Изобретение относится к области химии и медицины, а именно к соединениям, обладающим одновременной рибонуклеазной, мембранолитической и противовирусной активностями.
Вирусные заболевания представляют собой общую угрозу здоровью населения во всем мире. Среди вирусов вирусы, содержащие геном в виде РНК, вызывают целый ряд серьезных заболеваний животных (домашних и диких) и являются одними из наиболее опасных для человека. Высокопатогенные РНК-вирусы, такие как вирус гриппа птиц (Avian Influenza Virus), клещевой энцефалит (Tick-borne enchefalitis), вирус Денге (Denge virus), коронавирус (SARS) и т.д., являются постоянной угрозой для здоровья населения. На сегодняшний день особую опасность представляют вирусы птиц и животных, способных заражать людей (Lipsitch, M. et al. Science, 2006, 312, 845; Krug, R.M. Science, 2006, 311, 1562-1563; Chen, H. et al. Nature, 2005, 436, 191-192). Например, долгое время считалось, что вирус гриппа птиц не может переходить к людям, однако в последние годы было зарегистрировано большое число передач вируса от птиц к человеку, что вызывало смертельные исходы у заразившихся людей (Subbarao, K. et al. Science, 1998, 279, 393-396; Fouchier, R.A. et al. Proc. Natl. Acad. Sci., USA, 2004, 101, 1356-1361). Вспышки высокопатогенного вируса гиппа H5N1 в некоторых азиатских и европейских странах вызвало серьезное опасение со стороны мирового сообщества (Koopmans, M. et al. Lancet, 2004, 363, 587-593; Enserink, M., Science, 2006, 311, 932; Fabain, N.J. Environ. Health 2006, 68, 46-63).
Опасность эпидемий и пандемий, вызванных РНК-вирусами, делает разработку новых противовирусных препаратов и новых средств и способов их инактивации одной из наиболее актуальных задач сегодняшнего дня.
В настоящее время в качестве средств инактивации вирусов используют физические методы (облучение ультрафиолетовым светом, ионизирующее излучение, нагревание при повышенном давлении) и целый ряд химических соединений (формалин, β-пропиолактон, этиленимин, ряд детергентов), которые различаются механизмом действия и, следовательно, областью применения, токсичностью, эффективностью инактивации вируса и стоимостью (De Benedictis, P. et al. Zoonoses Public Health. 2007, 54(2), 51-68).
Спектр соединений, позволяющих инактивировать вирус, в том числе и для получения цельновирионных вакцин, ограничен несколькими соединениями: это формальдегид, β-пропиолактон (Goldstein M. and Tauraso N., Applied Microbiol., 1970, 19, 290-294), этиленимин и его производные (Budowsky E.I., et al, Vaccine, 1991, 9, 473-476; Aarthi d., et al, Biologicals, 2004, 32, 153-156), облучение УФ-светом (Budowsky E.I., et al, Arch. Virol., 1981, 68-239-246) или УФ-светом в присутствии соединений, повышающих фоточувствительность вируса (Kasermann F and Kempf С., Antiviral Res., 1997, 34, 65-70), окисление кислородом воздуха на свету (Kasermann F. and Kempf С., Rev. Med. Virol, 1998, 8, 143-151). Известно, что обработка вируса такими соединениями приводит к частичному разрушению вирусных антигенных детерминант и тем самым к снижению иммуногенных свойств вакцин, а выборочное разрушение формалином антигенных детерминант поверхностных гликопротеидов вирусов может индуцировать развитие несбалансированного иммунного ответа (Kasermann F., et al., Antiviral Res., 2001, 52, 33-41).
Идеальной мишенью для инактивации вируса гриппа и других РНК-содержащих вирусов (далее по тексту РНК-вирусы) является геномная РНК вируса. После ее разрушения вирус теряет способность к репликации и, таким образом, не может размножаться.
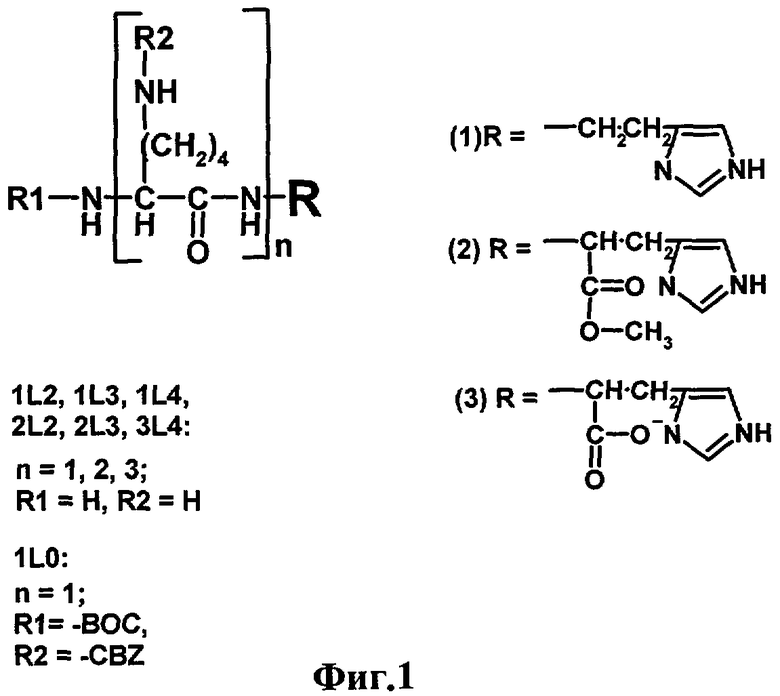
Наиболее близкими к заявляемым соединениям - прототипом являются производные лизина общей формулы nLm, обладающие рибонуклеазной активностью (Ждан Н.С. и др., Биоорган. химия. 1999, 25, 723-732). На фиг.1 представлена структурная формула соединений nLm, представляющих собой неприродные пептиды на основе лизина, где n - число остатков лизина, m - суммарный положительный заряд соединения при нейтральном рН. Было показано, что хотя эти соединения и проявляют высокую рибонуклеазную активность, эффективность расщепления ими РНК зависела от пространственной структуры этой РНК, не коррелировала с суммарным положительным зарядом молекулы (определяет эффективность связывания с РНК) и мало зависела от числа остатков лизина в молекуле.
Недостатками известных соединений nLm являются относительно невысокая рибонуклеазная активность и низкая эффективность инактивации РНК-вирусов.
Технической задачей изобретения является получение эффективных, низкотоксичных синтетических соединений (искусственных рибонуклеаз), обладающих одновременной рибонуклеазной, мембранолитической и противовирусной активностями.
Поставленная техническая задача решается предлагаемым средством для инактивации вирусов, обладающим одновременной рибонуклеазной, мембранолитической и противовирусной активностями, представляющим собой производные нескольких аминокислот (АК), связанных гидрофобным линкером (L) общей формулы
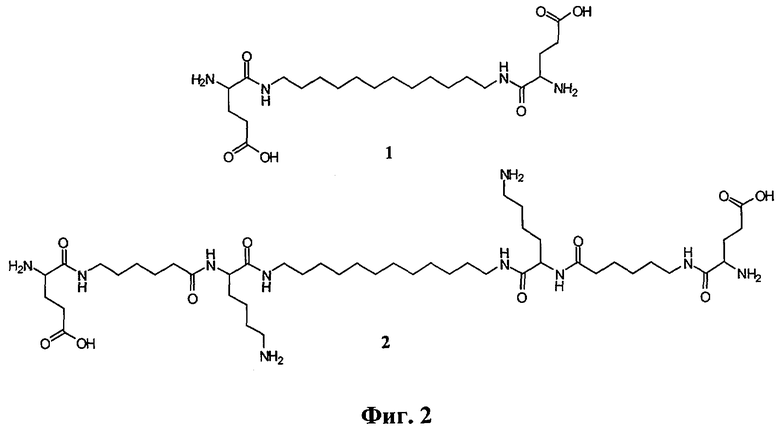
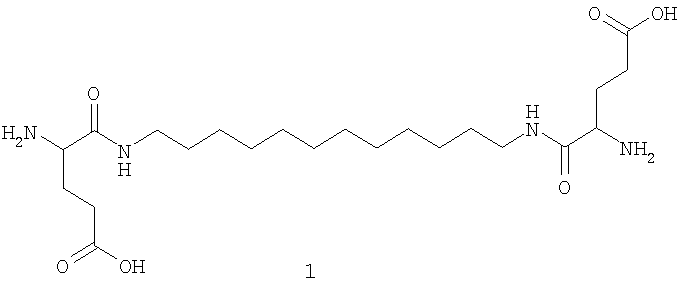
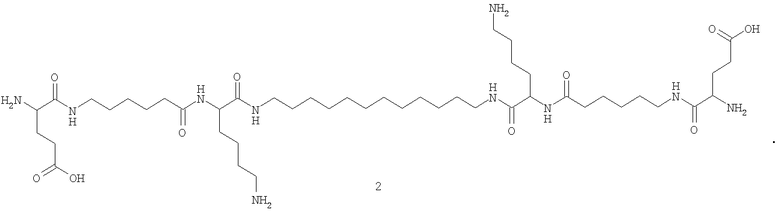
[nAK]2L, где n - число разных аминокислот в молекуле соединения, равное единице для соединения (1) или трем для соединения (2); L - додецильный линкер, АК - глутаминовая кислота для (1) и лизин, 6-аминогексановая и глутаминовая кислоты для (2). На фигуре 2 представлены структурные формулы соединений (1) и (2).
Для повышения рибонуклеазной активности соединений (1) и (2), по сравнению с прототипом, в их структуру была добавлена новая каталитически активная группа (карбоксигруппа), которая присутствует в активном центре многих природных рибонуклеаз. Присутствие в молекуле как отрицательно, так и положительно заряженных функциональных групп позволяет повысить эффективность реакции расщепления РНК. Кроме того, в молекулу был введен протяженный гидрофобный линкер, соединяющий два аминокислотных фрагмента, что позволяет усилить рибонуклеазную активность соединений.
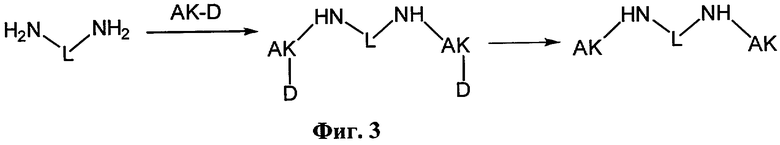
Соединения (1) и (2) общей формулы [nAK]2L были получены по общей схеме, представленной на фиг.3.
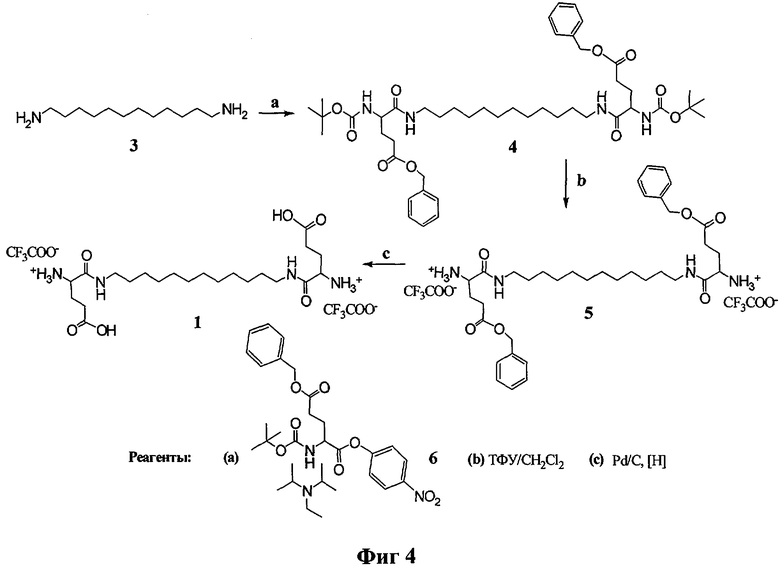
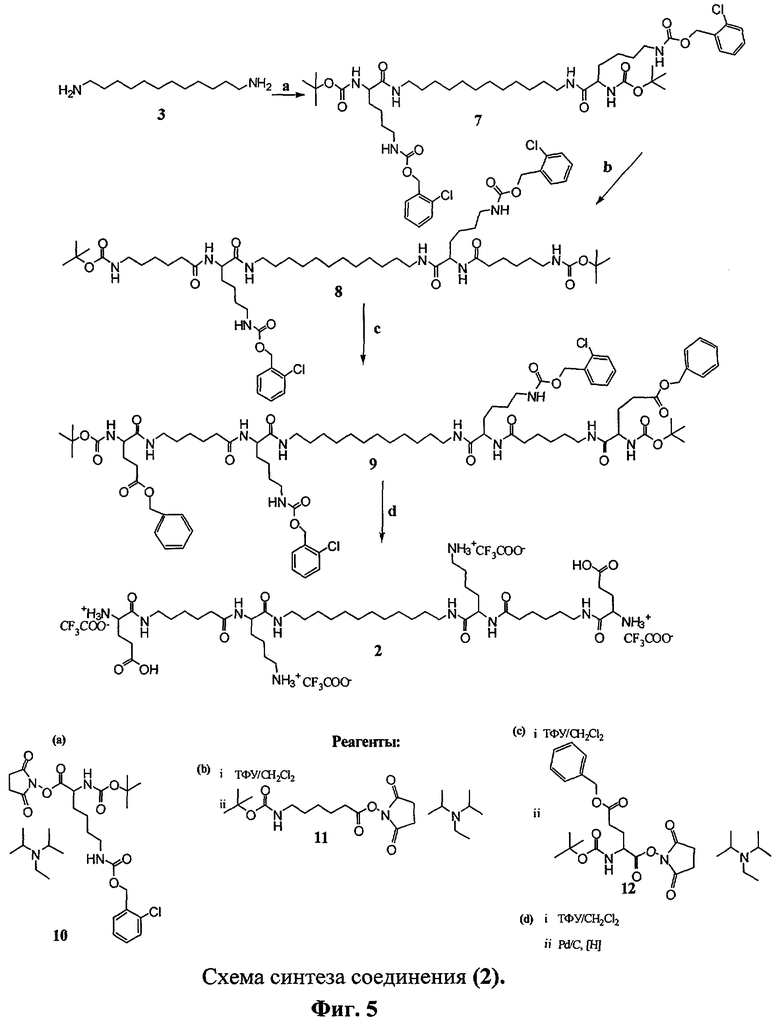
Процесс получения соединений (1) и (2) включает в себя реакцию коммерчески доступных активированных эфиров (N-гидроксисукцинимидные и n-нитрофениловые) аминокислот (соединения 6, 10, 11, 12). Схема синтеза соединений (1) и (2) представлена на фиг.4, 5, где L - 1,12-додекандиамин, AHA - 6-аминогексановая кислота, Glu - глутаминовая кислота, Lys - лизин, Вос - трет-бутилоксикарбонильная защитная группа, Z(2C1) - 2-хлорбензилоксикарбонильная защитная группа, Bzl - бензильная защитная группа, ТФУ - трифторуксусная кислота.
Введение трет-бутилоксикарбонильной защиты по аминогруппе 6-аминогексановой кислоты и синтез активированных эфиров проводили согласно стандартным методикам (А.А.Гершкович, В.К.Кибирев. Синтез пептидов. Реагенты и методы. Киев. Наукова думка. 1987).
Данная схема включает в себя реакцию коммерчески доступного 1,12-додекандиамина (3) с защищенной аминокислотой (AK-D), с образованием дизамещенного производного защищенных аминокислот (D-AK)2L, которое далее освобождали от защитных групп. В случае соединения (2) получающиеся концевые аминогруппы первой аминокислоты использовались для введения второй и третьей аминокислоты по такой же схеме. В результате реакции с высокими выходами образуется продукт, содержащий одну (три) аминокислоты, димеризованные с помощью додецильного линкера (L).
Спектры ЯМР записаны на спектрометрах «Bruker AM-400» и «Bruker AV-300» (Германия)» в CD3OD, (CD3)2СО («Aldrich», США). Для 1Н ЯМР-спектров использовали тетраметилсилан или сигналы остаточных протонов растворителей в качестве внутреннего стандарта. Химические сдвиги приведены в шкале δ, м.д.
Масс-спектры высокого разрешения получали с помощью MALDI-TOF (time-of-flight) масс-спектрометра Bruker reflex III MALDI TOF (Bruker Analytical Systems, Inc., Германия).
Для колоночной хроматографии использовали силикагель Kieselgel 60 (63-100 мкм, «Merck», Германия).
Для изучения рибонуклеазной активности соединений использовали 96-звенный фрагмент РНК вируса иммунодефицита человека HIV1 и синтетический олигорибонуклеотид ON21, r(UCGAAUUUCCACAGAAUUCGU), который был синтезирован фосфитамидным методом на синтезаторе ASM-102U (ТОО "БИОССЕТ", Новосибирск) и очищен ионообменной и обращеннофазовой хроматографией или препаративным электрофорезом в денатурирующих условиях. Чистоту олигонуклеотида проверяли с помощью электрофореза в 15% ПААГ в денатурирующих условиях, визуализацию олигонуклеотида в геле проводили с помощью окраски Stains-All. Фрагмент РНК был получен с помощью реакции транскрипции in vitro согласно опубликованному протоколу (Maniatis Т., Fritsch E. F., Sambrook J. Molecular cloning. A Laboratory Manual. Cold Spring Harbor Laboratory Press, 1989).
Рибонуклеазная активность соединений (1) и (2) была исследована в реакции расщепления in vitro двух РНК-субстратов - олигорибонуклеотида ON21 и РНК96, а антивирусная и мембранолитическая активности этих соединений - в экспериментах по инактивации РНК содержащих вирусов (вирус гриппа и вирус клещевого энцефалита) и в экспериментах по репродукции этих вирусов в культуре эукариотических клеток in vitro.
Сопоставительный анализ заявляемых соединений с известными и широко используемыми соединениями, такими как формалин, УФ-свет или мономерный или бинарный этиленимин (Budowsky. E.I., et al., Vac. Res., 1996, 5, 29-39), показал, что предлагаемые соединения обладают следующими преимуществами.
1) Заявляемые соединения обладают одновременно рибонуклеазной, мембранолитической и антивирусной активностями, что позволяет рассматривать их как перспективные лекарственные агенты.
2) Заявляемые соединения малотоксичны для человека и не требуют особых мер предосторожности (противогаз, перчатки, работа под вытяжкой) при работе с ними. Кроме того, эти соединения стабильны при хранении в виде концентрированных водных растворов или растворов в апротонных растворителях, таких как диметилсульфоксид.
3) Заявляемые соединения не оказывают побочного действия на белковые компоненты вирионов, что не снижает иммуногенные свойства инактивированных вирусов и улучшает свойства получаемых на их основе вакцин.
Поиск по источникам научно-технической и патентной литературы показал, что производные лизина, глутаминовой и 6-аминогексановой кислот общей формулы [nAK]2L, обладающие одновременно рибонуклеазной, мембранолитической и противовирусной активностями, в известных источниках не описаны.
Изобретение иллюстрируется следующими примерами.
Пример 1. Получение N,N'-бис(глутамил)-1,12-додекандиамина (1).
Схема синтеза соединения (1) представлена на фиг.4.
К раствору 1,12-додекандиамина (3) (2.1 ммоль, 0.42 г) и этилдиизопропиламина (4.4 ммоль, 0.75 мл) в 10 мл этилацетата добавляли при перемешивании раствор n-нитрофенилового эфира Nα-трет-бутилоксикарбонил-О-бензил-L-глутаминовой кислоты (6) (4.4 ммоль, 2.01 г) в 5 мл этилацетата. Реакционную смесь перемешивали 24 ч при 22°С. Затем в реакционную смесь добавляли N,N-диметилэтилендиамин (1 ммоль, 110 мкл) и перемешивали 30 мин. Реакционную смесь промывали последовательно 2%-ным раствором лимонной кислоты (20 мл), водой (20 мл), насыщенным раствором NaHCO3 (20 мл) и водой (20 мл). Органический слой сушили безводным Na2SO4. Растворитель удаляли в вакууме. Остаток сушили в вакууме. Очистку проводили с помощью хроматографии на силикагеле, элюируя продукт в градиенте концентрации этанола в хлористом метилене от 0 до 5%. Фракции, содержащие целевое соединение, упаривали и сушили в вакууме. Выход N,N'-бис((Nα-трет-бутилоксикарбонил-O-бензил)глутамил)-1,12- додекандиамина (4) 0.86 г, 49.0%.
Данные 1Н ЯМР-спектра для N,N'-бис((Nα-трет-бутилоксикар6онил-O-бензил)глутамил)-1,12-додекандиамина ((CD3)2СО) δ, м.д. (J/Гц): 1.27 (м, 16Н, [-NH-(CH2)2-(CH2)8-(CH2)2-NH-]L); 1.41 (с, 18Н, [CH3]Boc); 1.49 (м, 4Н, [-NH-CH2CH2-]L); 1.86-2.17 (м, 4Н, [CHCH2CH2]Glu, Glu'); 2.48 (т, 4Н, [CHCH2CH2]Glu, Glu', J=7.5); 3.22 (м, 4Н, [-NH-CH2CH2-]L); 4.13 (м, 2Н, CHCH2CH2]Glu, Glu'); 5.13 (с, 4Н, [CH2]Bzl); 7.37 (м, 10Н,
[CH]аром).
N,N'-бис ((Nα-трет-бутилоксикарбонил-О-бензил) глутамил)-1,12-додекандиамин (4) растворяли в смеси ТФУ:CH2Cl2=1:1 (5 мл) и выдерживали реакционную смесь 1 ч при комнатной температуре. Растворитель и ТФУ удаляли в вакууме, остаток упаривали с этанолом (3×10 мл), растворяли в CH2Cl2 (5 мл) и высаживали 10-кратным объемом серного эфира. Выдерживали ночь в холодильнике. Эфир декантировали, твердый остаток сушили в вакууме. Выход трифторацетата N,N'-бис((γ-бензил)-глутамил)-1,12-додекандиамина (5) 0.82 г, 92.0%. Данные 1Н ЯМР- спектра для N,N'-бис((γ-бензил)-глутамил)-1,12-додекандиамина (5) (CDCl3) δ, м.д. (J/Гц): 1.23 (м, 16Н, [-NH-(CH2)2-(CH2)8-(CH2)2-NH-]L); 1.49 (м, 4Н, [-NH-CH2CH2-]L); 2.28-2.38 (м, 4Н, [CHCH2CH2]Glu, Glu'); 2.48 (м, 4Н, [CHCH2CH2]Glu, Glu'); 3.28 (м, 4Н, [-NH-CH2CH2-]L); 4.16 (м, 2Н, CHCH2CH2]Glu, Glu'); 5.11 (с, 4Н, [CH2]Bzl); 7.34 (м, 10Н, [(CH]аром). MALDI-MS, m/z найдено 639.42 [M+H]+, вычислено 638.40.
Трифторацетат N,N'-бис((γ-бензил)-глутамил)-1,12-додекандиамина (5) (0.46 ммоль, 400 мг) растворяли в 5 мл метанола и подвергали гидрогенолизу в присутствии 200 мг 5%-ного Pd/C. По окончании реакции катализатор отфильтровывали, растворитель упаривали в вакууме. Выход N,N'-бис(глутамил)-1,12-додекандиамина (1) после гидрирования 292 мг, 92.2%.
Общий выход N,N'-бис(глутамил)-1,12-додекандиамина (1) в расчете на 1,12-додекандиамин (3) 41.6%.
Данные 1H ЯМР-спектра для N,N'-бис(глутамил)-1,12-додекандиамин трифторацетата (1) ((CD3)2СО) δ, м.д. (J/Гц): 1.33 (м, 16Н, [-NH-(CH2)2-(CH2)8-(CH2)2-NH-]L2); 1.55 (м, 4Н, [-NH-CH2CH2-]L2); 2.15 (м, 4Н, [CHCH2CH2]Glu, Glu'); 2.49 (м, 4Н, [CHCH2CH2 Glu, Glu'); 3.20-3.36 (м, 4Н, [-NH-CH2CH2-]L2); 3.90 (т, 2Н, [CHCH2CH2]Glu, Glu', J=3.6). MALDI-MS, m/z найдено 459.28 [M+H]+, вычислено 458.31.
Пример 2. Получение N,N'-бис(глутамил-6-аминогексаноил-лизил)-1,12-додекандиамина (2).
Схема синтеза соединения (2) представлена на фиг.5.
К раствору 1,12-додекандиамина (3) (2.4 ммоль, 0.48 г) и этилдиизопропиламина (5 ммоль, 0.85 мл) в 10 мл этилацетата добавляли при перемешивании раствор N-гидроксисукцинимидного эфира Nα-трет-бутилоксикарбонил-Nε-2-хлорбензилоксикарбониллизина (10) (5 ммоль, 2.6 г) в 5 мл этилацетата. Реакционную смесь перемешивали 24 ч при 22°С. Затем в реакционную смесь добавляли N,N-диметилэтилендиамин (1 ммоль, 110 мкл) и перемешивали 30 мин. Реакционную смесь промывали последовательно 2%-ным раствором лимонной кислоты (20 мл), водой (20 мл), насыщенным раствором NaHCO3 (20 мл) и водой (20 мл). Органический слой сушили безводным Na2SO4. Растворитель удаляли в вакууме. Полученную пену сушили в вакууме. Выход N,N'-бис((Nα-трет-бутилоксикарбонил-Nε-2-хлорбензилоксикарбонил)лизил)-1,12-додекандиамина (7) 1.38 г, 57.9%. Данные 1Н ЯМР-спектра для N,N'-бис((Nα-трет-бутилоксикарбонил-Nε-2-хлорбензилоксикарбонил)лизил)-1,12-додекандиамина (7) ((CD3)2СО) δ, м.д. (J/Гц): 1.29 (м, 16Н, [-NH-(CH2)2-(CH2)8-(CH2)2-NH-]L); 1.41 (с, 18Н, [CH3]Boc); 1.57 (м, 8Н, [-NH- CH2CH2-(СН2)8-]L, [-CHCH2CH2(СН2)2]Lys, Lys'); 1.64 (м, 4Н, [-СН(СН2)2CH2CH2]Lys, Lys'); 1.79 (м, 4Н, [-CHCH2(CH2)2CH2]Lys, Lys'); 3.18 (м, 8Н, [-СН(СН2)3CH2]Lys, Lys', 4Н, [-NH-CH2CH2-(CH2)8-]L); 4.05 (м, 2Н, [-CH(СН2)4]Lys, Lys'); 5.17 (с, 4Н, [CH2]Z(2Cl); 7.35 (м, 4Н, [CH]аром); 7.44 (м, 2Н, [CH]аром.); 7.50 (м, 2Н, [CH]аром.).
N,N'-бис((Nα-трет-бутилоксикарбонил-Nε-2-хлорбензилоксикарбонил) лизил)-1,12-додекандиамин (7) растворяли в смеси ТФУ:CH2Cl2=1:1 (5 мл) и выдерживали реакционную смесь 1 ч при комнатной температуре. Растворитель и ТФУ удаляли в вакууме, остаток упаривали с этанолом (3×10 мл), растворяли в этилацетате (15 мл) и промывали насыщенным раствором NaHCO3 (10 мл) и водой (10 мл), сушили над безводным Na2SO4. Растворитель удаляли в вакууме. Полученный маслообразный остаток сушили в вакууме и использовали в дальнейшем без дополнительной очистки.
Затем аналогично повторяли процедуру конденсации аминокомпоненты последовательно с N-гидроксисукцинимидным эфиром Nα-трет-бутилоксикарбонил-6-аминогексановой кислоты (11) и Nα-трет-бутилоксикарбонил-О-бензил-L-глутаминовой кислоты (12) в ДМФА.
Выход N,N'-бис(Nα-трет-бутилоксикарбонил-6-аминогексаноил-Nε-2-хлорбензилоксикарбонил)лизил)-1,12-додекандиамина (8) 0.54 г, 49.0%. Данные 1Н ЯМР-спектра для ((8) (CD3OD) δ, м.д. (J/Гц): 1.30 (м, 16Н, [-NH-(CH2)2-(CN2)8-(CH2)2-NH-]L); 1.44 (с, 18Н, [CH3]Boc); 1.50 (м, 12Н, [-NH- CH2CH2-(СН2)8-]L, [-CHCH2CH2CH2)2]Lys, Lys', [-С(O)-(СН2)2-CH2-(СН2)2-NH]AHA, AHA'); 1.63 (м, 8H, [-СН(СН2)2CH2CH2]Lys, Lys', [-С(O)-(СН2)3-CH2-СН2- NH]AHA, AHA'); 1.73 (м, 4H, [-С(O)-СН2-CH2-(СН2)3-NH]AHA, AHA'); 1.87 (м, 4Н, [-CHCH2(СН2)3]Lys, Lys'); 2.25 (т, 4Н, [-C(O)-CH2-(CH2)4-NH]AHA, AHA', J=7.4); 3.03 (м, 4Н, [-C(O)-(CH2)4-CH2-NH]AHA, AHA'); 3.15 (м, 8Н, [-CH(СН2)3CH2 Les, Lys', 4Н, [-NH-CH2CH2-(CH2)8-]L); 4.27 (м, 2Н, [-CH(СН2)4]Lys, Lys'); 5.19 (с, 4Н, [CH2]Z(2Cl)); 7.33 (м, 4Н, [CH]аром.); 7.41 (м, 2Н, [CH]аром.); 7.45 (м, 2Н, [CH]аром.).
Выход N,N'-бис((Nα-трет-6утилоксикарбонил-O-бензил)глутамил-аминогексаноил-Nε-2-хлорбензилоксикарбонил)лизил)-1,12-додекандиамина (9) 0.07 г, 6.1%. Данные 1H ЯМР-спектра для ((9) (CD3OD) δ, м.д. (J/Гц): 1.33 (м, 16Н, [-NH-(CH2)2-(CH2)8-(CH2)2-NH-]L); 1.44 (м, 22Н, [CH3]Boc, [-NH-CH2CH2-(CH2)8-]L); 1.53 (м, 12Н, [-CH(CH2)2CH2CH2]Lys, Les', [-С(О)- (CH2)2-CH2-(CH2)2-NH]AHA, AHA'), [-C(O)-(CH2)3-CH2-CH2-NH]AHA, AHA'); 1.64 (м, 12Н, [-CHCH2CH2(СН2)2]Lys, Les', [-C(O)-CH2-CH2-(CH2)3-NH]AHA, AHA', [-CHCH2CH2]Glu, Glu'); 1.92 (м, 4Н, [-CHCH2(CH2)3]Lys, Lus'); 2.13 (м, 4Н, [-С(О)-CH2-(CH2)4-NH]AHA, AHA'; 2.25 (м, 4Н, [-CHCH2CH2]Glu, Glu'); 2.50 (м, 4Н, [-СН(СН2)3CH2]Lys, Lys'); 3.17 (м, 4Н, [-C(O)-(CH2)4-CH2-NH]AHA, AHA'); 3.40 (м, 4Н, [-NH-CH2CH2-(CH2)8-]L); 4.05 (м, 2Н, [-CHCH2CH2]Glu, Glu'); 4.16 (м, 2Н, [-CH(СН2)4]Lys, Lus'); 5.12 (с, 4Н, [CH2]Z(2Cl); 7.35 (м, 18Н, [CH]аром.).
N,N'-бис((O-бензил)глутамил-аминогексаноил-(Nε-2-хлорбензилоксикарбонил)лизил)-1,12-додекандиамин (0.042 ммоль, 70 мг) растворяли в 5 мл метанола и подвергали гидрогенолизу в присутствии 50 мг 5%-ного Pd/C. По окончании реакции катализатор отфильтровывали, растворитель упаривали в вакууме. Выход N,N'-глутамил-аминогексаноил-лизил-1,12-додекандиамин трифторацетата (2) после гидрирования 38.4 мг, 66.2%.
Общий выход N,N'-бис(глутамил-аминогексаноил-лизил)-1,12-додекандиамин трифторацетата (2) в расчете на 1,12-додекандиамин составил 1.7%.
Данные 1Н ЯМР-спектра для N,N'-бис(глутамил-аминогексаноил-лизил)-1,12-додекандиамин трифторацетата (2) (CD3OD) δ, м.д. (J/Гц): 1.33 (м, 24Н, [-NH-(CH2)2-(CH2)8-(CH2)2-NH-]L, [-CHCH2CH2(CH2)2]Lys, Lys', [-C(O)-(CH2)2-CH2-(CH2)2-NH]AHA, AHA'); 1.44 (м, 4Н, [-NH-CH2CN2-(CH2)8-]L; 1.58 (м, 8Н, [-CH(СН2)2CH2CH2]Lys, Lys' [-C(O)-(CH2)3-CH2-CH2-NH]AHA, AHA'); 1.66 (м, 8Н, [-C(O)-CH2-CH2-(CH2)3-NH]AHA, AHA'), [-CHCH2CH2]Gly, Glu'); 1.84 (м, 4Н, [-CHCH2(СН2)3]Lys, Lys'); 2.17 (м, 4Н, [-C(O)-CH2-(CH2)4-NH]AHA, AHA'); 2.37 (м, 4Н, [-CHCH2CH2]Glu, Glu'); 2.47 (м, 4Н, [-СН(СН2)3CH2]Lys, Lys'); 2.56 (м, 4Н, [-С(O)-(СН2)4-CH2-NH]AHA, AHA'); 2.94 (м, 4Н, [-NH-CH2CH2-(CH2)8-]L; 3.88 (м, 2Н, [-CHCH2CH2]Glu, Glu'); 4.10 (м, 2Н, [-CH(CH2)4]Lys, Lys'). MALDI-MS, m/z найдено 963.27 [M+Na]+, вычислено 940.67.
Пример 3. Расщепление РНК соединениями (1) и (2).
Эксперименты по расщеплению РНК данными соединениями проводили в следующих условиях: стандартная реакционная смесь объемом 10 мкл содержала 10000 имп/мин по Черенкову [5'-32Р]-меченого ON21 (стабилизированного немеченым ON21 в концентрации 5×10-6 М) или [5'-32Р]-РНК96, одно из соединений (1) или (2) в концентрациях от 10-6 до 10-3 М и 50 мМ Tris-HCl рН 7.0, содержащий 0.2 М KCl, 0.5 мМ EDTA.
Реакции проводили при 37°С в течение 1 - 20 ч и останавливали путем осаждения продуктов реакции 10 объемами (100 мкл) 2%-ного раствора LiClO4 в ацетоне. Осадок РНК и продуктов ее расщепления отделяли центрифугированием, растворили в 5 мкл буфера для нанесения на гель. Продукты расщепления РНК анализировали электрофорезом в 15%-ном ПААГ, содержащем 8 М мочевину. Гель после электрофореза высушивали, и анализировали с помощью прибора фосфоримиджер "Molecular Imager FX" Степень расщепления РНК определяли как отношение количества радиоактивности в полосе, соответствующей продукту расщепления, к общей радиоактивности, нанесенной на дорожку.
При установлении зависимости степени расщепления РНК от концентрации соединений (1) и (2) было обнаружено, что для обоих соединений степень расщепления повышается при повышении концентрации соединений. Оба соединения проявляли наибольшую рибонуклеазную активность при концентрации 1 мМ. В таблице 1 представлены данные по влиянию концентрации соединений (1) и (2) на степень расщепления РНК.
При установлении зависимости степени расщепления РНК соединениями (1) и (2) от времени выяснилось, что степень расщепления линейно возрастает на участке от 1 до 5 ч, а через 7 ч инкубации в расщепляемой смеси остается не более 5-10% исходной РНК (Таблица 2). На линейном участке кинетической кривой по уравнению [P]t=[Р]∝×(1-e-Kobs×t), где [P]t и [Р]∝ - степени расщепления субстрата за время t и за бесконечное время (можно принять, что за бесконечное время субстрат будет расщеплен на 100%), были найдены константы скорости реакции kobs, равные 0.273 и 0.408 ч-1 для соединений (1) и (2) соответственно.
Приведенные примеры подтверждают высокую рибонуклеазную активность соединений (1) и (2).
Пример 4. Влияние соединений (1) и (2) на жизнеспособность клеток MDCK
Клетки линии MDCK (клетки почек собаки) культивировали в среде IMDM, содержащей 10%-ную эмбриональную телячью сыворотку, антибиотики (100 ед./мл пенициллина и 0.1 мг/мл стрептомицина) и антимикотик амфотерицин (0.25 мкг/мл), в атмосфере 5%-ного CO2 при 37°С.
Жизнеспособность клеток после инкубации с соединениями (1) или (2) определяли с помощью МТТ теста, который основан на способности живых клеток превращать соединения на основе тетразола (МТТ) в ярко окрашенные кристаллы формазана, что позволяет спектрофотометрически оценивать количество живых клеток в препарате. Для этого клетки высаживали в 96-луночные планшеты (104 клеток на лунку). Через 24 ч в лунках меняли среду и к клеткам добавляли водный раствор соединения (1) или (2) в воде до конечной концентрации в среде от 10-8 до 10-4 М. Клетки инкубировали в присутствии соединений еще в течение суток в тех же условиях. По окончании инкубации без смены среды к клеткам добавляли раствор МТТ (5 мг/мл) в фосфатно-солевом буфере до концентрации 0.5 мг/мл и инкубировали в течение 3 ч в тех же условиях. Среду удаляли, к клеткам добавляли по 100 мкл диметилсульфоксида, в котором происходит растворение образовавшихся в клетках кристаллов формазана, и измеряли оптическую плотность на многоканальном спектрофотометре на длинах волн 570 и 630 нм, где А570 - поглощение формазана, а А630 - фон клеток.
Из экспериментальных данных вычисляли значение IC50 -концентрацию соединений, при которой наблюдается гибель 50% клеток. Значения IC50 приведены в таблице 3.
Из приведенных данных видно, что обработка клеток соединениями (1) или (2) вызывает их эффективную гибель только при концентрациях соединений выше 5 мМ, что свидетельствует о чрезвычайно низкой токсичности соединений.
Пример 5. Инактивация вируса гриппа соединениями (1) и (2)
Вирулицидную активность искусственных рибонуклеаз (1), (2) изучали на модели вируса гриппа A/WSN/33, (H1N1). В этих экспериментах использовали вирус гриппа A/WSN/33, (H1N1) с инфекционным титром 106 фокус-образующих единиц (ФОЕ) вируссодержащего материала. Инфекционную активность вируса определяли методом ФОЕ на клетках MDCK. В основе метода лежит модифицированная иммуноферментная реакция: вирусные белки, экспрессирующиеся в инфицированных вирусом клетках, специфически связываются с моноклональными антителами, мечеными ферментом. Далее фермент взаимодействует с субстратом, что приводит к окрашиванию инфицированной клетки. Инактивацию вируса гриппа соединениями (1), (2) проводили при 37°С в 50 мМ буфере Трис-HCl, рН 7.0, содержащем 0.2 M KCl и 2 мМ EDTA, в течение 18 ч при 37°С при концентрациях соединений (1), (2) от 200 мкМ до 4 мМ.
Соединения (1), (2) эффективно инактивируют вирус гриппа, причем степень инактивации вируса увеличивается с повышением концентрации соединений, при которой проводится инактивация вируса. Так, инкубация вируса в течение 18 ч при 37°С с соединениями (1), (2) в концентрации 200 мкМ, снижает титр вируса на 0.8 lg по сравнению с контролем. При увеличении концентрации соединений (1), (2) до 2 мМ наблюдается полная инактивация вируса гриппа (таблица 4). Следует отметить, что концентрация соединений (1) и (2), при которой наблюдается полная инактивация вируса гриппа, значительно ниже, чем значение IC50 для этих соединений.
Пример 6. Инактивация вируса клещевого энцефалита (КЭ) соединениями (1) и (2).
Вирулицидную активность соединений (1), (2) изучали на модели вируса КЭ (штамм Софьин) с инфекционным титром 106 бляшко-образующих единиц (БОЕ). Инфекционную активность вируса КЭ определяли на клетках СПЭВ. Инактивацию вируса КЭ соединениями (1), (2) проводили при концентрации соединений 2 мМ в 50 мМ буфере Трис-HCl, рН 7.0, содержащем 0.2 M KCl и 2 мМ EDTA, в течение 18 ч при 37°С.
Соединения (1), (2) в этих условиях (концентрация 2 мМ, 18 ч, 37°С) полностью инактивируют вирус КЭ (таблица 4).
Инкубация клеток, инфицированных вирусом КЭ с соединением (2) в концентрации 1 мМ, приводила к снижению репродукции вируса на порядок, по сравнению с контролем.
Полученные результаты однозначно свидетельствуют о высокой антивирусной активности соединений (1), (2).
Пример 7. Исследование мембранолитической активности соединений (1) и (2)
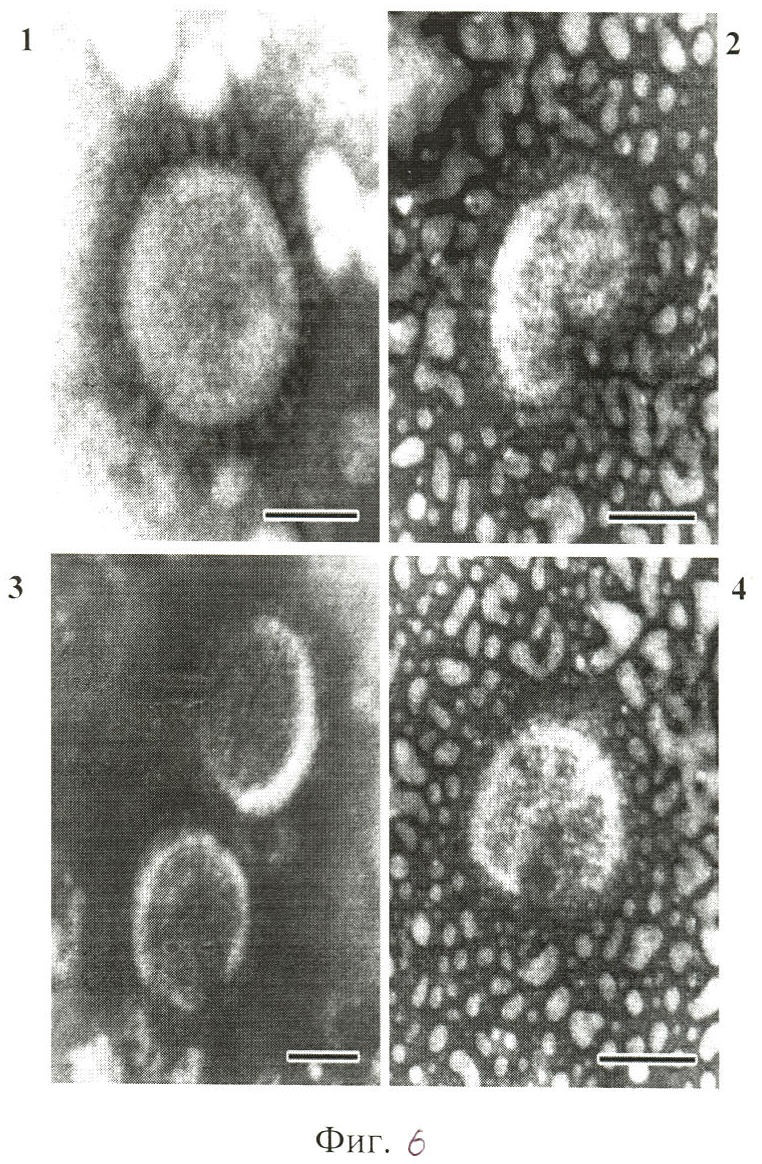
Исследование морфологии вирусных частиц после инкубации с соединениями (1) и (2) проводили с помощью электронной микроскопии методом негативного контрастирования. Исследуемые образцы сорбировали на опорные электронно-микроскопические сетки, покрытые формоваровой пленкой, в течение 1.5 минут, остаток жидкости убирали, сетку высушивали и помещали в контрастирующий раствор (2%-ный раствор фосфорно-вольфрамовой кислоты в дистиллированной воде) на 45 сек, остаток раствора убирали, сетку высушивали. Образцы (4-6 сеток) изучали в трансмиссионном электронном микроскопе Hitachi-H800 при увеличениях от 10000 до 200000.
Электронно-микроскопическое исследование морфологии вирусных частиц, инактивированных соединениями (1) и (2) выявило вирионы с поврежденной липидной оболочкой, у небольшой части вирусных частиц также наблюдали изменение толщины липидного слоя, пепломеры на поверхности вирусной оболочки были сохранены. Степень повреждения структуры вирионов прямо зависела от концентрации соединений (1) и (2). По мере возрастания концентрации соединений до 10-3 М происходило развитие повреждений их структуры.
На фиг.6 представлены электронные микрофотографии частиц вируса гриппа, где на снимке 1 - интактный вирус (на поверхности сферической частицы видны пепломеры, оболочка не повреждена), на снимках 2-4 - вирус гриппа, обработанный искусственной рибонуклеазой в стандартных условиях (на поверхности частиц видны пепломеры, разрывы оболочки, частицы деформированы) Негативное контрастирование ФВК. Длина масштабной линии 50 нм.
На основании результатов электронно-микроскопических исследований можно предполагать, что соединения (1) и (2) дезинтегрируют липидные оболочки вирионов, что способствует быстрой инактивации вируса.
Таким образом, полученные данные однозначно указывают на высокую рибонуклеазную, мембранолитическую и антивирусную активность соединений (1) и (2), что позволяет использовать их в качестве активных компонентов для разработки лекарственных форм препаратов, предназначенных для лечения вирусных заболеваний.
| название | год | авторы | номер документа |
|---|---|---|---|
| СРЕДСТВО, ОБЛАДАЮЩЕЕ ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ | 2008 |
|
RU2402563C2 |
| СРЕДСТВО ДЛЯ ИНАКТИВАЦИИ ДНК-ВИРУСОВ | 2012 |
|
RU2480478C1 |
| ПРОИЗВОДНЫЕ N-ЗАМЕЩЕННОГО 1,4-ДИАЗАБИЦИКЛО-[2.2.2]-ОКТАНА, ПРОЯВЛЯЮЩИЕ ПРОТИВОВИРУСНУЮ АКТИВНОСТЬ В ОТНОШЕНИИ РНК-ВИРУСОВ | 2008 |
|
RU2399669C2 |
| СРЕДСТВО, ПРОЯВЛЯЮЩЕЕ ПРОТИВИРУСНУЮ АКТИВНОСТЬ В ОТНОШЕНИИ ДНК-ВИРУСОВ | 2012 |
|
RU2487876C1 |
| ЛИПОТЕТРАПЕПТИДЫ НА ОСНОВЕ ДИЭФИРОВ L-ГЛУТАМИНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2013 |
|
RU2533554C1 |
| ПЕПТИДОПОДОБНОЕ СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2010 |
|
RU2450806C2 |
| КАТИОННЫЕ ДИМЕРНЫЕ АМФИФИЛЫ В КАЧЕСТВЕ АГЕНТОВ ТРАНСФЕКЦИИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2233834C2 |
| ЗАМЕЩЕННЫЙ БИСДИПЕПТИД С НЕЙРОПРОТЕКТИВНЫМ И АНТИДЕПРЕССИВНЫМ ЭФФЕКТОМ | 2014 |
|
RU2559880C1 |
| ДИМЕРНЫЕ ДИПЕПТИДНЫЕ МИМЕТИКИ НЕЙРОТРОФИНА-3 | 2022 |
|
RU2800369C1 |
| ЛИПОТРИПЕПТИДЫ НА ОСНОВЕ ДИЭФИРОВ L-ГЛУТАМИНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2575851C1 |
Изобретение относится к области химии и медицины. Охарактеризовано средство для инактивации вирусов, представляющее собой производные лизина, глутаминовой и 6-аминогексановой кислот. Представленное средство обладает рибонуклеазной, мембранолитической и противовирусной активностями и может быть использовано в медицине как активные компоненты для разработки лекарственных форм препаратов, используемых для лечения вирусных заболеваний. 6 ил., 4 табл.
Средство для инактивации вирусов, обладающее одновременной рибонуклеазной, мембранолитической и противовирусной активностями, представляющее собой производные лизина, глутаминовой и 6-аминогексановой кислот, связанные линкером гидрофобной природы, со структурными формулами (соединения 1 и 2):
| КОРОЛЕВА Л.С | |||
| Конструирование и синтез искусственных рибонуклеаз на основе коротких пептидов, Автореферат диссертации | |||
| - Новосибирск, 2007 | |||
| ЖДАН Н.С | |||
| Синтетические рибонуклеазы | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Синтез и свойства конъюгатов, содержащих РНК - связывающий фрагмент на основе остатков лизина и РНК - гидролизующий фрагмент, несущий остаток имидазола, Биоорганическая химия, 1999, т.25, №10, с.723-732 | |||
| Приспособление к хронографу | 1928 |
|
SU25766A1 |

