Изобретение относится к способам получения частично фторированных ароматических аминов, содержащих хотя бы один атом водорода в орто-положении к аминогруппе, общей формулы 1,
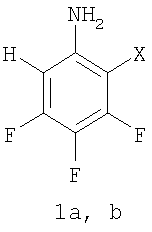
где: Х=F (1а) или Н (1b), которые могут использоваться в качестве исходных веществ в синтезе фторированных гетероциклических соединений, проявляющих широкий спектр биологической активности (L. A. Mitscher, Chem. Rev. 2005, Vol.105, p.559-592). Использование неполностью фторированных ароматических аминов позволяет уменьшить количество стадий в процессе получения препаратов фторхинолонового ряда (см. например, A. Jackson, О. Meth-Cohn. J. Chem. Soc. - Chem. Commun. 1995, Vol.13, p.1319) по сравнению с другими методами получения аналогичных соединений (L.A.Mitscher, Chem. Rev. 2005, Vol.105, №2, p.559-592).
Известен способ получения полифторированных анилинов общей формулы FnC6H(5-n)NH2, где: n = от 1 до 4, восстановлением соединений типа XmFnC6H(5-m-n)NO2, где: Х - атом хлора или брома, m = от 1 до 4, под действием водорода в присутствии палладиевого катализатора, амина, нерастворимого в воде и не образующего водорастворимых солей с галогеноводородными кислотами, и, если необходимо, инертного растворителя (US 5498794, C07D 213/73, 12.03.1999).
Также описан метод восстановления аналогичных соединений водородом в растворе, содержащем палладиевый, никелевый или платиновый катализатор (US 5856577, С07В 61/00, С07С 209/36, 05.01.1999). Недостатком обоих описанных методов является низкая доступность исходных соединений XmFnC6H(5-m-n)NO2.
Помимо этого известен метод получения 3,4,5-трифторанилина 1b взаимодействием 1,3,4,5-тетрафторбензола с амидом натрия в жидком аммиаке при температуре не выше - 33°С (А.А.Штарк, Т.В.Чуйкова, Г.А.Селиванова, В.Д.Штейнгарц. Журн. орган. химии, 1987, Т.23, стр.2574-2577). Недостатками данного способа являются: низкая доступность исходного соединения; необходимость проведения реакции при низких температурах, а также необходимость утилизации жидкого аммиака после проведения реакции.
Известен способ получения частично фторированных ацетанилидов путем гидродефторирования пентафторацетанилида (S.S.Laev, L.Yu.Gurskaya, G.A.Selivanova, I.V.Beregovaya, L.N.Shchegoleva, N.V.Vasil'eva, M.M.Shakirov, V.D.Shteingarts; Eur. J. Org. Chem., 2007, Vol.2, p.306-316). Процесс осуществляется под действием цинка в водном аммиаке в присутствии стехиометрических количеств солей Zn (II) или Cu (II). Основным недостатком данного способа является то обстоятельство, что авторам не удалось достичь гидродефторирования субстрата исключительно по орто-положениям к ацетанилидной группе. Кроме того, не было зафиксировано образование 3,4,5-трифторацетанилида - предшественника соединения 1b.
Предлагаемый способ отличается от известного тем, что соединения 1а и 1b получаются в результате трехстадийного процесса (схема 1), включающего функционализацию исходного пентафторанилина по аминогруппе, восстановительное каталитическое гидродефторирование N-ацилпроизводных пентафторанилина по одному или обоим орто-положениям к аминогруппе под действием металла-восстановителя в присутствии источников протонов при нагревании с последующим щелочным или кислотным гидролизом образующегося продукта с образованием соответствующего амина в условиях, указанных в пп.1-7 формулы изобретения. Выход 2,3,4,5-тетрафторанилина 1а и 3,4,5-трифторанилина 1b составляет 25-99%. Предлагаемый способ позволяет повысить селективность процесса гидродефторирования по oртo-положению к амино-группе. Кроме того, при получении соединений 1а и 1b данным способом в качестве исходных соединений используются производные пентафторанилина, являющегося доступным соединением.
Функционализация пентафторанилина осуществляется известными способами, а именно взаимодействием с соответствующими ангидридами, хлорангидридами или карбоновыми кислотами. Гидрогенолиз ароматических C-F связей протекает под действием восстановителя, в роли которого выступает цинк или магний, в присутствии каталитических количеств комплексных соединений никеля и/или кобальта, в среде апротонных диполярных растворителей, в присутствии источников протонов. Реакция может проводиться при температурах от 20 до 150°С. Нижний предел температуры определяется тем, что при температуре ниже 20°С сильно возрастает время, необходимое для протекания реакции. Верхний предел определяется стабильностью никелевого комплекса. Оптимальная температура для проведения реакции находится в промежутке между 35 и 85°С.

В качестве заместителей R1 и R2 могут выступать:
1. Нециклические ацильные заместители: R1=H, R2=-С(O)R3, где: R3 - алкильная группа от C1 до С10, которая может содержать различные заместители или ненасыщенные фрагменты, такие как двойные или тройные С-С-связи, или арильная группа; R1=-C(O)R3, R2=-C(O)R3, где: R3 - заместитель, описанный ранее.
2. Циклические ацильные заместители: (R1R2)=(-С(O)-(СН2)n-С(O)-), где: n = от 1 до 10; (R1R2)=(-C(O)-CH=CH-C(O)-); (R1R2)=(-C(O)(o-Ar)-C(O)-), где: o-Ar - о-фениленовая, 1,2-нафтилиденовая или 2,3-нафтилиденовая группа, которая может содержать различные заместители в других положениях ароматического кольца.
MLn - комплексные соединения никеля (II) или кобальта (II), используемые в виде готовых соединений или приготовляемые in situ. В качестве лигандов L могут использоваться азотсодержащие соединения, такие как 2,2'-бипиридил (Вру) или 1,10-фенантролин (Phen), алкил- или арилфосфины, бидентатные фосфорсодержащие лиганды Ph2P(CH2)nPPh2, где: n = от 1 до 4, или смешанные бидентатные лиганды, содержащие фосфор и азот. Также возможно использование смешанных комплексов, включающих в качестве лигандов и азот- и фосфорсодержащие соединения.
Каталитические комплексы можно получать in situ из солей никеля или кобальта и соответствующего лиганда или использовать готовые комплексные соединения. Количество каталитического комплекса по отношению к субстрату может составлять от 0.001 до 1 (по молям). Оптимальное количество катализатора составляет от 0.01 до 0.05 (по молям). При использовании меньшего количества катализатора требуется неоправданно большое время для протекания реакции, а верхний предел загрузки определяется из соображений рациональности расхода катализатора.
В качестве восстановителя используются цинк или магний в количестве от 1 до 15 эквивалентов по отношению к субстрату. Наиболее приемлемым является использование восстановителя в количестве от 3 до 10 эквивалентов по отношению к субстрату, поскольку использование меньшего количества резко снижает конверсию субстрата 2 в дефторированные продукты 3, а использование большего количества восстановителя ведет к излишнему расходу реагента.
Реакция протекает в среде апротонных диполярных растворителей, таких как N,N-диметилформамид (ДМФ), N,N-диметилацетамид (ДМА), N-метилпирролидон (МП), гексаметилфосфотриамид (ГМФА) или диметилсульфоксид (ДМСО).
Оптимальная температура проведения реакции составляет от 35 до 85°С и определяется тем, что при более низкой температуре возрастает время, необходимое для протекания реакции, а при температуре выше 85°С неоправданно возрастает энергоемкость процесса.
В качестве источников протонов могут использоваться вода, хлорид аммония, спирт, а также органические и неорганические кислоты.
Гидролиз производных 3, а также выделение и очистка целевых продуктов 1а и 1b осуществляются известными способами.
Неочевидность предлагаемого решения поставленной задачи иллюстрируется тем, что на сегодняшний день в литературе не встречается описания методов каталитической активации C-F связи комплексными соединениями никеля или кобальта в производных фторированных ароматических аминов.
В отличие от метода получения частично фторированных бензойных кислот (RU 2155185, С07С 63/70, 27.08.2000) в данном случае необходимой стадией является предварительная функционализация аминогруппы в молекуле пентафторанилина и гидролиз продукта гидродефторирования 3. Кроме того, априорно не было известно, как отразится на каталитической активности комплекса изменение природы субстрата и переходного металла.
Сущность изобретения иллюстрируется следующими примерами.
Пример 0. Функционализация пентафторанилина по аминогруппе. В колбу, снабженную обратным холодильником, магнитной мешалкой и масляной баней с терморегулятором, помещают 5 г (27 ммоль) пентафторанилина и 21.6 г (20 мл, 212 ммоль) уксусного ангидрида. Полученную смесь кипятят при перемешивании с обратным холодильником в течение 5 ч. После этого реакционную смесь разбавляют 50 мл воды, продукт отфильтровывают, промывают 2 раза по 10 мл холодной воды и сушат в вакуум-эксикаторе над гидроксидом калия. Получают 6.1 г (90%) пентафторацетанилида.
Пример 1. Получение 2,3,4,5-тетрафторанилина.
В колбу, снабженную термометром, газоотводной трубкой, магнитной мешалкой и масляной баней с терморегулятором помещают 30 мг (0.125 ммоль) NiCl2·6H2O, 50 мг (0.250 ммоль) Phen·Н2О, 2.5 мл ДМФ и 0.5 мл воды. Реакционную смесь перемешивают при температуре 70°С в течение 1 ч, затем добавляют 1.64 г (25 ммоль) цинковой пыли, перемешивают еще 10 мин и прибавляют 563 мг (2.5 ммоль) пентафторацетанилида. Реакционную смесь перемешивают в течение 6 ч при температуре 70°С, затем выливают в 10 мл воды, твердую часть отфильтровывают, промывают еще 10 мл воды. Продукт вымывают из твердой фазы 10 мл ацетонитрила. Растворитель упаривают, к остатку добавляют 10 мл воды и гидроксид натрия до рН 13-14, полученную смесь перемешивают в течение 1 ч при комнатной температуре. Продукт отгоняют с водяным паром, экстрагируют серным эфиром, экстракт сушат сульфатом магния, эфир отгоняют в вакууме. Получают 400 мг вещества, содержание 2,3,4,5-тетрафторанилина составляет 95% (степень конверсии пентафторацетанилида составляет 98%).
Примеры 2-16. Получение 2,3,4,5-тетрафторанилина.
Варьируется состав каталитического комплекса и природа растворителя. В колбу, снабженную термометром, газоотводной трубкой, магнитной мешалкой и масляной баней с терморегулятором, помещают 30 мг (0.125 ммоль) NiCl2·6H2O (опыты 2-5) или 30 мг (0.125 ммоль) CoCl2·6H2O (примеры 6-8), необходимое количество (1, 2 или 3 эквивалента по отношению к соли никеля или кобальта) Вру или Phen·H2O, 2.5 мл соответствующего растворителя и 0.5 мл воды. Реакционную смесь перемешивают при температуре 70°С в течение 1 ч, затем добавляют 1.64 г (25 ммоль) цинковой пыли, перемешивают еще 10 мин и прибавляют 563 мг (2.5 ммоль) пентафторацетанилида. Реакционную смесь перемешивают в течение 6 ч при температуре 70°С. Обработка реакционной смеси, гидролиз 2,3,4,5-тетрафторацетанилида и выделение продукта производятся аналогично методике, описанной в примере 1.
Результаты приведены в таблице 1.
Примеры 17-23. Получение 2,3,4,5-тетрафторанилина.
Реакции проводят с использованием готовых комплексов никеля или кобальта. Варьируют состав каталитического комплекса и природу растворителя.
В колбу, снабженную термометром, газоотводной трубкой, магнитной мешалкой и масляной баней с терморегулятором, помещают 0.125 ммоль соответствующего комплекса никеля или кобальта, 2.5 мл ДМФ и 0.5 мл воды и 1.64 г (25 ммоль) цинковой пыли. Смесь перемешивают еще 10 мин и прибавляют 563 мг (2.5 ммоль) пентафторацетанилида. Реакционную смесь перемешивают в течение 6 ч при температуре 70°С. Обработка реакционной смеси, гидролиз 2,3,4,5-тетрафторацетанилида и выделение продукта производят аналогично методике, описанной в примере 1.
Результаты экспериментов приведены в таблице 2.
Пример 24. Получение 2,3,4,5-тетрафторанилина.
В колбу помещают 82 мг (0.125 ммоль) Ni(PPh3)2Cl2, 20 мг (0.125 ммоль) Вру, 1635 мг (25 ммоль) цинковой пыли и 563 мг (2.5 ммоль) пентафторацетанилида. Колбу вакуумируют, затем заполняют аргоном. С помощью шприца добавляют смесь 2.5 мл ДМФ и 0.5 мл воды. Реакционную массу перемешивают при комнатной температуре в течение 1 ч, затем температуру поднимают до 70°С и перемешивают еще 6 ч. Обработку реакционной смеси, гидролиз 2,3,4,5-тетрафторацетанилида и выделение продукта производят аналогично методике, описанной в примере 1. Получают 360 мг вещества, содержание 2,3,4,5-тетрафторанилина составляет 80% (степень конверсии пентафторацетанилида составляет 100%).
Примеры 25-27. Получение 2,3,4,5-тетрафторанилина.
Варьируют состав каталитического комплекса и природу растворителя.
В колбу помещают 82 мг (0.125 ммоль) Ni(PPh3)2Cl2, 20 мг (0.125 ммоль) Вру или 25 мг (0.125 ммоль) Phen·H2O, 1635 мг (25 ммоль) цинковой пыли и 563 мг (2.5 ммоль) пентафторацетанилида. Колбу вакуумируют, затем заполняют аргоном. С помощью шприца добавляют смесь 2.5 мл ДМФ и 0.5 мл воды. Реакционную массу перемешивают при комнатной температуре в течение 1 ч, затем температуру поднимают до 70°С и перемешивают еще 6 ч. Обработка реакционной смеси, гидролиз 2,3,4,5-тетрафторацетанилида и выделение продукта производят аналогично методике, описанной в примере 1.
Результаты приведены в таблице 3.
Пример 28. Получение 3,4,5-трифторанилина.
В колбу, снабженную термометром, газоотводной трубкой, магнитной мешалкой и масляной баней с терморегулятором, помещают 30 мг (0.125 ммоль) NiCl2·6Н2О, 50 мг (0.250 ммоль) Phen·H2O, 2.5 мл МП и 0.5 мл воды. Реакционную смесь перемешивают при температуре 70°С в течение 1 ч, затем добавляют 1635 мг (25 ммоль) цинковой пыли, перемешивают еще 10 мин и прибавляют 563 мг (2.5 ммоль) пентафторацетанилида. Реакционную смесь перемешивают в течение 6 ч при температуре 70°С, затем выливают в 10 мл воды, твердую часть отфильтровывают, промывают еще 10 мл воды. Продукт вымывают из твердой фазы 10 мл ацетонитрила. Растворитель упаривают, к остатку добавляют 10 мл воды и гидроксид натрия до рН 13-14, полученную смесь перемешивают в течение 1 ч при комнатной температуре. Продукт отгоняют с водяным паром, экстрагируют серным эфиром, экстракт сушат сульфатом магния, эфир отгоняют в вакууме. Получают 305 мг вещества, содержание 3,4,5-тетрафторанилина составляет 92% (степень конверсии пентафтор- и 2,3,4,5-тетрафторацетанилидов составляет 100%).
Примеры 29-32. Получение 3,4,5-трифторанилина.
Варьируют температуру проведения реакции.
В колбу, снабженную термометром, газоотводной трубкой, магнитной мешалкой и масляной баней с терморегулятором, помещают 30 мг (0.125 ммоль) NiCl2·6H2O, 50 мг (0.250 ммоль) Phen·H2O, 2.5 мл МП и 0.5 мл воды. Реакционную смесь перемешивают при комнатной температуре в течение 1 ч, затем добавляют 1635 мг (25 ммоль) цинковой пыли, перемешивают еще 10 мин и прибавляют 563 мг (2.5 ммоль) пентафторацетанилида. Реакционную смесь перемешивают при соответствующей температуре в течение 7 ч. Обработка реакционной смеси, гидролиз 3,4,5-трифторацетанилида и выделение продукта производятся аналогично методике, описанной в примере 28.
Результаты приведены в таблице 4.
Примеры 33-41. Получение 3,4,5-трифторанилина.
Варьируют время реакции и соотношение восстановитель/субстрат.
В колбу, снабженную термометром, газоотводной трубкой, магнитной мешалкой и масляной баней с терморегулятором, помещают 30 мг (0.125 ммоль) NiCl2·6H2O, 50 мг (0.250 ммоль) Phen·H2O, 2.5 мл МП и 0.5 мл воды. Реакционную смесь перемешивают при температуре 70°С в течение 1 ч, затем добавляют соответствующее количество цинковой пыли, перемешивают еще 10 мин и прибавляют 563 мг (2.5 ммоль) пентафторацетанилида. Реакционную смесь перемешивают при температуре 70°С в течение требуемого времени. Обработка реакционной смеси, гидролиз 3,4,5-трифторацетанилида и выделение продукта производят аналогично методике, описанной в примере 28.
Результаты приведены в таблице 5.
Примеры 42-44. Гидродефторирование различных производных пентафторанилина.
В колбу, снабженную термометром, газоотводной трубкой, магнитной мешалкой и масляной баней с терморегулятором, помещают 30 мг (0.125 ммоль) NiCl2·6H2O, 50 мг (0.250 ммоль) Phen·H2O, 2.5 мл МП и 0.5 мл воды. Реакционную смесь перемешивают при температуре 70°С в течение 1 ч, затем добавляют соответствующее количество цинковой пыли, перемешивают еще 10 мин и прибавляют 2.5 ммоль соответствующего субстрата. Реакционную смесь перемешивают в течение требуемого времени при температуре 70°С. Обработка реакционной массы, гидролиз смеси N-ацилполифторанилинов и выделение продукта производят аналогично методике, описанной в примере 28.
Результаты приведены в таблице 6.
Таким образом, вышеописанный способ позволяет получать частично фторированные анилины с высокими выходами в мягких условиях, используя доступные реагенты.



| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 3,4,5-ТРИФТОРАНИЛИНА | 2009 |
|
RU2420515C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЧАСТИЧНО ФТОРИРОВАННЫХ БЕНЗОЙНЫХ КИСЛОТ | 1999 |
|
RU2155185C1 |
| СПОСОБ ПОЛУЧЕНИЯ КУБОВЫХ КРАСИТЕЛЕЙ И ПИГМЕНТОВ, СОДЕРЖАЩИХ ПЕРИЛЕНОВЫЙ ФРАГМЕНТ | 1997 |
|
RU2128200C1 |
| ПРОИЗВОДНЫЕ 2-ДЕЗОКСИ-2,3-ДИДЕГИДРО-N-АЦЕТИЛНЕУРАМИНОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ | 1991 |
|
RU2119487C1 |
| КАТАЛИЗАТОР ДЛЯ ПОЛУЧЕНИЯ 1-ДИАЛКИЛАМИН-2,3-ДИФЕНИЛАЛЮМАЦИКЛОПРОПЕНОВ | 2000 |
|
RU2175889C2 |
| СПОСОБ ПОЛУЧЕНИЯ 1-ФЕНИЛ-2,3,4,5-ТЕТРААЛКИЛБОРОЦИКЛОПЕНТА-2,4-ДИЕНОВ | 2008 |
|
RU2376310C2 |
| Комплексный никелевый катализатор гидрирования | 2022 |
|
RU2790674C1 |
| НИКЕЛЕВЫЙ КАТАЛИЗАТОР ГИДРИРОВАНИЯ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2009 |
|
RU2411228C1 |
| ОПРЕДЕЛЕННЫЕ 5-АЛКИЛ-2-АРИЛАМИНОФЕНИЛУКСУСНЫЕ КИСЛОТЫ И ИХ ПРОИЗВОДНЫЕ | 1998 |
|
RU2186762C2 |
| СПОСОБ СОВМЕСТНОГО ПОЛУЧЕНИЯ 1-ЭТИЛ-2,3-ДИАЛКИЛ(ФЕНИЛ)АЛЮМАЦИКЛОПРОПЕНОВ, 1-ЭТИЛ-2,3-ДИАЛКИЛ(ФЕНИЛ)АЛЮМАЦИКЛОПЕНТ-2-ЕНОВ И 1-ЭТИЛ-2,3,4,5-ТЕТРААЛКИЛАЛЮМАЦИКЛОПЕНТАДИЕНОВ | 1999 |
|
RU2160269C1 |
Изобретение относится к новому улучшенному способу получения частично фторированных ароматических аминов, содержащих хотя бы один атом водорода в орто-положении к аминогруппе, общей формулы 1,

где: Х=F (1а) или Н (1b), отличающемуся тем, что проводят функционализацию пентафторанилина по аминогруппе обработкой производным алифатической или ароматической моно- или дикарбоновой кислоты с получением соответствующего производного пентафторанилина в качестве субстрата, который подвергают восстановительному гидродефторированию под действием металла-восстановителя в присутствии источника протонов и в присутствии катализатора - комплексного соединения никеля и/или кобальта с лигандами, выбранными из гетероциклических азотсодержащих соединений или фосфорсодержащих соединений, в среде апротонного диполярного растворителя с последующим щелочным или кислотным гидролизом реакционной смеси с образованием соответствующего амина. 6 з.п. ф-лы, 6 табл.
1. Способ получения частично фторированных ароматических аминов, содержащих хотя бы один атом водорода в орто-положении к аминогруппе, общей формулы 1,
где Х=F (1а) или Н (1b), отличающийся тем, что проводят функционализацию пентафторанилина по аминогруппе обработкой производным алифатической или ароматической моно- или дикарбоновой кислоты с получением соответствующего производного пентафторанилина в качестве субстрата, который подвергают восстановительному гидродефторированию под действием металла-восстановителя в присутствии источника протонов и в присутствии катализатора - комплексного соединения никеля и/или кобальта с лигандами, выбранными из гетероциклических азотсодержащих соединений или фосфорсодержащих соединений, в среде апротонного диполярного растворителя с последующим щелочным или кислотным гидролизом реакционной смеси с образованием соответствующего амина.
2. Способ по п.1, отличающийся тем, что молярное соотношение катализатор/субстрат может составлять от 0,001-1, предпочтительно, 0,01-0,05 и восстановитель/субстрат 1-15, предпочтительно 3-10.
3. Способ по п.1, отличающийся тем, что в качестве лигандов используют такие, как 2,2'-бипиридил или 1,10-фенантролин, алкил- или арилфосфины, бидентатные фосфорсодержащие лиганды или смешанные бидентатные лиганды, содержащие фосфор и/или азот.
4. Способ по п.1, отличающийся тем, что в качестве источника протонов используют воду, соли аммония, спирты, а также органические и неорганические кислоты.
5. Способ по п.1, отличающийся тем, что в качестве апротонных диполярных растворителей используют такие, как N,N-диметилформамид (ДМФ), N,N-диметилацетамид (ДМА), N-метилпирролидон (МП), гексаметилфосфотриамид (ГМФА) или диметилсульфоксид (ДМСО).
6. Способ по п.1, отличающийся тем, что восстановительное гидродефторирование проводят при температуре 20-150°С, предпочтительно 35-85°С.
7. Способ по п.1, отличающийся тем, что в качестве металла-восстановителя используют цинк или магний.
| G.A.Selivanova et all, Selective hydrodechlorination of fluorinated arylamines | |||
| Journal of Fluorine Chemistry, 2004, 125, 1829-1834 | |||
| S.S.Laev et all, N-Acetilation as a means to activate polyfluorylamines for selective orthohydrodefluorination by zinc in aqueous ammonia: a concise route to polyfluorobenzo azaheterocycles, Eur | |||
| J | |||
| Org | |||
| Chem., 2007, |

