Изобретение относится к области биоорганической химии, в частности к производным аминокислот и пептидов, принадлежащих к классу алифатических диэфиров, содержащих три аминокислотных остатка.
Липосомы активно привлекаются для доставки в клетки биологически активных соединений, диагностических веществ и генетического материала, не способных самостоятельно преодолевать клеточную мембрану (белки, нуклеиновые кислоты, противоопухолевые препараты и многие другие). Отличительными особенностями таких систем являются неиммуногенность, биодеградируемость и биосовместимость. Сконструированные на данный момент липосомальные формы лекарственных препаратов содержат в качестве основного компонента фосфатидилхолин - природный липид. Однако каждый клеточный тип отличается по мембранному составу, поэтому для разных клеточных линий в опытах in vitro необходимы различные липидные агрегаты, способные контактировать и сливаться непосредственно с плазматической клеточной мембраной или взаимодействовать с внутриклеточными мембранами. Использование алифатических производных аминокислот и пептидов может послужить альтернативой применения «традиционных» фосфатидилхолиновых липосом.
Наиболее близким к заявленному техническому решению является ряд катионных липидов, содержащих тетрапептиды. Наличие четырех аминокислотных остатков в полярной части амфифила позволяет липотетрапептидам формировать в водной среде частицы с небольшим диаметром, эффективно компактезировать нуклеиновые кислоты и с высокой эффективностью трансфицировать эукариотические клетки [Себякин Ю.Л., Буданова У.А., Колоскова О.О. Заявка на патент РФ №2013116689, дата публ. 20.10.2014].
Данный вид полярной части молекулы приводит к тому, что наиболее вероятной формой структурной организации дисперсий на основе этих соединений являются не только мелкие липосомы, но и мицеллы, а также возможен смешанный состав. Подобные дисперсии не подходят для использования в качестве переносчиков биологически активных веществ, таких как, например, противоопухолевые препараты, ввиду невозможности достижения эффективной нагрузки транспортного средства.
Снижение количества аминокислотных остатков в полярном блоке амфифила по сравнению с липотетрапептидами позволяет увеличить диаметр формируемых в водной среде липосом и более эффективно встраивать в их внутренний объем биологически активные вещества, а также получать более устойчивые во времени дисперсии.
Предлагаемые в настоящем изобретении соединения ранее в литературе не описаны и аналогов не имеют.
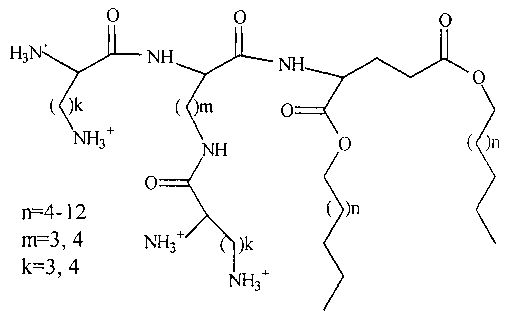
Техническим результатом предлагаемого изобретения является синтез ряда новых алифатических производных трипептидов, полярная часть которых состоит из аминокислотных последовательностей OrnOrnGlu, LysLysGlu, OrnLysGlu, LysOrnGlu, а гидрофобная часть представлена остатками спиртов с длиной цепи С8, С10, С16:
Для достижения указанного технического результата разработана схема получения липотрипептидов, включающая следующие этапы: синтез этерифицированных остатками жирных спиртов производных L-глутаминовой кислоты, активация карбоксильных групп Fmoc-Lys(Boc) или Fmoc-Orn(Boc) и образование пептидной связи между этими компонентами, удаление Fmoc защитных групп с полученного дипептидного производного, активация карбоксильных групп Boc-Lys(Boc) или Вос(Orn)Вос и образование пептидной связи с получением производных трипептидов, удаление Boc защитных групп с получением липотрипептидов.
Реализация данного изобретения подтверждается примерами.
Пример 1.
Синтез дигексадецил-N-(N-L-лизил)-L-лизил-L-глутамата.
Смесь 2,0 г (0,0136 моль) L-глутаминовой кислоты, 7,0 г (0,0292 моль) гексадецилового спирта и 3,1 г (0,0163 моль) n-толуолсульфокислоты нагревали на масляной бане при 130°C в течение 4 ч. После окончания реакции реакционную массу охлаждали до комнатной температуры и перекристаллизовывали из ацетона.
Для удаления тозильной группы 1,5 г соли растворяли в 50 мл хлороформа, промывали 5%-ным раствором гидрокарбоната натрия (2×80 мл), водой до pH 7, сушили сульфатом натрия. Растворитель отгоняли в вакууме. Получали 1,1 г (73%) аморфного вещества, Rf=0,47 (толуол-ацетонитрил, 3:1).
ИК-спектр (в пленке, νmax, см-1): 3395 (NH2), 2945 (C-H), 1725 (C=O), 1632 (NH2), 1482 (CH2), 1381 (CN), 1281 (CH3), 1202 (C-O-C), 1126 (O-C-C), 753 (NH2).
К раствору Fmoc-Lys(Boc) 0,941 г (2 ммоль) в 4 мл N,N-диметилформамида (ДМФА) добавляли 0,271 г (2 ммоль) N-оксибензотриазола (НОВТ) в 2 мл ДМФА и при активном перемешивании охлажденный раствор 0,412 г (2 ммоль) дициклогексилкарбодиимида (DCC) в 4 мл хлороформа. Смесь выдерживали 1 ч при охлаждении, выпавший осадок отфильтровывали.
К полученному раствору добавляли 0,795 г (1,3 ммоль) дигексадецил-L-глутамата в 4 мл хлороформа. Смесь перемешивали при комнатной температуре 5 ч, растворитель отгоняли в вакууме. Продукт очищали препаративной тонкослойной хроматографией в системе толуол-хлороформ-метилэтилкетон-изопропанол, 10:6:3:1 с предварительной отмывкой непрореагировавшего избытка Fmoc-Lys-(Boc) переосаждением из хлороформа. Выход дигексадецил-N-(Fmoc-L-лизил-Boc)-L-глутамата составил 0,965 г (71%), Rf=0,8 (толуол-хлороформ-метилэтилкетон-изопропанол, 10:6:3:1).
1H-ЯМР-спектр (DMSO-D6, δ, м.д.): 0,88 (6Н, т, CH3), 1,24 (~56H, м, CH2), 1,57 (4Н, м, СООСН2СН2), 4,06 (2Н, т, CH2 (Fmoc)), 4,11 (4Н, м, COOCH2), 4,57 (1Н, м, СН (Glu)), 5,72 (1H, д, NH), 7,62 (8Н, м, CH (Fmoc)).
Растворяли 0,965 г (1,3 ммоль) полученного соединения в 2 мл 20% раствора пиперидина в ДМФА. Раствор выдерживали при комнатной температуре 20 мин. Продукт NH2Lys(Boc)Glu(C16)2 выделяли препаративной ТСХ в системе (толуол-хлороформ-метилэтилкетон-изопропанол, 10:6:3:1). Rf=0,2 (толуол-хлороформ-метилэтилкетон-изопропанол, 10:6:3:1).
Масс-спектр [M]+: 824.
К раствору Nα,Nε-(ди-трет-бутилоксикарбонил)-L-лизина 0,265 г (0,728 ммоль) в 4 мл хлороформа добавляли 0,96 г (0,728 ммоль) оксибензотриазола (НОВТ) в 2 мл N,N-диметилформамида (ДМФА) и при активном перемешивании охлажденный раствор 0,150 г (0,728 ммоль) дициклогексилкарбодиимида (DCC) в 4 мл хлороформа. Смесь выдерживали 1 ч при охлаждении, выпавший осадок отфильтровывали. К полученному раствору добавляли 0.300 г (0.364 ммоль) NH2Lys(Boc)Glu(C16)2 в 2 мл хлороформа. Смесь перемешивали при комнатной температуре 10 ч, растворитель отгоняли в вакууме. Продукт выделяли препаративной тонкослойной хроматографией в системе толуол-хлороформ-метилэтилкетон-изопропанол, 10:6:3:1. Полученное соединение 0,2 г растворяли в 1 мл хлороформа и 2 мл безводной трифторуксусной кислоте. Раствор выдерживали при комнатной температуре 3 ч. Растворитель отгоняли в вакууме. Продукт дигексадецил-N-L-лизил-N-L-лизил-L-глутамат тристрифторацетат LysLysGlu(C16)2 выделяли переосаждением из эфира. Выход 143 мг (46%), Rf=0,2 (хлороформ-метанол, 1:2).
1H-ЯМР-спектр (DMSO-D6, δ, м.д.): 0,89 (6 Н, т, 2 СН3), 1,27 (~48 Н, с, 24 СН2), 1,37 (2 Н, м, γСН2), 1,55 (2 Н, м, δСН2), 1,59 (4 Н, м, 2 СООСН2СН2), 1,6 (2 Н, м, δСН2), 1,64 (2 Н, м, γСН2), 1,73-1,76 (4 Н, м, 2 βСН2), 1,95 (2 Н, м, СНСН2), 2,46 (2 Н, м, СН2СОО), 2,91-3,05 (4 Н, м, 2CH2NH2), 4,05-4,10 (4 Н, м, 2 СООСН2), 4,29-4,41 (3 Н, м, СН), 4,76 (2Н, д, 2NH), 7,35 (6 Н, с, 3NH3 +).
Масс-спектр [М]+: 852.
Пример 2.
Синтез дигексадецил-N-L-орнитил-N-L-лизил-L-глутамата.
Аналогично из 2,0 г (0.0136 моль) L-глутаминовой кислоты, 7,0 г (0.0292 моль) гексадецилового спирта и 0.941 г (2 ммоль) Fmoc-Lys(Boc) получали NH2Lys(Boc)Glu(C16)2. Масс-спектр [М]+: 824.
К раствору Nα,Nε-(ди-трет-бутилоксикарбонил)-L-орнитина 0,216 г (0,68 ммоль) в 4 мл хлороформа добавляли 0,92 г (0,68 ммоль) N-оксибензотриазола (НОВТ) в 2 мл N,N-диметилформамида (ДМФА) и при активном перемешивании охлажденный раствор 0,140 г (0,68 ммоль) дициклогексилкарбодиимида (DCC) в 4 мл хлороформа. Смесь выдерживали 1 ч при охлаждении, выпавший осадок отфильтровывали. К полученному раствору добавляли 0,280 г (0,34 ммоль) NH2Lys(Boc)Glu(C16)2 в 2 мл хлороформа. Смесь перемешивали при комнатной температуре 10 ч, растворитель отгоняли в вакууме. Продукт выделяли препаративной тонкослойной хроматографией в системе толуол-хлороформ-метилэтилкетон-изопропанол, 10:6:3:1. Полученное соединение 0,2 г растворяли в 1 мл хлороформа и 2 мл безводной трифторуксусной кислоте. Раствор выдерживали при комнатной температуре 3 ч. Растворитель отгоняли в вакууме. Продукт дигексадецил-N-L-орнитил-N-L-лизил-L-глутамат тристрифторацетат OrnLysGlu(C16)2 выделяли переосаждением из эфира. Выход 151 мг (53%), Rf=0,18 (хлороформ-метанол, 1:2).
1Н-ЯМР-спектр (DMSO-D6, δ, м. д.): 0,89 (6 Н, т, 2 СН3), 1,27 (~48 Н, с, 24 СН2), 1,33 (2 Н, м, γСН2 (Lys)), 1,50 (2 Н, м, βCH2 (Orn)), 1,55 (2 Н, м, δСН2 (Lys)), 1,57 (2 Н, м, γСН2 (Orn)), 1,76 (2 Н, м, βCH2 (Lys)), 2,15 (2 Н, м, СНСН2), 2,46 (2 Н, м, СН2СОО), 2,92 (4 Н, м, 2CH2NH2), 4,05-4,10 (4 Н, м, 2 СООСН2), 4,3-4,4 (3 Н, м, СН), 4,46 (2Н, д, 2NH), 7,58 (6 Н, с, 3NH3 +).
Масс-спектр [М]+: 838.
Пример 3.
Синтез дигексадецил-N-L-орнитил-N-L-орнитил-L-глутамата.
Аналогично из 2,0 г (0,0136 моль) L-глутаминовой кислоты, 7,0 г (0,0292 моль) гексадецилового спирта получали Glu(C16)2. ИК-спектр (в пленке, νmax, см-1): 3395 (NH2), 2945 (С-Н), 1725 (С=O), 1632 (NH2), 1482 (СН2), 1381 (CN), 1281 (СН3), 1202 (С-О-С), 1126 (О-С-С), 753 (NH2).
К раствору Fmoc-Orn(Boc) 0,534 г (1,174 ммоль) в 5 мл N,N-диметилформамида (ДМФА) добавляли 0,159 г (1.174 ммоль) N-оксибензотриазола (НОВТ) в 2 мл ДМФА и при активном перемешивании охлажденный раствор 0,242 г (1,174 ммоль) дициклогексилкарбодиимида (DCC) в 2 мл хлороформа. Смесь выдерживали 1,5 ч при охлаждении, выпавший осадок отфильтровывали.
К полученному раствору добавляли 0,350 г (0,587 ммоль) дигексадецил-L-глутамата в 5 мл хлороформа. Смесь перемешивали при комнатной температуре 7 ч, растворитель отгоняли в вакууме. Продукт очищали препаративной тонкослойной хроматографией в системе толуол-хлороформ-метилэтилкетон-изопропанол, 10:6:3:1. Выход дигексадецил-N-(Fmoc-L-орнитил-Boc)-L-глутамата составил 0,465 г (77%), Rf=0.7 (толуол-хлороформ-метилэтилкетон-изопропанол, 10:6:3:1).
1H-ЯМР-спектр (DMSO-D6, δ, м.д.): 0,9 (6Н, т, CH3), 1,25 (~48Н, м, CH2), 1,5 (2H, м, δCH2), 1,6 (4Н, м, COOCH2CH2), 1,76 (2H, м, βCH2), 2,0 (1Н, с, СН (Fmoc)), 2,15 (2H, м, CHCH2), 2,46 (2H, м, CH2COO), 3,06 (2H, м, CH2NH2), 3,9 (2Н, т, CH2 (Fmoc)), 4,10 (4Н, м, СООСН2), 4,41 (1Н, м, СН (Glu)), 5,95 (1H, д, NH), 7,5-7,8 (8Н, м, 8CH (Fmoc)).
Растворяли 0,465 г (0,587 ммоль) полученного соединения в 1 мл 20% раствора пиперидина в ДМФА. Раствор выдерживали при комнатной температуре 20 мин. Продукт NH2Orn(Boc)Glu(C16)2 выделяли препаративной ТСХ в системе (толуол-хлороформ-метилэтилкетон-изопропанол, 10:6:3:1). Rf=0,23 (толуол-хлороформ-метилэтилкетон-изопропанол, 10:6:3:1)
Масс-спектр [M]+: 810.
Аналогично к раствору Nα,Nε-(ди-трет-бутилоксикарбонил)-L-орнитина 0,159 г (0,501 ммоль) в 2 мл хлороформа добавляли 0,68 г (0,501 ммоль) N-оксибензотриазола (НОВТ) в 2 мл N,N-диметилформамида (ДМФА) и при активном перемешивании охлажденный раствор 0,103 г (0,501 ммоль) дициклогексилкарбодиимида (DCC) в 2 мл хлороформа. Смесь выдерживали 2 ч при охлаждении, выпавший осадок отфильтровывали. К полученному раствору добавляли 0,133 г (0,167 ммоль) NH2Orn(Boc)Glu(C16)2 в 2 мл хлороформа. Смесь перемешивали при комнатной температуре 7 ч, растворитель отгоняли в вакууме. Продукт выделяли препаративной тонкослойной хроматографией в системе толуол-хлороформ-метилэтилкетон-изопропанол, 10:6:3:1. Полученное соединение 0,2 г растворяли в 1 мл хлороформа и 2 мл безводной трифторуксусной кислоте. Раствор выдерживали при комнатной температуре 5 ч. Растворитель отгоняли в вакууме. Продукт дигексадецил-N-L-орнитил-N-L-орнитил-L-глутамат тристрифторацетат OrnOrnGlu(C16)2 выделяли переосаждением из эфира. Выход 72 мг (52%), Rf=0,2 (хлороформ-метанол, 1:2).
Масс-спектр [М]+: 824.
Синтезированные соединения могут быть использованы для получения липосомальных водных дисперсий.
По данным фотонно-коррелляционной спектроскопии размер частиц на основе липотрипептидов (анализатор размера частиц серии LSTM 13320 (Beckman Coulter, USA)) составляет от 80 нм (в случае OrnOrnGlu(C8)2) до 100 нм (в случае LysLysGlu(C16)2).
Липосомы на основе синтезированных липотрипептидов проявляют высокую устойчивость при хранении. Стабильность липосомальных растворов во времени исследовали спектрофотометрически. Показано, что липосомы стабильны в течение 8 месяцев, при этом не наблюдалось заметных отклонений оптических свойств дисперсий.
Форма, размер и устойчивость частиц, формируемых липотрипептидами в водной среде, позволяют эффективно встраивать в них различные биологически активные вещества. Например, в липосомы на основе полученных амфифилов возможно введение противоопухолевого препарата доксорубицина в количестве 40% от массы липотрипептида, при этом процент включения активного компонента во внутренний объем составляет более 90%.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛИПОТЕТРАПЕПТИДЫ НА ОСНОВЕ ДИЭФИРОВ L-ГЛУТАМИНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2013 |
|
RU2533554C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЛИПОДИПЕПТИДОВ | 2011 |
|
RU2463307C1 |
| 1-ГЕКСАДЕЦИЛ-5-(1-ПИРЕНБУТИЛ)-N-(L-ОРНИТИЛ)-L-ГЛУТАМАТ БИСХЛОРИД | 2009 |
|
RU2409587C2 |
| Способ получения высокоочищенного тетрадекапептида | 2020 |
|
RU2759377C1 |
| ТЕТРАВАЛЕНТНЫЕ НЕОГЛИКОКОНЪЮГАТЫ С УГЛЕВОДНЫМ РАЗВЕТВЛЯЮЩИМ ЯДРОМ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2575925C1 |
| ПЕПТИД И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1994 |
|
RU2067000C1 |
| ФОЛАТ-ПОЛИЭТИЛЕНГЛИКОЛЬ-ДИГЕКСАДЕЦИЛ-L-ГЛУТАМАТ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2011 |
|
RU2472796C1 |
| БИВАЛЕНТНЫЕ НЕОГЛИКОКОНЪЮГАТЫ НА ОСНОВЕ ДИЭФИРА L-ГЛУТАМИНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2462471C1 |
| КАТИОННЫЕ ДИМЕРНЫЕ АМФИФИЛЫ В КАЧЕСТВЕ АГЕНТОВ ТРАНСФЕКЦИИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2233834C2 |
| ПРОИЗВОДНЫЕ КАХАЛАЛИДА F | 2004 |
|
RU2395520C2 |
Изобретение относится к области биоорганической химии, в частности к алифатическим производным трипептидов, полярная часть которых состоит из аминокислотных последовательностей OrnOrnGlu, LysLysGlu, OrnLysGlu, LysOrnGlu, а гидрофобная часть представлена остатками спиртов с длиной цепи C8-C16, и способу их получения. Изобретение обеспечивает эффективное встраивание и перенос биологически активных веществ. 2 н.п. ф-лы, 3 пр.
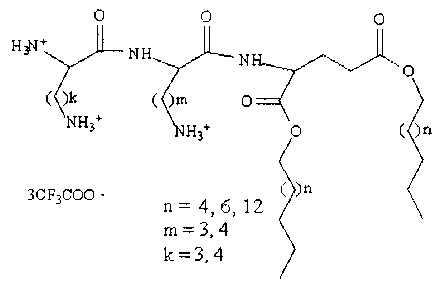
1. Алифатические производные трипептидов, полярная часть которых состоит из аминокислотных последовательностей OrnOrnGlu, LysLysGlu, OrnLysGlu, LysOrnGlu, гидрофобная часть представлена остатками спиртов с длиной цепи С8, С10, C16.
2. Способ получения липотрипептидов, охарактеризованных в п. 1, включающий следующие этапы: синтез этерифицированных остатками жирных спиртов производных L-глутаминовой кислоты, активация карбоксильных групп Fmoc-Lys(Boc) или Fmoc-Orn(Boc) и образование пептидной связи между этими компонентами, удаление Fmoc защитных групп с полученного дипептидного производного, активация карбоксильных групп Boc-Lys(Boc) или Вос(Orn)Вос и образование пептидной связи с получением производных трипептидов, удаление Boc защитных групп с получением липотрипептидов.
| ЛИПОТЕТРАПЕПТИДЫ НА ОСНОВЕ ДИЭФИРОВ L-ГЛУТАМИНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2013 |
|
RU2533554C1 |
| Gopal Vijaya et al, "Synthesis and Transfection Efficiency of Cationic Oligopeptide Lipids: Role of Linker.", Bioconjugate Chemistry, 2011, 22(11), 2244-2254. | |||

