Изобретение относится к биотехнологии и медицине, в частности к иммунологии, и предназначено для связывания иммуноглобулинов различных подклассов. Изобретение может использоваться как в биотехнологии для выделения иммуноглобулинов из различных биологических жидкостей или растворов, содержащих иммуноглобулины, так и в медицине для удаления иммуноглобулинов из плазмы/крови больных тяжелыми формами аутоиммунных патологий, например, таких как системная красная волчанка, ревматоидный артрит, идиоматическая тромбоцитоменическая пурпура [1], синдром Гильена-Барре, миастения, дилатационная кардиомиопатия [2, 3], когда лекарственная терапия не эффективна или не может быть применена [4-7].
Выделение иммуноглобулинов из различных биологических жидкостей является одной из актуальных задач современной биотехнологии и препаративной биохимии. Среди сорбентов, используемых для выделения иммуноглобулинов, ведущее место занимают сорбенты с иммобилизованным белком A из клеточной стенки Staphylococcus aureus. Основным недостатком данных лигандов является высокая стоимость, отсутствие стабильности в условиях стерилизации и ограниченная способность связывания отдельных подклассов иммуноглобулинов G.
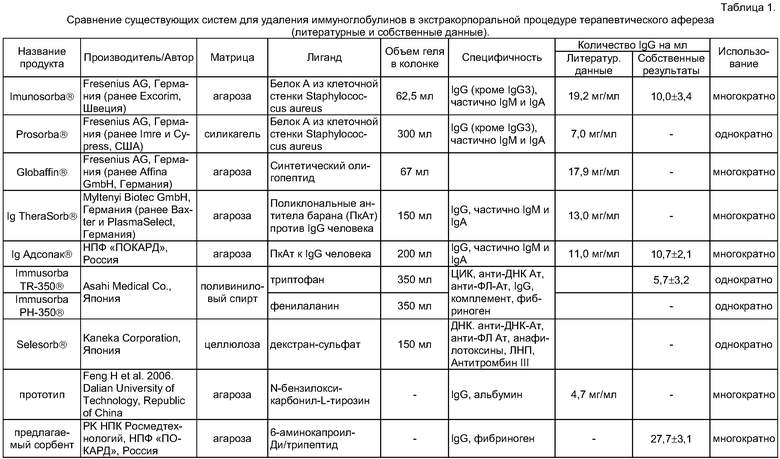
В клинической практике в настоящее время для Ig афереза (удаления иммуноглобулинов) наиболее широко применяют сорбенты с иммобилизованными поликлональными антителами к иммуноглобулинам человека («Ig Адсопак»®, НПФ «ПОКАРД», Россия; «Ig Therasorb»®, «Miltenyi Biotec»®, Германия) [5, 8], колонки с иммобилизованным белком A («Immunosorba»® и «Prosorba»®, «Fresenius HemoCare», Германия) [9].
Основные недостатки данных сорбентов: 1) использование в процессе производства продуктов животного или бактериального происхождения (для всех перечисленных сорбентов), 2) невозможность стерилизации продукта, содержащего протеин, на последних стадиях производства, что требует использования асептических условий на протяжении практически всего процесса производства колонок (также для всех перечисленных сорбентов), 3) для некоторых из вышеназванных сорбентов - только однократное применение [9], 4) утечка биологически активного лиганда, что может привести к побочным эффектам (в первую очередь для сорбентов с иммобилизованным протеином A).
Описана колонка с иммобилизованными пептидами из 11-13 аминокислотных остатков, способная связывать иммуноглобулины G «Globaffin», («Fresenius», Германия), содержащая 67 мл агарозного геля с иммобилизованным пептидом PGAM146 [10]. Колонки «Globaffin» удаляют широкий спектр иммуноглобулинов человека и разработаны для многократного применения. Сорбционная емкость сорбента составляет в среднем 17 мг IgG на мл геля [10]. Несмотря на то, что колонки были разработаны в 2003 г., новых данных об использовании колонок в клинической практике за последние 5 лет не появилось.
Близким аналогом предлагаемого изобретения является сорбент с иммобилизованным триптофаном, используемый в колонках «TR-350» Asahi Medicals [11]. Колонки содержат 350 мл геля, представляющего собой триптофан, иммобилизованный на полимерную матрицу на основе поливинилового спирта. Основным недостатком данного сорбента является ограниченная специфичность, например высокий уровень сорбции фибриногена.
Прототипом настоящего изобретения является сорбент с иммобилизованным на сефарозу производным L-тирозина (N-бензилоксикарбонил-L-тирозин) [12]. Концентрация лиганда на матрице составляет 35 мкмоль/мл (15 мг/мл геля). Сорбент способен селективно связывать иммуноглобулины G из плазмы крови человека в ряду нескольких хроматографических циклов. Максимальная сорбционная емкость сорбента составляла 17 мг IgG на мл геля. Соотношения иммуноглобулинов и альбумина в элюате с колонки составляет 33,7 и 64,5% соответственно, а количество иммуноглобулинов G, сорбированных из 10 мл плазмы крови, - человека 9,5 мг на 2 мл геля [12].
Основные недостатки прототипа: 1) сложность синтеза сорбента (так, предложенная в прототипе схема синтеза включает 4 стадии, на каждой из которых используются органические растворители); 2) высокий уровень сорбции сывороточного альбумина - транспортного белка; 3) в случае утечки лиганда неизвестна токсичность N-карбобензокситирозина и его метаболитов; 4) не было проведено исследований по термостабильности сорбента (возможности стерилизации автоклавированием).
Задачей изобретения является упрощение схемы синтеза сорбента, минимизация сорбции альбумина сорбентом, повышение безопасности и емкости сорбента.
Поставленная задача решается использованием ковалентно иммобилизованных на нерастворимую полимерную матрицу синтетических низкомолекулярных пептидов из ряда:
тирозил-триптофан (I),
триптофил-тирозин (II),
тирозил-глицил-триптофан (III),
триптофил-треонил-тирозин (IV).
Пептиды состоят из аминокислот белкового происхождения и имеют сродство к иммуноглобулинам человека и животных. Каждый пептид имеет в своем составе остатки двух ароматических аминокислот. Пептиды в случае утечки лиганда распадаются до аминокислот белкового происхождения, широко представленных в организме человека и являющихся компонентами растворов для парентерального питания [Регистр лекарственных средств России].
Пептиды I-IV были синтезированы классическими методами пептидной химии в растворе. Для блокирования α-аминогруппы использовали бензилоксикарбонильную или трет-бутилоксикарбонильную защиту, для защиты боковой функции тирозина применяли трет-бутиловый эфир. Конденсацию осуществляли методом активированных эфиров или смешанных ангидридов. После деблокирования конечных продуктов трифторуксусной кислотой пептиды очищали с помощью ВЭЖХ до 96-98% чистоты и характеризовали данными ЯМР-спектроскопии. Ковалентную иммобилизацию пептидов на сорбенте осуществляли двумя основными способами: 1) присоединением содержащего аминогруппы лиганда к полисахаридной матрице, предварительно активированной бромцианом; 2) присоединением лиганда, содержащего аминогруппу, к аминированной полисахаридной матрице конденсацией с глутаровым альдегидом, с предварительным аминированием полисахаридной матрицы через стадии окисления полимера периодатом натрия и последующего присоединения диаминопроизводных.
Сопоставление заявляемого сорбента с литературными данными аналогов и прототипа приведены в таблице 1, там же содержатся собственные данные сравнения сорбционной емкости предлагаемого сорбента с коммерчески доступными аналогами и прототипом.
Список сокращений
Вос - трет-бутилоксикарбонил;
Bu1 - трет-бутил;
Bzl - бензил;
DMF - N,N′-диметилформамид;
NMM - N-метилморфолин;
ONSu - N-оксисукцинимидный эфир;
ONp - п-нитрофениловый эфир;
Z - бензилоксикарбонил;
Tos - n-толуолсульфонропил;
DCHA - дициклогексиламин;
ТСХ - тонкослойная хроматография;
ВЭЖХ - высокоэффективная жидкостная хроматография
Экспериментальная часть
В синтезе использованы производные L-аминокислот фирмы Bachem (Швейцария). DMF перегоняли над нингидрином и окисью бария. Для экстракции и кристаллизации пептидов применяли растворители марки «ч» и «хч».
Индивидуальность промежуточных и конечных соединений подтверждали с помощью ТСХ на хроматографических пластинках Kiselgel 60 (Merck, Германия) в следующих системах растворителей:
Вещества на хроматограммах детектировали хлор-бензидиновым реагентом и нингидрином.
Гидрирование пептидов проводили в присутствии 5% палладия на угле (Pd/C, Merck, Германия).
Очистку пептидов проводили методом ВЭЖХ па колонке (25×250 мм) с диасорбом С16Т. Элюцию осуществляли градиентом концентрации (0,5% в мин) CH3CN в 0,1% трифторуксусной кислоте, скорость потока 9 мл в минуту, детекция при 226 нм. Фракции, соответствующие целевым продуктам, объединяли, упаривали. Остаток растворяли в воде и лиофилизировали.
Для иммобилизации на нерастворимую матрицу использовали 6-аминокапроил-производные пептидов. 6-аминокапроновая кислота добавлена в структуру в качестве вспомогательного соединения (спейсера), связывающего лиганд с матрицей.
Для аналитической ВЭЖХ использовали колонку Ultrasphere ODS, 5 мкм (4,6×200 мм) Beckman (США), в качестве элюентов применяли буфер А - 0,1% трифторуксусная кислота и буфер Б - 80% ацетонитрила в буфере А. Скорость потока 1 мл/мин, детекция при 220 нм. Градиент от 10% до 70% Б в А за 30 минут.
Пример 1. 6-аминокапроил-триптофил-тирозин (I)
1,65 г (5 ммоль) Z-Trp-OH растворяли в 25 мл этилацетата, добавляли 0,55 мл (5 ммоль) NMM, охлаждали до -30°С, добавляли 0,65 мл (5 ммоль) изобутилхлорформиата. Реакционную смесь выдерживали в течение 10 минут при -30°С, добавляли 1,65 г (5 ммоль) HCl-Tyr·(But)OBut и 0,55 мл (5 ммоль) NMM. Реакционную смесь выдерживали в течение 30 минут при комнатной температуре (полноту протекания реакции контролировали с помощью ТСХ; Rf целевого продукта 0,85 (А), 0,55 (B)). Реакционную смесь промывали в делительной воронке 5% NaHCO3, водой, 5% раствором лимонной кислоты, водой, упаривали. Остаток растворяли в 50 мл этанола и гидрировали. Катализатор отфильтровывали, промывали на фильтре этанолом, фильтрат упаривали, остаток растворяли в 10 мл этилацетата, охлаждали до -30°С (раствор 1).
1,16 г (5 ммоль) Вос-ε-аминокапроновой кислоты растворяли в 25 мл этилацетата, добавляли 0,55 мл (5 ммоль) NMM, охлаждали до -30°С, добавляли 0,65 мл (5 ммоль) изобутилхлорформиата. Реакционную смесь выдерживали в течение 10 минут при -30°С и добавляли к раствору 1, полученному ранее. Реакционную смесь выдерживали в течение 30 минут при комнатной температуре, добавляли 10 мл н-бутанола, промывали в делительной воронке водой, упаривали, остаток кристаллизовали из этилацетата, выпавший осадок отфильтровывали, промывали на фильтре этилацетатом, гексаном, сушили. Получено 2,14 г (выход 58%) продукта. Полученный продукт растворяли в смеси 25 мл трифторуксусной кислоты и 0,25 мл меркаптоэтанола, выдерживали в течение 1 часа при комнатной температуре, упаривали, остаток обрабатывали эфиром, выпавший осадок отфильтровывали, промывали на фильтре эфиром, сушили. Полученный продукт подвергали очистке методом ВЭЖХ, как описано выше. Получали 1,28 г (выход 43% в расчете на Z-Trp-OH) соединения (I) 98% чистоты по данным аналитической ВЭЖХ. Rt 18,15; Rf 0,24 (A).
1Н-ЯМР - спектр (DMSO-d6, δ, м.д.):
6-аминокапроновая к-та - 2,00 (α-CH2, 2H); 1,35 (β-СН2, 2H); 1,11 (γ-CH2, 2Н); 1,43 (δ-СН2, 2H); 2,68 (ε-CH2, 2H); 7,60 (NH, 1H)
Trp - 7,87 (NH, 1H); 4,57 (α-CH, 1H); 3,10/2,85 (β-CH2, 2H)
Tyr - 8,1 (NH, 1H); 4,36 (α-СН, 1H); 2,95/2,81 (β-CH2, 2H); 9,13 (ОН)
Пример 2. 6-аминокапроил-тирозил-триптофан 7 (II)
2,04 г (10 ммоль) триптофана растворяли в смеси 30 мл ДМФ и 5 мл 40% тритона В (10 ммоль) в метаноле. Метанол упаривали, остаток охлаждали до -5°С (раствор 2).
3,72 г (10 ммоль) растворяли в 25 мл ДМФ, добавляли 1,11 мл (10 ммоль) NMM, охлаждали до -30°С, добавляли 1,3 мл (10 ммоль) изобутилхлорформиата. Реакционную смесь выдерживали в течение 10 минут при -10°С, добавляли раствор 2. Реакционную смесь выдерживали в течение 30 минут при комнатной температуре (полноту протекания реакции контролировали с помощью ТСХ; Rf целевого продукта 0,7 (В)), упаривали, остаток растворяли в воде, подкисляли 15% лимонной кислотой до рН 3,0, продукт экстрагировали 200 мл этилацетета, промывали в делительной воронке водой, упаривали. Остаток кристаллизовали в гексане, отфильтровывали, промывали на фильтре гексаном, сушили. Получено 5,4 г (выход 97%). Полученный продукт растворяли в 100 мл этанола и гидрировали в присутствии 5% Pd/C. После завершения гидрирования (ТСХ: Rf 0,25 (А)), к реакционной смеси добавляли 5 мл 40% раствора тритона В в метаноле. Катализатор отфильтровывали, промывали на фильтре этанолом, упаривали, остаток растворяли в 50 мл ДМФА, упаривали наполовину. К полученному раствору добавляли 2,6 г (8 ммоль) N-гидроксисукцинимидного эфира Вос-6-аминокапроновой кислоты. Реакционную смесь выдерживали в течение 10 часов при комнатной температуре, упаривали, остаток растворяли в 100 мл воды, экстрагировали эфиром, водный слой подкисляли 15% лимонной кислотой до pH 3,0, экстрагировали этилацетатом, промывали водой до нейтральной реакции, упаривали, остаток обрабатывали гексаном. Выпавший осадок отфильтровывали, промывали на фильтре гексаном, сушили. Полученный продукт растворяли в смеси 25 мл трифторуксусной кислоты и 0,25 мл меркаптоэтанола, выдерживали в течение 1 часа при комнатной температуре, упаривали, остаток обрабатывали эфиром, выпавший осадок отфильтровывали, промывали на фильтре эфиром, сушили. Полученный продукт подвергали очистке методом ВЭЖХ, как описано выше. Получено 3,65 г (60% в расчете на первую стадию) соединения (II) 97% чистоты по данным аналитической ВЭЖХ. Rt 18,52; Rf 0,21 (А).
1Н-ЯМР - спектр (DMSO-d6, δ, м.д.):
6-аминокапроновая к-та - 1.99 (α-CH2, 2H); 1.36 (β-CH2, 2H); 1,12 (γ-CH2, 2H); 1,44 (δ-CH2, 2Н); 2,70 (ε-CH2, 2H); 7.61 (NH, 1H)
Tyr - 7,86 (NH, 1H); 4,47 (α-СН, 1H); 2,89/2,58 (β-CH2, 2H); 9,13 (ОН)
Trp - 8,80 (NH, 1H); 4,47 (α-CH, 1H); 3,18/3,06 (β-CH2, 2H)
Пример 3. 6-аминокапроил-тирозил-глицил-триптофан (III)
0,55 г (1 ммоль) Z-Tyr(But)-OH·DCHA суспендировали в 50 мл этилацетата, органический слой промывали в делительной воронке 2% серной кислотой, водой, упаривали досуха. Остаток растворяли в 5 мл ДМФА, добавляли 0,11 мл (1 ммоль) NMM, охлаждали до -30°С, добавляли 0,13 мл (1 ммоль) изобутилхлорформиата. Реакционную смесь выдерживали в течение 3 минут при -10°С, добавляли 0,38 г (1 ммоль) Tos·H-Gly-OBzl и NMM. Реакционную смесь выдерживали в течение 1 часа при комнатной температуре, упаривали, остаток растворяли в 25 мл этилацетата, промывали в делительной воронке 5% NaHCO3, водой, 2% раствором серной кислоты, водой, упаривали. Остаток растворяли в 20 мл этанола и гидрировали. Катализатор отфильтровывали, промывали на фильтре этанолом, упаривали, остаток растворяли в 10 мл ДМФА, добавляли к нему 0,11 мл (1 ммоль) NMM и 0,35 г (1 ммоль) N-гидроксисукцинимидного эфира Вос-ε-аминокапроновой кислоты и выдерживали при комнатной температуре в течение 18 часов.
Затем реакционную смесь упаривали, остаток растворяли в этилацетате, органический слой подкисляли 2% серной кислотой, промывали водой до нейтральной реакции, упаривали досуха. Остаток растворяли в 25 мл этилацетата, полученный раствор наносили на колонку 1,6 см2×20 см с силикагелем 60, продукт элюировали этилацетатом. После упаривания фракций, соответствующих целевому продукту, и кристализации остатка в гексане получено 0,45 г (выход 86%) продукта.
Полученный продукт растворяли в 10 мл ДМФА, добавляли 0,11 мл (1 ммоль) NMM, охлаждали до -20°С, добавляли 0,13 мл (1 ммоль) изобутилхлорформиата. Реакционную смесь выдерживали в течение 3 минут при -5°С, добавляли охлажденный до -5°С раствор тритоновой соли триптофана, полученной из 0,21 г триптофана, как описано выше. Реакционную смесь выдерживали в течение 1 часа при комнатной температуре, упаривали, остаток растворяли в 25 мл этилацетата, промывали в делительной воронке, 15% раствором лимонной кислоты, водой до нейтральной реакции, упаривали досуха. Получали 0,57 г продукта в виде аморфного порошка. Полученный продукт растворяли в 25 мл трифторуксусной кислоты и 0,25 мл меркаптоэтанола, выдерживали в течение 1 часа при комнатной температуре, упаривали, остаток обрабатывали эфиром, выпавший осадок отфильтровывали, промывали на фильтре эфиром, сушили. Полученный продукт подвергали очистке методом ВЭЖХ, как описано выше. Получено 0,36 г (выход 53,8%) соединения (III) 97% чистоты по данным аналитической ВЭЖХ. Rt 17,74; Rf 0,18 (A).
1Н-ЯМР - спектр (DMSO-d6, δ, м.д.):
6-аминокапроновая к-та - 2,03 (α-CH2, 2H); 1,38 (β-CH2, 2Н); 1,13 (γ-СН2, 2H); 1,44 (δ-СН2, 2H); 2,70 (ε-CH2, 2H); 7,60 (NH, 1H)
Tyr - 7,96 (NH, 1H); 4,41 (α-СН, 1H); 2,88/2,61 (β-СН2, 2H); 9,14 (ОН)
Gly - 8,18 (NH, 1H); 3,78/3,63 (α-CH2, 2H);
Trp - 8,09 (NH, 1H); 4,48 (α-СН, 1H); 3,18/3,04 (β-CH2, 2H)
Пример 4. 6-аминокапроил-триптофил-треонил-тирозин (IV)
0,12 г (1 ммоль) треонина растворяли в 1 мл 1 М NaOH, охлаждали до 0°С, к полученному раствору добавляли раствор 0,46 г Z-Trp-ONp в 5 мл ДМФА, охлажденный до 0°С. Реакционную смесь выдерживали в течение 18 часов при комнатной температуре, упаривали, остаток растворяли в 50 мл воды, экстрагировали смесью этилацетат-гексан 1:1 (2×25 мл), водный слой подкисляли 15% лимонной кислотой до рН 3,5, экстрагировали этилацетатом, органический слой промывали водой до нейтральной реакции, упаривали досуха, остаток растворяли в 10 мл ДМФА, добавляли 0,11 мл (1 ммоль) NMM, охлаждали до -20°С, добавляли 0,13 мл (1 ммоль) изобутилхлорформиата. Реакционную смесь выдерживали в течение 5 минут при -10°С, добавляли 0,33 г (1 ммоль) HCl Tyr(But)OBut и NMM. Реакционную смесь выдерживали в течение 1 часа при комнатной температуре, упаривали, остаток растворяли в 25 мл этилацетата, промывали в делительной воронке 5% NaHCO3, водой, 5% раствором лимонной кислоты, водой, упаривали. Остаток растворяли в 20 мл этанола и гидрировали в присутствии 5% Pd/C (Rf 0,45 (А)). Катализатор отфильтровывали, промывали на фильтре этанолом, упаривали, остаток растворяли в 10 мл ДМФА, добавляли к нему 0,3 г (0,86 ммоль) N-гидроксисукцинимидного эфира Вос-ε-аминокапроновой кислоты и выдерживали при комнатной температуре в течение 18 часов.
Затем реакционную смесь упаривали, остаток растворяли в этилацетате, органический слой промывали в делительной воронке 5% NaHCO3, водой, 15% лимонной кислотой, упаривали, остаток растворяли в смеси 10 мл трифторуксусной кислоты и 0,1 мл меркаптоэтанола, выдерживали в течение 1 часа при комнатной температуре, упаривали, остаток обрабатывали эфиром, выпавший осадок отфильтровывали, промывали на фильтре эфиром, сушили. Полученный продукт подвергали очистке методом ВЭЖХ, как описано выше. Получено 0,3 г (выход 42%) целевого продукта 97% чистоты по данным аналитической ВЭЖХ. Rt 17,66; Rf - 0,17 (А).
1Н-ЯМР - спектр (DMSO-d6, δ, м.д.):
6-аминокапроновая к-та - 2,04 (α-CH2, 2H); 1,44 (β-СН2, 2Н); 1,14 (γ-СН2, 2Н); 1,38 (δ-CH2, 2H); 2,69 (ε-CH2, 2Н); 7,60 (NH, 1H)
Tyr - 7,87 (NH, 1H); 4,40 (α-CH, 1H); 2,93/2,84 (β-CH2, 2H); 9,20 (OH)
Thr - 7,77 (NH, 1Н); 4,24 (α-CH, 1H); 3,96 (β-СН, 1H); 1,01 (γ-СН3, 3Н)
Trp - 8,01 (NH, 1Н); 4,60 (α-CH, 1H); 3,13/2,93 (β-СН2, 2H)
Пример 5. Синтез сорбентов с иммобилизованными пептидами. Синтез сорбента на матрице, активированной бромцианом.
В качестве агарозной матрицы использовали коммерческие матрицы Sepharose 4 Fast Flow и матрицу, пригодную для перфузии клеток Sepharose 6МВ. В 15 мл 30% суспензии агарозной матрицы в растворе, содержащем 4 М КОН и 1,6 М KH2PO4, при температуре 5° добавляли 1 мл раствора 0,6 г/мл BrCN в диоксане. Смесь инкубировали в течение 10 мин при той же температуре, постоянно помешивая. Затем гель промывали десятикратным объемом дистиллированной воды на стеклянном фильтре при помощи колбы Бунзена с водоструйным насосом, затем готовили суспензию геля в буферной смеси 0,2 М H3BO3-NaOH рН 8,0. В полученную суспензию добавляли раствор лиганда из расчета 20-30 мкмоль на мл осевшего геля. Иммобилизацию проводили при непрерывном перемешивании при температуре 20°С. Через 2 часа количество связавшегося лиганда составляло 65-70% от количества, взятого в иммобилизацию. Затем добавляли 1 мл 1 М этаноламина рН 8,0 и через 30 мин гель промывали десятикратным объемом воды и затем пятикратным объемом буферной смеси 0,1 М KH2PO4-NaOH с рН 7,0. Количество иммобилизованного лиганда оценивали по разнице между добавленным количеством и количеством в первых смывах с полученного сорбента.
Пример 6. Синтез сорбентов с иммобилизованными пептидами. Синтез сорбента на матрице, активированной периодатом натрия с последующим аминированием и конденсацией лиганда глутаровым альдегидом.
Приготовленную 30% суспензию промытой агарозной матрицы в 1% водном растворе NaIO4 инкубировали при перемешивании в течение 2 часов при 20°С. Гель промывали 20-кратным объемом воды на стеклянном фильтре. В отжатый гель добавляли равный объем 2 М водного раствора диаминогексана/диаминоэтана. Инкубировали 2 часа при 20°С. Через 2 часа несвязавшийся лиганд удаляли на стеклянном фильтре и промывали сорбент 20-кратным объемом дистиллированной воды. К отжатому гелю дважды добавляли 2-кратный объем 1,2% свежеприготовленного водного раствора NaBH4 и инкубировали дважды по 10 минут. Затем гель промывали 10-кратным объемом воды, 2-кратным объемом буферной смеси 0,1 М Na2CO3-NaHCO3 pH 9,0 и добавляли равный объем раствора пептида в том же буфере и 25% раствор глутарового альдегида из расчета 30 мкл на 1 мл геля. Инкубировали суспензию при перемешивании в течение 2 часов при 20°С. Затем добавляли двукратный объем 1,2% свежеприготовленного водного раствора NaBH4. Через час промывали 20-кратным раствором дистиллированной воды. Количество иммобилизованного лиганда контролировали также, как описано в п.5.
Следует подчеркнуть, что в приведенном примере 6 синтез сорбента проводится преимущественно в водных растворах, а в процессе синтеза практически отсутствуют токсические вещества и органические растворители.
Пример 7. Иммобилизация пептида на Toyopearl-AF65.
Эпоксиактивированный гель Toyopearl-AF65 (Tosoh Bioscience, Япония) промывали водой (100 мл на 5 мл геля). К 1 г осевшего геля добавляли 1 мл 2 М раствора 1,6-диаминогексана и перемешивали смесь в течение 2 часов при температуре 50°С. Затем полученный сорбент (Toyopearl-AF65-NH-C6H12-NH2) промывали 50 мл воды. Готовили раствор пептида 6-аминокапроил-Trp-Thr-Tyr концентрации 17 мг/мл в буферной смеси 0,1 М NaHCO3-NaOH с pH 9,6. Для присоединения пептида к 1 мл осевшего геля Toyopearl-AF65-NH-C6H12-NH2 добавляли 1 мл раствора пептида, 1 мл диметилсульфоксида и 0,1 мл 2,6 М раствора глутарового альдегида. Перемешивали смесь в течение 2 часа при температуре 20°С. Через 2 часа в смесь добавляли 1 мл свежеприготовленного 1% раствора NaBH4, перемешивали 30 минут, после этого гель промывали 5 мл воды и затем 1 мл 0,1 М КН2РО4-NaOH pH 7,0. Количество иммобилизованного лиганда составило 30,2 мг на мл геля.
Сорбционная емкость сорбента на основе Sepharose с иммобилизованным пептидом.
При нанесении раствора, содержащего иммуноглобулины G на колонку с сорбентом с иммобилизованным в концентрации 30 мкмоль/мл геля (14,6 мг/мл геля) пептидом триптофил-тирозин (Trp-Tyr) количество связавшегося IgG составляет 19±1 мг IgG на мл геля, что превышает аналогичный показатель для прототипа на 12%.
При нанесении плазмы крови человека на сорбент с иммобилизованными пептидами в условиях, аналогичных описанному, для прототипа [12] количество связавшихся иммуноглобулинов G составляет от 18,3 до 27,7 мг/мл геля, в отличие от прототипа - 4,7 мг/мл геля (см. таблицу 2).
При пропускании 5 мл крови человека, стабилизированной цитратом натрия в конечной концентрации 0,6% через хроматографическую колонку, сечением 1 см2, содержащую 1 мл геля Sepharose 6MB-6-аминокапроил-TrpThrTyr, при скорости тока 0,2 мл/мин сорбционная емкость сорбента составила 11,5±1,6 мг IgG (таблица 2а).
Данные о гемосовместимости сорбента на основе Sepharose 6МВ в опытах in vitro при перфузии цельной крови приведены в таблице 2б. Некоторое снижение концентрации форменных элементов (кроме тромбоцитов) относительно исходного происходит за счет эффекта разведения крови физиологическим раствором в процессе проведения хроматографического цикла.
Сорбционная емкость сорбента на основе Toyopearl-AF65.
Сорбционная емкость сорбента с лигандом 6-аминокапроил-Trp-Thr-Tyr на основе матрицы Toyopearl-AF65 составила 8,3±0,4 мг IgG человека на 1 мл геля, что почти в 2 раза превышает аналогичный показатель для прототипа. Использование в качестве матрицы геля на основе сшитого поливинилового спирта подтверждает возможность использования полимерных матриц, отличных по своей природе от агарозной матрицы (Sepharose) для синтеза сорбентов, содержащих в качестве лиганда заявленные пептиды.
Связывание иммуноглобулинов различного видового происхождения из белковых растворов.
При нанесении на сорбент с 6-аминокапроил-Trp-Tyr-Tyr, синтезированный, как описано в примере 6, растворов IgG различного видового происхождения (концентрация IgG 5 мг/мл в физиологическом растворе) количество связавшихся иммуноглобулинов составило 23,4 мг IgG козы, 13,9 мг IgG барана и 10,5 мг IgG быка на мл геля. В качестве контроля специфичности связывания иммуноглобулинов проверяли также сорбцию бычьего сывороточного альбумина (БСА) и человеческого сывороточного альбумина (ЧСА) из растворов с концентрацией белка 5 мг/мл в физиологическом растворе. Связывания сорбентом БСЛ и ЧСА не наблюдалось. Следовательно, показана способность сорбентов на основе заявленных пептидов избирательно связывать иммуноглобулины различного видового происхождения из белковых растворов.
Специфичность связывания сорбентов с иммобилизованными дипептидами.
Специфичность взаимодействия пептидов с иммуноглобулинами, т.е. способность специфически связывать иммуноглобулины G из смеси белков (плазмы крови человека), определяли методом «сорбции в объеме» с плазмой крови человека.
Присутствие в элюатах иммуноглобулинов G человека подтверждено исследованием состава элюатов методами твердофазного иммуноферментного анализа и вертикального электрофореза в градиенте полиакриламидного геля (градиент полиакриламида от 4 до 15%) в денатурирующих условиях. Анализ электрофореграммы показал, что основным компонентом элюата с сорбентом являются иммуноглобулины класса G. Элюат не содержит значительных количеств сывороточного альбумина и других примесных белков, тогда как в прототипе количество сорбированного сывороточного альбумина составляет 64% от общего количества связавшегося с сорбентом белка [21].
Стабильность сорбентов при автоклавировании.
Стабильность сорбентов в условиях стерилизации методом автоклавирования подтверждена измерениями сорбционной емкости сорбентов на модельных растворах иммуноглобулинов G человека с образцами сорбентов до и после стерилизации в автоклаве в стандартных для стерилизации жидких сред условиях (121°С, 1,1 Бар, 30 мин).
Количество иммуноглобулинов, связавшихся с неавтоклавированным и автоклавированным образцами сорбентов с иммобилизованным пептидом триптофил-тирозин (Trp-Tyr) для периодатного метода иммобилизации, составляет 19,5±0,7 мг/мл геля и 15,2±1,9 мг/мл геля, соответственно; падение сорбционной емкости при автоклавировании не превышало 22%.
Основными преимуществами предлагаемого сорбента относительно прототипа являются:
1) упрощение схемы синтеза сорбентов,
2) минимизация использования токсических веществ и органических растворителей в процессе синтеза сорбентов,
3) использование биологически совместимых природных аминокислот,
4) отсутствие сорбции транспортного белка альбумина,
5) более высокая сорбционная емкость по сравнению с прототипом,
6) возможность стерилизации сорбента.
Список использованных источников
1. McMillan R. // Autoantibodies and autoantigens in chronic immune thrombocytopenic purpura. / Semin. Hematol. - 2000. - V.37 (3). - P.239-248.
2. Schulze K., Becker B.F., Schauer R., Schultheiss H.P. // Antibodies to ADP-ATP carrier - an autoantigen in myocarditis and dilated cardiomyopathy - impair cardiac function. / Circulation. - 1990 - V.81. - P.959-969.
3. Magnusson Y., Wallukat G., Waagstein F., Hjalmarson A., Hoebeke J. // Autoimmunity in idiopathic dilated cardiomyopathy. Characterization of antibodies against the β1-adrenoceptor with positive chronotropic effect. / Circulation. - 1994 - V.89. - P.2760-2767.
4. Muller J., Wallukat G., Dandel M., Bieda H., Brandes K., Spiegelsberger S., Nissen E., Kunze R., Hetzer R. / Immunoglobulin adsorption in patients with idiopathic dilated cardiomyopathy // Circulation. - 2000 - V.101. - P.385-391.
5. Коновалов Г.А., Беленков Ю.Н., Звездкин П.В., Чебышев A.M., Семин С.Н., Кузнецова Ю.В., Адамова И.Ю., Кипор С.Г., Покровский С.Н. / Аферез иммуноглобулинов - новый подход к лечению тяжелых форм дилатационной кардиомиопатии. // Кардиология. - 2002. - Т.6. - С.92-96.
6. Moreso F., Poveda R., Gil-Vernet S., Carreras L., Garcia-Osuna R., Grino J.M., Alsina J. // Therapeutic immunoadsorption in Goodpasture disease. / Med. Clin. (Barc) - 1995 Jun. 10. - V.105 (2) - P.59-61.
7. Robinson J.A. // Apheresis in thoracic organ transplantation. / Ther. Apher. - 1999 Feb. - V.3 (1) - P.34-39.
8. Koll R.A. // Ig-Therasorb immunoadsorption for selective removal of human immunoglobulins in diseases associated with pathogenic antibodies of all classes and IgG subclasses, immune complexes, and fragments of immunoglobulins. / Ther. Apher. - 1998 May. - V.2 (2). - P.147-152.
9. Euler H.H., Schwab U.M., Schroeder J.O., Hasford J. // The Lupus Plasmapheresis Study Group: rationale and updated interim report. / Artif Organs. - 1996. - V.20 (4). - P.356-359.
10. Ronspeck W., Brinekmann R., Egner R., et. al. // Peptide based adsorbers for therapeutic immunoadsorption. / Ther Apher Dial. - 2003. - V.7 (1). - P.91-97.
11. Hirata N., Kuriyama T., Yamawaki N. Immusorba TR and PH. Ther Apher Dial. 2003 Feb; 7 (1): 85-90.
12. Feng H., Jia L., Li H., Wang X. // Screening and chromatographic assessing of a novel IgG biomimetic ligand. / Biomed Chromatogr. 2006; 20 (10): 1109-15.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИНТЕТИЧЕСКИЙ АНТИГЕН, ОБЛАДАЮЩИЙ СПОСОБНОСТЬЮ СВЯЗЫВАТЬ АУТОАНТИТЕЛА К β-АДРЕНОРЕЦЕПТОРУ | 2007 |
|
RU2356576C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДОДЕКАПЕПТИДА И ТРИПЕПТИД ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2007 |
|
RU2340626C1 |
| СПОСОБ ИММУНОФЕРМЕНТНОГО АНАЛИЗА ДЛЯ ОПРЕДЕЛЕНИЯ АУТОАНТИТЕЛ К β-АДРЕНОРЕЦЕПТОРУ В ПЛАЗМЕ И СЫВОРОТКЕ КРОВИ ЧЕЛОВЕКА | 2011 |
|
RU2452964C1 |
| СОРБЕНТ ДЛЯ УДАЛЕНИЯ ИММУНОГЛОБУЛИНОВ | 2006 |
|
RU2325172C2 |
| КОНЪЮГАТ АНТИТЕЛА К TROP-2 И АНАЛОГА ЭКЗАТЕКАНА И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2830167C1 |
| КОНЪЮГАТ АНТИТЕЛО К B7H3-АНАЛОГ ЭКЗАТЕКАНА И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2019 |
|
RU2785664C2 |
| ПОЛУЧЕНИЕ ПЕПТИДОВ СОМАТОСТАТИНА | 2004 |
|
RU2360921C2 |
| СПОСОБ ПОЛУЧЕНИЯ НОНАПЕПТИДЭТИЛАМИДА | 1995 |
|
RU2086561C1 |
| Способ получения полипептидов или их солей | 1977 |
|
SU910116A3 |
| АМИД ОКТАПЕПТИДА, ОБЛАДАЮЩИЙ СПОСОБНОСТЬЮ ПОВЫШАТЬ АРТЕРИАЛЬНОЕ ДАВЛЕНИЕ И ЧАСТОТУ СЕРДЕЧНЫХ СОКРАЩЕНИЙ | 2007 |
|
RU2346001C1 |
Изобретение относится к иммунологии, в частности к сорбенту для удаления иммуноглобулинов класса G из биологических жидкостей и растворов, содержащих иммуноглобулины, в том числе из плазмы и крови человека. Сорбент представляет собой полимерную матрицу, ковалентно связанную через спейсер с лигандом, в качестве спейсера используют вещества из ряда: 6-аминокапроновая кислота, глутаровый альдегид, остаток 6-аминокапроила, сконденсированного с глутаровым альдегидом, а в качестве лиганда используют синтетические ди- и трипептиды, содержащие два остатка ароматических аминокислот. Сорбент представляет собой полимерную матрицу, ковалентно связанную с лигандом, представленным синтетическими ди- и трипептидами, содержащими два остатка ароматических аминокислот. Предлагаемый сорбент имеет упрощенную схему синтеза, с минимальным использованием токсических веществ и органических растворителей, обеспечивает снижение сорбции сывороточного альбумина и других примесных белков, повышение сорбционной емкости и специфичности сорбента. Кроме этого предлагаемый сорбент может быть подвергнут стерилизации. 2 н.п. ф-лы, 4 табл.
1. Сорбент для удаления иммуноглобулинов класса G из биологических жидкостей и растворов, содержащих иммуноглобулины, в том числе из плазмы или крови человека, представляющий собой полимерную матрицу, ковалентно связанную через спейсер с лигандом, отличающийся тем, что в качестве спейсера используют вещества из ряда: 6-аминокапроновая кислота, глутаровый альдегид, остаток 6-аминокапроила, сконденсированного с глутаровым альдегидом, а в качестве лиганда используют синтетические пептиды из ряда: тирозил-триптофан, триптофил-тирозин, тирозил-глицил-триптофан, триптофил-треонил-тирозин.
2. Сорбент для удаления иммуноглобулинов класса G из биологических жидкостей и растворов, содержащих иммуноглобулины, в том числе из плазмы или крови человека, представляющий собой полимерную матрицу, ковалентно связанную с лигандом, отличающийся тем, что в качестве лиганда используют синтетические пептиды из ряда: тирозил-триптофан, триптофил-тирозин, тирозил-глицил-триптофан, триптофил-треонил-тирозин.
| Feng Н | |||
| et al | |||
| Screening and chromatographic assessing of a novel IgG biomimetic ligand | |||
| Biomed Chromatogr | |||
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| JP 2004301759 A, 28.10.2004 | |||
| WO 9717980 A1, 22.05.1997 | |||
| СПОСОБ ОЧИСТКИ КРОВИ ОТ РЕВМАТОИДНОГО ФАКТОРА | 1991 |
|
RU2027192C1 |

