Изобретение относится к химии органических соединений, а именно к способу разделения на отдельные энантиомеры рацемического 1-карбамоилокси-2-гидрокси-3-(2-метоксифенокси)пропана. Изобретение может быть использовано в фармацевтической промышленности при получении нерацемических лекарственных препаратов.
Исследуемое соединение формулы (1) зарегистрировано в качестве субстанции лекарственного средства метокарбамол (международное название "Methocarbamol") в рацемическом виде и применяется в основном как скелетный мышечный релаксант и спазмолитик при заболеваниях и травмах опорно-двигательного аппарата [Negwer М., Scharnow H.-G. Organic-Chemical Drugs and Their Synonyms. Eighth Edition; Wiley-VCH: Weinheim, 2001, 2900; The Merck Index. 14th ed., Ed. M.J. O′Neil, Merck and Co., Inc., Whitehouse Station, NJ, USA; 2006, 5980].
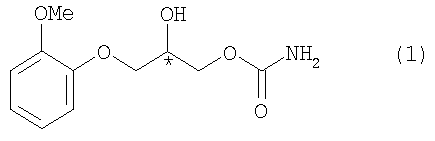
Недавние исследования показали, что две трети рецептов на мышечные релаксанты, выписываемых лицам, испытывающим боли в спине, приходится на метокарбамол (наряду с циклобензаприном и каризопродолом) [Luo X., Pietrobon R., Curtis L.H., Hey L.A. Spine. 2004, 29, E531-537]. В настоящее время метокарбамол используется в виде рацемата, хотя в экспериментах на мышах было показано, что активность рацемического и чистого (R)-1 энантиомера различны, при этом (R)-изомер проявляет большую физиологическую активность, чем рацемический или (S)-метокарбамол [Souri Е., Sharifzadeh М., Farsam Н., Gharavi N.J. Pharm. Pharmacol. 1999, 51, 853-855]. Поскольку энантиомеры метокарбамола обладают различной биологической активностью, целесообразно использование индивидуальных энантиомеров по отдельности. Кроме того, имея ввиду общую тенденцию к замещению рацемических лекарств на их энантиочистые аналоги [Murakami Н. From racemates to single enantiomers - chiral synthetic drugs over the last 20 years. Top. Curr. Chem. 2007, 269, 273-299], актуальна задача замены рацемической субстанции метокарбамола индивидуальным (R)-энантиомером.
Известны три общих способа получения нерацемических соединений: трансформация исходных энантиочистых соединений, энантиоселективный синтез, разделение рацемических веществ. Все существующие лабораторные и промышленные методы получения скалемических соединений относятся к одной из этих категорий или являются их комбинацией [Crosby J. Tetrahedron. 1991, 47, N 27, 4789-4846; Sheldon R.A. Chirotechnology: Industrial Synthesis of Optically Active Compounds. N.Y.: M. Deccer, 1993, 416 pp.]. Разделение рацемического вещества на энантиомеры методом вовлечения (resolution by entrainment), по-видимому, является самым эффективным из существующих способов разделения рацемических смесей [a) Collet A., Jacques J., Brienne М.J. Chem. Rev., 1980, 80, 215-230; б) Jacques J., Collet A., Wilen S.H. Enantiomers, Racemates, and Resolutions, Krieger Publishing Co. Malabar, FL., 1994, 217-368; в) Coquerel G. Preferential crystallization. Top. Curr. Chem. 2007, 269, 1-51]. Необходимой предпосылкой разделения рацемического вещества на энантиомеры методом вовлечения является кристаллизация его из раствора в виде рацемического конгломерата, т.е. механической смеси отдельных кристаллов, каждый из которых образован молекулами одного энантиомера. Методом вовлечения, например, проведено разделение D,L-треонина [Amiard G. Bull. Soc. Chim. France, 1956, 447], гидрохлорида D,L-валина [U.S. Patent 3182079, 1965; Chem. Abstr. 1965, 63, 5740c], солей рацемической глутаминовой кислоты [French Patent 1389840, 1965; Chem. Abstr. 1965, 63, 5740f]. Описано разделение (R,S)-бромянтарной кислоты [Shiraiwa Т., Ohkubo М., Miyazaki H., Kubo M., Nishigawa H., Tsujimoto Т., Kurokawa H. Bull. Chem. Soc. Jpn. 1998, 71, 735-739], разделение соли рацемического фенилэтиламина с итаконовой кислотой [Bocskey Z., Kassai С., Simon K., Fogassy Е., Kozma D.J. Chem. Soc., Perkin Trans. 2. 1996, 1511-1515], разделение соли диэтиламина и рацемического напроксена [Eur. Patent 298395; Chem. Abstr. 1989, 111, 7085a]. Авторами подаваемой заявки ранее описано разделение рацемического пропранолола гидрофторида [Бредихин А.А., Бредихина З.А., Диева С.А., Синяшин О.Г. Патент РФ №2245868, 2005], разделение лекарственных средств гвайфенезин и мефенезин [Бредихин А.А., Бредихина З.А., Лазарев С.Н., Синяшин О.Г. Патент РФ №2213724, 2003].
Для лекарственного средства метокарбамол (1) в литературе описана схема синтеза рацемата [Baizer М.М., Clark J.R., Swidinsky J.J. Org. Chem. 1957, 22, 1595-1599] и обоих энантиомеров метокарбамола (1) [Bredikhin А.А., Bredikhina Z.A., Zakharychev D.V., Pashagin A.V. Tetrahedron: Asymmetry. 2007, 18, 1239-1244]. В качестве исходного соединения при синтезе рацемата используется рацемический гвайфенезин (2), который при нагревании с диэтилкарбонатом приводит к образованию циклического карбоната (3) с последующим превращением последнего в метокарбамол (1) при взаимодействии с аммиаком. Для получения энантиочистых производных (1) используется методика спонтанного разделения на энантиомеры при кристаллизации рацемического гвайфенезина, разработанная Бредихиным А. с сотр. [а) Бредихин А.А., Бредихина З.А., Лазарев С.Н., Синяшин О.Г. Патент РФ №2213724, 2003; б) Bredikhina Z.A., Novikova V.G., Zakharychev D.V., Bredikhin A.A. Tetrahedron: Asymmetry, 2006, 17, 3015-3020]. Для полученных таким способом энантиочистых (S)- и (R)-2 вся цепочка превращений повторяется с каждым энантиомером по отдельности, давая соответственно (R)- и (S)-l (схема 1) [Bredikhin А.А., Bredikhina Z.A., Zakharychev D.V., Pashagin A.V. Tetrahedron: Asymmetry. 2007, 18, 1239-1244].
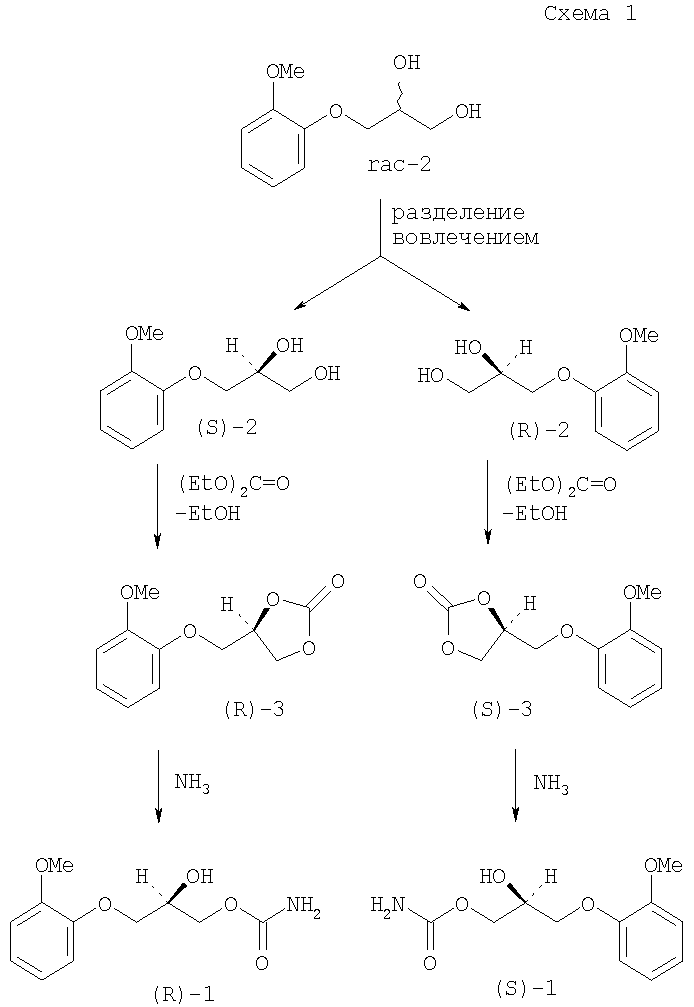
Следует отметить, что способ с проведением процесса разделения исходного вещества явно проигрывает по сравнению с разделением конечного продукта, так как энантиочистое сырье дороже рацемического и неизбежны потери на двух стадиях синтеза. Поэтому экономически целесообразнее, если это возможно, проводить процесс разделения конечного продукта, полученного в рацемическом виде.
В работе Бредихина с сотр. [Bredikhin А.А., Bredikhina Z.A., Zakharychev D.V., Pashagin A.V. Tetrahedron: Asymmetry. 2007, 18, 1239-1244] впервые показано, что метокарбамол кристаллизуется из раствора в виде рацемического конгломерата, что дает возможность разделить его на энантиомеры методом вовлечения. Ранее методы, основанные на явлении самопроизвольного разделения энантиомеров при кристаллизации для метокарбамола, не были разработаны, при этом способность его кристаллизоваться в виде конгломерата является необходимым, но не достаточным условием для успешной реализации препаративного метода разделения. Так, в работах Дж.Кокуреля показано, что примерно половина конгломератообразующих соединений демонстрируют плохую эффективность при разделении [Coquerel G. Top. Curr. Chem., 2007, 269, 1-51; Dufour F., Perez G., Coquerel G. Bull. Chem. Soc. Jpn. 2004, 77, 79-86].
Мы впервые предлагаем способ разделения метокарбамола на отдельные энантиомеры методом вовлечения. Ранее таким способом нерацемическое соединение (1) не получалось. К тому же было неочевидно, что использование свойства самопроизвольного разделения приведет к получению нерацемического соединения (1) с высокой эффективностью.
Задача предлагаемого изобретения - создание способа, позволяющего получить известное лекарственное средство метокарбамол в нерацемическом виде из рацемата путем его разделения при кристаллизации методом вовлечения.
Технический результат - возможность получения энантиомеров метокарбамола высокой оптической чистоты разделением рацемического метокарбамола методом вовлечения.
Технический результат достигается тем, что насыщенный водный раствор рацемического метокарбамола (1), обогащенный одним из энантиомеров, готовят при температуре 50-55°С. Полученный раствор перемешивают, постепенно охлаждают до комнатной температуры, добиваясь его пересыщения, и вносят кристаллическую затравку того же энантиомера, который добавлен для обогащения. Раствор выдерживают при перемешивании и термостатировании до достижения максимального значения энантиомерного избытка другого энантиомера в маточном растворе. Затем отфильтровывают скалемический (энантиообогащенный) метокарбамол, обладающий той же конфигурацией, что и образец внесенной затравки. После отделения осадка в маточный раствор, обогащенный противоположным преобладающему в отделенном твердом осадке энантиомером, добавляют твердый рацемический метокарбамол, в количестве, равном по весу отделенному на первой стадии осадку, и нагревают до 50-55°С. Достигнув полного растворения внесенного твердого вещества, раствор охлаждают, снова добиваясь пересыщения, и вносят кристаллическую затравку преобладающего в растворе энантиомера. Раствор перемешивают, охлаждают и отделяют выпавший осадок второго энантиомера. Описанный цикл повторяют несколько раз, тщательно анализируя энантиомерную чистоту и количество раствора и выделенного осадка.
Определение энантиомерной чистоты нерацемического метокарбамола
В процессе кристаллизации через определенные промежутки времени отбирают аликвоту (10 мкл) отфильтрованного через мембранный фильтр (фирмы MillexR) маточного раствора и смешивают с 1 мл воды. Порцию 20 мкл раствора анализируют методом ВЭЖХ на хроматографе Shimadzu LC-20AD с УФ-спектрофотометрическим детектором при длине волны 275 нм. Используют стальные насадочные колонки Chiralcel OD-RH (0.46 см×15 см) фирмы Daicel, Inc. Элюирование проводят в изократическом режиме (элюэнт: изо-пропанол-вода, 1:3) с термостатированием (Т=40°С). Скорость подачи элюэнта 0.4 мл/мин; tR (S)=9.2 мин, tR (R)=10.8 мин. Для анализа твердого осадка ~1 мг вещества растворяют в 10 мл горячей воды и анализируют порцию 20 мкл методом ВЭЖХ. Энантиомерный избыток, ее рассчитывают по формуле ее=(R-S/R+S)×l00%, где R и S - содержание соответствующего энантиомера в смеси.
Рацемический 1-карбамоилокси-2-гидрокси-3-(2-метоксифенокси)пропан [(R,S)-метокарбамол] использован производства фирмы Sigma-Aldrich. Энантиомеры (R)-метокарбамола и (S)-метокарбамола (ее 99%) получены по методике, приведенной в работе [Bredikhin A.A., Bredikhina Z.A., Zakharychev D.V., Pashagin A.V. Tetrahedron: Asymmetry. 2007, 18, 1239-1244].
Заявляемый способ разделения рацемического метокарбамола на энантиомеры иллюстрируется следующими примерами.
ПРИМЕР 1.
10 г (R,S)-метокарбамола (1), обогащенного (R)-энантиомером, растворяют в 320 мл воды при 50-55°С и охлаждают при перемешивании до 28°С (ее вещества в растворе 9.2%, концентрация раствора 3.0% по весу), затем вносят мелкоизмельченную затравку (R)-метокарбамола (0.025 г). Раствор охлаждают до 25.5°С, термостатируют и перемешивают при этой температуре, и через 80 минут с момента внесения затравки отфильтровывают 2.89 г (R)-метокарбамола, ее 60%. Ее маточного раствора 12.6% (с преобладанием S-энантиомера). К фильтрату, оставшемуся после отделения осадка, добавляют 2.89 г (R,S)-метокарбамола, растворяют при 50-55°С, охлаждают до 28°С, вносят затравку измельченного (S)-метокарбамола (0.025 г). Раствор охлаждают до 25.5°С, термостатируют и перемешивают при этой температуре, и через 70 минут с момента внесения затравки отфильтровывают 3.36 г (S)-метокарбамола, ее 46%; ее маточного раствора 7.4% (с преобладанием R-энантиомера).
ПРИМЕР 2.
2.76 г (R,S)-метокарбамола (1), обогащенного (R)-энантиомером, растворяют в 115 мл воды при 50-55°С, раствор охлаждают при перемешивании до 29°С (ее раствора 13.4%, концентрация 2.3%) и вносят мелкоизмельченную затравку (R)-метокарбамола (0.014 г). Раствор охлаждают до 27.5°С, термостатируют и перемешивают при этой температуре, и через 45 минут с момента внесения затравки отфильтровывают 0.90 г (R)-метокарбамола, ее 72%; ее маточного раствора 15.6% (S) (операция №1 в табл.1). К фильтрату, оставшемуся после отделения осадка, добавляют 0.90 г (R,S)-метокарбамола, растворяют твердую компоненту при 50-55°С, охлаждают до 29°С, вносят затравку измельченного (S)-метокарбамола (0.014 г). Затем раствор охлаждают до 27°С, термостатируют и перемешивают при этой температуре, и через 50 минут с момента внесения затравки отфильтровывают 0.96 г (S)-метокарбамола, ее 50%; ее маточного раствора 11.5% (R) (операция №2 в табл.1). Таким же образом цикл повторяют несколько раз, добавляя каждый раз недостающее количество рацемического метокарбамола. Детали описаны в таблице 1 для операций №3-6.
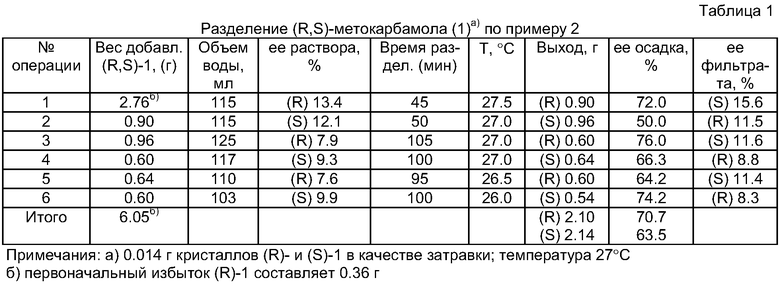
ПРИМЕР 3.
8.17 г (R,S)-метокарбамола (1), обогащенного (R)-энантиомером, растворяют в 355 мл воды при 50-55°С и охлаждают при перемешивании до 29°С (ее раствора 10.9%, концентрация 2.25%), затем вносят мелкоизмельченную затравку (R)-метокарбамола (0.020 г). Раствор охлаждают до 27°С, термостатируют и перемешивают при этой температуре, и через 85 минут с момента внесения затравки отфильтровывают 2.20 г (R)-метокарбамола, ее 75.5%; ее маточного раствора 13.0% (S). Операция №1 в табл.2.
К фильтрату, оставшемуся после отделения осадка, добавляют 2.20 г (R,S)-метокарбамола и растворяют при 50-55°С, раствор охлаждают до 29°С, вносят затравку измельченного (S)-метокарбамола (0.020 г). Затем раствор охлаждают до 27°С, термостатируют и перемешивают при этой температуре, и через 65 минут с момента внесения затравки отфильтровывают 1.97 г (S)-метокарбамола, ее 72%; ее маточного раствора 8.4% (R) (операция №2 в табл.2). Таким же образом цикл повторяют несколько раз, добавляя каждый раз недостающее количество рацемического метокарбамола. Детали описаны в таблице 2 для операций №3-4.

Очистка (R) и (S)-метокарбамола
Повышение энантиомерной чистоты полученных таким образом нерацемических образцов (R) и (S)-метокарбамола возможно путем дополнительной перекристаллизации из подходящего растворителя, например этилацетата. (R)-метокарбамол (2.20 г, ее 75.5%) растворяют при кипячении в 28 мл этилацетата и охлаждают до 13-15°С, получают 1.83 г кристаллов (выход 83%, ее 87.9%). Полученный осадок растворяют при кипячении в 35 мл этилацетата и охлаждают до 13-15°С. Отфильтровывают 1.58 г кристаллов (выход 87%, ее 96.5%). Полученный осадок вновь перекристаллизовывают из 20 мл этилацетата. Получают 1.46 г энантиочистого (R)-1: Т.пл. °С; ее 99.8%; [α]D 20=-0.8 (с 1.0, МеОН); лит.: [α]D 22=-0.8 (с 1, МеОН) [Demian I. Chirality 1993, 5, 238-240].
По аналогии перекристаллизацией скалемических образцов (S)-метокарбамола получен энантиочистый (S)-1. Например, скалемический (S)-метокарбамол в количестве 3,72 г (полученный объединением образцов с ее 76,7% 1.95 г и 72% 1.77 г) растворяют при кипячении в этилацетате (45 мл) и охлаждают до 13-15°С. Отфильтровывают 3.0 г бесцветных кристаллов (выход 80%, ее 75.5%). После двух последующих перекристаллизаций получают 2.16 г энантиочистого (S)-1: Т.пл. 113-114°С; ее 99.5%. [α]D 20=+0.8 (с 1.1, МеОН); лит.: [α]D 22=+0.5 (с 1, МеОН)) [Demian I. Chirality 1993, 5, 238-240].
Маточные растворы, полученные после перекристализаций, упаривают до нужной для повторных перекристаллизации объемов и объединяют с аналогичными по оптической чистоте растворами, полученными из других опытов для проведения укрупненных перекристаллизаций.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ РАЗДЕЛЕНИЯ НА ЭНАНТИОМЕРЫ РАЦЕМИЧЕСКИХ 3-(2-МЕТОКСИФЕНОКСИ)-1,2-ПРОПАНДИОЛА И 3-(2-МЕТИЛФЕНОКСИ)-1,2-ПРОПАНДИОЛА | 2002 |
|
RU2213724C1 |
| СПОСОБ РАЗДЕЛЕНИЯ НА ЭНАНТИОМЕРЫ РАЦЕМИЧЕСКОГО 1-(ИЗОПРОПИЛАМИНО)-3-(1-НАФТИЛОКСИ)-2-ПРОПАНОЛА | 2003 |
|
RU2245868C2 |
| РАЗДЕЛЕНИЕ 3-АМИНОАЛКИЛНИТРИЛОВ | 2004 |
|
RU2309145C1 |
| ДИАЛКИЛ(АРИЛ)-ЦИС-2-(2-ГИДРОКСИАРИЛ)-2-АЛКИЛ(АРИЛ)ЭТЕНИЛФОСФИНОКСИДЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2007 |
|
RU2329271C1 |
| ХИРАЛЬНОЕ РАЗДЕЛЕНИЕ СМЕСИ ЭНАНТИОМЕРОВ НИКОТИНА | 2018 |
|
RU2753492C1 |
| СПОСОБ ПРОИЗВОДСТВА НИКОТИНА | 2018 |
|
RU2753548C1 |
| СИНТЕЗ ОПТИЧЕСКИ ЧИСТОГО (R)-5-(2-АМИНОПРОПИЛ)-2-МЕТОКСИБЕНЗОЛСУЛЬФОНАМИДА | 2004 |
|
RU2419605C2 |
| СПОСОБ РАЦЕМИЗАЦИИ ОПТИЧЕСКИ ЧИСТОГО ИЛИ ОБОГАЩЕННОГО ПИПЕРАЗИН-2- ТРЕТ.БУТИЛКАРБОКСАМИДНОГО СУБСТРАТА И РАЦЕМИЧЕСКИЙ 2-ТРЕТ-БУТИЛКАРБОКСАМИД-4-(3- ПИКОЛИЛ)ПИПЕРАЗИН | 1995 |
|
RU2135482C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФЕНИЛПРОПИОНОВОЙ КИСЛОТЫ | 1993 |
|
RU2133733C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-(ОКСИРАН-2-ИЛ)-ЭТАНОЛА | 2008 |
|
RU2384577C2 |
Изобретение относится к химии органических соединений, а именно к способу разделения на отдельные энантиомеры рацемического 1-карбамоилокси-2-гидрокси-3-(2-метоксифенокси)пропана (1), и заключается в том, что готовят при 50-55°С насыщенный водный раствор рацемического 1-карбамоилокси-2-гидрокси-3-(2-метоксифенокси)пропана, обогащенного одним из энантиомеров, перемешивают, постепенно охлаждая до комнатной температуры, добиваясь его пересыщения; затем вносят кристаллическую затравку того же энантиомера, который добавлен для обогащения, выдерживают при перемешивании и термостатировании до достижения максимального значения энантиомерного избытка маточного раствора, отфильтровывают скалемический 1-карбамоилокси-2-гидрокси-3-(2-метоксифенокси)пропан, обладающий той же конфигурацией, что и образец внесенной затравки; добавляют в маточный раствор, обогащенный вторым энантиомером, рацемическое соединение в количестве, компенсирующем отделенный осадок, нагревают до полного растворения при 50-55°С, затем охлаждают до комнатной температуры, добиваясь пересыщения; вносят кристаллическую затравку преобладающего в растворе энантиомера, выдерживают при перемешивании и термостатировании и отделяют осадок, обогащенный вторым энантиомером, с последующим повторением указанного процесса несколько раз, тщательно анализируя энантиомерную чистоту целевого соединения. Технический результат состоит в возможности получения энантиомеров метокарбамола(1-карбамоилокси-2-гидрокси-3-(2-метоксифенокси)пропана высокой оптической чистоты из рацемической смеси. 2 табл.

Способ разделения на энантиомеры рацемического 1-карбамоилокси-2-гидрокси-3-(2-метоксифенокси)пропана, заключающийся в том, что:
готовят при 50-55°С насыщенный водный раствор рацемического 1-карбамоилокси-2-гидрокси-3-(2-метоксифенокси)пропана, обогащенного одним из энантиомеров, перемешивают, постепенно охлаждая до комнатной температуры, добиваясь его пересыщения;
вносят кристаллическую затравку того же энантиомера, который добавлен для обогащения, выдерживают при перемешивании и термостатировании до достижения максимального значения энантиомерного избытка маточного раствора, отфильтровывают скалемический 1-карбамоилокси-2-гидрокси-3-(2-метоксифенокси)пропан, обладающий той же конфигурацией, что и образец внесенной затравки;
добавляют в маточный раствор, обогащенный вторым энантиомером, рацемическое соединение в количестве, компенсирующем отделенный осадок, нагревают до полного растворения при 50-55°С, затем охлаждают до комнатной температуры, добиваясь пересыщения;
вносят кристаллическую затравку преобладающего в растворе энантиомера, выдерживают при перемешивании и термостатировании и отделяют осадок, обогащенный вторым энантиомером, с последующим повторением указанного процесса несколько раз.
| СПОСОБ РАЗДЕЛЕНИЯ НА ЭНАНТИОМЕРЫ РАЦЕМИЧЕСКИХ 3-(2-МЕТОКСИФЕНОКСИ)-1,2-ПРОПАНДИОЛА И 3-(2-МЕТИЛФЕНОКСИ)-1,2-ПРОПАНДИОЛА | 2002 |
|
RU2213724C1 |
| Bredikhin A.A | |||
| et al | |||
| "Chiral drugs related to guaifenesin: synthesis and phase properties of methocarbamol and methenoxalone" Tetrahedron: Asymmetry | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Farsam, H | |||
| et al | |||
| «A new HPLC technique for the separation of methocarbamol enantiomers.» Journal of Pharmacy and Pharmacology, 51 (7), | |||

