Изобретение относится к области химии и медицины, а именно к новым соединениям, обладающим противовирусной активностью.
Вирусные заболевания представляют собой общую угрозу здоровью населения во всем мире. Среди вирусов вирусы, содержащие геном в виде РНК, вызывают целый ряд серьезных заболеваний животных (домашних и диких) и являются одними из наиболее опасных для человека. Высоко патогенные РНК-вирусы, такие как вирус гриппа птиц (Avian Influenza Virus), клещевой энцефалит (Tick-borne enchefalitis), вирус Денге (Denge virus), коронавирус (SARS) и т.д., являются постоянной угрозой для здоровья населения. На сегодняшний день особую опасность представляют вирусы птиц и животных, способных заражать людей (Lipsitch, M. et al. Science, 2006, 312, 845; Krag, R.M.Science, 2006, 311, 1562-1563; Chen, H. et al. Nature, 2005, 436, 191-192). Например, долгое время считалось, что вирус гриппа птиц не может переходить к людям, однако в последние годы было зарегистрировано большое число передач вируса от птиц к человеку, что вызывало смертельные исходы у заразившихся людей (Subbarao, К. et al. Science, 1998, 279, 393-396; Fouchier, R.A. et al. Proc. Natl. Acad. Sci., USA, 2004, 101, 1356-1361). Вспышки высокопатогенного вируса гиппа H5N1 в некоторых азиатских и европейских странах вызвали серьезное опасение со стороны мирового сообщества (Koopmans, M. et al. Lancet, 2004, 363, 587-593; Enserink, M., Science, 2006, 311, 932; Fabain, N.J.Environ. Health 2006, 68, 46-63).
Опасность эпидемий и пандемий, вызванных РНК-вирусами, делает разработку новых противовирусных препаратов и новых средств и способов их инактивации одной из наиболее актуальных задач сегодняшнего дня.
В настоящее время в качестве средств инактивации вирусов используют физические методы (облучение ультрафиолетовым светом, ионизирующее излучение, нагревание при повышенном давлении) и целый ряд химических соединений (формалин, β-пропиолактон, этиленимин, ряд детергентов), которые различаются механизмом действия и, следовательно, областью применения, токсичностью, эффективностью инактивации вируса и стоимостью (De Benedictis; P. et al. Zoonoses Public Health. 2007, 54(2), 51-68).
Спектр соединений, позволяющих инактивировать вирус, в том числе и для получения цельновирионных вакцин, ограничен несколькими соединениями: это формальдегид, β-пропиолактон (Goldstein M. and Tauraso N., Applied Microbiol, 1970, 19, 290-294), этиленимин и его производные (Budowsky E.I., et al, Vaccine, 1991, 9, 473-476; Aarthi d., et al, Biologicals, 2004, 32, 153-156), облучение УФ-светом (Budowsky E.I., et al, Arch. Virol., 1981, 68-239-246) или УФ-светом в присутствии соединений, повышающих фоточувствительность вируса (Kasermann F and Kempf С., Antiviral Res., 1997, 34, 65-70), окисление кислородом воздуха на свету (Kasermann F. and Kempf С., Rev. Med. Virol., 1998, 8, 143-151). Известно, что обработка вируса такими соединениями приводит к частичному разрушению вирусных антигенных детерминант и тем самым к снижению иммуногенных свойств вакцин, а выборочное разрушение формалином антигенных детерминант поверхностных гликопротеидов вирусов может индуцировать развитие несбалансированного иммунного ответа (Kasermann F., et al., Antiviral Res., 2001, 52, 33-41).
Идеальной мишенью для инактивации вируса гриппа является его геномная РНК. После разрушения геномной РНК вирус теряет способность к репликации и, таким образом, не может размножаться.
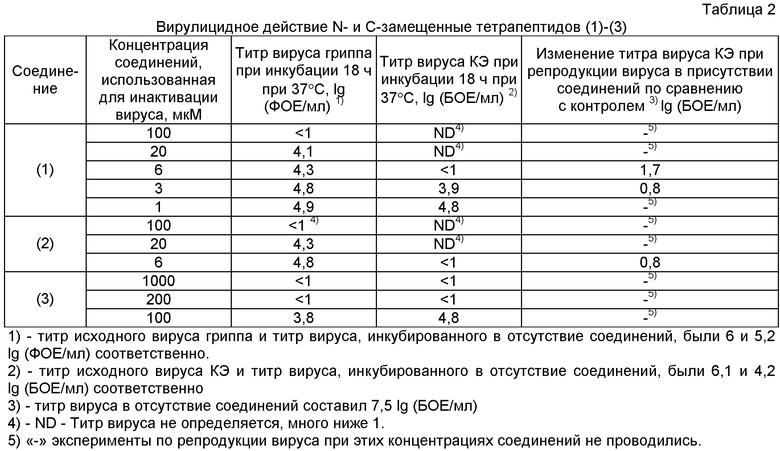
Наиболее близкими к заявляемым соединениям - прототипам являются производные лизина общей формулы nLm, обладающие, согласно литературным данным, высокой рибонуклеазной активностью (Ждан Н.С., и др., Биоорган, химия. 1999, 25, 723-732). На фиг.1 представлена общая структурная формула соединений nLm, представляющих собой неприродные пептиды на основе лизина, где n -число остатков лизина, m - суммарный положительный заряд соединения при нейтральном рН. Было показано, что хотя эти соединения и проявляют высокую рибонуклеазную активность, эффективность расщепления ими РНК не кореллировала с суммарным положительным зарядом молекулы.
Недостатками известных соединений nLm является их низкая противовирусная активность.
Технической задачей изобретения является создание средства на основе синтетических соединений, обладающих высокой противовирусной активностью.
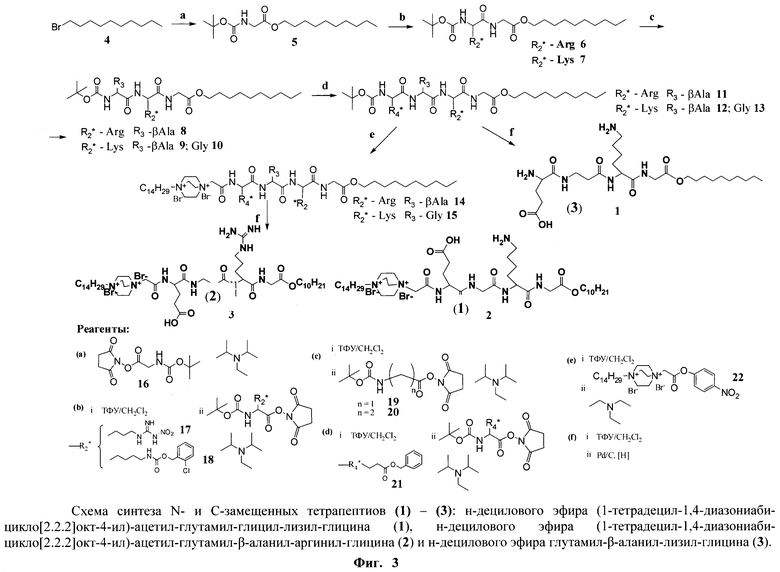
Поставленная техническая задача решается предлагаемыми соединениями, представляющими собой N- и С-замещенные тетрапептиды общей формулы A1D-Glu-AK1-AK2-GlyA2 (соединения 1, 2 и 3), где A1D=1-тетрадецил-1,4-диазониабицикло[2.2.2]октан-4-ил для 1 и 2 и = Н для 3; АК1-=глицин для 1 и β-аланин для 2 и 3; АК2 = лизин для 1 и 3 и аргинин для 2; GlyA2 = н-дециловый эфир глицина.
На фиг.2 представлена общая структурная формула соединений (1), (2) и (3).
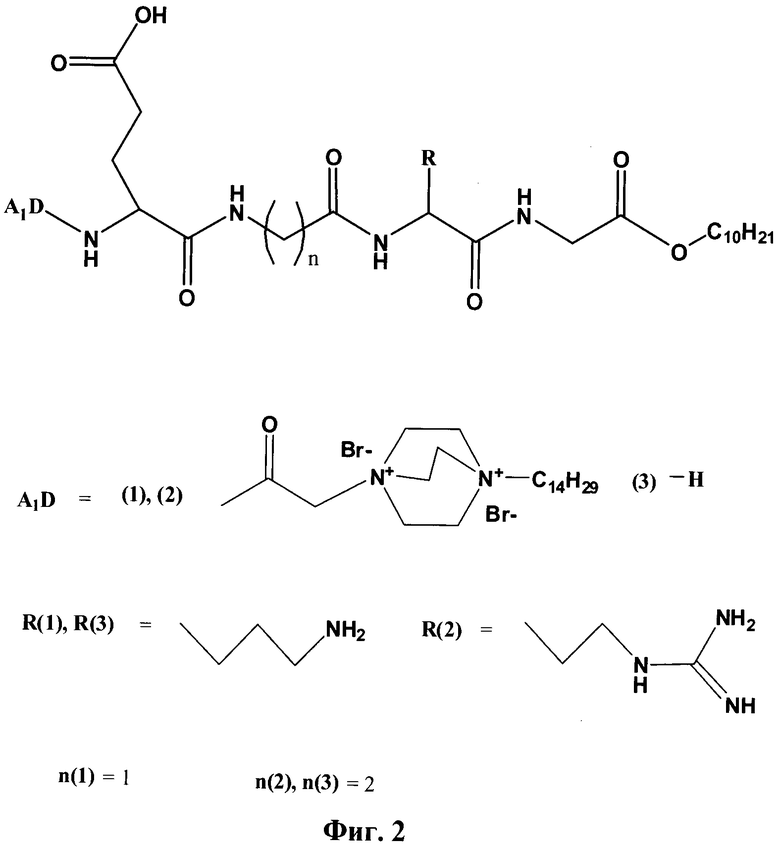
Заявляемые соединения (1)-(3) общей формулы A1D-Glu-AK1-AK2-GlyA2 были получены по общей схеме, представленной на фиг.3. В синтезе пептидов использовались активированные N-гидроксисукцинимидные эфиры аминокислот. Введение трет-бутилоксикарбонильной защиты и синтез активированных эфиров проводили согласно стандартным методикам [А.А.Гершкович, В.К.Кибирев. Синтез пептидов. Реагенты и методы. // Киев. Наукова думка. 1987]. На фиг.3 использованы следующие сокращения: β-Ala - β - аланин, Gly - глицин, Arg - аргинин, Glu - глутаминовая кислота, Lys - лизин, Boc - трет-бутилоксикарбонильная защитная группа, Z(2Cl) - 2-хлорбензилоксикарбонильная защитная группа, Bzl - бензильная защитная группа, ТФУ - трифторуксусная кислота, ДМФА - диметилформамид, Et3N - триэтиламин, DABCO - 1,4-диазабицикло[2.2.2]октан.
Спектры ЯМР записаны на спектрометрах «Bruker AM-400» и «Bruker AV-300» (Германия)» в CD3OD, (СО3)2СО («Aldrich», США). Для 1Н ЯМР-спектров использовали тетраметилсилан или сигналы остаточных протонов растворителей в качестве внутреннего стандарта. Химические сдвиги приведены в шкале δ, м.д.
Масс-спектры высокого разрешения получали с помощью MALDI time-of-flight масс-спектрометра Bruker reflex III MALDI TOP (Bruker Analytical Systems, Inc., Германия), Agilent 1100 series LC/MSD (Agilent, Германия) электрораспылением.
Для колоночной хроматографии использовали силикагель Kieselgel 60 (63-100 мкм, «Merck», Германия).
Для повышения противовирусной активности заявляемых соединений была увеличена гидрофобность соединений путем присоединения к аминогруппе N-концевого глутамина тетрадецил-1,4-диазониабицикло-[2.2.2]октана, а карбоксильная группа С-концевого остатка глицина была этерифицирована н-дециловым спиртом. Для оптимизации геометрии молекулы и взаиморасположения функциональных групп боковых радикалов аминокислот в центр молекулы были помещены остатки лизина (1 и 3) или аргинина (2).
Была исследована антивирусная активность этих соединений в экспериментах по инактивации РНК содержащих вирусов (вирус гриппа и вирус клещевого энцефалита) и в экспериментах по подавлению репродукции этих вирусов в культуре эукариотических клеток in vitro.
Сопоставительный анализ заявляемых соединений с известными и широко используемыми для инактивации вирусов соединениями, такими как формалин, УФ-свет или мономерный или бинарный этиленимина (Budowsky. E.I., et al., Vac. Res., 1996, 5, 29-39), показал, что предлагаемые соединения обладают следующими преимуществами.
1) Заявляемые соединения обладают антивирусной активностью, что позволяет рассматривать их как перспективные лекарственные агенты.
2) Заявляемые соединения малотоксичны для человека и не требуют особых мер предосторожности (противогаз, перчатки, работа под вытяжкой) при работе с ними. Кроме того, эти соединения стабильны при хранении в виде концентрированных водных растворов или растворов в апротонных растворителях, таких как диметилсульфоксид.
3) Заявляемые соединения не оказывают побочного действия на белковые компоненты вирионов, что не снижает иммуногенные свойства инактивированных вирусов и улучшает свойства получаемых на их основе вакцин.
Поиск по источникам научно-технической и патентной литературы показал, что N- и С-замещенные тетрапептиды, обладающие противовирусной активностью, в известных источниках не описаны.
Изобретение иллюстрируется следующими примерами.
Пример 1. Получение н-децилового эфира глутамил-β-аланил-лизил-глицина (3)
Nα-Вос-глицин (16) (28 ммоль, 5 г) растворяли в 15 мл этилацетата, добавляли N-этилдиизопропиламин (34 ммоль, 5.8 мл) и 1-бромдекан (4) (28 ммоль, 5.9 мл). Реакционную смесь кипятили с обратным холодильником в течение суток. Выпавший бромгидрат N-этилдиизопропиламина отфильтровывали. Полученный раствор промывали последовательно 2%-ным раствором лимонной кислоты (20 мл), водой (20 мл), насыщенным раствором NaHCO3 (20 мл) и водой (20 мл). Органический слой сушили безводным Na2SO4. Растворитель удаляли в вакууме. Полученный Nα-Boc-Gly-OC10H21 растворяли в смеси TFA:CH2Cl2=1:1 (5 мл) и выдерживали реакционную смесь 1.5 ч при комнатной температуре. Растворитель и избыток TFA удаляли в вакууме, остаток упаривали с этанолом (3×10 мл). Полученное масло растирали с диэтиловым эфиром. Образовавшийся осадок белого цвета отфильтровывали, сушили в вакууме. Выход трифторацетата децилового эфира глицина 4.41 г, 47%.
К раствору трифторацетата децилового эфира глицина (3 ммоль, 1 г) и N-этилдиизопропиламина (7.6 ммоль, 1.3 мл) в 10 мл этилацетата добавляли при перемешивании раствор N-гидроксисукцинимидного эфира Nα-Вос-Lys(Z(2Cl)) (3.6 ммоль, 1.9 г) в 5 мл этилацетата. Через 2 часа в реакционную смесь добавляли N,N-диметилэтилендиамин (1 ммоль, 110 мкл) и перемешивали 30 мин. Реакционную смесь промывали последовательно 2%-ным раствором лимонной кислоты (20 мл), водой (20 мл), насыщенным раствором NaHCO3 (20 мл) и водой (20 мл). Органический слой сушили безводным Na2SO4. Растворитель удаляли в вакууме. Полученное масло растворяли в смеси TFA:CH2Cl2=1:1 (5 мл) и выдерживали реакционную смесь 1 ч при комнатной температуре. Растворитель и TFA удаляли в вакууме, остаток упаривали с этанолом (3×10 мл), растворяли в этилацетате (15 мл) и промывали насыщенным раствором NaHCO3 (10 мл) и водой (10 мл), сушили над безводным Na2SO4. Растворитель удаляли в вакууме. Полученное вещество в виде пены сушили в вакууме. Выход децилового эфира Z(2Cl)-лизил-глицина 1.47 г, 95.0%.
Далее повторяли процедуру конденсации. Последовательно проводили реакцию децилового эфира Z(2Cl)-лизил-глицина (0.78 ммоль, 400 мг) с N-гидроксисукцинимидным эфиром Вос-β-аланина (20) (0.86 ммоль, 246 мг), затем с N-гидроксисукцинимидным эфиром Nα-Вос-Glu(Bzl) (21) (0.82 ммоль, 356 мг). Раствор защищенного тетрапептида (12) в этилацетате (15 мл) после удаления Вос-защиты промывали насыщенным раствором NaHCO3 (10 мл) и водой (10 мл), сушили безводным Na2SO4. Растворитель удаляли в вакууме. Полученную пену сушили в вакууме. Выход Glu(Bzl)-β-Ala-Lys(Z(2Cl))-Gly-OC10H21 в расчете на дециловый эфир Nε-Z(2Cl)-лизил-глицина 463 мг, 74%.
Glu(Bzl)-β-Ala-Lys(Z(2Cl))-Gly-OC10H21 (0.29 ммоль, 230 мг) после удаления Вос-защиты растворили в 10 мл метанола и подвергали гидрогенолизу в присутствии 80 мг 5%-ного Pd/C. По окончании реакции катализатор отфильтровывали, растворитель упаривали в вакууме. Выход н-децилового эфира глутамил-β-аланил-лизил-глицина после гидрирования 133 мг, 85.2%.
Общий выход н-децилового эфира глутамил-β-аланил-лизил-глицина (3) в расчете на трифторацетат децилового эфира глицина 61.5%. Данные 1Н ЯМР-спектра для н-децилового эфира глутамил-β-аланил-лизил-глицина (1) (CD3OD) δ, м.д. (J/Гц): 0.89 (т, 3Н, СН3, J=6.5); 1.32 (м, 16Н, СН2(CH2)8СН3); 1.71 (м, 8Н, [CHCH2CH2]Glu, [CH(СН2)3СН2)Lys); 2.52 (м, 4Н, [CH2CH2]βAla); 3.00 (т, 2Н, [CHCH2CH2]Glu, J=7.5); 3.56 (т, 2Н, [CH(CH2)3CH2]Lys, J=6.0); 4.01 (м, 3Н, [CHCH2CH2]Glu, [CH2]Gly); 4.15 (т, 2Н, OCH2(СН2)8СН3, J=6.5); 4.40 (т, 1Н, [CH(CH2)4]Lys, J=7.0). MALDI-MS, m/z найдено 544.12 [М+Н]+, вычислено 543.36.
Пример 2. Получение н-децилового эфира (1-тетрадецил-1,4-диазониабицикло[2.2.2]октан-4-ил)-ацетил-глутамил-глицил-лизил-глицина (1)
Синтез н-децилового эфира глутамил-глицил-лизил-глицина проводили аналогично процедуре, описанной в примере 1, используя вместо Вос-β-аланина Вос-глицин. Выход Glu(Bzl)-Gly-Lys(Z(2Cl))-Gly-OC10H21 в расчете на дециловый эфир Nε-Z(2Cl)-лизил-глицина 370 мг, 55.8%.
Н-дециловый эфир тетрапептида Glu(Bzl)-Gly-Lys(Z(2Cl))-Gly (0.1 ммоль, 80.0 мг) растворяли в 1 мл сухого ДМФА, добавляли триэтиламин (0.1 ммоль, 14.2 мкл). В реакционную смесь добавляли суспензию дибромида 1-тетрадецил-4-(4-нитрофеноксикарбонил)метил-1,4-диазониабицикло[2.2.2]октана (22) (0.1 ммоля, 66.0 мг) в 1 мл ДМФА. Реакционную смесь перемешивали при комнатной температуре в течение 48 ч. При этом наблюдается гомогенизация реакционной смеси. Продукт осаждали из реакционной смеси 10-кратным избытком диэтилового эфира. Выпавший белый хлопьевидный осадок отделяли центрифугированием, промывали эфиром и сушили в вакууме. Выход защищенного конъюгата (15) в реакции конденсации 47 мг, 35.8%.
Защищенный конъюгат (15) (0.036 ммоль, 47 мг) растворяли в 10 мл метанола и подвергали гидрогенолизу в присутствии 40 мг 5%-ного Pd/C. По окончании реакции катализатор отфильтровывали, растворитель упаривали в вакууме. Выход в реакции гидрирования 29.5 мг, 78.4%.
Общий выход н-децилового эфира (1-тетрадецил-1,4-диазониабицикло[2.2.2]октан-4-ил)-ацетил-глутамил-глицил-лизил-глицина (2) в расчете на тетрапептид (с защищенными функциональными группами без Вос-защиты) 28.1%. Данные 1Н ЯМР-спектра для н-децилового эфира (1-тетрадецил-1,4-диазониабицикло[2.2.2]октан-4-ил)-ацетил-глутамил-глицил-лизил-глицина (2) (CD3OD) δ, м.д. (J/Гц): 0.92 (т, 6Н, 2 CH3, J=6.5); 1.33 (м, 40Н, СН2(CH2)8СН3, СН2(CH2)12CH3); 1.70 (м, 4Н, [CHCH2CH2]Glu, [CHCH2CH2CH2CH2]Lys); 1.88 (м, 4Н, [CHCH2CH2CH2CH2]Lys); 2.52 (т, 2Н, [CHCH2CH2]Glu, J=7.5); 3.00 (м, 2Н, [CH(CH2)3CH2]Lys); 3.64 (м, 2Н, [СН2]Glu); 3.98 (м, 3Н, Glu-CHCH2CH2, -CH2C(О)-OAlk); 4.08-4.50 (м, 16Н, DABCO (12H), OCH2(СН2)8СН3, NCH2(СН2)12СН3); 4.78 (м, 1Н, [CH(СН2)4]Lys); 4.90 (м, 2Н, N-CH2-С(О)- Glu). ESI-MS, m/z найдено 531.75 [M+Na]2+, вычислено 1037.51.
Пример 3. Получение н-децилового эфира (1-тетрадецил-1,4-диазониабицикло[2.2.2]октан-4-ил)-ацетил-глутамил-β-аланил-аргинил-глицина (2)
Синтез н-децилового эфира глутамил-β-аланил-аргинил-глицина проводили аналогично процедуре, описанной в примере 1. Выход Glu(Bzl)-β-Ala-Arg(NO2)-Gly-OC10H21 в расчете на дециловый эфир Nω-NO2-аргинил-глицина 148 мг, 43.7%.
Синтез н-децилового эфира (1-тетрадецил-1,4-диазониабицикло[2.2.2]октан-4-ил)-ацетил-глутамил-β-аланил-аргинил-глицина проводили аналогично процедуре, описанной в примере 2. Выход защищенного конъюгата (14) в реакции конденсации 45 мг, 61.6%. Выход продукта в реакции гидрирования 17.9 мг, 44.9%.
Общий выход н-децилового эфира (1-тетрадецил-1,4-диазониабицикло[2.2.2]октан-4-ил)-ацетил-глутамил-β-аланил-аргинил-глицина (3) в расчете на тетрапептид (с защищенными функциональными группами без Вос-защиты) 27.6%. Данные 1Н ЯМР-спектра для н-децилового эфира (1-тетрадецил-1,4-диазониабицикло[2.2.2]октан-4-ил)-ацетил-глутамил-β-аланил-аргинил-глицина (3) (CD3OD) δ, м.д. (J/Гц): 0.92 (т, 6Н, 2 CH3, J=6.5); 1.35 (м, 40Н, СН2(CH2)8СН3, СН2(CH2)12СН3); 1.70 (м, 2Н, [CHCH2CH2]Glu); 1.88 (м, 4Н,); 2.54 (м, 4Н, [CHCH2CH2]Glu, [CH(CH2)2CH2]Arg); 3.60 (м, 2Н, [CH2CH2]βAla); 3.99 (м, 3Н, Glu-CHCH2CH2, [CH2]Gly); 4.03-4.50 (м, 16Н, DABCO (12H), OCH2(CH2)8СН3, NCH2(СН2)12СН3); 4.60 (м, 1Н, [CH(СН2)3]Arg); 4.90 (м, 2Н, N-CH2-C(O)-Glu). ESI-MS, m/z найдено 552.37 [M+Na]2+, вычислено 1079.54.
Пример 4. Влияние соединений (1)-(3) на жизнеспособность клеток MDCK
Клетки линии MDCK (клетки почек собаки) культивировали в среде IMDM, содержащей 10%-ную эмбриональную телячью сыворотку, антибиотики (100 ед./мл пенициллина и 0.1 мг/мл стрептомицина) и антимикотик амфотерицин (0.25 мкг/мл), в атмосфере 5%-ного CO2 при 37°С.
Жизнеспособность клеток после инкубации с соединениями (1), (2) или (3) определяли с помощью МТТ теста, который основан на способности живых клеток превращать соединения на основе тетразола (МТТ) в ярко окрашенные кристаллы формазана, что позволяет спектрофотометрически оценивать количество живых клеток в препарате. Для этого клетки высаживали в 96-луночные планшеты (10 клеток на лунку). Через 24 ч в лунках меняли среду и к клеткам добавляли водный раствор соединения (1), (2) или (3) в воде до конечной концентрации в среде от 10-8 до 10-3 М. Клетки инкубировали в присутствии соединений еще в течение суток в тех же условиях. По окончании инкубации без смены среды к клеткам добавляли раствор МТТ (5 мг/мл) в фосфатно-солевом буфере до концентрации 0.5 мг/мл и инкубировали в течение 3 ч в тех же условиях. Среду удаляли, к клеткам добавляли по 100 мкл диметилсульфоксида, в котором происходит растворение образовавшихся в клетках кристаллов формазана, и измеряли оптическую плотность на многоканальном спектрофотометре на длинах волн 570 и 630 нм, где А570 - поглощение формазана, а А630 - фон клеток.
Из экспериментальных данных вычисляли значение IC50 - концентрацию соединений, при которой наблюдается гибель 50% клеток. Значения IC50 приведены в таблице 1.
Из приведенных данных видно, что обработка клеток соединениями (1)-(3) вызывает их эффективную гибель только при концентрациях соединений выше 130 мкМ, что свидетельствует о низкой токсичности соединений. При этом соединение (3) наименее токсично для клеток (IC50=2,5 мМ)
Пример 5. Инактивация вируса гриппа соединениями (1)-(3)
Вирулицидную активность соединений (1)-(3) изучали на модели вируса гриппа A/WSN/33, (H1N1). В этих экспериментах использовали вирус гриппа A/WSN/33, (H1N1) с инфекционным титром 106 фокус-образующих единиц (ФОЕ) вируссодержащего материала. Инфекционную активность вируса определяли методом ФОЕ на клетках MDCK. В основе метода лежит модифицированная иммуноферментная реакция: вирусные белки, экспрессирующиеся в инфицированных вирусом клетках, специфически связываются с моноклональными антителами, мечеными ферментом. Далее фермент взаимодействует с субстратом, что приводит к окрашиванию инфицированной клетки. Инактивацию вируса гриппа соединениями (1)-(3) проводили при 37°С в 50 мМ буфере Трис-HCl, рН 7.0, содержащем 0.2 М KCl и 2 мМ EDTA, в течение 18 ч при 37°С при концентрациях соединений (1)-(3) от 1 мкМ до 1 мМ.
Соединения (1)-(3) эффективно инактивируют вирус гриппа, причем степень инактивации вируса увеличивается с повышением концентрации соединений, при которой проводится инактивация вируса. Так, инкубация вируса в течение 18 ч при 37°С с соединениями (1), (2) в концентрации 20 мкМ снижает титр вируса на 1,1 lg по сравнению с контролем. При увеличении концентрации соединений (1), (2) до 100 мкМ наблюдается полная инактивация вируса гриппа (таблица 2). В случае соединения (3) полная инактивация вируса наблюдается при концентрации соединения 0,2 мМ. Следует отметить, что концентрация соединений (1)-(3), при которой наблюдается полная инактивация вируса гриппа, ниже, чем значение IC50 для этих соединений.
Пример 6. Инактивация вируса клещевого энцефалита (КЭ) соединениями (1)-(3)
Вирулицидную активность соединений (1)-(3) изучали на модели вируса КЭ (штамм Софьин) с инфекционным титром 106 бляшко-образующих единиц (БОЕ). Инфекционную активность вируса КЭ определяли на клетках СПЭВ. Инактивацию вируса КЭ соединениями (1)-(3) проводили при концентрациях соединений от 3 мкМ до 1 мМ в 50 мМ буфере Трис-HCl, рН 7.0, содержащем 0.2 М KCl и 2 мМ EDTA, в течение 18 ч при 37°С.
Соединения (1), (2) в этих условиях уже при концентрация 6 мкМ (18 ч, 37°С) полностью инактивируют вирус КЭ. Для соединения (3) полная инактивання вируса наблюдается при концентрации 1 мМ (таблица 2).
Инкубация клеток, инфицированных вирусом КЭ с соединением (1) в концентрации 6 и 20 мкМ, приводила к снижению репродукции вируса почти в 100 раз по сравнению с контролем.
Полученные результаты однозначно свидетельствуют о высокой антивирусной активности соединений (1)-(3).
Таким образом, приведенные примеры однозначно указывают на высокую антивирусную активность соединений, что позволяет использовать их в качестве активного компонента для разработки лекарственных форм препаратов, предназначенных для лечения вирусных заболеваний.
| название | год | авторы | номер документа |
|---|---|---|---|
| СРЕДСТВО ДЛЯ ИНАКТИВАЦИИ ДНК-ВИРУСОВ | 2012 |
|
RU2480478C1 |
| СРЕДСТВО ДЛЯ ИНАКТИВАЦИИ ВИРУСОВ, ОБЛАДАЮЩЕЕ ОДНОВРЕМЕННОЙ РИБОНУКЛЕАЗНОЙ, МЕМБРАНОЛИТИЧЕСКОЙ И ПРОТИВОВИРУСНОЙ АКТИВНОСТЯМИ | 2008 |
|
RU2399388C2 |
| ПРОИЗВОДНЫЕ N-ЗАМЕЩЕННОГО 1,4-ДИАЗАБИЦИКЛО-[2.2.2]-ОКТАНА, ПРОЯВЛЯЮЩИЕ ПРОТИВОВИРУСНУЮ АКТИВНОСТЬ В ОТНОШЕНИИ РНК-ВИРУСОВ | 2008 |
|
RU2399669C2 |
| СРЕДСТВО, ОБЛАДАЮЩЕЕ АНТИБАКТЕРИАЛЬНОЙ АКТИВНОСТЬЮ | 2010 |
|
RU2443705C1 |
| ПОЛИКАТИОННОЕ СОЕДИНЕНИЕ "ТРИВИРОН (TRIVIRON)" И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2527256C1 |
| СПОСОБ ЛЕЧЕНИЯ С.-Х. ПТИЦ С ПОМОЩЬЮ ПОЛИКАТИОННОГО СОЕДИНЕНИЯ "ТРИВИРОН" (TRIVIRON) | 2013 |
|
RU2532355C1 |
| СРЕДСТВО, ПРОЯВЛЯЮЩЕЕ ПРОТИВИРУСНУЮ АКТИВНОСТЬ В ОТНОШЕНИИ ДНК-ВИРУСОВ | 2012 |
|
RU2487876C1 |
| Способ получения пептидов с последовательностью актг-человека,содержащих в -конечном положении аминооксикислоту | 1973 |
|
SU490284A3 |
| Способ получения -норлейцин13-мотилина | 1974 |
|
SU562193A3 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕПТАПЕПТИДА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2303603C2 |
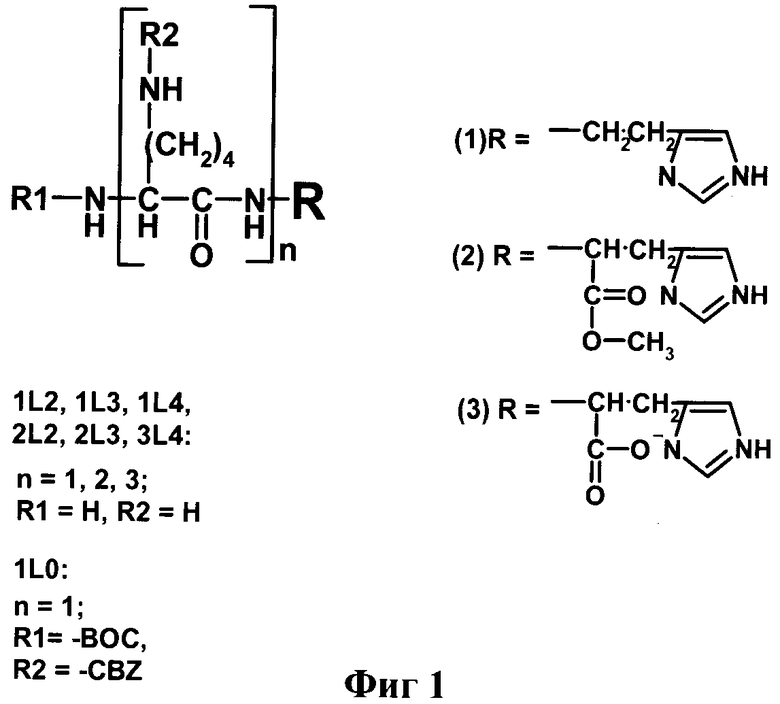
Изобретение относится к средству, обладающему противовирусной активностью, которое представляет собой N- и С-замещенный пептид, выбранный из н-децилового эфира (1-тетрадецил-1,4-диазониабицикло[2.2.2.]октан-4-ил)-ацетил-глутамил-глицил-лизил-глицина (1), н-децилового эфира (1-тетрадецил-1,4-диазониабицикло[2.2.2.]октан-4-ил)-ацетил-глутамил-β-аланил-аргинил-глицина (2) и н-децилового эфира глутамил-β-аланил-лизил-глицина (3). 3 ил., 2 табл.
Средство, обладающее противовирусной активностью, которое представляет собой N- и С-замещенный пептид, выбранный из н-децилового эфира (1-тетрадецил-1,4-диазониабицикло[2.2.2.]октан-4-ил)-ацетил-глутамил-глицил-лизил-глицина (1), н-децилового эфира (1-тетрадецил-1,4-диазониабицикло[2.2.2.]октан-4-ил)-ацетил-глутамил-β-аланил-аргинил-глицина (2) и н-децилового эфира глутамил-β-аланил-лизил-глицина (3).
| Королева Л.С | |||
| и др | |||
| Искусственные рибонуклеазы | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Рибонуклеазная активность тетрапептидов на основе аминокислот, формирующих каталитический центр РНКазы Т1 | |||
| Известия Академии Наук | |||
| Серия Химическая, 2005, 11, с.2596-2604. | |||

