Изобретение относится к области химии и биомедицины, а именно к средствам, проявляющим противовирусную активность в отношении ДНК-вирусов.
Вирусные заболевания представляют собой общую угрозу здоровью населения во всем мире. Вирусы, содержащие геном в виде ДНК, вызывают целый ряд серьезных заболеваний животных (домашних и диких) и являются одними из наиболее опасных для человека. ДНК-вирусы обладают способностью трансформировать клетки, которые они заражают. Так, вирусы папиллом являются этиологическим фактором опухолей шейки матки (Adams, М. et. al, Vaccine, 2007, 25(16), 3007-3013), вирус гепатита В является ассоциированным с опухолями печени (Chu, СМ., J Gastroenterol Hepatol., 2000, Е25-30), вирус Эпштейна-Барра ассоциирован с раком носоглотки и лимфомой Беркитта (Moormann, AM. et al, Curr Opin Infect Dis., 2011, 24(5), 435-41), а вирус герпеса типа 8 - связан с саркомой Капоши (Cai, Q et al., Adv Virus Res. 2010, 78, 87-142).
Опасность заболеваний, вызванных ДНК-вирусами, делает разработку новых противовирусных препаратов и новых средств и способов их инактивации одной из наиболее актуальных задач сегодняшнего дня.
В настоящее время в качестве средств инактивации вирусов используют физические методы (облучение ультрафиолетовым светом, ионизирующее излучение, нагревание при повышенном давлении) и целый ряд химических соединений (формалин, β-пропиолактон, этиленимин, ряд детергентов), которые различаются механизмом действия и, следовательно, областью применения, токсичностью, эффективностью инактивации вируса и стоимостью (De Benedictis, P. et al. Zoonoses Public Health. 2007, 54(2), 51-68).
Спектр соединений, позволяющих инактивировать вирус, в том числе и для получения цельновирионных вакцин, ограничен несколькими соединениями: это формальдегид, вторичный этиленимин (Hulskotte E.G.J., Vaccine, 1997, 15, 1839-1845), каприлат (Korneyeva M et.al, Biologicals, 2002, 30, 153-162), окисленные полиамины (Bachrach U., Amino Acids, 2007, 33(2), 267-272), облучение длинноволновым УФ-светом в присутствии соединений, повышающих фоточувствительность вируса (Hanson C.V., et.al., J. gen. Virol., 1978, 40, 345-358; Specht, K.G., Photochem Photobiol., 1994, 59, 506-514). Известно, что обработка вируса такими соединениями приводит к частичному разрушению вирусных антигенных детерминант и тем самым к снижению иммуногенных свойств вакцин, а выборочное разрушение формалином антигенных детерминант поверхностных гликопротеидов вирусов может индуцировать развитие несбалансированного иммунного ответа (Kasermann F., et al., Antiviral Res., 2001,52, 33-41).
Наиболее ближайшим к заявляемому средству прототипом является средство, представляющее собой 1,4-диазабицикло[2.2.2.]октана общей формулы Dxn, обладающее высокой рибонуклеазной активностью (N.Koval'ov, et al., Nucleosides, Nucleotides and Nucleic Acids. 2004, 32, 981-985.) На Фиг.1 представлена структурная формула соединений Dxn, где n=1, 4, 6 или 12, x - положение в бензольном кольце - орто, мета или пара.
Однако в доступной литературе отсутствует информация о противовирусной активности известного средства в отношении ДНК-вирусов.
Задачей изобретения является получение эффективного, низкотоксичного средства, обладающего противовирусной активностью в отношении ДНК-вирусов.
Поставленная задача решается применением известных производных N-замещенного 1,4-диазабицикло[2.2.2.]октана общей формулы (I):
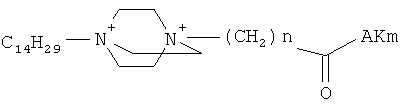 ,
,
где n=3 для (1, 2), n=1 для (3); AKm: гистамин для (1), гистидин для (2), дециловый эфир глутамил-глицил-лизил-глицина для (3), у которых выявлена новая биологическая активность, заключающаяся в противовирусном действии в отношении ДНК-вирусов.
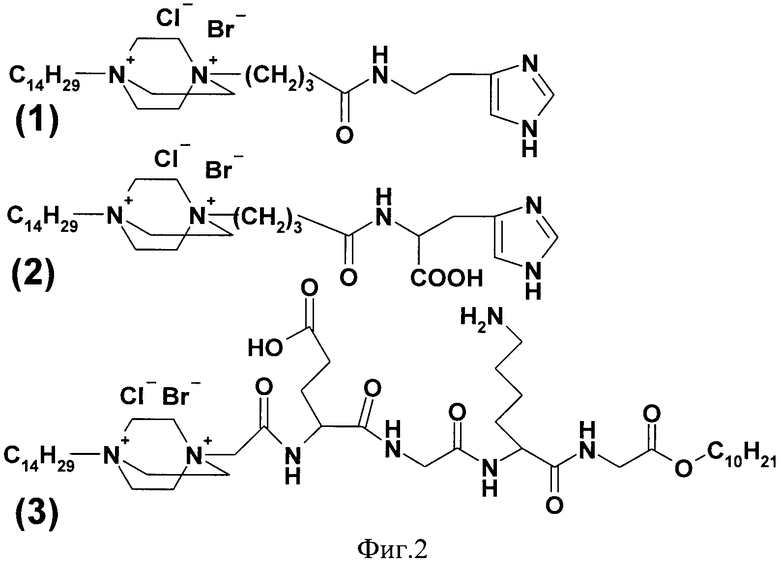
На фигуре 2 представлены структурные формулы соединений (1), (2), (3).
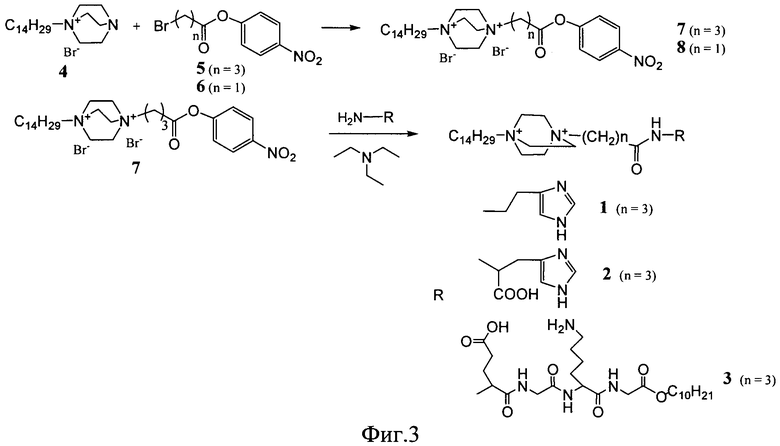
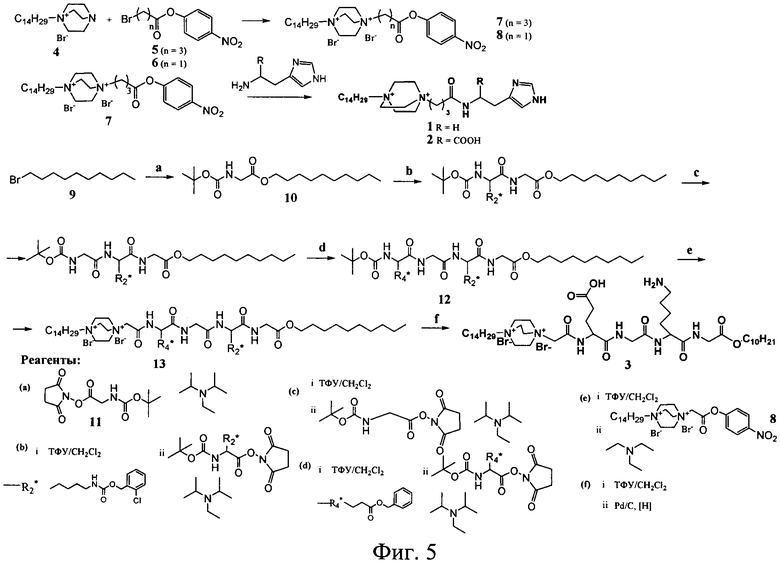
Известные соединения (1-3) общей формулы (I) были получены по общей схеме, представленной на фигуре 3.
Процесс получения заявляемых известных соединений включает в себя реакцию монозамещенного 1,4-диазабицикло-[2.2.2]октана (4) с 4-нитрофениловыми эфирами бромкарбоновых кислот с образованием активированного нитрофенилового эфира производного 1,4-диазабицикло-[2.2.2]октана, которое далее вступает в реакцию либо с коммерчески доступными гистамином и гистидином (для 1 и 2), либо с дециловым эфиром тетрапептида Glu-Gly-Lys-Gly (Konevets DA, et al. 1999, Tetrahedron, 55, 503-512). В синтезе пептида Glu-Gly-Lys-GlyA2 используют активированные N-гидроксисукцинимидные эфиры аминокислот. Введение трет-бутилоксикарбонильной защиты и синтез активированных эфиров проводят согласно стандартным методикам (А.А.Гершкович, В.К.Кибирев. Синтез пептидов. Реагенты и методы. Киев: Наукова думка, 1987).
Сопоставительный анализ заявляемого средства, представляющего собой производные N-замещенного 1,4-диазабицикло[2.2.2.]октана общей формулы (I), и известных соединений с широко используемыми соединениями, такими как формалин, УФ-свет или мономерный или вторичный этиленимин (Hulskotte E.G.J., Vaccine, 1997, 15, 1839-1845), показал, что предлагаемые соединения обладают следующими преимуществами:
1) Заявляемое средство обладает противовирусной активностью в отношении ДНК-вирусов, что позволяет рассматривать его как перспективный обеззараживающий агент для применения в ветеринарии и здравоохранении.
2) Заявляемое средство малотоксично для человека и не требует особых мер предосторожности (противогаз, перчатки, работа под вытяжкой) при работе с ним. Кроме того, средство стабильно при хранении в виде концентрированного водного раствора или раствора в апротонных растворителях, таких как диметилсульфоксид.
3) Заявляемое средство обладает мембранолитической активностью, что позволяет рассматривать его как перспективный агент против ДНК-вирусов. Под «мембранолитической» активностью понимается «способность соединения дезинтегрировать липидные мембраны, включая оболочки вирионов».
Поиск по источникам научно-технической и патентной литературы показал, что противовирусная активность в отношении ДНК-содержащих вирусов для производных N-замещенного 1,4-диазабицикло[2.2.2.]октана общей формулы (I) в известных источниках не описана.
Изобретение иллюстрируется следующими примерами.
Пример 1. Получение 1-тетрадецил-4-γ-[N-(2-(имидазол-4-ил)-этил)-карбамил]-пропил-1,4-диазониабицикло[2.2.2]октана дибромида (соединение 1)
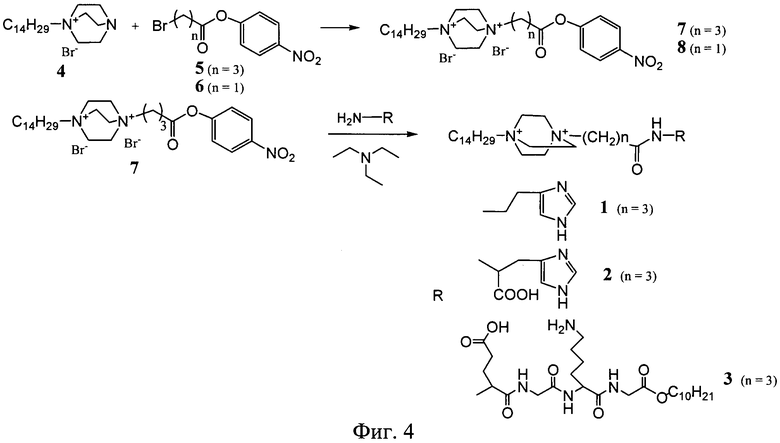
Схема синтеза соединения (1) представлена на фигуре 4. Бромид 1-тетрадецил-4-аза-1-азониабицикло[2.2.2]октана (4) (1.95 г, 5 ммоль) суспендировали в растворе 1.51 г (5.25 ммоль) 4-нитрофенилового эфира γ-броммасляной кислоты (5) в 10 мл ацетона. Смесь встряхивали при 50°C в течение 70 ч. После охлаждения осадок отфильтовали, промыли ацетоном и высушили в вакууме. Выход дибромида 1-тетрадецил-4-(4-нитрофеноксикарбонил)пропил-1,4-диазониабицикло[2.2.2]октана 3.08 г (91%). Раствор 12 мг (0.11 ммоль) гистамина и 11.1 мг (0.11 ммоль) триэтиламина прибавили к раствору 69.3 мг (0.1 ммоль) дибромида 1-тетрадецил-4-(4-нитрофеноксикарбонил)пропил-1,4-диазониабицикло[2.2.2]-октана в 1 мл ДМФА. Смесь выдерживали при 45°C в течение 12 ч, упаривали в вакууме до объема 0.2 мл и осаждали продукт ацетоном. Для очистки соединение (1) переосаждали ацетоном из этанола, осадок отфильтровали, промыли ацетоном и сушили в вакууме. Выход целевого продукта 30 мг (45%).

ES-TOF-MS: m/z найдено 244.76 [M-2Br]2+, вычислено 244.72.
Пример 2. Получение 1-тетрадецил-4-[N-(1-карбокси-2-(имидазол-4-ил)-этил)-карбамил]-пропил-1,4-диазониабицикло[2.2.2]октана дибромида (2)
Соединение (2) получали способом, описанным в примере 1, используя вместо гистамина метиловый эфир гистидина (37 мг, 0.22 ммоль) и 138 мг (0.2 ммоль) активированного эфира (5). Выход 66 мг (47%).
Далее полученный конъюгат (2) растворяли в смеси этанол/вода/триэтиламин 4:5:1 (v/v/v) и выдерживали при 50°C в течение 12 ч. Раствор упарили досуха, остаток растирали с ацетоном. Выход количественный.
Общий выход 1-тетрадецил-4-[N-(1-карбокси-2-(имидазол-4-ил)-этил)-карбамил]-пропил-1,4-диазониабицикло[2.2.2]октана дибромида составил 47%.

ES-TOF-MS: m/z найдено 532.46 [M-2Br]+, вычислено 532.42.
Пример 3. Получение н-децилового эфира (1-тетрадецил-1,4-диазониабицикло[2.2.2]окт-4-ил)-ацетил-глутамил-глицил-лизил-глицина (3)
Схема синтеза соединения (3) представлена на фигуре 5. Nα-Boc-глицин (11) (28 ммоль, 5 г) растворяли в 15 мл этилацетата, добавляли N-этилдиизопропиламин (34 ммоль, 5.8 мл) и 1-бромдекан (9) (28 ммоль, 5.9 мл). Реакционную смесь кипятили с обратным холодильником в течение суток. Выпавший бромгидрат N-этилдиизопропиламина отфильтровывали. Полученный раствор промывали последовательно 2%-ным раствором лимонной кислоты (20 мл), водой (20 мл), насыщенным раствором NaHCO3 (20 мл) и водой (20 мл). Органический слой сушили безводным Na2SO4. Растворитель удаляли в вакууме. Полученный Nα-Boc-Gly-OC10H21 растворяли в смеси TFA:CH2Cl2=1:1 (5 мл) и выдерживали реакционную смесь 1.5 ч при комнатной температуре. Растворитель и избыток TFA удаляли в вакууме, остаток упаривали с этанолом (3×10 мл). Полученное масло растирали с диэтиловым эфиром. Образовавшийся осадок белого цвета отфильтровывали, сушили в вакууме. Выход трифторацетата децилового эфира глицина 4.41 г, 47%.
К раствору трифторацетата децилового эфира глицина (3 ммоль, 1 г) и N-этилдиизопропиламина (7.6 ммоль, 1.3 мл) в 10 мл этилацетата добавляли при перемешивании раствор N-гидроксисукцинимидного эфира Nα-Boc-Lys(Z(2Cl)) (3.6 ммоль, 1.9 г) в 5 мл этилацетата. Через 2 часа в реакционную смесь добавляли N,N-диметилэтилендиамин (1 ммоль, 110 мкл) и перемешивали 30 мин. Реакционную смесь промывали последовательно 2%-ным раствором лимонной кислоты (20 мл), водой (20 мл), насыщенным раствором NaHCO3 (20 мл) и водой (20 мл). Органический слой сушили безводным Na2SO4. Растворитель удаляли в вакууме. Полученное масло растворяли в смеси TFA:CH2Cl2=1:1 (5 мл) и выдерживали реакционную смесь 1 ч при комнатной температуре. Растворитель и TFA удаляли в вакууме, остаток упаривали с этанолом (3×10 мл), растворяли в этилацетате (15 мл) и промывали насыщенным раствором NaHCO3 (10 мл) и водой (10 мл), сушили над безводным Na2SO4. Растворитель удаляли в вакууме. Полученное вещество в виде пены сушили в вакууме. Выход децилового эфира Z(2Cl)-лизил-глицина 1.47 г, 95.0%.
Далее повторяли процедуру конденсации. Последовательно проводили реакцию децилового эфира Z(2Cl)-лизил-глицина (0.78 ммоль, 400 мг) с N-гидроксисукцинимидным эфиром Boc-β-аланина (0.86 ммоль, 246 мг), затем с N-гидроксисукцинимидным эфиром Nα-Boc-Glu(Bzl) (0.82 ммоль, 356 мг). Раствор защищенного тетрапептида (12) в этилацетате (15 мл) после удаления Boc-защиты промывали насыщенным раствором NaHCO3 (10 мл) и водой (10 мл), сушили безводным Na2SO4. Растворитель удаляли в вакууме. Полученную пену сушили в вакууме. Выход Glu(Bzl)-β-Ala-Lys(Z(2Cl))-Gly-OC10H21 в расчете на дециловый эфир Nε-Z(2Cl)-лизил-глицина 463 мг, 74%.
H-дециловый эфир тетрапептида Glu(Bzl)-Gly-Lys(Z(2Cl))-Gly (0.1 ммоль, 80.0 мг) растворяли в 1 мл сухого ДМФА, добавляли триэтиламин (0.1 ммоль, 14.2 мкл). В реакционную смесь добавляли суспензию дибромида 1-тетрадецил-4-(4-нитрофеноксикарбонил)метил-1,4-диазониабицикло[2.2.2]-октана (8) (0.1 ммоль, 66.0 мг) в 1 мл ДМФА. Реакционную смесь перемешивали при комнатной температуре в течение 48 ч. Продукт осаждали из реакционной смеси 10-кратным избытком диэтилового эфира. Выпавший белый хлопьевидный осадок отделяли центрифугированием, промывали эфиром и сушили в вакууме. Выход защищенного конъюгата (13) в реакции конденсации 47 мг, 35.8%.
Защищенный конъюгат (13) (0.036 ммоль, 47 мг) растворяли в 10 мл метанола и подвергали гидрогенолизу в присутствии 40 мг 5%-ного Pd/C. По окончании реакции катализатор отфильтровывали, растворитель упаривали в вакууме. Выход в реакции гидрирования 29.5 мг, 78.4%.
Общий выход н-децилового эфира (1-тетрадецил-1,4-диазониабицикло[2.2.2]октан-4-ил)-ацетил-глутамил-глицил-лизил-глицина (3) в расчете на тетрапептид (с защищенными функциональными группами без Вос-защиты) 28.1%. Данные 1H ЯМР-спектра для н-децилового эфира (1-тетрадецил-1,4-диазониабицикло[2.2.2]октан-4-ил)-ацетил-глутамил-глицил-лизил-глицина (3) (CD3OD) δ, м.д. (J/Гц): 0.92 (т, 6Н, 2 CH3, J=6.5); 1.33 (м, 40Н, СН2(CH2)8CH3, СН2(CH2)12СН3); 1.70 (м, 4Н, [CHCH2CH2]Glu, [CHCH2CH2CH2CH2]Lys); 1.88 (м, 4Н, [CHCH2CH2CH2CH2]Lys); 2.52 (т, 2Н, [CHCH2CH2]Glu, J=7.5); 3.00 (м, 2Н, [СН(СН2)3CH2]Lys); 3.64 (м, 2Н, [CH2]Gly); 3.98 (м, 3H, Glu-CHCH2CH2, -CH2C(O)-OAlk); 4.08-4.50 (м, 16H, DABCO (12Н), OCH2(СН2)8CH3, NCH2(СН2)12CH3); 4.78 (м, 1Н, [CH(СН2)4]Lys); 4.90 (м, 2Н, N-CH2-C(O)-Glu). ESI-MS, m/z найдено 531.75 [M+Na]2+, вычислено 1037.51.
Пример 4. Инактивация вируса осповакцины соединениями (1), (2), (3).
Противовирусную активность соединений (1), (2), (3) изучали на модели вируса осповакцины штамм ЛИ ВП.
Использовали вирус осповакцины (ВОВ) с инфекционным титром 105 бляшкообразующих единиц (БОЕ) вируссодержащего материала. Инфекционную активность вируса определяли методом подсчета БОЕ на клетках CV-1 (клетки почки обезьян). В основе метода лежит способность вируса образовывать единичные участки лизированных клеток (бляшек) на клеточном монослое после заражения одной клетки определенным количеством вирусных частиц, минимально необходимых для инфицирования одной клетки. При заражении клеточного монослоя последовательными разведениями вируса и последующем окрашивании живых клеток красителем «гентиановым фиолетовым» происходит визуализация числа лизированных бляшек, являющихся мерой количества вируса. Инактивацию вируса осповакцины соединениями (1), (2), (3) проводили при 37°C в 50 мМ Трис-HCl буфере, рН 7.0, содержащем 0.2 M KCl и 2 мМ EDTA, в течение 24 ч при 37°C при концентрациях соединений (1), (2), (3) от 0,04 мкМ до 1 мМ.
В таблице 1 представлены данные по противовирусному действию соединений (1), (2), (3).
Соединения (1), (2), (3) эффективно инактивируют ВОВ, причем уровень инактивации вируса повышается с увеличением концентрации соединений, при которой проводили инактивацию вируса. Так, инкубация вируса в течение 24 ч при 37°C с соединениями (1) в концентрации 0,2 мМ, (2) - 0,2 мМ, (3) - 0,25 мМ снижает титр вируса приблизительно на 1 порядок по сравнению с контролем. При увеличении концентрации соединений (1), (2), (3) до 0,3, 0,4 и 0,5 мМ соответственно наблюдается полная инактивация ВОВ.
Полученные результаты однозначно свидетельствуют о высокой противовирусной активности соединений (1), (2), (3).
Пример 5. Исследование мембранолитической активности соединений (1), (2), (3).
Мембранолитическую активность соединений исследовали в экспериментах с эритроцитами барана. Способность соединений вызывать лизис мембран эритроцитов оценивали после инкубации эритроцитов барана в концентрации 15·106 в присутствии одного из соединений (1), (2), (3) в диапазоне концентраций от 0.001 до 2 мМ в течение 120 минут при 37°C. После инкубации эффективность лизиса оценивали по изменению окрашивания фосфатного буфера, измеренному на спектрофотометре на длине волны 550 нм. В качестве положительного контроля за 100% принимали мембранолитическую активность равного объема дистиллированной воды. В качестве негативного контроля использовали 100 мМ фосфатный буфер, эффективность лизиса мембран эритроцитов в присутствии которого в обозначенных условиях не превышала 2%.
В таблице 2 представлены значения эффективности лизиса мембран эритроцитов соединениями (1), (2), (3).
Соединения (1), (2), (3) вызывают лизис мембран эритроцитов, причем его эффективность возрастает с увеличением концентрации соединений, при которой проводили инкубацию суспензии эритроцитов. Так, инкубация вируса в течение 2 ч при 37°C с соединениями (1) и (2) в концентрации 0,5 мМ, (3) - 0,05 мМ приводит к полному лизису мембран эритроцитов, сопоставимому с лизисом эритроцитов, инкубированных в присутствии дистиллированной воды. Снижение концентрации соединений до 0,05 мМ в случае (1) и (2), 0,01 мМ - (3) приводило к полной потере мембранолитической активности данными соединениями. Таким образом, было продемонстрировано, что соединения (1), (2), (3) обладают высокой мембранолитической активностью.
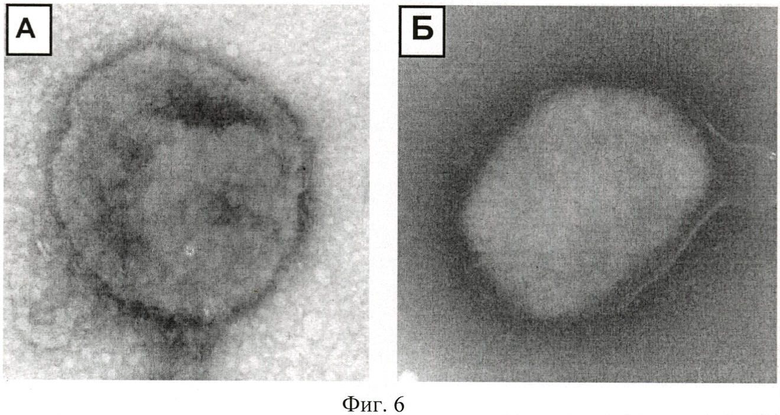
Пример 6. Исследование морфологии вирусных частиц в процессе инактивации с помощью соединения (2)
Исследование морфологии вирусных частиц после инкубации с соединением (2) проводили с помощью электронной микроскопии методом негативного контрастирования. Исследуемые образцы сорбировали на опорные электронно-микроскопические сетки, покрытые формоваровой пленкой, в течение 30 секунд, остаток жидкости убирали, сетку высушивали и помещали в контрастирующий раствор (2% раствор фосфорно-вольфрамовой кислоты в дистиллированной воде) на 30 сек, остаток раствора убирали, сетку высушивали. Образцы (4-6 сеток) изучали в трансмиссионном электронном микроскопе Jem 1400 при увеличениях от 10000 до 200000.
Электронно-микроскопическое исследование морфологии вирусных частиц, инактивированных соединением (2), выявило вирионы с поврежденной липидной оболочкой, у небольшой части вирусных частиц также наблюдали изменение толщины липидного слоя (Фиг.6А), контроль представлен на фиг.6Б. Степень повреждения структуры вирионов прямо зависела от концентрации соединения (2). По мере возрастания концентрации соединения с 0,05 до 0,5 мМ происходило развитие повреждений их структуры. На основании результатов электронно-микроскопических исследований можно заключить, что соединение (2) дезинтегрирует липидные оболочки вирионов, то есть обладает мембранолитической активностью, что приводит к инактивации вируса.
Пример 7. Влияние соединений (1), (2), (3) на жизнеспособность клеток CV-1
Клетки линии CV-1 (клетки почки африканской зеленой мартышки) культивировали в среде DMEM, содержащей 5%-ную эмбриональную телячью сыворотку, антибиотики (100 ед./мл пенициллина и 0.1 мг/мл стрептомицина) и антимикотик амфотерицин (0.25 мкг/мл) в атмосфере 5%-ного CO2 при 37°C.
Жизнеспособность клеток после инкубации с соединениями (1), (2), (3) определяли с помощью МТТ теста, который основан на способности живых клеток превращать соединения на основе тетразола (МТТ) в ярко окрашенные кристаллы формазана, что позволяет спектрофотометрически оценивать количество живых клеток в препарате. Для этого клетки высаживали в 96-луночные планшеты (15×103 клеток на лунку клеток на лунку). Через 48 ч в лунках меняли среду и к клеткам добавляли равный объем водного раствора соединения (1), (2) или (3) до конечной концентрации в среде от 10-7 до 10-3 M. Клетки инкубировали в присутствии соединений еще в течение суток в тех же условиях. По окончании инкубации без смены среды к клеткам добавляли раствор МТТ (5 мг/мл) в фосфатно-солевом буфере до концентрации 0.5 мг/мл и инкубировали в течение 3 ч в тех же условиях. Среду удаляли, к клеткам добавляли по 100 мкл диметилсульфоксида, в котором происходит растворение образовавшихся в клетках кристаллов формазана, и измеряли оптическую плотность на многоканальном спектрофотометре на длинах волн 570 и 630 нм, где А570 - поглощение формазана, а A630 - фон клеток.
Из экспериментальных данных вычисляли значение IC50, концентрацию соединений, при которой наблюдается гибель 50% клеток. Значения IC50 соединений (1), (2), (3) для клеток CV-1 приведены в таблице 3.
Из приведенных данных видно, что обработка клеток соединениями (1), (2), (3) вызывает их эффективную гибель только при концентрациях соединений выше 0.05 мМ, что свидетельствует о низкой токсичности данных соединений.
Таким образом, приведенные примеры однозначно указывают на высокую мембранолитическую и противовирусную активность в отношении ДНК-вирусов соединений (1), (2), (3), что позволяет использовать их в качестве перспективных компонентов для разработки лекарственных форм препаратов, предназначенных для лечения вирусных заболеваний.
| название | год | авторы | номер документа |
|---|---|---|---|
| СРЕДСТВО, ОБЛАДАЮЩЕЕ ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ | 2008 |
|
RU2402563C2 |
| СРЕДСТВО, ПРОЯВЛЯЮЩЕЕ ПРОТИВИРУСНУЮ АКТИВНОСТЬ В ОТНОШЕНИИ ДНК-ВИРУСОВ | 2012 |
|
RU2487876C1 |
| ПРОИЗВОДНЫЕ N-ЗАМЕЩЕННОГО 1,4-ДИАЗАБИЦИКЛО-[2.2.2]-ОКТАНА, ПРОЯВЛЯЮЩИЕ ПРОТИВОВИРУСНУЮ АКТИВНОСТЬ В ОТНОШЕНИИ РНК-ВИРУСОВ | 2008 |
|
RU2399669C2 |
| ПОЛИКАТИОННОЕ СОЕДИНЕНИЕ "ТРИВИРОН (TRIVIRON)" И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2527256C1 |
| СПОСОБ ЛЕЧЕНИЯ С.-Х. ПТИЦ С ПОМОЩЬЮ ПОЛИКАТИОННОГО СОЕДИНЕНИЯ "ТРИВИРОН" (TRIVIRON) | 2013 |
|
RU2532355C1 |
| СРЕДСТВО, ОБЛАДАЮЩЕЕ АНТИБАКТЕРИАЛЬНОЙ АКТИВНОСТЬЮ | 2010 |
|
RU2443705C1 |
| СРЕДСТВО ДЛЯ ИНАКТИВАЦИИ ВИРУСОВ, ОБЛАДАЮЩЕЕ ОДНОВРЕМЕННОЙ РИБОНУКЛЕАЗНОЙ, МЕМБРАНОЛИТИЧЕСКОЙ И ПРОТИВОВИРУСНОЙ АКТИВНОСТЯМИ | 2008 |
|
RU2399388C2 |
| Производное ципрофлоксацина, обладающее антибактериальной активностью в отношении антибиотикоустойчивых штаммов микроорганизмов | 2021 |
|
RU2757741C1 |
| ПРОИЗВОДНЫЕ ГЕМИНА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ, ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2238950C2 |
| ГЕМИНПЕПТИД, ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВОВИРУСНОГО И ВИРУЛЕЦИДНОГО АГЕНТА | 2004 |
|
RU2296131C2 |
Изобретение относится к химии и биомедицине. Предложено средство, представляющее собой одно из производных N-замещенного 1,4-диазабицикло[2.2.2.]октана, проявляющее противовирусную активность в отношении ДНК-вирусов. Предложенное средство может найти применение в ветеринарии и здравоохранении в качестве агента, обладающего противовирусной активностью в отношении ДНК-вирусов. 6 ил., 3 табл., 7 пр.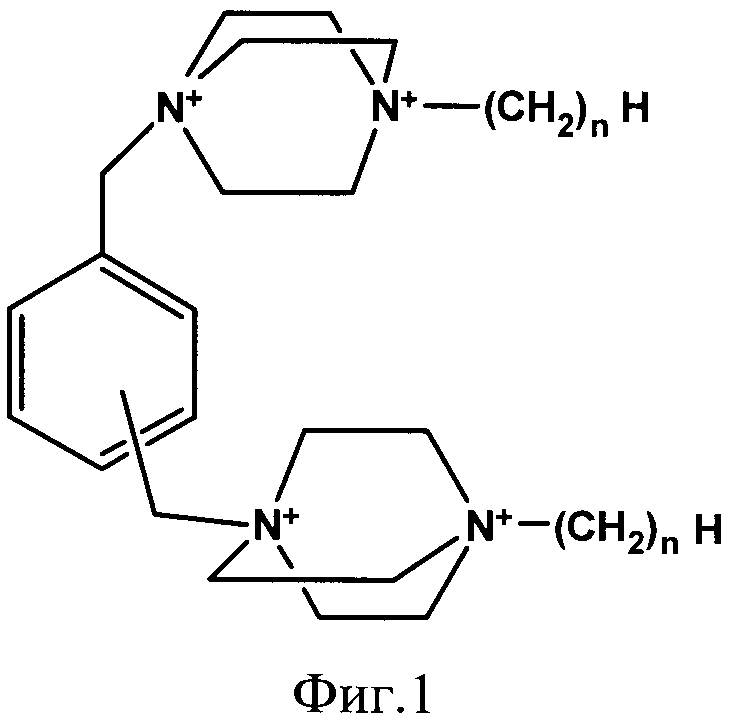
Средство, представляющее собой одно из производных N-замещенного 1,4-диазабицикло[2.2.2.]октана общей формулы (I):
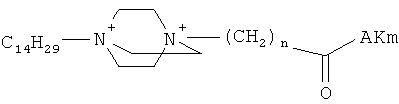
где AKm выбирают из
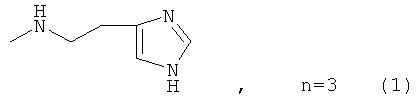

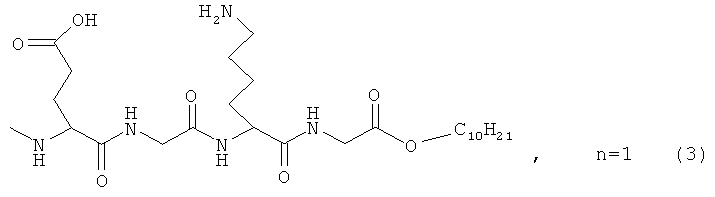
для инактивации ДНК-вирусов.
| СРЕДСТВО, ОБЛАДАЮЩЕЕ ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ | 2008 |
|
RU2402563C2 |
| Регистр Лекарственных Средств | |||
| Энциклопедия лекарств | |||
| - М.: Издательство «РЛС», 2007 (15), с.1488 (с.758) | |||
| Тамкович Н.В | |||
| и др | |||
| ХИМИЧЕСКИЕ РИБОНУКЛЕАЗЫ | |||
| VII | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

