Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к способу получения соединения формулы I-а, включающему способ разделения смеси энантиомеров, представленных формулой I-а, на энантиомерно чистые вещества, представленные формулой I-b и формулой I-c.

В частности, настоящее изобретение относится к экологичному и эффективному способу получения никотина формулы I-а. Никотин получают в чистом виде, избегая использования вредных химических соединений при синтезе.
Настоящее изобретение также относится к способу получения рацемических смесей (R/S)-никотина, энантиомерно чистого (R)-никотина и энантиомерно чистого (S)-никотина, в котором энантиомеры обычно сложно разделяются.
Предпосылки настоящего изобретения
Никотин является природным алкалоидом, используемым в различных применениях. В частности, (S)-никотин используют в качестве активного фармацевтического ингредиента для лечения злоупотребления никотином и никотиновой зависимости. Также сообщалось об успехе при лечении синдрома Туретта, болезни Альцгеймера, шизофрении и других заболеваний, связанных с расстройствами нервной системы. Обычными путями введения являются жевательные резинки, крема, транедермальные пластыри, таблетки, назальные спреи и электронные сигареты.
Значительные количества никотина также используются в сельском хозяйстве в качестве защитного средства для растений или пестицида против тли.
Натуральный никотин экстрагируют из растений табака способом, который требует эффективных стадий очистки для удаления нежелательных, вредных примесей. Повышение спроса на никотин создает необходимость в предложении экологичных и рентабельных путей для получения синтетического никотина в очень чистом виде.
Предшествующий уровень техники настоящего изобретения
Никотин ((S)-3-(1-метилпирролидин-2-ил)пиридин) и его энантиомеры получали многие годы различными, неудовлетворительными способами. Известные синтезы обычно являются дорогостоящими и в них используют средства, которые создают проблемы или даже являются токсичными для окружающей среды.
В 1904 г. Pictet А. уже сообщал о синтезе никотина, предусматривающем использование виннокаменной кислоты для разделения энантиомеров (Berichte der deutschen chemischen Gesellschaft, vol. 37, 1904, pages 1225-1235). После этого виннокаменную кислоту затем использовали десятилетиями (см., например: Aceto М.D., et al. (J. Med. Chem., 1979, vol. 22, 17 4-177)).
Позднее Chavdarian C.G. и соавт. раскрыли более новые идеи для синтеза оптически активных никотиноидов (J. Org. Chem., 1982, vol. 41, 1069-1073).
Katsuyama А. и соавт. сообщали о пути синтеза никотина при помощи трет-бутанолата калия для рацемизации никотина с получением исходного материала для дальнейшего разделения энантиомеров (Bull. Spec. CORESTA Symposium, Winston-Salem, 1982, p. 15, S05, ISSN 0525-6240).
Кроме того, в документе ЕР 4487172 описан путь синтеза из 5 различных стадий, предлагающий чистый выход 37,7%.
В документе WO 2017/117575 раскрыт синтез никотина при помощи гидрида калия (KH) или гидрида натрия (NaH) в качестве сильного основания в тетрагидрофуране (THF) в качестве растворителя с получением никотиноил-1-винилпирролидин-2-она. Выход (R/S)-никотина составляет приблизительно 31%. Подобные процедуры, а также процедуры для разделения энантиомеров никотина были раскрыты Wang J. и соавт. (Wang J. et al., E. J Med. Chem., 2017, vol. 130, 15-25), Desai D. и соавт. (Desai D. et al., J. Labeled Compd. Radiopharm, 2008, vol. 51, 226-230), Aceto M.D. и соавт. (Aceto M.D. et al., J. Med. Chem., 1979, vol. 2, 174-177) или Bowman E.R. и соавт. (Bowman E.R. et al., Synthetic Comm., 1982, vol. 12, 11, 871-879).
В документе US 2016/0326134 описан синтез, предусматривающий конденсацию 1-метилпирролидин-2-она и метилникотината в присутствии сильного основания (такого как трет-бутоксид K) при нагревании с обратным холодильником в промежуточный 1-метил-3-никотиноил-4,5-дигидро-1Н-пиррол-2-олат калия, который затем можно превращать в рацемическую смесь R/S-никотина. Ди-пара-толуоил-L-виннокаменная кислота служит в качестве средства расщепления.
В основе документа ЕР 2484673 (US 8378111) лежат хорошо известные пути синтеза, и он раскрывает D-DBTA (D-дибензоиловый сложный эфир виннокаменной кислоты) в качестве средства разделения энантиомеров.
Документ WO 2016/065209 (ЕР 3209653, US 9556142) раскрывает препаративный путь, предусматривающий 3 стадии до промежуточного миосмина, включая конденсацию N-винилпирролидинона и сложного эфира никотината в присутствии гидрида металла.
В последние годы исследования главным образом направлены на очистку и оптимизацию стадии расщепления оптически активных энантиомеров. Но существует потребность в усовершенствовании с получением более эффективного, более экологичного синтеза никотина и использования экологичных средств и растворителей.
Краткое раскрытие настоящего изобретения
Настоящее изобретение предлагает новый способ получения никотина, предусматривающий специфичный процесс для разделения полученных энантиомеров. Авторы настоящего изобретения обнаружили способ с меньшим числом стадий синтеза на основе легкодоступных исходных материалов и менее токсичных средств по сравнению с раскрытыми в литературе. Весь синтез можно проводить как однореакторный синтез, в частности, без изменения растворителя на различных стадиях. В то же время в заключительном отчете определили увеличенный выход и высокую чистоту. В общем, новый способ экономически и экологически лучше по сравнению со способами, известными в данной области.
В первом аспекте настоящее изобретение относится к способу получения соединения формулы I-а, предусматривающему

(i) реакцию этилникотината и N-винилпирролидона в присутствии основания-алкоголята с получением 3-никотиноил-1-винилпирролидин-2-она;
(ii) реакцию 3-никотиноил-1-винилпирролидин-2-она с кислотой с получением миосмина;
(iii) восстановление миосмина до норникотина при помощи восстанавливающего средства и
(iv) метилирование норникотина с получением соединения формулы I-а.
Дополнительные варианты осуществления раскрыты в зависимых пунктах формулы изобретения и могут быть взяты из следующего описания и примеров, но не ограничиваются ими.
Подробное раскрытие настоящего изобретения
Если не определено иное, технические и научные термины имеют такое же значение, как обычно понимается специалистом в области настоящего изобретения.
Все диапазоны, раскрытые в настоящем документе, следует рассматривать как дополненные термином «приблизительно», если явно не определено обратное или иным образом не ясно из контекста.
Все численные значения или проценты, относящиеся к количествам вещества в настоящей заявке, даны в масс. %, если явно не определено обратное или иным образом не ясно из контекста.
Настоящее изобретение предлагает новый способ производства никотина удобным путем.
Кроме того, настоящее изобретение в дополнительном аспекте относится к применению соединения, представленного формулой I-а, формулой I-b или формулой I-c, полученного способом согласно настоящему изобретению, в фармацевтическом составе.
В первом аспекте настоящее изобретение относится к способу получения соединения формулы I-а, предусматривающему
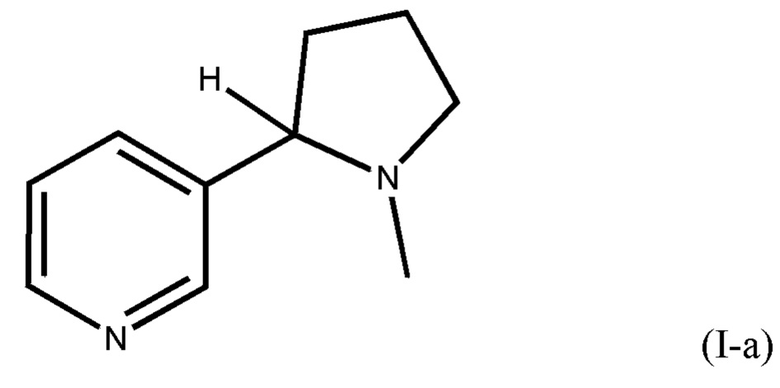
(i) реакцию этилникотината и N-винилпирролидона (NVP) в присутствии основания-алкоголята с получением 3-никотиноил-1-винилпирролидин-2-она;
(ii) реакцию 3-никотиноил-1-винилпирролидин-2-она с кислотой с получением миосмина;
(iii) восстановление миосмина до норникотина при помощи восстанавливающего средства и
(iv) метилирование норникотина с получением соединения формулы Ia.
Стадия (i) здесь является следующей:

Основание-алкоголят на стадии (i) конкретно не ограничено. Оно может быть алкоголятом (также известным как алкоксид) с 1-20 атомами углерода и может быть получено из первичного, вторичного или третичного алкилового, алкенилового и/или алкинилового спирта и/или ароматического спирта. Отмечая, что исходные материалы, такие как N-винилпирролидон, являются средними-сильными основаниями (типичные значения pKa от 20 до 26, в некоторых случаях до 35), авторы настоящего изобретения неожиданно обнаружили, что относительно мягкие основные алкоголяты (типичные значения pKa от 15 до 17) обеспечивают такую же селективную химическую реакцию, что и более агрессивные основания, такие как, например, NaH или KH (значения pKa приблизительно 35). Согласно некоторым вариантам осуществления основание-алкоголят получено из алкилового спирта с 1-20 атомами углерода, предпочтительно 1-10 атомами углерода, более предпочтительно 1-6 атомами углерода, в частности 1-4 атомами углерода, причем алкил может быть линейным или разветвленным. Например, оно может быть метанолятом, этанолятом, н-пропанолятом, изопропанолятом, н-бутанолятом, изобутанолятом, втор-бутанолятом, трет-бутанолятом и пр., предпочтительно метанолятом и этанолятом, более предпочтительно этанолятом (также известным как этоксид). Алкоголят может иметь подходящий катион, который конкретно не ограничен и может быть, например, выбран из одновалентных и двухвалентных катионов, например, из катионов щелочных и щелочноземельных металлов, предпочтительно катионов щелочных металлов, более предпочтительно Li+, Na+, K+ и их смесей. Подходящие предпочтительные алкоголяты предпочтительно представляют собой алкоголяты щелочных металлов, предпочтительно этаноляты щелочных металлов. Предпочтительно алкоголят щелочного металла выбирают из этанолята натрия, этанолята калия и их смесей. В одном аспекте обнаружили, что этанолят натрия является подходящим основанием, которое особенно предпочтительно.
Стадию (i) можно проводить в подходящем растворителе, который конкретно не ограничен. Согласно некоторым вариантам осуществления стадию (i) проводят в присутствии ароматического растворителя. Предпочтительный ароматический растворитель представляет собой бензол, толуол или их смесь. В предпочтительном аспекте настоящего изобретения толуол выбирают как растворитель. Также пригодны смеси ароматических растворителей с неароматическими растворителями. Например, можно использовать смеси ароматического растворителя с 15 масс. % или менее, по меньшей мере, спирта, например, одноатомного спирта с 1-10 атомами углерода, например, этанола, н-пропанола и/или изопропанола и пр.
Согласно некоторым вариантам осуществления ароматический растворитель, используемый на стадии (i), присутствует на всех стадиях (i), (ii), (iii) и (iv) способа настоящего изобретения, по меньшей мере, в некотором количестве.
Согласно некоторым вариантам осуществления стадию (i) проводят в безводных условиях, т.е. в отсутствие воды.
Согласно некоторым вариантам осуществления стадию (i) проводят при температуре от 50 до 150°С, предпочтительно от 80 до 120°С, более предпочтительно от 90 до 110°С, например, при приблизительно 100°С.
Согласно некоторым вариантам осуществления основание-алкоголят добавляют на стадии (i) в избытке относительно этилникотината, предпочтительно в количестве от 1,4 до 2 эквивалентов, более предпочтительно в количестве от 1,5 до 17 эквивалентов, особенно предпочтительно 1,6 эквивалентов, в пересчете на 1 эквивалент этилникотината. Согласно некоторым вариантам осуществления, альтернативно или в дополнение, NVP добавляют на стадии (i) в избытке относительно этилникотината, предпочтительно в количестве от 1,05 до 1,4 эквивалентов, более предпочтительно в количестве от 1,1 до 1,3 эквивалентов, особенно предпочтительно 1,2 эквивалентов, в пересчете на 1 эквивалент этилникотината.
Стадия (ii) является следующей:

На стадии (ii) образуется 1-пирролиновое кольцо, происходит снятие защитных групп с амида, и никотиноил-1-винилпирролидин-2-он декарбоксилируется.
Согласно некоторым вариантам осуществления стадию (ii) проводят при повышенной температуре в присутствии кислоты, предпочтительно сильной кислоты. В некоторых аспектах температура составляет от 90 до 115°С, предпочтительно от 100°С до 105°С. Понятно, что при повышенной температуре часть растворителя, например, органического растворителя стадии (i) и, например, часть воды, добавленной с кислотой, может удаляться дистилляцией и может быть извлечена и повторно использована. Также низкокипящие компоненты, такие как ацетальдегид, этанол и газообразный СО2, можно удалять.
На этой стадии кислота конкретно не ограничена. Согласно некоторым вариантам осуществления кислота представляет собой неорганическую кислоту, предпочтительно минеральную кислоту, более предпочтительно HCl и/или H2SO4, особенно предпочтительно HCl. Кислоту можно разбавлять водой, например, она может присутствовать как НСlводн. Согласно некоторым вариантам осуществления кислоту добавляют по каплям в охлажденный раствор, полученный на стадии (i), например, охлажденный до температуры от 20 до 40°С, например, до 30°С. Согласно некоторым вариантам осуществления смесь, полученную на стадии (i), или никотиноил-1-винилпирролидин-2-он - необязательно в подходящем растворителе, как указано выше, - добавляют по каплям в кислоту, предпочтительно неорганическую кислоту, более предпочтительно минеральную кислоту, еще более предпочтительно HCl и/или H2SO4, особенно предпочтительно HCl, например, НСlводн., что может давать увеличенный выход.
Согласно некоторым вариантам осуществления стадию (ii) проводят с использованием неорганической кислоты, особенно предпочтительно НСlводн., при температуре от 90 до 115°С, предпочтительно от 100 до 105°С.
После окончания реакции на стадии (ii), по меньшей мере, часть растворителей можно удалять дистилляцией согласно некоторым вариантам осуществления.
Стадия (iii) является следующей:
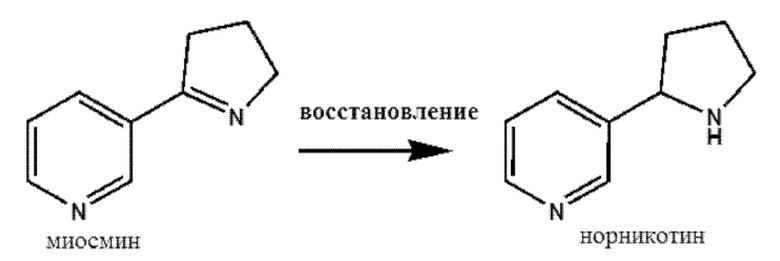
На стадии (iii) 1-пирролиновое кольцо миосмина восстанавливают подходящим восстанавливающим средством, которое конкретно не ограничено. Согласно некоторым вариантам осуществления восстановление 1-пирролинового кольца проводят при помощи стандартного способа. Согласно некоторым вариантам осуществления стадию (iii) проводят при помощи NaBH4 в качестве восстанавливающего средства. Поскольку существуют различные способы, авторы настоящего изобретения обнаружили, что согласно некоторым аспектам настоящего изобретения NaBH4, особенно в изопропаноле, предлагал возможность следовать идее однореакторной реакции. Использование этого восстанавливающего средства, таким образом, является особенно предпочтительным, если способ настоящего изобретения проводят как однореакторный процесс без каких-либо стадий очистки промежуточных соединений. Согласно некоторым вариантам осуществления реакцию проводят при температурах от 18°С до 30°С, предпочтительно при температурах от 20°С до 25°С.
Стадия (iv) является следующей:

На стадии (iv) пирролидиновое кольцо метилируется на N-атоме подходящим метилирующим средством, которое также конкретно не ограничено. На этой стадии никотин может образовываться в виде смеси его (R)- и (S)-энантиомеров.
Согласно некоторым вариантам осуществления стадию (iv) проводят с использованием муравьиной кислоты и параформальдегида или муравьиной кислоты и формальдегида, предпочтительно при температуре от 40 до 95°С, более предпочтительно от 60 до 85°С, еще более предпочтительно при температуре от 60 до 70°С, еще более предпочтительно при 65±2°С.
Один аспект настоящего изобретения состоит в том, что никаких дополнительных растворителей не требуется добавлять в реакционную смесь на этой стадии, не увеличивая количество растворителя, уже присутствующего в смеси, и/или не добавляя другой растворитель.
Согласно некоторым вариантам осуществления реакция протекает при предпочтительно повышенных температурах, температурах выше комнатной температуры. Обнаружили, что температуры предпочтительно составляют от 40 до 95°С, более предпочтительно от 60°С до 85°С, еще более предпочтительно от 60°С до 70°С, и наиболее предпочтительно температура составляет 65°С±2°С, чтобы получить желаемые результаты.
Согласно некоторым вариантам осуществления способ настоящего изобретения можно проводить как однореакторный процесс. Это, конечно, исключает дальнейшие стадии разделения и экономит растворители, энергию и время. В частности, однореакторный процесс можно обеспечивать при помощи предпочтительных стадий, указанных выше. Неожиданно обнаружили, что весь синтез можно проводить как однореакторный процесс без каких-либо стадий очистки промежуточных соединений в таких вариантах осуществления. Одним конкретным дополнительным преимуществом настоящего изобретения является то, что однореакторный синтез обеспечивает прямую последовательность реакций и использование минимальных количеств и типов растворителей. Согласно некоторым вариантам осуществления нет необходимости в изменении растворителя в таком однореакторном процессе.
Согласно некоторым вариантам осуществления соединение формулы I-а или соединение формулы I-c, т.е. смесь энантиомеров никотина или никотин в энантиочистой (R)-форме, затем приводят в реакцию с органическим основанием при температуре от 140 до 160°С. Таким образом, может происходить «рацемизация» полученной смеси, т.е. она может сдвигаться в направлении рацемической смеси. Согласно некоторым вариантам осуществления (R)-никотин можно сдвигать к (S)-никотину на этой стадии. Эта реакция происходит с образованной смесью, соединением формулы I-а, а также, например, с по существу чистым (R)-никотином, который можно, например, получать после выделения (S)-никотина из смеси энантиомеров никотина, как описано ниже.
Органическое основание конкретно не ограничено. Оно может быть алкоголятом с 1-20 атомами углерода и может быть получено из первичного, вторичного или третичного алкилового, алкенилового и/или алкинилового спирта и/или ароматического спирта. Согласно некоторым вариантам осуществления его получают из алкилового спирта с 1-20 атомами углерода, предпочтительно 1-10 атомами углерода, более предпочтительно 1-6 атомами углерода, в частности 1-4 атомами углерода, причем алкил может быть линейным или разветвленным. Например, оно может быть метанолятом, этанолятом, н-пропанолятом, изопропанолятом, н-бутанолятом, изобутанолятом, втор-бутанолятом, трет-бутанолятом и пр., предпочтительно бутанолятом, более предпочтительно трет-бутанолятом. Алкоголят может иметь подходящий катион, который конкретно не ограничен и может быть, например, выбран из одновалентных и двухвалентных катионов, например, из катионов щелочных и щелочноземельных металлов, предпочтительно катионов щелочных металлов, более предпочтительно Li+, Na+, K+ и их смесей. Подходящие предпочтительные алкоголяты предпочтительно представляют собой алкоголяты щелочных металлов, предпочтительно трет-бутаноляты щелочных металлов. Предпочтительно алкоголят щелочного металла выбирают из трет-бутанолята натрия, трет-бутанолята калия и их смесей. В одном аспекте обнаружили, что трет-бутанолят калия является подходящим основанием на этой стадии, которое особенно предпочтительно.
Органическое основание можно поставлять или чистым, т.е. без дополнительного растворителя, особенно если уже содержится растворитель из однореакторного процесса, или в подходящем растворителе, который конкретно не ограничен. Ароматические растворители, такие как бензол и толуол, предпочтительны, и толуол является особенно предпочтительным в качестве растворителя. Также являются подходящими ароматические растворители, содержащие неароматические растворители.
Согласно некоторым аспектам настоящего изобретения было обнаружено, что «рацемизация» может происходить при умеренных температурах, т.е. без нагревания до кипения никотина. Авторы настоящего изобретения обнаружили, что использование трет-бутоксида в толуоле, рацемизируя (R)-никотин в смесь (S)- и (K)-никотинас отношением от 45:55 до 55:45, можно проводить при температуре от 130 до 180°С, предпочтительно от 140 до 170°С, более предпочтительно от 140 до 160°С.
Согласно некоторым вариантам осуществления смесь энантиомеров никотина, например, рацемический никотин, можно разделять при помощи способа настоящего изобретения, используя экономичные и экологичные предпочтительные средства.
Согласно некоторым вариантам осуществления способ настоящего изобретения, таким образом, также предусматривает разделение энантиомеров соединения формулы I-а путем добавления хиральной O,O'-дизамещенной виннокаменной кислоты, предпочтительно дибензоилвиннокаменной кислоты или дитолуоилвиннокаменной кислоты или их смесей.
Никотин формулы I-а конкретно не ограничен и может быть получен вышеуказанным способом. Он представляет собой смесь (R)- и (S)-энантиомеров никотина, которая конкретно не ограничена и которая может содержать два энантиомера в любом отношении при условии, что оба энантиомера присутствуют. Она может быть рацемической смесью, т.е. смесью с мольным отношением 50:50, но она также может быть смесью с отношением (S)-энантиомера к (R)-энантиомеру в диапазоне, например, от 1:99 до 99:1, например, от 10:90 до 90:10, например, от 20:80 до 80:20, например, от 30:70 до 70:30, например, от 40:60 до 60:40, например, от 45:55 до 55:45, или любым другим отношением между этими отношениями. Способ настоящего изобретения позволяет выделять (S)-энантиомер из этой смеси.
Хиральная O,O'-дизамещенная виннокаменная кислота конкретно не ограничена при условии, что она является хиральной, т.е. оптически активной, и она не должна быть энантиочистой. Два заместителя на кислороде гидроксигрупп конкретно не ограничены и могут быть одинаковыми или различными. Согласно некоторым вариантам осуществления их выбирают из алкильных групп с 1-20 С-атомами, алкенильных и/или алкинильных групп с 2-20 С-атомами, арильных групп с 6-20 С-атомами и/или алкиларильных и/или арилалкильных групп с 7-20 С-атомами, все из которых могут быть замещенными или незамещенными функциональными группами, такими как галогенсодержащие группы, нитрогруппы, аминогруппы, сложноэфирные группы, амидные группы и пр., и все из которых предпочтительно являются незамещенными. Предпочтительные заместители в хиральной O,O'-дизамещенной виннокаменной кислоте представляют собой арильные группы с 6-20 С-атомами и/или алкиларильные и/или арилалкильные группы с 7-20 С-атомами, которые являются незамещенными.
Согласно некоторым вариантам осуществления хиральную O,O'-дизамещенную виннокаменную кислоту выбирают из O,O'-дибензоилвиннокаменной кислоты и O,O'-дитолуоилвиннокаменной кислоты, например, O,O'-ди-о-толуоилвиннокаменной кислоты, O,O'-ди-м-толуоилвиннокаменной кислоты и/или O,O'-ди-п-толуоилвиннокаменной кислоты, и/или их смесей, предпочтительно O,O'-дибензоилвиннокаменной кислоты. Согласно некоторым вариантам осуществления ее добавляют в этанол как растворитель.
В способе настоящего изобретения хиральная O,O'-дизамещенная виннокаменная кислота содержит предпочтительно L-энантиомер. O,O'-дизамещенная виннокаменная кислота может в этом случае или состоять из L-энантиомера, или содержать L-энантиомер и D-энантиомер в виде смеси. В последнем случае предпочтительно, чтобы L-энантиомер содержался в избытке относительно D-энантиомера, например, в мольном отношении L-энантиомера к D-энантиомеру по меньшей мере 80:20, предпочтительно по меньшей мере 90:10, причем это отношение можно также описать как энантиомерный избыток (ее) по меньшей мере 60%, предпочтительно по меньшей мере 80%.
Согласно некоторым вариантам осуществления хиральная O,O'-дизамещенная виннокаменная кислота представляет собой O,O'-дибензоил-L-виннокаменную кислоту, т.е. имеет ее 100%. Согласно некоторым вариантам осуществления ее добавляют в этанол как растворитель.
Хотя обнаружили, что разделение (R)- и (S)-никотина можно обеспечивать при помощи разделяющего средства, которое является чистым энантиомером, т.е. L-энантиомером хиральной O,O'-дизамещеннной виннокаменной кислоты, если (S)-никотин необходимо получить, также неожиданно обнаружили, что то же самое также достигается, если используют не чистое разделяющее средство, также называемое средством расщепления, а также когда используют смесь энантиомеров хиральной O,O'-дизамещеннной виннокаменной кислоты, что неожиданно обеспечивает эффект разделения. Хотя доступны чистые разделяющие средства/средства расщепления, преимуществом является использование экономически и экологически более легкодоступных средств в виде смесей с избытком одного энантиомера, например, L-энантиомера, если необходимо получить (S)-никотин (соединение формулы I-b).
Согласно некоторым вариантам осуществления O,O'-дизамещеннная виннокаменная кислота представляет собой смесь L-энантиомера и D-энантиомера, причем L-энантиомер содержится в избытке относительно D-энантиомера, причем предпочтительно мольное отношение L-энантиомера к D-энантиомеру составляет 80:20 или более, предпочтительно 90:10 или более. Согласно некоторым вариантам осуществления ее добавляют в этанол как растворитель.
Согласно некоторым вариантам осуществления O,O'-дизамещеннная виннокаменная кислота представляет собой смесь O,O'-дибензоил-L-виннокаменной кислоты (L-DBTA) и O,O'-дибензоил-D-виннокаменной кислоты (D-DBTA) с мольным отношением L-DBTA к D-DBTA 80:20 или более, предпочтительно 90:10 или более. Согласно некоторым вариантам осуществления ее добавляют в этанол как растворитель.
В способе настоящего изобретения растворитель, используемый для добавления O,O'-дизамещеннной виннокаменной кислоты, конкретно не ограничен и может быть любым подходящим растворителем, в котором O,O'-дизамещеннная виннокаменная кислота может быть растворена. Согласно некоторым вариантам осуществления растворителем является этанол. Для разделения смесь, полученную добавлением O,O'-дизамещеннной виннокаменной кислоты к соединению формулы I-а, можно, например, нагревать с обратным холодильником в течение некоторого периода времени для прохождения реакции в смеси.
После этой стадии соединение формулы I-b можно получать из этой прореагировавшей смеси. Получение соединения формулы I-b конкретно не ограничено и может проводиться подходящими способами, например, гидролизом полученной соли (S)-никотина с разделяющим средством и водой в щелочной среде, экстракцией органическим растворителем, таким как толуол, и дистилляцией растворителя. Для получения соли (S)-никотина с разделяющим средством ее можно предварительно осаждать, отфильтровывать и необязательно промывать, например, этанолом. Стадии осаждения, фильтрации и промывки здесь можно проводить повторно, например, два, три, четыре или более раз.
Типичная схема реакции для способа настоящего изобретения представлена ниже:

Согласно этой схеме смесь энантиомеров никотина можно синтезировать в однореакторном процессе (стадии 1a-d), начиная с конденсации этилникотината и 1-винил-2-пирролидона в присутствии основания, например, EtONa (стадия 1а). В присутствии сильной кислоты, такой как HCl, например, НСlводн., с азота амида снимают защитные группы, и происходит декарбоксилирование (стадия 1b). Восстановление пирролинового кольца в пирролидиновое кольцо выполняют, например, при помощи NaBH4 в изопропаноле (стадия 1с), с последующим метилированием в никотин, например, при помощи муравьиной кислоты и параформальдегида (стадия 1d) или муравьиной кислоты и формальдегида. Рацемическую смесь энантиомеров никотина, например, рацемическую смесь, можно расщеплять при помощи средства расщепления, такого как L-DBTA, с получением целевого продукта (S)-никотина (стадия 2). Полученный (R)-никотин можно рециркулировать путем рацемизации при помощи основания (стадия 3) и подвергания дополнительной стадии расщепления.
Вышеуказанные варианты осуществления можно произвольно объединять при необходимости. Дополнительные варианты осуществления и реализации настоящего изобретения включают также явно не указанные комбинации признаков, упомянутых выше или ниже в отношении примеров настоящего изобретения. В частности, специалист в данной области также добавит отдельные аспекты в качестве усовершенствований или дополнений к соответствующей основной форме настоящего изобретения.
Примеры
Настоящее изобретение будет теперь описано более подробно со ссылкой на несколько его примеров. Однако эти примеры являются иллюстративными и не ограничивают объем настоящего изобретения.
Пример 1
Общая процедура 1 для получения рацемического никотина
1,0 экв. этилникотината, толуол (50,0 г или 4,4 массовые части относительно этилникотината), необязательно этанол (1 г, 0,09 массовые части относительно этилникотината) и 1,3 экв. этоксида натрия нагревали при перемешивании до 80-85°С. 1,2 экв. 1-винил-2-пирролидона (NVP) загружали в реакционную смесь при 80-85°С в течение 1,5-2,0 часов в безводных условиях. Реакция протекала при 95-100°С в течение 3 часов. Затем реакция завершалась, и реакционную смесь дистиллировали в течение 2 часов для удаления некоторого количества растворителей (азеотроп, содержащий 68 масс. % этанола и 32 масс. % толуола). Оставшуюся реакционную смесь выливали в HCl (водн., 30% в воде; 58,0 г или 4,4 экв.). Низкокипящие компоненты, такие как ацетальдегид (из «винилового» компонента), этанол и газообразный СО2, удаляли дистилляцией. Когда температура реакции достигала 105°С, дистилляцию останавливали и реакционную смесь перемешивали при температуре от 90°С до 94°С в течение ночи. После завершения реакции рН доводили до значения от 9,5 до 10,0 при помощи NaOH (30 масс. % в воде). Изопропанол (29,0 г, как указано в таблице 1а) и 1,0 экв. NaBH4 (относительно этилникотината) загружали порциями в течение 1 часа в реакционную емкость. Реакция протекала при приблизительно 10°С в течение более чем 3 часов (в этот момент содержание миосмина было ниже 3,0 масс. %). Муравьиную кислоту (НСООН) добавляли, реакционную смесь нагревали и формальдегид (Н2СНО) медленно добавляли и смесь перемешивали при 60°С. После завершения реакции (в этот момент содержание миосмина было ниже 0,5 масс. %), рН смеси медленно доводили до значения 10,5-11,0 при помощи NaOH (30 масс. % в воде) и перемешивание проводили при 55°С в течение 30 минут, пока не наблюдали разделение фаз. Смесь экстрагировали дважды толуолом. Объединенные органические фазы концентрировали с получением неочищенного продукта. Дистилляцией неочищенного продукта получали рацемический никотин в виде бесцветного масла, как показано в таблице 1а.
Общая процедура 2 для получения рацемического никотина
1,0 экв. этилникотината, толуол, 1,6 экв. этоксида натрия и 1,2 экв. 1-винил-2-пирролидина (NVP) загружали в колбу при комнатной температуре приблизительно 20°С в безводных условиях. Затем реакция протекала при 100°С в течение 3 часов. Реакция завершалась, и после охлаждения смеси до 30°С добавляли по каплям HCl (36 масс. % в воде). Низкокипящие компоненты, такие как ацетальдегид (из «винила»), этанол и газообразный СО2, удаляли дистилляцией вместе с частями толуола и воды. Когда температура реакции достигала 105°С, дистилляцию останавливали и реакционную смесь перемешивали при температуре от 100°С до 105°С в течение ночи. После завершения реакции рН доводили до значения от 9,5 до 10,5 при помощи NaOH (30 масс. % в воде). Изопропанол и 1,0 экв. NaBH4 (относительно этилникотината) загружали в реакционную емкость. Реакция протекала при комнатной температуре (приблизительно 20°С) в течение более чем 3 часов (в этот момент содержание миосмина составляло менее 3,0 масс. %). Муравьиную кислоту (НСООН) и параформальдегид ((НСНО)n) добавляли и смесь перемешивали при 65°С в течение по меньшей мере 3 часов. После завершения реакции (в этот момент содержание миосмина было ниже 0,5 масс. %) рН смеси доводили до значения 13-14,0 при помощи NaOH (30 масс. % в воде). Воду добавляли, пока все неорганические твердые вещества не растворялись. Смесь экстрагировали дважды толуолом. Объединенные органические фазы концентрировали с получением неочищенного продукта. Дистилляцией неочищенного продукта получали рацемический никотин в виде бесцветного масла, как показано в таблице 1b.

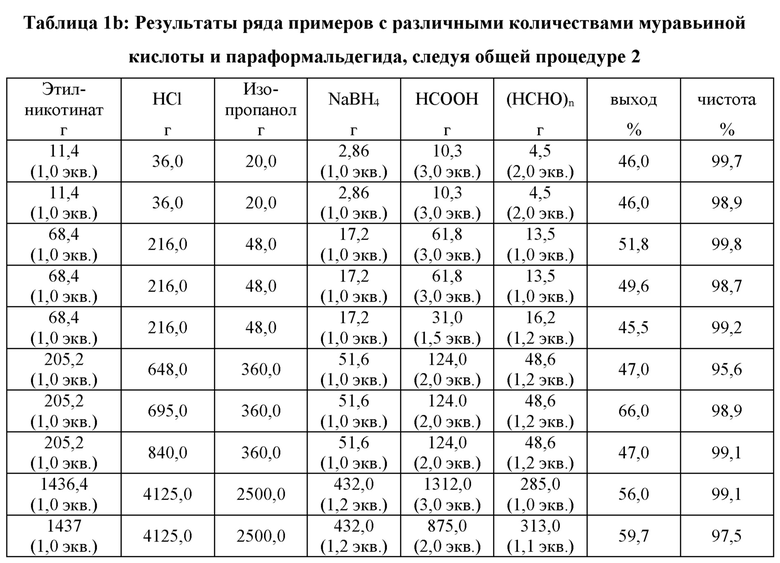
Во всех случаях использовали точно 1,2 экв. NVP относительно этилникотината, точно 1,6 экв. EtONa относительно этилникотината и 60,0 г толуола на 11,4 г этилникотината.
В седьмом примере общей процедуры 2 с выходом 66,0%, отмеченным в таблице 1b, следует отметить, что смесь, полученную реакцией этилникотината и NVP в толуоле и этоксиде натрия, добавляли по каплям в водную HCl, а не как указано выше.
Пример 2: Стадия расщепления
1,0 г рацемического никотина, полученного в предыдущих примерах, смешивали при комнатной температуре с этанолом (1) и 2,2 г виннокаменной кислоты (DBTA) (1 эквивалент). Смесь нагревали с обратным холодильником в течение нескольких минут и охлаждали до комнатной температуры (приблизительно 20°С). Начиналось осаждение, и смесь перемешивали в течение ночи (10-12 часов) при 20°С. Осадок, который образовался, отфильтровывали, промывали этанолом (2). Неочищенный продукт растворяли в этаноле (3). Смесь нагревали с обратным холодильником в течение нескольких минут и охлаждали до комнатной температуры. Начиналось осаждение, и смесь перемешивали в течение ночи (10-12 часов) при 20°С. Осадок отфильтровывали и промывали этанолом (4). Продукт сушили и чистый продукт получали.
3,2 г никотин-L-DBTA, полученной в примере 1b, образец 1, суспендировали в 7,2 г воды и 7,2 г толуола. Водный аммиак (25 масс. %) добавляли, пока рН не становился от 9,8 до 10,4. Фазы разделяли, и водную фазу экстрагировали дважды при помощи 2,4 г толуола. Толуоловые фазы объединяли, и толуол удаляли дистилляцией. Осадок дистиллировали под вакуумом, получая 0,93 г чистого (S)-никотина. Энантиочистоту определяли хиральной ВЭЖХ.
Используя различные количества, указанные в таблице 2, проводили подобные эксперименты по расщеплению/разделению.

Использовали мольные эквиваленты средства расщепления и рацемического никотина. Количество этанола выбирали как кратное массе рацемического никотина.
Кроме того, обнаружили, что увеличенный выход и чистоту можно получать с введением затравки при 40°С.
Пример 3
Эквивалентные количества дибензоил-D-виннокаменной кислоты (23,2 г) и рацемического никотина (10,0 г) растворяли в этаноле и перемешивали в течение 1 часа, нагревали с обратным холодильником в течение 15 минут, охлаждали до комнатной температуры и перемешивали в течение еще одного часа. Получали дибензоил-D-тартрат (R)-никотина. После повторной кристаллизации в смеси изопропанола-метанола (1,0:0,3) получали (R)-никотин. Результаты даны в следующей таблице 3.

Пример 4: Стадия рацемизации
Никотин, рециркулированный из маточных жидкостей, полученных в примере 2 после выделения (S)-никотина, был обогащен (R)-никотином и обычно показывал мольное отношение 70:30 (R:S) и, таким образом, был «рацемизирован», как описано ниже.
Однако процедура рацемизации, описанная в следующем параграфе, применима для любой смеси (R)-никотина и (S)-никотина (R:S никотин). Количество никотина можно или определять аналитическими методами (например, количественной ВЭЖХ), или оценивать по массовому балансу чистого (S)-никотина относительно никотина, введенного в эксперимент по расщеплению рацемической смеси, как описано выше.
Собирали все маточные жидкости из эксперимента по расщеплению и растворители удаляли вакуумной дистилляцией. Остаток подщелачивали (рН обычно >12) путем добавления водного NaOH (30 масс. %).
Смесь экстрагировали толуолом - дважды 7 объемами относительно введенного никотина. Толуоловые фазы объединяли и растворитель удаляли дистилляцией под давлением окружающей среды.
Затем 5 масс. % KO-tBu добавляли (относительно введенного никотина) и смесь нагревали до 160°С в течение 1 ч. После этой тепловой обработки никотин отгоняли вакуумной дистилляцией. Восстановленный никотин показывает энантиомерный избыток >90%, т.е. имеет мольное отношение или отношение (R)-никотина к (S)-никотину от 55:45 до 45:55.
Смесь (R)- и (S)-никотина можно снова расщеплять при помощи способов, раскрытых в соответствующих примерах.
| название | год | авторы | номер документа |
|---|---|---|---|
| ХИРАЛЬНОЕ РАЗДЕЛЕНИЕ СМЕСИ ЭНАНТИОМЕРОВ НИКОТИНА | 2018 |
|
RU2753492C1 |
| СПОСОБ ПОЛУЧЕНИЯ L-НИКОТИНА | 2022 |
|
RU2836890C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХИРАЛЬНОГО СИНТЕТИЧЕСКОГО НИКОТИНА | 2021 |
|
RU2780283C1 |
| Способ получения дексмедетомидина и его фармацевтически приемлемых солей | 2023 |
|
RU2824994C1 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ ОБРАТНОГО ЗАХВАТА ДОПАМИНА, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2004 |
|
RU2358719C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ ОБРАТНОГО ЗАХВАТА ДОПАМИНА, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 1999 |
|
RU2238084C2 |
| ПРОЛЕКАРСТВА МЕТИЛФЕНИДАТА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2012 |
|
RU2573835C2 |
| СПОСОБ ПОЛУЧЕНИЯ РАЦЕМИЧЕСКОГО НЕБИВОЛОЛА | 2006 |
|
RU2392277C2 |
| СПОСОБ СТЕРЕОСЕЛЕКТИВНОГО ПОЛУЧЕНИЯ АСИММЕТРИЧНО АЛКИЛИРОВАННЫХ ПРОИЗВОДНЫХ ОКСИНДОЛА | 1991 |
|
RU2072354C1 |
| СПОСОБ СИНТЕЗА (1S,2R)-МИЛНАЦИПРАНА | 2010 |
|
RU2521342C2 |
Изобретение относится к способу получения соединения формулы I-a, в котором проводят (i) реакцию этилникотината и N-винилпирролидона в присутствии алкоголята-основания с получением 3-никотиноил-1-винилпирролидин-2-она; (ii) реакцию 3-никотиноил-1-винилпирролидин-2-она с кислотой с получением миосмина; (iii) восстановление миосмина до норникотина при помощи восстанавливающего средства и (iv) метилирование норникотина с получением соединения формулы Ia, где стадию (ii) проводят с использованием неорганической кислоты, и где способ представляет собой однореакторный процесс. Технический результат: разработан способ получения соединения формулы I-a, представляющий однореакторный процесс, с высоким выходом и чистотой целевого соединения. 14 з.п. ф-лы, 4 табл., 4 пр.

1. Способ получения соединения формулы I-a, предусматривающий

(i) реакцию этилникотината и N-винилпирролидона в присутствии алкоголята-основания с получением 3-никотиноил-1-винилпирролидин-2-она;
(ii) реакцию 3-никотиноил-1-винилпирролидин-2-она с кислотой с получением миосмина;
(iii) восстановление миосмина до норникотина при помощи восстанавливающего средства и
(iv) метилирование норникотина с получением соединения формулы Ia,
где стадию (ii) проводят с использованием неорганической кислоты, и где способ представляет собой однореакторный процесс.
2. Способ по п. 1, в котором стадию (i) проводят в присутствии ароматического растворителя.
3. Способ по п. 2, в котором ароматический растворитель представляет собой бензол, толуол или их смесь.
4. Способ по любому из предшествующих пунктов, в котором основание-алкоголят на стадии (i) представляет собой алкоголят щелочного металла.
5. Способ по п. 4, в котором алкоголят щелочного металла выбирают из этанолята натрия, этанолята калия и их смесей.
6. Способ по любому из предшествующих пунктов, в котором основание-алкоголят добавляют на стадии (i) в количестве от 1,4 до 2,0 эквивалентов в пересчете на 1 эквивалент этилникотината.
7. Способ по любому из предшествующих пунктов, в котором стадию (ii) проводят при температуре от 90 до 115°C.
8. Способ по п. 7, в котором неорганическая кислота представляет собой HCl.
9. Способ по любому из предшествующих пунктов, в котором стадию (iv) проводят при помощи муравьиной кислоты и параформальдегида, или муравьиной кислоты и формальдегида.
10. Способ по п. 9, в котором стадию (iv) проводят при температуре от 40 до 95°C.
11. Способ по п. 10, в котором стадию (iii) проводят при помощи NaBH4 в качестве восстанавливающего средства.
12. Способ по любому из предшествующих пунктов, дополнительно предусматривающий реакцию соединения формулы I-a с органическим основанием при температуре от 140 до 160°C.
13. Способ по любому из предшествующих пунктов, дополнительно предусматривающий разделение энантиомеров соединения формулы I-a путем добавления хиральной O,O’-дизамещеннной виннокаменной кислоты.
14. Способ по п. 13, в котором дибензоилвиннокаменная кислота представляет собой O,O’-дибензоил-L-виннокаменную кислоту.
15. Способ по п. 13, в котором дибензоилвиннокаменная кислота представляет собой смесь O,O’-дибензоил-L-виннокаменной кислоты (L-DBTA) и O,O’-дибензоил-D-виннокаменной кислоты (D-DBTA) с мольным отношением L-DBTA к D-DBTA 80:20 или более.
| WO 2016065209 A2, 28.04.2016 | |||
| В.Г | |||
| Ненайденко и др | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Известия Академии наук | |||
| Серия химическая, 2003, N11, 2338-2347 | |||
| US 8884021 B2, 11.11.2014 | |||
| СПОСОБ ПРОИЗВОДСТВА КОНСЕРВОВ "ЯЗЫК С КАПУСТОЙ И СОУСОМ СМЕТАННЫМ С ХРЕНОМ" | 2012 |
|
RU2484673C1 |

