Изобретение относится к химии органических соединений, а именно к способу разделения на отдельные энантиомеры рацемических 3-(2-метоксифенокси)-1,2-пропандиола (I), международное непатентованное название гвайфенезин (guaiphenesin), и 3-(2-метилфенокси)-1,2-пропандиола (II), международное непатентованное название мефенезин (mephenesin). Изобретение может быть использовано в фармацевтической промышленности при получении нерацемических (скалемических, энантиочистых) лекарственных препаратов.
В настоящее время названные соединения зарегистрированы в качестве субстанций лекарственных средств в рацемическом виде.
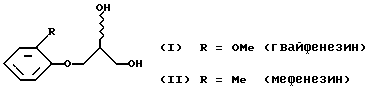
Гвайфенезин применяется как мышечный релаксант и отхаркивающее средство, мефенезин используется как релаксант скелетных мышц. Зарегистрированными лекарственными средствами являются и простейшие производные рацемических диолов (I) и (II), а именно, их 1-карбаматы, так же являющиеся релаксантами скелетных мышц и спазмолитиками. Химическую структуру (I) и (II) можно модифицировать и в другие физиологически активные производные с хиральным С3-фрагментом, например (Бредихина З.А., Савельев Д. В., Бредихин А.А. // Ж. орган, химии. 2002. Т. 38. 2. С. 233-239) через циклические сульфиты в 1-алкиламино-3-арилокси-2-пропанолы, многие из которых являются хиральными лекарственными средствами класса бета-адреноблокаторов.
Рацемические лекарственные средства (ЛС) представляют собой смесь равных количеств противоположных энантиомеров конституционно тождественных соединений. В хирально поляризованной среде живого организма энантиомеры проявляют различную физиологическую активность. В то время как один из энантиомеров обладает полезной, целевой для данного ЛС активностью (такой энантиомер называют эутомером), его антипод (называемый дистомером) может оказаться физиологически нейтральным, выступать синергистом или антагонистом эутомера, обладать совершенно иным спектром физиологической активности, обладать иной, часто большей, чем эутомер, токсичностью, тератогенностью и т.д.. В частности, для метокарбамола, 1-карбамата гвайфенезина, показано, что (+)-(R)-изомер проявляет большую физиологическую активность, чем рацемический или (-)-(S)-метокарбамол (Souri Е., Sharifzadeh M., Farsam H., Gharavi N. // J. Pharm. Pharmacol., 1999. Vol. 51. N 7. Р. 853-855).
В настоящее время твердо установлено, что всегда (за исключением случаев, когда ЛС быстро рацемизуется при хранении или в физиологических средах живого организма) скалемические (обогащенные одним из энантиомеров или энантиочистые) субстанции ЛС обладают значительными терапевтическими преимуществами перед рацемическими (Lehmann Р.А. II Trends Pharmacol. Sci., 1986. Vol. 7, N 7. Р. 281-285). Различия в физиологическом действии энантиомеров тем значительнее, чем выше физиологическая активность (меньше физиологическая доза) данного ЛС (Pfeiffer С. С. // Science, 1956. N 124. Р. 29-41). Сегодняшние ЛС обладают высокой активностью, поэтому основной тенденцией современной медицинской химии и фармацевтической промышленности является замена там, где это целесообразно, рацемических субстанций скалемическими (Deutsch D. H. // Chemtech. 1991. Vol. 21. N 3. Р. 157-159; Stinson S.C. // Chem. Eng. News. 1998. Vol. 76. N 38. P. 83; Aboul-Enein H.Y. Impact of Stereochemistry on Drug Development and Use. N. Y.: Wiley, 1997. 736 pp.). В связи с этим проблема получения скалемических арилоксипропандиолов, являющихся субстанциями лекарственных средств, является востребованной.
Известно три общих способа получения скалемических соединений. Во-первых, это химическая трансформация исходных энантиочистых соединений традиционными методами. Во-вторых, это модификация прохиральных исходных соединений методами энантиоселективного синтеза. В-третьих, это разделение на отдельные энантиомеры рацемических целевых (или специально полученных промежуточных) веществ. Все существующие лабораторные и промышленные методы получения скалемических соединений относятся к одной из этих категорий или являются их комбинацией (Crosby J. // Tetrahedron. 1991. Vol. 47. N 27. Р. 4789-4846; Sheldon R.A. Chirotechnology: Industrial Synthesis of Optically Active Compounds. N.Y.: M. Deccer, 1993. 416 pp.).
Применительно к получению скалемических 3-арилоксипропандиолов в качестве описанных примеров первого подхода можно привести присоединение фенолов АrОН к скалемическому глицидолу, региоселективно протекающее в присутствии катализаторов, таких как Ti(OAlk)4 (Klunder J.M., Ко S. Y., Sharpless KB. // J. Org. Chem. 1986. Vol. 51.N 19. P. 3710-3712), Alk3N (Chen J., Shum W. // Tetrahedron Lett. 1995. Vol. 36. N. 14. P. 2379-2380; Бредихина З.А., Савельев Д. В. , Бредихин А.А. // ЖОрХ. 2002. Т. 38. Вып.2. С. 233-239) или CsF (Kitaori К., Furukawa Y., Yoshimoto H., Otera J. // Tetrahedron, 1999. Vol. 55. N 50. P. 14381-14390), и приводящее к скалемическим 3-арилокси-пропан-1,2-диолам: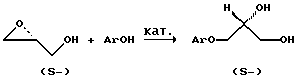
Этот способ достаточно прост и хорошо воспроизводится, однако для реализации он нуждается в наличии дорогостоящего энантиочистого глицидола.
Примером второго подхода является способ получения энантиочистых 3-арилокси-пропан-1,2-диолов асимметрическим дигидроксилированием по Шарплессу аллиловых эфиров фенолов (Wang Z.-M., Zhang X.-L., Sharpless K.B. // Tetrahedron Lett. 1993. Vol. 34, 2267-2270; Kolb H.C., VanNieuwenhze M.S., Sharpless K.B. // Chem.Rev. 1994. Vol. 94. N 8. P.2483-2547).
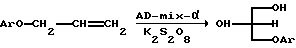
На схеме AD-mix-α - торговое название смеси, содержащей хиральный лиганд (DHQ)2PHAL, К3Fе(СN)6, K2CO3, K2OsO4•2H2O; здесь (DHQ)2PHAL:
Для проведения этого процесса используются экологически вредные производные тяжелого металла осмия и дорогостоящие энантиочистые катализаторы. Кроме того, полученные этим способом аналоги (I) и (II) имеют невысокий энантиомерный избыток (в пределах 60%).
В качестве примера третьего подхода укажем описанный многостадийный способ получения диолов (I) и (II) с умеренной энантиомерной чистотой путем кинетического разделения рацемических 3-арилокси-1,2-пропандиолов с использованием катализируемого биологическими ферментами липазами процесса переэтерификации (Theil F., Weidner J., Ballschuh S., Kunath A., Schick H. // J. Org. Chem. 1994. Vol. 59. N 2. P. 388-393) (прототип).
На первой стадии rас-1,2-диолы с помощью липаз региоселективно ацилируются винилацетатом по первичной гидроксигруппе без проявления энантиоселективности. Последующая стадия катализируемого липазами ацилирования вторичной гидроксигруппы протекает с удовлетворительной энантиоселективностью (для R= OMe, ee= 87%; R= Me, ee=80%). Моноацетат отделяется от диацетата флеш-хроматографией и затем после последовательного удаления ацильных групп выделяются целевые (S)- и (R)-диолы.
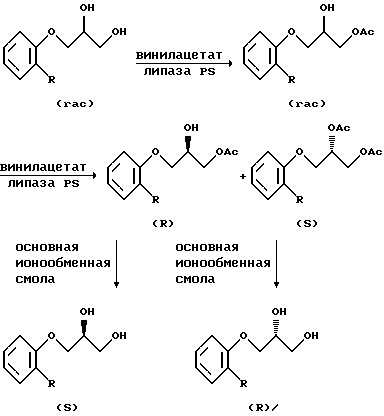
Недостатком этого способа является то, что результаты использования биоэнзимов сильно зависят от условий проведения процесса и от характера заместителей в ароматическом кольце. Метод нуждается в значительном ассортименте вспомогательных реактивов, при разработке используется хроматография, процесс трудно масштабируется.
Известно получение некоторых хиральных соединений в нерацемическом виде путем разделения рацемического вещества на обогащенные противоположными энантиомерами фракции при кристаллизации методом вовлечения. Применение избирательной кристаллизации ("preferential crystallization") или метода разделения вовлечением ("resolution by entrainment") для оптического расщепления рацемических соединений описано в литературе (Jacques J., Collet A., Wilen S. H. "Enantiomers, Racemates, and Resolutions", Krieger Publishing Co. Malabar, FL. , 1994. P. 217-368; Collet A., Brienne M-J., Jacques J. // Chem. Rev. 1980. Vol. 80. N 3. P. 215-230).
Как правило, рацемические вещества кристаллизуются в виде рацемических соединений, рацемических конгломератов и рацемических твердых растворов. Методом избирательной кристаллизации могут быть разделены только вещества, проявляющие свойство самопроизвольного разделения на энантиомеры при кристаллизации, то есть образующие рацемические конгломераты. Избирательная кристаллизация достигается внесением небольшого количества затравки чистого энантиомера в пересыщенный раствор рацемического или слегка обогащенного тем же энантиомером вещества. Таким способом, например, проведено разделение D, L-треонина (Amiard G. // Bull. Soc. Chim. France. 1956. P. 447), гидрохлорида D,L-валина (U.S. Patent 3 182 079, 1965; Chem. Abstr. 1965, Vol. 63, 5740 с.), солей рацемической глутаминовой кислоты (French Patent 1 389 840, 1965; Chem. Abstr. 1965, Vol. 63, 5740f). Описано разделение(R; S)-бромянтарной кислоты (Shiraiwa Т., Ohkubo M., Miyazaki H., Kubo M., Nishigawa H., Tsujimoto Т., Kurokawa Н. //Bull. Chem. Soc. Jpn. 1998. Vol. 71. N 3. Р. 735-739), разделение соли рацемического фенилэтиламина с итаконовой кислотой (Bocskey Z. , Kassai С., Simon К., Fogassy E., Kozma D. // J. Chem. Soc., Perkin Trans. 2. 1996. P. 1511-1515), разделение соли диэтиламина и рацемического напроксена (Eur. Patent 298 395; Chem. Abstr. 1989, Vol. 111, 7085a).
Нами впервые обнаружено, что рацемические диолы (I) и (II) кристаллизуются в виде рацемических конгломератов без какого-либо предварительного превращения их в другие химические производные, например соли, как в большинстве известных случаев, и мы предлагаем способ их разделения на отдельные энантиомеры по схеме избирательной кристаллизации. Ранее свойство самопроизвольного расщепления при кристаллизации для соединений (I) и (II) не было известно, и таким способом нерацемические соединения (I) и (II) не получались. К тому же было неочевидно, что использование этого свойства приведет к получению нерацемических соединений (I) и (II).
Цель предлагаемого изобретения - создание способа, позволяющего получить известные лекарственные средства - гвайфенезин и мефенезин в нерацемическом виде из дешевых рацемических субстратов путем их разделения при кристаллизации методом вовлечения.
Поставленная цель достигается тем, что рацемических диол слегка обогащают одним из энантиомеров в подходящем растворителе при температуре 40-60oС с получением концентрированного раствора и перемешивают, постепенно охлаждая до 10-40oС, добиваясь его пересыщения, после чего вносят кристаллическую затравку того же энантиомера, который добавлен для обогащения, выдерживают при перемешивании при температуре кристаллизации в интервале 5-33oС в течение 30-60 мин, затем отфильтровывают твердый осадок скалемического диола, обладающего той же конфигурацией, что и образец внесенной затравки. В маточный раствор, обогащенный вторым энантиомером, добавляют рацемическое соединение в количестве, компенсирующем отделенный осадок, нагревают до полного растворения при 40-60oС, затем охлаждают до 10-40oС, вносят кристаллическую затравку преобладающего в растворе энантиомера, выдерживают при перемешивании при температуре кристаллизации в интервале 5-33oС, отделяют осадок, обогащенный вторым энантиомером, с последующим повторением указанного процесса несколько раз, тщательно анализируя оптическую чистоту и количество выделенного осадка.
Разделение на энантиомеры проводили для соединений (I) и (II) в бензоле, водном этаноле, воде, четыреххлористом углероде или его смеси с хлористым метиленом (1: 1). Заявляемый способ разделения диолов (I) и (II) иллюстрируется следующими примерами.
ПРИМЕР 1
4.56 г (R, S)-гвайфенезина (I) (Т. пл. 76-78oС) и 0.50 г (S)-гвайфенезина (Т. пл. 97oС, [α]
Повышение энантиомерной чистоты полученных таким образом скалемических (R)- и (S)-гвайфенезина возможно путем дополнительной перекристаллизации из органических или смешанных растворителей. Например, 0.8 г (S)-гвайфенезина (ор=69.1%) растворяют в 3.5 мл 40% (по объему) водного этанола при 50oС. После охлаждения раствора до 10oС и внесения затравки (S)-гвайфенезина (0.008 г) смесь перемешивают при 5oС 5 ч. После фильтрования и высушивания собирают (5)-гвайфенезин: выход 0.45 г, [α]
ПРИМЕР 2
5.00 г (R,S)-гвайфенезина (I) и 0.20 г (R)- гвайфенезина растворяют в 20 мл 40% водного этанола при 40oС и охлаждают при перемешивании до 10oС, затем вносят мелкоизмельченную затравку (R)-гвайфенезина (0.05 г, op>99%) и в течение 60 мин перемешивают раствор при 5oС. После фильтрования и высушивания собирают 0.45 г (R)-гвайфенезина с [α]
ПРИМЕР 3
2.55 г (R, S)-гвайфенезина (I) и 0.25 г (R)-гвайфенезина (ор=90.0%) растворяют в 20 мл воды при 60oС и охлаждают при перемешивании до 30oС, затем вносят мелкоизмельченную затравку (R)-гвайфенезина (0.02 г, op>99%) и в течение 60 мин перемешивают раствор при 20oС. После фильтрования и высушивания собирают 0.44 г (R)-гвайфенезина с [α]
ПРИМЕР 4
0.4 г (R, S)-мефенезина (II) (Т. пл. 70-71 oС) и 0.04 г (S)-мефенезина (Т. пл. 90-92 oС, [α]
ПРИМЕР 5
2.00 г (R, S)-мефенезина (II) и 0.20 г (R)- мефенезина растворяют в 24 мл 40% (по объему) водного этанола при 40oС и охлаждают при перемешивании до 10oС, затем вносят мелкоизмельченную затравку (R)-мефенезина (0.01 г), op>99%, и в течение 60 мин перемешивают раствор при +5oС. Затем отфильтровывают 0.50 г (R)- мефенезина с [α]
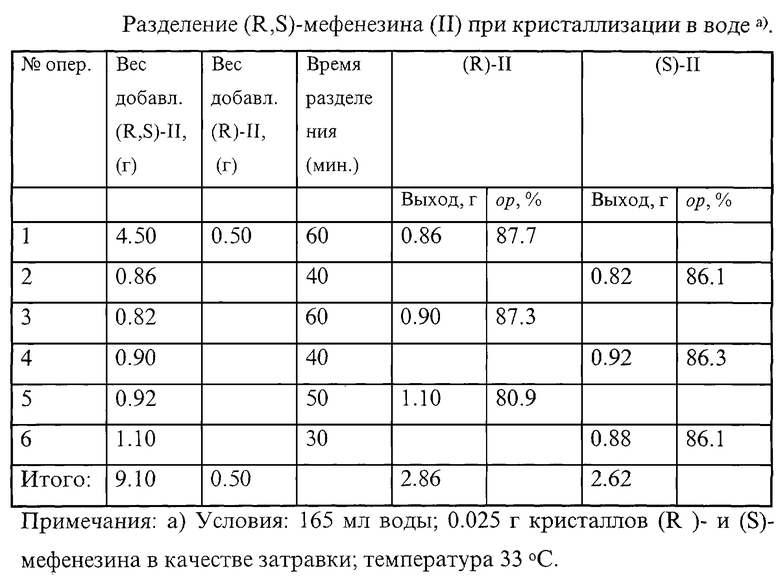
ПРИМЕР 6
4.50 г (R, S)-мефенезина (II) и 0.50 г (R)- мефенезина растворяют в 165 мл воды при 60oС и охлаждают при перемешивании до 40oС, затем вносят мелкоизмельченную затравку (R)-мефенезина (0.025 г), op>99%, охлаждают раствор до 33oС (7 мин) и в течение 60 мин перемешивают раствор при этой температуре. Затем отфильтровывают 0.86 г (R)- мефенезина с [α]
Повышение энантиомерной чистоты полученных таким образом скалемических (R)- и (S)-мефенезина возможно путем дополнительной перекристаллизации из органических или смешанных растворителей. Например, 0.47 г (R)-мефенезина (ор 81.8%) растворяют в 7.0 мл 40%-ного водного этанола при 50oС, затем охлаждают до 20oС, вносят затравку (0.004 г) (R)-мефенезина (oр>99%), охлаждают до 8oС, перемешивают 30 мин, отфильтровывают 0.21 г (R)-мефенезина [α]
ПРИМЕР 7.
0.53 г (R,S)-гвайфенезина (I) и 0.06 г (S)-гвайфенезина растворяют в 14 мл смеси четыреххлористого углерода и хлористого метилена 1:1 при 40oС и охлаждают при перемешивании до 25oС, затем вносят мелкоизмельченную затравку (S)-гвайфенезина (0.002 г) ор=99.5%) и в течение 30 мин перемешивают раствор при 20oС. После фильтрования и высушивания собирают 0.08 г (S)-гвайфенезина с [α]
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ РАЗДЕЛЕНИЯ НА ЭНАНТИОМЕРЫ РАЦЕМИЧЕСКОГО 1-КАРБАМОИЛОКСИ-2-ГИДРОКСИ-3-(2-МЕТОКСИФЕНОКСИ)ПРОПАНА | 2008 |
|
RU2396247C2 |
| СПОСОБ РАЗДЕЛЕНИЯ НА ЭНАНТИОМЕРЫ РАЦЕМИЧЕСКОГО 1-(ИЗОПРОПИЛАМИНО)-3-(1-НАФТИЛОКСИ)-2-ПРОПАНОЛА | 2003 |
|
RU2245868C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФОСФОРИСТОЙ КИСЛОТЫ (ВАРИАНТЫ) | 2000 |
|
RU2179152C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЙМАЛИНА | 2000 |
|
RU2174555C1 |
| МЕЛАМИНОВАЯ СОЛЬ БИС(ОКСИМЕТИЛ)ФОСФИНОВОЙ КИСЛОТЫ(МЕЛАФЕН) В КАЧЕСТВЕ РЕГУЛЯТОРА РОСТА И РАЗВИТИЯ РАСТЕНИЙ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1999 |
|
RU2158735C1 |
| СПОСОБ ИНГИБИРОВАНИЯ КОРРОЗИИ И КОМПОЗИЦИИ ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2001 |
|
RU2198245C2 |
| СПОСОБ ВЫДЕЛЕНИЯ НИЗКОМОЛЕКУЛЯРНЫХ АЛИФАТИЧЕСКИХ КИСЛОТ ИЗ ВОДНЫХ РАСТВОРОВ, СОДЕРЖАЩИХ МУРАВЬИНУЮ КИСЛОТУ | 2001 |
|
RU2197471C1 |
| СПОСОБ КОРМЛЕНИЯ МОЛОДНЯКА КУР | 1999 |
|
RU2160994C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕКТИНА ИЗ ЗЕЛЕНОЙ БИОМАССЫ ЛЮПИНА | 1998 |
|
RU2157380C2 |
| ПРЕПАРАТ ДЛЯ ЛЕЧЕНИЯ ДЛИТЕЛЬНО НЕ ЗАЖИВАЮЩИХ РАН И ТРОФИЧЕСКИХ ЯЗВ (ВАРИАНТЫ) | 1998 |
|
RU2148989C1 |
Изобретение относится к способу разделения на энантиомеры рацемических 3-(2-метоксифенокси)-1,2-пропандиола и 3-(2-метилфенокси)-1,2-пропандиола, который может быть использован в фармацевтической промышленности при получении нерацемических лекарственных препаратов. Способ заключается в том, что рацемический диол слегка обогащают одним из энантиомеров в подходящем растворителе при 40-60oС с получением концентрированного раствора и перемешивают, постепенно охлаждая до 10-40oС, добиваясь его пересыщения, после чего вносят кристаллическую затравку того же энантиомера, который добавлен для обогащения, выдерживают при перемешивании при температуре кристаллизации в интервале 5-33oС в течение 30-60 мин, отфильтровывают твердый осадок скалемического диола, обладающего той же конфигурацией, что и образец внесенной затравки, затем в маточный раствор, обогащенный вторым энантиомером, добавляют рацемическое соединение в количестве, компенсирующем отделенный осадок, нагревают до полного растворения при 40-60oС, затем охлаждают до 10-40oС, вносят кристаллическую затравку преобладающего в растворе энантиомера, выдерживают при перемешивании при температуре кристаллизации в интервале 5-33oС, отделяют осадок, обогащенный вторым энантиомером, с последующим повторением указанного процесса несколько раз. Как правило, в качестве растворителя используют бензол, четыреххлористый углерод или его смесь с хлористым метиленом 1: 1, 40% водный раствор этанола, воду. Способ позволяет получить известные лекарственные средства в нерацемическом виде из дешевых рацемических субстратов. 1 з.п.ф-лы, 1 табл.
| Способ разделения D, L-трео-1-(n-нитрофенил)-2-амино-1,3-пропандиола | 1961 |
|
SU145233A1 |
| СПОСОБ РАЗДЕЛЕНИЯ ЭНАНТИОМЕРОВ d- и /-ПАНТОТЕНАТА КАЛЬЦИЯ | 0 |
|
SU262009A1 |
| US 5210288 А, 11.05.1993 | |||
| GB 1210495 A, 28.10.1967 | |||
| US 3182079 A, 04.05.1965. | |||

