Перекрестные ссылки на родственные заявки
Настоящее изобретение испрашивает приоритет патентной заявки "Бициклические гетероциклические ингибиторы FGFR4, фармацевтические композиции и препараты, содержащие их, и их применение", поданной в Китае 26 марта 2021 г., заявка № 202110326618.7, все содержание которой включено в настоящий документ посредством ссылки. Все содержание данной патентной заявки включено в настоящий документ посредством ссылки.
Техническая область
Настоящее изобретение относится к области медицинской химии и касается, в частности, бициклических гетероциклических ингибиторов FGFR4, фармацевтических композиций и препаратов, содержащих их, а также их применения.
Фоновая технология
Рецептор фактора роста фибробластов (fibroblast growth factor receptor, FGFR) относится к тирозинкиназам рецепторного типа (RTKs), в его семейство входят четыре белка FGFR: FGFR1, FGFR2, FGFR3 и FGFR4, которые участвуют в различных этапах эмбрионального развития, формирования органов, тканевого динамического гомеостаза и воспаления. динамического гомеостаза, регенерации сосудов и воспаления. При связывании с лигандами фактора роста фибробластов (FGF) FGFR подвергается димеризации и фосфорилированию, что приводит к стимуляции протеинкиназной активности и привлечению многих внутриклеточных стыковочных белков, и эти взаимодействия влияют на рост, пролиферацию, дифференцировку и другие функции клеток через активацию ряда внутриклеточных сигнальных путей, включая Ras-MAPK, AKT-PI3K и фосфолипазу С. (Eswarakumar V P et, al. Cytokine Growth Factor Reviews, 2005, 16 (2): 139-149).
FGFR4 играет важную роль в эмбриональном развитии, центральном нервном контроле, восстановлении тканей, а также в опухолевой инвазии и ангиогенезе (Ho, H. K. et, al. Journal of Hepatology, 2009, 50: 118-127). Кроме того, сверхэкспрессия FGFR4 наблюдается в различных типах опухолей, среди которых гепатоцеллюлярная карцинома, рак желудка, почечно-клеточная карцинома, колоректальный рак, рак молочной железы, рак поджелудочной железы, рак простаты, рак легкого, рак яичников и другие. В настоящее время считается, что FGFR4 является единственным рецептором для FGF19, проявляющим специфичность, и что FGF19 проявляет свою активность путем связывания с FGFR4 и его активации. В патологических условиях чрезмерная активность MAPK- и PI3K/AKT-путей, обусловленная сверхэкспрессией FGF19, FGFR4 или мутациями в активированном FGFR4, приводит к развитию опухоли, ее прогрессии и устойчивости к традиционным методам лечения рака (Heinzle et. al. Cur. Pharm. Des. 2014, 20: 2881).
Исследование показывает, что моноклональное антитело FGF19 способно избирательно блокировать взаимодействие FGF19 с FGFR4, что подавляет рост трансплантированных опухолей у мышей nude с карциномой толстой кишки человека и эффективно предотвращает развитие гепатоцеллюлярной карциномы у трансгенных мышей с FGF19 (Desnoyers, L. R. et al. R. et al. Oncogene, 2008, 27: 85-97), что подтверждает целесообразность использования маломолекулярных ингибиторов FGFR4 для блокирования связывания молекул внеклеточного лиганда с FGFR4 или внутриклеточной киназы для ингибирования FGFR4-опосредованной сигнализации для лечения злокачественных опухолей, таких как гепатоцеллюлярная карцинома. В настоящее время в клинических исследованиях находится несколько ингибиторов FGFR4, таких как FGF401, разработанный компанией Novartis, который способен селективно ингибировать FGFR4 и демонстрирует хорошие перспективы; BLU554, ингибитор, специфичный для FGFR4, разработанный компанией Фарматика Blueprint , который обладает сильной противоопухолевой активностью и селективностью, а также демонстрирует хорошую безопасность; H3B6527, ингибитор, специфичный для FGFR4, разработанный компанией H3 Биомедика. Ингибитор H3B6527, разработанный компанией H3 Биомедицина, также показал хорошую противоопухолевую активность.
На основе углубления понимания исследователями структуры и функции FGFR4, механизма его действия, а также взаимодействия с другими киназами разработка ингибиторов FGFR4 с высокой специфичностью, хорошим терапевтическим эффектом и низким уровнем побочных эффектов будет иметь большое значение для профилактики и/или лечения FGFR4-связанных опухолей и других заболеваний.
Содержание изобретения
Задача, решаемая изобретением
Целью настоящего изобретения является создание серии новых соединений, оказывающих ингибирующее действие на активность FGFR4, фармацевтических композиций и препаратов, включающих эти соединения, и способов фармацевтического применения этих соединений.
Программы для решения проблем
<Первый аспект>
В настоящем изобретении представлены соединения, имеющие структуру формулы (I), или фармацевтически приемлемые соли, сложные эфиры, стереоизомеры, таутомеры, сольваты, хелаты, нековалентные комплексы или пролекарства этих соединений, причем соединения формулы (I) имеют общую формулу:
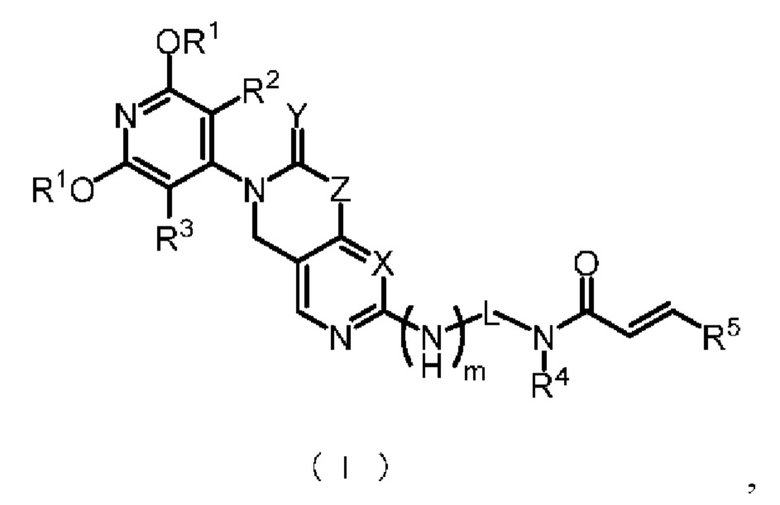
где:
X представляет собой CH или N;
Y представляет собой 2 H или 1 O;
Z представляет собой C(R6)2 или N(R6);
m представляет собой 0 или 1;
L представляет собой -C(R7)2- или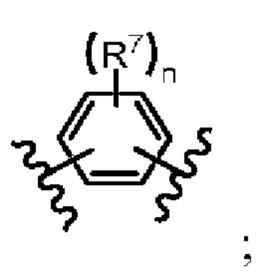
n представляет собой 0, 1 или 2;
каждый R1 независимо представляет собой C1-3 алкил или C1-3 галоалкил;
R2 и R3 каждый независимо представляет собой водород, галоген, С1-3 алкил, С1-3 галоалкил, циано или С1-3 алкокси;
R4 представляет собой водород, С1-6 алкил или С6-10 арил;
R5 представляет собой водород, галоген, циано, нитро, трифторметил, амино, C1-6 алкил, C2-8 алкенил, C6-10 арил, Cl-8 алкиламино, ди(C2-8 алкил)амино, C2-8 алкинил, C1-8 галоалкил или C3-8 циклоалкил;
каждый R6 независимо представляет собой водород, C1-8 алкил, C2-8 алкенил, C2-8 алкинил, C1-8 галоалкил, C6-10 арил, C6-10 арил, содержащий заместитель, C3-10 циклоалкил, 5-10-членный гетероарил или 4-10-членный гетероциклоалкил; при этом каждый из указанных 5-10-членных гетероарилов и 4-10-членных гетероциклоалкилов независимо включает от 1 до 3 кольцеобразующих гетероатомов, и каждый из указанных гетероатомов независимо является N, O или S;
или два R6 и атомы углерода, к которым они присоединены, вместе образуют C3-8 циклоалкил или 4-10-членный гетероциклоалкил; где 4-10-членный гетероциклоалкил включает от 1 до 3 кольцеобразующих гетероатомов, причем гетероатомами являются N, O или S; и где каждый из C3-8 циклоалкила и 4-10-членного гетероциклоалкила независимо по выбору замещен от 1 до 4 групп, каждая из которых независимо представляет собой галоген, циано, гидроксил, аминогруппу, C1-8 карбоксиамидо, C1-8 карбоксамоил, C1-8 алкил или C1-8 алкокси группу;
каждый R7 независимо представляет собой водород, галоген, амино, циано, C1-8 алкил, C1-8 алкил, содержащий заместитель, C1-8 алкокси, C1-8 алкокси, содержащий заместитель, C1-8 алкиламино, ди(C2-8 алкил)амино, C2-8 алкенил, C2-8 алкенил, содержащий заместитель, C2-8 алкинил, C2-8 алкинил, содержащий заместитель, C6-10 арил, C6-10 арил, содержащий заместитель, C3-8 циклоалкил, C3-8 циклоалкил, содержащий заместитель, 3-10-членный гетероциклоалкил, 3-10-членный гетероциклоалкил, содержащий заместитель, 5-10-членный гетероарил или 5-10-членный гетероарил, содержащий заместитель; при этом указанные 3-10-членный гетероциклоалкил и 5-10-членный гетероарил независимо включают от 1 до 3 кольцеобразующих гетероатомов и каждый из указанных гетероатомов независимо является N, O или S.
Предпочтительно указанное соединение имеет структуру, представленную в формуле (II):
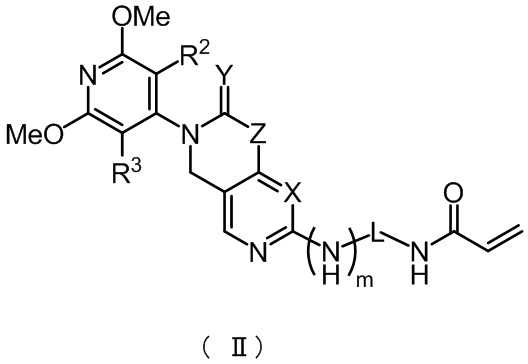 ,
,
где X, Y, Z, m, L, R2 и R3 определены, как указано выше.
Предпочтительно, указанное соединение имеет структуру, представленную в формуле (III):
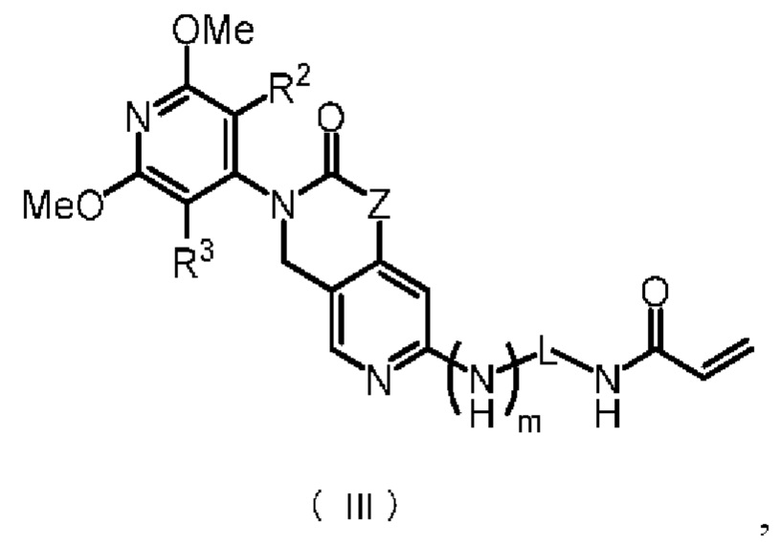
где Z, m, L, R2 и R3 определены так, как определено выше.
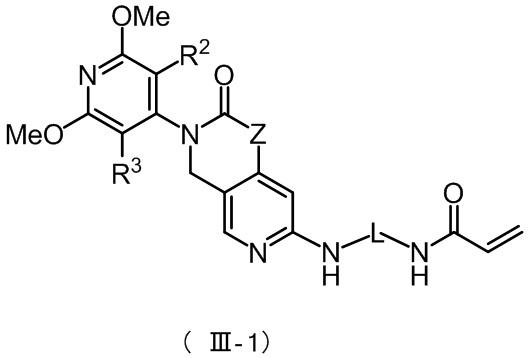
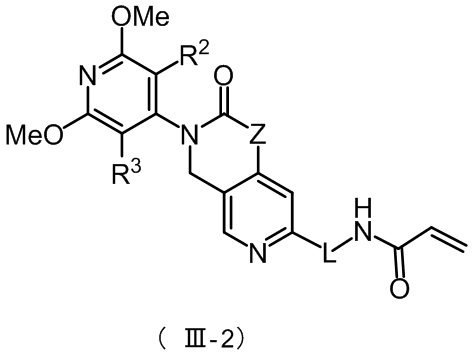
Более предпочтительно, указанное соединение имеет структуру, представленную в формуле (III-1) или формуле (III-2):
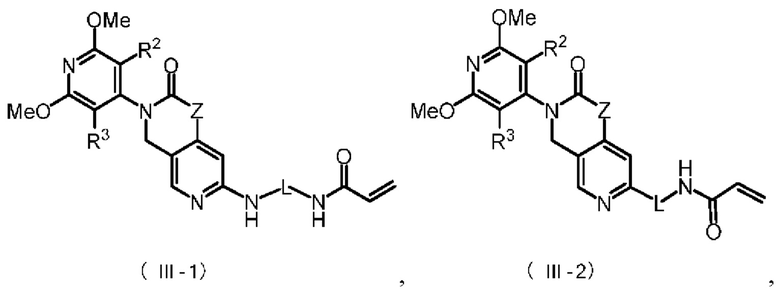
где Z, L, R2 и R3 определены так, как определено выше.
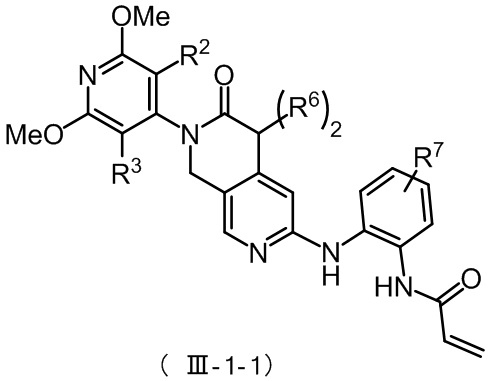
Более предпочтительно, указанное соединение формулы (III-1) имеет структуру, представленную в формуле (III-1-1):
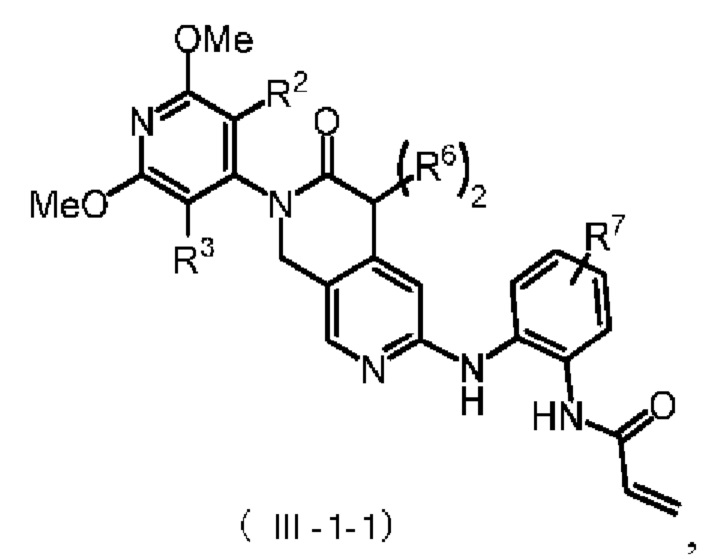
где R2, R3 и R6 определены, как определено выше, R7 представляет собой водород, C1-4 алкил, пиперазинил, пиперидинил или морфолинил, причем указанные C1-4 алкил, пиперазинил, пиперидинил и морфолинил по выбору замещены по меньшей мере одним R8, каждый R8 независимо представляет собой водород, C1-4 алкил (предпочтительно метил или этил), морфолинил, мостиковый морфолинил, пиперазинил, пиперазинил, содержащий заместитель, мостиковый пиперазинил, мостиковый пиперазинил, содержащий заместитель, оксетанил или оксетанил, содержащий заместитель, причем этот заместитель представляет собой C1-4 алкил (предпочтительно метил или
Предпочтительно R2, R3 и R6 определены, как определено выше, а R7 представляет собой водород, C1-4 алкил, пиперазинил, пиперидинил или морфолинил, причем указанные C1-4 алкил, пиперазинил, пиперидинил и морфолинил по выбору замещены по меньшей мере одним R8, каждый из которых независимо друг от друга представляет собой водород, морфолинил, мостиковый морфолинил, пиперазинил, пиперазинил, содержащий заместитель, мостиковый пиперазинил, мостиковый пиперазинил, содержащий заместитель, оксетанил или оксетанил, содержащий заместитель, причем указанный заместитель представляет собой C1-4 алкил;
Более предпочтительно, R2, R3 и R6 определены, как определено выше, а R7 представляет собой один из следующих фрагментов:
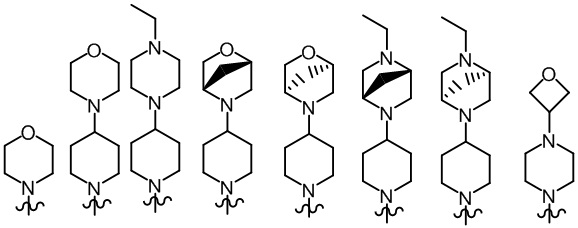 ;
;
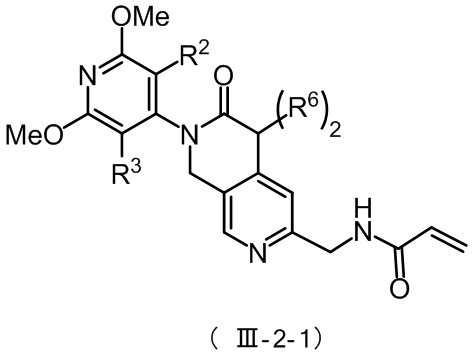
Более предпочтительно, соединение формулы (III-2) имеет структуру, представленную в формуле (III-2-1) или формуле (III-2-2):
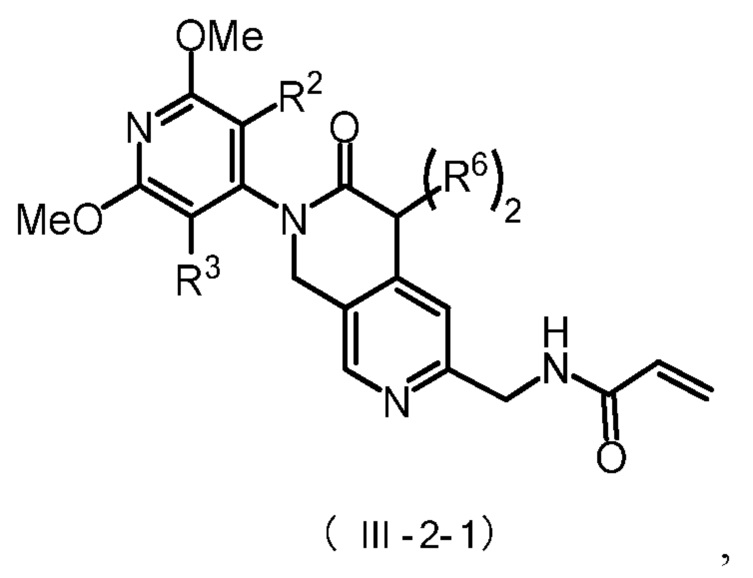
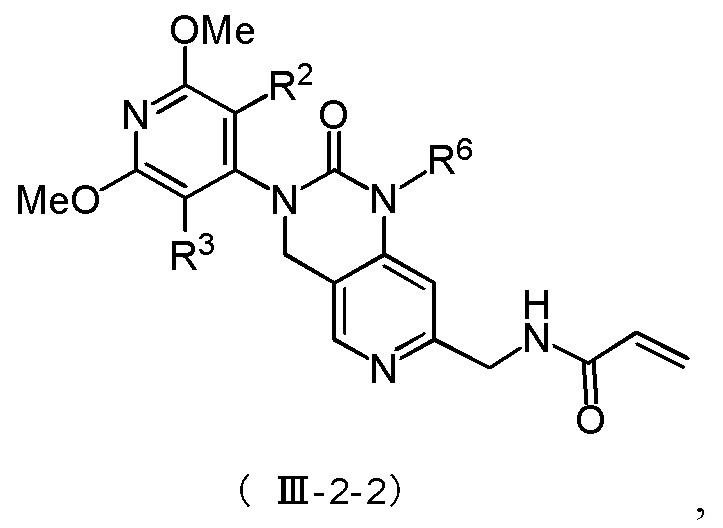
где R2, R3 и R6 определены, как указано выше.
Предпочтительно, указанное соединение имеет структуру, представленную в формуле (IV):
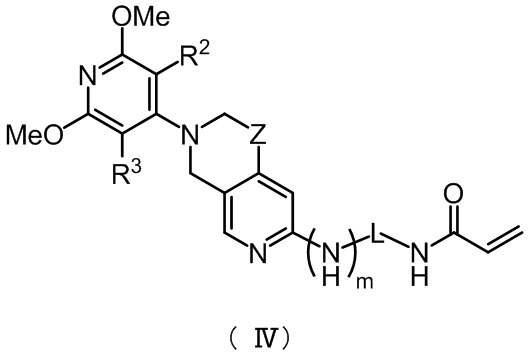 ,
,
где Z, m, L, R2 и R3 определены так, как определено выше.
Более предпочтительно, указанное соединение имеет структуру, представленную в формуле (IV-1) или формуле (IV-2):
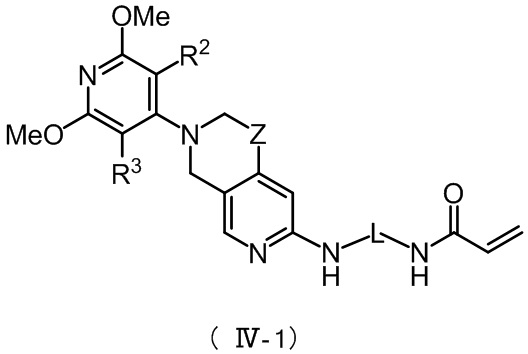 ,
,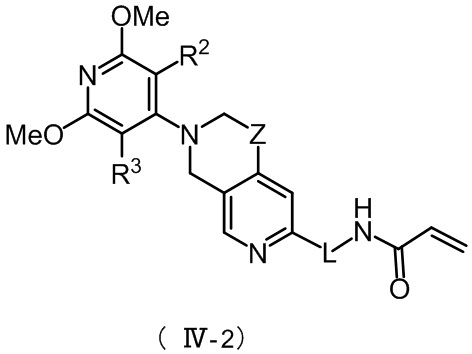 ,
,
где Z, L, R2 и R3 определены так, как определено выше;
Более предпочтительно, указанное соединение формулы (IV-1) имеет структуру, представленную в формуле (IV-1-1):
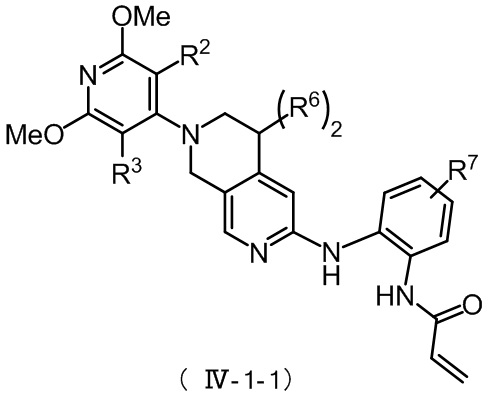 ,
,
где R2, R3 и R6 определены, как определено выше, а R7 представляет собой водород, C1-4 алкил, пиперазинил, пиперидинил или морфолинил, причем указанные C1-4 алкил, пиперазинил, пиперидинил и морфолинил по выбору замещены по меньшей мере одним R8, каждый из которых независимо друг от друга представляет собой водород, морфолинил, мостиковый морфолинил, пиперазинил, пиперазинил, содержащий заместитель, мостиковый пиперазинил, мостиковый пиперазинил, содержащий заместитель, оксетанил или оксетанил, содержащий заместитель, причем указанный заместитель представляет собой C1-4 алкил;
Более предпочтительно, R2, R3 и R6 определены, как определено выше, а R7 представляет собой один из следующих фрагментов:
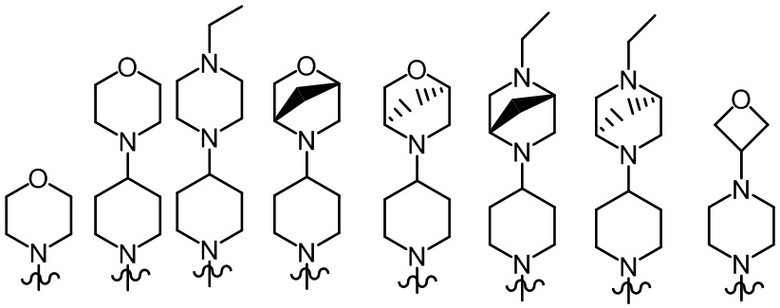 ;
;
Более предпочтительно, соединение формулы (IV-2) имеет структуру, представленную в формуле (IV-2-1) или формуле (IV-2-2):
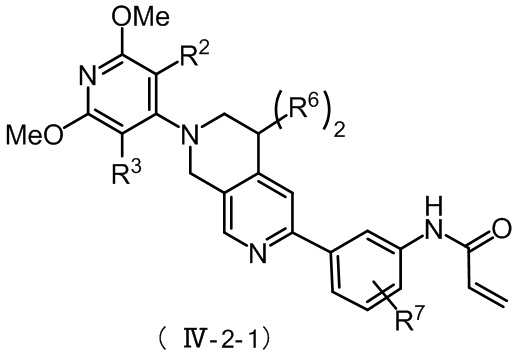 ,
,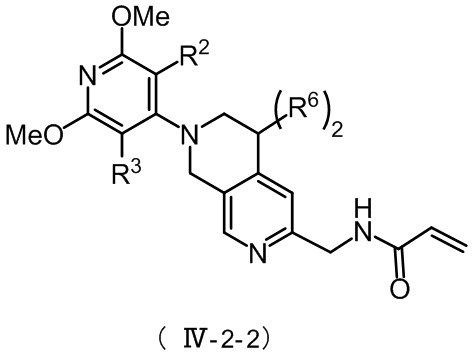 ,
,
где R2, R3 и R6 в формуле (IV-2-1) определены, как определено выше, а R7 представляет собой водород, C1-4 алкил или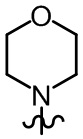 ;
;
R2, R3 и R6 в формуле (IV-2-2) определяются так, как определено выше.
<Второй аспект>
Настоящее изобретение предоставляет следующие соединения или их фармацевтически приемлемые соли, сложные эфиры, стереоизомеры, таутомеры, сольваты, хелаты, нековалентные комплексы или пролекарства:
 ,
, ,
, ,
, ,
,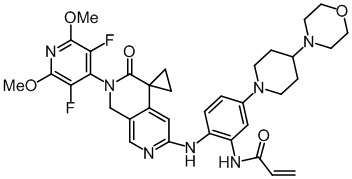 ,
,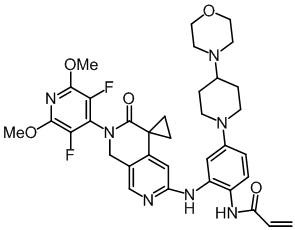 ,
,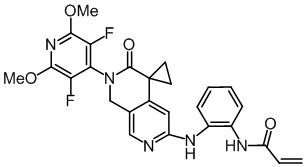 ,
,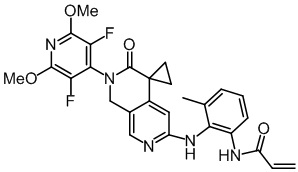 ,
,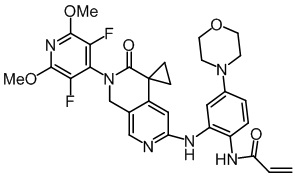 ,
,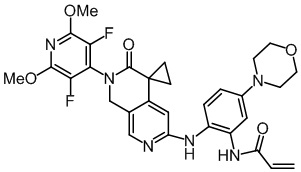 ,
,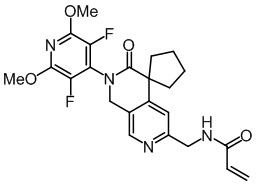 ,
,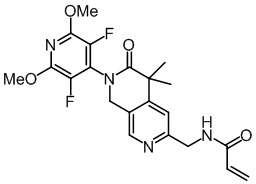 ,
, ,
, ,
, ,
, ,
,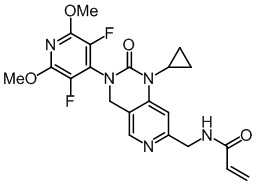 ,
,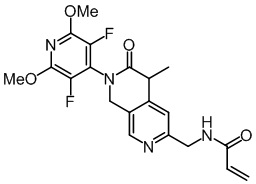 ,
,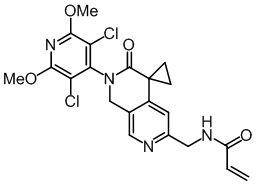 ,
,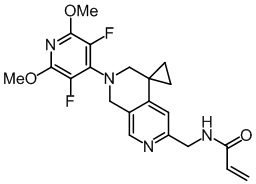 ,
, ,
,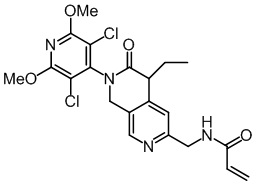 ,
,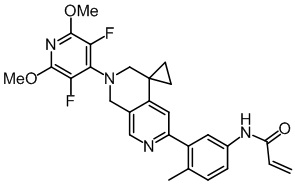 ,
,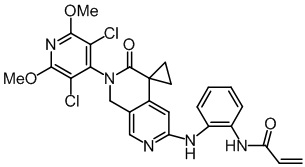 ,
, ,
,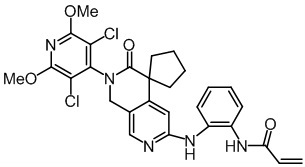 ,
,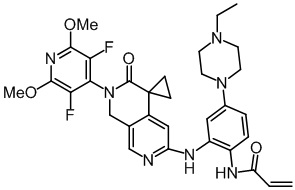 ,
,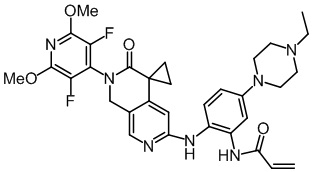 ,
,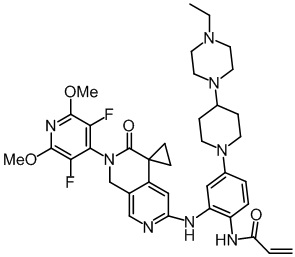 ,
,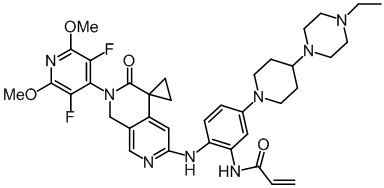 и
и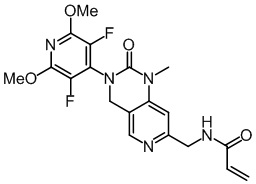 .
.
<третий аспект>
Настоящее изобретение представляет собой фармацевтическую композицию, включающую соединение согласно <Первый аспект>, <Второй аспект> или фармацевтически приемлемую соль, сложный эфир, стереоизомер, таутомер, сольват, хелат, нековалентный комплекс или пролекарство этого соединения.
<четвертый аспект>
Настоящее изобретение представляет собой фармацевтический препарат, включающий соединение согласно <Первый аспект>, <Второй аспект> или его фармацевтически приемлемую соль, сложный эфир, стереоизомер, таутомер, сольват, хелат, нековалентный комплекс или пролекарство, или фармацевтическую композицию согласно <третий аспект>, причем указанный фармацевтический препарат представляет собой таблетки, капсулы, инъекции, гранулы, порошок, суппозитории, пилюли, гели, дисперсии, растворы для перорального применения, лекарственные формы для ингаляции, суспензии или сухие суспензии.
<пятый аспект>
Настоящее изобретение предоставляет соединения согласно <Первый аспект>, <Второй аспект> или его фармацевтически приемлемые соли, сложные эфиры, стереоизомеры, таутомеры, сольваты, хелаты, нековалентные комплексы или пролекарства, или фармацевтические композиции согласно <третий аспект>, или фармацевтические препараты согласно <четвертый аспект> для получения лекарственных средств для профилактики и/или лечения заболеваний, опосредованных, по крайней мере частично, FGFR4.
Предпочтительно, указанное заболевание, по крайней мере частично опосредованное FGFR4, включает рак.
Более предпочтительно, рак выбран из гепатоцеллюлярной карциномы, рака мочевого пузыря, рака молочной железы, рака шейки матки, колоректального рака, рака эндометрия, рака желудка, рака головы и шеи, рака почек, гепатоцеллюлярной карциномы, рака легких, рака яичников, рака простаты, рака пищевода, рака желчного пузыря, рака поджелудочной железы, рака щитовидной железы, рака кожи, лейкоза, множественной миеломы, хронической лимфобластной лимфомы, Т-клеточного лейкоза взрослых, В-клеточной лимфомы, острого миелоидного лейкоза миелоидный лейкоз, лимфома Ходжкина или неходжкинская лимфома, макроглобулинемия Вальденстрема, волосатоклеточная лимфома, лимфома Буркитта, нейроглиобластома, меланома, мезотелиома, нейробластома у взрослых, рак яичка, плоскоклеточная карцинома, глиобластома и рабдомиосаркома.
<Шестой аспект>
Настоящее изобретение предоставляет соединения согласно <Первый аспект>, <Второй аспект> или их фармацевтически приемлемые соли, сложные эфиры, стереоизомеры, таутомеры, сольваты, хелаты, нековалентные комплексы или пролекарства, или фармацевтические композиции согласно <третий аспект>, или фармацевтические препараты согласно <четвертый аспект>, которые используются в качестве ингибиторов EGFR4.
<седьмой аспект>
Настоящее изобретение предоставляет способ профилактики и/или лечения заболевания, опосредованного, по крайней мере частично, FGFR4, включающий стадии: введение нуждающемуся в этом пациенту профилактически и/или терапевтически эффективного количества соединения согласно <Первый аспект>, соединения согласно <Второй аспект>, соединения согласно <Первый аспект>, соединения согласно <Первый аспект>, или фармацевтической композиции согласно <третий аспект>, или фармацевтического препарата согласно <четвертый аспект>, вводимая нуждающемуся в ней пациенту.
<Восьмой аспект>
Настоящее изобретение представляет собой способ профилактики и/или лечения рака, включающий следующие стадии: введение профилактически и/или терапевтически эффективного количества соединения согласно <Первый аспект>, <Второй аспект> или его фармацевтически приемлемой соли, сложного эфира, стереоизомера, таутомера, сольвата, хелата, нековалентного комплекса или пролекарства, или фармацевтической композиции согласно <третий аспект>, или фармацевтического препарата согласно <четвертый аспект>, и по меньшей мере одного дополнительного терапевтического агента для лечения рака, вводимое нуждающемуся в нем пациенту.
Предпочтительно, указанный рак выбирается из гепатоцеллюлярной карциномы, рака мочевого пузыря, рака молочной железы, рака шейки матки, колоректального рака, рака эндометрия, рака желудка, рака головы и шеи, рака почек, гепатоцеллюлярной карциномы, рака легких, рака яичников, рака простаты, рака пищевода, рака желчного пузыря, рака поджелудочной железы, рака щитовидной железы, рака кожи, лейкоза, множественной миеломы, хронической лимфобластной лимфомы, Т-клеточного лейкоза взрослых, В-клеточной лимфомы, острого миелоидного лейкоза миелоидный лейкоз, лимфома Ходжкина или неходжкинская лимфома, макроглобулинемия Вальденстрема, волосатоклеточная лимфома, лимфома Буркитта, нейроглиобластома, меланома, мезотелиома, нейробластома у взрослых, рак яичка, плоскоклеточная карцинома, глиобластома и рабдомиосаркома.
<Девятый аспект>
Настоящее изобретение предоставляет комбинированную лекарственную форму, включающую соединение согласно <Первый аспект>, <Второй аспект> или его фармацевтически приемлемую соль, сложный эфир, стереоизомер, таутомер, сольват, хелат, нековалентный комплекс или пролекарство, или фармацевтическую композицию согласно <третий аспект>, или фармацевтический препарат согласно <четвертый аспект>, и по меньшей мере один дополнительный терапевтический агент для лечения рака.
Эффект изобретения
Настоящее изобретение предоставляет структурно новое соединение формулы (I), которое может быть использовано для приготовления фармацевтических композиций и фармацевтических препаратов и т.п. для применения в человеческой или ветеринарной медицине для профилактики и/или лечения заболеваний (например, опухолей и т.п.), опосредованных, по крайней мере частично, FGFR4, с высокой степенью специфичности, терапевтической эффективностью и низким уровнем побочных эффектов.
конкретные варианты осуществления изобретения
Перед дальнейшим описанием изобретения следует понимать, что изобретение не ограничивается конкретными вариантами, описанными в настоящем документе; также следует понимать, что термины, используемые в настоящем документе, используются только для описания, но не для ограничения конкретных вариантов реализации.
[Определение терминов]
Если не указано иное, следующие термины имеют следующие значения:
Термин "фармацевтически приемлемая соль" представляет собой соль соединения изобретения, которая по существу нетоксична для организмов. Фармацевтически приемлемые соли обычно включают, но не ограничиваются ими, соли, образующиеся в результате реакции соединений изобретения с фармацевтически приемлемыми неорганическими/органическими кислотами или неорганическими/органическими основаниями, которые также называются солями присоединения кислот или солями присоединения оснований. Обычные неорганические кислоты включают (но не ограничиваются ими) соляную, бромистоводородную, серную, фосфорную и т.д., обычные органические кислоты включают (но не ограничиваются ими) трифторуксусную, лимонную, малеиновую, фумаровую, янтарную, винную, молочную, пировиноградную, щавелевую, муравьиную, уксусную, бензойную, метансульфоновую, бензенсульфоновую, п-толуиленсульфоновую и т.д., а обычные неорганические основания включают (но не ограничиваются ими) гидроксид натрия, гидроксид калия, гидроксид кальция. Распространенные неорганические основания включают (но не ограничиваются ими) гидроксид натрия, гидроксид калия, гидроксид кальция, гидроксид бария и т.д. Распространенные органические основания включают (но не ограничиваются ими) диэтиламин, триэтиламин, этамбутол и т.д.
Термин "сложный эфир" используется для обозначения органических эфиров, в том числе моно-, ди-, три- и, в более общем случае, полиэфиров.
Термин "стереоизомер" (или "спиновый изомер") относится к стабильному изомеру с перпендикулярной плоскостью асимметрии, обусловленной наличием хотя бы одного хирального элемента (включая хиральные центры, хиральные оси, хиральные поверхности и т.д.) , который способен вращать плоскополяризованный свет. Изомеры. Поскольку в соединениях настоящего изобретения имеются асимметрические центры, а также другие химические структуры, которые могут приводить к стереоизомерии, настоящее изобретение также включает эти стереоизомеры и их смеси. Поскольку соединения настоящего изобретения и их соли включают асимметрические атомы углерода, они способны существовать в виде отдельных стереоизомеров, рацематов, энантиомеров и смесей диастереомеров. Как правило, эти соединения могут быть получены в виде рацемических смесей. Однако при желании такие соединения могут быть приготовлены или выделены с получением чистых стереоизомеров, т.е. одиночных энантиомеров или диастереоизомеров, или смесей одиночных стереоизомеров с обогащенной (≥98%, ≥95%, ≥93%, ≥90%, ≥88%, ≥85% или ≥80% чистотой). Единичный стереоизомер соединения получают синтетически из спинового исходного материала, содержащего желаемый хиральный центр, или получают путем приготовления смеси энантиомерных продуктов с последующим выделением или расщеплением, например, превращением в смесь диастереоизомеров с последующим выделением или перекристаллизацией, хроматографической обработкой, использованием хиральных расщепляющих реагентов или прямым энантиомерным разделением на хиральной колонке. Разделение. Исходные соединения с определенной стереохимией либо коммерчески доступны, либо могут быть приготовлены, как описано ниже, и затем разделены методами, хорошо известными в данной области.
Термин "изомер" (или "изомерная форма") относится к структурным изомерам с различными энергиями, которые могут превращаться друг в друга через низкоэнергетические барьеры. Если изомерия возможна (например, в растворе), то можно достичь химического равновесия между изомерами. Например, изомеры протонной интеркаляции (или изомеры интеркаляции с переносом протона) включают (но не ограничиваются ими) взаимопревращения через миграцию протонов, такие как изомеризация кето-енола, изомеризация имино-енамина, изомеризация амида-иминола и т.п. Если не указано иное, все межконверсионные изомерные формы соединений настоящего изобретения входят в объем настоящего изобретения.
Термин "сольват" относится к веществу, образующемуся при соединении соединения изобретения или его фармацевтически приемлемой соли по меньшей мере с одной молекулой растворителя под действием нековалентных межмолекулярных сил. Обычные соединения растворителя включают, но не ограничиваются ими, гидраты, соединения этанола, ацетона и т.п.
Термин "хелат" представляет собой комплекс с циклической структурой, полученный путем хелатирования двух или более лигандов с одним ионом металла с образованием хелатного кольца.
Термин "нековалентный комплекс" образуется при взаимодействии соединения с другой молекулой, при этом между соединением и молекулой не образуется ковалентной связи. Комплексы могут образовываться, например, за счет ван-дер-ваальсовых взаимодействий, водородных связей и электростатических взаимодействий (также известных как ионная связь).
Термин "пролекарство" относится к производному соединению, которое прямо или косвенно доставляет соединение изобретения после его введения пациенту. Особенно предпочтительными производными соединениями или пролекарствами являются соединения, которые при введении пациенту повышают биодоступность соединений изобретения (например, легче всасываются в кровоток), или соединения, которые облегчают доставку исходного соединения к месту действия (например, в лимфатическую систему). Если не указано иное, все пролекарственные формы соединений настоящего изобретения входят в объем настоящего изобретения, а различные пролекарственные формы хорошо известны в данной области.
Термин "независимо друг от друга" означает, что по крайней мере две группы (или кольцевые системы), присутствующие в структуре в одинаковом или близком диапазоне значений, могут иметь одинаковые или разные значения в данной ситуации. Например, если заместитель X и заместитель Y независимо друг от друга являются водородом, галогеном, гидроксилом, циано, алкилом или арилом, то заместитель Y может быть водородом, галогеном, гидроксилом, циано, алкилом или арилом, когда заместитель X - водород; аналогично, заместитель X может быть водородом, галогеном, гидроксилом, циано, алкилом или арилом, когда заместитель Y - водород.
Термин "галоген" означает четыре атома: фтор (F), хлор (Cl), бром (Br) и йод (I).
Термин "алкил" включает прямые и разветвленные насыщенные алкильные группы. Например, алкильные группы включают, но не ограничиваются ими, метил, этил, пропил, н-бутил, изобутил, сек-бутил, трет-бутил, н-пентил и аналогичные группы. Термин "C1-8" в "C1-8 алкил" означает прямую или разветвленную группу, содержащую 1, 2, 3, 4, 5, 6, 7 или 8 атомов углерода.
Термин "алкенил" используется в настоящем документе для обозначения группы, имеющей и имеющей по меньшей мере один ненасыщенный участок алкенильной группы. Например, алкенильные группы включают, но не ограничиваются ими, винил, пропенил, аллил, изопропенил, бутенил, изобутенил и тому подобное. Термин "C2-8 алкенил" относится к алкенильной группе, содержащей 2, 3, 4, 5, 6, 7 или 8 атомов углерода в виде прямой, разветвленной или циклической цепи соответственно.
Термин "алкинил" относится в данном случае к группе, имеющей и имеющей по меньшей мере один ненасыщенный участок алкинильной группы. Например, алкильные группы включают, но не ограничиваются ими, этинил, пропаргил-1-ил, пропаргил-2-ил и тому подобные. "C2-8 алкинил" означает алкинильную группу, содержащую 2, 3, 4, 5, 6, 7 или 8 атомов углерода в виде прямой цепи, разветвленной цепи или циклической формы, соответственно.
Термин "алкилгалогенид" относится к алкильной группе, имеющей до полных валентных заместителей у атома галогена, которые могут быть одинаковыми или разными. Неограничивающие примеры галоалкилов включают -CF3, -C2F5, -CHF2, -CCL3, -CHCl2, -C2Cl5 и т.п. Термин "галоалкил C1-8" означает алкильную, алкенильную или алкинильную группу с 1, 2, 3, 4, 5, 6, 7 или 8 атомами углерода в прямой, разветвленной или циклической форме, содержащую до полного числа заместителей из атомов галогена.
Термин "арил" относится в данном случае к незамещенной или замещенной моноциклической или полициклической ароматической группе, состоящей из атомов углерода от 6 до 10 цельноуглеродных моноциклических или плотноупакованных полициклических (т.е. кольцо, имеющее общие соседние пары атомов углерода) групп. Например, гетероарильные группы включают, но не ограничиваются ими, фенил, 1-нафтил, 2-нафтил и подобные группы.
Термин "гетероарил" относится в данном случае к моноциклической или полициклической (например, имеющей два или три толстых кольца) ароматической части, которая является незамещенной или замещенной и имеет один или несколько гетероатомных членов кольца, независимо выбранных из N, S и O. Например, гетероарильные группы включают, но не ограничиваются ими, пиридинил, пиримидинил, пиразинил, пиридазинил, триазинил, фуранил, тиенил, имидазолил, тиазолил, индолил, пирролилокси, оксазолил, бензофуранил, бензотиофенил, бензотиазолил, изоксазолил, пиразолил, триазолил, тетразолил, индазолил, 1,2,4-тиадиазолил, изотиазолил, пуринил, карбазолил, бензимидазолил, индолинил, пирролил, азолил, хинолинил, изохинолинил, бензизоксазолил, имидазо[1,2-b]тиазолил и подобные группы.
Термин "циклоалкил" относится к неароматическим циклическим алкильным группам, имеющим моноциклическое или полициклическое кольцо (включая плотные, мостиковые и спирокольцевые системы), в том числе к циклизованным алкильным и алкенильным группам. Например, циклоалкил включает такие группы, как циклопропил, циклобутил, циклопентил, циклогексилгептил, циклопентенил, циклогексенил, циклогексадиенил, циклогептатриенил, норборнил, тетрагидронафталенил, октагидронафталенил, дигидронафталенил, норборнинил и т.п., но не ограничивается ими.
Термин "гетероциклоалкил" в настоящем документе относится к неароматическому кольцу или кольцевой системе, содержащей по меньшей мере один гетероатом, выбранный из группы, состоящей из O, N и S, и дополнительно содержащей одну или несколько алкенилиденовых или алкинилиденовых групп в составе кольцевой структуры. Гетероциклоалкильная группа в целом может иметь от 3 до 10 кольцевых атомов. Гетероциклоалкильная группа может быть ковалентно связана с определенной химической структурой у любого гетероатома или атома углерода, образующего стабильную структуру. Например, гетероциклоалкильные группы включают, но не ограничиваются ими: пирролидинил, пиперидинил, пиперазинил, тетрагидрофуранил, тетрагидропиранил, морфолинил, пиранил и тому подобное. Один или несколько атомов N или S в гетероциклоалкильной группе могут быть окислены (например, N-оксид морфолина, S-оксид тиоморфолина, S,S-диоксид тиоморфолина). Гетероциклическая алкильная группа может также содержать одну или несколько оксо-групп, например, о-фенилендиамин, пиперидинон, оксазолидинон, 2,4(1H,3H)-диоксо-пиримидинил, пиридин-2(1H)-он и другие группы.
Термин "алкиламино" относится к группе, имеющей формулу -NH(алкил). В некоторых вариантах осуществления изобретения алкиламиногруппа имеет от 1 до 8 атомов углерода. Неограничивающие примеры алкиламиногрупп включают метиламиногруппы, этиламиногруппы, пропиламиногруппы (например, н-пропиламиногруппы и изопропиламиногруппы) и т.п.
Термин "диалкиламино" относится к группе, имеющей формулу -N(алкил)2. Неограничивающие примеры диалкиламиногрупп включают диметиламиногруппы, диэтиламиногруппы, дипропиламиногруппы (например, ди(н-пропил)аминогруппы и ди(изопропил)аминогруппы) и т.п.
Термин "алкокси" используется в настоящем документе для обозначения алкильной группы, связанной с остальной частью молекулы атомом кислорода (-O-алкил), где указанная алкильная группа является такой, как определено в настоящем документе. Неограничивающие примеры алкокси включают метокси, этокси, трифторметокси, дифторметокси, н-пропокси, изопропокси, н-бутокси, трет-бутокси, н-пентилокси и тому подобное.
Термин "заместитель" в необязательной группе "C6-10 арилсодержащий заместитель" для R6 и термин "заместитель" в необязательной группе для R7 включают нециклические и циклические, разветвленные и неразветвленные, карбоциклические и гетероциклические, арильные и неароматические заместители органического соединения. Циклические и циклические, разветвленные и неразветвленные, карбоциклические и гетероциклические, ароматические и неароматические заместители органических соединений. Для подходящих органических соединений допускается наличие одного или нескольких заместителей, которые могут быть одинаковыми или разными. Неограничивающие примеры заместителей включают любой из описанных здесь заместителей, например, галоген, гидроксил, карбонил (например, карбоксил, алкоксикарбонил, формил или ацил), карбонил серы (например, тиоэфир, тиоацетат или тиокарбонат), алкил, алкенил, алкинил, алкокси, алкилгалоалкил, фосфорил, фосфатный эфир, фосфонат, фосфит, амидо, алкиламино, диалкиламино, амидо, амидино, имино, циано, нитро, азидо, меркапто, алкилсульфанил, сульфат, сульфонил, сульфаниламид, сульфонил, циклоалкил, гетероцикло, арил, арил, арил или гетероарил порции. Специалистам в данной области должно быть понятно, что замещенная часть углеводородной цепи сама может быть замещена при необходимости. Например, циклоалкил может быть дополнительно замещен алкилом, алкенилом, алкокси, алкилтио, аминоалкилом, углеродзамещенным алкилом, циано и т.п.; например, алкенил и алкинил могут быть аналогично замещены с получением аминоалкенила, аминоалкинила, амидоалкенила, амидоалкинила, иминоалкенила, иминоалкинила, тиоалкенила, тиоалкинила, углеродзамещенного алкенила или алкинила.
Термин "защитная группа" в "Защитная группа (PG), связанная с гидроксилом, амино, меркапто, карбоксилом и т.д." относится к защите гидроксила, амино, меркапто, карбоксила и т.д. от нежелательных реакций с помощью функциональной группы, причем в качестве защитных групп используются группы, известные специалистам в данной области, например, указанные в книге Protective Groups in Organic Synthesis (John Wiley & Sons, New York, 3rd ed. 1999). Термин "профилактика" относится к защите гидроксильных групп, аминогрупп, сульфгидрильных групп, карбоксильных групп и т.д. от нежелательных реакций с помощью функциональных групп, при этом используемые защитные группы известны специалистам в данной области, например, упомянутые в Protective Groups in Organic Synthesis (John Wiley & Sons, New York, 3rd ed., 1999).
Термин "профилактика" означает, например, полное или почти полное предотвращение развития заболевания или состояния (например, инфекции, ишемии или реперфузионного повреждения), когда пациент или субъект восприимчив или подвержен риску развития заболевания или состояния; профилактика может также включать ингибирование, т.е. остановку прогрессирования заболевания.
Термин "лечение" означает: 1) подавление заболевания; например, подавление заболевания, состояния или состояния у индивидуума, который испытывает или проявляет патологию или симптоматику заболевания, состояния или состояния (т.е. предотвращение дальнейшего прогрессирования патологии и/или симптоматики); или 2) облегчение заболевания; например, улучшение заболевания, состояния или состояния у индивидуума, который испытывает или проявляет патологию или симптоматику заболевания, состояния или состояния (т.е. обращение вспять патологии и/или симптоматики). или симптоматики заболевания, состояния или болезни у индивидуума, который испытывает или проявляет заболевание, состояние или болезнь (т.е. обращение вспять патологии и/или симптоматики).
Термин "терапевтически эффективное количество" означает количество активного соединения или агента, которое требуется исследователю, ветеринару, врачу или другому клиницисту, чтобы вызвать биологический или медицинский ответ в ткани, системе, животном, индивидууме или человеке.
В настоящем изобретении могут быть использованы следующие сокращения: (Boc)2O (ди-терт-бутил дикарбонат); DCM (дихлорметан); tBuOH (трет-бутанол); NaBH(OAc)3 (триацетоксиборогидрид натрия); DIPEA (N,N-диизопропилэтиламин); DMAP (4-диметиламинопиридин); DMF (N,N-диметилформамид); DMSO ( (диметилсульфоксид); EA или EtOAc (этилацетат); HOAc (уксусная кислота); LCMS или LC-MS (жидкостная хромато-масс-спектрометрия); TLC (тонкослойная хроматография); MeOH (метанол); NaOMe (метанол-натрий); NaH (гидрид натрия); Pd(dcpf)Cl2 (дихлоро[1,1'-бис(дициклогексилфосфино)ферроцен]палладий(II)); Pd2(dba)3 (трис(дибензилиденацетон)дипалладий); PdCl2(dppf)CH2Cl= ([1,1'-бис(дициклогексилфосфино)ферроцен]палладий(II) дихлорметановый комплекс); Pd(OAc)2 (ацетат палладия(II)); Pd(PPh3)4 (тетракис(трифенилфосфин)палладий); Pd(OH)2 (гидроксид палладия); DPPF (1,. 1'-бис(дифенилфосфино)ферроцен); Rt, r.t. или RT (комнатная температура); h, hr или hrs (часы); min (минуты); BnNH2 (бензиламин); TEA (триэтиламин): TFA (трифторуксусная кислота); BH3-THF (тетрагидрофурановый раствор борана); THF (тетрагидрофуран); NaOtBu (терт-бутанолят натрия); Cs2CO3 ( карбонат цезия); BrettPhos (дициклогексил[3,6-диметокси-2',4',6'-триизопропил[1,1'-бифенил]-2-ил]фосфин).
Исходные материалы для вариантов осуществления настоящего изобретения обычно являются коммерчески доступными, например, приобретаются у таких компаний, как Ballantine Vickers, Anergy Chemical, Aladdin, Bidet и т.д., или готовятся методами, хорошо известными специалистам в данной области. Замены, не соответствующие условиям реакции, очевидны для специалиста в данной области, поэтому альтернативные варианты указаны в тексте.
Коммерческие растворители и реагенты, используемые в исследованиях, применяются непосредственно без дополнительной очистки или обработки после приобретения, если не указано иное. Условия реакции (температура реакции, реакционные растворители, молярные соотношения реактантов и/или продолжительность реакции) могут варьироваться, если речь идет о других вариантах реализации или синтетических методах. В общем случае ход реакции может контролироваться методом ТЛК, что позволяет выбрать подходящее время для завершения реакции и последующей обработки. Условия очистки соединений также могут варьироваться. В общем случае выбор подходящего элюента для колоночной хроматографии основывается на значении Rf при ТЛХ или очистке соответствующих соединений методом препаративной ТЛХ.
[родовое соединение].
В настоящем изобретении представлены соединения формулы (I) или фармацевтически приемлемые соли, сложные эфиры, стереоизомеры, таутомеры, сольваты, хелаты, нековалентные комплексы или пролекарства, причем соединения формулы (I) имеют структурную формулу:
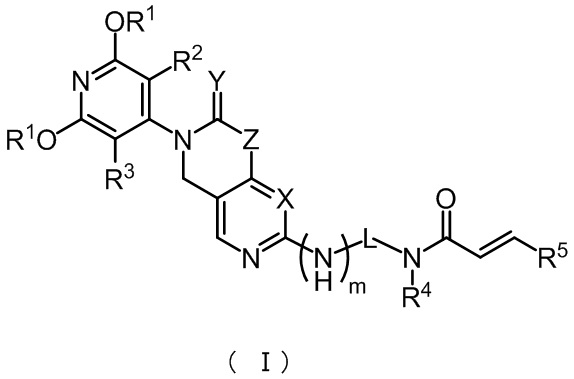 ,
,
где:
X представляет собой CH или N;
Y представляет собой 2 H или 1 O;
Z представляет собой C(R6)2 или N(R6);
m представляет собой 0 или 1;
L представляет собой -C(R7)2- или  ;
;
n представляет собой 0, 1 или 2;
Каждый R1 независимо представляет собой C1-3 алкил или C1-3 галоалкил;
R2 и R3 каждый независимо представляет собой водород, галоген, С1-3 алкил, С1-3 галоалкил, циано или С1-3 алкокси;
R4 представляет собой водород, С1-6 алкил или С6-10 арил;
R5 представляет собой водород, галоген, циано, нитро, трифторметил, амино, C1-6 алкил, C2-8 алкенил, C6-10 арил, Cl-8 алкиламино, ди(C2-8 алкил)амино, C2-8 алкинил, C1-8 галоалкил или C3-8 циклоалкил;
Каждый R6 независимо представляет собой водород, C1-8 алкил, C2-8 алкенил, C2-8 алкинил, C1-8 галоалкил, C6-10 арил, C6-10 арил, содержащий заместитель, C3-10 циклоалкил, 5-10-членный гетероарил или 4-10-членный гетероциклоалкил; при этом каждый из указанных 5-10-членных гетероарилов и 4-10-членных гетероциклоалкилов независимо включает от 1 до 3 кольцеобразующих гетероатомов, и каждый из указанных гетероатомов независимо является N, O или S;
Или два R6 и атомы углерода, к которым они присоединены, вместе образуют C3-8 циклоалкил или 4-10-членный гетероциклоалкил; где 4-10-членный гетероциклоалкил включает от 1 до 3 кольцеобразующих гетероатомов, причем гетероатомами являются N, O или S; и где каждый из C3-8 циклоалкила и 4-10-членного гетероциклоалкила независимо по выбору замещен от 1 до 4 групп, каждая из которых независимо представляет собой галоген, циано, гидроксил, аминогруппу, C1-8 карбоксиамидо, C1-8 карбоксамоил, C1-8 алкил или C1-8 алкокси группу;
Каждый R7 независимо представляет собой водород, галоген, амино, циано, C1-8 алкил, C1-8 алкил, содержащий заместитель, C1-8 алкокси, C1-8 алкокси, содержащий заместитель, C1-8 алкиламино, ди(C2-8 алкил)амино, C2-8 алкенил, C2-8 алкенил, содержащий заместитель, C2-8 алкинил, C2-8 алкинил, содержащий заместитель, C6-10 арил, C6-10 арил, содержащий заместитель, C3-8 циклоалкил, C3-8 циклоалкил, содержащий заместитель, 3-10-членный гетероциклоалкил, 3-10-членный гетероциклоалкил, содержащий заместитель, 5-10-членный гетероарил или 5-10-членный гетероарил, содержащий заместитель; при этом указанные 3-10-членный гетероциклоалкил и 5-10-членный гетероарил независимо включают от 1 до 3 кольцеобразующих гетероатомов и каждый из указанных гетероатомов независимо является N, O или S.
В некоторых конкретных вариантах осуществления настоящего изобретения каждое из указанных выше соединений формулы (I) R1 независимо представляет собой C1-3 алкил или C1-3 галоалкан, предпочтительно C1-3 алкил. При этом C1-3 алкил представляет собой метил, этил или пропил, предпочтительно метил; C1-3 галоалкан представляет собой -CF3, -C2F5, -CHF2, -CCL3, -CHCl2 или -C2Cl5.
В некоторых конкретных вариантах осуществления настоящего изобретения в указанных выше соединениях формулы (I) R2 и R3 независимо друг от друга представляют собой водород, галоген, C1-3 алкил, C1-3 галоалкил, циано или C1-3 алкокси, предпочтительно галоген. При этом галоген представляет собой -F, -Cl или -Br, предпочтительно -F или -Cl; C1-3 алкил представляет собой метил, этил или пропил; C1-3 галоалкил представляет собой -CF3, -C2F5, -CHF2, -CCl3, -CHCl2 или -C2Cl5; и C1-3 алкокси представляет собой метокси, этокси, н-пропилокси или изопропокси.
В некоторых конкретных вариантах осуществления настоящего изобретения в указанных соединениях формулы (I) R4 представляет собой водород, C1-6 алкил или C6-10 арил, предпочтительно H. При этом C1-6 алкил представляет собой метил, этил или пропил, а C6-10 арил представляет собой фенил, 1-нафтил или 2-нафтил.
В некоторых конкретных вариантах осуществления настоящего изобретения в указанных соединениях формулы (I) R5 представляет собой водород, галоген, циано, нитро, трифторметил, амино, C1-6 алкил, C2-8 алкенил, C6-10 арил, Cl-8 алкиламино, ди(C2-8 алкил)амино, C2-8 алкинил, C1-8 галоалкил или C3-8 циклоалкил, и предпочтительно водород. где галоген представляет собой -F, -Cl или -Br; C1-6 алкил - метил, этил или пропил; C2-8 алкенил - винил, пропенил, аллил или изопропенил; C6-10 арил - фенил, 1-нафталенил или 2-нафталенил; Cl-8 алкиламино группа - метиламино группа, этиламино группа или пропиламино группа; ди-(C2-8 алкил)амино группа - диметиламино группа, диэтиламино группа или дипропил амино; C2-8 алкинил - этинил, пропаргил-1-ил или пропаргил-2-ил; C1-8 галоалкил - -CF3, -C2F5, -CHF2, -CCl3, -CHCl2 или -C2Cl5; C3-8 циклоалкил - циклопропил, циклобутил, циклопентил или циклопентенил.
В некоторых конкретных вариантах осуществления настоящего изобретения каждый R6 в указанных соединениях формулы (I) независимо представляет собой водород, C1-8 алкил, C2-8 алкенил, C2-8 алкинил, C1-8 галоалкил, C6-10 арил, C6-10 арил, C3-10 циклоалкил, 5-10-членный гетероарил или 4-10-членный гетероциклоалкил, предпочтительно водород, C1-8 алкил, C6-10 арил или C3-10 циклоалкил. где C1-8 алкильная группа представляет собой метил, этил или пропил, предпочтительно метил или этил; C2-8 алкенильная группа представляет собой винил, пропенил, аллил или изопропенил; C2-8 алкинильная группа представляет собой этинил, пропаргил-1-ил или пропаргил-2-ил; C1-8 галоалкильная группа представляет собой -CF3, -C2F5, -CHF2, -CCL3, -CHCl2 или -C2Cl5; C6-10 арильная группа представляет собой фенил, 1-нафтил или 2-нафтил, предпочтительно фенил; С6-10 арил, содержащий заместитель, представляет собой замещенную фенильную, 1-нафтильную или 2-нафтильную группу, заместителем которой может быть галоген, гидроксил, карбонил (например, карбоксил, алкоксилкарбонил, карбонил или ацил), сероуглерод (например, серный эфир, сульфоацетил или сульфокарбоксилат), алкил, алкенил, алкинил, алкокси, галоалкил, фосфорил, фосфат, фосфонат, фосфит, амино, алкиламино, диалкиламино, амидо, амидино, имино, циано, нитро, азидо, меркапто, алкилтио, сульфат, сульфонат, сульфонил, сульфонамидо, сульфонил, циклоалкил, гетероцикло, арил, арилы или гетероарильные фрагменты; C3-10 циклоалкил - циклопропил, циклобутил или циклопентил, предпочтительно циклопропил; 5-10-членный гетероарил - пиридинил, пиримидинил, пиразинил, пиридазинил, триазинил, фуранил, тиенил, имидазолил, тиазолил, индолил, пирролил или оксазолил; 4-10-членный гетероциклический циклоалкил - пирролидинил, пиперидинил, пиперазинил, тетрагидрофурил, тетрагидропиранил, морфолино, пиранил, морфолин N -оксид, тиоморфолин S-оксид, тиоморфолин S,S-диоксид, фталимидо, пиперидинон-ил, оксазолидинон-ил, 2,4-(1H,3H)-диоксо-пиримидинил или пиридин-2(1H)-он-ил.
В некоторых вариантах осуществления настоящего изобретения в указанных соединениях формулы (I) два R6 и присоединенные к ним атомы углерода вместе образуют C3-8 циклоалкил или 4-10-членный гетероциклоалкил, предпочтительно C3-8 циклоалкил. где 4-10-членная гетероциклоалкильная группа включает от 1 до 3 кольцеобразующих гетероатомов, причем гетероатомами являются N, O или S. С3-8 циклоалкильная группа и 4-10-членная гетероциклоалкильная группа независимо друг от друга могут быть по выбору замещены от 1 до 4 заместителями, причем заместители независимо друг от друга являются галогеном, циано, гидрокси, амино, С1-8 карбоксамидо, С1-8 карбоксаноилом, С1-8 алкилом или С1-8 алкокси; где С3-8 циклоалкил представляет собой циклопропил, циклобутил или циклопентил, предпочтительно циклопропил; 4-10-членные гетероциклические алкильные группы - пирролидинил, пиперидинил, пиперазинил, тетрагидрофуранил, тетрагидропиранил, морфолинил, пиранил, морфолин N-оксид, тиоморфолин S-оксид, тиоморфолин S,S-диоксид, о-фенилендиамин, пиперидиновая группа, оксазолидиноновая группа, 2,4-(1H,3H)-диоксопиримидинил или пиридин-2(1H)-он; C1-8 карбоксамид С1-8 карбоксамидо - формамидо или ацетамидо; С1-8 карбоксил - формил или ацетил; С= алкил - метил, этил или пропил; С1-8 алкокси - метокси, этокси, трифторметокси, дифторметокси, н-пропокси, изопропокси, н-бутокси, трет-бутокси или н-пентилокси.
В некоторых вариантах осуществления настоящего изобретения каждый R7 в указанных соединениях формулы (I) представляет собой водород, галоген, амино, циано, C1-8 алкил, C1-8 алкил, содержащий заместители, C1-8 алкокси, Cl-8 алкокси, содержащий заместители, Cl-8 алкиламино, ди(C2-8 алкил)амино, C2-8 алкенил, C2-8 алкенил содержащий заместители, C2-8 алкинил, C2-8 алкинил содержащий заместители, C6-10 арил, C6-10 арил содержащий заместители, C3-8 циклоалкил, C3-8 циклоалкил содержащий заместители и 3- 10-членный гетероциклический алкил, заменитель, содержащий от 3 до 10-членного гетероциклоалкила, от 5 до 10-членного гетероарила или заместитель, содержащий от 5 до 10-членного гетероарила, предпочтительно водород, галоген, C1-8 алкил, C6-10 арил, заместитель, содержащий C6-10 арил, от 3 до 10-членного гетероциклоалкила или заместитель, содержащий от 3 до 10-членного гетероциклоалкила, более предпочтительно водород, C6-10 арил, заменитель, содержащий C6-10 арил, от 3 до 10-членного гетероциклоалкил или 3-10-членный гетероциклоалкил, содержащий заместители. где галоген представляет собой -F, -Cl или -Br; C1-8 алкил представляет собой метил, этил или пропил; а C1-8 алкокси представляет собой метокси, этокси, н-пропокси, изопропокси, н-бутокси, трет-бутокси или н-пентил; Cl-8 алкиламино - метиламино, этиламино или пропиламино; ди(C2-8 алкил)амино - диметиламино, диэтиламино или дипропиламино; C2-8 алкенил - винил, пропенил, аллил или изопропенил; C2-8 алкинил - этенил, пропаргил-1-ил или пропаргил-2-ил; C6-10 арил - фенил, 1-нафтил или 2-нафтил; C3-8 циклоалкил - циклопропил, циклобутил, циклопентил или циклопентенил; От 3 до 10-членных гетероциклоалкильных групп - пирролидинил, пиперидинил, пиперазинил, тетрагидрофуранил, тетрагидропиранил, морфолинил или пиранил; от 5 до 10-членных гетероарильных групп - пиридинил, пиримидинил, пиразинил, пиридазинил, триазинил, фуранил, тиофенил, имидазолил, тиазолил, индолил, пирролоксилил или оксазолил; где заместитель-содержащий C1-8 алкил, заместитель-содержащий Cl-8 алкокси, заместитель-содержащий C2-8 алкенил, заместитель-содержащий C2-8 алкинил, заместитель-содержащий C6-10 арил, заместитель-содержащий C3-8 циклоалкил, замещенный 3-10-членный гетероциклоалкил и замещенный 5-10-членный гетероарил представляют собой вышеупомянутые C1-8 алкил, Cl-8 алкокси, C2-8 алкенил, C2-8 алкинил, C6-10 арил, C3-8 циклоалкил, 3-10-членный гетероциклоалкил и 5-10-членный гетероарил, замещенные заместителем, соответственно, заместителем может быть галоген, гидроксил, карбонил (например, карбоксил, алкоксикарбон, формил или ацил), тиокарбонил (например, тиоэфир, тиоацетат или тиокарбонат), алкил, алкенил, алкинил, алкокси, галоалкил, фосфорил, фосфатно-эфирная группа, фосфонатно-эфирная группа, фосфитно-эфирная группа, амино, алкиламино, диалкиламино, амидо, амидино, имино, циано, нитро, азидо, меркапто, алкилтио, сульфат, сульфонат, аминосульфонил, сульфонамидо, сульфонил, циклоалкильная, гетероциклическая, арильная, арильная или гетероарильная часть, предпочтительно алкильная или гетероциклическая группа.
В некоторых конкретных вариантах осуществления настоящего изобретения указанное соединение формулы (I) имеет структуру, представленную в формуле (II):
,
где X, Y, Z, m, L, R2 и R3 определены, как определено в формуле (I).
В некоторых конкретных вариантах осуществления настоящего изобретения указанное соединение формулы (II) имеет структуру, представленную в формуле (III):
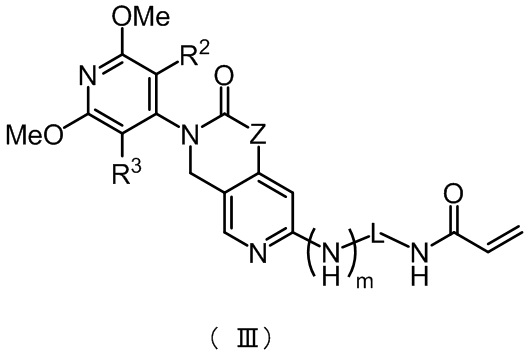 ,
,
где Z, m, L, R2 и R3 определены, как определено в формуле (II).
В некоторых конкретных вариантах осуществления настоящего изобретения указанное соединение формулы (III) имеет структуру, представленную в формуле (III-1):
 ,
,
где Z, L, R2 и R3 определены, как определено в формуле (III).
В некоторых конкретных вариантах осуществления настоящего изобретения указанное соединение формулы (III-1) имеет структуру, представленную в формуле (III-1-1):
 ,
,
где R2, R3 и R6 определены, как определено в формуле (III-1). а R7 представляет собой водород, C1-4 алкил, пиперазинил, пиперидинил или морфолинил, причем указанные C1-4 алкил, пиперазинил, пиперидинил и морфолинил по выбору замещены по меньшей мере одним R8, каждый из которых независимо друг от друга представляет собой водород, морфолинил, мостиковый морфолинил, пиперазинил, заместитель, содержащий пиперазинил, мостиковый пиперазинил, заместитель, содержащий пиперазинил, мостиковый пиперазинил, заместитель, содержащий морфолинил. Мостиковый пиперазинил, оксетанил или заместитель, содержащий оксетанил, причем указанный заместитель представляет собой C1-4 алкил.
В некоторых конкретных вариантах осуществления настоящего изобретения соединения формулы (III-1-1), где R2, R3 и R6 определены так, как определено в формуле (III-1), являются соединениями формулы (III-1).а R7 представляет собой водород, C1-4 алкил, пиперазинил, пиперидинил или морфолинил, причем указанные C1-4 алкил, пиперазинил, пиперидинил и морфолинил по выбору замещены по меньшей мере одним R8, каждый из которых независимо друг от друга представляет собой водород, морфолинил, мостиковый морфолинил, пиперазинил, заместитель, содержащий пиперазинил, мостиковый пиперазинил, заместитель, содержащий пиперазинил, мостиковый пиперазинил, заместитель, содержащий морфолинил. Мостиковый пиперазинил, оксетанил или заместитель, содержащий оксетанил, причем указанный заместитель представляет собой C1-4 алкил.
В некоторых конкретных вариантах осуществления настоящего изобретения соединения формулы (III-1-1), R+, R3 и R6 определены, как определено в формуле (III-1), и R7 представляет собой один из следующих фрагментов:
.
В некоторых конкретных вариантах осуществления настоящего изобретения указанное соединение формулы (III) имеет структуру, представленную в формуле (III-2):
 ,
,
где Z, L, R2 и R3 определены, как определено в формуле (III).
В некоторых конкретных вариантах осуществления настоящего изобретения указанное соединение формулы (III-2) имеет структуру, представленную в формуле (III-2-1):
 ,
,
где R2, R3 и R6 определены, как определено в формуле (III-2).
В некоторых конкретных вариантах осуществления настоящего изобретения указанное соединение формулы (III-2) имеет структуру, представленную в формуле (III-2-2):
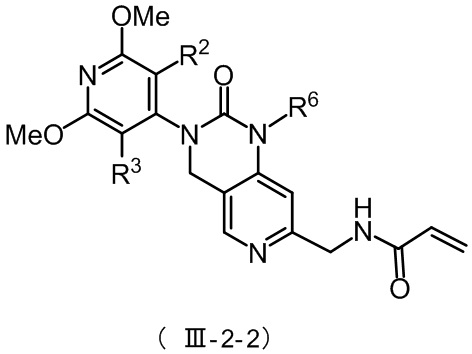 ,
,
где R2, R3 и R6 определены, как определено в формуле (III-2).
В некоторых конкретных вариантах осуществления настоящего изобретения указанное выше соединение формулы (II) имеет структуру, представленную в формуле (IV):
,
где Z, m, L, R2 и R3 определены, как определено в формуле (II).
В некоторых конкретных вариантах осуществления настоящего изобретения указанное соединение формулы (IV) имеет структуру, представленную в формуле (IV-1):
,
где Z, L, R2 и R3 определены, как определено в формуле (IV).
В некоторых конкретных вариантах осуществления настоящего изобретения соединение формулы (IV-1) имеет структуру, представленную в формуле (IV-1-1):
,
где R2, R3 и R6 определены, как определено в формуле (IV-1), а R7 представляет собой водород, C1-4 алкил, пиперазинил, пиперидинил или морфолинил, причем указанные C1-4 алкил, пиперазинил, пиперидинил и морфолинил по выбору замещены по меньшей мере одним R8, каждый из которых независимо друг от друга представляет собой водород, морфолинил, мостиковый морфолинил, пиперазинил, заместитель, содержащий пиперазинил, мостиковый пиперазинил, заместитель, содержащий пиперазинил, мостиковый пиперазинил, заместитель, содержащий морфолинил. Мостиковый пиперазинил, оксетанил или заместитель, содержащий оксетанил, причем указанный заместитель представляет собой C1-4 алкил.
В некоторых конкретных вариантах осуществления настоящего изобретения соединения формулы (IV-1-1), где R2, R3 и R6 определены, как определено в формуле (IV-1), а R7 представляет собой один из следующих фрагментов:
.
В некоторых конкретных вариантах осуществления настоящего изобретения указанное соединение формулы (IV) имеет структуру, представленную в формуле (IV-2):
,
где Z, L, R2 и R3 определены, как определено в формуле (IV).
В некоторых конкретных вариантах осуществления настоящего изобретения указанное соединение формулы (IV-2) имеет структуру, представленную в формуле (IV-2-1):
,
где R2, R3 и R6 определены, как определено в формуле (IV-2), а R7 представляет собой водород, C1-4 алкил или.
В некоторых конкретных вариантах осуществления настоящего изобретения указанное соединение формулы (IV-2) имеет структуру, представленную в формуле (IV-2-2):
,
где R2, R3 и R6 определены, как определено в формуле (IV-2).
В некоторых более предпочтительных вариантах осуществления настоящего изобретения соединение представляет собой любое из следующих:
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,и。
[фармацевтическая композиция]
Настоящее изобретение также предоставляет фармацевтическую композицию, включающую описанное выше соединение или фармацевтически приемлемую соль, сложный эфир, стереоизомер, таутомер, сольват, хелат, нековалентный комплекс или пролекарство.
В некоторых вариантах осуществления настоящего изобретения описанная выше фармацевтическая композиция дополнительно включает фармацевтически приемлемый носитель или разбавитель.
В некоторых предпочтительных вариантах осуществления настоящего изобретения указанная фармацевтическая композиция дополнительно включает:
-фармацевтически приемлемый носитель; и/или
-эксципиент.
Термин "фармацевтически приемлемый носитель" означает фармацевтический эксципиент, совместимый с активным ингредиентом лекарственного средства и не представляющий опасности для субъекта, включая, но не ограничиваясь этим, разбавители (или наполнители), связующие вещества, дезинтегранты, смазывающие вещества, смачивающие агенты, загустители, вспомогательные вещества, усилители вкуса, усилители запаха, консерванты, антиоксиданты, pH-адаптеры, растворители, сорастворители и поверхностно-активные вещества. , сорастворители и поверхностно-активные вещества и т.д.
Некоторые примеры подходящих вспомогательных веществ включают лактозу, декстрозу, сахарозу, сорбит, маннит, крахмал, камедь арабика, фосфат кальция, альгинат, камедь астрагала, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, сироп и метилцеллюлозу.
[Фармацевтический препарат].
Настоящее изобретение представляет собой фармацевтический препарат, включающий вышеуказанные соединения или фармацевтически приемлемые соли, сложные эфиры, стереоизомеры, таутомеры, сольваты, хелаты, нековалентные комплексы или пролекарства, или фармацевтические композиции из них.
В некоторых вариантах осуществления настоящего изобретения фармацевтический препарат представляет собой любое из таблеток, капсул, инъекций, гранул, порошка, суппозиториев, пилюль, гелей, дисперсий, растворов для перорального применения, лекарственных форм для ингаляции, суспензий или сухих суспензий.
[Фармацевтическое применение]
Либо соединения, описанные выше, либо фармацевтически приемлемые соли, сложные эфиры, стереоизомеры, таутомеры, сольваты, хелаты, нековалентные комплексы или пролекарства, либо фармацевтические композиции, либо фармацевтические препараты, как описано выше, которые проявляют ингибирующую активность в отношении FGFR4, Настоящее изобретение предоставляет соединения, описанные выше, либо фармацевтически приемлемые соли, стереоизомеры, таутомеры, сольваты, хелаты, из указанных выше, нековалентные комплексы или пролекарства, или фармацевтические композиции, как описано выше, или фармацевтические препараты, как описано выше, которые используются в качестве ингибиторов FGFR4.
Настоящее изобретение также предусматривает применение указанных соединений или фармацевтически приемлемых солей, сложных эфиров, стереоизомеров, таутомеров, сольватов, хелатов, нековалентных комплексов или пролекарств, или фармацевтических композиций, как описано выше, или фармацевтических препаратов, как описано выше, для получения препаратов для профилактики и/или лечения заболеваний, опосредованных, по крайней мере частично, FGFR4.
В некоторых вариантах осуществления настоящего изобретения заболевание, по крайней мере частично опосредованное FGFR4, включает рак.
В некоторых вариантах осуществления настоящего изобретения рак выбран из гепатоцеллюлярной карциномы, рака мочевого пузыря, рака молочной железы, рака шейки матки, колоректального рака, рака эндометрия, рака желудка, рака головы и шеи, рака почек, гепатоцеллюлярной карциномы, рака легких, рака яичников, рака простаты, рака пищевода, рака желчного пузыря, рака поджелудочной железы, рака щитовидной железы, рака кожи, лейкоза, множественной миеломы, хронической лимфобластной лимфомы, Т-клеточного лейкоза взрослых, В-клеточной лимфомы, острого миелоидного лейкоза миелоидный лейкоз, лимфома Ходжкина или неходжкинская лимфома, макроглобулинемия Вальденстрема, волосатоклеточная лимфома, лимфома Буркитта, нейроглиобластома, меланома, мезотелиома, нейробластома у взрослых, рак яичка, плоскоклеточная карцинома, глиобластома и рабдомиосаркома.
[Способ лечения]
Настоящее изобретение также предоставляет способ профилактики и/или лечения заболевания, опосредованного, по крайней мере частично, FGFR4, включающий стадии: введение профилактически и/или терапевтически эффективного количества соединения, описанного выше, или фармацевтически приемлемой соли, сложного эфира, стереоизомера, таутомера, сольвата, хелата, нековалентного комплекса или пролекарства, описанного выше, или фармацевтической композиции, описанной выше, или фармацевтического препарата, описанного выше, пациенту, нуждающемуся в этом.
Количество соединения, фармацевтической композиции или фармацевтического препарата, вводимого пациенту, зависит от вводимого препарата, цели введения (например, профилактической или терапевтической), состояния пациента, способа введения и т.п. При терапевтическом применении композиция может быть назначена пациенту, уже страдающему от заболевания, в количестве, достаточном для лечения или, по крайней мере, частичного подавления симптомов заболевания и его осложнений. Терапевтически эффективное количество зависит от состояния заболевания, которое лечится, и, по мнению лечащего врача, зависит от таких факторов, как тяжесть заболевания, возраст, вес и общее состояние пациента.
Настоящее изобретение также предоставляет способ профилактики и/или лечения рака, включающий стадии введения профилактически и/или терапевтически эффективного количества описанного выше соединения или фармацевтически приемлемой соли, сложного эфира, стереоизомера, таутомера, сольвата, хелата, нековалентного комплекса или пролекарства описанного выше соединения или описанных выше фармацевтических композиций или описанных выше фармацевтических препаратов, а также по меньшей мере одного дополнительного терапевтического агента для лечения рака нуждающемуся в этом пациенту.
В некоторых вариантах осуществления настоящего изобретения рак выбран из гепатоцеллюлярной карциномы, рака мочевого пузыря, рака молочной железы, рака шейки матки, колоректального рака, рака эндометрия, рака желудка, рака головы и шеи, рака почек, гепатоцеллюлярной карциномы, рака легких, рака яичников, рака простаты, рака пищевода, рака желчного пузыря, рака поджелудочной железы, рака щитовидной железы, рака кожи, лейкоза, множественной миеломы, хронической лимфобластной лимфомы, Т-клеточного лейкоза взрослых, В-клеточной лимфомы, острого миелоидного лейкоза миелоидный лейкоз, лимфома Ходжкина или неходжкинская лимфома, макроглобулинемия Вальденстрема, волосатоклеточная лимфома, лимфома Буркитта, нейроглиобластома, меланома, мезотелиома, нейробластома у взрослых, рак яичка, плоскоклеточная карцинома, глиобластома и рабдомиосаркома.
[комбинированная лекарственная форма]
Настоящее изобретение предоставляет комбинированную лекарственную форму, включающую вышеуказанные соединения или фармацевтически приемлемые соли, сложные эфиры, стереоизомеры, таутомеры, сольваты, хелаты, нековалентные комплексы или пролекарства вышеуказанных соединений, или фармацевтическую композицию, включающую вышеуказанные соединения, или фармацевтический препарат, включающий вышеуказанные соединения, а также по меньшей мере один дополнительный терапевтический агент для терапии рака.
[Способ получения]
Настоящее изобретение предлагает способ получения указанных соединений или фармацевтически приемлемых солей, сложных эфиров, стереоизомеров, таутомеров, сольватов, хелатов, нековалентных комплексов или пролекарств, а примерный синтетический маршрут для соединений общей формулы (I) описан ниже в порядке дальнейшего описания технического воплощения настоящего изобретения.
Схема получения промежуточного продукта A5
Промежуточное соединение A5 может быть получено по следующей схеме, конкретный синтетический маршрут выглядит следующим образом:
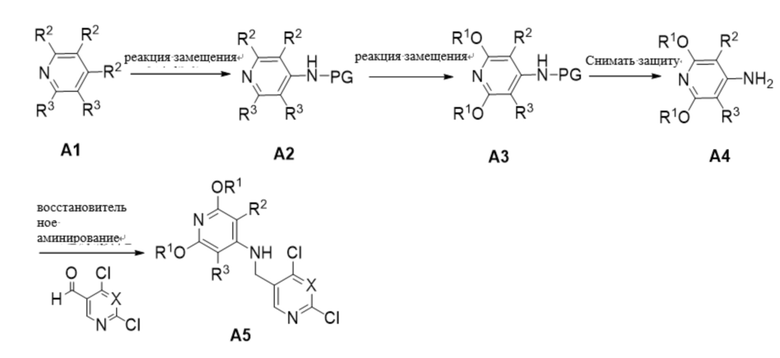
В приведенной схеме исходный материал А1 реагирует с первичным амином с защитной группой с получением промежуточного продукта А2, который затем замещается алкоксигруппой с получением промежуточного продукта А3, который депротеируется с получением промежуточного продукта А4, который реагирует с 4,6-дихлорникотинальдегидом (Х - CH) или 2,4-дихлорпиримидин-5-карбоксальдегидом (Х - N) с получением промежуточного продукта А5, где исходный материал А1 является коммерчески доступным.
Схема получения промежуточного продукта A8
Промежуточное соединение A8 было синтезировано по следующей схеме:

Промежуточное соединение А6 может быть получено обработкой промежуточного соединения А5 этилмалонилхлоридом в щелочных условиях. В сильных щелочных условиях промежуточное соединение A6 подвергается внутримолекулярному кольцеобразованию с образованием промежуточного соединения A7, а затем декарбоксилированию под действием кислоты (например, HCl) с получением промежуточного соединения A8.
Схема получения соединения A13
Соединение А13 было синтезировано по следующей схеме:
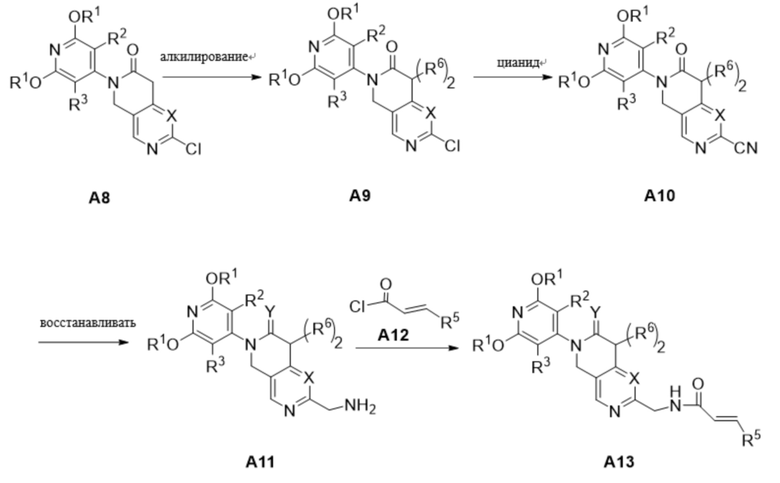
В основных условиях промежуточный продукт A8 может быть прореагирован с R6X (X - галогенированная группа, например Cl, Br или I) с получением R6-содержащего промежуточного продукта A9 (в зависимости от синтетических потребностей решается, требуется ли алкилирование, если алкилирование не требуется, то следующая шаг реакции проводится напрямую). При использовании Zn(CN)2 в качестве реактива и палладия в качестве катализатора промежуточный продукт A9 превращается в промежуточный продукт A10. Цианогруппа в промежуточном продукте A10 может быть восстановлена в условиях гексагидрата хлорида никеля/NaBH4 для получения промежуточного продукта A11, в котором Y представляет собой атом кислорода, или при обработке промежуточного продукта A10 с BH3-THF углеродная группа и цианогруппа восстанавливаются одновременно для получения промежуточного продукта A11, в котором Y представляет собой два атома водорода. Промежуточное соединение A11 вступало в реакцию с ацилхлоридом A12 при низкой температуре с получением соединения A13.
Схема получения соединения A18
Соединение A18 было синтезировано по следующей схеме:
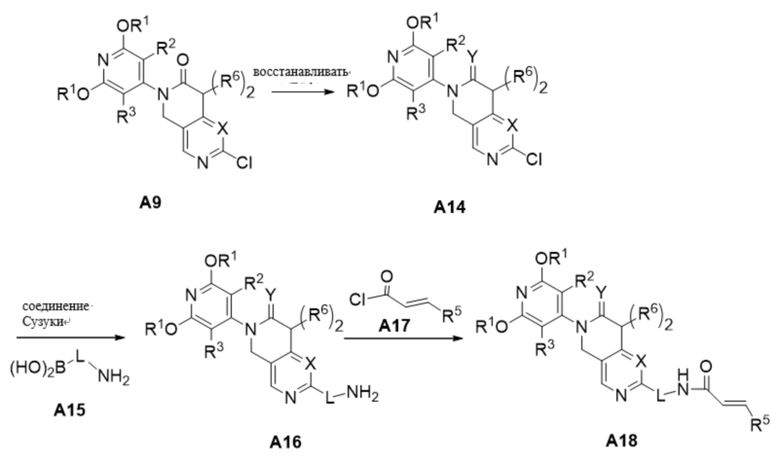
Соединение A9 обрабатывали BH3-THF с получением промежуточного продукта A14 (в зависимости от необходимости синтеза решали, нужна ли реакция восстановления, если реакция восстановления не нужна, то следующую реакцию проводили напрямую). Промежуточное соединение А14 соединяли по Сузуки с промежуточным соединением А15 при использовании [1,1'-бис(дифенилфосфино)ферроцен]дихлорида палладия в качестве катализатора с получением промежуточного соединения А16, которое при низкой температуре реагировало с ацилхлоридом А17 с получением соединения А18.
Схема получения соединения A25
Соединение A25 было синтезировано по следующей схеме:
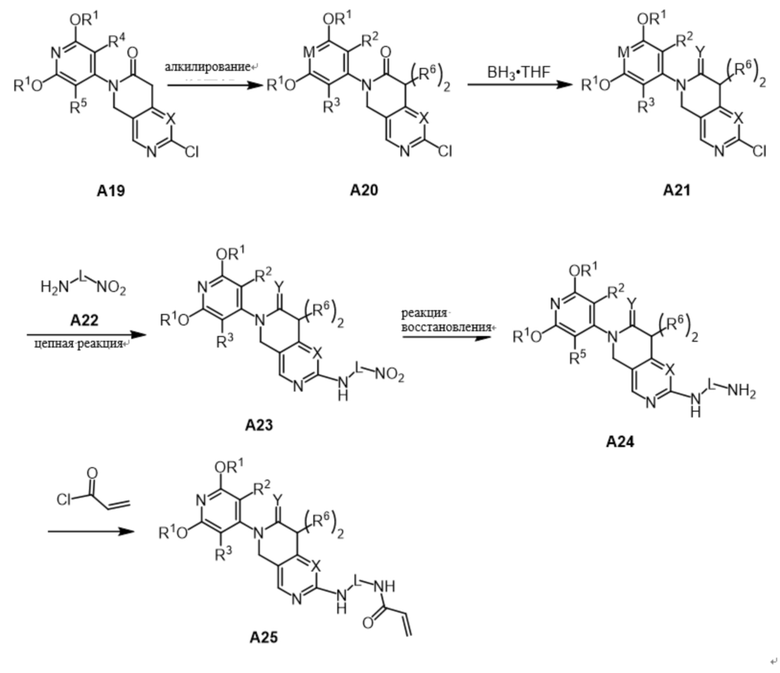
В основных условиях R6X (X - галогенированная группа, например Cl, Br или I) добавляют к раствору A19 для получения R6-содержащего соединения A20 (в зависимости от синтетических потребностей решают, требуется ли реакция алкилирования, и если реакция алкилирования не требуется, то следующую реакцию проводят непосредственно). При использовании BH3-THF в качестве восстановителя карбонильная группа A20 может быть восстановлена до метиленовой с получением соединения A21 (если реакция восстановления не требуется, то этот этап пропускается и следующая реакция проводится напрямую). Соединение A21 соединяется с A22 с получением соединения A23, а нитрогруппа в соединении A23 может быть восстановлена с получением A24, которое может быть получено капельным добавлением ацилхлорида к раствору A24 при низкой температуре.
Схема получения соединения A30
Соединение A30 было синтезировано по следующей схеме:
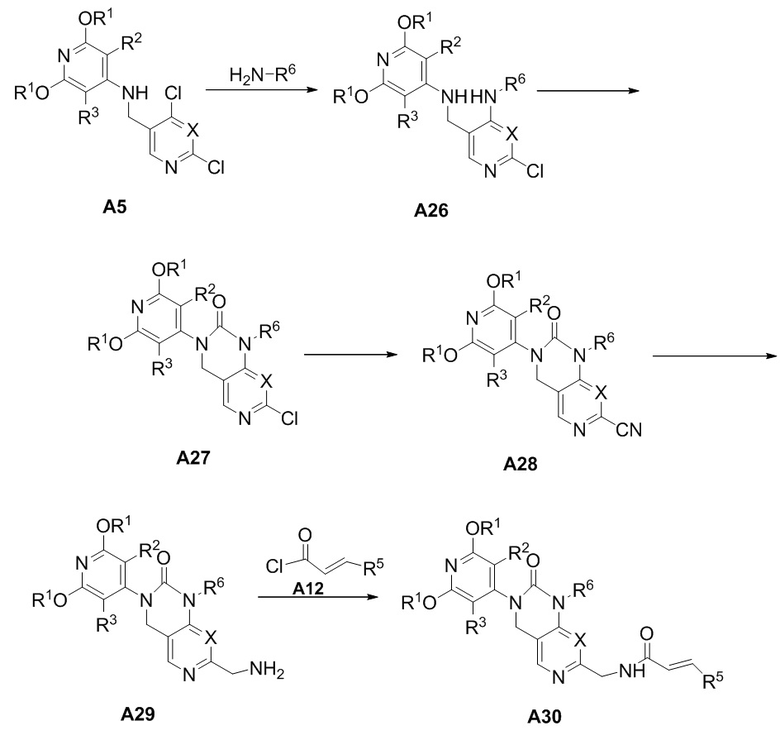
В присутствии палладия в качестве катализатора A5 соединялся с амином с получением соединения A26, которое под действием трифосгена образовывало внутримолекулярное кольцо с получением соединения A27, которое под действием Zn(CN)2 в качестве реагента и палладия в качестве катализатора превращалось в промежуточное соединение A28, промежуточное соединение A28 восстанавливается до промежуточного соединения A29, которое при низкой температуре реагирует с ацилхлоридом A12 и дает соединение A30.
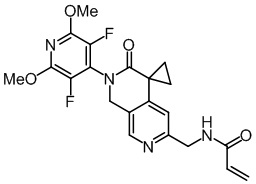
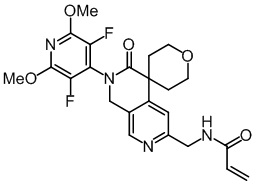
Пример 1: Синтез N-((2'-(3,5-дифтор-2,6-диметоксипиридин-4-ил)-3'-оксо-2',3'-дигидро-1'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-6'-ил)метил)акриламида (Соединение 1)
Синтетический маршрут был следующим:
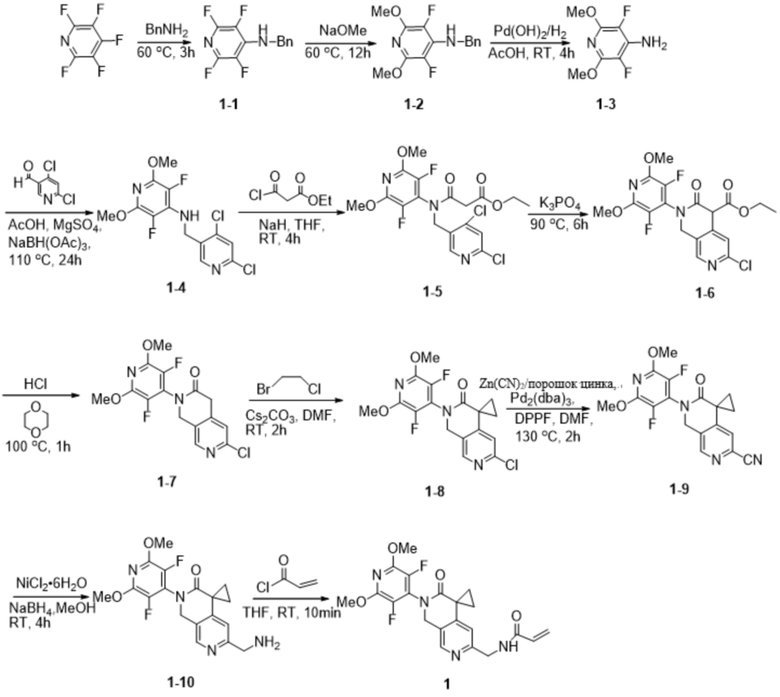
Шаг 1: Синтез промежуточного соединения 1-1
К ацетонитрилу (150 мл) при перемешивании при комнатной температуре добавили пентафторпиридин (3,6 г, 21,30 ммоль), затем бензиламин (4,5 г, 42,00 ммоль) и продолжали перемешивание при комнатной температуре в течение 1 ч. Реакцию нагрели до 60 °С. Через 3 ч реакцию охладили до комнатной температуры и сконцентрировали под пониженным давлением. К концентрату добавляли DCM (100 мл) и воду (100 мл), растворяли при перемешивании, разделяли и собирали органическую фазу. Органическую фазу однократно промывали насыщенным раствором NaCl (100 мл), сушили безводным Na2SO4 и концентрировали с получением белого твердого продукта (5,3 г, 20,69 ммоль), выход: 97,1%.
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 257,1 [M+H]+ (расчетное значение 257,1, C12H8F4N2).
Шаг 2: Синтез промежуточных соединений 1-2
В безводный метанол (250 мл) при комнатной температуре при перемешивании добавили метанол натрия (28,5 г, 527,54 ммоль), затем промежуточное соединение 1-1 (9,0 г, 35,13 ммоль) и нагрели полученную смесь до 60 °C для проведения реакции, через 12 ч охладили до комнатной температуры, погасили ледяной уксусной кислотой (31,6 г, 526,23 ммоль) и провели реакцию. Реакционный раствор концентрировали, к остатку добавляли воду (600 мл) и DCM (300 мл), растворяли при перемешивании, разделяли, собирали органическую фазу, водную фазу снова экстрагировали DCM (300 мл), органические фазы объединяли, промывали водой (3 х 300 мл), сушили безводным Na2SO4 и концентрировали с получением желаемого продукта (9,5 г, 33,89 ммоль) с выходом 96,5%. 96.5%.
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 281,1 [M+H]+ (расчетное значение 281,1, C14H14F2N2O2).
Шаг 3: Синтез промежуточных соединений 1-3
Промежуточные соединения 1-2 (3,0 г, 10,71 ммоль) добавляли к уксусной кислоте (60 мл) и перемешивали при комнатной температуре, затем добавляли Pd(OH)2 (0,6 г, 4,28 ммоль), который трижды вытесняли H2, защищали шаром H2 и перемешивали в течение 4 часов при комнатной температуре. Реакционный раствор фильтровали и концентрировали, получая желаемый продукт в виде ацетата (2,1 г, 8,60 ммоль), выход: 80,3%.
Идентификационные данные конкретного соединения приведены ниже:
LC-MS: m/z 191,1 [M+H]+ (расчетное значение 191,1, C7H8F2N2O2).
Шаг 4: Синтез промежуточных продуктов 1-4
При комнатной температуре к толуолу (70 мл) при перемешивании добавили ацетат (10,6 г, 42,37 ммоль) и 4,6-дихлорникотинальдегид (6,0 г, 34,09 ммоль) промежуточных соединений 1-3, затем добавили ледяную уксусную кислоту (3 мл) и безводный сульфат магния (24,0 г, 199,40 ммоль) и нагрели полученную смесь до 110°С для проведения реакции. Через 24 ч реакцию охлаждали до комнатной температуры, фильтровали и фильтрат концентрировали при пониженном давлении. К полученному концентрату добавили DCM (70 мл), перемешали до растворения, затем добавили трифторуксусную кислоту (11,7 г, 102,61 ммоль) и понизили температуру до -5~0°C. Затем реакционный раствор охладили до -5~0°C. В реакционный раствор медленно добавили триацетоксиборогидрид натрия (26,5 г, 125,04 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 4 ч. После завершения реакции ее гасили насыщенным водным раствором NH4Cl и экстрагировали DCM (3 х 100 мл), органические слои объединяли, сушили над Na2SO4 и концентрировали. Остаток очищали на силикагеле (элюируя гексаном, содержащим 0-5% (v/v) EtOAc) с получением желаемого продукта (10,3 г, 29,42 ммоль) с выходом 69,4%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 349,9 [M+H]+ (расчетное значение 350,0, C13H11Cl2F2N3O2).
Шаг 5: Синтез промежуточных соединений 1-5
Промежуточные соединения 1-4 (5,0 г, 14,28 ммоль) добавляли в тетрагидрофуран (25 мл) при перемешивании при комнатной температуре, затем добавляли NaH (60% масс. в минеральном масле, 600,0 мг, 15,0 ммоль). Через 10 мин по каплям добавили этилмалонилхлорид (2,64 мл, 18,68 ммоль). После завершения капельного добавления реакцию проводили в течение 4 ч. Реакцию гасили насыщенным водным NH4Cl и экстрагировали ЭА (2 х 100 мл), органические слои объединяли, сушили над Na2SO4 и концентрировали. Остаток очищали на силикагеле (элюируя гексаном, содержащим 0-20% (v/v) EtOAc) с получением желаемого продукта (5,7 г, 12,28 ммоль), выход: 86,0%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 463,9 [M+H]+ (расчетное значение 464,1, C18H17Cl2F2N3O5).
Шаг 6: Синтез промежуточных соединений 1-6
К промежуточным соединениям 1-5 (3,0 г, 6,46 ммоль) при комнатной температуре добавляли DMF (100 мл) и растворяли при перемешивании, затем добавляли фосфат калия (9,0 г, 42,40 ммоль), после чего полученную смесь нагревали до 90 °C. Через 6 ч реакцию охлаждали до комнатной температуры, гасили добавлением 2%-ной водной уксусной кислоты (100 мл) и экстрагировали ЭА (2 х 100 мл), органические слои объединяли, сушили над Na2SO4 и концентрировали. Остаток очищали на силикагеле (элюируя гексаном, содержащим 0-20% (v/v) EtOAc) с получением желаемого продукта (1,5 г, 3,51 ммоль) с выходом: 54,3%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 428,0 [M+H]+ (расчетное значение 428,1, C18H16ClF2N3O5).
Шаг 7: Синтез промежуточных соединений 1-7
Промежуточные соединения 1-6 (120 мг, 0,28 ммоль) добавляли в 1,4-диоксан (6 мл) при перемешивании при комнатной температуре, затем добавляли концентрированную соляную кислоту (4 мл) и нагревали полученную смесь до 100 °С. Через 1 ч реактивы охлаждали до комнатной температуры, гасили насыщенным водным NaHCO3 и экстрагировали ЭА (2 х 100 мл), органические слои объединяли, сушили при помощи Na2SO4, сушили и концентрировали. Остаток очищали на силикагеле (элюируя гексаном, содержащим 0-25% (v/v) EtOAc) с получением желаемого продукта (62,3 мг, 0,18 ммоль) с выходом: 62,5%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 356,0 [M+H]+ (расчетное значение 356,1, C15H12ClF2N3O3).
Шаг 8: Синтез промежуточных соединений 1-8
Промежуточные соединения 1-7 (2,6 г, 7,31 ммоль) добавляли в DMF (45 мл) при перемешивании при комнатной температуре, затем последовательно добавляли карбонат цезия (5,2 г, 15,95 ммоль) и 1-бром-2-хлорэтан (12,1 мл, 140,48 ммоль). Через 2 ч реакцию гасили насыщенным водным NH4Cl и органический слой экстрагировали EA (2 × 100 мл). 100 мл), органические слои объединили, высушили над Na2SO4 и сконцентрировали. Остаток очищали на силикагеле (элюируя гексаном, содержащим 0-20% (v/v) EtOAc) с получением желаемого продукта (2,0 г, 5,23 ммоль) с выходом: 71,7%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 382,0 [M+H]+ (расчетное значение 382,1, C17H14ClF2N3O3).
Шаг 9: Синтез промежуточных соединений 1-9
Промежуточные соединения 1-8 (150 мг, 0,39 ммоль), цианид цинка (100 мг, 0,85 ммоль), порошок цинка (14,3 мг, 0,22 ммоль), Pd2(dba)3 (40 мг, 0,04 ммоль) смешивали с DPPF (47 мг, 0,08 ммоль) при 130 °С в атмосфере N2 в N,N-. Диметилформамиде (3 мл) в реакционной смеси перемешивали в течение 2 часов. Реакцию охлаждали до комнатной температуры, гасили насыщенным водным NaHCO3 и экстрагировали этилацетатом (3 х 50 мл), объединенные органические слои промывали рассолом, сушили Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали на силикагеле (элюируя гексаном, содержащим 0-20% (v/v) EtOAc) с получением желаемого продукта (115 мг, 0,31 ммоль) с выходом 79,2%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 373,0 [M+H]+ (расчетное значение 373,1, C18H14F2N4O3).
Шаг 10: Синтез промежуточных продуктов 1-10
Промежуточные соединения 1-9 (60 мг, 0,16 ммоль), гексагидрат хлорида никеля (7,7 мг, 0,03 ммоль) с трифторуксусной кислотой (147 мг, 1,29 ммоль) добавляли в безводный метанол (6 мл) под атмосферой N2 при комнатной температуре, затем медленно добавляли борогидрид натрия (182,9 мг, 4,83 ммоль). После перемешивания в течение 4 часов при комнатной температуре реакционный раствор был отфильтрован и сконцентрирован с получением желаемого продукта (54,0 мг, 0,14 ммоль), выход: 87,5%.
Идентификационные данные конкретного соединения приведены ниже:
LC-MS: m/z 377,1 [M+H]+ (расчетное значение 377,1, C18H18F2N4O3).
Шаг 11: Синтез соединения 1
К перемешиваемому раствору промежуточных соединений 1-10 (54,0 мг, 0,14 ммоль) и тетрагидрофурана (6 мл) при 0~5°С в атмосфере N2 добавили акрилоилхлорид (12,6 мг, 0,14 ммоль), нагрели до комнатной температуры, перемешивали в течение десяти минут, затем промыли насыщенным водным раствором NaHCO3 и экстрагировали этилацетатом (3 х 50 мл), объединенные органические слои промыли рассолом, высушили над Na2SO4 и отфильтровали. Объединенные органические слои промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали на силикагеле (элюируя гексаном, содержащим 0-75% (v/v) EtOAc) с получением желаемого продукта (27 мг, 0,06 ммоль) с выходом 44,8%.
Идентификационные данные конкретных соединений следующие:
1Н NMR (500 МHz, DMSO-d6): δ 8.60 (t, J = 5.8 Hz, 1H), 8.37 (s, 1H), 6.93 (s, 1H), 6.33 (dd, J = 17.1, 10.2 Hz, 1H), 6.12 (dd, J = 17.1, 2.1 Hz, 1H), 5.62 (dd, J = 10.2, 2.2 Hz, 1H), 5.01 (s, 2H), 4.42 (d, J = 5.8 Hz, 2H), 3.99 (s, 6H), 1.77 (q, J = 4.0 Hz, 2H), 1.48 (q, J = 4.1 Hz, 2H).
LC -МС: m/z 431,0 [M+H]+ (расчетное значение 431,2, C21H20F2N4O4).
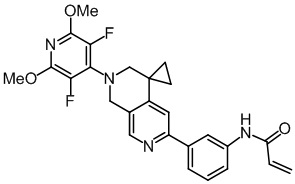
Пример 2: Синтез N-(3-(2'-(3,5-дифтор-2,6-диметоксипиридин-4-ил)-2',3'-дигидро-1'H-спиро[циклопропил-1,4'-[2,7]нафтиридин]-6'-ил)фенил)акриламида (соединение 2)
Синтетический маршрут был следующим:
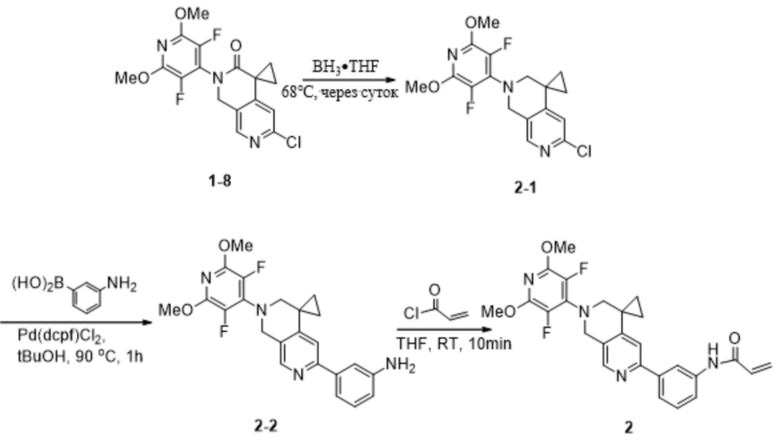
Шаг 1: Синтез промежуточного продукта 2-1
Промежуточное соединение 1-8 (300 мг, 0,79 ммоль) добавляли к боран-тетрагидрофурановому комплексу (7 мл) при 68 °С и перемешивали в течение ночи. Реакцию охлаждали до комнатной температуры, гасили метанолом, добавляли водную соляную кислоту (1,3 мл, 1 моль/л), вновь нагревали до 68 °C и перемешивали в течение 1 часа. Реакцию охлаждали до комнатной температуры, гасили насыщенным водным раствором NaHCO3 и экстрагировали этилацетатом (2 х 15 мл), органические фазы объединяли, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали на силикагеле (элюируя гексаном, содержащим 0-10% (v/v) EtOAc) с получением желаемого продукта (110 мг, 0,30 ммоль) с выходом 38,0%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 368,0 [M+H]+ (расчетное значение 368,1, C17H16ClF2N3O2).
Шаг 2: Синтез промежуточного продукта 2-2
Промежуточное соединение 2-1 (60 мг, 0,16 ммоль), 3-аминофенилбороновую кислоту (27 мг, 0,20 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорид палладия (18 мг, 0,02 ммоль) добавляли к трет-бутанолу (6 мл) и воде ( 6 мл) и перемешивали в течение 1 часа. Реакционный раствор гасили насыщенным водным NH4Cl и экстрагировали DCM (3 х 10 мл), органические слои объединяли, сушили над Na2SO4 и концентрировали с получением желаемого продукта (55,0 мг, 0,13 ммоль) с выходом 81,3%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 425,1 [M+H]+ (расчетное значение 425,2, C23H22F2N4O2).
Шаг 3: Синтез соединения 2
Промежуточное соединение 2-2 (55,0 мг, 0,13 ммоль) добавляли в тетрагидрофуран (10 мл) в атмосфере N2 при 0~5°C при перемешивании, затем добавляли акрилоилхлорид (11,1 мг, 0,12 ммоль), нагревали до комнатной температуры, перемешивали в течение десяти минут, гасили насыщенным водным раствором NaHCO3 и экстрагировали DCM (2×20 мл), объединенные органические слои промывали рассолом, сушили Na2SO4, фильтровали и концентрировали при пониженном давлении. Объединенные органические слои промывали рассолом, сушили Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали препаративным выделением с получением желаемого продукта (44 мг, 0,09 ммоль) с выходом 70,7%.
Идентификационные данные конкретных соединений приведены ниже:
1H NMR (500 MHz, DMSO-d6): δ 10.26 (s, 1H), 8.41 (s, 1H), 8.29 (t, J = 2.0 Hz, 1H), 7.87-7.83 (m, 1H), 7.75 (dt, J = 8.0, 1.3 Hz, 1H), 7.41 (t, J = 7.9 Hz, 1H), 7.24 (s, 1H), 6.46 (dd, J = 17.0, 10.1 Hz, 1H), 6.28 (dd, J = 17.0, 2.0 Hz, 1H), 5.77 (dd, J = 10.1, 2.0 Hz, 1H), 4.70 (s, 2H), 3.90 (s, 6H), 3.48 (s, 2H), 1.23 (t, J = 4.5 Hz, 2H), 1.07 (q, J = 4.5 Hz, 2H).
LC-MS: m/z 479,1 [M+H]+ (расчетное значение 479,2, C26H24F2N4O3).
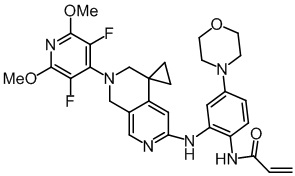
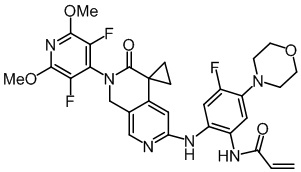
Пример 3: Синтез N-(2-((2'-(3,5-дифтор-2,6-диметоксипиридин-4-ил)-2',3'-дигидро-1'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-6'-ил)амино)-5-морфолинофенил)акриламида (соединение 3)
Синтетический маршрут был следующим:
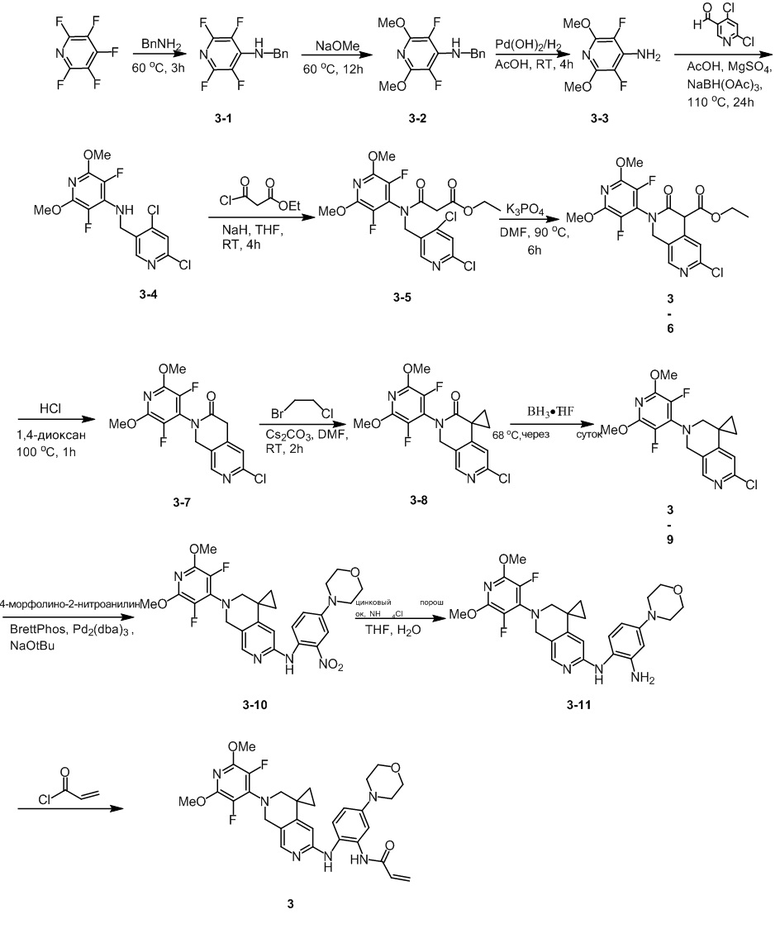
Шаг 1: Синтез промежуточного соединения 3-1
К ацетонитрилу (300 мл) при перемешивании при комнатной температуре добавили пентафторпиридин (7,2 г, 42,59 ммоль), затем бензиламин (9,0 г, 83,99 ммоль) и перемешивали при комнатной температуре в течение 1 ч, после чего нагрели до 60 °С. Через 3 ч реакцию охладили до комнатной температуры и сконцентрировали под пониженным давлением. К концентрату добавляли DCM (200 мл) и воду (200 мл), растворяли при перемешивании, наслаивали и собирали органическую фазу. Органическую фазу однократно промывали насыщенным раствором NaCl (200 мл), сушили безводным Na2SO4 и концентрировали с получением белого твердого продукта (10,6 г, 41,37 ммоль), выход: 97,1%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 257,1 [M+H]+ (расчетное значение 257,1, C12H8F4N2).
Шаг 2: Синтез промежуточного соединения 3-2
В безводный метанол (250 мл) при комнатной температуре при перемешивании добавили метанол натрия (28,5 г, 527,54 ммоль), затем соединение 3-1 (9,0 г, 35,13 ммоль) и нагрели полученную смесь до 60 °С. Через 12 ч реакцию охладили до комнатной температуры, погасили ледяной уксусной кислотой (31,6 г, 526,23 ммоль) и реакционный раствор сконцентрировали, к остатку добавили воду (600 мл) и DCM (300 мл). концентрировали, к остатку добавляли воду (600 мл) и DCM (300 мл), растворяли при перемешивании, разделяли, органическую фазу собирали, водную фазу экстрагировали еще раз DCM (300 мл), органические фазы объединяли, промывали водой (3 х 300 мл), затем сушили безводным Na2SO4 и концентрировали с получением желаемого продукта (9,5 г, 33,89 ммоль) с выходом 96,5%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 281,1 [M+H]+ (расчетное значение 281,1, C14H14F2N2O2).
Шаг 3: Синтез промежуточного соединения 3-3
Промежуточное соединение 3-2 (6,0 г, 21,41 ммоль) добавляли к уксусной кислоте (120 мл) и перемешивали при комнатной температуре, затем добавляли Pd(OH)2 (1,2 г, 8,55 ммоль), трижды заменяли его H2, защищали шаром H2 и перемешивали в течение 4 ч при комнатной температуре. Реакционный раствор отфильтровывали и концентрировали, получая желаемый продукт в виде ацетата (4,3 г, 17,19 ммоль) с выходом 80,3%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 191,1 [M+H]+ (расчетное значение 191,1, C7H8F2N2O2).
Шаг 4: Синтез промежуточных соединений 3-4
Ацетат Промежуточного соединения 3-3 (10,6 г, 42,37 ммоль) и 4,6-дихлорникотинальдегид (6,0 г, 34,09 ммоль) добавили в толуол (70 мл) и перемешивали при комнатной температуре, затем добавили ледяную уксусную кислоту (3 мл) и безводный сульфат магния (24,0 г, 199,40 ммоль) и нагрели полученную смесь до 110 °C. Через 24 ч охладили реакцию до комнатной температуры, отфильтровали и фильтрат сконцентрировали при пониженном давлении. К полученному концентрату добавили DCM (70 мл), перемешали до растворения, затем добавили трифторуксусную кислоту (11,7 г, 102,61 ммоль) и понизили температуру до -5~0°C. Затем реакцию охладили до 110°C. Через 24 ч реакцию отфильтровали, фильтрат сконцентрировали под пониженным давлением. В реакционный раствор медленно добавляли триацетоксиборогидрид натрия (26,5 г, 125,04 ммоль). Реакционный раствор перемешивали и проводили реакцию при комнатной температуре в течение 4 ч. Реакционный раствор гасили насыщенным водным раствором NH4Cl и экстрагировали DCM (3 х 100 мл). Органические слои объединили, высушили над Na2SO4 и сконцентрировали. Остаток очищали на силикагеле (элюируя гексаном, содержащим 0-5% (v/v) EtOAc) с получением желаемого продукта (10,3 г, 29,42 ммоль) с выходом 69,4%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 349,9 [M+H]+ (расчетное значение 350,0, C13H11Cl2F2N3O2).
Шаг 5: Синтез промежуточных соединений 3-5
Промежуточное соединение 3-4 (5,0 г, 14,28 ммоль) добавляли в тетрагидрофуран (25 мл) при комнатной температуре при перемешивании, затем добавляли NaH (60% масс. в минеральном масле, 600,0 мг, 15,0 ммоль). Через 10 мин по каплям добавляли этилмалонилхлорид (2,64 мл, 18,68 ммоль). Через 4 ч реакцию гасили насыщенным водным NH4Cl и экстрагировали ЭА (2 х 100 мл), органические слои объединяли, сушили над Na2SO4 и концентрировали. Остаток очищали на силикагеле (элюируя гексаном, содержащим 0-20% (v/v) EtOAc) с получением желаемого продукта (5,7 г, 12,28 ммоль) с выходом: 86,0%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 463,9 [M+H]+ (расчетное значение 464,1, C18H17Cl2F2N3O5).
Шаг 6: Синтез промежуточных соединений 3-6
Промежуточные соединения 3-5 (3,0 г, 6,46 ммоль) добавляли в DMF (100 мл) при перемешивании при комнатной температуре, затем добавляли фосфат калия (9,0 г, 42,40 ммоль) и нагревали полученную смесь до 90 °С. Через 6 ч реакцию охлаждали до комнатной температуры, гасили добавлением 2%-ной водной уксусной кислоты (100 мл) и экстрагировали ЭА (2 х 100 мл), органические слои объединяли, сушили Na2SO4 и концентрировали. Остаток очищали на силикагеле (элюируя гексаном, содержащим 0-20% (v/v) EtOAc) с получением желаемого продукта (1,5 г, 3,51 ммоль) с выходом: 54,3%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 428,0 [M+H]+ (расчетное значение 428,1, C18H16ClF2N3O5).
Шаг 7: Синтез промежуточных соединений 3-7
Промежуточные соединения 3-6 (120 мг, 0,28 ммоль) перемешивали в 1,4-диоксане (6 мл) при комнатной температуре, затем добавляли концентрированную соляную кислоту (4 мл) и нагревали полученную смесь до 100 °С. Через 1 ч реактивы охлаждали до комнатной температуры, гасили насыщенным водным NaHCO3 и экстрагировали ЭА (2 х 100 мл), органические слои объединяли, сушили при помощи Na2SO4, сушили и концентрировали. Остаток очищали на силикагеле (элюируя гексаном, содержащим 0-25% (v/v) EtOAc) с получением желаемого продукта (62,3 мг, 0,18 ммоль) с выходом: 62,5%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 356,0 [M+H]+ (расчетное значение 356,1, C15H12ClF2N3O3).
Шаг 8: Синтез промежуточных соединений 3-8
Промежуточное соединение 3-7 (2,6 г, 7,31 ммоль) добавляли в DMF (45 мл) при перемешивании при комнатной температуре, затем последовательно добавляли карбонат цезия (5,2 г, 15,95 ммоль) и 1-бром-2-хлорэтан (12,1 мл, 140,48 ммоль). Через 2 ч реакцию гасили насыщенным водным NH4Cl и органический слой экстрагировали EA (2 × 100 мл). 100 мл), органические слои объединили, высушили над Na2SO4 и сконцентрировали. Остаток очищали на силикагеле (элюируя гексаном, содержащим 0-20% (v/v) EtOAc) с получением желаемого продукта (2,0 г, 5,23 ммоль) с выходом: 71,7%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 382,0 [M+H]+ (расчетное значение 382,1, C17H14ClF2N3O3).
Шаг 9: Синтез промежуточных соединений 3-9
В закрытую пробирку, содержащую промежуточное соединение 3-8 (250 мг, 0,65 ммоль), при комнатной температуре вводили 1,0 моль/л боран-тетрагидрофуранового комплекса (5,3 мл, 5,3 ммоль). Полученную смесь нагревали до 67°С, выдерживали и перемешивали в течение 12 часов. Смесь гасили метанолом, добавляли раствор соляной кислоты (1 мл, 1 моль/л), нагревали до 67°С, выдерживали и перемешивали в течение 1 ч, затем охлаждали до комнатной температуры. pH доводили до pH ≥ 7 насыщенным водным NaHCO3 и экстрагировали ЭА (2 х 15 мл), органические слои объединяли, сушили Na2SO4 и концентрировали. Остаток очищали на силикагеле (элюируя гексаном, содержащим 0-10% (v/v) EtOAc) с получением желаемого продукта (134 мг, 0,36 ммоль) с выходом 55,6%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 368,0 [M+H]+ (расчетное значение 368,1, C17H16ClF2N3O2).
Шаг 10: Синтез промежуточных продуктов 3-10
Промежуточные продукты 3-9 (60 мг, 0,16 ммоль), 4-морфолино-2-нитроанилин (43,8 мг, 0,20 ммоль), 2-(дициклогексилфосфино)-3,6-диметокси-2'-4'-6'-триизопропил-11'-бифенил (9,0 мг, 0,02 ммоль), Pd2(dba)3 (15,0 мг, 0,02 ммоль) с трет-бутоксидом натрия (31,3 мг, 0,33 ммоль) добавляли в толуол. (15,0 мг, 0,02 ммоль) с трет-бутоксидом натрия (31,3 мг, 0,33 ммоль) добавляли в толуол (6 мл) и реакцию перемешивали в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры, фильтровали и концентрировали, остаток очищали на силикагеле (элюируя гексаном, содержащим 0-20% (v/v) EtOAc) с получением желаемого продукта (40 мг, 0,07 ммоль) с выходом 45,0%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 555,2 [M+H]+ (расчетное значение 555,2, C27H28F2N6O5).
Шаг 11: Синтез промежуточных соединений 3-11
Промежуточное соединение 3-10 (34 мг, 0,06 ммоль) добавляли в THF (4 мл) при перемешивании при комнатной температуре в атмосфере N2, затем последовательно добавляли порошок цинка (260,6 мг, 3,99 ммоль) и 9%-ный водный раствор хлорида аммония (2,1 мл). Через 1 ч реакционный раствор фильтровали, фильтрат сушили и концентрировали с Na2SO4 с получением желаемого продукта (27,2 мг, 0,05 ммоль). , 0,05 ммоль).
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 525,3 [M+H]+ (расчетное значение 525,2, C27H30F2N6O3).
Шаг 12: Синтез соединения 3
Промежуточное соединение 3-11 (27,2 мг, 0,05 ммоль) добавляли в тетрагидрофуран (3 мл) при 0~5°С в атмосфере N2 при перемешивании, затем добавляли акрилоилхлорид (4,53 мг, 0,05 ммоль), нагревали до комнатной температуры и перемешивали в течение 10 мин, затем гасили насыщенным водным раствором NaHCO3 и экстрагировали этилацетатом (3 х 50 мл), объединяли. Органический слой промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали на силикагеле (элюируя гексаном, содержащим 0-50% (v/v) EtOAc) с получением желаемого продукта (7 мг, 0,01 ммоль) с выходом 24,2%.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 579,2 [M+H]+ (расчетное значение 579,3, C30H32F2N6O4).
Пример 4: Синтез N-(2-((2'-(3,5-дифтор-2,6-диметоксипиридин-4-ил)-2',3'-дигидро-1'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-6'-ил)амино)-4-морфолинофенил)акриламида (Соединение 4)
Синтетический маршрут был следующим:
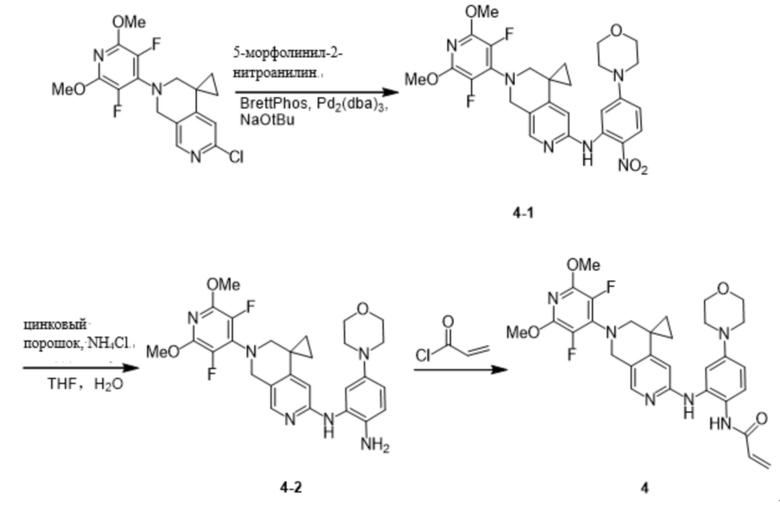
Шаг 1: Синтез промежуточного продукта 4-1
Аналогично методике Шага 10 Примера 3, было получено промежуточное соединение 4-1.
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 555,2 [M+H]+ (расчетное значение 555,2, C27H28F2N6O5).
Шаг 2: Синтез промежуточного продукта 4-2
Аналогично методике Шага 11 Примера 3 был получен промежуточный продукт 4-2.
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 525,3 [M+H]+ (расчетное значение 525,2, C27H30F2N6O3).
Шаг 3: Синтез соединения 4
Аналогично методике шага 12 примера 3 было получено соединение 4.
Идентификационные данные конкретных соединений приведены ниже:
1H NMR (500 MHz, DMSO-d6): δ 9.64 (s, 1H), 7.98 – 7.94 (m, 2H), 7.39 (d, J = 8.8 Hz, 1H), 7.24 (d, J = 2.8 Hz, 1H), 6.71 (dd, J = 8.8, 2.8 Hz, 1H), 6.47 (dd, J = 17.0, 10.2 Hz, 1H), 6.26 (dd, J = 17.0, 2.0 Hz, 1H), 6.23 (s, 1H), 5.75 (dd, J = 10.2, 2.0 Hz, 1H), 4.59 (s, 2H), 3.95 (s, 6H), 3.79 (d, J = 4.4 Hz, 4H), 3.46 (s, 2H), 3.14 – 3.08 (m, 4H), 1.08 – 1.03 (m, 2H), 0.99 – 0.94 (m, 2H).
LC-MS: m/z 579.2 [M+H]+ (расчетное значение 579.3, C30H32F2N6O4).
Пример 5: Синтез N-(2-((2'-(3,5-дифтор-2,6-диметоксипиридин-4-ил)-3'-оксо-2',3'-дигидро-1'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-6'-ил)-амино)-5-(4-морфолинопиперидин-1-ил)фенил)акриламида (соединение 5)
Синтетический маршрут следующий:
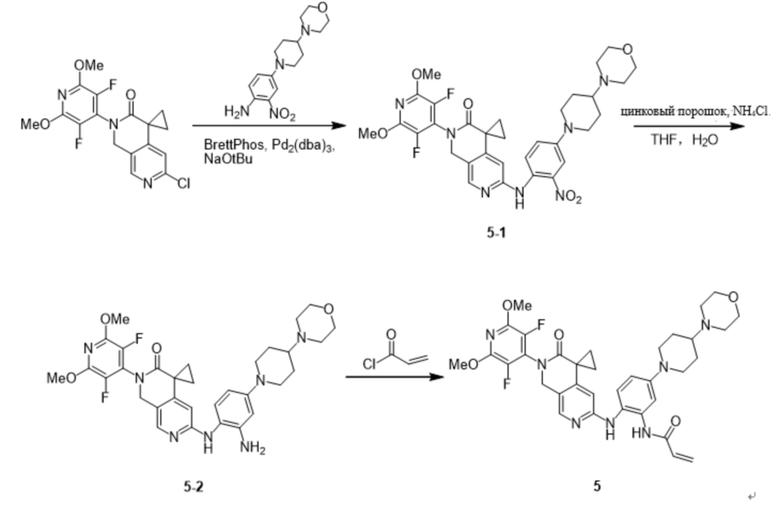
Шаг 1: Синтез промежуточного продукта 5-1
Аналогично методике примера 3, шаг 10, был получен промежуточный продукт 5-1.
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 652,2 [M+H]+ (расчетное значение 652,3, C32H35F2N7O6).
Шаг 2: Синтез промежуточного продукта 5-2
Аналогично методике Шага 11 Примера 3 был получен промежуточный продукт 5-2.
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 622,3 [M+H]+ (расчетное значение 622,3, C32H37F2N7O4).
Шаг 3: Синтез соединения 5
Аналогично методике шага 12 примера 3 было получено соединение 5.
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 676,2 [M+H]+ (расчетное значение 676,3, C35H39F2N7O5).
Пример 6: Синтез N-(2-((2'-(3,5-дифтор-2,6-диметоксипиридин-4-ил)-3'-оксо-2',3'-дигидро-1'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-6'-ил)-амино)-4-(4-морфолинопиперидин-1-ил)фенил)акриламида (соединение 6)
Синтетический маршрут следующий:
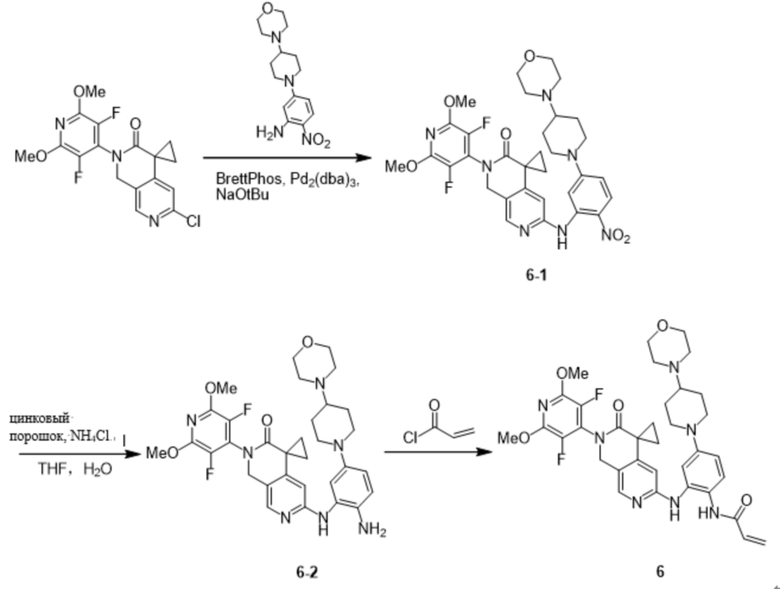
Шаг 1: Синтез промежуточного продукта 6-1
Аналогично методике примера 3, шаг 10, был получен промежуточный продукт 6-1.
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 652,2 [M+H]+ (расчетное значение 652,3, C32H35F2N7O6).
Шаг 2: Синтез промежуточного продукта 6-2
Аналогично методике Шага 11 Примера 3 был получен промежуточный продукт 6-2.
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 622,2 [M+H]+ (расчетное значение 622,3, C32H37F2N7O4).
Шаг 3: Синтез соединения 6
Аналогично методике Шага 12 Примера 3 было получено соединение 6.
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 676,2 [M+H]+ (расчетное значение 676,3, C35H39F2N7O5)
Пример 7: Синтез N-(2-((2'-(3,5-дифтор-2,6-диметоксипиридин-4-ил)-3'-оксо-2',3'-дигидро-1'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-6'-ил)амино)фенил)акриламида (Соединение 7)
Синтетический маршрут выглядит следующим образом:
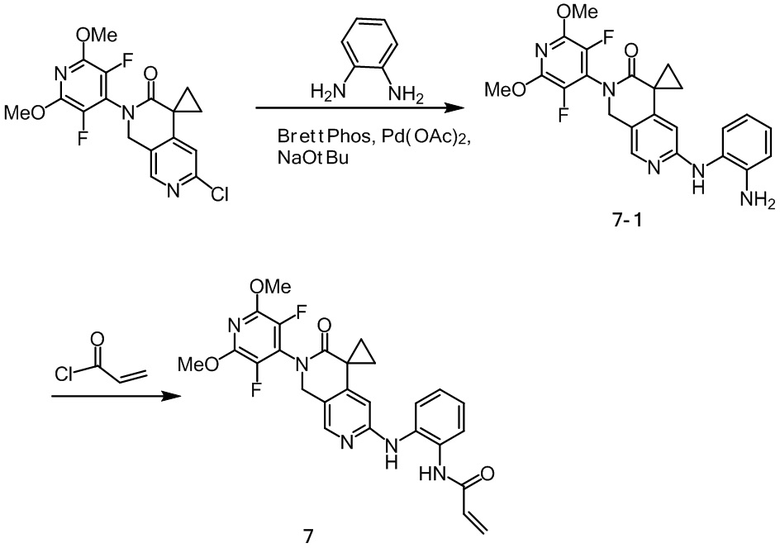
Шаг 1: Синтез промежуточного продукта 7-1
Под защитой N2 к 6'-хлор-2'-(2,6-дифтор-3,5-диметоксипиридин)-1',2'-дигидро-3'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-3'-ону (200 мг, 0,52 ммоль), 1,2-фенилендиамину (60 мг, 0,55 ммоль), ацетату палладия (25 мг, 0,11 ммоль), бретфосу (59 мг, 0,11 ммоль) и трет-бутоксиду натрия (200 мг, 2,08 ммоль) добавляли, бретфос (59 мг, 0.11 ммоль) и трет-бутоксид натрия (200 мг, 2.08 ммоль) добавляли в безводный диоксан (10 мл), и реакцию нагревали в микроволновой печи при 110°С в течение 1 ч. После завершения реакции отбирали, фильтровали, промывали ЭА (5 мл) 2 раза, собирали фильтрат, концентрировали и разделяли тонкослойной хроматографией с получением олеилидена 7-1 (80 мг).
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 454,1 [M+H]+ (расчетное значение 454,2, C23H21F2N5O3).
Шаг 2: Синтез соединения 7
Промежуточное соединение 7-1 (80 мг) растворяли в 3 мл сухого THF и при охлаждении до 0 °С по каплям добавляли 10 мкл акрилоилхлорида, реакцию проводили в течение 10 мин, гасили добавлением 0,1 мл воды и после выпаривания растворителя остаток подвергали препаративному выделению и очистке с получением желаемого продукта 7 (5,6 мг).
Идентификационные данные конкретных соединений приведены ниже:
1H NMR (500 MHz, DMSO-d6): δ 9.71 (s, 1H), 8.22 (s, 1H), 7.99 (s, 1H), 7.66 (d, J = 8.1 Hz, 1H), 7.57 (d, J = 7.9 Hz, 1H), 7.15 (td, J = 7.7, 1.6 Hz, 1H), 7.05 (td, J = 7.6, 1.5 Hz, 1H), 6.48 (dd, J = 17.0, 10.2 Hz, 1H), 6.30 (s, 1H), 6.24 (dd, J = 17.0, 2.0 Hz, 1H), 5.73 (dd, J = 10.2, 2.0 Hz, 1H), 4.88 (s, 2H), 3.98 (s, 6H), 1.69 (q, J = 4.0 Hz, 2H), 1.38 (q, J = 4.1 Hz, 2H).
LC-MS: m/z 508,2 [M+H]+ (расчетное значение 508,2, C26H23F2N5O4).
Пример 8: Синтез N-(2-((2'-(3,5-дифтор-2,6-диметоксипиридин-4-ил)-3'-оксо-2',3'-дигидро-1'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-6'-ил)амино)-3-метилфенил)акриламида (Соединение 8)
Синтетический маршрут был следующим:
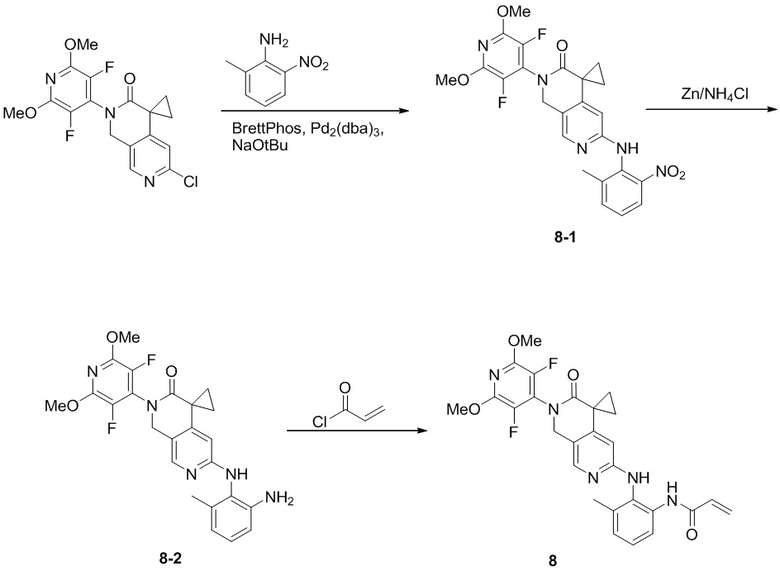
Шаг 1: Синтез промежуточного продукта 8-1
В атмосфере N2 в качестве исходного вещества использовали 6'-хлор-2'-(2,6-дифтор-3,5-диметоксипиридин)-1',2'-дигидро-3'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-3'-он (100 мг, 0,26 ммоль), 2-метил-6-нитроанилин (47 мг, 0,31 ммоль), трет-бутанолат натрия (50 мг. 0,52 ммоль), трет-бутоксид натрия (50 мг, 0,52 ммоль) и смесь бретфоса (14 мг, 0,026 ммоль) и Pd2(dba)3 (24 мг, 0,026 ммоль) в ультрасухом толуоле (5 мл) перемешивали при 110 °С в течение 1 ч. После завершения реакции по данным ТСХ температуру понизили, раствор разбавили добавлением ЭА (5 мл), отфильтровали через кизельгур, промыли ЭА (5 мл). 2 раза, фильтрат собирали и концентрировали досуха при пониженном давлении с получением красновато-коричневого масла, остаток очищали на силикагеле (элюируя гексаном, содержащим от 0 до 0-20% EtOAc) с получением Промежуточного продукта 8-1 (80 мг).
Идентификационные данные для конкретных соединений приведены ниже:
LC-MS: m/z 498,2 [M+H]+ (расчетное значение 498,2, C24H21F2N5O5).
Шаг 2: Синтез промежуточного продукта 8-2
К смеси растворителей тетрагидрофурана (6 мл) и воды (3 мл) при комнатной температуре добавляли промежуточное соединение 8-1 (80 мг, 0,16 ммоль), хлорид аммония (500 мг, 9,35 ммоль), порошок цинка (600 мг, 9,17 ммоль) и проводили реакцию при комнатной температуре в течение 1 ч. После завершения реакции с помощью TLC планшет фильтровали через кизельгур, фильтрат промывали THF (5 мл). Фильтрат собирали, подщелачивали насыщенным водным NaHCO3 и экстрагировали этилацетатом (3 х 10 мл). Органические фазы объединили, высушили и сконцентрировали с получением Промежуточного соединения 8-2 (60 мг).
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 468,3 [M+H]+ (расчетное значение 468,2, C24H23F2N5O3).
Шаг 3: Синтез соединения 8
К перемешиваемому раствору промежуточного соединения 8-2 (80 мг, 0,17 ммоль) в тетрагидрофуране (4,0 мл) при температуре 0-5 °С по каплям добавляли акрилоилхлорид (14 мкл, 0,17 ммоль). Через 5 мин реакционный раствор гасили насыщенным водным раствором NaHCO3 и экстрагировали дихлорметаном. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали до сухого состояния при пониженном давлении. Неочищенный продукт был очищен препаративным выделением с получением желаемого продукта 8 (40 мг).
Идентификационные данные конкретных соединений приведены ниже:
1H NMR (500 MHz, DMSO-d6): δ 9.43 (s, 1H), 7.87 (d, J = 4.3 Hz, 2H), 7.65 (d, J = 8.1 Hz, 1H), 7.15 (t, J = 7.8 Hz, 1H), 7.07 (d, J = 7.3 Hz, 1H), 6.45 (dd, J = 17.0, 10.2 Hz, 1H), 6.18 (dd, J = 17.0, 2.0 Hz, 1H), 5.90 (s, 1H), 5.67 (dd, J = 10.2, 2.0 Hz, 1H), 4.83 (s, 2H), 3.97 (s, 6H), 2.13 (s, 3H), 1.66 (q, J = 4.0 Hz, 2H), 1.27 (q, J = 4.1 Hz, 2H).
LC-MS: m/z 522,3 [M+H]+ (расчетное значение 522,2, C27H25F2N5O4).
Пример 9: Синтез N-(2-((2'-(3,5-дифтор-2,6-диметоксипиридин-4-ил)-3'-оксо-2',3'-дигидро-1'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-6'-ил)амино)-4-морфолинофенил)акриламида (соединение 9)
Синтетический маршрут был следующим:

Шаг 1: Синтез промежуточного продукта 9-1
В атмосфере N2 из смеси 6'-хлор-2'-(2,6-дифтор-3,5-диметоксипиридина)-1',2'-дигидро-3'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-3'-она (100 мг, 0,26 ммоль), 5-морфолино-2-нитроанилина (69 мг, 0,31 ммоль), трет-бутанолата натрия (50 мг. 0,52 ммоль) и бретфос (14 мг, 0,026 ммоль), Pd2(dba)3 (24 мг, 0,026 ммоль) в смеси с ультрасухим толуолом (5 мл) перемешивали при 110 °С в течение 1 ч. После завершения реакции, обнаруженного TLC, температуру понижали, разбавляли добавлением ЕА (5 мл), отфильтровывали кусочки кизельгура, фильтрат промывали ЕА (5 мл) 2 раза, фильтрат собирали и концентрировали до сухости при пониженном давлении с получением красновато-коричневого масла, которое очищали колоночной хроматографией (градиентное элюирование раствором н-гексана в 0-20% EtOAc) с получением промежуточного соединения 9-1 (80 мг).
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 569,2 [M+H]+ (расчетное значение 569,2, C27H26F2N6O6)
Шаг 2: Синтез промежуточного продукта 9-2
Промежуточное соединение 9-1 (80 мг, 0,14 ммоль) добавляли к смешанному растворителю тетрагидрофурана (6 мл) и воды (3 мл) при комнатной температуре, хлориду аммония (500 мг, 9,35 ммоль), порошку цинка (600 мг, 9,17 ммоль) и реагировали в течение 1 ч при комнатной температуре, затем отфильтровывали кусочки кизельгура, фильтрат дважды промывали THF (5 мл) и собирали фильтрат. Фильтрат собирали, подщелачивали насыщенным водным раствором NaHCO3, экстрагировали этилацетатом (3 х 10 мл), сушили и концентрировали, получая промежуточное соединение 9-2 (60 мг).
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 539,3 [M+H]+ (расчетное значение 539,2, C27H28F2N6O4).
Шаг 3: Синтез соединения 9
Аналогично методике шага 3 примера 8, идентификационные данные конкретных соединений для получения соединения 9 приведены ниже:
1H NMR (500 MHz, DMSO-d6): δ 9.60 (s, 1H), 8.15 (s, 1H), 8.00 (s, 1H), 7.34 (d, J = 8.8 Hz, 1H), 7.23 (d, J = 2.7 Hz, 1H), 6.68 (dd, J = 8.9, 2.7 Hz, 1H), 6.43 (dd, J = 17.0, 10.1 Hz, 1H), 6.30 (s, 1H), 6.21 (dd, J = 17.0, 2.1 Hz, 1H), 5.69 (dd, J = 10.1, 2.1 Hz, 1H), 4.87 (s, 2H), 3.98 (s, 6H), 3.73 (t, J = 4.7 Hz, 4H), 3.06 (t, J = 4.8 Hz, 4H), 1.69 (q, J = 4.0 Hz, 2H), 1.36 (q, J = 4.1 Hz, 2H).
LC-MS: m/z 593,3 [M+H]+ (расчетное значение 593,2, C30H30F2N6O5).
Пример 10: Синтез N-(2-((2'-(3,5-дифтор-2,6-диметоксипиридин-4-ил)-3'-оксо-2',3'-дигидро-1'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-6'-ил)амино)-5-морфолинофенил)акриламида (соединение 10)
Синтетический маршрут был следующим:
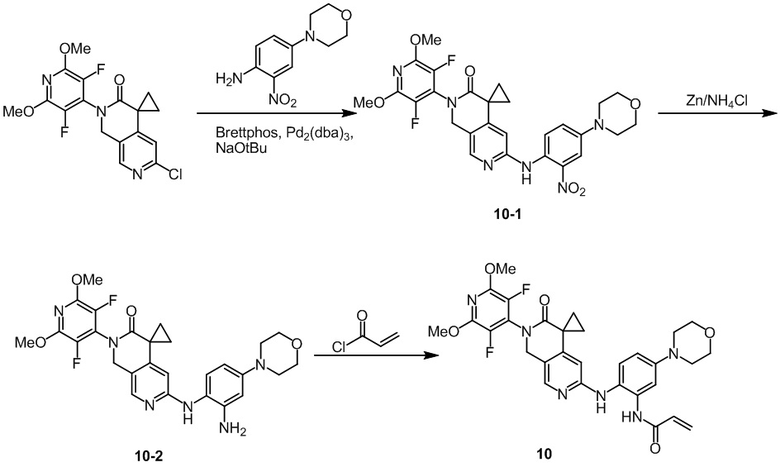
Шаг 1: Синтез промежуточного продукта 10-1
Под защитой азотом синтезировали 6'-хлор-2'-(2,6-дифтор-3,5-диметоксипиридин)-1',2'-дигидро-3'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-3'-он (130 мг, 0,35 ммоль), 4-морфолино-2-нитроанилин (100 мг, 0,45 ммоль), ацетат палладия (11,8 мг. 0,05 ммоль), ацетат палладия (11,8 мг, 0,05 ммоль), бретфос (28,1 мг, 0,05 ммоль) и трет-бутоксид натрия (134 мг, 1,4 ммоль) в ультрасухом толуоле (10 мл) реагировали в микроволновой печи при 130°С в течение 30 мин. После завершения реакции фильтрат отбирали и промывали дважды ЭА (10 мл), фильтрат собирали, концентрировали до сухости при пониженном давлении и очищали хроматографией. Получено промежуточное соединение 10-1 (47 мг, 0,08 ммоль).
Идентификационные данные конкретных соединений были следующими:
LC-MS m/z:569,3 [M+H]+ (расчетное значение 569,2, C27H26F2N6O6).
Шаг 2: Синтез промежуточного соединения 10-2
При комнатной температуре к метанолу (3 мл) добавили промежуточное соединение 10-2 (47 мг, 0,08 ммоль), порошок цинка (250 мг, 3,83 ммоль) и хлорид аммония (300 мг, 5,61 ммоль) и перемешивали в течение 2 ч. Отфильтровали, растворитель выпарили под пониженным давлением, добавили THF (3 мл) и снова выпарили под пониженным давлением, получив твердый неочищенный продукт промежуточного соединения 10-2 (55 мг).
Идентификационные данные конкретных соединений были следующими:
LC-MS m/z: 539,3 [M+H]+ (расчетное значение 539,2, C27H28F2N6O4).
Шаг3: Синтез соединения 10
Твердый остаток 10-2 (55 мг) растворяли в сухом THF (5 мл), охлаждали на ледяной бане, капельно добавляли 7 мкл акрилоилхлорида, реакцию проводили в течение 10 мин, гасили добавлением 0,1 мл воды и после выпаривания растворителя остаток использовали для получения 10 (13,62 мг), продукта, необходимого для выделения и очистки.
Идентификационные данные конкретных соединений приведены ниже:
1H NMR (500 MHz, DMSO-d6): δ 9.61 (s, 1H), 7.95 (s, 1H), 7.92 (s, 1H), 7.34 (d, J = 8.8 Hz, 1H), 7.27 (d, J = 2.9 Hz, 1H), 6.79 (dd, J = 8.9, 2.9 Hz, 1H), 6.45 (dd, J = 17.0, 10.2 Hz, 1H), 6.22 (dd, J = 17.0, 2.0 Hz, 1H), 6.10 (s, 1H), 5.71 (dd, J = 10.1, 2.0 Hz, 1H), 4.84 (s, 2H), 3.98 (s, 6H), 3.74 (t, J = 4.7 Hz, 4H), 3.06 (t, J = 4.8 Hz, 4H), 1.66 (q, J = 4.0 Hz, 2H), 1.32 (q, J = 4.2 Hz, 2H).
LC-MS: m/z 593,3 [M+H]+ (расчетное значение 593,2, C30H30F2N6O5).
Пример 11: Синтез N-((2'-(3,5-дифтор-2,6-диметоксипиридин-4-ил)-3'-оксо-2',3'-дигидро-1'H-спиро[циклопентан-1,4'-[2,7]нафтиридин]-6'-ил)метил)акриламида (соединение 11)
Синтетический маршрут был следующим:
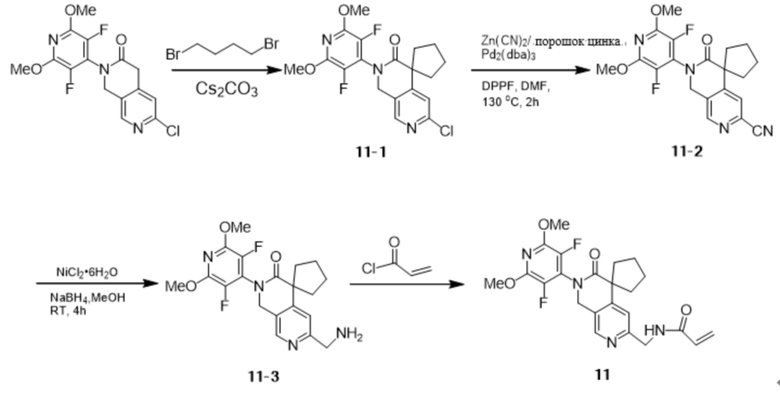
Шаг 1: Синтез промежуточного продукта 11-1
К смеси 6'-хлор-2'-(2,6-дифтор-3,5-диметоксипиридина)-1',2'-дигидро-3'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-3'-она (355 мг, 1,00 ммоль), 1,4-дибромбутана (432 мг, 2,00 ммоль), карбоната цезия (651 мг, 2 ммоль) в DMF (5 Смесь перемешивали при комнатной температуре в течение 2 ч. После завершения реакции на точечной пластинке TLC к реакционному раствору добавляли 20 мл воды и экстрагировали ЭА (3х20 мл), органические фазы объединяли и промывали один раз 20 мл воды и один раз 20 мл насыщенного солевого раствора. Органическую фазу высушили безводным MgSO4, затем отфильтровали и сконцентрировали при пониженном давлении, колоночной хроматографией получили Промежуточное соединение 11-1 (350 мг, 0,85 ммоль).
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 410,1 [M+H]+ (расчетное значение 410,1, C19H18ClF2N3O3).
Шаг 2: Синтез промежуточного продукта 11-2
Промежуточное соединение 11-1 (330 мг, 0,80 ммоль), Pd2(dba)3 (110 мг, 0,12 ммоль), DPPF (67 мг, 0,12 ммоль), Zn(CN)2 (140 мг, 1,2 ммоль) и порошок Zn (20 мг, 0,30 ммоль) помещали в реакционную колбу под защитой N2, добавляли DMF (6 мл) и проводили реакцию в микроволновых условиях при 140°С в течение 45 мин. DMF (6 мл) и проводили реакцию в микроволновых условиях при 140 °С в течение 45 мин. После завершения реакции добавляли 20 мл воды и экстрагировали ЭА (3х20 мл) для объединения органических фаз. Органическую фазу промывали 20 мл воды и 20 мл насыщенного солевого раствора каждый, сушили безводным MgSO4, затем отфильтровывали и концентрировали при пониженном давлении, остаток очищали колоночной хроматографией с получением Промежуточного соединения 11-2 (190 мг, 0,48 ммоль).
Идентификационные данные конкретных соединений следующие:
LC-MS: m/z 401,1 [M+H]+ (расчетное значение 401,1, C20H18F2N4O3).
Шаг 3: Синтез промежуточного продукта 11-3
Под защитой N2 в безводный метанол (5 мл) добавили промежуточное соединение 11-2 (90 мг, 0,23 ммоль), гексагидрат хлорида никеля (9 мг, 0,04 ммоль), по каплям добавили 0,1 мл трифторуксусной кислоты и перемешивали в течение 10 мин, затем тремя порциями добавили борогидрид натрия (90 мг, 2,4 ммоль) и проводили реакцию в течение 90 мин. После завершения реакции твердое вещество отфильтровали. Метанол удаляли отгонкой при пониженном давлении, добавляли 5 мл безводного THF и снова выпаривали досуха. Реакцию проводили непосредственно без дополнительной очистки. Масса остатка составила около 100 мг.
Идентификация конкретных соединений проводилась следующим образом:
LC-MS: m/z 405.2 [M+H]+ (расчетное значение 405.2, C20H22F2N4O3).
Шаг 4: Синтез соединения 11
Аналогично методике примера 10, шаг 3, было получено соединение 11.
Идентификационные данные конкретных соединений приведены ниже:
1H NMR (500 MHz, DMSO-d6): δ 8.71 (t, J = 5.9 Hz, 1H), 8.45 (s, 1H), 7.30 (s, 1H), 6.34 (dd, J = 17.1, 10.2 Hz, 1H), 6.13 (dd, J = 17.1, 2.1 Hz, 1H), 5.64 (dd, J = 10.3, 2.1 Hz, 1H), 4.92 (s, 2H), 4.48 (d, J = 5.9 Hz, 2H), 3.98 (s, 6H), 2.37 (dt, J = 13.1, 6.5 Hz, 2H), 1.98 (dt, J = 12.4, 5.6 Hz, 2H), 1.81-1.78 (m, 2H), 1.78 -1.69 (m, 2H).
LC-MS: m/z 459,2 [M+H]+ (расчетное значение 459,2, C23H24F2N4O4).
Пример 12: Синтез N-((7-(3,5-дифтор-2,6-диметоксипиридин-4-ил)-5,5-диметил-6-оксо-5,6,7,8тетрагидро-2,7-нафтиридин-3-ил)метил)акриламида (соединение 12)
Синтетический маршрут следующий:
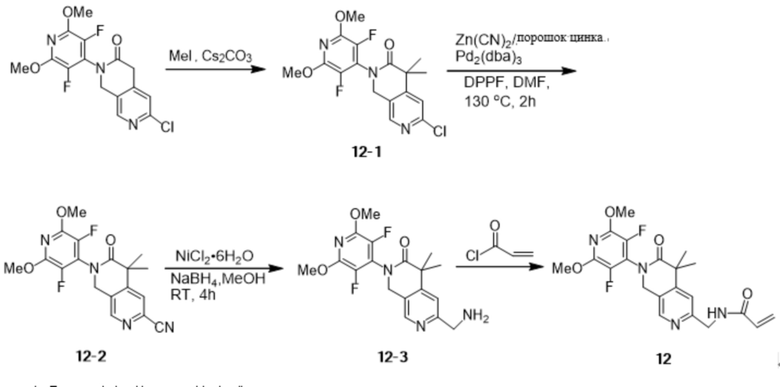
Шаг 1: Синтез промежуточного продукта 12-1
К смеси 6'-хлор-2'-(2,6-дифтор-3,5-диметоксипиридина)-1',2'-дигидро-3'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-3'-она (500 мг, 1,41 ммоль), йодометана (700 мг, 5,00 ммоль), карбоната цезия (1,10 г, 3,38 ммоль) в DMF ( 10 мл) смесь перемешивали при комнатной температуре в течение 2ч. После завершения реакции по данным TLC к реакционному раствору добавили 40 мл воды и трижды экстрагировали ЭА (3х40 мл), органические фазы объединили и промыли один раз 40 мл воды и один раз 40 мл насыщенного солевого раствора. Органическую фазу сушили безводным MgSO4, затем фильтровали и концентрировали при пониженном давлении с получением неочищенного промежуточного соединения 12-1 (460 мг, 1,20 ммоль).
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 384,0 [M+H]+ (расчетное значение 384,1, C17H16ClF2N3O3)
Шаг 2: Синтез промежуточного соединения 12-2
Под защитой N2 промежуточное соединение 12-1 (400 мг, 1,04 ммоль), Pd2(dba)3 (140 мг, 0,15 ммоль), DPPF (83,1 мг, 0,15 ммоль), Zn(CN)2 (176 мг, 1,5 ммоль) и порошок Zn (30 мг, 0,46 ммоль) помещали в DMF (6 мл). Реакцию проводили в микроволновой печи при 140 °С в течение 45 мин. После завершения реакции реакцию гасили 20мл воды, трижды экстрагировали ЭА (3х20 мл), органические фазы объединяли, промывали один раз 20мл воды и один раз 20 мл насыщенного солевого раствора. Органическую фазу сушили безводным MgSO4, затем фильтровали и концентрировали при пониженном давлении, остаток очищали колоночной хроматографией с получением Промежуточного соединения 12-2 (210 мг, 0,56 ммоль).
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 375,1 [M+H]+ (расчетное значение 375,1, C18H16F2N4O3).
Шаг 3: Синтез промежуточного соединения 12-3
Промежуточное соединение 12-2 (150 мг, 0,40 ммоль), гексагидрат хлорида никеля (14,2 мг, 0,06 ммоль) добавили в 5 мл безводного метанола под защитой N2, по каплям добавили 0,2 мл трифторуксусной кислоты, перемешивали в течение 10 мин, затем тремя порциями добавили борогидрид натрия (151 мг, 4,0 ммоль) и проводили реакцию в течение 90 мин. после завершения реакции твердое вещество удалили фильтрованием. После завершения реакции твердое вещество удаляли фильтрацией, метанол отгоняли при пониженном давлении, добавляли 5 мл безводного THF и снова выпаривали досуха. Остаток Промежуточный продукт 12-3 неочищенный (160 мг) был получен без дополнительной очистки и непосредственно перешел к следующей стадии реакции.
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 379,1 [M+H]+ (расчетное значение 379,2, C18H20F2N4O3).
Шаг 4: Синтез соединения 12
Остаток 12-3 (160 мг), полученный на предыдущей стадии, растворяли в 5 мл THF, охлаждали на ледяной бане до 0°С, по каплям добавляли 15 мкл акрилоилхлорида и перемешивали в течение 10 мин. После завершения реакции ее гасили добавлением 0,1 мл воды. Реакцию отгоняли при пониженном давлении до полного исчезновения остатков растворителя и остаточное твердое вещество очищали препаративным выделением с получением желаемого продукта 12 (20,29 мг).
Идентификационные данные конкретных соединений приведены ниже:
1H NMR (500 MHz, DMSO-d6): δ 8.71 (t, J = 5.9 Hz, 1H), 8.46 (s, 1H), 7.41 (s, 1H), 6.34 (dd, J = 17.1, 10.2 Hz, 1H), 6.13 (dd, J = 17.1, 2.1 Hz, 1H), 5.63 (dd, J = 10.2, 2.1 Hz, 1H), 4.95 (s, 2H), 4.48 (d, J = 5.8 Hz, 2H), 3.99 (s, 6H), 1.49 (s, 6H).
LC-MS: m/z 433,2 [M+H]+ (расчетное значение 433,2, C21H22F2N4O4).
Пример 13: Синтез N-((2'-(3,5-дифтор-2,6-диметоксипиридин-4-ил)-3'-оксо-2,2',3,3',5,6-гексагидро-1'H-спиро[пиран-4,4'-[2,7]нафтиридин]-6'-ил)метил)акриламида (соединение 13)
Синтетический маршрут был следующим:
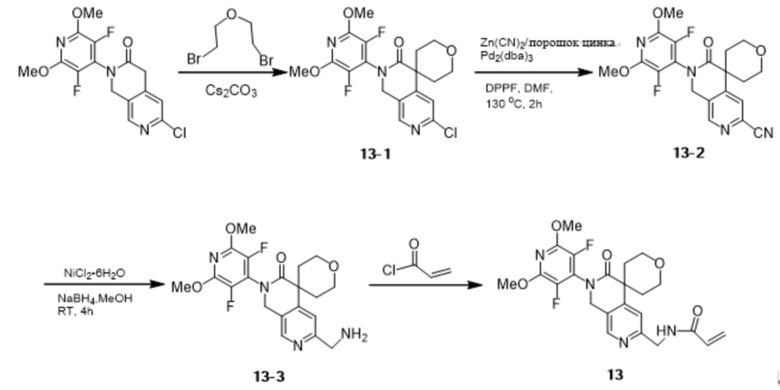
Шаг 1: Синтез промежуточного продукта 13-1
6'-Хлор-2'-(2,6-дифтор-3,5-диметоксипиридин)-1',2'-дигидро-3'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-3'-он (150 мг, 0,42 ммоль), бис(2-бромэтиловый) эфир (195 мг, 0,84 ммоль), карбонат цезия (390 мг, 1,20 ммоль) в смеси DMF (5 мл) и перемешивали при комнатной температуре в течение 2ч. После завершения реакции по данным TLC к реакционному раствору добавили 20 мл воды и трижды экстрагировали EA (3x20 мл), органические фазы объединили и промыли 20 мл воды и 20 мл насыщенного солевого раствора каждая. Органические фазы сушили безводным MgSO4, затем фильтровали и концентрировали при пониженном давлении с получением неочищенного промежуточного продукта 13-1 (153 мг, 0,36 ммоль).
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 426,0 [M+H]+ (расчетное значение 426,1, C19H18ClF2N3O4).
Шаг 2: Синтез промежуточного продукта 13-2
Аналогично методике шага 2 примера 12 был получен промежуточный продукт 13-2.
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 417,1 [M+H]+ (расчетное значение 417,1, C20H18F2N4O4).
Шаг 3: Синтез промежуточного продукта 13-3
Аналогично методике шага 3 примера 11 был получен промежуточный продукт 13-3.
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 421,1 [M+H]+ (расчетное значение 421,2, C20H22F2N4O4).
Шаг 4: Синтез соединения 13
Аналогично методике примера 10, шаг 3, было получено соединение 13.
Идентификационные данные конкретных соединений приведены ниже:
1H NMR (500 MHz, DMSO-d6): δ 8.70 (t, J = 5.9 Hz, 1H), 8.50 (s, 1H), 7.50 (s, 1H), 6.34 (dd, J = 17.1, 10.2 Hz, 1H), 6.14 (dd, J = 17.1, 2.1 Hz, 1H), 5.64 (dd, J = 10.2, 2.1 Hz, 1H), 4.96 (s, 2H), 4.50 (d, J = 5.8 Hz, 2H), 3.99 (s, 6H), 3.90 - 3.77 (m, 4H), 2.18 - 2.08 (m, 2H), 2.01 - 1.92 (m, 2H).
LC-MS: m/z 475,2 [M+H]+ (расчетное значение 475,2, C23H24F2N4O5).
Пример 14: Синтез N-(2-((2'-(3,5-дифтор-2,6-диметоксипиридин-4-ил)-3'-оксо-2',3'-дигидро-1'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-6'-ил)амино)-4-фтор-5-морфолинофенил)акриламида (Соединение 14)
Синтетический маршрут был следующим:
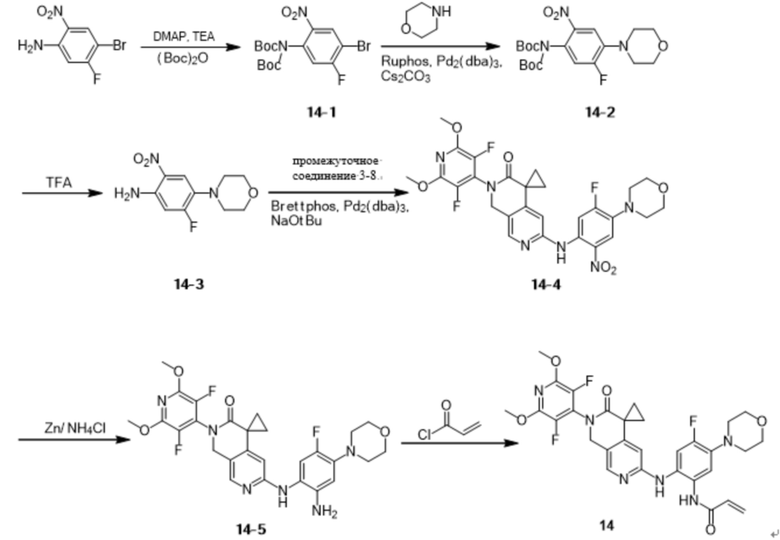
Шаг 1: Синтез промежуточного соединения 14-1
4-Бром-5-фтор-2-нитроанилин (4,70 г, 20 ммоль), DMAP (732 мг, 6 ммоль) и TEA (4 мл) растворяли в DCM (60 мл), затем по каплям добавляли ангидрид Boc (21,8 г, 100 ммоль), и реакцию проводили при комнатной температуре в течение 1 ч. После завершения реакции ее гасили добавлением насыщенного раствора хлорида аммония, а органический слой отбирали и промывали по одному разу 30 мл воды и промывали по одному разу 30 мл насыщенного солевого раствора. Органическую фазу сушили безводным сульфатом магния, фильтровали, и после вычитательного испарения получали желтое твердое вещество 14-1 (5,22 г, 15,5 ммоль).
Идентификационные данные для конкретных соединений приведены ниже:
LC-MS: m/z 335,0 [M+H]+ (расчетное значение 335,0, C16H20BrFN2O6).
Шаг 2: Синтез промежуточного соединения 14-2
Под защитой азота к сухому диоксану (15 мл) добавляли промежуточное соединение 14-1 (2,51 г, 7,50 ммоль), морфолин (1,26 г, 15,0 ммоль), Руфос (0,50 г, 1,14 ммоль), Pd2(dba)3 (1,00 г, 1,14 ммоль) и карбонат цезия (7,3 г, 23 ммоль) и проводили реакцию при 110°С в течение 30 мин. Реакцию проводили при 110°С в течение 30 мин. После завершения реакции продукт отфильтровывали, перегоняли досуха при пониженном давлении и остаток очищали колоночной хроматографией с получением промежуточного соединения 14-2 (2,00 г, 5,86 ммоль).
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 442,2 [M+Na]+ (расчетное значение 442,2, C20H28FN3O7).
Шаг 3: Синтез промежуточного соединения 14-3
Промежуточное соединение 14-2 (1,50 г, 4,4 ммоль) растворяли в 20 мл DCM при комнатной температуре при перемешивании, затем по каплям добавляли трифторуксусную кислоту в количестве 12 мл и проводили реакцию в течение 1 ч. После завершения реакции и полного испарения растворителя получали промежуточное соединение 14-3 (942 мг, 4,01 ммоль).
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 242,2 [M+H]+ (расчетное значение 242,1, C10H12FN3O3).
Шаг 4: Синтез промежуточного продукта 14-4
В 10 мл толуола под защитой азота добавляли Промежуточное соединение 3-8 (178 мг, 0,5 ммоль, см. Пример 3), Промежуточное соединение 14-3 (144 мг, 0,6 ммоль), ацетат палладия (18,6 мг, 0,08 ммоль), Руфос (37,3 мг, 0,08 ммоль) и трет-бутоксид натрия (192 мг, 2 ммоль). в микроволновом реакторе при 130°С в течение 30 мин, после завершения реакции отгоняли досуха при пониженном давлении, остаток очищали колоночной хроматографией с получением промежуточного соединения 14-4 (96 мг, 0,17 ммоль).
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 587,3 [M+H]+ (расчетное значение 587,2, C27H25F3N6O6).
Шаг 5: Синтез промежуточного соединения 14-5
Смесь промежуточного соединения 14-4 (96 мг, 0,17 ммоль), порошка цинка (500 мг, 7,65 ммоль), хлорида аммония (600 мг, 9,18 ммоль) в метаноле (10 мл) перемешивали и проводили реакцию в течение 2 ч при комнатной температуре, отфильтровали, отогнали досуха при пониженном давлении, добавили 10 мл THF и снова выпарили досуха, получив твердый остаток 14-5 (108 мг).
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 557,4 [M+H]+ (расчетное значение 557,2, C27H27F3N6O4).
Шаг 6: Синтез соединения 14
Аналогично методике примера 10, шаг 3, было получено соединение 14.
Идентификационные данные конкретных соединений приведены ниже:
1HNMR (500 MHz, DMSO-d6): δ 9.63 (s, 1H), 8.12 (s, 1H), 7.99 (s, 1H), 7.61 (d, J = 14.7 Hz, 1H), 7.22 (d, J = 9.3 Hz, 1H), 6.47 (dd, J = 17.0, 10.2 Hz, 1H), 6.33 (s, 1H), 6.24 (dd, J = 17.0, 2.0 Hz, 1H), 5.77-5.71 (m, 1H), 4.88 (s, 2H), 3.98 (s, 6H), 3.74 (t, J = 4.6 Hz, 4H), 2.95 (t, J = 4.6 Hz, 4H), 1.70 (q, J = 4.0 Hz, 2H), 1.39 (q, J = 4.2 Hz, 2H).
LC-MS: m/z 611,3 [M+H]+ (расчетное значение 611,2, C30H29F3N6O5).
Пример 15: Синтез N-(2-((2'-(3,5-дифтор-2,6-диметоксипиридин-4-ил)-3'-оксо-2',3'-дигидро-1'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-6'-ил)амино)-5-(3-метилморфолино)фенил)акриламида (Соединение 15)
Синтетический маршрут был следующим:
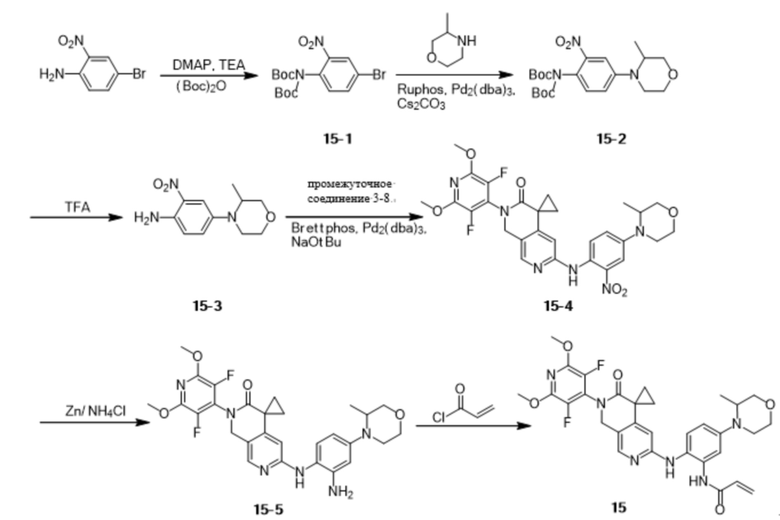
Шаг 1: Синтез промежуточного соединения 15-1
Аналогично методике шага 1 примера 14, соединение было приготовлено с получением желтого твердого вещества 15-1 (5,12 г, 16,2 ммоль).
Идентификационные данные конкретного соединения были следующими:
LC-MS: m/z 439,0 [M+Na]+ (расчетное значение 439,0, C16H21BrN2O6).
Шаг 2: Синтез промежуточного соединения 15-2
Соединение было получено заменой морфолина на 3-метилморфолин, аналогично методике шага 2 примера 14, с получением желтого твердого вещества 15-2 (2,45 г, 5,86 ммоль).
Идентификационные данные конкретного соединения приведены ниже:
LC-MS: m/z 438,3 [M+H]+ (расчетное значение 438,2, C21H31N3O7).
Шаг 3: Синтез промежуточного соединения 15-3
Аналогично методике шага 3 примера 14 было получено промежуточное соединение 15-3 (1104 мг, 4,65 ммоль).
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 238,2 [M+H]+ (расчетное значение 238,1, C11H15N3O3)
Шаг 4: Синтез промежуточного соединения 15-4
Аналогично методике шага 4 примера 14 был получен промежуточный продукт 15-4 (482 мг, 0,83 ммоль).
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 583,2 [M+H]+ (расчетное значение 583,2, C28H28F2N6O6).
Шаг 5: Синтез промежуточного продукта 15-5
Аналогично методике шага 5 примера 14 был получен твердый остаток 15-5 (508 мг).
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 553,3 [M+H]+ (расчетное значение 553,2, C28H30F2N6O4).
Шаг 6: Синтез соединения 15
Соединение 15 (256,48 мг, 0,42 ммоль) было приготовлено аналогично методике примера 10, шаг 3.
Идентификационные данные конкретных соединений были следующими.
1H NMR (500 MHz, DMSO-d6): δ 9.59 (s, 1H), 7.92 (d, J = 4.4 Hz, 2H), 7.33 (d, J = 8.8 Hz, 1H), 7.24 (d, J = 2.9 Hz, 1H), 6.75 (dd, J = 8.9, 2.8 Hz, 1H), 6.45 (dd, J = 16.9, 10.2 Hz, 1H), 6.22 (dd, J = 17.0, 2.0 Hz, 1H), 6.09 (s, 1H), 5.71 (dd, J = 10.1, 2.0 Hz, 1H), 4.85 (s, 2H), 3.98 (s, 6H), 3.92-3.86 (m, 1H), 3.76-3.69 (m, 2H), 3.67-3.61 (m, 1H), 3.58 (td, J = 10.8, 3.2 Hz, 1H), 3.10-3.04 (m, 1H), 3.00 (td, J = 11.9, 11.2, 3.5 Hz, 1H), 1.66 (q, J = 3.9 Hz, 2H), 1.32 (q, J = 4.1 Hz, 2H), 1.00 (d, J = 6.3 Hz, 3H).
LC-MS: m/z 607,3 [M+H]+ (расчетное значение 607,2, C31H32F2N6O5).
Пример 16: Синтез N-((3-(3,5-дифтор-2,6-диметоксипиридин-4-ил)-2-оксо-1-фенил-1,2,3,4-тетрагидропиридо[4,3-d]пиримидин-7-ил)метил)акриламида (соединение 16)
Синтетический маршрут следующий:
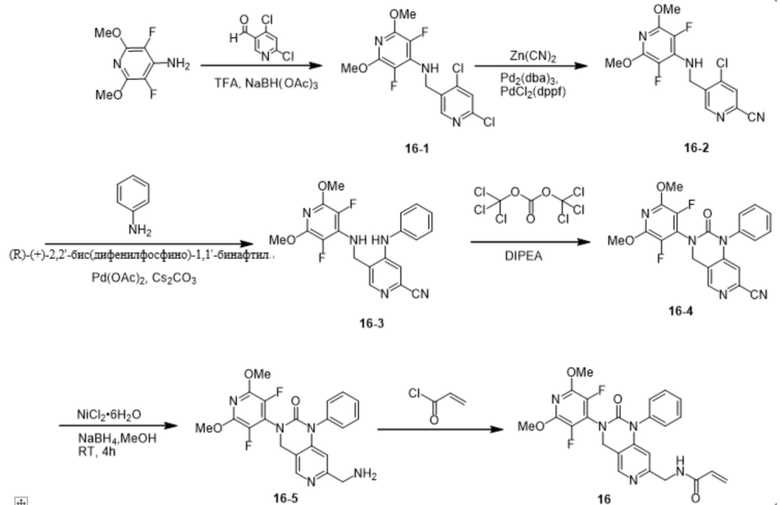
Шаг 1: Синтез промежуточного продукта 16-1
Триацетоксиборогидрид натрия (635 мг, 3,00 ммоль) добавляли к перемешиваемой смеси дихлорметана (5 мл)/трифторуксусной кислоты (600 мкл), состоящей из 3,5-дифтор-2,6-диметоксипиридин-4-амина (200 мг, 1,05 ммоль), 4,6-дихлорникотинового альдегида (185 мг, 1,05 ммоль), в течение 1 ч при 0-5 °С. После этого ледяную баню убирали и проводили реакцию при комнатной температуре. После завершения реакции по данным ТЛК реакцию гасили добавлением насыщенного водного раствора хлорида аммония и экстрагировали дихлорметаном. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали до сухого состояния при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией с получением Промежуточного продукта 16-1 (220 мг).
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 350,1 [M+H]+ (расчетное значение 350,0, C13H11Cl2F2N3O2).
Шаг 2: Синтез промежуточного продукта 16-2
Смесь Промежуточного соединения 16-1 (200 мг, 0,57 ммоль), цианида цинка (45 мг, 0,38 ммоль), трис(дибензилиденацетон)дипалладия(0) (52 мг, 0,057 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладия(II) дихлорида, комплексообразованного с дихлорметаном (1. 1) (47 мг, 0,057 ммоль) в смеси с N,N-диметилформамидом (5 мл) нагревали в течение 1,5 часов. Затем реакционную смесь охлаждали до комнатной температуры, гасили насыщенным водным раствором NaHCO3 и экстрагировали этилацетатом (3 х 10 мл). Объединенные органические слои промывали рассолом, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали на силикагеле (элюируя гексаном, содержащим 0-25% EtOAc) с получением Промежуточного соединения 16-2 (130 мг).
Идентификационные данные для конкретных соединений приведены ниже:
LC-MS: m/z 340,2 [M+H]+ (расчетное значение 340,1, C14H11ClF2N4O2).
Шаг 3: Синтез промежуточного соединения 16-3
В герметичную пробирку добавили промежуточное соединение 16-2 (100 мг, 0,29 ммоль), ацетат палладия (20 мг, 0,09 ммоль), (R)-(+)-2,2′-бис(дифенилфосфино)-1,1′-бинафталин (40 мг, 0,06 ммоль), карбонат цезия (300 мг, 0,92 ммоль) и 3 мл ультрасухого диоксана, герметично закрыли и перемешивали магнитом. Азот был полностью вытеснен, и, наконец, анилин (54 мкл, 0,58 ммоль) был добавлен с помощью шприца, помещен в микроволновой реактор, перемешивание, 130 ℃, реакция длилась около 1 ч. После завершения реакции температура была охлаждена, разбавлена ЭА, подушка была отфильтрована кизельгуром, промыта ЭА и откачана до сухости, фильтрат был собран и сконцентрирован до сухости при пониженном давлении, с получением красновато-коричневого маслянистого материала в качестве сырого продукта Промежуточного продукта 16-3 (60 мг). Неочищенный продукт был использован непосредственно на следующей стадии без дополнительной очистки.
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 398,2 [M+H]+ (расчетное значение 398,1, C20H17F2N5O2).
Шаг 4: Синтез промежуточного соединения 16-4
К перемешиваемому раствору промежуточного соединения 16-3 (55 мг, 0,14 ммоль) в тетрагидрофуране (2,0 мл) при 0-5 °С добавляли N,N-диизопропилэтиламин (116 мкл, 0,70 ммоль), затем медленно добавляли трифосген (42 мг, 0,14 ммоль (растворен в 2 мл THF и добавлен)). Через 5 мин ледяная баня была снята, и реакцию проводили при комнатной температуре. После завершения реакции ее гасили добавлением насыщенного водного NaHCO3 и экстрагировали этилацетатом. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали до сухого состояния при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией с получением Промежуточного продукта 16-4 (30 мг).
Идентификационные данные конкретных соединений приведены ниже:
LC-MS: m/z 424,1 [M+H]+ (расчетное значение 424,1, C21H15F2N5O3)
Шаг 5: Синтез промежуточного соединения 16-5
Аналогично методике шага 3 примера 11 было получено соединение 16-5 (40 мг).
Идентификационные данные конкретных соединений были следующими.
LC-MS: m/z 428,2 [M+H]+ (расчетное значение 428,2, C21H19F2N5O3)
Шаг 6: Синтез соединения 16
Соединение 16 (40 мг) было получено аналогично методике примера 10, стадия 3.
Идентификационные данные конкретных соединений приведены ниже.
1H NMR (500 MHz, DMSO-d6): δ 8.56 (t, J = 5.9 Hz, 1H), 8.31 (s, 1H), 7.55 (t, J = 7.5 Hz, 2H), 7.50 (t, J = 7.3 Hz, 1H), 7.34 (d, J = 7.6 Hz, 2H), 6.14 (dd, J = 17.1, 10.2 Hz, 1H), 6.07 (s, 1H), 6.02-5.94 (m, 1H), 5.57-5.51 (m, 1H), 5.02 (s, 2H), 4.23 (d, J = 6.0 Hz, 2H), 3.98 (s, 6H).
LC-MS: m/z 482,2 [M+H]+ (расчетное значение 482,2, C24H21F2N5O4).
Пример 17: Синтез N-((1-циклопропил-3-(3,5-дифтор-2,6-диметоксипиридин-4-ил)-2-оксо-1,2,3,4-тетрагидропиридо[4,3-d]пиримидин-7-ил)метил)акриламида (соединение 17)
Синтетический маршрут следующий:
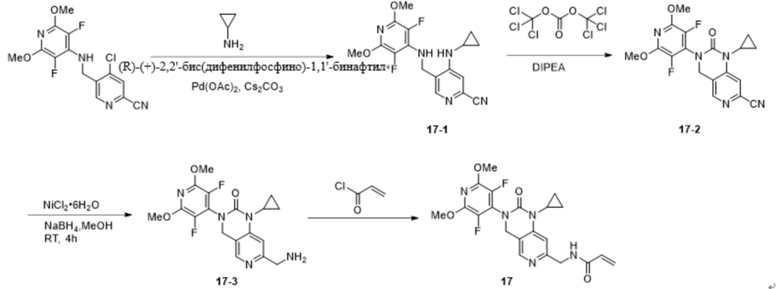
Шаг 1: Синтез промежуточного продукта 17-1
В закрытую пробирку добавляли 6'-хлор-2'-(2,6-дифтор-3,5-диметоксипиридин)-1',2'-дигидро-3'H-спиро[циклопропан-1,4'-[2,7]нафтиридин]-3'-он (100 мг, 0,29 ммоль), ацетат палладия (20 мг, 0,09 ммоль), (R)-(+)-2,2′-бис(дифенилфосфинил)-1, 1′-бинафтил (40 мг, 0,06 ммоль), карбонат цезия (300 мг, 0,92 ммоль) и ультрасухой диоксан (3 мл), герметично закрытый, магнитное перемешивание, и азот был полностью вытеснен, и наконец циклопропиламин (40 мкл, 0,58 ммоль) был добавлен с помощью шприца, и помещен в микроволновой реактор, и реакция проводилась в течение примерно 1 ч при 130 ℃, и после завершения реакции, температура была снижена, EA был разбавлен После завершения реакции реакцию охлаждали, разбавляли ЭА, фильтровали с кизельгуром, промывали ЭА, сливали, фильтрат собирали и концентрировали до сухости при пониженном давлении с получением красновато-коричневого масла, которое и представляло собой сырой продукт Промежуточного соединения 17-1 (60 мг). Неочищенный продукт был использован непосредственно на следующей стадии без дополнительной очистки.
Идентификационные данные конкретных соединений были следующими:
LC-MS: m/z 362,1 [M+H]+ (расчетное значение 362,1, C17H17F2N5O2).
Шаг 2: Синтез промежуточного продукта 17-2
Аналогично методике примера 16, шаг 4, было получено Промежуточное соединение 17-2 (30 мг).
Идентификационные данные конкретного соединения были следующими:
LC-MS: m/z 388,2 [M+H]+ (расчетное значение 388,1, C18H15F2N5O3).
Шаг 3: Синтез промежуточного продукта 17-3
Аналогично методике шага 3 примера 11 был получен Промежуточный продукт 17-3 (40 мг).
Идентификационные данные конкретного соединения были следующими:
LC-MS: m/z 392,2 [M+H]+ (расчетное значение 392,2, C18H19F2N5O3).
Шаг 4: Синтез соединения 17
Аналогично методике примера 10, шаг 3, было получено соединение 17 (40 мг).
Идентификационные данные конкретных соединений следующие:
1H NMR (500 MHz, DMSO-d6): δ 8.73 (t, J = 5.9 Hz, 1H), 8.24 (s, 1H), 7.23 (s, 1H), 6.35 (dd, J = 17.1, 10.2 Hz, 1H), 6.14 (dd, J = 17.1, 2.1 Hz, 1H), 5.64 (dd, J = 10.2, 2.1 Hz, 1H), 4.73 (s, 2H), 4.44 (d, J = 5.9 Hz, 2H), 3.97 (s, 6H), 2.79 (tt, J = 6.9, 3.7 Hz, 1H), 1.04 (td, J = 7.3, 5.4 Hz, 2H), 0.64-0.55 (m, 2H).
LC-MS: m/z 446,2 [M+H]+ (расчетное значение 446,2, C21H21F2N5O4).
Фармакологические испытания
Пример А: киназный анализ
Влияние соединений изобретения на активность тирозинкиназ FGFR1 и FGFR4 оценивалось с помощью теста киназного анализа in vitro. В качестве метода анализа использовался метод гомогенной флуоресценции с временным разрешением (HTRF).
Приготовление соединения: 45мкл DMSO добавляли к 5мкл резервного раствора с концентрацией 10 мМ для приготовления раствора LY2874455 с концентрацией 1000 мкМ, затем 12 мкл соединения с концентрацией 1000 мкМ добавляли к 88 мкл DMSO для приготовления раствора LY2874455 с концентрацией 120 мкМ в качестве исходной концентрации; 2 мкл соединения с концентрацией 10 мМ добавляли к 2 мкл резервного раствора с концентрацией 10 мМ; 2 мкл соединения с концентрацией 10 мкМ добавляли к 2 мкл резервного раствора с концентрацией 10 мМ; 2 мкл соединения с концентрацией 10 мкМ добавляли к 2 мкл резервного раствора с концентрацией 10 мМ. раствор был приготовлен путем добавления 48 мкл DMSO к 2 мкл резервного раствора с концентрацией 10 мМ для приготовления раствора BLU554 с концентрацией 400 мкМ в качестве начальной концентрации; 15 мкл DMSO было добавлено к 10 мкл резервного раствора с концентрацией 10 мМ соединений образца для приготовления раствора соединений образца с концентрацией 4 мМ в качестве начальной концентрации; и раствор соединений был перенесен на планшет назначения с помощью дозатора Echo.
Киназная реакция: в каждую лунку добавляли по 5 мкл раствора киназы, соединения и киназу инкубировали в течение 60 мин при комнатной температуре, затем инициировали реакцию добавлением 5 мкл смеси субстрата и АТФ, проводили реакцию в течение определенного времени (30 мин для киназного анализа FGFR1 и 40 мин для киназного анализа FGFR4) при 37°С, останавливали реакцию добавлением 10 мкл Xl665 и антитела. Реакцию останавливали добавлением 10 мкл Xl665 и смеси реагентов для детекции и после инкубации в течение 60 мин при комнатной температуре считывали сигнал TR-FRET при длинах волн эмиссии 665 нМ/612 нМ на планшетном ридере SPARK 10M. Активность фермента определяли в 10 концентрациях для каждого соединения в отдельности и рассчитывали данные с помощью аналитического программного обеспечения для получения значения IC50 соединения, как показано в табл. 1.
Таблица 1 Значения IC50 и селективное ингибирование соединений
Примечание: BLU554 - соединение № 40, раскрытое компанией Blueprint Medicines Corporation (Blueprint) в WO2015061572.
Как видно из табл. 1, соединения настоящего изобретения оказывают ингибирующее действие на киназу FGFR4, причем ингибирующее действие соединений на FGFR4 значительно сильнее, чем ингибирующее действие на FGFR1, с очень хорошей селективностью.
Пример B: Анализ пролиферации клеток
Для оценки влияния соединений настоящего изобретения на пролиферацию клеток гепатоцеллюлярной карциномы человека, клеток Hep3B, был использован клеточный анализ in vitro. Для анализа использовался метод люминесцентного анализа CELL TITER-GLO (CTG).
Распространение клеток: клетки Hep3B в логарифмической фазе роста отбирали, переваривали и собирали центрифугированием при 1000 об/мин в течение 5 мин при комнатной температуре, затем клетки подсчитывали после ресуспендирования в среде EMEM с 10% FBS и инокулировали в 384-луночные планшеты (#3765) при плотности 800 клеток/лунку, по 20 мкл на лунку.
Приготовление соединений: соединения растворяли в ДМСО для получения исходного раствора с концентрацией 10 мМ, затем разбавляли исходный раствор образцового соединения до 2 мМ (6 мкл исходного раствора плюс 24 мкл ДМСО), используя ДМСО в качестве исходной концентрации, и исходный раствор контрольного соединения до 200 мкМ (2 мкл исходного раствора плюс 98 мкл ДМСО), используя ДМСО в качестве исходной концентрации; соединения разводили 9 раз в последовательных 3-кратных градиентах с ДМСО, а затем разводили до 40-кратной конечной концентрационной дозы (2 мкл соединения плюс 78 мкл ДМСО) с помощью клеточной культуральной среды. ДМСО для 9 последовательных 3-кратных градиентных разведений, затем соединения разбавляли до 40-кратной конечной концентрационной дозы клеточной культуральной средой (2 мкл соединения плюс 78 мкл ДМСО).
Введение в клетки: 5 мкл (5×) соединения добавляли в соответствующие лунки, конечная концентрация образцов соединений составляла 10000 нМ, 3333 нМ, 1111 нМ, 370,37 нМ, 123,46 нМ, 41,15 нМ, 13,72 нМ, 4,57 нМ, 1,52 нМ и 0,50 нМ в порядке убывания, в качестве отрицательного контроля использовали 0,5% ДМСО. Планшеты инкубировали при 37°C и 5% CO2 в течение 3 дней.
После 3 дней инкубации в каждую лунку добавляли по 5 мкл реагента CellTiter-Glo®, инкубировали 60 мин при 300 об/мин на шейкере в условиях защиты от света и комнатной температуры, считывали сигналы люминесценции на планшетном ридере Spark 10M и рассчитывали полуингибирующую концентрацию соединений на пролиферацию клеток, т.е. значение IC50 с помощью аналитического программного обеспечения, как показано в табл. 2.
Таблица 2 Значения IC50 соединений на клетках Hep3B
Как видно из табл. 2, соединения настоящего изобретения обладают хорошим ингибирующим действием на пролиферацию клеток Hep3B.
Настоящее изобретение относится к области медицинской химии и касается, в частности, соединения формулы (I) или его фармацевтически приемлемой соли или пролекарства. В формуле (I): X представляет собой CH; Y представляет собой 1 О; Z представляет собой C(R6)2; m представляет собой 0 или 1; L представляет собой -C(R7)2- или  n представляет собой 0, 1 или 2; каждый R1 независимо представляет собой C1-3 алкил или С1-3 галоалкил; R2 и R3 каждый независимо представляет собой галоген; R4 представляет собой водород; R5 представляет собой водород; два R6 и атомы углерода, к которым они присоединены, вместе образуют С3-8 циклоалкил или 4-10-членный гетероциклоалкил; где 4-10-членный гетероциклоалкил включает 1 кольцеобразующий гетероатом О; каждый R7 независимо представляет собой водород, галоген, C1-8 алкил, 3-10-членный гетероциклоалкил или 3-10-членный гетероциклоалкил, замещенный С1-4 алкилом или морфолинилом; при этом указанный 3-10-членный гетероциклоалкил независимо включает от 1 до 2 кольцеобразующих гетероатомов, и каждый из указанных гетероатомов независимо является N, О. Также предложены фармацевтическая композиция и фармацевтический препарат, содержащие указанное соединение, и их применение. Предложенное соединение может быть использовано в качестве ингибитора FGFR4 для профилактики и/или лечения заболеваний, опосредованных, по крайней мере частично, FGFR4. 7 н. и 4 з.п. ф-лы, 2 табл., 19 пр.
n представляет собой 0, 1 или 2; каждый R1 независимо представляет собой C1-3 алкил или С1-3 галоалкил; R2 и R3 каждый независимо представляет собой галоген; R4 представляет собой водород; R5 представляет собой водород; два R6 и атомы углерода, к которым они присоединены, вместе образуют С3-8 циклоалкил или 4-10-членный гетероциклоалкил; где 4-10-членный гетероциклоалкил включает 1 кольцеобразующий гетероатом О; каждый R7 независимо представляет собой водород, галоген, C1-8 алкил, 3-10-членный гетероциклоалкил или 3-10-членный гетероциклоалкил, замещенный С1-4 алкилом или морфолинилом; при этом указанный 3-10-членный гетероциклоалкил независимо включает от 1 до 2 кольцеобразующих гетероатомов, и каждый из указанных гетероатомов независимо является N, О. Также предложены фармацевтическая композиция и фармацевтический препарат, содержащие указанное соединение, и их применение. Предложенное соединение может быть использовано в качестве ингибитора FGFR4 для профилактики и/или лечения заболеваний, опосредованных, по крайней мере частично, FGFR4. 7 н. и 4 з.п. ф-лы, 2 табл., 19 пр.
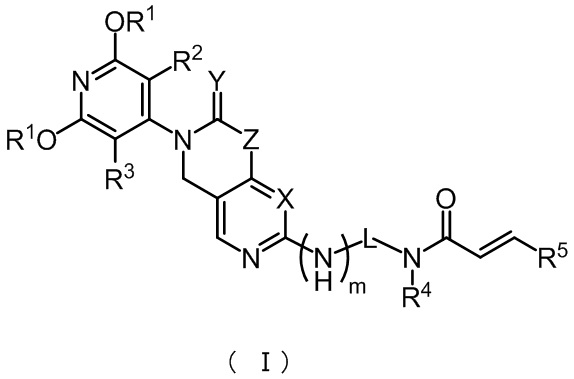
1. Соединение формулы (I) или его фармацевтически приемлемая соль или пролекарство
где X представляет собой CH;
Y представляет собой 1 О;
Z представляет собой C(R6)2;
m представляет собой 0 или 1;
L представляет собой -C(R7)2- или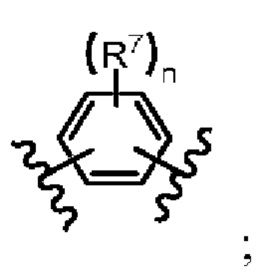
n представляет собой 0, 1 или 2;
каждый R1 независимо представляет собой C1-3 алкил или С1-3 галоалкил;
R2 и R3 каждый независимо представляет собой галоген;
R4 представляет собой водород;
R5 представляет собой водород;
два R6 и атомы углерода, к которым они присоединены, вместе образуют С3-8 циклоалкил или 4-10-членный гетероциклоалкил; где 4-10-членный гетероциклоалкил включает 1 кольцеобразующий гетероатом О;
каждый R7 независимо представляет собой водород, галоген, C1-8 алкил, 3-10-членный гетероциклоалкил или 3-10-членный гетероциклоалкил, замещенный С1-4 алкилом или морфолинилом; при этом указанный 3-10-членный гетероциклоалкил независимо включает от 1 до 2 кольцеобразующих гетероатомов, и каждый из указанных гетероатомов независимо является N, О.
2. Соединение по п. 1 или его фармацевтически приемлемая соль или пролекарство, характеризующееся тем, что указанное соединение имеет структуру, представленную в формуле (III)
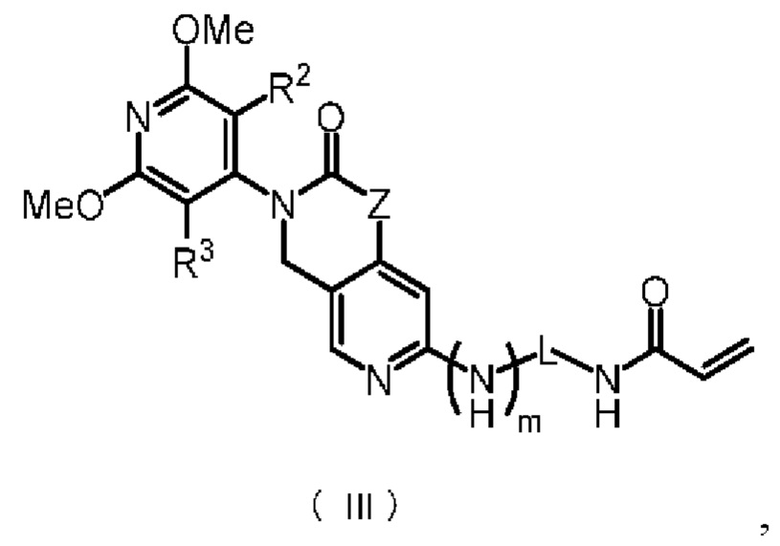
где Z, m, L, R2 и R3 определены так, как определено в п. 1.
3. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль или пролекарство, характеризующееся тем, что указанное соединение имеет структуру, представленную формулой (III-1) или формулой (III-2)
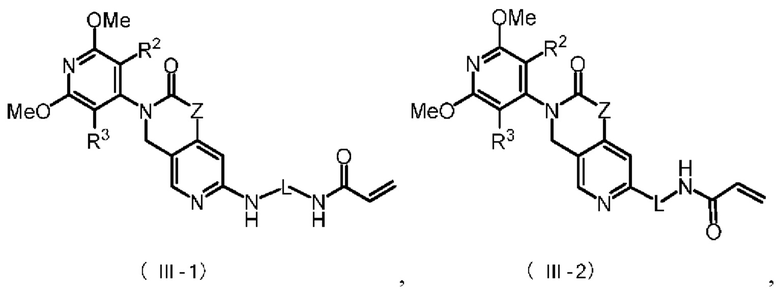
где Z, L, R2 и R3 определены так, как определено в п. 1;
предпочтительно, указанное соединение формулы (III-1) имеет структуру, представленную в формуле (III-1-1)
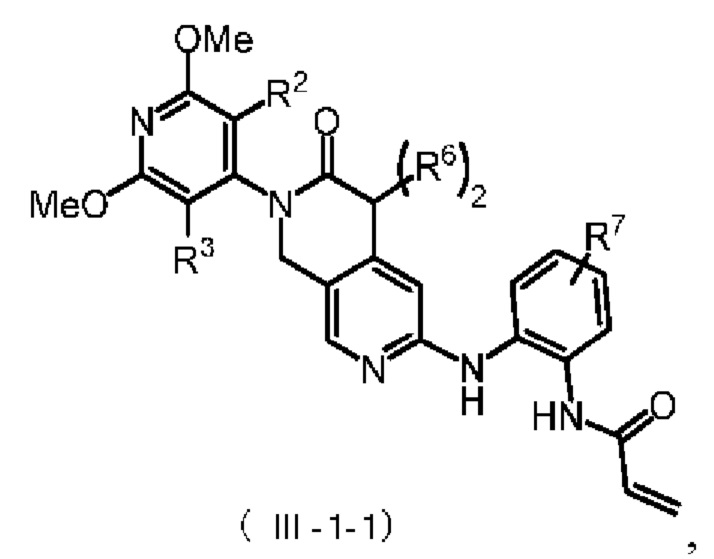
где R2, R3 и R6 определены, как определено в п. 1, R7 представляет собой водород, С1-4 алкил, пиперазинил, пиперидинил или морфолинил, где указанные пиперазинил, пиперидинил и морфолинил по выбору замещены по меньшей мере одним R8, причем каждый R8 независимо от другого представляет собой C1-4 алкил или морфолинил;
предпочтительно, R2, R3 и R6 определены, как определено в п. 1; a R7 представляет собой водород, С1-4 алкил, пиперазинил, пиперидинил или морфолинил, причем указанные пиперазинил, пиперидинил и морфолинил по выбору замещены по меньшей мере одним R8, который представляет собой морфолинил;
более предпочтительно, R2, R3 и R6 определены, как определено в п. 1, a R7 представляет собой один из следующих фрагментов:
предпочтительно, соединение формулы (III-2) имеет структуру, представленную формулой (III-2-1)
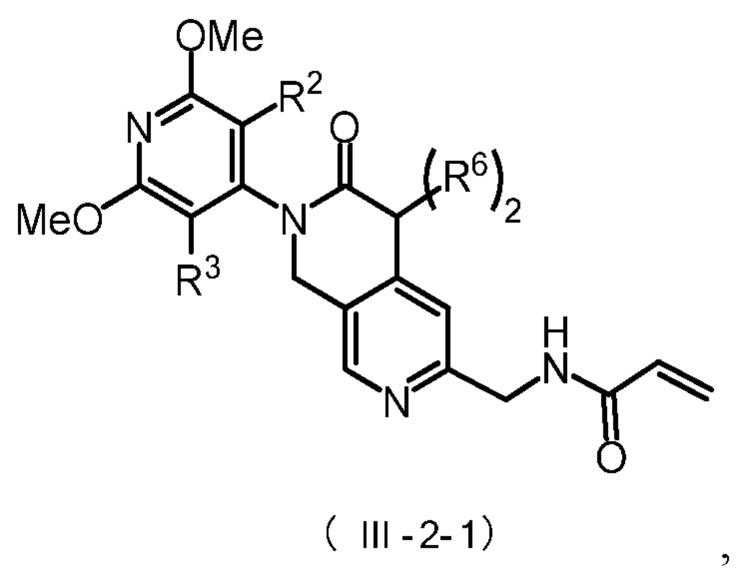
где R2, R3 и R6 определены так, как определено в п. 1.
4. Соединение или его фармацевтически приемлемая соль или пролекарство, выбранное из следующих соединений:
5. Фармацевтическая композиция, оказывающая ингибирующее действие на активность FGFR4, включающая соединение по любому из пп. 1-4 или его фармацевтически приемлемую соль или пролекарство.
6. Фармацевтический препарат, оказывающий ингибирующее действие на активность FGFR4, включающий соединение по любому из пп. 1-4 или его фармацевтически приемлемую соль или пролекарство, или фармацевтическую композицию по п. 5, причем указанный фармацевтический препарат представляет собой любое из таблетки, капсулы, инъекции, гранулы, порошка, суппозитория, пилюли, геля, дисперсии, раствора для перорального применения, лекарственной формы для ингаляции, суспензии или сухой суспензии.
7. Применение соединения по любому из пп. 1-4 или его фармацевтически приемлемой соли или пролекарства, или фармацевтической композиции по п. 5, или фармацевтического препарата по п. 6, в получении лекарственного средства для профилактики и/или лечения заболевания, опосредованного, по крайней мере частично, FGFR4.
8. Применение по п. 7, где указанное заболевание, по крайней мере частично опосредованное FGFR4, включает рак;
предпочтительно указанный рак выбран из гепатоцеллюлярной карциномы, рака мочевого пузыря, рака молочной железы, рака шейки матки, колоректального рака, рака эндометрия, рака желудка, рака головы и шеи, рака почек, гепатоцеллюлярной карциномы, рака легких, рака яичников, рака простаты, рака пищевода, рака желчного пузыря, рака поджелудочной железы, рака щитовидной железы, рака кожи, лейкоза, множественной миеломы, хронической лимфобластной лимфомы, Т-клеточного лейкоза взрослых, В-клеточной лимфомы, острого миелоидного лейкоза, лимфомы Ходжкина или неходжкинской лимфомы, макроглобулинемии Вальденстрема, волосатоклеточной лимфомы, лимфомы Буркитта, нейроглиобластомы, меланомы, мезотелиомы, нейробластомы у взрослых, рака яичка, плоскоклеточной карциномы, глиобластомы и рабдомиосаркомы.
9. Применение соединения по любому из пп. 1-4 или его фармацевтически приемлемой соли или пролекарства, или фармацевтической композиции по п. 5, или фармацевтического препарата по п. 6, в качестве ингибитора EGFR4.
10. Применение соединения по любому из пп. 1-4 или его фармацевтически приемлемой соли или пролекарства, или фармацевтической композиции по п. 5, или фармацевтического препарата по п. 6 для профилактики и/или лечения заболевания, опосредованного, по крайней мере частично, FGFR4.
11. Применение по п. 10, где указанное заболевание, по крайней мере частично опосредованное FGFR4, включает рак;
предпочтительно указанный рак выбран из гепатоцеллюлярной карциномы, рака мочевого пузыря, рака молочной железы, рака шейки матки, колоректального рака, рака эндометрия, рака желудка, рака головы и шеи, рака почек, гепатоцеллюлярной карциномы, рака легких, рака яичников, рака простаты, рака пищевода, рака желчного пузыря, рака поджелудочной железы, рака щитовидной железы, рака кожи, лейкоза, множественной миеломы, хронической лимфобластной лимфомы, Т-клеточного лейкоза взрослых, В-клеточной лимфомы, острого миелоидного лейкоза, лимфомы Ходжкина или неходжкинской лимфомы, макроглобулинемии Вальденстрема, волосатоклеточной лимфомы, лимфомы Буркитта, нейроглиобластомы, меланомы, мезотелиомы, нейробластомы у взрослых, рака яичка, плоскоклеточной карциномы, глиобластомы и рабдомиосаркомы.
| CN 107286130 А, 24.10.2017 | |||
| EA 201791867 A1, 19.12.2017 | |||
| US 20180008610 A1, 11.01.2018 | |||
| US 20160115164 A1, 28.04.2016 | |||
| US 20170253593 A1, 07.09.2017 | |||
| Подставка указатель для всякого рода съемных предметов | 1933 |
|
SU36160A1 |
| ИНГИБИТОРЫ РЕЦЕПТОРА ФАКТОРА РОСТА ФИБРОБЛАСТОВ | 2013 |
|
RU2679130C2 |
| CN 110272415 A, 24.09.2019. | |||

