Настоящее изобретение относится к новым соединениям, ингибирующим серин-протеазу (MASP), ассоциированную со связывающим маннозу лектином (MBL), а также их аналогами и производными, к способам их получения, к их применению отдельно или в комбинациях для лечения и/или профилактики заболеваний и к их использованию для получения лекарственных средств для лечения и/или профилактики заболеваний, особенно для лечения и/или профилактики почечных и сердечно-сосудистых заболеваний и ишемически-реперфузионных повреждений.
Система комплемента состоит из сложной каскадной сети белков, рецепторов и ферментов, многие из которых циркулируют в кровотоке. Система комплемента является важным компонентом врожденного иммунитета и необходима для защиты от вторжения патогенов и уничтожения мертвых и инфицированных вирусом клеток. Она образует мостик между врожденными и адаптивными иммунными реакциями. Активация системы комплемента также вовлечена в патологию сепсиса и ишемически-реперфузионных повреждений, т.е. после инфаркта миокарда, ишемического повреждения почек или трансплантации органов. Выявлены три ветви системы комплемента: лектиновый путь, классический и альтернативный пути (Dunkelberger and Song, Complement and its role in innate and adaptive immune responses. Cell Res. 2010; 20(1): 34-50). Лектиновый путь активируется за счет отложения лектинов, которые циркулируют в кровотоке и в нормальных условиях выполняют охраную функцию против вторгшихся патогенов и мертвых клеток, распознавая чужеродные и измененные углеводные модели поверхности, соответственно, и декорируя их поверхности. Основными представителями лектинов, которые продуцируются в печени, почках и других органах, являются связывающий маннозу лектин (MBL), фиколины и коллектины (Garred et al, A journey through the lectin pathway of complement-MBL and beyond. Immunol Rev. 2016;274(1): 74-97). После их отложения происходит дальнейшее рекрутирование зимогенов двух по существу близкородственных сериновых протеаз из кровеносной системы, серинпротеаз, ассоциированных со связывающим маннозу лектином 1 и 2 (MASP-1 и MASP-2), образующих комплекс, в котором зимогены находятся в непосредственной близости друг от друга. Текущая концепция состоит в том, что в условиях in vivo зимоген MASP-1 после рекрутирования самоактивируется, а затем активирует зимоген MASP-2 путем расщепления. Кроме того, активированный MASP-1 расщепляет фактор комплемента С2 на С2а и C2b. Активированный MASP-2 также расщепляет С2 и фактор комплемента С4 на С4а и C4b, которые вместе с С2а образуют комплекс C4bC2a, выполняющий роль конвертазы фактора комплемента СЗ. Конституция С3-конвертазной активности и последовательное отложение СЗ на поверхности клеток-мишеней представляет собой точку схождения всех трех путей комплемента, активирующих общий каскад ниже по ходу потока, который приводит к образованию медиаторов воспаления и лизису клеток-мишеней. В сыворотке интактного человека активность обоих MASP-ферментов необходима для образования С3-конвертазы (Heja et al, Revised mechanism of complement lectin-pathway activation revealing the role of serine protease MASP-1 as the exclusive activator of MASP-2. P roc Natl Acad Sci USA. 2012; 109(26): 10498-503).
Микрососудистая система играет решающую роль при воспалительных и ишемических поражениях барьерной функции органов, перенос лейкоцитов и контроль коагуляции тесно зависят от целостности люминальной поверхности эндотелиальных клеток в мелких кровеносных сосудах. Люминальная эндотелиальная поверхность выстлана плотным слоем гликозилированных отростков из интегрированных в мембрану гликопротеи-нов, протеогликанов и гликолипидов, которые в совокупности называются гликокалик-сом. Электронно-микроскопические анализы образцов из экспериментов на животных и патологий человека показали, что, в частности, эндотелиальный гликокаликс быстро и основательно разрушается при ишемическом воздействии, а также при воспалительных состояниях, таких как сепсис.Эти изменения приводят к попаданию в кровоток остатков углеводов, которые в нормальных условиях не обнаруживаются (для обзора см.: Sieve et at, Regulation and function of endothelial glycocalyx layer in vascular diseases. Vascul Pharmacol. 2018; 100: 26-33). Помимо других изменений клеточной поверхности, в частности, полагают, что измененная углеводная модель активирует лектиновый путь, вызывая отложение лектинов модели распознавания, последующее отложение С3 и инициацию клеточного лизиса. Было показано, что отложение MBL и С3 происходит после ишемии и острого повреждения почек у разных видов, включая человека. Активация лектинового пути имела особое значение при реперфузионных повреждениях, поскольку направленная делеция MBL и MASP-2 защищала мышей от ишемических реперфузионных повреждений в почках, сердце и кишечнике (Moller-Kristensen et al, Mannan-binding lectin recognizes structures on ischaemic reperfused mouse kidneys and is implicated in tissue injury. Scand J Immunol. 2005; 61(5): 426-34; Schwaeble et al., Targeting of mannan-binding lectin-associated serine protease-2 confers protection from myocardial and gastrointestinal ischemia/reperfusion injury. Proc Natl Acad Sci USA. 2011; 108(18): 7523-8). Более того, делеция коллектина 11, другого лектина, активирующего MASP, который преимущественно экспрессируется в почках, сделала мышей устойчивыми к острому ишемическому повреждению почек (Farrar et al, Collectin-11 detects stress-induced L-fucose pattern to trigger renal epithelial injury. J Clin Invest. 2016; 126(5): 1911-1925). Селективные пептидные ингибиторы MASP 1 и MASP 2 были идентифицированы из библиотек фагового дисплея, используя в качестве отправной точки природные ингибиторы трипсина из подсолнечников или кузнечиков. Было показано, что эти пептиды ингибируют зависимое от лектинового пути образование С3-конвертазы in vitro (Kocsis et al., Selective inhibition of the lectin pathway of complement with phage display selected peptides against mannose-binding lectin-associated serine protease (MASP)-l and -2: significant contribution of MASP-1 to lectin pathway activation. J Immunol. 2010; 185(7): 4169-78; He]a et al., Monospecific inhibitors show that both mannan-binding lectin-associated serine protease-1 (MASP-1) and are essential for lectin pathway activation and reveal structural plasticity of MASP-2. J Biol Chem. 2012; 287(24): 20290-300). Однако пока нет доказательств фармацевтической полезности и эффективности этих пептидных ингибиторов in vivo. Точно так же антитела, направленные против MASP 2, которые препятствуют взаимодействию зимогена MASP, были идентифицированы и доведены до клинической разработки для атипичного гемолитико-уремического синдрома и других воспалительных заболеваний почек (ClinicalTrials.gov Identifier: NCT03205995; NCT 02682407; NCT 03608033). Однако клинические доказательства полезности для профилактики или лечения острых, в частности, ишемических поражений органов до сих пор отсутствуют.
В WO 2004/075837 описаны антитела против MASP, их функционально эквивалентные фрагменты и пептиды, связывающие MASP, для снижения заболеваемости и смертности, вызванных повреждением ткани, связанным с ишемически-реперфузионным повреждением, или восстановления ТААА путем ингибирования системы комплемента. Малые пептиды, такие как ингибитор MASP-1 (SFMI-1) подсолнечника и MASP-ингибитор-2 (SFMI-2) подсолнечника, а также их производные для лечения заболеваний, связанных с системой комплемента, прежде всего лектиновым путем, описаны в WO 2010/136831.
WO 2015/054298 раскрывает способы сохранения зрения или уменьшения потери зрения у субъекта и ингибирования или уменьшения гибели фоторецепторных клеток у субъекта путем снижения активности MASP-1, MASP-2 или MASP-3. WO 2004/106384, WO 2005/123128, WO 2007/117996 и WO 2014/144542 раскрывают антитела против MASP-2 для терапии заболеваний, связанных с MASP-2-зависимой активацией комплемента.
Задачей настоящего изобретения было создание новых пептидов, обладающих ин-гибирующим действием на ферменты MASP-1 и/или MASP-2 и другими полезными свойствами, делающими их подходящими в качестве эффективных и безопасных альтернатив для профилактики и/или лечения нарушений, связанных с MASP-1 и/или MASP-2, как определено ниже. Еще одной задачей было создание новых пептидов, обладающих улучшенным ингибирующим действием на ферменты MASP-1 и/или MASP-2 человека и/или ферменты MASP-1 крысы и/или MASP-2 крысы.
Настоящее изобретение в основном относится к пептидам, действующим как ингибиторы ферментов MASP-1 и/или MASP-2, а также к способам их получения и применения.
В частности, настоящее изобретение обеспечивает соединения, содержащие пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из следующей структурной формулы (I):
(Х0)р (Х1)q (X2)r X3 X4 X5 X6 X7 X8 X9 X10 X11 X12 (X13)s (X14)t (X15)u (I),
или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где
Х0 представляет собой группу формулы (IIa)
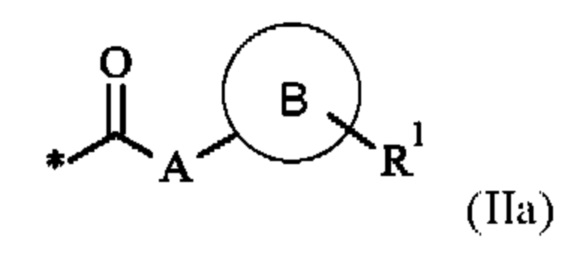
где
* указывает связь с концевой аминогруппой соседней аминокислоты,
А представляет собой связь или C1-С6-алкилен, где одна СН2 группа в C1-С6-алкилене может быть заменена на -О- или -S-, и где C1-С6-алкилен имеет до трех заместителей, которые представляют собой одинаковые или разные радикалы, выбранные из группы, состоящей из гидроксила, метокси, этокси, карбокси, амино и галогена,
В отсутствует, представляет собой арил, гетероарил, С3-С8-циклоалкил или С3-С7гетероциклоалкил,
где арил, гетероарил, С3-С8-циклоалкил и С3-С7-гетероциклоалкил может иметь до трех заместителей, которые представляют собой одинаковые или разные радикалы, выбранные из группы, состоящей из С1-С4-алкила, гидроксила, метокси, этокси, карбонила, карбокси, амино и галогена, и
R1 представляет собой водород, галоген, амино, гидроксил или С1-С20-алкил, где С1-С20-алкил имеет до трех заместителей, которые представляют собой одинаковые или разные радикалы, выбранные из группы, состоящей из гидроксила, карбокси, амино и галогена,
р представляет собой целое число 0 или 1,
X1 представляет собой любую природную аминокислоту или не встречающуюся в природе аминокислоту, где любая природная аминокислота и/или не встречающаяся в природе аминокислота может иметь D- или L-стереоконфигурацию,
И, когда р представляет собой 0, и q отличен от 0, концевая аминогруппа X1 является незамещенной, ацетилированной или моно- или дизамещенной С^-Сго-алкилом,
q представляет собой целое число от 0 до 5,
X2 представляет собой природную аминокислоту, выбранную из перечня, состоящего из I, L, М, V и А, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N-метилизолейцина ((N-Me)I), алло-Ь-изолейцина (алло-1), L-циклобутилаланина (Cba), L-норвалина (Nva), L-2-аминомасляной кислоты (Abu), (2S,3S)-2-[(3R)-3-амино-2-оксопирролидин-1-ил]-3-метилпентановой кислоты, (2S,3S)-2-[2-оксопиперазин-1-ил]-3-метилпентановой кислоты, (2S,3S)-2-[(3S)-2-оксопиперазин-1-ил]-3-метилпентановой кислоты, L-метионин-L-сульфоксида, L-метионин-сульфона и L-трет-бутилглицина,
где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации,
и, когда р и q оба представляют собой 0, и г представляет собой 1, концевая аминогруппа X2 является незамещенной, ацетилированной или моно- или дизамещенной С1-С20-алкилом,
r представляет собой целое число 0 или 1,
X3 представляет собой природную аминокислоту С, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-пеницилламина (Pen) и L-N-метилцистеина ((N-Me)C),
И, когда р и q и r все представляют собой 0, концевая аминогруппа X3 является незамещенной, ацетилированной или моно- или дизамещенной С1-С20-алкилом,
X4 представляет собой природную аминокислоту, выбранную из перечня, состоящего из S, С, Т, R или K, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из алло-L-треонина (алло-Т), L-гомосерина (hSer) и L-орнитина (Orn),
X5 представляет собой природную аминокислоту R или N(5)-метил-L-аргинин ((Me)R),
X6 представляет собой природную аминокислоту, выбранную из перечня, состоящего из S, С или Т, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из алло-L-треонина (алло-Т) и L-2,3-диаминопропионовой кислоты (Dap),
X7 представляет собой природную аминокислоту, выбранную из перечня, состоящего из L, F или N или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 2,3,3а, 4,5,6,7,7а-октагидроиндол-2-карбоновой кислоты (Oic), L-4-бромфенилаланина ((4-бромо)F), 2,5-дифтор-L-фенилаланина ((2,5-дифтор)Р), L-трет-бутилаланина ((tBu)A), 2-хлор-L-фенилаланина ((2-хлор)F), L-2-бромфенилаланина ((2-бромо)F), (S)-2-(амино)-1,6-гександионовой кислоты (AAD), (2S)-2-амино-4,4,4-трифторбутановой кислоты, Ь-2-амино-4-цианомасляной кислоты (Cnba), 4-фтор-лейцина ((4-фтор)L), (S)-(трифторметил)-Ь-цистеина, (2S)-2-амино-3-(1-метилциклопропил)пропановой кислоты, L-трет-бутил глицина ((tBu)G), 3-(триметилсилил)-Ь-аланина, 2,5-дифтор-L-фенилаланина, 2-амино-7-(трет-бутокси)-7-оксогептановой кислоты, 5,5,5-трифтор-L-лейцина ((трифтор)F), 2-метил-L-фенилаланина ((2-Me)F), L-циклобутилаланина (Cba), L-циклопентилаланина (Сра), L-циклопропилметилаланина, L-трифторметилаланина, L-дифторметилаланина, 2-фтор-L-фенилаланина ((2-фтор)F), (2S)-3-(2,3-дифторфенил)-2-аминопропановой кислоты, (2S)-3-(3-цианофенил)-2-аминопропановой кислоты, и (2S)-3-(индол-4-ил)-2-(амино)пропановой кислоты,
X8 представляет собой природную аминокислоту Р, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из (1S,2S,5R)-3-азабицикло[3.1.0]гексан-2-карбоновой кислоты, L-гидроксипролина (Hyp), (3S)-морфолин-3-карбоновой кислоты (морфолин-3-карбоновой кислоты), L-пипеколиновой кислоты (Pip), (4aR,6aR,9S,11aS)-11-оксо-2,3,4,4а,6а,7,8,9,11,11а-декагидро-1Н-пиридо[3,2-е]пирроло[1,2-а]азепин-9-карбоновой кислоты, транс-4-фторпролина ((транс-4-фтор)Р) и (1R,2S,5S)-3-азабицикло[3.1.0]гексан-2-карбоновой кислоты,
X9 представляет собой природную аминокислоту Р, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 2,3,За,4,5,6,7,7а-октагидроиндол-2-карбоновой кислоты (Oic), L-гидроксипролина (Hyp), (2S,4S)-4-трифторметил-пирролидин-2-карбоновой кислоты ((4-CF3)P), (2S,4S)-4-фторпролина ((цис-4-фтор)Р), транс-4-фторпролина ((транс-4-фтор)Р), (2S)-2-амино-4,4,4-трифторбутановой кислоты, L-транс-3-гидроксипролина ((3S-OH)P, (1R,3S,5R)-2-азабицикло[3.1.0]гексан-3-карбоновой кислоты, (68)-5-азаспиро[2.4]гептан-6-карбоновой кислоты, rel-(1R,3R,5R,6R)-6-(трифторметил)-2-азабицикло[3.1.0]гексан-3-карбоновой кислоты, (2S)-2-амино-4,4,4-трифторбутановой кислоты, (2S,3aS,6aS)-октагидроциклопента[b]пиррол-2-карбоновой кислоты, транс-4-фторпролина ((транс-4-фтор)Р), (2S,4S)-4-фторпролина ((цис-4-фтор)Р), L-4,4-дифторпролина ((дифтор)Р), rel-(3R,6R)-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновой кислоты (энантиомер 1) и rel-(311,611)-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновой кислоты (энантиомер 2),
X10 представляет собой природную аминокислоту I, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-циклопентил глицин a (Cpg), L-циклогексилглицина (Chg), (S)-2-амино-3-этил-пентановой кислоты, 3-хлор фенил глицина ((3-хлор-Ph)G), L-трет-бутилглицина, алло-L-изолейцина (алло-I), L-циклобутилглицина, L-норвалина (Nva) и (2S)-2-(амино)-2-[(1S,3R)-3-гидроксициклогексил]уксусной кислоты,
X11 представляет собой природную аминокислоту С, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N-метилцистеина ((N-Me)C) и L-пеницилламина (Реп),
X12 представляет собой природную аминокислоту I, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из алло-Ь-изолейцина (алло-1), (S)-2-амино-2-циклобутилуксусной кислоты (Cbg), (2S,3S)-2-((амино)метил)-3-метилпентановой кислоты, L-фенилглицина (Phg), 2-[(1S,2S)-1-(амино)-2-метилбутил]-1,3-оксазол-4-карбоновой кислоты, 2-метил-В-аллоизолейцина, L-норвалина (Nva), L-2-аминомасляной кислоты (Abu), L-трет-бутилглицина и Аминоизомасляной кислоты (Aib),
И, когда и и t и s все представляют собой 0, концевая карбоксильная группа X является незамещенной или амидированной,
X13 представляет собой природную аминокислоту, выбранную из перечня, состоящего из Р, A, S, Т, r, D, Е, Q или N или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из N-метил-глицина ((N-Me)G), 5-азаспиро[2.4]гептан-6-карбоновой кислоту, L-2-аминомасляной кислоты (Abu), 2-аминоизомасляной кислоты (Aib), 2-метил-Ь-пролина (2-Ме)Р, гидроксипролина (Hyp), 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновой кислоты (Oic), транс-4-фторпролина ((транс-4-фтор)Р), (2S,4S)-4-фторпролина ((цис-4-фтор)Р), L-4,4-дифторпролина ((дифтор)Р), L-циклопентилглицина (Cpg), (S)-2-амино-2-циклобутилуксусной кислоты (Cbg) и (2S)-пирролидин-2-илуксусной кислоты (бета-гомо-Р),
где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации,
и, когда u и t оба представляют собой 0, и s отличен от 0, концевая карбоксильная группа X13 является незамещенной или амидированной,
s представляет собой целое число от 0 до 3,
X14 представляет собой любую природную аминокислоту или не встречающуюся в природе аминокислоту, где любая природная аминокислота и/или не встречающаяся в природе аминокислота может быть в D- или L-стереоконфигурации,
и, когда и представляет собой 0 и t отличен от 0, концевая карбоксильная группа X14 является незамещенной или амидированной,
t представляет собой целое число от 0 до 4,
X15 представляет собой группу формулы (IIb)
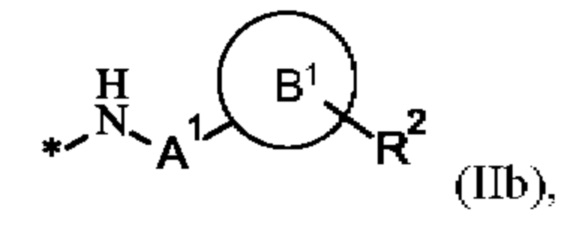
где
* показывает связь с терминальной карбоксильной группой соседней аминокислоты,
А1 представляет собой связь или C1-С6-алкилен, где одна СН2 группа в C1-С6-алкилене может быть заменена на -О- или -S-, и где C1-С6-алкилен имеет до трех заместителей, которые представляют собой одинаковые или разные радикалы, выбранные из группы, состоящей из гидроксила, метокси, этокси, карбокси, амино и галогена,
В1 отсутствует, представляет собой арил, гетероарил, С3-С8-циклоалкил или С3-С7-гетероциклоалкил,
где арил, гетероарил, С3-С8-циклоалкил и С3-С7гетероциклоалкил может иметь до трех заместителей, которые представляют собой одинаковые или разные радикалы, выбранные из группы, состоящей из С1-С4-алкила, гидроксила, метокси, этокси, карбонила, карбокси, амино и галогена, и
R2 представляет собой водород, галоген, амино, гидроксил или С1-С20-алкил, где С1-С20-алкил имеет до трех заместителей, которые представляют собой одинаковые или разные радикалы, выбранные из группы, состоящей из гидроксила, карбокси, амино и галогена,
u представляет собой целое число 0 или 1,
при условии, что по меньшей мере один из X1-X14 представляет собой не встречающуюся в природе аминокислоту.
Кроме того, настоящее изобретение относится к соединениям, содержащим пептид следующей формулы (II),
(X1)q(X2)rX3X4X5X6X7X8X9X10X11X12(Х13)s(X14)t (II),
или его производное, пролекарство, аналог, фармацевтически приемлемая соль, сольват или сольват соли, где
X1 представляет собой природную аминокислоту или не встречающуюся в природе аминокислоту,
q представляет собой целое число 0 или 1,
X2 представляет собой природную аминокислоту I,
r представляет собой целое число 0 или 1,
X3 представляет собой природную аминокислоту С или не встречающуюся в природе аминокислоту L-пеницилламин (Pen),
X4 представляет собой природную аминокислоту S,
X5 представляет собой природную аминокислоту R или не встречающуюся в природе аминокислоту N(5)-метил-L-аргинин ((Me)R),
X6 представляет собой природную аминокислоту S,
X7 представляет собой природную аминокислоту L или не встречающуюся в природе аминокислоту L-трет-бутилаланин ((tBu)A),
X8 представляет собой природную аминокислоту Р или не встречающуюся в природе аминокислоту L-пролин (3,4-2Н),
X9 представляет собой природную аминокислоту Р или не встречающуюся в природе аминокислоту 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновую кислоту (Oic),
X10 представляет собой природную аминокислоту I,
X11 представляет собой не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N-метилцистеина ((N-Me)C) и L-пеницилламина (Pen)
X12 представляет собой природную аминокислоту I,
X13 представляет собой природную аминокислоту Р,
s представляет собой целое число 0 или 1,
X14 представляет собой природную аминокислоту, выбранную из перечня, состоящего из D, Q и Е,
t представляет собой целое число 0 или 1,
где N-конец пептида является незамещенным, ацетилированным или моно- или ди-замещенным С1-С20-алкилом,
где С-конец пептида является незамещенным или амидированным, и
где пептид циклизован, предпочтительно посредством связи, содержащей X3 и X11.
Если иное не определено в настоящем документе, научные и технические термины, используемые в данной заявке, должны иметь значения, которые обычно понятны специалистам в данной области техники. Как правило, номенклатура и методы, используемые в связи с химией, молекулярной биологией, клеточной биологией и биологией рака, иммунологией, микробиологией, фармакологией, а также химией белков и нуклеиновых кислот, описанные в настоящем документе, хорошо известны и обычно используются в данной области техники.
В данном описании под словом «содержать» или его вариантами, такими как «содержит» или «содержащий», подразумевается включение указанного целого (или компонентов) или группы целых (или компонентов), но не исключение любого другого целого (или компонентов) или группы целых (или компонентов). Формы единственного числа включают формы множественного числа, если из контекста явно не следует иное. Термин «включающий» и «содержащий» используется для обозначения «включая без ограничения», эти выражения могут использоваться взаимозаменяемо. В частности, выражение «соединение, содержащее пептид» означает соединение, которое содержит пептид определенной последовательности и может необязательно содержать дополнительные химические группы или заместители, ковалентно связанные с пептидом, например, аминокислоты, жирные кислоты, химические группы для усиления фармакодинамических или фарма-кокинетических свойств пептида или любые другие химические группы. Также следует понимать, что выражение «соединение, содержащее пептид» явно включает определенную последовательность пептида без каких-либо дополнительных химических групп или заместителей, ковалентно связанных с этим пептидом.
Используемые здесь следующие термины имеют приписанные им значения, если не указано иное. «По существу состоящий из» понимается как пептид, на по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95%, по меньшей мере 96%, по меньшей мере 97%, по меньшей мере 98% или по меньшей мере 99%. % идентичный пептиду, с которым он сравнивается.
Термины «белок», «полипептид» и «пептид» используются взаимозаменяемо для обозначения в широком смысле последовательности двух или более аминокислот, связанных вместе, предпочтительно пептидными (амидными) связями. Пептидные (амидные) связи образуются, когда карбоксильная группа одной аминокислоты взаимодействует с аминогруппой другой. Кроме того, следует понимать, что термины «белок», «полипептид» и «пептид» не указывают на конкретную длину полимера аминокислот, а также не предназначены для того, чтобы подразумевать или различать, получен ли полипептид с использованием рекомбинантных методов, химического или ферментативного синтеза, или встречается в природе. Далее следует понимать, что пептид может содержать одну или несколько частей, которые не являются аминокислотами в соответствии с определением настоящей заявки. Эти участки обычно присутствуют на N- и С-концах пептида.
Термин «аминокислота» или «любая аминокислота», используемый в настоящем документе, относится к органическим соединениям, содержащим функциональные группы амина (-NH2) и карбоксила (-СООН) вместе с боковой цепью, и относится ко всем или любым аминокислотам, включая природные аминокислоты (например, a-L-аминокислоты), не встречающиеся в природе аминокислоты, модифицированные аминокислоты и неприродные аминокислоты. «Природные аминокислоты» включают те, которые встречаются в природе, такие как, например, 23 аминокислоты, которые объединяются в пептидные цепи, образуя строительные блоки огромного количества белков. В основном это L-стереоизомеры, хотя некоторые D-аминокислоты встречаются в бактериальных оболочках и некоторых антибиотиках. 20 протеиногенных природных аминокислот в стандартном генетическом коде перечислены в Таблице 2. «Нестандартными» природными аминокислотами являются пирролизин (обнаружен в метаногенных организмах и других эукариотах), селеноцистеин (присутствует во многих неэукариотических, а также большинстве эукариот), и N-формилметионин (кодируется стартовым кодоном AUG в бактериях, митохондриях и хлоропластах).
«Неприродные» или «не встречающиеся в природе» аминокислоты представляют собой непротеиногенные аминокислоты (т.е. те, которые не кодируются естественным путем или не обнаруживаются в генетическом коде), которые либо существуют в природе, либо синтезируются химическим путем. Известно более 140 природных аминокислот и возможны еще тысячи комбинаций. Примеры «не встречающихся в природе» аминокислот включают β-аминокислоты (β3 и β2), гомоаминокислоты, производные пролина и пи-ровиноградной кислоты, 3-замещенные производные аланина, производные глицина, производные фенилаланина и тирозина с замещенным кольцом, аминокислоты с линейным ядром, диаминокислоты, D-аминокислоты и N-метиламинокислоты. К не встеречающимся в природе или неприродным аминокислотам относятся также модифицированные аминокислоты. «Модифицированные» аминокислоты включают аминокислоты (например, природные аминокислоты), которые были химически модифицированы для включения группы, групп или химического фрагмента, не присутствующего в природе в аминокислоте. Согласно настоящему изобретению предпочтительные не встречающиеся в природе аминокислоты перечислены в Таблице 1. Таблица 1 отображает не встречающиеся в природе аминокислоты в виде D- и/или L-стереоизомеров, однако предпочтительными не встречающимися в природе аминокислотами согласно изобретению являются как D-, так и L-стереоизомеры не встречающихся в природе аминокислот, перечисленных в Таблице.






Более предпочтительно не встречающаяся в природе аминокислота выбрана из перечня, состоящего из N-метил-аланина (N-Me)A, N-метил-глицина ((N-Me)G), (1R,3S,4S)-2-азабицикло[2.2.1]гептан-3-карбоновой кислоты, L-3-бромфенилаланина ((3-бромо)F), L-N,N-диметилаланина ((N,N-диМе)A), N,N-диметилглицина ((N,N-диМе)G), N-фенилглицина ((N-Ph)G), (R)-пиперидин-3-карбоновой кислоты, (S)-пиперидин-3-карбоновой кислоты, L-трет-бутилаланина ((tBu)A), L-2-пиридилаланина (2-Pal), L-3-пиридилаланина (3-Pal), L-4-пиридилаланина (4-Pal), 3-(аминометил)бензойной кислоты, 3-амино-2,2-диметилпропионовой кислоты, 3-амино-3-метилмасляной кислоты, 4-(аминометил)бензойной кислоты, L-2-аминомасляной кислоты (Abu), 1-аминоциклобутан-1-карбоновой кислоты (АСВА), 6-аминогексановой кислоты (Ahx), 2-аминоизомасляной кислоты (Aib), L-2-тиенилаланина (бета-2-тиенилаланин), бета-аланина (бета-А), бета-пролина (бета-Р), L-цитруллина (Cit), L-2,4-диаминомасляной кислоты (Dab), L-2,3-диаминопропионовой кислоты (Dap), Гамма-аминомасляной кислоты (Гамма-Abu), L-3-метилгистидина (3-Ме)Н), L-дигидрооротовой кислоты (Ноо), L-норлейцина (Nle), N-метил-L-пролина ((N-Me)P), L-норвалина (Nva), L-орнитина (Orn), L-пипеколиновой кислоты (Pip), (2S)-2[(амино)-2-(тетрагидро-2Н-пиран-4-ил)]уксусной кислоты; 2,3,3а, 4,5,6,7,7а-октагидроиндол-2-карбоновой кислоты (Oic), L-N-метилцистеина ((N-Me)C), N(5)-метил-L-аргинина ((Me)R), L-пеницилламина (Pen) и транексамовой кислоты (транексамовой кислоты).
Наиболее предпочтительные не встречающиеся в природе аминокислоты выбраны из перечня, состоящего из N-метил-L-аланина (N-Me)A, N-метил-глицина ((N-Me)G), L-норлейцина (Nle), L-норвалина (Nva), L-орнитина (Orn), N(5)- метил-L-аргенина ((Me)R), L-трет-бутилаланина ((tBu)A), 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновой кислоты (Oic), L-N-метилцистеина ((N-Me)C) и L-пеницилламина (Pen).
Далее следует понимать, что пептид согласно настоящему изобретению может содержать одну или несколько химических групп, которые не являются аминокислотами согласно определению настоящего изобретения. Эти химические группы могут находиться на N- и/или С-концах пептида и представлены формулами Х0 и X15. Следует понимать, что все аминокислоты и химические группы пептидов согласно настоящему изобретению связаны пептидными (амидными) связями. Обычно пептиды образуются путем связывания α-амино- и карбоксигрупп α-аминокислот, которые затем соединяются α-пептидными связями. Согласно настоящему изобретению пептидная связь может быть образована любой карбокси- и аминогруппой, присутствующей в соответствующей природной или не встречающейся в природе аминокислоте. Например, α-аминокислоты, которые содержат вторую аминогруппу в дополнение к α-аминогруппе (например, L-лизин), или α-аминокислоты, которые, помимо α-карбоксигруппы содержат вторую карбоксигруппу (например, L-аспарагиновая кислота и L-глутаминовая кислота), могут быть присоединены через дополнительную амино-или карбоксигруппу.
В соответствии с пониманием специалиста в данной области техники, раскрытые в настоящем документе пептидные последовательности представляют собой последовательности аминокислот, которые соединены через α-пептидные связи. Аминокислота, связанная пептидной связью, которая не является α-пептидной связью, отмечена знаком «*». «*» находится либо с левой, либо с правой стороны аминокислоты, чтобы показать, используется ли дополнительная аминогруппа или дополнительная карбоксигруппа этой аминокислоты для пептидной связи с соседней аминокислотой (например, (*L), (Е*), (*Dap) и т.д.).
В соответствии с пониманием специалиста в данной области техники, раскрытые в настоящем документе пептидные последовательности показаны слева направо, причем левый конец последовательности представляет собой «N-конец» («аминоконец», «N-концевой конец») пептида, а правый конец последовательности представляет собой «С-конец» («карбоксиконец», «С-конец») пептида. Эта терминология «N-конец (аминоконец, N-конец)» применяется независимо от того, действительно ли пептид содержит аминогруппу на N-конце. Эта терминология С-конец (карбокси-конец, С-конец) применяется независимо от того, действительно ли пептид содержит карбоксигруппу на С-конце. Термин «концевая аминогруппа» относится к любой аминогруппе, присутствующей в N-конце. Термин «концевая карбоксильная группа» относится к любой карбоксильной группе, присутствующей в С-конце.
Согласно настоящему изобретению N-конец может быть образован Х0, в случае, если р представляет собой 1. В качестве альтернативы N-конец может быть образован X1, если q представляет собой по меньшей мере 1, а р представляет собой 0. В качестве альтернативы N-конец может быть образован Х2 X2 в случае, если г представляет 1, а р и q оба представляют собой 0. В случае, если р, q и r все представляют 0, N-конец образован X3 в случае линейного пептида. В случае, если пептид согласно настоящему изобретению циклизуется посредством связи, соединяющей X3 и X11, и р, q и r все представляют собой 0, пептид не содержит N-конец.
Согласно настоящему изобретению С-концы могут быть образованы X15, в случае, если и представляет собой 1. В качестве альтернативы С-концы могут быть образованы X14, в случае, если t представляет собой целое число по меньшей мере 1, а и представляет собой 0. В качестве альтернативы С-концы могут быть образованы X14, если s представляет собой 1, a t и и оба представляют собой 0. В случае, если s, t и и все представляют 0, С-конец образован X12.
В настоящем изобретении названия встречающихся в природе и не встречающихся в природе аминоацильных остатков, используемых в настоящем документе, следуют соглашениям об именах, предложенным Комиссией ИЮПАК по номенклатуре органической химии и Комиссией ИЮПАК-IUB по биохимической номенклатуре, изложенным в Nomenclature of a-Amino Acids (Recommendations, 1974), Biochemistry, 14(2), (1975).
В настоящем описании встречающиеся в природе протеиногенные аминокислоты обычно обозначаются их обычными однобуквенными сокращениями. В качестве альтернативы они также могут называться трехбуквенными аббревиатурами (например, в частности, в перечне последовательностей) или полными именами, как показано в Таблице 2 ниже:

В случае непротеиногенных или не встречающихся в природе аминокислот, если они не упоминаются по их полному названию (например, орнитин и т.д.), для их остатков часто используются трех- или шестизначные коды, включая те сокращения, которые указаны в списке сокращений ниже (таблица 3).
Термин «L-аминокислота», используемый в настоящем документе, относится к «Ь»-изомерной форме аминокислоты, и, наоборот, термин «D-аминокислота» относится к «0»-изомерной форме аминокислоты. Кроме того, общепринято обозначать L-аминокислоту заглавными буквами, такими как Ala/A, Arg/R и т.д., a D-аминокислоту строчными буквами, такими как ala/a, arg/r и т.д.
Трехбуквенный код в форме, указанной в Таблице 2 выше, т.е. Ala, Arg, Asn и т.д., и как в общем используется в настоящем описании, обычно включает формы D- и L-, а также гомо- и нор-формы, если прямо не указано иное. Приставка «нор» относится к структурному аналогу, который может быть получен из исходного соединения путем удаления одного атома углерода вместе с сопутствующими атомами водорода. Приставка «гомо» указывает на следующего старшего члена в гомологическом ряду. Ссылка на конкретную изомерную форму будет обозначаться заглавной буквой L- или D-, как описано выше (например, D-Arg, L-Arg и т.д.). Соответственно, конкретная ссылка на гомо- или нор-формы будет явно указана соответствующим префиксом (например, гомо-Arg, гомо-R, нор-Arg, нор-R, гомо-Cys, гомо-С и т.д.).
Термин "C1-С6-алкил" означает линейную или разветвленную, насыщенную, моновалентную углеводородную группу, имеющую 1, 2, 3, 4, 5 или 6 атомов углерода, например, группу метил, этил, пропил, изопропил, бутил, втор-бутил, изобутил, трет-бутил, пентил, изопентил, 2-метилбутил, 1-метилбутил, 1-этилпропил, 1,2-диметилпропил, не-опентил, 1,1-диметилпропил, гексил, 1-метил пентил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 1-этилбутил, 2-этилбутил, 1,1-диметилбутил, 2,2-диметилбутил, 3,3-диметилбутил, 2,3-диметилбутил, 1,2-диметилбутил или 1,3-диметилбутил или ее изомер. В частности, указанная группа имеет 1, 2, 3 или 4 атома углерода ("С1-С4-алкил"), например, группу метил, этил, пропил, изопропил, бутил, втор-бутил изобутил или трет-бутил, более конкретно 1, 2 или 3 атома углерода ("C1-С3-алкил"), например, группу метил, этил, н-пропил или изопропил. Особенно предпочтительным является метил, этил, н-пропил. Наиболее предпочтительным является метил.
Термин "С1-С20-алкил" означает линейную или разветвленную, насыщенную, моновалентную углеводородную группу, имеющую от 1 до 20 атомов углерода, например, метил, этил, пропил, изопропил, бутил, втор-бутил, изобутил, трет-бутил или пентил, изопентил, гексил, изогексил, гептил, изогептил, октил и изооктил, нонил, децил, додецил или эйкозил.
Термин "С1-С4-алкилен" означает неразветвленную цепь или разветвленный углеводородный мостик, имеющий от 1 до 4 атомов углерода, например, метилен, этилен, пропилен, (α-метилэтилен, β-метилэтилен, α-этилэтилен, β-этилэтилен, бутилен, α-метилпропилен, β-метилпропилен и γ-метилпропилен.
Термин "C1-С6-алкилен" означает неразветвленную цепь или разветвленный углеводородный мостик, имеющий от 1 до 6 атомов углерода, например, метилен, этилен, пропилен, (α-метилэтилен, β-метилэтилен, α-этилэтилен, β-этилэтилен, бутилен, α-метилпропилен, β-метилпропилен, γ-метилпропилен, α-этилпропилен, β-этилпропилен, γ-этилпропилен, пентилен и гексилен.
Термин "С3-С8-циклоалкил" означает насыщенное углеводородное кольцо, которое содержит 3, 4, 5, 6, 7 или 8 атомов углерода. Указанная С3-С8-циклоалкилбная группа представляет собой, например, моноциклическое углеводородное кольцо, т.е. группу цик-лопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил, бициклическое углеводородное кольцо, например, бицикло[4.2.0]октил или октагидропентале-нил, или насыщенные кольцевые группы с мостиков ой или клеткой, такие как нор бор ан или адамантан, и кубан.
Термин "С3-С7-гетероциклоалкил" означает насыщенный гетероцикл с 4, 5, 6 или 7 атомами, который содержит один или два одинаковых или разных кольцевых гетероатома из ряда N, О и S, причем указанная гетероциклоалкильная группа может быть присоединена к остальной части молекулы через любой атом углерода или, если он присутствует, атом азота. Указанная С3-С7-гетероциклоалкильная группа без ограничения может быть 4-членным кольцом, таким как азетидинил, оксетанил или тиетанил, например, или 5-членным кольцом, таким как тетрагидрофуранил, 1,3-диоксоланил, тиоланил, пирролидинил, имидазолидинил, пиразолидинил, 1,1-диоксидотиоланил, 1,2-оксазолидинил, 1,3-оксазолидинил или 1,3-тиазолидинил, для примера; или 6-членное кольцо, такое как тетра-гидропиранил, тетрагидротиопиранил, пиперидинил, морфолинил, дитианил, тиоморфолинил, пиперазинил, гексагидропиримидинил, 1,3-диоксанил, 1,4-диоксанил или 1,2-оксазинанил, например, или 7-членное кольцо, такое как азепанил, 1,4-диазепанил или 1,4-оксазепанил, например.
Термин «арил» означает ненасыщенный или частично ненасыщенный цикл, содержащий от 6 до 10 атомов углерода. Предпочтительными ар ильными остатками являются фенил и нафтил.
Термин «гетероарил» означает моновалентное, моноциклическое, бициклическое или трициклическое ароматическое кольцо, содержащее 5, 6, 8, 9, 10, 11, 12, 13 или 14 атомов в кольце (группа «5-14-членный гетероарил»), в частности 5, 6, 9 или 10 кольцевых атомов, которое содержит по меньшей мере один кольцевой гетероатом и необязательно один, два или три дополнительных кольцевых гетероатома из ряда: N, О и/или S, и которое связано через кольцевой атом углерода или необязательно через кольцевой атом азота (если это разрешено валентностью). Указанная гетероарильная группа может представлять собой 5-членную гетероарильную группу, такую как, например, тиенил, фуранил, пирролил, оксазолил, тиазолил, имидазолил, пиразолил, изоксазолил, изотиазолил, оксадиазолил, триазолил, тиадиазолил или тетразолил; или 6-членную гетероарильную группу, такую как, например, пиридинил, пиридазинил, пиримидинил, пиразинил или триазинил; или трициклическую гетероарильную группу, такую как, например, карбазолил, акридинил или феназинил; или 9-членную гетероарильную группу, такую как, например, бензофуранил, бензотиенил, бензоксазолил, бензизоксазолил, бензимидазолил, бензотиазолил, бензотриазолил, индазолил, индолил, изоиндолил, индолизинил или пуринил; или 10- членную гетероарильную группу, такую как, например, хинолинил, хиназолинил, изохинолинил, циннолинил, фталазинил, хиноксалинил или птеридинил.
В общем, если не указано иное, гетероарильные или гетероариленовые группы включают все их возможные изомерные формы, например, таутомеры и изомеры в положении по отношению к месту соединения с остальной частью молекулы. Таким образом, для некоторых иллюстративных неограничивающих примеров термин пиридинил включает пиридин-2-ил, пиридин-3-ил и пиридин-4-ил; или термин тиенил включает тиен-2-ил- и тиен-3-ил.
Среди раскрываемых в настоящем документе последовательностей последовательности представлены последовательности, включающие либо фрагмент «-ОН», либо фрагмент «-NH2» на карбокси-конце (С-конец) последовательности. Фрагмент «-ОН» или «NH2» на С-конце последовательности указывает на гидроксигруппу или аминогруппу, что соответствует присутствию карбоксигруппы или амидогруппы (-(С=O)- NH2) на С-конце, соответственно. В каждой последовательности настоящего изобретения С-концевой фрагмент «-ОН» может быть заменен на С-концевой фрагмент «-NH2», который также упоминается как «амидированный С-конец» в настоящем изобретении, и наоборот.Однако среди указанных альтернатив предпочтительным является С-концевой фрагмент «-ОН».
Термин «ацетилированный» (также сокращенно «Ас») относится к защите ацетилом N-концевой части посредством ацетилирования N-конца пептида (N-конец пептида ацетилирован).
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из формулы (I) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где
Х0 представляет собой химическую группу, выбранную из перечня, состоящего из (1S,2S,4S)-6huhigto[2.2.1]гепт-5-ен-2-илуксусной кислоты, (2,4-диоксоимидазолидин-1-ил)уксусной кислоты, 2-(тиоморфолин)уксусной кислоты ((2-тиоморфолин)ацетил), 2-(N-изопропил-М-метиламино)уксусной кислоты, (S)-3-метилвалерьяновой кислоты ((S)-3-метилпентановой кислоты), 2-(3-пиридил)уксусной кислоты, 2-(циклогексиламино)уксусной кислоты (2-(циклогексиламино)ацетила), 2-(диэтиламино)уксусной кислоты (2-(диэтиламино)ацетила), 2-(морфолин)уксусной кислоты (2-(морфолин)ацетила), 2-(1Ч-метил-М-циклопропиламино)уксусной кислоты (2-(N-метил-К-циклопропиламино)ацетила 2-(пиперидин)уксусной кислоты (2-(пиперидин)ацетила), 2-(пирролидин)уксусной кислоты (2-(пирролидин)ацетила), 2-гидроксиуксусной кислоты (2-гидроксиацетила), 2-гидроксиизомасляной кислоты (2-гидроксиизо масляной кислоты), 3-(аминометил)бензойной кислоты, 3-метоксипропионовой кислоты, 4-(аминометил)бензойной кислоты, 4-метилпентановой кислоты (4-метилвалерьяновой кислоты), 5-хлортиофен-карбоновой кислоты, 1-(аминометил)-цикло пропил-1-карбоновой кислоты (АСМР), адипиновой кислоты, (S)-азетидин-2-карбоновой кислоты, бензойной кислоты (бензойная), 4-(3,5-диметил-1,2-оксазол-4-ил)-L-фенилаланина, циклобутанкарбоновой кислоты (циклобутанкарбоновой кислоты), 2-(циклобутил)уксусной кислоты (циклобутилуксусной кислоты), циклобутилуксусной кислоты (циклобутилуксусной кислоты), циклогексилуксусной кислоты (циклогексилуксусной кислоты), циклогексанкарбоновой кислоты (циклогексилкарбоновой кислоты), циклопентан-карбоновой кислоты, циклопентилуксусной кислоты (циклопентилуксусной кислоты), цик-лопропанкарбоновой кислоты (циклопропанкарбоновой кислоты), циклопропилуксусной кислоты, 0-(+)биотина, фумаровой кислоты, 3-фенилпропановой кислоты (гидрокоричной кислоты), изомасляной кислоты, изовалериановой кислоты (изовалериановая кислота), L-(+)-молочной кислоты (молочная кислота), фенилуксусной кислоты, пиперидин-4-илуксусной кислоты, пивалевой кислотыа (пивалевая кислота), пробковой кислоты, трет-бутилуксусной кислоты, тетрагидропиранил-4-уксусной кислоты, тетрагидро-2Н-пиран-3-илуксусной кислоты и транс-2-(3-((1-бутокси)карбониламино)циклогексил)уксусной кислоты), р представляет собой целое число 0 или 1,
X1 представляет собой природную аминокислоту, выбранную из перечня, состоящего из A, F, G, Н, I, К, L, Р, R, S, Т, V, W и Y или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из N-метил-аланина (N-Me)A, N-метил-глицина ((N-Me)G), (1R,3S,4S)-2-азабицикло[2.2.1]гептан-3-карбоновой кислоты, L-3-бромфенилаланина ((3-бромо)F), L-N,N-диметилаланина ((N,N-диМе)А), N,N-диметилглицина ((N,N-диМе)G), N-фенилглицина ((N-Ph)G), (Рч.)-пиперидин-3-карбоновой кислоты, (S)-пиперидин-3-карбоновой кислоты, L-трет-бутилаланина ((tBu)A), L-2-пиридилаланина (2-Pal), L-3-пиридилаланина (3-Pal), L-4-пиридилаланина (4-Pal), 3-(аминометил)бензойной кислоты, 3-амино-2,2-диметилпропионовой кислоты, 3-амино-3-метилмасляной кислоты, 4-(аминометил)бензойной кислоты, L-2-аминомасляной кислоты (Abu), 1-аминоциклобутан-1-карбоновой кислоты (АСВА), 6-аминогексановой кислоты (Ahx), 2-аминоизомасляной кислоты (Aib), L-2-тиенилаланина (бета-2-тиенилаланина), бета-аланина (бета-А), бета-пролина (бета-Р), L-цитруллина (Cit), L-2,4-диаминомасляной кислоты (Dab), L-2,3-диаминопропионовой кислоты (Dap), Гамма-аминомасляной кислоты (Гамма-Abu), L-3-метилгистидина (3-Ме)Н), L-дигидрооротовой кислоты (Ноо), L-норлейцина (Nle), N-метил-L-пролина ((N-Me)P), L-норвалина (Nva), L-орнитина (Orn), L-пипеколиновой кислоты (Pip), (2S)-2[(амино)-2-(тетрагидро-2Н-пиран-4-ил)]уксусной кислоты и транексамовой кислоты (транексамовой кислоты),
где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации,
q представляет собой целое число от 0 до 5,
X2 представляет собой природную аминокислоту I или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N-метилизолейцина ((N-Me)I), алло-L-изолейцина (алло-1), L-циклобутилаланина (Cba), L-норвалин (Nva), L-2-аминомасляной кислоты (Abu), (28,38)-2-[(311)-3-амино-2-оксопирролидин-1-ил]-3-метил-пентановой кислоты, (2S,3S)-2-[2-оксопиперазин-1-ил]-3-метилпентановой кислоты и (2S,3S)-2-[(3S)-2-оксопиперазин-1-ил]-3-метилпентановой кислоты,
где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации,
r представляет собой целое число 0 или 1,
X3 представляет собой природную аминокислоту С или не встречающуюся в природе аминокислоту L-пеницилламин (Реп),
X4 представляет собой природную аминокислоту, выбранную из перечня, состоящего из S, С, Т, R и K,
X5 представляет собой природную аминокислоту R или не встречающуюся в природе аминокислоту N(5)-метил-L-аргинин ((Me)R),
X6 представляет собой природную аминокислоту S, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из алло-L-треонина (алло-Т) и L-2,3-диаминопропионовой кислоты (Dap),
X7 представляет собой природную аминокислоту, выбранную из перечня, состоящего из L и N, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 2,5-дифтор-L-фенилаланина ((2,5-дифтор)F), L-2-бромфенилаланина ((2-бромо)F), 2-хлор-L-фенилаланина ((2-хлор)F), L-4-бромфенилаланина ((4-бромо)Б), 4-фтор-L-лейцина ((4-фтор)L), L-трет-бутилаланина ((tBu)A), L-трет-бутилглицина ((tBu)G), 5,5,5-трифтор-L-лейцина ((трифтор)L), (S)-2-(амино)-1,6-гександионовой кислоты (AAD), (2S)-2-амино-4,4,4-трифторбутановой кислоты, Ь-2-амино-4-цианомасляной кислоты (Cnba), (S)-(трифторметил)-L-цистеина, (2S)-2-амино-3-(1-метилциклопропил)пропановой кислоты, (2S)-3-(2,3-дифторфенил)-2-аминопропановой кислоты, 2,3,3а, 4,5,6,7,7а-октагидроиндол-2-карбоновой кислоты (Oic), 3-(триметилсилил)-L-аланина и 2-амино-7-(трет-бутокси)-7-оксогептановой кислоты,
X8 представляет собой природную аминокислоту Р, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из (1S,2S,5R)-3-азабицикло[3.1.0]гексан-2-карбоновой кислоты, (1R,2S,5S)-3-азабицикло[3.1.0]гексан-2-карбоновой кислоты, транс-4-фторпролина ((транс-4-фтор)Р), L-гидроксипролина (Hyp), (3S)-морфолин-3-карбоновой кислоты (морфолин-3-карбоновой), L-пипеколиновой кислоты (Pip) и (4aR,6aR,9S,11aS)-11-оксо-2,3,4,4a,6a,7,8,9,11,11a-декагидро-1H-пиридо[3,2-е]пирроло[1,2-а]азепин-9-карбоновой кислоты,
X9 представляет собой природную аминокислоту Р, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновой кислоты (Oic), (6S)-5-азаспиро[2.4]гептан-6-карбоновой кислоты, rel-(1R,3R,5R,6R)-6-(трифторметил)-2-азабицикло[3.1.0]гексан-3-карбоновой кислоты, (2S,4S)-4-трифторметил-пирролидин-2-карбоновой кислоты ((4-CF3)P), (2S,3aS,6aS)-октагидроциклопента[b]пиррол-2-карбоновой кислоты, (1R,3S,5R)-2-абазицикло[3.1.0]гексан-3-карбоновой кислоты, (2S)-2-амино-4,4,4-трифторбутановой кислоты, L-транс-3-гидроксипролина ((3S-OH)P, транс-4-фторпролина ((транс-4-фтор)Р), L-гидроксипролина (Hyp), (2S,4S)-4-фторпролина ((цис-4-фтор)Р), (3R,6R)-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновой кислоты (энантиомер 1) и (3R,6R)-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновой кислоты (энантиомер 2),
X10 представляет собой природную аминокислоту I, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из (2S)-2-(амино)-2-[(1S,3R)-3-гидроксициклогексил] уксус ной кислоты, 3-хлорфенилглицина ((3-хлор-Рр)О), (S)-2-амино-3-этил-пентановой кислоты, алло-L-изолейцина (алло-1), L-циклогексилглицина (Chg), L-циклопентилглицина (Cpg), L-циклобутилглицина и L-норвалина (Nva),
X11 представляет собой природную аминокислоту С, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N-метилцистеина ((N-Me)C) и L-пеницилламина (Pen),
X12 представляет собой природную аминокислоту I, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 2-[(1S,2S)-1-(амино)-2-метилбутил]-1,3-оксазол-4-карбоновой кислоты, (S)-2-амино-2-циклобутилуксусной кислоты (Cbg), алло-L-изо лейцина (алло-I), L- фенил глицина (Phg), 2-метил-В-аллоизолейцина и (2S,3S)-2-((амино)метил)-3-метилпентановой кислоты,
И, когда u и t и s все представляют собой 0, концевая карбоксильная группа X12 является незамещенной или амидированной,
X13 представляет собой природную аминокислоту, выбранную из перечня, состоящего из Р, А и D, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 2-метил-Ь-пролина (2-Ме)Р, N-метил-глицина ((N-Me)G), транс-4-фторпролина ((транс-4-фтор)Р), L-2-аминомасляной кислоты (Abu), 2-аминоизомасляной кислоты (Aib), гидроксипролина (Hyp) и 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновой кислоты (Oic),
где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации,
и когда u и t оба представляют собой 0, и s отличен от 0, концевая карбоксильная группа X13 является незамещенной или амидированной,
s представляет собой целое число от 0 до 3,
X14 представляет собой природную аминокислоту, выбранную из перечня, состоящего из D, Е, G, К, N, Р и Q, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 3-гарбоксифенилаланина ((3-Карбокси)F), (2S)-2-амино-4-(бензиламино)-4-оксобутанкарбоновой кислоты ((N-бензил)О), N-метил-глицина ((N-Me)G), 6-аминогексановой кислоты (Ahx), (2S)-пирролидин-2-илуксусной кислоты (бета-гомо-Р), L-2,3-диаминопропионовой кислоты (Dap), L-орнитин (Orn) и Транексамовой кислоты (транексамовой кислоты),
и когда u представляет собой 0 и t отличен от 0, концевая карбоксильная группа X14 является незамещенной или амидированной,
где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации,
t представляет собой целое число от 0 до 4,
X15 представляет собой химическую группу, выбранную из перечня, состоящего из (1R,3S)-3-(амино)циклопентанкарбоновой кислоты, (1S,3R)-3-(амино)циклопентанкарбоновой кислоты, (R)-4-амино-6-метилгептановой кислоты, (S)-(1-пиперидин-3-ил)-уксусной кислоты, (S)-3-(1-пирролидин-2-ил)-пропионовой кислоты, (S)-3-(2Н-тетразол-5-ил)пропановой кислоты, (S)-пирролидин-3-карбоновой кислоты, 5-азаспиро[2.4]гептан-1-карбоновой кислоты и (2S)-3-(триазол-1-ил)-2-(амино)пропановой кислоты,
u представляет собой целое число 0 или 1,
при условии, что пептид содержит по меньшей мере одну не встречающуюся в природе аминокислоту.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из формулы (I) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где
Х0 представляет собой химическую группу, выбранную из перечня, состоящего из (1R,3S,4S)-2-азабицикло[2.2.1]гептан-3-карбоновой кислоты, (2-тиоморфолин)уксусной кислоты, (N-изопропил-N-метиламино)уксусной кислоты, 2-(3-пиридил)уксусной кислоты, 2-(циклогексиламино)уксусной кислоты, 2-(диэтиламино)уксусной кислоты, 2-(морфолин)уксусной кислоты, 2-(пиперидин)уксусной кислоты, 2-(пирролидин)уксусной кислоты, 3-(аминометил)бензойной кислоты, 4-(аминометил)бензойной кислоты, 1-(аминометил)-циклопропил-1-карбоновой кислоты (АСМР), азетидин-2-карбоновой кислоты, бензойной кислоты, циклобутилуксусной кислоты, циклопропилуксусной кислоты, фенилуксусной кислоты, пиперидин-4-илуксусной кислоты, тетрагидро-2Н-пиран-3-илуксусной кислоты, транексамовой кислоты и транс-2-(3-((t-бутокси)карбониламино)циклогексил)уксусной кислоты,
р представляет собой целое число 0 или 1,
X1 представляет собой природную аминокислоту, выбранную из перечня, состоящего из A, F, г, Н, I, K, L, Р, R, S, Т, V, W и Y или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N,N-диметилаланина ((N,N-диМе)A), N-метилаланина (N-Me)A, N-метилглицина ((N-Me)G), L-2-пиридилаланина (2-Pal), L-3-пиридилаланина (3-Pal), L-4-пиридилаланина (4-Pal), L-2-аминомасляной кислоты (Abu), 6-аминогексановой кислоты (Ahx), 2-аминоизомасляной кислоты (Aib), 3-(аминометил)бензойной кислоты, 3-амино-2,2-диметилпропионовой кислоты, 3-амино-3-метилмасляной кислоты, 4-(аминометил)бензойной кислоты, бета-2-тиенилаланина, (S)-пирролидин-2-карбоновой кислоты (бета-Р), L-цитруллина (Cit), Ь-2,4-диаминомасляной кислоты (Dab), L-2,3-диаминопропионовой кислоты (Dap), Гамма-аминомасляной кислоты (Гамма-Abu), L-3-метилгистидина (Н(3-Ме)), L-дигидрооротовой кислоты (Ноо), L-норлейцина (Ме), L-норвалина (Nva), L-орнитина (Orn), L-пипеколиновой кислоты (Pip), N-метил-L-пролина ((N-Me)P), транексамовой кислоты (транексамовой кислоты), (1R,3S,4S)-2-азабицикло[2.2.1]гептан-3-карбоновой кислоты и (2S)-2[(амино)-2-(тетрагидро-2Н-пиран-4-ил)]уксусной кислоты,
Где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации,
q представляет собой целое число от 0 до 5,
X2 представляет собой природную аминокислоту I или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N-метилизолейцина ((N-Me)I), L-циклобутилаланина (Cba), L-норвалина (Nva) и L-2-аминомасляной кислоты (Abu),
где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурациию,
r представляет собой целое число 0 или 1,
X3 представляет собой природную аминокислоту С или не встречающуюся в природе аминокислоту L-пеницилламин (Pen),
X4 представляет собой природную аминокислоту, выбранную из перечня, состоящего из S и Т,
X5 представляет собой природную аминокислоту R,
X6 представляет собой природную аминокислоту S или не встречающуюся в природе аминокислоту алло-L-треонин (алло-Т),
X7 представляет собой природную аминокислоту, выбранную из перечня, состоящего из N и L, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 2,5-дифтор-L-фенилаланина ((2,5-дифтор)Р), L-2-бромфенилаланина ((2-бромо)F), 2-хлор-L-фенилаланина ((2-хлор)F), 4-фтор-L-лейцина ((4-фтор)L), L-трет-бутилаланина ((tBu)A), (S)-(трифторметил)-L-цистеина, (2S)-2-амино-3-(1-метилциклопропил)пропановой кислоты и 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновой кислоты (Oic), 3-(триметилсилил)-L-аланина,
X8 представляет собой природную аминокислоту Р, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из (1S,2S,5R)-3-азабицикло[3.1.0]гексан-2-карбоновой кислоты и (4aR,6aR,9S,11aS)-11-оксо-2,3,4,4а,6а,7,8,9,11,11а-декагидро-1Н-пиридо[3,2-е]пирроло[1,2-а]азепин-9-карбоновой кислоты,
X9 представляет собой природную аминокислоту Р, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновой кислоты (Oic), (2S,4S)-4-трифторметил-пирролидин-2-карбоновой кислоты ((4-CF3)P), (6S)-5-азаспиро[2.4]гептан-6-карбоновой кислоты, rel-(1R,3R,5R,6R)-6-(трифторметил)-2-азабицикло[3.1.0]гексан-3-карбоновой кислоты, (2S,3aS,6aS)-октагидроциклопента[b]пиррол-2-карбоновой кислоты, rel-(3R,6R)-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновой кислоты (энантиомер 1) и rel-(3R,6R)-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновой кислоты (энантиомер 2),
X10 представляет собой природную аминокислоту I, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из (2S)-2-(амино)-2-[(1S,3R)-3-гидроксициклогексил]уксусной кислоты, (S)-2-амино-3-этил-пентановой кислоты, L-циклогексилглицина (Chg) и L-циклопентилглицина (Cpg),
X11 представляет собой природную аминокислоту С, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N-метилцистеина ((N-Me)C) и L-пеницилламина (Pen),
X12 представляет собой природную аминокислоту I, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из алло-Е-изолейцина (алло-1), (S)-2-амино-2-циклобутилуксусной кислоты (Cbg) и 2-[(1S,2S)-1-(амино)-2-метилбутил]-1,3-оксазол-4-карбоновой кислоты,
и когда u и t и s все представляют собой 0, концевая карбоксильная группа X12 является незамещенной или амидированной,
X13 представляет собой природную аминокислоту, выбранную из перечня, состоящего из Р и D или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 2-аминоизомасляной кислоты (Aib), 2-метил-L-пролина (2-Ме)Р и транс-4-фторпролина ((транс-4-фтор)Р),
где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации,
и, когда u и t оба представляют собой 0, и s отличен от 0, концевая карбоксильная группа X13 является незамещенной или амидированной,
s представляет собой целое число от 0 до 3,
X14 представляет собой природную аминокислоту, выбранную из перечня, состоящего из D, Q, N, Е и Р, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 3-карбоксифенилаланина ((3-карбокси)Е), N-метил-глицина ((N-Me)G), (2S)-пирролидин-2-илуксусной кислоты (бета-гомо-Р), Е-2,3-диаминопропионовой кислоты (Dap), L-орнитин (Orn) и транексамовой кислоты (транексамовой кислоты),
где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации,
и когда и представляет собой 0, и t отличен от 0, концевая карбоксильная группа X14 является незамещенной или амидированной,
t представляет собой целое число от 0 до 4,
X15 представляет собой химическую группу, выбранную из перечня, состоящего из (1R,3S)-3-(амино)циклопентанкарбоновой кислоты, (1S,3R)-3-(амино)циклопентанкарбоновой кислоты, (R)-4-амино-6-метилгептановой кислоты, (S)-(1-пиперидин-3-ил)-уксусной кислоты, (S)-3-(1-пирролидин-2-ил)-пропионовой кислоты, (S)-3-(2Н-тетразол-5-ил)пропановой кислоты, (S)-пирролидин-3-карбоновой кислоты, 5-азаспиро[2.4]гептан-1-карбоновой кислоты и (2S)-3-(триазол-1-ил)-2-(амино)пропановой кислоты,
u представляет собой целое число 0 или 1,
при условии, что пептид содержит по меньшей мере одну не встречающуюся в природе аминокислоту.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из формулы (I) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где
р представляет собой целое число О,
X1 представляет собой природную аминокислоту, выбранную из перечня, состоящего из А и G, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из N-метил-Ь-аланина (N-Me)A, N-метил-глицина ((N-Me)G), L-норлейцина (Nle), L-норвалина (Nva) и L-орнитина (Orn),
q представляет собой целое число 0 или 1,
X2 представляет собой природную аминокислоту I,
r представляет собой целое число 0 или 1,
X3 представляет собой природную аминокислоту С или не встречающуюся в природе аминокислоту L-пеницилламин (Pen),
X4 представляет собой природную аминокислоту S,
X5 представляет собой природную аминокислоту R или не встречающуюся в природе аминокислоту N(5)-метил-L-аргинин ((Me)R),
X6 представляет собой природную аминокислоту S,
X7 представляет собой природную аминокислоту L или не встречающуюся в природе аминокислоту L-трет-бутилаланин ((tBu)A),
X8 представляет собой природную аминокислоту Р,
X9 представляет собой природную аминокислоту Р или не встречающуюся в природе аминокислоту 2,3,За,4,5,6,7,7а-октагидроиндол-2-карбоновую кислоту (Oic),
X10 представляет собой природную аминокислоту I,
X11 представляет собой природную аминокислоту С, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N-метилцистеина ((N-Me)C) и L-пеницилламина (Pen),
X12 представляет собой природную аминокислоту I,
и, когда u и t и s все представляют собой 0, концевая карбоксильная группа X12 является незамещенной или амидированной,
X13 представляет собой природную аминокислоту Р,
и, когда u и t оба представляют собой 0, и s отличен от 0, концевая карбоксильная группа X13 является незамещенной или амидированной,
s представляет собой целое число 0 или 1,
X14 представляет собой природную аминокислоту, выбранную из перечня, состоящего из D, Q и Е,
и, когда u представляет собой 0, и t отличен от 0, концевая карбоксильная группа X14 является незамещенной или амидированной,
t представляет собой целое число 0 или 1,
u представляет собой целое число 0,
при условии, что пептид содержит по меньшей мере одну не встречающуюся в природе аминокислоту.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид следующей формулы (II),
(X1)q(X2)rX3X4X5X6X7X8X9X10X11X12(Х13)s(X14)t (II),
или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где
X1 представляет собой природную аминокислоту, выбранную из перечня, состоящего из А и G, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из N-метил-L-аланина (N-Me)A, N-метил-глицина ((N-Me)G), L-норлейцина (Nle), L-норвалина (Nva) и L-орнитина (Orn),
q представляет собой целое число 0 или 1,
X2 представляет собой природную аминокислоту I,
r представляет собой целое число 0 или 1,
X3 представляет собой природную аминокислоту С или не встречающуюся в природе аминокислоту L-пеницилламин (Реп),
X4 представляет собой природную аминокислоту S,
X5 представляет собой природную аминокислоту R или не встречающуюся в природе аминокислоту N(5)-метил-L-аргинин ((Me)R),
X6 представляет собой природную аминокислоту S,
X7 представляет собой природную аминокислоту L или не встречающуюся в природе аминокислоту L-трет-бутилаланин ((tBu)A),
X8 представляет собой природную аминокислоту Р или не встречающуюся в природе аминокислоту L-пролин (3,4-2Н),
X9 представляет собой природную аминокислоту Р или не встречающуюся в природе аминокислоту 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновую кислоту (Oic),
X10 представляет собой природную аминокислоту I,
X11 представляет собой не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N-метилцистеина ((N-Me)C) и L-пеницилламина (Pen),
X12 представляет собой природную аминокислоту I,
X13 представляет собой природную аминокислоту Р, s представляет собой целое число 0 или 1,
X14 представляет собой природную аминокислоту, выбранную из перечня, состоящего из D, Q и Е,
t представляет собой целое число 0 или 1,
где N-конец пептида является незамещенным, ацетилированным или моно- или ди-замещенным С1-С20-алкилом,
где С-конец пептида является незамещенным или амидированным, и
где пептид циклизован посредством связи, соединяющей X3 и X11.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид формулы (II) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где
X1 представляет собой природную аминокислоту или не встречающуюся в природе аминокислоту,
q представляет собой целое число 0 или 1,
X2 представляет собой природную аминокислоту I,
r представляет собой целое число 0 или 1,
X3 представляет собой природную аминокислоту С или не встречающуюся в природе аминокислоту L-пеницилламин (Pen),
X4 представляет собой природную аминокислоту S,
X5 представляет собой природную аминокислоту R или не встречающуюся в природе аминокислоту N(5)-метил-Е-аргинин ((Me)R),
X6 представляет собой природную аминокислоту S,
X7 представляет собой природную аминокислоту L или не встречающуюся в природе аминокислоту L-трет-бутилаланин ((tBu)A),
X8 представляет собой природную аминокислоту Р или не встречающуюся в природе аминокислоту L-пролин (3,4-2Н),
X9 представляет собой не встречающуюся в природе аминокислоту 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновую кислоту (Oic),
X10 представляет собой природную аминокислоту I,
X11 представляет собой природную аминокислоту С, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N-метилцистеина ((N-Me)C) и L-пеницилламина (Pen),
X12 представляет собой природную аминокислоту I,
X13 представляет собой природную аминокислоту Р,
s представляет собой целое число 0 или 1,
X14 представляет собой природную аминокислоту, выбранную из перечня, состоящего из D, Q и Е,
t представляет собой целое число 0 или 1,
где N-конец пептида является незамещенным, ацетилированным или моно- или ди-замещенным С1-С20-алкилом,
где С-конец пептида является незамещенным или амидированным, и
где пептид циклизован посредством связи, соединяющей X3 и X11.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид формулы (II) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где
X1 представляет собой природную аминокислоту, выбранную из перечня, состоящего из А и G, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из N-метил-L-аланина (N-Me)A, N-метил-глицина ((N-Me)G), L-норлейцина (Nle), L-норвалина (Nva) и L-орнитина (Orn),
q представляет собой целое число 0 или 1,
X2 представляет собой природную аминокислоту I,
r представляет собой целое число 0 или 1,
X3 представляет собой природную аминокислоту С или не встречающуюся в природе аминокислоту L-пеницилламин (Pen),
X4 представляет собой природную аминокислоту S,
X5 представляет собой природную аминокислоту R или не встречающуюся в природе аминокислоту N(5)-метил-L-аргинин ((Me)R),
X6 представляет собой природную аминокислоту S,
X7 представляет собой природную аминокислоту L или не встречающуюся в природе аминокислоту L-трет-бутилаланин ((tBu)A),
X8 представляет собой природную аминокислоту Р или не встречающуюся в природе аминокислоту L-пролин (3,4-2Н),
X9 представляет собой не встречающуюся в природе аминокислоту 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновую кислоту (Oic),
X10 представляет собой природную аминокислоту I,
X11 представляет собой природную аминокислоту С, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N-метилцистеина ((N-Me)C) и L-пеницилламина (Pen),
X12 представляет собой природную аминокислоту I,
X13 представляет собой природную аминокислоту Р,
s представляет собой целое число 0 или 1,
X14 представляет собой природную аминокислоту, выбранную из перечня, состоящего из D, Q и Е,
t представляет собой целое число 0 или 1,
где N-конец пептида является незамещенным или ацетилированным,
где С-конец пептида является незамещенным или амидированным, и
где пептид циклизован посредством связи, соединяющей X3 и X11.
В частности, настоящее изобретения обеспечивают соединения, содержащие пептид формулы (I), или соединения, содержащие пептид формулы (II).
Согласно настоящему изобретению, следующие определения X1-X14 относятся к пептидам формулы (I) и пептидам формулы (II), определения положений Х0 и X15 относятся к пептидам формулы (I).
Согласно настоящему изобретению Х0, если присутствует, может представлять собой химическую группу, которой не является аминокислота, согласно определению в контексте настоящего изобретения.
Согласно другому варианту осуществления настоящего изобретения Х0 представляет собой химическую группу, выбранную из перечня, состоящего из 2-цианобензойной кислоты, (1S,2S,4S)-бицикло[2.2.1]гепт-5-ен-2-илуксусной кислоты, (2,4-
диоксоимидазолидин-1-ил)уксусной кислоты, 2-(тиоморфолин)уксусной кислоты ((2-тиоморфолин)ацетил), 2-(N-изопропил-N-метиламино)уксусной кислоты, (Заметил валерьяновой кислоты ((S)-3-метилпентановой кислоты), 2-(3-пиридил)уксусной кислоты, 2-(циклогексиламино)уксусной кислоты (2-(циклогексиламино)ацетил), 2-(диэтиламино)уксусной кислоты (2-(диэтиламино)ацетил), 2-(морфолин)уксусной кислоты (2-(морфолин)ацетил), 2-(N-метил-N-циклопропиламино)уксусной кислоты (2-(N-метил-N-циклопропиламино)ацетила), 2-(пиперидин)уксусной кислоты (2-(пиперидин)ацетила), 2-(пирролидин)уксусной кислоты (2-(пирролидин)ацетила), 2-гидроксиуксусной кислоты (2-гидроксиацетила), 2-гидроксиизомасляной кислоты (2-гидроксиизомасляной кислоты), 3-(аминометил)бензойной кислоты, 3-метоксипропионовой кислоты, 4-(аминометил)бензойной кислоты, 4-метилпентановой кислоты (4-метилвалерьяновой кислоты), 5-хлортиофен-карбоновой кислоты, 1-(аминометил)-циклопропил-1-карбоновой кислоты (АСМР), адипиновой кислоты, (S)-азетидин-2-карбоновой кислоты, бензойной кислоты (бензойной), 4-(3,5-диметил-1,2-оксазол-4-ил)-L-фенилаланина, циклобутанкарбоновой кислоты (циклобутанкарбоновой), 2-(циклобутил)уксусной кислоты (циклобутилуксусной кислоты), циклобутилуксусной кислоты (циклобутилуксусной кислоты), циклогексилуксусной кислоты (циклогексилуксусной кислоты), циклогексанкарбоновой кислоты (циклогексилкарбоновой), циклопен-танкарбоновой кислоты, циклопентилуксусной кислоты (циклопентилуксусной кислоты), циклопропанкарбоновой кислоты (циклопропанкарбоновой), циклопропилуксусной кислоты, В-(+)биотина, фумаровой кислоты, 3-фенилпропановой кислоты (гидрокоричной кислоты), изомасляной кислоты, изовалериановой кислоты (изовалериановая), L-(+)-молочной кислоты (молочной кислоты), фенилуксусной кислоты, пиперидин-4-илуксусной кислоты, пивалевой кислоты (пивалевой кислоты), пробковой кислоты, трет-бутилуксусной кислоты, тетрагидропиранил-4-уксусной кислоты, тетрагидро-2Н-пиран-3-илуксусной кислоты и транс-2-(3-((т-бутокси)карбониламино)циклогексил)уксусной кислоты).
Согласно другому варианту осуществления настоящего изобретения Х0 представляет собой химическую группу, выбранную из перечня, состоящего из 2-цианобензойной кислоты, (1R,3S,4S)-2-азабицикло[2.2.1]гептан-3-карбоновой кислоты, (2-тиоморфолин)уксусной кислоты, (N-изопропил-N-метиламино)уксусной кислоты, 2-(3-пиридил)уксусной кислоты, 2-(циклогексиламино)уксусной кислоты, 2-(диэтиламино)уксусной кислоты, 2-(морфолин)уксусной кислоты, 2-(пиперидин)уксусной кислоты, 2-(пирролидин)уксусной кислоты, 3-(аминометил)бензойной кислоты, 4-(аминометил)бензойной кислоты, 1-(аминометил)-циклопропил-1-карбоновой кислоты (АСМР), азетидин-2-карбоновой кислоты, бензойной кислоты, циклобутилуксусной кислоты, циклопропилуксусной кислоты, фенилуксусной кислоты, пиперидин-4-илуксусной кислоты, тетрагидро-2Н-пиран-3-илуксусной кислоты, транексамовой кислоты и транс-2-(3-((1-бутокси)карбониламино)циклогексил)уксусной кислоты.
Согласно другому варианту осуществления настоящего изобретения Х0 представляет собой химическую группу, выбранную из не встречающейся в природе аминокислоты, как определено выше, 2-цианобензойной кислоты, замещенной бензойной кислоты или фенилуксусных кислот.
Согласно другому варианту осуществления настоящего изобретения Х0 представляет собой Ноо, ODD или Ahx.
Согласно другому варианту осуществления настоящего изобретения р представляет собой целое число 1.
Согласно другому варианту осуществления настоящего изобретения р представляет собой целое число 0.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из формулы (I) или формулы (II) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где X1 представляет собой любую природную аминокислоту или не встречающуюся в природе аминокислоту, предпочтительно выбранную из перечня, состоящего из не встречающихся в природе аминокислот, перечисленных в Таблице 1, где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации.
Согласно другому варианту осуществления настоящего изобретения X1 представляет собой природную аминокислоту, выбранную из перечня, состоящего из A, F, r, Н, I, K, L, Р, R, S, Т, V, W и Y, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из N-метил-аланина (N-Me)A, N-метил-глицина ((N-Me)G), (1R,3S,4S)-2-азабицикло[2.2.1]гептан-3-карбоновой кислоты, L-3-бромфенилаланина ((3-бромо)F), L-N,N-диметилаланина ((N,N-диМе)A), N,N-диметилглицина ((N,N-диМе)G), N-фенилглицина ((N-Ph)G), (R)-пиперидин-3-карбоновой кислоты, (S)-пиперидин-3-карбоновой кислоты, L-трет-бутилаланина ((tBu)A), L-2-пиридилаланина (2-Pal), L-3-пиридилаланина (3-Pal), L-4-пиридилаланина (4-Pal), 3-(аминометил)бензойной кислоты, 3-амино-2,2-диметилпропионовой кислоты, 3-амино-3-метилмасляной кислоты, 4-(аминометил)бензойной кислоты, L-2-аминомасляной кислоты (Abu), 1-аминоциклобутан-1-карбоновой кислоты (АСВА), 6-аминогексановой кислоты (Ahx), 2-аминоизомасляной кислоты (Aib), L-2-тиенилаланина (бета-2-тиенилаланин), бета-аланина (бета-А), бета-пролина (бета-Р), L-цитруллина (Cit), Ь-2,4-диаминомасляной кислоты (Dab), L-2,3-диаминопропионовой кислоты (Dap), гамма-аминомасляной кислоты (Гамма-Abu), L-3-метилгистидина (3-Ме)Н), L-дигидрооротовой кислоты (Ноо), L-норлейцина (Nle), N-метил-L-пролина ((N-Me)P), L-норвалина (Nva), L-орнитина (Orn), L-пипеколиновой кислоты (Pip), (2S)-2[(амино)-2-(тетрагидро-2Н-пиран-4-ил)]уксусной кислоты и транексамовой кислоты (транексамовой кислоты), где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации.
Согласно другому варианту осуществления настоящего изобретения X1 представляет собой природную аминокислоту, выбранную из перечня, состоящего из А и G, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из N-метил-L-аланина (N-Me)A, N-метил-глицина ((N-Me)G), L-норлейцина (Nle), L-норвалина (Nva) и L-орнитина (Orn), где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации.
Согласно другому варианту осуществления настоящего изобретения X1 представляет собой природную аминокислоту, выбранную из перечня, состоящего из А и G, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из N-метил-L-аланина (N-Me)A, N-метил-глицина ((N-Me)G), L-норлейцина (Nle), L-норвалина (Nva) и L-орнитина (Orn).
Согласно другому варианту осуществления настоящего изобретения X1 представляет собой природную аминокислоту А или не встречающуюся в природе аминокислоту N-метил-глицин ((N-Me)G).
Согласно другому варианту осуществления настоящего изобретения, когда р представляет собой 0, и q отличен от 0, концевая аминогруппа X1 является незамещенной, аце-тилированной или моно- или дизамещенной С1-С4-алкилом, предпочтительно монозаме-щенной, С1-С20-алкилом.
Согласно другому варианту осуществления настоящего изобретения X1 является метилированным.
Согласно другому варианту осуществления настоящего изобретения X1 является ацетилированным.
Согласно другому варианту осуществления настоящего изобретения q представляет собой целое число 0.
Согласно другому варианту осуществления настоящего изобретения q представляет собой целое число 1.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из формулы (I) или формулы (II) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где X2 представляет собой природную аминокислоту, выбранную из перечня, состоящего из I, L, М, V и А, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N-метил изо лейцина ((N-Me)I), алло-L-изолейцина (алло-I), L-цикло бутил ал анина (Cba), L-норвалина (Nva), L-2-аминомасляной кислоты (Abu), (2S,3S)-2-[(3R)-3-амино-2-оксопирролидин-1-ил]-3-метилпентановой кислоты, (2S,3S)-2-[2-оксопиперазин-1-ил]-3-метилпентановой кислоты, (2S,3S)-2-[(3S)-2-оксопиперазин-1-ил]-3-метилпентановой кислоты, L-метионин-L-сульфоксида, L-метионин-сульфона и L-трет-бутилглицина, где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации.
Согласно другому варианту осуществления настоящего изобретения X представляет собой природную аминокислоту I или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N-метилизолейцина ((N-Me)I), алло-L-изолейцина (алло-I), L-циклобутилаланина (Cba), L-норвалина (Nva), L-2-аминомасляной кислоты (Abu), (2S,3S)-2-[(3R)-3-амино-2-оксопирролидин-1-ил]-3-метилпентановой кислоты, (2S,3S)-2-[2-оксопиперазин-1-ил]-3-метилпентановой кислоты и (2S,3S)-2-[(3S)-2-оксопиперазин-1-ил]-3-метилпентановой кислоты, где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации.
Согласно другому варианту осуществления настоящего изобретения X представляет собой природную аминокислоту I или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N-метилизолейцина ((N-Me)I), L-циклобутилаланина (Cba), L-норвалин (Nva) и L-2-аминомасляной кислоты (Abu), где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации.
Согласно другому варианту осуществления настоящего изобретения X представляет собой природную аминокислоту I.
Согласно другому варианту осуществления настоящего изобретения, когда р и q оба представляют собой 0, и r представляет собой 1, концевая аминогруппа X2 является незамещенной, ацетилированной или моно- или дизамещенной С1-С20-алкилом, предпочтительно монозамещенной, С1-С4-алкилом.
Согласно другому варианту осуществления настоящего изобретения r представляет собой целое число 0.
Согласно другому варианту осуществления настоящего изобретения r представляет собой целое число 1.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из формулы (I) или формулы (II) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где X3 представляет собой природную аминокислоту С, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-пеницилламина (Pen) и L-N-метилцистеина ((N-Me)C).
Согласно другому варианту осуществления настоящего изобретения X3 представляет собой природную аминокислоту С или не встречающуюся в природе аминокислоту L-пеницилламин (Pen).
Согласно другому варианту осуществления настоящего изобретения X3 представляет собой природную аминокислоту С.
Согласно другому варианту осуществления настоящего изобретения, когда р и q и r все представляют собой 0, концевая аминогруппа X является незамещенной, ацетилированной или моно- или дизамещенной С1-С4-алкилом, предпочтительно монозамещенной С1-С20-алкилом.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из формулы (I) или формулы (II) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где X4 представляет собой природную аминокислоту, выбранную из перечня, состоящего из S, С, Т, R или K, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из алло-L-треонина (алло-Т), L- Гомосерина (hSer) и L-орнитина (Orn).
Согласно другому варианту осуществления настоящего изобретения X4 представляет собой природную аминокислоту, выбранную из перечня, состоящего из S, С, Т, R и K.
Согласно другому варианту осуществления настоящего изобретения X4 представляет собой природную аминокислоту, выбранную из перечня, состоящего из S или Т.
Согласно другому варианту осуществления настоящего изобретения X4 представляет собой природную аминокислоту S.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из формулы (I) или формулы (II) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где X5 представляет собой природную аминокислоту R или не встречающуюся в природе аминокислоту N(5)-метил-L-аргинин ((Me)R).
Согласно другому варианту осуществления настоящего изобретения X5 представляет собой природную аминокислоту R или не встречающуюся в природе аминокислоту N(5)-метил-L-аргинин ((Me)R).
Согласно другому варианту осуществления настоящего изобретения X5 представляет собой природную аминокислоту R.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из формулы (I) или формулу (II) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где X6 представляет собой природную аминокислоту, выбранную из перечня, состоящего из S, С или Т, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из алло-L-треонина (алло-Т) и L-2,3-диаминопропионовой кислоты (Dap).
Согласно другому варианту осуществления настоящего изобретения X6 представляет собой природную аминокислоту S или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из алло-L-треонина (алло-Т) и L-2,3-диаминопропионовой кислоты (Dap).
Согласно другому варианту осуществления настоящего изобретения X6 представляет собой природную аминокислоту S или не встречающуюся в природе аминокислоту алло-L-треонин (алло-Т).
Согласно другому варианту осуществления настоящего изобретения X6 представляет собой природную аминокислоту S.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из формулы (I) или формулу (II) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где X7 представляет собой природную аминокислоту, выбранную из перечня, состоящего из L, F или N, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновой кислоты (Oic), L-4-бромфенилаланина ((4-бромо)F), 2,5-дифтор-L-фенилаланина ((2,5-дифтор)F), L-трет-бутилаланина ((tBu)A), 2-хлор-L-фенилаланина ((2-хлор)F), L-2-бромфенилаланина ((2-бромо)F), (S)-2-(амино)-1,6-гександионовой кислоты (AAD), (2S)-2-амино-4,4,4-трифторбутановой кислоты, L-2-амино-4-цианомасляной кислоты (Cnba), 4-фтор-лейцина ((4-фтор)L), (S)-(трифторметил)-L-цистеина, (2S)-2-амино-3-(1-метилциклопропил)пропановой кислоты, L-трет-бутил глицина ((tBu)G), 3-(триметилсилил)-L-аланина, 2,5-дифтор-L-фенилаланина, 2-амино-7-(трет-бутокси)-7-оксогептановой кислоты, 5,5,5-трифтор-L-лейцина ((трифтор)L), 2-метил-L-фенилаланина ((2-Me)F), L-циклобутилаланина (Cba), L-циклопе нтилаланина (Сра), L-циклопропилметилаланина, L-трифторметилаланина, L-дифторметилаланина, 2-фтор-L-фенилаланина ((2-фтор)F), (2S)-3-(2,3-дифторфенил)-2-аминопропановой кислоты, (2S)-3-(3-цианофенил)-2-аминопропановой кислоты, 2-амино-5,5,5-трифтор-4-метил-пентановой кислоты, (2S)-2-амино-5-метил-гексановой кислоты и (2S)-3-(индол-4-ил)-2-(амино)пропановой кислоты.
Согласно другому варианту осуществления настоящего изобретения X7 представляет собой природную аминокислоту, выбранную из перечня, состоящего из L и N, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 2,5-дифтор-L-фенилаланина ((2,5-дифтор)F), L-2-бромфенилаланина ((2-бромо)F), 2-хлор-L-фенилаланина ((2-хлор)F), L-4-бромфенилаланина ((4-бромо)F), 4-фтор-L-лейцина ((4-фтор)L), L-трет-бутил ал анина ((tBu)A), L-трет-бутилглицина ((tBu)G), 5,5,5-трифтор-L-лейцина ((трифтор)L), (S)-2-(амино)-1,6-гександионовой кислоты (AAD), (2S)-2-амино-4,4,4-трифторбутановой кислоты, L-2-амино-4-цианомасляной кислоты (Cnba), (S)-(трифторметил)-L-цистеина, (2S)-2-амино-3-(1-метилциклопропил)пропановой кислоты, (2S)-3-(2,3-дифторфенил)-2-аминопропановой кислоты, 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновой кислоты (Oic), 3-(триметилсилил)-L-аланина и 2-амино-7-(трет-бутокси)-7-оксогептановой кислоты.
Согласно другому варианту осуществления настоящего изобретения X7 представляет собой природную аминокислоту, выбранную из перечня, состоящего из N или L, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 2,5-дифтор-L-фенилаланина ((2,5-дифтор)F), L-2-бромфенилаланина ((2-бромо)F), 2-хлор-L-фенилаланина ((2-хлор)F), 4-фтор-L-лейцина ((4-фтор)L), L-трет-бутилаланина ((tBu)A), (S)-(трифторметил)-L-цистеина, (2S)-2-амино-3-(1-метилциклопропил)пропановой кислоты и 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновой кислоты (Oic), 3-(триметилсилил)-L-аланина.
Согласно другому варианту осуществления настоящего изобретения X7 представляет собой природную аминокислоту L или не встречающуюся в природе аминокислоту L-трет-бутилаланин ((tBu)A).
Согласно другому варианту осуществления настоящего изобретения X7 представляет собой не встречающуюся в природе аминокислоту L-трет-бутилаланин ((tBu)A).
Согласно другому варианту осуществления настоящего изобретения X7 представляет собой природную аминокислоту L.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из формулы (I) или формулу (II) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где X8 представляет собой природную аминокислоту Р, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-пролина (3,4-2Н), (1S,2S,5R)-3-азабицикло[3.1.0]гексан-2-карбоновой кислоты, (1R,2S,5S)-3-азабицикло[3.1.0]гексан-2-карбоновой кислоты, L-гидроксипролина (Hyp), (3S)-морфолин-3-карбоновой кислоты (мор-фолин-3-карбоновой кислоты), L-пипеколиновой кислоты (Pip), (4aR,6aR,9S,11aS)-11-оксо-2,3,4,4а,6а,7,8,9,11,11а-декагидро-1Н-пиридо[3,2-е]пирроло[1,2-а]азепин-9-карбоновой кислоты, транс-4-фторпролина ((транс-4-фтор)Р) и (1R,2S,5S)-3-азабицикло[3.1.0]гексан-2-карбоновой кислоты.
Согласно другому варианту осуществления настоящего изобретения X8 представляет собой природную аминокислоту Р, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-пролина (3,4-2Н), (1S,2S,5R)-3-азабицикло[3.1.0]гексан-2-карбоновой кислоты, транс-4-фторпролина ((транс-4-фтор)Р), L-гидроксипролина (Hyp), (3S)-морфолин-3-карбоновой кислоты (морфолин-3-карбоновой), L-пипеколиновой кислоты (Pip) и (4aR,6aR,9S,11aS)-11-оксо-2,3,4,4a,6a,7,8,9,11,11а-декагидро-1Н-пиридо[3,2-е]пирроло[1,2-а]азепин-9-карбоновой кислоты.
Согласно другому варианту осуществления настоящего изобретения X8 представляет собой природную аминокислоту Р, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-пролина (3,4-2Н), (1S,2S,5R)-3-азабицикло[3.1.0]гексан-2-карбоновой кислоты и (4aR,6aR,9S,11aS)-11-оксо-2,3,4,4а,6а,7,8,9,11,11а-декагидро-1Н-пиридо[3,2-е]пирроло[1,2-а]азепин-9-карбоновой кислоты.
Согласно другому варианту осуществления настоящего изобретения X представляет собой природную аминокислоту Р или не встречающуюся в природе аминокислоту L-пролин (3,4-2Н).
Согласно другому варианту осуществления настоящего изобретения X8 представляет собой природную аминокислоту Р.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из формулы (I) или формулу (II) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где X9 представляет собой природную аминокислоту Р, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновой кислоты (Oic), L-гидроксипролина (Hyp), (2S,4S)-4-трифторметил-пирролидин-2-карбоновой кислоты ((4-CF3)P), (2S,4S)-4-фторпролина ((цис-4-фтор)Р), транс-4-фторпролина ((транс-4-фтор)Р), (2S)-2-амино-4,4,4-трифторбутановой кислоты, L-транс-3-гидроксипролина ((3S-OH)P, (1R,3S,5R)-2-азабицикло[3.1.0]гексан-3-карбоновой кислоты, (6S)-5-азаспиро[2.4]гептан-6-карбоновой кислоты, rel-(1R,3R,5R,6R)-6-(трифторметил)-2-азабицикло[3.1.0]гексан-3-карбоновой кислоты, (2S)-2-амино-4,4,4-трифторбутановой кислоты, (2S,3aS,6aS)-октагидроциклопента[b]пиррол-2-карбоновой кислоты, транс-4-фторпролина ((транс-4-фтор)Р), (2S,4S)-4-фторпролина ((цис-4-фтор)Р), L-4,4-дифторпролина ((дифтор)Р), rel-(3R,6R)-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновой кислоты (энантиомер 1) и rel-(3R,6R)-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновой кислоты (энантиомер 2).
Согласно другому варианту осуществления настоящего изобретения X9 представляет собой природную аминокислоту Р, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновой кислоты (Oic), (6S)-5-азаспиро[2.4]гептан-6-карбоновой кислоты, rel-(1R,3R,5R,6R)-6-(трифторметил)-2-азабицикло[3.1.0]гексан-3-карбоновой кислоты, (2S,4S)-4-трифторметил-пирролидин-2-карбоновой кислоты ((4-CF3)P), (2S,3aS,6aS)-октагидроциклопента[b]пиррол-2-карбоновой кислоты, (1R,3S,5R)-2-азабицикло[3.1.0]гексан-3-карбоновой кислоты, (2S)-2-амино-4,4,4-трифторбутановой кислоты, L-транс-3-гидроксипролина ((3S-OH)P, транс-4-фторпролина ((транс-4-фтор)Р), L- Гидроксипролина (Hyp), (2S,4S)-4-фторпролина ((цис-4-фтор)Р), (3R,6R)-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновой кислоты (энантиомер 1) и (3R,6R)-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновой кислоты (энантиомер 2).
Согласно другому варианту осуществления настоящего изобретения X9 представляет собой природную аминокислоту Р, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновой кислоты (Oic), (2S,4S)-4-трифторметил-пирролидин-2-карбоновой кислоты ((4-CF3)P), (6S)-5-азаспиро[2.4]гептан-6-карбоновой кислоты, rel-(1R,3R,5R,6R)-6-(трифторметил)-2-азабицикло[3.1.0]гексан-3-карбоновой кислоты, (2S,3aS,6aS)-октагидроциклопента[b]пиррол-2-карбоновой кислоты, rel-(3R,6R)-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновой кислоты (энантиомер 1) и rel-(3R,6R)-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновой кислоты (энантиомер 2).
Согласно другому варианту осуществления настоящего изобретения X9 представляет собой природную аминокислоту Р или не встречающуюся в природе аминокислоту 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновую кислоту (Oic).
Согласно другому варианту осуществления настоящего изобретения X9 представляет собой не встречающуюся в природе аминокислоту 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновую кислоту (Oic).
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из формулы (I) или формулу (II) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где X10 представляет собой природную аминокислоту I, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-циклопентилглицина (Cpg), L-циклогексилглицина (Chg), (S)-2-амино-3-этил-пентановой кислоты, 3-хлорфенилглицина ((3-хлор-Ph)G), L-трет-бутилглицина, алло-L-изолейцина (алло-1), L-циклобутил глицина, L-норвалина (Nva) и (2S)-2-(амино)-2-[(1S,3R)-3-гидроксициклогексил]уксусной кислоты.
Согласно другому варианту осуществления настоящего изобретения X10 представляет собой природную аминокислоту I, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из (2S)-2-(амино)-2-[(1S,3R)-3-гидроксициклогексил]уксусной кислоты, 3-хлорфенилглицина ((3-хлор-Ph)G), (S)-2-амино-3-этил-пентановой кислоты, алло-L-изолейцина (алло-1), L-циклогексилглицина (Chg), L-циклопентилглицина (Cpg), L-циклобутилглицина и L-норвалина (Nva).
Согласно другому варианту осуществления настоящего изобретения X10 представляет собой природную аминокислоту I, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из (2S)-2-(амино)-2-[(1S,3R)-3-гидроксициклогексил]уксусной кислоты, (S)-2-амино-3-этил-пентановой кислоты, L-циклогексилглицина (Chg) и L-циклопентилглицина (Cpg).
Согласно другому варианту осуществления настоящего изобретения X10 представляет собой природную аминокислоту I.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из формулы (I) или формулу (II) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где X11 представляет собой природную аминокислоту С, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N-метилцистеина ((N-Me)C) и L-пеницилламина (Pen).
Согласно другому варианту осуществления настоящего изобретения X11 представляет собой природную аминокислоту С, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N-метилцистеина ((N-Me)C) и L-пеницилламина (Pen).
Согласно другому варианту осуществления настоящего изобретения X11 представляет собой не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N-метилцистеина ((N-Me)C) и L-пеницилламина (Pen).
Согласно другому варианту осуществления настоящего изобретения X11 представляет собой не встречающуюся в природе аминокислоту L-пеницилламин (Pen).
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из формулы (I) или формулу (II) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где X9 представляет собой не встречающуюся в природе аминокислоту 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновую кислоту (Oic), и X11 представляет собой не встречающуюся в природе аминокислоту L-N-метилцистеин ((N-Me)C) или не встречающуюся в природе аминокислоту L-пеницилламин (Pen).
Согласно другому варианту осуществления настоящего изобретения X9 представляет собой не встречающуюся в природе аминокислоту 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновую кислоту (Oic), и X11 представляет собой не встречающуюся в природе аминокислоту L-пеницилламин (Pen).
Согласно другому варианту осуществления настоящего изобретения X9 представляет собой не встречающуюся в природе аминокислоту 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновую кислоту (Oic), и X11 представляет собой не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из L-N-метилцистеина ((N-Me)C) и L-пеницилламина (Pen).
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из формулы (I) или формулу (II) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где X12 представляет собой природную аминокислоту I, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из алло-L-изолейцина (алло-I), (S)-2-амино-2-циклобутилуксусной кислоты (Cbg), (2S,3S)-2-((амино)метил)-3-метилпентановой кислоты, L-фенилглицина (Phg), 2-[(1S,2S)-1-(амино)-2-метилбутил]-1,3-оксазол-4-карбоновой кислоты, 2-метил-D-аллоизолейцина, L-норвалина (Nva), L-2-аминомасляной кислоты (Abu), L-трет-бутил глицина и Аминоизо-масляной кислоты (Aib).
Согласно другому варианту осуществления настоящего изобретения X12 представляет собой природную аминокислоту I, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 2-[(1S,2S)-1-(амино)-2-метилбутил]-1,3-оксазол-4-карбоновой кислоты, (S)-2-амино-2-циклобутилуксусной кислоты (Cbg), алло-L-изолейцина (алло-1), L-фенилглицина (Phg), 2-метил-D-аллоизолейцина и (2S,3S)-2-((амино)метил)-3-метилпентановой кислоты.
Согласно другому варианту осуществления настоящего изобретения X12 представляет собой природную аминокислоту I, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из алло-L-изолейцина (алло-1), (S)-2-амино-2-циклобутилуксусной кислоты (Cbg) и 2-[(1S,2S)-1-(амино)-2-метилбутил]-1,3-оксазол-4-карбоновой кислоты.
Согласно другому варианту осуществления настоящего изобретения X12 представляет собой природную аминокислоту I.
Согласно другому варианту осуществления настоящего изобретения, когда u и t и s все представляют собой 0, концевая карбоксильная группа X12 является незамещенной или амидированной.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из формулы (I) или формулу (II) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где X13 представляет собой природную аминокислоту, выбранную из перечня, состоящего из Р, A, S, Т, г, D, Е, Q или N или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из N-метил-глицина ((N-Me)G), 5-азаспиро[2.4]гептан-6-карбоновой кислоты, L-2-аминомасляной кислоты (Abu), 2-аминоизомасляной кислоты (Aib), 2-метил-L-пролина (2-Ме)Р, гидроксипролина (Hyp), 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновой кислоты (Oic), транс-4-фторпролина ((транс-4-фтор)Р), (2S,4S)-4-фторпролина ((цис-4-фтор)Р), L-4,4-дифторпролина ((дифтор)Р), L-циклопентилглицина (Cpg), (S)-2-амино-2-циклобутилуксусной кислоты (Cbg) и (2S)-пирролидин-2-илуксусной кислоты (бета-гомо-Р), где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации.
Согласно другому варианту осуществления настоящего изобретения X13 представляет собой природную аминокислоту, выбранную из перечня, состоящего из Р, А и D, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 2-метил-L-пролина (2-Ме)Р, N-метил-глицина ((N-Me)G), транс-4-фторпролина ((транс-4-фтор)Р), L-2-аминомасляной кислоты (Abu), 2-аминоизомасляной кислоты (Aib), Гидро-ксипролина (Hyp) и 2,3,3а,4,5,6,7,7а-октагидроиндол-2-карбоновой кислоты (Oic), где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации.
Согласно другому варианту осуществления настоящего изобретения X13 представляет собой природную аминокислоту, выбранную из перечня, состоящего из Р или D, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 2-аминоизомасляной кислоты (Aib), 2-метил-L-пролинв (2-Ме)Р и транс-4-фторпролинв ((транс-4-фтор)Р), где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации.
Согласно другому варианту осуществления настоящего изобретения X13 представляет собой природную аминокислоту Р.
Согласно другому варианту осуществления настоящего изобретения s представляет собой целое число 0.
Согласно другому варианту осуществления настоящего изобретения s представляет собой целое число 1.
Согласно другому варианту осуществления настоящего изобретения, когда u и t оба представляют собой 0, и s отличен от 0, концевая карбоксильная группа X13 является незамещенной или амидированной.
Согласно другому варианту осуществления настоящего изобретения, когда t представляет собой 0, и s представляет собой 1, концевая карбоксильная группа X13 является незамещенной или амидированной.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из формулы (I) или формулу (II) или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, где X14 представляет собой любую природную аминокислоту или не встречающуюся в природе аминокислоту, где любая природная аминокислота и/или не встречающаяся в природе аминокислота может быть в D- или L-стереоконфигурации.
Согласно другому варианту осуществления настоящего изобретения X14 представляет собой природную аминокислоту, выбранную из перечня, состоящего из D, Е, G, K, N, Р и Q, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 3-карбоксифенилаланина ((3-карбокси)F), (2S)-2-амино-4-(бензиламино)-4-оксобутанкарбоновой кислоты ((N-бензил)D), N-метил-глицина ((N-Me)G), 6-аминогексановой кислоты (Ahx), (2S)-пирролидин-2-илуксусной кислоты (бета-гомо-Р), L-2,3-диаминопропионовой кислоты (Dap), L-орнитина (Orn) и транексамовой кислоты (транексамовой кислоты), где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации.
Согласно другому варианту осуществления настоящего изобретения X14 представляет собой природную аминокислоту, выбранную из перечня, состоящего из D, Q, N, Е и Р, или не встречающуюся в природе аминокислоту, выбранную из перечня, состоящего из 3-карбоксифенилаланина ((3-карбокси)Е), N-метил-глицина ((N-Me)G), (2S)-пирролидин-2-илуксусной кислоты (бета-гомо-Р), L-2,3-диаминопропионовой кислоты (Dap), L-орнитина (Orn) и транексамовой кислоты (транексамовой кислоты), где каждая природная аминокислота и/или не встречающаяся в природе аминокислота в L-стереоконфигурации может быть заменена стереоизомером в D-стереоконфигурации.
Согласно другому варианту осуществления настоящего изобретения X14 представляет собой природную аминокислоту, выбранную из перечня, состоящего из D, Q или I.
Согласно другому варианту осуществления настоящего изобретения X14 представляет собой природную аминокислоту D.
Согласно другому варианту осуществления настоящего изобретения С-концевой "-ОН" фрагмент при X14 замещен С-терминальным "-NH2" фрагментом.
Согласно другому варианту осуществления настоящего изобретения q представляет собой целое число 0.
Согласно другому варианту осуществления настоящего изобретения q представляет собой целое число 1.
Согласно другому варианту осуществления настоящего изобретения, когда s и t представляет собой целое число 1, концевая карбоксильная группа X14 является незамещенной или амидированной.
Согласно другому варианту осуществления настоящего изобретения, когда s и t представляет собой целое число 1, концевая карбоксильная группа X14 является незамещенной.
Согласно настоящему изобретению X15, если присутствует, может представлять собой химическую группу, которая не является аминокислотой.
Согласно другому варианту осуществления настоящего изобретения X15 представляет собой химическую группу, выбранную из перечня, состоящего из -NH2, -NH-C1-С6-алкила, (1R,3S)-3-(амино)циклопентанкарбоновой кислоты, (1S,3R)-3-(амино)циклопентанкарбоновой кислоты, (R)-4-амино-6-метилгептановой кислоты, (S)-(l-пиперидин-3-ил)-уксусной кислоты, (S)-3-(1-пирролидин-2-ил)-пропио новой кислоты, (S)-3-(2Н-тетразол-5-ил)пропановой кислоты, (S)-пирролидин-3-карбоновой кислоты, 5-азаспиро[2.4]гептан-1-карбоновой кислоты и (2S)-3-(триазол-1-ил)-2-(амино)пропановой кислоты.
Согласно другому варианту осуществления настоящего изобретения X15 представляет собой химическую группу, выбранную из -NH2 или -NH-C1-С6-алкила, предпочтительно -NH2.
Согласно другому варианту осуществления настоящего изобретения q представляет собой целое число 0.
Согласно другому варианту осуществления настоящего изобретения q представляет собой целое число 1.
Согласно другому варианту осуществления настоящего изобретения N-конец пептида является незамещенным, ацетилированным или моно- или дизамещенным С1-С20-алкилом.
Согласно другому варианту осуществления настоящего изобретения N-конец пептида является незамещенным, ацетилированным или моно- или дизамещенным С1-С4-алкилом.
Согласно другому варианту осуществления настоящего изобретения N-конец пептида является незамещенным или ацетилированным.
Согласно другому варианту осуществления настоящего изобретения С-конец является незамещенным или амидированным.
Согласно другому варианту осуществления настоящего изобретения N-конец и С-конец является незамещенным.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из пептида формулы (I) или формулы (II) или его производное, пролекарство, аналог, фармацевтически приемлемая соль, сольват или сольват соли, которые выбраны из группы, состоящей из Примера 29, 44, 45, 67, 75, 109, 166, 185, 443 и 466, или которые выбраны из группы, состоящей из Примера 29, 44, 45, 67, 75, 109, 166, 185, 443, 466 и 499.
Пептид формулы (I) или (II) может быть линейным или циклическим. Используемый в настоящем изобретении термин «циклизованный» относится к реакции, в которой одна часть молекулы полипептида (например, Cys, (N-Me)Cys или Pen) становится связанной с другой частью молекулы полипептида (например, другой Cys, (N-Me)Cys или Pen) с образованием замкнутого кольца, например, путем образования дисульфидного мостика (-S-S-) или других подобных связей, таких как связь углерод-сера (например, (-CH2-S-) или (-(CH2)2-S-)), связь сера-углерод (например, (-S-CH2-) или (-S-(CH2)2-)), связь углерод-сера-углерод (-CH2-S-CH2-) или связь углерод-углерод (например, (-СН2-СН2-). Согласно настоящему изобретению связь между положениями X3 и X11 пептида является предпочтительной. Аминокислота, за которой следует «+» в последовательности пептида согласно настоящему изобретению, относится к циклическому пептиду, где аминокислота связана с другой аминокислотой, за которой следует «+», дисульфидным мостиком, образующим замкнутое кольцо. В качестве альтернативы две связанные химические группы полипептидной молекулы могут быть изображены соединительной линией.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из пептида формулы (I) или формулы (II), или его производное, пролекарство, аналог, фармацевтически приемлемая соль, сольват или сольват соли, где пептид является циклизованным.
Согласно другому варианту осуществления настоящего изобретения пептид формулы (I) или формулы (II) циклизован посредством связи, соединяющей X3 и X11.
Согласно другому варианту осуществления настоящего изобретения пептид формулы (I) или формулы (II) циклизован посредством связи, соединяющей X3 и X11, и где X3 и X11 соединены линкером формулы -S-S-, -CH2-S-, -S-CH2-, -СН2-СН2-, -S-(CH2)2-, -(СН2)2-S- или -CH2-S-CH2-.
Согласно другому варианту осуществления настоящего пептид формулы (I) или формулы (II) циклизован посредством связи, соединяющей X3 и X11, и где X3 и X11 соединены дисульфидной связью формулы -S-S-.
Пептид согласно настоящему изобретению может быть заменен подходящим водорастворимым полимером, характеризующимся повторяющимися звеньями. Полимеры подходящих полимеров могут быть выбраны из группы, состоящей из полиалкилоксиполимеров, гиалуроновой кислоты и ее производных, поливиниловых спиртов, полиоксазолинов, полиангидридов, поли(ортоэфиров), поликарбонатов, полиуретанов, полиакриловых кислот, полиакриламидов, полиакрилатов, полиметакрилатов, полиорганофосфазенов, полисилоксанов, поливинилпирролидона, полицианоакрилатов и сложных полиэфиров.
Пептиды согласно настоящему изобретению могут быть замещены по меньшей мере одной полиэтиленовой группой (группой PEG). По меньшей мере одна группа PEG предпочтительно связана с N-концом и/или С-концом. Группа PEG может быть связана с любой подходящей функциональной группой химической группы и/или аминокислоты пептида, например, гидроксильная группа, карбоксильная группа, аминогруппа, тиоловая группа, предпочтительно амино- или карбоксигруппа. Предпочтительно пептид согласно настоящему изобретению содержит одну группу PEG, связанную с N-концом, или одну группу PEG, связанную с С-концом. Более предпочтительно, одна группа PEG связана с N-концом или С-концом через амидную связь.
Пэгилирование пептидов может повышать их растворимость, снижать иммуноген-ность, улучшать стабильность и/или увеличивать период полувыведения за счет снижения почечного клиренса, что является хорошо известной концепцией с начала 1980-х годов (Caliceti P.,Veronese F.M., Adv. Drug Deliv. Rev. 2003, 55, 1261-1277). Для некоторых лекарственных средств это было успешно используется, но во многих случаях пегилирова-ние снижает эффективность лекарственного средства до такой степени, что эта концепция более не подходит (Т. Peleg-Shulman et al., J. Med.Chem., 2004, 47, 4897-4904).
Группа PEG согласно настоящему изобретению представляет собой любую группу, содержащую по меньшей мере два звенья этиленоксида с образованием олигомера или полимера этиленоксида. Группа PEG ковалентно связана с пептидом согласно настоящему изобретению (Пэгилирование), который затем называют Пэгилированным пептидом. Как правило, этот тип модификации молекулы хорошо известен в данной области техники.
Группа PEG может состоять из связующего фрагмента, полимерного фрагмента и концевой группы. Соединяющиеся фрагменты могут состоять из С1-С20 алкила, который необязательно прерывается или оканчивается гетероатомами или функциональными группами, выбранными из группы, состоящей из -О-, -S-, N(RX), С(О), C(O)RX, C(O)N(RX), N(RX)C(O), одно или нескольких С3-С8-циклоалкилов, С3-С7-гетероциклоалкилов, арилов или гетероарилов, где RX представляет собой водород или C1-С6-алкил, который необязательно прерывается или оканчивается одним или несколькими из вышеупомянутых атомов или групп, которые дополнительно содержат водород в качестве концевого атома. Полимерные фрагменты состоят по меньшей мере из двух звеньев этиленоксида, необязательно прерываемых или заканчивающихся одним или несколькими гетероатомами или функциональными группами, выбранными из группы, состоящей из -О-, -S-, N(RX), С(О), C(Q)RX, C(Q)N(RX), N(RX)C(Q), C1-C6-алкила, С3-С8-циклоалкила, С3-С7-гетероциклоалкила, арила или гетероарила, где RX представляет собой водород или C1-C6-алкил. Концевая группа может состоять из гетероатомов или функциональных групп, выбранных из ОН, SH, N(RX)2, C(O)RX; CORX, COORX, C(O)N(RX), N(RX)C(O)NH2, где RX представляет собой водород или C1-С6-алкил. Предпочтительными концевыми группами являются СООН и C(O)NH2.
Примеры спейсера PEG согласно настоящему изобретению включают PEG 1 (10 атомов), имеющий 1 звено этиленгликоля (n=1; 10 атомов), PEG 2 (13 атомов), имеющий 2 звена этиленгликоля (n=2; 13 атомов), PEG 3 (16 атомов) с 3 звеньями этиленгликоля (n=3; 16 атомов), PEG 4(19 атомов) с 4 звеньями этиленгликоля (n=4; 19 атомов), PEG5(22 атома) с 5 звеньями этиленгликоля (n=5; 22 атомов), PEG 6 (25 атомов) с 6 звеньями этиленгликоля (n=6; 25 атомов) и т.д.
Следует отметить, что группы PEG, как определено в настоящем документе, могут быть указаны коммерческими поставщиками под разными названиями, что не должно исключать такие идентичные соединения с разными названиями из настоящего изобретения.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделенным и/или очищенным, содержащим, по существу состоящим из или состоящим из пептида формулы (I) или формулы (II), или его производное, пролекарство, аналог, фармацевтически приемлемая соль, сольват или сольват соли, где пептид замещен по меньшей мере одной группой PEG.
Согласно другому варианту осуществления настоящего изобретения пептид замещен одной группой PEG.
Согласно другому варианту осуществления настоящего изобретения пептид замещен одной группой PEG, где группа PEG связана с С-концом, предпочтительно через амидную связь.
Согласно другому варианту осуществления настоящего изобретения пептид замещен по меньшей мере одной группой PEG, где по меньшей мере одна или несколько групп PEG представляет собой группу формулы (IIIa)
где
* означает присоединение азота к атому кислорода,
D представляет собой С1-С4-алкилен,
Y выбран из группы, состоящей из гидроксила, метокси, этокси, карбокси, карбоксамида или амино, и
n представляет собой целое число 2-15, или группу формулы (IIIb)
где
* означает связь с карбонильной группой, D1 представляет собой С1-С4-алкилен,
Y1 выбран из группы, состоящей из гидроксила, метокси, этокси, карбокси, кар-боксамида и амино, и
m представляет собой целое число 2-15,
или TTDS.
Согласно другому варианту осуществления настоящего изобретения пептид замещен по меньшей мере одной группой PEG, где по меньшей мере одна или несколько групп PEG выбраны из перечня, состоящего из PEG1(10 атомов), PEG2(13 атомов), PEG3(16 атомов), PEG4(19 атомов), PEG4-CH2CO2H (15 атомов), PEG5(22 атомов), PEG5-CH2CO2H (18 атомов), PEG7(25 атомов), PEG8(28 атомов), PEG9(31 атомов) и TTDS.
Пептид согласно настоящему изобретению может содержать по меньшей мере одну жирную кислоту С8-С20. В общем, такая жирная кислота может быть разветвленной или циклической. По меньшей мере одна жирная кислота C8-C20 предпочтительно связана с N-концом и/или С-концом. Жирная кислота C8-C20 может быть связана с любой подходящей функциональной группой химической группы и/или аминокислоты пептида, например, гидроксильной группой, карбоксильной группой, аминогруппой, тиольной группой, предпочтительно амино или карбоксильной группой. Предпочтительно пептид согласно настоящему изобретению содержит одну C8-C20 жирную кислоту, связанную с N-концом, или одну С8-С20 жирную кислоту, связанную с С-концом. Более предпочтительно одна С8-С20 жирная кислота связана с N- концом или С- концом через амидную связь. Предпочтительно боковая цепь жирной кислоты, образованная Х0 или X15, представляет собой жирную кислоту > С8, более предпочтительно жирную кислоту ≥ C12, более предпочтительно жирную кислоту ≥ С14. Кроме того, предпочтительно, чтобы боковая цепь жирной кислоты, образованная Х0 или X15, представляет собой жирную кислоту C12-C18, предпочтительно Наиболее предпочтительной является C12-C16 жирная кислота или С14-С18 жирная кислота, или С14-С16 жирная кислота. Наиболее предпочтительной является жирная кислота, такая как пальмитиновая кислота (пальмитоил, Palm) и a C18 жирная кислота, такая как 1,18-октадекандиовая кислота (ODD).
Также следует понимать, что фрагмент на N-конце или С-конце может быть связью, например, ковалентной связью, особенно в ситуациях, когда амино-конец или карбокси-конец связан с линкером или с другим химическим фрагментом.
Химические группы, не встречающиеся в природе аминокислоты или фрагменты могут быть обозначены сокращенно, как показано в Таблице 3.
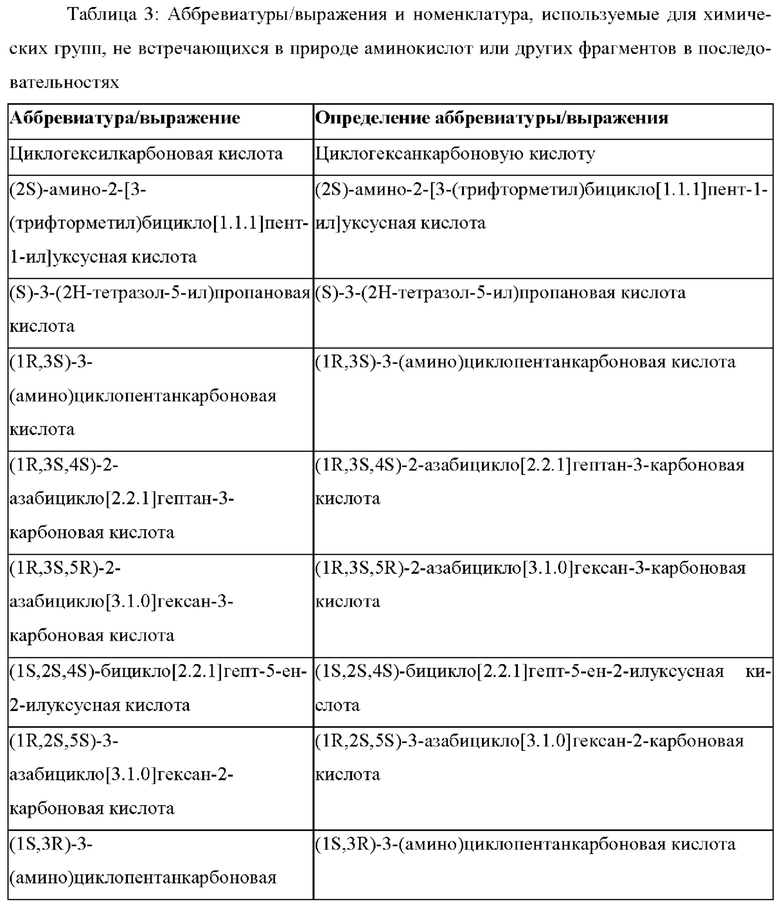
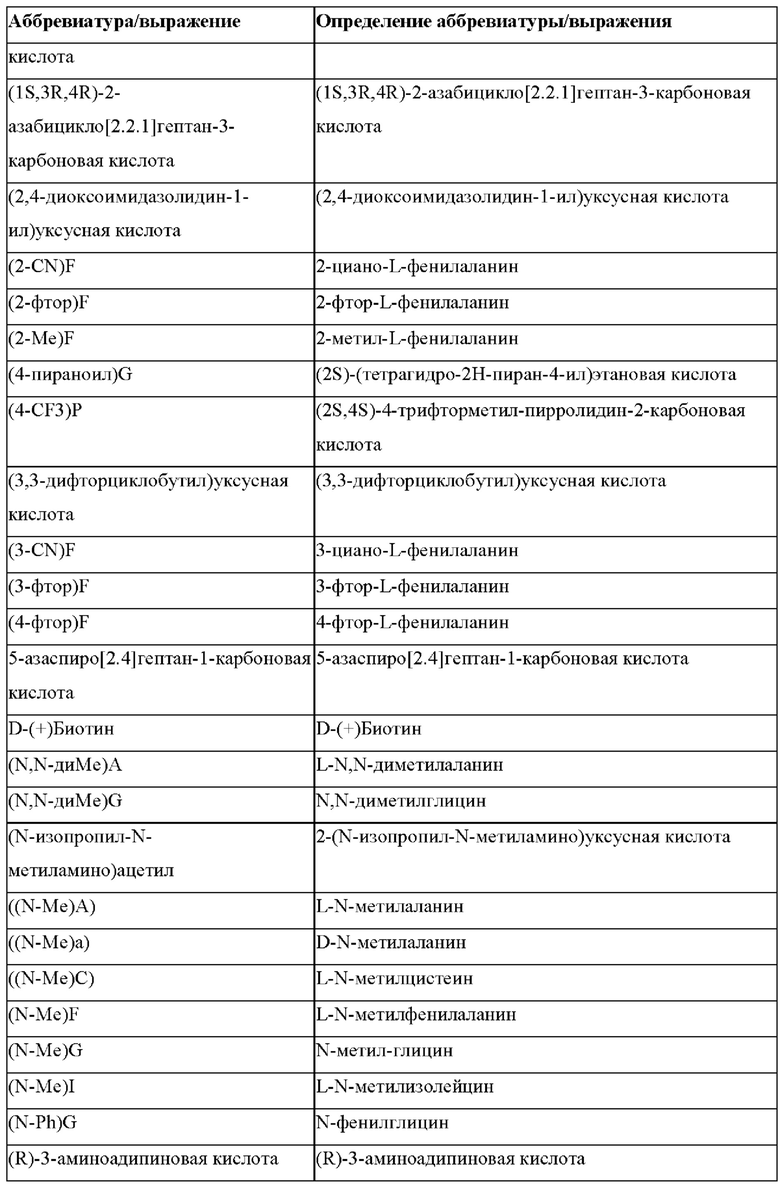


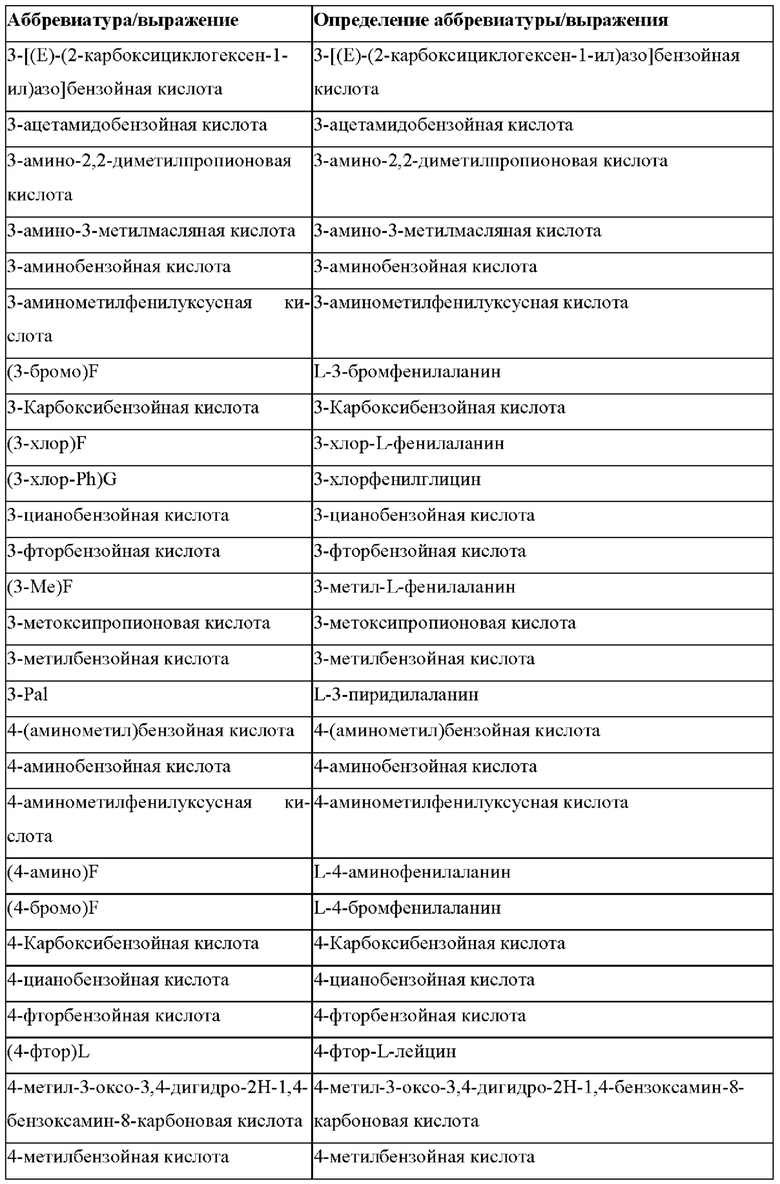
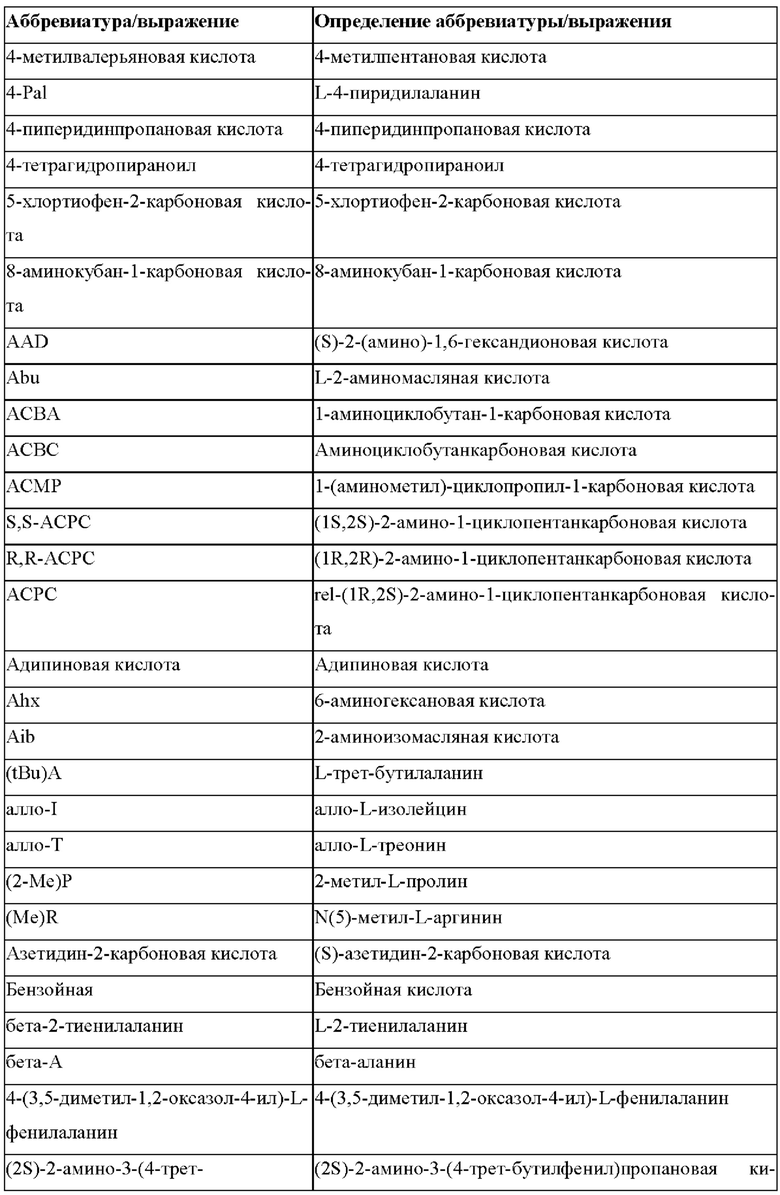
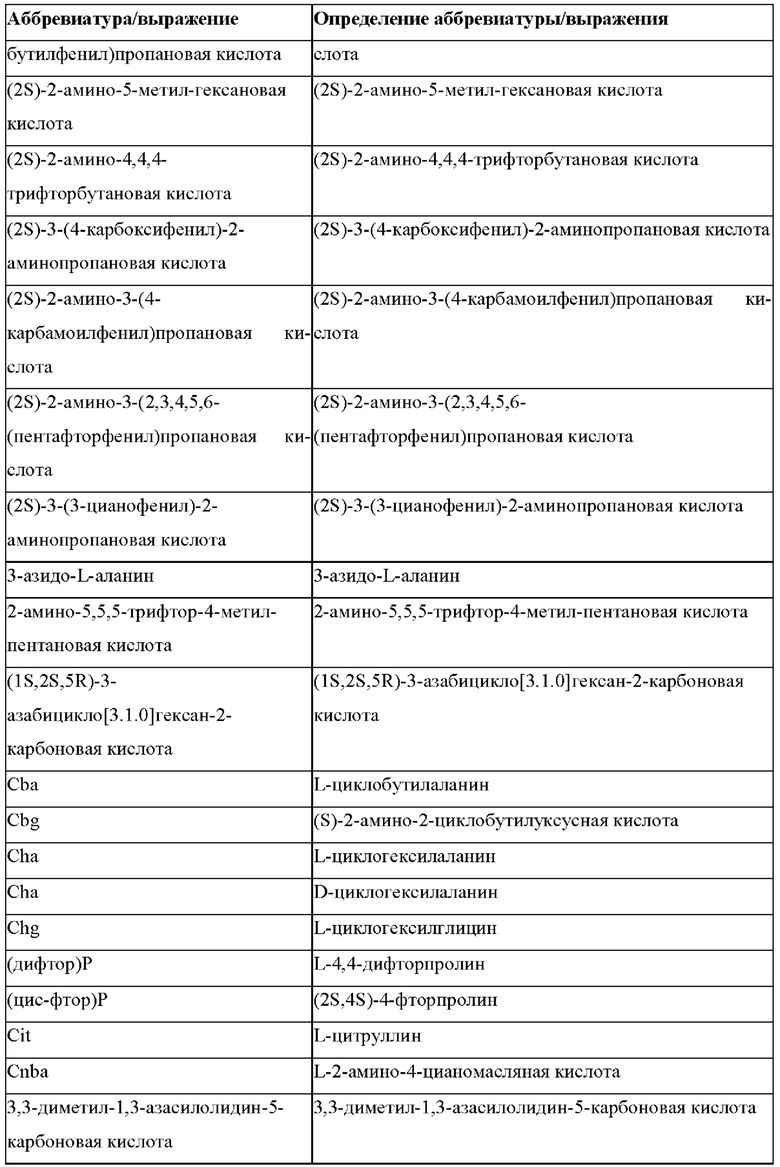

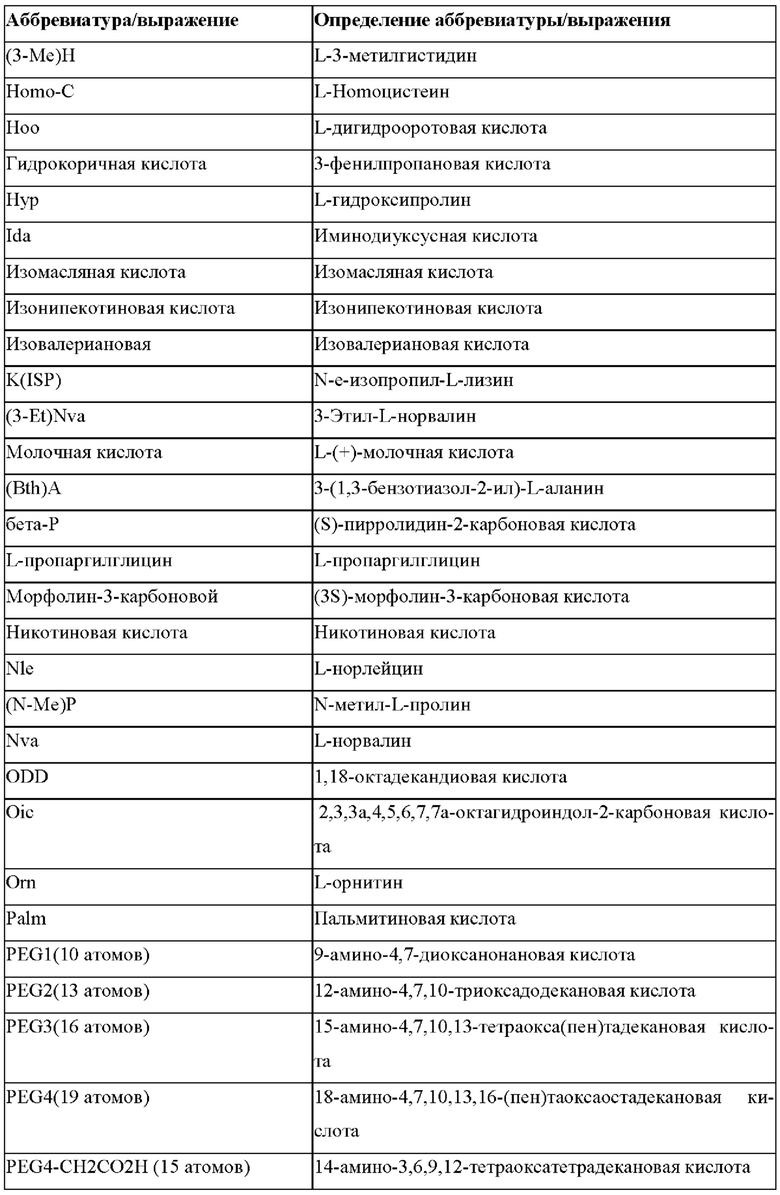
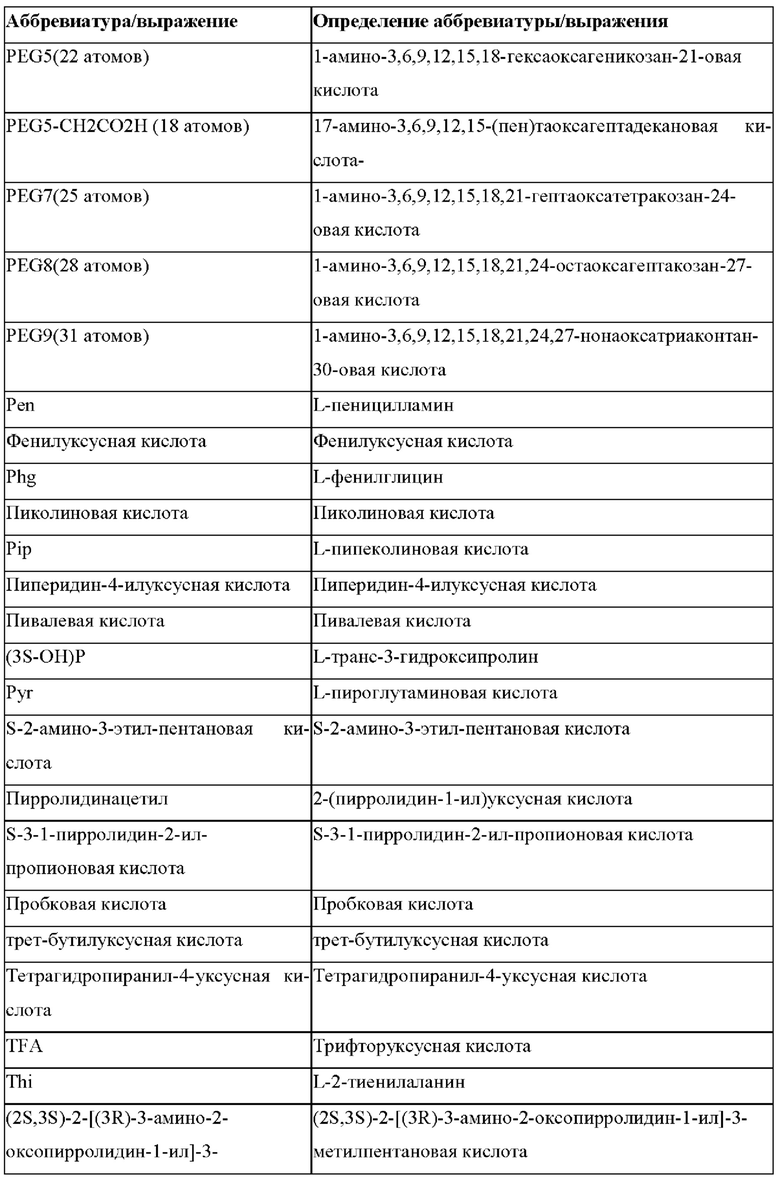
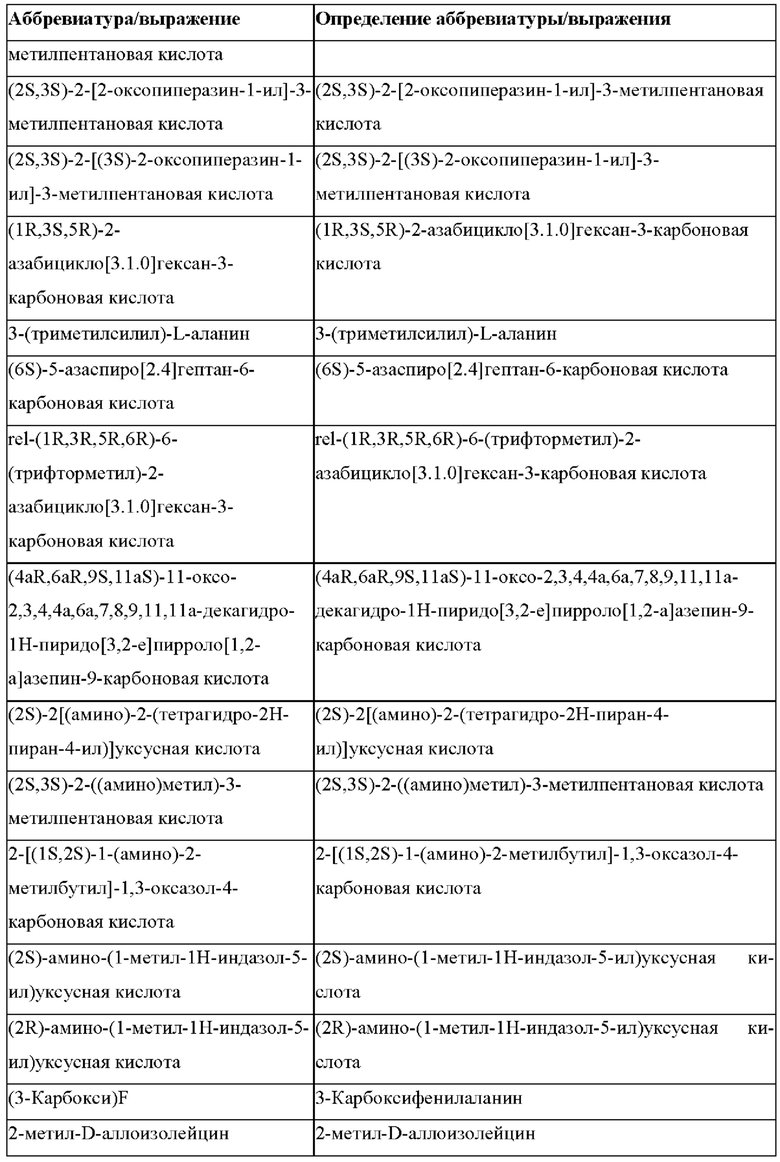
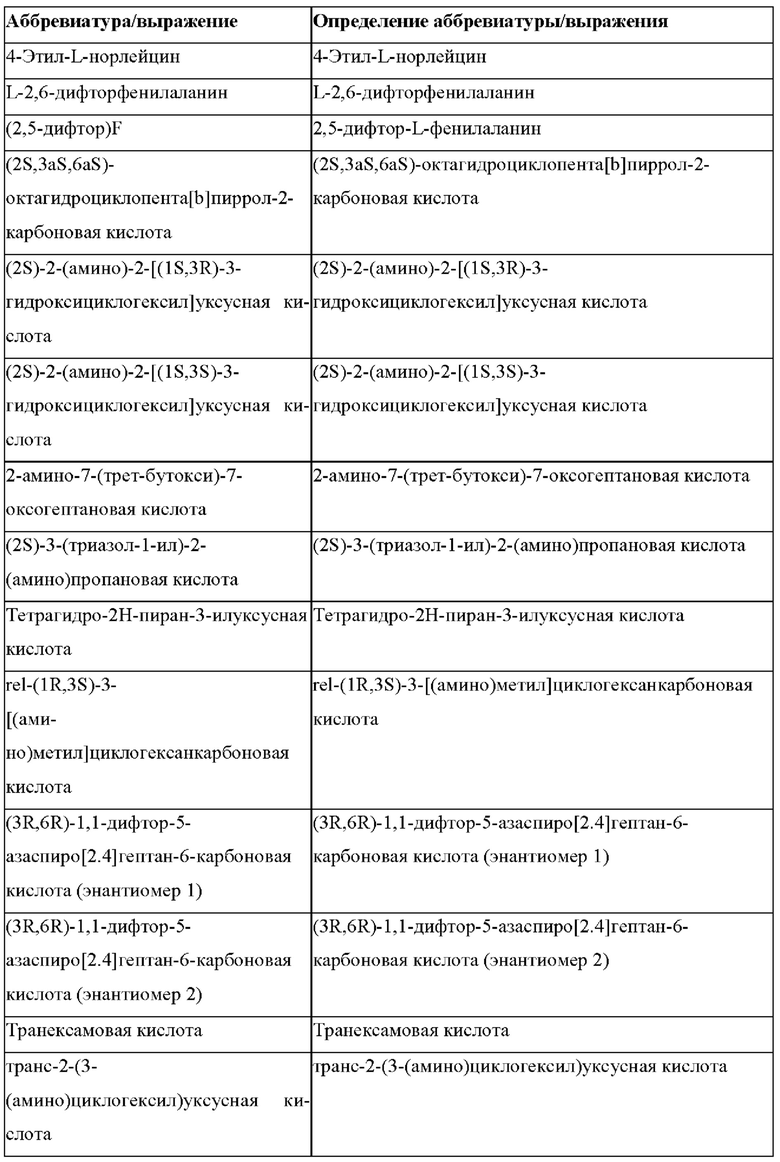
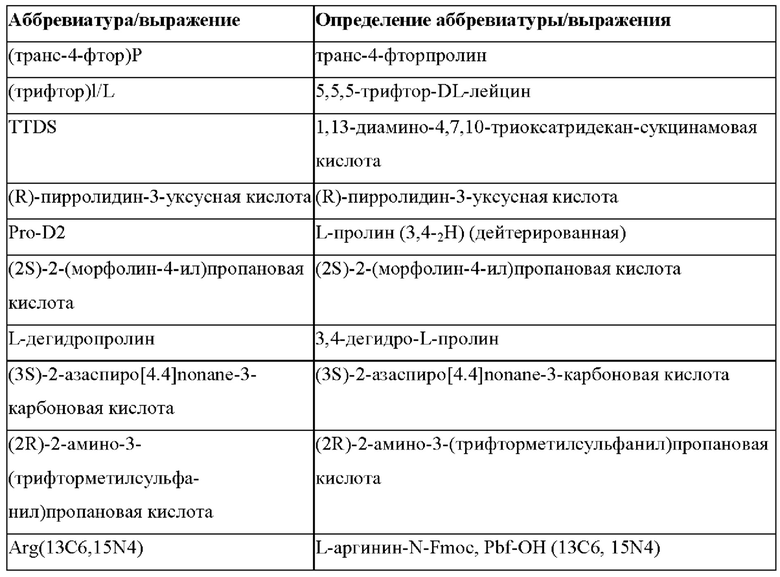
Термин «миметик», используемый в контексте с некоторыми аминокислотами в определении нескольких фрагментов пептида в соответствии с формулой (I) или формулой (II) согласно настоящему изобретению, представляет собой соответствующий миметик аминокислоты, такой как, например, миметик аргинина, миметик изолейцина или миметик пролина. Как правило, «миметик белка» означает молекулу, такую как пептид, модифицированный пептид или любую другую молекулу, которая биологически имитирует действие или активность какого-либо другого белка. В контексте использования термина «миметик» в связи с определенной аминокислотой указанный термин «миметик» аналогично означает любую другую аминокислоту, аналог аминокислоты, производное аминокислоты, конъюгат аминокислоты или тому подобное, которые биологически имитируют действие или активность соответствующей аминокислоты.
Миметики пролина согласно настоящему изобретению содержат, в частности (1S,2S,5R)-3-азабицикло[3.1.0]гексан-2-карбоновую кислоту, Hyp, морфолин-3-карбоновой, Pip, (4aR,6aR,9S,11aS)-11-оксо-2,3,4,4а,6а,7,8,9,11,11а-декагидро-1H-пиридо[3,2-е]пирроло[1,2-а]азепин-9-карбоновую кислоту или (транс-4-фтор)Р, (1R,2S,5S)-3-азабицикло[3.1.0]гексан-2-карбоновую кислоту, Oic, Hyp, (4-CF3)P, (цис-4-фтор)Р, 3,3-диметил-1,3-азасилолидин-5-карбоновую кислоту, (3S-OH)P, (1R,3S,5R)-2-азабицикло[3.1.0]гексан-3-карбоновую кислоту, (6S)-5-азаспиро[2.4]гептан-6-карбоновую кислоту, rel-(1R,3R,5R,6R)-6-(трифторметил)-2-азабицикло[3.1.0]гексан-3-карбоновую кислоту, (2S,3aS,6aS)-октагидроциклопента[b]пиррол-2-карбоновую кислоту или дифтор-пролин, (3R,6R)-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновую кислоту (энантиомер 1), (3R,6R)-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновую кислоту (энантиомер 2) и замещенные пролины.
Миметики изолейцина согласно настоящему изобретению содержат, в частности, (N-метил)-I, алло-Ile, Cba, Nva, Abu, Leu, Cpg, циклогексил-Gly, (S)-2-амино-3-этил-пентановую кислоту, 3-хлор-Phg, алло-Ile, Chg, циклобутилглицин, алло-Ile, Cbg, (2S,3S)-2-((амино)метил)-3-метилпентановую кислоту, Phg, 2-[(1S,2S)-1-(амино)-2-метилбутил]-1,3-оксазол-4-карбоновую кислоту, 2-метил-D-аллоизолейцин, Nva, Abu или Ala.
Миметики лейцина согласно настоящему изобретению содержат в частности (tBu)A, (2-хлор)F, (2-бромо)F, AAD, (2S)-2-амино-4,4,4-трифторбутановой кислоты, Cnba, (4-фтор)L, (S)-(трифторметил)-L-цистеин, (2S)-2-амино-3-(1-метилциклопропил)пропановую кислоту, Gly(tBu), 3-(триметилсилил)-L-аланин, 2,5-дифтор-L-фенилаланин, 2-амино-7-(трет-бутокси)-7-оксогептановую кислоту, 5,5,5-трифтор-L-лейцин ((трифтор)L), (2-Me)F, Cba, Сра, циклопропилметилаланин, трифтор-метилаланин или дифторметилаланин, (2-фтор)F, (2S)-3-(2,3-дифторфенил)-2-аминопропановую кислоту, (2S)-3-(3-цианофенил)-2-аминопропановую кислоту, 2-амино-5,5,5-трифтор-4-метил-пентановую кислоту, (2S)-2-амино-5-метил-гексановую кислоту или (2S)-3-(индол-4-ил)-2-(амино)пропановую кислоту.
Настоящее изобретение дополнительно включает аналоги и производные описанных пептидов. Термин «аналог» или «производное» пептида или аминокислотной последовательности согласно настоящему изобретению включает, в частности, любую аминокислотную последовательность, имеющую последовательность, на по меньшей мере 80% или по меньшей мере 85%, предпочтительно по меньшей мере 90%, более предпочтительно по меньшей мере 95%, и даже более предпочтительно по меньшей мере 99% идентичную указанной последовательности, и или имеющую некоторые или сопоставимые свойства или активность. Идентичность последовательности может быть определена с помощью обычных методов, таких как визуальное сравнение, или с помощью любого компьютерного инструмента, обычно используемого в данной области. Примеры включают программы BLAST, используемые с параметрами по умолчанию.
Аналог или производное последовательности пептида или аминокислоты согласно настоящему изобретению может быть результатом изменений, происходящих в результате мутации или изменения последовательностей пептидов согласно изобретению, включая делецию или вставку одной или нескольких аминокислот или замену одной или нескольких аминокислот или даже к альтернативному сплайсингу. Некоторые из этих модификаций могут быть объединены. Предпочтительно, аналог последовательности аминокислот согласно настоящему изобретению содержит консервативные замены относительно последовательности аминокислот.
Используемый в настоящем документе термин «консервативная замена» означает, что одна или несколько аминокислот заменены другим биологически сходным остатком. Примеры включают замену аминокислотных остатков с аналогичными характеристиками, например, небольшие аминокислоты, кислотные аминокислоты, полярные аминокислоты, основные аминокислоты, гидрофобные аминокислоты и ароматические аминокислоты. См., например, схему в Таблице 4 ниже, где консервативные замены аминокислот сгруппированы по физико-химическим свойствам. I: нейтральные, гидрофильные; II: кислоты и амиды; III: основные; IV: гидрофобные; V: ароматические, объемные аминокислоты, VI: нейтральные или гидрофобные; VII: кислотный; VIII: полярный.
Аналог или производное пептида также может включать одну или несколько дополнительных модификаций, таких как, например, конъюгация с другим соединением с образованием аминокислотного конъюгата.
Такая модификация может, альтернативно или дополнительно, быть результатом конъюгации с боковыми цепями одного или нескольких аминокислотных остатков в пептиде согласно настоящему изобретению, например, химической группы, как определено выше в контексте с химической группой Х0 или полимерного фрагмента, такого как группа PEG. Такая модификация может, например, повышать растворимость и/или период полувыведения in vivo (например, в плазме) и/или биодоступность пептида, а также известно, что она снижают клиренс (например, почечный клиренс) терапевтических белков и пептидов. Подобные модификации хорошо известны специалистам в данной области техники и включают, в частности, без ограничения пэгилирование одной или нескольких боковых цепей пептида согласно настоящему изобретению. Здесь «пэгилирование» представляет собой событие связывания (например, ковалентно) структуры полиэтиленгликоля (PEG) с пептидом согласно настоящему изобретению. Специалисту в данной области техники хорошо известны возможные PEG для связывания с боковыми цепями аминокислот малых пептидов для образования соответствующего конъюгата, например, из WO 2015/200916 А1, который включен в настоящее описание посредством ссылки.
Все пептиды по данному изобретению, если не указано иное, представляют собой соли TFA. Настоящее изобретение включает дополнительные фармацевтически приемлемые соли пептидов, как они определены в настоящем документе, и бессолевые формы. При этом фармацевтически приемлемые соли представляют собой соли или цвиттерионные формы пептидов или соединений согласно настоящему изобретению, которые являются растворимыми или диспергируемыми в воде или масле, которые подходят для лечения заболеваний без чрезмерной токсичности, раздражения и аллергической реакции; которые соответствуют разумному соотношению польза/риск и эффективны при использовании по назначению. Соль можно получить при окончательном выделении и очистке соединений или отдельно путем взаимодействия аминогруппы с подходящей кислотой. Типичные кислотно-аддитивные соли включают ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат, карбонат, диглюконат, глицерофосфат, гемисульфат, гептаноат, гексаноат, формиат, фумарат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат (изетионат), лактат, малеат, мезитиленсульфонат, мэтансульфонат, нафтиленсульфонат, никотинат, 2-нафталенсульфонат, оксалат, памоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, сульфат, тартрат, трихлорацетат, трифторацетат, фосфат, глутамат, бикарбонат, пара-толуолсульфонат и ундеканоат. Предпочтительные кислотно-аддитивные соли включают трифторацетат, формиат, гидрохлорид и ацетат.
Кроме того, аминогруппы в соединениях согласно настоящему изобретению могут быть кватернизованы метил-, этил-, пропил- и бутилхлоридами, бромидами и йодидами; ди-метил, диэтил, дибутил и диамилсульфатами; децил-, лаурил-, миристил- и стерилхлоридами, бромидами и йодидами; и бензил и фенэтил бромидами. Примеры кислот, которые можно использовать для образования терапевтически активных кислотно-аддитивных, включают неорганические кислоты, такие как соляная, бромистоводородная, серная и фосфорная, и органические кислоты, такие как щавелевая, малеиновая, янтарная и лимонная. Фармацевтически приемлемая соль соли может представлять собой соль, выбранную, например, из кислот но-аддитивных солей и основных солей. Примеры кислотно-аддитивных солей включают хлоридные соли, нитратные соли и ацетатные соли.
Примеры основных солей включают соли, в которых катион выбран из катионов щелочных металлов, таких как ионы натрия или калия, катионов щелочноземельных металлов, таких как ионы кальция или магния, а также замещенных ионов аммония, таких как ионы типа N(R1)(R2)(R3)(R4)+, где R1, R2, R3 и R4 независимо друг от друга обычно обозначают водород, необязательно замещенный С1-6-алкил или необязательно замещенный С2-6-алкенил. Примеры соответствующих C1-6-алкильных групп включают метил, этил, 1-пропил и 2-пропил группы. Примеры С2-6-алкенильных групп, которые могут иметь значение, включают этенил, 1-пропенил и 2-пропенил. При этом предпочтительны соли, в которых катион выбран из натрия, калия и кальция.
Другие примеры фармацевтически приемлемых солей описаны в "Remington's Pharmaceutical Sciences", 17th edition, Alfonso R. Gennaro (Ed.), Mark Publishing Company, Easton, PA, USA, 1985 (and more recent editions thereof), in the "Encyclopaedia of Pharmaceutical Technology", 3rd edition, James Swarbrick (Ed.), Informa Healthcare USA (Inc.), NY, USA, 2007. Также для обзора подходящих солей см. Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (Wiley- VCH, 2002). Другие подходящие основные соли образуются из оснований, образующих нетоксичные соли. Репрезентативные примеры включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина, глицина, лизина, магния, меглумина, оламина, калия, натрия, трометамина и цинка, предпочтительно холина. Также могут образовываться гемисоли кислот и оснований, например, геми-сульфатные и гемикальциевые соли.
Настоящее изобретение дополнительно включает сольваты пептидов, как определено в настоящем документе. В контексте настоящего документа термин «сольват» относится к комплексу определенной стехиометрии, образованному между растворенным веществом (например, пептидом согласно изобретению или его фармацевтически приемлемой солью) и растворителем. Растворителем в этой связи может быть, например, вода, этанол или другие фармацевтически приемлемые, как правило, низкомолекулярные органические соединения, такие как без ограничения уксусная кислота или молочная кислота. Когда рассматриваемым растворителем является вода, такой сольват обычно называют гидратом.
Соединения согласно настоящему изобретению обладают полезными фармакологическими свойствами и могут быть использованы для профилактики и лечения заболеваний у людей и животных.
В контексте настоящего изобретения термин «лечение» или «лечить» включает торможение, отсрочку, остановку, улучшение, ослабление, ограничение, уменьшение, подавление, обращение вспять или излечение заболевания, состояния, расстройства, травмы или нарушения здоровья, развития, течения или прогрессирования таких состояний и/или симптомов таких состояний. Здесь термин «терапия» понимается как синоним термина «лечение».
В контексте настоящего изобретения термины «предупреждение», «профилактика» или «предостережение» используются как синонимы и относятся к предотвращению или уменьшению риска получить, приобрести, пострадать или иметь болезнь, состояние, нарушение, травму или нарушение здоровья, развитие или прогрессирование таких состояний и/или симптомы таких состояний.
Лечение или предупреждение заболевания, состояния, нарушения, травмы или нарушения здоровья может иметь место частично или полностью.
Соединения согласно настоящему изобретению особенно подходят для лечения и/или профилактики сердечно-сосудистых, сердечно-легочных, почечных, легочных, фиброзных, тромбоэмболических и воспалительных заболеваний.
Следовательно, соединения согласно изобретению могут применяться для лечения и/или профилактики сердечно-сосудистых и сердечно-легочных заболеваний и их симптомов, таких как, например, воспалительные заболевания сердца, миокардит, эндокардит, перикардит, ревматическая лихорадка без и с поражением сердца, острый ревматический перикардит, острый ревматический эндокардит, острый ревматический миокардит, хронические ревматические пороки сердца с эндокардитом и без, вальвулит, перикардит, ишемическая болезнь сердца, например, нестабильная стенокардия и острый инфаркт миокарда, предсердные и желудочковые аритмии и нарушения проводимости, такие как, а также нарушения проводимости такие как, например, атриовентрикулярная блокада I-III степени, суправентрикулярная тахиаритмия, мерцательная аритмия, трепетание предсердий, фибрилляция желудочков, трепетание желудочков, вентрикулярная тахиаритмия, пируэтная тахикардия, экстрасистолы предсердий и желудочков, преждевременная деполяризация, исходящая из АВ соединения, синдром слабости синусового узла, инсульт вследствие окклюзии и стеноза церебральных артерий (инфаркт головного мозга после, например, тромбоэмболических, атеросклеротических, инфекционных и воспалительных поражений сосудов), для лечения и/или профилактики инсульта вследствие внутримозгового или внутричерепного кровоизлияния, периферической ишемии тканей (например, атеросклеротическая гангрена) вследствие заболеваний артерий, артериол и капилляров (например, тромбоэмболические, атеросклеротические, инфекционные и воспалительные поражения сосудов, деформирующий или облитерирующий эндартериит, расслоение аневризмы), флебиты и тромбофлебиты, для профилактики постпроцедурных нарушений кровеносной системы, например, синдрома системного воспалительного ответа, послеоперационной вазоплегии, посткардиотомного синдрома, постпроцедурной гипотензии и сердечной недостаточности, для профилактики или лечения ишемии, реперфузионного повреждения и дисфункции органов, например, после тромболитической терапии, чрескожно-транслюминальной ангиопластики (РТА), чрескожно-транслюминальной коронарной ангиопластики (РТСА), операций по шунтированию, трансплантаций сердца, легкого, печени и почек, и для профилактики и лечения замедленной функции трансплантата после трансплантации почки.
Соединения согласно настоящемк изобретению, кроме того, подходят для лечения шока, такого как кардиогенный шок, септический шок и анафимлический кислотный шок, путем предотвращения опосредованных MASP поражений органов-мишеней.
Кроме того, соединения согласно настоящему изобретению обладает противовоспалительным действием и, следовательно, может использоваться в качестве противовоспалительных средств для лечения и/или профилактики сепсиса (SIRS), полиорганной недостаточности (MODS, MOF), воспалительных заболеваний почек, хронические кишечные воспаления (IBD, болезнь Крона, UC), панкреатита, перитонита, ревматоидных заболеваний, воспалительных заболеваний кожи и воспалительных заболеваний глаз.
Благодаря своему профилю активности соединения согласно настоящему изобретению особенно подходят для лечения и/или профилактики сердечно-сосудистых, легочных, церебральных и почечных проявлений сепсиса и синдрома системной воспалительной реакции.
Соединения согласно настоящему изобретению особенно подходят для лечения и/или профилактики ишемии и/или связанных с реперфузией повреждений сердца, почек и других органов в контексте реанимационных и хирургических вмешательств, таких как без ограния операция по шунтирования, хирургия клапанов сердца, хирургия аневризмы аорты,
Соединения согласно настоящему изобретению дополнительно могут быть также использованы для предотвращения ишемического и/или реперфузионного повреждения органов или тканей, а также в качестве добавок к перфузионным и консервирующим растворам для органов, частей органов, тканей или частей тканей человека или животного происхождения, в частности для хирургических вмешательств или в области трансплантационной медицины.
Кроме того, соединения согласно настоящему изобретению подходят для лечения и/или профилактики заболеваний крови, кроветворных органов и иммунной системы, включая без ограничения приобретенную гемолитическую анемию, гемолитико-уремический синдром, пароксизмальную ночную гемоглобинурию [Marchiafava-Micheli], дефекты свертывания крови, пурпура и другие геморрагические состояния, диссеминиро-ванное внутрисосудистое свертывание [синдром дефибринации], эссенциальную (геморрагическую) тромбоцитемию, молниеносную пурпуру, тромботическую тромбоцитопеническую пурпуру, аллергическую пурпуру, аллергический васкулит, лимфопению и гранулоцитоз, и саркоидоз.
Кроме того, соединения по изобретению подходят для лечения и/или профилактики побочных эффектов сахарного диабета, таких как почечные осложнения сахарного диабета, диабетическая нефропатия, внутрикапиллярный гломерулонефроз, офтальмологические осложнения сахарного диабета, диабетическая ретинопатия, неврологические осложнения, диабетическая полиневропатия и сосудистые осложнения, такие как микроангиопатия и гангрена.
Более того, соединения по изобретению подходят для лечения и/или профилактики воспалительных заболеваний нервной системы, таких как рассеянный склероз, менингит и энцефалит, бактериальный и вирусный менингит и энцефалит, постиммунизационный энцефалит, воспалительная полинейропатия и полиневропатия при инфекционных и паразитарных заболеваниях.
Кроме того, соединения по изобретению подходят для лечения и/или профилактики офтальмологических заболеваний, такие как острый и неострый иридоциклит, хориоидальная дистрофия, хориоретинальное воспаление, хориоретинальное воспаление при инфекционных и паразитарных заболеваниях, фоновая ретинопатия и изменения сосудов сетчатки, пролифе-ративная ретинопатия, дегенерация желтого пятна и заднего полюса, периферическая дегенерация сетчатки, возрастная дегенерация желтого пятна (AMD), включая сухую (неэкссудативную) и влажную (экссудативную, неоваскулярную) AMD, хориоидальная неоваскуляризация (CNV), хориоидальные неоваскулярные мембраны (CNVM), цистоидный макулярный отек (СМЕ), эпиретинальные мембраны (ERM) и макулярный разрыв сетчатки, хориоидальная неоваскуляризация, ассоциированная с миопией, ангиоидные и сосудистые полосы, отслоение сетчатки, диабетическая ретинопатия, диабетический макулярный отек (DME), атрофические и гипертрофические изменения пигментного эпителия сетчатки, венозная окклюзия сетчатки, хориоидальная венозная окклюзия сетчатки макулярный отек, макулярный отек, ассоциированный с окклюзией вен сетчатки, постпроцедурные нарушения глаза и его придатков, в т.ч. кератопатия после операции по удалению катаракты.
Кроме того, соединения согласно настоящему изобретению подходят для лечения и/или профилактики заболеваний дыхательной системы, включая без ограничения вирусную, бактериальную и грибковую пневмонию, радиационный пневмонит, пневмокониоз, аллергический альвеолит, заболевания дыхательных путей из-за специфической органической пыли, например легкое фермера, бронхит, пневмонит и отек легких, вызванный химическими веществами, газами, дымами и парами, лекарственные интерстициальные заболевания легких, респираторный дистресс-синдром взрослых (ARDS) и острое повреждение легких (ALI), острый отек легких, интерстициальный заболевания легких с фиброзом, ревматоидные заболевания легких, нарушения дыхания при других диффузных заболеваниях соединительной ткани, таких как ассоциированные с системной красной волчанкой, склеродермией и гранулематозом Вегенера.
Кроме того, соединения согласно настоящему изобретению подходят для лечения и/или профилактики повреждения микрососудов, тромбоза и последовательных тромбо-эмболических явлений, вызванных вирусными инфекциями, такими как, но не ограничиваясь ими, вирусы гриппа (например, вызванные штаммами серотипов H1N1, H5N1, H7N9), и коронавирусы (например, SARS-CoV, возбудитель тяжелого острого респираторного синдрома (SARS), MERS-CoV, возбудитель ближневосточного респираторного синдрома (MERS), и SARS-CoV-2, возбудитель COVID -19 пандемический).
Кроме того, соединения согласно настоящему изобретению подходят для лечения и/или профилактики заболеваний пищеварительной системы, включая, но не ограничиваясь этим, неинфекционный энтерит и колит, такие как болезнь Крона и язвенный колит, панкреатит (включая острый алкогольный и медикаментозный панкреатит), холецистит, воспалительные заболевания печени, гепаторенальный синдром, постпроцедурные нарушения печени, например, после операции на печени.
Благодаря своему профилю активности соединения согласно настоящему изобретению особенно подходят для лечения и/или профилактики заболеваний мочеполовой системы, включая, но не ограничиваясь, острую почечную недостаточность, острую почечную недостаточность (AKI), связанную с хирургическим вмешательством AKI, AKI, связанную с сепсисом, AKI, индуцированную контрастными веществами и химиотерапией, ишемию и инфаркт почки, такие осложнения, как гиперчувствительность на фоне гемодиализа и гемодиафильтрации, цистит, лучевой цистит, воспалительные заболевания предстательной железы и эндометриоз.
Соединения согласно настоящему изобретению, кроме того, подходят для лечения и/или профилактики последствий ожогов и травм, включая, но не ограничиваясь ими, ранние осложнения травмы, травматическую анурию, синдром раздавливания, почечную недостаточность после раздавливания, травматическую ишемию мышцы, черепно-мозговую травму, поражение органов после воздействия электрического тока, радиации и экстремальных температур окружающего воздуха и давления, после воздействия дыма, огня и пламени, после контакта с ядовитыми животными и растениями.
Благодаря своему профилю активности соединения согласно настоящему изобретению, кроме того, подходят для лечения воспалительных заболеваний кожи, таких как кожная красная волчанка, буллезные заболевания и акантолитические кожные заболевания, такие как подтипы пузырчатки, папулосквамозные заболевания, такие как псориаз, дерматит и экзема, крапивница и эритема.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделен и/или очищен, содержащий, по существу состоящий из или состоящий из формулы (I) или формулы (II), или его производное, пролекарство, аналог или фармацевтически приемлемую соль или сольват, для применения для профилактики и/или лечения заболеваний.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделен и/или очищен, содержащий, по существу состоящий из или состоящий из формулы (I) или формулы (II), или его производное, пролекарство, аналог или фармацевтически приемлемую соль или сольват, для применения для профилактики и/или лечения MASP-связанных нарушений.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделен и/или очищен, содержащий, по существу состоящий из или состоящий из формулы (I) или формулы (II), или его производное, пролекарство, аналог или фармацевтически приемлемую соль или сольват, которое действует как ингибитор MASP-1 и/или MASP-2, и/или который ингибирует отложение C3, для применения для профилактики и/или лечения MASP-связанных.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделен и/или очищен, содержащий, по существу состоящий из или состоящий из формулы (I) или формулы (II), или его производное, пролекарство, аналог или фармацевтически приемлемую соль или сольват, для применения для профилактики и/или лечения сердечно-сосудистых и сердечно-легочных заболеваний, шока, воспалительных заболеваний, сердечно-сосудистых, легочных, церебральных и почечных последствий сепсиса, ишемии и/или реперфузионных повреждений, острой почечной недостаточности, защиты трансплантата и отсроченной функции трансплантата, заболевания крови и органов кроветворения и иммунной системы, последствий сахарного диабета, воспалительных заболеваний нервной системы, заболеваний глаз, заболеваний кожи, заболеваний дыхательной, пищеварительной или мочеполовой системы и заболеваний мочеполовой системы и последствий ожогов и травм.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее пептид, который может быть выделен и/или очищен, содержащий, по существу состоящий из или состоящий из формулы (I) или формулы (II), или его производное, пролекарство, аналог или фармацевтически приемлемую соль или сольват, для применения для профилактики и/или лечения заболеваний мочеполовой системы, включая, помимо прочего, острую почечную недостаточность, острую почечную недостаточность (AKI), связанную с хирургическим вмешательством AKI, AKI, связанную с сепсисом, AKI, индуцированную контрастными веществами и химиотерапией, ишемию и инфаркт почки, такие осложнения, как гиперчувствительность на фоне гемодиализа и гемодиафильтрации, цистит, лучевой цистит, воспалительные заболевания предстательной железы и эндометриоз.
Настоящее изобретение также относится к способу лечения или облегчения MASP-связанных нарушений, как определено выше, у субъекта или пациента путем введения по меньшей мере одного пептида, производного или аналога, как определено в настоящем документе, или его фармацевтически приемлемой соли или сольвата, комплекса или фармацевтической композиции, как определено выше, указанному субъекту или пациенту, нуждающемуся в этом.
В контексте настоящего изобретения термины «пациент», «субъект» или «индивидуум» могут использоваться взаимозаменяемо и относиться либо к человеку, либо к животному, отличному от человека. Эти термины включают млекопитающих, таких как люди, приматы, домашний скот (например, крупный рогатый скот, свиньи), домашние животные (например, псовые, кошачьи) и грызуны (например, мыши и крысы). Термин «млекопитающее» относится к любому виду млекопитающих, такому как человек, мышь, крыса, собака, кошка, хомяк, морская свинка, кролик, домашний скот и т.п.
В соответствии с настоящим изобретением по меньшей мере один пептид, производное или аналог, как определено в настоящем документе, или его фармацевтически приемлемая соль или сольват, или комплекс, как определено выше, вводят пациенту или субъекту в терапевтически эффективном количестве, где «терапевтически эффективное количество» соединения согласно настоящему изобретению означает достаточное количество соединения согласно настоящему изобретению для лечения MASP-связанного нарушения, как определено в настоящем документе. Согласно конкретным вариантам осуществления терапевтически эффективное количество обеспечивает желаемое соотношение польза/риск, применимое к любому медицинскому лечению.
Настоящее изобретение, в частности, включает следующие варианты осуществления, где заместители или фрагменты пептида формулы (I) или формулы (II), как определено ниже, могут независимо иметь значения, как описано ниже.
Пептид или соединение, содержащее пептид, или его производное, или аналог, как определено в настоящем документе, или его фармацевтически приемлемая соль или соль-ват, или комплекс, или фармацевтическая композиция (как определено ниже), в дальнейшем также обычно обозначаются как «MASP-ингибирующий пептид согласно настоящему изобретению».
Согласно некоторым вариантам осуществления MASP-ингибирующий пептид согласно настоящему изобретению связывается с MASP-1 и/или MASP-2, например, MASP-1 и/или MASP-2 человека. Согласно определенным вариантам осуществления MASP-ингибирующий пептид согласно настоящему изобретению специфически связывается с MASP-1 и/или MASP-2 человека. В контексте настоящего изобретения термин «специфически связывается» относится к предпочтительному взаимодействию специфического связывающего агента с данным лигандом по сравнению с другими агентами в образце. Например, специфический связывающий агент, который специфически связывает данный лиганд, связывает данный лиганд в конкретных условиях в количестве или степени, которые можно наблюдать по сравнению с любым неспецифическим взаимодействием с другими компонентами в образце. Подходящие условия представляют собой те, которые позволяют взаимодействовать между данным специфическим связывающим агентом и данным лигандом. Эти условия включают рН, температуру, концентрацию, растворитель, время инкубации и т.п., и могут различаться среди данных пар специфического связывающего агента и лиганда, но могут быть легко определены специалистами в данной области техники. Согласно некоторым вариантам осуществления MASP-ингибирующий пептид согласно настоящему изобретению связывается с MASP-1 и/или MASP-2 с большей специфичностью, чем эталонный MASP-ингибирующий пептид (например, любой из эталонных MASP-ингибирующих пептидных соединений, приведенных в настоящем документе).
Таким образом, настоящее изобретение дополнительно относится к комплексу, содержащему по меньшей мере один пептид или соединение, содержащее пептид, производное или аналог, как определено в настоящем документе, связывающееся с MASP-1 или MASP-2.
Согласно некоторым вариантам осуществления MASP-ингибирующий пептид согласно настоящему изобретению проявляет специфическое связывание с MASP-1 и/или MASP-2, особенно с MASP-1 и/или MASP-2 человека, которое на по меньшей мере около 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500%, 700%, 1000%, или 10000% выше, чем у выбранного ссылочного соединения MASP-ингибирующего пептида.
Согласно некоторым вариантам осуществления MASP-ингибирующий пептид согласно настоящему изобретению проявляет специфическое связывание с MASP-1 и/или MASP-2, особенно MASP-1 и/или MASP-2 человека, которое в по меньшей мере около 1, 2, 3, 4, 5 раз или в по меньшей мере около 10, 20, 50 или 100 раз выше, чем у выбранного ссылочного соединения MASP-ингибирующего пептида.
Согласно некоторым вариантам осуществления MASP-ингибирующий пептид согласно настоящему изобретению проявляет аффинность связывания с MASP-1 и/или MASP-2, которая на по меньшей мере около 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500%, 700%, 1000% или 10000% выше, чем у выбранного ссылочного соединения MASP-ингибирующего пептида.
Согласно некоторым вариантам осуществления MASP-ингибирующий пептид согласно настоящему изобретению проявляет аффинность связывания с MASP-1 и/или MASP-2, которая в по меньшей мере около 1, 2, 3, 4, 5 раз или в по меньшей мере около 10, 20, 50, 100 или 1000 раз выше, чем у выбранного ссылочного соединения MASP-ингибирующего пептида.
Согласно некоторым вариантам осуществления MASP-ингибирующий пептид согласно настоящему изобретению проявляет активность ингибирования MASP-1 и/или MASP-2 (например, MASP-1 и/или MASP-2 крысы или человека). Согласно некоторым вариантам осуществления активность представляет собой активность in vitro или in vivo, т.е. описанная в настоящем документе активность in vitro или in vivo. Согласно некоторым вариантам осуществления MASP-ингибирующий пептид согласно настоящему изобретению ингибирует по меньшей мере около 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500%, 700%, 1000% или 10000% активности MASP-1 и/или MASP-2, ингибируемой выбранным эталонным соединением пептидом, ингибирующим MASP.
Согласно определенному варианту осуществления MASP-ингибирующий пептид согласно настоящему изобретению проявляет 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 30, 40, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180 или 200-кратное ингибирова-ние MASP-1 и/или MASP-2 по сравнению с эталонным выбранным MASP-ингибирующим пептидом.
Согласно другим конкретным вариантам осуществления ингибирующая активность в отношении MASP-1 и/или MASP-2 MASP-ингибирующих пептидов согласно настоящему изобретению определяется посредством измерения их IC50 для MASP-1 и/или MASP-2 (на пример, MASP-1 и/или MASP-2 человека или крысы). Определение IC50 для MASP-1 и/или MASP-2 можно проводить биохимическим анализом, описанным в настоящем документе. Особенно предпочтительно MASP-ингибирующий пептид согласно настоящему изобретению проявляет IC50 для MASP-1 и/или MASP-2 ≤1,000 нм, предпочтительно ≤500 нм, более предпочтительно ≤300 нм, более предпочтительно ≤250 нм, более предпочтительно ≤200 нм, более предпочтительно ≤150 нм, более предпочтительно ≤100 нм, более предпочтительно ≤75 нм, более предпочтительно ≤50 нм, более предпочтительно ≤45 нм, более предпочтительно ≤40 нм, более предпочтительно ≤35 нм, более предпочтительно ≤30 нм.
Согласно некоторым вариантам осуществления MASP-ингибирующий пептид согласно настоящему изобретению имеет более низкую IC50 (т.е. более высокую аффинность связывания) для MASP-1 и/или MASP-2, (например, MASP-1 и/или MASP-2 крысы или человека) по сравнению с выбранным эталонным MASP-ингибирующим пептидом. Согласно некоторым вариантам осуществления MASP-ингибирующий пептид согласно настоящему изобретению имеет IC50 согласно анализу конкурентного связывания MASP-1 и/или MASP-2, которая на по меньшей мере около 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500%, 700%, 1000% или 10000% ниже, чем у выбранного эталонного MASP-ингибирующего пептидного соединения.
Согласно некоторым вариантам осуществления MASP-ингибирующий пептид согласно настоящему изобретению проявляет по меньшей мере на около 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 97%, 98%, 99% или более 99%, 100%, 200%, 300%, 400%, 500%, 700%, 1000% или 10000% более высокое ингибирование in vitro активности MASP-1 и/или MASP-2 человека, чем у выбранного эталонного MASP-ингибирующего пептидного соединения.
Согласно некоторым вариантам осуществления MASP-ингибирующий пептид согласно настоящему изобретению проявляет по меньшей мере на около 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 97%, 98%, 99%, или более 99%, 100%, 200%, 300%, 400%, 500%, 700%, 1000% или 10000% более высокое ингибирование in vivo активности MASP-1 и/или MASP-2 человека, чем у выбранного эталонного MASP-ингибирующего пептидного соединения.
Используемый здесь термин «MASP-ингибирующий пептид, обладающий ингиби-рующей активностью в отношении MASP-1 и/или MASP-2» в определенных вариантах осуществления означает, что соединение обладает способностью ингибировать отложение C3 in vitro или у субъектов (например, у мышей или людей), при введении им (например, парентеральным путем, например, путем инъекции или легочным, назальным, подъязычным, язычным, буккальным, дермальным, чрескожным, конъюнктивальным, глазным путем или в виде имплантата или стента при пероральном введении), в дозозависимым или зависимым от времени образом.
Согласно некоторым вариантам осуществления MASP-ингибирующий пептид согласно настоящему изобретению проявляет ингибирование отложения С3 (например, отложение С3 у человека). Согласно некоторым вариантам осуществления ингибирование отложения C3 определяется ингибированием in vitro или in vivo. Согласно некоторым вариантам осуществления MASP-ингибирующий пептид согласно настоящему изобретению ингибирует по меньшей мере около 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 200%, 300%, 400%, 500%, 700%, 1000% или 10000% отложения C3, ингибируемого выбранным эталонным MASP-ингибирующим пептидным соединением.
Согласно определенным вариантам осуществления MASP-ингибирующий пептид согласно настоящему изобретению проявляет 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 30, 40, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180 или 200-кратное ингибирование отложения C3 по сравнению с выбранным эталонным MASP-ингибирующим пептидным соединением.
Согласно другим конкретным вариантам осуществления ингибирующая активность в отношении MASP-1 и/или MASP-2 MASP-ингибирующих пептидов согласно настоящему изобретению определяется измерением их IC50 ингибирования отложения C3 in vitro или у субъектов (например, у мышей или людей). Определение IC50 для отложения C3 может быть сделано посредством анализа отложения C3 у человека, показанного в настоящем документе. Особенно предпочтительно MASP-ингибирующий пептид согласно настоящему изобретению проявляет IC50 для отложения C3 <1,000 нм, предпочтительно ≤500 нм, более предпочтительно ≤300 нм, более предпочтительно ≤250 нм, более предпочтительно ≤200 нм, более предпочтительно ≤150 нм, более предпочтительно ≤100 нм, более предпочтительно ≤75 нм, более предпочтительно ≤50 нм, более предпочтительно ≤45 нм, более предпочтительно ≤40 нм, более предпочтительно ≤35 нм, более предпочтительно ≤30 нм.
Согласно некоторым вариантам осуществления MASP-ингибирующий пептид согласно настоящему изобретению проявляет на по меньшей мере около 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 97%, 98%, 99%, или более 99%, 100%, 200% 300%, 400%, 500%, 700%, 1000% или 10000% более высокое ингибирование in vitro отложения C3 по сравнению с выбранным эталонным MASP-ингибирующим пептидным соединением.
Согласно некоторым вариантам осуществления MASP-ингибирующий пептид согласно настоящему изобретению проявляет на по меньшей мере около 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 97%, 98%, 99%, или более 99%, 100%, 200% 300%, 400%, 500%, 700%, 1000% или 10000% более высокое ингибирование in vivo отложения C3 по сравнению с выбранным эталонным MASP-ингибирующим пептидным соединением.
Особенно предпочтительно, чтобы пептид согласно настоящему изобретению действовал как MASP-ингибирующий пептид, при этом его активность определялась в соответствии по меньшей мере с одним из конкретных анализов и/или исследований in vivo в соответствии с примерами настоящего изобретения.
Из-за его вышеупомянутой ингибирующей активности в отношении MASP-1 и/или MASP-2 и ингибирующей активности в отношении С3-отложения соединение, содержащее пептид, или пептид согласно настоящему изобретению (включая аналоги, производные и фармацевтически приемлемые соли или сольваты, а также вышеуказанный комплекс) являются подходящими для применения для профилактики и/или лечении MASP-1 и/или MASP-2-связанных нарушений.
Согласно другому варианту осуществления настоящее изобретение обеспечивает соединение, содержащее, который может быть выделенным и/или очищенным, содержащий, по существу состоящий из или состоящий из формулы (I) или формулы (II), или его производное, пролекарство, аналог, фармацевтически приемлемая соль, сольват или сольват соли, которое действует как ингибитор MASP-1 и/или MASP-2 и/или которое ингибирует отложение C3.
Соединения согласно настоящему изобретению можно использовать отдельно или в комбинации с другими активными соединениями, если это необходимо. Настоящее изобретение дополнительно относится к лекарственным средствам, содержащим по меньшей мере одно из соединений согласно настоящему изобретению и одно или несколько дополнительных активных соединений, в частности, для лечения и/или профилактики вышеупомянутых заболеваний. В качестве подходящей комбинации активных соединений можно упомянуть например и предпочтительно:
• соединения, которые ингибируют расщепление циклического гуанозинмоно-фосфата (цГМФ) и/или циклического аденозинмонофосфата (цАМФ), например, ингибиторы фосфодиестеразы (ФДЭ) 1, 2, 3, 4 и/или 5, в частности, ингибиторы ФДЭ-4, такие как такие как рофлумиласт или ревамиласт, и ингибиторы ФДЭ-5, такие как силденафил, вар-денафил, тадалафил, уденафил, дасантафил, аванафил, мироденафил или лоденафил;
• NO-независимые, однако гем-зависимые стимуляторы гуанилатциклазы, в частности, риоцигуат, нелоцигуат, верицигуат, а также те, которые описаны в международных заявках WO 00/06568, WO 00/06569, WO 02/42301, WO 03/095451, WO 2011/147809, WO 2012/004258, WO 2012/028647 и WO 2012/059549;
• NO-независимые и гем-независимые стимуляторы гуанилатциклазы, в частности Рункацигуат, BI 703704 и соединения, которые описаны в международных заявках WO2012/139888, WO 2001/019780 и WO2014/012934;
• аналоги простациклина и агонисты IP-рецепторов, в качестве примера и предпочтительно, илопрост, берапрост, трепростинил, эпопростенол, NS-304, селексипаг или ралинепаг;
• антагонисты эндотелиновых рецепторов, например и предпочтительно, босен-тан, дарусентан, амбризентан, мацитентан или ситаксентан;
• антагонисты рецепторов вазопрессина, например, толваптан, кониваптан, релко-ваптан;
• ингибиторы человеческой нейтрофильной эластазы (ЧНЭ), например и предпочтительно, сивелестат или DX-890 (релтран);
• соединения, ингибирующие каскады трансдукционных сигналов, в частности, из группы ингибиторов тирозинкиназы, например и предпочтительно, дазатиниб, нилотиниб, бозутиниб, регорафениб, сорафениб, сунитиниб, седираниб, акситиниб, телатиниб, имати-ниб, бриваниб, пазопаниб, ваталаниб, гефетиниб, эрлотиниб, лапатиниб, канертиниб, леста-уртиниб, пелитиниб, семаксаниб, маситиниб или тандутиниб;
• модуляторы сигнальной трансдукции из группы ингибиторов киназы ASK1, например селонсертиб;
• ингибиторы Rho-киназы, например и предпочтительно, фазудил, Y-27632, SLx-2119, BF-66851, BF-66852, BF-66853, KI-23095 или ВА-1049;
• активные ингредиенты, снижающие проницаемость сосудистой стенки (образование отека), например и предпочтительно ингибиторы сигнального пути ALKl-Smadl/5, ингибиторы сигнального пути VEGF и/или PDGF, ингибиторы циклооксигеназы, ингибиторы калликреин-кининовой система или ингибиторы сигнальных путей сфингозин-1-фосфата;
• кортикостероиды, например, кортизон, кортизол, преднизолон, метилпреднизо-лон, триамцинолон или дексаметазон;
• активные ингредиенты, которые уменьшают повреждение органов при окислительном стрессе, например и предпочтительно ингибиторы системы комплемента, особенно антагонисты рецептора комплемента С5а, анти-С5-антитела или агонисты 5-НТ1А-рецептора;
• модуляторы, стимуляторы и энхансеры фактора транскрипции Nrf2, например, СХА-10, олтипраз, диметилфумарат или бардоксолон;
• адреномедуллин и производные адреномедуллина, например, пегилированный адреномдуллин, и стабилизаторы адреномедуллина, например, адрецизумаб;
• соединения, которые ингибируют индуцируемый гипоксией фактор пролилгид-роксилазу (ингибиторы HIF-PH), например, молидустат, вададустат, роксадустат, дапроду-стат или дезидустат;
• соединения, которые ингибируют индукцию пути клеточной гибели и апоптоза, пример, QPI-1002;
• агонисты C-Met и миметики фактора роста гепатоцитов, например, рефаналин;
• щелочная фосфатаза и рекомбинантная щелочная фосфатаза;
• соединения, которые ингибируют воспалительную реакцию и пролиферацию Т-клеток, например, соединения-антагонисты CD28, такие как Релтецимод;
• соединения, которые модулируют активацию Т-клеток Th17, например, модуляторы фактора транскрипции RORc/ROR-гамма;
• соединения, антагонизирующие Т-клеточный ответ Th17, например, антитела против IL-17 и антитела против IL-23, например, иксекизумаб, секукинумаб, бродалумаб, устекинумаб, гуселкумаб или PTG-200;
• антитромботические средства, например и предпочтительно из группы ингибиторов агрегации тромбоцитов, антикоагулянтов или профибринолитических веществ;
• Согласно предпочтительному варианту осуществления настоящего изобретения соединения согласно настоящему изобретению вводят в комбинации с ингибитором агрегации тромбоцитов, например и предпочтительно аспирином, клопидогрелем, тиклопидином или дипиридамолом.
• Согласно предпочтительному варианту осуществления настоящего изобретения соединения согласно настоящему изобретению вводят в комбинации с ингибитором тромбина, например и предпочтительно ксимелагатран, мелагатран, дабигатран, бивалирудин или клексан.
• Согласно предпочтительному варианту осуществления настоящего изобретения соединения согласно настоящему изобретению вводят в комбинации с антагонистом GPIIb/IIIa, например и предпочтительно тирофибан или абциксимаб.
• Согласно предпочтительному варианту осуществления настоящего изобретения соединения согласно настоящему изобретению вводят в комбинации с ингибитором фактора Ха, наприме и предпочтительно ривароксабан, апиксабан, фидексабан, разаксабан, фон-дапаринукс, идрапаринукс, DU-176b, PMD-3112, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR-126512 или SSR-128428.
• Согласно предпочтительному варианту осуществления настоящего изобретения соединения согласно настоящему изобретению вводят в комбинации с гепарином или низкомолекулярным (LMW) производным гепарина.
• Согласно предпочтительному варианту осуществления настоящего изобретения соединения согласно настоящему изобретению вводят в комбинации с прямыми ингибиторами фактора свертывания крови XI, ингибиторами экспрессии фактора свертывания крови XI и антителами к фактору свертывания крови XI, такими как ксисомаб 3G3.
• Согласно предпочтительному варианту осуществления настоящего изобретения соединения согласно настоящему изобретению вводят в комбинации с антагонистом мине-ралокортикоидных рецепторов, например и прелпочтительно спиронолактоном, эплерено-ном или финереноном.
• Согласно предпочтительному варианту осуществления настоящего изобретения соединения согласно настоящему изобретению вводят в комбинации с диуретиками, например и предпочтительно фуросемид, буметанид, торасемид, бендрофлуметиазид, хлор-тиазид, гидрохлортиазид, гидрофлуметиазид, метиклотиазид, политиазид, трихлорметиазид, хлорталидон, индапамид, метолазон, хинтазон, ацетазоламид, дихлорфенамид, метазоламид, глицерин амилорид или триамтерен.
• Согласно предпочтительному варианту осуществления настоящего изобретения соединения согласно настоящему изобретению вводят в комбинации с агонистом PPAR-гамма, например и предпочтительно пиоглитазон или розиглитазон.
• Согласно предпочтительному варианту осуществления настоящего изобретения соединения согласно настоящему изобретению вводят в комбинации с агонистом PPAR-дельта, например и предпочтительно GW 501516 или BAY 68-5042.
Согласно другому варианту осуществления настоящее изобретение обеспечивает фармацевтическую композицию, содержащую по меньшей мере одно соединение, содержащее пептид, который может быть выделен и/или очищен, содержащий, по существу состоящий из или состоящий из формулы (I) или формулы (II), или его производное, про-лекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, в комбинации с одним или несколькими дополнительными активными агентами, выбранными из группы, состоящей из ингибиторов фосфодиэстераз, стимуляторов или активаторов гуанилатциклазы, агонистов IP-рецепторов, антагонистов минералокортикоидных рецепторов, диуретиков, агонистов PPAR-гамма, агонистов PPAR-дельта, кортикостероидов, активных ингредиентов, уменьшающих повреждение органов при окислительном стрессе, соединений, которые ингибируют индукцию пути клеточной гибели и апоптоза, соединений, которые ингибируют воспалительный ответ и пролиферацию Т-клеток, антитромбо-тических средств, ингибиторов агрегации тромбоцитов, ингибиторов тромбина, антагонистов GPIIb/IIIa, ингибиторов фактора Ха, гепарина или низкомолекулярных производных гепарина (LMW) и ингибиторов фактора свертывания крови XI.
Настоящее изобретение также относится к комбинации в виде набора частей, содержащей по меньшей мере один пептид, производное или аналог, как определено в настоящем документе, или его фармацевтически приемлемую соль или сольват, комплекс или фармацевтическую композицию, как определено выше, и по меньшей мере одно, выбранное из реагента, медицинского устройства, письма с инструкциями или любой их комбинации.
Настоящее изобретение также относится к медицинскому устройству, содержащему по меньшей мере один пептид, производное или аналог, как определено в настоящем документе, или его фармацевтически приемлемую соль или сольват, комплекс или фармацевтическую композицию, как определено выше, для доставки пептида, производного, аналога или комплекса или фармацевтической композиции субъекту.
Фармацевтическая композиция, комбинация в виде набора частей или медицинское устройство, как определено выше, предназначены, в частности, для применения для профилактики и/или лечении нарушений или заболеваний, как определено в настоящем документе.
MASP-ингибирующий пептид согласно настоящему изобретению может обладать системной и/или местной активностью. Для этой цели его можно вводить особым образом, например, перорально, парентерально, легочно, назально, сублингвально, лингваль-но, буккально, ректально, вагинально, дермально, чрескожно, конъюнктивально, ушно или как имплантат или стент.
Для этих способов введения соединения согласно настоящему изобретению можно вводить в подходящих формах введения.
Для перорального введения можно получить соединения согласно настоящему изобретению в лекарственных формах, известных в данной области техники, которые доставляют соединения согласно настоящему изобретению быстро и/или модифицированным образом, таких как, например, таблетки (таблетки без покрытия или покрытые оболочкой таблетки, например с энтеросолюбильными покрытиями или покрытиями с контролируемым высвобождением, которые растворяются с задержкой или нерастворимы), перораль-но-распадающиеся таблетки, пленки/пластины, пленки/лиофилизаты, капсулы (например, твердые или мягкие желатиновые капсулы), таблетки с сахарным покрытием, гранулы, пеллеты, жевательные резинки (например мягкие жевательные резинки), порошки, эмульсии, суспензии, аэрозоли или растворы. Можно включать соединения согласно настоящему изобретению в кристаллической и/или аморфизированной и/или растворенной форме в указанные лекарственные формы.
Парентеральное введение может быть осуществлено с предотвращением стадии абсорбции (например, внутривенно, внутриартериально, внутрисердечно, внутриспинально или эндолюмбально) или с включением абсорбции (например, внутримышечно, подкожно, внутрикожно, чрескожно или внутрибрюшинно). Формы введения, которые подходят для парентерального введения, включают, среди прочего, препараты для инъекций и ин-фузии в виде растворов, суспензий, эмульсий, лиофилизатов или стерильных порошков.
Примерами, подходящими для других путей введения, являются фармацевтические формы для ингаляции (в том числе порошковые ингаляторы, небулайзеры), назальные капли, назальные растворы, назальные спреи; таблетки/пленки/пастилки/капсулы для лин-гвального, сублингвального или буккального введения; суппозитории; глазные капли, глазные мази, глазные ванны, линзы, ушные капли, ушные аэрозоли, ушные порошки, ушные протирки, ушные тампоны; вагинальные капсулы, водные суспензии (лосьоны, микстуры, требующие взбалтывания), липофильные суспензии, эмульсии, мази, кремы, трансдермальные терапевтические системы (такие как, например, пластыри), молоко, пасты, пены, точечно наносимые средства, пылевидные порошки, имплантаты или стенты.
Соединения согласно настоящему изобретению могут быть включены в установленные формы введения. Это может быть осуществлено известным образом путем смешивания с фармацевтически приемлемыми эксципиентами. Фармацевтически приемлемые эксципиенты включают, среди прочего,
• наполнители и носители (например, целлюлоза, микрокристаллическая целлюлоза (например, Avicel®), лактоза, маннит, крахмал, фосфат кальция (такой как, например, Di-Cafos®)),
• основания мази (например, вазелин, парафины, триглицериды, воски, шерстный воск, спирты шерстного воска, ланолин, гидрофильная мазь, полиэтиленгликоли),
• основания для суппозиторий (например, полиэтиленгликоль, кокосовое масло, твердый жир),
• растворители (например, вода, этанол, изопропанол, глицерин, пропиленгли-коль, жирные масла триглицеридов средней длины цепи, жидкие полиэтиленгликоли, парафины),
• поверхностно-активные вещества, эмульгаторы, диспергирующие средства или смачивающие средства (например, натрия додецилсульфат), лецитин, фосфолипиды, жирные спирты (такие как, например, Lanette®), сложные эфиры сорбитана и жирной кислоты (такие как, например, Span®), полиоксиэтиленовые сложные эфиры сорбитана и жирной кислоты (такие как, например, Tween®), полиоксиэтиленовые глицериды жирной кислоты (такие как, например, Cremophor®), полиоксиэтиленовые сложные эфиры жирной кислоты, полиоксиэтиленовые сложные эфиры жирного спирта, глицериновые сложные эфиры жирной кислоты, полоксамеры (такие как, например, Pluronic®),
• буферы, кислоты и основания (например, фосфаты, карбонаты, лимонная кислота, уксусная кислота, соляная кислота, раствор гидроксида натрия, аммония карбонат, трометамол, триэтаноламин),
• изотонические средства (например, глюкоза, хлорид натрия),
• адсорбенты (например, высоко диспергированные диоксиды кремния),
• повышающие вязкость средства, гелеобразоатели, загустители и/или связующие вещества (например, поливинилпирролидон, метилцеллюлоза, гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза, карбоксиметилцеллюлоза-натрий, крахмал, карбомеры, полиакриловые кислоты (такие как, например, Carbopol®); альгинаты, желатин),
• дезинтегрирующие средства (например, модифицированный крахмал, карбоксиметилцеллюлоза-натрий, натрия крахмала гликолят (как например, Explotab®), поперечно-сшитый поливинилпирролидон, кроскармеллоза-натрий (как например, AcDiSol®)),
• регуляторы скорости потока, смазывающие вещества, вещества, способствующие скольжению и смазки, облегчающие выемке изделий из форм (например, стеарат магния, стеариновая кислота, тальк, высоко диспергированные диоксиды кремния (как например, Aerosil®)),
• покрывающие вещества (например, сахара, шеллак) и пленкообразователи для пленок или диффузионные мембраны, которые растворяются быстро или модифицированным образом (например поливинипирролидоны (такие как, например, Kollidon®), поливиниловый спирт, гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза, этил-целлюлоза, гидроксипропилметилцеллюлоза фталат, целлюлозы ацетат, целлюлозы ацетат фталат, полиакрилаты, полиметакрилаты, такие как, например, Eudragit®)),
• материалы капсулы (например, желатин, гидроксипропилметилцеллюлоза),
• синтетические полимеры (например, полилактиды, полигликолиды, полиакрилаты, полиметакрилаты (такие как, например, Eudragit®), поливинипирролидоны (такие как, например, Kollidon®), поливиниловые спирты, поливинилацетаты, полиэтиленоксиды, полиэтиленгликоли и их сополимеры и блок-сополимеры),
• пластификаторы (например полиэтиленгликоли, пропиленгликоль, глицерин, триацетин, триацетилцитрат, дибутилфталат),
• усилители проникновения,
• стабилизаторы (например, антиоксиданты, такие как, например, аскорбиновая кислота, аскорбилпальмитат, натрия аскорбат, бутилгидроксианизол, бутилгидрокситолуол, пропилгаллат),
• консерванты (например, парабены, сорбиновая кислота, тиомерзал, бензалкония хлорид, хлоргексидина ацетат, натрия бензоат),
• красители (например, неорганические пигменты, такие как, например, оксиды железа, диоксид титана),
• ароматизаторы, подсластители, ароматизаторы и/или средства против запаха.
Кроме того, настоящее изобретение относится к фармацевтической композиции, которая содержит по меньшей мере один пептид, производное или аналог, как определено в настоящем документе, или его фармацевтиечски приемлемую соль или сольват, или комплекс, как опредлено выше.
В частности, настоящее изобретение относится к фармацевтической композиции, содержащей по меньшей мере один пептид, производное или аналог, как определено в настоящем документе, или его фармацевтически приемную соль или сольват, или комплекс, как определено выше, обычно вместе с одним или несколькими фармацевтически подходящими вспомогательными веществами, и их применению в соответствии с настоящим изобретением.
Фармацевтическая композиция согласно настоящему изобретению может содержать по меньшей мере один дополнительный активный ингредиент, такой как предпочтительно дополнительный активный ингредиент, который является активным при профилактике и/или лечении нарушений или заболеваний, как определено в настоящем документе.
По меньшей мере один пептид, производное или аналог, как определено в настоящем документе, или его фармацевтически приемлемую соль или сольват, или комплекс, или фармацевтическую композицию, как определено выше, можно вводить энтерально или парентерально, включая внутривенные, внутримышечные, внутрибрюшинные, интра-стернальные, подкожные, внутрикожные и внутрисуставные инъекции и инфузии, перорально, интравагинально, внутрибрюшинно, интраректально, местно или буккально. Подходящие составы для соответствующих путей введения хорошо известны специалисту в данной области и включают, не ограничиваясь этим: пилюли, таблетки, таблетки с энтеросолюбильным покрытием, таблетки с пленкой, таблетки с покрытием, составы с замедленным высвобождением или пролонгированным высвобождением для перорального введения, пластыри, препараты пролонгированного действия для местного применения, драже, пессарии, гели, мази, сиропы, гранулы, суппозитории, эмульсии, дисперсии, микрокапсулы, микропрепараты, нанопрепараты, липосомальные препараты, капсулы, капсулы с кишечнорастворимой оболочкой, порошки, порошки для ингаляций, микрокристаллические составы, спреи для ингаляций, порошки, капли, капли в нос, назальные спреи, аэрозоли, ампулы, растворы, соки, суспензии, инфузионные растворы или растворы для инъекций и т.д. Согласно другому варианту осуществления настоящее изобретение обеспечивает фармацевтическую композицию, содержащую по меньшей мере одно соединение, содержащее пептид, который может быть выделен и/или очищен, содержащий, по существу состоящий из или состоящий из формулы (I) или формулы (II), или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, в комбинации с одним или несколькими инертными, нетоксичными, фармацевтически пригодными вспомогательными веществами для применения для профилактики и/или лечения сердечно-сосудистых и сердечно-легочных заболеваний, шока, воспалительных заболеваний, сердечно-сосудистых, легочных, церебральных и почечных последствий сепсиса, ишемии и/или реперфузионного повреждения, острой почечной недостаточности, защиты трансплантата и отсроченной функции трансплантата, заболеваний крови и органов кроветворения и иммунной системы, последствий сахарного диабета, воспалительных заболеваний нервной системы, заболеваний глаз, заболеваний кожи, заболеваний дыхательной, пищеварительной или мочеполовой системы и последствий ожогов и травм.
Согласно другому варианту осуществления настоящее изобретение обеспечивает фармацевтическую композицию, содержащую по меньшей мере одно соединение, содержащее пептид, который может быть выделен и/или очищен, содержащий, по существу состоящий из или состоящий из формулы (I) или формулы (II), или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, в комбинации с одним или несколькими дополнительными активными агентами, выбранными из группы, состоящей из ингибиторов фосфодиэстераз, стимуляторов или активаторов гуанилатциклазы, агонистов IP-рецепторов, антагонистов минералокортикоидных рецепторов, диуретиков, агонистов PPAR-гамма, агонистов PPAR-дельта, кортикостероидов, активных ингредиентов, уменьшающих повреждение органов при окислительном стрессе, соединений, которые ингибируют индукцию пути клеточной гибели и апоптоза, соединений, которые ингибируют воспалительный ответ и пролиферацию Т-клеток, антитромботических средств, ингибиторов агрегации тромбоцитов, ингибиторов тромбина, антагонистов GPIIb/IIIa, ингибиторов фактора Ха, гепарина или низкомолекулярных производных гепарина (LMW) и ингибиторов фактора свертывания крови XI, для применения для профилактики и/или лечения сердечно-сосудистых и сердечно-легочных заболеваний, шока, воспалительных заболеваний, сердечно-сосудистых, легочных, церебральных и почечных последствий сепсиса, ишемии и/или реперфузионного повреждения, острой почечной недостаточности, защиты трансплантата и отсроченной функции трансплантата, заболеваний крови и органов кроветворения и иммунной системы, последствий сахарного диабета, воспалительных заболеваний нервной системы, заболеваний глаз, заболеваний кожи, заболеваний дыхательной, пищеварительной или мочеполовой системы и последствий ожогов и травм.
Согласно другому варианту осуществления настоящее изобретение относится к способу лечения и/или профилактики сердечно-сосудистых и сердечно-легочных заболеваний, шока, воспалительных заболеваний, сердечно-сосудистых, легочных, церебральных и почечных последствий сепсиса, ишемии и/или реперфузионного повреждения, острой почечной недостаточности, защиты трансплантата и отсроченной функции трансплантата, заболеваний крови и органов кроветворения и иммунной системы, последствий сахарного диабета, воспалительных заболеваний нервной системы, заболеваний глаз, заболеваний кожи, заболеваний дыхательной, пищеварительной или мочеполовой системы и последствий ожогов и травм у людей и животных, посредством введения эффективного количества фармацевтической композиции, содержащей по меньшей мере одно соединение, содержащее пептид, который может быть выделен и/или очищен, содержащий, по существу состоящий из или состоящий из формулы (I) или формулы (II), или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, или фармацевтической композиции, содержащей по меньшей мере одно соединение, содержащее пептид, который может быть выделен и/или очищен, содержащий, по существу состоящий из или состоящий из формулы (I) или формулы (II), или его производное, пролекарство, аналог, фармацевтически приемлемую соль, сольват или сольват соли, в комбинации с одним или несколькими инертными, нетоксичными, фармацевтически пригодными вспомогательными веществами и/или одним или несколькими дополнительными активными ингредиентами.
Подходящая доза MASP-ингибирующего пептида согласно настоящему изобретению может быть определена лечащим врачом в рамках здравого медицинского заключения. Конкретный терапевтически эффективный уровень дозы для любого конкретного субъекта будет зависеть от множества факторов, включая: а) подлежащее лечению нарушение и тяжесть нарушения; b) активность конкретного используемого соединения; с) конкретный применяемый состав, возраст, массу тела, общее состояние здоровья, пол и диету пациента; d) время введения, способ введения и скорость выведения используемого конкретного аналога гепсидина; е) продолжительность лечения; f) препараты, используемые в комбинации или одновременно с применяемым MASP-ингибирующим пептидом, и подобные факторы, хорошо известные в области медицины.
Согласно конкретным вариантам осуществления общая суточная доза MASP-ингибирующего пептида согласно изобретению, вводимая субъекту или пациенту в виде однократной или многократной дозы, может составлять, например, от 0,0001 до 300 мг/кг массы тела в день или 1 до 300 мг/кг массы тела в день или от около 0,0001 до около 100 мг/кг массы тела в день, например, от около 0,0005 до около 50 мг/кг массы тела в день, например, от около 0,001 до около 10 мг/ кг массы тела в день, например от около 0,01 до около 1 мг/кг массы тела в день, вводимых в виде одной или нескольких доз, таких как от одной до трех доз. Как правило, MASP-ингибирующий пептид согласно изобретению можно вводить непрерывно (например, путем внутривенного введения или другого способа непрерывного введения лекарственного средства) или его можно вводить субъекту через определенные промежутки времени, обычно через регулярные промежутки времени, в зависимости от желаемой дозы и фармацевтической композиции, выбранной специалистом для конкретного субъекта. Регулярные интервалы дозирования включают, например, один раз в день, два раза в день, один раз в два, три, четыре, пять или шесть дней, один или два раза в неделю, один или два раза в месяц и т.п.
Настоящее изобретение дополнительно включает применение MASP-ингибирующего пептида, описанного в настоящем документе, для получения лекарственного средства, в частности, для получения лекарственного средства для профилактики и/или лечения нарушения или заболевания, как определено в настоящем документе.
Настоящее изобретение дополнительно включает способ производства пептидов согласно настоящему изобретению, их производных или аналогов, или их фармацевтически приемлемых солей, сольватов или комплексов, каждый из которых описан в настоящем документе. Способ получения включает стадии, как показано в примерах согласно настоящему изобретению.
Как правило, MASP-ингибирующий пептид согласно настоящему изобретению может быть получен синтетическим или полурекомбинантным путем.
Согласно данному варианту осуществления изобретение обеспечивает способ получения соединения, содержащего пептид, который может быть выделен и/или очищен, содержащий, по существу состоящий из или состоящий из формулы (I) или формулы (II), или производного, пролекарства, аналога или фармацевтически приемлемой соли или сольвата, с использованием твердофазного пептидного синтеза.
Согласно другому варианту осуществления изобретение обеспечивает способ получения соединения, содержащего пептид, который может быть выделен и/или очищен, содержащий, по существу состоящий из или состоящий из формулы (I) или формулы (II), или производного, пролекарства, аналога или фармацевтически приемлемой соли или сольвата, или сольвата соли, включающий стадии
1. Использование смолы 2-хлортритилового типа с загрузкой 0,2-1,0 ммоль/г или смолы типа Ванга с загрузкой 0,2-1,0 ммоль/г,
2. Загрузку С-концевой аминокислоты последовательности на смолу,
3. Удаление защитной группы fmoc с 15-25% раствором пиперидина в DMF или NMP,
4. Связывание следующей аминокислоты в последовательности с реагентами связывания, такими как HBTU, HATU или DIC/Oxyma, используя стехиометрию между 3-8 экв.,
5. Повтор стадий 3 и 4 до завершения последовательности,
6. Расщепление пептида с твердой подложки с использованием расщепляющего коктейля, включающего TFA и поглотитель тиолов,
7. Циклизацию двух цистеинов в последовательности в окислительных условиях (воздух или 12),
8. Очистку расщепленного пептида с помощью ВЭЖХ с обращенной фазой.
Согласно другому варианту осуществления изобретение обеспечивает способ получения соединения, содержащего пептид, который может быть выделен и/или очищен, содержащий, по существу состоящий из или состоящий из формулы (I) или формулы (II), или производного, пролекарства, аналога или фармацевтически приемлемой соли или сольвата, или сольвата соли, включающий стадии
1. Использование смолы 2-хлортритилового типа с загрузкой 0,2 1,0 ммоль/г или смолы типа Ванга с загрузкой 0,2-1,0 ммоль/г,
2. Загрузку С-концевой аминокислоты последовательности на смолу,
3. Удаление защитной группы fmoc с 15-25% раствором пиперидина в DMF или NMP,
4. Связывание следующей аминокислоты в последовательности с реагентами связывания, такими как HBTU, HATU или DIC/Oxyma, используя стехиометрию между 3-8 экв.,
5. Повтор стадий 3 и 4 до завершения последовательности,
6. Расщепление пептида с твердой подложки с использованием расщепляющего коктейля, включающего TFA и поглотитель тиолов,
7. Циклизацию двух цистеинов в последовательности в окислительных условиях (воздух или 12),
8. Очистку расщепленного пептида с помощью ВЭЖХ с обращенной фазой.
9. Превращение в HCl соль.
По меньшей мере один пептид, производное или аналог, как определено в настоящем документе, или его фармацевтически приемлемая соль или сольват, или комплекс, как определено в настоящем документе, также могут быть использованы в качестве биохимического агента в биохимическом анализе, как например, в диагностическом анализе для измерения реакции на ингибиторы MASP или в любом биохимическом анализе, основанном на связывании ингибитора MASP.
Настоящее изобретение также включает полинуклеотиды, содержащие последовательность, кодирующую MASP-ингибирующий пептид, согласно настоящему изобретению, а также вектор, содержащий полинуклеотид, содержащий последовательность, кодирующую MASP-ингибирующий пептид, согласно настоящему изобретению.
Изобретение дополнительно проиллюстрировано следующими примерами, которые относятся к некоторым конкретным вариантам осуществления согласно настоящему изобретению. Примеры были выполнены с использованием хорошо известных стандартных методов в пределах рутинных процедур для специалистов в данной области техники, если не указано иное. Следующие примеры предназначены только для иллюстративных целей и не претендуют на то, чтобы быть полностью определяющими в отношении условий или объема изобретения. Как таковые, они никоим образом не должны толковаться как ограничивающие объем согласно настоящему изобретению.
Примеры
Перечень аббревиатур, применяемых в экспериментальной части:
Å ангстрем
вод. Водный, водный раствор
бар единицы давления
BPR регулятор обратного давления
конц концентрированный
d дублет (ЯМР)
dd дублет дублета (ЯМР)
DCM Дихлорметан
DEA диэтиламин
DIPEA N,N,-диизопропилэтиламин (основание Хунига)
DMAP 4-диметиламинопиридин
DMF N,N-диметилформамид
DMSO Диметилсульфоксид
dt дублет триплета (ЯМР)
ЕА Этилацетат
ее Энантиомерный избыток
ent Энантиомерный
экв эквивалент(ы)
эквив. эквивалент(ы) (ионная хроматография)
ESI ионизация электрораспылением (масс-спектроскопия)
Fmoc-OSu 1-{[(9Н-флуорен-9-илметокси)карбонил]окси}пирролидин-2,5-дион
GC-MS Газовая хроматография в сочетании с масс-спектрометрией
ч час(ы)
HATU O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония-гексафторфосфат
ВЭЖХ высокоэффективная жидкостная хроматография
IC ионная хроматография
ID внутренний диаметр
L литр
LC-MS жидкостная хроматография в сочетании с масс-спектрометрией
LiHMDS лития бис(триметилсилил)амид
lit. литеартура
m мультиплет (ЯМР)
MALDI матрично-активированная лазерная десорбция/ионизация (масс-спектрометрия)
Me Метил
минминута(минуты)
мм миллиметр
мкммикрометр (микрон)
М молярный
MPLC жидкостная хроматография среднего давления
MS масс-спектрометрия
МТВЕ трет-бутилметиловый простой эфир
МТР микротитровальный планшет
m/z соотношение массы и заряда (масс-спектрометрия)
нм нанометр
НМР N-метил-2-пирролидон
ЯМР ядерная магнитно-резонансная спектроскопия
PBS физиологический раствор с фосфатным буфером
РЕ простой петролейный эфир
PEG полиэтиленгликоль
pos позитивный
ppm частей на миллион
Pr Пропил
Psi фунты на квадратный дюйм (давление)
q / quart квартет (ЯМР)
qd квартет дублета (ЯМР)
quint квинтет (ЯМР)
гас рацемический
Rf фактор удерживания (TLC)
RP обращенная фаза (для жидкостной хроматографии)
Rt время удерживания (хроматография)
s синглет (ЯМР)
с секунды (время)
SPPS теврдофазный пептидный синтез
t триплет (ЯМР)
TBTU О (бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат
tBu трет-бутил
TEA триэтиламин
THF Тетрагидрофуран
TLC тонкослойная хроматография
UPLC сверхвысокоэффективная жидкостная хроматография
UV ультрафиолет
Другие аббревиатуры можно найти в Таблице 2 и 3.
Аналитические способы LC-MS
Способ 1
Тип устройства MS: ThermoFisherScientific LTQ-Orbitrap-XL; Тип устройства ВЭЖХ: Agilent 1200SL; Колонка: Agilent, POROSHELL 120, 3 × 150 мм, SB - С18 2.7 мкм; элюент А: 1 л воды + 0.1% трифторуксусной кислоты; элюент В: 1 л ацетонитрила + 0.1% трифторуксусной кислоты; градиент: 0.0 мин 2% В → 1.5 мин 2% В → 15.5 мин 98% В → 18.0 мин 98% В; печь: 40°С; скорость потока: 0.75 мл/мин; УФ-обнаружение: 210 нм
Способ 2
Тип устройства MS: ThermoFisherScientific LTQ-Orbitrap-XL; Тип устройства ВЭЖХ: Agilent 1200SL; Колонка: Agilent, POROSHELL 120; 3 × 150 мм, SB - С18 2.7 мкм; элюент А: 1 л воды + 0.1% трифторуксусной кислоты; элюент В: 1 л ацетонитрила + 0.1% трифторуксусной кислоты; градиент: 0.0 мин 5% В → 0.3 мин 5% В → 7.0 мин 98% В → 10 мин 98% В; печь: 40°С; скорость потока: 0.75 мл/мин; УФ-обнаружение: 210 нм
Способ 3
Тип устройства MS: Waters TOF устройство; Тип устройства UPLC: Waters Acquity I-CLASS; Колонка: YMC, TRIART C18, 75 × 1 мм, 3.0 мкм × 12 нм; элюент А: 1 л воды + 0.01% муравьиной кислоты; элюент В: 1 л ацетонитрила + 0.01% муравьиной кислоты; градиент: 0.0 мин 1% В → 2.0 мин 1% В → 8.0 мин 95% В → 10.0 мин 95% В; печь: 50°С; скорость потока: 0.63 мл/мин; УФ-обнаружение: 210 нм
Способ 4
Тип устройства MS: Waters TOF устройство; Тип устройства UPLC: Waters Acquity I-CLASS; Колонка: YMC, TRIART C18, 75 × 1 мм, 3.0 мкм 12 нм; элюент А: 1 л воды+0.01% муравьиной кислоты; элюент В: 1 л ацетонитрила + 0.01% муравьиной кислоты; градиент: 0.0 мин 1% В → 1.0 мин 1% В → 15.0 мин 50% В → 18.0 мин 95% В; печь: 50°С; скорость потока: 0.63 мл/мин; УФ-обнаружение: 210 нм
Способ 5
Тип устройства MS: Waters TOF устройство; Тип устройства UPLC: Waters Acquity I-CLASS; Колонка: Waters, HSST3, 2.1 × 50 мм, C18 1.8 мкм; элюент А: 1 л воды + 0.01% муравьиной кислоты; элюент В: 1 л ацетонитрила+0.01% муравьиной кислоты; градиент: 0.0 мин 2% В → 0.5 мин 2% В → 7.5 мин 95% В → 10.0 мин 95% В; печь: 50°С; скорость потока: 1.00 мл/мин; УФ-обнаружение: 210 нм
Способ б
Тип устройства MS: Waters Synapt G2S; Тип устройства UPLC: Waters Acquity I-CLASS; Колонка: Waters, BEH300, 2.1 × 150 мм, C18 1.7 мкм; элюент A: 1 л воды + 0.01% муравьиной кислоты; элюент В: 1 л ацетонитрила + 0.01% муравьиной кислоты; градиент: 0.0 мин 2% В → 1.5 мин 2% В → 8.5 мин 95% В → 10.0 мин 95% В; печь: 50°С; скорость потока: 0.50 мл/мин; УФ-обнаружение: 220 нм
Способ 7
Тип устройства MS: Agilent 6410 Triple Quad; Тип устройства ВЭЖХ: Agilent 1200; Колонка: Gemini-NX С18 5 мкм 110Å 150 × 4.6 мм; элюент A: 0.1%TFA в H2O; элюент В: 0.1%TFA в ACN; градиент: 0.0 мин 20% В → 20 мин 50% В → 20.1 мин 90% В → 23 мин 90% В; температура печи: 50°С; скорость потока: 1.0 мл/мин; УФ-обнаружение: 220 нм
Способ 8
Тип устройства MS: Agilent 6410 Triple Quad; Тип устройства ВЭЖХ: Agilent 1200; Колонка: Discovery BIO Wide Pore С18 5 мкм 300Å × 4.6 мм; элюент A: 0.1%TFA в H2O; элюент В: 0.1%TFA в ACN; градиент: 0.0 мин 10% В → 20 мин 80% В → 20.1 мин 90% В → 23 мин 90% В; температура печи: 50°С; скорость потока: 1.0 мл/мин; УФ-обнаружение: 220 нм
Способ 9
Устройство: Waters ACQUITY SQD UPLC System; колонка: Waters Acquity UPLC HSS T3 1.8 мкм 50 × 1 мм; элюент A: 1 л воды + 0.25 мл 99% муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.25 мл 99% муравьиной кислоты; градиент: 0.0 мин 90% А → 1.2 мин 5% А → 2.0 мин 5% А; печь: 50°С; поток: 0.40 мл/мин; УФ-обнаружение: 210 нм
Способ 10
Устройство MS: Thermo Scientific FT-MS; Устройство UBЭЖX: Thermo Scientific UltiMate 3000; Колонка: Waters, HSST3, 2.1 × 75 мм, C18 1.8 мкм; элюент A: 1 л воды + 0.01% муравьиной кислоты; элюент В: 1 л ацетонитрила + 0.01% муравьиной кислоты; градиент: 0.0 мин 10% В → 2.5 мин 95% В → 3.5 мин 95% В; печь: 50°С; скорость потока: 0.90 мл/мин; УФ-обнаружение: 210 нм/ Optimum Integration Path 210-300 нм
Способ 11
MS Устройство: Agilent MS Quad 6150; ВЭЖХ: Agilent 1290; Колонка: Waters Acquity UPLC HSS T3 1.8 мкм 50 × 2.1 мм; элюент A: 1 л воды + 0.25 мл муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.25 мл муравьиной кислоты; градиент: 0.0 мин 90% А → 0.3 мин 90% А → 1.7 мин 5% А → 3.0 мин 5% А печь: 50°С; скорость потока: 1.20 мл/мин; УФ-обнаружение: 205 - 305 нм.
Способ 12
Устройство: Waters ACQUITY SQD UPLC System; колонка: Waters Acquity UPLC HSST3 1.8 мкм 50 × 1 мм; элюент A: 1 л воды + 0.25 мл 99% муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.25 мл 99% муравьиной кислоты; градиент: 0.0 мин 95% А → 6.0 мин 5% А → 7.5 мин 5% А печь: 50°С; скорость потока: 0.35 мл/мин; УФ-обнаружение: 210 нм.
Способ 13
Устройство: Waters Single Quad MS System; Устройство Waters UPLC Acquity; колонка: Waters ВЕН C18 1.7 мкм 50 × 2.1 мм; элюент A: 1 л воды + 1.0 мл (25% аммиака)/л, элюент В: 1 л ацетонитрила; градиент: 0.0 мин 92% А → 0.1 мин 92% А → 1.8 мин 5% А → 3.5 мин 5% А; печь: 50°С; скорость потока: 0.45 мл/мин; УФ-обнаружение: 210 нм.
Способ 14
System MS: Waters TOF устройство; System UPLC: Waters Acquity I-CLASS; Колонка: Waters Acquity UPLC Peptide ВЕН C18 300Å, 1.7 мкм 150 × 2.1 мм; элюент А: 1 л воды + 0.100 мл 99% муравьиной кислоты, Элюент В: 1 л ацетонитрила + 0.100 мл 99% муравьиной кислоты; Градиент: 0.0 мин 90% А → 0.25 мин 90% А → 8.0 мин 45% А → 10.0 мин 2% → 12.0 мин 2% А Печь: 50°С; Поток: 0.475 мл/мин; УФ-обнаружение: 210 нм.
Способ 15
MS тип устройства: Agilent G6110A; ВЭЖХ тип устройства: Agilent 1200 Series LC; UV DAD; колонка: Chromolith Flash RP-18e 25 × 2.0 мм; подвижная фаза A: 0.0375% TFA в воде (v/v), подвижная фазае В: 0.01875% TFA в ацетонитриле (V/V); градиент: 0.01 мин 5% В → 0.80 мин 95% В → 1.20 мин 95% В → 1.21 мин 5% В → 1.5 мин 5% В; скорость потока: 1.50 мл/мин; температура печи: 50°С; UV обнаружение: 220 нм & 254 нм.
MALDI Способ
Точные массовые измерения были выполнены для выбранных пептидов с использованием метода масс-спектрометрии с матричной лазерной десорбцией/ионизацией (MALDI) на системе Bruker autoflex maX LRF MALDI MS Time-of-Flight (ToF-MS). Образцы готовили на планшете Bruker MALDI с использованием α-циано-4-гидроксикоричной кислоты (CAS 28166-41-8) в качестве матрицы. Образец пептида 0,1-1,0 мг растворяли в 1,0 мл ацетонитрила-воды (50/50 или 30/70) и готовили сток-раствор матрицы (10 мг/мл) в 50% ацетонитриле в воде, содержащей 0,05% трифторуксусной кислоты. 1,0 мкл каждого раствора помещали на планшет MALDI и давали высохнуть. После этого образец готовили к анализу. Рекомендуемые пробоподготовки для планшетов MALDI можно найти в документации, предоставленной Bruker.
Способ аналитической ионной хроматографии
Способ: IC - количественная
Количественное измерение катионов и анионов с использованием внешних стандартов; Устройство: Thermo Scientific ICS 5000+; Капиллярные ИС Колонки: IonPac AS11-НС и IonPac CS16; элюент: градиент элюента [Н]+[ОН]-; Детектор: обнаружение проводимости; возможны обычные анионы: ацетат, бромид, цитрат, хлорид, фторид, формиат, лактат, мезилат, фосфат, сульфат, тартрат, трифторацетат; возможны обычные катионы: аммоний, барий, кальций, калий, литий, натрий, магний, холин.
Аналитический способ газовой хроматографии масс-спектрометрии
GC-MS Способ 1
Устройство: Thermo Scientific DSQII, Thermo Scientific Trace GC Ultra; колонка: Restek RTX-35MS, 15 m × 200 мкм × 0.33 мкм; постоянный поток с Helium: 1.20 мл/мин; печь: 60°С; вход: 220°С; градиент: 60°С, 30°С/мин 300°С (3.33 мин, поддерживали).
ЯМР
Данные 1H-ЯМР выбранных соединений записаны в виде списков сигналов 1Н-ЯМР. Для каждого пика сигнала перечислены δ-значение в ppm с последующей интенсивностью сигнала в круглых скобках. Между парами δ-значений интенсивности различных пиков стоят точка с запятой в качестве разделителей. Поэтому перечень пиков описан общей формой: δ1 (интенсивность1), δ2 (интенсив-ность2), …, δi (интенсивность1), …, δn (интенсивностьn).
Интенсивность резкого сигнала коррелирует с высотой (в см) сигнала в печатном спектре ЯМР. При сравнении с другими сигналами эти данные могут быть соотнесены с реальными соотношениями интенсивностей сигналов. В случае широких сигналов отображаются более одного пика или центр сигнала вместе с их относительной интенсивностью по сравнению с наиболее интенсивным сигналом, отображаемым в спектре. Перечень пиков 1H-ЯМР аналогичен классическому показанию 1H-ЯМР и, таким образом, обычно содержит все пики, перечисленные в классической интерпретации ЯМР. Кроме того, как и в классических распечатках 1H-ЯМР, в перечнях пиков могут отображаться сигналы растворителя, сигналы, полученные от стереоизомеров конкретного целевого соединения, пики примесей, сателлитные пики 13С и/или боковые полосы вращения. Пики стереоизомеров целевых соединений и/или пики примесей обычно имеют в среднем более низкую интенсивность, чем пики целевых соединений (например, с чистотой >90%). Такие стереоизомеры и/или примеси могут быть типичными для конкретного процесса получения. Поэтому их пики могут помочь идентифицировать воспроизводимость нашего процесса получения с помощью "отпечатков пальцев побочных продуктов". Эксперт, который рассчитывает пики целевых соединений с помощью известных способов (MestreC, ACD-моделирования, но также и с эмпирически оцененных средних значений) может выделить пики целевых соединений при необходимости, используя дополнительные фильтры интенсивности. Такое выделение будет аналогично соответствующему пику классической интерпретации 1H-ЯМР. Дальнейшие подробности описания ЯМР-данных со списками пиков можно найти в публикации "Citation of NMR Peaklist Data within Patent Applications" (см. http://www.researchdisclosure.com/searching-disclosures, Research Disclosure Database Number 605005, 2014, 01 августа 2014). В ходе сбора пиков, как описано в Research Disclosure Database Number 605005, параметр "MinimumHeight" может быть установлен от 1% до 4%. Однако в зависимости от химической структуры и/или в зависимости от концентрации измеряемого соединения может быть целесообразно установить параметр "MinimumHeight" <1%.
Примеры получения
Общие способы синтеза
Твердофазный синтез пептидов (SPPS) проводили либо с помощью автоматического синтезатора пептидов, либо вручную. Синтез пептидов обычно проводили в масштабах от 0,1 до 1,0 ммоль. При ручном синтезе пептидов использовали общие процедуры Способов С, D и Е, описанные ниже. Автоматизированный синтез пептидов проводили на синтезаторе пептидов Symphony X (Gyros Protein Technologies).
Fmoc-защищенные аминокислоты (или промежуточные аминокислоты, используемые для получения Fmoc-аминокислот) приобретали у Novabiochem, Bachem, Iris Biotech, Sigma-Aldrich, Alfa, Enamine, Amatek, Ani-chem, ACBR, ABC Laboratory, AP Bioscience, Combiblocks., ArZa Bioscience, Ark Pharm, Acrotein-chem, Apollo Scientific, Biofine, Broadpharm, VWR или Gyros Protein Technologies (Флуоренилметоксикарбонил = Fmoc), GL Biochem (Shanghai) Ltd, Chengdu Aminotp Pharmaceu-tical Technology Ltd, Suzhou Highfine Biotech Co., Ltd, WuXi AppTec или других китайских поставщиков. Некоторые специальные fmoc-защищенные аминокислоты были синтезированы внутри, и эти способы синтеза описаны в настоящем документе. Некоторые аминокислоты fmoc, синтезированные внутри компании, также коммерчески доступны. В некоторых случаях, когда Fmoc-защищенная аминокислота не была коммерчески доступна, но Вос-защищенная (трет-бутилоксикарбонил = Boc), не встречающаяся в природе аминокислота, была коммерчески доступной, fmoc-защищенная аминокислота получали из Вос-защищенной аминокислоты путем удаления защитной группы и повторной защиты с использованием способов, обычно используемых в данной области техники. Номера CAS для коммерчески доступных, не встречающихся в природе аминокислот, используемых в синтезе пептидов согласно настоящему изобретению, в большинстве случаев были включены в Таблицу 5. В тех случаях, когда была приобретена рацемическая аминокислота (Fmoc или Вое), специалистам понятно, что энантиомеры можно разделить с помощью хиральной хроматографии, и это действительно иногда проводили для получения энантиомерно чистой аминокислоты перед синтезом пептида.
Следующие не встречающиеся в природе аминокислоты были использованы при получении пептидов согласно настоящему изобретению. Fmoc- или Вос-защищенные аминокислоты были либо получены из коммерческих источников (имеется номер CAS), либо синтезированы внутри компании способами, описанными в настоящем документе. В таблице 5 приведены номера CAS химических групп/аминокислот, использованных для синтеза пептида (правая колонка), и соответствующая химическая группа/аминокислота, присутствующая в пептидах (левая колонка).
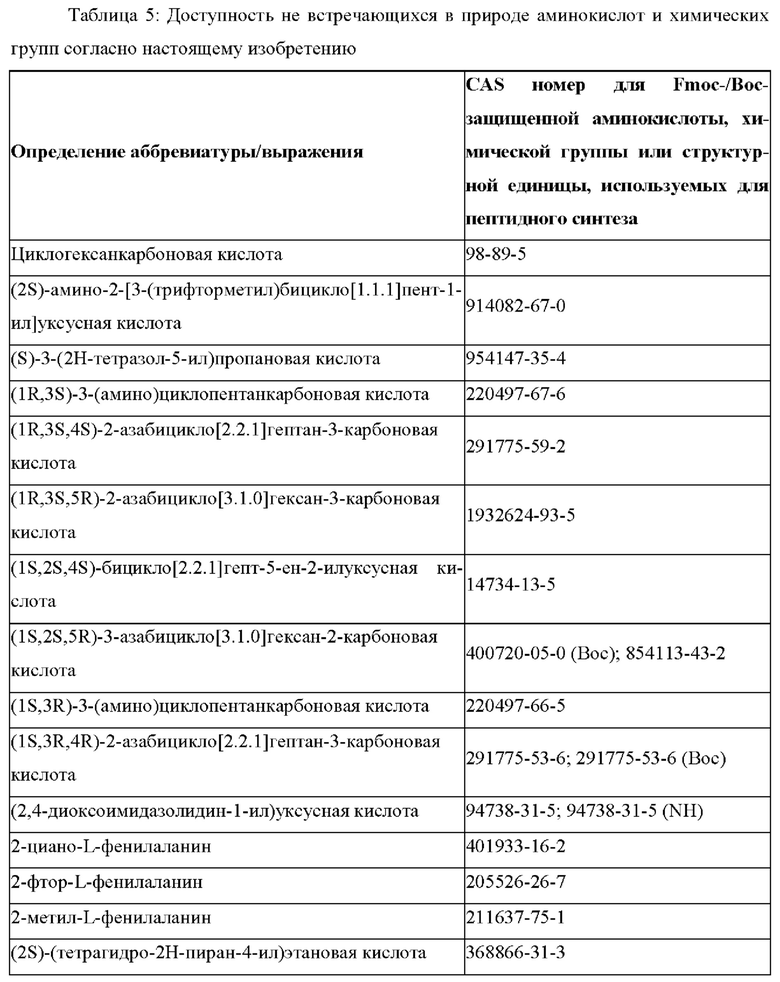
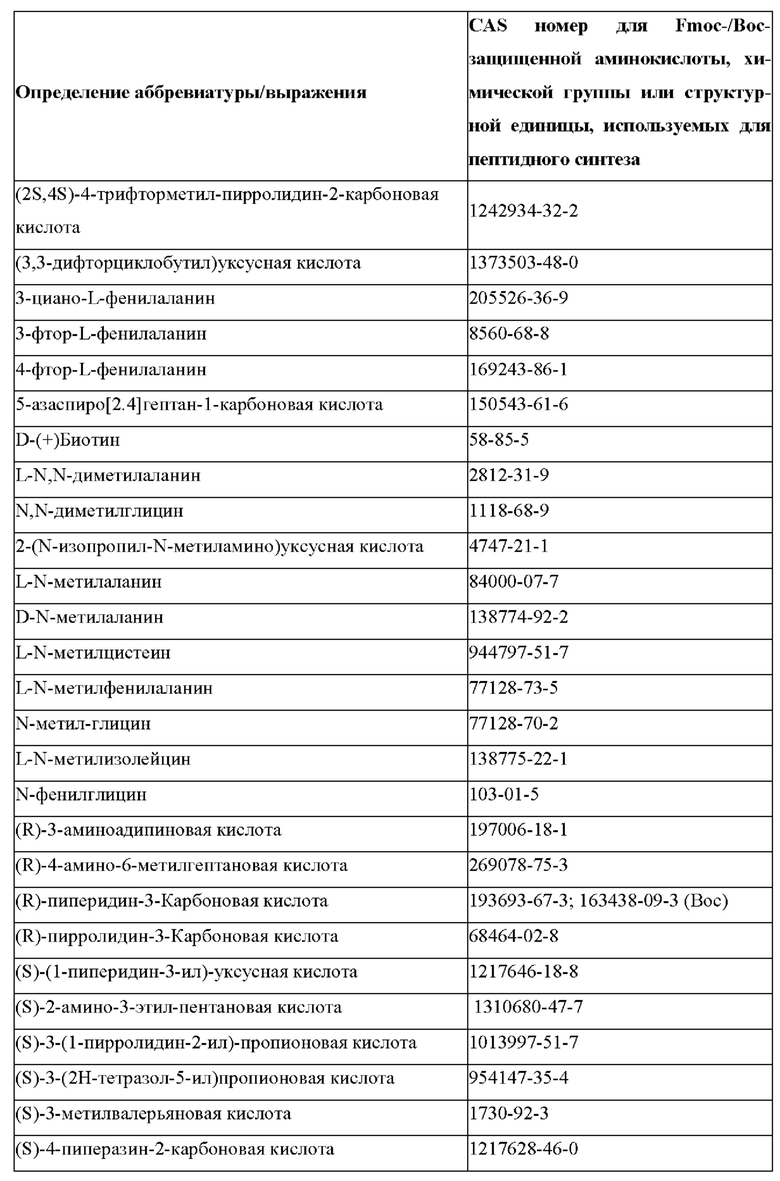

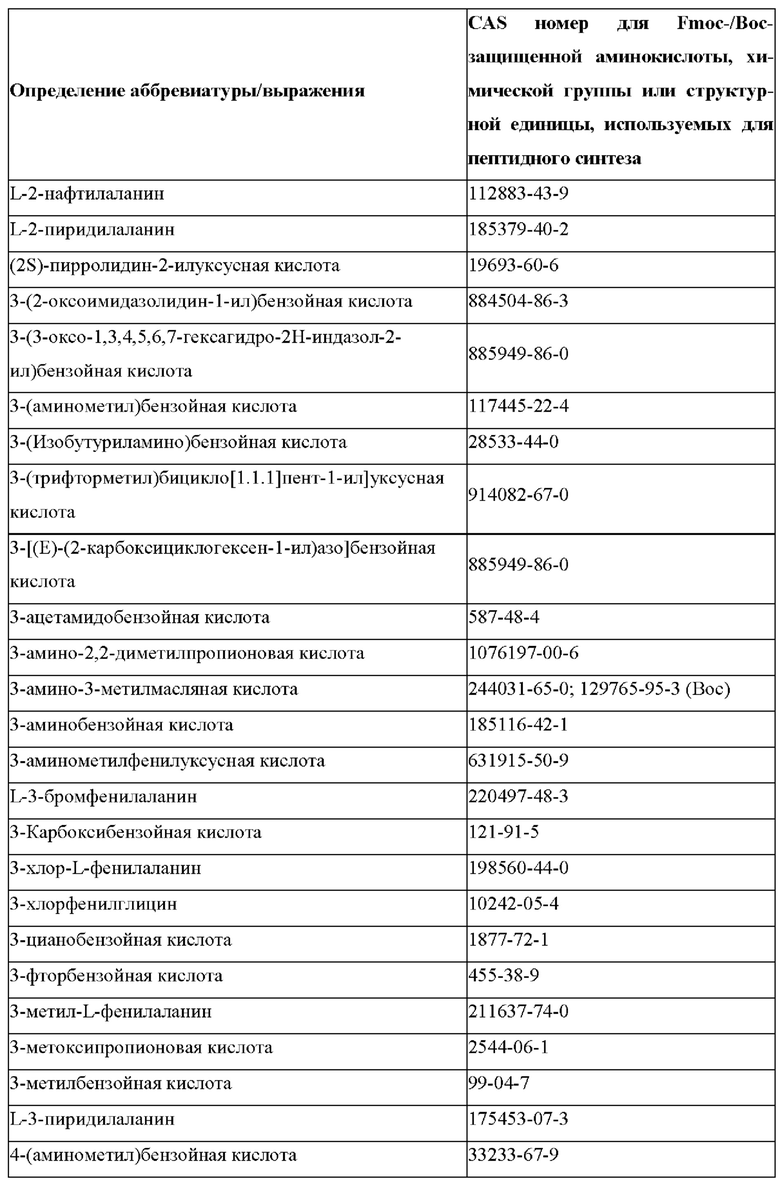
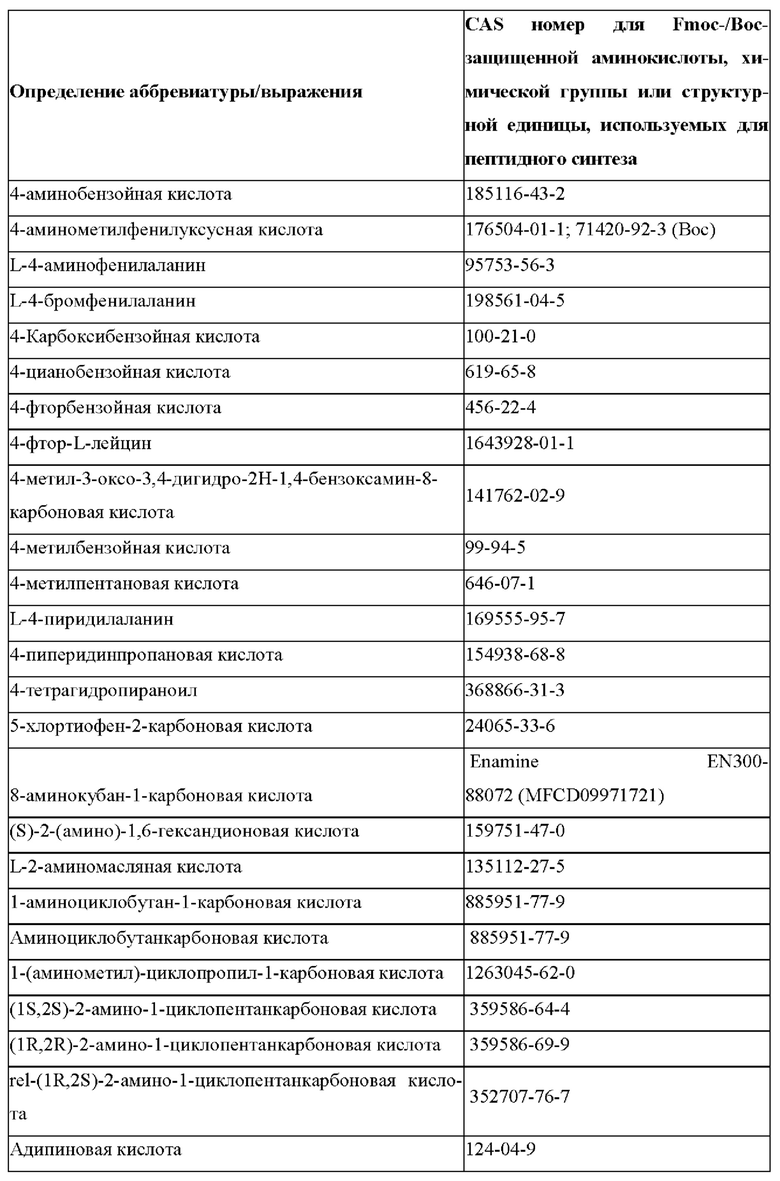
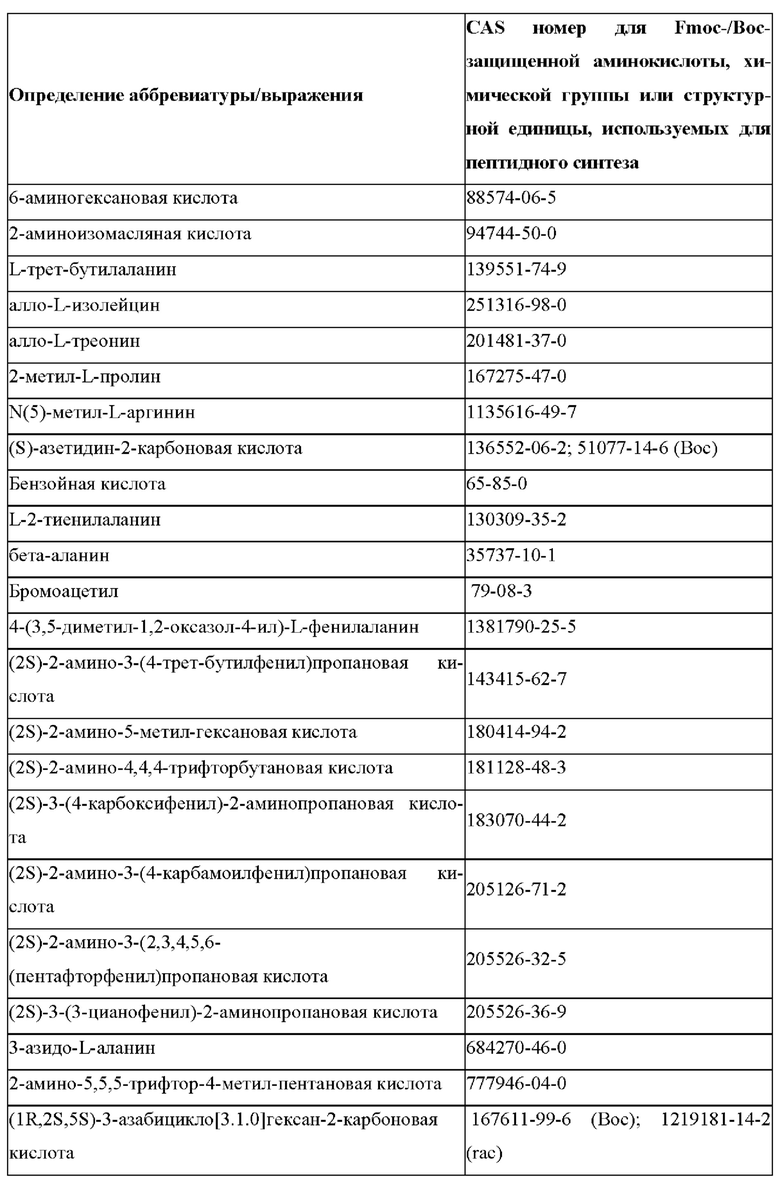
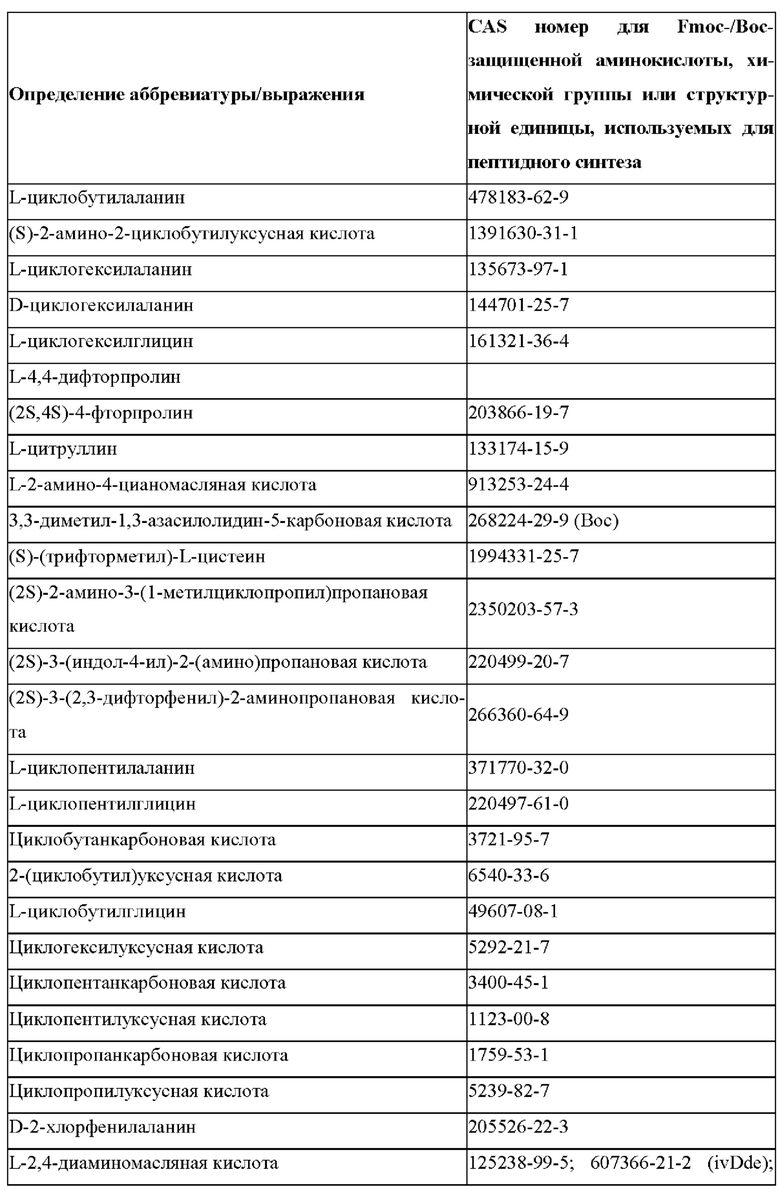
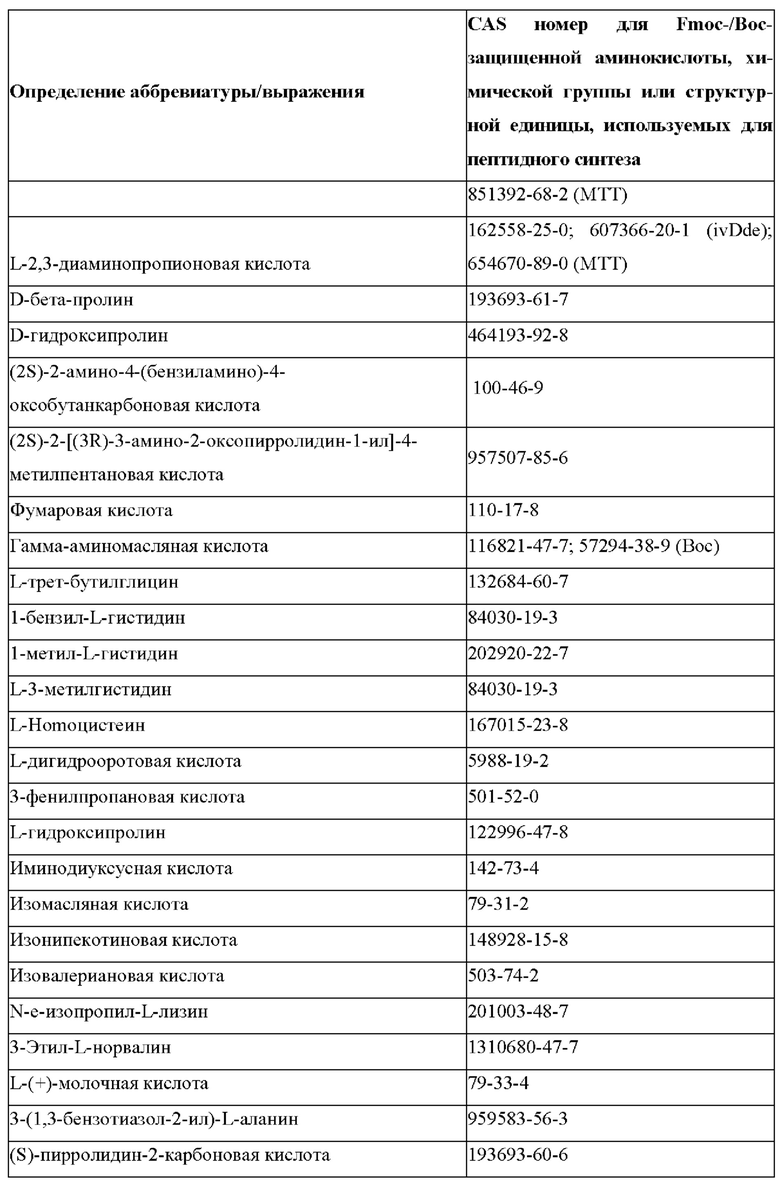
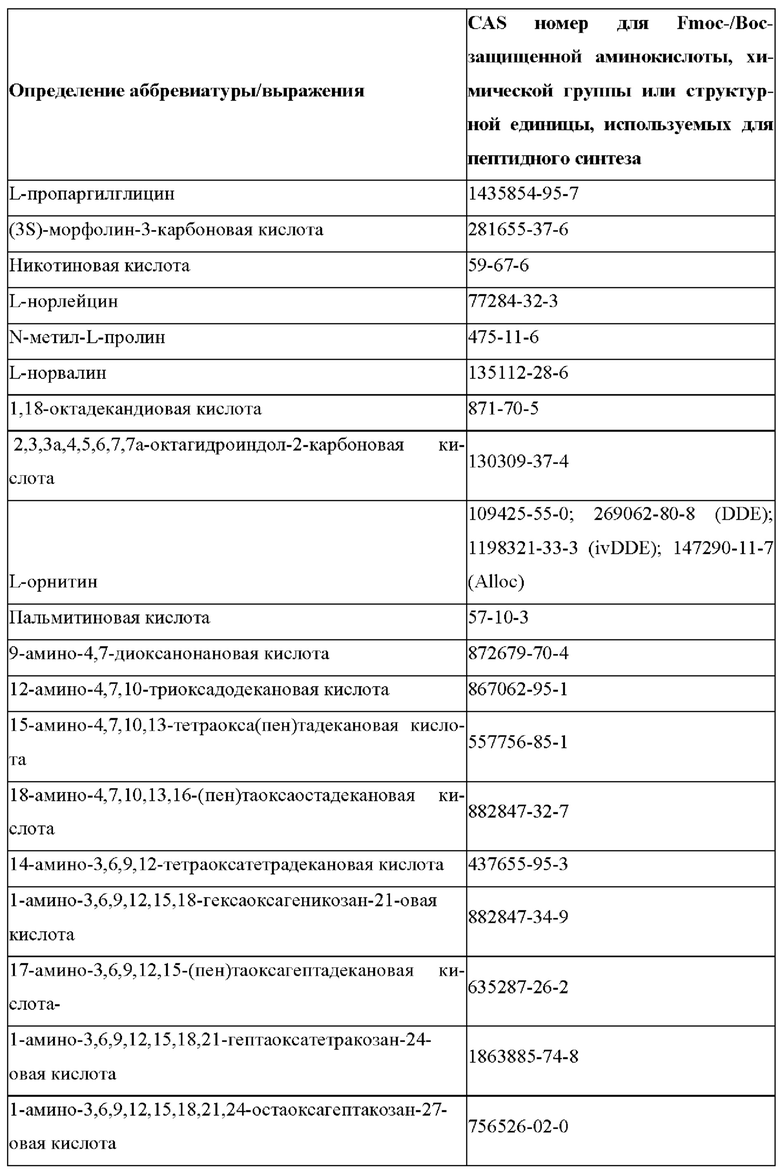
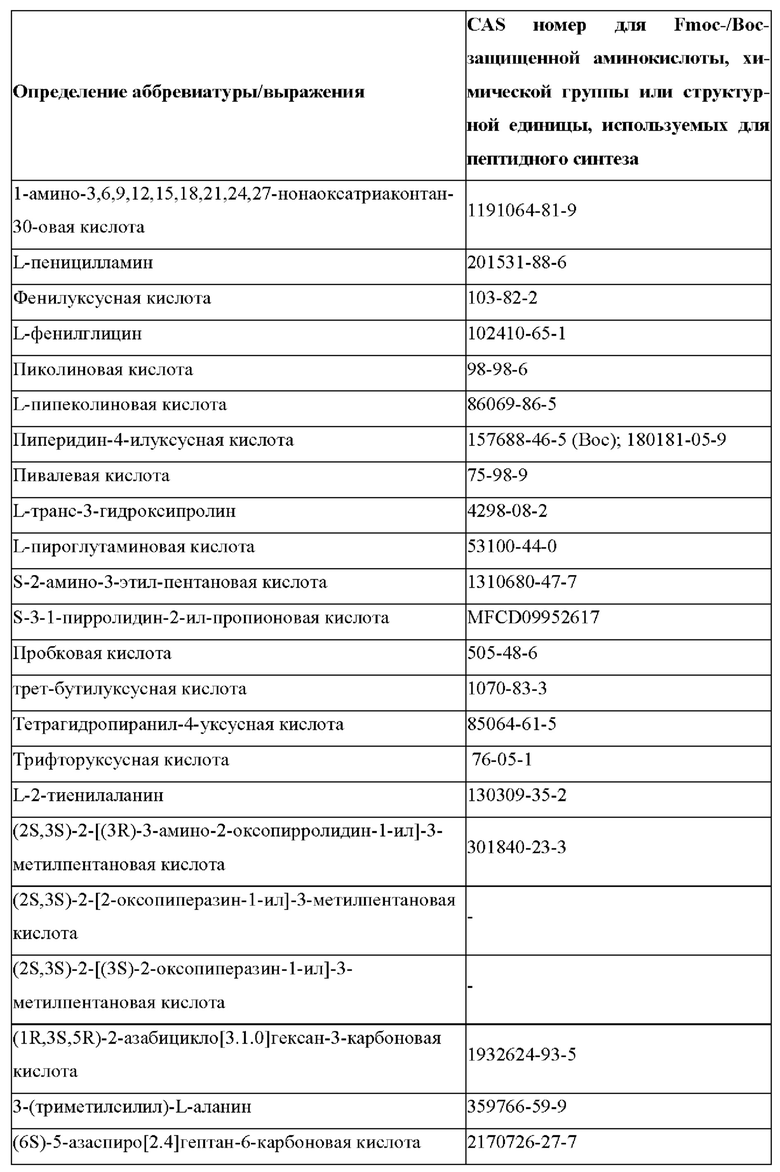
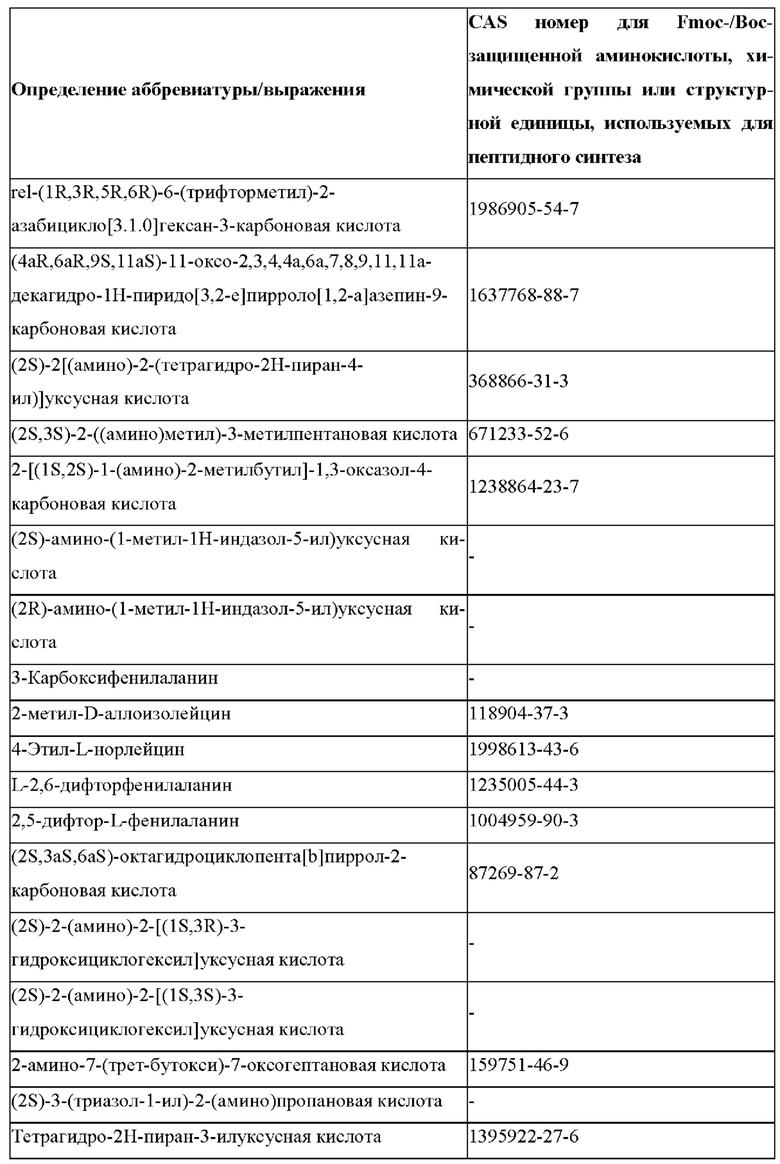
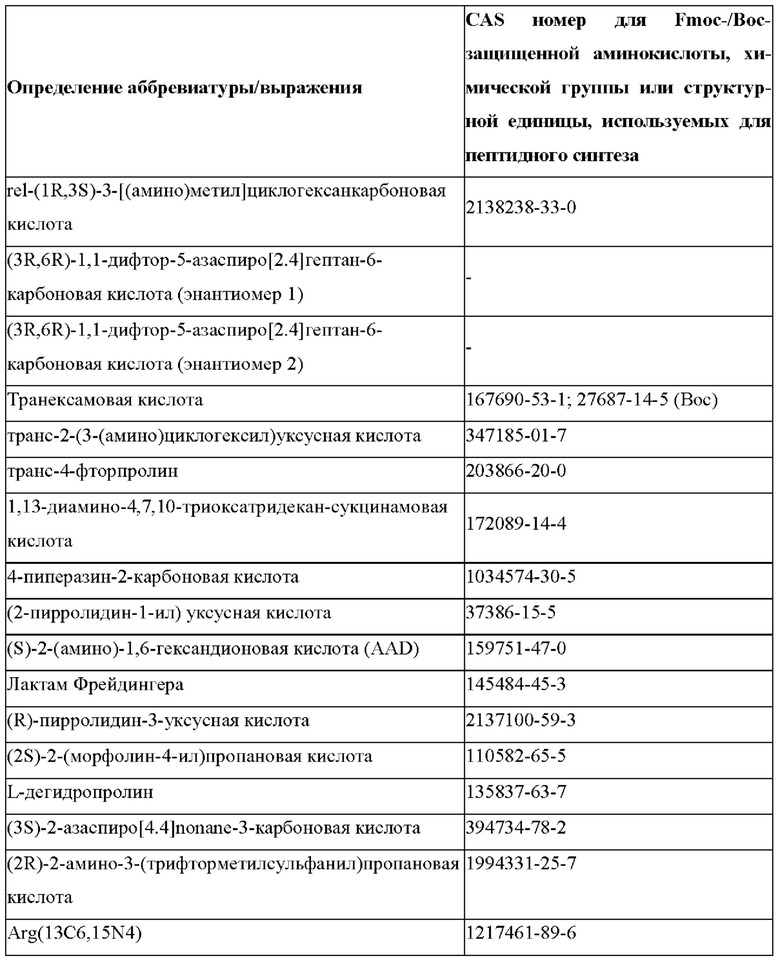
Твердофазные смолы были приобретены у Novabiochem, Bachem, Iris Biotech, Peas Biomatrix, GL Biochem (Shanghai) Ltd, СЕМ или Protein Technologies. Загрузка смолы составляла 0,3-1,0 ммоль/г. Пептиды синтезировали на 2-хлортритиловой смоле, на смоле Ванга или на смоле амидного типа Ринка в зависимости от желаемого С-конца. Расщепление защитной группы флуоренилметоксикарбонила (Fmoc) было достигнуто с использованием 20% пиперидина в диметилформамиде при температуре окружающей среды. Каждую стадию расщепления Fmoc проводили два раза. Аминокислоты связывали на автоматическом синтезаторе (Symphony X) с использованием 8 экв. Fmoc-аминокислоты, с 8 экв. DIC (диипропилкарбодиимид) (0,5 М в DMF) и 8 экв. Oxyma (этилцианогидроксииминоацетат) (0,5 М в DMF). Аминокислотные связывания проводили при температуре окружающей среды и в атмосфере азота, когда Symphony X применяли. При использовании дорогостоящих или самостоятельно приготовленных fmoc-аминокислот связывание проводили вручную с использованием 3 экв. Fmoc-аминокислоты, с 3 экв. DIC (0,5 М в DMF) и 3 экв. оксима (0,5 М в DMF). На каждой стадии связывание аминокислот проводили два раза (двойное связывание). Альтернативно, пептиды готовили с помощью SPPS вручную, используя 3 экв. Fmoc-аминокислоты, 2,85 экв. HBTU ((2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат, гексафторфосфат бензотриазола тетраметилурония) (0,5 М в DMF) и 6 экв. DIPEA (0,5 М в DMF) Реакцию связывания контролировали с помощью нингидринового теста.
У пептидов полностью удаляли защитную группу с использованием трифторуксусной кислоты (TFA)/тиоанизола (ТА)/1,2-этандитиола (EDT) (90:7:3) или с помощью 92,5% TFA/2,5% EDT/2,5% TIS (триизопропилсилан)/2,5% Н2О. Для пептидов, содержащих метионин, пептиды обрабатывали раствором 1,6% EDT и 1,2% триметилсилилбромида в TFA в течение 2 ч при температуре окружающей среды для снижения окисленного метионина.
Циклизация дисульфида:
Дисульфидные мостики формировали при встряхивании пептидов в 0,1 М аммиачно-бикарбонатном буфере (рН 7,83) при концентрации 0,5 мг/мл в течение ночи. Раствор затем лиофилизировали. Альтернативно, дисульфидные мостики образовывались при встряхивании пептидов в смеси ацетонитрил/вода (часто 3:7), доведенной до рН 9,0 твердым аммоний-бикарбонатным буфером при концентрации 1-3 мг/мл всю ночь. Альтернативно дисульфидные мостики готовили путем окисления йодом (I2) (0,1 М в МеОН) при концентрации 1-1,3 мг/мл в ацетонитриле/воде (1:1) при 20°С в течение 2 мин с последующей обработкой тиосульфатом натрия (0,1 М в воде) с последующей лиофилизацией.
Необязательное ацетилирование:
N-концевое ацетилирование проводили с использованием 10 экв. уксусного ангидрида в DMF (2 мл) и 2,5 экв. DIPEA посредством встряхивания суспензии при комнатной температуре в течение 1 ч на орбитальном шейкере. Растворитель удаляли и смолу промывали с помощью DMF (5х) и DCM (5х). Затем процедуру повторяли снова. В качестве альтернативы проводили N-концевое ацетилирование с использованием 10 мл кэппирующего раствора, состоящего из уксусного ангидрида/N-метилморфолина (НММ)/DMF (10:5:85), путем встряхивания суспензии при комнатной температуре в течение 30 мин на орбитальном шейкере.
Пептидное расщепление:
Готовили коктейль для расщепления, содержащий TFA/EDT/тиоанизол (90:3:7). Коктейль для расщепления (2 мл) добавляли к пептид содержащей смоле и суспензию встряхивали на орбитальном шейкере в течение 2,5 часов. Холодный эфир (-20°С) добавляли для осаждения пептида. Полученный раствор центрифугировали в атмосфере азота (Sigma 2-16KL), а полученное твердое вещество, полученное после декантации, промывали холодным простым эфиром еще 3 раза путем центрифугирования и декантации. Полученное твердое вещество очистили препаративной ВЭЖХ.
В качестве альтернативы получали коктейль для расщепления, содержащий TFA/EDT/TIS/H2O (92,5:2,5:2,5:2,5). Коктейль для расщепления (6 мл (масштаб 0,3 ммоль)) добавляли к пептидсодержащей смоле и суспензию встряхивали на орбитальном шейкере в течение 2,5 часов. Холодный трет-бутилметиловый эфир (-20°С) добавляли для осаждения пептида. Полученный раствор центрифугировали при 3000 об/мин в течение 3 мин, а полученное твердое вещество, полученное после декантации, промывали холодным трет-бутилметиловым эфиром еще 3 раза (20 мл×3), путем центрифугирования и декантации. Полученный неочищенный пептид сушили в вакууме в течение 2 часов, а затем очищали посредством препаративной ВЭЖХ.
Препаративная ВЭЖХ:
Agilent 1260 обращенно-фазовой препаративной ВЭЖХ или Knauer AZURA обращенно-фазовой препаративной ВЭЖХ применяли для очистки. Колонку выбирали по результатам скрининга колонки. Пептид растворяли в 10-30% ACN/воды (обычно начальная точка градиента). Вода и ацетонитрил оба содержат 0,1% TFA. Скорость потока 20 мл/мин, обычно использовали от 10-30% ACN/вода до 85-90% ACN/вода. Фракции анализировали с помощью ВЭЖХ (Agilent 1260 Infinity) с использованием колонки Chromolith Speedrod, градиент 5-95% ACN/вода в течение 8 мин) и одним или несколькими из следующих способов LC-MS: Способ 1, Способ 2, Способ 3, Способ 4, Способ 5, Способ 6.
В качестве альтернативы, Gilson GX-281 обращенно-фазовой препаративной ВЭЖХ применяли для очистки. Колонку выбирали по результатам скрининга колонок. Пептид растворяли в 10-30% ACN/воде (обычно начальная точка градиента). Вода содержала 0,075% TFA. Обычно применяли колонку Luna (25×200 мм, С18 10 мкм, 110  ) или колонку Gemini (30×150 мм, С18 5 мкм, 110 ). Условия препаративной ВЭЖХ: скорость потока 20 мл/мин, от 10-30% ACN/вода до 85-90% ACN/вода, длина волны 214/254 нм, температура печи 30°С. Фракции анализировали с помощью ВЭЖХ (Agilent 1260 Infinity) с использованием Способа W1 или Способа 7. После этого пептиды анализировали одним или несколькими из следующих способов: Способ 1, Способ 2, Способ 3, Способ 4, Способ 5, Способ 6.
) или колонку Gemini (30×150 мм, С18 5 мкм, 110 ). Условия препаративной ВЭЖХ: скорость потока 20 мл/мин, от 10-30% ACN/вода до 85-90% ACN/вода, длина волны 214/254 нм, температура печи 30°С. Фракции анализировали с помощью ВЭЖХ (Agilent 1260 Infinity) с использованием Способа W1 или Способа 7. После этого пептиды анализировали одним или несколькими из следующих способов: Способ 1, Способ 2, Способ 3, Способ 4, Способ 5, Способ 6.
Дисульфидные миметики, где дисульфидная связь -S-S- замещена на -CH2-S-, -S-СН2-, -СН2-СН2-, -S-(CH2)2-, -(CH2)2-S- или -CH2-S-CH2-, могут быть получены в соответствии с процедурой, описанной в следующих ссылках, в сочетании со способами, описанными в настоящем документе: (1) Hong-Kui Cui, Ye Guo, Yao He, Feng-Liang Wang, Hao-Nan Chang, Yu-Jia Wang, Fang-Ming Wu, Chang-Lin Tian, Lei Liu Angew. Chem. Int. Ed. 2013, 52, 9558-9562; (2) Ye Guo, De-Meng Sun, Feng-Liang Wang, Yao He, Lei Liu, Chang-Lin Tian Angew. Chem. Int. Ed. 2015, 54, 14276-14281; (3) Yang Xu, Tao Wang, Chao-Jian Guan, Yi-Ming Li, Lei Liu, Jing Shi, Donald Bierer Tetrahedron Letters 2017, 58, 1677-1680; (4) Tao Wang, Yi-Fu Kong, Yang Xu, Jian Fan, Hua-Jian Xu, Donald Bierer, Jun Wang, Jing Shi, Yi-Ming Li Tetrahedron Letters 2017, 58, 3970-3973; (5) Tao Wang, Jian Fan, Xiao-Xu Chen, Rui Zhao, Yang Xu, Donald Bierer, Lei Liu, Yi-Ming Li, Jing Shi, Ge-Min Fang Org. Lett. 2018, 20, 6074-6078; (6) Jan-Patrick Fischer, Ria  Sylvia Els-Heindl, Donald Bierer, Johannes Koebberling, Bernd Riedl, Annette G. Beck-Sickinger J Pep Sci. 2019; e3147; (7) Dong-Liang Huang, Jing-Si Bai, Meng Wu, Xia Wang, Bernd Riedl, Elisabeth Pook, Carsten Alt, Marion Erny, Yi-Ming Li, Donald Bierer, Jing Shi, Ge-Min Fang Chem. Commun., 2019, 55, 2821-2824; (8) Shuai-Shuai Sun, Junyou Chen, Rui Zhao, Donald Bierer, Jun Wang, Ge-Min Fang, Yi-Ming Li Tetrahedron Letters 2019, 60, 1197-1201; (9) С.M.В.K. Kourra and N. Cramer Chem. Sci., 2016, 7, 7007-7012.
Sylvia Els-Heindl, Donald Bierer, Johannes Koebberling, Bernd Riedl, Annette G. Beck-Sickinger J Pep Sci. 2019; e3147; (7) Dong-Liang Huang, Jing-Si Bai, Meng Wu, Xia Wang, Bernd Riedl, Elisabeth Pook, Carsten Alt, Marion Erny, Yi-Ming Li, Donald Bierer, Jing Shi, Ge-Min Fang Chem. Commun., 2019, 55, 2821-2824; (8) Shuai-Shuai Sun, Junyou Chen, Rui Zhao, Donald Bierer, Jun Wang, Ge-Min Fang, Yi-Ming Li Tetrahedron Letters 2019, 60, 1197-1201; (9) С.M.В.K. Kourra and N. Cramer Chem. Sci., 2016, 7, 7007-7012.
Все пептиды согласно настоящему изобретению, если не указано иное, представляют собой соли TFA.
Общий способ автоматизированного SPPS пептидов Masp (Способ А)
Синтез ((N-Me)G)-IC+SRSLP-(Oic)-I-(Pen)+IPD-NH2 (пример 165) представлен.
Пептид синтезировали с использованием стандартной химии Fmoc.
Автоматизированный SPPS проводили на синтезаторе пептидов Symphony X (Protein Technologies). Обычно использовали амидную смолу Ринка Chemmatrix (загрузка 0,5 ммоль/грамм) в масштабе 0,1 ммоль. Смолу помещали в сосуд и помещали на устройство. В ходе синтеза были приготовлены и использованы следующие растворы: Fmoc Амино Acids: 0.2 М (8 экв.)
1) Активатор 1: 0.5 М DIC в DMF (7,5 экв.)
2) Активатор 2: 0.5 М Oxyma в DMF (7,5 экв.)
3) Удаление защитной группы Fmoc: 20% пиперидин в DMF
4) Уксусный ангидрид (10 экв.) в 2 мл DMF
Двойные связывания обычно выполняли для каждой аминокислоты. Для дорогостоящих природных аминокислот Fmoc или Вос, синтезированных собственными силами аминокислот Fmoc или N-метилированной аминокислоты, последовательность прерывали, и эту аминокислоту связывали вручную (двойное связывание, но обычно с меньшим количеством реагента (3-5 экв.). После завершения связывания синтез обычно продолжали на синтезаторе. Если N-метиламинокислоту добавляли в последовательность, обычно следующую аминокислоту связывали также вручную. Все стадии выполняли при комнатной температуре в атмосфере азота. Fmoc-Pen и Fmoc-Oic обычно связывали с использованием автоматизированного SPPS. Fmoc(N-Me)G обычно связывали вручную. Ahx всегда связывали вручную.
Смолу подвергали набуханию и промывали DMF (3×3 мл, 10 мин). Если смола содержала Fmoc, затем Fmoc удаляли 20% раствором пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Asp(t-Bu) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Asp(Ot-Bu) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Pro (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Pro (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Ile (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Ile (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Pen(Trt) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Pen(Trt) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Ile (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Ile (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Oic (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Oic (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Pro (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Pro (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Leu (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Leu (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Ser(t-Bu) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Ser(t-Bu) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Arg(Pbf) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Arg(Pbf) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Ser(t-Bu) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Ser(t-Bu) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Cys(Trt) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Cys(Trt) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Ile (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Ile (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Ручное связывание в ходе автоматизированного SPPS:
Fmoc-(N-Me)G (0.2 М в DMF, 5 экв.) добавляли в смолу. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание протекало при встряхивании (Thermomixer, rt) в течение 2 часов. Раствор фильтровали и промывали с DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-(N-Me)G (0.2 М в DMF, 5 экв.) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание протекало при встряхивании (Thermomixer, rt) в течение 2 часов. Раствор фильтровали и промывали с DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Если в последовательности присутствовали дополнительные аминокислоты, их связывали с использованием описанных выше стадий.
Тестовое расщепление:
Когда связывания выполняли вручную, обычно выполняли тестовое расщепление для контрольных реакций. Коктейль для тестового расщепления представлял собой TFA/EDT/тиоанизол (90:3:7); 1,5 ч встряхивания на термосмесителе при температуре окружающей среды и 750 об/мин. Анализ проводили способом LC-MS с использованием одного из описанных выше способов.
Полное расщепление:
Смолу, содержащую пептид, помещали в шприц, и 3.0 мл буфера для расщепления TFA/EDT/Thioanisol (90:3:7) добавляли к смоле. Смесь встряхивали при комнатной температуре в течение 2.5 часов. Раствор собирали посредством фильтрации и пептид осаждали посредством добавления холодного простого диэтилового эфира (-20°С). Раствор центрифугировали (3000 об/мин) в пробирке Falcon (60 мл) в атмосфере аргона. Простой эфир декантировали и твердый остаток многократно промывали холодным диэтиловым эфиром (5×10 мл). Твердый остаток затем сушили.
Дисульфидная циклизация:
Неочищенный пептид растворяли в 0,1 М бикарбоната аммония (рН 7,8-8,2) при концентрации 1 мг/2 мл. Раствор встряхивали на орбитальном шейкере всю ночь в круглодонной колбе, открытой на воздух. Раствор затем лиофилизировали с получением порошка белого цвета.
Скрининг колонок для очистки посредством ВЭЖХ:
Пептид растворяли в 5% CH3CN и 95% воды. Скрининг колонок проводили на каждом пептиде, чтобы определить, какой способ препаративный ВЭЖХ использовать для очистки. Были проверены следующие аналитические колонки.
Для скрининга колонок доступны для способа:
1) 5-60% ACN_8 Мин_1 мл/мин_25°С
2) 30-85% ACN_8 Мин_1 мл/мин_25°С
Доступные колонки (50 мм×ID 4,6 мм) (доступны так же как препаративные колонки):
1) Aeris С18 (Phenomenex)
2) X-Bridge C18 (Waters)
3) Kinetex C18 (Coreshell Material) (Phenomenex)
4) YMC Triart C18 (возможна элюция 100% водой)
5) Kinetix Biphenyl (Phenomenex)
6) X-Select С18 (положительный заряд)
7) Jupiter Proteo C18 (Phenomenex)
8) Luna С18 (Phenomenex)
Для пептидов согласно настоящему изобретению применяли одну из следующих 5 препаративных колонок:
1) Колонка: Phenomenex, Aeris Peptide 5 мкм ХВ-С18, AXIA Packed, 21,2×250 мм+Картридж 5 мкм
2) Колонка: Phenomenex, Kinetex С18 5 мкм 21,5×250 мм+Картридж 5 мкм
3) Колонка: Phenomenex, Kinetex 5 мкм Biphenyl 100А, AXIA Packed, 21,2×250 мм+Картридж 5 мкм
4) Колонка: YMC Actus Triart Prep. С18 12 нм, S-10 мкм 250×20 мм+Картридж 3 мкм (10×4 мм)
5) Колонка: Waters, Xbridge Prep. С18 5 мкм OBD 19×250 мм+Картридж 10 мкм
Сразу после выбора колонки применяли следующие способы:
1) способ: Градиент 5-60% ACN в воде (0,10% TFA)
2) способ: Градиент 30-85% ACN в воде (0,10% TFA)
3) сфокусированный градиент по результатам скрининга колонки.
Скорость потока 20 мл/мин
Объединенные фракции анализировали с помощью ВЭЖХ (5-95 за 8 мин, Chromolith SpeedROD & YMC C18 5-95 за 18 мин) и с использованием одного из способов LC-MS, описанных выше.
Для примера 165 неочищенный пептид растворяли в CH3CN/воде (1:1) и очищали на колонке Phenomenex Kinetex C18 5 мкм, 21,5×250 мм+5 мкм картридж, Скорость потока: 20 мл/мин, Способ: 5-60% ACN в воде (каждый из которых содержит 0,10% TFA) в течение 48 мин. Объединенные фракции лиофилизировали с получением 39 мг Примера 165 (чистота 95%).
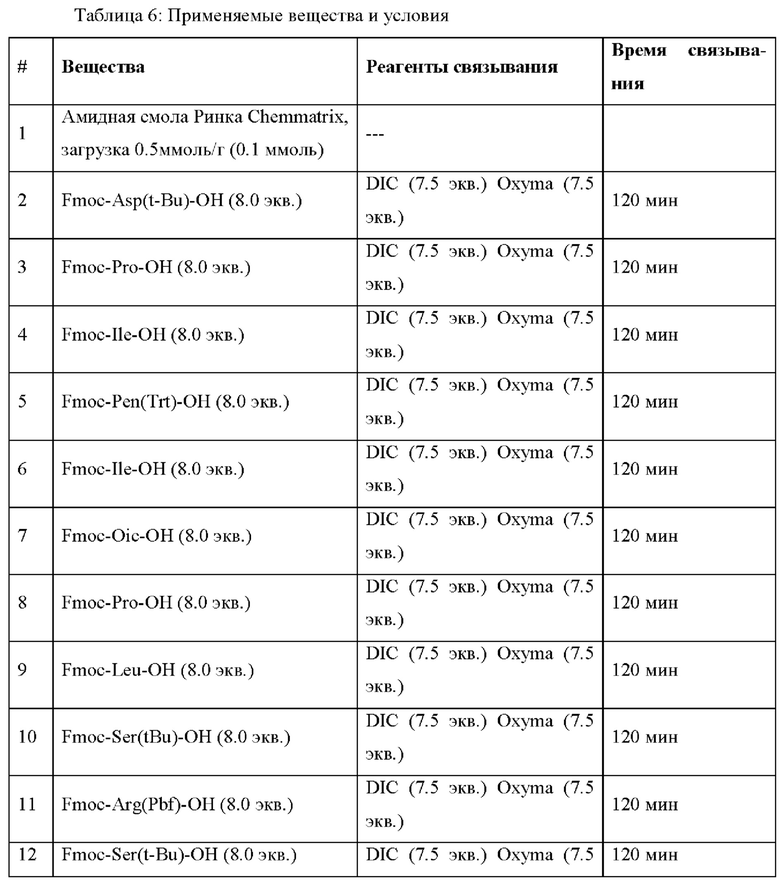

Общий способ автоматизированного SPPS пептидов Masp (Способ В)
Синтез ((N-Me)G)-IC+SRSLP-(Oic)-I-(Pen)+I-((5-азаспиро[2.4]гептан-1-карбоновая кислота)-ОН (пример 164) проиллюстрирован.
Пептид был синтезирован с использованием стандартной химии Fmoc.
По возможности использовалась смола Ванга, предварительно загруженная первой аминокислотой (например, предварительно загруженная Fmoc-Asp(t-Bu)-OH, Fmoc-Pro-ОН и т.д.), с типичным содержанием 0,55-0,7 ммоль/г. Когда первая аминокислота была не доступна, ее добавляли вручную.
Когда первая аминокислота была не доступна в виде предварительно загруженной в смолу, ее добавляли вручную и хлортритиловую смолу применяли для синтеза.
Загрузка первой аминокислоты на 2-хлортритиловую смолу:
2-хлортритиловую смолу (500 мг, 0.775 ммоль) оставляли набухать в 10 мл DCM в пробирке Falcon 50 мл на 15 мин. 5-[(9Н-флуорен-9-илметокси)карбонил]-5-азаспиро[2.4]гептан-1-карбоновую кислоту (281 мг, 0.775 ммоль) растворяли в DCM с добавлением DIEA (6 экв., 0.81 мл), и раствор добавляли в смолу. Раствор продували аргоном и встряхивали всю ночь при комнатной температуре. Смесь фильтровали и промывали с DMF (3×5 мл) и DCM (3×5 мл). Метанол добавляли (5 мл), смесь встряхивали в течение 30 минут, а затем фильтровали. Смолу промывали с DMF (3×5 мл) и DCM (3×5 мл). Метанол снова добавляли (5 мл), смесь встряхивали в течение 30 минут, а затем фильтровали. Смолу промывали с DMF (3×5 мл) и DCM (3×5 мл). Загрузка была определена равной 0,42 ммоль/г на основании процедуры определения загрузки, описанной ниже.
Затем смолу использовали для автоматизированного SPPS на синтезаторе Symphony X от Protein Technologies в масштабе 0,1 ммоль.
Определение загрузки смолы:
Для определения загрузки смолы защитную группу Fmoc отщепляли от определенного количества смолы и затем измеряли концентрацию полученного флуоренильного соединения в растворе супернатанта расщепления с помощью фотометрии при 301 нм. Это значительно коррелирует с количеством аминокислот, загруженных на смолу.
1) 1-3 мг смолы отвешивают в пробирку Эппендорфа объемом 2 мл или аналогичную (обратите внимание на точное количество).
2) Добавляют 1000 мкл раствора 20% пиперидин в DMF.
3) Смесь перемешивают в течение 30 минут, чтобы расщепить группу FMOC.
4) 100 мкл раствора супернатанта затем переносят в кювету из кварцевого стекла и разбавляют 900 мкл 20% пиперидина/DMF.
5) Во второй кювете готовят холостой образец 1000 мкл пиперидин/DMF.
6) После определения холостой пробы на эталонном образце затем измеряется экстинкция испытуемого образца при λ=301 нм на фотометре UV-Vis (Thermo Scientific Evolution 201).
7) Для большей точности можно изготовить несколько тестовых образцов (обычно два); затем для расчета используется среднее арифметическое измерений.
Вычисление загрузки смолы:
Загрузка смолы L301 в ммоль/г рассчитывается по следующей формуле:
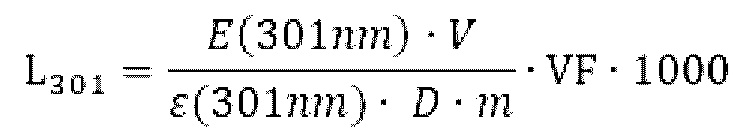
Е=экстинкция
ε=коэффициент экстинкции на длине волны 301 нм (7800 л/моль*см)
m=количество использованной смолы (г)
V=объем образца (л)
D=толщина слоя кюветы (см)
VF=коэффициент разбавления (=10)
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание последующих аминокислот Fmoc происходило, как описано в Способе А.
В ходе синтеза были приготовлены и использованы следующие растворы:
1) Fmoc аминокислоты: 0.2 М (8 экв.)
2) Активатор 1: 0.5 М DIC в DMF (7,5 экв.)
3) Активатор 2: 0.5 М Oxyma в DMF (7,5 экв.)
4) Удаление защитной группы Fmoc: 20% Пиперидин в DMF
5) Уксусный ангидрид (10 экв.) в 2 мл DMF
Двойное связывание обычно проводили для каждой аминокислоты. Для дорогостоящих не встречающихся в природе аминокислот Fmoc или Вос, аминокислот Fmoc собственного синтеза или N-метилированной аминокислоты последовательность прерывалась, и эту аминокислоту связывали вручную (двойное связывание, но обычно с меньшим количеством реагента (3-5 экв.). После того, как это связывание было завершено, синтез обычно продолжали на синтезаторе. Если N-метиламинокислоту добавляли в последовательность, обычно следующую аминокислоту также связывали вручную. Все стадии выполняли при температуре окружающей среды в атмосфере азота. Fmoc-Pen и Fmoc-Oic обычно связывали с использованием автоматизированного SPPS. Fmoc(N-Me)G обычно связывали вручную. Ahx всегда связывали вручную.
Fmoc расщепление:
Смолу подвергали набуханию и промывали DMF (3×3 мл, 10 мин). Если смола содержала Fmoc, затем Fmoc удаляли с 20% раствора пиперидина (2x3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Ile (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Ile (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Pen(Trt) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Pen(Trt) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Ile (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Ile (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Oic (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Oic (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Pro (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Pro (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Leu (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Leu (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Ser(t-Bu) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Ser(t-Bu) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Arg(Pbf) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Arg(Pbf) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Ser(t-Bu) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Ser(t-Bu) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Cys(Trt) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Cys(Trt) (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Связывание:
Fmoc-Ile (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-Ile (4.0 мл) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание продолжалось с барботированием азота в течение 2 часов. Раствор сливали и промывали DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Ручное связывание в ходе автоматизированного SPPS:
Fmoc-(N-Me)G (0.2 М в DMF, 5 экв.) добавляли в смолу. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание протекало при встряхивании (Thermomixer, rt) в течение 2 часов. Раствор фильтровали и промывали с DMF (1×3 мл, 30 с). Стадию связывания повторяли. Fmoc-(N-Me)G (0.2 М в DMF, 5 экв.) добавляли. Раствор активатора 1 (DIC, 1.5 мл) и раствор активатора 2 (Oxyma, 1.5 мл) добавляли и связывание протекало при встряхивании (Thermomixer, rt) в течение 2 часов. Раствор фильтровали и промывали с DMF (6×3 мл, 30 с).
Fmoc расщепление:
Защитную группу Fmoc удаляли посредством добавления 20% раствора пиперидина (2×3 мл, 10 мин). Смолу промывали с DMF (6×3 мл, 30 с).
Если в последовательности присутствовали дополнительные аминокислоты, их связывали с использованием описанных выше стадий.
Тестовое расщепление:
Когда связывания выполнялись вручную, обычно выполнялось тестовое расщепление для контрольных реакций. Тестовым коктейлем для расщепления был TFA/EDT/тиоанизол (90:3:7); 1,5 ч встряхивания на термосмесителе при температуре окружающей среды и 750 об/мин. Анализ, проведенный методом LC-MS с использованием одного из описанных выше способов.
Полное расщепление:
Смолу, содержащую пептид, помещали в шприц и добавляли к смоле 3,0 мл расщепляющего буфера TFA/EDT/тиоанизол (90:3:7). Смесь встряхивали при комнатной температуре в течение 2,5 часов. Раствор собирали посредством фильтрации, и пептид осаждали добавлением холодного диэтилового эфира (-20°С). Раствор центрифугировали (3000 об/мин) в пробирке Falcon (60 мл) в атмосфере азота. Эфир декантировали и твердый остаток многократно промывали холодным диэтиловым эфиром (5×10 мл). Твердый остаток затем сушили
Дисульфидная циклизация:
Неочищенный пептид растворяли в 0,1 М бикарбоната аммония (рН 7,8-8,2) при концентрации 1 мг/2 мл. Раствор встряхивали на орбитальном шейкере всю ночь в круглодонной колбе, открытой на воздух. Раствор затем лиофилизировали с получением белого порошка.
Скрининг колонок для очистки ВЭЖХ:
Пептид растворяли в 5% CH3CN и 95% воды. Скрининг колонок проводили на каждом пептиде, чтобы определить, какой препаративный ВЭЖХ способ использовать для очистки. Были проверены следующие аналитические колонки.
Два способа доступны для скрининга колонок:
1) 5-60% ACN_8 Мин_1 мл/мин_25°С
2) 30-85% ACN_8 Мин_1 мл/мин_25°С
Доступные колонки (50 мм×ID 4,6 мм) (доступны так же как препаративные колонки):
1) Aeris С18 (Phenomenex)
2) X-Bridge C18 (Waters)
3) Kinetex С18 (Coreshell Material) (Phenomenex)
4) YMC Triart C18 (возможна элюция 100% водой)
5) Kinetix Biphenyl (Phenomenex)
6) X-Select C18 (положительный заряд)
7) Jupiter Proteo C18 (Phenomenex)
8) Luna C18 (Phenomenex)
9) Для пептидов согласно настоящему изобретению применяли одну из следующих 5 препаративных колонок:
1) Колонка: Phenomenex, Aeris Peptide 5 мкм ХВ-С18, AXIA Packed, 21,2×250 мм+Картридж 5 мкм
2) Колонка: Phenomenex, Kinetex С18 5 мкм 21,5×250 мм+Картридж 5 мкм
3) Колонка: Phenomenex, Kinetex 5 мкм Biphenyl 100A, AXIA Packed, 21,2×250 мм+Картридж 5 мкм
4) Колонка: YMC Actus Triart Prep. С18 12 нм, S-10 мкм 250×20 мм+Картридж 3 мкм (10×4 мм)
5) Колонка: Waters, Xbridge Prep. С18 5 мкм OBD 19×250 мм+Картридж 10 мкм
Сразу после выбора колонки применяли один из следующих способов:
1) способ: Градиент 5-60% ACN в воде (0,10% TFA)
2) способ: Градиент 30-85% ACN в воде (0,10% TFA)
3) сфокусированный градиент по результатам скрининга колонок.
Скорость потока 20 мл/мин
Объединенные фракции анализировали с помощью ВЭЖХ (5-95 за 8 мин, Chromolith SpeedROD & YMC C18 5-95 за 18 мин) и с использованием одного из способов LC-MS, описанных выше.
Для примера 164 неочищенный пептид растворяли в CH3CN/вода (1:1) и очищали на колонке Phenomenex Kinetex C18 5 мкм, 21,5×250 мм+5 мкм картридж, Скорость потока: 20 мл/мин, Способ: 5-60% ACN в воде (каждый из которых содержит 0,10% TFA) в течение 48 мин. Объединенные фракции лиофилизировали с получением 29,8 мг примера 164 (чистота 98,3%).

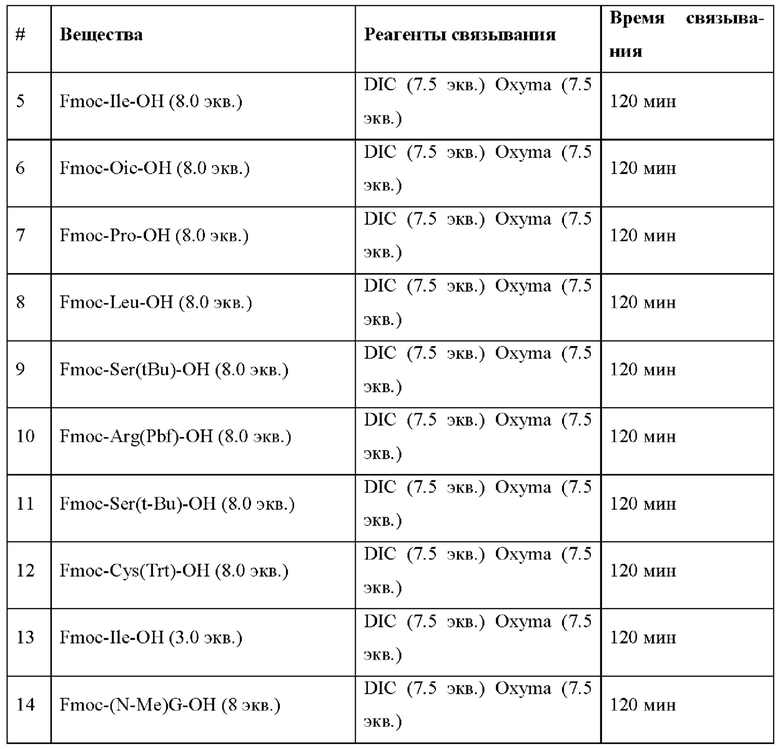
Общий способ ручного синтеза SPPS пептидов Masp (Способ С)
Синтез последовательности AIC+SRSLP-(Oic)-I-(Pen)+IPN-OH (пример 418) является иллюстративным.
Пептидный синтез:
Пептид синтезировали с применением стандартной химии Fmoc.
1) Приготовление смолы: К хлортритиловой смоле (СТС Resin) (0,3 ммоль, 0,3 г, 1,0 ммоль/г) добавляли Fmoc-Asn(Trt)-OH (0,3 ммоль, 0,18 г, 1,0 экв.) и DIEA (0,21 мл)., 1,2 ммоль, 4,0 экв.) в DCM (6,0 мл). Смесь перемешивали с N2 в течение 2 ч при 20°С, затем добавляли МеОН (0,3 мл) и перемешивали с N2 еще 30 мин. Смолу промывали с DMF (6,0 мл×3). Затем добавляли 20% пиперидин в DMF (6,0 мл) и смесь перемешивали с N2 в течение 20 мин при 20°С. Затем смесь фильтровали, чтобы получить смолу. Смолу промывали с DMF (6,0 мл×3) и фильтровали с получением смолы.
2) Связывание: Fmoc-Pro-OH (0.30 г, 0.9 ммоль, 3.0 экв.) HBTU (0.32 г, 0.85 ммоль, 2.85 экв.) и DIEA (1.80 ммоль, 0.32 мл, 6.00 экв.) в DMF (3.0 мл) добавляли в смолу и перемешивали с N2 в течение 30 мин при 20°С. Смолу затем промывали с DMF (6.0 мл×5).
3) Удаление защитной группы: 20% Пиперидин в DMF (6.0 мл) добавляли в смолу и смесь перемешивали с N2 в течение 20 мин при 20°С.
4) Повторение стадий 2-3 для всех других аминокислот:
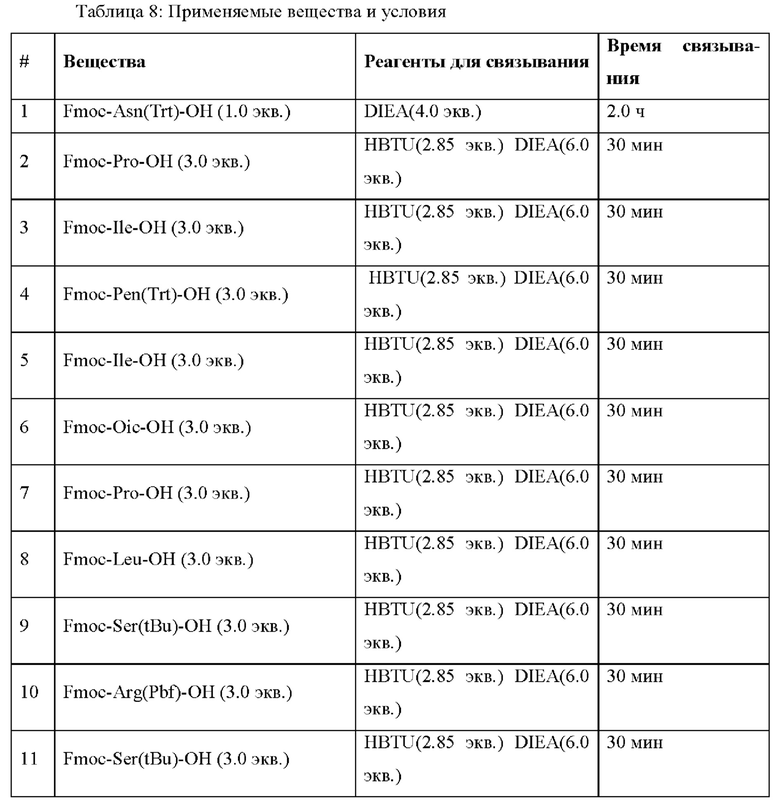
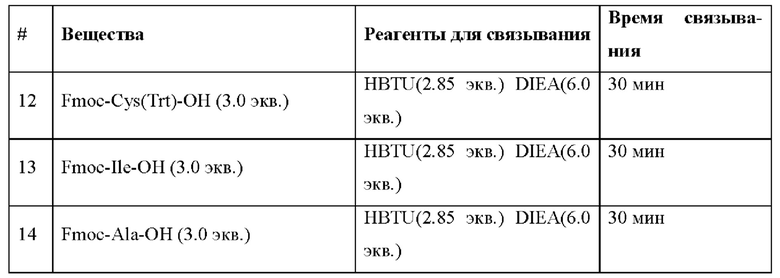
20% Пиперидин в DMF применяли для удаления защитной группы Fmoc в течение 30 мин. Реакцию связывания контролировали нингидриновым (все аминокислоты, кроме Pro) и хлораниловым тестом (Pro), и смолу промывали с DMF (5,0 мл) 5 раз.
Расщепление и очистка пептида:
1) Смолу промывали с МеОН (6.0 мл×5) и сушили под вакуумом с получением 0.62 г пептидной смолы. Затем 6.0 мл расщепляющего буфера (92.5%TFA/2.5%EDT/2.5%TIS/2.5%H2O) добавляли в колбу, содержащую смолу с пептидом с защищенной боковой цепью, при 20°С и смесь перемешивали в течение 2.5 ч.
2) Пептид осаждали холодным трет-бутилметиловым эфиром (50 мл) и центрифугировали (3 мин при 3000 об/мин) с получением твердого неочищенного вещества. Неочищенный пептидный осадок промывают трет-бутилметиловым эфиром в течение еще двух раз (20,0 мл×3). Неочищенный пептид сушили над вакуумом в течение 2,0 ч с получением 0,4 г неочищенного пептида.
3) Неочищенное вещество (0,4 г) растворяли в CH3CN (150 мл) и воде (150 мл). Йод (I2) (0,1 М в МеОН, 1,5 мл) добавляли при 20°С до тех пор, пока не сохранялась желтая окраска. Затем смесь перемешивали при 20°С в течение 2 мин. Затем тиосульфат натрия (0,1 М в воде, 0,05 мл) добавляли по каплям до исчезновения желтой окраски. Смесь лиофилизировали с получением неочищенного порошка (0.41 г)
4) Неочищенный пептид очищали посредством препаративной ВЭЖХ (условия: А: 0.075% TFA в воде В: CH3CN) и лиофилизировали с получением желаемого пептида (пример 418) (108.1 мг, 23.0% выход, 97.3% чистота согласно Способу W1; 97.4% чистота согласно Способу 7) в виде твердого вещества белого цвета и TFA соль. Условия очистки: пептид растворяли в TFA/H2O (7:3); скорость потока 20 мл/мин; градиент 12-42% за 60 мин; время удерживания=42 мин; очистка на колонке Luna 25×200 мм, С18 10 мкм, 110  затем снова на колонке Gemini 150×30 мм, С18 5 мкм, 110 с достижением желаемой чистоты.
затем снова на колонке Gemini 150×30 мм, С18 5 мкм, 110 с достижением желаемой чистоты.
Если С-концевые амиды получали этим способом, то синтез начинали с амидной смолы Ринка МВНА (загрузка обычно около 0,4-0,6 ммоль/г), при этом указанные выше стадии были такими же.
Общий способ ручного SPPS пептидов Masp (Способ D)
Если получали С-концевые амиды, синтез начинали со с амидной смолы Ринка МВНА (загрузка обычно около 0,4-0,6 ммоль/г). Синтез последовательности PIC+SRS-((tBu)A)-PPI-(Pen)+IPD-NH2 (пример 9) является иллюстративным.
В случае получения С-концевых кислот синтез начинали с 2-хлортритиловой смолы, при этом первую Fmoc-аминокислоту добавляли по способу, описанному в Способе С.
Синтез пептида:
Пептид синтезировали с использованием стандартной химии Fmoc.
1) Получение смолы: Амидную смолу Ринка МВНА (0.66 г, 0.30 ммоль, загрузка=0.45 ммоль/г) в DMF (10.0 мл) встряхивали с N2 в течение 0.5 ч при 20°С. Затем 20% Пиперидин в DMF (10 мл) добавляли и смесь встряхивали с N2 в течение 20 мин при 20°С. Затем смесь фильтровали с получением смолы. Смолу промывали с DMF (10×5 мл) и фильтровали с получением смолы.
2) Связывание: Fmoc-Asn(Trt)-OH (0.9 ммоль, 0.37 г, 3.0 экв.) HBTU (0.32 г, 0.85 ммоль, 2.95 экв.) и DIEA (1.80 ммоль, 0.32 мл, 6.00 экв.) в DMF (3.0 мл) добавляли в смолу и встряхивали с N2 в течение 30 мин при 20°С. Смолу затем промывали с DMF (10.0 мл×3).
3) Удаление защитной группы: 20% Пиперидин в DMF (10.0 мл) добавляли в смолу и смесь встряхивали с N2 в течение 20 мин при 20°С.
4) Повторяли стадии 2-3 для всех других аминокислот.
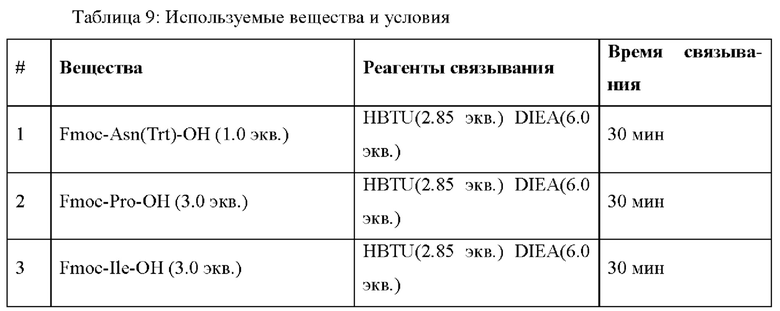
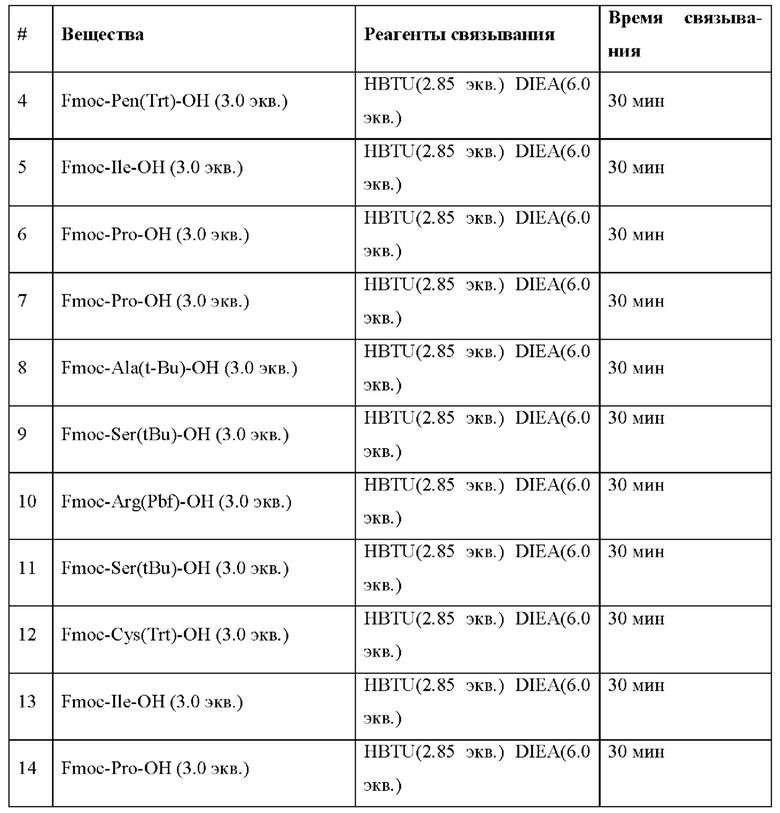
20% Пиперидин в DMF применяли для удаления защитной группы Fmoc в течение 30 мин. Реакцию связывания контролировали нингидриновым (все аминокислоты, кроме Pro) и хлораниловым тестом (Pro), и смолу промывали с DMF (5,0 мл) 5 раз.
Расщепление и очистка пептида:
1) Смолу промывали с МеОН (6.0 мл×5) и сушили под вакуумом с получением 0.66 г пептидной смолы. Затем 6.0 мл расщепляющего буфера (90%TFA/5%EDT/2.5%TIS/2.5%H2O) добавляли в колбу, содержащую смолу с пептидом с защищенной боковой цепью, при 20°С и смесь перемешивали в течение 2.5 ч.
2) Пептид осаждали холодным трет-бутилметиловым эфиром (50 мл) и центрифугировали (3 мин при 3000 об/мин) с получением твердого неочищенного вещества. Неочищенный пептидный осадок промывают трет-бутилметиловым эфиром в течение еще двух раз (20,0 мл×3). Неочищенный пептид сушили над вакуумом в течение 2,0 ч с получением 0,4 г неочищенного пептида.
3) Неочищенное вещество (0,4 г) растворяли в CH3CN (15 мл) и воде (15 мл) до достижения концентрации 0.1 мм. Раствор NH4HCO3 (1 М) добавляли для доведения рН до около 8-9. Раствор встряхивали при комнатной температуре в течение около 8 часов. Реакцию контролировали посредством ВЭЖХ. После того, как реакция была завершена, реакцию гасили уксусной кислотой, чтобы довести рН примерно до 6. Затем реакционную смесь лиофилизировали, а полученное твердое вещество очистили с помощью обращенно-фазовой ВЭЖХ.
4) Очищали неочищенный пептид посредством препаративной ВЭЖХ (условия: А: 0.075% TFA в воде В: CH3CN) и лиофилизировали с получением желаемого пептида (пример 9) (202.40 мг, 41.4.0% выход, 93.7% чистота согласно Способу W1; 95.1% чистота согласно Способу 7) в виде твердого вещества белого цвета и TFA соли. Условия очистки: пептид растворяли в TFA/H2O (7:3); скорость потока 20 мл/мин; градиент 12-42% за 60 мин; время удерживания=42 мин; очистка на колонке Luna 25×200 мм, С18 10 мкм, 110  колонка.
колонка.
Альтернативный общий способ ручного SPPS пептидов Masp (Способ Е)
Синтез последовательности ((2S)-2[(амино)-2-(тетрагидро-2Н-пиран-4-ил)]уксусная кислота)-IC+SRSLP-(Oic)-IC+I-OH (пример 184) является иллюстративным.
Пептидный синтез:
Пептид синтезировали с использованием стандартной химии Fmoc.
Темный шар в химических структурах ниже указывает на твердую полимерную подложку, используемую для твердофазного синтеза пептидов (SPPS), например, 2-хлортритиловая смола, амидная смола Ринка и т.д.
Пример 1А
(2S)-({[(9Н-флуорен-9-ил)метокси]карбонил}амино)(оксан-4-ил)уксусная кислота (Единственный стереоизомер)
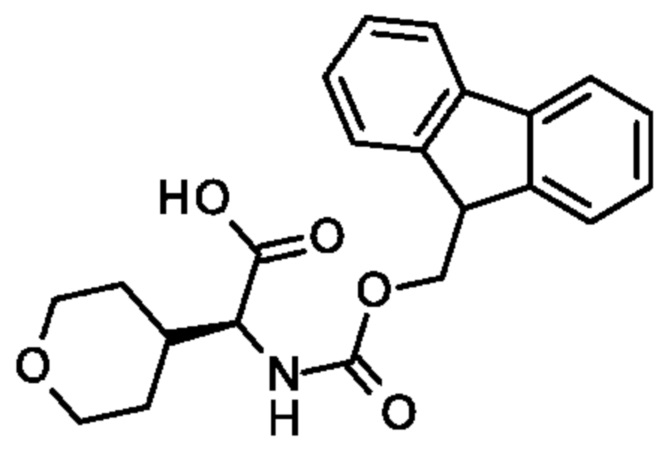
К (2S)-амино(оксан-4-ил)уксусной кислоте (950 мг, 5.97 ммоль) в ацетоне/воде (15 мл/10 мл) добавляли бикарбонат натрия (5.01 г, 59.7 ммоль) и 1-({[(9Н-флуорен-9-ил)метокси]карбонил}окси)пирролидин-2,5-дион (2.11 г, 6.27 ммоль). Смесь перемешивали в течение выходных при комнатной температуре. Суспензию обрабатывали водой и экстрагировали с МВТЕ еще два раза. Водную фазу подкисляли 1 М водной соляной кислотой и экстрагировали дихлорметаном еще три раза. Органическую фазу промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и выпаривали. Остаток очищали посредством хроматографии на силикагеле (Isolera; 50 г SNAP Ultra; градиент: циклогексан 50%/этилацетат 50%+1% уксусной кислоты до циклогексан 20%/этилацетат 80%+1% уксусной кислоты). Соответствующие фракции объединяли и выпаривали. Остаток растворяли в толуоле и выпаривали еще два раза, затем остаток сушили под вакуумом с получением 1.08 г (100% чистота, 47% выход) целевого соединения.
LC-MS (Способ 9): Rt=0.90 мин; MS (ESIpos): m/z=382 [М+Н]+
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: -0.150 (0.43), -0.008 (4.33), 0.008 (3.74), 0.146 (0.43), 1.280 (1.03), 1.300 (2.26), 1.309 (2.36), 1.331 (2.75), 1.341 (2.92), 1.371 (2.77), 1.401 (2.54), 1.411 (2.32), 1.430 (1.49), 1.464 (6.34), 1.496 (3.49), 1.932 (1.76), 1.942 (2.14), 1.951 (1.93), 1.960 (2.08), 2.327 (0.64), 2.366 (0.64), 2.670 (0.64), 2.710 (0.65), 3.169 (2.11), 3.212 (2.67), 3.237 (6.32), 3.265 (6.90), 3.704 (0.49), 3.715 (0.49), 3.840 (4.66), 3.854 (5.62), 3.867 (4.30), 3.879 (6.28), 3.899 (5.14), 3.918 (3.56), 4.201 (2.11), 4.218 (5.83), 4.237 (9.07), 4.263 (8.42), 4.274 (9.80), 4.293 (5.14), 4.318 (1.05), 7.309 (6.35), 7.328 (15.12), 7.347 (9.86), 7.400 (9.65), 7.419 (15.88), 7.437 (7.02), 7.652 (5.77), 7.674 (5.52), 7.735 (8.21), 7.740 (8.28), 7.754 (7.63), 7.883 (16.00), 7.902 (14.80), 12.609 (0.56).
Пример 139А
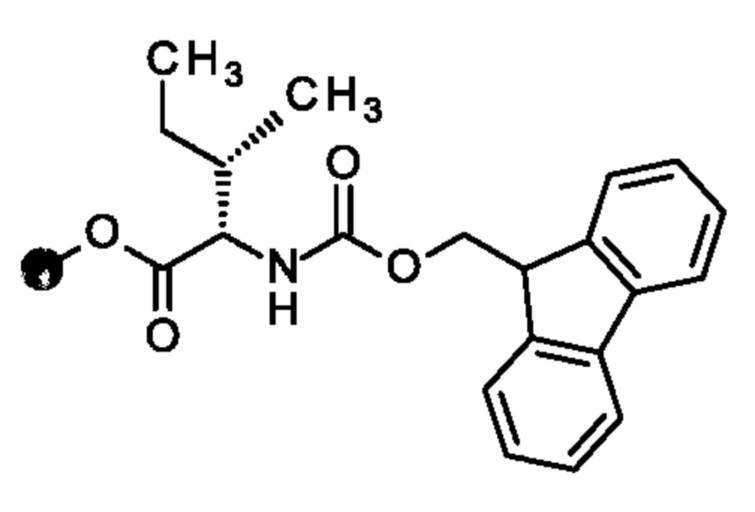
Реакцию проводили в атмосфере аргона. К N-{[(9Н-флуорен-9-ил)метокси]карбонил}-L-изолейцину (9.40 г, 26.6 ммоль) в 53 мл дихлорметана добавляли N,N-диизопропилэтиламин (19 мл, 110 ммоль) и 2-хлортритилхлоридную смолу (10.0 г, 13.3 ммоль). Реакционную смесь встряхивали всю ночь при комнатной температуре. Осадок собирали посредством фильтрации и промывали с DMF три раза. Собранную смолу встряхивали с дихлорметаном/метанолом 1:1 в течение 30 мин. Смолу собирали посредством фильтрации и промывали три раза при вращении метанолом и дихлорметан. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 3А
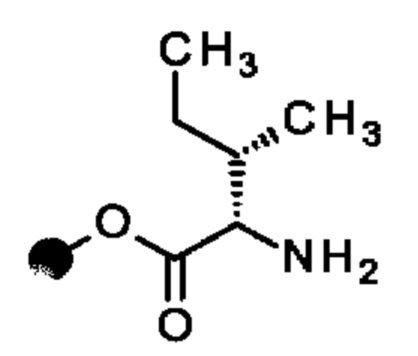
Пример 2А (15.2 г, 6.82 ммоль) в 200 мл DMF/Пиперидина 4:1 встряхивали в течение 15 мин при комнатной температуре. Смолу собирали посредством фильтрации и промывали с DMF три раза. Обе стадии повторяли и промывали три раза при вращении с 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 140А
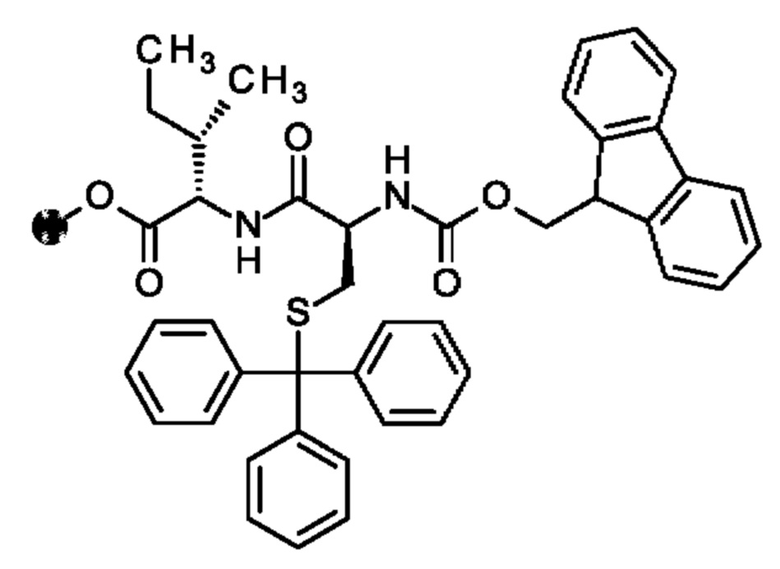
Пример 3А (11.4 г, 5.13 ммоль) разбухал в 100 мл DMF при комнатной температуре в течение 5 мин. Смесь N-{[(9Н-флуорен-9-ил)метокси]карбонил}-S-(трифенилметил)-L-цистеина (6.01 г, 10.3 ммоль) в 50 мл DMF, DIC (1.5 мл, 10 ммоль) и Oxyma (1.42 г, 10.0 ммоль) добавляли и реакционную смесь встряхивали в течение 2 ч при комнатной температуре. Смолу собирали посредством фильтрации и промывали с DMF три раза. Стадию связывания повторяли всю ночь. Смолу собирали посредством фильтрации и промывали с 150 мл DMF три раза, а затем еще три раза при вращении с 150 мл метанола и 150 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 5А
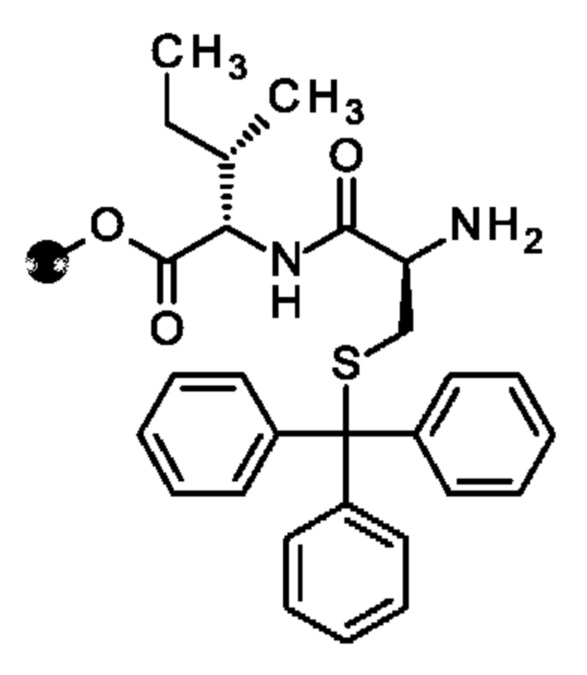
Пример 4А (18.9 г, 8.51 ммоль) в 200 мл DMF/Пиперидина 4:1 встряхивали в течение 15 мин при комнатной температуре. Смолу собирали посредством фильтрации и промывали с DMF три раза. Обе стадии повторяли и собранную смолу промывали три раза при вращении с 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 141А
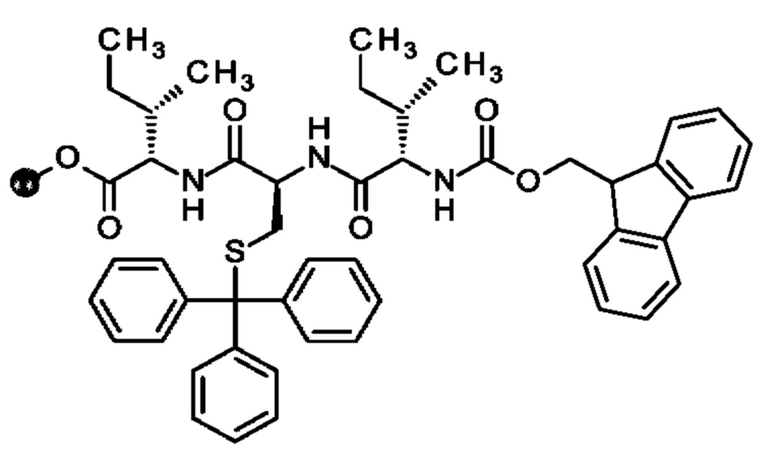
Пример 5А (16.3 г, 7.32 ммоль) разбухал в 150 мл DMF в течение 5 мин при комнатной температуре. Смесь N-{[(9Н-флуорен-9-ил)метокси]карбонил}-L-изолейцина (5.18 г, 14.6 ммоль) в 50 мл DMF, DIC (2.2 мл, 14 ммоль) и Oxyma (2.03 г, 14.3 ммоль) добавляли и реакционную смесь встряхивали в течение 2 ч при комнатной температуре. Смолу собирали посредством фильтрации и промывали с DMF три раза. Стадию связывания повторяли всю ночь. Смолу собирали посредством фильтрации и промывали с 200 мл DMF три раза, а затем еще три раза при вращении с 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 7А
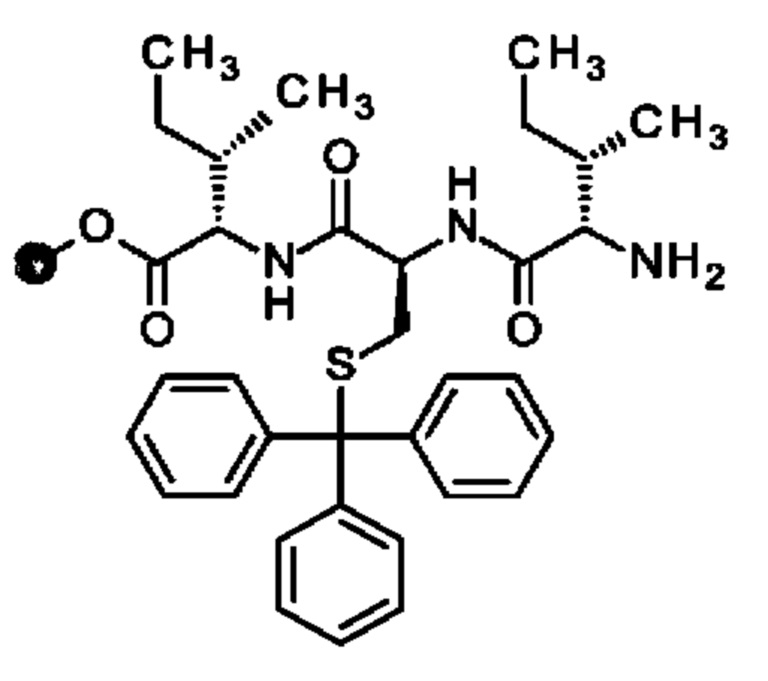
Пример 6А (20.0 г, 8.98 ммоль) в 200 мл DMF/Пиперидина 4:1 встряхивали в течение 15 мин при комнатной температуре. Смолу собирали посредством фильтрации и промывали с DMF три раза. Обе стадии повторяли и собранную смолу промывали три раза при вращении с 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 142А
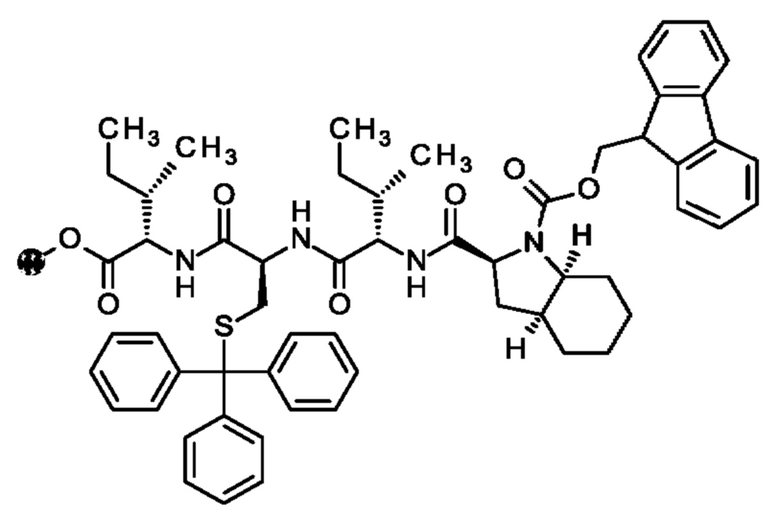
Пример 7А (17.8 г, 7.99 ммоль) разбухал в 150 мл DMF в течение 5 мин при комнатной температуре. Смесь (2S,3aS,7aS)-1-{[(9H-флуорен-9-ил)метокси]карбонил}октагидро-1Н-индол-2-карбоновой кислоты (6.25 г, 16.0 ммоль) в 50 мл DMF, DIC (2.4 мл, 16 ммоль) и Oxyma (2.21 г, 15.6 ммоль) добавляли и реакционную смесь встряхивали в течение 2 ч при комнатной температуре. Смолу собирали посредством фильтрации и промывали с DMF три раза. Стадию связывания повторяли в течение выходных. Смолу собирали посредством фильтрации и промывали с 200 мл DMF три раза, а затем еще три раза при вращении с 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 9А
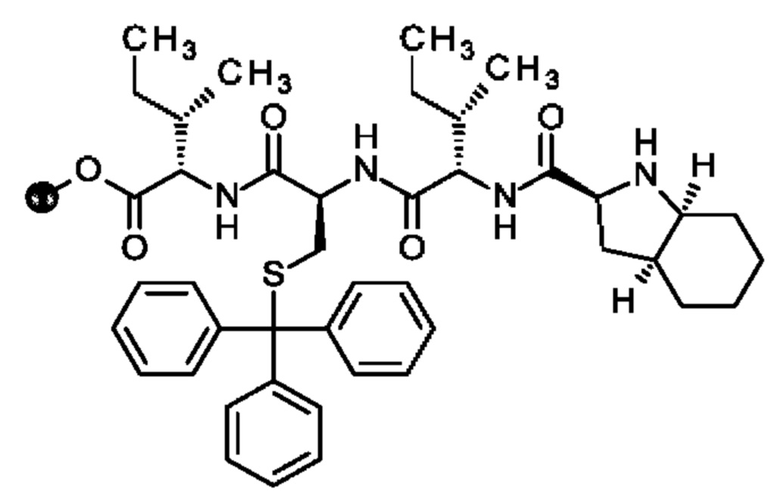
Пример 8А (21.5 г, 9.68 ммоль) в 200 мл DMF/Пиперидина 4:1 встряхивали в течение 30 мин при комнатной температуре. Смолу собирали посредством фильтрации, промывали с DMF три раза, а затем еще три раза при вращении с 200 мл метанола и 200 мл дихлорметана. В собранную смолу добавляли 200 мл DMF/Пиперидина 4:1 и встряхивали в течение 1 ч при комнатной температуре. Смолу собирали посредством фильтрации, промывали с DMF три раза, а затем еще три раза при вращении с 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 10А
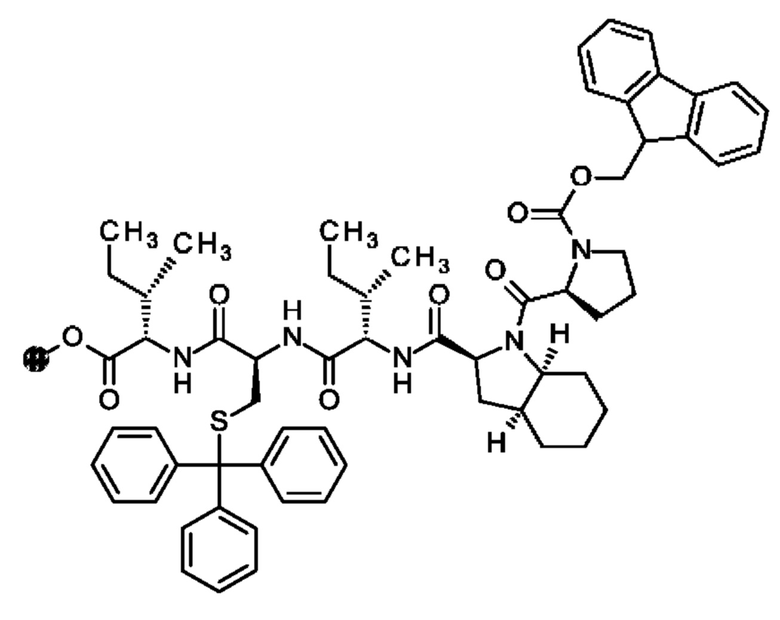
Пример 9А (18.1 г, 8.13 ммоль) разбухал в 150 мл DMF в течение 5 мин при комнатной температуре. Смесь 1-{[(9Н-флуорен-9-ил)метокси]карбонил}-L-пролина (11.0 г, 32.5 ммоль) в 50 мл DMF, DIC (4.9 мл, 32 ммоль) и Oxyma (4.51 г, 31.7 ммоль) добавляли и реакционную смесь встряхивали в течение выходных при комнатной температуре. Смолу собирали посредством фильтрации, промывали с 200 мл DMF три раза, а затем еще три раза при вращении с 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 11А
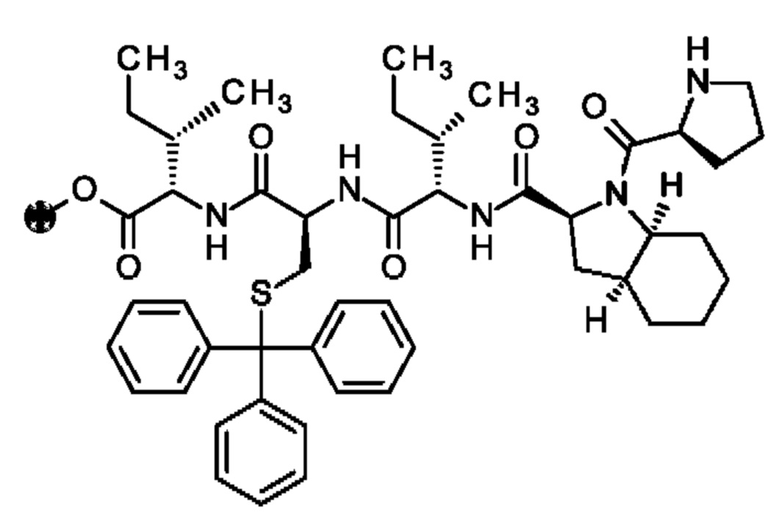
Пример 10А (20.4 г, 9.16 ммоль) в 150 мл DMF/Пиперидина 4:1 встряхивали в течение 30 мин при комнатной температуре. Смолу собирали посредством фильтрации и промывали с DMF три раза. Обе стадии повторяли и собранную смолу промывали три раза при вращении с 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 12А
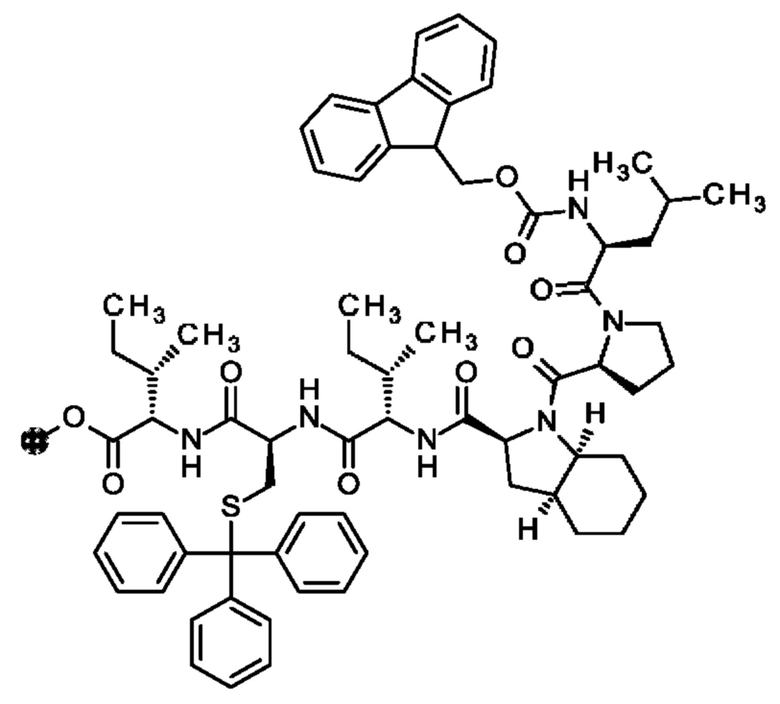
Пример 11А (18.2 г, 8.18 ммоль) разбухал в 150 мл DMF в течение 5 мин при комнатной температуре. Смесь N-{[(9Н-флуорен-9-ил)метокси]карбонил}-L-лейцина (11.6 г, 32.7 ммоль) в 50 мл DMF, DIC (4.9 мл, 32 ммоль) и Oxyma (4.53 г, 31.9 ммоль) добавляли и реакционную смесь встряхивали всю ночь при комнатной температуре. Смолу собирали посредством фильтрации, промывали с 200 мл DMF три раза, а затем еще три раза при вращении с 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 145А
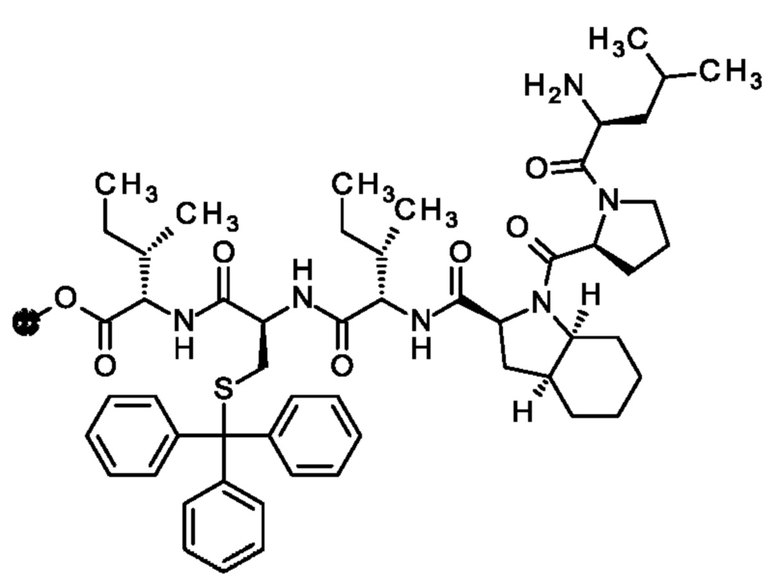
Пример 12А (20.9 г, 9.42 ммоль) в 150 мл DMF/Пиперидина 4:1 встряхивали в течение 30 мин при комнатной температуре. Смолу собирали посредством фильтрации и промывали с DMF три раза. Обе стадии повторяли и собранную смолу промывали три раза при вращении с 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 14А
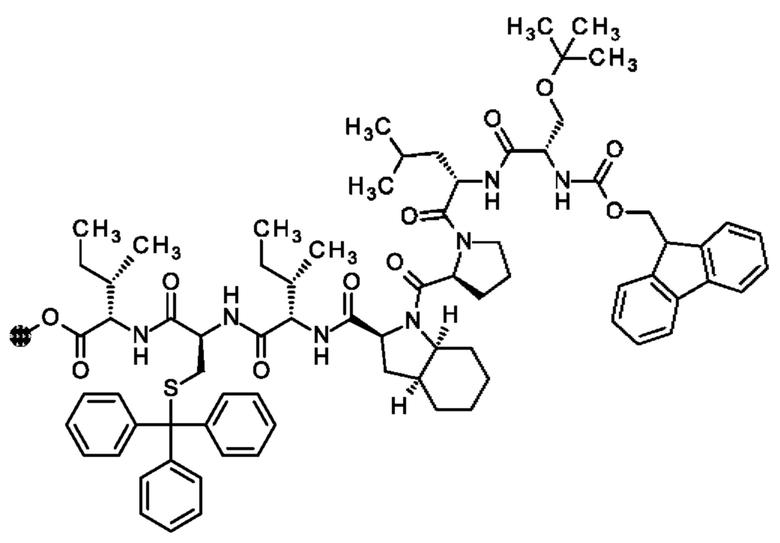
Пример 13А (18.0 г, 8.11 ммоль) разбухал в 150 мл DMF в течение 5 мин при комнатной температуре. Смесь O-трет-бутил-N-{[(9Н-флуорен-9-ил)метокси]карбонил}-L-серина (12.4 г, 32.4 ммоль) в 50 мл DMF, DIC (4.9 мл, 32 ммоль) и Oxyma (4.49 г, 31.6 ммоль) добавляли и реакционную смесь встряхивали всю ночь при комнатной температуре. Смолу собирали посредством фильтрации, промывали с 200 мл DMF три раза, а затем еще три раза при вращении с 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 15А
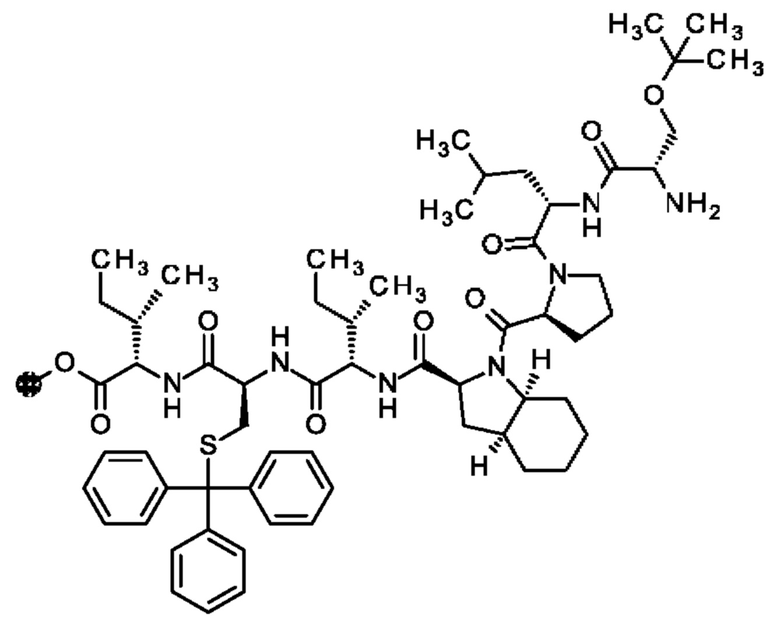
Пример 14А (21.4 г, 9.64 ммоль) в 200 мл DMF/Пиперидина 4:1 встряхивали в течение 30 мин при комнатной температуре. Смолу собирали посредством фильтрации и промывали с DMF три раза. Обе стадии повторяли и собранную смолу промывали три раза при вращении с 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 147А
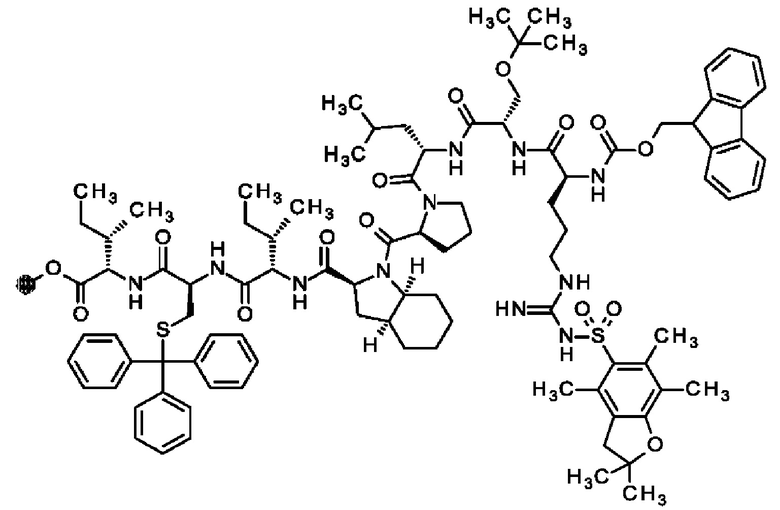
Пример 15А (18.9 г, 8.51 ммоль) разбухал в 150 мл DMF в течение 5 мин при комнатной температуре. Смесь N2-{[(9Н-флуорен-9-ил)метокси]карбонил}-N5-[N-(2,2,4,6,7-пентаметил-2,3-дигидро-1-бензофуран-5-сульфонил)карбамимидоил]-L-орнитина (22.1 г, 34.0 ммоль) в 50 мл DMF, DIC (5.1 мл, 33 ммоль) и Oxyma (4.72 г, 33.2 ммоль) добавляли и реакционную смесь встряхивали всю ночь при комнатной температуре. Смолу собирали посредством фильтрации, промывали с 200 мл DMF три раза, а затем еще три раза при вращении 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 148А
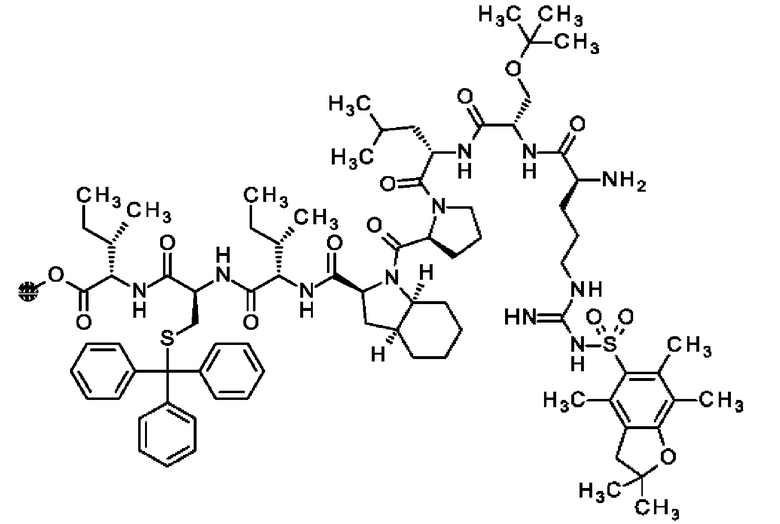
Пример 16А (23.8 г, 10.7 ммоль) в 200 мл DMF/Пиперидина 4:1 встряхивали в течение 30 мин при комнатной температуре. Смолу собирали посредством фильтрации и промывали с DMF три раза. Обе стадии повторяли и собранную смолу промывали три раза при вращении с 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 18А
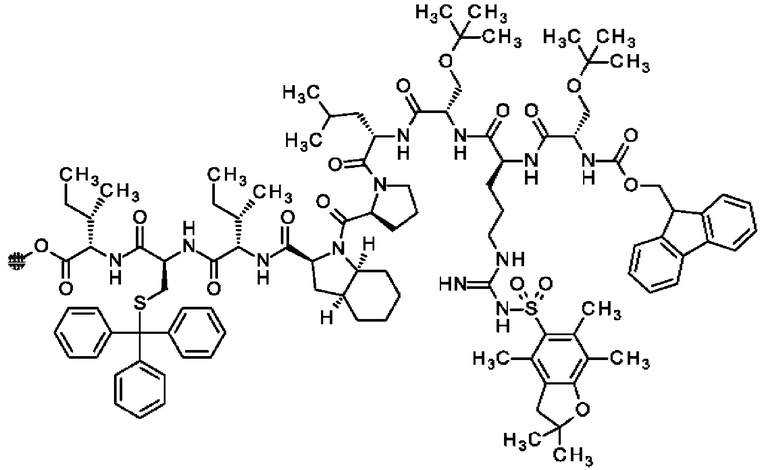
Пример 17А (22.1 г, 9.95 ммоль) разбухал в 150 мл DMF в течение 5 мин при комнатной температуре. Смесь O-трет-бутил-N-{[(9Н-флуорен-9-ил)метокси]карбонил}-L-серина (15.3 г, 39.8 ммоль) в 50 мл DMF, DIC (6.0 мл, 39 ммоль) и Oxyma (5.51 г, 38.8 ммоль) добавляли и реакционную смесь встряхивали всю ночь при комнатной температуре. Смолу собирали посредством фильтрации, промывали с 200 мл DMF три раза, а затем еще три раза при вращении с 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 19А
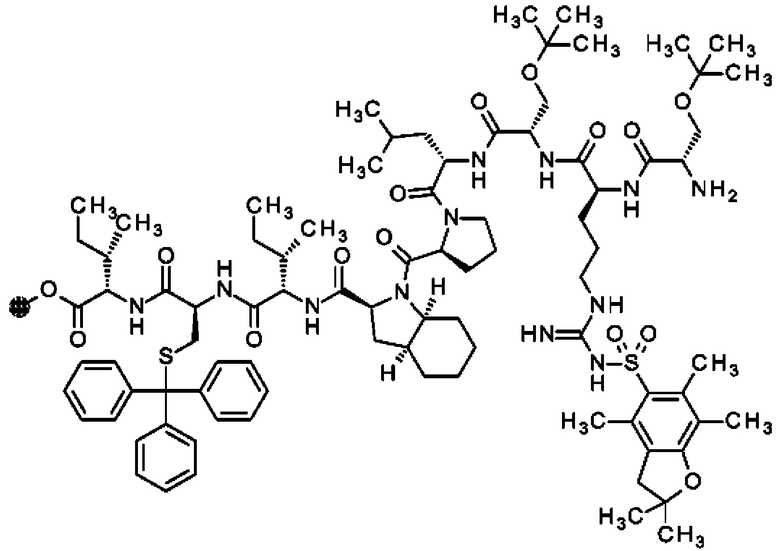
Пример 18А (24.7 г, 11.1 ммоль) в 200 мл DMF/Пиперидина 4:1 встряхивали в течение 30 мин при комнатной температуре. Смолу собирали посредством фильтрации и промывали с DMF три раза. Обе стадии повторяли и собранную смолу промывали три раза при вращении с 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 20А
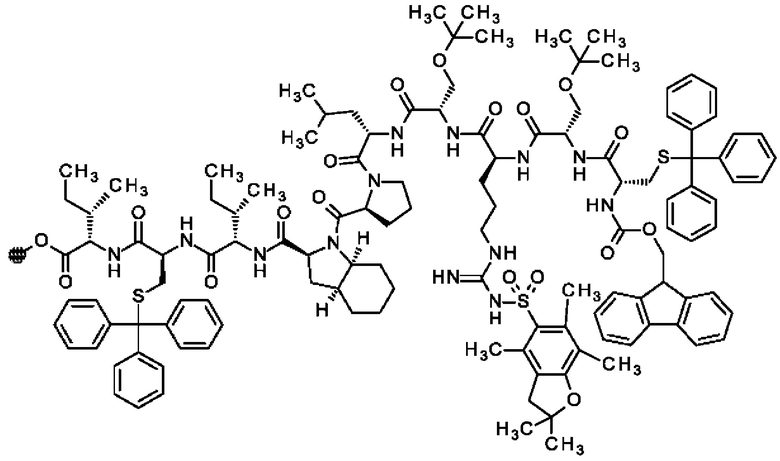
Пример 19А (22.2 г, 9.99 ммоль) разбухал в 150 мл DMF в течение 5 мин при комнатной температуре. Смесь N-{[(9Н-флуорен-9-ил)метокси]карбонил}-S-(трифенилметил)-L-цистеина (23.4 г, 40.0 ммоль) в 50 мл DMF, DIC (6.0 мл, 39 ммоль) и Oxyma (5.54 г, 39.0 ммоль) добавляли и реакционную смесь встряхивали всю ночь при комнатной температуре. Смолу собирали посредством фильтрации, промывали с 200 мл DMF три раза, а затем еще три раза при вращении с 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 21A
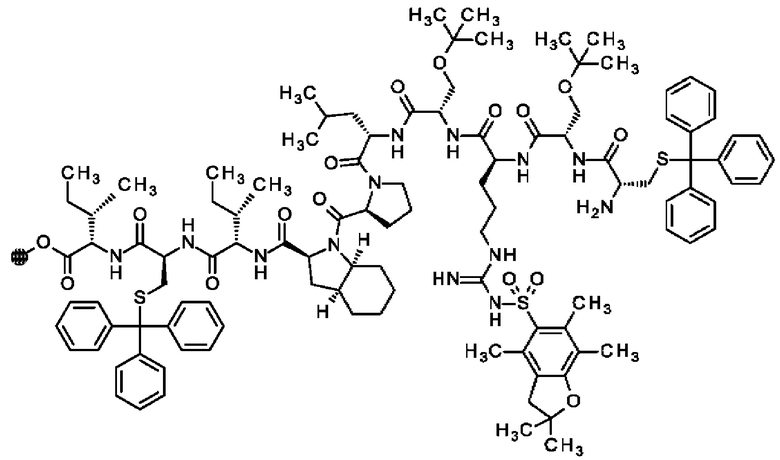
Пример 20А (24.5 г, 11.0 ммоль) в 200 мл DMF/Пиперидина 4:1 встряхивали в течение 30 мин при комнатной температуре. Смолу собирали посредством фильтрации и промывали с DMF три раза. Обе стадии повторяли и собранную смолу промывали три раза при вращении с 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 22А
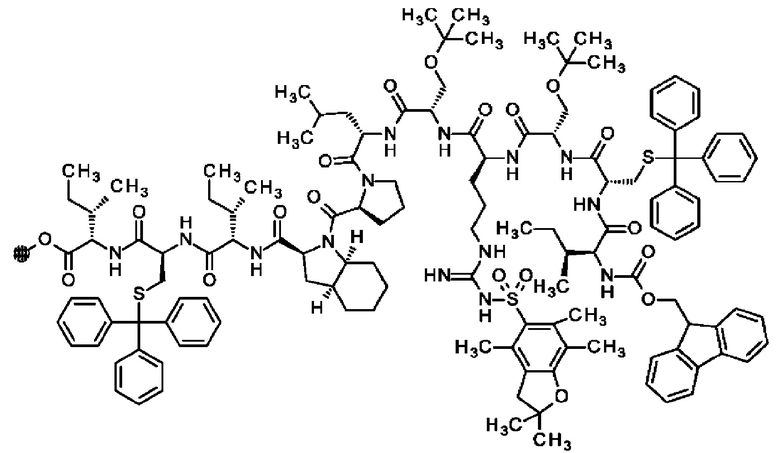
Пример 21А (22.3 г, 10.0 ммоль) разбухал в 150 мл DMF в течение 5 мин при комнатной температуре. Смесь N-{[(9Н-флуорен-9-ил)метокси]карбонил}-L-изолейцина (14.2 г, 40.1 ммоль) в 50 мл DMF, DIC (6.0 мл, 39 ммоль) и Oxyma (5.55 г, 39.0 ммоль) добавляли и реакционную смесь встряхивали в течение выходных при комнатной температуре. Смолу собирали посредством фильтрации, промывали с 200 мл DMF три раза, а затем еще три раза при вращении с 200 мл метанола и 200 мл дихлорметана. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 23А
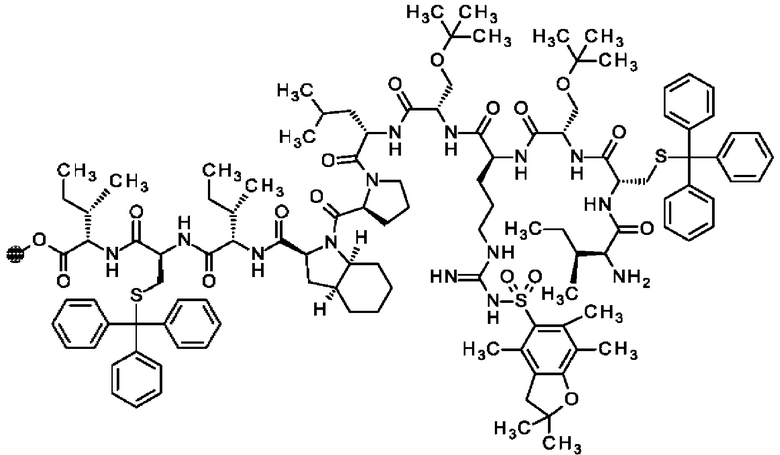
Пример 22А (1.00 г, 250 мкмоль) в 7.5 мл DMF/Пиперидина 4:1 встряхивали в течение 30 мин при комнатной температуре. Смолу собирали посредством фильтрации и промывали с DMF три раза. Обе стадии повторяли и собранную смолу промывали три раза при вращении метанолом и дихлорметаном. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 24А
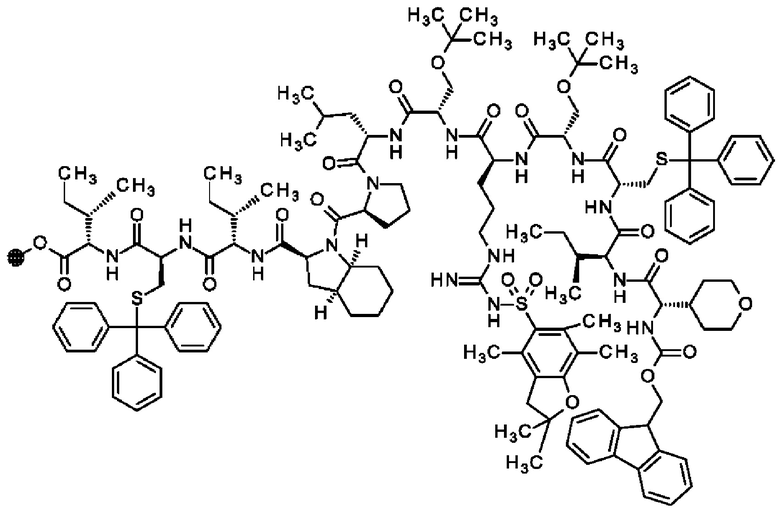
Пример 23А (1.00 г, 250 мкмоль) разбухал в 5 мл DMF в течение 5 мин при комнатной температуре. Смесь (2S)-({[(9Н-флуорен-9-ил)метокси]карбонил}амино)(оксан-4-ил)уксусной кислоты (381 мг, 1.00 ммоль) в 1 мл DMF, DIC (150 мкл, 980 мкмоль) и Oxyma (139 мг, 975 мкмоль) добавляли и реакционную смесь встряхивали в течение выходных при комнатной температуре. Смолу собирали посредством фильтрации, промывали с DMF три раза, а затем еще три раза при вращении метанолом и дихлорметан. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 25А
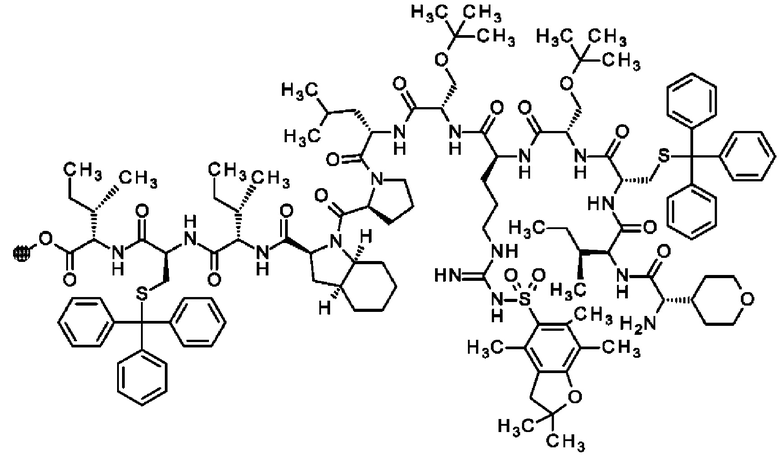
Пример 24А (1.00 г, 250 мкмоль) в 7.5 мл DMF/Пиперидина 4:1 встряхивали в течение 30 мин при комнатной температуре. Смолу собирали посредством фильтрации и промывали с DMF три раза. Обе стадии повторяли и собранную смолу промывали три раза при вращении метанолом и дихлорметаном. Собранную смолу сушили под вакуумом в ходе следующей стадии.
Пример 26А
(2S,3S)-2-{[(2R)-2-{[(2S,3S)-2-{[(2S,3aS,7aS)-1-{(2S)-1-[(2S,5S,8S,11S,14R,17S,20S)-20-амино-17-[(2S)-бутан-2-ил]-8-(3-карбамимидамидопропил)-5,11-бис(гидроксиметил)-2-(2-метилпропил)-20-(оксан-4-ил)-4,7,10,13,16,19-гексаоксо-14-(сульфанилметил)-3,6,9,12,15,18-гексаазаикозанан-1-оил]пирролидин-2-карбонил}октагидро-1Н-индол-2-карбонил]амино}-3-метилпентаноил]амино}-3-сульфанилпропаноил]амино}-3-метилпентановая кислота (Единственный стереоизомер)
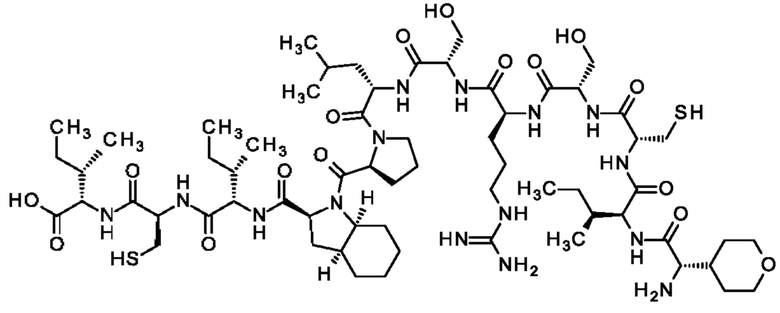
К Примеру 25А (1.00 г, 250 мкмоль) добавляли расщепляющий коктейль TFA/EDT/тиоанизол (90:3:7) и смесь встряхивали в течение 2 ч при комнатной температуре. Фильтрат собирали, и смолу промывали дихлорметаном. Объединенный фильтрат выпаривали. Остаток перемешивали диэтиловым простым эфиром и твердое вещество собирали посредством фильтрации. Твердое вещество промывали несколько раз диэтиловым простым эфиром и сушили под вакуумом в ходе следующей стадии.
Пример 184
((2S)-2[(амино)-2-(тетрагидро-2Н-пиран-4-ил)]уксусная кислота)-IC+SRSLP-(Oic)-IC+I-OH (пример 184)
N-[(6S,9S,12S,15S,18R,23R,26S,28aS,29aS,33aS,35aS)-18-({N-[(2S)-2-амино-2-(оксан-4-ил)ацетил]-L-изолейцил}амино)-26-[(2S)-бутан-2-ил]-12-(3-карбамимидамидопропил)-9,15-бис(гидроксиметил)-6-(2-метилпропил)-5,8,11,14,17,25,28,35-октаоксодотриаконтагидро-1Н,5Н,22Н-пирроло[2',1':13,14][1,2,5,8,11,14,17,20,23,26]дитиаоктаазациклононакозино[11,10-а]индол-23-карбонил]-L-изолейцин (Единственный стереоизомер)
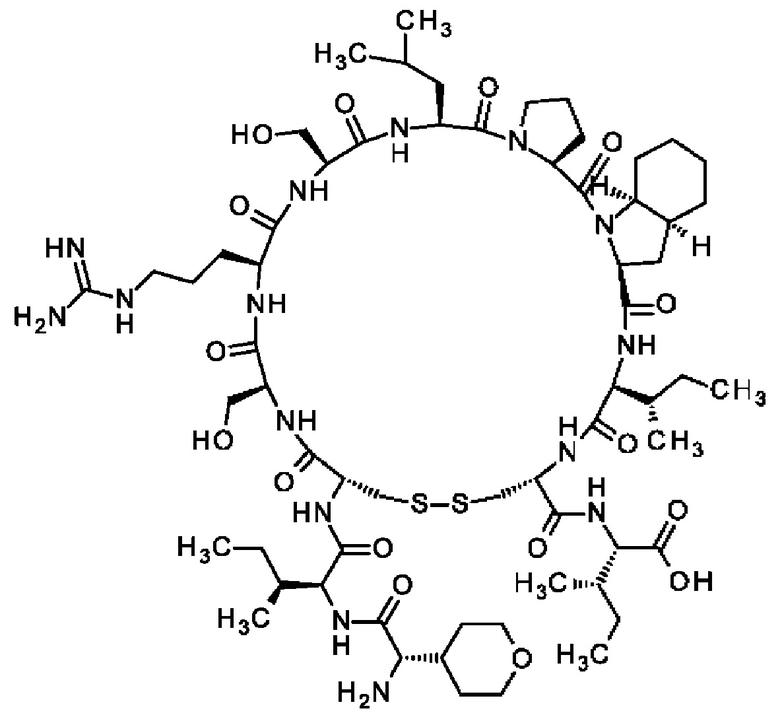
К Примеру 26А (425 мг, 304 мкмоль) добавляли 850 мл 0.1 М водного раствора бикарбоната аммония (рН 7.86). Воздух барбатировали через реакционную смесь в течение 5 мин. Реакционную смесь перемешивали в открытой колбе всю ночь при комнатной температуре. Растворители лиофилизировали с получением 580 мг твердого вещества белого цвета. Очистка с применением обращенно-фазовой ВЭЖХ, как описано выше (Способ В), дала три фракции 7.4 мг (>99%), 24.5 мг (98%) и 15 мг (95%) целевого Примера 184.
Общий способ F: Превращение TFA солей пептида в соли HCl
Синтез AIC+SRS-((tBu)A)-PPI-((N-Me)C)+IPD-NH2 (HCl Соль) (пример 30) является иллюстративным.
Процедура автоматической станции ионного обмена:
Перистальтический насос фирмы Hirschmann (Rotarus объем 50), Трубки: Tygon 2001 (ID 0,64 мм) Настройки:
Промывка Н2О: время работы 1200 с; 80 мин-1; 1 цикл (объем 35 мл)
Циркуляция образца с пептидом: время работы 1200 с; 80 мин-1; 1 цикл (объем 70 мл)
Промывка Н2О (или %ACN в Н2О: время работы 1200 с; 80 мин-1; 1 цикл (объем 35 мл)
Amberlite IRA 410 (форма HCl) применяли. 1500 мг смолы помещали в 2 картриджа фильтра и промывали деионизированной водой (10 раз).
Пептид, растворенный в 3 мл 5% раствора ACN/ H2O, загружали на колонку и циклически пропускали через колонку 10 раз. Колонку промывали водой, раствор собирали в пробирку Фалькона и лиофилизировали.
Получали 72,83 мг желаемого пептида в виде соли НС1: LC-MS (>99%); Ионно-хроматографический анализ: 3,7 мас. % Cl- (1,57 экв. Cl-), <1 мас. % TFA.
Процесс ионного обмена также может быть выполнен с использованием следующего протокола:
Смола Amberlite IRA 410 (форма HCl) (1-2 г) была помещена в 10 мл фритт-шприц (на 100 мг пептида требуется 1 г смолы IRA 410).
1) Смолу промывали водой (10 раз по 3 мл)
2) Смолу промывали 5% ACN в воде (1 раз × 3 мл)
3) Пептид растворяли в 5% ACN в воде.
4) Пептид добавляли в шприц и раствор пропускали через колонку 10-20 раз. Элюент собирали в пробирку Falcon.
5) Смолу промывали 5% ACN в воде (10 раз по 3 мл); этот раствор добавляли к раствору в пробирке Falkon
6) Объединенный раствор лиофилизировали.
Общий способ FA: превращение солей TFA пептида в другие соли
Другие солевые формы:
Противоион хлорида можно заменить другими противоионами, пропуская через колонку раствор желаемой солевой формы (например, ацетат натрия), затем многократно промывая колонку водой. Затем загружали пептид и выполняли описанные выше процедуры. Таким образом были получены пептидные соли ацетата, тартрата, цитрата и лактата. Следующие три примера являются репрезентативными:
Пример 249
Последовательность: ((N-Me)G)-IC+SRSLP-(Oic)-I-(Pen)+IP-NH2 (ацетат соль) (пример 249)
N-[(6S,9S,12S,15S,18R,23R,26S,28aS,29aS,33aS,35aS)-26-[(2S)-бутан-2ил]-12-(3-карбамимидамидопропил)-9,15-бис(гидроксиметил)-22,22-диметил-18-[(N-метилглицил-L-изолейцил)амино]-6-(2-метилпропил)-5,8,11,14,17,25,28,35-октаоксодотриаконтагидро-1Н,5Н,22Н-пирроло[2',1':13,14][1,2,5,8,11,14,17,20,23,26]дитиаоктаазациклононакозино[11,10-а]индол-23-карбонил]-L-изолейцил-L-пролинамид, соль уксусной кислоты
IRA 410 хлоридную смолу (12 г) помещали в пустую 20 мл колонку Biotage и смолу промывали с 5% раствором ACN-вода (10 раз). Картридж был присоединен к автоматической станции ионного обмена. Смолу промывали 20 объемами колонки 1 М раствора NaOH (2×1800 с, скорость вращения 80 мин-1). Колонку промывали водой до рН элюента <рН 9(10 объемов колонки). Затем колонку элюировали 1 М раствором уксусной кислоты (10 объемов колонок), затем промывали водой до тех пор, пока рН элюента не стал >5 (около 10 объемов колонок). Пептид (N-Me)GIC+SRSLP-(Oic)-I-Pen+IP- NH2 (соль TFA) (1200 мг) загружали на колонку в 12 мл 5% ACN/вода и оставляли циклически проходить через колонку 10 раз согласно общему способу F. Смолу затем промывали 5% раствором ACN/вода 10 раз и элюент собирали в пробирки Falcon объемом 50 мл. Настройки параметров, используемые для этой шкалы, были следующими:
Промывка с H2O: время работы 3085 с; 80 мин-1; 1 цикл (объем составляет 90 мл) Циркуляция образца с пептидом: время работы 3085 с; 80 мин-1; 1 цикл (объем составляет 180 мл)
Промывка с H2O (или с %ACN в H2O: время работы 3085 с; 80 мин-1; 1 цикл (объем составляет 90 мл)
Объединенный элюент лиофилизировали с получением 1120 мг (100% чистота, 97% выход) целевой ацетатной соли.
Ионная хроматография (содержание хлорида): <1% хлорид
Ионная хроматография (содержание TFA): <1% TFA
Ионная хроматография (содержание ацетата): 7.1% ацетат = 1.84 экв.
LC-MS (MCW-TOF-AQ-YMC-18мин): Rt=7.74 мин; MS (ESIpos): m/z=725 [М+2Н]+
Пример 264
Последовательность: ((N-Me)G)-IC+SRSLP-(Oic)-I-(Pen)+IP-NH2 (соль L-винной кислоты) (пример 264)
N-[(6S,9S,12S,15S,18R,23R,26S,28aS,29aS,33aS,35aS)-26-[(2S)-бутан-2ил]-12-(3-карбамимидамидопропил)-9,15-бис(гидроксиметил)-22,22-диметил-18-[(N-метилглицил-L-изолейцил)амино]-6-(2-метилпропил)-5,8,11,14,17,25,28,35-октаоксодотриаконтагидро-1Н,5Н,22Н-пирроло[2',1':13,14][1,2,5,8,11,14,17,20,23,26]дитиаоктаазациклононакозино[11,10-а]индол-23-карбонил]-L-изолейцил-L-пролинамид (2R,3R)-2,3-дигидроксибутандикислота соль (L-(+) винной кислоты)
IRA 410 хлоридную смолу (12,5 г) помещали в пустую 20 мл колонку Biotage и смолу промывали с 5% раствором ACN-вода (10 раз). Картридж был присоединен к автоматической станции ионного обмена. Смолу промывали 20 объемами колонки 1 М раствора NaOH (2×1800 с, скорость вращения 80 мин-1). Колонку промывали водой до рН элюента <рН 9 (10 объемов колонки). Затем колонку элюировали 1 М раствором уксусной кислоты (10 объемов колонок), затем промывали водой до тех пор, пока рН элюента не стал >5 (около 10 объемов колонок). Пептид ((N-Me)G)-IC+SRSLP-(Oic)-I-(Pen)+IP-NH2 (соль TFA) (1200 мг) загружали на колонку в 12 мл 5% ACN/вода и оставляли циклически проходить через колонку 10 раз согласно общему способу F. Смолу затем промывали 5% раствором ACN/вода 10 раз и элюент собирали в пробирки Falcon объемом 50 мл. Настройки параметров, используемые для этой шкалы, были следующими:
Промывка с Н2О: время работы 3085 с; 80 мин-1; 1 цикл (объем составляет 90 мл) Циркуляция образца с пептидом: время работы 3085 с; 80 мин-1; 1 цикл (объем составляет 180 мл)
Промывка с Н2О (или с %ACN в Н2О: время работы 3085 с; 80 мин-1; 1 цикл (объем составляет 90 мл)
Объединенный элюент лиофилизировали с получением 1170 мг (100% чистота, 91% выход) целевой ацетатной соли.
Ионная хроматография (содержание хлорида): <1% хлорид
Ионная хроматография (содержание TFA): <1% TFA
Ионная хроматография (содержание ацетата): 13,4% ацетат=1.49 экв.
LC-MS (MCW-TOF-AQ-YMC-18mhh): Rt=7.62 мин; MS (ESIpos): m/z=725 [М+2Н]+
Пример 376
Последовательность: ((N-Me)G)-IC+SRSLP-(Oic)-I-(Pen)+IP-NH2 (Соль лимонной кислоты) (пример 376)
N-[(6S,9S,12S,15S,18R,23R,26S,28aS,29aS,33aS,35aS)-26-[(2S)-бутан-2ил]-12-(3-карбамимидамидопропил)-9,15-бис(гидроксиметил)-22,22-диметил-18-[(N-метилглицил-L-изолейцил)амино]-6-(2-метилпропил)-5,8,11,14,17,25,28,35-октаоксодотриако нтагидро-1Н,5Н,22Н-пирроло[2',1':13,14][1,2,5,8,11,14,17,20,23,26]дитиаоктаазациклононакозино[11,10-а]индол-23-карбонил]-L-изолейцил-L-пролинамид, 2-гидроксипропан-1,2,3-трикарбоновой кислоты (лимонной кислоты) соль
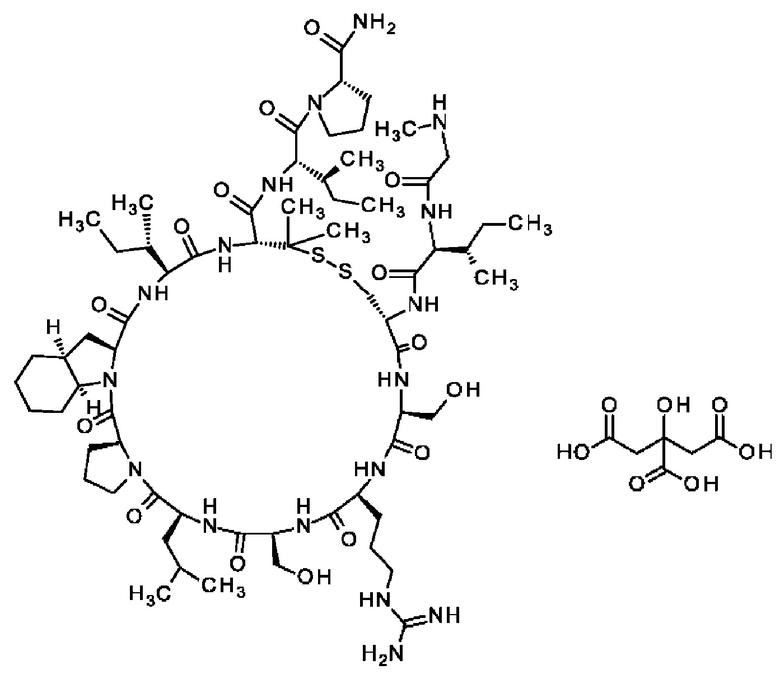
IRA 410 хлоридную смолу (26 г) помещали в пустую 20 мл колонку Biotage и смолу промывали с 5% раствором ACN-вода (10 раз). Картридж был присоединен к автоматической станции ионного обмена. Смолу промывали 20 объемами колонки 1 М раствора NaOH (2×1800 с, скорость вращения 80 мин-1). Колонку промывали водой до рН элюента <рН 9(10 объемов колонки). Затем колонку элюировали 1 М раствором уксусной кислоты (10 объемов колонок), затем промывали водой до тех пор, пока рН элюента не стал >5 (около 10 объемов колонок). Пептид (N-Me)GIC+SRSLP-(Oic)-I-Pen+IP-NH2 (соль TFA) (1200 мг) загружали на колонку в 12 мл 5% ACN/вода и оставляли циклически проходить через колонку 10 раз согласно общему способу F. Смолу затем промывали 5% раствором ACN/вода 10 раз и элюент собирали в пробирки Falcon объемом 50 мл. Настройки параметров, используемые для этой шкалы, были следующими:
Промывка с Н2О: время работы 3085 с; 80 мин-1; 1 цикл (объем составляет 90 мл) Циркуляция образца с пептидом: время работы 3085 с; 80 мин-1; 1 цикл (объем составляет 180 мл)
Промывка с Н2О (или с %ACN в Н2О: время работы 3085 с; 80 мин-1; 1 цикл (объем составляет 90 мл)
Объединенный элюент лиофилизировали с получением 1370 мг (100% чистота, 97% выход) целевой ацетатной соли.
Ионная хроматография (содержание хлорида): <1% хлорид
Ионная хроматография (содержание TFA): <1% TFA
Ионная хроматография (содержание ацетата): 19,0% ацетат = 1.77 экв.
LC-MS (MCW-TOF-AQ-YMC-18 мин): Rt=7.6 мин; MS (ESIpos): m/z=725 [М+2Н]+
Специалисту в данной области техники понятно, что, хотя стехиометрия по данным ионной хроматографии не всегда является стехиометрической (например, 1:2 или 1:1), содержание воды не принималось во внимание, которое в некоторых случаях измерения может быть между 8-15% содержания воды.
Общий способ G: Получение бессолевой формы
Получение AIC+SRSLP-(Oic)-I-(Pen)+IPD-NH2 (бессолевая форма) (пример 27) является иллюстративным.
5 граммов соли TFA (праймер 75) очищали обращенно-фазовой ВЭЖХ с использованием ацетонитрилового водного градиента при 70°С без кислотного модификатора. Нужные фракции объединяли и лиофилизировали. Было получено 2,2 г бессолевой формы (пример 27); LC-MS (чистота >99%); Ионная хроматография (<1% TFA)
Общий способ Н: Крупномасштабный синтез
Синтез AIC+SRS-((tBu)A)-PPI-((N-Me)C)+IPD-NH2 (пример 48) является иллюстративным
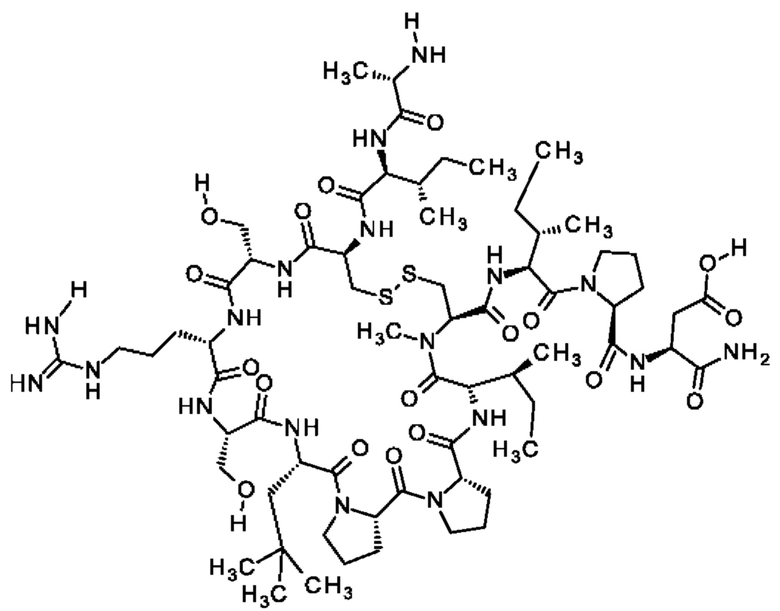
Пептид синтезировали с применением стандартной химии Fmoc.
1) DMF добавляли в сосуд, содержащий смолу МВНА (180 ммоль) и смолу оставляли набухать в течение 2 часов.
2) Линкер амидной смолы Ринка Fmoc добавляли и смешивали в течение 30 с, затем буфер активации добавляли. Смешивание продолжали с барбатированием N2 в течение 1 часа.
3) Раствор удаляли из смолы и смолу промывали с DMF (30 с × 3 раза).
4) Раствор 20% Пиперидин/DMF добавляли и смешивание продолжали с барбатированием N2 в течение 30 мин.
5) Раствор удаляли из смолы и смолу промывали с DMF (5 раз).
6) Раствор Fmoc-аминокислоты добавляли и смешивали в течение 30 с, затем буфер активации добавляли. Смешивание продолжали с барбатированием N2 в течение 1 часа.
7) Стадии 3-6 повторяли для каждого связывания аминокислоты.

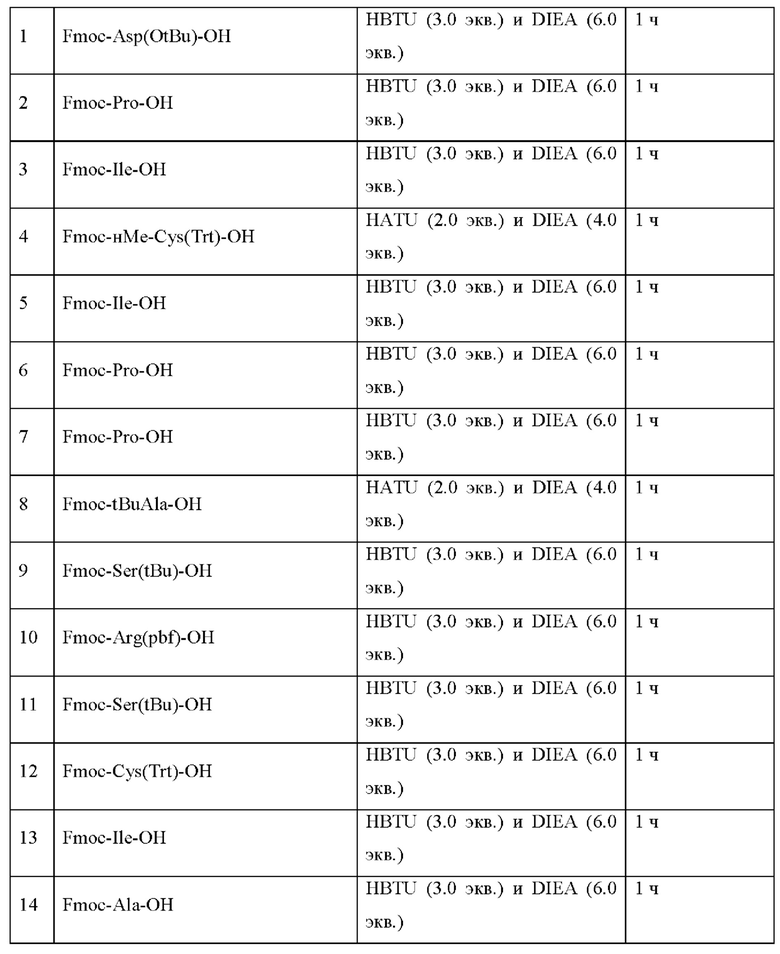
Раствор 20% Пиперидин в DMF применяли для удаления защитной группы Fmoc в течение 30 мин. Реакцию связывания контролировали нингидриновым тестом и смолу промывали с DMF 5 раз.
Расщепление и очистка пептида:
1) Расщепляющий буфер (92.5% TFA: 2.5% EDT: 2.5% TIS: 2.5% H2O) добавляли в колбу, содержащую пептид с защищенной боковой цепью при комнатной температуре и смесь перемешивали в течение 2 часов.
2) Пептид осаждали изопропиловым простым эфиром и центрифугировали (2 мин при 5000 об/мин).
3) Пептид промывали еще 2 раза изопропиловым простым эфиром.
4) Неочищенный пептид сушили под вакуумом в течение 2 часов.
Образование дисульфида
В раствор неочищенного линейного пептида (15 г, 9.92 ммоль, 1.0 экв.) (20 партий) в Н2О/I (3 L 2:1) добавляли NH4HCO3 (7.84 г, 99.2 ммоль, 10 экв.). Смесь перемешивали при 30°С в течение 16 ч, гасили добавлением HCl до РН~6, а затем смесь лиофилизировали. Лиофилизат очищали посредством препаративной ВЭЖХ с получением целевого Примера 48 (41.8 г, 95.8%, 13.3% выход) в виде твердого вещества белого цвета, 156.8 г в общем.
Условия очистки
Устройство: Shimadzu 10А; пептид растворяли в DMF/H2O. Подвижная фаза:, А (H2O (0.075% TFA в H2O), В EtOH; градиент: 10-50%-60 мин. Время удерживания: 47 мин; Колонка: Luna, С18, 10 мкм 100А, 25 см × 50 мм; скорость потока: 80 мл/мин; длина волны: 220/254 нм; температура печи: комнатная температура.
Способ I: получение ((N-Me)A)-IC+SRSLP-(Oic)-I-(Pen)+IP-((N-бензил)D)-NH2 (пример 462)
N-[(6S,9S,12S,15S,18R,23R,26S,28aS,29aS,33aS,35aS)-26-[(2S)-бутан-2ил]-12-(3-карбамимидамидопропил)-9,15-бис(гидроксиметил)-22,22-диметил-18-[(N-метил-L-аланил-L-изолейцил)амино]-6-(2-метилпропил)-5,8,11,14,17,25,28,35-октаоксодотриаконтагидро-1Н,5Н,22Н-пирроло[2',1':13,14][1,2,5,8,11,14,17,20,23,26]дитиаоктаазациклононакозино[11,10-а]индол-23-карбонил]-L-изолейцил-L-пролил-N4-бензил-L-аспартамид
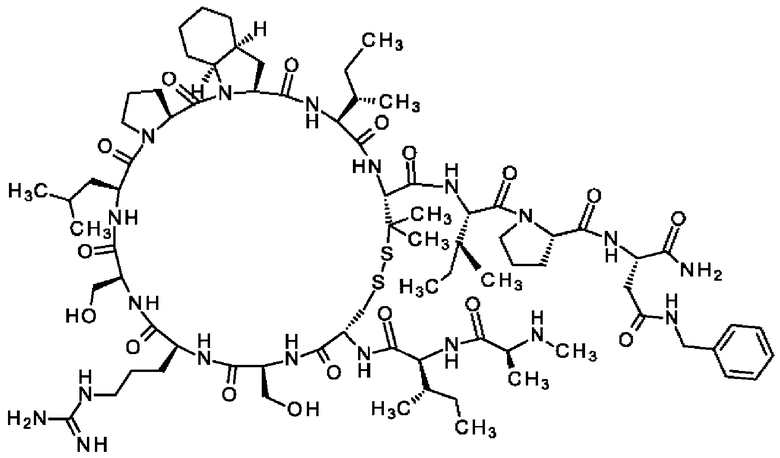
Пептид готовили на амидной смоле Ринка МВНА (загрузка обычно около 0,4-0,6 ммоль/г) в масштабе 0,3 ммоль. Первую аминокислоту Fmos-Asp(2-фенилизопропиловый эфир)-ОН (CAS 200336-86-3) добавляли в смолу, используя способ, описанный в Способе С. Пептидную последовательность конструировали, используя стадии, описанные в Способе С. После завершения последовательности, защитную группу 2-ОРР удаляли с помощью 1% TFA в DCM, 30 минут. N-бензиламин (3 экв.), DIC (3 экв.) и Oxyma (3 экв.) добавляли и смесь встряхивали в течение 2 часов. Пептид расщепляли, образовывалась ди-сульфидная связь, и пептид очищали по Способу С.
Общий способ J. Получение ((N-Me)G)-IC+SRSLP-(Oic)-I-(Pen)+IP-(*Dap)-OH (пример 467)
N-[(6S,9S,12S,15S,18R,23R,26S,28aS,29aS,33aS,35aS)-26-[(2S)-бутан-2ил]-12-(3-карбамимидамидопропил)-9,15-бис(гидроксиметил)-22,22-диметил-18-[(N-метилглицил-L-изолейцил)амино]-6-(2-метилпропил)-5,8,11,14,17,25,28,35-октаоксодотриако нтагидро-1Н,5Н,22Н-пирроло[2',1':13,14][1,2,5,8,11,14,17,20,23,26]дитиаоктаазациклононакозино[11,10-а]индол-23-карбонил]-L-изолейцил-N-[(2S)-2-амино-2-карбоксиэтил]-L-пролинамид
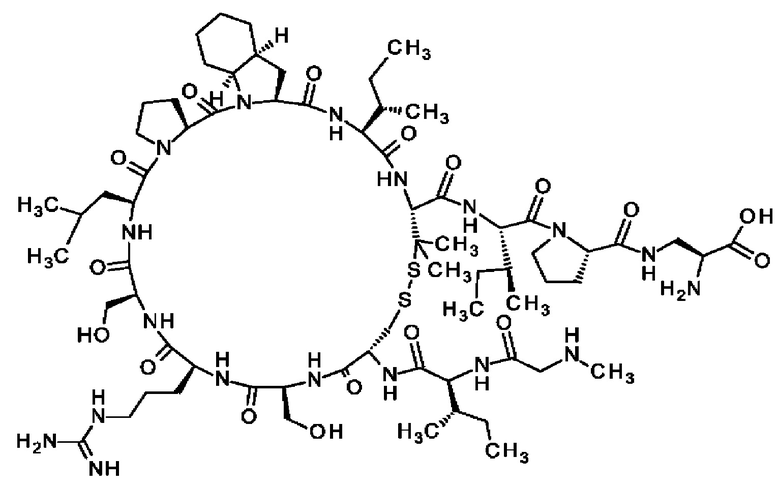
Пептид был приготовлен на 2-хлортритиловой смоле в соответствии со Способом С. BOC-Dap(Fmoc)-OH (CAS 122235-70-5) добавляли в смолу в соответствии со Способом С. Пептид был сконструирован и очищен в соответствии со Способом С.
Общий способ K. Получение (2-(морфолин)ацетил)-IC+SKS-((tBu)А)-PPI-(Pen)+IPD-NH2 (пример 283)
Пример 27А
N-(бромоацетил)-L-изолейцил-L-цистеинил-L-серил-L-аргинил-L-серил-4-метил-L-лейцил-L-пролил-L-пролил-L-изолейцил-3-сульфанил-L-валил-L-изолейцил-L-пролил-L-альфа-аспарагин
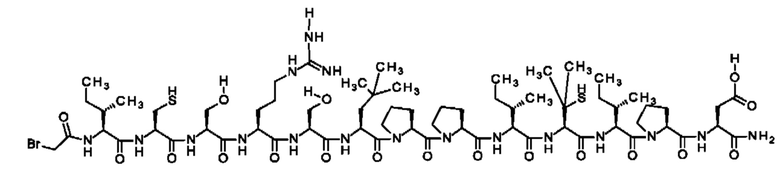
Последовательность IC+SRS-((tBu)A)-PPI-(Pen)+IPD-NH2 получали 12 раз на амидной смоле Ринка Chemmatrix в масштабе 0.1 ммоль согласно Способу А. К смоле, содержащей пептид, (200 мг 100 мкмоль) добавляли N,N'-диизопропилкарбодиимид (31 мкл, 200 мкмоль), этил(гидроксиимино)цианоацетат (28.4 мг, 200 мкмоль) и бромоуксусную кислоту (14 мкл, 200 мкмоль) и смесь встряхивали в 5 мл DMF всю ночь при комнатной температуре на шейкере термосмесителя. Смолу фильтровали, промывали с DMF (6×5 мл), промывали с DCM (6×5 мл), а затем сушили под вакуумом в ходе следующей стадии.
Пример 283
N-[(5aS,11S,14S,17S,20S,23R,28R,31S,33aS)-31-[(2S)-бутан-2ил]-17-(3-карбамимидамидопропил)-11-(2,2-диметилпропил)-14,20-бис(гидроксиметил)-27,27-диметил-23-({N-[(морфолин-4-ил)ацетил]-L-изолейцил}амино)-5,10,13,16,19,22,30,33-октаоксооктакозагидро-1Н,5Н,10Н-дипирроло[2,1-j:2',1'-m][1,2,5,8,11,14,17,20,23,26]дитиаоктаазациклононакзин-28-карбонил]-L-изолейцил-L-пролил-L-альфа-аспарагин
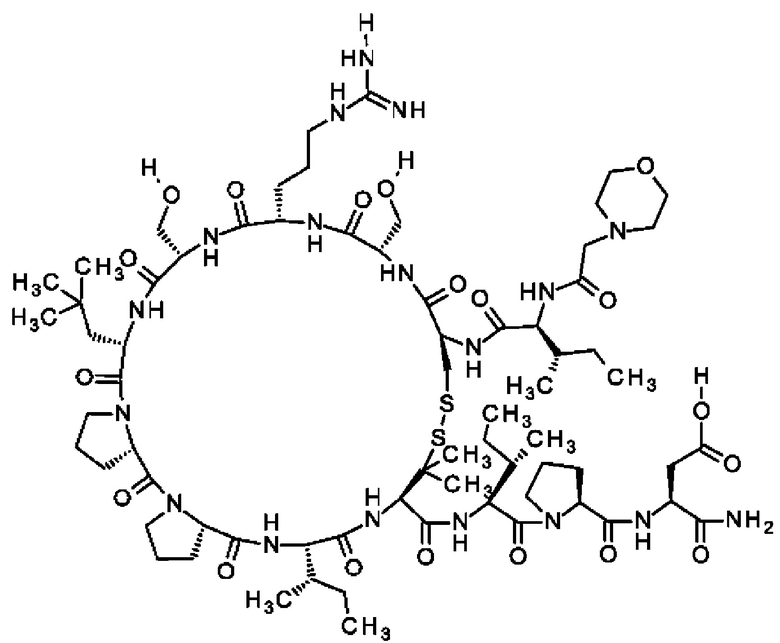
К примеру 27а (200 мг, 74% чистота, 77.7 мкмоль) в NMP (10 мл) добавляли морфолин (34 мкл, 390 мкмоль) и суспензию встряхивали при 37°С на термосместителе. Твердую смолу собирали посредством фильтрации, промывали с 10 мл DMF и 10 мл дихлорметан, в каждом случае два раза. Остаток обрабатывали с TFA/EDT/тиоанизол (90:3:7) и встряхивали 2 ч при комнатной температуре. Реакционную смесь обрабатывали холодным диэтиловым простым эфиром (-10°С), а затем суспензию центрифугировали в атмосфере азота и промывали три раза (3×30 мл) холодным диэтиловым простым эфиром.
К остатку добавляли 0,1 М водный раствор бикарбоната аммония (400 мл) и растворную смесь встряхивали всю ночь в открытой на воздухе круглодонной колбе. Раствор лиофилизировали, и лиофилизированный порошок очищали в соответствии со Способом А, с получением 20,4 мг (чистота 98%, выход 16%) Примера 283.
Пример 300 (2-гидроксиацетил)-IC+SRS-((tBu)А)-PPI-(Pen)+IPD-NH2 выделяли в качестве побочного продукта.
Общий способ L. Получение форм соли натрия и холина
Эти соли были получены из пептидов в бессолевой форме. Два примера ниже являются иллюстративными примерами общего способа:
Пример 431
N-[(6S,9S,12S,15S,18R,23R,26S,28aS,29aS,33aS,35aS)-18-[(L-аланил-L-изолейцил)амино]-26-[(2S)-бутан-2-ил]-12-(3-карбамимидамидопропил)-9,15-бис(гидроксиметил)-22,22-диметил-6-(2-метилпропил)-5,8,11,14,17,25,28,35-октаоксодотриаконтагидро-1Н,5Н,22Н-пирроло[2',1':13,14][1,2,5,8,11,14,17,20,23,26]дитиаоктаазациклононакозино[11,10-а]индол-23-карбонил]-L-изолейцил-N-[(1S)-1,2-дикарбоксилатоэтил]-L-пролинамид, натриевая соль
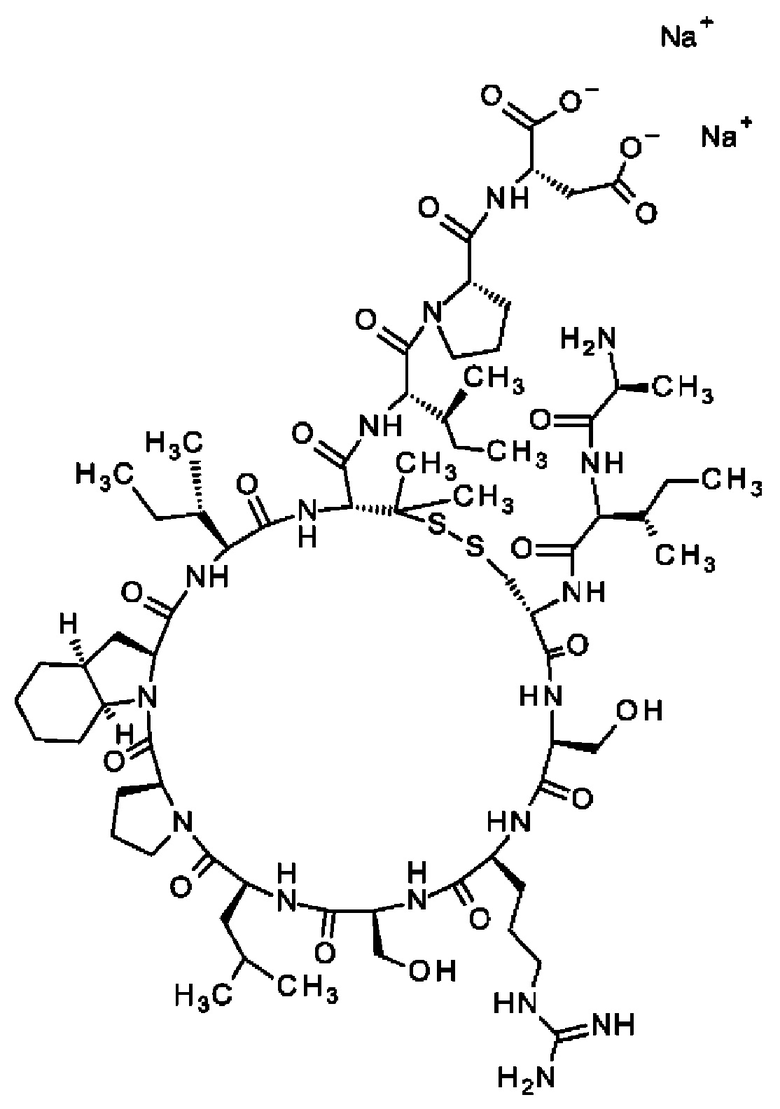
Последовательность: AIC+SRSLP-(Oic)-I-(Pen)+IPD-OH (натриевая соль) (пример 431)
В раствор AIC+SRSLP-(Oic)-I-(Pen)+IPD-OH (бессолевая форма) (100 мг) в 20 мл ацетонитрил/вода 1:1 добавляли 1.3 мл 0.1 М раствора гидроксида натрия. Смесь сушили под вакуумом с получением 109 мг (104% выход, 99% чистота) целевого соединения.
LC-MS (MCW-TOF-AQ-YMC-18 мин): Rt=8.62 мин; MS (ESIpos): m/z=783 [М+2Н]2+
Ионная хроматография (содержание натрия): 2.9% натрия = 2.03 экв.
Пример 432
N-[(6S,9S,12S,15S,18R,23R,26S,28aS,29aS,33aS,35aS)-18-[(L-аланил-L-изолейцил)амино]-26-[(2S)-бутан-2-ил]-12-(3-карбамимидамидопропил)-9,15-бис(гидроксиметил)-22,22-диметил-6-(2-метилпропил)-5,8,11,14,17,25,28,35-октаоксодотриаконтагидро-1Н,5Н,22Н-
пирроло[2',1':13,14][1,2,5,8,11,14,17,20,23,26]дитиаоктаазациклононакозино[11,10-а]индол-23-карбонил]-L-изолейцил-N-[(1S)-1,2-дикарбоксилатоэтил]-L-пролинамид, 2-гидрокси-N,N,N-триметилэтан-1-аммония) (холин) соль
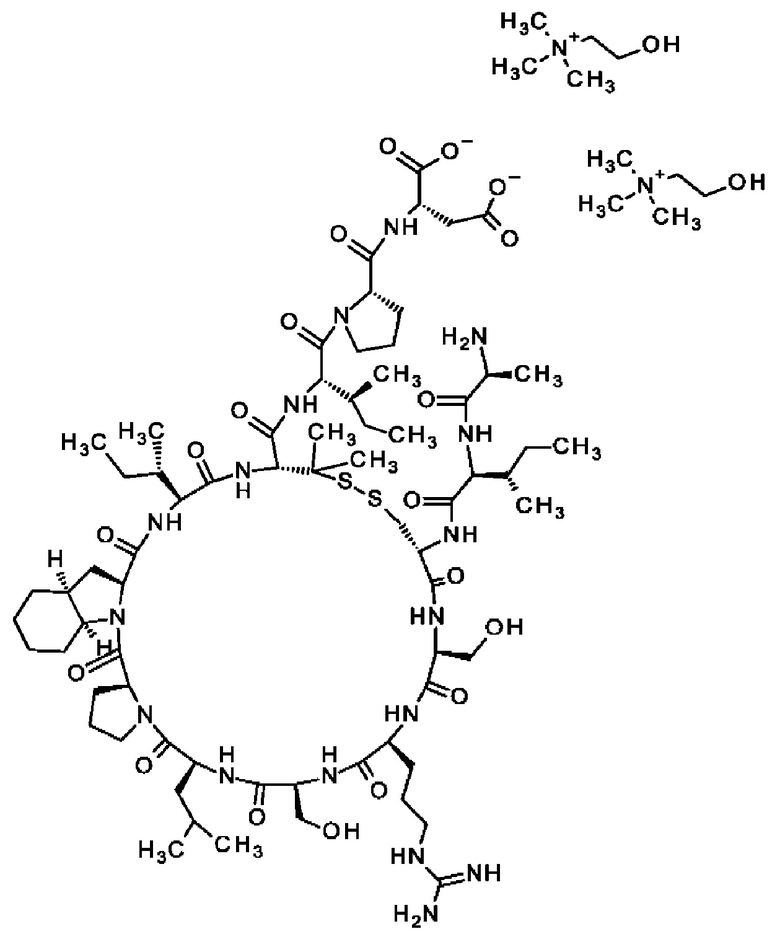
В раствор AIC+SRSLP-(Oic)-I-(Pen)+IPD-OH (бессолевая форма) (100 мг) в 20 мл ацетонитрил/вода 1:1 добавляли 511 мкл 0.25 М раствора гидроксида холина. Смесь сушили под вакуумом с получением 122 мг (105% выход, 99% чистота) целевого соединения.
LC-MS (MCW-TOF-AQ-YMC-18 мин): Rt=8.65 мин; MS (ESIpos): m/z=783 [М+2Н]2+
Ионная хроматография (содержание холина): 11.7% холина = 1.97 экв.
Пример 13
AIC+SRSLP-(Oic)-I-(Pen)+IPD-OH (пример 13)
(3S)-4-амино-3-({(2S)-1-[(2S,3S)-2-{[(2R)-2-{[(2S,3S)-2-{[(2S,3aS,7aS)-l-{(2S)-l-[(2S,5S,8S,11S,14R,17S,20S)-20-амино-17-[(2S)-бутан-2-ил]-8-(3-карбамимидамидопропил)-5,11-бис(гидроксиметил)-2-(2-метилпропил)-4,7,10,13,16,19-гексаоксо-14-(сульфанилметил)-3,6,9,12,15,18-гексаазагеникозанан-1-оил]пирролидин-2-карбонил}октагидро-1Н-индол-2-карбонил]амино}-3-метилпентаноил]амино}-3-метил-3-сульфанилбутаноил]амино}-3-метилпентаноил]пирролидин-2-карбонил}амино)-4-оксобутановая кислота
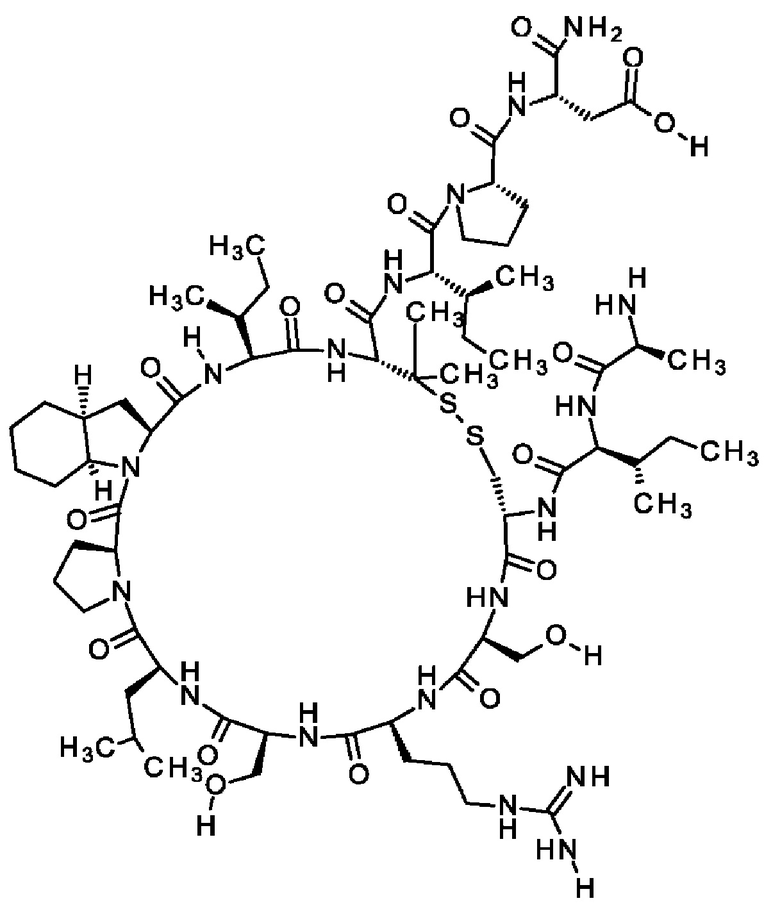
Пептидный синтез
Пептид синтезировали с применением стандартной химии Fmoc.
l) Получение смолы: к смоле СТС (15.0 ммоль, 15.0 г, 1.0 ммоль/г) добавляли Fmoc-Asp(OtBu)-OH (15.0 ммоль, 6.17 г, 1.0 экв.) и DIEA (10.4 мл, 60.0 ммоль, 4.0 экв.) в DCM (300 мл). Смесь встряхивали с N2 в течение 2 ч при 20°С. Метанол затем добавляли (15.0 мл) и смолу встряхивали с N2 в течение 30 мин. Смолу промывали с DMF (300 мл × 3). Затем раствор 20% Пиперидин в DMF (300 мл) добавляли и смесь w встряхивали с N2 в течение 20 мин при 20°С. Затем смесь фильтровали с получением смолу. Смолу промывали с DMF (300 мл × 3) и фильтровали с получением смолу.
2) Связывание: Fmoc-Pro-OH (15.2 г, 45.0 ммоль, 3.0 экв.), HBTU (16.2 г, 42.8 ммоль, 2.85 экв.) и DIEA (90.0 ммоль, 15.6 мл, 6.00 экв.) в DMF (100 мл) добавляли в смолу и смолу встряхивали с N2 в течение 30 мин при 20°С. Смолу затем промывали с DMF (300 мл × 5).
3) Удаление защитной группы: 20% Пиперидин в DMF (300 мл) добавляли в смолу и смесь встряхивали с N2 в течение 20 мин при 20°С.
4) Стадии 2 и 3 повторяли для всех других аминокислот:
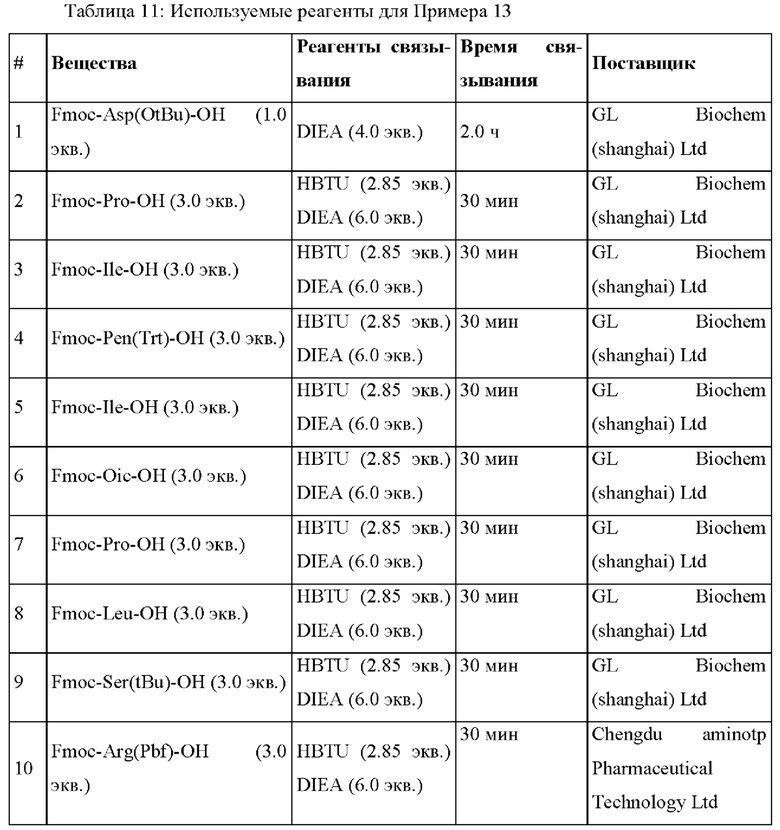
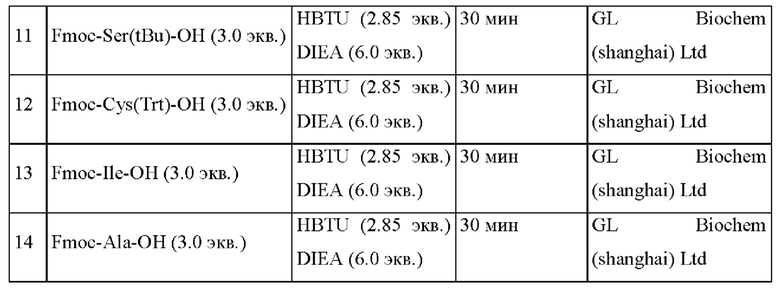
20% Пиперидин в DMF применяли для удаления защитной группы Fmoc в течение 30 мин. Реакцию связывания контролировали нингидриновым (все аминокислоты, кроме Pro) и хлораниловым тестом (Pro), и смолу промывали с DMF (300 мл) 5 раз.
HBTU, DIEA поставщик: Suzhou Highfine Biotech Co., Ltd
Расщепление и очистка пептида
1) Смолу промывали с Me ОН (300 мл × 5) и сушили под вакуумом с получением 41,5 г пептидной смолы. Затем 400 мл расщепляющего буфера (92.5%TFA/2.5%3-тегсар1опропионовая кислота/2.5%Т18/2.5%H2O) добавляли в колбу, содержащую смолу с пептидом с защищенной боковой цепью, при 20°С и смесь перемешивали в течение 2.5 ч, а затем фильтровали.
2) Пептид осаждали холодным трет-бутилметиловым эфиром (4000 мл) и центрифугировали (3 мин при 3000 об/мин) с получением твердого неочищенного вещества. Неочищенный пептидный осадок промывают трет-бутилметиловым эфиром в течение еще двух раз (1000 мл × 3). Неочищенный пептид сушили над вакуумом в течение 2,0 ч с получением 21,8 г неочищенного пептида.
3) Неочищенный пептид (21.8 г) растворяли в CH3CN (6.5 л) и воде (6.5 л), h (0.1 М в МеОН, 55 мл) добавляли при 20°С до установления желтого цвета. Затем смесь перемешивали при 20°С в течение 2 мин. Раствор тиосульфата натрия (0.1 М в воде, 15 мл) затем добавляли по каплям до исчезновения желтого цвета. Смесь затем лиофилизировали с получением неочищенного порошка (22.7 г).
4) Очистка неочищенного пептида посредством препаративной ВЭЖХ (условия: А: 0.075% TFA в воде В: CH3CN), сбор содержащих продукт фракций, и лиофилизация с получением целевого пептида (последовательность ID 13); (4305.1 мг, 18.3% выход, 99.1% посредством 110  С18 RP-ВЭЖХ колонка (Gemini С18 5 мкм 110 150 × 4.6 мм); 97.8% посредством 300
С18 RP-ВЭЖХ колонка (Gemini С18 5 мкм 110 150 × 4.6 мм); 97.8% посредством 300  С18 RP-ВЭЖХ колонка (Discovery BIO Wide Роге С18 5 мкм 300 150 × 4.6 мм), TFA соль) в виде твердого вещества белого цвета.
С18 RP-ВЭЖХ колонка (Discovery BIO Wide Роге С18 5 мкм 300 150 × 4.6 мм), TFA соль) в виде твердого вещества белого цвета.

LC-MS: Rt=1.369 мин; 1564 [М+2Н]+
Rt=8.72 мин (99.1% чистота) Gemini С18 5 мкм, 110 150×4.6 мм
Rt=9.42 мин (97.8%) Discovery BIO Wide Porei C18 5 мкм, 300 150 × 4.6 мм Пример 13 (TFA соль), пример 29 (HCl соль) может быть получен с применением общего способа F; Пример 5 (бессолевая форма) может быть получен с применением общего способа G. Получение Примера 431 (натриевая соль) и Примера 432 (холиновая соль) из Примера 5 показаны ниже как Пример 431 и Пример 432, соответственно.
Пример 237
AIC+SRSL-(L-дегидропролин)-(Oic)-I-(Pen)+IPD-OH (пример 237)
N-[(6S,9S,12S,15S,18R,23R,26S,28aS,35aS)-18-[(L-аланил-L-изолейцил)амино]-26-[(2S)-бутан-2-ил]-12-(3-карбамимидамидопропил)-9,15-бис(гидроксиметил)-22,22-диметил-6-(2-метилпропил)-5,8,11,14,17,25,28,35-октаоксо-6,7,8,9,10,11,12,13,14,15,16,17,18,19,23,24,25,26,27,28,28а,29,29а,30,31,32,33,33а,35,35а-триаконтагидро-3Н,5Н,22Н-пирроло[2',1':13,14][1,2,5,8,11,14,17,20,23,26]дитиаоктаазациклононакозино[11,10-а]индол-23-карбонил]-L-изолейцил-L-прлил-L-аспарагиновая кислота
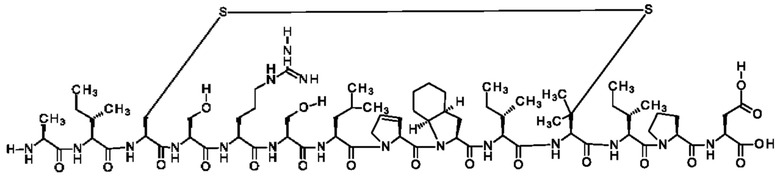
Пептид готовили согласно способу С (3,12 г). Аналитические данные для этого пептида представлены в Таблице 16 и Таблице 17.
Пример 142
AIC+SRSL-(Pro-D2)-(Oic)-I-(Pen)+IPD-OH (пример 142)
N-[(1R,2S,6S,9S,12S,15S,18R,23R,26S,28aS,35aS)-18-[(L-аланил-L-изолейцил)амино]-26-[(2S)-бутан-2-ил]-12-(3-карбамимидамидопропил)-9,15-бис(гидроксиметил)-22,22-диметил-6-(2-метилпропил)-5,8,11,14,17,25,28,35-octaoxo(1,2-2Н2)дотриаконтагидро-1Н,5Н,22Н-пирроло[2',1':13,14][1,2,5,8,11,14,17,20,23,26]дитиаоктаазациклононакозино[11,10-а]индол-23-карбонил]-L-изолейцил-L-пролил-L-аспарагиновая кислота
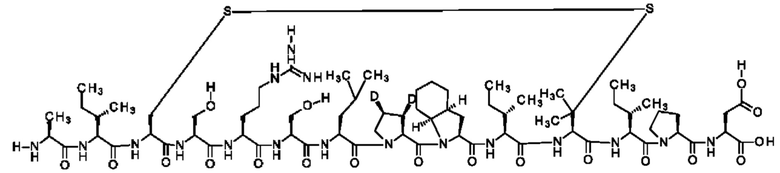
Пептид из Примера 237 (30 мг, 19,2 мкмоль) растворяли в метаноле-с14 (3 мл) (чистота D 99,8%). Родий черный (CAS 7440-16-6, ACBR, артикул № АВ25746) (10 мол.%, 1,9 мкмоль) добавляли в раствор в атмосфере аргона в перчаточном боксе M-Braun. Реакционную смесь в флаконе объемом 8 мл помещали в блок реактора под давлением МТР, который расположен на платформе Chemspeed Swing XL и заключен в перчаточный бокс М-Braun (Chemspeed Technologies AG,  8, 4414
8, 4414  Switzerland, chemspeed@chemspeed.com). Реакцию встряхивали с орбитальной скоростью 650 об/мин при нагрузке под давлением 3 бар газообразного дейтерия в течение 16 ч всю ночь. Блок давления продували аргоном, и флакон удалили из блока реактора. Метанол-с14 выпаривали на роторном испарителе и полученное твердое вещество растворяли в воде (2 мл) и перемешивали в течение 1 часа для обмена обменных атомов дейтерия на водород. Через 1 час добавляли ацетонитрил и продукт очищали обращенно-фазовой ВЭЖХ [колонка: Phenomenex Aeris Peptide 5 мкм ХВ-С18, AXIA Packed, 21.2×250 мм + Картридж 5 мкм, поток: 20 мл/мин, фокусированный градиент с использованием H2O / ACN с 0.1% TFA в качестве кислотного модификатора. FOK30-40: 0 мин 5%ACN, 0-13 мин до 30% ACN, 13-39 мин до 40% ACN, 39-46 мин изократно 40% ACN]. Хроматографические фракции, содержащие продукт, анализировали с помощью аналитического ВЭЖХ (сфокусированный градиент) и с помощью LC-MS, а затем объединяли соответственно для получения двух фракций чистого продукта: 11,4 мг (чистота 90%, выход 34%) и 4,2 мг (чистота 99%, 14% выход). Аналитические данные для этого пептида приведены в Таблице 16.
Switzerland, chemspeed@chemspeed.com). Реакцию встряхивали с орбитальной скоростью 650 об/мин при нагрузке под давлением 3 бар газообразного дейтерия в течение 16 ч всю ночь. Блок давления продували аргоном, и флакон удалили из блока реактора. Метанол-с14 выпаривали на роторном испарителе и полученное твердое вещество растворяли в воде (2 мл) и перемешивали в течение 1 часа для обмена обменных атомов дейтерия на водород. Через 1 час добавляли ацетонитрил и продукт очищали обращенно-фазовой ВЭЖХ [колонка: Phenomenex Aeris Peptide 5 мкм ХВ-С18, AXIA Packed, 21.2×250 мм + Картридж 5 мкм, поток: 20 мл/мин, фокусированный градиент с использованием H2O / ACN с 0.1% TFA в качестве кислотного модификатора. FOK30-40: 0 мин 5%ACN, 0-13 мин до 30% ACN, 13-39 мин до 40% ACN, 39-46 мин изократно 40% ACN]. Хроматографические фракции, содержащие продукт, анализировали с помощью аналитического ВЭЖХ (сфокусированный градиент) и с помощью LC-MS, а затем объединяли соответственно для получения двух фракций чистого продукта: 11,4 мг (чистота 90%, выход 34%) и 4,2 мг (чистота 99%, 14% выход). Аналитические данные для этого пептида приведены в Таблице 16.
Пептид-продукт подвергали расщеплению трипсином с последующей реакцией с дитиотреитолом, и полученные фрагменты анализировали методом TOF-MS-MS. Расщепление трипсином расщепило пептид между аргинином и серином (точная масса 1583,8384, найдено 1583,852 [M+2H]2+). Реакция с дитиотреитолом дала два фрагмента: AICSR-OH (точная масса 548,2741, найдено 548,283 [М+Н]+) и SL-(Pro-D2)-(Oic)-I-(Pen)+IPD-OH (точная масса 1037,5800, найдено 1038,587 [М+Н]+), с фрагментами при 838,47 а.е.м. ((Pro-D2)-(Oic)-I-(Pen)+IPD-OH) и при 738,40 а.е.м. SL-((Oic)-I-(Pen)+IPD-OH), проверяя положение атомов дейтерия.
Пример 28А
(2S,3aS,6aS)-октагидроциклопента[b]пиррол-2-карбоновая кислота-хлорид водорода (1/1) (единственный стереоизомер)
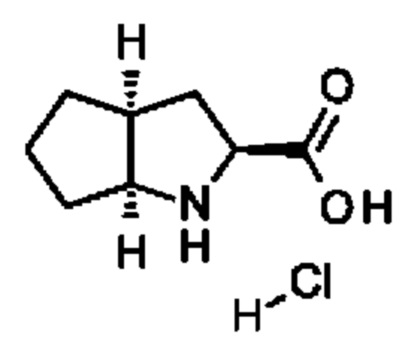
Реакцию проводили в атмосфере аргона. К палладию на углероде (200 мг, 10%) добавляли 20 мл метанола и бензил (2S,3aS,6aS)-октагидроциклопента[b]пиррол-2-карбоксилата-хлорида водорода (1/1) (1.00 г, 3.55 ммоль). Реакционную загружали газообразным водородом (1 атм) и реакционную смесь перемешивали в атмосфере водорода всю ночь при комнатной температуре. Реакционную смесь фильтровали над диатомовой землей и фильтровальную лепешку промывали метанолом. Фильтрат выпаривали и сушили под вакуумом с получением 698 мг указанного в названии соединения.
(2S,3aS,6aS)-октагидроциклопента[b]пиррол-2-карбоновая кислота также коммерчески доступна в бессолевой форме.
Пример 29А
(2S,3aS,6aS)-1-{[(9Н-флуорен-9-ил)метокси]карбонил}октагидроциклопента[b]пиррол-2-карбоновая кислота (единственный стереоизомер)
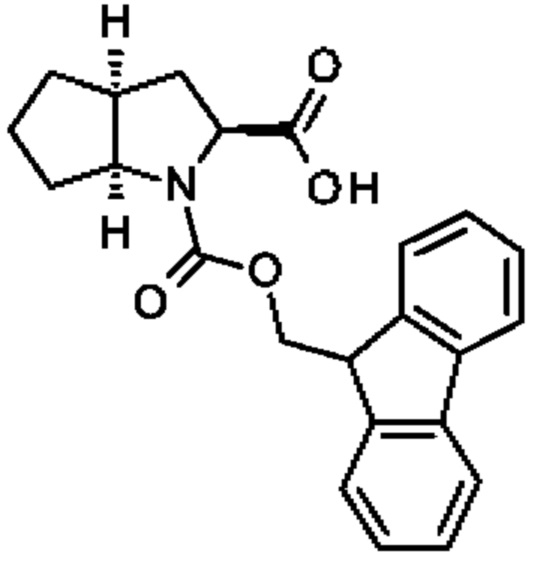
К (2S,3aS,6aS)-октагидроциклопента[b]пиррол-2-карбоновой кислоте-хлорид водорода (1/1) (680 мг, 3.55 ммоль) в 12 мл воды добавляли бикарбонат натрия (2.98 г, 35.5 ммоль) и раствор 1-({[(9Н-флуорен-9-ил)метокси]карбонил}окси)пирролидин-2,5-диона (Fmoc-OSu) (1.26 г, 3.73 ммоль) в 18 мл ацетона. Реакционную смесь перемешивали всю ночь при комнатной температуре. Реакционную смесь обрабатывали водой, подкисляли 1 М соляной кислотой и экстрагировали этилацетатом два раза. Органическую фазу промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали, а затем выпаривали. Остаток разделяли посредством хроматографии на силикагеле (Biotage Isolera, колонка: 50 г SNAP Ultra; элюент: дихлорметан/метанол 100:2 100:5). Соответствующие фракции объединяли, выпаривали и сушили под вакуумом с получением 1.27 г (99% чистота, 94% выход) целевого соединения.
LC-MS (Способ 10): Rt=2.08 мин; MS (ESI pos): m/z=378 [M+H]+
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 1.260 (1.76), 1.274 (1.85), 1.299 (1.78), 1.321 (2.09), 1.335 (2.38), 1.440 (0.92), 1.455 (1.10), 1.473 (0.87), 1.521 (3.61), 1.537 (3.86), 1.551 (2.28), 1.607 (4.53), 1.620 (3.68), 1.640 (3.08), 1.653 (1.66), 1.712 (2.02), 1.781 (1.38), 1.816 (2.30), 1.827 (2.26), 1.839 (2.37), 1.855 (1.68), 2.073 (1.20), 2.305 (0.83), 2.328 (1.99), 2.351 (1.35), 2.360 (1.64), 2.384 (0.92), 2.468 (0.60), 2.606 (1.50), 2.670 (1.11), 3.860 (0.94), 3.874 (1.94), 3.893 (1.95), 3.907 (0.92), 4.143 (2.61), 4.156 (3.68), 4.167 (5.16), 4.186 (5.89), 4.266 (1.43), 4.280 (3.20), 4.294 (2.11), 4.345 (1.31), 4.371 (3.19), 4.386 (3.83), 4.396 (4.39), 4.411 (3.74), 4.422 (2.40), 4.437 (0.98), 7.292 (0.97), 7.311 (3.89), 7.329 (6.99), 7.346 (6.08), 7.363 (2.49), 7.395 (9.91), 7.414 (16.00), 7.432 (6.98), 7.663 (9.73), 7.680 (6.46), 7.879 (12.31), 7.897 (11.49), 12.586 (0.80).
Пример 30А
(1R,3S,5R)-2-азабициюто[3.1.0]гексан-3-карбоновая кислота (единственный стерео-изомер)
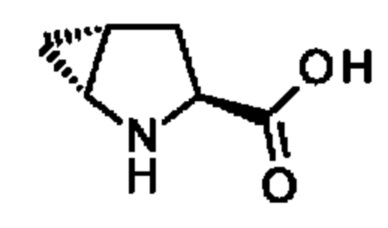
(1H,3S,5R)-2-(трет-бутоксикарбонил)-2-азабицикло[3.1.0]гексан-3-карбоновую кислоту (CAS RN: 197142-34-0, 500 мг) растворяли в 4М растворе HCl-диоксана (3 мл) и перемешивали в течение 8 ч при комнатной температуре. Растворитель выпаривали и неочищенный продукт применяли непосредственно в ходе следующей стадии
Пример 31А
(1H,3S,5R)-2-{[(9Н-флуорен-9-ил)метокси]карбонил}-2-азабицикло[3.1.0]гексан-3-карбоновая кислота (единственный стереоизомер)
В раствор (1R,3S,5R)-2-азабицикло[3.1.0]гексан-3-карбоновой кислоты (пример 30А, 280 мг, 2.20 ммоль) в 3 мл воды добавляли бикарбонат натрия (1.85 г, 22.0 ммоль) и раствор 1-({[(9Н-флуорен-9-ил)метокси]карбонил}окси)пирролидин-2,5-диона (0.78 г, 2.31 ммоль) в 4.5 мл ацетона. Реакционную смесь перемешивали всю ночь при комнатной температуре. Реакционную смесь обрабатывали водой и экстрагировали два раза этилацетатом. Органическую фазу промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали, а затем концентрировали. Неочищенный продукт очищали посредством препаративной обращенно-фазовой ВЭЖХ (Колонка: Reprosil; C18; 10 мкм; 125×30 мм; элюент A: CAN, элюент В: вода с 0.1% муравьиной кислоты), и фракции, содержащие продукт, объединяли и лиофилизировали с получением 88.0 мг (96% чистота, 11% выход) целевого соединения.
LC-MS (Способ 11): Rt=1.32 мин; MS (ESI pos): m/z=372 [M+Na]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: -0.149 (1.15), -0.008 (10.73), 0.008 (9.13), 0.146 (1.21), 0.468 (1.54), 0.534 (3.66), 0.803 (4.38), 1.682 (2.68), 2.072 (0.75), 2.128 (2.19), 2.142 (2.32), 2.223 (1.05), 2.293 (2.19), 2.327 (4.45), 2.349 (2.68), 2.366 (2.75), 2.523 (3.89), 2.670 (1.44), 2.710 (1.31), 2.816 (0.82), 3.413 (3.93), 3.757 (1.83), 4.010 (2.91), 4.140 (2.39), 4.211 (3.11), 4.285 (12.30), 6.929 (0.43), 6.950 (0.43), 7.340 (9.91), 7.358 (6.02), 7.406 (10.08), 7.425 (16.00), 7.443 (7.10), 7.650 (2.85), 7.702 (4.52), 7.724 (5.53), 7.746 (3.40), 7.892 (12.40), 7.910 (9.85), 12.680 (0.65).
Эту аминокислоту применяли в Fmoc SPPS. Эта fmoc аминокислота также является коммерчески доступной.
Пример 32А
Бензил (2R,4R)-4-[(2S)-бутан-2-ил]-2-трет-бутил-5-оксо-l,3-оксазолидин-3-карбоксилат (единственный стереоизомер)
К суспензии D-аллоизолейцина (8.06 г, 61.4 ммоль) в 120 мл этанола, добавляли раствор гидроксида натрия, полученный из 2.45 г гидроксида натрия в 11 мл воды. Реакционную смесь перемешивали 90 мин при комнатной температуре, а затем выпаривали. Остаток обрабатывали 195 мл пентана, а затем 2,2-диметилпропанол (10 мл, 92 ммоль) добавляли. Реакционную смесь перемешивали 24 ч при 45°С с ловушкой Дина-Старка, соединенной с круглодонной колбой. После охлаждения растворители выпаривали, и остаток сушили несколько раз толуолом (азеотропная дистилляция). Остаток обрабатывали с 165 мл дихлорметана и охлаждали до 0°С. Бензилхлорформиат (13 мл, 92 ммоль) добавляли, и реакционную смесь перемешивали в течение одной недели при 0-4°С. 4-диметиламинопиридин (375 мг, 3.1 ммоль) и 65 мл воды добавляли в реакционную смесь, реакционную смесь оставляли дойти до комнатной температуры, а затем перемешивали всю ночь при комнатной температуре. Реакционную смесь обрабатывали этилацетатом несколько раз и экстрагировали. Объединенную органическую фазу промывали два раза 10% водной лимонной кислотой, а затем два раза насыщенным водным раствором бикарбоната натрия. Водную фазу повторно экстрагировали этилацетатом. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали, а затем выпаривали. Остаток очищали посредством хроматографии на силикагеле (Isolera; элюент: циклогексан/этилацетат 100:3-100:7). Фракции, содержащие продукт, объединяли, выпаривали, а затем сушили под вакуумом с получением 8.71 г указанного в названии соединения. Пример ЗЗА
Бензил (2R,4R-4-[(2S)-бутан-2-ил]-2-трет-бутил-4-метил-5-оксо-1,3-оксазолидин-3-карбоксилат (единственный стереоизомер)
Реакцию проводили в атмосфере аргона. Бензил (2R,4R)-4-[(2S)-бутан-2-ил]-2-трет-бутил-5-оксо-1,3-оксазолидин-3-карбоксилат (пример 32А, 8.12 г, 24.4 ммоль) в 100 мл сухого THF охлаждали до -78°С. Калия гексаметилдисилазид (44 мл, 29 ммоль) добавляли по каплям и реакционную смесь перемешивали в течение 2 ч при -78°С. Иодметан (2.3 мл, 37 ммоль) добавляли по каплям в реакционную смесь и реакционную смесь перемешивали в течение 2.5 ч при комнатной температуре. Насыщенный водный раствор хлорида аммония (20 мл) добавляли в реакционную смесь, реакционную смесь оставляли дойти до комнатной температуры, а затем продукт экстрагировали в этилацетат. Объединенные органические фазы промывали насыщенным водным раствором бикарбоната натрия, сушили над сульфатом магния, фильтровали, а затем концентрировали. Остаток очищали посредством хроматографии на силикагеле (Isolera, колонка: 100 г SNAP Ultra; элюент: циклогексан/этилацетат 100:3 100:7). Фракции, содержащие продукт, объединяли, выпаривали, а затем сушили под вакуумом с получением 3.2 г указанного в названии соединения.
Пример 34А
N-[(Бензилокси)карбонил]-2-метил-D-аллоизолейцин (единственный стереоизомер)
В раствор бензил (2R,4R)-4-[(2S)-бутан-2-ил]-2-трет-бутил-4-метил-5-оксо-1,3-оксазолидин-3-карбоксилата (пример 33А, 3.20 г, 9.21 ммоль) в метаноле (40 мл) добавляли 1 М раствор гидроксида натрия (40 мл). Реакционную смесь перемешивали в течение 2 ч при возврате флегмы. После охлаждения, метанол выпаривали. Остаток обрабатывали 10% раствором лимонной кислоты и продукт экстрагировали в этилацетат. Водную фазу экстрагировали этилацетатом еще два раза. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали, выпаривали, а затем сушили под вакуумом с получением 2.77 г указанного в названии соединения.
Пример 35А
2-метил-D-аллоизолейцин (единственный стереоизомер)
Реакцию проводили в атмосфере аргона. В N-[(бензилокси)карбонил]-2-метил-D-аллоизолейцин (пример 34А, 2.57 г, 9.20 ммоль) в 190 мл метанола добавляли палладий на углероде (390 мг, 10%). Реакционную смесь перемешивали в атмосфере водорода в течение 4 ч при комнатной температуре и нормальном давлении. Реакционную смесь фильтровали над диатомовой землей и фильтрат концентрировали, затем сушили под вакуумом с получением 1.32 г (98% выход) целевого соединения.
Пример 36А
N-{[(9Н-флуорен-9-ил)метокси]карбонил}-2-метил-D-аллоизолейцин (единственный стереоизомер)
В раствор 2-метил-D-аллоизолейцина (пример 35 А, 1.32 г, 9.09 ммоль) в смеси воды (20 мл) и ацетона (29 мл) добавляли бикарбонат натрия (7.64 г, 90.9 ммоль), а затем раствор 1-({[(9Н-флуорен-9-ил)метокси]карбонил}окси)пирролидин-2,5-диона (3.22 г, 9.55 ммоль) в ацетоне (20 мл) добавляли. Реакционную смесь перемешивали всю ночь при комнатной температуре. Реакционную смесь обрабатывали МТВЕ и экстрагировали. Водную фазу подкисляли концентрированной соляной кислотой (рН 1) и экстрагировали этилацетатом еще три раза. Объединенные органические фазы сушили над сульфатом магния, фильтровали и выпаривали. Остаток разделяли посредством хроматографии на силикагеле (Isolera, колонка: 50 г SNAP Ultra; элюент: дихлорметан/метанол 100:3 -100:5). Соответствующие фракции объединяли, выпаривали, а затем сушили под вакуумом.
Фазу МТВЕ затем обрабатывали 5% раствором бикарбоната натрия и экстрагировали этилацетатом (600 мл). Водную фазу подкисляли концентрированной соляной кислотой (рН 1) и экстрагировали этилацетатом каждый раз. Объединенные органические фазы сушили над сульфатом магния, фильтровали, выпаривали, а затем сушили под вакуумом. Объединенный неочищенный продукт выход 2.53 г.
Остаток очищали посредством хроматографии на силикагеле (Isolera, колонка: 50 г SNAP Ultra; элюент: дихлорметан/метанол 97:3 - 97:5). Фракции, содержащие продукт, объединяли, выпаривали, а затем сушили под вакуумом с получением 967.4 мг указанного в названии соединения. Продукт далее растворяли в воде/ацетонитриле и лиофилизировали перед применением.
LC-MS (Способ 10): Rt=2.10 мин; MS (ESI pos): m/z=368.1857 [M+H]+
Эту аминокислоту применяли в Fmoc SPPS.
Пример 37А
(6S)-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновя кислота (смесь стереоизомеров)
В (6S)-5-(трет-бутоксикарбонил)-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновую кислоту (Omega, CAS 1357482-03-1) (2.50 г, 9.02 ммоль) в 85 мл дихлорметана добавляли трифторуксусной кислоты (15 мл, 250 ммоль). Реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Реакционную смесь концентрировали и сушили под вакуумом в ходе следующей стадии.
Пример 38А
(6S)-5-{[(9Н-флуорен-9-ил)метокси]карбонил}-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновая кислота (смесь стереоизомеров)
В раствор (6S)-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновой кислоты (смесь стереоизомеров) (пример 37А, 1.60 г, 9.03 ммоль) в воде (13 мл) добавляли бикарбонат натрия (7.59 г, 90.3 ммоль), а затем раствор 1-({[(9Н-флуорен-9-ил)метокси]карбонил}окси)пирролидин-2,5-диона (3.20 г, 9.48 ммоль) в ацетоне (20 мл). Реакционную смесь перемешивали всю ночь при комнатной температуре. Реакционную смесь обрабатывали водой и экстрагировали два раза с МТВЕ. Водную фазу подкисляли соляной кислотой до рН3 и экстрагировали дихлорметаном три раза. Органическую фазу промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали, выпаривали и сушили под вакуумом с получением 3.31 г (92% выход) целевого соединения.
LC-MS (Способ 10): Rt=1.99 мин; MS (ESI pos): m/z=400 [M+H]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 1.106 (16.00), 1.589 (0.92), 1.605 (1.39), 1.619 (1.06), 1.627 (1.11), 1.655 (0.62), 1.682 (1.25), 1.703 (1.21), 1.916 (0.49), 1.949 (0.86), 1.978 (0.91), 2.012 (0.85), 2.042 (0.52), 2.086 (11.35), 2.558 (0.83), 2.568 (0.67), 2.591 (0.67), 2.645 (0.52), 2.670 (0.56), 3.077 (5.29), 3.442 (0.43), 3.452 (0.50), 3.471 (1.17), 3.479 (1.13), 3.500 (1.39), 3.527 (0.51), 3.562 (1.21), 3.627 (1.20), 3.652 (1.65), 3.677 (0.78), 4.186 (1.02), 4.200 (2.72), 4.223 (2.09), 4.238 (2.18), 4.257 (1.63), 4.279 (2.63), 4.294 (2.46), 4.306 (1.36), 4.322 (1.56), 4.348 (2.13), 4.359 (1.17), 4.372 (1.33), 4.382 (0.84), 4.477 (0.57), 4.499 (0.61), 4.510 (0.69), 4.518 (0.72), 4.533 (0.73), 4.541 (0.64), 5.754 (0.94), 7.300 (0.77), 7.316 (3.26), 7.334 (5.65), 7.353 (3.32), 7.406 (4.71), 7.425 (7.78), 7.443 (3.45), 7.640 (4.69), 7.659 (4.35), 7.683 (0.73), 7.884 (5.08), 7.903 (4.79).
Пример 39А
(6S)-5-{[(9Н-флуорен-9-ил)метокси]карбонил}-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновая кислота (энантиомер 1) и (энантиомер 2)
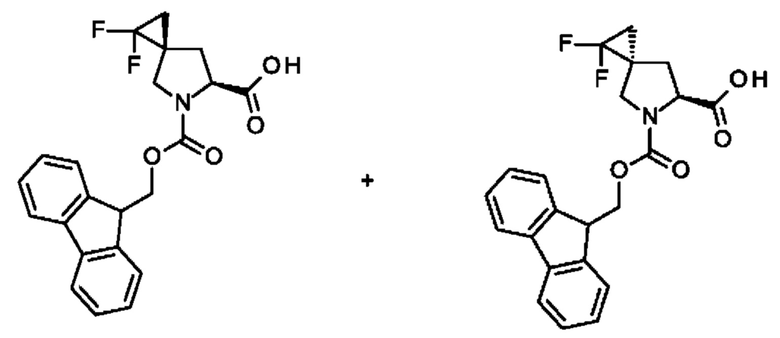
Диастереомерную смесь разделяли с применением THAR сверхкритической жидкостной хроматографии, Prep 200 устройство с применением Chiralpcel AD-H (SFC) 5 мкм, 250 × 30 мм колонка; элюент: СО2/изопропанол (83:17); давление: 135 бар; температура элюента: 38°С; температура циклона: 40°С; давление циклона: 24 бар; скорость потока: 100 г/мин; обнаружение: УФ 210 нм; объем впрыска 0.4 мл/впрыск; при общих рабочих условиях 1 г/мин CO2 приблизительно эквивалентен 1 мл/мин. Установки последовательности: время цикла = 11,2 мин
Фракцию 1 собирали между 5.85 мин и 8.0 мин (энантиомер 1)
Фракцию 2 собирали между 8.4 мин и 10.25 мин (энантиомер 2)
Фракции концентрировали.
Фракции анализировали с применением аналитической SFC-MS (Agilent, колонка: Chiralpak AD-3 3 мкм 100 × 4,6 мм, элюент: СО2/изопропанол (85:15); BPR давление: 130 бар; BPR температура: 60°С; температура колонки: 40°С; скорость потока: 3 мл/мин; UV 210 нм.
Фракцию 1 (rt=2.081 мин) обозначали дополнительно "(энантиомер 1)".
Фракцию 2 (rt=2.838 мин) обозначали дополнительно "(энантиомер 2)". Стереохимия двух энантиомеров не установлена.
Пример 40А
(3R*,6S)-5-{[(9Н-флуорен-9-ил)метокси]карбонил}-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновая кислота (единственный энантиомер, энантиомер 1, стереохимия в положении 2 не определена)
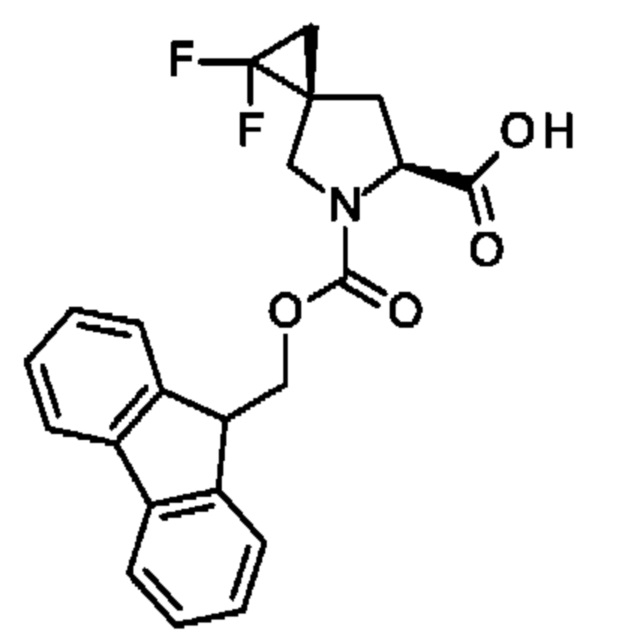
Посредством хиральной препаративной очистки SFC получали 2,015 г указанного в названии соединения, 100% ее
LC-MS (Способ 10): Rt=1.99 мин; MS (ESI pos): m/z=400 [M+H]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: -0.008 (0.70), 0.008 (0.74), 1.032 (15.85), 1.047 (16.00), 1.285 (1.59), 1.568 (0.69), 1.589 (1.52), 1.617 (1.57), 1.635 (0.96), 1.656 (0.63), 1.686 (1.36), 1.702 (1.37), 1.941 (0.90), 1.974 (1.05), 2.008 (0.87), 2.041 (0.90), 2.073 (1.11), 2.557 (1.08), 2.566 (0.95), 2.591 (0.80), 2.645 (0.71), 2.670 (0.95), 2.701 (0.65), 3.451 (0.87), 3.468 (1.64), 3.479 (1.88), 3.495 (1.12), 3.505 (1.17), 3.626 (2.11), 3.651 (2.91), 3.677 (1.38), 3.770 (0.41), 3.778 (0.42), 4.169 (0.43), 4.186 (1.17), 4.200 (3.11), 4.221 (2.05), 4.238 (1.40), 4.256 (1.57), 4.279 (3.79), 4.293 (3.74), 4.304 (1.62), 4.322 (2.70), 4.336 (1.12), 4.348 (2.93), 4.358 (1.92), 4.371 (1.64), 4.381 (1.52), 4.509 (1.14), 4.517 (1.25), 4.531 (1.28), 4.540 (1.12), 7.300 (0.91), 7.315 (3.87), 7.334 (7.04), 7.350 (3.82), 7.352 (4.21), 7.406 (5.88), 7.424 (9.76), 7.443 (4.35), 7.640 (7.54), 7.658 (6.74), 7.885 (6.80), 7.904 (6.34), 10.194 (0.55), 10.991 (0.42).
Эту аминокислоту применяли в Fmoc SPPS.
Пример 41А
(3S*,6S)-5-{[(9Н-флуорен-9-ил)метокси]карбонил}-1,1-дифтор-5-азаспиро[2.4]гептан-6-карбоновая кислота (единственный энантиомер, энантиомер 2, стереохимия при циклопропильном центре не определена)
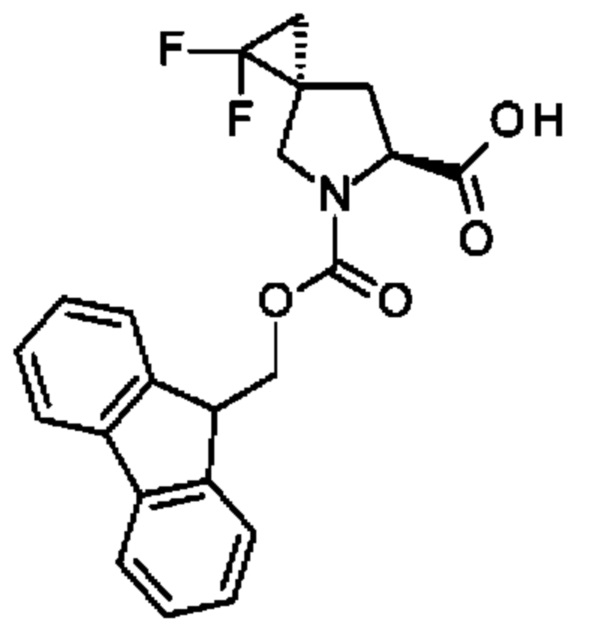
Посредством хиральной препаративной очистки SFC получали 1.041 г указанного в названии соединения, 94.58% ее
LC-MS (Способ 10): Rt=1.93 мин; MS (ESI pos): m/z=400 [M+H]+
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: -0.008 (1.19), 0.008 (1.31), 1.031 (0.55), 1.047 (0.56), 1.285 (0.60), 1.583 (1.53), 1.604 (4.87), 1.627 (5.45), 1.650 (3.25), 1.673 (2.78), 1.682 (3.76), 1.704 (3.11), 1.718 (0.95), 1.915 (3.17), 1.949 (3.55), 1.979 (3.13), 2.012 (3.33), 2.073 (3.85), 2.526 (1.60), 2.590 (1.44), 2.626 (1.61), 2.649 (0.89), 2.659 (0.80), 3.471 (2.04), 3.498 (5.50), 3.527 (3.26), 3.561 (7.96), 3.570 (4.68), 3.598 (0.84), 4.167 (0.94), 4.183 (2.75), 4.199 (6.46), 4.223 (7.75), 4.238 (9.60), 4.258 (5.95), 4.277 (4.06), 4.290 (3.98), 4.306 (3.80), 4.319 (4.63), 4.340 (4.17), 4.346 (4.47), 4.354 (2.34), 4.367 (3.96), 4.477 (3.66), 4.497 (3.66), 7.300 (1.97), 7.318 (8.34), 7.334(11.70), 7.337 (12.42), 7.352 (6.35), 7.355 (6.41), 7.407 (9.62), 7.425 (16.00), 7.444 (7.11), 7.633 (6.46), 7.651 (10.65), 7.666 (6.62), 7.683 (4.36), 7.883 (9.71), 7.893 (10.32), 7.901 (9.57), 7.912 (8.99), 12.802 (0.79), 12.984 (0.67).
Эту аминокислоту применяли в Fmoc SPPS.
Пример 42А
3-[4-(трет-бутоксикарбонил)-1Н-1,2,3-триазол-1-ил]-N-{[(9Н-флуорен-9-ил)метокси]карбонил}-L-аланин (единственный стереоизомер)
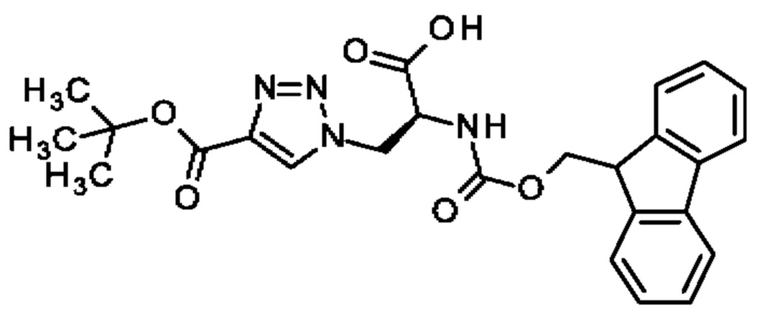
К 3-азидо-N-{[(9Н-флуорен-9-ил)метокси]карбонил}-L-аланину (1.00 г, 2.84 ммоль) в смеси 27 мл воды/трет, бутанола/дихлорметана (1:1:1) добавляли трет-бутил проп-2-иноат (580 мкл, 4.3 ммоль), сульфат меди(II) пентагидрат (709 мг, 2.84 ммоль) и раствор аскорбата натрия (1.41 г, 7.10 ммоль) в 14 мл воды. Реакционную смесь перемешивали всю ночь при комнатной температуре. Реакционную смесь обрабатывали водой, подкисляли разбавленной лимонной кислотой и экстрагировали дихлорметаном три раза. Органическую фазу сушили над сульфатом натрия, фильтровали, а затем выпаривали. Остаток растворяли в ацетонитриле/воде и разделяли на 2 порции посредством препаративной ВЭЖХ (колонка: Chromatorex С18 10 мкм 125 × 40 мм; вода-ацетонитрил-градиент с 0.05% TFA: 0-2 мин 25% ацетонитрила, 2-22 мин 25-75% ацетонитрила, 23-26 мин 75% ацетонитрила, 26-28 мин обратно до 25% ацетонитрила; поток 100 мл/мин). Фракции, содержащие продукт, объединяли, выпаривали и сушили под вакуумом с получением 1.02 г (100% чистота, 75% выход) целевого соединения.
LC-MS (Способ 11): Rt=1.30 мин; MS (ESI neg): m/z=477 [M-H]-
Эту аминокислоту применяли в Fmoc SPPS.
Пример 43А
5-азаспиро[2.4]гептан-1-карбоновую кислоту-трифторуксусная кислота (1/1)
В раствор 5-(трет-бутоксикарбонил)-5-азаспиро[2.4]гептан-1-карбоновой кислоты (1.00 г, 4.14 ммоль) в 25 мл дихлорметана добавляли TFA (960 мкл, 12 ммоль). Реакционную смесь перемешивали 2 ч при комнатной температуре. Реакционную смесь выпаривали и сушили под вакуумом с получением 0.80 г (76% выход) целевого соединения.
LC-MS (Способ 9): Rt=0.18 мин; MS (ESI pos): m/z=142 [M+H]+
Пример 44А
5-{[(9Н-флуорен-9-ил)метокси]карбонил}-5-азаспиро[2.4]гептан-1-карбоновая кислота
В раствор 5-азаспиро[2.4]гептан-1-карбоновой кислоты-трифторуксусной кислоты (1/1) (пример 43А, 994 мг, 3.90 ммоль) в смеси 5 мл ацетонитрила и 5 мл воды добавляли 1-({[(9Н-флуорен-9-ил)метокси]карбонил}окси)пирролидин-2,5-дион (2.63 г, 7.79 ммоль) и триэтиламин (2.2 мл, 16 ммоль). Реакционную смесь перемешивали всю ночь при комнатной температуре. Реакционную смесь обрабатывали водой, подкисляли 1 М соляной кислотой до рН 4, а затем экстрагировали дихлорметаном три раза. Объединенные органические фазы сушили над сульфатом натрия, фильтровали и выпаривали. Остаток растирали с ацетонитрилом, затем осадок собирали посредством фильтрации и промывали ацетонитрилом. Фильтрат очищали в виде 3 частей посредством препаративной ВЭЖХ (колонка: Chromatorex С18 10 мкм 125×40 мм; вода-ацетонитрил-градиент с 0.05% TFA: 0-2 мин 25% ацетонитрила, 2-22 мин 25-75% ацетонитрила, 23-26 мин 75% ацетонитрила, 26-28 мин обратно до 25% ацетонитрила; поток 100 мл/мин). Соответствующие фракции объединяли, выпаривали и сушили под вакуумом с получением 1.16 г (95% чистота, 78% выход) целевого соединения.
LC-MS (Способ 9): Rt=0.99 мин; MS (ESI pos): m/z=364 [M+H]+
Эту аминокислоту применяли в Fmoc SPPS.
Пример 45А
трет-бутил (3S,5S,6R)-3-(2-метилаллил)-2-оксо-5,6-дифенил-морфолин-4-карбоксилат
Два партии проводили параллельно: В раствор трет-бутил (2R,3S)-6-оксо-2,3-дифенил-морфолин-4-карбоксилата (90.0 г, 255 ммоль) и 3-бромо-2-метил-проп-1-ена (37.0 г, 27.6 мл, 274 ммоль) в THF (1.8 L) и НМРА (180 мл) добавляли натрия бис(триметилсилил)амид (279 мл, 1 M в THF) по каплям при -70°С за 60 мин в атмосфере аргона. Затем раствор перемешивали при -70°С в течение 30 мин. Реакцию объединяли и гасили насыщенным водным раствором хлорида аммония (2.5 L), затем экстрагировали этилацетатом (2.0 л × 2). Объединенную органическую фазу промывали насыщенным водным раствором хлорида натрия (1.5 л × 2), сушили над сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (простой петролейный эфир/этилацетат = 1/0, 10/1) с получением остатка, смесь остатка и простого петролейного эфира/EtOAc (2.0 л/0.2 л) перемешивали в течение 0.5 ч при 25°С, затем фильтровали с получением 145 г (89% чистота, 62% выход) целевого соединения.
LC-MS: Rt=1.027 мин, MS+Na=430.2
1H ЯМР: CDCl3: 7.35-7.00 (m, 8Н), 6.52 (m, 2Н), 6.35-6.28 (m, 1Н), 5.15-4.82 (m, 4H), 2.95-2.70 (m, 2H), 1.87 (s, 3Н), 1.41 (s, 3Н), 1.03 (s, 6H)
Пример 46А
(3S,5S,6R)-3-[(1-метилциклопропил)метил]-5,6-дифенил-морфолин-2-он
Три партии проводили параллельно. В раствор трет-бутил (3S,5S,6R)-3-(2-метилаллил)-2-оксо-5,6-дифенил-морфолин-4-карбоксилата (пример 45А, 35.0 г, 76 ммоль) в 1.3 л дихлорметана добавляли диэтилцинк (400 мл, 1 М в толуоле) при 0°С за 30 мин, затем хлор (иод)метан (140 г, 793 ммоль, 57.6 мл) добавляли при 0°С за 30 мин. Реакцию перемешивали в течение 1.5 ч при 0°С. Реакцию объединяли и переливали в 2.0 л насыщенного водного раствора бикарбоната натрия, раствор фильтровали, а затем фильтрат экстрагировали дихлорметаном (1.0 L × 2). Объединенную органическую фазу промывали л 1.5 л насыщенного водного раствора хлорида натрия, сушили над сульфатом натрия, фильтровали через короткий кусок кремнезема. Затем фильтрат концентрировали с получением остатка. Смесь остатка и простого петролейнго эфира/этилацетата/дихлорметана (10/1/1, 400 мл) перемешивали в течение 0.5 ч при 25°С и фильтровали. Фильтрат концентрировали до около 120 мл, затем фильтровали с получением 20 г (92% чистота, 25% выход) целевого соединения.
LC/MS: Rt=0.909 мин, М+Н+=322.2
Пример 47А
(2S)-2-(трет-бутоксикарбониламино)-3-(1-метилциклопропил)пропановая кислота
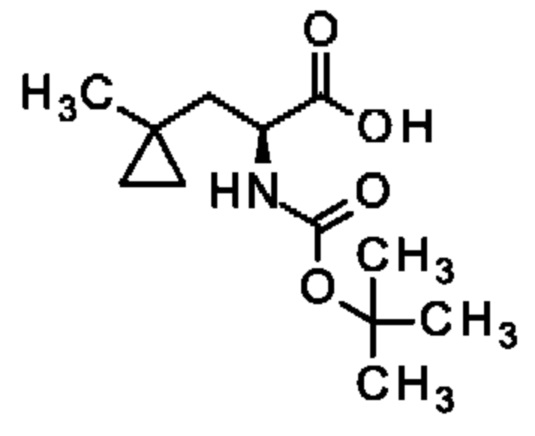
К перемешанному раствору, содержащему этанол (50 мл), жидкий аммиак (600 мл), THF (500 мл) и (3S,5S,6R-3-[(1-метилциклопропил)метил]-5,6-дифенил-морфолин-2-он (пример 46А, 57.4 г), который охлаждали до -70°С, добавляли литий (8 г, 1.15 моль) небольшими частями за 0.5 ч при -70°С пока темно-синий цвет не сохранялся в течение 10 мин при -70°С. Реакцию гасили осторожным добавлением 50 мл 20% водного раствора хлорида аммония, холодную баню удаляли, и реакцию нагревали до 25°С. Реакционную смесь разбавляли 300 мл воды и экстрагировали с МТВЕ (200 мл × 2). Водную фазу охлаждали на льду, подкисляли до рН=1 с 1 М водной соляной кислотой, и немедленно экстрагировали этилацетатом (800 мл × 2). Объединенные фазы этилацетата промывали 300 мл насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и концентрировали при пониженном давление с получением 24 г (99 ммоль) целевого соединения.
Пример 48А
(2S)-2-амино-3-(1-метилциклопропил)пропановой кислоты гидрохлорид
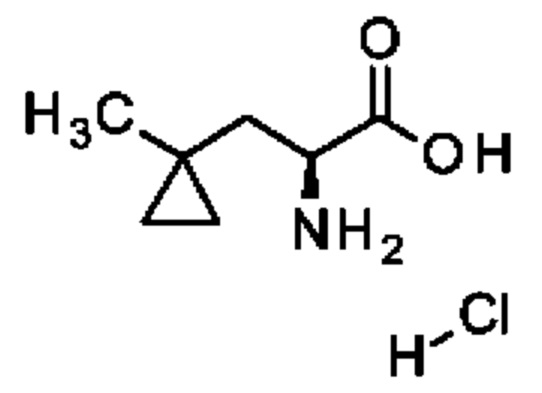
К раствору (2S)-2-(трет-бутоксикарбониламино)-3-(1-метилциклопропил)пропановой кислоты (пример 47А, 24.0 г, 99 ммоль) в дихлорметане (60 мл) добавляли соляную кислоту/диоксан (200 мл, 4М) за 15 мин при 25°С, затем раствор перемешивали в течение 0.5 ч при 25°С. Реакцию концентрировали с получением остатка. Остаток обрабатывали с 150 мл МТВЕ и перемешивали в течение 15 мин при 25°С, затем фильтровали с получением 16 г (90% выход) целевого соединения.
1H ЯМР: DMSO-d6: 0.21-0.33 (3Н, m), 0.37-0.51 (1Н, m), 1.64-1.81 (2Н, d), 3.88-3.92 (1Н, t), 8.41 (3Н, s), 13.73 (1Н, s)
Пример 49А
(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-(1-метилциклопропил)пропановая кислота
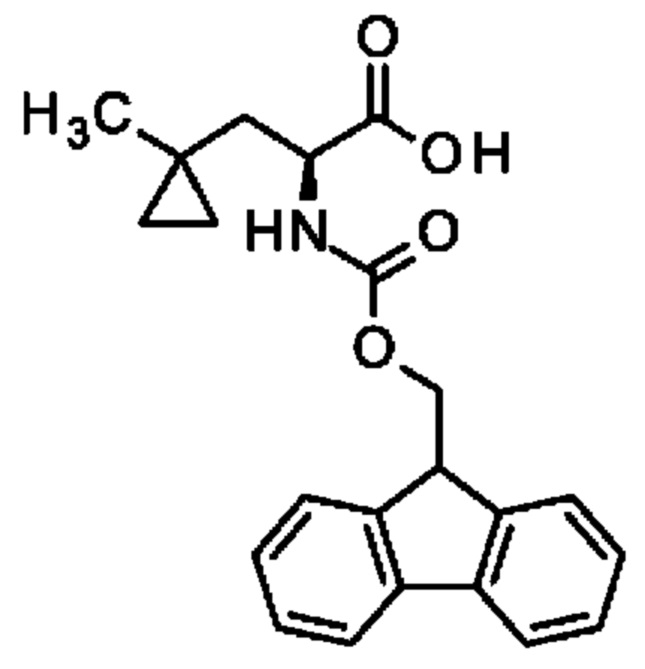
В раствор ((2S)-2-амино-3-(1-метилциклопропил)пропановой кислоты гидрохлорида (пример 48А, 14 г, 77.9 ммоль) и карбоната натрия (25 г, 236 ммоль) в THF (200 мл) и воде (200 мл) добавляли 1-({[(9Н-флуорен-9-ил)метокси]карбонил}окси)пирролидин-2,5-дион (25 г, 74.1 ммоль) за 30 мин при 0°С, затем раствор перемешивали в течение 2 ч при 0°С. 100 мл воды добавляли. Раствор экстрагировали с МТВЕ (300 мл × 2), перед этим рН водной фазы доводили до 2 с помощью 2 М водной соляной кислоты. Объединенную органическую фазу промывали 150 мл насыщенного водного раствора хлорида натрия, сушили над сульфатом натрия, и фильтровали через небольшую колонку диоксида кремния. Фильтрат концентрировали с получением остатка. Объединенный остаток (из предшествующих циклов) очищали посредством хроматографии на силикагеле (петролейный простой эфир / этилацетат (10:1, 3:1) с получением получистого остатка. Остаток далее очищали посредством препаративной ВЭЖХ (колонка: Phenomenex Luna CI8 250 × 50 мм, 10 мкм; подвижная фазае: [вода (0.05% HCl)-ACN]; В%: 38% - 68%, 31min, 50% мин) с получением 24.4 г (97% чистота, 74% выход) целевого соединения.
LC-MS: Rt=0.914 мин, [М+Н]+=366.1
1H ЯМР: CDCl3: δ ppm 0.23-0.40 (4Н, m), 1.02-1.20 (3Н, m), 1.48-1.95 (2Н, m), 4.22-4.33 (1Н, m), 4.39-4.61 (3Н, m), 5.26-5.33 (1Н, m), 7.31-7.49 (4Н, m), 7.56-7.81 (4Н, m).
Эту аминокислоту применяли в Fmoc SPPS.
Пример 50А
Метил N-(трет-бутоксикарбонил)-D-метионил-L-изолейцинат
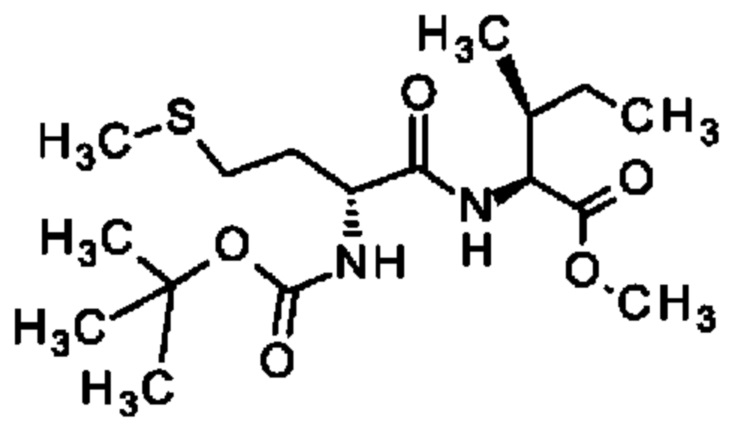
N-(трет-бутоксикарбонил)-D-метионин (2.42 г, 9.71 ммоль), метил L-изолейцинат-хлорид водорода (1/1) (1.76 г, 9.71 ммоль), 1H-бензотриазол-1-ол (1.78 г, 11.6 ммоль), 3-{[(Этилимино)метилен]амино}-N,N-диметилпропан-1-амингидрохлорид (1:1) (2.23 г, 11.6 ммоль) и N,N-диизопропилэтиламин (3.5 мл, 20 ммоль) растворяли в DMF (30 мл). Смесь перемешивали при температуре окружающей среды в течение 16 ч. Воду (100 мл) затем добавляли, смесь экстрагировали двумя порциями этилацетат. Объединенный органический экстракт промывали последовательно соляной кислотой, водой (10 мл), насыщенным водным раствором бикарбоната натрия (10 мл) и солевым раствором, а затем сушили над безводным сульфатом натрия. Органический слой фильтровали и фильтрат концентрировали. Остаток растворяли в простом эфире и продукт кристаллизовали. Твердое вещество фильтровали и промывали последовательно простым эфиром и пентаном. 3.13 г (99% чистота, 86% выход) указанного в названии соединения получали.
Пример 51А
Метил (2S,3S)-2-{(3R)-3-[(трет-бутоксикарбонил)амино]-2-оксопирролидин-1-ил}-3-метилпентаноат
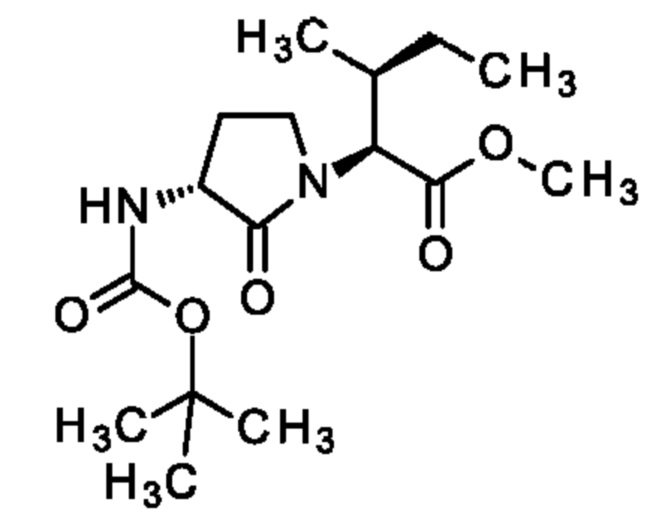
Метил N-(трет-бутоксикарбонил)-D-метионил-L-изолейцинат (пример 50А, 2.85 г, 7.57 ммоль) растворяли в дихлорметане (40 мл) и раствор охлаждали до 0°С. Триметилоксония тетрафторборат (1.12 г, 7.57 ммоль) добавляли и смесь перемешивали при комнатной температуре в течение 3 ч. Карбонат калия (3.13 г, 22.71 ммоль) добавляли. Затем смесь нагревали с возвратом флегмы в течение 16 ч. Реакционную смесь переливали в 50 мл дихлорметана и промывали пятью порциями воды. Органический слой промывали солевым раствором, сушили над безводном сульфатом магния, а затем фильтровали. Фильтрат концентрировали и кристаллизовали из гексана с получением 2.01 г (100% чистота, 81% выход) указанного в названии соединения.
Пример 52А
(2S,3S)-2-{(3R)-3-[(трет-бутоксикарбонил)амино]-2-оксопирролидин-1-ил}-3-метилпентановая кислота
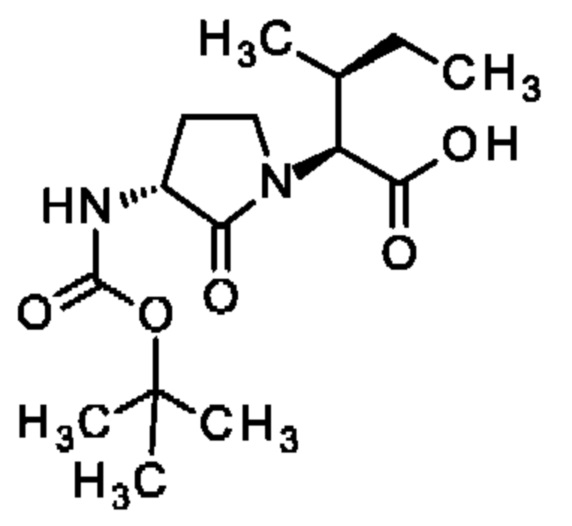
Метил (2S,3S)-2-{(3R)-3-[(трет-бутоксикарбонил)амино]-2-оксопирролидин-1-ил}-3-метилпентаноат (пример 51А, 1.96 г, 5.97 ммоль) растворяли в THF (7.8 мл) и метаноле (7.8 мл). Раствор гидроксида лития (300 мг, 12.5 ммоль) в 8 мл воды добавляли и смесь перемешивали в течение 1 ч при комнатной температуре. Затем реакционную смесь концентрировали, белый осадок растворяли в воде (40 мл), промывали с DCM (6 мл), а затем подкисляли 1 М соляной кислотой (12 мл). Смесь экстрагировали тремя частями этилацетата. Объединенный органический экстракт сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали. Остаток кристаллизовали из этилацетата и гексана с получением 1.3 г (100% чистота, 67% выход) указанного в названии соединения.
LC-MS (Способ 11): Rt=1.09 мин; MS (ESIneg): m/z=313 [М-Н]-
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 0.810 (0.96), 0.829 (2.41), 0.847 (1.25), 0.889 (2.53), 0.906 (2.59), 1.387 (16.00), 3.260 (0.71), 3.274 (0.56), 3.283 (0.75), 3.298 (0.64), 4.200 (0.97), 4.225 (0.94).
Пример 53А
Метил N-(трет-бутоксикарбонил)глицил-L-изолейцинат
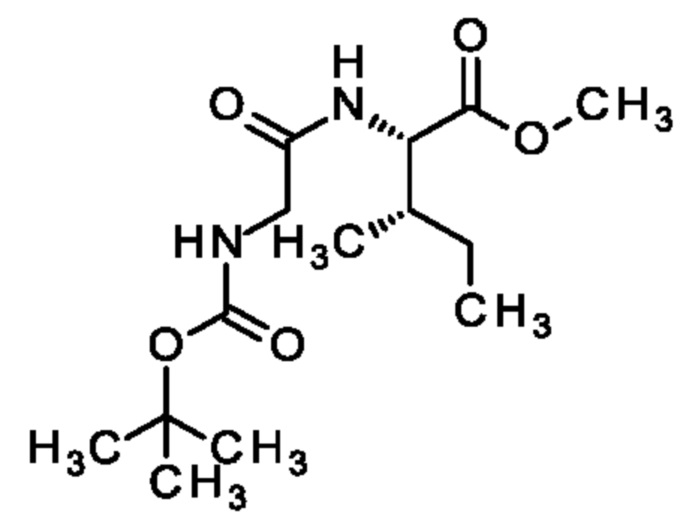
Метил L-изолейцинат (3.00 г, 20.7 ммоль) и N-(трет-бутоксикарбонил)глицин (3.98 г, 22.7 ммоль) растворяли в DMF (20 мл). Этот раствор охлаждали до 0-5°С на ледяной бане. N,N-диизопропилэтиламин (3.6 мл, 21 ммоль), 1H-бензотриазол-1-ол гидрат (316 мг, 2.07 ммоль) и 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид (4.36 г, 22.7 ммоль) добавляли и смесь перемешивали при комнатной температуре. Реакционную смесь гасили концентрированным водным хлоридом аммония и экстрагировали тремя частями этилацетата. Объединенные органические экстракты сушили над безводном сульфатом магния и концентрировали. Неочищенный продукт очищали посредством хроматографии на силикагеле (циклогексан / этилацетат 0% -10%) с получением указанного в названии соединения, 5.40 г (90% чистота, 78% выход).
LC-MS (Способ 11): Rt=1.13 мин; MS (ESI pos): m/z=247 [M-tBu]+
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 0.819 (4.90), 0.835 (8.09), 0.853 (2.63), 1.152 (0.40), 1.174 (0.45), 1.354 (1.52), 1.375 (16.00), 1.459 (0.46), 3.561 (0.84), 3.571 (0.98), 3.577 (1.45), 3.594 (0.81), 3.630 (11.35), 4.220 (0.63), 4.237 (0.78), 4.240 (0.82), 4.257
Пример 54A
Метил глицил-L-изолейцинат
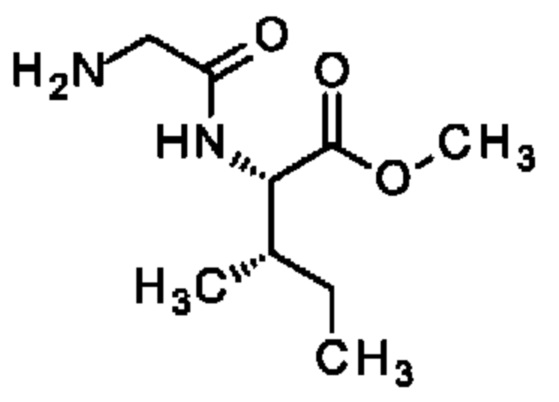
Метил N-(трет-бутоксикарбонил)глицил-L-изолейцинат (пример 53А, 1.50 г, 4.96 ммоль) растворяли в DCM (27 мл). Трифторуксусную кислоту (3.0 мл, 39 ммоль) добавляли и реакционную смесь перемешивали в течение 30 мин при комнатной температуре. Реакционную смесь концентрировали с получением неочищенного продукта, который применяли непосредственно в ходе следующей стадии.
Пример 55А
Метил N-(4-нитробензол-1-сульфонил)глицил-L-изолейцинат
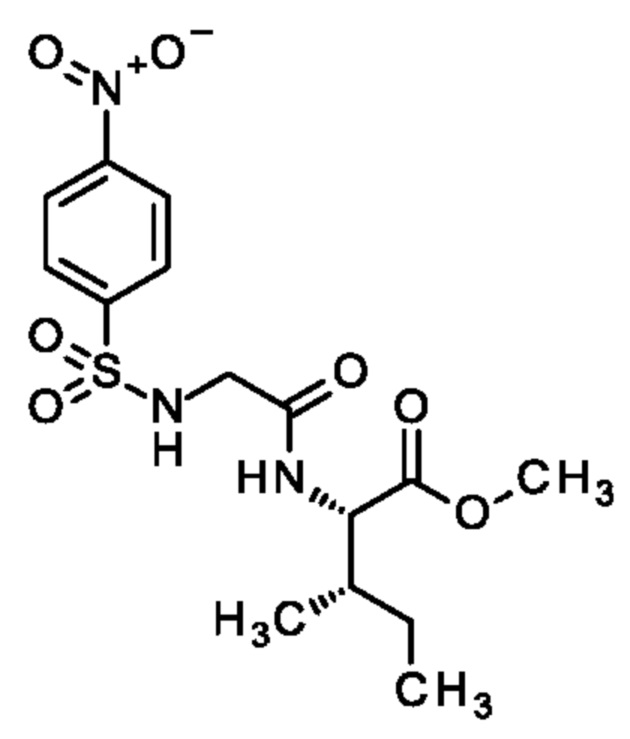
Метил глицил-L-изолейцинат (пример 54А, 1.00 г, 4.96 ммоль) растворяли в дихлорметане (40 мл, 620 ммоль), триэтиламин (2.4 мл, 17 ммоль) добавляли, а затем раствор охлаждали до 0°С. 4-нитробензол-1-сульфонил хлорид (1.21 г, 5.46 ммоль) добавляли и реакционную смесь перемешивали в течение 1 ч. Реакцию затем гасили концентрированным водным хлоридом аммония и экстрагировали тремя частями этилацетата. Объединенные органические экстракты сушили над безводном сульфатом магния, фильтровали, а затем концентрировали, с получением 1.70 г (71% выход) указанного в названии соединения. Продукт применяли непосредственно в ходе следующей стадии.
Пример 56А
Метил (2S,3S)-3-метил-2-[4-(4-нитробензол-1-сульфонил)-2-оксопиперазин-1-ил]пентаноат
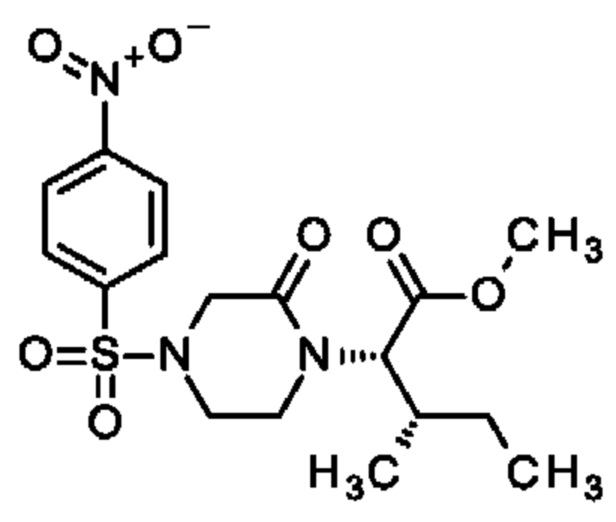
Метил N-(4-нитробензол-1-сульфонил)глицил-L-изолейцинат (пример 55А, 1.92 г, 4.96 ммоль) и 1,2-дибромэтан (4.3 мл, 50 ммоль) растворяли в DMF. Карбонат калия (6.86 г, 49.6 ммоль) добавляли и смесь нагревали до 60°С в течение 3 ч. Реакцию затем гасили концентрированным водным хлоридом аммония и экстрагировали тремя частями простого эфира. Объединенные органические экстракты сушили над безводном сульфатом магния, фильтровали, а затем выпаривали. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (циклогексан / этилацетат 6:1) с получением 1.40 г указанного в названии соединения (97% чистота, 66% выход).
LC-MS (Способ 11): Rt=1.30 мин; MS (ESI pos): m/z=414 [M+H]+
Пример 57А
Метил (2S, 3S)-3-метил-2-(2-оксопиперазин-1-ил)пентаноат
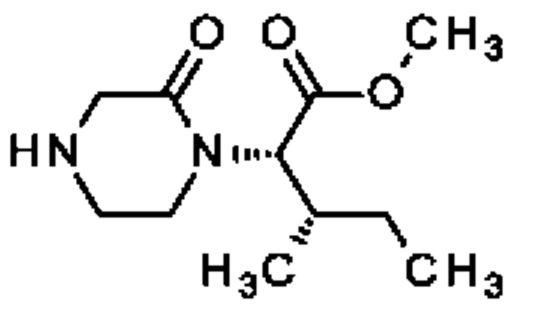
Метил (2S,3S)-3-метил-2-[4-(4-нитробензол-1-сульфонил)-2-оксопиперазин-1-ил]пентаноат (пример 56А, 1.40 г, 3.39 ммоль) и бензолтиол (1.0 мл, 10 ммоль) растворяли в ацетонитриле (20 мл). Карбонат калия (936 мг, 6.77 ммоль) добавляли и смесь перемешивали при комнатной температуре. Смесь фильтровали и выпаривали. Остаток очищали посредством флэш-хроматографии на силикагеле (градиент дихлорметан / метанол 100:0 до 95:5) с получением 310 мг (40% выход) указанного в названии соединения, которое применяли непосредственно в ходе следующей стадии.
Пример 58А
трет-бутил 4-[(2S,3S)-1-метокси-3-метил-1-оксопентан-2-ил]-3-оксопиперазин-1-карбоксилат
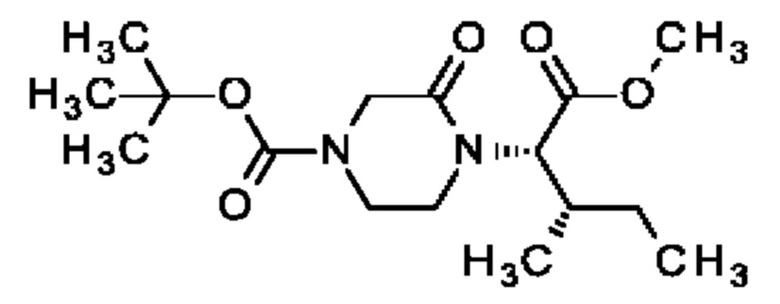
Метил (2S,3S)-3-метил-2-(2-оксопиперазин-1-ил)пентаноат (пример 57А, 310 мг, 1.36 ммоль) растворяли в дихлорметане (1.7 мл, 26 ммоль). Ди-трет-бутил дикарбонат (370 мкл, 1.6 ммоль) добавляли и смесь перемешивали при комнатной температуре в течение 1 ч. Реакцию затем гасили концентрированным водным хлоридом аммония, а затем экстрагировали тремя частями DCM (20 мл). Объединенные органические экстракты сушили над безводном сульфатом магния, фильтровали, а затем концентрировали, с получением 440 мг (99% чистота, 98% выход) указанного в названии соединения.
LC-MS (§Способ 11): Rt=1.27 мин; MS (ESI pos): m/z=273 [M+H-tBu]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 0.807 (0.86), 0.826 (2.27), 0.844 (1.20), 0.870 (2.25), 0.887 (2.30), 1.410 (16.00), 3.645 (6.00), 3.963 (0.63), 3.970 (0.62), 4.766 (0.82), 4.792 (0.80), 5.754 (0.87).
Пример 59A
(2S,3S)-2-[4-(трет-бутоксикарбонил)-2-оксопиперазин-1-ил]-3-метилпентановая кислота
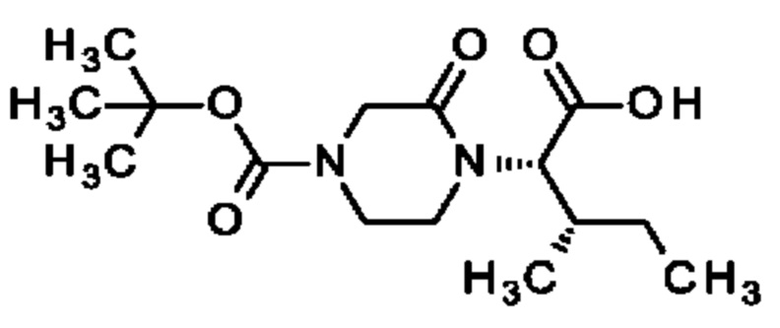
трет-бутил 4-[(2S,3S)-1-метокси-3-метил-1-оксопентан-2-ил]-3-оксопиперазин-1-карбоксилат (пример 58А, 440 мг, 80% чистота, 1.07 ммоль) растворяли в THF (1.4 мл) и метанол (1.4 мл). Раствор гидроксида лития (53.9 мг, 2.25 ммоль) в воде (1.4 мл) добавляли и смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали и остаток растворяли в воде, а затем промывали с DCM. Водный слой разделяли, подкисляли до рН 3-4 с 1М соляной кислотой, а затем экстрагировали три раза этилацетатом. Объединенный органический слой сушили над безводном сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении с получением 310 мг (95% чистота, 87% выход) указанного в названии соединения в виде бесцветного масла.
LC-MS (Способ 11): Rt=1.11 мин; MS (ESIneg): m/z=313 [М-Н]-
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 0.809 (0.81), 0.827 (2.06), 0.845 (1.12), 0.908 (2.11), 0.924 (2.09), 1.411 (16.00), 3.438 (0.43), 3.968 (0.78), 4.688 (0.79), 4.713 (0.77).
Пример 60А
Метил N-(трет-бутоксикарбонил)-L-аланил-L-изолейцинат
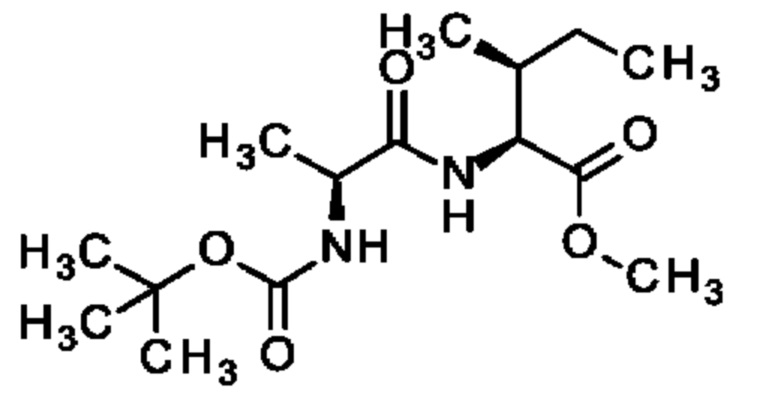
N-(трет-бутоксикарбонил)-L-аланин (1.50 г, 7.93 ммоль) и метил L-изолейцинат-хлорид водорода (1/1) (1.58 г, 8.72 ммоль) растворяли в DMF (7.7 мл). Этот раствор охлаждали до 0-5°С на ледяной бане. N,N-диизопропилэтиламин (1.4 мл, 7.9 ммоль), 1Н-бензотриазол-1-олгидрат (121 мг, 793 мкмоль) и 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид (1.67 г, 8.72 ммоль) добавляли и смесь перемешивали при комнатной температуре. Реакцию затем гасили концентрированным водным хлоридом аммония и экстрагировали тремя частями этилацетата. Объединенные органические экстракты сушили над безводном сульфатом магния, фильтровали, а затем концентрировали. Неочищенный продукт очищали посредством хроматографии на силикагеле (циклогексан / этилацетат 0% - 15%) с получением 2.12 г (100% чистота, 84% выход) указанного в названии соединения.
LC LC-MS (Способ 10): Rt=1.70 мин; MS (ESI pos): m/z=317 [M+H]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: -0.008 (1.00), 0.008 (0.77), 0.818 (3.17), 0.825 (4.25), 0.836 (7.51), 0.842 (4.72), 0.855 (3.30), 1.144 (4.07), 1.161 (4.10), 1.179 (0.54), 1.200 (0.53), 1.371 (16.00), 1.398 (13.44), 1.775 (0.49), 2.523 (0.85), 3.621 (15.73), 4.021 (0.46), 4.039 (0.60), 4.207 (1.06), 4.223 (1.19), 4.228 (1.22), 4.244 (1.06), 6.908 (0.61), 6.928 (0.58), 7.886 (0.57), 7.907 (0.60).
Пример 61А
Метил L-аланил-L-изолейцинат
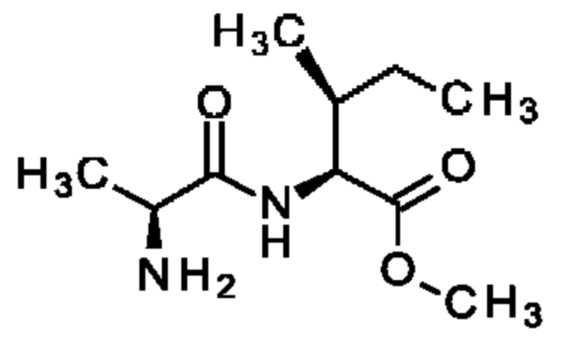
Метил N-(трет-бутоксикарбонил)-L-аланил-L-изолейцинат (пример 60А, 2.10 г, 6.64 ммоль) растворяли в DCM (20 мл). Трифторуксусную кислоту (20 мл, 260 ммоль) добавляли и реакционную смесь перемешивали в течение 30 мин при комнатной температуре. Реакционную смесь концентрировали и неочищенный дипептид применяли непосредственно в ходе следующей стадии.
Пример 62А
Метил N-(4-нитробензол-1-сульфонил)-L-аланил-L-изолейцинат
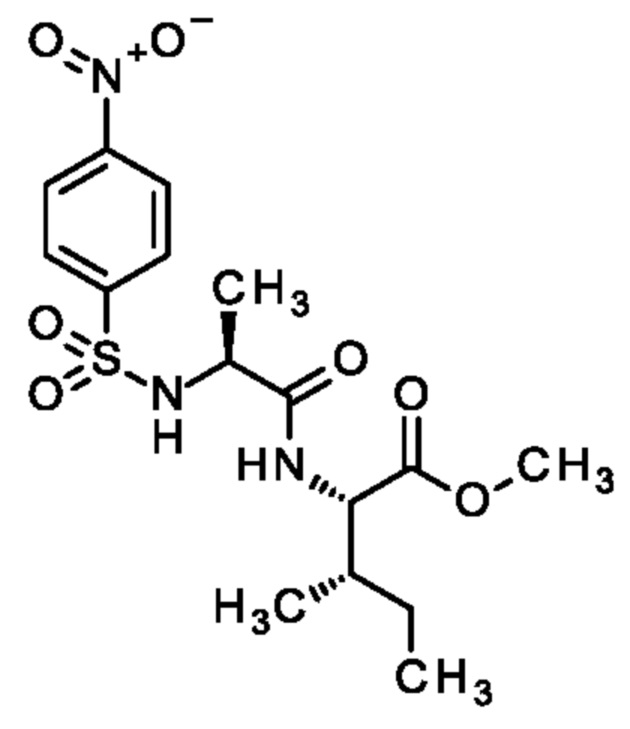
Метил L-аланил-L-изолейцинат (пример 61А, 1.44 г, 6.64 ммоль) растворяли в дихлорметане (54 мл), триэтиламин (3.2 мл, 23 ммоль) добавляли, а затем раствор охлаждали до 0°С. 4-нитробензол-1-сульфонил хлорид (1.62 г, 7.30 ммоль) добавляли и реакционную смесь перемешивали. Реакцию затем гасили концентрированным водным хлоридом аммония и экстрагировали двумя частями дихлорметана. Объединенные органические экстракты промывали насыщенным водным раствором хлорида натрия, сушили над безводном сульфатом магния, фильтровали, и концентрировали с получением 1.65 г (95% чистота, 59% выход) указанного в названии соединения.
LC-MS (Способ 11): Rt=1.18 мин; MS (ESI neg): m/z=400 [M-H]-
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 0.008 (0.40), 0.676 (6.59), 0.693 (6.80), 0.740 (2.79), 0.758 (6.99), 0.777 (3.46), 1.013 (0.52), 1.025 (0.62), 1.047 (0.69), 1.066 (0.50), 1.117 (6.09), 1.135 (6.12), 1.175 (0.51), 1.192 (0.64), 1.203 (0.60), 1.210 (0.56), 1.222 (0.67), 1.237 (0.48), 1.243 (0.42), 1.551 (0.53), 1.568 (0.61), 1.578 (0.45), 1.584 (0.47), 1.988 (0.64), 3.577 (16.00), 3.933 (1.14), 3.948 (1.32), 3.953 (1.31), 3.968 (1.12), 4.056 (0.61), 4.073 (0.82), 4.091 (0.54), 7.994 (4.08), 7.999 (1.44), 8.012 (1.63), 8.016 (4.58), 8.135 (1.41), 8.155 (1.39), 8.365 (4.39), 8.383 (1.48), 8.388 (3.90), 8.463 (1.04), 8.482 (0.99).
Пример 63А
Метил (2S,3S)-3-метил-2-[(3S)-3-метил-4-(4-нитробензол-1-сульфонил)-2-оксопиперазин-1-ил]пентаноат
Метил N-(4-нитробензол-1-сульфонил)-L-аланил-L-изолейцинат (пример 62А, 1.65 г, 4.11 ммоль) и 1,2-дибромэтан (3.5 мл, 41 ммоль) растворяли в DMF (35 мл). Карбонат калия (5.68 г, 41.1 ммоль) добавляли и реакционную смесь нагревали до 60°С в течение 3 ч. Реакцию затем гасили концентрированным водным хлоридом аммония и экстрагировали тремя частями простого эфира. Объединенные органические экстракты сушили над безводном сульфатом магния, фильтровали, а затем концентрировали. Неочищенный продукт очищали посредством хроматографии на силикагеле (циклогексан / этилацетат 6:1) с получением 1.60 г (70% чистота, 64% выход) указанного в названии соединения.
LC-MS (Способ 11): Rt=1.30 мин; MS (ESI pos): m/z=428 [M+H]+
Пример 64А
Метил (2S,3S)-3-метил-2-[(3S)-3-метил-2-оксопиперазин-1-ил]пентаноат
Метил (2S,3S)-3-метил-2-[(3S)-3-метил-4-(4-нитробензол-1-сульфонил)-2-оксопиперазин-1-ил]пентаноат (пример 63А, 900 мг, 2.11 ммоль) и бензолтиол (650 мкл, 6.3 ммоль) растворяли в ацетонитриле (12 мл). Карбонат калия (582 мг, 4.21 ммоль) добавляли и реакционную смесь перемешивали при комнатной температуре всю ночь. Смесь фильтровали и концентрировали. Остаток очищали посредством флэш-хроматографии на силикагеле (градиент дихлорметан / метанол 100:0 до 95:5) с получением 336 мг (100% чистота, 66% выход) указанного в названии соединения.
LC-MS (Способ 13): Rt=1.09 мин; MS (ESI pos): m/z=243 [M+H]+
Пример 65А
трет-бутил (2S)-4-[(2S,3S)-1-метокси-3-метил-1-оксопентан-2-ил]-2-метил-3-оксопиперазин-1-карбоксилат
Метил (2S,3S)-3-метил-2-[(3S)-3-метил-2-оксопиперазин-1-ил]пентаноат (пример 64А, 736 мг, 3.04 ммоль) растворяли в дихлорметан (8 мл). Ди-трет-бутил дикарбонат (1.0 мл, 4.6 ммоль) добавляли, и реакционную смесь перемешивали при комнатной температуре в течение выходных. Реакцию затем гасили концентрированным водным хлоридом аммония и экстрагировали двумя порциями DCM. Объединенные органические экстракты промывали раствором хлорида натрия, сушили над безводном сульфатом магния, фильтровали, а затем концентрировали. Неочищенный продукт очищали посредством хроматографии на силикагеле (циклогексан / этилацетат 3:1) с получением 1.08 г (100% чистота, 104% выход) указанного в названии соединения.
LC-MS (Способ 10): Rt=1.98 мин; MS (ESI pos): m/z=343 [M+H]+
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 0.800 (0.95), 0.818 (2.41), 0.837 (1.25), 0.875 (2.38), 0.892 (2.43), 1.305 (2.02), 1.323 (2.06), 1.414 (16.00), 3.645 (6.08), 4.769 (0.88), 4.795 (0.86).
Пример 66А
(2S,3S)-2-[(3S)-4-(трет-бутоксикарбонил)-3-метил-2-оксопиперазин-1-ил]-3-метилпентановая кислота
трет-бутил (2S)-4-[(2S,3S)-1-метокси-3-метил-1-оксопентан-2-ил]-2-метил-3-оксопиперазин-1-карбоксилат (пример 65А, 1.08 г, 3.15 ммоль) растворяли в смеси THF (4.5 мл) и метанола (4.5 мл). Раствор гидроксида лития (159 мг, 6.62 ммоль) в воде (4.5 мл) добавляли и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали и остаток растворяли в концентрированном водном хлориде аммония. Водный слой подкисляли до рН 3-4 1М соляной кислотой и экстрагировали три раза с DCM. Объединенный органический слой сушили над безводном сульфатом магния, фильтровали, а затем концентрировали с получением 952 мг (100% чистота, 92% выход) указанного в названии соединения.
LC-MS (Способ 9): Rt=0.87 мин; MS (ESI pos): m/z=329 [M+H]+
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 0.802 (1.09), 0.820 (2.24), 0.838 (1.18), 0.911 (2.22), 0.927 (2.28), 1.295 (0.50), 1.308 (2.03), 1.326 (1.99), 1.415 (16.00), 3.342 (0.65), 4.691 (0.84), 4.717(0.82).
Пример 67 А
(2R,5R)-3,6-диметокси-2-(пропан-2-ил)-5-[(триметилсилил)метил]-2,5-дигидропиразин
(2R)-3,6-диметокси-2-(пропан-2-ил)-2,5-дигидропиразин (2.9 мл, 16 ммоль) растворяли в THF (58 мл) и раствор охлаждали до -78°С, н-бутиллитий (10 мл, 1.6 М, 16 ммоль) добавляли по каплям при перемешивании. Перемешивание продолжали в течение 15 мин при -78°С. (Хлорметил)(триметил)силан (5.0 мл, 36 ммоль) добавляли и полученную смесь перемешивали при температуре окружающей среды всю ночь. Реакционную смесь разбавляли водой и экстрагировали диэтиловым простым эфиром. Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали. Неочищенный продукт очищали посредством хроматографии на силикагеле (циклогексан / этилацетат 2%) с получением 3.0 г (87% чистота, 60% выход) указанного в названии соединения.
LC-MS (Способ 9): Rt=1.47 мин; MS (ESI pos): m/z=271 [M+H]+
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: -0.036 (0.45), -0.028 (9.14), -0.020 (0.46), -0.008 (1.38), 0.580 (3.69), 0.597 (3.75), 0.737 (0.61), 0.762 (0.63), 0.773 (0.73), 0.798 (0.73), 0.959 (3.60), 0.977 (3.69), 1.107 (0.73), 1.119 (0.75), 1.143 (0.66), 1.148 (0.47), 1.155 (0.63), 1.370 (1.30), 2.512 (16.00), 3.574 (8.95), 3.582 (8.85), 3.885 (0.64), 3.894 (1.26), 3.903 (0.72), 3.999 (0.49), 4.023 (0.47).
Пример 68А
Метил 3-(триметилсилил)-L-аланинат
(2R, 5R)-3,6-диметокси-2-(пропан-2-ил)-5-[(триметилсилил)метил]-2,5-дигидропиразин (пример 67 А, 3.00 г, 11.1 ммоль) растворяли в метаноле (30 мл, 740 ммоль) и раствор охлаждали до 0°С. Соляную кислоту (10 мл, 10%) добавляли и раствор перемешивали в течение 2 ч. Раствор выпаривали и остаток растворяли в DCM и растворе карбоната натрия (100 мл, 2.0 М, 200 ммоль). Водный слой экстрагировали двумя порциями DCM. Экстрат DCM сушили над безводном сульфатом магния, фильтровали, а затем концентрировали. Неочищенный продукт очищали посредством хроматографии на силикагеле (2% метанол: (1% N,N-диэтилэтанамин в DCM)) с получением 2.83 г (68% чистота, 99% выход) указанного в названии соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: -0.010 (3.10), 0.008 (1.81), 0.736 (0.85), 0.759 (0.89), 0.772 (1.48), 0.795 (1.56), 0.803 (1.89), 0.820 (1.90), 0.841 (1.84), 0.858 (2.43), 0.875 (1.64), 0.900 (4.19), 0.911 (1.07), 0.918 (8.65), 0.936 (4.28), 1.682 (1.42), 2.384 (1.35), 2.402 (4.11), 2.420 (4.02), 2.438 (1.25), 3.088 (0.55), 3.102 (0.54), 3.302 (1.19), 3.306 (1.81), 3.321 (1.27), 3.330 (1.15), 3.345 (1.03), 3.583 (16.00), 3.591 (0.48), 3.605 (4.33), 5.742 (4.37).
Пример 69А
N-{[(9H-флуорен-9-ил)метокси]карбонил}-3-(триметилсилил)-L-аланин
Метил 3-(триметилсилил)-L-аланинат (пример 68А, 2.83 г, 68% чистота, 11.0 ммоль) растворяли в метаноле (44 мл) и раствор охлаждали до 0°С. Раствор гидроксида натрия (11 мл, 1.0 М, 11 ммоль) добавляли и реакционную смесь перемешивали всю ночь при комнатной температуре. Воду (40 мл), гидрокарбонат натрия (922 мг, 11.0 ммоль) и карбонат натрия (1.53 г, 14.4 ммоль) добавляли. Метанол удаляли при пониженном давлении, а затем 1,4-диоксан (32 мл) добавляли в водный раствор. Этот раствор охлаждали до 0°С на ледяной бане. (9H-флуорен-9-ил)метил хлорформиат (Fmoc-Cl) (4.26 г, 16.5 ммоль) затем добавляли и реакционную смесь перемешивали в течение 5 ч при комнатной температуре. Дополнительный Fmoc-Cl (1.42 г, 5.5 ммоль) добавляли и реакционную смесь перемешивали в течение еще 2 ч. Эквивалентные порции воды и трет-бутилметилового простого эфира добавляли в реакционную смесь. Водную фазу разделяли и промывали еще трет-бутилметиловым простым эфиром, затем подкисляли 1 М соляной кислотой (рН 3-4). Подкисленную смесь экстрагировали тремя частями МТВЕ. Объединенный органический слой сушили над безводном сульфатом магния, фильтровали, а затем концентрировали. Неочищенный продукт очищали посредством препаративной ВЭЖХ (колонка: Reprosil; С18; 10 мкм; 125 × 40 мм; элюент: AcCN/H2O с 0,1% НСООН; скорость потока: 100 мл/мин; градиент: 0-5.50 мин 10:90; впрыск образца при 3.00 мин; 5.50 - 17.65 мин до 95:5; 17.65-19.48 мин 95:5; 19.48-19.66 мин - 10:90; 19.66-20.72 мин 10:90;) с получением 1.65 г (99% чистота, 39% выход) указанного в названии соединения.
LC-MS (Способ 10): Rt=2.20 мин; MS (ESI pos): m/z=384 [M+H]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: -0.006 (16.00), 0.987 (3.81), 1.006 (3.64), 3.309 (2.95), 3.950 (0.67), 3.970 (1.71), 3.991 (1.59), 4.010 (0.53), 4.182 (0.67), 4.199 (1.66), 4.217 (1.67), 4.228 (1.08), 4.253 (2.79), 4.268 (3.86), 4.287 (1.60), 4.293 (0.85), 4.313 (0.41), 7.286 (1.24), 7.291 (1.27), 7.293 (1.31), 7.302 (2.68), 7.304 (2.70), 7.309 (2.55), 7.321 (1.75), 7.323 (1.70), 7.328 (1.57), 7.389 (2.90), 7.408 (4.73), 7.426 (2.11), 7.566 (1.81), 7.587 (1.74), 7.697 (2.14), 7.714 (3.62), 7.731 (1.81), 7.872 (4.43), 7.891 (4.02), 12.451 (0.54).
Эту аминокислоту применяли в Fmoc SPPS.
Пример 70А
Ди-трет-бутил (2S)-5-гидроксипирролидин-1,2-дикарбоксилат (смесь изомеров)
Ди-трет-бутил (2S)-5-оксопирролидин-1,2-дикарбоксилат (CAS-RN: 91229-91-3, 21.0 г, 73.6 ммоль) растворяли в 250 мл THF и раствор охлаждали до -78°С. Диизобутилалюминия гидрид (160 мл, 1.0 М, 160 ммоль) добавляли по каплям и смесь перемешивали в течение 1 ч при -78°С. Реакцию затем гасили 2-пропанолом (25 мл). Раствор калия натрия (2R,3R)-2,3-дигидроксибутандиоата (200 мл) добавляли, смесь перемешивали при комнатной температуре, а затем реакционную смесь разбавляли МТВЕ и водой. Водный слой разделяли и экстрагировали еще двумя порциями МТВЕ. Объединенный органический экстракт сушили над безводном сульфатом магния, фильтровали, и остаток концентрировали при пониженном давлении с получением 19.53 г (79% чистота, 73% выход) указанного в названии соединения в виде бесцветного масла.
Пример 71А
Ди-трет-бутил (2S)-5-метоксипирролидин-1,2-дикарбоксилат (смесь изомеров)
Ди-трет-бутил (2S)-5-гидроксипирролидин-1,2-дикарбоксилат (пример 70А, 19.5 г, 67.9 ммоль) растворяли в метаноле (160 мл). Пиридиния пара-толуолсульфонат (341 мг, 1.36 ммоль) добавляли в раствор и реакционную смесь перемешивали всю ночь при комнатной температуре. Реакционную смесь концентрировали и неочищенный продукт очищали посредством флэш-хроматографии на силикагеле (градиент циклогексан - циклогексан этилацетат 7:3) с получением 14.4 г (90% чистота, 64% выход) указанного в названии соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 1.363 (9.51), 1.375 (8.10), 1.393 (2.67), 1.419 (16.00), 1.756 (0.58), 1.775 (0.93), 1.781 (0.88), 1.807 (0.46), 3.225 (3.77), 3.240 (1.93), 3.261 (2.02), 3.307 (4.01), 4.059 (0.64), 4.081 (0.82), 5.128 (0.56).
Пример 72А
Ди-трет-бутил (2S)-5-цианопирролидин-1,2-дикарбоксилат (смесь изомеров)
Ди-трет-бутил (2S,)-5-метоксипирролидин-1,2-дикарбоксилат (пример 71А, 14.4 г, 47.8 ммоль) растворяли в DCM (125 мл) и раствор охлаждали до -40°С. Триметилсилил трифторметансульфонат (1.3 мл) (1 об. %) добавляли, затем триметилсиланкарбонитрил (7.0 мл, 53 ммоль) добавляли по каплям за 40 мин. Смесь затем перемешивали в течение еще 1.5 ч при -40°С. Реакционную смесь гасили метанолом (19 мл), а затем концентрировали с выделением 14.87 г (86% выход) указанного в названии соединения.
GC-MS (GC-MS Способ 1): 195.2 (М+-Вос), 139.1 (-Boc, -t-Bu)
Пример 73А
Ди-трет-бутил (2S)-5-формилпирролидин-1,2-дикарбоксилат (смесь изомеров)
Ди-трет-бутил (2S)-5-цианопирролидин-1,2-дикарбоксилат (пример 72А, 14.0 г, 47.2 ммоль) растворяли в смеси пиридина (250 мл), уксусной кислоты (130 мл) и воды (130 мл). Суспензию никеля Ренея в воде (70.0 г, 50% чистота) добавляли и смесь гидрогенировали за 7 ч при давлении окружающей среды при 80°С. Затем катализатор удаляли фильтрованием через слой диатомовой земли, и растворитель промывали 250 мл воды. Водную фазу экстрагировали три раза с МТВЕ (650 мл). Объединенный органический слой промывали водой три раза, промывали солевым раствором, сушили над безводном сульфатом магния, а затем фильтровали. Фильтрат выпаривали при пониженном давлении и остаток очищали посредством флэш-хроматографии на силикагеле (градиент циклогексан - циклогексан/этилацетат 7:3) с получением 7.40 г (85% чистота, 44% выход) вязкого масла.
GC-MS (GC-MS Способ 1): (М+- СО); (М+- СО, - Boc)
Пример 74А
Ди-трет-бутил (2S)-5-этенилпирролидин-1,2-дикарбоксилат (смесь изомеров)
Метил(трифенил)фосфония бромид (17.7 г, 49.4 ммоль) суспендировали в 180 мл THF. Раствор калия бис(триметилсилил)амида (1 М в толуоле, 75 мл, 15% раствор, 49 ммоль) добавляли по каплям при комнатной температуре и перемешивали в течение 1 ч. Смесь охлаждали до -78°С. Ди-трет-бутил (2S)-5-формилпирролидин-1,2-дикарбоксилат (пример 73А, 7.40 г, 24.7 ммоль) растворяли в THF (60 мл). Раствор добавляли и перемешивали в течение 2 ч при комнатной температуре. Реакционную смесь переливали в раствор калия натрия (2R,3R)-2,3-дигидроксибутандиоата (140 мл) и воды (85 мл) и экстрагировали тремя частями МТВЕ (210 мл). Объединенный органический экстракт промывали раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле (градиент циклогексан - циклогексан этилацетат (9:1)) с выделением 4.25 г (100% чистота, 58% выход) указанного в названии соединения.
GC-MS (GC-MS Способ 1): Rt=5.07 мин, 5.17 мин; 197.3 (М+-Вос)
Пример 75А
трет-бутил 5-винил-L-пролинат (смесь изомеров)
Ди-трет-бутил (2S)-5-этенилпирролидин-1,2-дикарбоксилат (пример 74А, 4.25 г, 14.3 ммоль) растворяли в 45 мл DCM и раствор охлаждали до 0°С. Триметилсилил трифтортэтансульфонат (2.6 мл, 14 ммоль) добавляли по каплям и полученную смесь перемешивали в течение 5 мин при 0°С. Реакцию затем гасили концентрированным водным раствором гидрокарбоната натрия (21 мл) и экстрагировали двумя порциями DCM. Объединенные органические слои сушили над сульфатом магния, фильтровали, а затем концентрировали. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле (градиент метанол - дихлорметан (100:2)) с получением 2.09 г (100% чистота, 74% выход) указанного в названии соединения.
GC-MS (GC-MS Способ 1): Rt=3.37 мин, 3.50 мин; 197.3 (М+)
Пример 76А
трет-бутил (5R)-5-этенил-L-пролинат (единственный стереоизомер)
Изомерное разделение трет-бутил-5-этенил-L-пролината. (диастереомерная смесь) (пример 75А, 2.55 г, 12.9 ммоль) посредством флэш-хроматографии на силикагеле (градиент циклогексан - этилацетат - триметиламин (70:30:1)) обеспечило 1.35 г (100% чистота, 53% выход) указанного в названии соединения.
GC-MS (GC-MS Способ 1): Rt=3.50 мин; 197.3 (М+)
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 1.3-1.4 (1H), 1.40 (9Н), 2.4 (1Н), 3.5 (2Н), 4.9 (1H), 5.15 (1H), 5.8 (1Н).
Пример 77А
1-трет-бутил 2-метил-3-этенилпиперидин-1,2-дикарбоксилат (смесь изомеров)
Раствор этенилмагния бромида (39 мл, 0.70 М, 27 ммоль) в безводном THF (20 мл) охлаждали до -35°С, а затем раствор бромида меди(I) - диметилсульфида (1:1) (741 мг, 3.61 ммоль) добавляли. Реакционную смесь перемешивали в течение 1 ч при -35°С. Раствор 1-трет-бутил 2-метил 5,6-дигидропиридин-1,2(4H)-дикарбоксилата (CAS-RN: 155905-80-9, 4.35 г, 18.0 ммоль) в THF (10 мл) добавляли в смесь, а затем реакционную смесь перемешивали в течение 6 ч при -35°С. Смесь гасили смесью водного хлорида аммония (85 мл) и водным аммиаком (85 мл) и экстрагировали двумя порциями МТВЕ (285 мл). Объединенные органические слои промывали последовательно водным хлоридом аммония (570 мл) и солевым раствором (570 мл), сушили над сульфатом магния, фильтровали, а затем концентрировали. Неочищенный продукт очищали посредством хроматографии на силикагеле (градиент: циклогексан - этилацетат (2%-20%)) с получением 1.59 г (100% чистота, 33% выход) указанного в названии соединения.
LC-MS (Способ 12): Rt=3.33 мин; MS (ESI pos): m/z=270 [M+H]+
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 1.038 (0.51), 1.087 (0.77), 1.382 (16.00), 1.419 (0.83), 1.429 (0.59), 1.448 (0.73), 1.459 (0.80), 1.466 (0.54), 1.479 (1.01), 1.485 (1.09), 1.496 (0.73), 1.502 (0.66), 1.506 (0.59), 1.514 (0.47), 1.532 (0.47), 1.544 (0.42), 1.600 (0.64), 1.627 (0.61), 2.899 (0.88), 3.690 (13.11), 3.817 (0.47), 3.849 (0.45), 5.120 (2.03), 5.148 (0.90), 5.164 (1.49), 5.869 (0.57).
Пример 78А
1-(трет-бутоксикарбонил)-3-этенилпиперидин-2-карбоновая кислота (смесь изомеров)
1-трет-бутил 2-метил 3-этенилпиперидин-1,2-дикарбоксилат (пример 77А, 1.59 г, 5.90 ммоль) растворяли в смеси метанола (5.9 мл) и THF (17.7 мл). Раствор гидроксида лития (5.9 мл, 2.0 М, 12 ммоль) добавляли и смесь перемешивали в течение 5 ч при 50°С, затем охлаждали до комнатной температуры. Реакционную смесь промывали с МТВЕ (7.5 мл), охлаждали до 0°С, подкисляли 1 М соляной кислотой (14.5 мл), затем экстрагировали пять раз с DCM (15 мл). Объединенный органический экстракт сушили над сульфатом магния, фильтровали, а затем концентрировали с получением 1.39 г (100% чистота, 92% выход) указанного в названии соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 1.381 (16.00), 1.423 (0.70), 1.434 (0.57), 1.455 (0.77), 1.465 (1.32), 1.473 (0.93), 1.488 (1.66), 1.495 (1.29), 1.519 (0.46), 1.530 (0.40), 1.608 (0.80), 1.632 (0.75), 2.911 (0.96), 3.815 (0.50), 3.842 (0.47), 5.113 (1.95), 5.140 (0.73), 5.156 (1.59), 5.753 (0.50), 5.874 (0.51), 12.910 (0.69).
Пример 79А
трет-бутил 2 - {[(2S,5R)-2-(трет-бутоксикарбонил)-5-винилпирролидин-1-ил]карбонил}-3-винилпиперидин-карбоксилат (смесь изомеров)
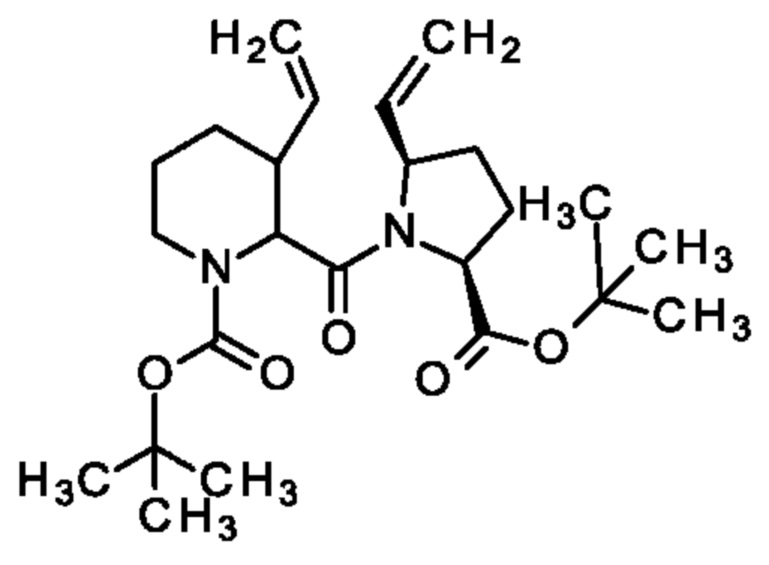
1-(трет-бутоксикарбонил)-3-этенилпиперидин-2-карбоновую кислоту (рацемат) (пример 78А, 1.35 г, 5.30 ммоль) и трет-бутил (5R)-5-этенил-L-пролинат (пример 76А, 950 мг, 4.82 ммоль) растворяли в ацетонитриле (60 мл). N,N-диизопропилэтиламин (2.5 мл, 14 ммоль) добавляли и реакционную смесь охлаждали до 0°С. Раствор 1H-бензотриазол-1-илокси)(трипирролидин-1-ил)фосфония гексафторфосфат (3.26 г, 6.26 ммоль) в ацетонитриле (30 мл) добавляли в смесь, и реакционную смесь перемешивали в течение 20 ч при комнатной температуре. Реакционную смесь концентрировали, повторно растворяли в МТВЕ и промывали водой. Водную фазу повторно экстрагировали с МТВЕ. Объединенный органический слой промывали солевым раствором, сушили над сульфатом магния, фильтровали, а затем концентрировали. Неочищенный продукт очищали посредством хроматографии на силикагеле (циклогексан / этилацетат (85:15)) с получением 2.03 г (88% выход) указанного в названии соединения.
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: -0.008 (0.60), 1.373 (10.15), 1.392 (16.00), 1.396 (14.53), 1.458 (0.57), 1.489 (0.46), 1.763 (0.46), 1.780 (0.56), 2.105 (0.42), 2.122 (0.42), 2.524 (0.41), 5.050 (0.44), 5.090 (0.70), 5.116 (0.79), 5.754 (1.04).
Пример 80А
ди-трет-бутил (4aR,6aR,9S,11aS)-11-оксо-2,3,4,4а,6а,7,8,9,11,11а-декагидро-1H-пиридо[3,2-е]пирроло[1,2-а]азепин-1,9-дикарбоксилат (единственный стереоизомер)
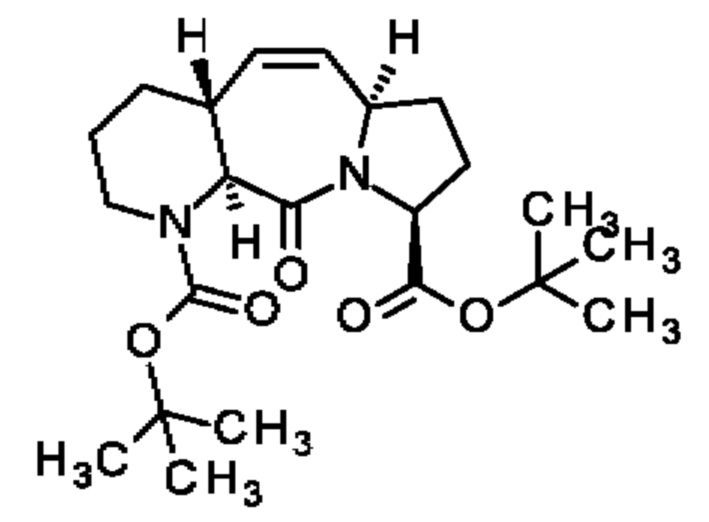
трет-бутил 2-{[(2S,5R)-2-(трет-бутоксикарбонил)-5-винилпирролидин-1-ил]карбонил}-3-винилпиперидин-карбоксилат (пример 79А, 2.00 г, 4.60 ммоль) растворяли в DCM (100 мл) и [1,3-бис(2,4,6-триметилфенил)имидазолидин-2-илиден](дихлор)(фенилметилиден)рутения-трициклогексилфосфан (1:1) (781 мг, 920 мкмоль) добавляли. Реакционную смесь перемешивали в течение 48 ч при возврате флегмы, затем охлаждали до комнатной температуры. DMSO (2.0 мл) добавляли и реакционную смесь перемешивали всю ночь при комнатной температуре. Реакционную смесь концентрировали и неочищенный продукт очищали посредством хроматографии на силикагеле (циклогексан / этилацетат (25%-50%)) с получением 805 мг (99% чистота, 42% выход) указанного в названии соединения.
LC-MS (Способ 9): Rt=1.12 мин; MS (ESI pos): m/z=407 [M+H]+
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 1.296 (5.30), 1.361 (16.00), 1.378 (2.93), 5.540 (0.72), 5.562 (0.51).
Пример 81А
(4aR,6aR,9S,11aS)-1-{[(9H-флуорен-9-ил)метокси]карбонил}-11-оксо-2,3,4,4a,6a,7,8,9,11,11а-декагидро-1Н-пиридо[3,2-е]пирроло[1,2-a]азепин-9-карбоновая кислота (единственный стереоизомер)
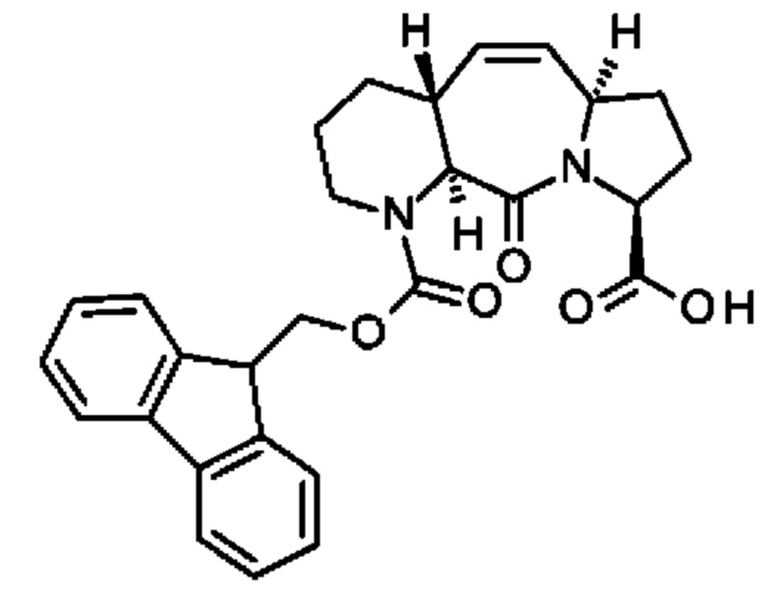
Ди-трет-бутил-(4aR,6aR,9S,11aS)-11-оксо-2,3,4,4а,6а,7,8,9,11,11а-декагидро-1H-пиридо[3,2-е]пирроло[1,2-а]азепин-1,9-дикарбоксилат (пример 80А, 800 мг, 1.97 ммоль) растворяли в 8 мл DCM и раствор охлаждали до 0°С. Трифторуксусную кислоту (8.0 мл, 100 ммоль) добавляли и реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Реакционную смесь концентрировали и остаток разбавляли раствором гидрокарбоната натрия (20 мл, рН=8). Раствор (9H-флуорен-9-ил)метил хлорформиата (CAS-RN: 28920-43-6,764 мг, 2.95 ммоль) в THF (24 мл) добавляли и реакционную смесь перемешивали в течение 16 ч при комнатной температуре. Реакционную смесь разбавляли с DCM (100 мл) и подкисляли 1 М соляной кислотой (40 мл) (рН=1). Раствор экстрагировали два раза с DCM (50 мл) и объединенный органический экстракт сушили над сульфатом магния, фильтровали, а затем концентрировали. Очистка неочищенного продукта посредством колоночной хроматографии на силикагеле (DCM/MeOH 95/5) дала 676 мг (100% чистота, 73% выход) указанного в названии соединения.
LC-MS (Способ 10): Rt=1.86 мин; MS (ESI pos): m/z=473 [M+H]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: -0.008 (2.87), 0.008 (2.54), 0.778 (0.66), 0.800 (1.51), 0.808 (1.58), 0.832 (2.85), 0.856 (1.93), 0.865 (2.01), 0.889 (0.79), 1.125 (0.77), 1.157 (1.38), 1.182 (0.90), 1.215 (0.50), 1.235 (1.06), 1.517 (2.61), 1.538 (3.44), 1.563 (6.49), 1.593 (6.17), 1.610 (5.67), 1.636 (4.98), 1.660 (3.92), 1.684 (2.46), 1.719 (2.25), 1.747 (2.01), 1.846 (4.49), 1.858 (7.62), 1.868 (6.96), 1.901 (3.19), 2.010 (0.57), 2.073 (9.83), 2.189 (2.95), 2.200 (3.74), 2.241 (3.30), 2.276 (2.71), 2.307 (1.21), 2.327 (1.10), 2.366 (0.52), 2.398 (0.80), 2.428 (1.41), 2.456 (0.84), 2.670 (0.63), 2.710 (0.50), 2.816 (1.60), 2.833 (1.66), 2.849 (2.51), 2.862 (2.28), 2.877 (2.34), 2.895 (1.47), 3.040 (0.69), 3.075 (1.10), 3.086 (1.04), 3.101 (1.03), 3.121 (0.69), 3.690 (1.24), 3.724 (4.64), 3.744 (6.62), 3.756 (6.47), 3.777 (4.44), 3.823 (8.20), 3.852 (7.84), 4.120 (4.36), 4.137 (4.86), 4.148 (4.24), 4.191 (3.57), 4.202 (7.64), 4.212 (4.19), 4.261 (1.02), 4.276 (2.33), 4.291 (4.52), 4.316 (4.20), 4.328 (5.37), 4.346 (4.61), 4.370 (3.31), 4.383 (5.08), 4.395 (4.54), 4.411 (5.53), 4.422 (5.83), 4.456 (3.22), 4.595 (1.63), 4.761 (4.95), 4.773 (5.04), 4.788 (4.59), 4.800 (4.19), 5.364 (2.72), 5.393 (7.67), 5.422 (7.48), 5.451 (2.73), 5.510 (0.98), 5.540 (4.23), 5.553 (4.14), 5.585 (0.91), 7.282 (3.12), 7.300 (7.84), 7.319 (10.54), 7.337 (13.57), 7.355 (9.71), 7.366 (2.95), 7.377 (5.06), 7.399 (12.17), 7.420 (16.00), 7.438 (6.68), 7.521 (8.10), 7.539 (6.98), 7.613 (12.53), 7.631 (11.16), 7.646 (3.71), 7.665 (3.16), 7.847 (8.31), 7.865 (15.04), 7.884 (10.42), 7.908 (4.87), 12.429 (2.52).
Эту аминокислоту применяли в Fmoc SPPS.
Пример 82А
(6S)-5-{[(9H-флуорен-9-ил)метокси]карбонил}-5-азаспиро[2.4]гептан-6-карбоновая кислота
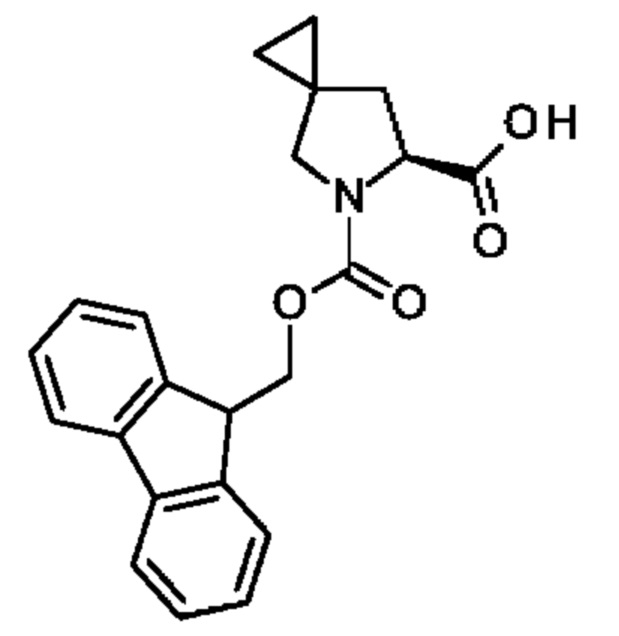
(6S)-5-азаспиро[2.4]гептан-6-карбоновую кислоту (CAS-RN: 152723-57-4, 1.38 г, 9.79 ммоль) растворяли в воде (14 мл), и гидрокарбонат натрия (8.23 г, 97.9 ммоль) добавляли. Затем раствор 1-({[(9H-флуорен-9-ил)метокси]карбонил}окси)пирролидин-2,5-диона (CAS-RN:82911-69-l, 3.47 г, 10.3 ммоль) в ацетоне (21 мл) добавляли и смесь перемешивали всю ночь при комнатной температуре. Реакционную смесь разбавляли водой и экстрагировали два раза с МТВЕ. Водный слой подкисляли до рН 3-4 с 1М соляной кислотой и экстрагировали три раза с DCM. Объединенную органическую фазу сушили над безводном сульфатом магния, фильтровали, а затем концентрировали, с получением 3.3 г (100% чистота, 93% выход) указанного в названии соединения.
LC-MS (Способ 9): Rt=1.04 мин; MS (ESI pos): m/z=364 [M+H]+
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 0.497 (1.67), 0.510 (2.34), 0.527 (5.55), 0.559 (3.82), 0.567 (5.51), 0.590 (9.33), 0.598 (11.21), 0.619 (1.87), 0.636 (0.58), 1.106 (9.23), 1.714 (2.14), 1.723 (2.15), 1.745 (2.40), 1.755 (2.50), 1.764 (1.94), 1.772 (1.85), 1.796 (2.02), 1.804 (1.93), 2.086 (7.36), 2.280 (2.12), 2.301 (2.47), 2.311 (2.21), 2.333 (2.24), 2.390 (1.75), 2.412 (2.07), 2.421 (1.87), 2.443 (1.71), 2.524 (0.88), 2.594 (3.10), 3.076 (2.73), 3.219 (3.24), 3.245 (4.13), 3.283 (3.41), 3.309 (5.19), 3.385 (5.05), 3.403 (4.32), 3.410 (3.56), 3.429 (3.16), 3.747 (1.01), 4.160 (0.68), 4.175 (1.57), 4.187 (3.09), 4.192 (3.69), 4.209 (6.08), 4.218 (4.18), 4.224 (3.09), 4.237 (2.43), 4.247 (1.22), 4.254 (2.15), 4.272 (16.00), 4.284 (3.83), 4.297 (4.75), 4.306 (2.72), 4.427 (2.10), 4.435 (2.31), 4.449 (2.34), 4.457 (2.02), 5.754 (0.45), 7.301 (1.47), 7.318 (5.18), 7.320 (5.43), 7.325 (2.74), 7.336 (7.24), 7.339 (7.37), 7.344 (4.81), 7.348 (3.25), 7.352 (3.50), 7.354 (3.84), 7.357 (3.67), 7.403 (9.03), 7.422 (14.80), 7.440 (6.64), 7.640 (8.10), 7.647 (7.82), 7.658 (7.43), 7.666 (6.87), 7.884 (10.09), 7.902 (9.58).
Эту аминокислоту применяли в Fmoc SPPS.
Эту Fmoc аминокислоту также получали из (6S)-5-азаспиро[2.4]гептан-6-карбоновой кислоты, полученной в Пример 124А.
Пример 83A
(1S,3S,5S,6S)-2-{[(9H-флуорен-9-ил)метокси]карбонил}-6-(трифторметил)-2-азабицикло[3.1.0]гексан-3-карбоновая кислота и rel-(1R,3R,5R,6R)-2-{[(9H-флуорен-9-ил)метокси]карбонил}-6-(трифторметил)-2-азабицикло[3.1.0]гексан-3-карбоновая кислота
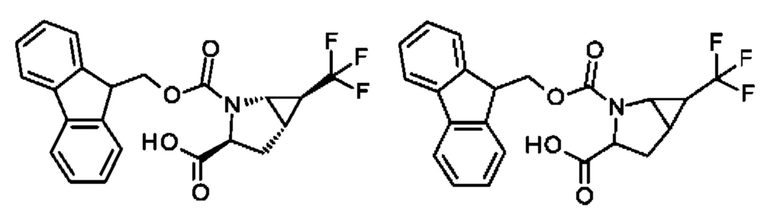
(1S,3S,5S,6S)-6-(трифторметил)-2-азабицикло[3.1.0]гексан-3-карбоновую кислоту (450 мг, 2.31 ммоль) растворяли в воде (6 мл), и гидрокарбонат натрия (1.94 г, 23.1 ммоль) добавляли. Затем раствор 1-({[(9H-флуорен-9-ил)метокси]карбонил}окси)пирролидин-2,5-диона (CAS-RN: 82911-69-1, 817 мг, 2.42 ммоль) в ацетоне (9 мл) добавляли и полученную суспензию перемешивали всю ночь при комнатной температуре. Реакционную смесь разбавляли водой и экстрагировали два раза с МТВЕ. Водный слой подкисляли до рН 3-4 с 1М соляной кислотой и экстрагировали три раза с DCM. Объединенную органическую фазу сушили над безводном сульфатом магния, фильтровали, а затем концентрировали. Неочищенный продукт очищали посредством препаративной ВЭЖХ (колонка: Reprosil; С18; 10 мкм; 125 × 40 мм; растворитель: AcCN/H2O с содержанием 0,1% НСООН; скорость потока: 100 мл/мин; градиент: 0-5.50 мин 10:90; впрыск образца при 3.00 мин; 5.50-17.65 мин градиент до 95:5; 17.65-19.48 мин при 95:5; 19.48-19.66 мин - 10:90; 19.66-20.72 мин 10:90;), с получением 571 мг (96% чистота, 57% выход) указанного в названии соединения.
LC-MS (Способ 9): Rt=1.06 мин; MS (ESI pos): m/z=418 [M+H]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 2.073 (0.99), 2.203 (4.88), 2.345 (2.00), 2.366 (1.94), 3.755 (2.94), 4.078 (1.74), 4.221 (1.50), 4.253 (4.04), 4.270 (5.55), 4.290 (4.95), 4.337 (2.21), 7.293 (3.93), 7.301 (4.07), 7.312 (9.32), 7.319 (8.88), 7.330 (6.19), 7.337 (5.57), 7.407 (9.40), 7.425 (16.00), 7.444 (7.29), 7.695 (5.70), 7.887 (13.98), 7.906 (12.87), 12.866 (0.63).
Вещество также коммерчески доступно (CAS RN: 1986905-54-7)
Подобным образом получали рацемическую аминокислоту с применением rel-(1R,3R,5R,6R)-6-(трифторметил)-2-азабицикло[3.1.0]гексан-3-карбоновой кислоты (Enamine). Эти аминокислоты применяли в SPPS.
Пример 84А
Метил N-(трет-бутоксикарбонил)-L-изолейцилсеринат (смесь диастереомеров)
N-(трет-бутоксикарбонил)-L-изолейцин (7.50 г, 32.4 ммоль) и Метил DL-серинат-хлорид водорода (1/1) (5.05 г, 32.4 ммоль) растворяли в дихлорметане (65 мл). (Бензотриазол-1-илокси)трис(диметиламино)фосфония гексафторфосфат (14.9 г, 33.6 ммоль) и N,N-диизопропилэтиламин (11 мл, 65 ммоль) добавляли и реакционную смесь перемешивали всю ночь при комнатной температуре. Дихлорметан (250 мл) добавляли и реакционную смесь промывали последовательно водой, водной соляной кислотой (1М) и водным раствором гидрокарбоната натрия. Объединенный органический экстракт сушили над сульфатом магния, фильтровали, а затем концентрировали. Неочищенный продукт растворяли в DCM и тщательно промывали водой, еще два раза водной соляной кислотой (1М), а затем еще два раза водным раствором гидрокарбоната натрия. Затем объединенную органическую фазу сушили над сульфатом магния, фильтровали, а затем концентрировали. Неочищенный продукт применяли непосредственно в ходе следующей стадии.
LC-MS (Способ 11): 355.2 (M+Na+)
Пример 85А
Метил 2-{(1S,2S)-1-[(трет-бутоксикарбонил)амино]-2-метилбутил}-4,5-дигидро-1,3-оксазол-4-карбоксилат (смесь диастереомеров)
Метил N-(трет-бутоксикарбонил)-L-изолейцилсеринат (пример 84А, 4.62 г, 13.9 ммоль) растворяли в дихлорметане (51 мл) и раствор охлаждали до -78°С. N-Этил-N-(трифтор-лямбда4-сульфанил)этанамин (2.2 мл, 17 ммоль) добавляли и реакционную смесь перемешивали при -78°С в течение 1.5 ч. Реакцию гасили карбонатом калия (7.68 г, 55.6 ммоль), а затем позволяли охладиться до комнатной температуры. Раствор перемешивали в течение 1 ч при комнатной температуре. Затем раствор разбавляли с DCM (50 мл) и промывали три раза насыщенным водным раствором гидрокарбоната натрия. Водную фазу далее экстрагировали с DCM. Объединенную органическую фазу промывали солевым раствором, сушили над безводном сульфатом магния, фильтровали, а затем концентрировали с получением 5.75 г целевого продукта.
LC-MS (Способ 11): 315.2 (М+Н+), 337.2 (M+Na+)
Пример 86А
Метил 2-{(1S,2S)-1-[(трет-бутоксикарбонил)амино]-2-метилбутил}-1,3-оксазол-4-карбоксилат
Метил 2-{(1S,2S)-1-[(трет-бутоксикарбонил)амино]-2-метилбутил}-4,5-дигидро-1,3-оксазол-4-карбоксилат (пример 85А, 4.37 г, 13.9 ммоль) растворяли в дихлорметане (35 мл, 540 ммоль) и раствор охлаждали при -40°С. Бромо(трихлор)метан (2.7 мл, 27 ммоль) добавляли, затем 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-а]азепин (4.6 мл, 31 ммоль) также добавляли. Реакционный раствор перемешивали в течение 2.5 ч при 0°С, а затем разбавляли с DCM. Реакционную смесь промывали пять аз с 10% раствором лимонной кислоты. Объединенный водный слой экстрагировали с DCM. Объединенный органический слой промывали солевым раствором, сушили над безводном сульфатом магния, фильтровали и концентрировали. Неочищенный продукт очищали посредством хроматографии на силикагеле (градиент:циклогексан / этилацетат (4:1)) с получением 2.2 г (50.7% выход) указанного в названии соединения.
LC-MS (Способ 10): 335.15 (M+Na+)
Пример 87А
2-{(1S,2S)-1-[(трет-бутоксикарбонил)амино]-2-метилбутил}-1,3-оксазол-4-карбоновая кислота
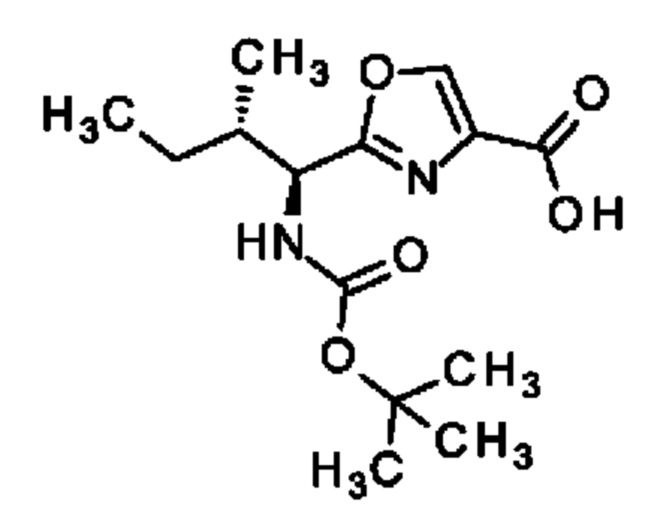
Метил 2-{(1S,2S)-1-[(трет-бутоксикарбонил)амино]-2-метилбутил}-1,3-оксазол-4-карбоксилат (пример 86А, 2.21 г, 7.06 ммоль) растворяли в метаноле (13.5 мл). Водный раствор гидроксида натрия (4.3 мл, 3М) добавляли и реакционную смесь перемешивали в течение 40 мин, а затем концентрировали для удаления метанола. Водный раствор подкисляли водной 1М соляной кислотой (14 мл) и экстрагировали этилацетатом. Объединенный органический слой сушили над безводном сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении с получением 12.0 г (95% чистота, 51% выход) указанного в названии соединения.
LC-MS (MCW_OA_S1): Rt=1.13 мин; MS (ESI neg): m/z=297 [M+H]-
Пример 88А
(4R)-3-[(3S)-3-метилпентаноил]-5,5-дифенил-4-(пропан-2-ил)-1,3-оксазолидин-2-он
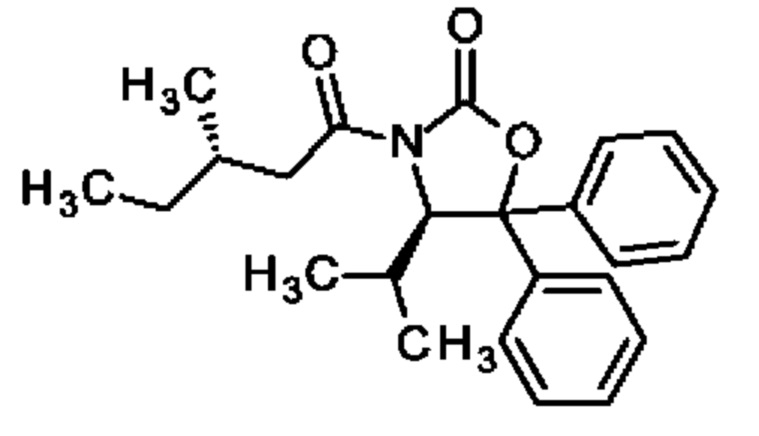
Раствор (3S)-3-метилпентановой кислоты (867 мг, 7.46 ммоль) в безводном THF (50 мл) охлаждали до -30°С. 2,2-диметилпропаноил хлорид (920 мкл, 7.5 ммоль) добавляли по каплям и реакционную смесь перемешивали в течение 1.5 ч при -30°С. Сухой лития хлорид (347 мг, 8.17 ммоль) и (4R)-5,5-дифенил-4-(пропан-2-ил)-1,3-оксазолидин-2-он (2.00 г, 7.11 ммоль) ((R)-диOZ) затем добавляли и реакционную смесь оставляли медленной дойти до комнатной температуры и перемешивали всю ночь. Реакционную смесь разбавляли этилацетатом и водой. Водный слой разделяли и экстрагировали этилацетатом. Объединенный слой этилацетата промывали водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали, а затем концентрировали, с получением 2.87 г твердого вещества белого цвета. Неочищенный продукт растворяли в DCM небольшим количеством метанола и очищали посредством колоночной хроматографии через силикагель (Biotage Isolera, 50 г картридж) с циклогексаном / этилацетатом (80:20) в качестве элюента. Фракции, содержащие продукт, объединяли, концентрировали и сушили с получением 2.25 г (98% чистота, 82% выход) целевого соединения.
LC-MS (Способ 10): Rt=2.69 мин; MS (ESI pos): m/z=380 [M+H]+
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 0.616 (13.33), 0.633 (13.58), 0.650 (7.84), 0.660 (16.00), 0.668 (15.61), 0.676 (15.27), 0.686 (7.40), 0.719 (0.40), 0.875 (12.87), 0.893 (12.59), 0.940 (0.92), 0.958 (1.50), 0.975 (2.08), 0.993 (2.23), 1.011 (1.38), 1.028 (0.84), 1.045 (1.41), 1.061 (1.93), 1.079 (1.89), 1.095 (1.16), 1.112 (0.62), 1.541 (0.55), 1.557 (1.29), 1.574 (1.91), 1.590 (1.82), 1.607 (1.10), 1.623 (0.40), 2.020 (0.71), 2.036 (1.44), 2.049 (1.78), 2.065 (1.28), 2.082 (0.50), 2.449 (2.60), 2.468 (2.84), 2.755 (2.63), 2.771 (2.50), 2.793 (2.05), 2.808 (1.92), 5.557 (5.98), 5.563 (5.64), 7.271 (2.61), 7.289 (6.37), 7.307 (5.02), 7.352 (5.04), 7.368 (10.74), 7.385 (9.60), 7.403 (3.47), 7.557 (8.45), 7.577 (6.54), 7.639 (8.45), 7.659 (6.92).
Пример 89А
Бензил {(2S,3S)-3-метил-2-[(4R)-2-оксо-5,5-дифенил-4-(пропан-2-ил)-1,3-оксазолидин-3-карбонил]пентил}карбамат
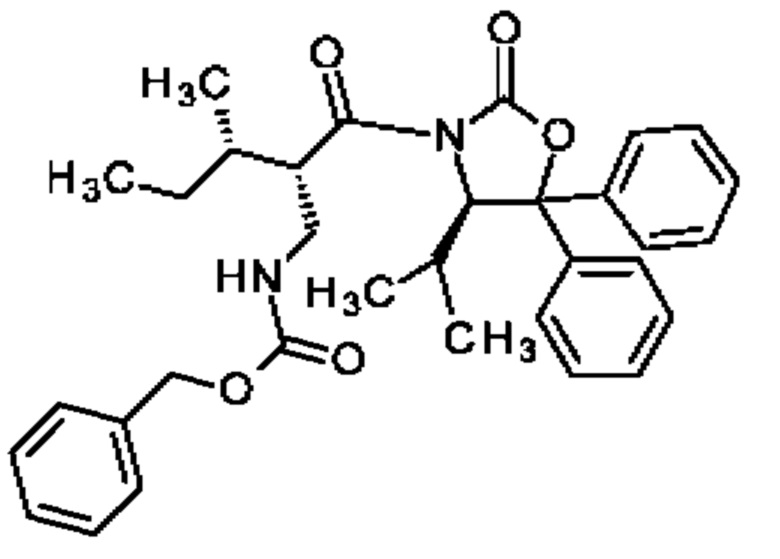
Описанную в литературе процедуру (Sebesta snd Seebach, Helv. Chim. Acta 2003, 86, 4061-4072) проводили. (4R-3-[(3S)-3-метилпентаноил]-5,5-дифенил-4-(пропан-2-ил)-1,3-оксазолидин-2-он (пример 88A, 2.20 г, 5.80 ммоль) растворяли в DCM (20 мл) и раствор охлаждали до -15°С. Раствор хлорида титана(IV) (6.1 мл, 1.0 М в DCM, 6.1 ммоль) добавляли по каплям при поддержании температуры при -15°С. Триэтиламин (890 мкл, 6.4 ммоль) затем добавляли и реакционную смесь перемешивали в течение 30 мин при -15°С.
В отдельной круглодонной колбе раствор бензил (метоксиметил)карбамата (1.24 г, 6.38 ммоль, CAS RN: 94471-35-9) в DCM (10 мл) охлаждали до 0°С. Раствор хлорида титана(IV) (6.4 мл, 1.0 М в DCM, 6.4 ммоль) добавляли по каплям, а затем весь раствор добавляли по каплям в колбу, содержащую (4R)-3-[(3S)-3-метилпентаноил]-5,5-дифенил-4-(пропан-2-ил)-1,3-оксазолидин-2-он. Реакционную смесь перемешивали в течение 4 ч при 0°С, затем перемешивали при комнатной температуре всю ночь. Реакцию гасили посредством добавления водного раствора хлорида аммония, а затем разбавляли дополнительным DCM. Отделенный органический слой промывали водным 1М раствором HCl, водным 1М раствором NaOH, а затем водным раствором хлорида натрия. Органический слой фильтровали, а затем концентрировали, с получением 3.39 г масла желтого цвета. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (Biotage Isolera 100g картридж) с циклогексаном/этилацетатом (85:15) элюентом. Фракции, содержащие продукт, объединяли, концентрировали, а затем сушили с получением 1.50 г (96% чистота, 46% выход, селективно) указанного в названии соединения с диастереоселективностью 96,3% (лит. 96%).
LC-MS (Способ 10): Rt=2.63 мин; MS (ESI pos): m/z=543 [M+H]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 0.429 (3.22), 0.446 (3.32), 0.480 (1.44), 0.498 (3.18), 0.516 (1.79), 0.595 (4.27), 0.611 (4.65), 0.643 (0.49), 0.663 (0.46), 0.768 (0.45), 0.786 (0.46), 0.874 (3.86), 0.891 (4.01), 1.098 (0.48), 1.397 (16.00), 1.993 (0.45), 2.010 (0.57), 2.023 (0.45), 3.146 (0.44), 3.169 (0.66), 3.180 (1.02), 3.192 (0.63), 3.206 (0.44), 3.222 (0.53), 3.243 (0.48), 3.816 (0.57), 4.920 (0.59), 4.952 (1.85), 4.978 (2.04), 5.010 (0.65), 5.539 (1.70), 5.544 (1.73), 7.187 (0.53), 7.201 (0.87), 7.282 (3.41), 7.289 (5.28), 7.306 (4.14), 7.322 (1.84), 7.342 (2.69), 7.362 (3.73), 7.385 (3.47), 7.405 (1.52), 7.566 (3.08), 7.584 (2.46), 7.662 (2.88), 7.681 (2.48).
Пример 90А
(2S,3S)-2-({[(Бензилокси)карбонил]амино}метил)-3-метилпентановая кислота
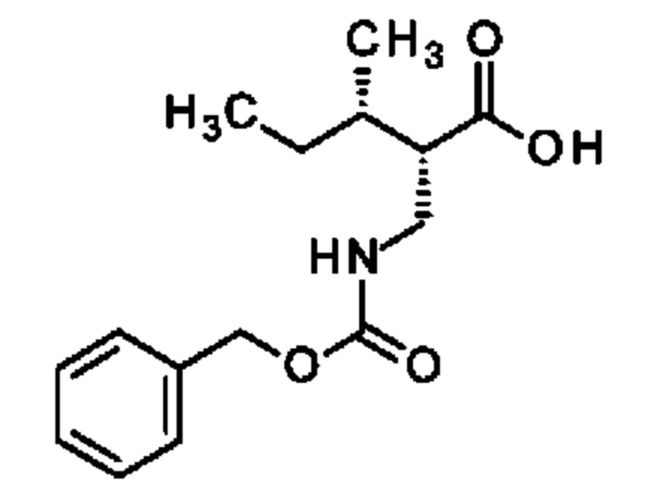
Раствор бензил {(2S,3S)-3-метил-2-[(4R-2-оксо-5,5-дифенил-4-(пропан-2-ил)-1,3-оксазолидин-3-карбонил]пентил}карбамата (пример 89А, 1.49 г, 2.75 ммоль) в THF (15 мл) получали. Раствор LiOH (105 мг, 4.39 ммоль) в воде (5 мл) добавляли и реакционную смесь перемешивали при комнатной температуре в течение 24 ч. ВЭЖХ анализ показал, что реакция не завершена, поэтому добавили дополнительный эквивалент LiOH (65.8 мг, 2.75 ммоль) в воде (3 мл). Реакционную смесь перемешивали при комнатной температуре всю ночь. Реакционную смесь разбавляли диэтиловым простым эфиром, смесь перемешивали в течение 5 мин, а затем фильтровали. Фильтровальную лепешку промывали водой и простым эфиром, а затем слои разделили. Водный слой подкисляли водным 1М раствором HCl до рН 1 и экстрагировали три раза диэтиловым простым эфиром. Объединенную фазу простого эфира промывали раствором хлорида натрия, сушили над сульфатом магния, фильтровали, а затем концентрировали. После сушки под высоким вакуумом получали 715 мг (100% чистота, 93% выход) указанного в названии соединения.
LC-MS (Способ 10): Rt=1.71 мин; MS (ESI pos): m/z=280 [M+H]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 0.837 (6.34), 0.856 (14.23), 0.863 (13.72), 0.880 (12.65), 1.098 (0.91), 1.117 (1.35), 1.133 (1.47), 1.151 (1.49), 1.170 (0.95), 1.348 (0.42), 1.367 (1.02), 1.380 (1.33), 1.398 (1.53), 1.414 (1.09), 1.431 (0.72), 1.596 (0.82), 1.611 (1.42), 1.627 (1.61), 1.644 (1.16), 1.660 (0.53), 1.908 (3.41), 2.396 (1.17), 2.413 (2.79), 2.429 (2.67), 2.445 (1.07), 2.501 (16.00), 3.139 (4.27), 3.155 (7.41), 3.171 (4.60), 5.000 (15.52), 7.221 (1.46), 7.234 (2.66), 7.248 (1.41), 7.283 (0.89), 7.290 (0.97), 7.300 (2.98), 7.318 (5.22), 7.332 (11.34), 7.339 (12.29), 7.356 (5.30), 7.375 (1.45), 12.135 (3.58).
Пример 91А
(2S,3S)-2-(аминометил)-3-метилпентановаяа кислота
10% Палладий на углероде (100 мг) отвешивали в круглодонную колбу и колбу продували аргоном. Раствор (2S,3S)-2-({[(бензилокси)карбонил]амино}метил)-3-метилпентановой кислоты (пример 90А, 710 мг, 2.54 ммоль) в метаноле (40 мл) добавляли и реакционную смесь перемешивали при комнатной температуре при нормальном давлении газообразного Н2. Анализ ВЭЖХ показал, что через 2 часов при комнатной температуре реакция завершилась. Реакционную смесь фильтровали через слой силикагеля и фильтровальную лепешку промывали несколько раз метанолом. Объединенный фильтрат концентрировали при пониженном давлении с получением 345 мг (93%) указанного в названии соединения. Соединение применяли непосредственно в ходе следующей стадии.
Пример 92А
(2S,3S)-2-[({[(9H-флуорен-9-ил)метокси]карбонил}амино)метил]-3-метилпентановая кислота
(2S,3S)-2-(аминометил)-3-метилпентановую кислоту (пример 91А, 344 мг, 2.37 ммоль) растворяли в водном растворе карбоната натрия (32 мл, 0.15 М, 4.7 ммоль). 1-({[(9H-флуорен-9-ил)метокси]карбонил}окси)пирролидин-2,5-дион (959 мг, 2.84 ммоль) растворяли в ацетоне (30 мл) и этот раствор добавляли в предшествующий раствор и реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Реакционную смесь концентрировали с удалением ацетона. Водный раствор разбавляли водой и экстрагировали этилацетатом. Водный слой подкисляли до рН 3-4 с 1М водной соляной кислотой и экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором, сушили над безводном сульфатом магния, фильтровали, а затем концентрировали. Неочищенный продукт очищали посредством препаративной ВЭЖХ (колонка: Reprosil; С18; 10 мкм; 125 × 30 мм; растворители: AcCN/H2O с 0,1% НСООН; скорость потока: 75 мл/мин; градиент: 0-5.50 мин (10:90); впрыск образца при 3.00 мин; 5.50-17.65 мин градиент до 95:5; 17.65-19.48 мин (95:5); 19.48-19.66 мин градиент до 10:90; 19.66-20.72 мин (10:90) с получением 795 мг (96% чистота, 88% выход) указанного в названии соединения.
LC-MS (Способ 9): Rt = 1.06 мин; MS (ESIpos): m/z = 368 [М+Н]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: -0.008 (1.04), 0.008 (0.54), 0.752 (0.73), 0.841 (7.69), 0.860 (16.00), 0.870 (15.28), 0.878 (10.54), 0.887 (13.22), 1.085 (0.41), 1.103 (0.99), 1.123 (1.45), 1.139 (1.58), 1.157 (1.60), 1.176 (1.00), 1.371 (1.16), 1.384 (1.50), 1.403 (1.71), 1.418 (1.23), 1.435 (0.83), 1.604 (0.88), 1.619 (1.50), 1.636 (1.70), 1.651 (1.23), 2.408 (1.27), 2.425 (2.91), 2.441 (3.13), 2.458 (1.88), 2.524 (0.87), 2.749 (0.82), 3.143 (4.18), 3.158 (6.08), 3.174 (3.40), 4.176 (1.25), 4.194 (3.04), 4.211 (4.04), 4.241 (8.74), 4.255 (4.27), 4.263 (3.35), 7.303 (5.42), 7.322 (12.94), 7.340 (10.81), 7.353 (1.98), 7.394 (7.49), 7.413 (11.83), 7.431 (5.34), 7.673 (5.88), 7.683 (5.84), 7.691 (5.19), 7.701 (4.30), 7.876 (12.54), 7.895 (11.30), 12.176 (1.21).
Эту аминокислоту применяли в SPPS.
Пример 93А
2-[(1S,2S)-1-({[(9Н-флуорен-9-ил)метокси]карбонил}амино)-2-метилбутил]-1,3-оксазол-4-карбоновая кислота
2-{(1S,2S)-1-[(трет-бутоксикарбонил)амино]-2-метилбутил}-1,3-оксазол-4-карбоновую кислоту (пример 87А, 1.85 г, 6.21 ммоль) растворяли в трифторуксусной кислоты (7.6 мл, 99 ммоль) и раствор перемешивали в течение 15 мин. Затем раствор разбавляли толуолом и выпаривали. Эту стадию снова повторяли. Неочищенный продукт растворяли в 1,4-диоксане (20 мл, 230 ммоль) и воде (20 мл). Карбонат калия (1.60 г, 11.6 ммоль) и 1-{[(9Н-флуорен-9-илметокси)карбонил]окси}пирролидин-2,5-дион (2.35 г, 6.96 ммоль) добавляли и реакционную смесь перемешивали всю ночь при комнатной температуре. Реакционную смесь разбавляли водой (65 мл) и подкисляли до рН 2-3 с 10% водным раствором лимонной кислоты. Реакционную смесь экстрагировали этилацетатом (три раза), и объединенный органический слой промывали солевым раствором, сушили над сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (дихлорметан-метанол (3-7% + 1% уксусной кислоты), и фракции, содержащие продукт, объединяли, разбавляли толуолом, а затем концентрировали. Толуол затем добавляли снова, а затем раствор концентрировали, с получением 2.09 г (100% чистота, 80% выход) указанного в названии соединения.
LC-MS (MCWFTMSM1): Rt = 2.01 мин; MS (ESI pos): m/z = 421 [M+H]+
Эту аминокислоту применяли в SPPS.
Пример 94А
ди(проп-2-ен-1-ил)амино](1-метил-1Н-индазол-5-ил)уксусная кислота (рацемат)
(1-метил-1Н-индазол-5-ил)бороновую кислоту (CAS-RN: 590418-08-9, 500 мг, 2.84 ммоль), оксоуксусную кислоту (CAS-RN: 298-12-4, 310 μl, 50% чистота, 2.8 ммоль) и N-(проп-2-ен-1-ил)проп-2-ен-1-амин (CAS-RN: 124-02-7, 350 ц1, 2.8 ммоль) растворяли в аце-тонитриле (7.5 мл) и реакционную смесь перемешивали в течение 40 ч при 60°С. После охлаждения полученное твердое вещество фильтровали, промывали этилацетатом и сушили с получением 740 мг (100% чистота, 91% выход) указанного в названии соединения.
LC-MS (Способ 9): Rt = 0.44 мин; MS (ESI neg): m/z = 284 [M-H]-
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 0.008 (1.87), 2.073 (0.51), 3.110 (0.68), 3.125 (0.77), 3.147 (2.14), 3.161 (2.16), 3.176 (2.32), 3.193 (2.28), 3.213 (0.90), 3.230 (0.96), 4.030 (16.00), 4.531 (3.61), 5.110 (2.47), 5.134 (4.58), 5.174 (2.56), 5.752 (0.60), 5.768 (1.07), 5.778 (0.73), 5.783 (0.73), 5.794 (1.49), 5.810 (1.35), 5.820 (0.63), 5.826 (0.67), 5.837 (0.87), 5.852 (0.49), 6.514 (1.52), 7.432 (1.39), 7.435 (1.47), 7.454 (1.77), 7.457 (1.87), 7.602 (2.26), 7.624 (1.75), 7.699 (3.10), 8.041 (4.18).
Пример 95А
({[(9H-флуорен-9-ил)метокси]карбонил}амино)(1-метил-1H-индазол-5-ил)уксусная кислота (рацемат)
1,3-диметил-1,3-диазинан-2,4,6-трион (CAS-RN: 769-42-6,1.97 г, 12.6 ммоль) и тет-ракис(трифенилфосфин)палладий(0) (48.6 мг, 42.1 мкмоль) помещали в круглодонную колбу в атмосфере аргона. Раствор [ди(проп-2-ен-1-ил)амино](1-метил-1H-индазол-5-ил)уксусной кислоты (пример 94А, 600 мг, 2.10 ммоль) в сухом DCM затем добавляли, и реакционную смесь перемешивали в течение 2 ч при 35°С. Суспензию разбавляли водой и ацетоном, а затем реакционную смесь концентрировали для удаления DCM. Гидрокарбонат натрия (2.65 г, 31.5 ммоль) добавляли (образование газа). 1-({[(9Н-флуорен-9-ил)метокси]карбонил}окси)пирролидин-2,5-дион (780 мг, 2.31 ммоль) добавляли в суспензию и реакционную смесь перемешивали всю ночь при комнатной температуре. Воду и водный раствор карбоната натрия (10%) добавляли и раствор экстрагировали два раза с МТВЕ. Водную фазу подкисляли водным раствором лимонной кислоты, а затем экстрагировали три раза с DCM. Объединенные органические слои промывали солевым раствором, сушили над сульфатом магния, фильтровали, а затем концентрировали. Остаток очищали посредством препаративной ВЭЖХ (колонка: Reprosil; С18; 10 мкм; 125 × 40 мм; растворитель: AcCN/H2O с 0.1% НСООН; скорость потока: 100 мл/мин; градиент: 0-5.50 мин (10:90); впрыск образца при 3.00 мин; 5.50-17.65 мин градиент до 95: 5; 17.65-19.48 мин (95:5); 19.48-19.66 мин градиент до 10:90; 19.66-20.72 мин (10:90) с получением 422 мг (100% чистота, 47% выход) указанного в названии соединения.
LC-MS (Способ 9): Rt = 0.94 мин; MS (ESI pos): m/z = 428 [M+H]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 2.073 (0.59), 4.041 (16.00), 4.198 (0.63), 4.215 (1.87), 4.231 (3.51), 4.253 (1.84), 4.269 (1.50), 4.280 (1.92), 4.298 (1.70), 4.319 (0.59), 5.256 (1.81), 5.276 (1.80), 7.275 (0.90), 7.293 (2.03), 7.320 (2.08), 7.339 (1.26), 7.388 (1.98), 7.406 (3.38), 7.424 (1.73), 7.445 (1.60), 7.467 (1.96), 7.621 (2.30), 7.643 (1.76), 7.744 (3.56), 7.763 (3.31), 7.792 (3.19), 7.875 (4.81), 7.893 (4.48), 8.063 (4.53), 8.213 (1.70), 8.232 (1.65), 12.835 (1.03).
Пример 96А
({[(9Н-флуорен-9-ил)метокси]карбонил}амино)(1-метил-1Н-индазол-5-ил)уксусная кислота(энантиомер 1)
Диастереомерную смесь (пример 95А, 600 мг) растворяли в метаноле / ацетонитри-ле (60 мл) и разделяли с применением THAR сверхкритической жидкостной хроматографии Prep 200 устройство с применением Chiralpak AD-H (SFC) 5 мкм, 250 × 30 мм колонка; элюент: CO2/изопропанол (80:20); давление: 135 бар; температура элюента: 38°С; скорость потока: 100 г/мин; обнаружение: UV 210 нм; объем впрыска 0.4 мл/впрыск; при общих рабочих условиях 1 г/мин CO2 приблизительно эквивалентен 1 мл/мин.
Установки последовательности: время цикла = 11.2 мин
Фракцию 1 собирали при 5.8 мин (энантиомер 1); Фракцию 2 собирали между 7.7 мин (энантиомер 2). Фракции концентрировали.
Фракции анализировали с применением аналитической SFC-MS (Agilent, колонка: Daicel AD-3 3 мкм 100 × 4,6 мм, элюент: CO2/метанол (80:20); BPR давление: 130 бар; температура BPR: 60°С; температура колонки: 40°С; скорость потока: 3 мл/мин; UV 210 нм.
Стереохимию двух энантиомеров не оценивали.
185 мг энантиомера 1 (100% чистота, 99.5% ее) получали (стереохимию не определяли)
LC-MS (Способ 10): Rt = 1.80 мин; MS (ESI pos): m/z = 428 [M+H]+
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 4.040 (16.00), 4.197 (0.54), 4.214 (1.44), 4.230 (2.80), 4.251 (1.59), 4.268 (1.38), 4.278 (1.68), 4.295 (1.46), 4.316 (0.48), 5.241 (1.20), 5.261 (1.19), 7.275 (0.78), 7.294 (1.73), 7.321 (1.78), 7.339 (1.11), 7.386 (1.77), 7.394 (1.54), 7.405 (2.97), 7.412 (2.26), 7.423 (1.63), 7.444 (1.36), 7.466 (1.53), 7.617 (1.85), 7.638 (1.42), 7.742 (3.01), 7.761 (2.84), 7.787 (2.53), 7.874 (4.50), 7.893 (4.19), 8.058 (4.56), 8.183 (0.93), 8.201 (0.92).
Эту аминокислоту применяли в SPPS.
Пример 97А
({[(9Н-флуорен-9-ил)метокси]карбонил}амино)(1-метил-1Н-индазол-5-ил)уксусная кислота (энантиомер 2)
Применяя способ хирального разделения, описанный в Примере 96А, 145 мг (100% чистота, 94.3%) энантиомера 2 получали (стереохимию не определяли).
LC-MS (Способ 10): Rt = 1.80 мин; MS (ESI pos): m/z = 428 [M+H]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: -0.008 (0.78), 0.008 (0.70), 4.040 (16.00), 4.198 (0.41), 4.214 (1.16), 4.230 (2.33), 4.252 (1.27), 4.268 (1.03), 4.278 (1.36), 4.295 (1.14), 4.300 (0.99), 5.243 (0.98), 5.262 (0.98), 7.276 (0.59), 7.294 (1.34), 7.313 (1.11), 7.321 (1.38), 7.340 (0.86), 7.387 (1.36), 7.394 (1.12), 7.406 (2.32), 7.413 (1.70), 7.424 (1.22), 7.431 (0.89), 7.444 (1.06), 7.466 (1.21), 7.618 (1.52), 7.640 (1.16), 7.743 (2.38), 7.762 (2.23), 7.787 (2.02), 7.875 (3.68), 7.894 (3.45), 8.059 (3.94), 8.188 (0.73), 8.207 (0.72).
Эту аминокислоту применяли в SPPS.
Пример 98А
Бензил N-[(бензилокси)карбонил]-3-бромо-L-фенилаланинат
N-[(Бензилокси)карбонил]-3-бромо-L-фенилаланин (15.3 г, 40.5 ммоль) растворяли в метаноле (113 мл). Карбонат цезия (6.59 г, 20.2 ммоль) добавляли и смесь перемешивали пока газ не прекратил выделение. Раствор выпаривали сухости. Остаток растворяли в DMF (140 мл, 1.8 моль) и (бромометил)бензол (5.3 мл, 44 ммоль) добавляли по каплям. Через короткое время выпало твердое вещество. Суспензию разбавляли простым эфиром и водой. Органическую фазу разделяли, и водную фазу экстрагировали два раза диэтиловым простым эфиром. Объединенные органические слои промывали водой, промывали солевым раствором, а затем сушили над сульфатом магния. Кристаллический остаток растворяли в эфире и после добавления пентана образовывался осадок. Твердое вещество фильтровали и сушили. Раствор эфир/пентан выпаривали и растворяли в эфире; после добавления пентана выпадал осадок. Твердое вещество фильтровали и сушили, с получением 16.0 г (100% чистота, 84% выход) указанного в названии соединения.
LC-MS (Способ 10): Rt = 2.42 мин; MS (ESI pos): m/z = 468 [M+H]+
1Н-ЯМР (500 МГц, DMSO-d6) δ [ppm]: 2.874 (2.41), 2.895 (2.86), 2.902 (3.10), 2.923 (2.76), 3.071 (2.84), 3.082 (2.96), 3.099 (2.26), 3.109 (2.09), 3.636 (0.82), 4.336 (1.73), 4.347 (2.20), 4.353 (2.43), 4.357 (2.48), 4.363 (2.48), 4.373 (1.83), 4.383 (1.37), 4.921 (0.55), 4.963 (1.81), 4.989 (12.84), 4.994 (11.01), 5.020 (1.18), 5.043 (0.96), 5.093 (1.11), 5.119 (12.74), 5.139 (0.55), 5.147 (0.97), 7.174 (0.85), 7.211 (3.13), 7.226 (9.23), 7.241 (11.35), 7.248 (16.00), 7.263 (11.82), 7.298 (10.22), 7.313 (14.37), 7.316 (14.35), 7.321 (12.62), 7.332 (10.08), 7.336 (11.10), 7.344 (8.08), 7.351 (11.64), 7.366 (7.19), 7.379 (2.35), 7.414 (5.80), 7.429 (4.73), 7.499 (7.87), 7.880 (4.47), 7.896 (4.30).
Пример 99А
3-[(2S)-3-(Бензилокси)-2-{[(бензилокси)карбонил]амино}-3-оксопропил]бензойная кислота
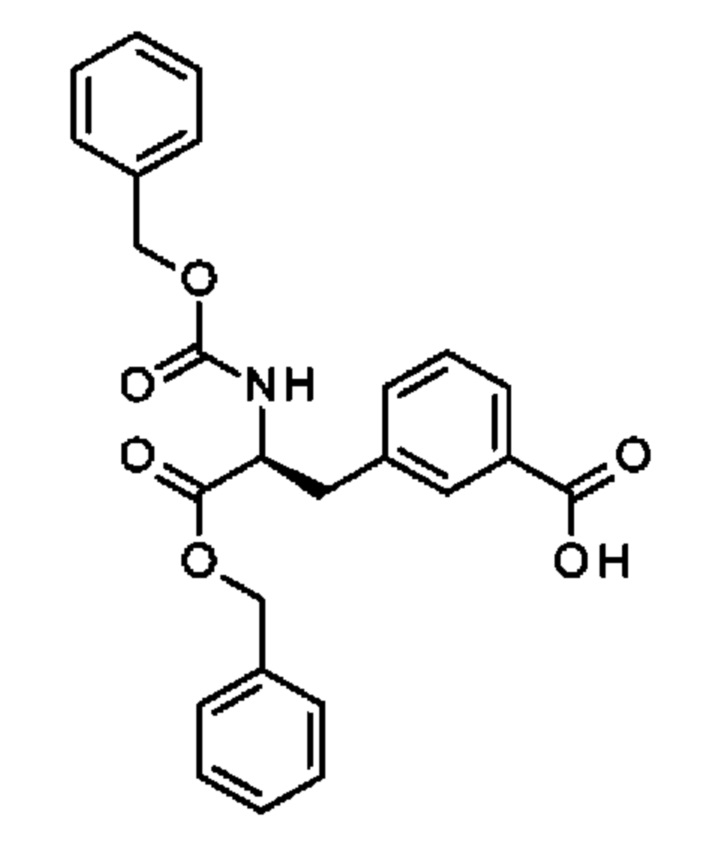
Реакцию проводили в атмосфере аргона. В автоклав объемом 2 л (скорость перемешивания 750 об/мин) добавляли палладия(II)ацетат (2.16 г, 9.61 ммоль), 1,1'-бис(дифенилфосфино)ферроцен (10.63 г, 19.22 ммоль) и калия ацетат (47.1 г, 480 ммоль). Автоклав продували аргоном и реагенты растворяли посредством добавления сухого DMF (450 мл). Раствор бензил N-[(бензилокси)карбонил]-3-бромо-1,-фенилаланината (пример 98А, 45.0 г, 96.1 ммоль) в сухом DMF (930 мл) добавляли в автоклаве, с последующим добавлением воды (18 мл). Автоклав герметизировали, повторно продували азотом и позволяли кратко перемешиваться. Автоклав находился под давлением угарного газа (3 бар), а затем его нагревали до 80°С и 3 бар в течение 960 мин. После завершения реакции, автоклав продували, открывали, а затем содержимое растворяли в воде (13 L). Реакционную смесь экстрагировали 3 раза этилацетатом (7 L) и объединенный органический слой промывали солевым раствором, а затем концентрировали. Неочищенный продукт очищали посредством препаративной нормально-фазовой хроматографии (10 кг силикагеля) с DCM/MeOH (95/5), а затем далее очищали посредством препаративной хроматографии (DCM/MeOH градиент, Biotage Isolera) с получением 10.7 г (92% чистота) указанного в названии соединения.
LC-MS (Способ 10): Rt = 1.92 мин; MS (ESI neg): m/z = 432.14 [M-H]-
Пример 100А
Бензил N-[(бензилокси)карбонил]-3-(трет-бутоксикарбонил)-L-фенилаланинат
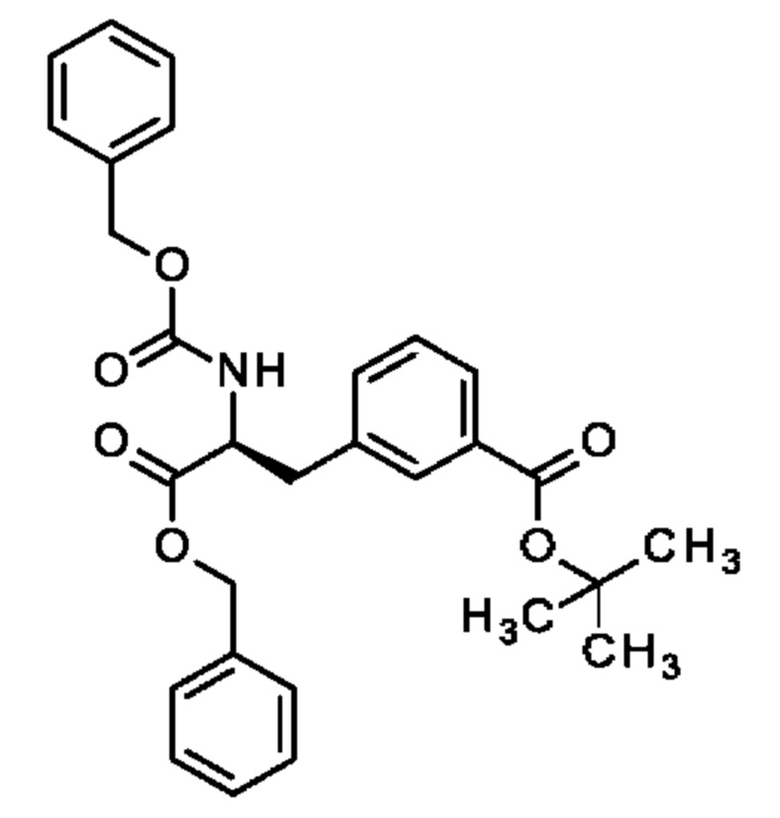
К 3-[(2S)-3-(Бензилокси)-2-{[(бензилокси)карбонил]амино}-3-оксопропил]бензойной кислоте (пример 99А 1,8.0 г, 41.5 ммоль) добавляли дихлорметан (280 мл, 4.4 моль). Циклогексан (140 мл, 1.3 моль), трет-бутил 2,2,2-трихлорэтанимидат (36.3 г, 166 ммоль) и (диэтиловый эфир)(трифтор)борон (1.6 мл, 12 ммоль) добавляли и реакционную смесь перемешивали при комнатной температуре. Через 1 ч еще (диэтиловый эфир)(трифтор)борон (1.6 мл, 12 ммоль) добавляли и реакционную смесь перемешивали всю ночь при комнатной температуре, (диэтиловый эфир)(трифтор)борон (1 мл, 7.5 ммоль) добавляли и реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Еще трет-бутил 2,2,2- трихлорэтанимидат (9.0 г, 40.5 ммоль) и еще (диэтиловый эфир)(трифтор)борон (1 мл, 7.5 ммоль) добавляли и реакционную смесь перемешивали всю ночь при комнатной температуре. Суспензию фильтровали и осадок промывали с DCM: циклогексан (2:1). Раствор разбавляли концентрированным водным раствором гидрокарбоната натрия. Объединенные водные слои экстрагировали два раза с DCM. Объединенные органические слои затем сушили над сульфатом магния, фильтровали, а затем выпаривали. Полученное масло черного цвета перемешивали с циклогексаном и этилацетатом (2%), осадок фильтровали, и фильтрат выпаривали. Полученное масло черного цвета очищали посредством колоночной хроматографии на силикагеле (циклогексан этил-ацетат (9:1)) с выходом 12.1 г (100% чистота, 60% выход) указанного в названии соединения.
LC-MS (Способ 9): Rt = 1.34 мин; MS (ESIpos): m/z = [М+Н]+490
Пример 101А
3-(трет-бутоксмкарбонил)-L-фенилаланин
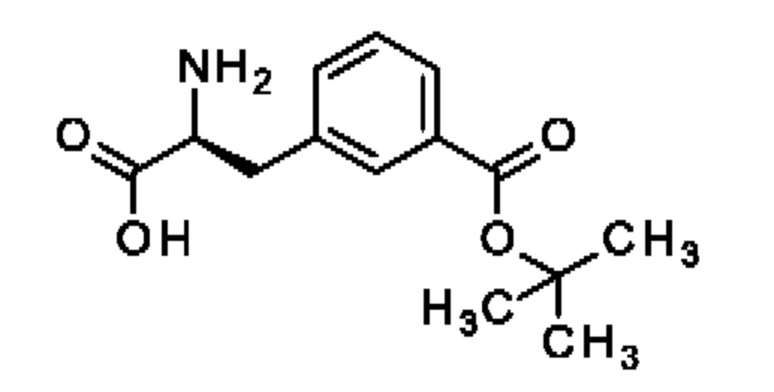
Бензил N-[(бензилокси)карбонил]-3-(трет-бутоксикарбонил)-L-фенилаланинат (пример 100А, 13.0 г, 26.6 ммоль) растворяли в метаноле (250 мл). 10% палладий на активированном угле (1.54 г) добавляли и смесь гидрогенировали при давлении окружающей среды в течение 4 часов при комнатной температуре. Образовался осадок. Суспензию растворяли в метаноле и перемешивали при возврате флегмы. Теплый раствор фильтровали над слоем диатомовой земли и фильтровальную лепешку промывали метанолом (около 1 л). Объединенный фильтрат концентрировали и сушили, с получением 6.10 г (94% чистота, 81% выход) указанного в названии соединения.
LC-MS (Способ 9): Rt = 0.59 мин; MS (ESI neg): m/z = 264 [M-H]-
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 1.525 (5.74), 1.546 (16.00), 2.326 (0.50), 2.366 (0.48), 2.670 (0.50), 2.709 (0.48), 3.169 (0.57), 7.400 (0.89), 7.419 (0.66), 7.501 (0.76), 7.739 (0.84), 7.759 (0.85), 7.796 (1.02).
Пример 102 А
3-(трет-бутоксмкарбонил)-N-{[(9H-флуорен-9-ил)метокси]карбонил}-L-фенилаланин
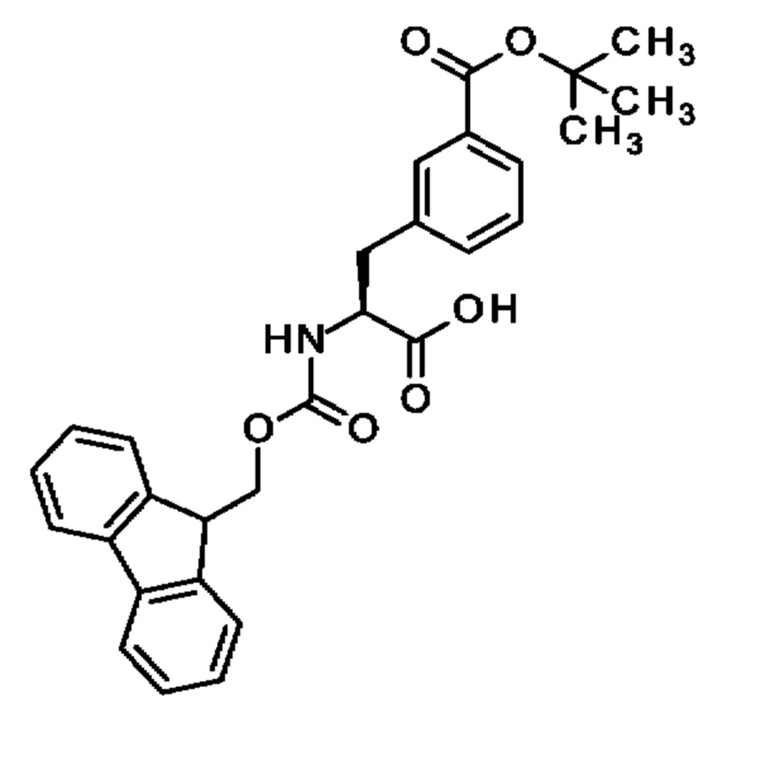
3-(трет-бутоксмкарбонил)-L-фенилаланин (пример 101 А, 6.10 г, 23.0 ммоль) растворяли в воде (100 мл) и ацетоне (100 мл). Гидрокарбонат натрия (19.3 г, 230 ммоль) добавляли. Раствор 1-({[(9H-флуорен-9-ил)метокси]карбонил}окси)пирролидин-2,5-диона (8.14 г, 24.1 ммоль) растворяли в ацетоне (100 мл) добавляли и реакционную смесь перемешивали всю ночь при комнатной температуре. Смесь разбавляли водой и экстрагировали одной порцией МТВЕ. Водный слой подкисляли с 1М соляной кислотой и экстрагировали с DCM. Объединенный органический слой промывали солевым раствором, сушили над безводном сульфатом магния, фильтровали, а затем концентрировали с получением 8.90 г (100% чистота, 79% выход) указанного в названии соединения. Продукт также присутствовал в органическом слое. Органический слой концентрировали. DCM и 1М соляную кислоту добавляли в остаток и смесь перемешивали в течение 5 мин. Остаток фильтровали и промывали водой и DCM. Слои разделяли, и водный слой экстрагировали с DCM. Объединенный органический слой затем сушили над безводном сульфатом магния, фильтровали, а затем концентрировали. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле (градиент DCM: метанол (0-10%)). Остаток растворяли в ацетонитриле и очищали посредством препаративной ВЭЖХ (колонка: Reprosil; С18; 10 мкм; 125 × 40 мм; растворитель: AcCN/H2O с 0,1% НСООН; скорость потока: 100 мл/мин; градиент: 0-5.50 мин (10:90); впрыск образца при 3.00 мин; 5.50-17.65 мин градиент до 95:5; 17.65-19.48 мин (95:5); 19.48-19.66 мин градиент до 10:90; 19.66-20.72 мин (10:90). В общем, 1.58 г (100% чистота, 14% выход) указанного в названии соединения получали.
LC-MS (Способ 10): Rt = 2.27 мин; MS (ESI neg): m/z = 486 [M-H]-
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 1.106 (1.34), 1.144 (4.63), 1.519 (16.00), 1.544 (0.64), 2.086 (1.34), 2.119 (1.97), 2.483 (1.16), 2.595 (0.68), 3.077 (0.43), 3.319 (1.89), 4.158 (0.42), 4.169 (0.81), 4.181 (2.03), 4.195 (0.76), 4.203 (0.48), 4.541 (0.43), 7.259 (0.80), 7.277 (0.76), 7.293 (0.68), 7.312 (0.44), 7.378 (0.62), 7.384 (0.86), 7.397 (1.11), 7.403 (1.62), 7.415 (0.62), 7.422 (0.90), 7.521 (0.63), 7.540 (0.45), 7.583 (0.68), 7.605 (0.86), 7.626 (0.62), 7.747 (0.59), 7.766 (0.57), 7.784 (0.68), 7.806 (0.63), 7.847 (1.02), 7.864 (1.58), 7.883 (1.42).
Эту аминокислоту применяли в SPPS.
Пример 103А
(2S,5R)-2-(2-Этилбутил)-3,6-диметокси-5-(пропан-2-ил)-2,5-дигидропиразин
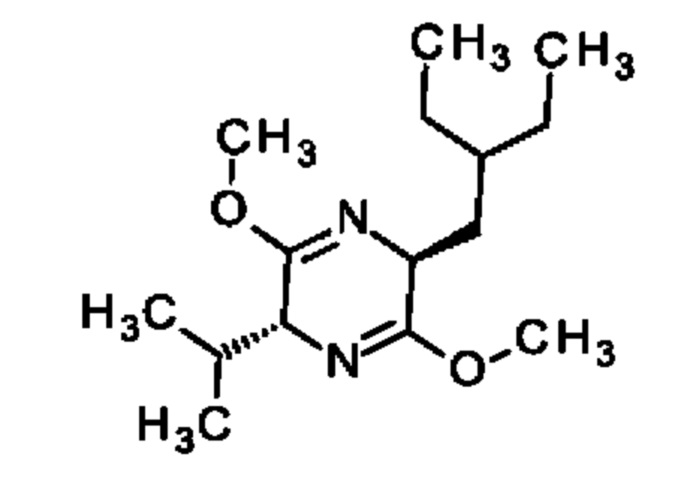
(2R)-3,6-диметокси-2-(пропан-2-ил)-2,5-дигидропиразин (CAS-RN: 109838-859, 2.9 мл, 16 ммоль) растворяли в безводном THF (60 мл, 740 ммоль) и раствор охлаждали до -78°С. н-бутиллитий (10 мл, 1.6 М, 16 ммоль) добавляли по каплям медленно и по каплям за 15 минут при перемешивании при -78°С. 3-(Бромометил)пентан (5.91 г, 35.8 ммоль) добавляли и реакционную смесь перемешивали в течение 2 ч при -78°С, затем позволяли дойти до комнатной температуры всю ночь. Раствор охлаждали до 0°С и медленно гасили водой. Диэтиловый простой эфир добавляли и после перемешивания в течение нескольких минут органический слой разделяли. Водную фазу экстрагировали два раза диэтиловым простым эфиром. Объединенные органические слои промывали солевым раствором, сушили над сульфатом магния, фильтровали и выпаривали. Неочищенный продукт очищали посредством хроматографии на силикагеле (циклогексан / этилацетат 2%) с получением 2.60 г (95% чистота, 57% выход) указанного в названии соединения.
LC-MS (Способ 10): Rt = 2.90 мин; MS (ESI pos): m/z = 269 [M+H]+
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: -0.008 (0.51), 0.008 (0.45), 0.608 (6.77), 0.624 (6.91), 0.771 (3.04), 0.790 (7.57), 0.796 (3.51), 0.808 (4.20), 0.815 (7.45), 0.833 (3.54), 0.999 (6.56), 1.016 (6.71), 1.179 (0.65), 1.197 (0.97), 1.215 (0.95), 1.223 (0.51), 1.232 (0.73), 1.240 (0.81), 1.257 (1.26), 1.275 (1.64), 1.294 (1.28), 1.310 (0.57), 1.343 (0.55), 1.356 (1.67), 1.364 (0.66), 1.369 (0.83), 1.376 (1.52), 1.388 (1.40), 1.398 (0.66), 1.403 (0.63), 1.409 (1.04), 1.422 (0.43), 1.459 (0.56), 1.474 (0.63), 1.489 (0.45), 1.637 (0.66), 1.647 (0.68), 1.656 (0.56), 1.667 (0.69), 1.670 (0.69), 1.681 (0.54), 1.690 (0.48), 1.700 (0.44), 2.191 (0.54), 2.199 (0.55), 2.208 (0.71), 2.216 (0.71), 2.225 (0.52), 2.233 (0.51), 3.597 (16.00), 3.616 (15.85), 3.916 (1.08), 3.925 (2.24), 3.933 (1.46), 3.959 (0.67), 3.969 (1.06), 3.980 (0.99), 3.990 (1.00), 4.000 (0.49). Пример 104А
Метил 4-этил-L-норлейцинант
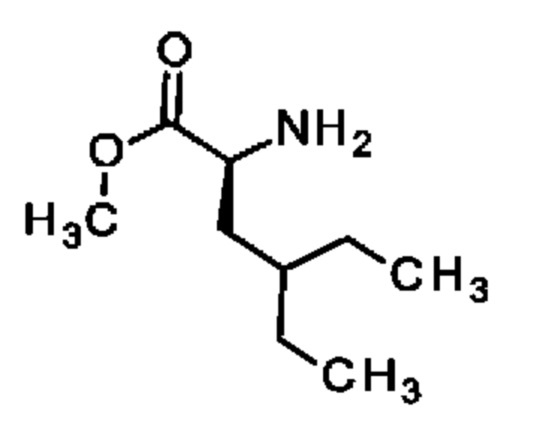
Раствор (2S,5R)-2-(2-Этилбутил)-3,6-диметокси-5-(пропан-2-ил)-2,5-дигидропиразина (пример 103А, 2.60 г, 9.69 ммоль) в метаноле (30 мл) охлаждали до 0°С и 10% водный раствор соляной кислоты (10 мл) добавляли, а затем реакционную смесь перемешивали в течение 2 ч. Раствор выпаривали и остаток разбавляли с DCM и раствор карбоната натрия (100 мл, 2.0 М). Отделенный водный слой экстрагировали два раза с DCM. Объединенный органический слой сушили над сульфатом магния, фильтровали, а затем концентрировали с получением 3.49 г (48% чистота, 100% выход) указанного в названии соединения.
GC-MS (GC-MS Способ 1): Rt = 3.18 мин, m/z = 173 [М+]
Пример 105 А
4-Этил-N-{[(9H-флуорен-9-ил)метокси]карбонил}-L-норлейцин
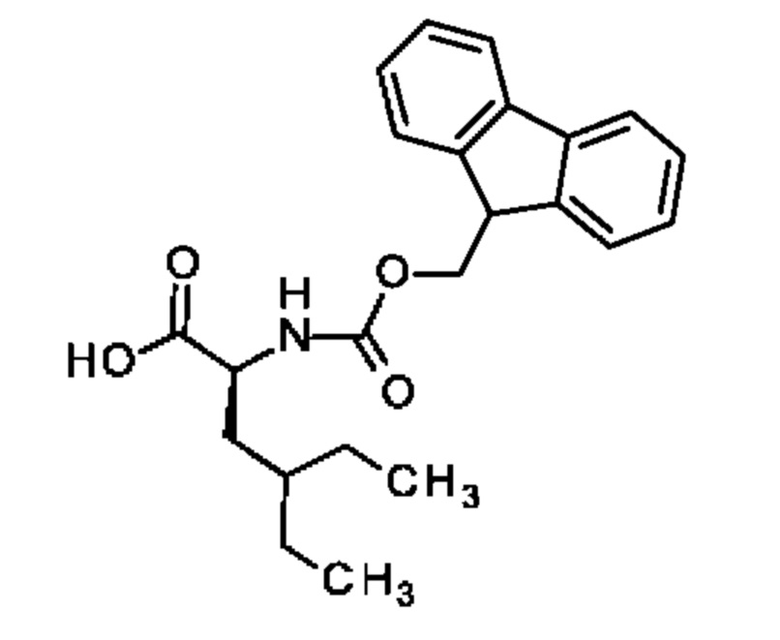
К метил 4-этил-L-норлейцинанту (пример 104А, 3.97 г, 48% чистота, 11.0 ммоль) добавляли метанол (100 мл) и полученный раствор охлаждали до 0°С. Раствор гидроксида натрия (24 мл, 1.0 М) добавляли и реакционную смесь перемешивали в течение 1 ч при 0°С, затем нагревали до комнатной температуры и перемешивали еще в течение 4 ч. Воду (80 мл), гидрокарбонат натрия (2.03 г, 24.2 ммоль) и карбонат натрия (4.00 г, 37.7 ммоль) последовательно добавляли в раствор. Метанол удаляли из реакционной смеси посредством роторного испарителя, и водный раствор разбавляли 1,4-диоксаном (60 мл). Суспензию охлаждали до 0°С и (9H-флуорен-9-ил)метил хлорформиат (CAS-RN: 28920-43-6, 8.54 г, 33.0 ммоль) добавляли. Реакционную смесь перемешивали всю ночь при комнатной температуре. Воду и МТВЕ добавляли, быстро перемешивали, а затем органический слой разделяли. Водный слой экстрагировали с МТВЕ, а затем объединенный органический слой промывали с 5% раствором карбоната натрия. Объединенный водный слой подкисляли до рН 1 гидросульфатом калия и водную суспензию экстрагировали три раза с DCM. Объединенный органический слой промывали солевым раствором, сушили над безводном сульфатом магния, фильтровали, а затем концентрировали. Неочищенный продукт очищали посредством препаративной ВЭЖХ (колонка: Reprosil; С18; 10 мкм; 125 × 40 мм; растворитель: AcCN/H2O с содержанием 0,1% НСООН; скорость потока: 100 мл/мин; градиент: 0-5.50 мин (10:90); впрыск образца при 3.00 мин; 5.50-17.65 мин градиент до 95:5; 17.65-19.48 мин (95:5); 19.48-19.66 мин градиент до 10:90; 19.66-20.72 мин (10:90), с получением 1.57 г (100% чистота, 37% выход) указанного в названии соединения.
LC-MS (Способ 11): Rt = 1.49 мин; MS (ESI pos): m/z = 404 [M+Na]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: -0.008 (3.19), 0.008 (3.01), 0.774 (6.73), 0.792 (16.00), 0.809 (13.36), 0.826 (14.04), 0.844 (7.32), 1.181 (0.92), 1.197 (1.90), 1.216 (3.12), 1.234 (3.84), 1.253 (2.82), 1.273 (1.79), 1.293 (2.23), 1.308 (3.41), 1.331 (4.60), 1.339 (4.12), 1.505 (0.45), 1.557 (3.55), 1.575 (5.01), 1.590 (3.09), 3.950 (1.22), 3.970 (2.67), 3.989 (2.69), 4.009 (1.11), 4.204 (1.35), 4.220 (3.90), 4.238 (7.46), 4.263 (5.22), 4.279 (3.57), 4.286 (5.10), 4.304 (3.43), 4.310 (2.32), 4.328 (1.06), 7.299 (2.60), 7.306 (2.93), 7.317 (5.73), 7.322 (5.59), 7.336 (3.69), 7.341 (3.48), 7.400 (6.17), 7.419 (10.33), 7.437 (4.62), 7.626 (4.21), 7.647 (3.80), 7.711 (8.98), 7.729 (8.16), 7.884 (9.94), 7.903 (9.15), 12.540 (0.98). Эту аминокислоту применяли в SPPS.
Пример 106 А
N-{[(9H-флуорен-9-ил)метокси]карбонил}-2,6-дифтор-L-фенилаланин
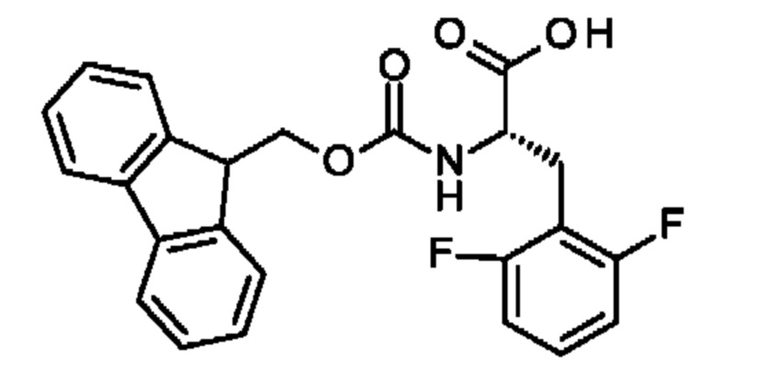
К метил 2,6-дифтор-Ь-фенилаланинату (CAS-RN: 1192057-28-5, 1.03 г, 4.79 ммоль) добавляли метанол (18.5 мл) и раствор охлаждали до 0°С. Раствор гидроксида натрия (4.8 мл, 1.0 М) добавляли и реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Воду (17 мл), гидрокарбонат натрия (402 мг, 4.79 ммоль) и карбонат натрия (667 мг, 6.29 ммоль) добавляли в раствор. Метанол удаляли посредством роторного испарителя и водный слой разбавляли с 1,4-диоксаном (14 мл). Суспензию охлаждали до 0°С и (9H-флуорен-9-ил)метил хлорформиат (CAS-RN: 28920-43-6, 1.86 г, 7.18 ммоль) добавляли. Смесь перемешивали в течение 2 ч при комнатной температуре. Реакционную смесь разбавляли водой и промывали с МТВЕ. Водный слой подкисляли до рН 1 с 1 М соляной кислотой и экстрагировали три раза этилацетатом. Объединенные органические слои промывали солевым раствором, сушили над безводном сульфатом магния, фильтровали и концентрировали, с получением 1.74 г (100% чистота, 86% выход) указанного в названии соединения.
LC-MS (Способ 10): Rt = 2.00 мин; MS (ESI pos): m/z = 424 [M+H]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 1.157 (0.74), 1.175 (1.50), 1.193 (0.76), 1.988 (2.79), 2.992 (0.74), 3.014 (0.87), 3.025 (1.22), 3.048 (1.17), 3.118 (1.24), 3.134 (1.32), 3.153 (0.81), 3.169 (0.76), 3.316 (16.00), 4.021 (0.71), 4.039 (0.70), 4.131 (0.95), 4.149 (1.39), 4.166 (2.72), 4.194 (4.15), 4.213 (2.65), 4.235 (0.72), 7.013 (1.71), 7.033 (3.28), 7.053 (2.07), 7.290 (1.33), 7.307 (3.44), 7.323 (4.18), 7.341 (2.36), 7.357 (0.48), 7.397 (2.41), 7.415 (4.00), 7.434 (1.83), 7.639 (2.22), 7.659 (3.87), 7.679 (1.99), 7.818 (1.97), 7.839 (1.95), 7.876 (4.80), 7.894 (4.40), 12.793 (0.81).
Эту аминокислоту применяли в SPPS.
Пример 107А
N-{[(9H-флуорен-9-ил)метокси]карбонил}-2,5-дифтор-L-фенилаланин
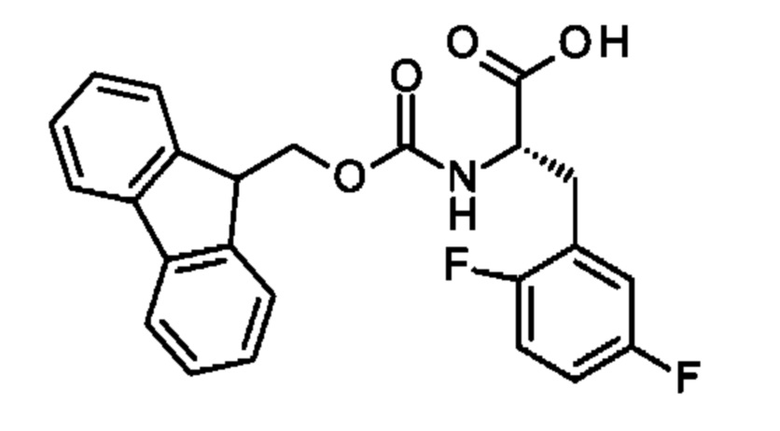
К метил 2,5-дифтор-£-фенилаланинату (CAS-RN: 1213491-95-2) (2.27 г, 10.5 ммоль) добавляли метанол (40 мл) и полученный раствор охлаждали до 0°С. Раствор гидроксида натрия (11 мл, 1.0 М) добавляли и реакционную смесь перемешивали в течение 1.5 ч при комнатной температуре. Воду (35 мл), гидрокарбонат натрия (886 мг, 10.5 ммоль) и карбонат натрия (1.47 г, 13.9 ммоль) добавляли в раствор. Метанол удаляли посредством роторного испарителя и водный слой разбавляли с 1,4-диоксаном (30 мл). Суспензию охлаждали до 0°С и (9H-флуорен-9-ил)метил хлорформиат (CAS-RN: 28920-43-6, 4.09 г, 15.8 ммоль) добавляли, и реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Смесь разбавляли водой и промывали с МТВЕ. Водный слой подкисляли до рН 1 1 М раствором соляной кислоты и экстрагировали три раза этилацетатом. Объединенные органические слои промывали солевым раствором, сушили над безводном сульфатом магния, фильтровали и концентрировали. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле (градиент DCM: метанол 0-5%), затем остаток растворяли в ацетонитриле и снова очищали посредством препаративной ВЭЖХ (колонка: Reprosil; С18; 10 мкм; 125 × 40 мм; растворитель: AcCN/H2O с содержанием 0,1% НСООН; скорость потока: 100 мл/мин; градиент: 0-5.50 мин (10: 90); впрыск образца при 3.00 мин; 5.50-17.65 мин градиент до 95: 5; 17.65-19.48 мин (95: 5); 19.48-19.66 мин до 10: 90; 19.66-20.72 мин (10:90) с получением 1.82 г (100% чистота, 41% выход) указанного в названии соединения.
LC-MS (Способ 9): Rt = 1.04 мин; MS (ESI pos): m/z = 424 [M+H]+
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: -0.008 (2.88), 0.008 (2.75), 1.234 (0.68), 2.329 (0.47), 2.671 (0.46), 2.843 (2.48), 2.870 (3.13), 2.877 (3.47), 2.904 (3.10), 3.166 (3.44), 3.177 (3.74), 3.200 (2.97), 3.212 (2.73), 4.141 (1.90), 4.158 (5.16), 4.173 (9.08), 4.196 (12.08), 4.201 (12.22), 4.211 (8.55), 4.221 (7.38), 4.233 (3.76), 4.246 (2.97), 4.259 (2.15), 7.084 (0.87), 7.093 (1.61), 7.106 (2.33), 7.114 (3.81), 7.124 (3.21), 7.135 (2.97), 7.144 (2.17), 7.175 (3.13), 7.187 (4.77), 7.197 (6.58), 7.210 (7.69), 7.220 (4.45), 7.232 (3.61), 7.263 (4.47), 7.282 (9.79), 7.301 (7.22), 7.305 (7.95), 7.325 (4.82), 7.349 (0.45), 7.389 (6.78), 7.408 (11.73), 7.426 (5.46), 7.489 (0.48), 7.509 (0.67), 7.532 (0.46), 7.610 (7.40), 7.628 (12.92), 7.646 (6.48), 7.791 (6.23), 7.812 (6.10), 7.871 (16.00), 7.890 (14.50), 12.862 (3.43).
Эту аминокислоту применяли в SPPS.
Эту аминокислоту также получали из метил-2,5-дифтор-L-фенилаланината, полученного в Примере 126А. Аминокислота также может быть получена из 2,5-дифтор-L-фенилаланината (CAS RN: 31105-92-7) с использованием процедуры, описанной в Примере 106А.
Пример 108А
Бензил тетрагидро-2H-пиран-3-илацетат (рацемат)
Тетрагидро-2H-пиран-3-илуксусную кислоту (1.00 г, 6.94 ммоль) растворяли в метаноле (10 мл, 250 ммоль). Карбонат цезия (1.13 г, 3.47 ммоль) добавляли и реакционную смесь перемешивали при комнатной температуре пока не прекратится выделение газа. Раствор затем выпаривали. Остаток растворяли в DMF (10 мл), (бромометил)бензол (1.3 мл, 11 ммоль) добавляли, и реакционную смесь перемешивали всю ночь при комнатной температуре. Еще (бромометил)бензол (325 мкл, 2.75 ммоль) добавляли и реакционную смесь перемешивали в течение 3 ч при комнатной температуре. Суспензию затем разбавляли водой и простым эфиром. После перемешивания в течение нескольких минут, органический слой разделяли. Водный слой экстрагировали три раза диэтиловым простым эфиром. Объединенный органический слой промывали солевым раствором, сушили над безводном сульфатом магния, фильтровали, а затем концентрировали. Неочищенный продукт очищали посредством хроматографии на силикагеле (циклогексан / этилацетат (85/15)) с получением 1.50 г (100% чистота, 92% выход) указанного в названии соединения.
LC-MS (Способ 10): Rt = 1.84 мин; MS (ESI pos): m/z = 235 [M+H]+
1Н-ЯМР (500 МГц, DMSO-d6) δ [ppm]: 1.161 (0.45), 1.170 (0.51), 1.183 (0.89), 1.187 (0.68), 1.191 (0.98), 1.196 (0.60), 1.204 (0.68), 1.208 (1.03), 1.212 (0.77), 1.217 (1.00), 1.229 (0.62), 1.238 (0.63), 1.428 (0.47), 1.447 (0.62), 1.449 (0.74), 1.455 (0.95), 1.463 (0.63), 1.468 (0.83), 1.476 (1.42), 1.484 (0.80), 1.489 (0.56), 1.498 (0.98), 1.501 (0.53), 1.506 (1.11), 1.509 (1.00), 1.516 (1.65), 1.523 (1.08), 1.532 (0.56), 1.543 (0.70), 1.550 (0.40), 1.747 (0.90), 1.751 (0.77), 1.755 (0.89), 1.764 (0.62), 1.773 (0.82), 1.777 (0.69), 1.781 (0.77), 1.887 (0.41), 1.894 (0.68), 1.901 (0.68), 1.908 (0.76), 1.914 (0.90), 1.921 (0.74), 1.927 (0.68), 1.934 (0.63), 2.194 (1.30), 2.208 (1.18), 2.225 (3.67), 2.239 (3.41), 2.255 (3.55), 2.270 (3.23), 2.286 (1.23), 2.301 (1.12), 3.018 (2.02), 3.038 (2.30), 3.041 (2.34), 3.059 (2.06), 3.246 (1.05), 3.252 (1.07), 3.267 (1.61), 3.273 (1.61), 3.289 (1.21), 3.295 (1.19), 3.685 (0.75), 3.692 (1.34), 3.705 (1.77), 3.708 (1.89), 3.713 (2.24), 3.716 (2.16), 3.726 (1.17), 3.730 (1.14), 3.734 (1.08), 3.738 (0.97), 5.089 (16.00), 7.310 (0.41), 7.315 (0.73), 7.320 (0.71), 7.327 (2.21), 7.334 (1.16), 7.339 (1.36), 7.342 (1.95), 7.345 (2.49), 7.348 (2.36), 7.351 (1.63), 7.360 (9.48), 7.364 (11.33), 7.371 (1.08), 7.376 (3.55), 7.378 (3.74), 7.382 (1.62), 7.390 (0.55), 7.393 (1.20), 7.395 (0.74).
Пример 109А
Бензил тетрагидро-2H-пиран-3-ил ацетат
Диастереомерную смесь (пример 108А, 1.5 г) растворяли в метаноле / ацетонитриле (30 мл) и разделяли с применением THAR сверхкритической жидкостной хроматографии Prep 200 устройство с применением Chiralpak AZ-H (SFC) 5 мкм, 250 × 30 мм колонка; элюент: CO2/этанол (90:10); давление: 135 бар; температура элюента: 35°С; скорость потока: 100 г/мин; обнаружение: UV 210 нм; объем впрыска 0.4 мл/впрыск; при общих рабочих условиях 1 г/мин CO2 приблизительно эквивалентен 1 мл/мин.
Установки последовательности: время цикла = 11,2 мин
Фракцию 1 собирали при 1.48 мин (энантиомер 1)
Фракцию 2 собирали при 1.89 мин (энантиомер 2)
Фракции, содержащие продукт, объединяли и концентрировали.
Фракции анализировали с помощью аналитической SFC-MS (Agilent, колонка: Daicel AZ-3 3 мкм 100 × 4,6 мм, элюент: CO2/этанол (90:10), давление BPR: 130 бар, температура BPR: 60°С, температура колонки: 40°С; скорость потока: 3 мл/мин; УФ 210 нм.
Стереохимия двух энантиомеров не установлена.
Бензил тетрагидро-2H-пиран-3-илацетат (энантиомер 1) получали с 99.5% ее (641 мг (100% чистота).
LC-MS (Способ 9): Rt = 0.98 мин; MS (ESI pos): m/z = 235 [M+H]+
1H-ЯМР (500 МГц, DMSO-d6) δ [ppm]: 1.161 (0.45), 1.170 (0.51), 1.183 (0.89), 1.187 (0.67), 1.191 (0.98), 1.196 (0.61), 1.204 (0.67), 1.209 (1.03), 1.213 (0.77), 1.217 (1.00), 1.230 (0.61), 1.238 (0.62), 1.428 (0.48), 1.447 (0.61), 1.449 (0.75), 1.455 (0.95), 1.463 (0.64), 1.468 (0.82), 1.476 (1.41), 1.484 (0.79), 1.489 (0.56), 1.498 (0.98), 1.501 (0.52), 1.507 (1.05), 1.510 (1.00), 1.516 (1.65), 1.523 (1.07), 1.532 (0.55), 1.543 (0.70), 1.747 (0.90), 1.751 (0.78), 1.755 (0.89), 1.764 (0.63), 1.773 (0.81), 1.777 (0.69), 1.781 (0.78), 1.887 (0.40), 1.894 (0.69), 1.901 (0.68), 1.908 (0.76), 1.914 (0.91), 1.921 (0.74), 1.927 (0.67), 1.934 (0.64), 2.194 (1.30), 2.208 (1.18), 2.225 (3.69), 2.239 (3.42), 2.255 (3.56), 2.270 (3.24), 2.286 (1.23), 2.301 (1.12), 3.019 (2.02), 3.038 (2.29), 3.041 (2.36), 3.060 (2.07), 3.246 (1.05), 3.252 (1.08), 3.268 (1.63), 3.273 (1.63), 3.289 (1.24), 3.295 (1.23), 3.686 (0.75), 3.692 (1.34), 3.705 (1.73), 3.708 (1.90), 3.713 (2.19), 3.715 (2.20), 3.726 (1.19), 3.730 (1.13), 3.734 (1.08), 3.738 (0.97), 5.089 (16.00), 7.310 (0.40), 7.315 (0.74), 7.321 (0.69), 7.328 (2.23), 7.334 (1.13), 7.339 (1.36), 7.342 (1.99), 7.345 (2.52), 7.347 (2.34), 7.350 (1.66), 7.360 (9.33), 7.364 (11.16), 7.371 (1.09), 7.376 (3.58), 7.378 (3.72), 7.382 (1.63), 7.390 (0.54), 7.393 (1.20), 7.396 (0.75).
Бензил тетрагидро-2H-пиран-3-илацетат (энантиомер 2) также получали с 99.5% ее (611 мг (100% чистота).
Пример 110А
Тетрагидро-2H-пиран-3-илуксусная кислота (энантиомер 1)
10% палладий на угле (84,0 мг) увлажняли метанолом в атмосфере аргона. Добавляли раствор бензил тетра-гидро-2Н-пиран-3-илацетата (пример 109А, 641 мг, 2,74 ммоль). Затем реакционную смесь гидрировали в течение 2 часов при давлении окружающей среды при температуре окружающей среды. Катализатор удаляли путем фильтрации над слоем диатомовой земли, фильтровальную лепешку промывали метанолом, и растворитель удаляли посредством роторной дистилляции, с получением 364 мг (100% чистота, 92% выход) указанного в названии соединения.
GC-MS (GC-MS Способ 1): Rt = 3.57 мин, m/z = 144 [М+]
1H-ЯМР (500 МГц, DMSO-d6) δ [ppm]: 1.145 (1.98), 1.154 (2.21), 1.166 (4.03), 1.171 (3.11), 1.175 (4.45), 1.180 (2.85), 1.188 (3.11), 1.192 (4.69), 1.196 (3.55), 1.201 (4.57), 1.213 (2.68), 1.222 (2.75), 1.428 (1.03), 1.436 (1.93), 1.445 (1.35), 1.449 (1.65), 1.455 (2.72), 1.458 (3.25), 1.463 (4.18), 1.471 (2.92), 1.476 (3.82), 1.484 (6.50), 1.493 (3.67), 1.498 (2.58), 1.506 (5.73), 1.514 (6.62), 1.521 (7.99), 1.528 (5.13), 1.537 (2.67), 1.548 (3.18), 1.554 (1.80), 1.563 (0.64), 1.759 (1.78), 1.767 (4.22), 1.771 (3.85), 1.775 (4.26), 1.784 (2.89), 1.793 (3.85), 1.797 (3.49), 1.801 (3.85), 1.809 (1.56), 1.824 (0.81), 1.832 (1.31), 1.839 (1.87), 1.846 (3.17), 1.853 (3.16), 1.859 (3.72), 1.866 (4.34), 1.873 (3.58), 1.879 (3.34), 1.886 (3.00), 1.893 (1.65), 1.900 (1.44), 1.908 (0.69), 2.023 (6.18), 2.036 (5.11), 2.054 (16.00), 2.068 (14.33), 2.087 (15.54), 2.101 (13.64), 2.118 (5.69), 2.133 (5.09), 2.999 (9.23), 3.018 (10.90), 3.021 (11.02), 3.040 (9.47), 3.134 (0.64), 3.247 (6.14), 3.253 (6.29), 3.269 (9.57), 3.275 (9.49), 3.291 (7.06), 3.297 (6.90), 3.586 (0.59), 3.696 (3.77), 3.703 (6.42), 3.709 (4.14), 3.717 (8.63), 3.725 (11.12), 3.739 (5.37), 3.743 (5.40), 3.747 (5.21), 3.750 (4.68).
Это вещество применяли в SPPS.
Пример 111А
(3S)-3-[(2S,5R)-3,6-диметокси-5-(пропан-2-ил)-2,5-дигидропиразин-2-ил]циклогексан-1-он
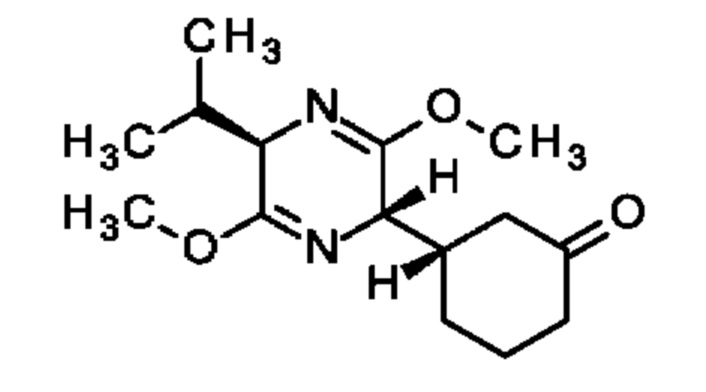
(2R)-3,6-диметокси-2-(пропан-2-ил)-2,5-дигидропиразин (4.61 г, 25.0 ммоль) растворяли в THF (20 мл) и охлаждали до -78°С. Бутиллитий (16 мл, 1.6 М, 25 ммоль) добавляли по каплям при перемешивании. Перемешивание продолжали в течение 15 мин при -78°С. Лития (азанидилиенметилиден)(тиофен-2-ил)медь (100 мл, 0.25 М, 25 ммоль) добавляли и смесь перемешивали в течение 15 мин при -78°С. Раствор циклогексен-2-ен-1-она (2.4 мл, 25 ммоль) в THF (10 мл) добавляли и реакционную смесь перемешивали в течение 10 мин при -78°С, затем далее перемешивали при температуре окружающей среды в течение 2 ч. Реакцию гасили водой и экстрагировали два раза этилацетатом. Объединенный органический слой промывали раствором гидрокарбоната натрия и солевым раствором, сушили над безводном сульфатом магния, фильтровали, а затем остаток концентрировали при пониженном давлении с получением 6.8 г бесцветного масла. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле (градиент циклогексан-этил-ацетат 85-15) с получением 5.7 г (90% чистота, 64% выход) указанного в названии соединения.
LC-MS (Способ 12): Rt = 3.14 мин; MS (ESIpos): m/z = 281.2 [М+Н]+
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 0.587 (0.94), 0.604 (1.17), 0.617 (6.04), 0.634 (6.04), 0.971 (0.51), 0.988 (5.87), 0.995 (1.32), 1.005 (5.67), 1.012 (0.98), 1.032 (0.88), 1.049 (0.91), 1.575 (0.55), 1.596 (0.42), 1.607 (0.61), 1.750 (0.61), 1.760 (0.78), 1.784 (1.05), 1.794 (1.28), 1.816 (0.50), 1.837 (0.50), 1.843 (0.57), 1.874 (0.43), 1.979 (0.82), 2.012 (1.26), 2.024 (0.52), 2.032 (0.55), 2.044 (0.95), 2.057 (0.40), 2.122 (0.46), 2.148 (0.80), 2.156 (1.00), 2.164 (1.00), 2.173 (0.67), 2.181 (0.70), 2.190 (0.69), 2.199 (0.54), 2.207 (0.59), 2.225 (0.71), 2.242 (0.64), 2.259 (0.74), 2.275 (0.64), 2.294 (0.45), 2.310 (0.65), 2.319 (0.47), 2.329 (0.48), 2.338 (0.58), 2.348 (0.54), 3.602 (0.89), 3.631 (16.00), 3.642 (4.12), 3.652 (14.26), 3.902 (1.14), 3.911 (2.10), 3.920 (1.33), 3.943 (0.45), 4.069 (1.14), 4.078 (1.82), 4.086 (0.98), 4.492 (0.55), 5.674 (0.91).
Пример 112А
(1R,3S)-3-[(2S,5R)-3,6-диметокси-5-(пропан-2-ил)-2,5-дигидропиразин-2-ил]циклогексан-1-ол (2 стереоизомера присутствуют)
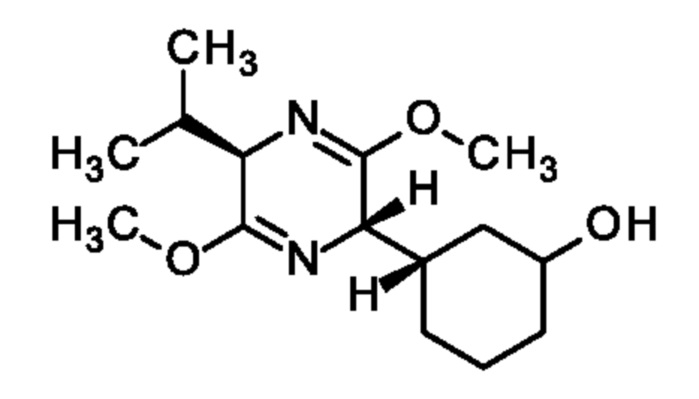
(3S)-3-[(2S,5R)-3,6-диметокси-5-(пропан-2-ил)-2,5-дигидропиразин-2-ил]циклогексан-1-он (пример 111А, 5.70 г, 20.3 ммоль) растворяли в метаноле (50 мл) и охлаждали до 0°С. Натрия тетрагидроборат (769 мг, 20.3 ммоль) добавляли и реакционную смесь перемешивали всю ночь при комнатной температуре. Еще натрия тетрагидроборат (769 мг, 20.3 ммоль) добавляли и перемешивание продолжали при комнатной температуре. Эту стадию повторяли еще 2 раза с 0.5 экв. Тетрагидробората натрия. Метанол выпаривали и остаток разбавляли водой и этилацетатом. После перемешивания в течение нескольких минут, слои разделяли. Водный слой экстрагировали этилацетатом. Объединенный органический слой промывали солевым раствором, сушили над безводном сульфатом магния, фильтровали, и остаток концентрировали при пониженном давлении с получением 5.7 г желтого масла. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле (градиент циклогексан-циклогексан этилацетат 5% - 30%) с получением 4.95 г (86% выход) указанного в названии соединения.
LC-MS (Способ 10): Rt = 1.87 мин; MS (ESI pos): m/z = 283.2 [M+H]+; Rt = 1.91 мин; MS (ESI pos): m/z = 283.2 [M+H]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 0.589 (1.83), 0.600 (6.10), 0.605 (2.52), 0.617 (6.02), 0.739 (0.91), 0.768 (0.96), 0.798 (0.42), 0.898 (0.47), 0.934 (0.50), 0.997 (6.80), 1.014 (7.00), 1.157 (0.53), 1.175 (0.92), 1.193 (0.73), 1.203 (0.72), 1.211 (0.59), 1.227 (0.47), 1.235 (0.72), 1.243 (0.51), 1.313 (0.52), 1.321 (0.63), 1.352 (0.99), 1.398 (2.43), 1.485 (0.59), 1.515 (0.56), 1.668 (0.44), 1.676 (0.56), 1.684 (0.44), 1.708 (0.51), 1.745 (0.57), 1.774 (0.54), 1.879 (0.58), 1.887 (0.56), 1.988 (1.39), 2.185 (0.54), 2.193 (0.57), 2.202 (0.73), 2.210 (0.75), 2.219 (0.55), 2.227 (0.55), 3.611 (7.06), 3.617 (3.03), 3.622 (16.00), 3.629 (14.28), 3.845 (0.55), 3.854 (0.50), 3.862 (1.17), 3.871 (2.39), 3.880 (1.79), 3.933 (1.11), 3.942 (1.74), 3.951 (0.86), 4.215 (0.59), 4.223 (0.57), 4.431 (2.16), 4.443 (2.10).
Пример 113A
Метил (2S)-амино[(1S)-3-гидроксициклогексил]ацетат (2 стереоизомера присутствуют)
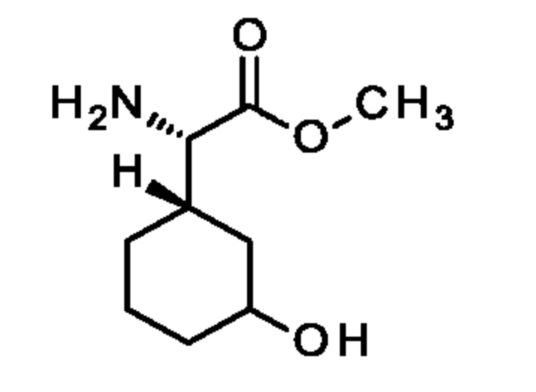
(3S)-3-[(2S,5R)-5-изопропил-3,6-диметокси-2,5-дигидропиразин-2-ил]циклогексанол (пример 112А, 4.95 г, 17.5 ммоль) растворяли в метаноле (45 мл) и раствор охлаждали до 0°С. А 10% водный раствор соляной кислоты (15 мл) добавляли и реакционную смесь перемешивали в течение короткого времени при 0°С, затем перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь концентрировали, остаток разбавляли с DCM, а затем водный раствор карбоната натрия (150 мл, 2.0 М, 300 ммоль) добавляли. Смесь перемешивали, а затем слои разделяли. Водный слой экстрагировали два раза с DCM. Объединенный органический слой сушили над безводном сульфатом магния, фильтровали, а затем концентрировали. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле (DCM: метанол 95+5) с получением 1.92 г (58% выход) указанного в названии соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: -0.008 (1.04), 0.008 (0.93), 0.760 (0.69), 0.777 (0.71), 0.834 (0.61), 0.867 (1.92), 0.875 (1.16), 0.885 (1.54), 0.892 (1.18), 0.910 (0.45), 0.922 (0.61), 0.942 (1.05), 0.951 (0.95), 0.973 (1.17), 0.981 (1.06), 1.003 (0.54), 1.012 (0.50), 1.157 (0.67), 1.165 (0.41), 1.190 (0.65), 1.198 (0.44), 1.223 (0.42), 1.375 (0.69), 1.407 (0.79), 1.498 (0.52), 1.513 (0.89), 1.519 (0.95), 1.527 (0.74), 1.549 (0.69), 1.633 (2.72), 1.671 (0.69), 1.680 (0.78), 1.753 (1.26), 1.783 (1.67), 1.959 (0.49), 2.086 (0.85), 3.096 (0.46), 3.110 (0.48), 3.138 (1.12), 3.151 (1.10), 3.162 (1.07), 3.175 (1.02), 3.276 (0.43), 3.288 (0.55), 3.593 (0.71), 3.603 (4.85), 3.612 (16.00), 3.626 (1.87), 3.633 (0.96), 3.637 (1.57), 4.232 (0.60), 4.240 (0.55), 4.481 (1.83), 4.492 (1.73), 5.754 (0.68).
Пример 114А
(2S)-2-{[(9H-флуорен-9-илметокси)карбонил]амино}-2-[(1S)-3-гидроксициклогексил]уксусная кислота (2 стереоизомера присутствуют)
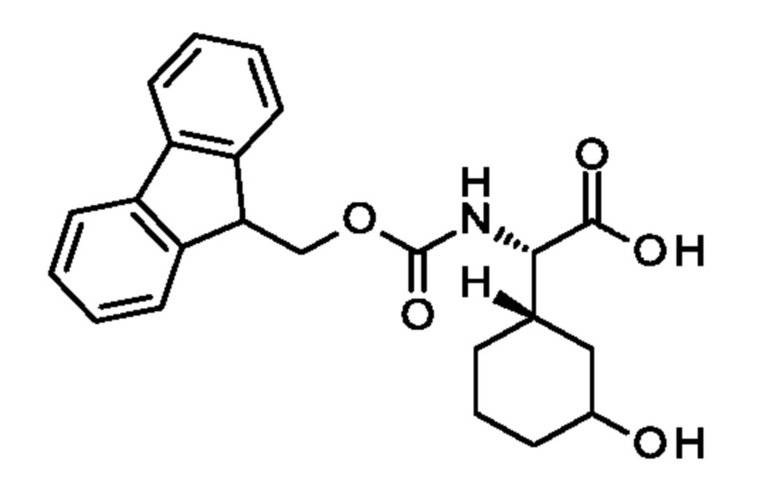
Метил (2S)-амино[(1S)-3-гидроксициклогексил]ацетат (пример 113А, 1.92 г, 10.3 ммоль) добавляли в метанол (40 мл) и раствор охлаждали до 0°С. Раствор гидроксида натрия (10 мл, 1.0 М, 10 ммоль) добавляли и реакционную смесь перемешивали в течение 1.5 ч при комнатной температуре, а затем в течение 1 ч при 50°С. Воду (37 мл), гидрокарбонат натрия (861 мг, 10.3 ммоль) и карбонат натрия (1.43 г, 13.5 ммоль) добавляли в раствор. Метанол выпаривали и водный слой разбавляли 1,4-диоксаном (30 мл). Суспензию охлаждали до 0°С и (9H-флуорен-9-ил)метил хлорформиат (CAS-RN: 28920-43-6, 3.98 г, 15.4 ммоль) добавляли. Смесь перемешивали всю ночь при комнатной температуре. Воду и МТВЕ добавляли и органический слой разделяли. Водный слой подкисляли и экстрагировали три раза с DCM. Объединенные органические слои промывали солевым раствором, сушили над безводном сульфатом магния, и фильтровали. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле (DCM: метанол/НОАс 95+5+1) с получением 2.22 г (55% выход) указанного в названии соединения.
LC-MS (Способ 10): Rt = 1.62 мин; MS (ESI pos): m/z = 396.18 [M+H]+; Rt = 1.67 мин; MS (ESI pos): m/z = 396.18 [M+H]+
Пример 115A
(2S)-2-({[(9H-флуорен-9-ил)метокси]карбонил}амино)-2-[(1S,3R)-3-гидроксициклогексил]уксусная кислота (диастереомер 1)
Диастереомер 1
(2S)-{[(9H-флуорен-9-илметокси)карбонил]амино}[(1S)-3-гидроксициклогексил]уксусную кислоту (пример 114А, 2.22 г) очищали посредством хиральной хроматографии с применением способа, описанного в Примере 96 А, с получением 890 мг (100% чистота, 40% выход, 99% ее) диастереомера 1.
LC-MS (Способ 10): Rt = 1.62 мин; MS (ESI pos): m/z = 396.2 [M+H]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 0.878 (0.88), 0.909 (1.80), 0.940 (2.23), 0.962 (1.87), 0.986 (2.99), 1.025 (4.63), 1.053 (3.69), 1.083 (1.14), 1.151 (1.18), 1.184 (2.16), 1.217 (1.71), 1.249 (0.53), 1.492 (2.55), 1.523 (2.32), 1.668 (2.86), 1.701 (2.42), 1.779 (6.54), 1.802 (4.90), 2.073 (4.22), 3.875 (0.46), 3.900 (3.08), 3.916 (3.51), 3.921 (3.52), 3.936 (2.67), 4.197 (1.31), 4.211 (3.35), 4.223 (9.23), 4.238 (12.01), 4.242 (10.59), 4.253 (6.34), 4.270 (3.91), 4.296 (0.72), 4.563 (2.04), 7.312 (6.21), 7.331 (14.61), 7.349 (9.50), 7.402 (9.30), 7.420 (15.31), 7.439 (6.83), 7.603 (4.81), 7.624 (4.95), 7.752 (11.74), 7.770 (10.65), 7.884 (16.00), 7.903 (14.82), 8.155 (0.69).
Эту аминокислоту применяли в SPPS.
Пример 116А
(2S)-2-({[(9Н-флуорен-9-ил)метокси]карбонил}амино)-2-[(1S,3S)-3-гидроксициклогексил]уксусная кислота (диастереомер 2)
В ходе хиральной хроматографии диастереомерной смеси (2S)-({[(9Н-флуорен-9-ил)метокси]карбонил}амино)[(1S,3S)-3-гидроксициклогексил]уксусной кислоты (пример 114А) также выделили 280 мг (95% чистота, 12% выход) диастереомер 2.
LC-MS (Способ 10): Rt = 1.67 мин; MS (ESI pos): m/z = 396.2 [M+H]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 0.862 (0.81), 0.879 (0.71), 1.024 (0.76), 1.055 (1.68), 1.084 (1.84), 1.113 (1.00), 1.274 (1.39), 1.308 (2.53), 1.339 (1.79), 1.404 (4.60), 1.430 (3.90), 1.498 (3.23), 1.531 (6.31), 1.566 (4.20), 1.609 (2.04), 1.640 (1.55), 2.179 (1.80), 2.328 (0.51), 2.670 (0.47), 3.820 (0.62), 3.839 (0.86), 3.858 (0.76), 3.904 (4.71), 3.920 (3.46), 3.925 (3.34), 3.941 (2.46), 4.195 (1.38), 4.210 (3.69), 4.223 (9.20), 4.238 (10.72), 4.244 (8.66), 4.254 (7.18), 4.272 (6.16), 7.309 (5.80), 7.328 (13.47), 7.346 (8.67), 7.400 (9.17), 7.419 (15.07), 7.437 (6.77), 7.487 (3.68), 7.508 (3.60), 7.546 (0.67), 7.637 (0.61), 7.748 (11.33), 7.767 (10.18), 7.883 (16.00), 7.901 (14.74).
Эту аминокислоту применяли в SPPS.
Пример 117А
3-({[(9Н-флуорен-9-илметокси)карбонил]амино}метил)циклогексанкарбоновая кислота (рацемат)
3-{[(трет-бутоксикарбонил)амино]метил}циклогексанкарбоновую кислоту (рацемат) (CAS RN: 145149-55-9, 1.00 г, 3.89 ммоль) растворяли в дихлорметане (20 мл, 310 ммоль), трифторуксусную кислоту (20 мл, 260 ммоль) добавляли, и реакционную смесь перемешивали в течение 30 мин при комнатной температуре. Раствор выпаривали и остаток растворяли в ацетоне (15 мл). Воду (10 мл), гидрокарбонат натрия (6.53 г, 77.7 ммоль) и 1-({[(9Н-флуорен-9-ил)метокси]карбонил}окси)пирролидин-2,5-дион (CAS-RN: 82911-69-11.38 г, 4.08 ммоль) добавляли. Суспензию перемешивали всю ночь при комнатной температуре. Гидрокарбонат натрия (6.53 г, 77.7 ммоль) добавляли и реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Суспензию разбавляли водой и экстрагировали с МТВЕ. Водный слой подкисляли водной соляной кислотой (1М), а затем экстрагировали три раза этилацетатом. Объединенные органические слои промывали солевым раствором, сушили над сульфатом магния, фильтровали, а затем концентрировали и выпаривали, с получением трех содержащих продукт фракций: 130 мг (96% чистота, 8% выход), 127 мг (96% чистота, 8% выход), 51.0 мг (3% выход).
LC-MS (Способ 9): Rt = 1.01 мин; MS (ESI pos): m/z = 380 [M+H]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 0.736 (0.49), 0.759 (1.44), 0.790 (1.73), 0.821 (0.80), 0.851 (0.96), 0.882 (2.75), 0.913 (2.99), 0.944 (1.23), 1.083 (0.45), 1.120 (0.52), 1.153 (1.38), 1.182 (3.81), 1.206 (3.18), 1.235 (1.28), 1.418 (1.48), 1.598 (1.97), 1.630 (2.23), 1.717 (2.24), 1.739 (1.48), 1.747 (1.53), 1.835 (1.93), 1.862 (3.95), 1.897 (2.04), 2.074 (2.00), 2.123 (1.19), 2.130 (1.10), 2.152 (2.02), 2.174 (0.85), 2.181 (0.95), 2.524 (1.06), 2.785 (0.60), 2.801 (1.06), 2.818 (2.22), 2.835 (3.08), 2.849 (2.89), 2.862 (3.29), 2.878 (2.21), 2.895 (1.16), 2.911 (0.57), 4.188 (1.46), 4.206 (3.84), 4.223 (3.22), 4.238 (0.88), 4.253 (0.91), 4.287 (11.07), 4.304 (6.87), 4.333 (0.64), 4.351 (0.40), 4.451 (0.68), 7.306 (6.09), 7.324 (16.00), 7.341 (10.19), 7.392 (6.85), 7.410 (10.96), 7.429 (4.86), 7.459 (0.43), 7.626 (0.73), 7.643 (0.75), 7.683 (9.67), 7.702 (8.66), 7.875 (11.34), 7.894 (10.10), 12.028 (1.95).
Эту аминокислоту применяли в SPPS.
Пример 118А
Бензил (2S)-2-(морфолин-4-ил)пропаноат
L-аланин (589 мг, 6.61 ммоль), 1-бромо-2-(2-бромоэтокси)этан (910 μL, 7.3 ммоль) и карбонат натрия растворяли в этаноле и раствор нагревали с возвратом флегмы в течение 2 дней. Суспензию оставляли дойти до комнатной температуры и суспензию фильтровали. Фильтрат выпаривали, суспендировали в этаноле (30 мл) и 3М соляную кислоту добавляли для установления рН (до рН=3). Суспензию фильтровали, и фильтрат выпаривали. Остаток растворяли в DMF (20 мл). Карбонат цезия (1.08 г, 3.30 ммоль) и (бромометил)бензол (1.6 мл, 13 ммоль) добавляли и смесь перемешивали в течение 2 дней при комнатной температуре. Затем воду добавляли, а затем раствор очищали посредством обращенофазовой ВЭЖХ (колонка: Reprosil; С18; 10 мкм; 125 × 30 мм; элюент ACN/H2O с 0.1% НСООН модификатора; поток: 75 мл/мин; градиент: 0-5.50 мин 10/ 90; Впрыск образца до 3.00 мин; 5.50-17.65 мин градиент до 95/5; 17.65-19.48 мин 95/5; 19.48-19.66 мин градиент до 10/90; 19.66-20.72 мин 10/90;). Желаемые фракции объединяли и лиофилизировали с получением 800 мг (98% чистота, 48% выход) указанного в названии соединения в виде бесцветного масла; ее >99%.
LC-MS (Способ 9): Rt = 0.51 мин; MS (ESI pos): m/z = 250 [M+H]+
1Н-ЯМР (500 МГц, DMSO-d6) δ [ppm]: 1.191 (10.46), 1.205 (10.52), 2.462 (0.66), 2.469 (0.77), 2.473 (0.76), 2.485 (1.55), 2.521 (1.43), 2.562 (0.63), 3.330 (0.93), 3.344 (2.66), 3.358 (2.62), 3.372 (0.86), 3.499 (0.46), 3.506 (0.63), 3.511 (0.45), 3.521 (2.34), 3.530 (3.52), 3.532 (3.50), 3.538 (3.54), 3.549 (2.18), 3.564 (0.56), 5.101 (0.60), 5.126 (5.99), 5.131 (5.75), 5.156 (0.57), 7.323 (0.69), 7.327 (0.52), 7.332 (1.27), 7.341 (1.23), 7.343 (0.94), 7.350 (1.14), 7.358 (0.50), 7.369 (1.15), 7.377 (16.00), 7.386 (6.48), 7.392 (0.46).
Пример 119А
(2S)-2-(морфолин-4-ил)пропановая кислота
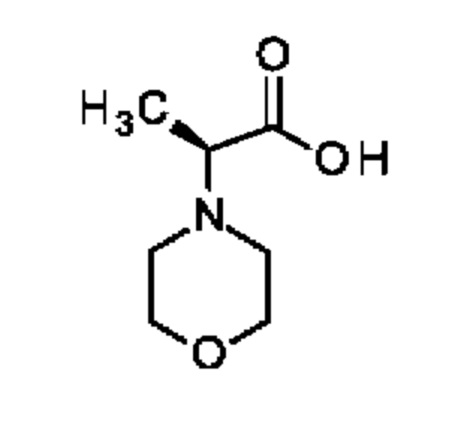
Палладий (10% на активированном угле) (100 мг) смачивали метанолом в атмосфере аргона. Затем бензил (2S)-2-(морфолин-4-ил)пропаноат (пример 118А, 800 мг, 3.21 ммоль) растворяли в метаноле (100 мл) и смоченный палладий на активированном угле добавляли в раствор в атмосфере аргона. Суспензию затем гидрогенировали в течение 2 часов при давлении окружающей среды в атмосфере водорода. Сосуд продували водородом с аргоном и суспензию фильтровали через целит. Фильтрат выпаривали, и остаток растворяли в MeCN/H2O и лиофилизировали с получением 501 мг (>99% чистота, 98% выход) указанного в названии соединения в виде твердого вещества светло-коричневого цвета.
LC-MS (Способ 9): Rt = 0.16 мин; MS (ESI pos): m/z = 160 [M+H]+
1H-ЯМР (500 МГц, оксид дейтерия) δ [ррт]: 1.519 (16.00), 1.534 (15.96), 3.695 (1.39), 3.709 (4.02), 3.724 (3.89), 3.738 (1.30).
Пример 120А
1-(трет-бутоксикарбонил)-4-этилиден-L-пролин
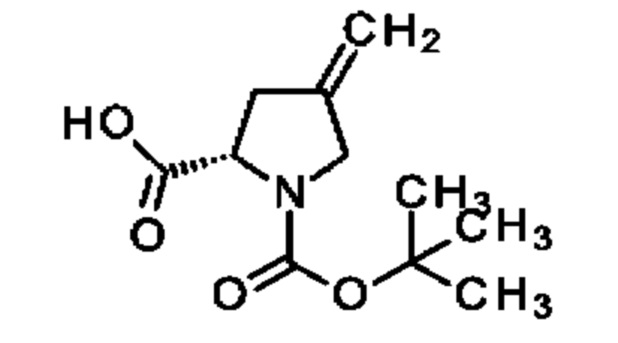
Метил(трифенил)фосфония бромид (34.7 г, 97.1 ммоль) суспендировали в 400 мл THF. Раствор KHMDS в толуоле (146.6 мл, 15% мас/об, 97.1 ммоль) добавляли по каплям при комнатной температуре и перемешивали в течение одного часа при комнатной температуре. Затем раствор охлаждали до 0°С, и 1-(трет-бутоксикарбонил)-4-оксо-L-пролин (11.1 г, 48.6 ммоль) добавляли по частям. Смесь перемешивали 2.5 часов при комнатной температуре. Воду добавляли в реакционную смесь. Смесь слегка подкисляли 10% раствором лимонной кислоты и экстрагировали три раза этилацетатом. Объединенный слой этилацетата промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, а затем сушили под вакуумом с густой смолы оранжевого цвета. Неочищенный продукт применяли в ходе следующей стадии без дальнейшей очистки.
Пример 121А
1-трет-бутил 2-метил (2S)-4-метилиденпирролидин-1,2-дикарбоксилат
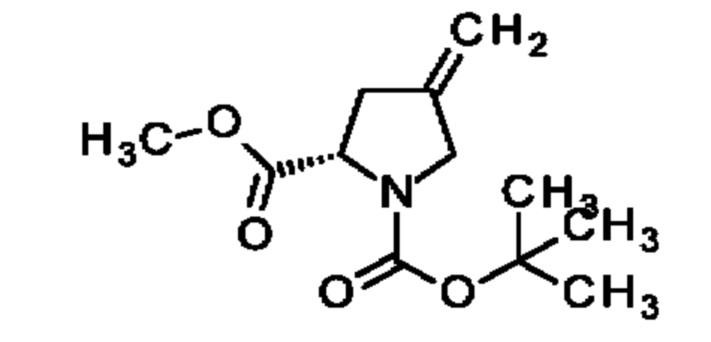
Неочищенный 1-(трет-бутоксикарбонил)-4-этилиден-L-пролин (пример 120А) (11.0 г, 48.6 ммоль) растворяли в DMF (60 мл). Карбонат калия (10.1 г, 72.9 ммоль) добавляли, затем иодметан (4.5 мл, 73 ммоль) добавляли, а затем смесь перемешивали всю ночь при комнатной температуре. DMF удаляли на роторном испарителе, этилацетат добавляли, а затем смесь промывали один раз водой. Водную фазу один раз повторно экстрагировали этилацетатом. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, а затем сушили под вакуумом с получением неочищенного продукта. Неочищенный продукт растворяли в DCM и очищали посредством нормально-фазовой хроматографии с применением системы для флэш-хроматографии Biotage Isolara, с применением элюента циклогексан-этилацетат (85/15) с получением 10.2 г (80%) указанного в названии соединения в виде желтого масла.
LC-MS (Способ 10): Rt = 1.77 мин; MS (ESI pos): m/z = 186 M-C4H8+
1Н-ЯМР (500 МГц, DMSO-d6) δ [ppm]: 1.339 (16.00), 1.405 (10.53), 3.313 (10.02), 3.627 (3.90), 3.938 (2.21), 3.967 (0.60), 4.331 (0.51), 4.337 (0.54), 4.350 (0.57), 4.356 (0.59), 4.363 (0.47), 4.367 (0.42), 4.992 (1.33), 5.019 (1.17).
Пример 122А
5-трет-бутил 6-метил (6S)-5-азаспиро[2.4]гептан-5,6-дикарбоксилат
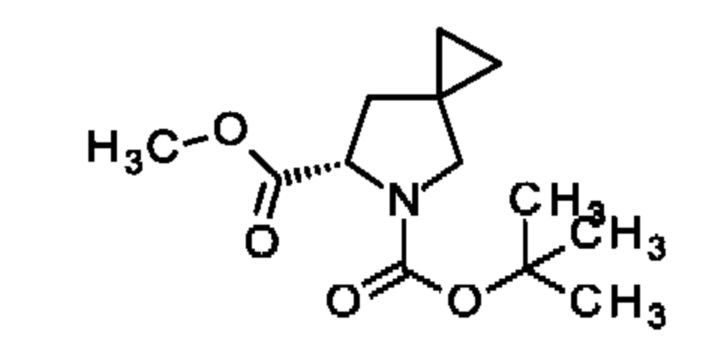
В атмосфере аргона, диэтилцинк (22 мл, 1.0 М в гексане, 22 ммоль) растворяли в безводном DCM (27 мл). Раствор охлаждали до 5°С, а затем раствор TFA (1.7 мл, 22 ммоль) в DCM (17 мл) добавляли по каплям за 1 час. Раствор перемешивали при 5°С в течение 30 мин. Раствор дииодметана (1.8 мл, 22 ммоль) в безводном DCM (5 мл) добавляли по каплям быстро, и полученный раствор перемешивали при 5°С в течение 30 мин. После этого раствор 1-трет-бутил 2-метил (2S)-4-метилиденпирролидин-1,2-дикарбоксилата (пример 121А) (2.70 г, 11.2 ммоль) в безводном DCM (5 мл) добавляли в реакционную смесь. Реакционную смесь перемешивали на тающей ледяной бане в течение 90 часов, затем гасили насыщенным водным раствором хлорида аммония. Фазы разделяли, и водную фазу повторно экстрагировали с DCM один раз. Органические фазы объединяли, сушили над сульфатом магния, а затем выпаривали с получением 1.57 г неочищенного продукта в виде желтого масла. Масло растворяли в этилацетате (54 мл), ди-трет-бутилдикарбонат (2.6 мл, 11 ммоль) и триэтиламин (3.1 мл, 22 ммоль) последовательно добавляли, а затем реакционную смесь перемешивали всю ночь при комнатной температуре. Реакцию контролировали с применением TLC (петролейный простой эфир/этилацетат 10/1; окрашивание нингидрином). Неочищенный продукт очищали посредством нормально-фазовой хроматографии с применением системы для флэш-хроматографии Biotage Isolara (100 г картридж Biotage Ultra), с применением градиента элюента петролейный простой эфир-этилацетат (ЕА) (2% - 18% ЕА) с получением 1.02 г указанного в названии соединения в виде бесцветного масла.
Пример 123А
(6S)-5-(трет-бутоксикарбонил)-5-азаспиро[2.4]гептан-6-карбоновая кислота
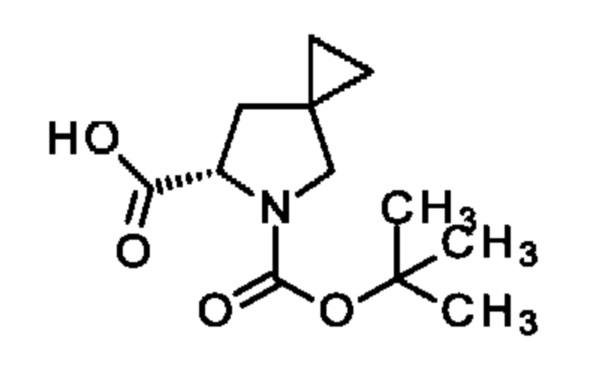
5-трет-бутил 6-метил (6S)-5-азаспиро[2.4]гептан-5,6-дикарбоксилат (пример 122А) (2.50 г, 9.79 ммоль) растворяли в 50% THF/вода (40 мл). Гидроксида лития (703 мг, 29.4 ммоль) добавляли, и реакционную смесь перемешивали всю ночь при комнатной температуре. THF удаляли с применением роторного испарителя, и водный раствор разбавляли дополнительной водой (20 мл) и промывали один раз МТВЕ. Отделенную водную фазу подкисляли 1М соляной кислотой (30 мл), а затем экстрагировали два раза с DCM. Объединенную органическую фазу сушили над сульфатом магния, фильтровали, а затем концентрировали с получением 2.4 г указанного в названии соединения в виде бесцветного масла.
Пример 124А
(6S)-5-азаспиро[2.4]гептан-6-карбоновая кислота
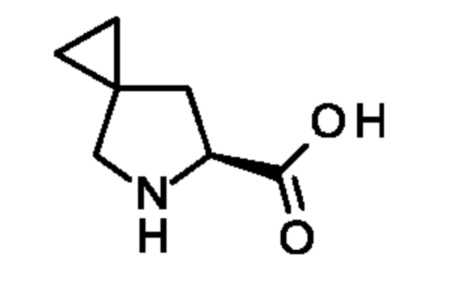
(6S)-5-(трет-бутоксикарбонил)-5-азаспиро[2.4]гептан-6-карбоновую кислоту (пример 123А) (2.36 г, 9.79 ммоль) растворяли в DCM (50 мл). Трифторуксусную кислоту (10 мл, 250 ммоль) добавляли, и реакционную смесь перемешивали 2 часов при комнатной температуре. Реакцию контролировали посредством TLC (DCM/MeOH 9/1). Раствор выпаривали и неочищенный продукт сушили под вакуумом с получением указанного в названии соединения в виде остатка коричневого цвета. Его применяли непосредственно в ходе следующей стадии (Fmoc защита, см. Пример 82А).
Пример 15А
(2S,5R)-2-[(2,5-дифторфенил)метил]-3,6-диметокси-5-(пропан-2-ил)-2,5-дигидропиразин
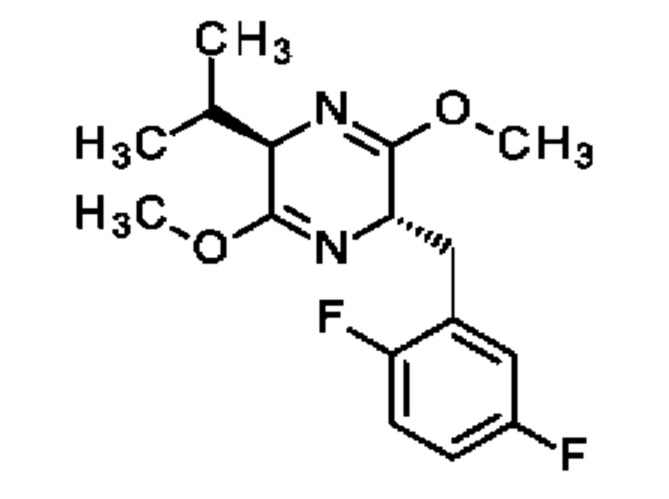
Реакцию проводили в атмосфере аргона. (2R)-3,6-диметокси-2-(пропан-2-ил)-2,5-дигидропиразин (CAS-RN: 109838-85-9) (2.9 мл, 16 ммоль) растворяли в безводном THF (40 мл) и раствор охлаждали до -78°С. При этой температуре раствор н-бутиллития (10 мл, 1.6 М в гексане, 16 ммоль) медленно добавляли по каплям, и полученный раствор перемешивали 15 мин при -78°С. Затем раствор 2-(бромометил)-1,4-дифторбензола (4.05 г, 19.5 ммоль) в безводном THF (15 мл) добавляли и перемешивание продолжали, пока реакционная смесь не достигла комнатной температуры. Реакционную смесь перемешивали в течение еще одного часа при комнатной температуре, при контроле развития реакции посредством ВЭЖХ. Смесь гасили водой и экстрагировали два раза этилацетатом. Объединенные органические фазы промывали один раз насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, а затем концентрировали с получением 5.46 г неочищенного масла желтого цвета. Масло очищали посредством нормально-фазовой хроматографии с применением системы для флэш-хроматографии Biotage Isolara (100 г Biotage картридж Ultra), используя элюент циклогексан-этилацетат (ЕА) (95/5) с получением 4.16 г (82%) указанного в названии соединения в виде масла.
LC-MS (Способ 10): Rt = 2.44 мин; MS (ESI pos): m/z = 311 [M+H]+
1H-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 0.563 (6.79), 0.580 (6.97), 0.933 (6.65), 0.951 (6.88), 2.103 (0.58), 2.111 (0.61), 2.120 (0.78), 2.129 (0.79), 2.137 (0.59), 2.145 (0.57), 2.864 (0.79), 2.882 (0.83), 2.898 (1.08), 2.915 (1.09), 3.089 (1.06), 3.101 (1.10), 3.123 (0.82), 3.134 (0.81), 3.313 (7.20), 3.518 (1.28), 3.527 (2.46), 3.535 (1.37), 3.566 (16.00), 4.293 (0.58), 4.304 (1.07), 4.313 (1.08), 4.321 (1.01), 4.330 (0.58), 6.992 (0.47), 7.000 (0.66), 7.005 (0.63), 7.015 (1.06), 7.023 (0.71), 7.028 (0.64), 7.037 (0.63), 7.075 (0.63), 7.084 (0.91), 7.095 (0.74), 7.105 (0.66), 7.131 (0.68), 7.143 (0.73), 7.154 (1.05), 7.166 (1.04), 7.177 (0.45), 7.189 (0.40).
Пример 126А
Метил 2,5-дифтор-L-фенилаланинат
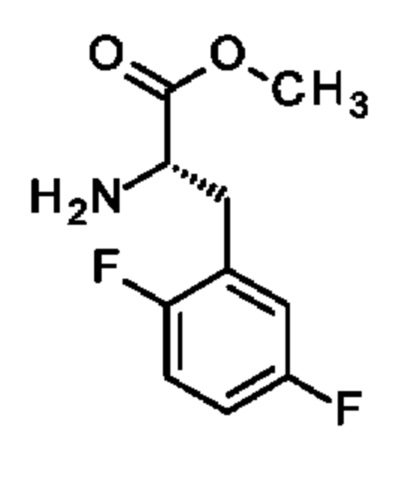
(2S,5R)-2-[(2,5-дифторфенил)метил]-3,6-диметокси-5-(пропан-2-ил)-2,5-дигидропиразин (пример 125А) (4.15 г, 13.4 ммоль) растворяли в метаноле (35 мл) и раствор охлаждали до 0°С. Раствор водной 10% соляной кислоты (12 мл) добавляли и раствор перемешивали 0°С в течение нескольких минут. Затем реакционный раствор оставляли дойти до комнатной температуры, а затем перемешивали три часа при этой температуре. Метанол выпаривали. Остаток разбавляли с DCM, 2М водный раствор карбоната натрия (120 мл) добавляли и поученную смесь перемешивали интенсивно в течение нескольких минут. Слои разделяли, и водную фазу экстрагировали два раза с DCM. Объединенные органические фазы сушили над сульфатом магния, фильтровали, а затем концентрировали с получением 3.97 г бесцветной жидкости. Неочищенную жидкость очищали посредством нормально-фазовой хроматографии с применением системы для флэш-хроматографии Biotage Isolara (100 г Biotage картридж Ultra), с применением градиента элюента циклогексан-этилацетат (ЕА) (10% - 75% ЕА) с получением 2.27 г (79%) указанного в названии соединения в виде бесцветного масла.
LC-MS (Способ 13): Rt = 1.20 мин; MS (ESI pos): m/z = 216 [M+H]+
1Н-ЯМР (400 МГц, DMSO-d6) δ [ppm]: 1.833 (16.00), 2.733 (3.07), 2.753 (3.26), 2.767 (4.81), 2.787 (5.01), 2.889 (4.71), 2.904 (4.91), 2.923 (3.10), 2.938 (3.19), 3.315 (15.20), 3.401 (0.42), 3.553 (5.22), 3.568 (6.29), 3.572 (6.60), 3.768 (0.41), 7.060 (0.89), 7.069 (1.71), 7.081 (2.21), 7.090 (4.07), 7.101 (3.20), 7.111 (2.84), 7.120 (1.89), 7.151 (5.97), 7.163 (5.82), 7.174 (9.84), 7.186 (7.52), 7.196 (4.42), 7.208 (2.10).
Пример 127А
трет-бутил (2R)-2-амино-3-[[(2R)-2-амино-3-трет-бутокси-3-оксо-пропил]дисульфанил] пропаноат
В суспензию L-цистеина (CAS RN: 56-89-3) (10.00 г, 41.61 ммоль) в трет-бутил ацетата (63.22 г, 544.24 ммоль, 73 мл) добавляли HClO4 (18.26 г, 127.23 ммоль, 11 мл, 70% чистота) (70% в Н2О) по каплям при 0°С за 0.5 ч, затем раствор перемешивали в течение 17 ч при 20°С. Лед добавляли и раствор применяли в ходе следующей стадии без дальнейшей очистки (14.67 г теоретический выход).
Пример 128А
трет-бутил (2R)-3-[[(2R)-3-трет-бутокси-2-(трет-бутоксикарбониламино)-3-оксо-пропил]дисульфанил]-2-(трет-бутоксикарбониламино)пропаноат
К смеси трет-бутил (2R)-2-амино-3-[[(2R)-2-амино-3-трет-бутокси-3-оксо0-пропил]дисульфанил]пропаноата (14.67 г теоретический выход, из Примера 127А) в воде (200 мл) и ЕА (200 мл) добавляли Na2CO3 (30.00 г, 283.08 ммоль) и Boc2O (27.25 г, 124.85 ммоль) при 0°С, и смесь (рН 8-9) перемешивали в течение 17 ч при 20°С. Смесь разбавляли с ЕА (100 мл) и водой (200 мл). Органический слой разделяли, и водный слой экстрагировали с ЕА (200 мл). Объединенный органический слой промывали солевым раствором (200 мл), концентрировали с получением остатка, остаток очищали посредством хроматографии на силикагеле (РЕ/ЕА = 1/0-1/1) с получением двух партий указанного в названии соединения: 8.5 г неочищенного продукта в виде масла светло-коричневого цвета и 6.5 г в виде масла светло-коричневого цвета.
1Н-ЯМР (400 МГц, CDCl3) δ [ppm]: 1.51, 1.61, 1.62. 1.64, 3.11, 3.12, 3.14, 3.16, 3.19, 3.203, 3.23, 3.24, 4.44, 4.46, 4.48, 4.49, 5.35, 5.37.
Пример 129А
трет-бутил (2R)-2-(трет-бутоксикарбониламино)-3-сульфанил-пропаноат
К раствору трет-бутил (2R)-3-[[(2R)-3-трет-бутокси-2-(трет-бутоксикарбониламино)-3-оксо-пропил]дисульфанил]-2-(трет-бутоксикарбониламино)пропаноата (8.5 г, неочищенный, из Примера 128А) в THF (150 мл) и воде (15 мл) добавляли трибутилфосфан (3.77 г, 4.6 мл) при 0°С за 0.5 ч, затем раствор перемешивали в течение 2 ч при 20°С. Смесь обрабатывали другой партией, приготовленной из 6,5 г материала (из Примера 128А). Обе реакционные смеси выливали в воду (300 мл) и экстрагировали этилацетатом (150 мл × 2). Объединенную органическую фазу промывали солевым раствором (300 мл), сушили безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (РЕ/ЕА = 40/1-10/1) с получением 12.5 г указанного в названии соединения в виде бесцветного масла, которое применяли как чистое. 6.5 г нечистой партии, содержащей остаточный трибутилфосфин, также были получены, но отброшены.
1H-ЯМР (400 МГц, CDCl3) δ [ppm]: 1.45, 2.94, 2.95, 2.96, 2.97, 4.46, 4.47, 4.48, 4.49, 5.39, 5.41
Пример 130А
трет-бутил (2R)-2-(трет-бутоксикарбониламино)-3-(трифторметилсульфанил)пропаноат
В раствор 3,3-диметил-1-(трифторметил)-1,2-бензиодксола (18 г, 54.53 ммоль) в МеОН (150 мл) добавляли раствор трет-бутил (2R)-2-(трет-бутоксикарбониламино)-3-сульфанил-пропаноата (14 г, 50.47 ммоль, из Примера 129А) в МеОН (20 мл) при -78°С в атмосфере N2 за 30 мин, и реакцию перемешивали в течение 1 ч при этой температуре, при которой образовался бесцветный раствор. Реакционную смесь нагревали до 20°С, перемешивали в течение 1 ч, и концентрировали с получением остатка, который очищали посредством хроматографии на силикагеле (РЕ/ЕА = 1/0-40/1) два раза с получением 13.2 г указанного в названии соединения в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ [ppm]: 1.47, 3.26, 3.28, 3.30, 3.31, 3.45, 3.46, 3.48, 3.50, 4.47, 4.48, 5.34, 5.35.
Пример 131А
трет-бутил (2R)-2-амино-3-(трифторметилсульфанил)пропаноат гидрохлорид
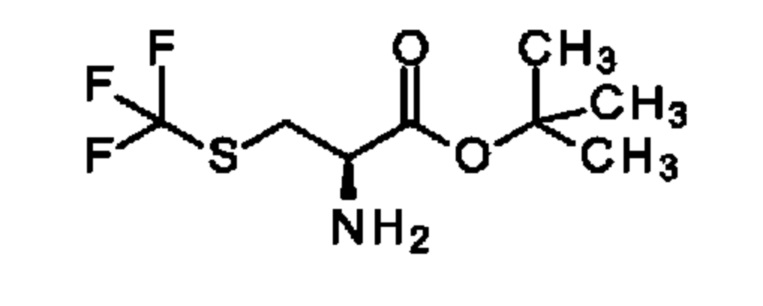
В раствор трет-бутил (2R)-2-(трет-бутоксикарбониламино)-3-(трифторметилсульфанил)-пропаноата (12.20 г, 35.32 ммоль, из Примера 130А) в DCM (10 мл) добавляли раствор HCl/диоксан (4 М, 50 мл) и реакцию перемешивали в течение 3 ч при 10°С. Смесь концентрировали с получением стеклообразного твердого вещества белого цвета, которое применяли непосредственно на следующей стадии.
Пример 132А
трет-бутил (2R)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-(трифторметилсульфанил)-пропаноат
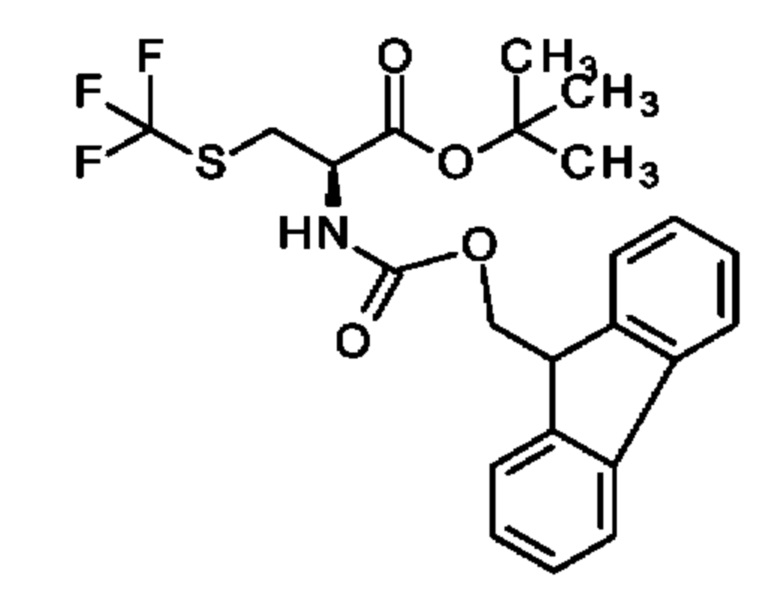
В смесь трет-бутил (2R)-2-амино-3-(трифторметилсульфанил)пропаноат гидрохлорида (неочищенный, из Примера 131А) в диоксане (80 мл) добавляли насыщенный NaHCO3 (80 мл), и Fmoc-OSu (19 г, 56.33 ммоль), и реакцию (рН ~7-8) перемешивали в течение 17 ч при 10°С. Смесь концентрировали, и остаток разбавляли водой (100 мл), затем экстрагировали с ЕА (100 мл × 2). Органический слой промывали солевым раствором (100 мл), концентрировали, а затем очищали посредством хроматографии на силикагеле (РЕ/ЕА = 40/1~10/1) с получением 13 г указанного в названии соединения в виде бесцветного масла.
LC-MS (Способ 15): Rt = 1.028 мин; MS (ESI pos): m/z = 490.2 [M+Na]+
1Н-ЯМР (400 МГц, CDC13) δ [ppm]: 1.45, 3.32, 3.47, 3.36, 3,49, 3.50, 4.16, 4.22, 4.24, 4.26, 4.37, 4.44, 4.46, 4.58, 5.62, 5.64, 7.33, 7.35, 7.40, 7.42, 7.44, 7.56, 7.61, 7.77, 7.79.
19F-ЯМР: (376 МГц, CDCl3) δ [ppm]: -40.85.
Пример 133A
2R)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-(трифторметилсульфанил)пропановая кислота

В раствор трет-бутил (2R)-2-(9H-флуорен-9-илметоксикарбониламино)-3-(трифторметилсульфанил)-пропаноата (13 г, 27.81 ммоль) в DCM (15 мл) добавляли TFA (30 мл) и тиоанизол (15.75 г, 126.81 ммоль, 15 мл), и реакционную смесь перемешивали в течение 17 ч при 10°С. Смесь концентрировали с получением остатка, который разбавляли насыщенным раствором NaHCO3 (100 мл), а затем экстрагировали с ЕА (50 мл × 2). Органический слой промывали с 1М HCl (50 мл), концентрировали, а затем очищали посредством хроматографии на силикагеле (РЕ/ЕА = 10/1, затем с DCM/MeOH = 40/1 - 5/1) с получением 10 г указанного в названии соединения в виде стеклообразного твердого вещества белого цвета, чистота: 98.8%. 1Н-ЯМР показала, что продукт, содержал этилацетат, поэтому твердое вещество повторно растворяли в DCM (80 мл), концентрировали, а затем сушили под высоким вакуумом с получением 9.67 г (23.22 ммоль, 83.5% выход, 98.8% чистота) указанного в названии соединения в виде стеклообразного твердого вещества белого цвета.
LC-MS (Способ 15): Rt = 0.895 мин; MS (ESI pos): m/z = 434.1 [M+Na]+
1Н-ЯМР (400 МГц, CDCl3) δ [ppm]: 3.25, 3.26, 3.32, 3.33, 3.93, 3.95, 4.22, 4.24, 4.26, 4.28, 4.30, 4.31, 6.95, 6.98, 7.31, 7.32, 7.33, 7.34, 7.39, 7,41, 7.42, 7.68, 7.70, 7.87, 7.89.
19F-ЯМР: (376 МГц, CDCl3) δ [ppm]: -40.89
Хиральная SFC: Rt = 2.49 мин, 99% ее
Условия хиральной SFC: AD-3_5СМ_IPA (DEA)_10_20_3ML_T35 Колонка: Chiralpak AD-3 50 × 4.6 мм I.D., 3 мкм, подвижная фазае: изопропанол (0.05% DEA) в CO2 от 10% до 20%, Скорость потока: 3 мл/мин, длина волны: 220 нм.
Эту аминокислоту применяли в SPPS. Эта аминокислота также является коммерчески доступной (CAS-RN: 1994331-25-7.
Пример 134А
Ди-трет-бутил (2S)-5-оксопирролидин-1,2-дикарбоксилат
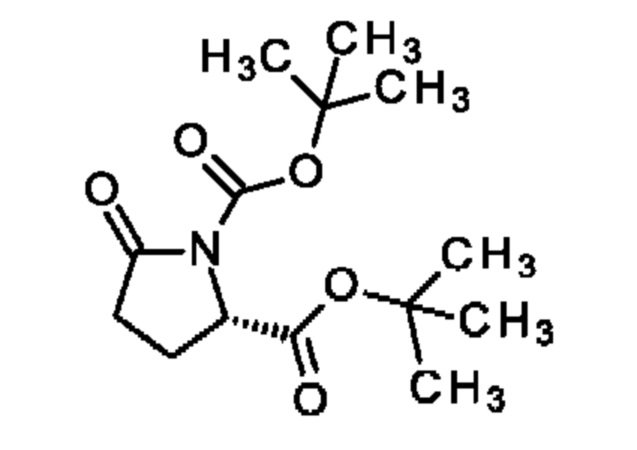
В раствор соединения трет-бутил (2S)-5-оксопирролидин-1,2-дикарбоксилата (CAS-RN: 35418-16-7) (290 г, 1.57 моль, 1.00 экв.) и DMAP (1.91 г, 15.66 ммоль, 0.01 экв.) в CH3CN (2.00 L), добавляли Вос2О (341 г, 1.57 моль, 359 мл, 1.00 экв.) при 5°С. После добавления смесь перемешивали при 20°С в течение 12 ч. TLC показало, что исходное вещество полностью израсходовано, и два новых пятна (TLC) образовались (РЕ/ЕА = 3/1, Rf = 0.43, Rf = 0.9). Реакционную смесь концентрировали при пониженном давлении для удаления CH3CN, и остаток распределили между DCM (1000 мл) и водой (1000 мл). Органический слой промывали солевым раствором (300 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Остаток очищали посредством колоночной хроматографии (SiO2, РЕ/ЕА = 100/1 - 10/1) с получением указанного в названии соединения (260.00 г, 874.75 ммоль, 55.72% выход, 96% чистота) в виде твердого вещества грязновато-белого цвета.
LC-MS (Способ 15): Rt = 0.873 мин; MS (ESI pos): m/z = 308.0 [M+Na]+, 331 [M+2Na]+
1H-ЯМР (400 МГц, CDCl3) δ [ppm]: 1.44-1.42 (d, J=8.4, 8H), 1.97-1.92 (m, 1H), 2.22-2.19 (m, 1H), 2.43-2.40 (m, 1H), 2.58-2.49 (m, 1H), 4.42-4.39 (m, 1H).
Пример 135A
Ди-трет-бутил (2S)-4,4-диаллил-5-оксо-пирролидин-1,2-дикарбоксилат
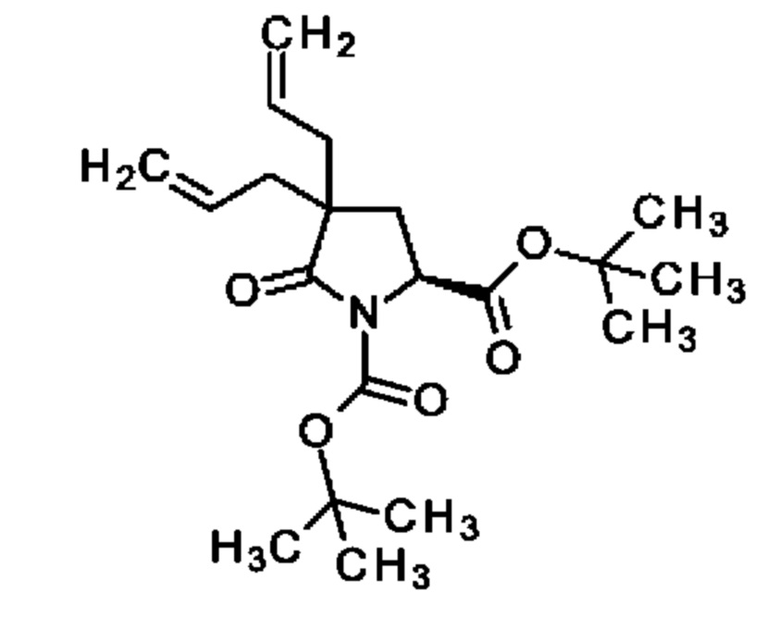
В раствор ди-трет-бутил (28)-5-оксопирролидин-1,2-дикарбоксилата (61.9 г, 208 ммоль, 1.00 экв., из Примера 134А) в THF (40.0 мл) добавляли LiHMDS (1 М, 437 мл, 2.10 экв.) по каплям при -73°С, а затем перемешивали при -73°С в течение 1 ч. В смесь добавляли аллилбромид (50.4 г, 416 ммоль, 2.00 экв.) по каплям. После добавления смесь перемешивали при 15°С в течение 2 ч. TLC показала, что исходное вещество было полостью израсходовано, и образовалось четыре новых пятна (РЕ/ЕА = 3/1, Rf = 0.9, Rf = 0.8, Rf = 0.7, Rf = 0.1). Реакционную смесь гасили насыщенным водным раствором хлорида аммония (1000 мл), экстрагировали с DCM (500 мл × 3), сушили над сульфатом натрия, фильтровали, а затем концентрировали при пониженном давлении с получением неочищенного остатка. Остаток очищали посредством колоночной хроматографии (SiO2, РЕ/ЕА=100/1 до 30/1) с получением указанного в названии соединения (100 г, 38% выход, 86% чистота) в виде бесцветного масла.
LC-MS (Способ 15): Rt = 1.044 мин; MS (ESI pos): m/z = 411.4 [M+2Na]+
1Н-ЯМР (400 МГц, CDCl3) δ [ppm]: 1.49-1.43 (d, J=8.4, 8H), 1.87-1.82 (m, 1H), 2.22-2.18 (m, 3H),
Пример 136А
Ди-трет-бутил (2S)-4,4-диаллилпирролидин-1,2-дикарбоксилат
В раствор ди-трет-бутил (2S)-4,4-диаллил-5-оксо-пирролидин-1,2-дикарбоксилата (80.0 г, 188 ммоль, 1.00 экв., из Примера 135А) в THF (300 мл) добавляли LiBHEt3 (1 М, 225 мл, 1.20 экв.) при -78°С. После добавления смесь перемешивали при -78°С в течение 3 ч. LCMS показала, что желаемая MS обнаружена. TLC показала два новых пятна (РЕ/ЕА=3/1, Rf=0.65, Rf=0.8). Реакционную смесь гасили насыщенным водным карбонатом натрия (50 мл) при 0°С, а затем 30% Н2О2 (1 мл) добавляли по каплям при 0°С. Смесь концентрировали при пониженном давлении с удалением THF, а затем остаток разбавляли водой (1000 мл) и DCM (1000 мл). Водную фазу экстрагировали с DCM (300 мл × 3). Объединенный органический слой сушили над сульфатом натрия, фильтровали, а затем концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный остаток очищали посредством колоночной хроматографии (SiO2, РЕ/ЕА = 80/1 до 30/1) с получением указанного в названии соединения (56.0 г, 69% выход, 85% чистота) в виде бесцветного масла.
LC-MS (Способ 15): Rt = 1.192 мин; MS (ESI pos): m/z = 390.3 [M+K]+
1Н-ЯМР (400 МГц, CDCl3) δ [ppm]: 1.53-1.47 (m, 18Н), 2.13-1.98 (m, 2Н), 2.22-2.15 (m, 3Н), 2.39-2.24 (m, 1H), 2.92-2.91 (m, 1H), 4.22-4.08 (m, 1H ), 5.18-5.06 (m, 5Н), 5.85-5.77 (m, 2Н).
Пример 137А
Ди-трет-бутил (3S)-2-азаспиро[4.4]поп-7-ене-2,3-дикарбоксилат
В раствор катализатора Граббса 1-го поколения (CAS RN: 172222-30-9) (2.85 г, 3.47 ммоль, 0.05 экв.) в DCM (400.00 мл) добавляли ди-трет-бутил (2S)-4,4-диаллилпирролидин-1,2-дикарбоксилат (25.0 г, 69.4 ммоль, 1.00 экв., из Пример а136А) по каплям при 34°С. После завершения добавления смесь перемешивали при 34°С в атмосфере N2 в течение 1 ч. TLC показала два новых пятна (РЕ/ЕА = 5/1, Rf = 0.6, Rf = 0.7). Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Остаток очищали посредством колоночной хроматографии (SiO2, РЕ/ЕА = 1/0 - 80/1) с получением указанного в названии соединения (12.0 г) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ [ppm]: 1.46-1.43 (m, 18Н), 1.92-1.87 (m, 1Н), 2.45-2.22 (m, 5Н), 3.49-3.29 (m, 2Н), 4.21-4.19 (m, 1Н), 5.64 (s, 2H).
Пример 138A
Ди-трет-бутил (3S)-2-азаспиро[4.4]попапе-2,3-дикарбоксилат

В раствор ди-трет-бутил (3S)-2-азаспиро[4.4]нон-7-ене-2,3-дикарбоксилата (12.0 г, 37.1 ммоль, 1.0 экв., из Примера 137А) в EtOH (100 мл) добавляли Pd/C (10%, 0.1 г) в атмосфере N2. Суспензию дегазировали и продували 3 раза газом Н2. Смесь перемешивали в атмосфере Н2 (15 Пси) при 25°С в течение 16 ч. Часть суспензии фильтровали, и фильтрат концентрировали, и применяли протонную ЯМР, которая показала что желаемый продукт образован. Реакционную смесь фильтровали, и фильтрат концентрировали с получением 10.0 г (неочищенного) указанного в названии соединения в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ [ppm]: 4.12-4.02 (m, 1H), 3.65-3.60 (m, 2Н), 3.12-3.38 (m, 5Н), 2.11-2.06 (m, 1H), 1.80-1.77 (m, 1H), 1.57-1.56 (m, 6Н), 1.40-1.36 (m, 18Н).
Пример 139А
(3S)-2-азаспиро[4.4]нонан-3-карбоновая кислота
Соединение ди-трет-бутил (3S)-2-азаспиро[4.4]нонан-2,3-дикарбоксилат (12 г, 36.8 ммоль, 1 экв., из Примера 138А) добавляли в раствор HCl/EtOAc (150 мл), а затем смесь перемешивали при 15°C в течение 1 ч. LC-MS анализ показал, что исходное вещество было израсходовано, и желаемая целевая масса обнаружена. Реакционную смесь концентрировали в вакууме для удаления растворителя, получая 7.5 г (36.4 ммоль, 99% выход) неочищенного указанного в названии соединения в виде твердого вещества белого цвета в виде соли гидрохлорида.
1H-ЯМР (400 МГц, DMSO) δ [ppm]: 10.07 (s, 1H), 8.81 (s, 1H), 4.38-4.27 (m, 1H), 3.16-3.02 (m, 2Н), 2.22-2.21 (m, 1Н), 1.96-1.90 (m, 1H), 1.63-1.56 (m, 8Н).
Пример 140А
(3S)-2-{[(9Н-флуорен-9-ил)метокси]карбонил}-2-азаспиро[4.4]нонан-3-карбоновая кислота
В раствор (3S)-2-азаспиро[4.4]попапе-3-карбоновой кислоты (5.1 г, 24.8 ммоль, из Примера 139А) и Fmoc-OSu (10.0 г, 29.7 ммоль, 1.2 экв.) в THF (70 мл) и H2O (70 мл) добавляли NaHCO3 (8.5 г, 101 ммоль, 3.94, 4.08 экв.). Смесь перемешивали при 15°C в течение 1 ч. LCMS анализ показал, что исходное вещество было израсходовано, и желаемая целевая масса обнаружена. Воду (200 мл) добавляли, а затем смесь экстрагировали с DCM (200 мл × 2). Объединенный органический слой промывали солевым раствором (100 мл), сушили над Na2SO4, фильтровали, а затем концентрировали. Неочищенный продукт очищали посредством обращенно-фазовой MPLC (ЕА) с получением 6.9 г (17.5 ммоль, 70% выход, 99% чистота) указанного в названии соединения в виде твердого вещества желтого цвета.
LC-MS (Способ 15): Rt=2.501 мин; MS (ESI pos): m/z=392.2 [M+H]+
1Н-ЯМР (400 МГц, CDCl3) δ [ppm]: 7.80-7.79 (d, J=7.2, 2H), 7.59-7.54 (m, 2H), 7.44-7.42 (m, 2H), 7.36-7.35 (m, 2H), 4.55-4.52 (m, 2H), 4.42-4.38 (m, 1H), 4.31-4.20 (m, 1H), 3.34-3.17 (m, 2H), 2.33-2.28 (m, 1H), 2.13-2.08 (m, 1H), 1.73-1.50 (m, 8H).
Эту аминокислоту применяли в SPPS. Эта аминокислота также является коммерчески доступной (CAS-RN: 394734-78-2).
Пример 141А
(2S,4R)-4-(трифторметил)пирролидин-2-карбоновая кислота
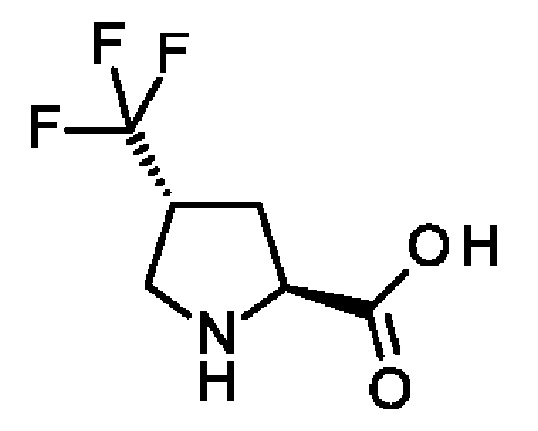
В раствор (2S,4R)-1-трет-бутоксикарбонил-4-(трифторметил)пирролидин-2-карбоновой кислоты (7 г, 24.7 ммоль, 1 экв.) в DCM (70 мл) добавляли TFA (8.00 мл, 108 ммоль, 4.37 экв.). Смесь перемешивали при 10°C в течение 4 ч. LCMS анализ показал, что исходное вещество было израсходовано, и желаемая целевая масса обнаружена. Реакционную смесь концентрировали под вакуумом и неочищенный продукт применяли непосредственно в ходе следующей стадии. Указанное в названии соединение (7.34 г, 24.7 ммоль, 100% выход, TFA соль) получали в виде твердого вещества белого цвета.
LC-MS (Способ 15): Rt=0.18 мин; MS (ESI pos): m/z=184.1 [M+H]+
1Н-ЯМР (400 МГц, CDCl3) δ [ppm]: 4.51-4.48 (m, 1H), 3.82 (m, 1H), 3.57-3.55 (m, 1H), 2.65-2.62 (m, 2H), 2.53-2.49 (m, 1H).
Пример 142A
(2S,4R)-1-(9Н-флуорен-9-илметоксикарбонил)-4-(трифторметил)пирролидин-2-карбоновая кислота
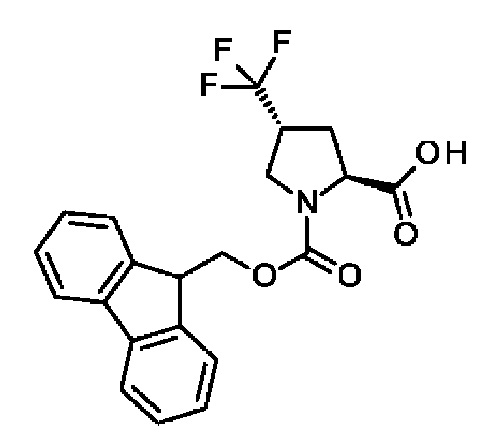
В раствор неочищенной (2S,4R)-4-(трифторметил)пирролидин-2-карбоновой кислоты (2 г, 6.73 ммоль, 1 экв., TFA соль, из Примера 141А) в Н2О (70 мл) и ацетоне (70 мл) добавляли Na2CO3 (2.85 г, 26.9 ммоль, 4 экв.) и Fmoc-OSu (2.72 г, 8.08 ммоль, 1.2 экв.). Смесь перемешивали при 10°C в течение 12 ч. LCMS анализ показал только один пик и обнаружение желаемой массы. TLC анализ (DCM/MeOH=10/1) показал, что исходное вещество было израсходовано, и что основное пятно (Rf=0.4) проявилось. Раствор HCl (1М, 30 мл) добавляли в смесь до рН=2. Смесь экстрагировали с DCM (70 мл × 2), и объединенный органический слой промывали солевым раствором (70 мл), сушили над Na2SO4, фильтровали, а затем концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, РЕ/ ЕА=10/1 - 5/1), с получением 1.7 г (61%, 97% чистота) указанного в названии соединения в виде твердого вещества желтого цвета.
LC-MS (Способ 15): Rt=2.425 мин; MS (ESI pos): m/z=406.2 [M+H]+, 406.2 [M+Na]+
1Н-ЯМР (400 МГц, DMSO) δ [ppm]: 7.92-7.89 (m, 2H), 7.66-7.64 (m, 2H), 7.42-7.41 (m, 2H), 7.35-7.32 (m, 2H), 4.16-4.19 (m, 4H), 3.72-3.50 (m, 3H), 2.38-2.30 (m, 2H), 2.18 (s, 1H).
19F-ЯМР (400 МГц, DMSO) δ [ppm]: -70.128, -70.275.
Хиральная SFC: Rt=2.33 мин, 99% ее
Условия хиральной SFC: AD-35CMIPA (DEA)_10_20_3ML_T35 Колонка: Chiralpak AD-3 50 × 4.6 мм ID., 3 мкм, Подвижная фаза: изопропанол (0.05% DEA) в СО2 от 10% до 20%, Скорость потока: 3 мл/мин, длина волны: 220 нм.
Эту аминокислоту применяли для SPPS.
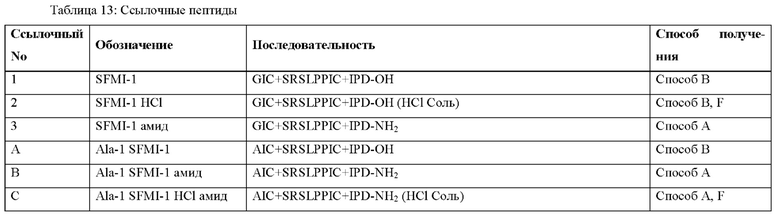





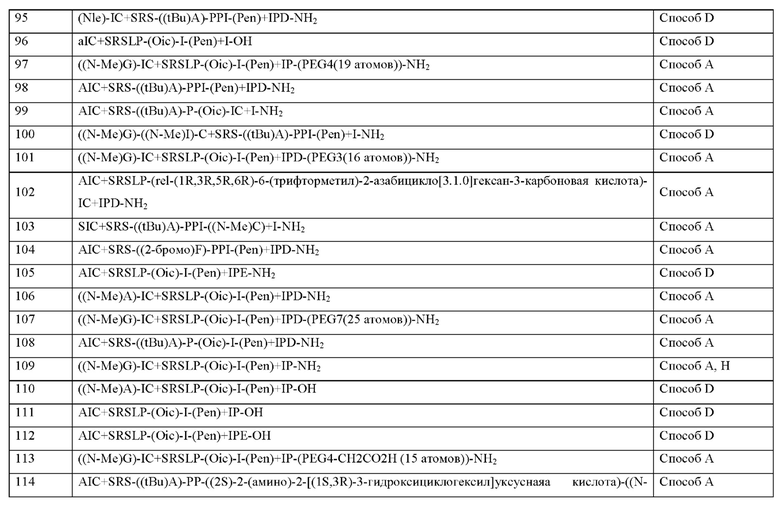
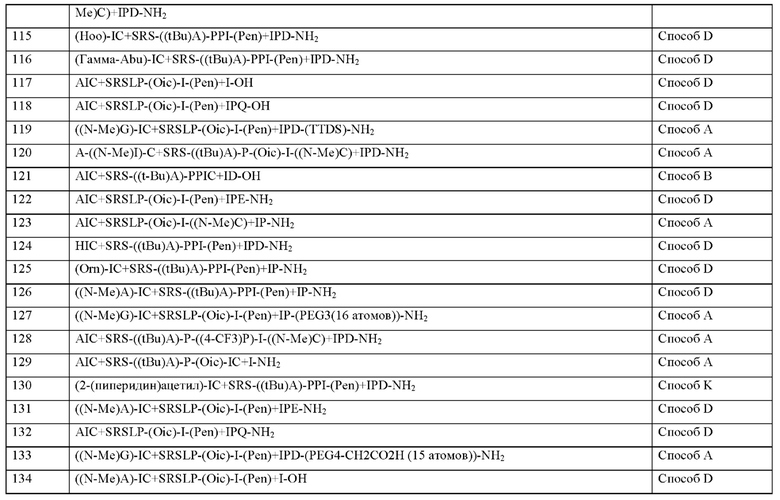
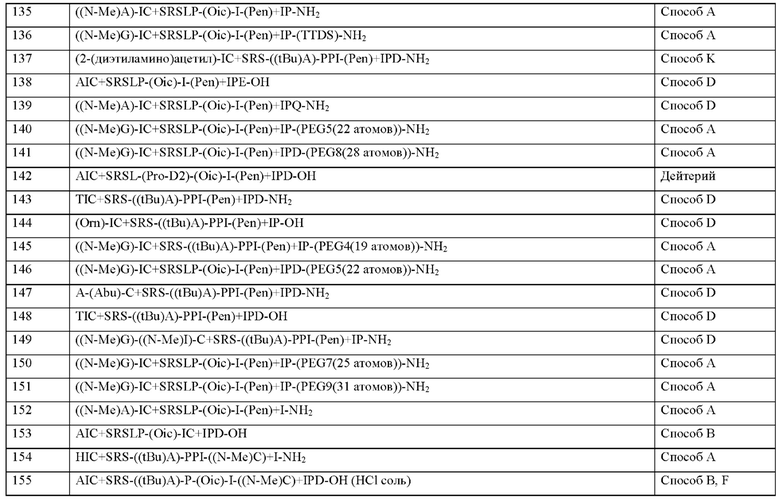
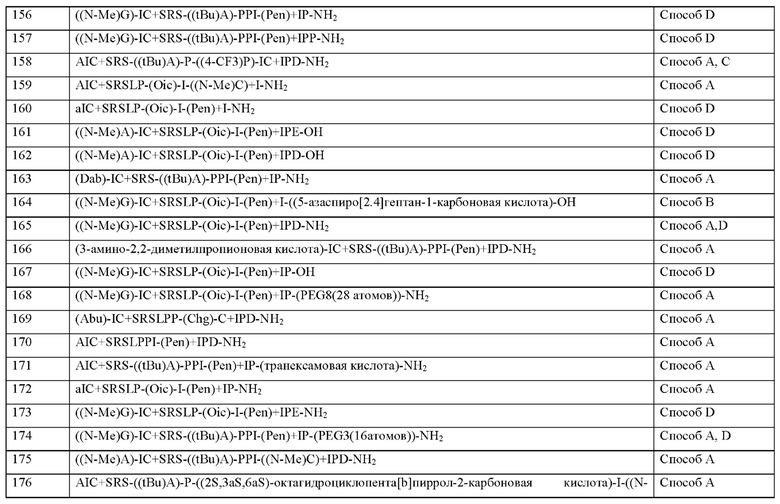
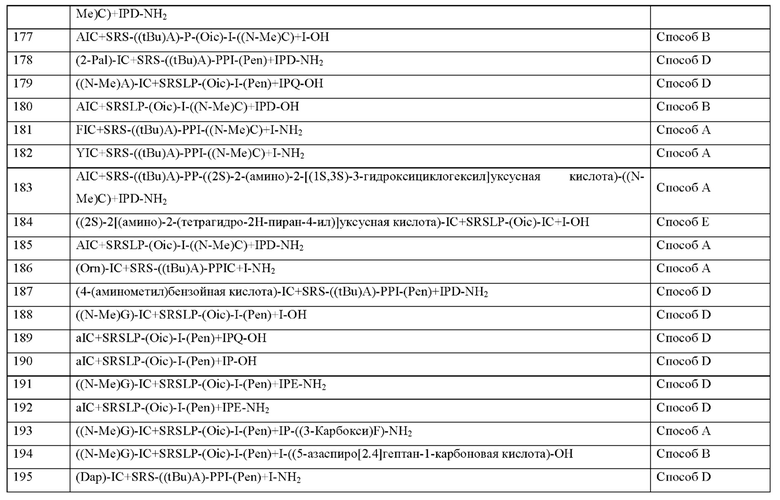
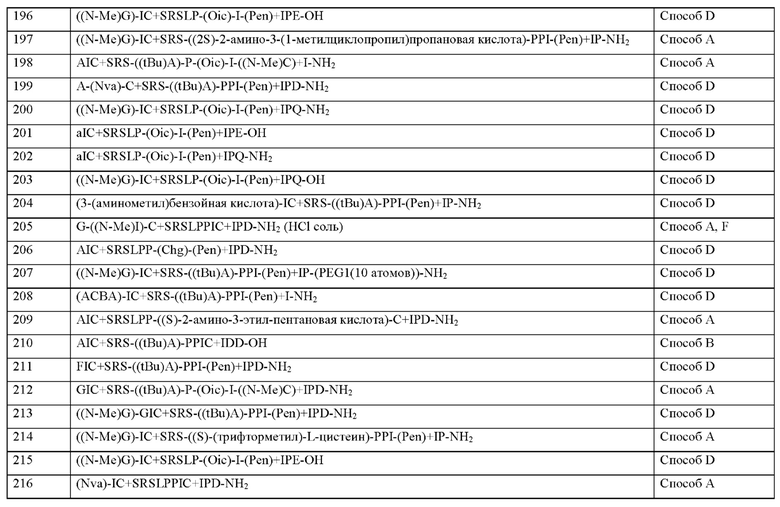
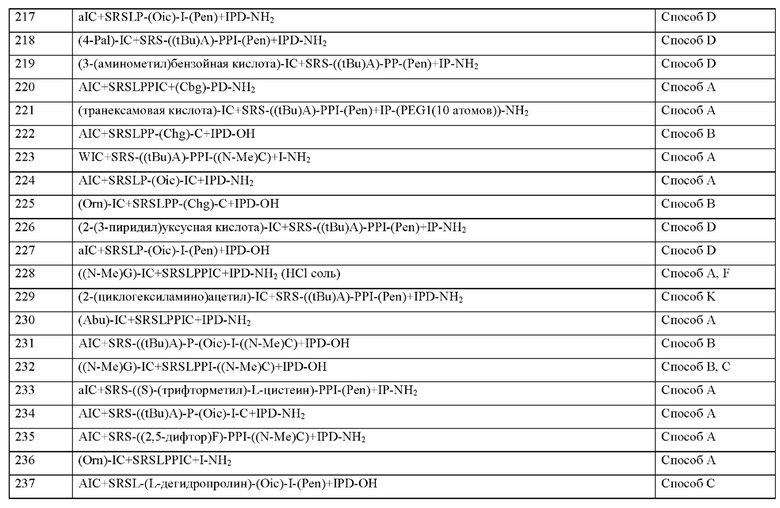
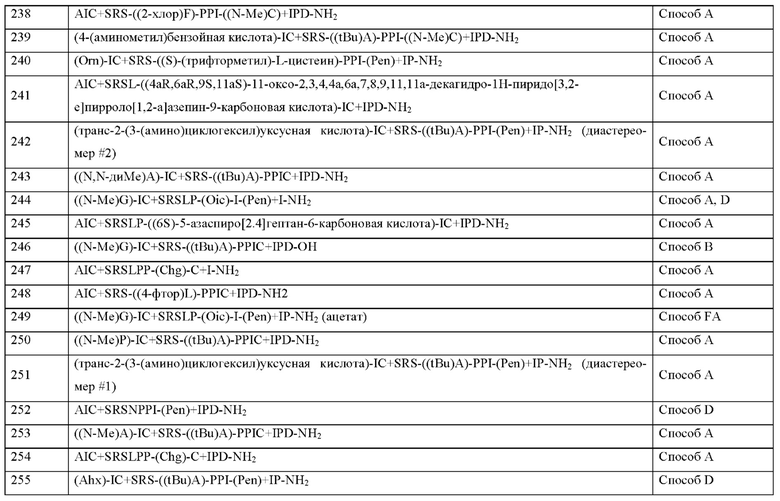
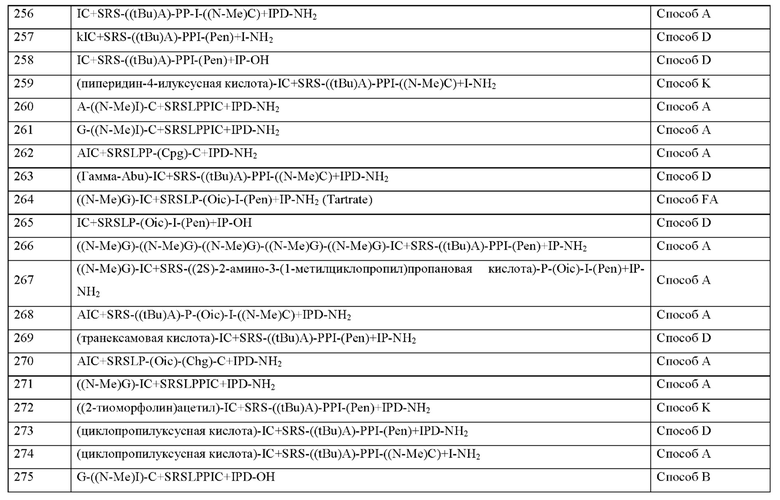
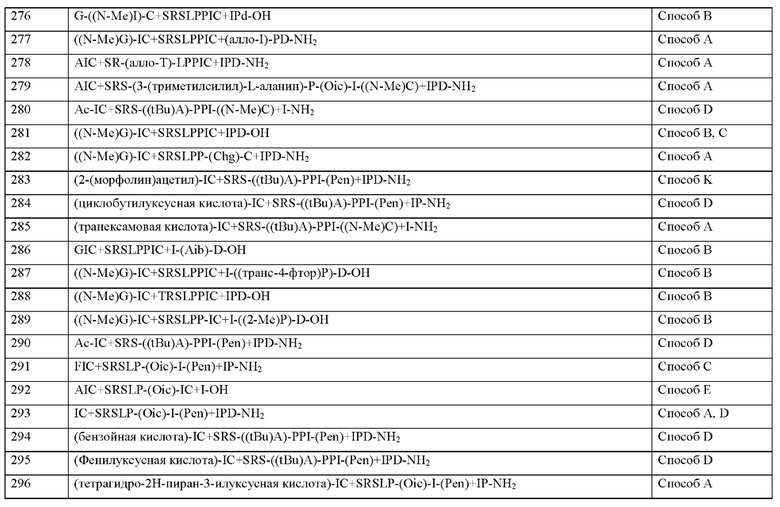
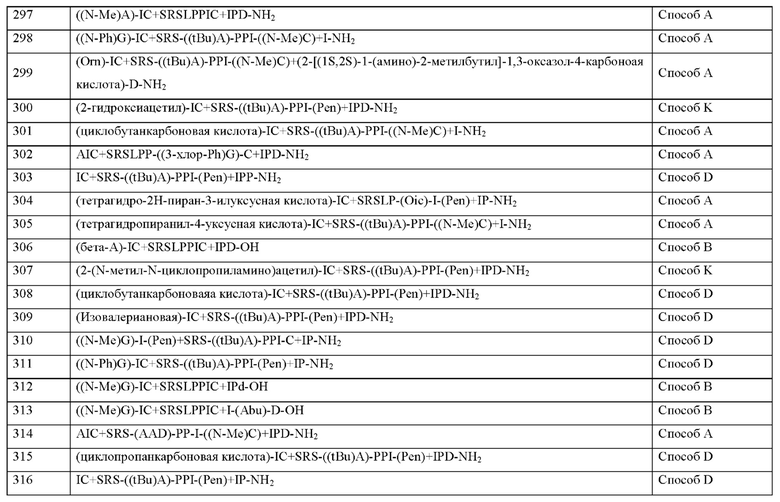
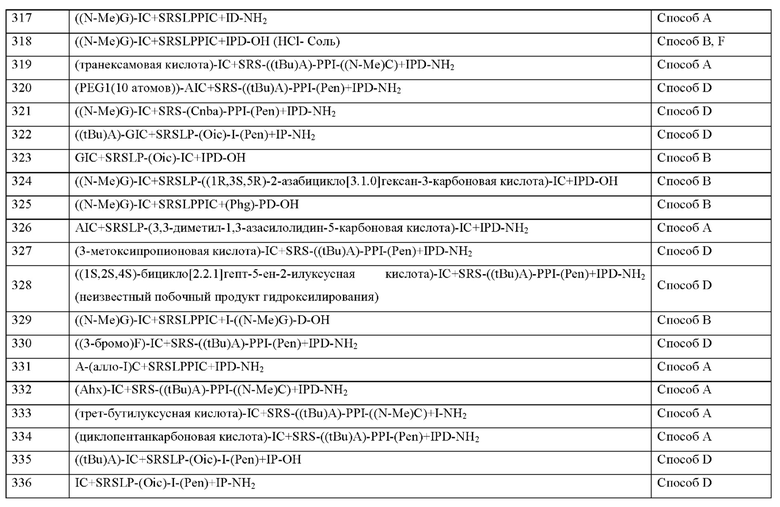
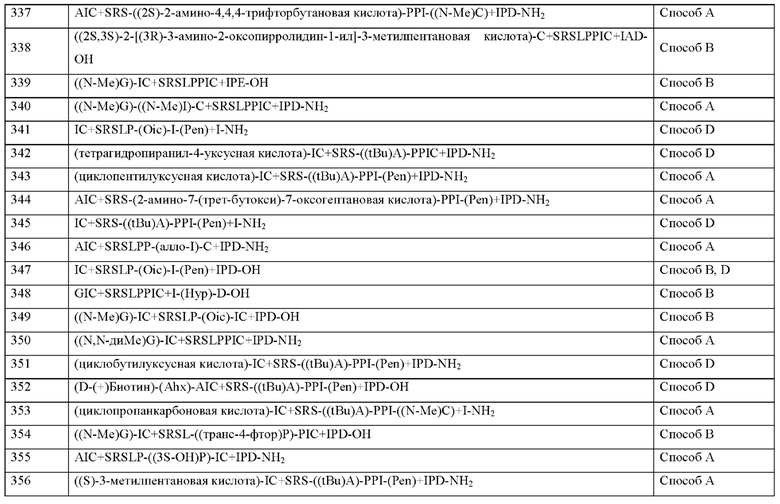
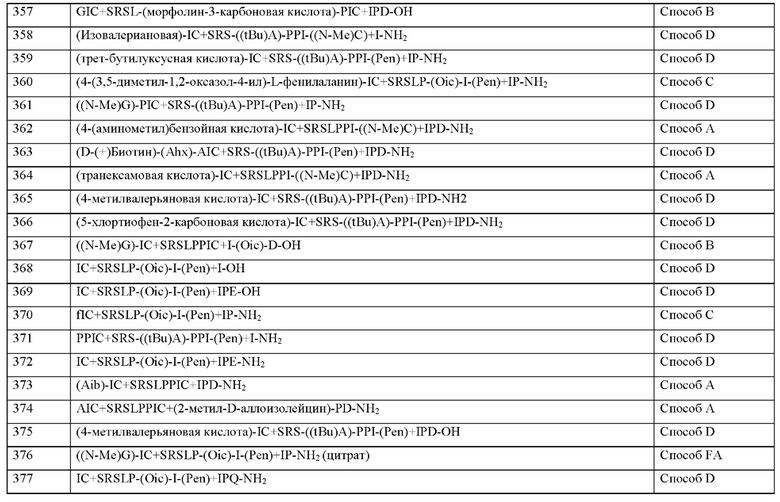
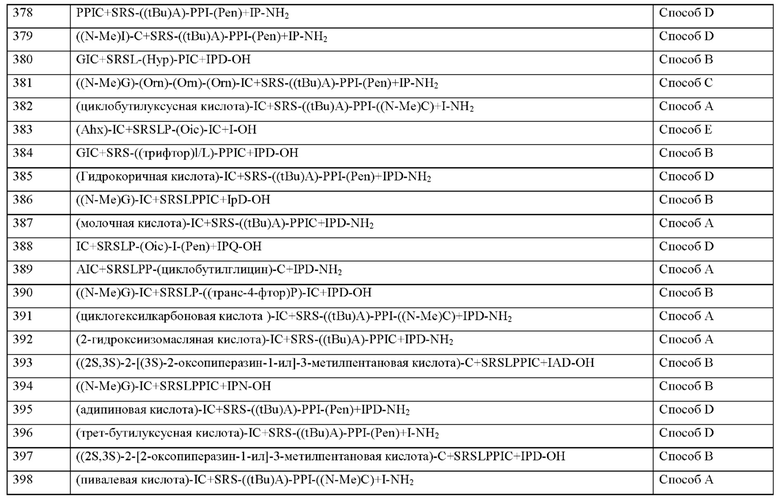
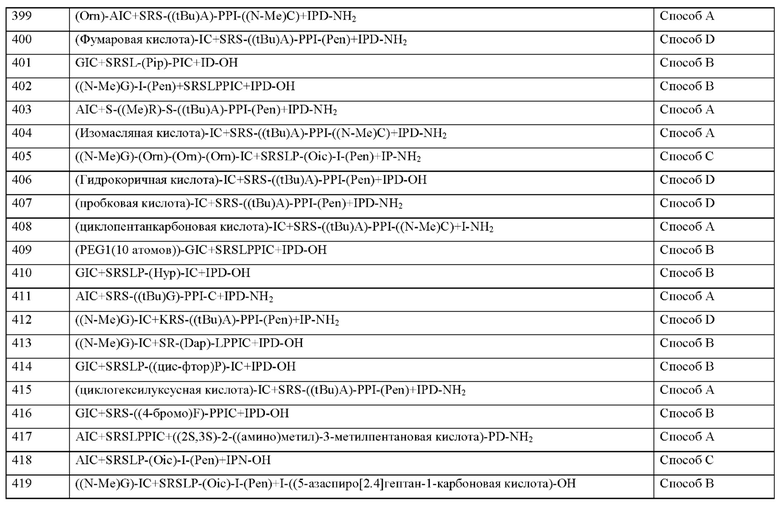


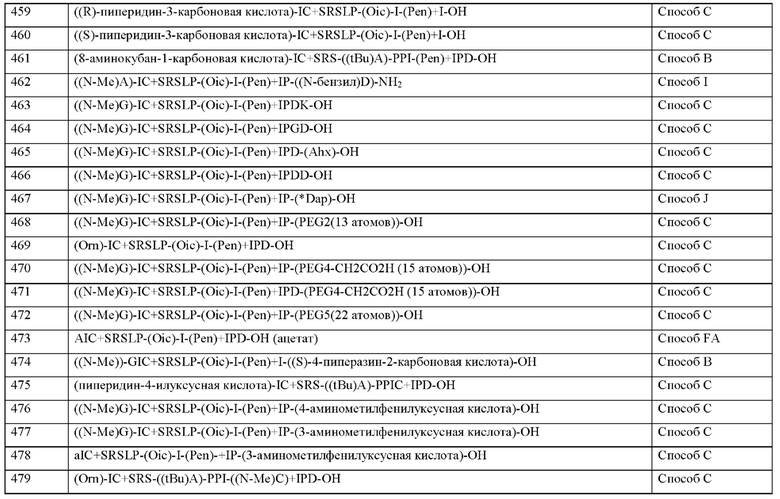
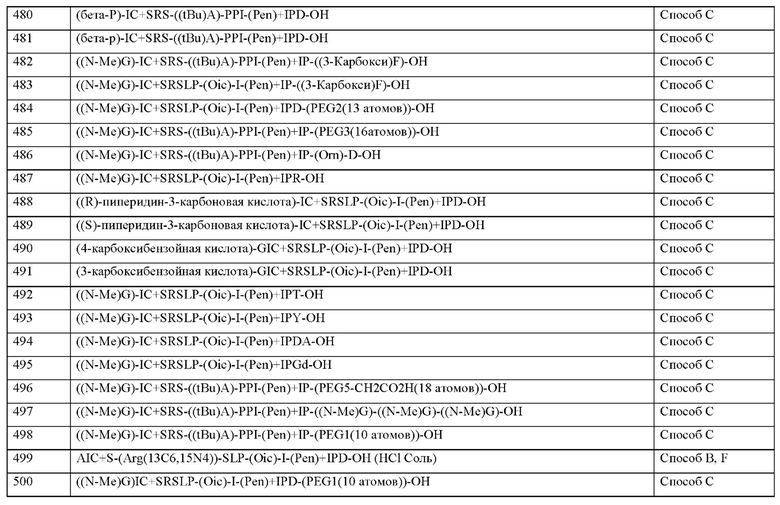
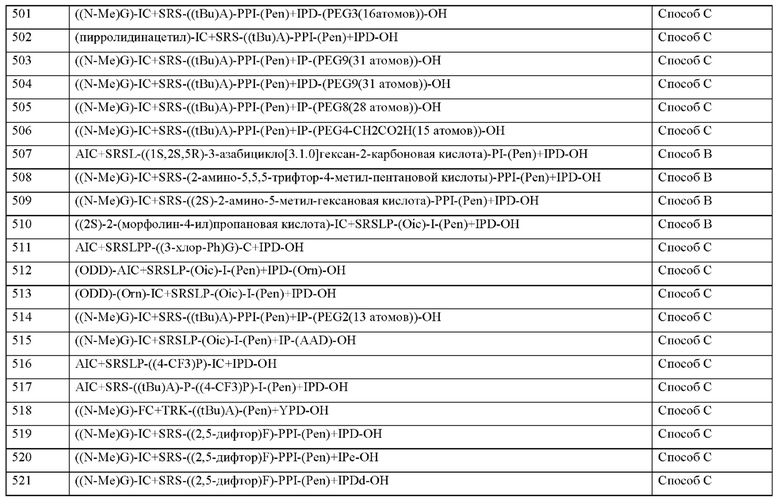

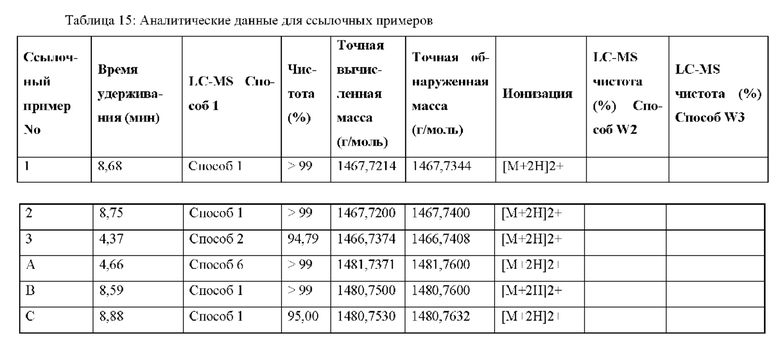
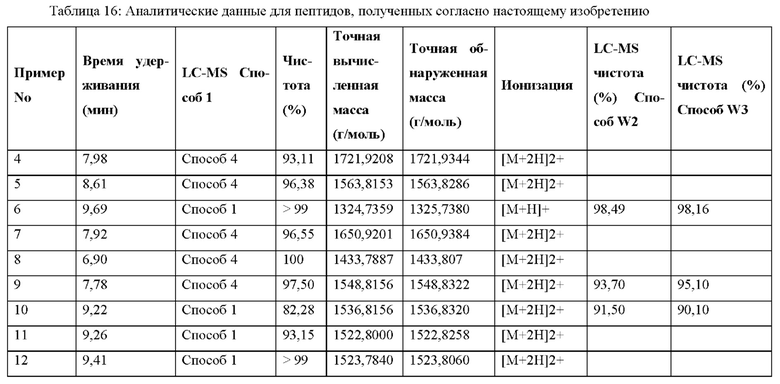
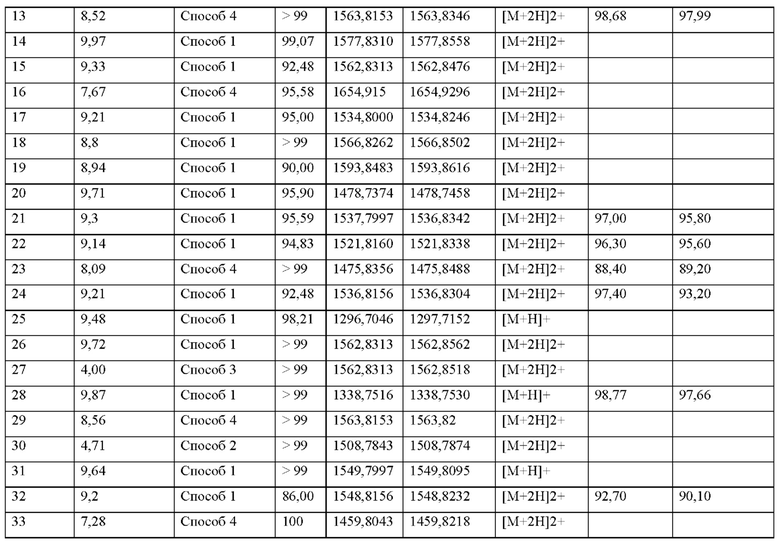
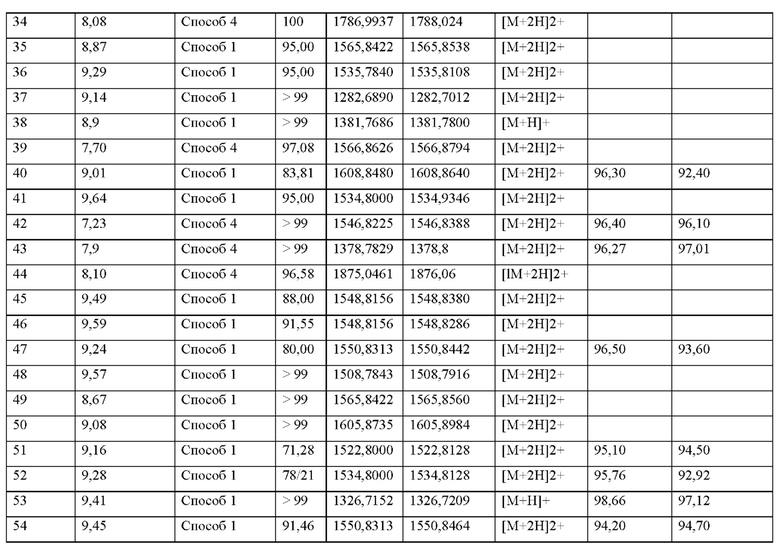
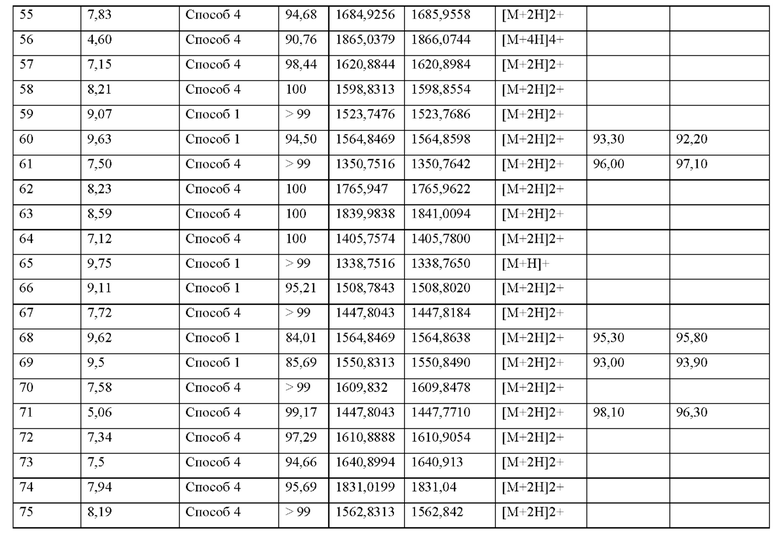
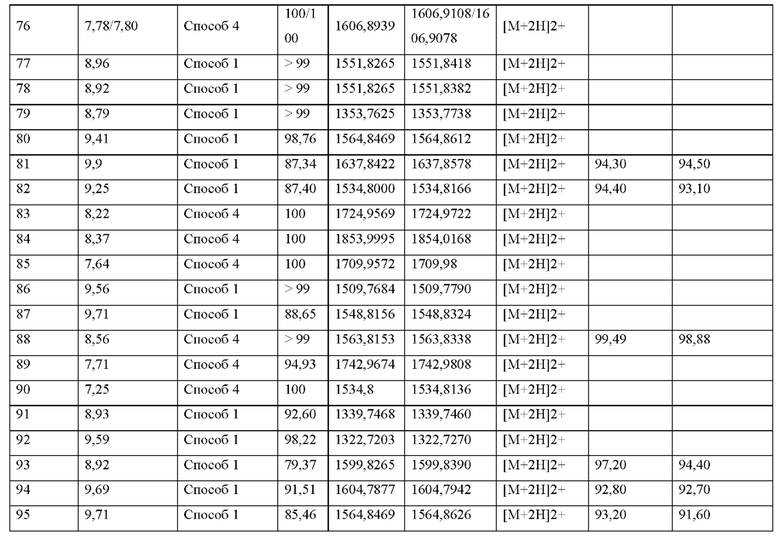
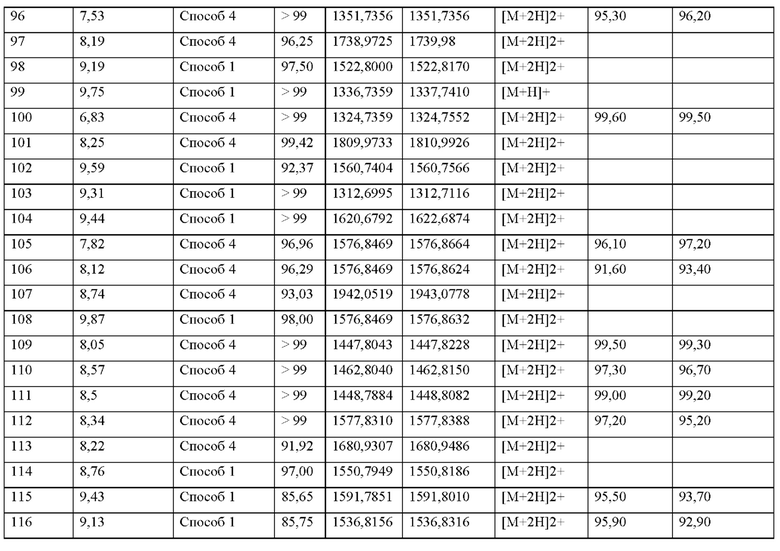
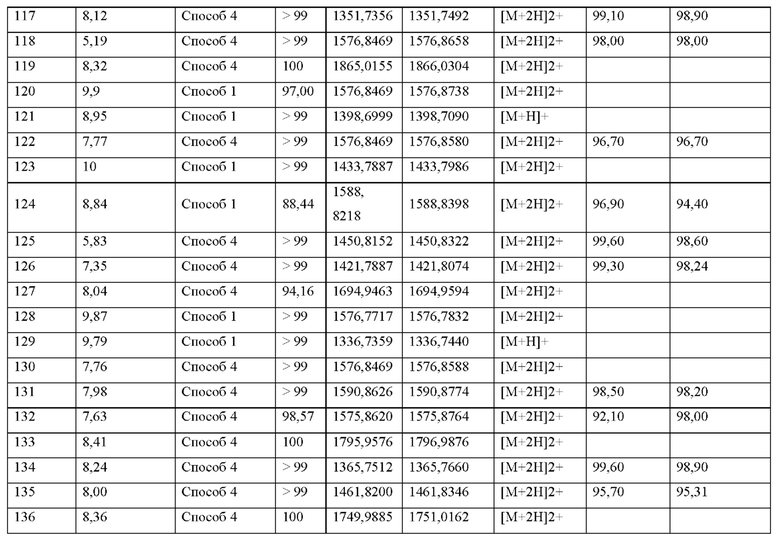
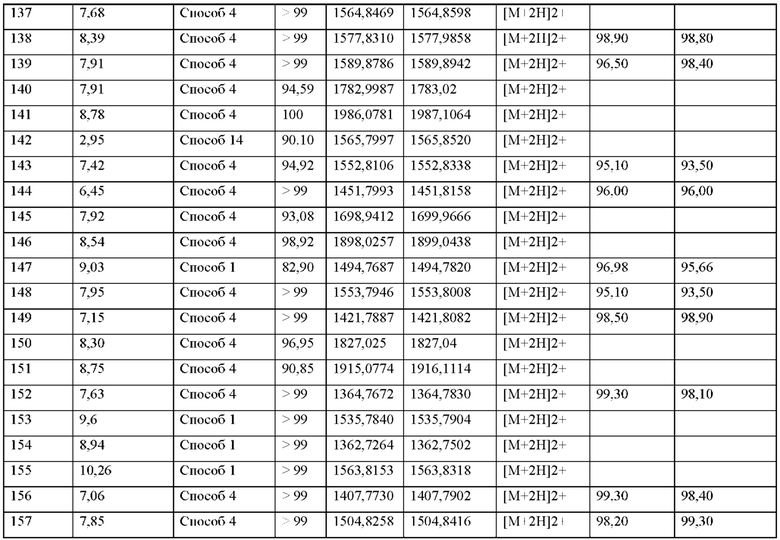
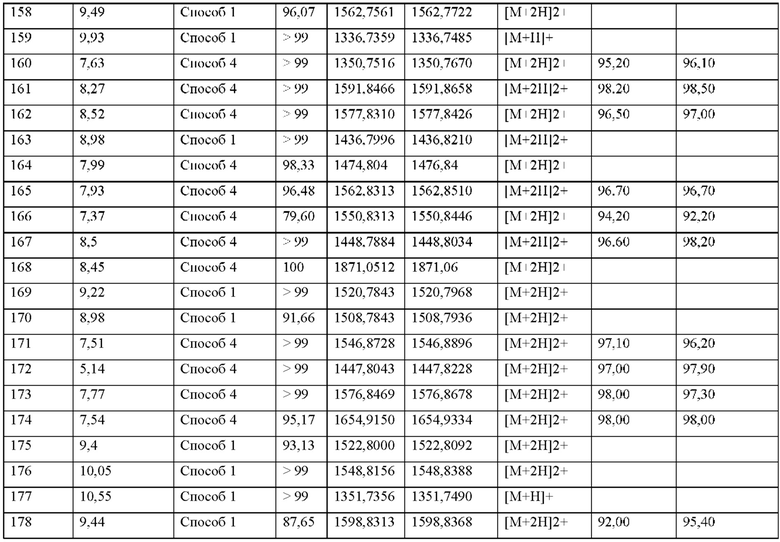
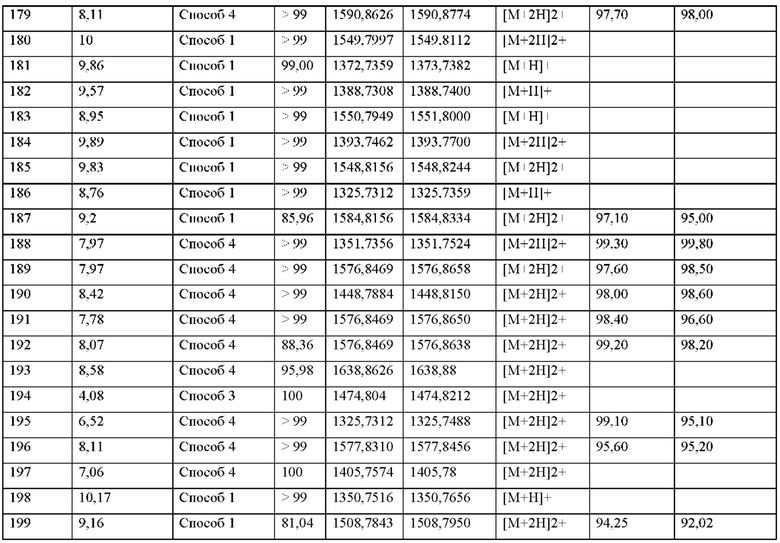


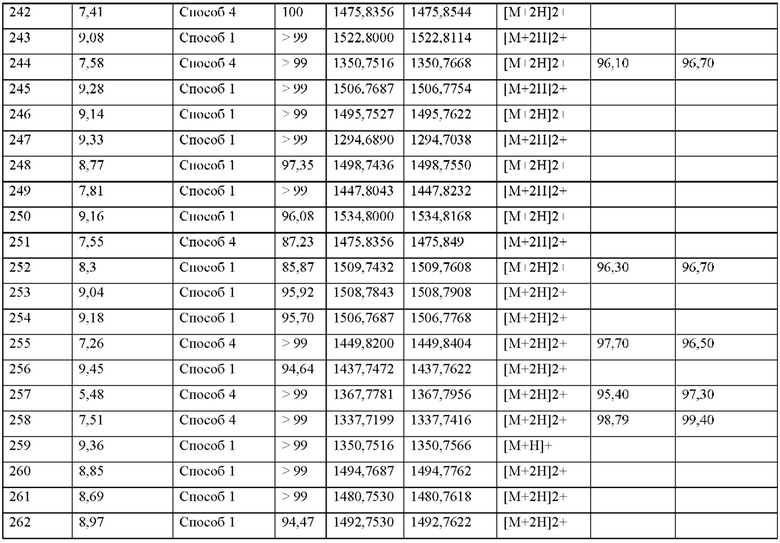

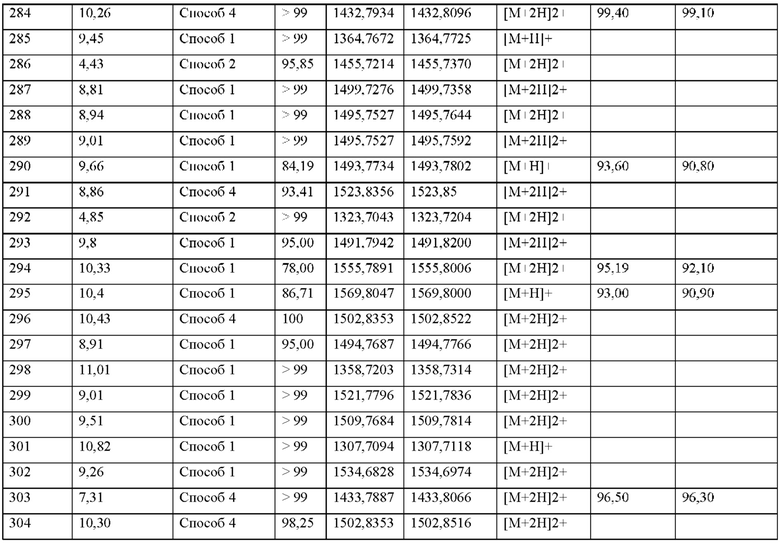
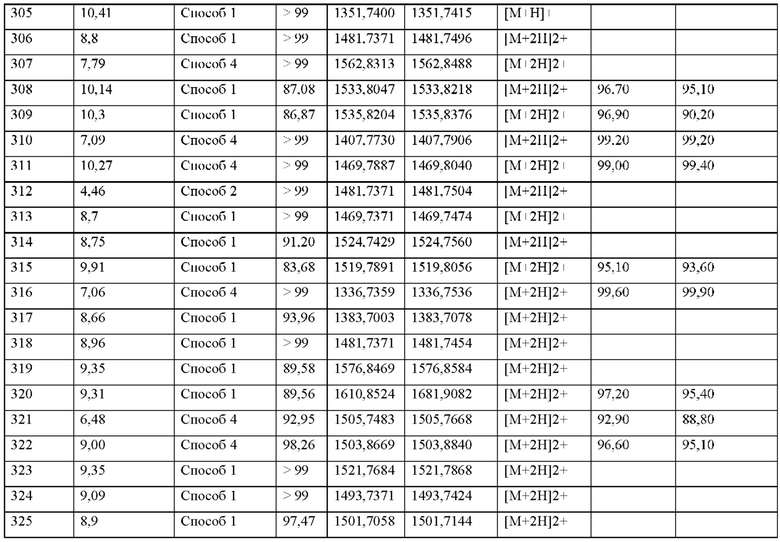
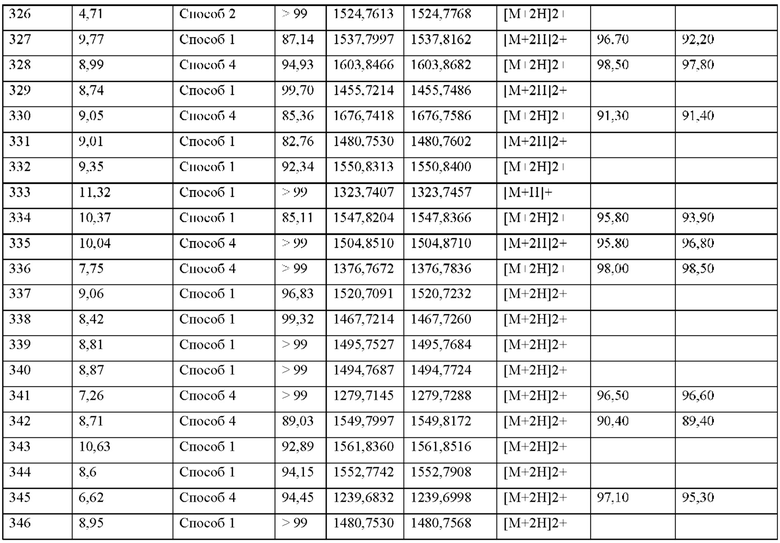
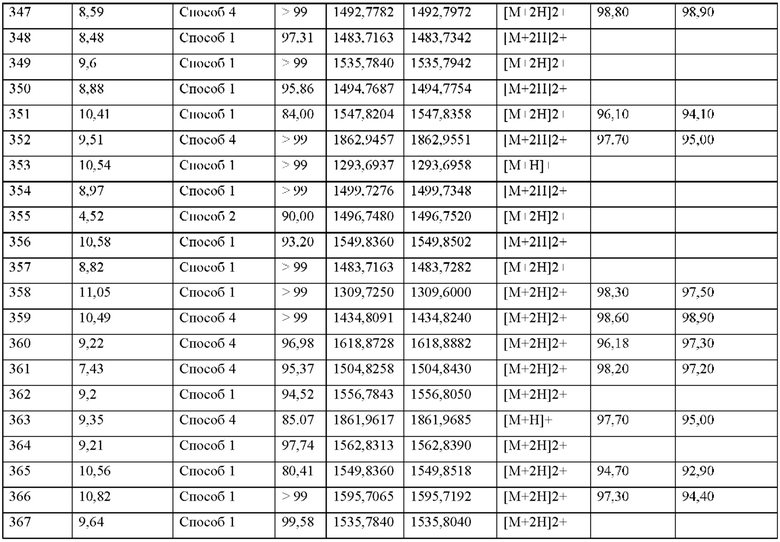
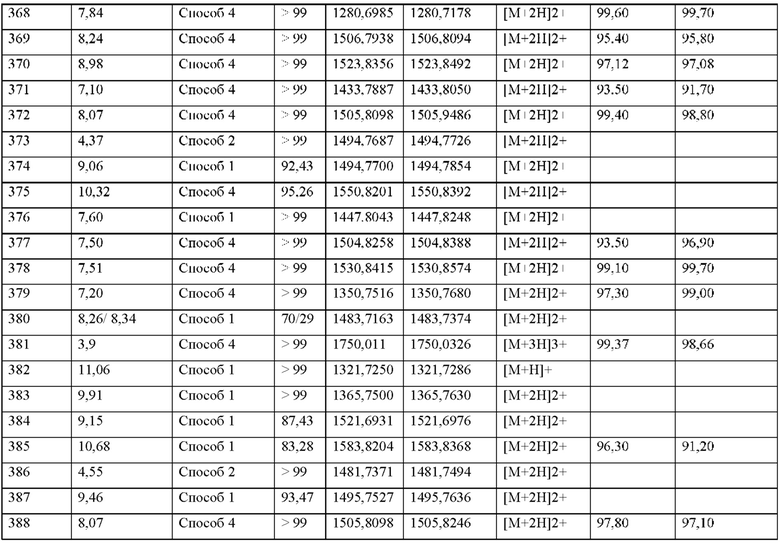
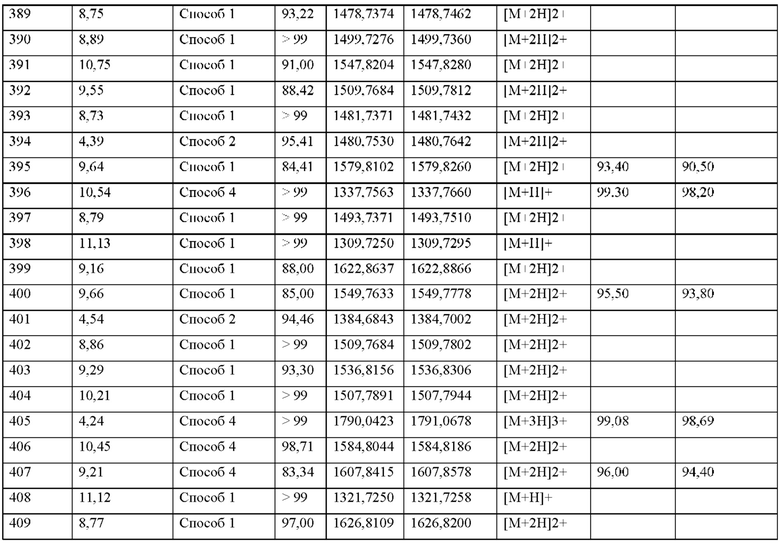
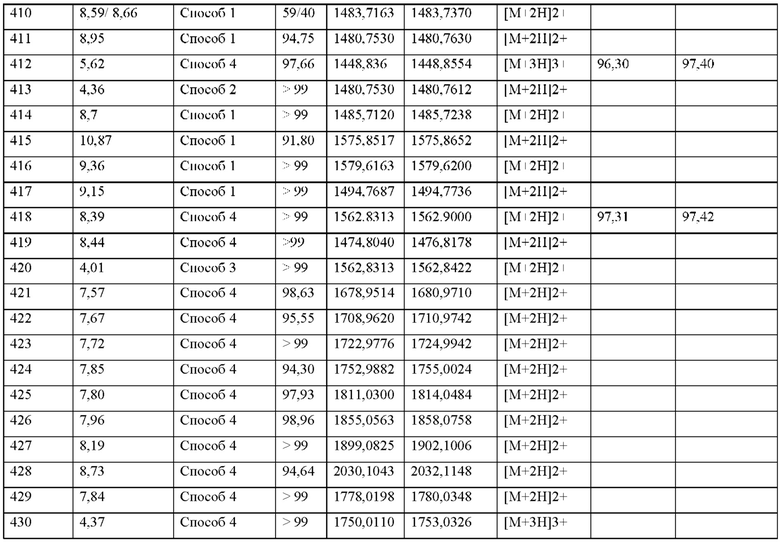
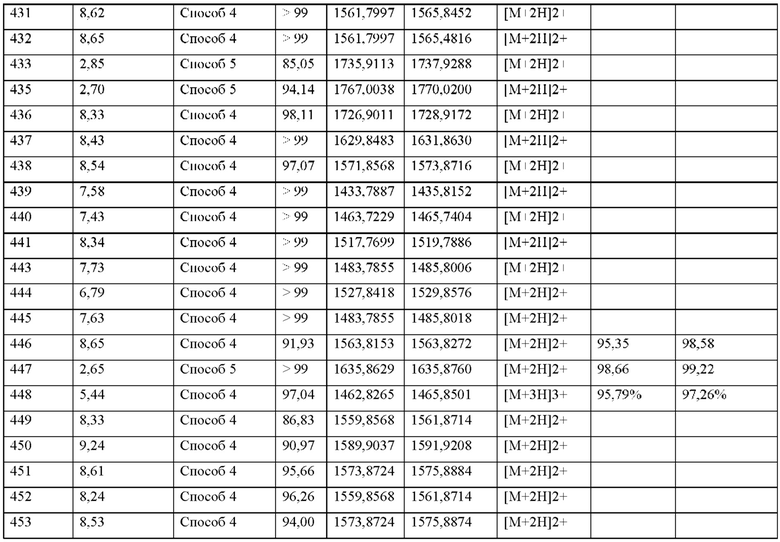
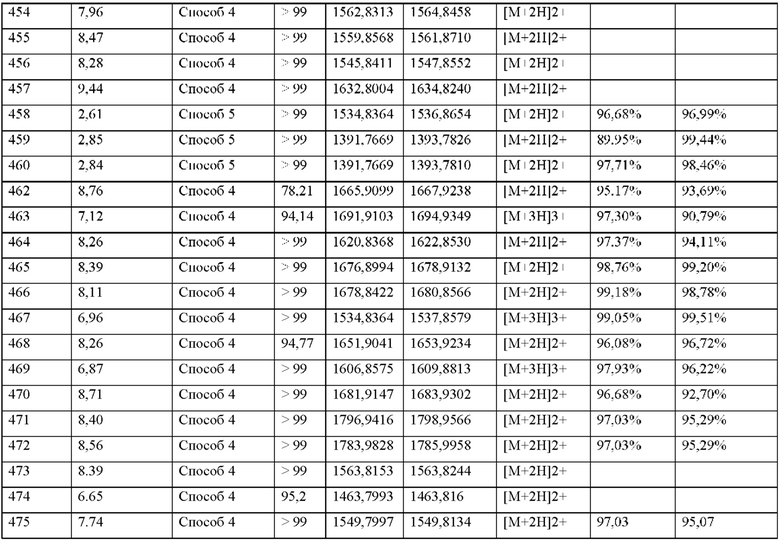
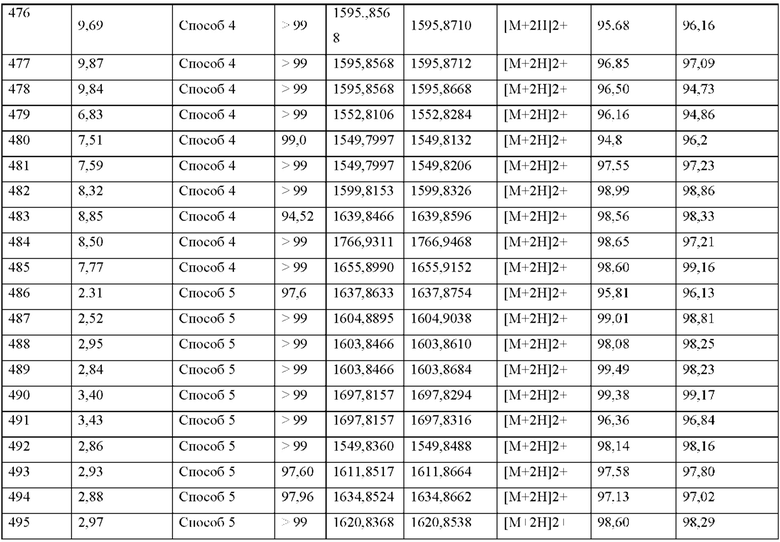
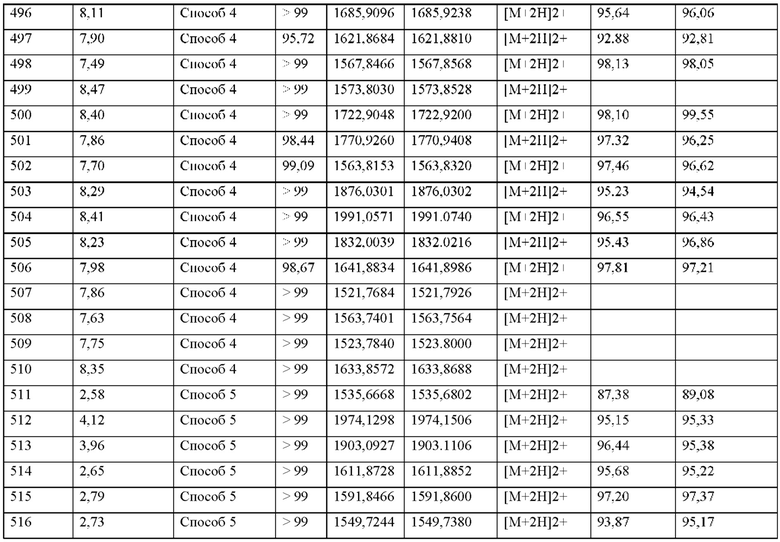
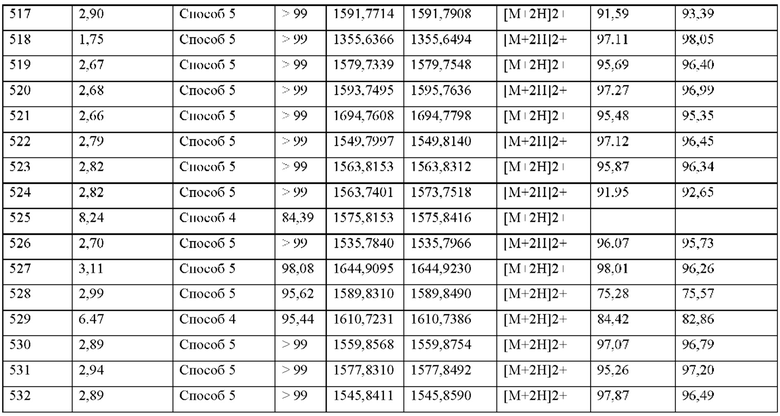
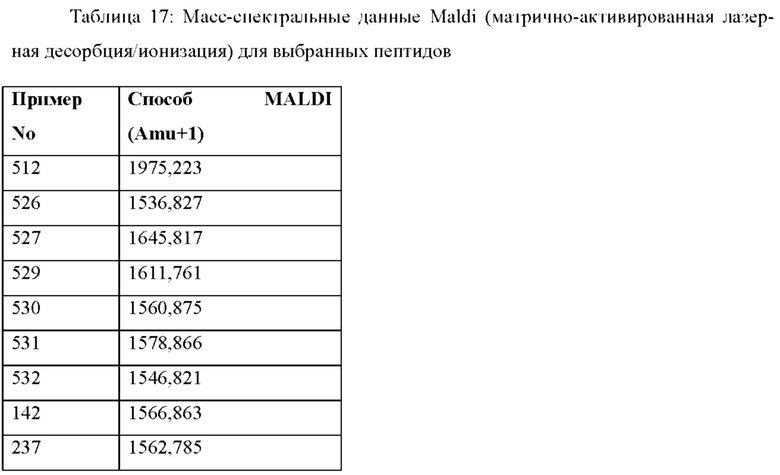
Далее Примеры 29, 44, 45, 67, 75, 109, 166, 185, 347, 443, 466 и 499 проиллюстрированы их химической структурой. Настоящее изобретение включает фармацевтически приемлемые соли, сольваты или сольваты солей этих примеров. Химические структуры приведены в виде бессолевых форм.
Пример 29, последовательность: AIC+SRSLP-(Oic)-I-(Pen)+IPD-OH (HCl соль)
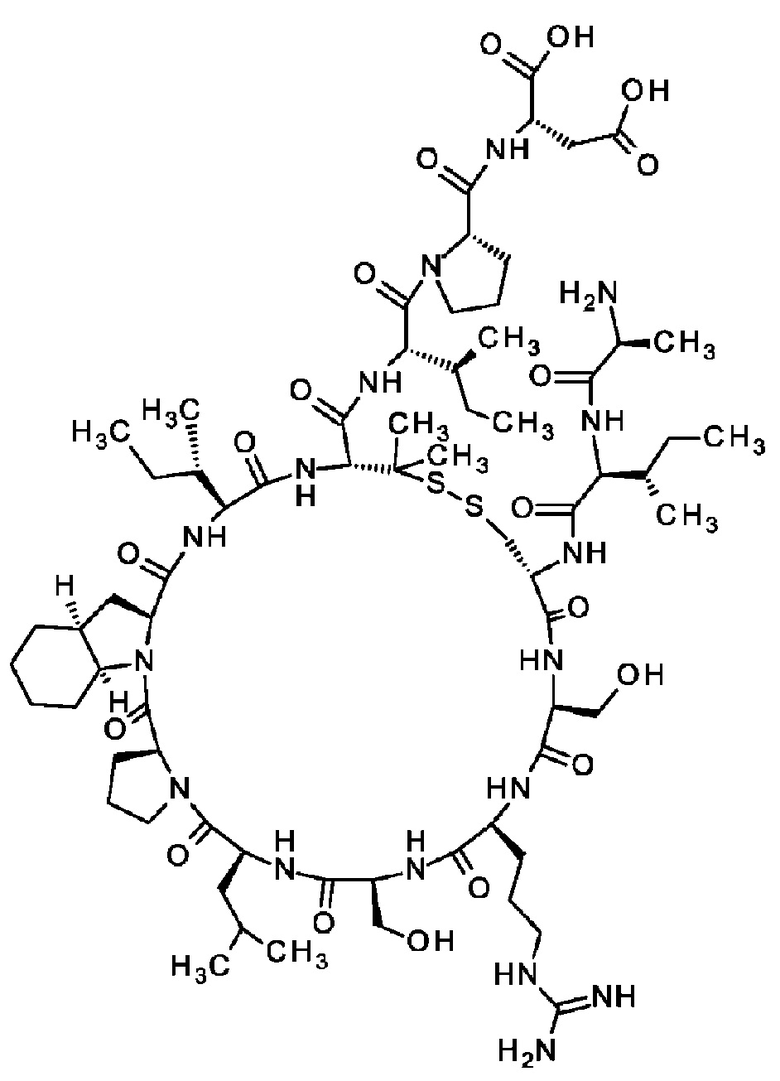
Пример 44, последовательность: ((N-Me)G)-IC+SRS-((tBu)A)-PPI-(Pen)+IP-(PEG9(31 aTOM))-NH2
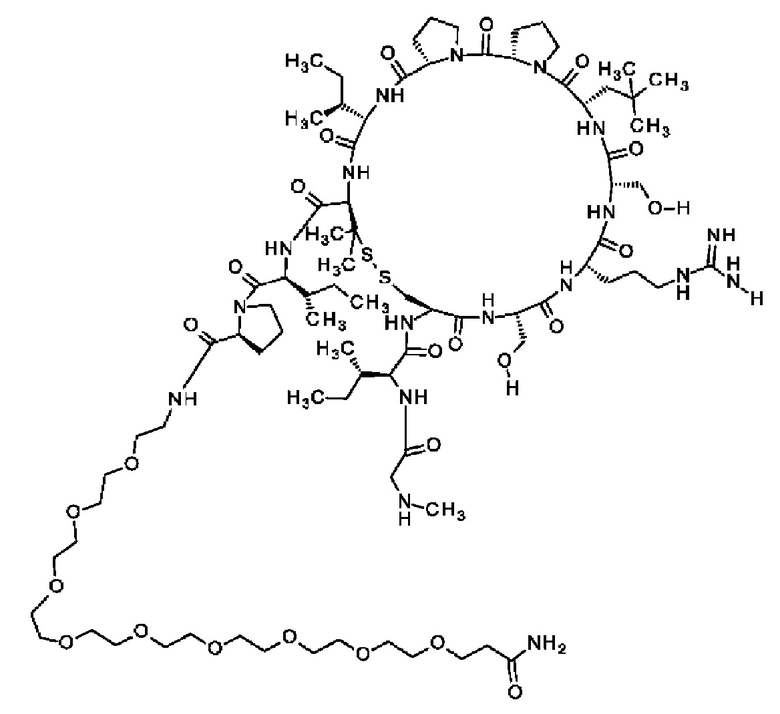
Пример 45, последовательность: AI-C+SRS-((tBu)A)-P-((6S)-5-азаспиро[2.4]гептан-6-карбоновая кислота)-I-(Pen)+IPD-NH2
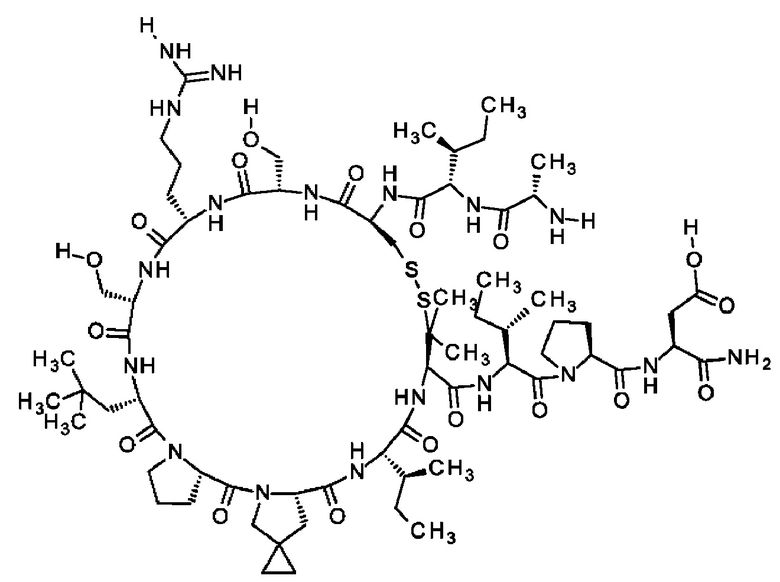
Пример 67, последовательность: ((N-Me)G)-IC+SRSLP-(Oic)-I-(Pen)+IP-NH2 (HCl соль)
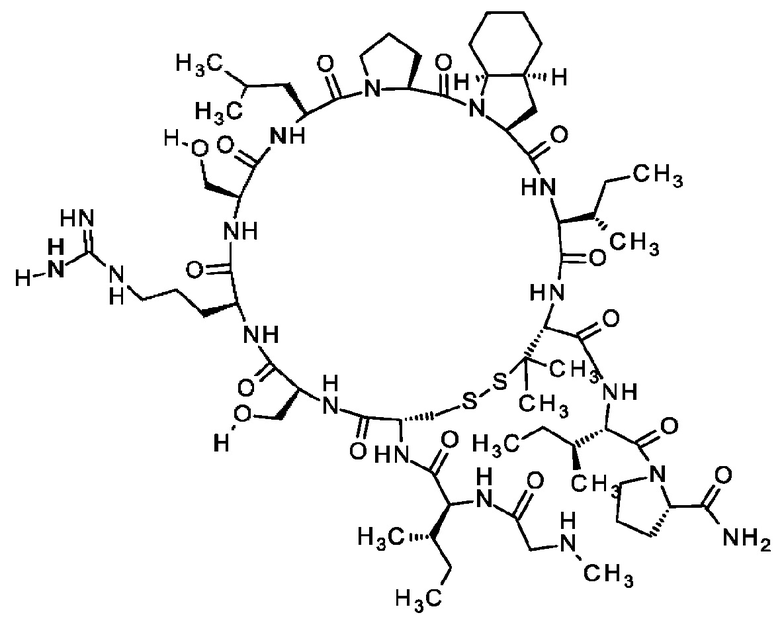
Пример 75, последовательность: AIC+SRSLP-(Oic)-I-(Pen)+IPD-NH2

Пример 109, последовательность: ((N-Me)G)-IC+SRSLP-(Oic)-I-(Pen)+IP-NH2
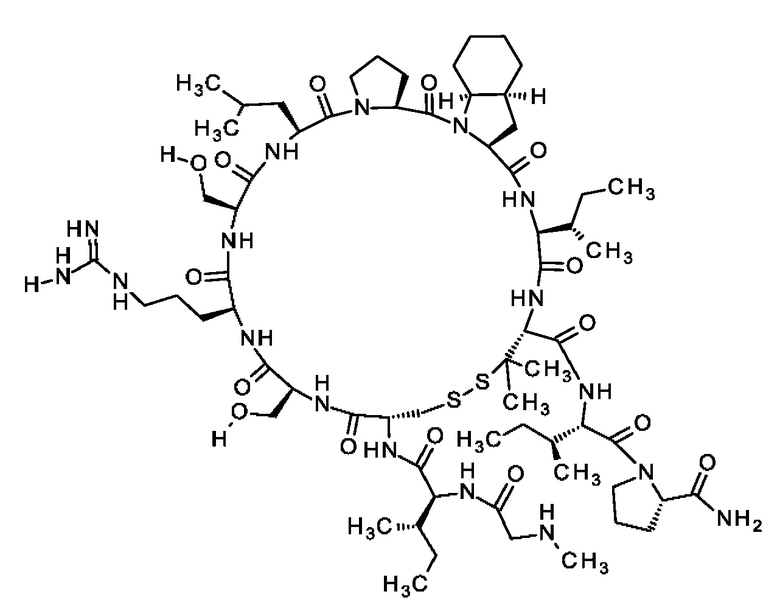
Пример 166, последовательность: (3-амино-2,2-диметилпропионовая кислота)-IC+SRS-((tBu)A)-PPI-(Pen)+IPD-NH2
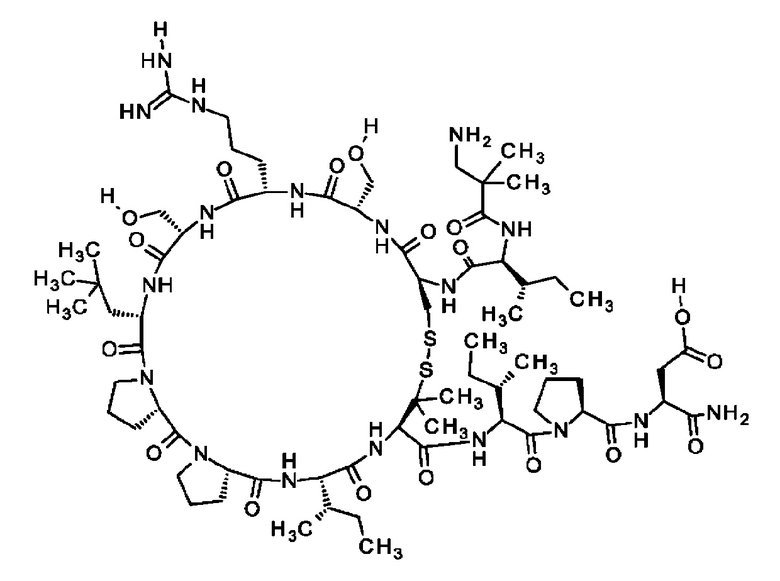
Пример 185, последовательность: AIC+SRSLP-(Oic)-I-((N-Me)C)+IPD-NH2
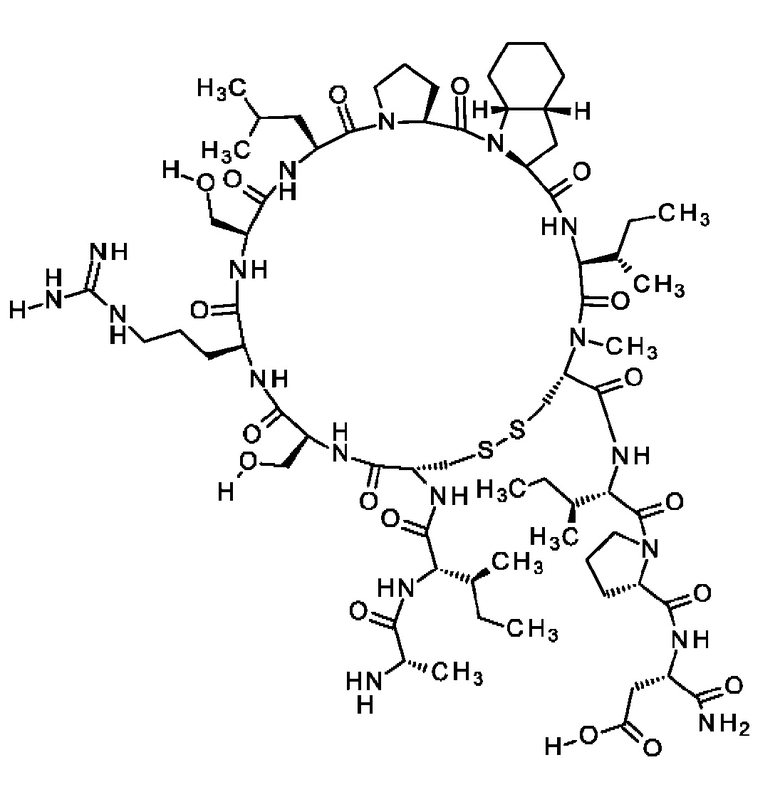
Пример 347, последовательность: IC+SRSLP-(Oic)-I-(Pen)+IPD-OH

Пример 443, последовательность: ((N-Me)A)-IC+SRS-((tBu)A)-P-((3R,6R)-l,l-дифтор-5-азаспиро[2.4]гептан-6-карбоновая кислота (энантиомер l))-I-(Pen)+IP-NH2

Пример 466, последовательность: ((N-Me)G)-IC+SRSLP-(Oic)-I-(Pen)+IPDD-OH
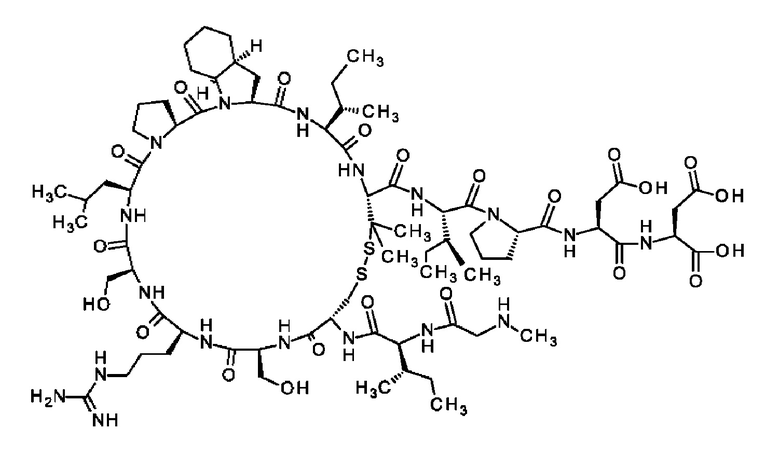
Пример 499, последовательность: AIC+S-(Arg(13C6,15N4))-SLP-(Oic)-I-(Pen)+IPD-ОН
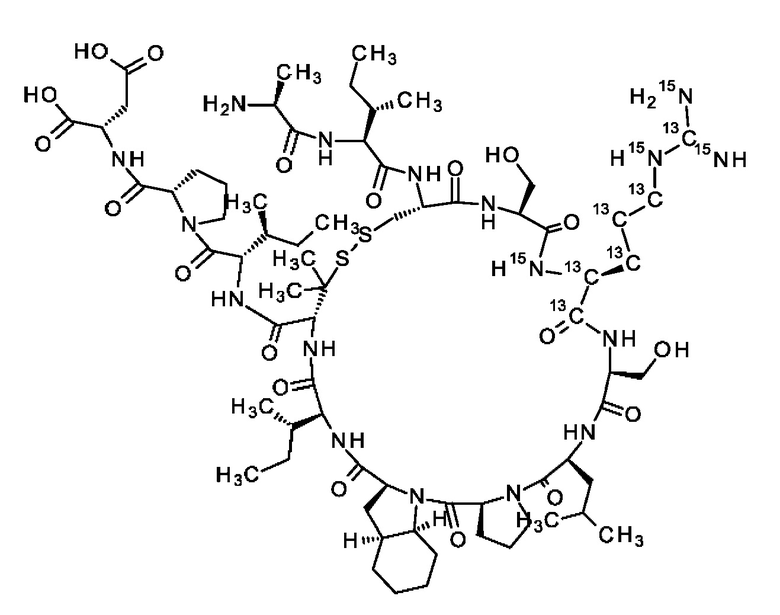
Биологические исследования in vitro
1. Серинпротеазный профиль тестируемых соединений
Тестируемые соединения тестировали на протеазной панели, состоящей из различных сериновых протеаз человека, включая калликреин, плазмин, FXIa, тромбин, фактор Ха, tPA и трипсин.
Описание теста
Определяли ингибирующую активность и/или селективность тестируемых соединений. Анализы основаны на флуоресцентном обнаружении аминометилкумарина (АМС), высвобождаемого из субстратов фторогенных пептидных протеаз при расщеплении, катализируемом протеазами. Активные протеазы или зимогены, обычно очищенные из плазмы человека или для трипсина из поджелудочной железы человека, и соответствующие субстраты являются коммерчески доступными.
Анализы сериновых протеазы включают следующие ферменты и субстраты. Все ферменты и субстраты разводят в буфере для анализа (50 мМ Трис/HCl рН7,4, 100 мМ NaCl, 5 мМ CaCl2, 0,1% БСА). Конечные концентрации анализа приведены:
• калликреин (Kordia; 0.2 нм), H-Pro-Phe-Arg-AMC (Bachem I-1295; 5 мкм)
• плазмин (Kordia; 0.1 мкг/мл, 1.2 нм), MeOSuc-Ala-Phe-Lys-AMC (Bachem I-1275; 50 мкм)
• фактор XIa (Kordia; 0.15 нм), Boc-Glu(OBzl)-Ala-Arg-AMC (Bachem 1-1575; 5
мкм)
• тромбин (Kordia; 0.02 нм), Boc-Asp(OBzl)-Pro-Arg-AMC (Bachem 1-1560; 5
мкм)
• фактор Ха (Kordia; 1.3 нм), Boc-Ile-Glu-Gly-Arg-AMC (Bachem I-1100; 5 мкм)
• Активатор тканевого плазминогена (tPA, Loxo; 2 нм), CH3SO2-D-Phe-Gly-Arg-AMC (Pentapharm 091-06; 5 мкм)
• трипсин (Sigma; 0.042 Ед/мл), субстрат Boc-Ile-Glu-Gly-Arg-AMC (Bachem I-1100; 5 мкм)
Для определения эффективности тестируемого соединения используют фермент и соответствующие разведения субстрата для проведения анализов протеазы:
На 384-луночные микротитровальные планшеты (белые, Greiner), содержащие 1 мкл/лунку серийных разведений тестируемых или эталонных соединений, добавляли 20 мкл буфера для анализа, 20 мкл разведения фермента и 20 мкл субстрата. Контрольные реакции не содержат тестируемого соединения (только DMSO). После инкубации в течение обычно 30 мин (линейная кинетика реакции) при комнатной температуре измеряют флуоресценцию (например, ех 360 нм, em 465 нм) на микротитровальном планшете для считывания флуоресценции (например, Tecan Safire II). Значения IC50 определяют путем построения логарифмической зависимости концентрации тестируемого соединения от процентной активности протеазы.
2. Биохимический анализ MASP-1 и MASP-2 человека
2.1 Рекомбинантная экспрессия и продукция белка рекомбинантными активными протеазами MASP1 и MASP2 человека.
Укороченную кДНК последовательность MASP1 человека, кодирующую фрагмент, соответствующий аминокислотам 297-699, с С-концевой His-меткой и N-концевым сигналом секреции Ig-каппа (последовательность ID No: 2), субклонировали в вектор экспрессии млекопитающих pcDNA3.1 (Invitrogen).
Укороченную кДНК последовательность MASP1 человека, кодирующую фрагмент, соответствующий аминокислотам 297-686, с С-концевой His-меткой и N-концевым сигналом секреции Ig-каппа (последовательность ID No: 3), субклонировали в вектор экспрессии млекопитающих pcDNA3.1 (Invitrogen).
Векторы экспрессии MASP1 или MASP2 трансфицировали в клеточную линию HEK293 (АТСС No. CRL-1573) с использованием реагента Lipofectamin LTX® (Thermo-Fischer), как описано производителем. Зрелую форму рекомбинантных протеаз MASP1 и MASP2 человека секретировали в культуральную среду. Белки MASP1 и MASP2 очищали из кондиционированных сред методом аффинной хроматографии на смоле Ni-NTA Superpotok (Qiagen), как описано производителем.
2.2 Биохимический анализ MASP1 человека
Рекомбинантный человеческий фермент MASP1, полученный в клетках HEK 293, разбавляли в реакционном буфере (50 мм HEPES рН 8,0; 100 мм NaCl; 0,01% CHAPS; 0,5 мм Глутатиона) до концентрации 20 нм и переносили 25 мкл в каждую отдельную лунку 384-луночного микротитровального планшета (Greiner Bio One 781075). 1 мкл раствора соединения-ингибитора (растворенного в DMSO при соответствующей концентрации) или чистого DMSO в качестве контроля добавляли в те же лунки. Ферментативную реакцию инициировали добавлением 25 мкл 20 мкм раствора субстрата FRET ABZ-MYGGARRL-Lys(Dnp)-NH2; (ABZ - 2-аминобензоил; DNP - 2,4-динитрофенил; заказной синтез Jerini Peptide Technologies, Берлин) в реакционном буфере. Микротитровальный планшет инкубировали в течение 60-120 мин при температуре 32°С.Увеличение интенсивности флуоресценции измеряли в соответствующем считывателе флуоресцентных планшетов (например, TECAN Ultra) с использованием длины волны возбуждения 320 нм и длины волны излучения 420 нм. Значения IC50 рассчитывали из процента ингиби-рования активности MASP1 человека в зависимости от концентрации тестируемого соединения.
2.3 Биохимический анализ MASP2
Рекомбинантный человеческий фермент MASP2, полученный в клетках HEK 293, разбавляли в реакционном буфере (50 мм HEPES рН 8,0; 100 мм NaCl; 0,01% CHAPS; 0,5 мм Глутатиона) до концентрации 20 нм и переносили 25 мкл в каждую отдельную лунку 384-луночного белого микротитровального планшета (Greiner Bio One 781075). 1 мкл раствора соединения-ингибитора (растворенного в DMSO при соответствующей концентрации) или чистого DMSO в качестве контроля добавляли в те же лунки. Ферментативную реакцию инициировали добавлением 25 мкл 60 мкм раствора субстрата FRET DABCYL-KISPQGYGRR-Glu(EDANS)-NH2; (Dabcyl -4-((4-(диметиламино)фенил)азо)бензойная кислота; Edans -5-[(2-аминоэтил)амино]нафталин-1-сульфонил; заказной синтез Jerini Peptide Technologies, Берлин) в реакционном буфере. Микротитровальный планшет инкубировали в течение 60-120 мин при температуре 32°С. Увеличение интенсивности флуоресценции измеряли в соответствующем считывателе флуоресцентных планшетов (например, TECAN Ultra) с использованием длины волны возбуждения 340 нм и длины волны излучения 490 нм. Значения IC50 рассчитывали из процента ингибирования активности MASP2 человека в зависимости от концентрации тестируемого соединения.
3. Анализ отложений С3 (человек, крыса, мышь, собака, мини-свинья) Анализ отложения С3 проводили в основном так, как описано (ссылка). Многолуночные планшеты (Greiner-Nunc 384 Maxi Sorp #464718) всю ночь покрывали маннаном от Sac-charomyces cerevisiae (Sigma М7504, 10 мкг/мл в 0,05 М карбонатно-бикарбонатном буфере, рН 9,6) при 4°С.Лунки трижды промывали TBS и инкубировали субэквивалентно в течение 2 часов с 50 мкл 1% бычьего сывороточного альбумина (BSA) в трис-буферизованном солевом растворе (TBS) при 37°С для блокирования неспецифического связывания. После этой стадии и каждой из следующих стадий инкубации лунки промывали трижды промывочным буфером С3 (TBS; 0,05% Tween 20; 5 мм CaCl2). Далее лунки инкубировали в течение 30 мин при 37°С с 50 мкл смеси тестируемых соединений с разведенной сывороткой в буфере Veronal (Veronal Puffer (Lonza 12624E). Сыворотка применялась при концентрациях, при которых в предварительных тестах не обнаруживалось отложение С3 на непокрытых планшетах. Было обнаружено, что соответствующие разведения находятся в диапазоне 1:100-1:200 для сыворотки человека, крысы, мыши и собаки и 1:20-1:100 для сыворотки мини-свиньи, соответственно. В типичных экспериментах соединения тестировались в диапазоне концентраций от 1×10-9 до 5×10-5 моль/л. После промывки отложение С3 выявляли путем инкубации с поликлональным кроличьим антителом против С3 человека (Dako (Biozol) А0062) в течение 1 ч с последующей промывкой и инкубацией с конъюгированным с пероксидазой анти-кроличьим IgG (Sigma А1949) в течение 30 мин при 37°С, с последующей промывкой и инкубацией с раствором субстрата ТМБ в темноте. При соответствующем проявлении цветную реакцию останавливали добавлением 25 мкл стоп-раствора (Sigma S5814) и количественно определяли на фотометре, измеряя поглощение при длине волны 450 нм. Антитела разводили в промывочном буфере С3 с добавлением 0,5% BSA. 4. Биохимический анализ MASP-1 и MASP-2 крыс
4.1 Рекомбинантная экспрессия и продукция белка рекомбинантных активных протеаз MASP1 и MASP2 крысы и человека.
Укороченную кДНК последовательность MASP1 крысы, кодирующую фрагмент, соответствующий аминокислотам 302-704, с С-концевой His-меткой и N-концевым сигналом секреции Ig-каппа (последовательность ID No: 4), субклонировали в вектор экспрессии млекопитающих pcDNA3.1 (Invitrogen).
Укороченную кДНК последовательность MASP1 крысы, кодирующую фрагмент, соответствующий аминокислотам 296-685, с С-концевой His-меткой и N-концевым сигналом секреции Ig-каппа (последовательность ID No: 5), субклонировали в вектор экспрессии млекопитающих pcDNA3.1 (Invitrogen).
Векторы экспрессии MASP1 или MASP2 трансфицировали в клеточную линию HEK293 (АТСС No. CRL-1573) с использованием реагента Lipofectamin LTX® (Thermo-Fischer), как описано производителем. Зрелую форму рекомбинантных протеаз MASP1 и MASP2 крысы секретировали в культуральную среду. Белки MASP1 и MASP2 очищали из кондиционированных сред методом аффинной хроматографии на смоле Ni-NTA Superpotok (Qiagen), как описано производителем.
4.2 Биохимический анализ MASP1 крыс.
Рекомбинантный крысиный фермент MASP1, полученный в клетках HEK 293, разбавляли в реакционном буфере (50 мм HEPES рН 8,0; 100 мм NaCl; 0,01% CHAPS; 0,5 мм Глутатиона) до концентрации 4 нм и переносили 25 мкл в каждую отдельную лунку 384-луночного микротитровального планшета (Greiner Bio One 781075). 1 мкл раствора соединения-ингибитора (растворенного в DMSO при соответствующей концентрации) или чистого DMSO в качестве контроля добавляли в те же лунки. Ферментативную реакцию инициировали добавлением 25 мкл 40 мкм раствора субстрата FRET ABZ- Dabcyl-MYGGARRL-Glu(Edans)-NH2; (Dabcyl -4-((4-(диметиламино)фенил)азо)бензойная кислота; Edans -5-[(2-аминоэтил)амино]нафталин-1-сульфонил; заказной синтез Jerini Peptide Technologies, Берлин) в реакционном буфере. Микротитровальный планшет инкубировали в течение 60-120 мин при температуре 32°С. Увеличение интенсивности флуоресценции измеряли в соответствующем считывателе флуоресцентных планшетов (например, TECAN Ultra) с использованием длины волны возбуждения 340 нм и длины волны излучения 490 нм. Значения IC50 рассчитывали из процента ингибирования активности MASP1 крысы в зависимости от концентрации тестируемого соединения. 4.3 Биохимический анализ MASP2 крысы.
Рекомбинантный фермент MASP1 крысы, полученный в клетках HEK 293, разбавляли в реакционном буфере (50 мм HEPES рН 8,0; 100 мм NaCl; 0,01% CHAPS; 0,5 мм Глутатиона) до концентрации 20 нм и переносили 25 мкл в каждую отдельную лунку 384-луночного микротитровального планшета (Greiner Bio One 781075). 1 мкл раствора соединения-ингибитора (растворенного в DMSO при соответствующей концентрации) или чистого DMSO в качестве контроля добавляли в те же лунки. Ферментативную реакцию инициировали добавлением 25 мкл 30 мкм раствора субстрата FRET ABZ-MYGGARRL-Lys(Dnp)-NH2; (ABZ - 2-аминобензоил; DNP - 2,4-динитрофенил; заказной синтез Jerini Peptide Technologies, Берлин) в реакционном буфере. Микротитровальный планшет инкубировали в течение 60-120 мин при температуре 32°С. Увеличение интенсивности флуоресценции измеряли в соответствующем считывателе флуоресцентных планшетов (например, TECAN Ultra) с использованием длины волны возбуждения 320 нм и длины волны излучения 420 нм. Значения IC50 рассчитывали из процента ингибирования активности MASP1 крысы в зависимости от концентрации тестируемого соединения.
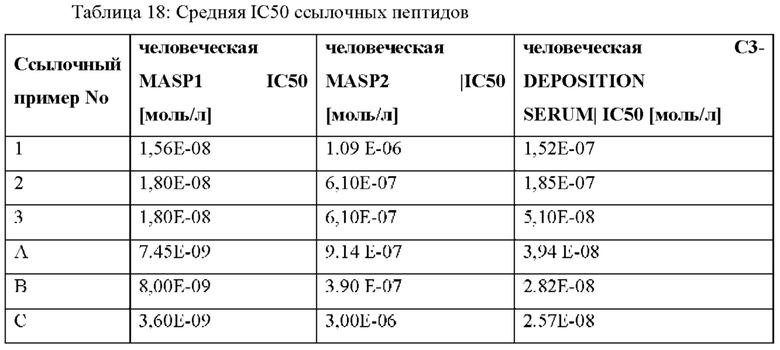

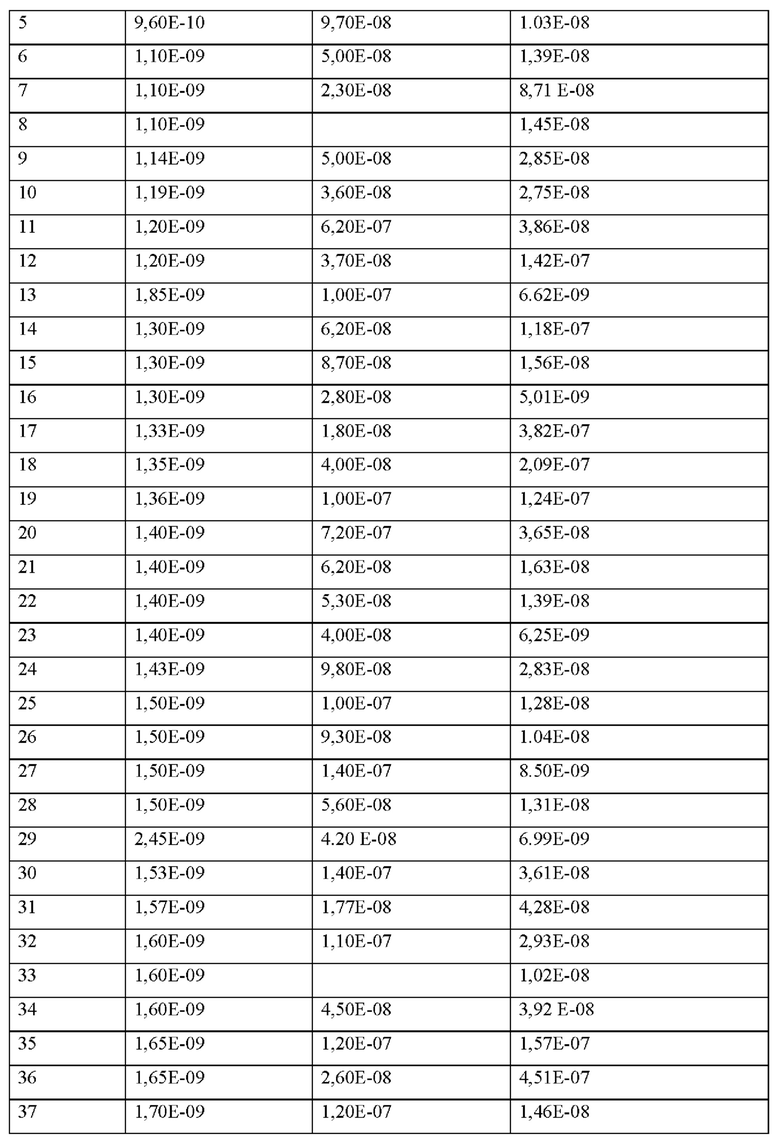
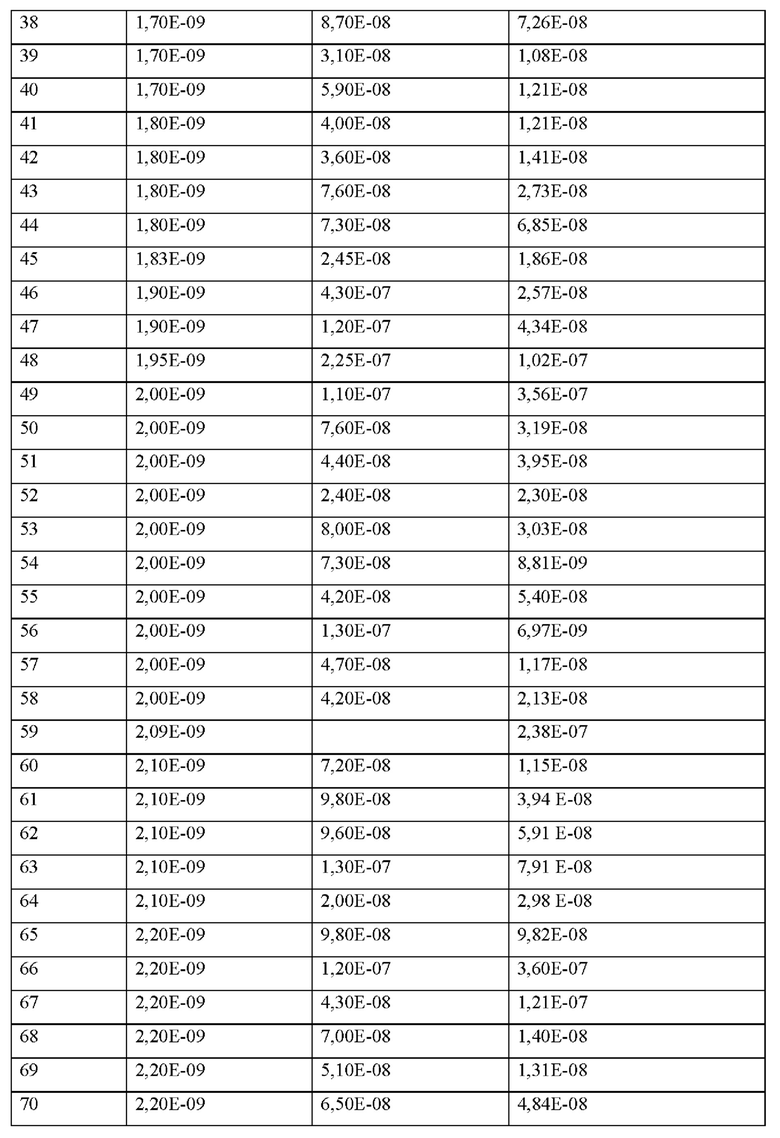
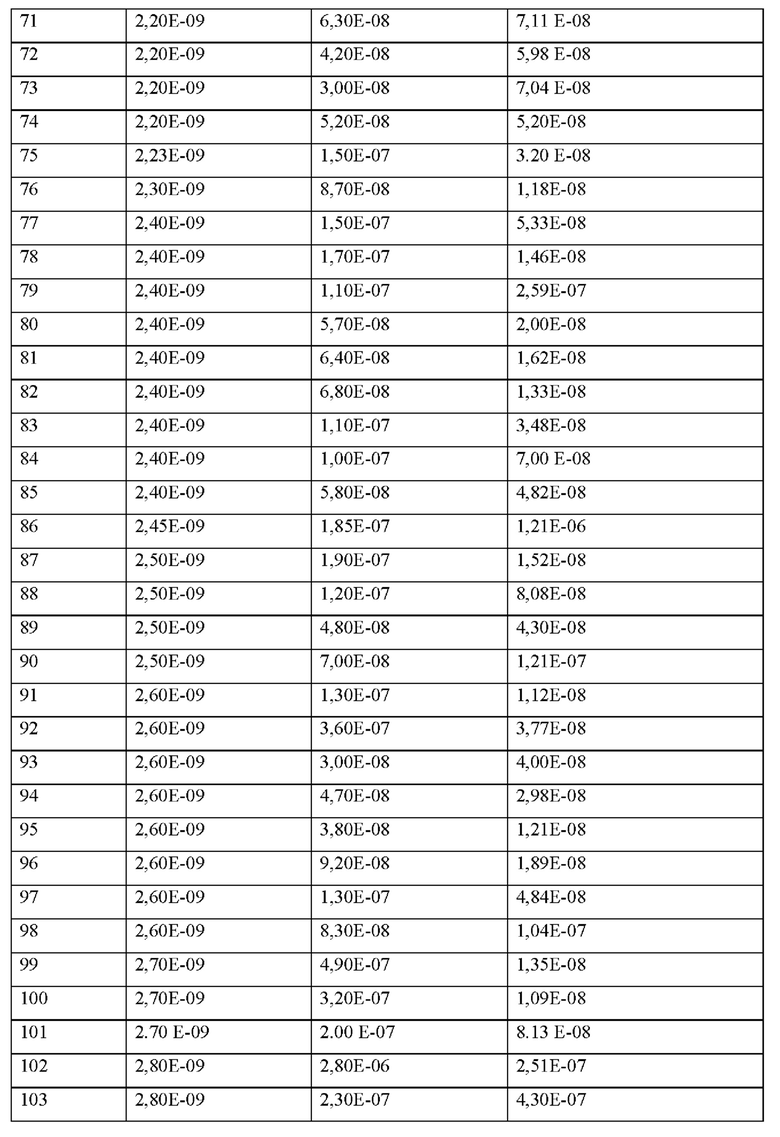
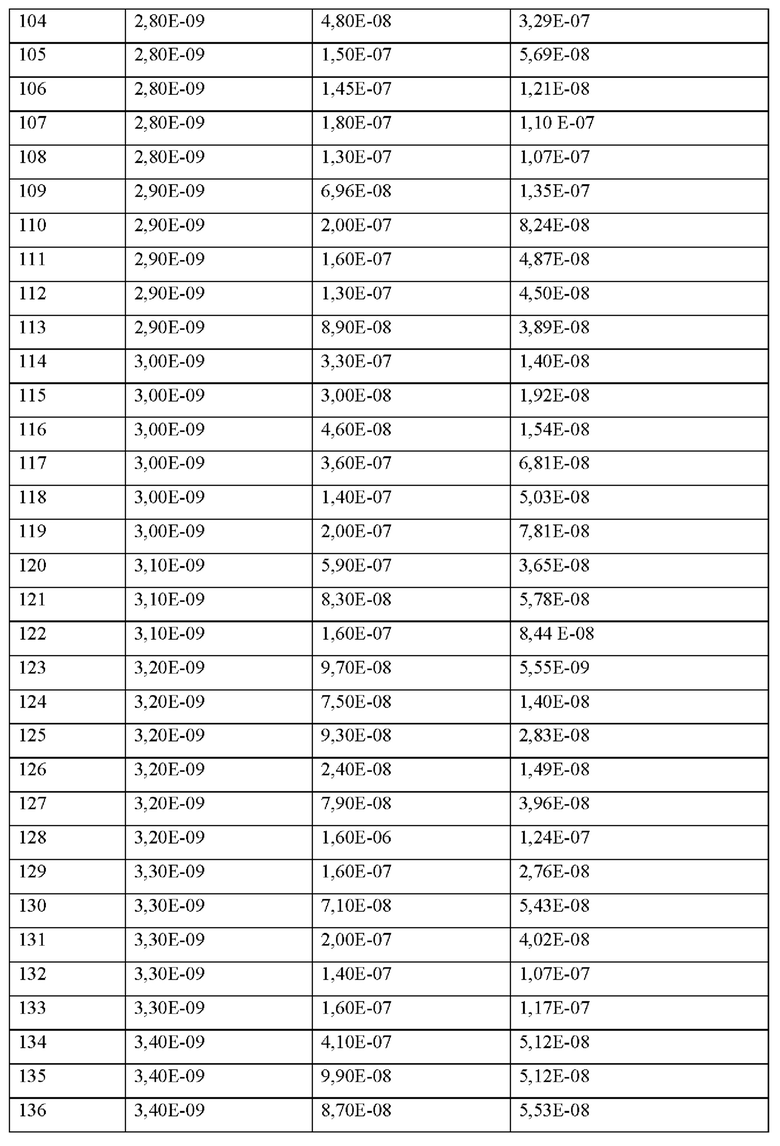
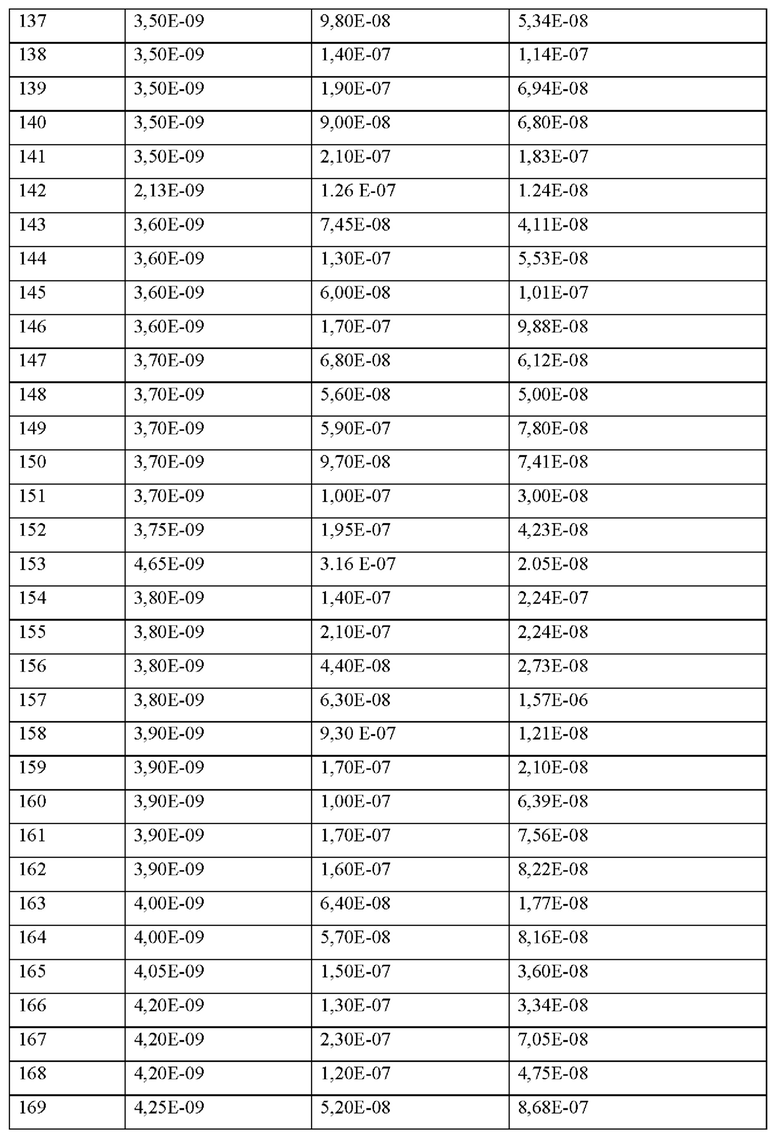
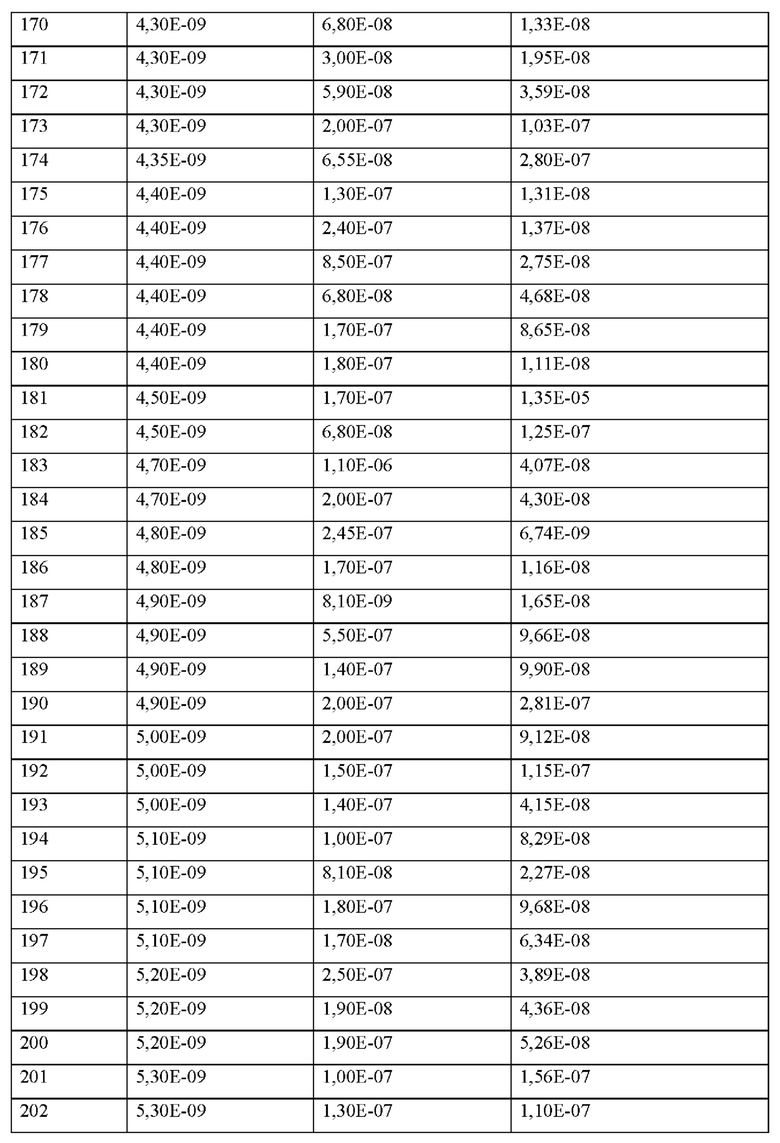
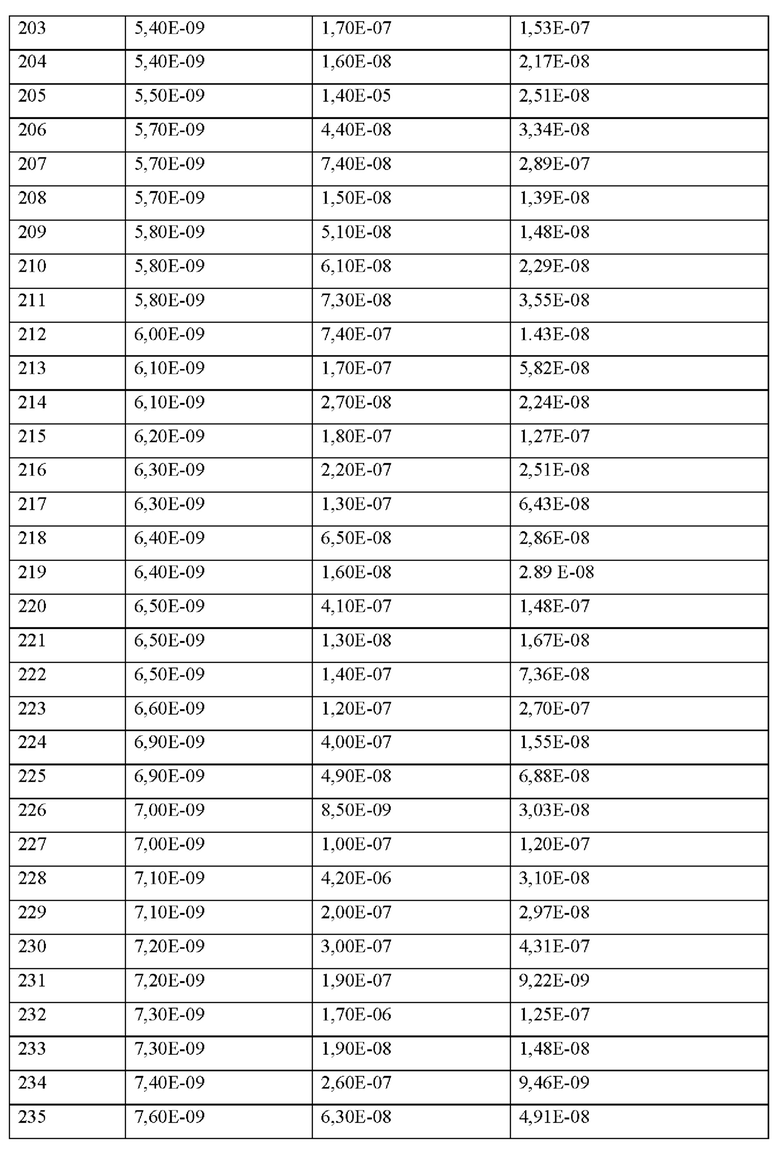
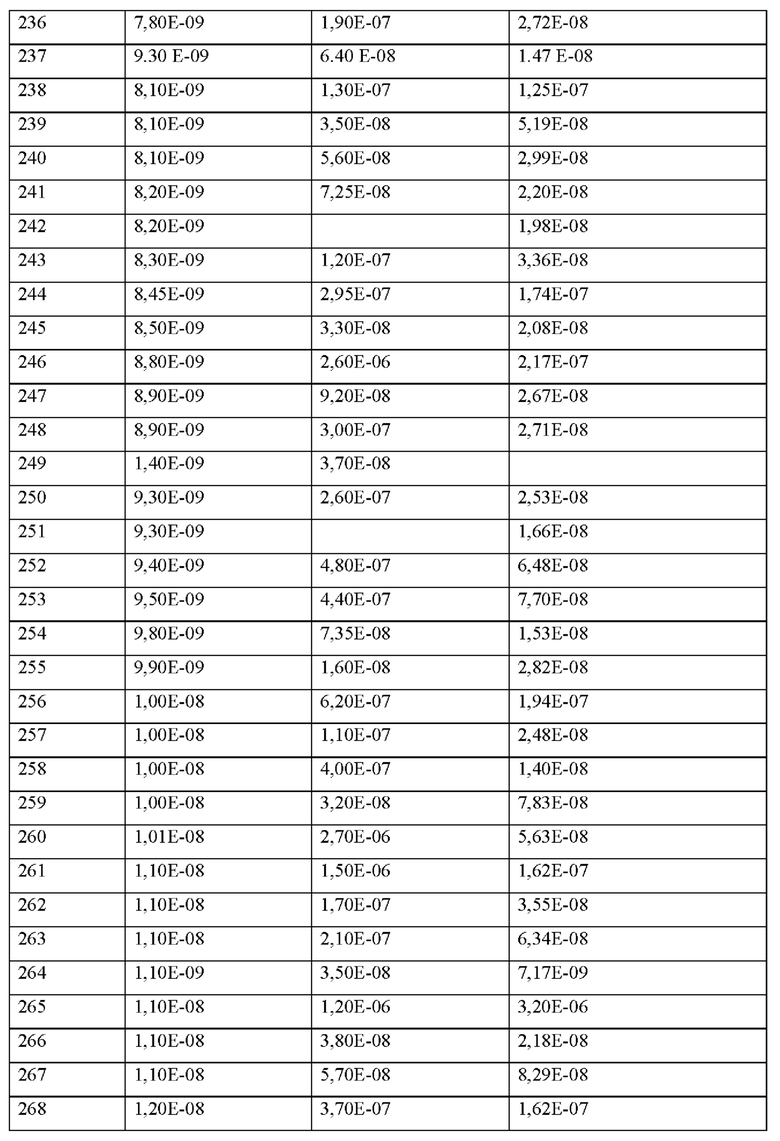
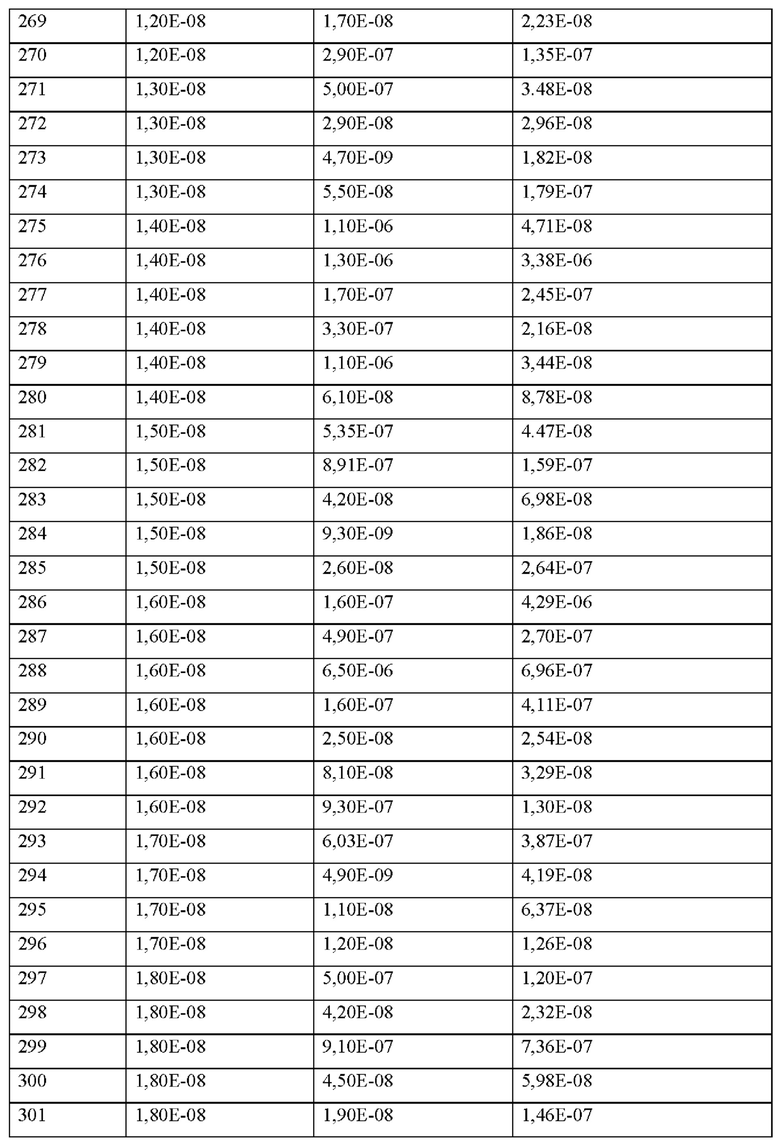
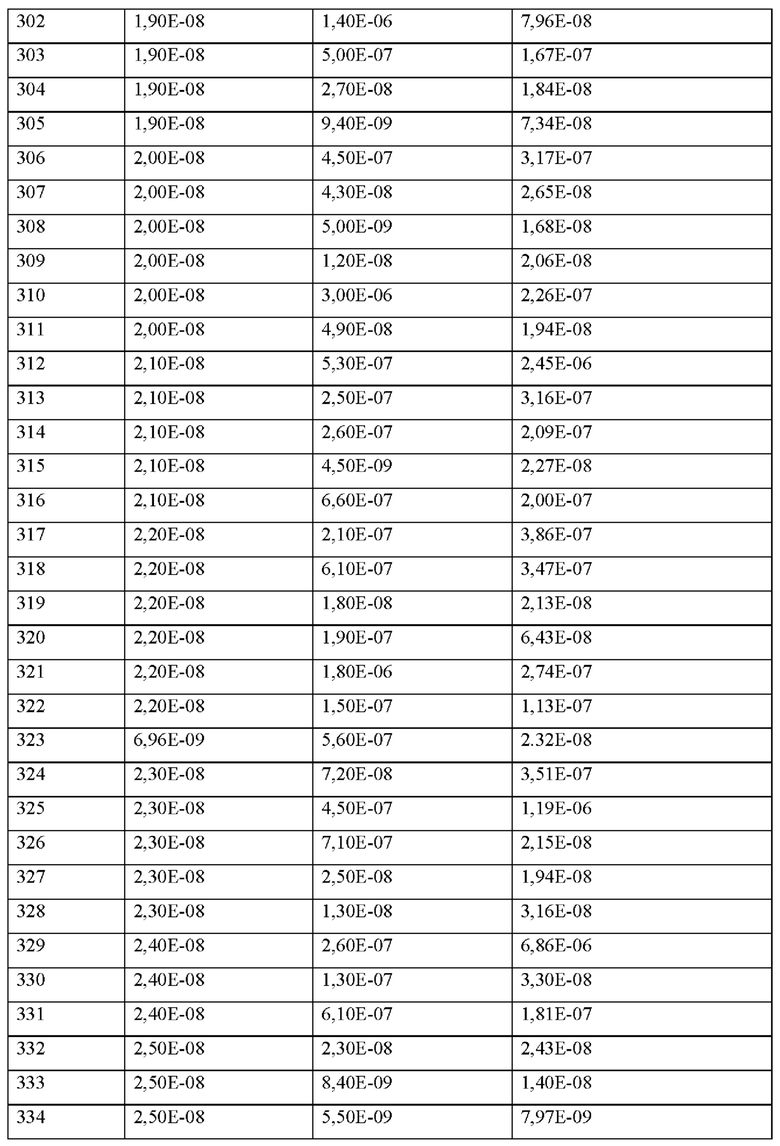
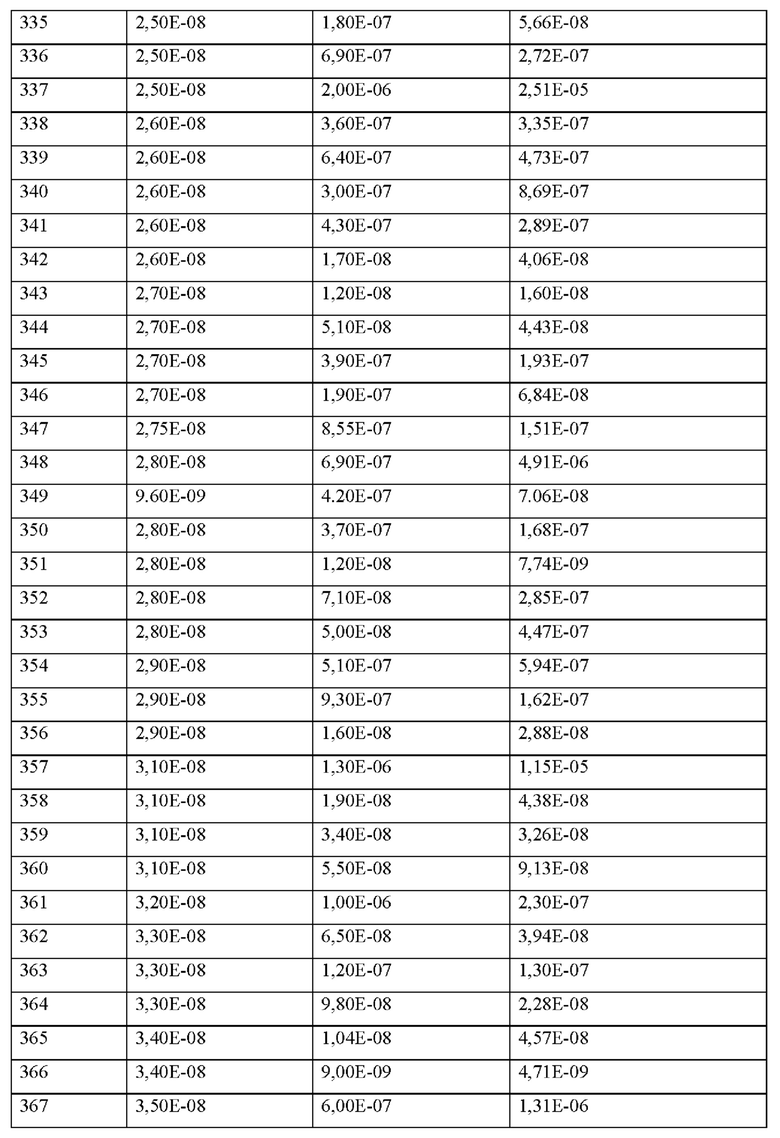
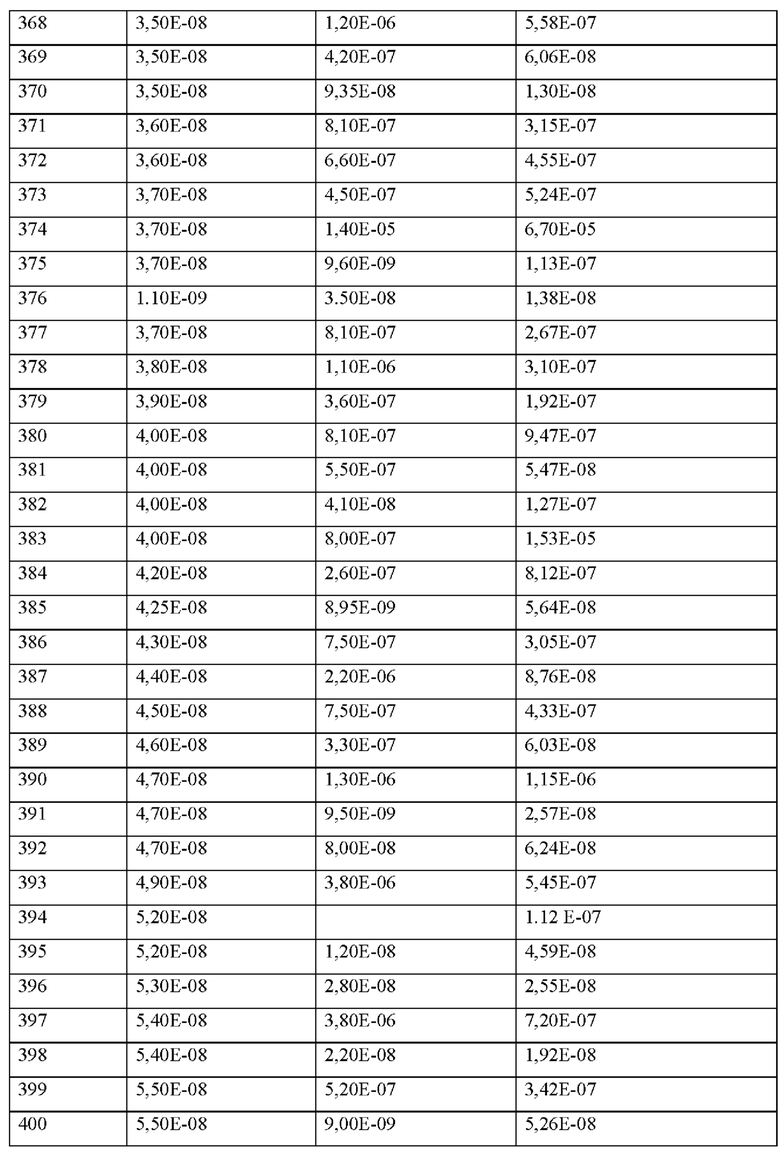
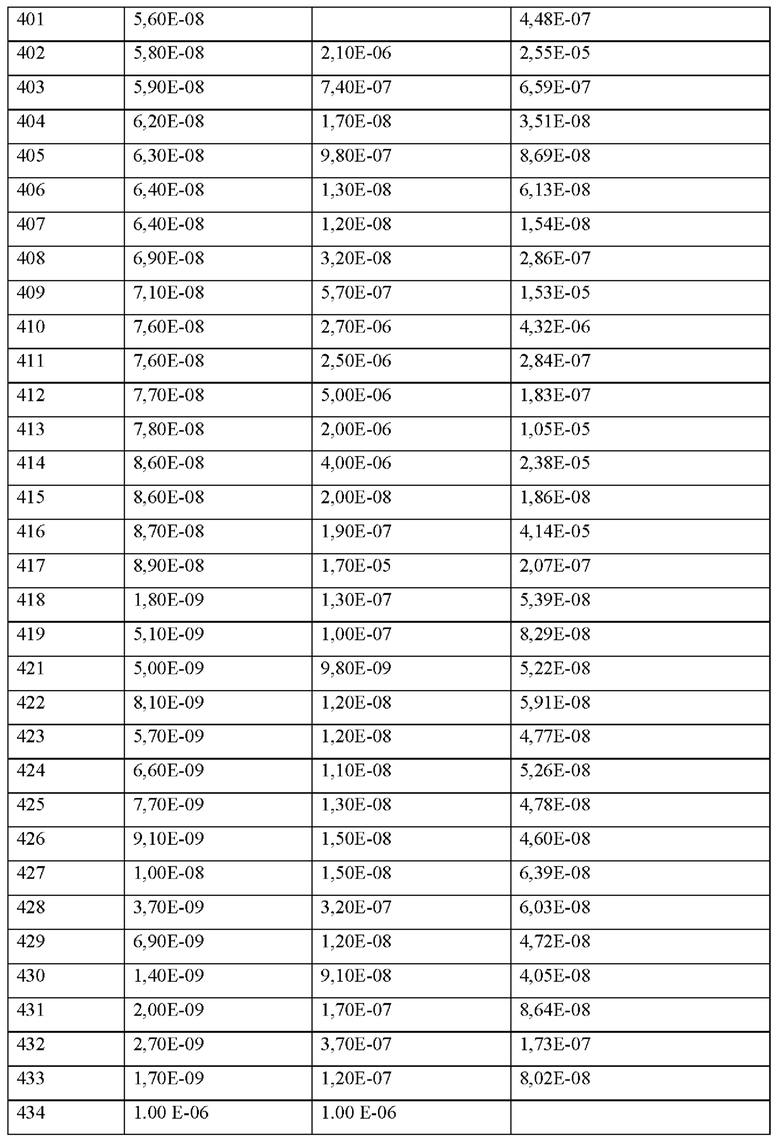
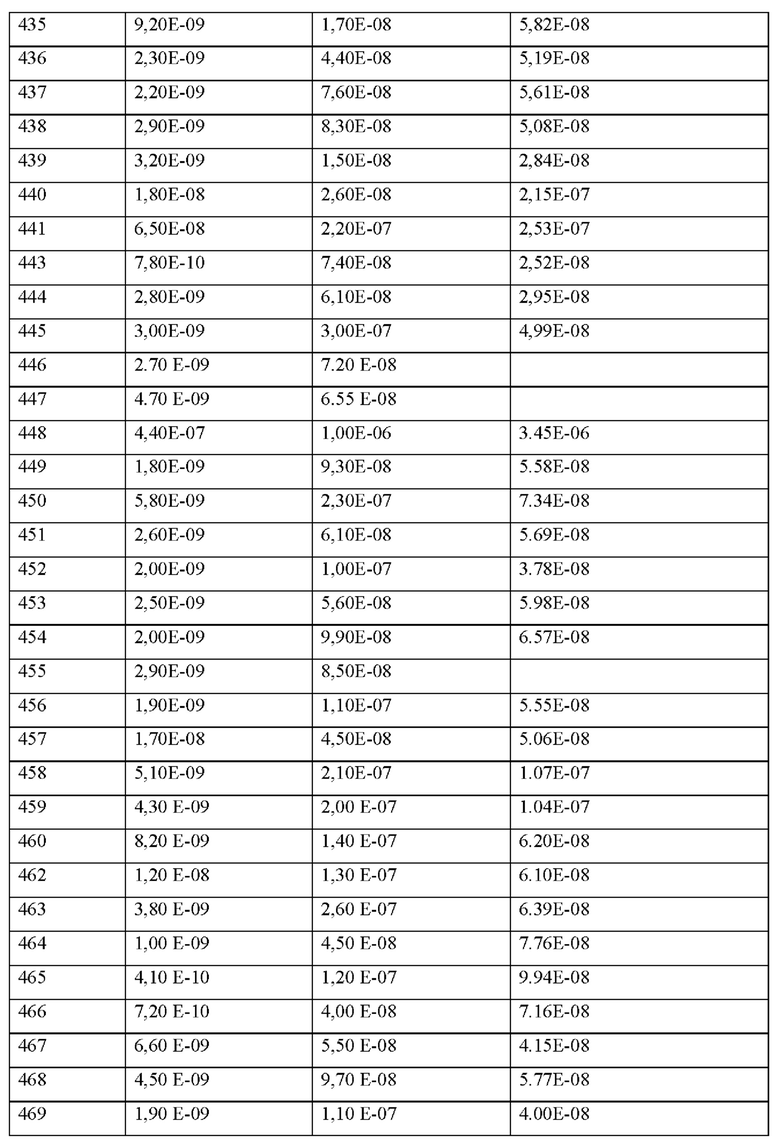
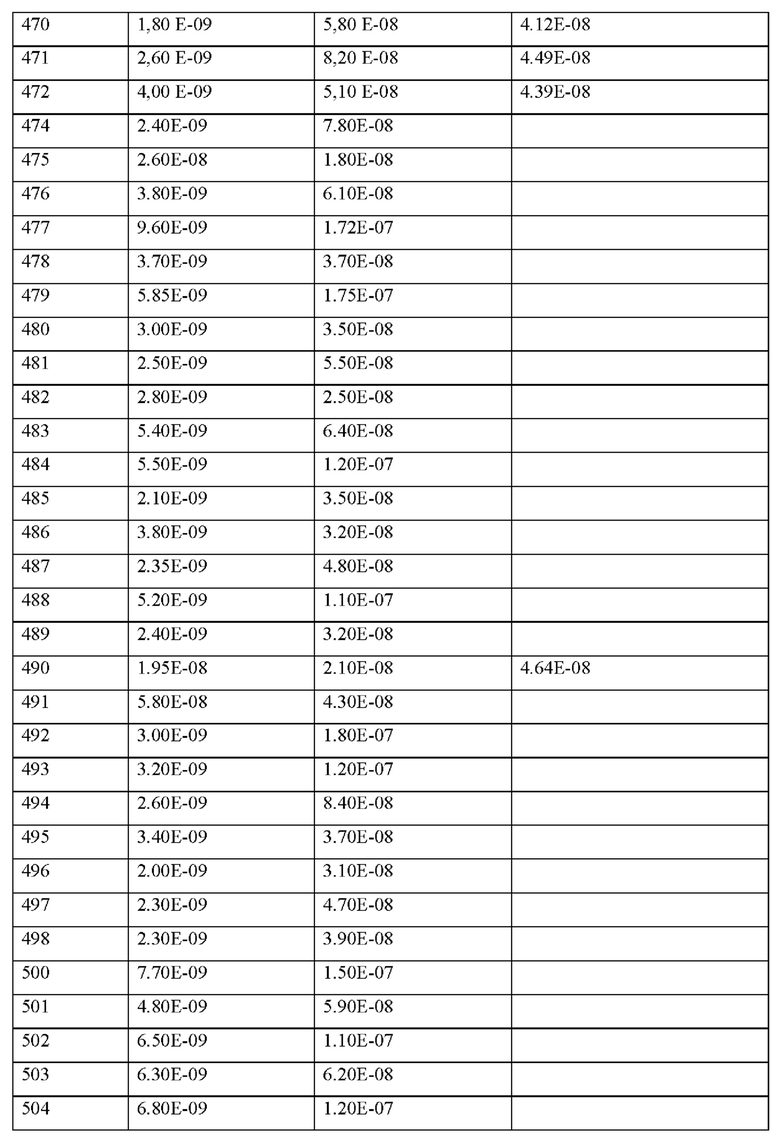
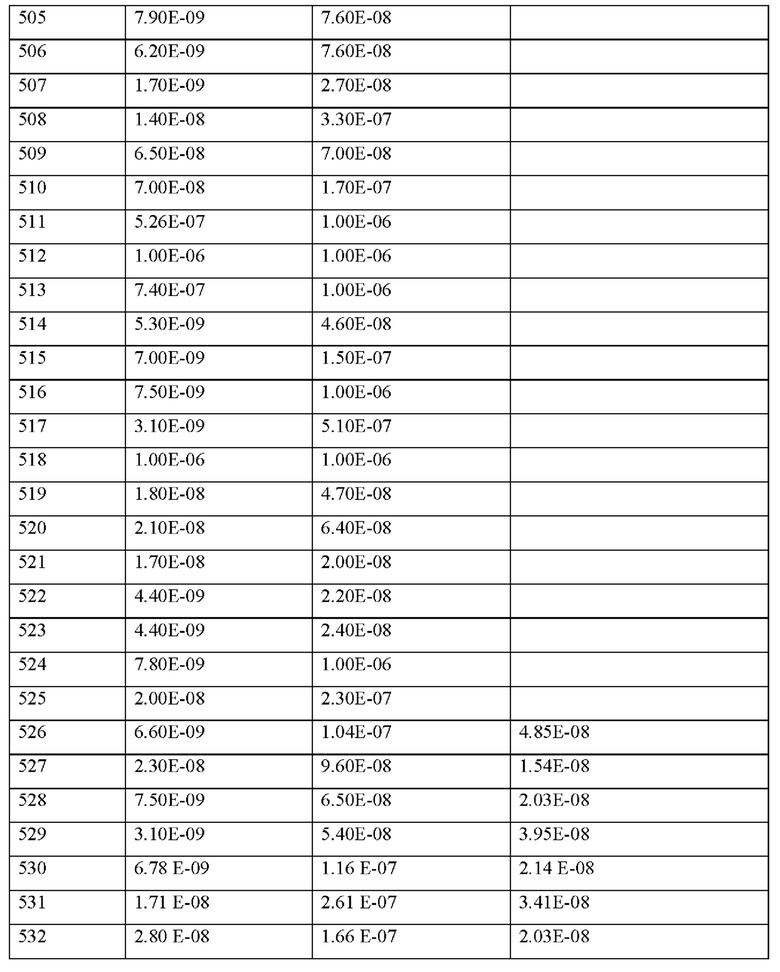


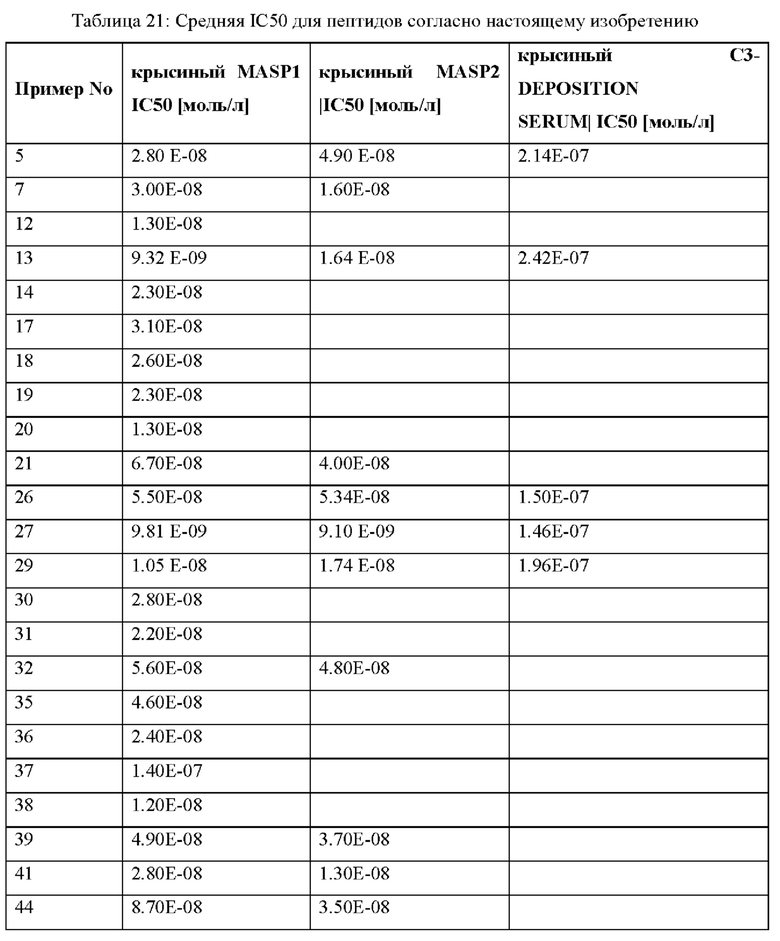
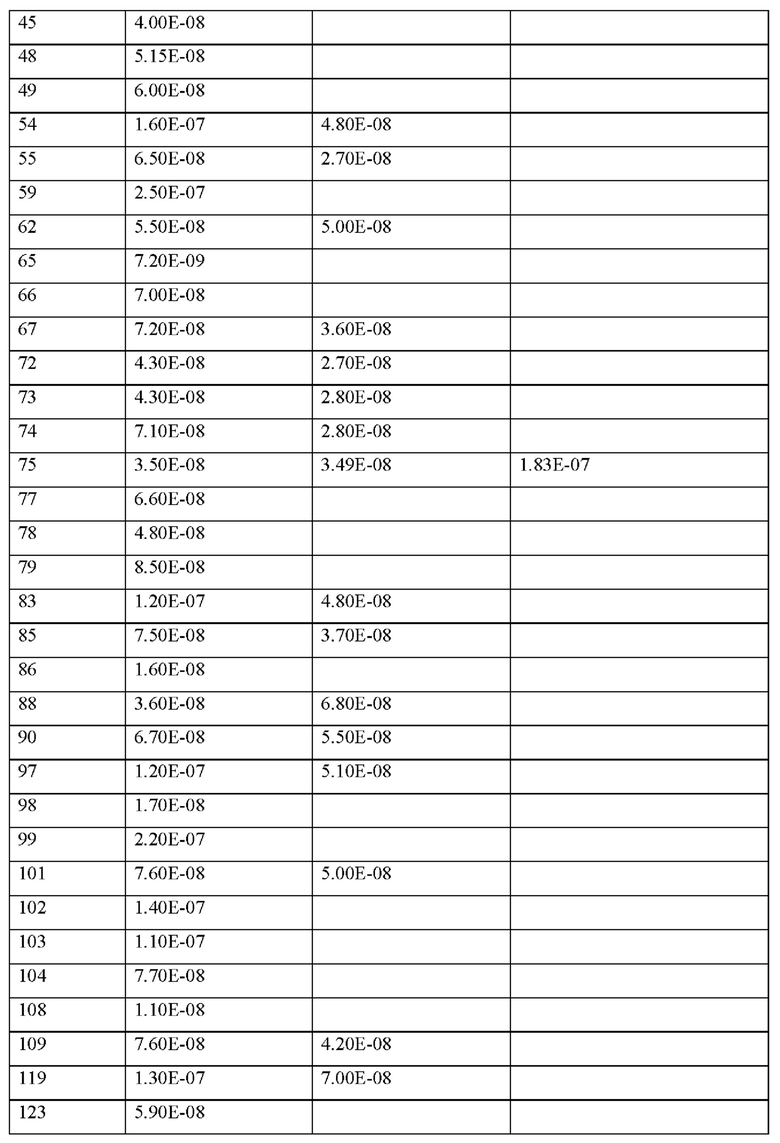
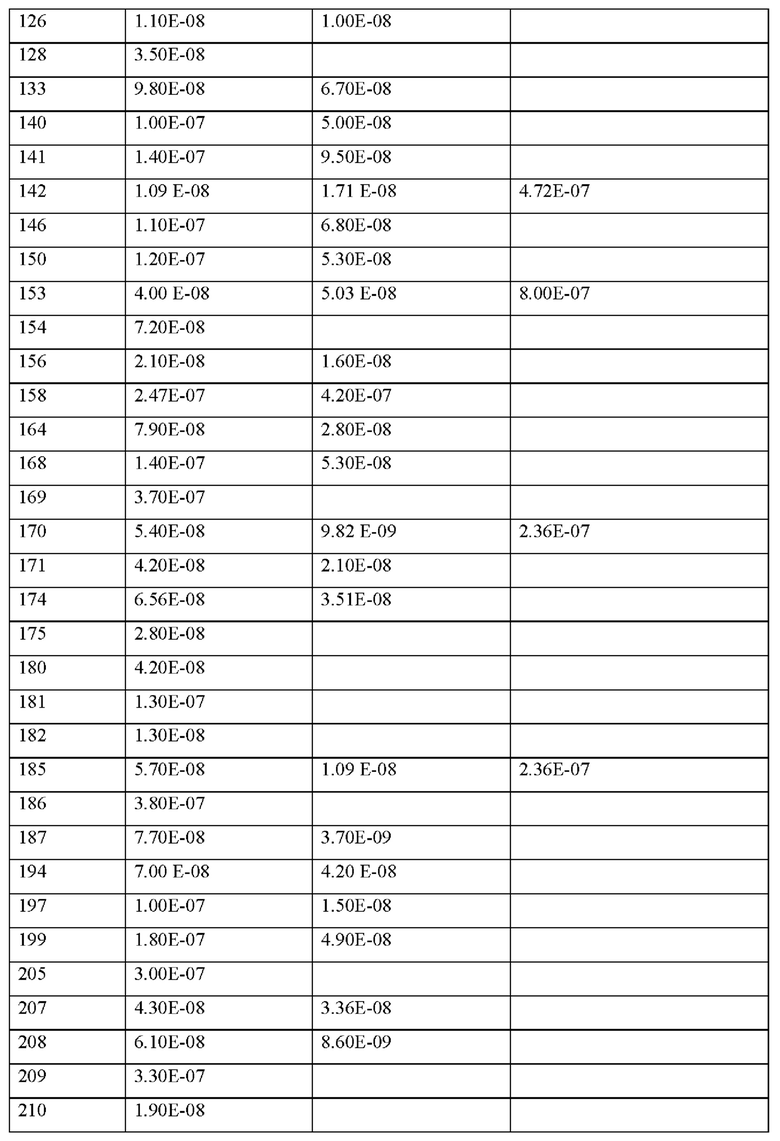

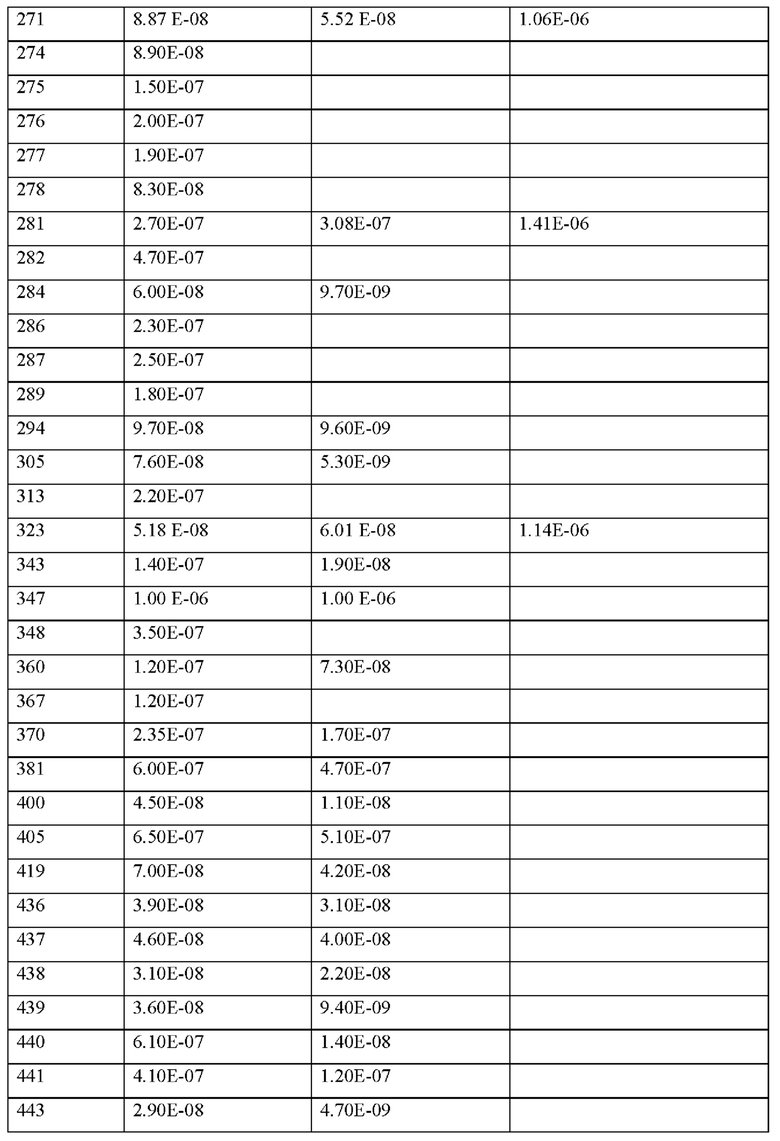
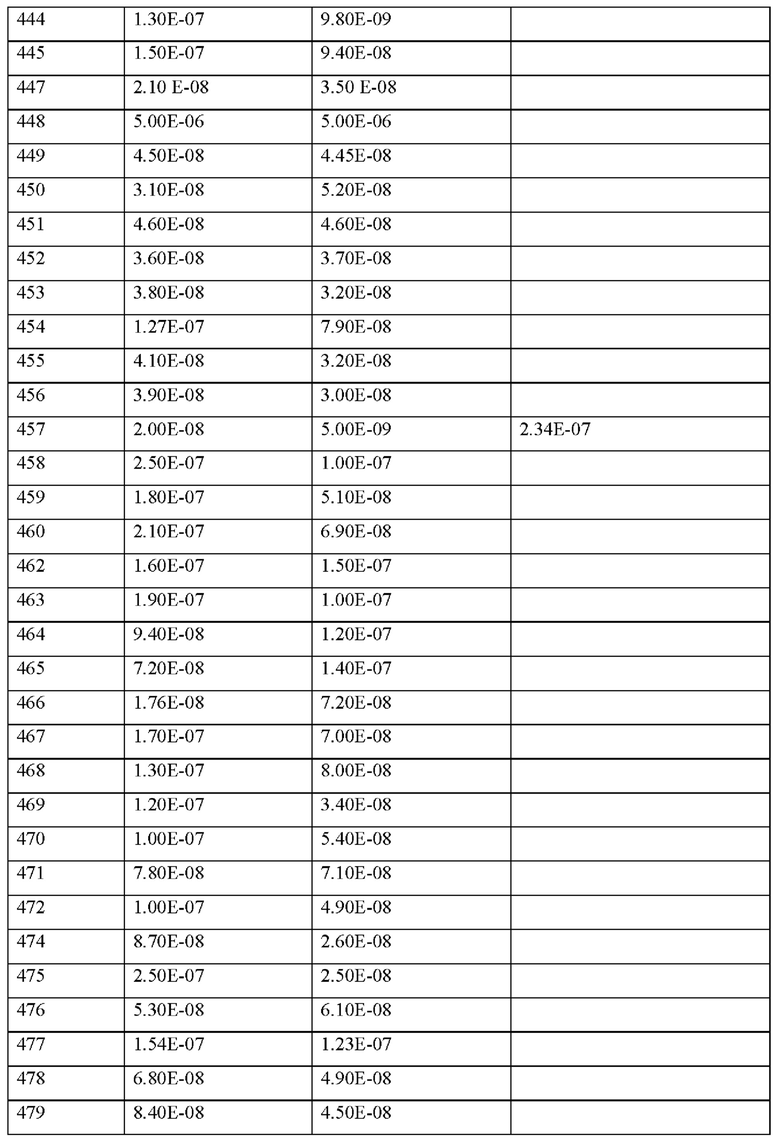
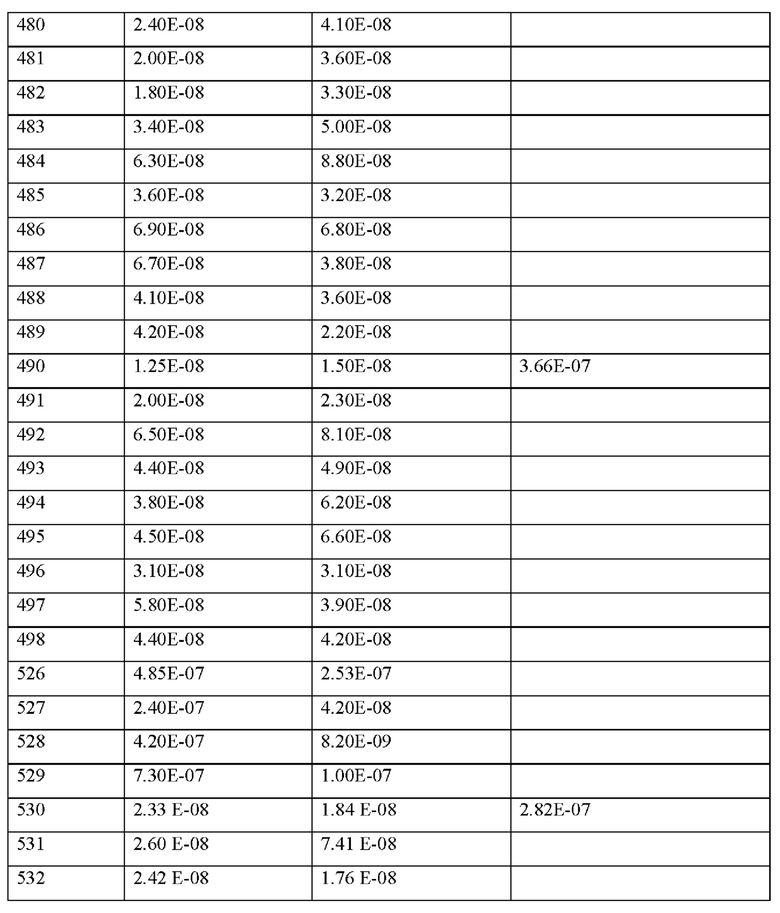
4. Реперфузионное повреждение почек (IRI) у крыс после односторонней нефрэктомии
Все процедуры соответствуют национальному законодательству (dt. Tierschutzgesetz) и директивам ЕС по использованию животных в научных целях и были одобрены ведомством по уходу за животными Bayer AG и компетентным региональным органом (LANUV Recklinghausen). Стандартный лабораторный рацион и водопроводная вода были доступны без ограничений. В типичном эксперименте количество используемых животных составляло от n=6-12. Животных случайным образом распределяли по экспериментальным группам. Реперфузионное повреждение почки (IRI) проводили у самцов крыс Вистар после односторонней нефрэктомии с предпочтительной массой тела в диапазоне от 250 до 350 г. Для односторонней нефрэктомии крыс содержали под наркозом при ингаляции 2% изофлурана в воздухе. Обезболивание осуществлялось подкожным введением 400 мкл/кг смеси 25% Кетавет и 8% Ромпун в 0,9 NaCl. Одностороннюю нефрэктомию проводят после выпячивания правой почки через небольшой разрез дорсо-латеральной брюшной стенки и перевязки ее ножки. После односторонней нефрэктомии разрез брюшной полости закрывали хирургическими швами послойно и животным давали возможность восстановиться в течение 7-8 дней перед IRI. IRI проводили под анестезией и обезболиванием, как описано выше. Остаток левой почки выпячивали через небольшой разрез брюшной стенки и сосуды ножки почки пережимали атравмагическим микрососудистым зажимом на 45 минут в типичной постановке. За это время почка вместе с зажимом in situ была перемещена в брюшную полость для обеспечения теплой ишемии. Через 45 мин зажим был открыт и снят, а разрез закрыт швами, как описано выше. Тестируемое соединение или носитель вводили внутривенно через полиэтиленовый катетер, помещенный перед операцией в яремную вену.
Соединения растворяли в соответствующем носителе и вводили либо профилактически перед IRI, либо терапевтически после завершения IRI. Типичный диапазон применяемых доз составлял 0,1-30 мг/кг внутривенно. Животным, которые служили в качестве контроля, вводили носитель без соединения. Имитация контрольных животных проводилась посредством всей процедуры, описанной выше, без закрытия зажима для индукции ишемии.
Образцы крови брали под наркозом на день 1 и 8 после IRI. В типичных условиях животных умерщвляли через 8 дней после IRI, а почки брали и замораживали в жидком азоте. В других типичных условиях животных умерщвляли через 1 день после IRI.
Типичными лабораторными параметрами, измеряемыми в образцах плазмы для оценки функции почек, были креатинин и мочевина. Для определения клиренса креатинина животных содержали в метаболических клетках и собирали мочу не менее 16 часов. После определения объемного потока мочи (VU) и определения концентрации креатинина в моче и плазме ([Crea]U и [Crea]Pl, соответственно), клиренс криатинина (ClCrea) вычисляли согласно стандартной формуле: ClCrea=VU*[Crea]U/[Crea]Pl
Экстракция РНК и количественная полимеразная цепная реакция в реальном времени: полную РНК экстрагировали из образцов тканей с помощью триазольного способа. Целостность полученной РНК проверяли на биоанализаторе (Agilent). Для обратной транскрипции 1 мкг полной РНК сначала расщепляли ДНКазой I, свободной от РНКаз (Gibco), в течение 15 мин при температуре окружающей среды, затем обратно транскрибировали с помощью Promiscript (Promega) в общем реакционном объеме 40 мкл по стандартному протоколу поставщика набора. После инактивации фермента нагреванием в течение 15 мин до 65°С полученную кДНК разбавляли до конечного объема 150 мкл с помощью бидест. воды и 4 мкл отбирали на реакцию ПЦР. ПЦР в реальном времени, включая нормализацию необработанных данных по цитозольному бета-актину в качестве гена домашнего хозяйства, проводили, как описано (Ellinghaus et al., 2005). Полученное выражение приведено в условных единицах. Последовательности используемых олигонуклеотидных праймеров и зондов приведены в Таблице 1.
5. Реперфузионное повреждение почек (IRI) у свиней после окклюзии аортального баллона
Все процедуры соответствуют национальному законодательству (dt. Tierschutzgesetz) и директивам ЕС по использованию животных в научных целях и были одобрены ведомством по уходу за животными Bayer AG и компетентным региональным органом (LANUV Recklinghausen). В опытах использовали самок геттингенских мини-свиней (Ellegaard, Дания) с живой массой от 12 до 16 кг. Животные были случайным образом распределены по экспериментальным группам.
Применялись малоинвазивные способы с модификациями, как описано (Simon et al., Effects of intravenous sulfide during porcine aortic occlusion-induced kidney ischemia/reperfusion injury. Shock. 2011; 35: 156-163; Matejkova et al., Carbamylated erythropoietin-FC fusion protein and recombinant human erythropoietin during porcine kidney ischemia/reperfusion injury. Intensive Care Med. 2011; 39: 497-510). Кратко: Свиней анестезировали путем непрерывного внутривенного введения Кетавет®, Дормикум® и Панкурониум® после премедикации внутримышечной инъекцией Кетавет®/Стреснил®. После интратрахеальной интубации животным проводилась искусственная вентиляция легких с помощью детского респиратора (Avance CS2, GE Healthcare) кислородно-воздушной смесью в дыхательном объеме от 6 до 8 мл/кг при постоянном положительном давлении в конце выдоха (ПДКВ) 3-4 см вод. ст. и частотой от 13 до 20 мин-1. Вентиляцию регулировали так, чтобы поддерживать артериальное PaCO2 на исходном уровне около 40 мм рт.ст. В правую яремную вену устанавливали катетер для введения лекарств и жидкости. Раствор Рингера-лактата вводили внутривенно с постоянной скоростью 10 мл/кг/ч. Животным вводили гепарин при дозе 50 МЕ/кг внутривенно После установки необходимых зондов и катетеров, подогнанных к соответствующим датчикам давления, и записывающего оборудования измеряли следующие сердечно-сосудистые и респираторные параметры: центральное венозное давление (через левую яремную вену), артериальное кровяное давление и частота сердечных сокращений (BP и HR; через левую сонную артерию) и сердечный выброс (СО) и системное сосудистое сопротивление (SVR) с помощью системы PiCCO® (Pulsion, Германия), подключенной к термодилюциоиному катетеру Pulsion 4F (PV2014L08N), помещенному в правую сонную артерию. Катетеры для измерения CVP, BP и HR устанавливали на регистрирующую систему Ponemah через датчики Combitrans (Braun, REF 5203660). Окклюзионный катетер Fogarty (8F/14F, Edwards Lifesiences, REF 6208014F) вводили в брюшную аорту через левую бедренную артерию таким образом, чтобы наконечник с баллоном располагался выше по ходу потока от почечных артерий. Катетер вводили в мочевой пузырь через небольшой разрез в брюшной полости и непрерывно собирали мочу. Через равные промежутки времени отбирали образцы артериальной крови, в которых определяли концентрацию креатинина, мочевины, печеночных ферментов, клеток крови и соединений. Артериальные рО2, рСО2 и рН определяли на анализаторе газов крови Stat Profile® PRIME® (Nova Biomedical) через равные промежутки времени в образцах артериальной крови. Перфузию почек оценивали через регулярные промежутки времени с помощью ультразвуковой допплерографии с определением резистивного индекса на ветеринарном ультразвуковом аппарате LOGIQ е (General Electrics), оснащенном широкоспектральным конвексным датчиком от 2,0 до 5,0 МГц (C1-5-RS, REF 5384874). Почечный резистивный индекс (RRI) является подходящим параметром для оценки тяжести острого повреждения почек у пациентов (Darmon et al., Diagnostic accuracy of Doppler renal resistive index for reversibility of acute kidney injury in critically ill patients. Intensive Care Med. 2011; 37 (1): 68-76)
Когда сердечно-сосудистые параметры показывали стабильный базовый уровень (что обычно имело место через 60 минут после операции), начинали регистрацию и собирали образцы для исходных параметров. HR и МАВР измеряли непрерывно и для записи усредняли за 2-минутные интервалы. По окончании эксперимента свиней умерщвляли обескровливанием.
Повреждение почек было вызвано раздуванием баллона баллонного катетера Фогарти физиологическим раствором, что немедленно прерывало кровоток к почкам и органам брюшной полости и приводило к резкому увеличению аортального кровяного давления выше по ходу потока от баллона. Остановка кровотока дополнительно подтверждалась ультразвуковой допплерографией сосудов почек. В типичных экспериментах аорту окклюзируют на 90-120 мин до реперфузии путем сдувания баллона. После реперфузии скорость инфузии Рингера-лактата удваивают до 20 мл/кг/ч для стабилизации артериального давления и обеспечения диуреза. Все параметры контролировались в течение 6 часов после реперфузии.
Соединения растворяли в соответствующем носителе и вводили либо профилактически перед IRI, либо терапевтически после завершения IRI. Типичный диапазон применяемых доз составлял 0,1-10 мг/кг внутривенно. В типичном эксперименте тестировали до трех доз у 6 животных в каждой дозовой группе. Носитель без соединения вводили животным, которые служили контролем. Имитация контрольных животных проводилась по всей процедуре, описанной выше, без индукции ишемии.
В качестве показателя функции почек после реперфузии использовали наблюдаемые, но не исключительно, изменения диуреза, креатинина сыворотки, калия сыворотки, бикарбоната сыворотки и резистивного индекса, определяемые с помощью ультразвуковой допплерографии.



Ссылочные источники
• Caliceti P., Veronese F.M., Adv. DrugDeliv. Rev. 2003, 55, 1261-1277
• Darmon et al., Diagnostic accuracy of Doppler renal resistive index for reversibility of acute kidney injury in critically ill patients. Intensive Care Med. 2011; 37 (1): 68-76
• Dong-Liang Huang, Jing-Si Bai, Meng Wu, Xia Wang, Bernd Riedl, Elisabeth Pook, Carsten Alt, Marion Erny, Yi-Ming Li, Donald Bierer, Jing Shi, Ge-Min Fang Chem. Commun, 2019, 55, 2821-2824
• Dunkelberger and Song, Complement and its role in innate and adaptive immune responses. Cell Res. 2010; 20 (1): 34-50
• Ellinghaus et al., J Thorac Cardiovasc Surg. 2005 Jun; 129 (6): 1383-90
• Encyclopaedia of Pharmaceutical Technology", 3rd edition, James Swarbrick (Ed.), pp. 3177-3187
• Farrar et al., Collectin-11 detects stress-induced L-fucose pattern to trigger renal epithelial injury. J Clin Invest. 2016; 126 (5): 1911-1925
• Garred et al., A journey through the lectin pathway of complement-MBL and beyond. Immunol Rev. 2016; 274 (1): 74-97
• Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (Wiley- VCH, 2002)
• Héja et al, Revised mechanism of complement lectin-pathway activation revealing the role of serine protease MASP-1 as the exclusive activator of MASP-2. Proc Natl Acad Sci USA. 2012; 109 (26): 10498-503
• Héja et al., Monospecific inhibitors show that both mannan-binding lectin-associated serine protease-1 (MASP-1) and are essential for lectin pathway activation and reveal structural plasticity of MASP-2. J Biol Chem. 2012; 287 (24):20290-300
• Hong-Kui Cui, Ye Guo, Yao He, Feng-Liang Wang, Hao-Nan Chang, Yu-Jia Wang, Fang-Ming Wu, Chang-Lin Tian, Lei Liu Angew. Chem. Int. Ed. 2013, 52, 9558-9562
• http://www.researchdisclosure.com/searching-disclosures, Research Disclosure Database Number 605005, 2014, 01 Aug 2014
• Jan-Patrick Fischer, Ria Schonauer, Sylvia Els-Heindl, Donald Bierer, Johannes Koebberling, Bernd Riedl, Annette G. Beck-Sickinger J Pep Sci. 2019; e3147
• Kocsis et al., Selective inhibition of the lectin pathway of complement with phage display selected peptides against mannose-binding lectin-associated serine protease (MASP)-1 and -2: significant contribution of MASP-1 to lectin pathway activation. J Immunol. 2010; 185 (7): 4169-78
• Kourra CMBK and Cramer N; Chem. Sci., 2016, 7, 7007-7012
• Matejkova et al., Carbamylated erythropoietin-FC fusion protein and recombinant human erythropoietin during porcine kidney ischemia/reperfusion injury. Intensive Care Med. 2011; 39: 497-510
• Møller-Kristensen et al., Mannan-binding lectin recognizes structures on ischaemic reperfused mouse kidneys and is implicated in tissue injury. Scand J Immunol. 2005; 61 (5): 426-34
• Nomenclature of α-Amino Acids (Recommendations, 1974), Biochemistry, 14 (2), (1975)
• Remington's Pharmaceutical Sciences, 17th edition, Alfonso R. Gennaro (Ed.), Mark Publishing Company, Easton, PA, USA, 1985
• Schwaeble et al., Targeting of mannan-binding lectin-associated serine protease-2 confers protection from myocardial and gastrointestinal ischemia/reperfusion injury. Proc Natl Acad Sci USA. 2011; 108 (18): 7523-8
• Sebesta and Seebach, Helv. Chim. Acta 2003, 86, 4061-4072
• Shuai-Shuai Sun, Junyou Chen, Rui Zhao, Donald Bierer, Jun Wang, Ge-Min Fang, Yi-Ming Li Tetrahedron Letters 2019, 60, 1197-1201
• Sieve et al., Regulation and function of endothelial glycocalyx layer in vascular diseases. Vascul Pharmacol. 2018; 100: 26-33
• Simon et al., Effects of intravenous sulfide during porcine aortic occlusion-induced kidney ischemia/reperfusion injury. Shock. 2011; 35: 156-163
• T. Peleg-Shulman et al., J. Med.Chem., 2004, 47, 4897-4904
• Tao Wang, Jian Fan, Xiao-Xu Chen, Rui Zhao, Yang Xu, Donald Bierer, Lei Liu, Yi-Ming Li, Jing Shi, Ge-Min Fang Org. Lett. 2018, 20, 6074-6078
• Tao Wang, Yi-Fu Kong, Yang Xu, Jian Fan, Hua-Jian Xu, Donald Bierer, Jun Wang, Jing Shi, Yi-Ming Li Tetrahedron Letters 2017, 58, 3970-3973;
• Yang Xu, Tao Wang, Chao-Jian Guan, Yi-Ming Li, Lei Liu, Jing Shi, Donald Bierer Tetrahedron Letters 2017, 58, 1677 1680
• Ye Guo, De-Meng Sun, Feng-Liang Wang, Yao He, Lei Liu, Chang-Lin Tian Angew. Chem. Int. Ed. 2015, 54, 14276-14281.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> БАЙЕР АГ
BayБАЙЕР ФАРМА АГ
<120> СОЕДИНЕНИЯ, ИНГИБИРУЮЩИЕ MASP, И ИХ ПРИМЕНЕНИЯ
<130> BHC171044WO
<160> 10
<170> PatentIn version 3.5
<210> 1
<211> 14
<212> PRT
<213> Искусственная последовательность
<220>
<223> SFMI-1
<220>
<221> ДИСУЛЬФИД
<222> (3)..(11)
<223> дисульфидная связь;
<220>
<221> ДИСУЛЬФИД
<222> (3)..(11)
<223> дисульфидная связь
<400> 1
Gly Ile Cys Ser Arg Ser Leu Pro Pro Ile Cys Ile Pro Asp
1 5 10
<210> 2
<211> 430
<212> PRT
<213> Искусственная последовательность
<220>
<223> MASP-1_Human_Sequence fragment_C-terminal His Tag and N-terminal
Ig-kappa secretion signal
<400> 2
Met Glu Thr Asp Thr Leu Leu Leu Trp Val Leu Leu Leu Trp Val Pro
1 5 10 15
Gly Ser Thr Gly Asp Ala Gly Asn Glu Cys Pro Glu Leu Gln Pro Pro
20 25 30
Val His Gly Lys Ile Glu Pro Ser Gln Ala Lys Tyr Phe Phe Lys Asp
35 40 45
Gln Val Leu Val Ser Cys Asp Thr Gly Tyr Lys Val Leu Lys Asp Asn
50 55 60
Val Glu Met Asp Thr Phe Gln Ile Glu Cys Leu Lys Asp Gly Thr Trp
65 70 75 80
Ser Asn Lys Ile Pro Thr Cys Lys Ile Val Asp Cys Arg Ala Pro Gly
85 90 95
Glu Leu Glu His Gly Leu Ile Thr Phe Ser Thr Arg Asn Asn Leu Thr
100 105 110
Thr Tyr Lys Ser Glu Ile Lys Tyr Ser Cys Gln Glu Pro Tyr Tyr Lys
115 120 125
Met Leu Asn Asn Asn Thr Gly Ile Tyr Thr Cys Ser Ala Gln Gly Val
130 135 140
Trp Met Asn Lys Val Leu Gly Arg Ser Leu Pro Thr Cys Leu Pro Val
145 150 155 160
Cys Gly Leu Pro Lys Phe Ser Arg Lys Leu Met Ala Arg Ile Phe Asn
165 170 175
Gly Arg Pro Ala Gln Lys Gly Thr Thr Pro Trp Ile Ala Met Leu Ser
180 185 190
His Leu Asn Gly Gln Pro Phe Cys Gly Gly Ser Leu Leu Gly Ser Ser
195 200 205
Trp Ile Val Thr Ala Ala His Cys Leu His Gln Ser Leu Asp Pro Glu
210 215 220
Asp Pro Thr Leu Arg Asp Ser Asp Leu Leu Ser Pro Ser Asp Phe Lys
225 230 235 240
Ile Ile Leu Gly Lys His Trp Arg Leu Arg Ser Asp Glu Asn Glu Gln
245 250 255
His Leu Gly Val Lys His Thr Thr Leu His Pro Gln Tyr Asp Pro Asn
260 265 270
Thr Phe Glu Asn Asp Val Ala Leu Val Glu Leu Leu Glu Ser Pro Val
275 280 285
Leu Asn Ala Phe Val Met Pro Ile Cys Leu Pro Glu Gly Pro Gln Gln
290 295 300
Glu Gly Ala Met Val Ile Val Ser Gly Trp Gly Lys Gln Phe Leu Gln
305 310 315 320
Arg Phe Pro Glu Thr Leu Met Glu Ile Glu Ile Pro Ile Val Asp His
325 330 335
Ser Thr Cys Gln Lys Ala Tyr Ala Pro Leu Lys Lys Lys Val Thr Arg
340 345 350
Asp Met Ile Cys Ala Gly Glu Lys Glu Gly Gly Lys Asp Ala Cys Ala
355 360 365
Gly Asp Ser Gly Gly Pro Met Val Thr Leu Asn Arg Glu Arg Gly Gln
370 375 380
Trp Tyr Leu Val Gly Thr Val Ser Trp Gly Asp Asp Cys Gly Lys Lys
385 390 395 400
Asp Arg Tyr Gly Val Tyr Ser Tyr Ile His His Asn Lys Asp Trp Ile
405 410 415
Gln Arg Val Thr Gly Val Arg Asn His His His His His His
420 425 430
<210> 3
<211> 417
<212> PRT
<213> Искусственная последовательность
<220>
<223> MASP-2_Human_Sequence fragment_C-terminal His Tag and N-terminal
Ig-kappa secretion signal
<400> 3
Met Glu Thr Asp Thr Leu Leu Leu Trp Val Leu Leu Leu Trp Val Pro
1 5 10 15
Gly Ser Thr Gly Asp Ala Gln Pro Cys Pro Tyr Pro Met Ala Pro Pro
20 25 30
Asn Gly His Val Ser Pro Val Gln Ala Lys Tyr Ile Leu Lys Asp Ser
35 40 45
Phe Ser Ile Phe Cys Glu Thr Gly Tyr Glu Leu Leu Gln Gly His Leu
50 55 60
Pro Leu Lys Ser Phe Thr Ala Val Cys Gln Lys Asp Gly Ser Trp Asp
65 70 75 80
Arg Pro Met Pro Ala Cys Ser Ile Val Asp Cys Gly Pro Pro Asp Asp
85 90 95
Leu Pro Ser Gly Arg Val Glu Tyr Ile Thr Gly Pro Gly Val Thr Thr
100 105 110
Tyr Lys Ala Val Ile Gln Tyr Ser Cys Glu Glu Thr Phe Tyr Thr Met
115 120 125
Lys Val Asn Asp Gly Lys Tyr Val Cys Glu Ala Asp Gly Phe Trp Thr
130 135 140
Ser Ser Lys Gly Glu Lys Ser Leu Pro Val Cys Glu Pro Val Cys Gly
145 150 155 160
Leu Ser Ala Arg Thr Thr Gly Gly Arg Ile Tyr Gly Gly Gln Lys Ala
165 170 175
Lys Pro Gly Asp Phe Pro Trp Gln Val Leu Ile Leu Gly Gly Thr Thr
180 185 190
Ala Ala Gly Ala Leu Leu Tyr Asp Asn Trp Val Leu Thr Ala Ala His
195 200 205
Ala Val Tyr Glu Gln Lys His Asp Ala Ser Ala Leu Asp Ile Arg Met
210 215 220
Gly Thr Leu Lys Arg Leu Ser Pro His Tyr Thr Gln Ala Trp Ser Glu
225 230 235 240
Ala Val Phe Ile His Glu Gly Tyr Thr His Asp Ala Gly Phe Asp Asn
245 250 255
Asp Ile Ala Leu Ile Lys Leu Asn Asn Lys Val Val Ile Asn Ser Asn
260 265 270
Ile Thr Pro Ile Cys Leu Pro Arg Lys Glu Ala Glu Ser Phe Met Arg
275 280 285
Thr Asp Asp Ile Gly Thr Ala Ser Gly Trp Gly Leu Thr Gln Arg Gly
290 295 300
Phe Leu Ala Arg Asn Leu Met Tyr Val Asp Ile Pro Ile Val Asp His
305 310 315 320
Gln Lys Cys Thr Ala Ala Tyr Glu Lys Pro Pro Tyr Pro Arg Gly Ser
325 330 335
Val Thr Ala Asn Met Leu Cys Ala Gly Leu Glu Ser Gly Gly Lys Asp
340 345 350
Ser Cys Arg Gly Asp Ser Gly Gly Ala Leu Val Phe Leu Asp Ser Glu
355 360 365
Thr Glu Arg Trp Phe Val Gly Gly Ile Val Ser Trp Gly Ser Met Asn
370 375 380
Cys Gly Glu Ala Gly Gln Tyr Gly Val Tyr Thr Lys Val Ile Asn Tyr
385 390 395 400
Ile Pro Trp Ile Glu Asn Ile Ile Ser Asp Phe His His His His His
405 410 415
His
<210> 4
<211> 430
<212> PRT
<213> Искусственная последовательность
<220>
<223> MASP-1_Rat_Sequence fragment_C-terminal His Tag and N-terminal
Ig-kappa secretion signal
<400> 4
Met Glu Thr Asp Thr Leu Leu Leu Trp Val Leu Leu Leu Trp Val Pro
1 5 10 15
Gly Ser Thr Gly Asp Ala Gly Asn Glu Cys Pro Lys Leu Gln Pro Pro
20 25 30
Val Tyr Gly Lys Ile Glu Pro Ser Gln Ala Val Tyr Ser Phe Lys Asp
35 40 45
Gln Val Leu Ile Ser Cys Asp Thr Gly Tyr Lys Val Leu Lys Asp Asn
50 55 60
Glu Val Met Asp Thr Phe Gln Ile Glu Cys Leu Lys Asp Gly Ala Trp
65 70 75 80
Ser Asn Lys Ile Pro Thr Cys Lys Ile Val Asp Cys Gly Val Pro Ala
85 90 95
Val Leu Lys His Gly Leu Val Thr Phe Ser Thr Arg Asn Asn Leu Thr
100 105 110
Thr Tyr Lys Ser Glu Ile Arg Tyr Ser Cys Gln Gln Pro Tyr Tyr Lys
115 120 125
Met Leu His Asn Thr Thr Gly Val Tyr Thr Cys Ser Ala His Gly Thr
130 135 140
Trp Thr Asn Glu Val Leu Lys Arg Ser Leu Pro Thr Cys Leu Pro Val
145 150 155 160
Cys Gly Leu Pro Lys Phe Ser Arg Lys His Ile Ser Arg Ile Phe Asn
165 170 175
Gly Arg Pro Ala Gln Lys Gly Thr Thr Pro Trp Ile Ala Met Leu Ser
180 185 190
Gln Leu Asn Gly Gln Pro Phe Cys Gly Gly Ser Leu Leu Gly Ser Asn
195 200 205
Trp Val Leu Thr Ala Ala His Cys Leu His His Pro Leu Asp Pro Glu
210 215 220
Glu Pro Ile Leu His Asn Ser His Leu Leu Ser Pro Ser Asp Phe Lys
225 230 235 240
Ile Ile Met Gly Lys His Trp Arg Arg Arg Ser Asp Glu Asp Glu Gln
245 250 255
His Leu His Val Lys His Ile Met Leu His Pro Leu Tyr Asn Pro Ser
260 265 270
Thr Phe Glu Asn Asp Leu Gly Leu Val Glu Leu Ser Glu Ser Pro Arg
275 280 285
Leu Asn Asp Phe Val Met Pro Val Cys Leu Pro Glu His Pro Ser Thr
290 295 300
Glu Gly Thr Met Val Ile Val Ser Gly Trp Gly Lys Gln Phe Leu Gln
305 310 315 320
Arg Leu Pro Glu Asn Leu Met Glu Ile Glu Ile Pro Ile Val Asn Tyr
325 330 335
His Thr Cys Gln Glu Ala Tyr Thr Pro Leu Gly Lys Lys Val Thr Gln
340 345 350
Asp Met Ile Cys Ala Gly Glu Lys Glu Gly Gly Lys Asp Ala Cys Ala
355 360 365
Gly Asp Ser Gly Gly Pro Met Val Thr Lys Asp Ala Glu Arg Asp Gln
370 375 380
Trp Tyr Leu Val Gly Val Val Ser Trp Gly Glu Asp Cys Gly Lys Lys
385 390 395 400
Asp Arg Tyr Gly Val Tyr Ser Tyr Ile Tyr Pro Asn Lys Asp Trp Ile
405 410 415
Gln Arg Val Thr Gly Val Arg Asn His His His His His His
420 425 430
<210> 5
<211> 417
<212> PRT
<213> Искусственная последовательность
<220>
<223> MASP-2_Rat_Sequence fragment_C-terminal His Tag and N-terminal
Ig-kappa secretion signal
<400> 5
Met Glu Thr Asp Thr Leu Leu Leu Trp Val Leu Leu Leu Trp Val Pro
1 5 10 15
Gly Ser Thr Gly Asp Thr Ala Gln Pro Cys Pro Asp Pro Thr Ala Pro
20 25 30
Pro Asn Gly His Ile Ser Pro Val Gln Ala Thr Tyr Val Leu Lys Asp
35 40 45
Ser Phe Ser Val Phe Cys Lys Thr Gly Phe Glu Leu Leu Gln Gly Ser
50 55 60
Val Pro Leu Lys Ser Phe Thr Ala Val Cys Gln Lys Asp Gly Ser Trp
65 70 75 80
Asp Arg Pro Ile Pro Glu Cys Ser Ile Ile Asp Cys Gly Pro Pro Asp
85 90 95
Asp Leu Pro Asn Gly His Val Asp Tyr Ile Thr Gly Pro Glu Val Thr
100 105 110
Thr Tyr Lys Ala Val Ile Gln Tyr Ser Cys Glu Glu Thr Phe Tyr Thr
115 120 125
Met Ser Ser Asn Gly Lys Tyr Val Cys Glu Ala Asp Gly Phe Trp Thr
130 135 140
Ser Ser Lys Gly Glu Lys Ser Leu Pro Val Cys Lys Pro Val Cys Gly
145 150 155 160
Leu Ser Thr His Thr Ser Gly Gly Arg Ile Ile Gly Gly Gln Pro Ala
165 170 175
Lys Pro Gly Asp Phe Pro Trp Gln Val Leu Leu Leu Gly Glu Thr Thr
180 185 190
Ala Ala Gly Ala Leu Ile His Asp Asp Trp Val Leu Thr Ala Ala His
195 200 205
Ala Val Tyr Gly Lys Thr Glu Ala Met Ser Ser Leu Asp Ile Arg Met
210 215 220
Gly Ile Leu Lys Arg Leu Ser Leu Ile Tyr Thr Gln Ala Trp Pro Glu
225 230 235 240
Ala Val Phe Ile His Glu Gly Tyr Thr His Gly Ala Gly Phe Asp Asn
245 250 255
Asp Ile Ala Leu Ile Lys Leu Lys Asn Lys Val Thr Ile Asn Arg Asn
260 265 270
Ile Met Pro Ile Cys Leu Pro Arg Lys Glu Ala Ala Ser Leu Met Lys
275 280 285
Thr Asp Phe Val Gly Thr Val Ala Gly Trp Gly Leu Thr Gln Lys Gly
290 295 300
Phe Leu Ala Arg Asn Leu Met Phe Val Asp Ile Pro Ile Val Asp His
305 310 315 320
Gln Lys Cys Ala Thr Ala Tyr Thr Lys Gln Pro Tyr Pro Gly Ala Lys
325 330 335
Val Thr Val Asn Met Leu Cys Ala Gly Leu Asp Arg Gly Gly Lys Asp
340 345 350
Ser Cys Arg Gly Asp Ser Gly Gly Ala Leu Val Phe Leu Asp Asn Glu
355 360 365
Thr Gln Arg Trp Phe Val Gly Gly Ile Val Ser Trp Gly Ser Ile Asn
370 375 380
Cys Gly Gly Ser Glu Gln Tyr Gly Val Tyr Thr Lys Val Thr Asn Tyr
385 390 395 400
Ile Pro Trp Ile Glu Asn Ile Ile Asn Asn Phe His His His His His
405 410 415
His
<210> 6
<211> 11
<212> PRT
<213> Искусственная последовательность
<220>
<223> Fluorescence Resonance Energy Transfer substrate
<220>
<221> Xaa
<222> (1)..(1)
<223> Xaa is 2-aminobenzoyl
<220>
<221> MOD_RES
<222> (10)..(10)
<223> AMIDATION
<220>
<221> Xaa
<222> (11)..(11)
<223> Xaa is a non peptidic capping group (2,4-dinitrophenyl; bound to
lysine amino group on position 6)
<400> 6
Xaa Met Tyr Gly Gly Ala Arg Arg Leu Lys Xaa
1 5 10
<210> 7
<211> 13
<212> PRT
<213> Искусственная последовательность
<220>
<223> Fluorescence resonance energy transfer substrate
<220>
<221> Xaa
<222> (1)..(1)
<223> Xaa is 4-((4-(dimethylamino)phenyl)azo)benzoic acid
<220>
<221> MOD_RES
<222> (12)..(12)
<223> AMIDATION
<220>
<221> Xaa
<222> (13)..(13)
<223> Xaa is 5-[(2-Aminoethyl) amino]naphthalene-1-sulfonyl (bound to
Glutamic acid caboxylic acid group on position 5)
<400> 7
Xaa Lys Ile Ser Pro Gln Gly Tyr Gly Arg Arg Glu Xaa
1 5 10
<210> 8
<211> 11
<212> PRT
<213> Искусственная последовательность
<220>
<223> Fluorescence resonance energy transfer substrate
<220>
<221> Xaa
<222> (1)..(1)
<223> Xaa is 4-((4-(dimethylamino)phenyl)azo)benzoic acid
<220>
<221> MOD_RES
<222> (10)..(10)
<223> AMIDATION
<220>
<221> Xaa
<222> (11)..(11)
<223> Xaa is 5-[(2-Aminoethyl) amino]naphthalene-1-sulfonyl (bound to
Glutamic acid caboxylic acid group on position 5)
<400> 8
Xaa Met Tyr Gly Gly Ala Arg Arg Leu Glu Xaa
1 5 10
<210> 9
<211> 11
<212> PRT
<213> Искусственная последовательность
<220>
<223> Fluorescence resonance energy transfer substrate
<220>
<221> Xaa
<222> (1)..(1)
<223> Xaa is 2-aminobenzoyl
<220>
<221> MOD_RES
<222> (10)..(10)
<223> AMIDATION
<220>
<221> Xaa
<222> (11)..(11)
<223> Xaa is a non peptidic capping group (2,4-dinitrophenyl; bound to
lysine amino group on position 6)
<400> 9
Xaa Ile Glu Gly Arg Thr Ser Glu Asp Lys Xaa
1 5 10
<210> 10
<211> 14
<212> PRT
<213> Искусственная последовательность
<220>
<223> Ala-1 SFMI-1
<220>
<221> ДИСУЛЬФИД
<222> (3)..(11)
<220>
<221> ДИСУЛЬФИД
<222> (3)..(11)
<223> дисульфидная связь
<400> 10
Ala Ile Cys Ser Arg Ser Leu Pro Pro Ile Cys Ile Pro Asp
1 5 10
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТЫ АНТИТЕЛА И ЛЕКАРСТВЕННОГО СРЕДСТВА (ADC) И КОНЪЮГАТЫ АНТИТЕЛА И ПРОЛЕКАРСТВА (APDC), СОДЕРЖАЩИЕ ФЕРМЕНТАТИВНО РАСЩЕПЛЯЕМЫЕ ГРУППЫ | 2016 |
|
RU2751512C2 |
| ПРОЛЕКАРСТВА, СОДЕРЖАЩИЕ КОНЬЮГАТ ГИАЛУРОНОВОЙ КИСЛОТЫ, ЛИНКЕРА И ДВОЙНОГО АГОНИСТА GLP-1/ГЛЮКАГОНА | 2016 |
|
RU2719482C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ АНАЛОГОВ ОКСИТОЦИНА | 2015 |
|
RU2696276C2 |
| ПЕПТИДНЫЕ ВЕКТОРЫ | 2004 |
|
RU2361876C2 |
| АГОНИСТЫ ОКСИТОЦИНОВЫХ РЕЦЕПТОРОВ | 2010 |
|
RU2539692C2 |
| НОВЫЕ АНАЛОГИ ГЛЮКАГОН-ПОДОБНОГО ПЕПТИДА, КОМПОЗИЦИЯ И СПОСОБ ПРИМЕНЕНИЯ | 2010 |
|
RU2557301C2 |
| ПЕПТИД GLP-1 С ПРИСОЕДИНЕННОЙ ОЛИГОСАХАРИДНОЙ ЦЕПЬЮ | 2008 |
|
RU2539829C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДА ЭКСЕНАТИДА | 2011 |
|
RU2458066C1 |
| СПОСОБ ОТЩЕПЛЕНИЯ ПЕПТИДОВ, СВЯЗАННЫХ С ТВЕРДОЙ ФАЗОЙ, ОТ ТВЕРДОЙ ФАЗЫ | 2019 |
|
RU2771712C2 |
| СИНТЕЗ ЛИКСИСЕНАТИДА С КЭППИРОВАНИЕМ | 2019 |
|
RU2782772C2 |
Настоящее изобретение относится к области биотехнологии и может быть использовано в медицине. Раскрываются новые соединения, способные ингибировать ассоциированные со связывающим маннозу лектином (MBL) сериновые протеазы 1 и 2 (MASP1 и MASP2). Изобретение применимо для профилактики или при лечении различных патологий почек, в частности при острой почечной недостаточности. 7 н. и 2 з.п. ф-лы, 22 табл., 5 пр.
1. Соединение, обладающее свойствами ингибитора MASP-1 и ингибитора MASP-2, содержащее пептид следующей структуры
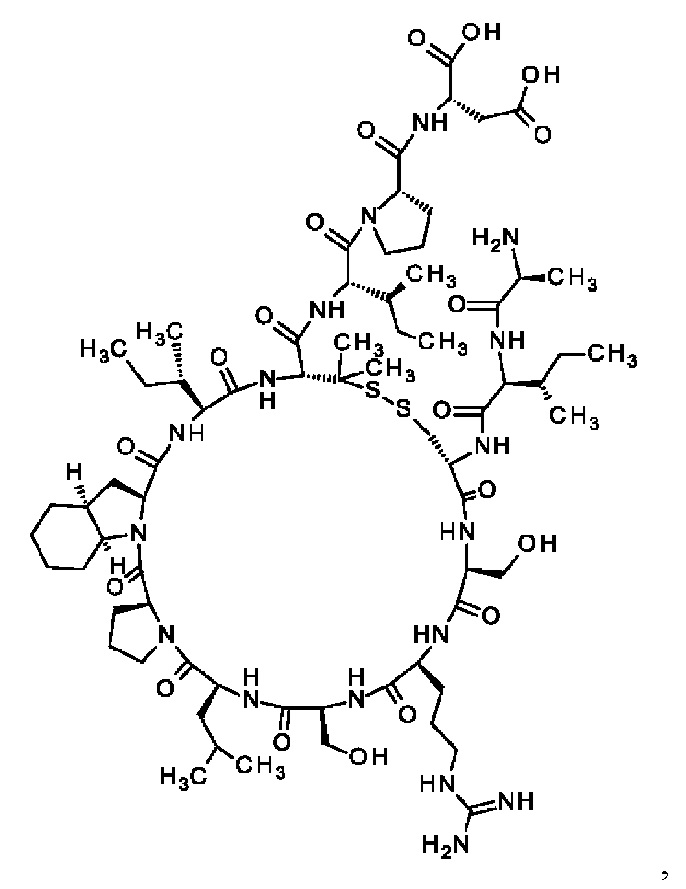
или его фармацевтически приемлемая соль, сольват или сольват соли.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, сольват или сольват соли, которое действует в качестве ингибитора MASP-1 и/или MASP-2 и/или которое ингибирует отложение С3.
3. Способ получения соединения, содержащего пептид, по любому из пп. 1-2 или его фармацевтически приемлемой соли, сольвата или сольвата соли с применением твердофазного пептидного синтеза.
4. Применение соединения, содержащего пептид, по любому из пп. 1-2 или его фармацевтически приемлемой соли, сольвата или сольвата соли для приготовления лекарственного средства, обладающего свойствами ингибитора MASP-1, ингибитора MASP-2 и/или ингибитора отложения С3.
5. Применение соединения, содержащего пептид, по любому из пп. 1-2 или его фармацевтически приемлемой соли, сольват или сольват соли для приготовления лекарственного средства для профилактики или лечения острого почечного повреждения.
6. Фармацевтическая композиция, обладающая свойствами ингибитора MASP-1 и ингибитора MASP-2, содержащая по меньшей мере одно соединение, содержащее пептид, по любому из пп. 1–2 или его фармацевтически приемлемую соль, сольват или сольват соли в комбинации с одним или несколькими инертными, нетоксичными, фармацевтически приемлемыми вспомогательными веществами.
7. Фармацевтическая композиция, обладающая свойствами ингибитора MASP-1 и ингибитора MASP-2, содержащая по меньшей мере одно соединение, содержащее пептид, по любому из пп. 1–2 или его фармацевтически приемлемую соль, сольват или сольват соли в комбинации с одним или несколькими дополнительными активными ингредиентами, выбранными из группы, состоящей из ингибиторов фосфодиэстераз, стимуляторов или активаторов гуанилатциклазы, агонистов IP-рецепторов, антагонистов минералокортикоидных рецепторов, диуретиков, агонистов PPAR-гамма, агонистов PPAR-дельта, кортикостероидов, активных ингредиентов, уменьшающих повреждение органов при окислительном стрессе, соединений, которые ингибируют индукцию пути клеточной гибели и апоптоза, соединений, которые ингибируют воспалительный ответ и пролиферацию Т-клеток, антитромботических средств, ингибиторов агрегации тромбоцитов, ингибиторов тромбина, антагонистов GPIIb/IIIa, ингибиторов фактора Ха, гепарина или низкомолекулярного производного гепарина (LMW) и ингибиторов фактора свертывания крови XI.
8. Фармацевтическая композиция по п. 6 или 7, причем свойства ингибитора MASP-1 и ингибитора MASP-2 придают композиции способность к профилактике или лечению острой почечной недостаточности.
9. Способ профилактики или лечения острой почечной недостаточности у людей и животных посредством введения эффективного количества по меньшей мере одного соединения, как определено в любом из пп. 1–2, или фармацевтической композиции по пп. 6-8.
| KOSTIS A | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| IMMUNOL., 2010, v | |||
| Способ укрепления под покрышкой пневматической шины предохранительного слоя или манжеты | 1917 |
|
SU185A1 |
| WO 2010136831 A1, 02.12.2010 | |||
| RU 2015101230 A1, 10.08.2016 | |||

