ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение в общем относится к каталитическим реакциям алкилирования ароматических соединений и, в частности, к катализаторам цеолитового типа, используемых в этих реакциях.
УРОВЕНЬ ТЕХНИКИ
Алкилароматические соединения представляют собой важное семейство веществ, которые используют в качестве сырьевых материалов во многих областях промышленности, таких как производство пластификаторов, полимерных материалов, инсектицидов, в сельском хозяйстве для предотвращения слипания удобрений, в производстве текстильных изделий и волокон, в кожевенной и скорняжной промышленности, при производстве гербицидов, в способах промышленной очистки, в фотографической промышленности, при производстве клеящих материалов и огнегасящих продуктов, таких как увлажняющие вещества, в электрохимических процессах для удаления загрязнения и смазочных веществ с поверхности подложки, и в биологически разлагаемых детергентах, причем в этом случае они являются линейными моноалкилароматическими соединениями (Surfactants in Consumers Products, Theory, Technology and Application, Edited by J.Falbe, Springer Verlag, 1987).
Стандартный способ, используемый в нефтехимической промышленности для получения линейных моноалкилароматических соединений, особенно для применений в детергентах, состоит в дегидрогенизации линейных парафинов для получения линейных моноолефинов и затем выполнения алкилирования бензола этими моноолефинами так, чтобы образовать линейную цепь моноалкилированного продукта (линейное моноалкилароматическое соединение), также называемого линейным алкилбензолом (ЛАБ). Линейный алкилсульфонат (ЛАС) является продуктом, который используют в конечных составах детергентов. ЛАС получают сульфонированием ЛАБ и последующей нейтрализацией соответствующих сульфокислот (Н-ЛАС) водными растворами щелочных или щелочноземельных гидроксидов согласно стандартным способам уровня техники. Линейные олефины, используемые в способе, имеют от девяти до шестнадцати атомов углерода. Стадию алкилирования проводят в жидкой фазе в присутствии катализаторов типа Фриделя-Крафтса, например плавиковой кислоты. Фтороводородный способ хорошо известен и его используют в промышленности (с его помощью получают примерно 75% от 3,3 млн метрических тонн ЛАБ, получаемых в год), получая высокий выход (>92 мас.%) ЛАБ с относительно низкой селективностью по отношению к 2-фенилизомерам, составляющей менее 20%. Объединенный способ получения ЛАБ описан в Handbook of Petroleum Refining Process, published by Robert A.Meyers, 1986, pp.1-23, включенном в качестве ссылки. В патенте US 5276231 описывают промежуточные стадии способа получения ЛАБ, такие как селективная гидрогенизация диолефиновых побочных продуктов, образующихся при дегидрогенизации парафинов, и отделение нелинейных побочных продуктов от потока стадии дегидрогенизации. Однако использование HF имеет некоторые недостатки на эксплуатационном уровне, так как она требует очень осторожного обращения и оборудования, изготовленного из специальных материалов, из-за ее высокой коррозионной способности, что приводит к высоким фиксированным и текущим расходам; поэтому осуществляют попытки разработать альтернативные катализаторы на основе твердых веществ с кислотной природой. В настоящее время единственным способом, внедренным на промышленном уровне, в котором используют гетерогенный катализ, является способ DETAL® (основанный на патентах PI 9204326-7, ES 2007545 и US 5146026), с помощью которого получают примерно 15% мирового производства ЛАБ. Он характеризуется использованием аморфных фторированных алюмосиликатов в качестве гетерогенного катализатора и этим способом получают примерно 30 мас.% 2-фенилизомеров.
Реакцию алкилирования можно охарактеризовать следующими показателями: конверсия, селективность по отношению к моноалкилбензолу и распределение изомера.
1) Конверсия алкилирования или, более конкретно, относительная конверсия:
В реакции алкилирования, рассматриваемой в данном изобретении, ароматические соединения всегда используют в стехиометрическом избытке по отношению к олефинам. Относительную конверсию можно определить как долю лимитирующего реагента, в данном случае олефина, которая потребляется для получения всех продуктов, таким образом:
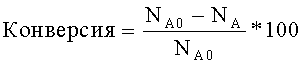
где NA0 представляет собой количество молей олефина на входе в реактор и NA представляет собой количество молей того же реагента на выходе реактора.
2) Селективность по отношению к моноалкилароматическим соединениям:
Ее определяют как
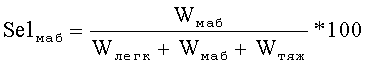
где Wмаб представляет собой массу моноалкилированного ароматического соединения (моноалкилароматического соединения), которое нужно получить, Wлегк представляет собой массу всех побочных продуктов, более легких, чем самое легкое представляющее интерес моноалкилароматическое соединение, и Wтяж представляет собой общую массу веществ, молекулярная масса которых выше, чем молекулярная масса получаемых моноалкилароматических соединений.
Группа тяжелых алкилатов включает все химические вещества с молекулярной массой выше, чем у моноалкилароматического соединения. Она обычно состоит из полиалкилароматических соединений (в основном диалкилароматических соединений), дифенилалканов, олигомеризованных олефинов и алкилатов этих олигомеризованных олефинов, образующихся в течение стадии алкилирования. Эти продукты в основном образуются в течение реакции алкилирования. Диалкилароматические соединения образуются путем алкилирования олефином ранее образованного моноалкилароматического соединения. Дифенилалканы образуются путем алкилирования бензола диолефином, который дегидроциклирован. Образование тяжелых побочных продуктов этого сорта в способе получения моноалкилароматического соединения нежелательно, так как эти побочные продукты не обладают никаким моющим действием (очищающей способностью) в процессе мытья вследствие их сильной липофильной природы. При их образовании, они уменьшают экономическую эффективность способа получения моноалкилароматического соединения, так как они не допускают интегрального использования сырьевых материалов. К тому же их необходимо отделить от моноалкилароматического соединения, чтобы они не влияли на поверхностно-активную силу конечного ЛАС, и их позиционируют на рынке как эмульгаторы, понижающие стоимость товара. К тому же среди тяжелых алкилатов существуют другие соединения, которые нужно принимать во внимание, такие как алкилполиароматические соединения, образующиеся путем алкилирования моноолефинами полиароматических соединений, полученных на стадии дегидрогенизации. Даже на уровне следов эти побочные продукты резко понижают качество конечного ЛАС, так как они значительно увеличивают его цветность после сульфонирования. Более того, их нельзя отделить от целевого продукта, так как они проявляются на уровне следов и их элюируют совместно с наиболее тяжелыми моноалкилированными ЛАБ из-за перекрывания диапазонов их температур перегонки.
3) Распределение изомеров:
Среди получаемых моноалкилароматических соединений распределение изомеров можно определить как массовое процентное содержание каждого типа полученных изомеров, таких как 2-фенил, 3-фенил…6-фенилизомеры, также как и разветвленного алкилата.

Распределение изомеров играет очень важную роль в растворимости и стабильности конечных детергентов, особенно жидких составов, так же как и в их поверхностной активности и в скорости их биологического разложения.
2-фенилизомеры представляют собой такие алкилированные молекулы, которые имеют ароматическое кольцо, связанное с алкильной цепью в положении 2 алкильной цепи. Содержание 2-фенилизомеров определяют как массовое процентное содержание 2-фенилизомеров в смеси ЛАБ или ЛАС и его вычисляют по следующей формуле:
2-фенилизомер [%]=(масса 2-фенилизомера)*100/(общая масса ЛАБ или ЛАС)
В настоящее время технологии, внедренные на промышленном уровне (HF и DETAL®), дают возможность получать ЛАБ только со средним содержанием 2-фенилизомеров 18 и 30% соответственно. В отношении растворимости и стабильности идеальный диапазон концентрации 2-фенилизомеров составляет от 25 до 30 мас.%. Однако смеси ЛАБ с содержанием внешнего изомера (2+3 фенил) более 60 мас.% характеризуются тем, что обеспечивают ЛАС с сильно увеличенной поверхностной активностью после сульфонирования и нейтрализации. Однако эти ЛАС обладают значительным недостатком, обусловленным их низкой растворимостью в холодной воде и высокой вязкостью. Смеси ЛАС, включающие более 60 мас.% внешних изомеров (2+3 фенил) имеют тенденцию к образованию сильно нерастворимых гелей (низкая температура помутнения при охлаждении) с высокой вязкостью, что затрудняет их использование и обработку. По этой причине было бы желательно включить гидротроп для улучшения растворимости конечного поверхностно-активного вещества, когда содержание 2-фенилизомеров превышает 60 мас.%. Хотя существует много патентов, связанных с использованием гидротропов, один из них рассматривают как наиболее рекомендуемый для этого способа. PCT/ES2005000169 относится к способу получения подходящего гидротропа из предварительно дегидрогенизированных парафинов, особенно из таких побочных продуктов, выделенных в течение стадии очистки моноолефина.
Наконец, разветвленные алкилароматические соединения (разветвленные алкилаты) можно определить как такие алкилароматические соединения, в которых алкильная цепь, связанная с ароматическим кольцом, является не линейной или нормальной, а разветвленной алкильной группой. Эти отличные от нормальных алкильные группы содержат радикалы, такие как метил, этил, пропил, изопропил, бутил, изобутил, третбутил и различные варианты гексила, гептила, октила, присоединенные в любом месте алкильной цепи за исключением концов цепи. Разветвленные алкилаты образуются путем алкилирования разветвленными олефинами, полученными из тех разветвленных парафинов, которые присутствуют в свежих исходных парафинах, или путем процессов алкильной перегруппировки, которые протекают в течение стадий дегидрогенизации и алкилирования.
Среди разветвленных алкилатов есть алкилаты, которые имеют один из атомов углерода алифатической алкильной группы в положении четвертичного углерода. Четвертичный атом углерода в алкильной цепи определяют как углерод, связанный с четырьмя другими атомами углерода, и один из них может быть связан с углеродным атомом в фенильной группе, образуя четвертичный алкилфенилалкан. Если четвертичный углерод является вторым атомом углерода в алкильной цепи, четвертичный углерод, присутствующий в полученном 2-алкил-2-фенилалкане, можно назвать «концевым четвертичным углеродом». Известно, что подобно алкилатам с одним разветвлением эти вещества имеют скорость биологического разложения, аналогичную линейному алкилбензолсульфонату. Однако, когда четвертичный углерод является другим атомом углерода в алкильной цепи, например 5-метил,5-фенилалканом, его называют «внутренним четвертичным углеродом», и соответствующий алкилбензолсульфонат обладает намного меньшей скоростью биологического разложения. В статьях "Iso-branching of LAS biodegradation study of two model compounds", L.Cavalli, G.Cassani, M.Lazzarin, C.Maraschin, G.Nuzzi, J.L.Berna, J.Bravo, J.Ferrer, A.Masno, Toxicology & Environmental Chemistry, vol.54, pp.167-186, 1966 и "Biodegradation of co-products of commercial LAS", A.M.Nielsen, L.N.Britton, L.Cavalli, J.L.Berna, The Cler Review, vol.2, №1, pp.14-27, 1996 представлено научное доказательство поведения при биологическом разложении этих разветвленных алкилбензольных производных.
Международная заявка на патент WO 2007/104805, рассматриваемая в качестве ближайшего аналога для этого изобретения, относится к способу получения линейных алкилбензолсульфонатов с регулируемым содержанием 2-фенилизомеров и очень низкой цветностью после сульфонирования, в котором используют каталитическую систему на основе твердых высокостабильных катализаторов с высокой селективностью по отношению к моноалкилатным соединениям. Однако посредством описанного в этом патенте способа получают содержание разветвленного алкилбензола (более 4 мас.%, хотя можно достичь до 10 мас.% при определенных рабочих условиях) значительно более высокое, чем это содержание, получаемое, как в стандартном промышленном способе, основанном на гомогенном катализе с HF (примерно 3,5 мас.%), так и в промышленном способе, основанном на гетерогенном катализе (способ DETAL®, в котором получают примерно 3 мас.% этих разветвленных продуктов). Так как было показано, что полученные из обоих этих промышленных способов ЛАС быстро и полно биологически разлагаются при этих содержаниях разветвленного алкилата (Berna J.L. et al. Tenside Surfactants Detergents, 26, 2, 1989), было бы целесообразно, чтобы новые способы алкилирования, основанные на гетерогенном катализаторе, обеспечивали возможность получения линейных моноалкилароматических соединений с предпочтительно более низким или по меньшей мере аналогичным количеством этих разветвленных побочных продуктов в сравнении с таким количеством, получаемым в используемых в настоящее время технологиях. Таким образом обеспечивают оптимальное экологическое поведение сульфонированного и нейтрализованного конечного продукта (ЛАС).
Более того, хотя катализаторы, предусматриваемые в международной заявке на патент WO 2007/104805, способны увеличивать продолжительность реакционных циклов по сравнению с гетерогенными катализаторами, используемыми в настоящее время в способе DETAL (60 часов против 24 часов с современной гетерогенной технологией), на промышленном уровне важно иметь возможность увеличивать эту продолжительность даже больше. Таким способом можно уменьшить частоту и количество циклов промывки катализатора. Это отражается на понижении текущих затрат на вспомогательное оборудование (более длительное чистое время эксплуатации реактора и уменьшение затрат на стадии регенерации катализатора), также как и на понижении потребления энергии (нагнетание и нагревание регенерирующего агента) и выбросов в атмосферу, связанных с этим процессом.
Наконец, хотя катализаторы, предусматриваемые в международной заявке на патент WO 2007/104805, способны уменьшать образование тяжелых побочных продуктов по сравнению с существующей технологией, было бы преимущественным иметь возможность снизить их образование еще больше, чтобы максимизировать экономическую эффективность способа получения ЛАБ (интегральная утилизация сырьевых материалов), так же как и улучшить качество продукта посредством уменьшения его цветности после сульфонирования.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложен способ получения моноалкилароматических соединений с высокой линейностью (их будем называть линейными моноалкилароматическими соединениями, хотя они могут содержать минимальные количества разветвленных алкилатов), минимальным содержанием тяжелых алкилатов и минимальной цветностью, в то же время также обладающих регулируемым содержанием 2-фенилизомеров, в котором используют новый катализатор, обеспечивающий низкое содержание 2-фенила, который может быть более селективным, активным и стабильным, чем катализаторы, предусматриваемые в предшествующем уровне техники. Благодаря большей селективности по отношению к моноалкилароматическим соединениям с этим новым катализатором можно получать конечный продукт с более низким содержанием тяжелых алкилатов, чем это содержание, обеспечиваемое в предшествующем уровне техники, что увеличивает экономическую эффективность способа через повышение утилизации сырьевых материалов и в то же время улучшает его качество из-за уменьшения цветности после сульфонирования получаемого ЛАС. Эту цветность дополнительно минимизируют посредством подходящей очистки как сырьевых материалов, так и получаемого линейного моноалкилароматического соединения. К тому же получают линейное моноалкилароматическое соединение с содержанием разветвленных алкилатов, равным этой величине для технологий, используемых в данной области, которое обеспечивает быстрое полное биологическое разложение получаемого ЛАС. Катализатор более устойчив к деактивации, чем катализаторы, предусматриваемые в предшествующем уровне техники, что обеспечивает большее количество реакционных циклов и меньшую частоту циклов промывки, в то время как сохраняется высокая активность, что приводит к более низким эксплуатационным затратам.
Этот способ включает способ утилизации примесей промежуточных потоков так, чтобы получать гидротроп, который, добавленный соответствующим образом, когда содержание 2-фенилизомеров в моноалкилароматическом соединении превышает 60 мас.%, дает возможность получить продукт с более высокой растворимостью, чем при добавлении других гидротропов, обычно применяемых в предшествующем уровне техники. К тому же, так как новый катализатор более устойчив к деактивации при загрязнении, длительность реакционных циклов существенно возрастает и частота циклов промывки уменьшается. Это приводит к более длительным циклам производства и уменьшению уровня потребления энергии, связанного со стадией регенерирующей промывки (нагнетание промывочного агента и нагревание).
Поэтому первый аспект изобретения относится к способу получения линейного моноалкилароматического соединения с содержанием 2-фенилизомеров от 18 до 70 мас.% посредством каталитического алкилирования ароматического соединения очищенным алкилирующим агентом, включающему следующие стадии:
1) каталитической дегидрогенизации подаваемого потока линейных парафинов с получением линейных моноолефинов, непреобразованных парафинов и определенного количества побочных продуктов, таких как диолефины и нелинейные соединения;
2) обработки выходящего потока стадии (1) для селективной гидрогенизации диолефинов, полученных в качестве побочных продуктов на стадии (1), до моноолефинов с получением таким образом исходного алкилирующего агента, включающего линейные моноолефины, непреобразованные парафины и нелинейные соединения;
3) очистки исходного алкилирующего агента путем отделения нелинейных продуктов, содержащихся в выходящем потоке стадии (2) так, что полученный очищенный алкилирующий агент содержит моноолефины и парафины;
4) обработки нелинейных продуктов, отделенных на стадии (3), с образованием гидротропного предшественника,
5) алкилирования ароматического углеводорода моноолефинами, присутствующими в очищенном алкилирующем агенте, посредством сочетания двух процессов алкилирования на основе:
а) процесса алкилирования с катализатором, который обеспечивает получение линейного алкилароматического соединения с максимальным содержанием 2-фенилизомеров, составляющим 20 мас.%,
б) процесса алкилирования с катализатором, который обеспечивает получение линейного алкилароматического соединения с минимальным содержанием 2-фенилизомеров, составляющим 20 мас.%, который включает цеолит типа MOR, от 0,01 мас.% до 0,20 мас.% по меньшей мере одного из металлов, выбираемых из группы, состоящей из Li, Na, K, Mg или Ca, при максимальном содержании Na 0,01%, и от 0 до 0,5 мас.% по меньшей мере одного из металлов, выбираемых из группы, состоящей из Ti, Zr, Hf;
6) фракционирования выходящего потока стадии (5) для отделения непрореагировавших ароматических соединений, парафинов и наиболее тяжелых побочных продуктов от целевых линейных моноалкилароматических соединений,
7) очистки фракции целевых линейных моноалкилароматических соединений, поступающих из стадии (6).
Данный способ отличается тем, что катализатор, который обеспечивает возможность получения максимально 20 мас.% 2-фенилизомеров, включает цеолит типа FAU, от 0,5 до 2 мас.% по меньшей мере одного из металлов, выбираемых из группы, состоящей из Li, Na, K, Mg или Ca, и от 8 до 16,5 мас.% по меньшей мере одного редкоземельного металла, выбираемого из группы, состоящей из La, Ce, Pr, Nd, Pm, Sm или Eu.
В настоящем изобретении, когда речь идет о цеолитах типа FAU или EMT-FAU, имеют ввиду группу цеолитов с изотипическими структурами, соответствующими структурному типу FAU, таких как цеолит Y, цеолит Na-X, кремнистый Na-Y, цеолит Линде X, цеолит Линде Y, цеолит ZSM-3 и цеолит ZSM-20, предпочтительно цеолит Линде или цеолит Y.
В настоящем изобретении, когда речь идет о цеолитах типа MOR, имеют ввиду группу цеолитов с изотипическими структурами, соответствующими структурному типу MOR, таких как морденит, цеолит Na-D и цеолит Ca-Q, наиболее предпочтительно морденит,
В конкретном воплощении настоящего изобретения катализатор, который обеспечивает получение максимально 20% 2-фенилизомеров, включает 0,9 мас.% Na.
В другом конкретном воплощении настоящего изобретения катализатор, который обеспечивает получение максимально 20% 2-фенилизомеров, включает от 4,5 до 10 мас.% La, от 1,2 до 4 мас.% Ce, от 0,5 до 1,5 мас.% Pr и от 2 до 3 мас.% Nd.
В другом конкретном воплощении настоящего изобретения катализатор, который обеспечивает получение максимально 20% 2-фенилизомеров, включает:
а) дифракционную рентгенограмму порошка, отличающуюся тем, что наиболее интенсивный дифракционный пик появляется при угле 2 тэта, соответствующем 6,2°, и другие основные пики появляются при углах дифракции 2 тэта, соответствующих 23,6°; 20,3°; 21,6°; 27,0°; 31,3°, расположенных в порядке убывания интенсивности соответствующих пиков;
б) общее молярное отношение кремний/алюминий от 0,5:1,0 до 3,0:1,0, предпочтительно от 0,5:1,0 до 2,0:1,0;
в) молярное отношение кремний/алюминий по структурной решетке от 1,5:1,0 до 2,5:1,0, предпочтительно от 2,1:1,0 до 2,3:1,0;
г) общую удельную площадь поверхности (БЭТ), составляющую от 500 до 1000 м2/г, предпочтительно от 600 до 700 м2/г;
д) площадь микропор, составляющую от 500 до 900 м2/г, предпочтительно от 500 до 600 м2/г,
е) удельный объем микропор, составляющий от 0,1 до 0,3 мл/г, предпочтительно от 0,19 до 0,22 мл/г,
ж) распределение макропор, в котором диаметр макропор составляет от 20 до 2000 ангстрем, предпочтительно 40 ангстрем.
В конкретном воплощении настоящего изобретения на стадии (1) парафины включают алканы с прямой цепью, содержащие от 8 до 30 атомов углерода, предпочтительно от 10 до 16 атомов углерода, более предпочтительно они содержат от 10 до 14 атомов углерода. Эти парафины можно дегидрогенизировать и очистить посредством любого способа, описанного в предшествующем уровне техники.
В конкретном воплощении настоящего изобретения ароматический углеводород выбирают из группы, состоящей из толуола, ксилола, бензола или их смесей, но предпочтительно он является бензолом.
В конкретном воплощении настоящего изобретения ароматический углеводород и олефины смешивают перед реакцией алкилирования стадии (5) в молярном отношении ароматический углеводород:олефин, составляющем от 5:1 до 70:1, предпочтительно от 10:1 до 30:1 и более предпочтительно от 10:1 до 15:1.
В конкретном воплощении настоящего изобретения смесь ароматического углеводорода и очищенного алкилирующего агента включает максимально 0,3 мас.% нелинейных соединений.
В конкретном воплощении настоящего изобретения смесь ароматического углеводорода и очищенного алкилирующего агента также включает от 0 до 0,1 мас.% воды.
В конкретном воплощении настоящего изобретения на стадии (5) реакции алкилирования осуществляют одновременно.
В конкретном воплощении настоящего изобретения на стадии (5) реакцию алкилирования осуществляют в реакторе с компоновкой катализатора, выбираемой из группы, состоящей из неподвижного слоя с одним катализатором, неподвижного слоя с двумя полностью перемешанными различными катализаторами, по меньшей мере двух различных неподвижных слоев с одним и тем же катализатором в каждом слое, по меньшей мере двух различных неподвижных слоев с различными катализаторами в каждом слое, псевдоожиженного слоя с одним или более различными катализаторами, суспензионного реактора с одним или более различными катализаторами.
В конкретном воплощении настоящего изобретения на стадии (5) реакцию алкилирования осуществляют в конфигурации реактора, которая включает по меньшей мере одну из конфигураций реактора, выбираемых из группы, состоящей из независимого реактора, по меньшей мере двух параллельных реакторов, по меньшей мере двух последовательных реакторов и сочетаний этих конфигураций.
В конкретном воплощении настоящего изобретения на стадии (3) способ очистки исходного алкилирующего агента осуществляют посредством технологий отделения нелинейных примесей, известных специалисту в данной области, таких как, например, гидрогенизация, фракционирование и селективная адсорбция. В случае селективной адсорбции слой адсорбента состоит по меньшей мере из одного материала, выбираемого из группы, состоящей из цеолитов, диоксида кремния, силикагеля, макропористого силиката магния, активированного оксида алюминия, оксида кремния-оксида алюминия, глин, молекулярных сит, ацетата целлюлозы, макропористого полистирольного геля, активированного угля и органоселективных полимерных мембран.
В конкретном воплощении настоящего изобретения обработка с образованием гидротропного предшественника на стадии (4) включает:
а) фракционирование нелинейных соединений, полученных на стадии (2), посредством перегонки при атмосферном давлении, причем диапазон температуры перегонки целевой фракции составляет от 195°С до 259°С;
б) селективную гидрогенизацию полиароматических веществ, содержащихся в целевой фракции, которую подвергли перегонке на предыдущей стадии.
В конкретном воплощении настоящего изобретения полученный на стадии (4) гидротропный предшественник включает:
- от 2 до 20 мас.% алкилароматических соединений с одной или более алкильными группами, которые в сумме содержат 4 атома углерода,
- от 5 до 40 мас.% алкилароматических соединений с одной или более алкильными группами, которые в сумме содержат 5 атомов углерода,
- от 15 до 30 мас.% алкилароматических соединений с одной или более алкильными группами, которые в сумме содержат 6 атомов углерода,
- от 0,5 до 50 мас.% алкилароматических соединений с одной или более алкильными группами, которые в сумме содержат 7 атомов углерода,
- от 0,01 до 10 мас.% алкилароматических соединений с одной или более алкильными группами, которые в сумме содержат 8 атомов углерода,
- от 0,5 до 10 мас.% алкилароматических соединений с одной или более алкильными группами, которые в сумме содержат 9 атомов углерода,
- от 0,5 до 10 мас.% алкилароматических соединений с одной или более алкильными группами, которые в сумме содержат 10 атомов углерода.
Гидротроп (гидротропный агент) как таковой образуют сульфонированием и нейтрализацией гидротропного предшественника либо отдельно, либо в смеси с линейным моноалкилароматическим соединением.
В конкретном воплощении настоящего изобретения способ очистки стадии (7) осуществляют посредством технологий удаления и/или отделения полиароматических и полиалкилароматических примесей, известных специалисту в данной области, таких как, например, гидрогенизация, фракционирование и адсорбция. В предпочтительном воплощении способ очистки стадии (7) осуществляют посредством селективной адсорбции, используя селективный адсорбент на основе глины, который включает:
а) общее молярное отношение кремний/алюминий от 3:1 до 5:1, предпочтительно 4,1:1,0;
б) от 1 до 4 мас.% Fe2O3, предпочтительно 2,9 мас.%,
в) от 0,5 до 2 мас.% K2O, предпочтительно 1,4 мас.%,
г) от 0,2 до 2 мас.% MgO, предпочтительно 1,2 мас.%,
д) от 0,1 до 1 мас.% TiO2, предпочтительно 0,45 мас.%,
е) от 1800 до 2500 частей на млн Na, предпочтительно 2200 мас. частей на млн,
ж) удельную площадь поверхности по БЭТ, составляющую от 150 до 500 м2/г, предпочтительно 260 м2/г,
з) суммарный объем пор от 0,1 до 2,0 мл/г, предпочтительно 0,42 мл/г,
и) распределение макропор, в котором диаметр макропор составляет от 20 до 800 ангстрем, предпочтительно от 20 до 200 ангстрем, более предпочтительно от 20 до 100 ангстрем при среднем диаметре исходя из объема пор, равного 34 ангстремам.
В конкретном воплощении настоящего изобретения гидротропный предшественник, полученный на стадии (4), добавляют в поток линейного моноалкилароматического соединения, когда содержание 2-фенилизомеров линейных моноалкилароматических соединений равно или более 60 мас.%, причем его добавляют перед стадией (7) очистки.
В конкретном воплощении настоящего изобретения процесс нейтрализации стадии (9) осуществляют посредством щелочного вещества, включающего один или более катионов, выбираемых из группы: Na, K, NH4 +, Mg, Ca, Ba, или посредством замещенных аммониевых щелочей.
В более конкретном воплощении настоящего изобретения олефины представляют собой α-олефины и включают от 9 до 30 атомов углерода.
В конкретном воплощении настоящего изобретения температура реакции составляет от 20 до 400°С.
В конкретном воплощении настоящего изобретения объемная скорость составляет от 1 час-1 до 15 час-1.
В конкретном воплощении способ, описанный в настоящем изобретении, включает дополнительную стадию (8) сульфонирования и нейтрализации соединения, полученного на стадии (7).
В конкретном воплощении настоящего изобретения гидротропный предшественник, полученный на стадии (4), сульфонируют и нейтрализуют отдельно и затем добавляют к продукту, полученному на стадии (8).
Другой аспект настоящего изобретения относится к линейному сульфонированному и нейтрализованному моноалкилароматическому соединению, полученному способом, описанным в настоящем изобретении.
Другой аспект настоящего изобретения относится к способу получения линейного моноалкилароматического соединения с содержанием 2-фенилизомеров по меньшей мере 18 мас.% посредством каталитического алкилирования ароматического соединения алкилирующим агентом, как описано ранее, где стадии (1), (2), (3) и (4) являются необязательными.
Другой аспект настоящего изобретения относится к соответствующим чистящим композициям для составов для мытья посуды, чистящих агентов для твердых поверхностей, жидких моющих средств, порошковых моющих средств, чистящих составов в форме пасты, гелей и моющих брусков, которые включают:
а) от 1 до 99 мас.% соединения, полученного на стадии (8),
б) от 99 до 1 мас.% других ингредиентов детергентов, выбранных из группы, состоящей из производных жирных спиртов, жирных кислот, алкилсульфатов, этаноламинов, аминоксидов, карбонатов щелочей, этанола, изопропанола, воды, хвойного масла, хлорида натрия, силиката натрия, полимеров, алкоксилатов спиртов, солей пербората, цеолитов, сульфатов щелочей, энзимов, гидротропов, красителей, ароматизаторов, консервантов, полировальных материалов, полиакрилатов, эфирных масел, щелочных гидроксидов, эфирсульфонатов, разветвленных алкилбензолсульфонатов, растворимых в воде, алкилфенолалкоксилатов, аминов жирных кислот, альфа-олефинсульфонатов, парафинсульфонатов, бетаинов, хелатирующих агентов, этоксилатов Wanin talo, этоксилатов полиэфираминов, блоксополимеров оксида этилена и оксида пропилена, этоксилированных спиртов, пропоксилированных спиртов, сложных метилэфирсульфонатов, алкилполисахаридов, н-метилглюкамидов, сульфонированного дифенилалкилоксида и полиэтиленгликоля.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На Фиг.1 показана схема способа, представляющего собой настоящее изобретение, в форме блок-схемы.
На Фиг.2 показана схема способа, представляющего собой настоящее изобретение, в форме блок-схемы, в которую включены стадии сульфонирования и нейтрализации.
На Фиг.3 показана схема реакции по настоящему изобретению в форме блок-схемы, где пропущены стадии каталитической дегидрогенизации парафина, гидрогенизации диолефина, очистки исходного алкилирующего агента и изолирования, очистки и добавления гидротропного предшественника,
ПОДРОБНОЕ ОПИСАНИЕ СПОСОБА РЕАЛИЗАЦИИ ВОПЛОЩЕНИЯ
На Фиг.1 представлена неограничивающая схема реализации данного изобретения.
Поток 15 представляет собой подачу линейных парафинов в блок дегидрогенизации и включает смесь свежих линейных парафинов, поток 10, с непревращенными линейными парафинами, отделенными в блоке очистки сырого алкилата, блок 350, и направляемых рециклом посредством потока 170. В блоке 300 дегидрогенизации подачу линейных парафинов превращают в смесь линейных моноолефинов, непрореагировавших парафинов и различных побочных продуктов, таких как диолефины и нелинейные соединения. В этом воплощении, в котором используют бензол в качестве ароматического углеводорода, сырой алкилат представляет собой смесь моноалкилбензолов, непрореагировавшего бензола, парафинов и легких и тяжелых побочных продуктов, которая составляет выходящий поток стадии алкилирования.
В блок очистки олефинов, блок 310, подают выходящий поток блока дегидрогенизации посредством потока 20, увеличивая чистое содержание моноолефинов путем конверсии некоторых побочных продуктов, полученных в блоке дегидрогенизации, в основном диолефинов, в моноолефины посредством реакции селективной гидрогенизации. Получаемый поток, поток 30, обрабатывают в блоке 320, который содержит селективный адсорбент для устранения нелинейных соединений, полученных в способе дегидрогенизации.
Свежий бензол нагнетают в процесс посредством потока 50 и его смешивают с непрореагировавшим бензолом, подаваемым рециклом (поток 160), который поступает из блока очистки сырого алкилата, блок 350. Смесь этих двух потоков образует подачу бензола (поток 55), которую смешивают с потоком (поток 40), выходящим из блока 320 селективной адсорбции, с образованием потока 60, состоящего из моноолефинов, бензола и парафинов, который подают в блоки алкилирования. Поток 60 разделяют на два одинаковых потока (по составу, но необязательно по скорости подачи), 60a и 60b, которые подают в два различных реактора алкилирования, блоки 330 и 340, соответственно; в реакторе алкилирования 330 используют катализатор, который обеспечивает получение выходящего потока (поток 70) с сырым алкилатом, в котором моноалкилбензолы имеют максимальное содержание 2-фенилизомеров, равное 20 мас.%, в то время как в реакторе 340 используют катализатор, который обеспечивает получение выходящего потока (поток 80) с сырым алкилатом, в котором моноалкилбензолы имеют содержание 2-фенилизомеров по меньшей мере 20 мас.%. Потоки 70 и 80 смешивают с образованием потока, поток 90, состоящего из сырого алкилата, в котором моноалкилбензолы имеют переменное содержание 2-фенила (в соответствии со скоростями подачи потоков 60а и 60b), непрореагировавшего бензола, парафинов и легких и тяжелых побочных продуктов. Поток 90 подают в блок очистки сырого алкилата, блок 350, в котором непрореагировавший бензол, парафины и побочные продукты, более легкие и более тяжелые, чем моноалкилбензолы, подвергают разгонке с получением относительно чистого линейного моноалкилбензола (поток 100). Парафины направляют рециклом в способ посредством потока 170, в то время как бензол направляют рециклом посредством потока 160. Затем поток 100 подают в блок очистки конечного линейного моноалкилбензола, блок 360, который содержит селективный адсорбент, для устранения ароматических соединений, которые, даже в небольших количествах, присутствуют в относительно чистом моноалкилбензоле из-за того, что диапазон их температуры перегонки перекрывается с диапазоном температуры перегонки целевого линейного моноалкилбензола.
Нелинейные соединения, извлеченные в блоке 320, нагнетают в блок 380 специальной обработки посредством потока 180. Блок 380 включает стадии фракционирования и гидрогенизации для изолирования и очистки соответственно фракции представляющих интерес нелинейных примесей, которая представляет собой гидротропный предшественник (поток 190). В зависимости от конкретных потребностей на выходе устройства в любое заданное время (продукт с содержанием 2-фенилизомеров, составляющим более или менее 60 мас.%, согласно требованию в данное время) поток 190 можно транспортировать посредством потоков 190а и 190b или его можно не использовать, если содержание 2-фенилизомеров линейного моноалкилбензола потока 100 составляет менее 60 мас.%. Таким образом, можно обойтись без потоков 190а и 190b, когда содержание 2-фенилизомеров в линейном алкилбензоле потока 100 ниже 60 мас.%. В этом случае относительно чистый линейный алкилбензол потоков 100-110 очищают (блок 360) индивидуально, причем выходящий из блока 360 поток (поток 120) включает очищенные линейные соединения. Когда содержание 2-фенилизомеров в линейном моноалкилбензоле превышает 60 мас.%, способ можно осуществлять по двум альтернативным каналам. С одной стороны, поток относительно чистого линейного моноалкилбензола, поток 100, и поток 190а гидротропного предшественника можно смешать с образованием потока 110, который подают в блок 360 для очистки. Выходящий из блока 360 поток (поток 120-130) содержит конечное линейное алкилароматическое соединение. С другой стороны, можно обойтись без потока 190а, так что относительно чистый линейный алкилбензол, который поступает из блока 350, поток 100-110, очищают отдельно в блоке 360 и после очистки (поток 120) его смешивают с гидротропным предшественником, подаваемым потоком 190b, получая поток 130, который включает конечное линейное моноалкилароматическое соединение.
На Фиг.2 представлена неограничивающая схема реализации этого изобретения, которая включает стадии сульфонирования и нейтрализации.
Поток 15 является подачей парафинов в блок дегидрогенизации и включает смесь свежих линейных парафинов, поток 10, с непревращенными линейными парафинами, отделенными в блоке очистки сырого алкилата, блок 350, и направляемыми рециклом посредством потока 170. В блоке 300 дегидрогенизации подачу линейных парафинов превращают в смесь моноолефинов, непрореагировавших парафинов и различных побочных продуктов, таких как диолефины и нелинейные соединения. Принимают, что сырой алкилат представляет собой смесь моноалкилбензолов, непрореагировавшего бензола, парафинов и легких и тяжелых побочных продуктов, которая составляет выходящий поток стадии алкилирования.
В блок очистки олефинов, блок 310, подают выходящий поток блока дегидрогенизации посредством потока 20, увеличивая чистое содержание моноолефинов путем конверсии некоторых побочных продуктов, полученных в блоке дегидрогенизации, в основном диолефинов, в моноолефины посредством реакции селективной гидрогенизации. Получаемый поток, поток 30, обрабатывают в блоке 320, который содержит селективный адсорбент для устранения нелинейных соединений, полученных в блоке дегидрогенизации.
Свежий бензол нагнетают в способ посредством потока 50 и его смешивают с непрореагировавшим бензолом, подаваемым рециклом (поток 160), который поступает из блока очистки сырого алкилата, блок 350. Смесь этих двух потоков образует подачу бензола (поток 55), которую смешивают с потоком (поток 40), выходящим из блока 320 селективной адсорбции, с образованием потока 60, состоящего из моноолефинов, бензола и парафинов, который подают в блоки алкилирования. Поток 60 разделяют на два одинаковых потока (по составу, но необязательно по скорости подачи) 60а и 60b, которые подают в два различных реактора алкилирования, блоки 330 и 340, соответственно; в реакторе алкилирования 330 используют катализатор, который обеспечивает получение выходящего потока (поток 70) с сырым алкилатом, в котором моноалкилбензолы имеют максимальное содержание 2-фенилизомеров, равное 20 мас.%, в то время как в реакторе 340 используют катализатор, который обеспечивает получение выходящего потока (поток 80) с сырым алкилатом, в котором моноалкилбензолы имеют содержание 2-фенилизомеров по меньшей мере 20 мас.%. Потоки 70 и 80 смешивают с образованием потока, поток 90, состоящего из сырого алкилата, в котором моноалкилбензолы имеют переменное содержание 2-фенилизомеров (в соответствии со скоростью подачи потоков 60а и 60b), непрореагировавшего бензола, парафинов и легких и тяжелых побочных продуктов. Поток 90 подают в блок очистки сырого алкилата, блок 350, в котором непрореагировавший бензол, парафины и побочные продукты, более легкие и более тяжелые, чем моноалкилбензолы, фракционируют с получением относительно чистого линейного моноалкилбензола (поток 100). Парафины направляют рециклом в способ посредством потока 170, в то время как бензол направляют рециклом посредством потока 160. Затем поток 100 подают в стадию очистки конечного линейного моноалкилбензола, блок 360, который содержит селективный адсорбент для устранения ароматических соединений, которые, даже в небольших количествах, присутствуют в относительно чистом моноалкилбензоле вследствие того, что диапазон их температуры перегонки перекрывается с диапазоном температуры перегонки целевого линейного моноалкилбензола.
Нелинейные соединения, извлеченные в блоке 320, нагнетают в блок 380 специальной обработки посредством потока 180. Блок 380 включает стадии фракционирования и гидрогенизации для отделения и очистки соответственно, представляющей интерес фракции нелинейных примесей, которая представляет собой гидротропный предшественник (поток 190). В зависимости от конкретных потребностей на выходе устройства в любое заданное время (продукт с содержанием 2-фенилизомеров, составляющим более или менее 60 мас.%, согласно требованию в данное время) поток 190 можно транспортировать посредством потоков 190а и 190b или его можно не использовать, если содержание 2-фенилизомеров линейного моноалкилбензола потока 100 составляет менее 60 мас.%. Если содержание 2-фенилизомеров в потоке 100 превышает 60 мас.%, поток относительно чистого моноалкилбензола, поток 100, и поток 190а гидротропного предшественника можно смешать с образованием потока 110 и их можно очистить совместно в блоке 360 и затем посредством потоков 120 и 130 их направляют в блок 370, где их сульфонируют/нейтрализуют совместно, получая конечный поток, поток 140-150. Без потока 190а также можно обойтись так, что относительно чистый линейный алкилбензол, который поступает из блока 350, поток 100-110, очищают отдельно в блоке 360, и после очистки (поток 120) его смешивают с гидротропным предшественником, подаваемым потоками 190b и 191а с получением потока 130, причем их сульфонируют и нейтрализуют совместно в блоке 370, таким образом получая конечный поток, поток 140-150. Без потоков 190а и 191а также можно обойтись так, что поток 100 относительно чистого алкилбензола очищают (блок 360), сульфонируют и нейтрализуют (блок 370) по отдельности. Выходящий из блока 370 поток (поток 140) затем смешивают с потоком 192, соответствующим гидротропному предшественнику, сульфонированному и нейтрализованному отдельно. Поток 192 получают при сульфонировании и нейтрализации гидротропного предшественника из блока 380 (через потоки 190, 190b и 191b) отдельно в блоке 390. Конечный поток 150 получают, когда смешивают потоки 140 и 192. Если содержание 2-фенилизомеров в потоке 100 составляет менее 60 мас.%, можно обойтись без потоков 190а и 190b, и линейный алкилбензол очищают отдельно в блоке 360 и транспортируют посредством потоков 120-130 в блок сульфонирования и нейтрализации (блок 370), выходящий поток которого (поток 140-150) является конечным потоком этого воплощения.
На фиг.3 представлена другая неограничивающая схема реализации этого изобретения, в которой стадии (1), (2), (3) и (4) необязательны.
Поток 10 представляет собой подачу свежих олефинов в способ. Поток 20 представляет собой свежую подачу ароматического соединения, такого как бензол, в способ. Поток 20 смешивают с потоком 60, который представляет собой рециркулируемый поток избытка ароматического соединения после отделения от продуктов алкилирования в перегонной колонне, блок 120. Смешиванием потоков 20 и 60 получают поток 30, который является подачей ароматического соединения. Поток 30 смешивают в смесительном блоке 100 с потоком 10 для получения смеси ароматического соединения и олефинов с требуемым молярным отношением олефинов к ароматическому соединению, поток 40. Этот поток подают в блок алкилирования, блок 110, состоящий из двух реакторов с неподвижным слоем, эксплуатируемых паралелльно, где один реактор загружают катализатором, который обеспечивает получение сырого алкилата, в котором алкилбензол содержит менее 20% 2-фенилизомеров, а другой реактор загружают катализатором, который обеспечивает получение сырого алкилата, в котором алкилбензол содержит более 20% 2-фенилизомеров. Подаваемый поток (поток 40) разделяют на два (имеющих одинаковый состав, но необязательно одинаковую скорость подачи) для подачи в оба реактора, в которых протекает алкилирование ароматического соединения олефинами. Выходящие из обоих реакторов потоки объединяют с получением выходящего потока блока алкилирования, потока 50, который состоит из непрореагировавшего ароматического соединения и алкилароматического соединения, полученного в течение алкилирования. Поток 50 фракционируют в перегонной колонне, блок 120, для отделения непрореагировавшего ароматического соединения в ее верхней части, поток 60, и алкилароматического соединения в ее нижней части, поток 70. Поток 60 направляют рециклом в способ, как упоминалось выше, в то время как поток 70 сульфонируют в реакторе с падающей пленкой газообразным SO3, блок 130. Сульфонированный продукт, поток 80, состоит из линейной алкилсульфоновой кислоты высокой чистоты с заданным содержанием 2-фенилизомера. Этот продукт можно затем нейтрализовать в блоке 140 щелочными солями кальция, бария, натрия, магния и аммония в присутствии высокоионизируемого соединения, такого как фенол, чтобы получить алкилароматический сульфонат высокой чистоты, как нейтральный, так и предпочтительно суперосновной, в зависимости от количества основания, используемого при нейтрализации.
Изобретение описано дополнительно, только в целях иллюстрации, посредством следующих примеров, которые не следует рассматривать как факторы, ограничивающие область настоящего изобретения.
ПРИМЕРЫ
Пример 1
Этот пример относится к способу получения очищенного алкилирующего агента и способу получения гидроторопного предшественника из примесей, выделенных в течение очистки указанного алкилирующего агента.
Способ начинают со смеси высокочистых линейных парафинов (обычно содержание парафинов должно быть выше 97 мас.% согласно методу UOP 411), распределение атомов углерода в парафинах должно быть таким, как показано в таблице 1.
Эти линейные парафины прошли обработку способом селективной по отношению к моноолефинам дегидрогенизации с использованием обычного промышленного катализатора для селективной дегидрогенизации парафинов группы детергентов. Условия дегидрогенизации сведены в таблице 2 и обеспечивают выход олефинов 14%.
Выходящий со стадии дегидрогенизации поток может содержать до 0,1 мас.% диолефинов, которые являются нежелательными побочными продуктами стадии дегидрогенизации. По этой причине выходящий со стадии дегидрогенизации поток подвергают процессу очистки. В течение такого процесса очистки выходящий со стадии дегидрогенизации поток подвергают селективной дегидрогенизации для превращения нежелательных диолефинов в требуемые моноолефины. Для этого используют промышленный катализатор Ni-Mo-типа. Условия селективной дегидрогенизации сведены в таблице 3.
Условия ЧОСЖ (объемное отношение жидкой загрузки к каталитическому слою) регулируют, чтобы получить конверсию диолефинов в моноолефины выше 99%, эту конверсию проверяют с помощью метода UOP 902-89. Полученная смесь моноолефинов и парафинов составляет неочищенный алкилирующий агент.
Стадию очистки алкилирующего агента осуществляют посредством слоя адсорбента, в котором размещено определенное количество конкретного молекулярного сита. Выбранное молекулярное сито является цеолитом типа 13Х, широко используемым в способах селективного удаления нелинейных компонентов из смеси олефинов и парафинов. Смесь олефинов и парафинов пропускают через слой, чтобы достичь селективной адсорбции нелинейных компонентов, переходящих из предыдущей стадии дегидрогенизации (или которые присутствуют в подаче свежих парафинов и/или в парафинах, направляемых рециклом). После насыщения слоя нелинейными компонентами слой промывают парафинами с короткой цепью для десорбции олефинов и парафинов, которые могут удерживаться в порах, и затем его промывают бензолом для десорбции ранее адсорбированных нелинейных компонентов, которые потом можно преобразовать в гидротропный предшественник. Рабочие условия опытной установки стадии очистки сведены в таблице 4.
Смесь олефинов и парафинов из селективной дегидрогенизации и гидрогенизации, т.е. неочищенный алкилирующий агент, содержит примерно 2 мас.% нелинейных компонентов. Состав алкилирующего агента, очищенного путем адсорбции нелинейных компонентов на цеолите 13Х, представлен в таблице 5.
Нелинейные компоненты, отделенные в течение стадии очистки алкилирующего агента, содержат сырьевой материал, необходимый для получения гидротропного предшественника. Далее следует описание способа трансформации указанных примесей в гидротропный предшественник. Промытые бензолом после очистки алкилирующего агента нелинейные примеси, десорбированные из цеолита 13Х, проходят стадию атмосферного фракционирования. В течение стадии фракционирования целью является отделение бензола, используемого при десорбции (потом бензол следует направлять рециклом в цикл десорбции с цеолитом 13Х), и примесей, которые не имеют гидротропного потенциала (полиароматические вещества с высокой молекулярной массой), от фракции веществ, которые имеют гидротропный потенциал. Было обнаружено, что фракция веществ с гидротропным потенциалом (те вещества, которые обеспечивают гидротропный эффект при сульфонировании и нейтрализации) для ЛАС, цели этого изобретения, является фракцией, которая имеет температуру перегонки от 195 до 259°С. После того как эту фракцию отделяют посредством атмосферной перегонки, она проходит процесс гидрогенизации, чтобы устранить компоненты, которые могут образовать красящие вещества при сульфонировании. Условия гидрогенизации приведены в таблице 6.
Полученный на этой стадии гидрогенизации продукт содержит гидротропный предшественник, состав которого описан в таблице 7.
Пример 2
Этот пример относится к преимуществам использования катализатора на основе цеолита Y с высоким содержанием редкоземельных элементов (таких, как La, Ce, Nd и Pd) и натрия по сравнению с катализатором на основе цеолита Y с низким содержанием редкоземельных элементов (таких, как La, Се, Nd и Pd) и натрия в способе алкилирования бензола очищенной смесью олефинов/парафинов группы детергентов. Конкретно сравнивают два катализатора. С одной стороны, катализатор (катализатор А) на основе цеолита Y с общим содержанием редкоземельных металлов, составляющим 7%, и низким содержанием натрия (0,1 мас.%), и, с другой стороны, катализатор (катализатор В) на основе цеолита Y с содержанием редкоземельных металлов, составляющим более 71% (12 мас.% редкоземельных металлов) и содержанием натрия более 90% (0,9 мас.%).
Оба катализатора испытывали, используя экструдированные частицы одинакового размера. Бензол сушили посредством молекулярного сита, чтобы минимизировать добавление воды, и после этого бензол смешивали со смесью очищенных моноолефинов и парафинов (очищенный алкилирующий агент, см. пример 1). Испытания по алкилированию на опытной установке проводили в изотермическом реакторе с неподвижным слоем с 24-часовыми реакционными циклами, за которыми следовали циклы регенерации катализатора путем промывки бензолом в течение такого же периода времени. Стандартный цикл состоял из 24-часового реакционного цикла при 140°С, ЧОСЖ=11 час-1 и молярном отношении бензол:олефин, равном 30:1, после чего следовал цикл промывки бензолом в течение такого же периода времени. Масса катализатора, условия промывки и т.п. сведены в таблице 8.
Состав исходного сырьевого материала, который относится к смеси олефинов и парафинов, представлен в таблице 9.
Эту очищенную смесь парафинов и олефинов (очищенный алкилирующий агент, см. пример 1) смешивали с высушенным бензолом до достижения требуемого молярного отношения бензол:олефин. Испытания катализатора проводили в четырех различных последовательностях реакционных циклов. После выполнения каждого реакционного цикла выходящий поток алкилирования (сырой алкилат, включающий образовавшийся алкилбензол, непрореагировавший бензол, парафины и тяжелый алкилат) перегоняли в три стадии, используя три последовательные перегонные колонны (причем первую из этих колонн эксплуатировали при атмосферном давлении, в то время как другие эксплуатировали в условиях вакуума). Первую колонну эксплуатировали при атмосферном давлении и отделяли непрореагировавший бензол в ее верхней части, в то время как соединения из ее нижней части подавали во вторую колонну. Во второй колонне отделяли парафины в ее верхней части, в то время как соединения из ее нижней части подавали в третью колонну. В третьей колонне отделяли моноалкилбензолы в ее верхней части и тяжелый алкилат в ее нижней части. Испытания (проводимые с газовым хроматографом с пламенно-ионизационным детектором (ГХ-ПИД)) относятся к соединениям, подаваемым в третью колонну. Во всех примерах, описанных в этом патенте, использовали хроматографический метод, ссылка U 698. В течение первой последовательности реакционных циклов оба катализатора (А и В) независимо испытывали при одинаковых рабочих условиях. Для каждого катализатора оценочная последовательность состояла из семи реакционных циклов, осуществляемых при ЧОСЖ 11 час-1 и температуре от 130 до 140°С, после чего следовали три цикла при 115°С при ЧОСЖ 4 час-1. Далее, выполняли стандартный цикл при 140°С и ЧОСЖ 11 час-1, чтобы проверить дезактивацию катализатора. Во всех этих циклах молярное отношение бензол:олефин поддерживали равным 30:1. За каждым их этих реакционных циклов следовал цикл регенерации (цикл промывки). Все условия и результаты представлены в таблице 10. Заметим, что различие до 100% добавления моноалкилбензолов и тяжелого алкилата соответствует легким побочным продуктам (низшим, чем 5-фенил-C10), в то время как различие до 100% добавления разветвленного алкилата и 2-фенилизомеров соответствует внутренним изомерам (3, 4, 5 и 6-фенил).
В течение второй последовательности реакционных циклов были проведены кинетические испытания катализаторов А и В для анализа их активности при более низких температурах. Эта последовательность включает три реакционных цикла, выполненных при ЧОСЖ 4 час-1, но в то же время при 100°С, за которыми следовал стандартный цикл (ЧОСЖ=11 час-1, Т=140°С). Во всех этих циклах молярное отношение бензол:олефин поддерживали равным 30. Все условия и результаты представлены в таблице 11. Заметим, что различие до 100% добавления моноалкилбензолов и тяжелого алкилата соответствует легким побочным продуктам (низшим, чем 5-фенил-C10), в то время как различие до 100% добавления разветвленного алкилата и 2-фенилизомеров соответствует внутренним изомерам (3, 4, 5 и 6-фенил).
Третья последовательность реакционных циклов включает шесть реакционных циклов, выполненных при изменяемых ЧОСЖ, температуре и молярном отношении бензол.-олефин для анализа влияния последней переменной на конверсию и селективность реакции. Условия и результаты приведены в таблице 12.
мол.%
В течение четвертой последовательности реакционных циклов оба катализатора испытывали для анализа скорости их дезактивации. Ставя своей целью стимулирование дезактивации катализатора, после начального стандартного цикла (ЧОСЖ=11 час-1, Т=140°С) обычный 24-часовой период реакции продлевали до 72 часов без стандартной промывки бензолом каждые 24 часа. В течение всех этих циклов молярное отношение бензол:олефин поддерживали равным 30. Активность катализатора анализировали в показателях конверсии. Все результаты и условия приведены в таблице 13.
Как можно наблюдать в таблице 10, активность катализатора В равна активности катализатора А во всех последовательностях 24-часовых реакционных циклов, так как, также как и катализатор А, катализатор В обеспечивает уровни конверсии от 99,9 до 100% в течение циклов при «высокой» температуре (от 115 до 140°С). В показателях конверсии можно наблюдать, что также в течение циклов при «низкой» температуре (100°С, таблица 11) указанный катализатор В может обеспечить активность 99,9%, которая равна активности катализатора А. Более того, в таблице 12 показано, что оба катализатора также дают степени конверсии 100%, когда ЧОСЖ, температура и молярное отношение бензол:олефин изменяют в пределах промышленных рабочих условий. В показателях каталитической активности большое преимущество катализатора В состоит в том, что он обеспечивает краткосрочный уровень активности (24-часовые циклы), равный этой величине для катализатора А, однако катализатор В также способен поддерживать высокие степени конверсии (выше 99,5%) в течение более долгих периодов времени (на 30% более продолжительное время) без каких-либо циклов промывки между этими периодами, как видно из испытаний стимулированной дезактивации, описанных в таблице 13. Более высокая стабильность катализатора В при дезактивации от загрязнений позволяет удлинить продолжительность циклов реакции и уменьшить количество циклов регенерации, необходимых для работы при высоких степенях конверсии, по отношению к катализатору А. Это обеспечивает для одинакового периода эксплуатации катализаторов А и В более длительное чистое время эксплуатации при использовании реакционного времени катализатора В (большая производительность катализатора В), так же как и уменьшение суммарных затрат на регенерацию (меньшее число циклов промывки за указанный период времени), в основном касающихся потребления энергии.
По сравнению с катализатором А, помимо эквивалентной активности и большей стабильности при дезактивации, катализатор В обеспечивает более высокую селективность по отношению к целевым линейным моноалкилированным продуктам. Таким образом, катализатор В обеспечивает получение в среднем на 30-35% меньше разветвленного алкилата и на 10-18% меньше тяжелого алкилата (в основном, диалкилбензолов), чем катализатор А. Уменьшение обоих нежелательных побочных продуктов влечет за собой два технологических преимущества по сравнению с катализатором А.
Основное преимущество катализатора В относится к уменьшению выработки разветвленного алкилата, поэтому происходит пропорциональное снижение «внутренних четвертичных атомов углерода». Второе преимущество, связанное с катализатором В по отношению к катализатору А, состоит в том, что катализатор В обеспечивает получение меньшего количества тяжелого алкилата. Путем снижения конечного содержания тяжелого алкилата происходит увеличение использования сырьевых материалов в течение способа образования моноалкилированных веществ (диалкилбензолы используют дополнительную олефиновую цепь по сравнению с молекулой моноалкилбензола). Более того, качество конечного продукта значительно улучшается, так как указанный тяжелый алкилат не проявляет себя так эффективно, как моноалкилированные вещества в процессе мытья, и тяжелый алкилат часто вмешивается в образование хромофорных побочных продуктов в течение конечной стадии сульфонирования, поэтому, как видно в примере 6, ЛАБ можно получить с более низкой цветностью после сульфонирования (по отношению к 3 единицам по шкале Клетта-Саммерсона).
Пример 3
Этот пример относится к преимуществам использования очищенного алкилирующего агента (очищенная смесь олефинов и парафинов по примеру 1) в течение стадии алкилирования. Более того, этот пример относится к преимуществам использования катализатора на основе цеолита Y с более высоким содержанием редкоземельных металлов (12 мас.% редкоземельных металлов в конечном катализаторе, катализатор В из примера 2) по сравнению с катализатором на основе цеолита Y с более низким содержанием редкоземельных металлов (7 мас.% редкоземельных металлов в конечном катализаторе, катализатор А из примера 2) при использовании очищенного алкилирующего агента.
Реакцию алкилирования выполняли с катализаторами А и В, описанными в примере 2. В этом случае использовали очищенные и неочищенные алкилирующие агенты для каждого катализатора с целью регистрации различия характеристик катализаторов. После очистки алкилирующего агента (и свыше 95% ароматических соединений было устранено) смесь олефинов и парафинов смешивали с высушенным бензолом, чтобы получить выбранное молярное отношение. Затем эту смесь использовали в качестве подачи в течение стадии алкилирования. Рабочие условия стадии алкилирования приведены в таблице 14.
Другое испытание проводили для анализа влияний, которые стадия очистки оказывает на распределение продуктов, сохраняя все рабочие параметры, но в то же время используя неочищенную смесь алкилирующего агента. С другой стороны, процесс перегонки сырого алкилата, который образует выходящий поток реактора алкилирования, выполняли для отделения бензола, парафинов, моноалкилбензолов и тяжелого алкилата, и этот процесс эквивалентен процессу, определенному в примере 2. Проводили ГХ анализ потока, который подавали в третью колонну (способ U 698). Легкие элементы анализировали посредством ГХ-ПИД на выходе первой колонны.
Распределение продукта в связи с алкилированием очищенной и неочищенной смесью с катализаторами А и В представлено в таблице 15.
Как можно видеть в таблице 15, по сравнению с неочищенными смесями количество тяжелого алкилата, получаемого при использовании очищенных смесей, уменьшают на 86% с катализатором А и уменьшают на 89% с катализатором В. Также видно, что очистка смеси парафин/олефин вносит вклад в уменьшение легких побочных продуктов, причем это уменьшение немного выше с катализатором А (уменьшение 44%), чем с катализатором В (уменьшение 30%). В любом случае показано, что очистка смеси парафин/олефин вызывает значительное уменьшение образования нежелательных побочных продуктов (выше 85% с обоими катализаторами). Помимо этого, как в очищенных, так и в неочищенных смесях катализатор В обеспечивает получение меньшего количества легких и тяжелых побочных продуктов, чем катализатор А.
Пример 4
Этот пример относится к более высокой стабильности, достигаемой в течение дезактивации катализатора на основе цеолита Y с добавлением более высокого содержания редкоземельных металлов (12 мас.% редкоземельных металлов в конечном катализаторе, катализатор В из примера 2) по сравнению с катализатором на основе цеолита Y с добавлением более низкого содержания редкоземельных металлов (7 мас.% редкоземельных металлов в конечном катализаторе, катализатор А из примера 2), когда первый используют в качестве катализатора алкилирования бензола очищенным алкилирующим агентом. Выполняли длительную последовательность реакционных циклов для обоих катализаторов А и В для анализа степени их дезактивации. В отличие от случая «обратимой» дезактивации (подразумевающей, что ее можно устранить промывкой), испытываемой в примере 2 (таблица 13), в этом случае целью является изучение «необратимой» дезактивации, т.е. дезактивации, которая не исчезает после регенерации катализатора. Все тридцать циклов выполняли при ЧОСЖ=11 час-1, Т=140°С и молярном отношении бензол:олефин, равном 30 (смесь очищенных олефинов и парафинов, как указано в примере 1, состав смеси описан в таблицах 5 и 9, остальные рабочие условия, как указано в таблице 8). После каждого 24-часового реакционного цикла выполняли цикл промывки бензолом в течение 24 часов. Результаты сведены в таблицу 16 и представляют собой среднее значение по учитываемым циклам.
Как можно видеть в таблице 16, для всей этой последовательности циклов реакции/регенерации средняя конверсия с катализатором В остается равномерной и постоянной (99,8%) наряду с тем, что ее величина выше, чем обеспечиваемая катализатором А. Ни один из катализаторов не показал какой-либо потери необратимой активности в течение тридцатого цикла по сравнению с начальными циклами. Благодаря тому, что катализатор В работает при более высоких показателях активности, чем катализатор А, и так как не зарегистрировано никакой необратимой дезактивации, можно подтвердить, что стойкость катализатора В к необратимой дезактивации по меньшей мере равна этой величине для катализатора А. Это означает, что катализатор В имеет время жизни, равное или даже большее, чем катализатор А.
Пример 5
Данный пример относится к преимуществам использования катализатора на основе цеолита Y, содержащего высокое содержание редкоземельных металлов и натрия (катализатор В из примера 2), по сравнению с использованием катализатора на основе цеолита Y, содержащего низкое содержание редкоземельных металлов и натрия (катализатор А из примера 2), при эксплуатации двух параллельных изотермических реакторов с неподвижным слоем, используемых для получения алкилата с регулируемым содержанием 2-фенилизомеров. Один из реакторов загружают одним из катализаторов А или В, а другой реактор загружают нефторированным промышленным кристаллическим морденитом, называемым катализатором С, регулируя распределение предварительно очищенной подачи (по примеру 1) между двумя реакторами и смешивая получаемые выходящие потоки для получения регулируемого содержания 2-фенилизомера в получаемом потоке.
Определенное количество катализатора А помещали в один из реакторов с неподвижным слоем (называемым слоем 1), в то время как другой слой (называемый слоем 2) загружали определенным количеством нефторированного промышленного кристаллического морденита (катализатор С). Состав подачи в оба реактора был одинаковым. Подаваемый поток образовывали путем смешивания очищенной смеси олефинов и парафинов (состав смеси, как описано в таблице 5, пример 1) с подходящим количеством высушенного бензола для получения требуемого молярного отношения бензол:олефин. Всегда поддерживали постоянный расход указанного исходного потока. После этого поток делили на два подпотока посредством трехходового клапана. Каждый поток подавали в реактор (после стадии предварительного нагревания), чтобы дозировать переменный расход в каждый реактор путем регулирования клапана (но поддерживая постоянным общий расход). Выходящий из каждого реактора поток (сырой алкилат) смешивали, получая таким образом конечный выходящий поток, который анализировали с помощью ГХ-ПИД (после отделения бензола, парафинов и тяжелого алкилата от моноалкилбензолов посредством процесса перегонки, как указано в примере 2). В этом примере оба состава подачи, так же как и давление реакции, поддерживали на постоянном уровне для обоих реакторов, однако температура реакции была различной в каждом реакторе (поскольку цеолиты Y являются более активными, чем морденит) и скорости подачи варьировали для изменения конечного содержания 2-фенилизомера. Такой же способ осуществляли, загружая слой 1 катализатором В (такая же масса катализатора В, как масса катализатора А, используемая в предыдущем испытании) и загружая слой 2 таким же количеством катализатора С, как количество, используемое в предыдущем испытании, чтобы увидеть различия в отношении характеристик обоих катализаторов на основе цеолитов Y (А и В). Рабочие условия приведены в таблице 17.
Заметим, что часовая объемная скорость жидкости (ЧОСЖ) для каждого реактора изменяется при изменении процента начальной подачи, дозируемого в каждый реактор, от 2,7 час-1 (когда 25% начальной подачи проходит через указанный реактор) до 11 час-1 (когда 100% начальной подачи проходит через один отдельный реактор).
Распределение продукта в конечном выходящем потоке, когда потоки исходных материалов являются измененными, приведено в таблице 18.
Как можно видеть в таблице 18, изменение расхода потока, подаваемого в каждый реактор, означает изменение конечного содержания 2-фенилизомера в моноалкилбензоле от 18 мас.% (полученных, когда вся начальная подача проходит через слой 1) до 70 мас.% (полученных, когда вся начальная подача проходит через слой морденита). Это подтверждает, что конфигурация способа, в которой используют катализатор В в слое 1, дает чистую конверсию, немного более высокую, чем чистая конверсия при использовании катализатора А, что обусловлено большей активностью катализатора В при низких температурах. Что касается образования 2-фенилизомеров, отсутствуют какие-либо существенные различия между результатами, полученными при использовании катализаторов А или В. Что касается образования тяжелого алкилата, видно, что для отношений слой 1/слой 2 дозирования подачи ниже или равны 50/50, при использовании катализатора В в слое 1 полученное конечное содержание тяжелого алкилата ниже (от 11 до 20%), чем количество тяжелого алкилата, полученное при использовании катализатора А в слое 1, что означает лучшее использование сырьевых материалов.
Наиболее значительный результат из таблицы 18 относится к получению разветвленных продуктов. В таблице 18 показано, что при использовании катализатора В в слое 1 конечное количество разветвленного алкилата в потоке, полученном смешиванием выходящих потоков из слоев 1 и 2, намного ниже, чем конечное количество разветвленного алкилата, полученное с использованием катализатора А. Такой же диапазон 2-фенилизомеров можно получить, используя катализатор А или В, но когда используют катализатор В, условия конверсии и селективности улучшаются и количество разветвленных алкилатов в конечном продукте (среднее содержание 3,1%) значительно уменьшается (до 60%), причем количество разветвленных алкилатов в конечном продукте не превышает 3,8% в любом случае. Процентное содержание разветвленного алкилата, полученного с применением В-С каталитической системы, находится в пределах того же уровня, что и процентное содержание разветвленного алкилата, полученного посредством существующих HF и DETAL технологий, с которыми, как отмечено в разделе уровня техники, получают ЛАБ, биологически разлагаемые быстро и полно.
Пример 6
Этот пример относится к преимуществам очистки моноалкилбензола, поступающего со стадии алкилирования и очистки сырого алкилата (описанной в примере 5) перед стадией сульфонирования для минимизации цветности после сульфонирования конечной сульфоновой кислоты. Этот пример также относится к преимуществу использования катализатора на основе цеолита Y с высоким содержанием редкоземельных металлов и натрия (катализатор В из примера 2) по сравнению с использованием катализатора на основе цеолита Y с низким содержанием редкоземельных металлов и натрия (катализатор А из примера 2), причем это сравнение основано на показателях более низких уровней конечной цветности после сульфонирования.
Как описано в примерах 2-5, очищенную смесь олефинов и парафинов, смешанную с высушенным бензолом, использовали в качестве подачи. Выбирали молярное отношение бензол:олефин, составляющее 20:1. Алкилирование выполняли посредством двух изотермических параллельных реакторов с неподвижным слоем, как описано в примере 5. Катализатор А или В загружали в один из слоев (слой 1), и катализатор С, описанный в примере 4, загружали в другой слой. Рабочие условия в течение стадии алкилирования были в точности такими же, как и рабочие условия, показанные в примере 5 (таблица 17). Выбирали дозирование подачи в 50% от начального потока для каждого реактора в конфигурации, в которой загрузку слоя 1 осуществляли катализатором А, так же как и в конфигурации, в которой в слой 1 загружали катализатор В. Сырой алкилат, образованный смешиванием выходящих потоков слоев 1 и 2, очищали путем фракционирования, чтобы отделить моноалкилбензолы. Перегонка (очистка) сырого алкилата слегка отличалась от очистки, используемой в предыдущих примерах. В этом примере использовали четыре перегонные колонны. Первая колонна работала при атмосферном давлении и в ней отделяли непрореагировавший бензол в ее верхней части, в то время как соединения из ее нижней части подавали во вторую колонну. Во второй колонне, которая работала при условиях вакуума, отделяли парафины в ее верхней части, в то время как соединения из ее нижней части подавали в третью колонну. В третьей колонне, которая работала при условиях вакуума, отделяли моноалкилбензол в ее верхней части, и отделяли тяжелый алкилат в ее нижней части. Полученный из верхней части третьей колонны моноалкилбензол, в котором были обнаружены следы тяжелого алкилата, подавали на очищающий слой, отвечающий за устранение предшественников хромофора. После выполнения очистки выходящий поток подавали в четвертую колонну перегонки. В этой колонне отделяли легкие побочные продукты, образовавшиеся в течение стадии очистки (в основном, бензол, полученный посредством реакции трансалкилирования, на уровне частей на млн), в ее верхней части, в то время как соединения, обнаруженные в нижней части, представляли собой высокочистые моноалкилбензолы. Указанные высокочистые моноалкилбензолы затем сульфонировали в однотрубном реакторе с падающей пленкой, причем сульфонирующий агент SO3 был растворен в азоте, и для завершения реакции эти моноалкилбензолы выдерживали и подвергали гидролизу.
Как указано в предыдущем абзаце, стадию очистки выполняли путем обработки моноалкилбензола (полученного при фракционировании сырого алкилата) в очистном устройстве (реакторе) с неподвижным слоем, в который помещали определенное количество промышленной кислой глины. В этом примере используемая глина характеризуется массовым отношением диоксида кремния к оксиду алюминия, равным 4,9:1,0, частично нейтрализована 1,4 мас.% K2O и 1,2 мас.% MgO и также характеризуется содержанием 2,9 мас.% Fe2O3 и 0,5 мас.% TiO2. Эту глину предварительно активировали, направляя поток горячего инертного газа для удаления адсорбированной воды. Условия активации и эксплуатации приведены в таблице 19.
Условия сульфонирования очищенных и неочищенных моноалкилбензолов, полученных сочетанием катализаторов А-С и В-С, приведены в таблице 20.
Конечная цветность после сульфонирования полученных линейных алкилбензолсульфонатов (ЛАС) (называемых ЛАС А-С, если моноалкилбензол поступает из алкилирования с катализаторами А-С, и ЛАС В-С, если моноалкилбензол поступает из алкилирования с сочетанием катализаторов В-С) анализировали, используя колориметр Клетта-Саммерсона. Для оценки влияния стадии очистки на конечную цветность после сульфонирования при использовании катализаторов А и В моноалкилбензол также сульфонировали при тех же условиях без стадии очистки глиной. Цветность сульфонированного неочищенного моноалкилбензола (ЛАС из неочищенного моноалкилбензола) и цветность сульфонированного очищенного моноалкилбензола (ЛАС из очищенного моноалкилбензола) в алкилатах, полученных с сочетанием катализаторов А-С и А-В, показана в таблице 21.
Как показано в таблице 21, очистка моноалкилбензола с использованием кислой глины позволяет значительно снизить цветность после сульфонирования ЛАС из очищенного алкилата моноалкилбензола по сравнению с ЛАС из неочищенного моноалкилбензола (по меньшей мере на 70-80% ниже, цветность после сульфонирования ниже 7 нельзя измерить посредством шкалы Клетта-Саммерсона). Заметим, что образец ЛАС из неочищенного моноалкилбензола, полученный при сочетании катализаторов В-С, показывает цветность после сульфонирования, значительно более низкую (на 17% ниже), чем цветность после сульфонирования, полученную при сочетании катализаторов А-С. Это означает, что в течение стадии алкилирования катализатор В обеспечивает получение меньшего количества предшественников хромофора, чем катализатор А. Уменьшая цветность после сульфонирования, увеличивают качество нейтрализованного конечного продукта, особенно, когда конечный продукт используют в жидком составе детергента, так как эта цветность может оказывать влияние на визуальный эффект красящего вещества, добавленного в состав детергента.
Пример 7
В этом примере показывают преимущества добавления гидротропного предшественника (полученного, как описано в примере 1) к моноалкилбензолу (полученному фракционированием сырого алкилата) перед стадией очистки указанного алкилбензола (поясняемой в примере 6), после которой получающийся алкилат отделяют и сульфонируют. Сам по себе гидротроп образуют сульфонированием (и последующей нейтрализацией) гидротропного предшественника на стадии сульфонирования, либо отделенного, либо смешанного с линейным моноалкилбензолом. Кроме того, сравнивали гидротропный эффект ЛАС, полученных с сочетанием катализаторов А-С и В-С.
Выходящие потоки (сырого алкилата) слоев 1 и 2 (используя А-С и В-С в различных операциях аналогично примеру 5) смешивали и затем перегоняли способом, эквивалентным тому, как указано в примерах 5 и 6, для отделения моноалкилбензолов. К этим моноалкилбензолам добавляли гидротропный предшественник, после чего полученную смесь очищали кислой глиной при тех же условиях, что и в примере 8, и полученные выходящие потоки перегоняли для отделения легких побочных продуктов конечного алкилата. После сульфонирования указанных алкилатов их окончательно нейтрализовали водным раствором гидроксида натрия в стехиометрическом количестве.
Для оценки гидротропного эффекта в зависимости от содержания 2-фенилизомера, было получено шесть образцов очищенного моноалкилбензола с различным содержанием 2-фенилизомеров на стадии алкилирования (как определено в примере 5) для каждого сочетания катализаторов, указанного в примерах 3 и 4 (А-С и В-С). Образцы обозначили S1 (А-С и В-С), S2 (А-С и В-С) и S3 (А-С и В-С) согласно содержанию в них 2-фенилизомеров и их происхождению. Расходы регулировали при работе с конфигурациями А-С и В-С для получения одинакового содержания 2-фенилизомеров. Контрольные испытания (без добавления гидротропа) и испытания с ксилолсульфонатом натрия (КСН) выполняли с алкилатом, полученным из А-С и В-С систем с таким же содержанием 2-фенилизомеров, как указанные выше S1, S2 и S3, так что одинаковое обозначение использовали, чтобы отличить содержание 2-фенилизомера. Результаты приведены ы таблице 22.
Эффект растворения гидротропа оценивали в показателях температуры помутнения при охлаждении (ТПО) конечного сульфонированного и нейтрализованного продукта. Указанный продукт разбавляли водой до получения обычных промышленных концентраций (20, 25, 30 мас.% в воде). Для наблюдения влияния гидротропного предшественника приготавливали образцы с 90 мас.% моноалкилбензола и 10 мас.% гидротропного предшественника. Помимо этого, приготавливали чистые образцы, то есть образцы без добавления гидротропного предшественника. Таким образом были приготовлены образцы со 100% продукта, полученного из системы А-В, и образцы со 100% побочного продукта вышеупомянутой системы В-С с таким же содержанием 2-фенилизомеров, как образцы, в которые был добавлен гидротропный предшественник. Другой хорошо известный гидротроп - ксилолсульфонат натрия (КСН) добавляли к образцам без гидротропного предшественника (90% чистого алкилбензолсульфоната натрия с содержанием 2-фенилизомеров, эквивалентным содержанию в других испытаниях, и 10% КСН) и после этого его разбавляли водой до тех же промышленных концентраций, что и предыдущие образцы. Результаты приведены в таблице 23.
Из таблицы 23 можно вывести два основных заключения. Во-первых, можно видеть, что для образцов с низким содержанием 2-фенила и без гидротропа (образцы 100% S1 и 100% S2) температуры помутнения, полученные для алкилата из системы А-С, слегка выше, чем температуры помутнения системы В-С, хотя добавление гидротропов дает тенденцию к понижению этих температур помутнения до одинаковых уровней (и при высоких концентрациях 2-фенилизомеров эта разница исчезает). Этот факт по-видимому связан с высоким содержанием разветвленных моноалкилсульфонатов (более растворимых, чем линейные эквивалентные соединения) в системе А-В. Второе заключение относится к тому факту, что способность предусматриваемого в этой заявке гидротропа к уменьшению температуры помутнения намного выше, чем у КСН, особенно для образцов с очень низким (18%) или очень высоким (70%)содержанием 2-фенилизомеров как для обоих продуктов каталитической системы А-С, так и для обоих продуктов системы В-С. Поэтому можно сделать вывод, что продукт системы В-С является практически настолько же растворимым, как и продукт системы А-В (фактически, одинаково растворимым для высоких концентраций 2-фенилизомеров), и что добавление предусматриваемого в этой заявке гидротропа уменьшает ТПО побочного продукта каталитических систем А-В и В-С до одинаковых уровней и намного ниже, чем величины, получаемые при использовании промышленного гидротропа, такого как КСН.
Пример 8
В этом примере иллюстрируют преимущества катализатора на основе цеолита Y, имеющего высокое содержание редкоземельных металлов и натрия (катализатор В в примере 2) по отношению к использованию катализатора на основе цеолита Y, имеющего низкое содержание редкоземельных металлов и натрия (катализатор А в примере 2) при выполнении алкилирования бензола длинноцепочечными линейными альфа-олефинами (диапазон C20-C22), по сравнению с результатами, полученными при использовании HF, катализатора, используемого длительное время в промышленном масштабе для этого способа.
Бензол сушили молекулярным ситом для минимизации добавления воды и затем его смешивали со смесью длинноцепочечных линейных альфа-олефинов. Испытания алкилирования на опытной установке в случае твердого катализатора выполняли в изотермическом реакторе с тонким слоем с 24-часовыми реакционными циклами, за которыми следовали циклы промывки бензолом в течение такого же периода. Стандартный цикл включает 24-часовой реакционный цикл с молярным отношением бензол:олефин, равным 50:1, за которым следует цикл промывки бензолом в течение такого же периода времени. Рабочие условия приведены в таблице 24.
Состав смеси альфа-олефинов, используемых в качестве подачи, представлен ниже в таблице 25.
Выполняли двенадцать реакционных циклов алкилирования, используя цеолиты Y А и В примера 2. Первые три цикла выполняли с ЧОСЖ 8 час-1 при Т=150°С (цикл 1). Затем три цикла с ЧОСЖ 6 час-1 при Т=150°С (цикл 2). Последние четыре цикла выполняли с ЧОСЖ 8 час-1, два из них при Т=150°С и последние два при более низкой температуре Т=140°С (цикл 3). Все эти циклы выполняли для анализа влияния температуры и объемной скорости на выход катализатора. Продукты анализировали с помощью газовой хроматографии (ГХ) с пламенно-ионизационным детектором (ПИД).
Алкилирование с HF выполняли периодическим способом в охлаждаемом реакторе с непрерывным перемешиванием, так как алкилирование является экзотермической реакцией и необходимо отводить тепло от реактора для поддержания постоянной требуемой температуры реакции, что типично для уровня техники. В реактор вводили определенное количество ранее рассмотренной смеси линейных альфа-олефинов (состав, отраженный в таблице 25), смешанных с сухим бензолом до получения требуемого молярного отношения бензол:олефин. Смесь бензола и олефинов предварительно нагревали до выбранной температуры реакции. Затем определенный объем жидкой HF вводили в реактор до достижения выбранного объемного отношения НF:олефин. Время реакции подбирали так, чтобы оно составляло 10 минут для получения ЧОСЖ=6 час-1, обычной для известных процессов алкилирования с HF. Рабочие условия реакции алкилирования с HF приведены в таблице 26.
Результаты, представленные в таблице 27, соответствуют среднему распределению продуктов (моноалкилбензолы и побочные продукты, включая легкие побочные продукты и тяжелые побочные продукты, после отделения непрореагировавшего бензола) таких циклов, в которых использовали твердые катализаторы А и В с различными условиями ЧОСЖ и температуры (называемые циклами 1, 2 и 3). Результаты, соответствующие технологии HF, включены в нижнюю часть таблицы 27 для сравнения выходов для HF и твердого катализатора.
Как можно видеть в таблице 27, активности цеолитов Y, содержащих в качестве добавок редкоземельные металлы А и В, очень похожи (слегка выше для катализатора В), так как конверсия олефина всегда выше 99,7%. При анализе полученных побочных продуктов оба катализатора А и В, как и HF, показали высокую селективность в отношении минимизации легких побочных продуктов и тяжелого алкилата. Количество всех побочных продуктов, полученных посредством твердых катализаторов А и В, ниже 0,4 мас.% в трех оцениваемых циклах, и в циклах 2 и 3 даже лучше, чем при использовании HF. Поэтому оба катализатора на основе цеолитов Y с редкоземельными металлами (А и В) эквивалентны катализатору, используемому в промышленности (HF), в показателях конверсии и селективности по отношению к моноалкилбензолам.
Распределение изомеров, соответствующее предыдущим экспериментам, представлено в таблице 28. Как и в таблице 27, строка, расположенная в нижней части, соответствует результатам, относящимся к реакции с HF при рабочих условиях, ранее определенных в таблице 26.
В таблице 28 распределение изомеров аналогично при сравнении продуктов цеолитов Y (А и В) с продуктами HF. Основное отличие относится к содержанию разветвленного алкилата. Как можно видеть, количество разветвленного алкилата, полученного при использовании цеолита с низким содержанием (7%) редкоземельных металлов, примерно на 27% выше, чем количество, полученное с использованием HF в качестве катализатора. Однако количество разветвленного алкилата, полученного с катализатором В, равно и даже ниже (цикл 3), чем полученное с HF. Поэтому, с этой точки зрения, с катализатором В можно получить меньше разветвленного алкилата, чем с катализатором А (на 25% меньше), и приблизительно такое же количество, что и с гомогенным катализатором, используемым в промышленности (HF). Что касается содержания 2-фенилизомеров, можно видеть, что с технологией HF получают немного меньшее количество 2-фенилизомеров, чем с цеолитами Y, модифицированными редкоземельными металлами (А и В).
Следовательно, видно, что катализатор В является катализатором, эквивалентным катализатору HF в способе алкилирования бензола чистыми альфа-олефинами (в показателях конверсии, селективности по отношению к моноалкилату и алкилатам), и, помимо этого, избегают сложностей обращения с такой коррозионной кислотой, как HF. Более того, катализатор В улучшает характеристику катализатора А в этом способе путем значительного понижения (на 25%) выработки разветвленных алкилатов.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКОРАСТВОРИМЫХ ЛИНЕЙНЫХ АЛКИЛБЕНЗОЛСУЛЬФОНАТОВ | 2006 |
|
RU2396254C2 |
| СПОСОБ ОЧИСТКИ АЛКИЛАРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 2006 |
|
RU2415834C2 |
| ДВУХСТАДИЙНЫЙ СПОСОБ АЛКИЛИРОВАНИЯ БЕНЗОЛА С ОБРАЗОВАНИЕМ ЛИНЕЙНЫХ АЛКИЛБЕНЗОЛОВ | 1997 |
|
RU2173677C2 |
| СПОСОБ СОВМЕСТНОГО ПОЛУЧЕНИЯ ЛИНЕЙНЫХ АЛКИЛБЕНЗОЛОВ И СИНТЕТИЧЕСКИХ МАСЕЛ | 2002 |
|
RU2209201C1 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛАРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 1973 |
|
SU398026A1 |
| СПОСОБ И УСТРОЙСТВО ДЛЯ ПОЛУЧЕНИЯ АЛКИЛБЕНЗОЛОВ, ПРИМЕНЯЕМЫХ В ПРОИЗВОДСТВЕ МОЮЩИХ СРЕДСТВ, С ПОМОЩЬЮ ТРАНСАЛКИЛИРОВАНИЯ | 2008 |
|
RU2453522C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОСНОВЫ ДЛЯ СМАЗОЧНЫХ МАТЕРИАЛОВ И ТЕПЛОНОСИТЕЛЕЙ | 2002 |
|
RU2231537C1 |
| КАТАЛИЗАТОР ДЛЯ ПОЛУЧЕНИЯ ЛИНЕЙНЫХ МОНОАЛКИЛБЕНЗОЛОВ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2009 |
|
RU2383387C1 |
| СПОСОБ И УСТРОЙСТВО ДЛЯ АЛКИЛИРОВАНИЯ БЕНЗОЛА | 1997 |
|
RU2179166C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ C-C-ОЛЕФИНОВ ИЗ С- И С-ПАРАФИНОВ | 2012 |
|
RU2564411C2 |

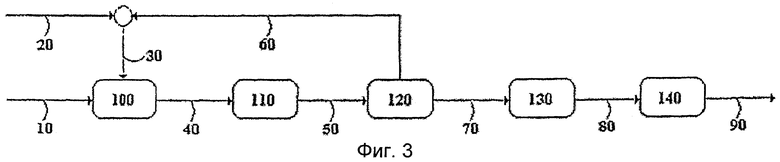
Изобретение относится к способу получения линейного моноалкилароматического соединения с регулируемой концентрацией 2-фенилизомеров и очень низкой цветностью после сульфонирования, включающему применение каталитической системы на основе высокостабильных твердых катализаторов, которые являются активными и имеют высокую селективность по отношению к линейным моноалкилированным соединениям. В процессе алкилирования осуществляют с катализатором, который обеспечивает получение линейного алкилароматического соединения с минимальным содержанием 2-фенил изомеров, составляющим 20 мас.%, включающим цеолит типа MOR, от 0,01 мас.% до 0,20 мас.% по меньшей мере одного из металлов, выбираемых из группы, состоящей из Li, Na, К, Mg или Са, при максимальном содержании Na 0,01%, и от 0 до 0,5 мас.% по меньшей мере одного из металлов, выбираемых из группы, состоящей из Ti, Zr, Hf, при этом также катализатор, обеспечивающий получение максимально 20 мас.% 2-фенилизомеров, включает цеолит типа FAU, от 0,5 до 2 мас.% по меньшей мере одного из металлов, выбираемых из группы, состоящей из Li, Na, К, Mg или Са, и редкоземельные металлы в количестве: от 4,5 до 10 мас.% La, от 1,2 до 4 мас.% Се, от 0,5 до 1,5 мас.% Рr и от 2 до 3 мас.% Nd. 2 н. и 17 з.п. ф-лы, 8 пр., 28 табл., 3 ил.
1. Способ получения линейного моноалкилароматического соединения с содержанием 2-фенилизомера от 18 до 70 мас.% посредством каталитического алкилирования ароматического соединения, выбираемого из группы, состоящей из толуола, ксилола, бензола или их смесей, очищенным алкилирующим агентом, включающим моноолефины; указанный способ включает следующие стадии:
1) алкилирования ароматического углеводорода моноолефинами, присутствующими в очищенном алкилирующем агенте, посредством сочетания следующих процессов:
а) процесса алкилирования с катализатором, который обеспечивает получение линейного алкилароматического соединения с максимальным содержанием 2-фенилизомеров, составляющим 20 мас.%,
б) процесса алкилирования с катализатором, который обеспечивает получение линейного алкилароматического соединения с минимальным содержанием 2-фенилизомеров, составляющим 20 мас.%, включающим цеолит типа MOR, от 0,01 мас.% до 0,20 мас.% по меньшей мере одного из металлов, выбираемых из группы, состоящей из Li, Na, К, Mg или Са, при максимальном содержании Na 0,01%, и от 0 до 0,5 мас.% по меньшей мере одного из металлов, выбираемых из группы, состоящей из Ti, Zr, Hf;
2) фракционирования выходящего потока стадии (1) для отделения непрореагировавших ароматических углеводородов, алканов с прямой цепью, содержащих от 8 до 30 атомов углерода, и наиболее легких и наиболее тяжелых побочных продуктов от целевых линейных моноалкилароматических соединений;
3) очистки фракции линейных моноалкилароматических соединений, поступающих со стадии (2),
отличающийся тем, что катализатор, обеспечивающий получение максимально 20 мас.% 2-фенилизомеров, включает цеолит типа FAU, от 0,5 до 2 мас.% по меньшей мере одного из металлов, выбираемых из группы, состоящей из Li, Na, К, Mg или Са, и редкоземельные металлы в количестве: от 4,5 до 10 мас.% La, от 1,2 до 4 мас.% Се, от 0,5 до 1,5 мас.% Рr и от 2 до 3 мас.% Nd.
2. Способ получения линейного моноалкилароматического соединения по п.1, включающий предварительные стадии:
каталитической дегидрогенизации алканов с прямой цепью, включающих от 8 до 30 атомов углерода;
селективной гидрогенизации диолефинов, полученных в качестве побочных продуктов на предшествующей стадии, до моноолефинов с получением исходного алкилирующего агента;
очистки исходного алкилирующего агента, полученного на предыдущей стадии, отделения нелинейных продуктов, содержащихся в выходящем потоке предыдущей стадии, с получением очищенного алкилирующего агента, и
обработки нелинейных продуктов, отделенных на предыдущей стадии, с образованием гидротропного предшественника.
3. Способ получения линейного моноалкилароматического соединения по п.1, в котором катализатор, обеспечивающий получение максимально 20% 2-фенилизомеров, включает 0,9 мас.% Na.
4. Способ получения линейного моноалкилароматического соединения по п.1, в котором катализатор, обеспечивающий получение максимально 20% 2-фенилизомеров, включает:
а) дифракционную рентгенограмму порошка, отличающуюся тем, что наиболее интенсивный дифракционный пик появляется при угле 2 θ, соответствующем 6,2°, а остальные основные пики появляются при углах дифракции 2 θ, соответствующих 23,6°; 20,3°; 21,6°; 27,0°; 31,3°, расположенных в порядке от наибольшей до наименьшей интенсивности соответствующих пиков;
б) общее молярное отношение кремний/алюминий от 0,5:1,0 до 3,0:1,0;
в) молярное отношение кремний/алюминий по структурной решетке от 1,5:1,0 до 2,5:1,0;
г) общую удельную площадь поверхности (БЭТ), составляющую от 500 дo 1000 м2/г;
д) площадь микропор, составляющую от 500 до 900 м2/г;
е) удельный объем микропор, составляющий от 0,1 до 0,3 мл/г;
ж) распределение макропор, в котором диаметр макропор составляет от 0,002 до 0,200 мкм.
5. Способ получения линейного моноалкилароматического соединения по п.1, в котором ароматический углеводород и очищенный алкилирующий агент смешивают перед реакцией алкилирования стадии (1) в молярном отношении ароматический углеводород:олефин, составляющем от 5:1 до 70:1.
6. Способ получения линейного моноалкилароматического соединения по п.1, в котором смесь ароматического углеводорода и очищенного алкилирующего агента включает максимально 0,3 мас.% нелинейных соединений, отличных от ароматического углеводорода.
7. Способ получения линейного моноалкилароматического соединения по п.1, в котором смесь ароматического углеводорода и очищенного алкилирующего агента включает от 0 до 0,1 мас.% воды.
8. Способ получения линейного моноалкилароматического соединения по п.1, в котором реакции алкилирования стадии (1) осуществляют одновременно.
9. Способ получения линейного моноалкилароматического соединения по п.1, в котором катализатор, используемый в реакции алкилирования, размещают в реакторе в компоновке, выбранной из группы, состоящей из неподвижного слоя с одним катализатором, неподвижного слоя с двумя полностью перемешанными различными катализаторами, по меньшей мере двух различных неподвижных слоев с одним и тем же катализатором в каждом слое, по меньшей мере двух различных неподвижных слоев с различными катализаторами в каждом слое, псевдоожиженного слоя с одним или более различными катализаторами, суспензионного реактора с одним или более различными катализаторами.
10. Способ получения линейного моноалкилароматического соединения по п.1, в котором процесс алкилирования осуществляют в конфигурации реактора, которая включает по меньшей мере одну из конфигураций реактора, выбираемых из группы, состоящей из независимого реактора, по меньшей мере двух параллельных реакторов, по меньшей мере двух последовательных реакторов и сочетаний указанных конфигураций.
11. Способ получения линейного моноалкилароматического соединения по п.1, в котором стадию (3) очистки выполняют посредством селективной абсорбции, и/или гидрогенизации, и/или фракционирования.
12. Способ получения линейного моноалкилароматического соединения по п.11, в котором селективную адсорбцию выполняют посредством селективного адсорбента на основе глины.
13. Способ получения линейного моноалкилароматического соединения по п.12, в котором глина включает:
а) общее молярное отношение кремний/алюминий от 3:1 до 5:1;
б) от 1 до 4 мас.% Fе2О3;
в) от 0,5 до 2 мас.% K2O;
г) от 0,2 до 2 мас.% MgO;
д) от 0,1 до 1 мас.% ТiO2;
е) от 1800 до 2500 ч./млн Na;
ж) удельную площадь поверхности, выраженную как площадь поверхности по БЭТ, составляющую от 150 до 500 м2/г;
з) суммарный объем пор от 0,1 до 2,0 мл/г;
и) распределение макропор, в котором диаметр макропор составляет от 0,002 до 0,080 мкм.
14. Способ получения линейного моноалкилароматического соединения по п.1, в котором олефины стадии (1) представляют собой линейные α-олефины.
15. Способ получения линейного моноалкилароматического соединения по п.14, в котором α-олефины стадии (1) включают от 9 до 30 атомов углерода.
16. Способ получения линейного моноалкилароматического соединения по п.1, в котором реакционная температура составляет от 20 до 400°С.
17. Способ получения линейного моноалкилароматического соединения по п.1, в котором объемная скорость составляет от 1 до 15 ч-1.
18. Способ получения линейного алкилсульфоната, включающий получение линейного моноалкилароматического соединения по любому из пп.1-17 и стадию сульфирования и нейтрализации полученного моноалкилароматического соединения.
19. Способ получения линейного алкилсульфоната по п.18, в котором указанную нейтрализацию выполняют в присутствии щелочного вещества, включающего один или более катионов, выбираемых из группы: Na, K, NH4 +, Mg, Ca, Ba или замещенной аммониевой щелочи.
| WO 2007104805 A1, 20.09.2007 | |||
| B.THOMAS et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| SCL, 2006, VOL.4, №5, PP.1611-1616 | |||
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРОВ ДЛЯ КОНВЕРСИИ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 0 |
|
SU187735A1 |
| US 2003166481 A1, 04.09.2003 | |||
| WO 2006108883 A1, 19.10.2006 | |||
| RU 98116813 A, 20.07.2000 | |||
| Способ приготовления катализатора на основе цеолита для алкилирования изобутана или бензола олефинами С @ -С @ | 1980 |
|
SU936991A1 |

