Настоящее изобретение относится к новым производным бензотиазина и их применению в качестве антибактериальных средств при инфекционных заболеваниях млекопитающих (людей и животных), вызываемых бактериями, особенно при таких заболеваниях, как туберкулез (ТБ) и проказа, вызываемых микобактериями.
Тиазиноны, их производные и их применение в качестве антибактериальных средств, особенно против микобактерий (ТБ), описаны, например, в AR 242567 А1, AU 3704400 A1, CA 1322551 C1 или ЕР 0245901 В1.
Как известно, во всем мире наблюдается угрожающий рост заболеваемости туберкулезом, вызываемого микобактериями, выработавшими устойчивость к имеющимся лекарствам (B.R.Bloom, J.L.Murray. Tuberculosis: commentary on a reemergent killer. Science 257, 1992, 1055-1064). Чрезвычайно опасным является распространение микобактерий, обладающих множественной лекарственной устойчивостью (МЛУ). Таковыми являются микобактерии, устойчивые по меньшей мере к двум из наиболее активных лекарств от туберкулеза, изониазиду и рифампицину, а также устойчивые к стрептомицину, пиранзинамиду и этамбутолу. Доля МЛУ-ТБ в некоторых странах уже превышает 20%. С учетом общего роста количества заболеваний туберкулезом это ежегодно служит причиной около 3000000 смертей по всему миру.
Для лечения таких заболеваний, как ТБ или проказа, существует острая необходимость в новых лекарствах с новыми механизмами действия, особенно для преодоления лекарственной устойчивости и для преодоления известных существенных побочных эффектов имеющихся лекарств.
Задача изобретения
Настоящее изобретение направлено на создание новых соединений, обладающих активностью против микобактерий, как потенциально новых лекарств от туберкулеза, призванных решить проблемы, связанные с устойчивостью и лекарственной непереносимостью.
Решение технической проблемы
Цель была достигнута получением соединений формулы I
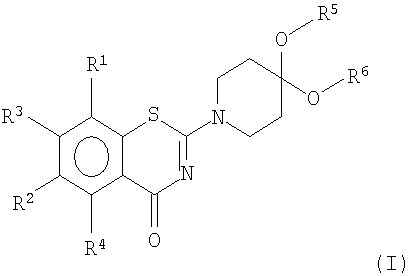
или его соли,
где R1 и R2 независимо друг от друга представляют собой NO2, CN, CONR7R8, COOR9, СНО, галоген, SO2NR7R8, OCF3, моно-, ди- или трифторметил;
R3 и R4 независимо друг от друга представляют собой Н или метил;
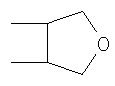
R5 и R6 независимо друг от друга представляют собой линейный или разветвленный алифатический радикал, имеющий 1-8 членов в цепи, или R5 и R6 вместе представляют собой бивалентный радикал -(CR9 2)m-, или R5 и R6 вместе представляют собой бивалентный радикал:
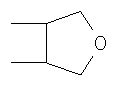
где m равен 1-4;
R7, R8 и R9 независимо представляют собой Н или линейный или разветвленный алифатический радикал, имеющий 1-7 членов в цепи, или фенил.
В предпочтительном варианте осуществления настоящее изобретение относится к соединениям формулы (I), выбранным из группы, состоящей из:
2-(4-R5-4-R6-пиперидин-1-ил)-8-нитро-6-трифторметил-1,3-бензотиазин-4-он,
6-циано-2-(4-R5-4-R6-пиперидин-1-ил)-8-нитро-1,3-бензотиазин-4-он,
6-амидо-2-(4-R5-4-R6-пиперидин-1-ил)-8-нитро-1,3-бензотиазин-4-он,
2-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-8-Rl-6-R2-1,3-бензотиазин-4-он,
2-(2-метил-1,4-диокса-8-азаспиро[4.5]дец-8-ил)-8-Rl-6-R2-1,3-бензотиазин-4-он,
2-[(2R)-2-метил-1,4-диокса-8-азаспиро[4.5]дец-8-ил]-8-Rl-6-R2-1,3-бензотиазин-4-он,
2-[(2S)-2-метил-1,4-диокса-8-азаспиро[4.5]дец-8-ил]-8-Rl-6-R2-l,3-бензотиазин-4-он,
2-(2,3-диметил-1,4-диокса-8-азаспиро[4.5]дец-8-ил)-8-Rl-6-R2-1,3-бензотиазин-4-он,
2-(1,5-диокса-9-азаспиро[5.5]ундец-9-ил)-8-Rl-6-R2-1,3-бензотиазин-4-он,
где R1, R2, R5 и R6 имеют значения, указанные выше.
Более конкретно, настоящее изобретение относится по меньшей мере к одному соединению, выбранному из группы, состоящей из:
2-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-8-нитро-6-(трифторметил)-1,3-бензотиазин-4-он,
2-(7,12-диокса-8-азаспиро[5.6]додец-3-ил)-6,8-динитро-1,3-бензотиазин-4-он,
2-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-7-метил-6,8-динитро-1,3-бензотиазин-4-он,
2-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-8-нитро-4-оксо-1,3-бензотиазин-6-карбонитрил,
2-(1,4-диокса-8-азампиро[4.5]дец-8-ил)-6,8-динитро-1,3-бензотиазин-4-он,
2-(2-метил-1,4-диокса-8-азаспиро[4.5]дец-8-ил)-6,8-динитро-1,3-бензотиазин-4-он,
2-(4,4-диэтоксипиперидин-1-ил)-6,8-динитро-1,3-бензотиазин-4-он,
7-метил-2-(2-метил-1,4-диокса-8-азаспиро[4.5]дец-8-ил)-6,8-динитро-1,3-бензотиазин-4-он,
2-(2-метил-1,4-диокса-8-азаспиро[4.5]дец-8-ил)-8-нитро-6-(трифторметил)-1,3-бензотиазин-4-он,
2-(2,3-диметил-1,4-диокса-8-азаспиро[4.5]дец-8-ил)-8-нитро-6-(трифторметил)-1,3-бензотиазин-4-он,
2-(1,5-диокса-9-азаспиро[5,.5]ундец-9-ил)-8-нитро-6-(трифторметил)-1,3-бензотиазин-4-он,
2-(1,5-диокса-9-азаспиро[5.5]ундец-9-ил)-8-нитро-4-оксо-1,3-бензотиазин-6-карбонитрил,
2-(2-метил-1,4-диокса-8-азаспиро[4.5]дец-8-ил)-8-нитро-4-оксо-1,3-бензотиазин-6-карбонитрил.
Для получения целевых соединений мы разработали оригинальный способ синтеза 1,3-бензотиазин-4-она с использованием производных дитиокарбамата в качестве промежуточного соединения (способ А). Классический способ синтеза 1,3-бензотиазин-4-она с использованием тиоцианатных солей (способ В) также пригоден. Оба способа представлены на схеме ниже.
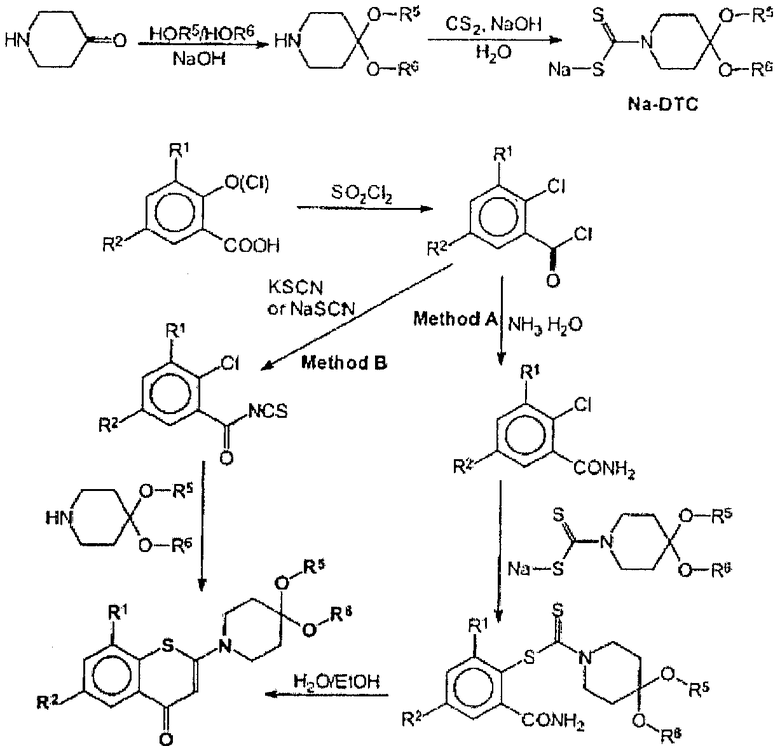
Соединения по настоящему изобретению неожиданно демонстрируют высокую антибактериальную активность, особенно против микобактерий, с величинами минимальных подавляющих концентраций (МПК) в диапазоне от 0,23 пг/мл до не более 10 мкг/мл для быстрорастущих микобактерий, в диапазоне 0,195-1,56 мкг/мл для М. tuberculosis, включая полирезистентные штаммы, определенными классическим способом, и величиной МПК 0,030 мкг/мл для М. tuberculosis H37Rv, определенной методом с использованием красителя Alamar Blue. Соединения по настоящему изобретению неожиданно демонстрируют высокий уровень селективности по отношению исключительно к микобактериям, что существенно снижает вероятность вредных побочных эффектов.
Соединения по настоящему изобретению не мутагенны при концентрации 5 мг/мл в SOS-хромотесте.
Соединения по настоящему изобретению терапевтически активны in vivo на мышиной модели туберкулезной инфекции, превосходя основное противотуберкулезное лекарство изониазид, использованное в качестве положительного контроля. Выжили 100% мышей. Все животные в контрольной группе умерли не позднее 33 дня.
Соединение по настоящему изобретению (особенно соединение №2 = пример 1 в примерах осуществления изобретения) не токсично при пероральном введении дозировок в диапазоне до 2000 мг/кг, и представляет собой соединение, хорошо переносимое животными в первый и последующие 24 часа после введения. В течение 7 дней исследований соединение 2 не вызвало изменений в общем состоянии и поведении мышей, не повлияло на двигательную и рефлекторную активность, циклы активности и спокойствия, чистку, потребление пищи, не было случаев смерти животных. LD50 для соединения 2 составляет >2000 мг/кг.
Таким образом, соединения по настоящему изобретению пригодны для лечения туберкулезной инфекции и других инфекций, вызванных микобактериями, у людей и животных.
Соответственно настоящее изобретение относится к фармацевтическим композициям, содержащим соединение формулы I.
Изобретение также относится к соединению формулы I для использования в способе лечения бактериальных инфекций у млекопитающих. Предпочтительными соединениями формулы I для использования в таком способе являются соединения, особо перечисленные выше.
Препараты соединений по настоящему изобретению получают приготовлением разбавленных растворов или суспензий в фармацевтически приемлемой водной, органической или водно-органической среде для местного или парентерального введения внутривенной, подкожной или внутримышечной инъекцией, или для интраназального применения; или выпускаются в форме таблетки, капсулы или водной суспензии с традиционными наполнителями для перорального введения или в форме суппозитория.
Соединения можно использовать в дозировках 0,001 -1000 мг/кг массы тела.
Описанные далее примеры в приведенной ниже экспериментальной части служат для иллюстрации изобретения, но не должны пониматься как ограничивающие данное изобретение.
Структуры соединений по настоящему изобретению устанавливали по способу синтеза и данным элементного анализа, а также с помощью метода ядерного магнитного резонанса и/или масс-спектрометрии, а также рентгеноструктурным анализом.
Примеры осуществления изобретения
Исходные материалы
Реагенты и растворители приобретали в Lancaster Synthesis (Lancashire, Англия) или в Aldrich (Sigma-Aldrich Company, St-Louis, США) и использовали в синтезе без дополнительной очистки. Температуры плавления определяли в соответствии с ВР процедурой и не корректировали (Electrothermal 9001, GB). Если в результатах анализа указаны только символы элементов, то экспериментально найденные значения находились в пределах ±0.3% от теоретически рассчитанных значений (Carlo-Erba 5500, Италия). ЯМР спектры записывали на приборе Varian Unity Plus 400 (США). Химические сдвиги для 1Н ЯМР выражены в м.д. в слабое поле относительно ТМС (δ). Масс-спектры записывали на приборе Finnigan SSQ-700 (США) с прямым вводом. Протекание реакций и чистоту соединений контролировали методом ТСХ с использованием алюминиевых пластинок Silicagel 60 F254 (Merck Co, Германия).
Пример 1
2-(1,4-Диокса-8-азаспиро[4.5]дец-8-ил)-8-нитро-6-(трифторметил)-1,3-бензотиазин-4-он, (соединение 1)
Способ А.
К 50 мл перемешиваемого 25%-ного водного раствора аммиака прикапывали раствор 5 г 2-хлор-3-нитро-5-трифторметилбензоилхлорида (D.E Welch, R.R.Baron, B.A.Burton, J. Med. Chem. 12; 2; 1969; 299-303) в ацетонитриле (10 мл) при -20°С. Через 10 мин добавляли 50 мл этилацетата. Органическую фазу отделяли, дважды промывали водой, высушивали над Na2SO4, обрабатывали активированным углем, фильтровали и концентрировали в вакууме. Сырой продукт очищали кристаллизацией из этанола. Выход 2-хлор-3-нитро-5-(трифторметил)бензамида составил 92%. Т.пл. 195-197°С (метанол). Вычислено для C8Н4ClFN2O3: С, 35,78; Н, 1,50; N, 10,43
Найдено: С, 36,01; Н, 1,53; N, 10,39
0,5 г 2,2-хлор-3-нитро-5-(трифторметил)бензамида растворяли в 25 мл этанола. Реакционную смесь обрабатывали 0,5 г дигидрата натриевой соли 1,4-диокса-8-азаспиро[4.5]декан-8-дитиокарбоновой кислоты (Z. Ge, R. Li, Т. Cheng, Synth. Commun., 29, 18, 1999, 3191 - 3196) и оставляли на 18 ч при комнатной температуре. Затем реакционную смесь выливали в 50 мл охлажденной воды и отфильтровывали полученный желтый осадок. Чистый конечный продукт получали после двукратной перекристаллизации из этанола. 2-(Аминокарбонил)-6-нитро-4-(трифторметил)фенил-1,4-диокса-8-азаспиро[4.5]декан-8-карбодитиоат представляет собой светло-желтое кристаллическое вещество. Выход 0,47 г.Т.пл. 138-140°С.
Вычислено для C11H12N4O2S2: С, 42,57; Н, 3,57; N, 9,31; S, 14,21
Найдено: С, 42,61; Н, 3,67; N, 9,22; S, 14,30
0,4 г 2-(аминокарбонил)-6-нитро-4-(трифторметил)фенил-1,4-диокса-8-азаспиро[4.5]декан-8-карбодитиоата растворяли в 25 мл этанола. Реакционную смесь обрабатывали 0,32 г Na2HPO4×12H2O и кипятили в течение 6 ч. Затем реакционную смесь охлаждали, светло-желтый осадок отфильтровывали и промывали 30 мл метанола. Чистый конечный продукт получали после двукратной перекристаллизации из этанола. 2 - представляет собой светло-желтое кристаллическое вещество. Выход 0,47 г. Т.пл. 211-212°С.
Rf (гексан-ацетон; 2/1) - 0,35
MC:m/z 417(M+).
1Н ЯМР (ДМСО-d6) δ 8,83 и 8,77 (два 1Н, два с, 2СН), 3,80 (8Н, уширенный с, N(CH2CH2)2C), 2,02 (4Н, уширенный с, OCH2CH2O) м.д.
Вычислено для C16H14F3N3O5S: С, 46,04; Н, 3,38; N, 10,07; S, 7,68
Найдено: С, 45,94; Н, 3,37; N, 10,09; S, 7,76
Способ В. Методика в точности повторяла методику, описанную в J.Imrich, P.Kristian, Coll. Czech. Chem. Commun., 47, 1982. 3268-3282; D.Koscik, P.Kristian, J.Gonda, E.Dandarova, Coll. Czech. Chem. Commun., 48, 1983, 3315-3328; D.Koscik, P.Kristian, O.Forgac, Coll. Czech. Chem. Commun., 48, 1983, 3427-3432; Т.Н.Cronin, Н. - J.E. Hess, Pat. US 3522247. Выход 2-(1,4-диокса-8-аза-спиро[4.5]дец-8-ил)-8-нитро-6-трифторметил)-1,3-бензо-тиазин-4-она составлял 0,21 г. Соединение по спектроскопическим данным идентично соединению, полученному способом А.
Пример 2
2-(2-Метил-1,4-диокса-8-азаспиро[4.5]дец-8-ил)-8-нитро-6-(трифторметил)-1,3-бензотиазин-4-он, (соединение 2)
Получено по методике, описанной в Примере 1. Светло-желтое кристаллическое вещество. Выход 54%. Т.пл. 192-3°С.
Rf (гексан-ацетон; 2/1) - 0,30.
MC: m/z 431 (M+).
1H ЯМР (ДМСО-d6) δ 8,81 и 8,77 (два 1Н, два с, 2СН), 4,24 (1Н, м, СН), 4,11 (1Н, м, СН), 4,06 (4Н, уширенный с, N(CH2)2), 3,47 (1Н, т, СН), 3,27 (1Н, с, СН), 1,80 (4Н, уширенный д, С(СН2)2), 1,23 (3Н, д, СН3) м.д.
Вычислено для C17H16N3O5S: С, 47,33; Н, 3,74; N, 9,74; S, 7,43
Найдено: С, 47,36; Н, 3,80; N, 9,87; S, 7,51
Пример 3
2-(1,4-Диокса-8-азаспиро[4.5]дец-8-ил)-6,8-динитро-1,3-бензотиазин-4-он, (соединение 4)
Получено по методике, описанной в Примере 1, с использованием 2-гидрокси-3,5-динитробензойной кислоты в качестве исходного материала. Светло-желтое кристаллическое вещество. Выход 43%. Т.пл. 271-3°С (EtOH/ДМФА).
Rf (гексан-ацетон; 2/1) - 0,25.
MC: m/z 394 (M+).
1H ЯМР (ДМСО-d6) δ 9,15 и 9,12 (два 1H, два с, 2СН), 3,86 (8Н, уширенный с, N(CH2CH2)2C), 2,97 (4Н, уширенный с, OCH2CH2O) м.д.
Вычислено для C15H14N4O7S: С, 45,68; Н, 3,58; N, 14,21; S, 8,13
Найдено: С, 45,34; Н, 3,56; N, 14,30; S, 7,98
Пример 4
2-(2-Метил-1,4-диокса-8-азаспиро[4.5]децил)-6,8-динитро-1,3-бензотиазин-4-он, (соединение 4)
Получено по методике, описанной в Примере 1, с использованием 2-гидрокси-3,5-динитробензойной кислоты в качестве исходного материала. Желтое кристаллическое вещество. Выход 57%. Т.пл. 139-142°С (EtOH/ДМФА).
Rf (гексан-ацетон; 2/1) - 0,50.
MC: m/z 408 (М+).
1Н ЯМР (ДМСО-d6) δ 9,08 и 9,11 (два 1H, два с, 2СН), 4,23 (1Н, м, СН), 4,10 (1Н, м, СН), 4,06 (4Н, уширенный с, N(HC2)2), 3,43 (1Н, т, СН), 3,27 (1Н, с, СН), 1,80 (4Н, уширенный д, С(СН2)2), 1,20 (3Н, д, СН3) м.д.
Вычислено для C16H16N4O7S: С, 47,06; Н, 3,95; N, 13,72; S, 7,85
Найдено: С, 46,87; Н, 3,91; N, 13,57; S, 7,83
Пример 5
2-(2,3-Диметил-1,4-диокса-8-азаспиро[4.5]дец-8-ил)-8-нитро-6-(трифторметил)-1,3-бензотиазин-4-он, (соединение 5)
Получено по методике, описанной в Примере 1, с использованием 2-гидрокси-3-нитро-5-трифторметилбензойной кислоты в качестве исходного материала. Светло-желтое кристаллическое вещество. Выход 58%. Т.пл. 205-207°С (EtOH/ДМФА).
Rf (гексан-ацетон; 2/1) - 0,55.
MC: m/z 44522 (M+).
1Н ЯМР (ДМСО-d6) δ 8,82 и 8,77 (два 1Н, два с, 2СН), 3,86 (4Н, уширенный с, N(CH2)2), 3,45-3,53 (2Н, м, 2СН), 2,41 (4Н, уширенный д, С(СН2)2), 1,13-1,17 (6Н, м, 2СН3) м.д.
Вычислено для C18H18F3N3O5S: С, 48,54; Н, 4,07; N, 9,43; S, 7,20
Найдено: С, 48,66; Н, 4,12; N, 9,32; S, 7,46
Пример 6
2-(4,4-Диэтоксипиперидин-1 -ил)-6,8-динитро-1,3-бензотиазин-4-он, (соединение 6)
Получено по методике, описанной в Примере 1, с использованием в качестве исходного материала 2-гидрокси-3,5-динитробензойной кислоты. Желтое кристаллическое вещество. Выход 32%. Т.пл. 179-181°С (i-PrOH).
Rf (гексан-ацетон; 2/1) - 0,30.
MC: m/z 424(M+).
1H ЯМР (ДМСО-d6) δ 9,08 и 9,11 (два 1Н, два с, 2СН), 3,60-3,67 (4Н, м, N(CH2)2) 2,11-2,08 (4Н, м, С(СН2)2), 3,47 и 3,57 (два 2Н, кв, 2OCH2), 1,16 (6Н, т, 2СН3), м.д.
Вычислено для C17H20N4O7S: С, 48,11; Н, 4,75; N, 13,20; S, 7,56
Найдено: С, 48,12; Н, 4,73; N, 13,41; S, 7,67
Пример 7
2-(7,12-Диокса-3-азаспиро[5.6]додец-3-ил)-6,8-динитро-1,3-бензотиазин-4-он, (соединение 7)
Получено по методике, описанной в Примере 1, с использованием в качестве исходного материала 2-гидрокси-3,5-динитробензойной кислоты. Желтое кристаллическое вещество. Выход 51%. Т.пл. 193-195°С (i-PrOH/ДМФА).
Rf (гексан-ацетон; 2/1) - 0,45.
MC: m/z 422(M+).
1H ЯМР (ДМСО-d6) δ 8,97 и 9,16 (два 1Н, два с, 2СН), 3,57-3,74 (8Н, м, 4СН2), 1,93-2,35 (8Н, м, 4СН2) м.д.
Вычислено для C17H18H4O7S: С, 48,34; Н, 4,30; N, 13,26; S, 7,56
Найдено: С, 48,21; Н, 4,43; N, 13,30; S, 7,66
Пример 8
2-(1,4-Диокса-8-азаспиро[4.5]дец-8-ил)-7-метил-6,8-динитро-1,3-бензотиазин-4-он, (соединение 8)
Получено по методике, описанной в Примере 1, с использованием в качестве исходного материала 2-гидрокси-4-метил-3,5-динитробензойной кислоты. Желтое кристаллическое вещество. Выход 51%. Т.пл. 207-210°С (i-PrOH/ДМФА).
Rf (гексан-ацетон; 2/1) - 0,30.
MC: m/z 408(M+).
1Н ЯМР (ДМСО-d6) δ 8,77 (1Н, с, СН), 3,86 (8Н уширенный с, N(CH2CH2)2C), 2,97 (4Н, уширенный с, OCH2CH2O), 2,79 (3Н с, СН3) м.д.
Вычислено для C16H16H4O7S: С, 47,06; Н, 3,95; N, 13,72; S, 7,85
Найдено: С, 47,12; Н, 4,01; N, 13,69; S, 7,94
Пример 9
2-(1,4-Диокса-8-азаспиро[4.5]дец-8-ил)-8-нитро-4-оксо-1,3-бензотиазин-6-карбонитрил, (соединение 9)
К перемешиваемому раствору 5 г (19 ммоль) 2-гидрокси-5-иодобензойной кислоты в 50 мл ДМФА добавляли небольшими порциями 2,5 г (22 ммоль) сухого CuCN (I). Реакционную смесь кипятили в течение 5 ч, добавляли 100 мл воды и 50 мл этилацетата. Затем при хорошей вентиляции осторожно добавляли концентрированную соляную кислоту, пока pH не достиг ~ 3. Органическую фазу отделяли, дважды промывали водой, высушивали над Na2SO4, обрабатывали активированным углем, фильтровали и концентрировали в вакууме. Сырой продукт очищали кристаллизацией из воды. Выход 5-циано-2-гидроксибензойной кислоты составил 71%. По методике, описанной в Примере 1. Выход 44%. Т.пл. 217-220°С (EtOH/ДМФА).
Rf (гексан-ацетон; 2/1) - 0,50.
MC: m/z 374(M+).
1H ЯМР (ДМСО-d6) δ 8,74 и 8,67 (два 1Н, два с, 2СН), 3,41 (8Н, уширенный с, N(CH2CH2)2C), 2,93 (4Н, уширенный с, OCH2CH2O) м.д.
Вычислено для C16H14N4O5S: С, 51,33; Н, 3,77; N, 14,97; S, 8,57
Найдено: С, 51,30; Н, 3,84; N, 14,89; S, 8,62
Пример 10
2-(2-Метил-1,4-диокса-8-азаспиро[4.5]дец-8-ил)-8-нитро-4-оксо-1,3-бензотиазин-6-карбонитрил, (соединение 10)
Получено по методике, описанной в Примере 9. Желтое кристаллическое вещество. Выход 34%. Т.пл. 251-253°С (EtOH/ДМФА).
Rf (гексан-ацетон; 2/1) - 0,40.
МС: m/z 388 (М+).
1Н ЯМР (ДМСО-d6) δ 8,73 и 8,61 (два 1Н, два с, 2СН), 4,23 (1Н, м, СН), 4,11 (1Н, м, СН), 4,07 (4Н, уширенный с N(CH2)2), 3,51 (1Н, т, СН), 3,27 (1Н, с, СН), 1,81 (4Н, уширенный д, С(СН2)2), 1,22 (3Н, д. СН3) м.д.
Вычислено для C17H16N4O5S: С, 52,57; Н, 4,15; N, 14,43; S, 8,26
Найдено: С, 52,42; Н, 4,08; N, 14,50; S, 8,27
Пример 11
2-(1,5-Диокса-9-азаспиро[5.5]ундец-9-ил)-8-нитро-4-оксо-1,3-бензотиазин-6-карбонитрил, (соединение 11)
Получено по методике, описанной в Примере 9. Желтое кристаллическое вещество. Выход 40%. Т.пл. 230-232°С (EtOH/ДМФА).
Rf (гексан-ацетон; 2/1) - 0,15.
МС: m/z 388 (М+).
1Н ЯМР (ДМСО-d6) 6 8,74 и 8,61 (два 1Н, два с, 2СН), 3,29-3,65 (6Н, м, 3СН2), 2,38 (4Н, уширенный с, 2СН2), 1,82-1,93 (4Н, м, 20%) м.д.
Вычислено для C17H16N4O5S: С, 52,57; Н, 4,15; N, 14,43; S, 8,26
Найдено: С, 52,52; Н, 4,11; N, 14,59; S, 8,13
Пример 12
Определение in vitro ингибирующей активности против микобактерий для соединений по настоящему изобретению.
Антибактериальную активность рассматриваемых соединений против Mycobacterium smegmatis SG 987, М. aureum SB66, М. vaccae IMET 1010670 и M.fortuitum В тестировали посредством определения минимальных подавляющих концентраций (МПК) методом микроразбавления в бульоне Мюллера-Хинтона (Difco) в соответствии с указаниями NCCLS [National Committee for Clinical Laboratory Standards: Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; 5th Ed.; Villanova, Ed.; Approved standard Document M7-A5. NCCLS, (2000)].
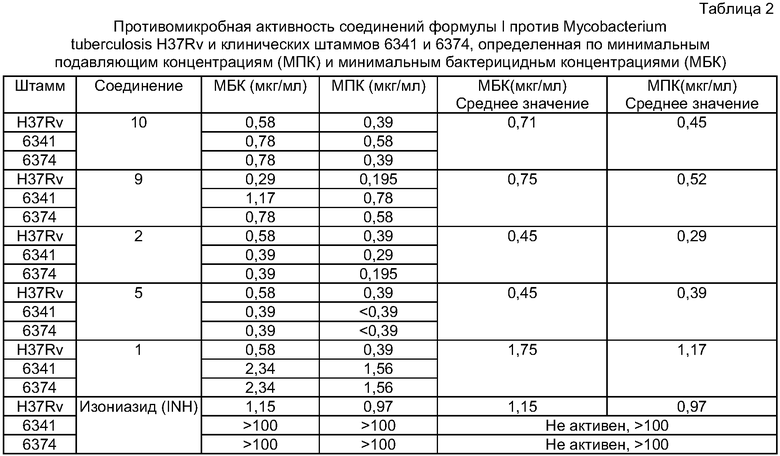
Активность против М. tuberculosis H37Rv тестировали следующим методом определения минимальных подавляющих концентраций (МПК) и минимальных бактерицидных концентраций (МБК).
Штаммы высевали на твердую среду Ловенштайна-Йенсена. Через 21 день выросшие культуры использовали для приготовления суспензий для посева, имеющих концентрацию 5×108 микробных клеток/мл. По 0,2 мл данной суспензии высевали в пробирки с 2 мл жидкой среды Школьниковой, содержащие соответствующие концентрации исследуемых соединений - от 100,0 до 0,195 мг/мл. После 14 дней инкубирования при 37°С, пробирки с жидкой средой центрифугировали 15 мин при 3000 об/мин. После удаления супернатанта осадок ресуспендировали в 0,8 мл стерильного 0,9%-ного раствора NaCl. 0,1 мл данной суспензии отбирали для приготовления мазков, которые затем окрашивали по методу Циль-Нильсена. Оставшийся осадок высевали в объемах 0,2 мл в три пробирки с твердой средой Ловенштайна-Йенсена, не содержащей лекарств, для определения минимальных бактерицидных концентраций (МБК). Результаты подводили после 21-28 дней культивирования при 37°С. Контролем служили пробирки с культурами тестовых штаммов, не подвергавшиеся обработке исследуемыми веществами.
Минимальными бактерицидными концентрациями лекарств (МБК) считали концентрацию лекарства, полностью подавляющую рост микобактерий на твердой среде. Бактериостатический эффект (МПК) характеризовался наличием в мазках всего лишь отдельных микобактерий и сильным уменьшением количества колоний, выросших на твердой среде, по сравнению с контрольными образцами.
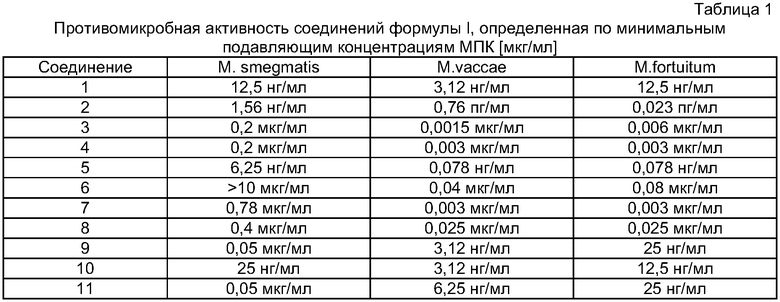
Результаты представлены в Таблицах 1 и 2.
Пример 13
Определение in vivo ингибирующей активности соединений по настоящему изобретению против Mycobacterium tuberculosis на мышиной модели ТБ
Для определения химиотерапевтической эффективности авторы использовали мышей линии BALB/c экспериментальным гематогенно-диссеминированным туберкулезом. Мыши были получены из Центрального Питомника Лабораторных Животных Российской Академии Медицинских Наук. В данное исследование авторы включали мышей после карантина, со стандартным весом (20-25 г) и только мужские особи. Мышей инфицировали 2-недельной вирулентной культурой Mycobacterium tuberculosis H37Rv посредством внутривенной инъекции (в хвостовую вену) микобактериальной суспензии в дозировке 5×106 КОЕ (колониеобразующих единиц) в 0,5 мл физиологического раствора. Всех экспериментальных животных разделяли на группы в зависимости от применяемых схем лечения (Таблица 3). Дозировки тестируемых лекарств выбирали исходя из литературных данных и результатов предшествующих исследований.
Лечение начинали на следующий день после инфицирования. Лекарства вводили перорально в виде суспензии в смеси карбоксиметилцеллюлоза/вода с небольшим количеством PEG-400.
Химиотерапию проводили ежедневно 6 раз в неделю (за исключением воскресенья).
Животных усыпляли эфирным наркозом. Для определения эффективности каждой схемы лечения авторы регистрировали макроскопические изменения в паренхиматозных органах мышей, рост микобактерий из патологического материала на твердой среде, а также бактериоскопический индекс поражения органов. Авторы проводили качественный и количественный анализ макроскопических изменений в печени, селезенке и легких и вычисляли индекс поражения (используя четырехбалльную шкалу).
Макроскопическое определение эффективности каждой схемы лечения выражали в индексе эффективности, вычисленном по формуле:
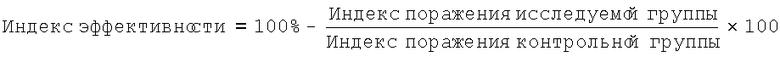
Микробиологическое исследование включало культуру для определения КОЕ в паренхиматозных органах. Для этого авторы гомогенизовали правое легкое и отдельно селезенку с 6%-ной серной кислотой, центрифугировали, промывали водой и солевым раствором. Полученное вещество (около 0,5 мл) разбавляли 1,0 мл солевого раствора и гомогенизовали. Данную суспензию (0,5 мл) исследуемых органов разбавляли в 100 и 1000 раз солевым раствором и распределяли на твердую среду Finn-2. Культуры инкубировали при 37°С в течение 1 месяца и контролировали еженедельно начиная с 10-го дня. Через 28 дней подсчитывали КОЕ.
Данные макроскопического и микробиологического исследований паренхиматозных органов мышей, умерших во время эксперимента, были также учтены в общей оценке результатов эксперимента, представленных в Таблицах 4-6.
Культура, без разбавления КОЕ
Культура, без разбавления КОЕ
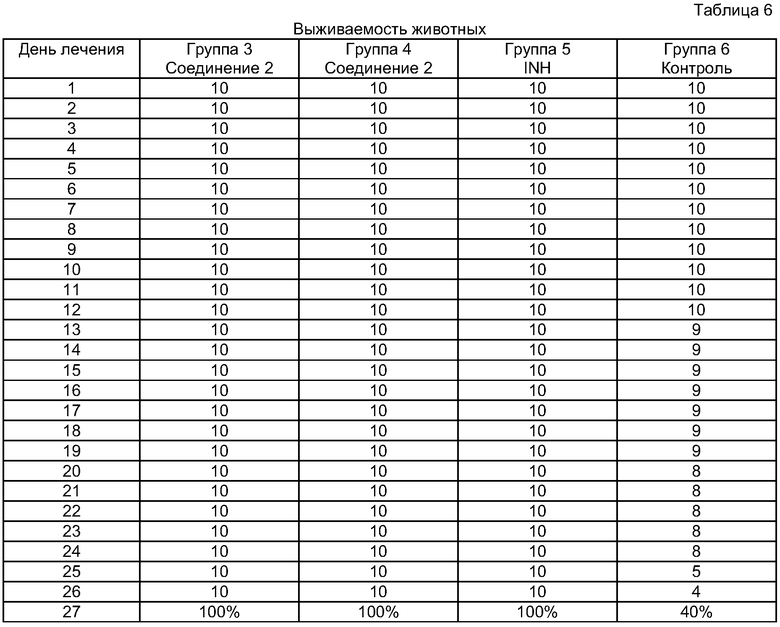
Все животные из контрольной группы умерли не позднее 33 дня.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ АНТИМИКРОБНЫЕ СОЕДИНЕНИЯ, ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ИНФЕКЦИЙ МЛЕКОПИТАЮЩИХ И НОВЫЙ МЕТАБОЛИЧЕСКИЙ МЕХАНИЗМ | 2017 |
|
RU2752568C2 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА И ПИРАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2687093C1 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2019 |
|
RU2809257C2 |
| ПРОИЗВОДНЫЕ ХИНОКСАЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СРЕДСТВО С КВИСКВАЛАТ-АНТАГОНИСТИЧЕСКИМ ДЕЙСТВИЕМ НА ИХ ОСНОВЕ | 1992 |
|
RU2117663C1 |
| ГЕРБИЦИДНЫЕ СОЕДИНЕНИЯ | 2020 |
|
RU2839723C2 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА И ПИРАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2686117C1 |
| ГЕРБИЦИДНЫЕ ЦИКЛОГЕКСАНДИОНОВЫЕ ПРОИЗВОДНЫЕ | 2020 |
|
RU2830286C2 |
| СПИРОАЗАЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИНГИБИРОВАНИЯ АКТИВНОСТИ ИЛИ АКТИВАЦИИ СЕРОТОНИНОВОГО 5-НТ2А РЕЦЕПТОРА, СПОСОБ ЛЕЧЕНИЯ БОЛЕЗНЕННОГО СОСТОЯНИЯ, СВЯЗАННОГО С СЕРОТОНИНОВЫМ 5-НТ2А РЕЦЕПТОРОМ, И ИХ ПРИМЕНЕНИЕ | 2002 |
|
RU2315051C2 |
| ЗАМЕЩЕННЫЕ БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ПРИМЕНЕНИЕ В КАЧЕСТВЕ СРЕДСТВ ПРОТИВ ОЖИРЕНИЯ И ГИПЕРХОЛЕСТЕРИНЕМИИ | 2000 |
|
RU2278114C2 |
| ПРОИЗВОДНЫЕ АРИЛКАРБОНОВЫХ КИСЛОТ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ И ПРЕДУПРЕЖДЕНИЯ РАЗЛИЧНЫХ ЗАБОЛЕВАНИЙ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1998 |
|
RU2247722C2 |
Описываются новые производные бензотиазинона формулы (I) и их использование в качестве антибактериальных средств при инфекционных заболеваниях, вызываемых бактериями, особенно вызываемых микобактериями туберкулеза (ТБ) и проказы, в которых R1 и R2 независимо друг от друга представляют собой NO2, CN, CONR7R8, COOR9, СНО, галоген, SO2NR7R8, OCF3, трифторметил; R3 и R4 независимо друг от друга представляют собой Н или метил; R5 и R6 независимо друг от друга представляют собой линейный или разветвленный алифатический радикал, имеющий 1-8 членов в цепи, или R5 и R6 вместе представляют собой бивалентный радикал -(CR9 2)m-, или R5 и R6 вместе представляют собой бивалентный радикал:
R7, R8 и R9 независимо представляют собой Н или линейный или разветвленный алифатический радикал, имеющий 1-7 членов в цепи, или фенил. Описывается также способ получения производных бензотиазинона, фармацевтическая композиция, обладающая активностью против микобактерий. 6 н. и 6 з.п. ф-лы, 6 табл.
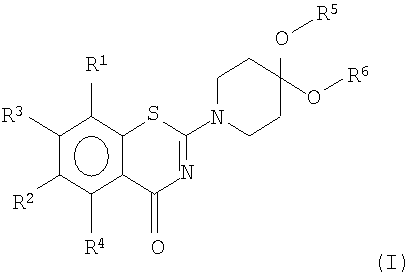
1. Соединение формулы I
или его соль,
где R1 и R2 независимо друг от друга представляют собой NO2, CN, CONR7R8, COOR9, СНО, галоген, SO2NR7R8, OCF3, или трифторметил;
R3 и R4 независимо друг от друга представляют собой Н или метил;
R5 и R6 независимо друг от друга представляют собой линейный или разветвленный алифатический радикал, имеющий 1-8 членов в цепи, или R5 и R6 вместе представляют собой бивалентный радикал -(CR9 2)m-, где m равен 1-4, или R5 и R6 вместе представляют собой бивалентный радикал:
R7, R8 и R9 независимо представляют собой Н или линейный или разветвленный алифатический радикал, имеющий 1-7 членов в цепи, или фенил.
2. Соединение формулы (I) по п.1, в котором R1 представляет собой NO2, R2 представляет собой CF3, R3 и R4 представляют собой Н, и R5 и R6 имеют значения, указанные в п.1.
3. Соединение формулы (I) по п.1, в котором R1 представляет собой NO2, R2 представляет собой CN, R3 и R4 представляют собой Н, и R5 и R6 имеют значения, указанные в п.1.
4. Соединение формулы (I) по п.1, в котором R1 и R2 представляет собой NO2, R3 и R4 представляют собой Н, и R5 и R6 имеют значения, указанные в п.1.
5. Соединение формулы (I) по п.1, в котором R5 и R6 независимо друг от друга представляют собой C1-8алкил.
6. Соединение формулы (I) по п.1, выбранное из группы, состоящей из следующих соединений:
2-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-8-нитро-6-(трифторметил)-1,3-бензотиазин-4-он,
2-(7,12-диокса-8-азаспиро[5,6]додец-3-ил)-6,8-динитро-1,3-бензотиазин-4-он,
2-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-7-метил-6,8-динитро-1,3-бензотиазин-4-он,
2-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-8-нитро-4-оксо-1,3-бензотиазин-6-карбонитрил,
2-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-6,8-динитро-1,3-бензотиазин-4-он,
2-(2-метил-1,4-диокса-8-азаспиро[4.5]дец-8-ил)-6,8-динитро-1,3-бензотиазин-4-он,
2-(4,4-диэтоксипиперидин-1-ил)-6,8-динитро-1,3-бензотиазин-4-он,
7-метил-2-(2-метил-1,4-диокса-8-азаспиро[4.5]дец-8-ил)-6,8-динитро-1,3-бензотиазин-4-он,
2-(2-метил-1,4-диокса-8-азаспиро[4.5]дец-8-ил)-8-нитро-6-(трифторметил)-1,3-бензотиазин-4-он,
2-(2,3-диметил-1,4-диокса-8-азаспиро[4.5]дец-8-ил)-8-нитро-6-(трифторметил)-1,3-бензотиазин-4-он,
2-(1,5-диокса-9-азаспиро[5.5]ундец-9-ил)-8-нитро-6-(трифторметил)-1,3-бензотиазин-4-он,
2-(1,5-диокса-9-азаспиро[5.5]ундец-9-ил)-8-нитро-4-оксо-1,3-бензотиазин-6-карбонитрил,
2-(2-метил-1,4-диокса-8-азаспиро[4.5]дец-8-ил)-8-нитро-4-оксо-1,3-бензотиазин-6-карбонитрил.
7. 2-(2-метил-1,4-диокса-8-азаспиро[4.5]дец-8-ил)-8-нитро-6-(трифторметил)-1,3-бензотиазин-4-он.
8. Применение соединения формулы (I) или его соли по любому из предшествующих пунктов для приготовления фармацевтической композиции, обладающей активностью против микобактерий.
9. Применение соединения по любому из пп.1-7 для приготовления лекарственного средства для терапевтического или профилактического лечения туберкулезной инфекции или инфекции проказы у млекопитающих.
10. Фармацевтическая композиция, обладающая активностью против микобактерий, включающая соединение по любому из пп.1-7.
11. Соединение по любому из пп.1-7 для использования в способе терапевтического или профилактического лечения туберкулезной инфекции или инфекции проказы у млекопитающих.
12. Способ получения соединения формулы (I), включающий стадию обработки соединения формулы:
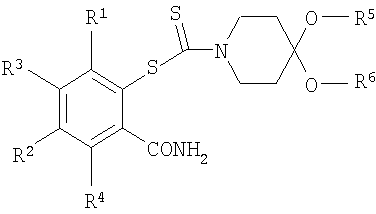
где заместители R1, R2, R5 и R6 такие, как они определены в п.1, и заместители R3 и R4 представляют собой водород, H2O/EtOH с получением соединения формулы (I).
| US 3522247, 28.07.1970 | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| 3-АЛКОКСИКАРБОНИЛ-ТИАДИАЗИНОНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2162087C2 |

