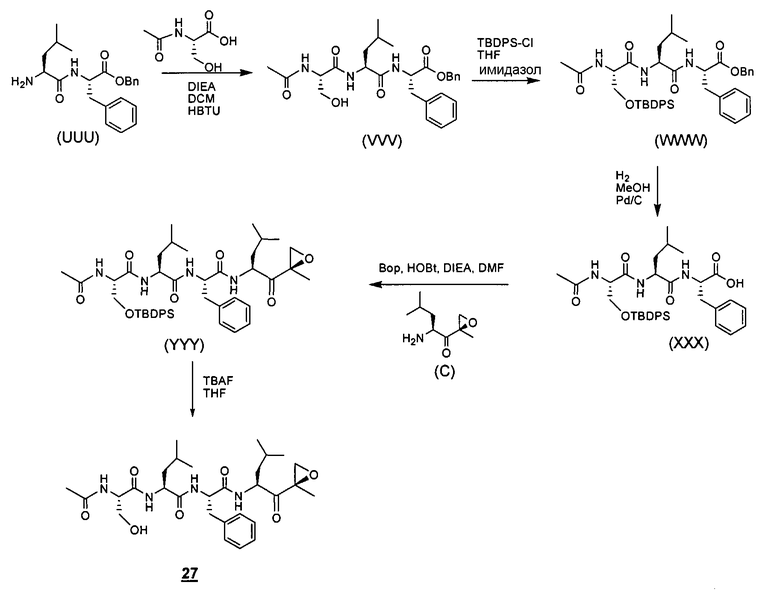
Синтез (VVV)
Целевое соединение получают по существу после процедуры превращения (TT) в (UU), за исключением того, что N-ацетилсерин используют вместо (TT) и (UUU) [полученный стандартными способами синтеза пептидов в растворах] используют вместо бензилового эфира серина.
Синтез (WWW)
Целевое соединение получают по существу после процедуры превращения (UU) в (VV), за исключением того, что (VVV) используют вместо (UU).
Синтез (XXX)
Целевое соединение получают по существу после процедуры превращения (VV) в (WW), за исключением того, что (WWW) используют вместо (VV).
Синтез (YYY)
Целевое соединение получают по существу после процедуры превращения (WW) в (XX), за исключением того, что (XXX) используют вместо (WW).
Синтез соединения 27
Целевое соединение получают по существу после процедуры превращения (XX) в 20, за исключением того, что (YYY) используют вместо (XX).
Схема 26: синтез по примеру 28
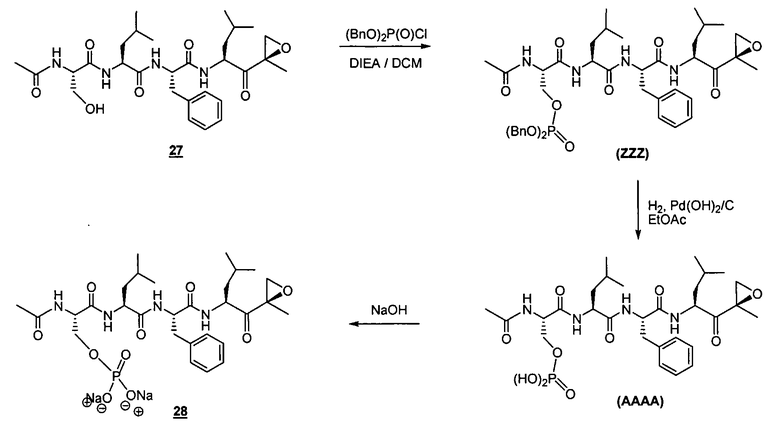
Синтез (ZZZ)
Целевое соединение получают по существу после процедуры превращения 2 в (RR), за исключением того, что 27 используют вместо 2.
Синтез (AAAA)
Целевое соединение получают по существу после процедуры превращения (RR) в (SS), за исключением того, что (ZZZ) используют вместо (RR).
Синтез соединения 28
Целевое соединение получают по существу после процедуры превращения (SS) в 19, за исключением того, что (AAAA) используют вместо (SS).
Схема 27: синтез по примеру 29
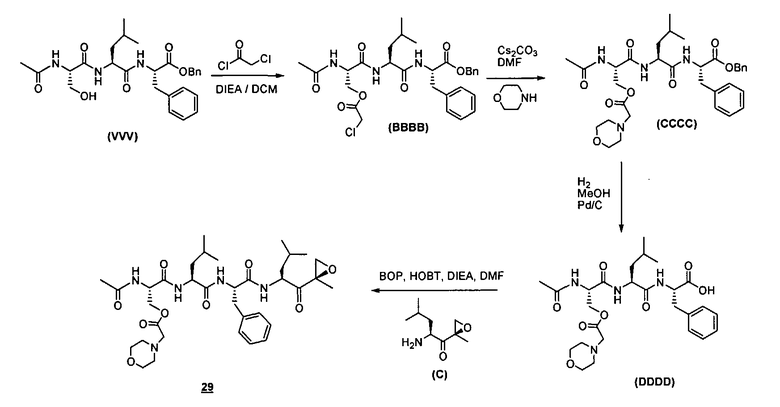
Синтез (BBBB)
Целевое соединение получают по существу после процедуры превращения (YY) в (LLL), за исключением того, что (VVV) используют вместо (SS).
Синтез (CCCC)
Целевое соединение получают по существу после процедуры превращения (LLL) в (MMM), за исключением того, что (BBBB) используют вместо (LLL) и морфолин используют вместо пиперидина.
Синтез (DDDD)
Целевое соединение получают по существу после процедуры превращения (MMM) в (NNN), за исключением того, что (CCCC) используют вместо (MMM).
Синтез соединения 29
Соединение (WW) получают по существу после процедуры превращения (WW) в (XX), за исключением того, что (DDDD) используют вместо (WW).
Эквиваленты
Специалисты в данной области узнают или смогут выявить, используя не более чем обычное экспериментирование, множество эквивалентов описанных здесь соединений и способов их использования. Предполагается, что такие эквиваленты входят в объем данного изобретения и охраняются в соответствии со следующей ниже формулой изобретения.
Все цитируемые выше ссылки и публикации тем самым включены сюда путем ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕПТИДНЫЕ ЭПОКСИКЕТОНЫ ДЛЯ ИНГИБИРОВАНИЯ ПРОТЕАСОМЫ | 2007 |
|
RU2450016C2 |
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ | 2015 |
|
RU2733405C2 |
| VEGF-НЕЙТРАЛИЗУЮЩИЕ ПРОЛЕКАРСТВА ДЛЯ ЛЕЧЕНИЯ ОФТАЛЬМОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2673881C2 |
| ИНГИБИТОРЫ MAGL | 2017 |
|
RU2754536C1 |
| ТИОФЕНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2709473C1 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ | 2013 |
|
RU2654692C2 |
| МОДУЛИРУЮЩИЕ JAK КИНАЗУ ХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2529019C2 |
| АНТИВИРУСНЫЕ СОЕДИНЕНИЯ | 2009 |
|
RU2505539C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ FGFR4, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СОСТАВЫ, СОДЕРЖАЩИЕ ИХ, И ИХ ПРИМЕНЕНИЕ | 2021 |
|
RU2827699C1 |
| ЗАМЕЩЕННЫЕ АМИНОМАСЛЯНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ НЕПРИЛИЗИНА | 2012 |
|
RU2604522C2 |
Изобретение относится к α',β'-эпоксидам пептидов формулы (III) и (IV), которые ингибируют химотрипсино-подобную активность 20S протеасомы. 2 н. и 17 з.п. ф-лы.
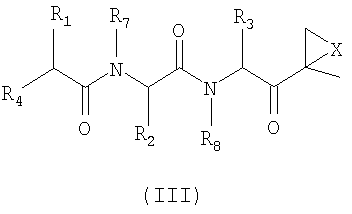
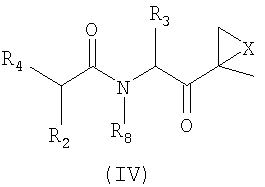
1. Соединение, имеющее структуру формулы (III), или его фармацевтически приемлемая соль
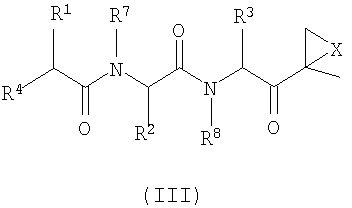
где L выбран из С=O и SO2;
Х представляет собой О;
Z отсутствует или представляет собой C1-6-алкил;
R1 и R3 оба представляют собой C1-6-алкил;
R2 представляет собой C1-6-аралкил;
R4 представляет собой N(R5)L-Z-R6;
R5 представляет собой водород;
R6 выбран из Ar и гетероциклила, представляющего собой неароматическое 3-7-членное кольцо, содержащее до двух гетероатомов, выбранных из азота и кислорода, и необязательно замещенное тиенилсульфонилом;
где Ar представляет собой фенил, необязательно замещенный метилом, или гетероарил, представляющий собой 5-6-членное кольцо, содержащее атом серы, и
R7 и R8 оба представляют собой водород.
2. Соединение по п.1, в котором R1 и R3 оба представляют собой изобутил и R2 представляет собой фенилметил.
3. Соединение по п.2, в котором R6 представляет собой Ar.
4. Соединение по п.3, в котором каждый Ar независимо выбран из фенила и тиенила.
5. Соединение по п.3, в котором Z представляет собой C1-6-алкил.
6. Соединение по п.2, в котором Z отсутствует.
7. Соединение по п.6, в котором R6 представляет собой Ar и Ar выбран из фенила и тиенила.
8. Соединение, имеющее структуру формулы (IV), или его фармацевтически приемлемая соль
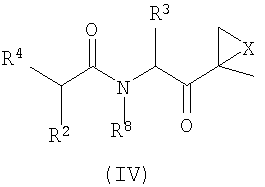
где L выбран из С=O и SO2;
Х представляет собой О;
Z отсутствует или представляет собой C1-6-алкил;
R2 и R3 каждый независимо выбраны из C1-6-алкила и C1-6-аралкила;
R4 представляет собой N(R5)L-Z-R6;
R5 представляет собой водород;
R6 выбран из Ar и гетероциклила, представляющего собой неароматическое 3-7-членное кольцо, содержащее до двух гетероатомов, выбранных из азота и кислорода, и необязательно замещенное тиенилсульфонилом;
где Ar представляет собой фенил, бифенил или гетероарил, представляющий собой 5-6-членное ароматическое кольцо, содержащее до двух гетероатомов, выбранных из азота и серы, и R7 и R8 оба представляют собой водород.
9. Соединение по п.8, в котором R3 представляет собой C1-6-алкил и R2 представляет собой C1-6-аралкил.
10. Соединение по п.9, в котором R3 представляет собой изобутил и R2 представляет собой фенилметил.
11. Соединение по п.10, в котором R6 представляет собой Ar.
12. Соединение по п.10, в котором каждый Ar независимо выбран из фенила и тиенила.
13. Соединение по п.11, в котором Z представляет собой C1-6-алкил.
14. Соединение по п.10, в котором Z отсутствует.
15. Соединение по п.14, в котором R6 представляет собой Ar и Ar выбран из фенила и тиенила.
| α-КЕТОАМИДНЫЕ ИНГИБИТОРЫ 20S ПРОТЕАСОМЫ | 1999 |
|
RU2192429C2 |
| 1971 |
|
SU411660A1 | |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US 6204257 B1, 20.03.2001 | |||
| ELOFSSON M; ET AL "Towards subunit-specific proteasome inhibitors: synthesis and evaluation of peptide α',β'-epoxyketones.", CHEMISTRY AND BIOLOGY, 1995 Vol:6, Nr:11, Page(s): 811-822 | |||
| Kim К.В.; Myung J.; Sin N.; Crews С.М. | |||

