Область изобретения
Настоящее изобретение относится к новым способам получения соединения формулы I (иклаприма), которое относится к ингибиторам дигидрофолатредуктазы, а также к промежуточным соединениям, использующимся в данных способах.
Предпосылки создания изобретения
Соединение формулы I обладает ценными антибиотическими свойствами. Данное соединение можно использовать для подавления или профилактики инфекционных заболеваний у млекопитающих, как у человека, так и у отличных от человека животных. В частности, оно обладает ярко выраженной антибактериальной активностью, даже в отношении полирезистентных грамположительных штаммов, а также в отношении таких условно-патогенных микроорганизмов, как, например, Pneumocystis carinii. Указанное соединение можно вводить в сочетании с известными веществами, обладающими антибактериальной активностью, причем некоторые из них оказывают синергическое действие по отношению к данному соединению.
Типичными агентами по сочетанию являются, например, сульфонамиды или другие ингибиторы ферментов, участвующих в биосинтезе фолевой кислоты, такие как, например, производные птеридина.
В синтезе соединения I, описанном в патенте США № 5773446 и патентной заявке PCT/EP 2004/008682, используются относительно дорогие исходные веществ, кроме того, последующие стадии синтеза трудно контролировать.
Следовательно, существует потребность в разработке способа получения соединения I с более высоким общим выходом, в котором меньше промежуточных соединений, требующих выделения. Целью данного изобретения является разработка способа, в котором все выделенные промежуточные соединения являются кристаллическими и не требуют хроматографических методов очистки. Кроме того, такой способ позволяет синтезировать соединения структуры I из простого промежуточного соединения и дешевого исходного вещества.
Краткое описание изобретения
Настоящее изобретение предлагает способ получения соединения формулы I из промежуточных соединений формул 6 и 12.
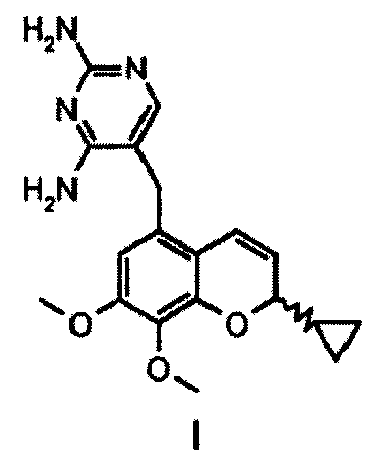
Промежуточное соединение формулы 3 синтезируют в 3 стадии из легкодоступного исходного вещества 1 (схема 1). Диаминопиримидиновый заместитель соединения 1 селективно защищают по способу R.J. Griffin et al., J.Chem.Soc. Perkin Trans I, 1811 (1992), получая соединение формулы 2, которое, в свою очередь, формилируют с получением соединения формулы 3 (схема 1).
Схема 1:
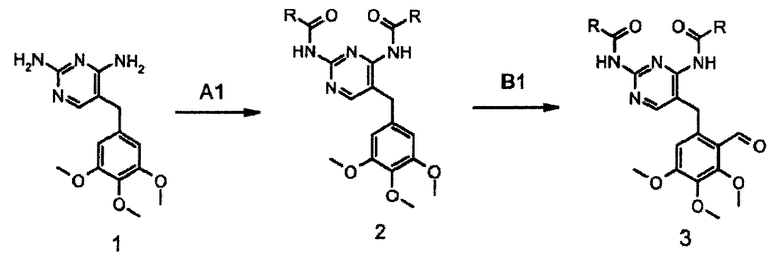
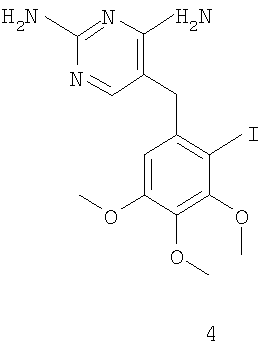
Другой способ получения соединения формулы 3 изображен на схеме 2. Соединение формулы 4 защищают по диаминопиримидиновой группе, получая соединения формулы 5, которое затем карбонилируют с получением промежуточного соединения структуры 3 (схема 2).
Схема 2:
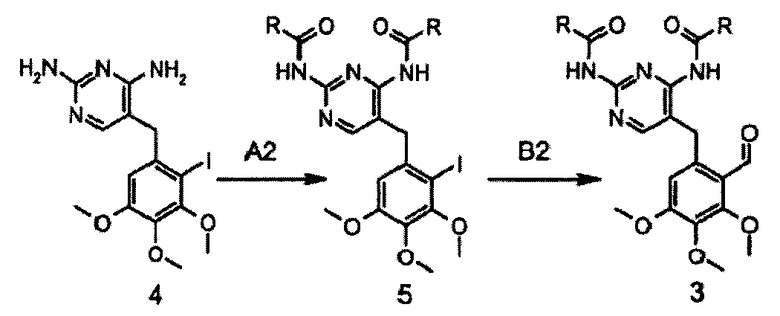
Путем селективного деметилирования соединение формулы 3 превращают в основное промежуточное соединение формулы 6 (схема 3).
Схема 3:
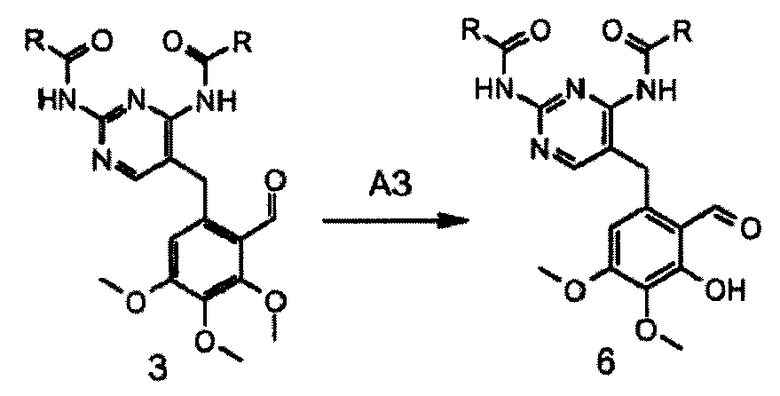
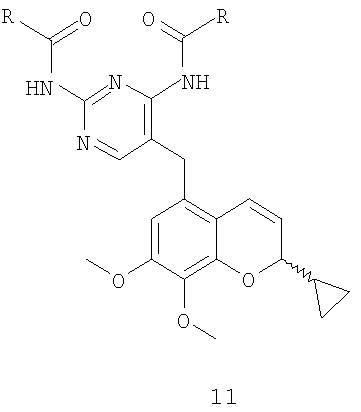
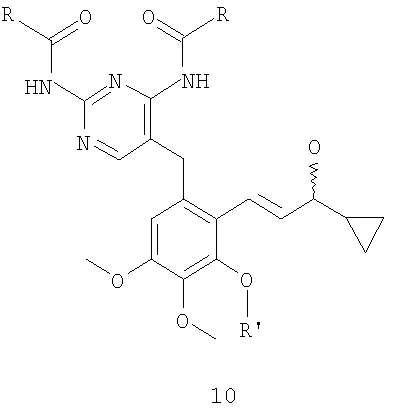
Конденсация соединения структуры 6, после защиты фенольного фрагмента (7), с кетоном 8 приводит к получению соединения формулы 9. После восстановления соединения структуры 10 и его циклизации, катализируемой кислотой или палладием, в зависимости от природы защитной группы, получают соединение формулы 11, в соответствии со схемой 4.
Схема 4:
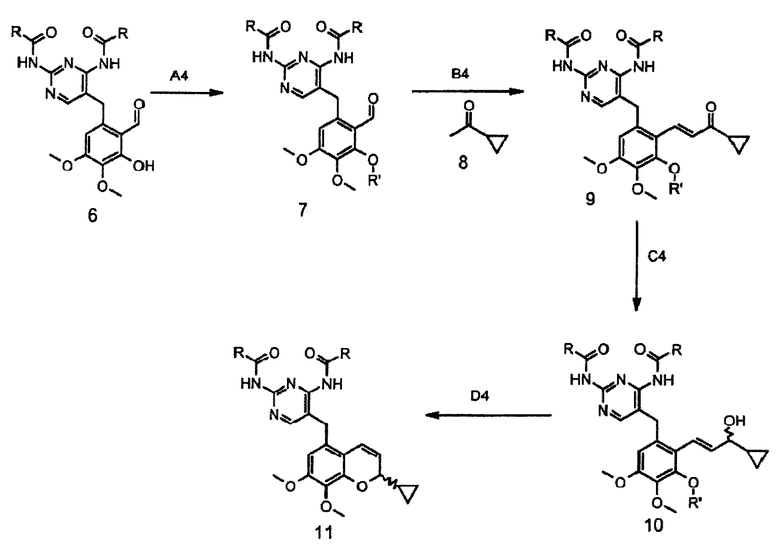
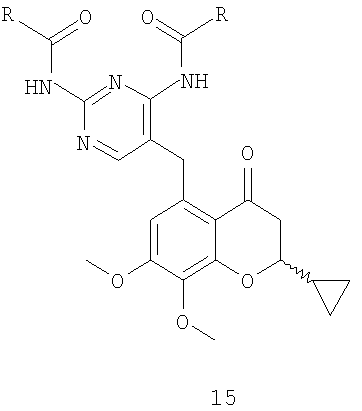
Альтернативно соединение структуры 2 ацилируют с получением соединения структуры 12, которое селективно деметилируют с получением соединения структуры 13 с последующим взаимодействием с соединением формулы 14 и получают соединение формулы 15 (схема 5).
Схема 5:
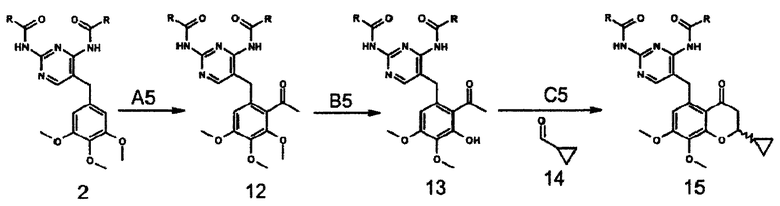
Соединение формулы 17 можно синтезировать либо ацилированием соединения формулы 2 соединением формулы 16, либо взаимодействием соединения формулы 12 с соединением формулы 14. Селективное деметилирование соединения формулы 17 обработкой йодидом натрия дает соединение формулы 18, а последующая циклизация приводит к получению соединения формулы 15 (схема 6).
Схема 6:
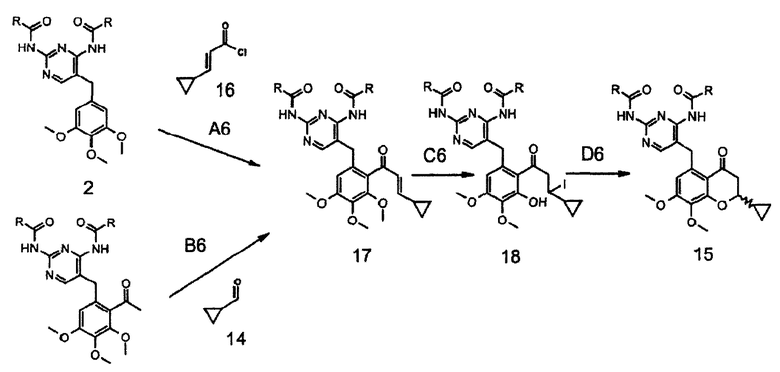
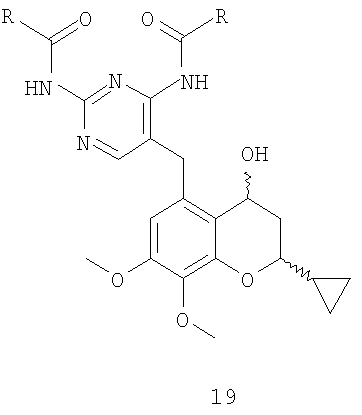
Соединение формулы 15 восстанавливают до соединения формулы 19 и превращают в соединение формулы 11, как показано на схеме 7.
Схема 7:
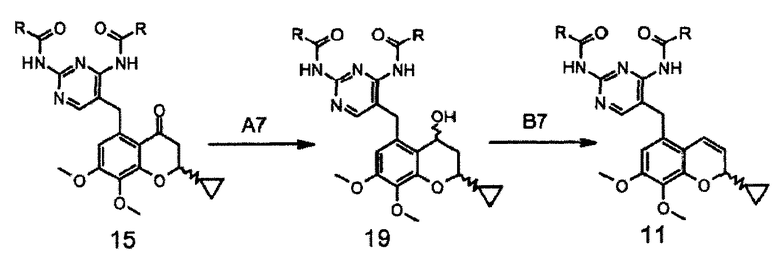
После удаления защитных групп с соединения формулы 11 получают целевое соединение формулы I, как показано на схеме 8.
Схема 8:
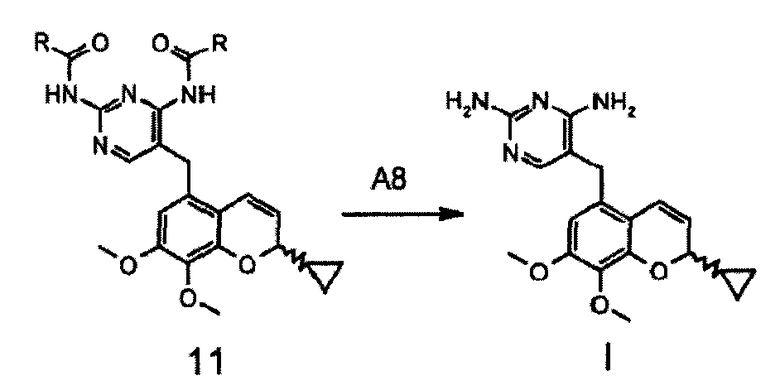
Соединение формулы I представляет собой основание и, при желании, может быть превращено взаимодействием с кислотой в фармацевтически приемлемые соли. Подходящими кислотами являются, например, соляная кислота, малеиновая кислота, янтарная кислота, L(+)-молочная кислота, DL-молочная кислота, гликолевая кислота, 1-гидроксинафталин-2-карбоновая кислота, винная кислота, лимонная кислота, метансульфоновая кислота. Наиболее предпочтительными являются карбоновые кислоты.
Подробное описание изобретения
Способ настоящего изобретения имеет много преимуществ и усовершенствований по сравнению с существующим в настоящее время способом синтеза соединения формулы I, описанным в патенте США № 5773446 и в патентной заявке PCT/EP 2004/008682. Соответствующие исходные вещества формул 1, 8 и 14 являются коммерчески доступными в больших количествах, а соединение формулы 16 можно получить из соединения формулы 14 и малоновой кислоты по способу, описанному в литературе, например в Z. Ma et al., Tetrahedron: Asymmetry 6 (6), 883 (1997). Основное промежуточное соединение формулы 6, используемое для синтеза соединения формулы I, можно получить с помощью ряда реакций, изображенных на схемах 1-3. Защиту фрагмента A1 в триметоприме 1 можно осуществить нагреванием соединения формулы 1 с таким ангидридом кислоты, как ангидрид уксусной кислоты, ангидрид изомасляной кислоты или ангидрид пивалоиловой кислоты, в таком инертном высококипящем растворителе, как толуол, п-ксилол, или в чистом ангидриде кислоты, приблизительно до 120-160°C. Структуру B1 защищенного триметоприма 2 можно получить в инертном растворителе, например в дихлорметане или дихлорэтане, предпочтительно в дихлорметане, с использованием дихлорметил-метилового эфира и такой кислоты Льюиса, как тетрахлорид олова, при температуре от 0°C до -30°C, предпочтительно при -10°C. Альтернативно соединение формулы 3 также можно синтезировать защитой фрагмента A2 соединения 4 такими ангидридами кислот, как уксусный ангидрид, ангидрид метилпропионовой кислоты или ангидрид пивалоиловой кислоты, в таком инертном высококипящем растворителе, как толуол, п-ксилол, или в чистом ангидриде кислоты, предпочтительно ангидриде метилпропионовой кислоты, приблизительно при 120-160°C. Карбонилирование фрагмента B2 соединения формулы 5 можно проводить в инертной атмосфере и в инертном растворителе, таком как тетрагидрофуран, с использованием тетракиспалладия в качестве катализатора, монооксида углерода и гидрида трибутилолова при 60-80°C. Селективное деметилирование A3 можно проводить в таком инертном растворителе, как дихлорметан или ацетонитрил, в присутствии такой кислоты Льюиса, как трихлорид алюминия, трихлорид бора, трибромид бора, дихлорид марганца, дийодид марганца, предпочтительно трихлорид алюминия, и такого нуклеофила, как йодид натрия, диметилсульфид, диэтилсульфид, тетрагидротиофен, предпочтительно йодид натрия, при комнатной температуре, до 40°C.
Синтез целевого соединения I с использованием триметоприма в качестве исходного соединения, показан на схемах 1-8. Вначале в промежуточном соединении формулы 6 защищают фенольную группу A4 взаимодействием, например, с хлорметил-метиловым эфиром, ди-трет-бутилметилхлорсиланом, аллилгалогенидами, предпочтительно с хлорметил-метиловым эфиром или 3-бром-1-пропеном, в таком инертном растворителе, как дихлорметан, тетрагидрофуран, диметоксиэтан, диметилформамид, предпочтительно тетрагидрофуран, в присутствии такого основания, как триэтиламин, гидрид натрия, трет-бутоксид калия, предпочтительно триэтиламин или трет-бутоксид калия, при температуре от 0 до 40°C, предпочтительно при 0°C. После конденсации фрагмента B4 соединения 7 с ацетилциклопропаном, например, в дихлорметане, тетрагидрофуране, диметоксиэтане, диметилформамиде, предпочтительно в тетрагидрофуране, в присутствии такого основания, как гидрид натрия, третичные амины, трет-бутоксид калия, предпочтительно трет-бутоксид калия, при температуре от 0 до 40°C, предпочтительно при температуре окружающей среды, получают соединение формулы 9. Восстановление фрагмента C4 соединения формулы 9 с получением соединения формулы 10 проводят, например, в изопропаноле, тетрагидрофуране, диметоксиэтане, предпочтительно в изопропаноле или тетрагидрофуране, в присутствии щелочи или боргидрида металла при температуре от -10°C до температуры окружающей среды, предпочтительно при температуре от -10 до 0°C, в присутствии боргидрида натрия. Удаление защитной группы фенола и последующую циклизацию фрагмента D4 соединения формулы 10 проводят в дихлорметане, тетрагидрофуране или ацетонитриле, в присутствии комплекса палладия или кислоты, например трифторуксусной кислоты, паратолуолсульфоновой кислоты, метансульфоновой кислоты, соляной кислоты, формиата аммония, предпочтительно в присутствии комплекса палладия и формиата аммония, или трифторуксусной кислоты, в зависимости от природы защитной группы, при комнатной температуре, или при температуре до 60°C, получая соединение формулы 11.
После ацилирования по Фриделю-Крафтсу фрагмента A5 соединения формулы 2 таким ацилирующим агентом, как ацетилхлорид или ангидрид уксусной кислоты, предпочтительно ангидрид уксусной кислоты, в присутствии кислоты Льюиса, например трихлорида алюминия, тетрахлорида титана или тетрахлорида олова, предпочтительно тетрахлорида олова, в инертном растворителе, например в дихлорметане или дихлорэтане, при температуре от -10 до 40°C, предпочтительно в интервале от 0°C до комнатной температуры, получают соединение 12, которое, в свою очередь, подвергают деметилированию по фрагменту B5 в инертном растворителе, например в дихлорметане или ацетонитриле, в присутствии такой кислоты Льюиса, как трихлорид алюминия, трихлорид бора, трибромид бора, предпочтительно трихлорид алюминия, и такого нуклеофила, как йодид натрия, диметилсульфид, тетрагидротиофен, предпочтительно йодид натрия, в интервале температур от комнатной до 40°C, получая соединение формулы 13. После конденсации фрагмента C5 соединения формулы 13 с соединением формулы 14 в ацетонитриле, в присутствии пирролидина в качестве основания, при температуре от комнатной до 50°C, предпочтительно при комнатной температуре, получают соединение формулы 15.
Ацилирование фрагмента A6 соединения формулы 2 соединением формулы 16 по Фриделю-Крафтцу, в присутствии кислоты Льюиса, например трихлорида алюминия, тетрахлорида титана или тетрахлорида олова, предпочтительно тетрахлорида олова, в инертном растворителе, например дихлорметане или дихлорэтане, при температуре от -10 до 40°C, предпочтительно в интервале от 0°C до комнатной температуры, приводит к получению соединения формулы 17, которое также можно синтезировать конденсацией фрагмента B6 соединения 12 с соединением формулы 14 в тетрагидрофуране, в присутствии трет-бутоксида калия в качестве основания, при температуре от комнатной до 50°C, предпочтительно при комнатной температуре. После деметилирования фрагмента C6 соединения 17 в инертном растворителе, например дихлорметане или ацетонитриле, в присутствии такой кислоты Льюиса, как трихлорид алюминия, трихлорид бора, трибромид бора, предпочтительно трихлорид алюминия, и нуклеофила, например йодида натрия, диметилсульфида, тетрагидротиофена, предпочтительно йодида натрия, при температуре от комнатной до 40°C получают соединение формулы 18. Циклизацию фрагмента D6 соединения формулы 18 можно проводить в таком инертном растворителе, как диметилформамид, в присутствии карбоната калия в качестве основания, при комнатной температуре, получая соединение формулы 15.
Восстановление фрагмента A7 соединения формулы 15 с получением соединения формулы 19 можно проводить, например, в изопропаноле, тетрагидрофуране, диметоксиэтане, предпочтительно в изопропаноле, в присутствии боргидрида натрия, в интервале температур от 0°C до температуры окружающей среды, предпочтительно при температуре окружающей среды. Отщепление молекулы воды из фрагмента B7 соединения формулы 19 в присутствии такой кислоты, как соляная кислота, метансульфоновая кислота, трифторуксусная кислота, предпочтительно трифторуксусная кислота, в толуоле, при температуре от комнатной до 100°C, предпочтительно при 80°C, приводит к получению соединения формулы 11.
После удаления защитной группы с фрагмента A8 соединения формулы 11, которое можно проводить в смеси таких органических растворителей, как тетрагидрофуран или метиловый спирт, предпочтительно тетрагидрофуран, с водой, в присутствии такого сильного основания, как гидроксид натрия или калия, предпочтительно гидроксид натрия, в интервале температур от 40 до 80°C, предпочтительно при 50°C, получают целевое соединение I.
Соединения формул 2, 3, 5, 6, 7, 9-13, 15 и 17-19 являются новыми и также составляют предмет данного изобретения. Их можно получить с помощью последовательностей реакций, изображенных на схемах 1-8. Кроме того, в нижеследующих примерах приведено более подробное описание способов получения соединений, изображенных на схемах 1-8.
Как указано, соединение формулы I или его фармацевтически приемлемые соли обладают ценными антибактериальными свойствами. Данные соединения активны в отношении широкого ряда таких патогенных микроорганизмов, как, например, S.aureus, P.carinii и др., благодаря их способности ингибировать бактериальную дигидрофолатредуктазу (DHFR). Активность соединения I более подробно описана в P.G. Hartmann et al. Abstracts, F2020, 42nd Interscience Conference on Antimicrobial Agents and Chemotherapy, San Diego, Ca, Sep 27-30, 2002; American Society for Microbiology: Washington, DC, 2002.
Другие цели, преимущества и новые признаки данного изобретения станут ясны специалистам в данной области после анализа нижеследующих примеров, которые не предназначены для ограничения объема изобретения.
Нижеследующие примеры более подробно иллюстрируют данное изобретение. В примерах 1-11 описан способ получения соединения 6, тогда как в примерах 12-19 описан способ получения соединения формулы 11 с использованием соединения формулы 6 в качестве исходного. В примерах 20-26 описан синтез соединения 15, превращение которого в соединение 11 описано в примерах 27 и 28, а в примерах 29 и 30 описано превращение соединения 11 в конечный продукт формулы I (иклаприм).
Примеры
Соединение формулы 4 можно получить, например, способом, описанным в M. Calas et al., Eur. J. Med. Chem. Chim. Ther., 17 (6), 497 (1982). Соединение 7 можно получить способом, аналогичным описанному, например, в W. B. Wright et al., J. Med. Chem., 11(6), 1164 (1968). Соединение формулы 27 можно синтезировать, например, способом, описанным в Z. Ma et al., Tetrahedron: Asymmetry 6 (6). 883 (1997).
Все другие реагенты и растворители являются коммерчески доступными, их можно получить, например, от Fluka или от других поставщиков. Температуры приведены в градусах Цельсия.
Система ЖХ-МС
Колонка ВЭЖХ 01: обращенная фаза Atlantis dC18 3 мкм, колонка 4,6×75 мм с предохранительным картриджем
Градиент 01:
Колонка ВЭЖХ 02: обращенная фаза Waters C18 3,5 мкм, колонка 3×20 мм
Градиент 02:
Растворитель A: 10 мM муравьиную кислоту (377 мкл муравьиной кислоты) добавляют к воде, чистой для ВЭЖХ (1 л, фильтрованной через Millipore)
Растворитель B: Ацетонитрил чистый для ВЭЖХ (Biosolve Ltd)
Длина волны: от 210 нм до 400 нм.
Аппаратура для ВЭЖХ: насос Finnigan Surveyor LC, детектор на фотодйодной матрице (PDA) Finnigan Surveyor
Аппаратура для МС: Finnigan Surveyor MSQ Plus (ION TRAP), способ ионизации ESI
Сокращения
Пример 1
Данный пример иллюстрирует получение N-[4-(2,2-диметилпропиониламино)-5-(3,4,5-триметоксибензил)пиримидин-2-ил]-2,2-диметилпропионамида 2 (R=C(CH3)3) (стадия A1).
Раствор триметоприма (5 г, 17,24 ммоль) в ангидриде пивалевой кислоты (8,74 мл, 43,10 ммоль, 2,5 экв.) нагревают в течение 2 ч при 150°C в атмосфере аргона. Добавляют горячий AcOEt, органические слои промывают 10%-ным водным раствором NaHCO3, водой и насыщенным раствором соли. Затем органические слои сушат над MgSO4, фильтруют и упаривают. После перекристаллизации из TBME получают 3,02 г соединения 2 (R=C(CH3)3).
1H-ЯМР (CDCl3, 400 MГц) δ: 8,35 (с, 1Н), 8,21 (шир.с, 1Н), 7,65 (шир.с, 1Н), 6,30 (с, 2H), 3,86 (с, 2Н), 3,79 (с, 3Н), 3,77 (с, 6Н), 1,31 (с, 9Н), 1,12 (с, 9Н). Т.пл.:130-133°С.
Пример 2
Данный пример иллюстрирует получение N-[4-изобутириламино-5-(3,4,5-триметоксибензил)пиримидин-2-ил]-изобутирамида 2 (R=CH(CH3)2) (стадия A1).
Раствор триметоприма (50 г, 172,4 ммоль) в изомасляном ангидриде (100 г, 105 мл, 632 ммоль, 3,6 экв.) нагревают в течение 2 ч при 150°C в атмосфере аргона. Теплый раствор выливают в 1 л циклогексана после чего происходит медленная кристаллизация. Продукт отфильтровывают и тщательно промывают циклогексаном (2×200 мл), получая 70 г соединения 2 (R=CH(CH3)2).
1H-ЯМР (D6-ДМСО, 400 MГц) δ: 10,42 (с, 1H, NH), 10,15 (с, 1H, NH), 8,41 (с, 1H, пиримидин), 6,41 (с, 2H, PhH), 3,81 (с, 2H, CH2), 3,70 (с, 6H, 2×OCH3), 3,59 (с, 3H, OCH3), 2,72-2,85 (м, 2H, CH), 1,06 (д, 6H, J=6,6Гц, 2×CH3), 1,01 (д, 6H, J=6,6Гц, 2×CH3). Т.пл.: 153-154°C, Rt(02)=1,65 минут.
Пример 3
Данный пример иллюстрирует получение N-[4-изобутириламино-5-(3,4,5-триметоксибензил)пиримидин-2-ил]-изобутирамида 2 (R=CH(CH3)2) (стадия A1).
Раствор триметоприма (50 г, 172,4 ммоль) в изомасляном ангидриде (62 г, 65,5 мл, 392 ммоль, 2,3 экв.) нагревают в течение 2 ч при 150°C в атмосфере аргона и перемешивают механической мешалкой. Раствор охлаждают до 130°C и добавляют 200 мл толуола (прозрачный раствор), затем медленно добавляют 1000 мл TBME (после добавления 500 мл начинается кристаллизация) при энергичном перемешивании. Густой кристаллический осадок перемешивают в течение 1 часа при окружающей температуре 100°C. Затем взвесь охлаждают до комнатной температуры и перемешивают в течение 2 часов. В конце взвесь охлаждают до 10°C и перемешивают в течение 2 часов. Кристаллы фильтруют и промывают 3 раза 90 мл TBME, чтобы удалить остаток изомасляной кислоты и ангидрида. Кристаллы сушат при HV/70°C в течение 8 часов и получают 70 г соединения 2 (R=CH(CH3)2).
1H-ЯМР (D6-ДМСО, 400 MГц) δ: 10,42 (с, 1H, NH), 10,15 (с, 1H, NH), 8,41 (с, 1H, пиримидин), 6,41 (с, 2H, PhH), 3,81 (с, 2H, CH2), 3,70 (с, 6H, 2×OCH3), 3,59 (с, 3H, OCH3), 2,72-2,85 (м, 2H, CH), 1,06 (д, 6H, J=6,6 Гц, 2×CH3), 1,01 (д, 6H, J=6,6 Гц, 2xCH3). Т.пл.: 153-154°C, Rt (02)=1,65 минут.
Пример 4
Данный пример иллюстрирует получение N-[4-(2,2-диметилпропиониламино)-5-(2-формил-3,4,5-триметоксибензил)пиримидин-2-ил]-2,2-диметилпропионамида 3 (R=C(CH3)3) (стадия B1).
К раствору соединения 2 (1 г, 2,18 ммоль, R=C(CH3)3) в DCM (5 мл), добавляют дихлорметил-метиловый эфир (0,58 мл, 6,54 ммоль). Раствор охлаждают до -30°C, после чего медленно добавляют хлорид олова (0,285 мл, 2,18 ммоль). Смесь перемешивают при температуре от -10 до -5°C. При 0°C реакционную смесь выливают в 1N раствор K3PO4. Затем смесь (pH 7-8) энергично перемешивают в течение 15 минут и дважды экстрагируют AcOEt. Органические слои промывают водой и насыщенным раствором соли, сушат над MgSO4, фильтруют и упаривают. Неочищенный продукт очищают флэш-хроматографией на силикагеле (AcOEt/циклогексан 7/3), получая 709 мг соединения 3 (R=C(CH3)3).
1H-ЯМР (CDCl3, 400 MГц) δ: 10,23 (с, 1Н), 8,44 (с, 1Н), 8,12 (с, 1Н), 8,09 (с, 1Н), 6,59 (с, 1Н), 4,10 (с, 2Н), 3,96 (с, 3Н), 3,89 (s, 3Н), 3,85 (с, 3Н), 1,29 (с, 9Н), 1,27 (с, 9Н). Т.пл.: 124-126°C.
Пример 5
Данный пример иллюстрирует получение N-[5-(2-формил-3,4,5-триметоксибензил)-4-изобутириламинопиримидин-2-ил]изобутирамида 3 (R=CH(CH3)2) (стадия B1).
К раствору соединения 2 (70 г, 162 ммоль, R=CH(CH3)2) в DCM (500 мл) добавляют дихлорметил-метиловый эфир (30 мл, 325 ммоль). Раствор охлаждают до -10°C, после чего медленно добавляют хлорид олова (35 мл, 300 ммоль). Смесь перемешивают при температуре от -10 до -5°C. В начале образуется смолистый осадок (требуется механическое перемешивание). После перемешивания в течение 1 ч при -5°C образуется "гомогенная" суспензия.
При 0°C реакционную смесь выливают в раствор, содержащий 300 мл 1N K3PO4 и 200 мл 1M тартрата Na/K, охлаждая на ледяной бане. Затем смесь (pH доводят до 7-8 с помощью 4N раствора NaOH) перемешивают в течение 15 минут до завершения гидролиза и затем экстрагируют DCM (300 мл) вместе с AcOEt (500 мл). Органический слой промывают 0,1н раствором HCl (2×200 мл) и насыщенным раствором соли (2×300 мл), сушат над MgSO4, фильтруют и упаривают. После концентрирования приблизительно до половины начального объема начинает осаждаться продукт, затем добавляют циклогексан для дополнительного осаждения продукта (200 мл), который отфильтровывают и получают 50 г соединения 3 (R=CH(CH3)2).
1H-ЯМР (D6-ДМСО, 400 MГц) δ: (с, 1H, NH), 10,27 (с, 1H, NH), 10,17 (с, 1H, CHO), 8,00 (с, 1H, пиримидин), 6,72 (с, 1H, PhH), 4,05 (с, 2H, CH2), 3,90 (с, 3H, OCH3), 3,85 (с, 3H, OCH3), 3,77 (с, 3H, OCH3), 2,75-2,85 (м, 2H, CH), 1,05 (д, 6H, J=6,6 Гц, 2×CH3), 1,01 (д, 6H, J=6,6Гц, 2×CH3). Т.пл.: 162-163°C, Rt(02)=1,85 минут.
Пример 6
Данный пример иллюстрирует получение N-[5-(2-формил-3,4,5-триметоксибензил)-4-изобутириламинопиримидин-2-ил]изобутирамида 3 (R=CH(CH3)2) (стадия B1).
Дихлорметил-метиловый эфир (4,3 мл, 46,4 ммоль, 2 экв.) растворяют в DCM (30 мл) и охлаждают до температуры от -15 до -20°C в реакторе, оборудованном механической мешалкой. К полученному раствору в течение 15 минут добавляют хлорид олова (5 мл, 42,8 ммоль, 1,8 экв.). Прозрачный раствор перемешивают при -18°C в течение 30 минут. Затем в течение 60 минут непрерывно добавляют раствор соединения 2 (10 г, 23,2 ммоль, кристаллизованный из толуола и TBME) в DCM (40 мл), при этом сразу начинает осаждаться твердое вещество желтого цвета, образующее в конце густую взвесь (желто/зеленую). Затем взвесь перемешивают два часа при -15°C, один час при -10°C и 30 минут при -5°C. Затем при -5°C добавляют 40 мл DCM и образовавшиеся кристаллы в верхней части слоя растворителя удаляют энергичным перемешиванием в течение 15 минут. Жидкую взвесь переносят в интенсивно перемешиваемую смесь, содержащую 35 г Na2CO3 (с однокристальной водой), растворенного в 100 мл воды и 35 мл DCM, при 10°C. Смесь перемешивают в течение 15 минут при комнатной температуре и затем снова переносят в реактор при непрерывном перемешивании при комнатной температуре, чтобы завершить обработку. После интенсивного перемешивания в течение 30 минут при комнатной температуре слои разделяют и органическую фазу дважды промывают смесью, содержащей 30 мл насыщенного раствора NaCl, 5 мл насыщенного раствора Na2CO3 и 40 мл воды (нужно несколько раз встряхнуть, водные фазы должны иметь pH 7-8). Мутные водные фазы промывают 50 мл DCM и отдельно 30 мл DCM.
Органические слои сушат над MgSO4, фильтруют и упаривают. Неочищенный кристаллический продукт белого цвета кристаллизуют из DCM и TBME. Получают взвесь 10,67 г неочищенного вещества в 25 мл DCM при 44°C при перемешивании в течение 30 минут, после чего медленно добавляют 100 мл TBME, взвесь перемешивают 30 минут при 44°C и затем охлаждают до комнатной температуры в течение 6 часов при перемешивании. Кристаллы фильтруют, промывают 40 мл TBME, сушат в высоком вакууме/КТ в течение 6 часов и получают 9,6 г соединения 3 (R=CH(CH3)2).
1H-ЯМР (D6-ДМСО, 400 MГц) δ: (с, 1H, NH), 10,27 (с, 1H, NH), 10,17 (с, 1H, CHO), 8,00 (с, 1H, пиримидин), 6,72 (с, 1H, PhH), 4,05 (с, 2H, CH2), 3,90 (с, 3H, OCH3), 3,85 (с, 3H, OCH3), 3,77 (с, 3H, OCH3), 2,75-2,85 (м, 2H, CH), 1,05 (д, 6H, J=6,6 Гц, 2×CH3), 1,01 (д, 6H, J=6,6Гц, 2×CH3). Т.пл.: 162-163°C, Rt (02)=1,85 минут.
Пример 7
Данный пример иллюстрирует получение N-[4-(2,2-диметилпропиониламино)-5-(2-йод-3,4,5-триметоксибензил)пиримидин-2-ил]-2,2-диметилпропионамида 5 (R=C(CH3)3) (стадия A2).
К раствору соединения 4 (5 г, 9,2 ммоль, R=C(CH3)3) в ангидриде пивалевой кислоты (4,1 мл, 20,24 ммоль) добавляют пиридин (1,65 мл, 20,24 ммоль). Смесь нагревают при 120°C в течение 12 ч. Добавляют 0,25н HCl (25 мл) и дважды экстрагируют AcOEt. Органические слои промывают последовательно водой, 10% NaHCO3, затем снова водой и насыщенным раствором соли, сушат над MgSO4, фильтруют и упаривают досуха. После флэш-хроматографии на силикагеле (AcOEt/циклогексан 1/1) получают 2,5 г конечного соединения 5 (R=C(CH3)3). УФ: 238 (282) нм.
Пример 8
Данный пример иллюстрирует получение N-[4-(2,2-диметилпропиониламино)-5-(2-формил-3,4,5-триметоксибензил)пиримидин-2-ил]-2,2-диметилпропионамида 3 (R=C(CH3)3) (стадия B2).
Соединение 5 (1 г, 1,71 ммоль, R=C(CH3)3) растворяют в THF (10 мл) и смесь дегазируют аргоном. Затем добавляют тетракис палладий (49,4 мг, 4 мол.%). Смесь нагревают до 70°C и начинают медленно пропускать газообразный монооксид углерода. В течение 2,5 ч медленно добавляют Bu3SnH (476 мкл, 1,05 экв.) в 5 мл THF. После выдерживания в течение 12 ч при 70°C соединение 3 (R=C(CH3)3) выделяют флэш-хроматографией. Аналитические данные аналогичны данным, полученным для соединения примера 4.
Пример 9
Данный пример иллюстрирует получение N-[4-(2,2-диметилпропиониламино)-5-(2-формил-3-гидрокси-4,5-диметоксибензил)пиримидин-2-ил]-2,2-диметилпропионамида 6 (R=C(CH3)3) (стадия A3).
В атмосфере аргона соединение 3 (2 г, 4,11 ммоль, R=C(CH3)3) растворяют в DCM (10 мл). К смеси при 0°C добавляют AlCl3 (823 мг, 6,17 ммоль). Перемешивание продолжают при комнатной температуре в течение 10 минут до полного растворения AlCl3. Добавляют йодид натрия (616 мг, 4,11 ммоль) и через 30 минут добавляют 0,5 мл ацетонитрила. За протеканием реакции следят с помощью ЖХ-МС, для завершения реакции добавляют 0,5 мл ацетонитрила. Затем реакционную смесь выливают в двухфазный раствор 1N K3PO4/DCM. Обе фазы разделяют. Водные слои дважды экстрагируют AcOEt, органические слои промывают водой и насыщенным раствором соли, затем сушат над MgSO4, фильтруют и упаривают. Соединение 6 (R='C(CH3)3) получают после очистки колоночной хроматографией на силикагеле с использованием в качестве элюента смеси AcOEt/циклогексан 6/4 (1,2 г).
1H-ЯМР (CDCl3, 400 MГц) δ: 12,07 (с, 1Н), 9,81 (с, 1Н), 8,94 (с, 1Н), 8,26 (с, 1Н), 7,95 (с, 1Н), 6,33 (с, 1Н), 4,07 (с, 2Н), 3,84 (с, 3Н), 3,80 (с, 3Н), 1,22 (с, 9Н), 1,20 (с, 9Н). Т.пл.: 110-112°C.
Пример 10
Данный пример иллюстрирует получение N-[5-(2-формил-3-гидрокси-4,5-диметоксибензил)-4-изобутириламинопиримидин-2-ил]изобутирамида 6 (R=CH(CH3)2) (стадия A3).
В атмосфере аргона соединение 3 (4 г, 8,7 ммоль, 1 экв., R=CH(CH3)2) растворяют в DCM (36 мл). К смеси при комнатной температуре добавляют трихлорид алюминия (3,48g, 26,1 ммоль, 3 экв.) и йодид натрия (2 г, 13,3 ммоль, 1,5 экв.). Смесь перемешивают в течение 20 минут, затем добавляют ацетонитрил (2,4 мл) и нагревают до 40°C. Перемешивание при 40°C продолжают в течение 3,5 ч. Смесь охлаждают до комнатной температуры, разбавляют 75 мл DCM и реакцию гасят добавлением 30 мл смеси воды со льдом, затем медленно добавляют 2,5 мл концентрированной HCl, которая способствует растворению осадка желтого цвета. Органический слой отделяют, а водный слой экстрагируют еще раз DCM (75 мл). Объединенные органические слои промывают насыщенным раствором соли (50 мл), дважды раствором бикарбоната натрия, полученным из 50 мл насыщенного раствора бикарбоната натрия (NaHCO3) и 150 мл воды (2×100 мл), 0,1н раствором HCl (50 мл) и снова насыщенным раствором соли (1×50 мл). Полученный раствор желтого цвета сушат над MgSO4 и концентрируют. Маслянистый остаток кристаллизуют из этилацетата (6 мл) и дихлорэтана (2,4 мл) путем нагревания до 50°C с последующим охлаждением до 4°C. После фильтрации маточный раствор концентрируют вдвое и выдерживают при 4°C, с получением второй порции кристаллов. Выделяют всего 2,48 г соединения 6 (R=CH(CH3)2).
1H-ЯМР (CDCl3, 400 MГц) δ: 11,95 (шир.с, 1H, PhOH), 9,9 (с, CHO, 1H), 8,04 (с, пиримидин, 1H), 6,44 (с, ArH, 1H), 4,15 (с, CH2, 2H), 3,93 (с, OCH3, 3H), 3,90 (с, OCH3, 3H), 2,7-2,8 (м, CH, 2H), 1,2-1,25 (м, CH3, 12H).
Пример 11
Данный пример иллюстрирует получение N-[5-(2-формил-3-гидрокси-4,5-диметоксибензил)-4-изобутириламинопиримидин-2-ил]изобутирамида 6 (R=CH(CH3)2) (стадия A3).
В атмосфере аргона соединение 3 (3 г, 6,54 ммоль) растворяют в DCM (28 мл) при комнатной температуре (15 минут). Раствор охлаждают до 0°C в течение 30 минут. К охлажденному раствору сразу добавляют AlCl3 (2,07 г, 15,52 ммоль, 2,3 экв.). Смесь перемешивают в течение 30 минут при 0°C, при этом цвет раствора меняется с желтого на темно-желтый и происходит растворение AlCl3. Затем добавляют NaI (1,5 г, 10 ммоль, 1,5 экв.) и смесь нагревают до 30°C. После перемешивания в течение 10 минут медленно добавляют ацетонитрил (1,6 мл). Перемешивают 2 часа при 30°С и добавляют еще 0,2 мл ацетонитрила. После 4 часов перемешивания при 30°С температуру реакции повышают до 35°C в течение 1 ч, при этом выделяются кристаллы. Взвесь охлаждают до комнатной температуры и затем выливают в интенсивно перемешиваемую смесь 20 мл DCM и 30 мл воды, содержащую 2 мл концентрированной HCl (охлажденной до 10°C). После перемешивания в течение 10 минут прозрачную смесь желтого цвета снова выливают в реакционный сосуд и перемешивание продолжают до полного растворения осадка (приблизительно 30 минут). Органическую фазу отделяют и промывают 25 мл смеси, содержащей 10 мл 1н HCl и 15 мл воды, и 25 мл смеси, содержащей 10 мл насыщенного раствора NaCl и 15 мл воды, водные слои экстрагируют 2 раза 20 мл DCM. Объединенный органический раствор сушат над MgSO4, фильтруют и упаривают. Желтый кристаллический остаток (2,62 г) кристаллизуют из DCM/TBME, получая 2,48 г соединения 6 (R=CH(CH3)2).
1H-ЯМР (CDCl3, 400 MГц) δ: 11,95 (шир.с, 1H, PhOH), 9,9 (с, CHO, 1H), 8,04 (с, пиримидин, 1H), 6,44 (с, ArH, 1H), 4,15 (с, CH2, 2H), 3,93 (с, OCH3, 3H), 3,90 (с, OCH3, 3H), 2,7-2,8 (м, CH, 2H), 1,2-1,25 (м, CH3, 12H).
Пример 12
Данный пример иллюстрирует получение N-[4-(2,2-диметилпропиониламино)-5-(2-формил-4,5-диметокси-3-метоксиметоксибензил)пиримидин-2-ил]-2,2-диметилпропионамида 7 (R=C(CH3)3, R'=метоксиметил) (стадия A4).
В атмосфере аргона соединение 6 (200 мг, 0,424 ммоль, R=C(CH3)3) растворяют в DCM (2 мл). К смеси при 0°C добавляют триэтиламин (59,6 мкл, 1 экв.). Медленно добавляют MOM-Cl (32,2 мкл, 1 экв.). После перемешивания в течение 1 ч для завершения реакции добавляют 1 эквивалент триэтиламина и 1 эквивалент MOM-Cl.
К реакционной смеси добавляют 0,25N HCl и смесь экстрагируют AcOEt. Органические слои промывают водой и насыщенным раствором соли, затем сушат над MgSO4, фильтруют и упаривают. Соединение 7 (R=C(CH3)3, R'=метоксиметил) используют сразу на следующей стадии. УФ: 234 (283) нм.
Пример 13
Данный пример иллюстрирует получение N-[-4-изобутириламинопиримидин-2-ил]изобутирамид 5-(2-формил-4,5-диметокси-3-аллилоксибензил)7 (R=CH(CH3)2, R*=аллил) (стадия A4).
Раствор соединения 6 (166,9 г, 0,35 моль, R=CH(CH3)2, R'=аллил) в DMF (800 мл) охлаждают до 0°C и добавляют tBuOK (47 г, 0,42 моль). К данному раствору медленно добавляют аллилбромид (41 мл, 0,49 моль) и перемешивают в течение 3 часов при 0°C и 16 часов при комнатной температуре. Реакционную смесь фильтруют и растворитель отгоняют при пониженном давлении (20 мбар) и 60°C. Остаток разбавляют DCM (850 мл), органическую фазу промывают 1N HCl (250 мл), насыщенным раствором бикарбоната натрия (500 мл) и водой (500 мл), затем сушат над MgSO4, фильтруют и упаривают, получая 175 г кристаллического вещества 7 (R=CH(CH3)2, R'=аллил).
1H-ЯМР (CDCl3, 400 MГц) δ: 10,34 (с, 1Н), 9,37 (широкий, NH), 8,64 (широкий, NH), 8,32 (с, 1Н), 6,53 (с, 1Н), 6,02 (м, 1Н), 5,31 (м, 2Н), 4,70 (м, 2Н), 4,12 (с, 2Н), 3,87 (с, 3Н), 3,84 (с, 3Н), 2,91 (широкий, 1H), 2,67 (м, 1Н), 1,22 (д, J=7,2 Гц, 6H), 1,17 (д, J=7,2 Гц, 6H).
Пример 14
Данный пример иллюстрирует получение N-{5-[2-(3-циклопропил-3-оксо-пропенил)-4,5-диметокси-3-метоксиметоксибензил]-4-(2,2-диметилпропиониламино)пиримидин-2-ил}-2,2-диметилпропионамида 9 (R=CH(CH3)3, R'=метоксиметил) (стадия B4).
В атмосфере аргона соединение 7 (140 мг, 0,27 ммоль, R=C(CH3)3, R'=метоксиметил) растворяют в THF (1 мл). Во второй колбе получают енолят ацетилциклопропана 8 (28 мкл, 1,1 экв.), добавляют tBuOK (34 мг, 1,1 экв.) в 1 мл THF и через 3 минуты енолят добавляют по каплям к раствору соединения 7 (R=CH(CH3)3, R'=метоксиметил) при комнатной температуре. Через 5 минут для завершения реакции добавляют еще эквивалент енолята 8. Через 1 ч к реакционной смеси добавляют 0,25н. HCl и смесь дважды экстрагируют AcOEt. Органические слои промывают водой и насыщенным раствором соли, затем сушат над MgSO4, фильтруют и упаривают. Неочищенное вещество очищают флэш-хроматографией на силикагеле (AcOEt/циклогексан 5/5→AcOEt/циклогексан 7/3) и получают 0,133 г соединения 9 (R=C(CH3)3, R'=метоксиметил).
1H-ЯМР (CDCl3, 400 MГц) δ: 8,22 (с, 1Н), 8,15 (с, 1Н), 8,08 (с, 1Н), 7,44 (д, J=16,4 Гц, 1Н), 6,78 (д, J=16,4 Гц, 1Н), 6,51 (с, 1Н), 5,06 (с, 2Н), 3,83 (с, 2Н), 3,77 (с, 3Н), 3,76 (с, 3Н), 3,43 (с, 3Н), 1,98-2,04 (м, 1Н), 1,22 (с, 9Н), 1,19 (с, 9Н), 1,01 (квинт, J=3,6 Гц, 2Н), 0,82-0,86 (м, 2Н).
Пример 15
Данный пример иллюстрирует получение N-{5-[2-(3-циклопропил-3-оксопропенил)-4,5-диметокси-3-аллилoксибензил]-4 изобутириламинопиримидин-2-ил]изобутирамид 9 (R=CH(CH3)2, R'=аллил) (стадия B4).
В атмосфере аргона соединение 7 (424 мг, 0,877 ммоль, R=CH(CH3)2, R*=аллил) растворяют в смеси THF/DMF (6 мл, 2:1). Во второй колбе получают енолят ацетилциклопропана 8 (109 мкл, 1,1 экв.), добавляют tBuOK (131 мг, 1,1 экв.) в 2 мл THF и через 3 минуты енолят добавляют по каплям к раствору соединения 7 (R=CH(CH3)3, R'=метоксиметил) при комнатной температуре. Через 5 минут для завершения реакции добавляют еще эквивалент енолята 8. Через 3 дня реакционную смесь разбавляют DCM и промывают три раза 5%-ным раствором гидрокарбоната натрия, затем сушат над MgSO4, фильтруют и упаривают. Неочищенное вещество очищают флэш-хроматографией на силикагеле (AcOEt/толуол 1/1), получая 0,340 г соединения 9 (R=CH(CH3)3, R'=аллил). Соединение кристаллизуют из TBME и получают 0,210 г кристаллов.
1H-ЯМР (CDCl3, 400 MГц) δ: 8,80 (широкий, 2 NH), 8,08 (с, 1Н), 7,45 (д, J=16,2 Гц, 1Н), 6,94 (д, J=16,2 Гц, 1Н), 6,61 (с, 1Н), 6,06 (м, 1H), 5,30 (м, 2Н), 4,53 (м, 2Н), 3,91 (с, 2Н), 3,87 (с, 3Н), 3,86 (с, 3Н), 3,13 (широкий, 1H), 2,27 (м, 1Н), 2,04 (м, 1Н), 1,22 (д, J=2,0 Гц, 6Н), 1,20 (д, J=2,5 Гц, 6Н), 1,06 (м, 2H), 0,91 (м, 2Н).
Пример 16
Данный пример иллюстрирует получение N-{5-[2-(3-циклопропил-3-гидроксипропенил)-4,5-диметокси-3-метоксиметоксибензил]-4-(2,2-диметилпропиониламино)пиримидин-2-ил}-2,2-диметилпропионамида 10 (R=C(CH3)3, R'=метоксиметил) (стадия C4).
В атмосфере аргона соединение 9 (418 мг, 0,718 ммоль, R=C(CH3)3, R'=метоксиметил) растворяют в iPrOH (5 мл). При комнатной температуре добавляют NaBH4 (54 мг, 1,436 ммоль). Через 2 ч реакцию останавливают. К реакционной смеси добавляют воду и дважды экстрагируют AcOEt. Органические слои промывают водой и насыщенным раствором соли, затем сушат над MgSO4, фильтруют и упаривают. Неочищенное вещество очищают флэш-хроматографией на силикагеле (AcOEt/циклогексан 8/2) и получают 0,29 г соединения 10 (R=C(CH3)3, R'=метоксиметил).
1H-ЯМР (CDCl3, 400 MГц) δ: 8,72 (с, 1Н), 8,15 (с, 1Н), 7,92 (с, 1Н), 6,46 (д, J=16,4 Гц, 1Н), 6,34 (с, 1Н), 6,05 (дд, J1=16,4 Гц, J2=6,2 Гц, 1Н), 5,04 (с, 2Н), 3,83 (с, 2Н), 3,78 (с, 5Н), 3,70 (с, 3Н), 3,57 (м, 1Н), 3,51 (с, 3Н), 1,29 (с, 9Н), 1,05 (с, 9Н), 037-0,56 (м, 2Н), 0,20-0,26 (м, 2Н).
Пример 17
Данный пример иллюстрирует получение N-{5-[2-(3-циклопропил-3-гидроксипропенил)-4,5-диметокси-3-аллилоксибензил]-4-изобутириламинопиримидин-2-ил}изобутирамид 10 (R=СН(СН3)2, R'=аллил) (стадия С4).
В атмосфере аргона соединение 9 (100 мг, 0,181 ммоль, R=CH(CH3)2, R'=аллил) растворяют в iPrOH (2 мл). При комнатной температуре добавляют NaBH4 (13 мг, 0,363 ммоль). В течение 2 часов реакционную смесь держат при комнатной температуре и в течение 16 часов при 4°C. К реакционной смеси добавляют воду и дважды экстрагируют AcOEt. Органические слои сушат над MgSO4, фильтруют и упаривают, получая 0,11 г соединения 10 (R=CH(CH3)2, R'=аллил). Неочищенное вещество используют на следующей стадии реакции без очистки. ЖХ-МС (градиент 01): Rt=8,43; [M+H]+=553.
Пример 18
Данный пример иллюстрирует получение N-[5-(2-циклопропил-7,8-диметокси-2H-хромен-5-илметил)-4-(2,2-диметилпропиониламино)пиримидин-2-ил]-2,2-диметилпропионамида 11 (R=C(CH3)3) (стадия D4).
В атмосфере аргона соединение 10 (180 мг, 0,308 ммоль, R=C(CH3)3, R'=метоксиметил) растворяют в DCM (2 мл). Добавляют TFA (24 мкл, 0,308 ммоль) и через 30 минут при комнатной температуре добавляют 2 эквивалент TFA. Через 5 минут добавляют 10%-ный раствор NaHCO3 (10 мл) и смесь экстрагируют AcOEt. Органические слои промывают водой и насыщенным раствором соли, затем сушат над MgSO4, фильтруют и упаривают. Неочищенное вещество очищают флэш-хроматографией на силикагеле (AcOEt/циклогексан 4/6→AcOEt/циклогексан 5/5) и получают 0,1 г соединения 11 (R=C(CH3)3).
1H-ЯМР (CDCl3, 400 MГц) δ: 8,19 (с, 1Н), 8,12 (с, 1Н), 8,00 (с, 1Н), 6,38 (д, J=10 Гц, 1Н), 6,16 (с, 1Н), 5,67 (дд, J1=10,2 Гц, J2=3,4 Гц, 1Н), 4,19 (дд, J1=8,4 Гц, J2=3,4 Гц, 1Н), 3,87 (с, 2Н), 3,80 (с, 2Н), 3,75 (с, 3Н), 1,30 (с, 9Н), 1,21 (с, 9Н), 0,43-0,61 (м, 3Н), 0,26-0,34 (м, 2Н).
Пример 19
Данный пример иллюстрирует получение N-[5-(2-циклопропил-7,8-диметокси-2Н-хромен-5-илметил)-4-изобутириламинопиримидин-2-ил]изобутирамид 11 (R=СН(СН3)2) (стадия D4).
В атмосфере аргона соединение 10 (110 мг, 0,18 ммоль, R=CH(CH3)2, R'=аллил) растворяют в ацетонитриле (2 мл). Добавляют тетракис(трифенилфосфин)палладий (46 мг, промытый ДМСО, EtOH, диэтиловым эфиром) и формиат аммония (57 мг), после чего взвесь нагревают до 70°C в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь разбавляют EtOAc (30 мл) и промывают водой. Органический слой промывают водой и насыщенным раствором соли, затем сушат над MgSO4, фильтруют и упаривают, получая масло, которое используют на следующей стадии реакции без очистки. ЖХ-МС (градиент 01): Rt=7,33 [M+H]+=495.
Пример 20
Данный пример иллюстрирует получение N-[5-(2-ацетил-3,4,5-триметоксибензил)-4-(2,2-диметилпропиониламино)пиримидин-2-ил]-2,2-диметилпропионамида 12 (R=C(CH3)3) (стадия A5).
В атмосфере аргона соединение 2 (2 г, 4,37 ммоль, R=C(CH3)3) растворяют в DCM (5 мл). При 0°C добавляют уксусный ангидрид (2,06 мл, 5 экв.) и затем SnCl4 (2,55 мл, 5 экв.). Перемешивание продолжают при комнатной температуре, через 4 ч реакционную смесь обрабатывают 10% NaHCO3, чтобы довести pH до 9, после чего дважды экстрагируют AcOEt. Органические слои промывают водой и насыщенным раствором соли, затем сушат над MgSO4, фильтруют и упаривают. Неочищенное вещество очищают флэш-хроматографией на силикагеле (AcOEt/циклогексан от 6/4 до 7/3) и получают 1,4 г соединения 12 (R=C(CH3)3).
1H-ЯМР (CDCl3, 400 MГц) δ: 8,28 (с, 1Н), 8,22 (с, 1Н), 8,14 (с, 1Н), 6,34 (с, 1Н), 3,89 (с, 3Н), 3,81 (с, 3Н), 3,80 (с, 2Н), 3,75 (с, 3Н), 2,41 (с, 3Н), 1,30 (с, 9Н), 1,18(с,9H). Т.пл.: 94-98°C.
Пример 21
Данный пример иллюстрирует получение N-[5-(2-ацетил-3-гидрокси-4,5-диметоксибензил)-4-(2,2-диметилпропиониламино)пиримидин-2-ил]-2,2-диметилпропионамида 13 (R=C(CH3)3) (стадия B5).
В атмосфере аргона соединение 12 (2,34 г, 4,68 ммоль, R=C(CH3)3) растворяют в DCM (20 мл). При 0°C добавляют AlCl3 (1,872 г, 14,04 ммоль) и перемешивание продолжают до завершения растворения. Затем добавляют йодид натрия (702 мг, 4,68 ммоль) и через 30 минут 3 мл CH3CN. Реакционную смесь перемешивают до тех пор, пока количество исходного вещества не перестанет уменьшаться (LCMS). К реакционной смеси добавляют AcOEt и 0,1н. HCl, после чего смесь дважды экстрагируют AcOEt. Органические слои промывают водой и насыщенным раствором соли, затем сушат над MgSO4, фильтруют и упаривают. Неочищенное вещество очищают флэш-хроматографией на силикагеле (AcOEt/циклогексан 8/2) и получают 1,5 г соединения 13 (R=C(CH3)3).
1H-ЯМР (CDCl3, 400 MГц) δ: 8,42 (с, 2Н), 8,22 (с, 1Н), 6,20 (с, 1Н), 3,92 (с, 2Н), 3,84 (с, 3Н), 3,77 (с, 3Н), 2,50 (с, 3Н), 1,29 (с, 9Н), 1,19 (с, 9Н).
Пример 22
Данный пример иллюстрирует получение N-[5-(2-циклопропил-7,8-диметокси-4-оксо-хроман-5-илметил)-4-(2,2-диметилпропиониламино)пиримидин-2-ил]-2,2-диметилпропионамида (R=C(CH3)3) (стадия C5)
Соединение 13 (432 мг, 0,889 ммоль, R=C(CH3)3) растворяют в CH3CN (10 мл). Добавляют пирролидин (81 мкл, 0,978 ммоль) и циклопропилкарбоксальдегид 14 (73 мкл, 0,978 ммоль), после чего реакционную смесь перемешивают в течение ночи при комнатной температуре. Добавляют AcOEt и 10%-ный раствор NaHCO3. Органические слои промывают 0,5н. HCl, водой и насыщенным раствором соли, затем сушат над MgSO4, фильтруют и упаривают досуха. Неочищенное вещество очищают флэш-хроматографией на силикагеле (AcOEt/циклогексан 7/3) и получают 0,24 г соединения 15 (R=C(CH3)3).
1H-ЯМР (CDCl3, 400 MГц) δ: 8,70 (шир.с, 1H), 8,37 (шир.с, 1Н), 8,04 (с, 1Н), 6,51 (с, 1Н), система АВ (δA 4,20, δB 4,07, JАВ=15,6 Гц, 2Н), 3,91 (с, 3Н), 3,85 (с, 3Н), 3,64-3,70 (м, 1Н), 2,61-2,76 (м, 2Н), 1,34 (с, 9Н), 1,29 (с, 9Н), 0,59-0,74 (м, 2Н), 0,50-0,57 (м, 1Н), 0,28-0,38 (м, 1Н).
Пример 23
Данный пример иллюстрирует получение N-{5-[2-(3-циклопропилакрилоил)-3,4,5-триметоксибензил]-4-(2,2-диметилпропиониламино)пиримидин-2-ил}-2,2-диметилпропионамида 17 (R=C(CH3)3) (стадия A6).
В атмосфере аргона соединение 2 (0,5 г, 1,09 ммоль, R=C(CH3)3) растворяют в DCM (5 мл), после чего при 0°C добавляют соединение 16 (0,71 г, 5 экв.) и затем SnCl4 (0,63 мл, 5 экв.). Перемешивание продолжают при комнатной температуре, через 4 ч реакционную смесь обрабатывают 10% NaHCO3, чтобы довести pH до 9, и дважды экстрагируют AcOEt. Органические слои промывают водой и насыщенным раствором соли, затем сушат над MgSO4, фильтруют и упаривают. Неочищенное вещество очищают флэш-хроматографией на силикагеле (AcOEt/циклогексан от 6/4 до 7/3) и получают соединение 17 (R=C(CH3)3).
1H-ЯМР (CDCl3, 400 MГц) δ: 0,58-0,61 (м, 2Н), 0,93-0,98 (м, 2Н), 1,16 (с, 9Н), 1,28 (с, 9Н), 1,51-1,60 (м, 1Н), 3,71 (с, 2Н), 3,74 (с, 3Н), 3,78 (с, 6Н), 6,02 (дд, J1=15,2 Гц, J2=10 Гц, 1Н), 6,35 (с, 1Н), 6,38 (д, J=15,2 Гц, 1Н), 8,15 (шир.с, 1Н), 8,28 (с, 1Н), 8,35 (шир.с, 1H).
Пример 24
Данный пример иллюстрирует получение N-{5-[2-(3-циклопропилакрилоил)-3,4,5-триметоксибензил]-4-(2,2-диметилпропиониламино)пиримидин-2-ил}-2,2-диметилпропионамида 17 (R=C(CH3)3) (стадия B6).
Соединение 12 (500 мг, 1 ммоль, R=C(CH3)3) растворяют в THF (10 мл). Добавляют tBuOK (168,3 мг, 1,5 ммоль) и смесь перемешивают 10 минут при комнатной температуре. Добавляют циклопропилкарбоксальдегид 14 (82 мкл, 1,1 ммоль) и затем реакционную смесь перемешивают в течение 30 минут при комнатной температуре. После добавления AcOEt и 0,1н. HCl органические слои промывают водой и насыщенным раствором соли, затем сушат над MgSO4, фильтруют и упаривают досуха. Неочищенное вещество очищают флэш-хроматографией на силикагеле (AcOEt/циклогексан 6/4) и получают 0,45 г соединения 17 (R=C(CH3)3), имеющего такой же спектр ЯМР, как в примере 23.
Пример 25
Данный пример иллюстрирует получение N-{5-[2-(3-циклопропил-3-йодпропионил)-3-гидрокси-4,5-диметоксибензил]-4-(2,2-диметилпропиониламино)пиримидин-2-ил}-2,2-диметилпропионамида 18 (R=C(CH3)3) (стадия C6).
Соединение 17 (152 мг, 0,275 ммоль, R=C(CH3)3) растворяют в DCM (2 мл). Добавляют йодид натрия (103 мг, 0,6875 ммоль) и смесь перемешивают в течение 5 минут, после чего добавляют трихлорид алюминия (55 мг, 0,413 ммоль). Перемешивание продолжают в течение 30 минут при комнатной температуре, затем добавляют 0,5 мл CH3CN. Через 2 ч добавляют AcOEt, органический слой промывают водой и насыщенным раствором соли, затем сушат над MgSO4, фильтруют и упаривают досуха. Неочищенное вещество очищают флэш-хроматографией на силикагеле (AcOEt/циклогексан 7/3) и получают 0,11 г соединения 18 (R=C(CH3)3).
1H-ЯМР (CDCl3, 400 MГц) δ: 8,6 (шир.с, 1Н), 8,46 (шир.с, 1Н), 8,30 (с, 1Н), 6,70 (тд, J=6,4 Гц, 1Н), 6,49 (д, J=16 Гц, 1Н), 6,21 (с, 1Н), 3,86 (с, 2Н), 3,85 (С, 3Н), 3,78 (с, 3Н), 3,17 (т, J=7 Гц, 2Н), 2,33 (квинт, J=6,9 Гц, 2Н), 1,95 (квинт, J=7,1 Гц, 2Н), 1,30 (с, 9H), 1,18 (с, 9Н).
Пример 26
Данный пример иллюстрирует получение N-[5-(2-циклопропил-7,8-диметокси-4-оксохроман-5-илметил)-4-(2,2-диметилпропиониламино)пиримидин-2-ил]-2,2-диметилпропионамида 15 (R=C(CH3)3) (стадия D6).
Соединение 18 (300 мг, 0,45 ммоль, R=C(CH3)3) растворяют в DMF (5 мл). Добавляют K2CO3 (124 мг, 0,9 ммоль) и смесь перемешивают 2 ч при комнатной температуре. Затем смесь перемешивают в течение 3 ч при 50°C. После добавления AcOEt органический слой промывают водой и насыщенным раствором соли, затем сушат над MgSO4, фильтруют и упаривают досуха. Неочищенное вещество очищают флэш-хроматографией на силикагеле (AcOEt/циклогексан 7/3) и получают 0,075 г соединения 15 (R=C(CH3)3), имеющего такой же спектр ЯМР, как и в примере 22.
Пример 27
Данный пример иллюстрирует получение N-[5-(2-циклопропил-4-гидрокси-7,8-диметоксихроман-5-илметил)-4-(2,2-диметилпропионилaминo)пиримидин-2-ил]-2,2-диметилпропионамида 19 (R=C(CH3)3) (стадия A7).
NaBH4 (11,6 мг, 1,5 экв.) добавляют при 0°C к раствору соединения 15 (110 мг, 0,205 ммоль, R=C(CH3)3) в 2 мл iPrOH. Смесь перемешивают 1 ч при комнатной температуре. Добавляют воду и смесь дважды экстрагируют AcOEt. Органические слои промывают насыщенным раствором соли, сушат над MgSO4, фильтруют и упаривают досуха. Неочищенное вещество очищают флэш-хроматографией на силикагеле (от AcOEt/циклогексан 8/2 до AcOEt 100%) и получают 0,073 г соединения 19 (R=C(CH3)3).
1H-ЯМР (CDCl3, 400 MГц) δ: 8,44 (шир.с, 1H), 8,39 (шир.с, 1Н), 8,11 (с, 1Н), 6,20 (с, 1Н), 4,92 (шир.с, 1Н), система AB (δA 4,02, δB 3,91, JAB=16 Гц, 2Н), 3,80 (с, 3Н), 3,72 (с, 3Н), 3,40 (тд, J1=8,6 Гц, J2=2,4 Гц, 1Н), 2,35-2,41 (м, 1Н), 2,1-2,19 (м, 1Н), 1,24 (с, 9Н), 1,13 (с, 9Н), 0,54-0,61 (м, 2Н), 0,43-0,48 (м, 1Н), 0,27-0,32 (м, 1Н).
Пример 28
Данный пример иллюстрирует получение N-[5-(2-циклопропил-7,8-диметокси-2H-хромен-5-илметил)-4-(2,2-диметилпропиониламино)пиримидин-2-ил]-2,2-диметилпропионамида 11 (R=C(CH3)3) (стадия B7).
К раствору соединения 19 (50 мг, 92,6 мкмоль, R=C(CH3)3) в толуоле (2 мл) добавляют TFA (36 мкл, 5 экв.). Смесь нагревают при 80°C в течение 12 ч. Затем смесь промывают 10% NaHCO3, дважды экстрагируют AcOEt. Органические слои промывают водой, насыщенным раствором соли, затем сушат над MgSO4, фильтруют и упаривают досуха, получая неочищенное соединение 11 (R=C(CH3)3), которое используют на следующей стадии без очистки.
Пример 29
Данный пример иллюстрирует получение 5-(2-циклопропил-7,8-диметокси-2H-хромен-5-илметил)пиримидин-2,4-диамин I (стадия A8).
В атмосфере аргона соединение 11 (100 мг, 0,192 ммоль, R=C(CH3)3) растворяют в метаноле (1 мл). Добавляют метоксид натрия (42 мг, 0,766 ммоль). Реакционную смесь нагревают при 50°C. Через 10 ч метанол удаляют при пониженном давлении, и твердое вещество желтого цвета перетирают с водой. Белый осадок отфильтровывают, промывают водой и сушат в высоком вакууме, получая 0,062 г соединения I.
1H-ЯМР (CDCl3, 400 MГц) δ: 7,06 (с, 1Н), 6,45 (д, J=10,4 Гц, 1Н), 6,42 (с, 1Н), 6,22 (шир.с, 2Н), 5,70-5,73 (м, 3Н), 4,25 (дд, J1=8,2 Гц, J2=2,6 Гц, 1H), 3,72 (с, 3Н), 3,70 (с, 3H), 3,52 (шир.с, 2H), 1,09-1,18 (м, 1H), 1,21 (с, 9Н), 0,29-0,53 (м, 4H).
Пример 30
Данный пример иллюстрирует получение 5-(2-циклопропил-7,8-диметокси-2H-хромен-5-илметил)пиримидин-2,4-диамина I (стадия A8)
В атмосфере аргона соединение 11 (неочищенное вещество, полученное в примере 19, R=CH(CH3)2) растворяют в THF (2 мл). Добавляют гидроксид натрия (0,6 мл, 4N) и реакционную смесь нагревают при 58°C в течение 2 часов. Реакционную смесь разбавляют смесью EtOAc/15% iPrOH, органический слой промывают 2 раза насыщенным раствором соли, затем сушат над MgSO4, фильтруют и упаривают досуха, получая неочищенное соединение I в виде масла. Аналитические данные аналогичны данным, полученным в примере 29.
ЖХ-МС (градиент 01): Rt=4,80 [M+H]+=355.
| название | год | авторы | номер документа |
|---|---|---|---|
| Производное 1'-бромо-2',3',4'-триметоксибензо[5',6':4,5]-(aR, 1S)-1-ацетамидо-6,7-дигидроциклогепта-[3,4-f]-1Н-индола и его применение | 2016 |
|
RU2630303C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ БЛОКАТОРОВ TTX-S | 2013 |
|
RU2652117C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ АКТИВАЦИИ PD1/PD-L1 | 2018 |
|
RU2783211C2 |
| ПРОИЗВОДНЫЕ АРИЛАМИДОВ В КАЧЕСТВЕ БЛОКАТОРОВ TTX-S | 2011 |
|
RU2535671C1 |
| ТИЕНО[2,3-d]ПИРИМИДИНЫ В КАЧЕСТВЕ АНТИМИКРОБНЫХ АГЕНТОВ | 2018 |
|
RU2803136C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИН-2,4-ДИАМИНА ДЛЯ ЛЕЧЕНИЯ РАКА | 2013 |
|
RU2672916C2 |
| ФТОРСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ТЕВИНОЛА И ОРВИНОЛА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 2012 |
|
RU2506265C1 |
| β-D-2'-ДЕЗОКСИ-2'-α-ФТОР-2'-β-С-ЗАМЕЩЕННЫЕ-2-МОДИФИЦИРОВАННЫЕ-N6-ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ НУКЛЕОТИДЫ ДЛЯ ЛЕЧЕНИЯ ВЫЗВАННЫХ HCV ЗАБОЛЕВАНИЙ | 2016 |
|
RU2764767C2 |
| ПРОИЗВОДНЫЕ 6-1Н-ИМИДАЗОХИНАЗОЛИНА И ХИНОЛИНА - ИНГИБИТОРЫ МАО ДЛЯ ЛЕЧЕНИЯ ДЕПРЕССИИ | 2008 |
|
RU2472508C2 |
| АНАЛОГ ПИРИДИНО[1,2-А]ПИРИМИДОНА, ИСПОЛЬЗУЕМЫЙ В КАЧЕСТВЕ ИНГИБИТОРА mTOR/PI3K | 2015 |
|
RU2658912C1 |
Настоящее изобретение относится к новым способам получения соединения формулы I (иклаприм), которое относится к ингибиторам дигидрофолатредуктазы

а также к промежуточным соединениям, использующимся в данных способах.
2 н. и 11 з.п. ф-лы., 2 табл.
1. Способ получения соединения формулы I

который включает в себя удаление защитных групп с соединения формулы 11 с получением целевого соединения I, где R представляет собой -С(СН3)3 или -С(СН3)2:
2. Способ по п.1, где удаление защитных групп с соединения формулы 11 осуществляют в смеси растворителей, содержащей тетрагидрофуран или метиловый спирт и воду, в присутствии сильного основания в интервале температур от 40 до 80°С с получением целевого соединения формулы I.
3. Способ по п.1 или 2, где соединение формулы 11 получают циклизацией с использованием в качестве катализатора комплекса палладия или/и кислоты соединения формулы 10
где R имеют значения, указанные в п.1, и R' представляет собой метоксиметил или аллил;
соединение формулы 10 получают восстановлением соединения формулы 9
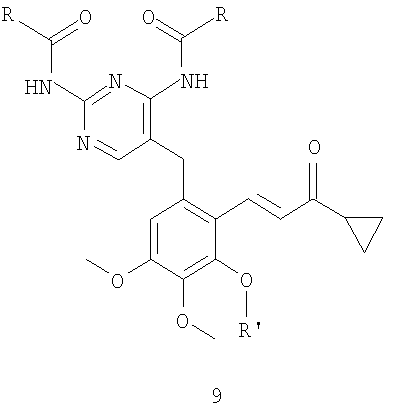
где R и R' имеют значения, указанные выше для формулы 10, соединение формулы 9 получают взаимодействием соединения формулы 7
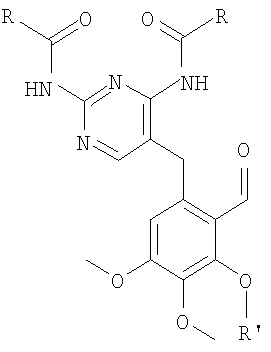
с кетоном формулы 8
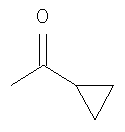
где R и R' имеют значения, указанные выше для формулы 10,
соединение формулы 7 получают защитой фенольной группы соединения формулы 6
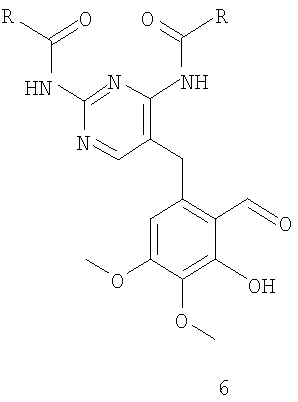
где R имеет значения, указанные выше для формулы 10.
4. Способ по п.3, где соединение формулы 6 получают селективным деметилированием соединения формулы 3

где R имеет значения, указанные выше в п.1.
5. Способ по п.4, где соединение формулы 3 получают формилированием соединения формулы 2

где R имеет значения, указанные выше в п.1;
соединение формулы 2 получают селективной защитой диаминопиримидинового заместителя соединения формулы 1
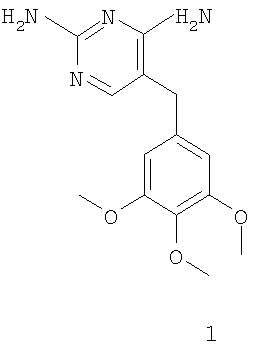 .
.
6. Способ по п.4, где соединение формулы 3 получают карбонилированием соединения формулы 5
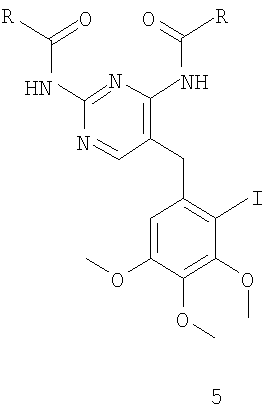
где R имеет значения, указанные выше в п.1;
соединение формулы 5 получают защитой диаминопиримидиновой группы соединения формулы 4
7. Способ по п.1 или 2, где соединение формулы 11 получают отщеплением воды от соединения формулы 19
кислотой при температуре от комнатной до 100°С, где R имеет значения, указанные выше в п.1.
8. Способ по п.1, где соединение формулы 19 получают восстановлением соединения формулы 15
где R имеет значения, указанные выше в п.1.
9. Способ по п.8, где соединение формулы 15 получают взаимодействием соединения формулы 13
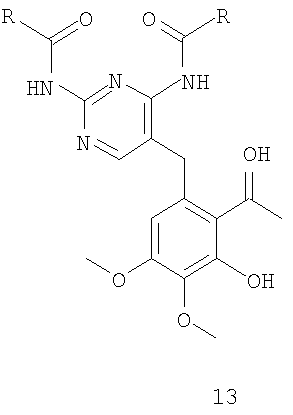
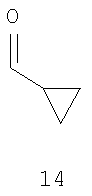
где R имеет значения, указанные выше в п.1 с соединением формулы 14
соединение формулы 13 получают селективным деметилированием соединения формулы 12

где R имеет значения, указанные выше для формулы 13, соединение формулы 12 получают ацилированием соединения формулы 2
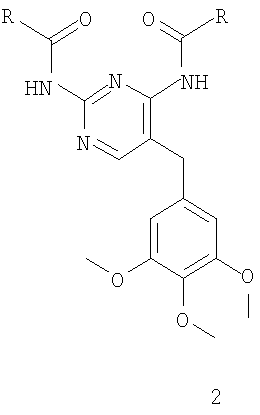
где R имеет значения, указанные выше для формулы 13.
10. Способ по п.8, где соединение формулы 15 получают циклизацией соединения формулы 18

где R имеет значения, указанные выше в п.1;
соединение формулы 18 получают селективным деметилированием соединения формулы 17
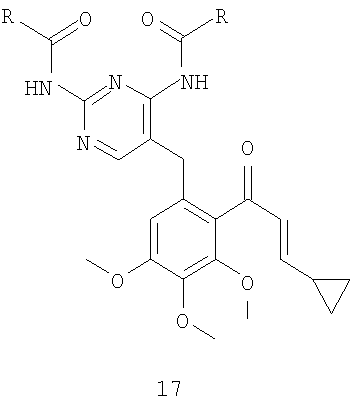
обработкой иодидом натрия, где R имеет значения, указанные выше для формулы 18.
11. Способ по п.10, где соединение формулы 17 получают ацилированием соединения формулы 2
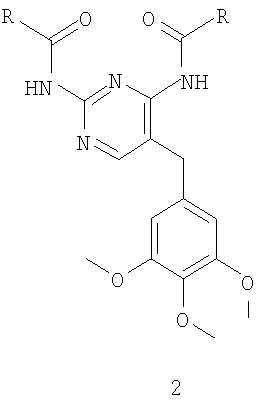
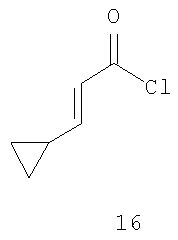
где R имеет значения, указанные выше в п.1, соединением формулы 16
12. Способ по п.10, где соединение формулы 17 получают взаимодействием соединения формулы 12
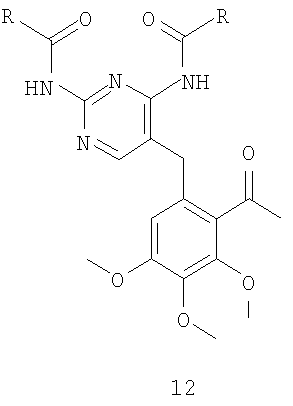
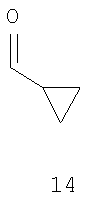
где R имеет значения, указанные выше в п.1, с соединением формулы 14
13. Промежуточные соединения, выбранные из группы, включающей в себя
N-[4-(2,2-диметилпропиониламино)-5-(3,4,5-триметоксибензил)пиримидин-2-ил]-2,2-диметилпропионамид формулы 2
(R=С(СН3)3)
N-[4-изобутириламино-5-(3,4,5-триметоксибензил)пиримидин-2-ил]изобутирамид формулы 2 (R=СН(СН3)2)
N-[4-(2,2-диметилпропиониламино)-5-(2-формил-3,4,5-триметоксибензил)пиримидин-2-ил]-2,2-диметилпропионамид формулы 3 (R=С(СН3)3)
N-[5-(2-формил-3,4,5-триметоксибензил)-4-изобутириламино-пиримидин-2-ил]изобутирамид формулы 3 (R=СН(СН3)2)
N-[4-(2,2-диметилпропиониламино)-5-(2-иод-3,4,5-триметоксибензил)пиримидин-2-ил]-2,2-диметилпропионамид формулы 5 (R=С(СН3)3)
N-[5-(2-иод-3,4,5-триметоксибензил)-4-изобутириламино-пиримидин-2-ил]изобутирамид формулы 5 (R=СН(СН3)2)
N-[4-(2,2-диметилпропиониламино)-5-(2-формил-3-гидрокси-4,5-диметоксибензил)пиримидин-2-ил]-2,2-диметилпропионамид формулы 6 (R=С(СН3)3)
N-[5-(2-формил-3-гидрокси-4,5-диметоксибензил)-4-изобутириламинопиримидин-2-ил]изобутирамид формулы 6 (N=СН(СН3)2)
N-[4-(2,2-диметилпропиониламино)-5-(2-формил-4,5-диметокси-3-метоксиметоксибензил)пиримидин-2-ил]-2,2-диметилпропионамид формулы 7 (R=С(СН3)3)
N-[5-(2-формил-4,5-диметокси-3-метоксиметоксибензил)-4-изобутириламинопиримидин-2-ил]-изобутирамид формулы 7 (R=СН(СН3)2)
N-[5-(2-формил-4,5-диметокси-3-аллилоксибензил)-4-изобутириламинопиримидин-2-ил]изобутирамид формулы 7 (R=СН(СН3)2)
N-{5-[2-(3-циклопропил-3-оксо-пропенил)-4,5-диметокси-3-метоксиметоксибензил]-4-(2,2-диметилпропиониламино)пиримидин-2-ил}-2,2-диметилпропионамид формулы 9 (R=С(СН3)3)
N-{5-[2-(3-циклопропил-3-оксопропенил)-4,5-диметокси-3-метоксиметоксибензил]-4-изобутириламинопиримидин-2-ил}изобутирамид формулы 9 (R=СН(СН3)2)
N-{5-[2-(3-циклопропил-3-оксопропенил)-4,5-диметокси-3-аллилоксибензил]-4-изобутириламинопиримидин-2-ил}изобутирамид формулы 9 (R=СН(СН3)2)
N-{5-[2-(3-циклопропил-3-гидроксипропенил)-4,5-диметокси-3-метоксиметоксибензил]-4-(2,2-диметилпропиониламино)пиримидин-2-ил}-2,2-диметилпропионамид формулы 10 (R=С(СН3)3)
N-{5-[2-(3-циклопропил-3-гидроксипропенил)-4,5-диметокси-3-метоксиметоксибензил]-4-изобутириламинопиримидин-2-ил}изобутирамид формулы 10 (R=СН(СН3)2)
N-{5-[2-(3-циклопропил-3-гидроксипропенил)-4,5-диметокси-3-аллилоксибензил]-4-изобутириламинопиримидин-2-ил}изобутирамид формулы 10 (R=СН(СН3)2)
N-[5-(2-циклопропил-7,8-диметокси-2Н-хромен-5-илметил)-4-(2,2-диметилпропиониламино)пиримидин-2-ил]-2,2-диметилпропионамид формулы 11 (R=С(СН3)3)
N-[5-(2-циклопропил-7,8-диметокси-2Н-хромен-5-илметил)-4-изобутириламинопиримидин-2-ил]изобутирамид формулы 11 (R=СН(СН3)2)
N-[5-(2-ацетил-3,4,5-триметоксибензил)-4-(2,2-диметилпропиониламино)пиримидин-2-ил]-2,2-диметилпропионамид формулы 12 (R=С(СH3)3)
N-[5-(2-ацетил-3,4,5-триметоксибензил)-4-изобутириламинопиримидин-2-ил]изобутирамид формулы 12 (R=СН(СН3)2)
N-[5-(2-ацетил-3-гидрокси-4,5-диметоксибензил)-4-(2,2-диметилпропиониламино)пиримидин-2-ил]-2,2-диметилпропионамид формулы 13 (R=С(СН3)3)
N-[5-(2-ацетил-3-гидрокси-4,5-диметоксибензил)-4-изобутириламинопиримидин-2-ил]изобутирамид формулы 13 (R=СН(СН3)2)
N-[5-(2-циклопропил-7,8-диметокси-4-оксохроман-5-илметил)-4-(2,2-диметилпропиониламино)пиримидин-2-ил]-2,2-диметилпропионамид формулы 15 (R=C(СН3)3)
N-[5-(2-циклопропил-7,8-диметокси-4-оксохроман-5-илметил)-4-изобутириламинопиримидин-2-ил]-изобутирамид формулы 15 (R=СН(СН3)2)
N-{5-[2-(3-циклопропилакрилоил)-3,4,5-триметоксибензил]-4-(2,2-диметилпропиониламино)пиримидин-2-ил}-2,2-диметилпропионамид формулы 17 (R=С(CH3)3)
N-{5-[2-(3-циклопропилакрилоил)-3,4,5-триметоксибензил]-4-изобутириламинопиримидин-2-ил}изобутирамид формулы 17 (R=СН(СН3)2)
N-{5-[2-(3-циклопропил-3-иодпропионил)-3-гидрокси-4,5-диметоксибензил]-4-(2,2-диметилпропиониламино)пиримидин-2-ил}-2,2-диметилпропионамид формулы 18 (R=С(СН3)3)
N-{5-[2-(3-циклопропил-3-иодпропионил)-3-гидрокси-4,5-диметоксибензил]-4-изобутириламинопиримидин-2-ил}-изобутирамид формулы 18 (R=СН(СН3)2)
N-[5-(2-циклопропил-4-гидрокси-7,8-диметоксихроман-5-илметил)-4-(2,2-диметилпропиониламино)пиримидин-2-ил]-2,2-диметилпропионамид формулы 19 (R=С(СН3)3)
N-[5-(2-циклопропил-4-гидрокси-7,8-диметоксихроман-5-илметил)-4-изобутириламино-пиримидин-2-ил]-изобутирамид формулы 19 (R=СН(СН3)2).
| ЕР 1149834 A1, 31.10.2001 | |||
| WO 200501487 A1, 17.02.2005 | |||
| US 5773446 A, 30.06.1998 | |||
| Способ получения 2,4-диамино-5-(замещенных) пиримидинов | 1986 |
|
SU1819264A3 |
| RU 2002107674 A, 10.10.2003. | |||

