Настоящее изобретение относится к 5-алкил-2-ариламинофенилуксусным кислотам и к их производным, как они определены в настоящем описании, которые являются особенно сильными и избирательными ингибиторами циклооксигеназы-2 (СОХ-2), к способам их получения, к фармацевтическим композициями, включающим эти соединения, к способам избирательного ингибирования активности СОХ-2 и к лечению у млекопитающих состояний, чувствительных к ингибированию СОХ-2, с помощью этих соединений или фармацевтических композиций, включающих соединения по изобретению.
Различные замещенные 2-ариламинофенилуксусные кислоты и их производные описаны, например, в J. Med. Chem. 33, 2358 (1990), в патентах US 3558690, US 3652762, US 4173577 и US 4548952, в DE 3445011 и в заявках WO 97/09977 и WO 96/00716 в качестве нестероидных противовоспалительных агентов и ингибиторов циклооксигеназы. Что касается 5-алкил-2-ариламинофенилуксусных кислот, то единственным известным из литературы примером является 5-метил-2-(2,6-диметиланилино)фенилуксусная кислота и ее натриевая соль (патент US 3558690), причем какие-либо данные о их биологической активности отсутствуют.
Нестероидные противовоспалительные агенты блокируют синтез простагландинов путем ингибирования фермента циклооксигеназы. Известно, что циклооксигеназа включает конститутивную изоформу (циклооксигеназа-1, СОХ-1) и индуцибельную изоформу (циклооксигеназа-2, СОХ-2). Вероятно, изоформа СОХ-1 отвечает за защитные полезные свойства простагландинов, например, в желудочно-кишечном тракте, в почке и т.д., в то время как индуцибельная изоформа СОХ-2, вероятно, отвечает за патологические состояния, связанные с простагландинами, такие как воспалительные состояния. Применение нестероидных противовоспалительных лекарств (НСПВЛ, в том числе диклофенака натрия, который представляет собой натриевую соль 2,6-дихлоранилинофенилуксусной кислоты) ограничено вследствие их токсичности в отношении желудочно-кишечного тракта, что в настоящее время связывают с ингибированием изоформы СОХ-1 циклооксигеназы. Установлено, что избирательное ингибирование индуцибельной изоформы СОХ-2 in vivo обусловливает противоспалительное и антиязвенное действие (Proc. Natl. Acad. Sci. (USA) 1994, 91: 3228-3232).
В настоящем изобретении предложены новые 5-алкилзамещенные 2- ариламинофенилуксусные кислоты и их производные, которые, как неожиданно было установлено при создании изобретения, ингибируют СОХ-2, но при этом не оказывают заметного ингибирующего действия в отношении СОХ-1. Таким образом, изобретение относится к новым нестероидным противовоспалительным агентам, которые, как неожиданно было установлено, лишены нежелательных побочных действий, обычно присущих классическим нестероидным противовоспалительным агентам, таким как побочные действия в отношении желудочно-кишечного тракта и почек.
Изобретение относится к соединениям формулы I
где
R обозначает метил или этил,
R1 обозначает хлор или фтор,
R2 обозначает водород или фтор,
R3 обозначает водород, фтор, хлор, метил, этил, метокси, этокси или гидрокси,
R4 обозначает водород или фтор,
R5 обозначает хлор, фтор, трифторметил или метил, к их фармацевтически приемлемым солям и
к их фармацевтически приемлемым пролекарствам в виде сложных эфиров.
В одном из вариантов в изобретении предлагаются, в частности, соединения формулы I, в которых R обозначает метил или этил, R1 обозначает хлор или в которых фтор, R2 обозначает водород, R3 обозначает водород, фтор, хлор, метил или гидрокси, R4 обозначает водород и R5 обозначает хлор, фтор или метил, их фармацевтически приемлемые соли и их фармацевтически приемлемые пролекарства в виде сложных эфиров.
В предпочтительном варианте осуществления изобретения предлагаются соединения формулы I, в которых R обозначает метил или этил, R1 обозначает фтор, R2 обозначает водород, R3 обозначает водород, фтор или гидрокси, R4 обозначает водород и R5 обозначает хлор, их фармацевтически приемлемые соли и их фармацевтически приемлемые пролекарства в виде сложных эфиров.
В другом предпочтительном варианте осуществления изобретения предлагаются соединения формулы I, в которых R обозначает этил или метил, R1 обозначает фтор, R2 обозначает водород или фтор, R3 обозначает водород, фтор, этокси или гидрокси, R4 обозначает водород или фтор и R5 обозначает хлор, фтор или метил, их фармацевтически приемлемые соли и их фармацевтически приемлемые пролекарства в виде сложных эфиров.
Также предпочтительными являются соединения формулы I, в которых R обозначает метил или этил, R1 обозначает фтор, R2-R4 обозначают водород или фтор и R5 обозначает хлор или фтор, их фармацевтически приемлемые соли и их фармацевтически приемлемые пролекарства в виде сложных эфиров.
В еще одном предпочтительном варианте осуществления изобретения предлагаются соединения формулы I, в которых R обозначает метил или этил, R1 обозначает фтор, R2 обозначает фтор, R3 обозначает водород, этокси или гидрокси, R4 обозначает фтор и R5 обозначает фтор, их фармацевтически приемлемые соли и их фармацевтически приемлемые пролекарства в виде сложных эфиров.
В еще одном предпочтительном варианте осуществления изобретения предлагаются соединения формулы I, в которых R обозначает метил, R1 обозначает фтор, R2 обозначает водород, R3 обозначает водород или фтор, R4 обозначает водород и R5 обозначает хлор, их фармацевтически приемлемые соли и их фармацевтически приемлемые пролекарства в виде сложных эфиров.
Особенно предпочтительными являются следующие соединения формулы I:
(а) в которых R обозначает метил, R1 обозначает фтор, R2 обозначает водород, R3 обозначает водород, R4 обозначает водород и R5 обозначает хлор, их фармацевтически приемлемые соли и их фармацевтически приемлемые пролекарства в виде сложных эфиров,
(б) в которых R обозначает метил, R1 обозначает фтор, R2 обозначает водород, R3 обозначает фтор, R4 обозначает водород и R5 обозначает хлор, их фармацевтически приемлемые соли и их фармацевтически приемлемые пролекарства в виде сложных эфиров,
(в) в которых R обозначает этил, R1 обозначает фтор, R2 обозначает фтор, R3 обозначает водород, R4 обозначает фтор и R5 обозначает фтор, их фармацевтически приемлемые соли и их фармацевтически приемлемые пролекарства в виде сложных эфиров,
(г) в которых R обозначает этил, R1 обозначает хлор, R2 обозначает водород, R3 обозначает хлор, R4 обозначает водород и R5 обозначает метил, их фармацевтически приемлемые соли и их фармацевтически приемлемые пролекарства в виде сложных эфиров.
В контексте настоящего описания основные понятия имеют следующие значения.
"Фармацевтически приемлемые пролекарства в виде сложных эфиров обозначают производные в виде сложных эфиров", которые обладают способностью превращаться путем сольволиза или в физиологических условиях в свободные карбоновые кислоты формулы I. Такие сложные эфиры представляют собой (низш. )алкиловые эфиры (такие как метиловый или этиловый эфиры), карбокси-(низш. )алкиловые эфиры, такие как карбоксиметиловый эфир, нитроокси-(низш. )алкиловые эфиры (такие как 4-нитроксибутиловый эфир) и т.п. Предпочтительными являются соединения формулы Iа
где R и R1-R5 имеют значения, указанные выше для соединений формулы I, и их фармацевтически приемлемые соли.
Фармацевтически приемлемые соли представляют собой соли металлов, такие как соли щелочных металлов, например соли натрия, калия, магния или кальция, а также соли аммония, которые образуются, например, с аммиаком и моно- или диалкиламинами, такие как соли диэтиламмония, и с аминокислотами, такие как соли аргинина и гистидина.
(Низш. )алкильная группа содержит до 7 атомов углерода, предпочтительно 1-4 атома углерода и представляет собой, например, метил, этил, пропил или бутил, и она может быть с прямой или разветвленной цепью.
Соединения по настоящему изобретению наиболее эффективны в качестве избирательных ингибиторов циклооксигеназы СОХ-2 или могут быть превращены в результате метаболизма в соединения, которые наиболее эффективны в качестве избирательных ингибиторов циклооксигеназы СОХ-2.
Таким образом, они пригодны для лечения у млекопитающих нарушений, связанных с циклооксигеназой, включая воспаление, повышенную температуру, боль, остеоартрит, ревматоидный артрит, связанную с мигренью головную боль, нейродегенеративные заболевания (такие как рассеянный склероз), болезнь Альцгеймера, остеопороз, астму, волчанку и псориаз.
Соединения по изобретению также пригодны для лечения неоплазии, особенно неоплазии, при которой продуцируются простагландины или происходит экспрессия циклооксигеназ, включая как доброкачественные, так и злокачественные опухоли, новообразования и полипы. Соединения по изобретению могут применяться для лечения любой неоплазии, например такой, которая указана в международной заявке WO 98/16222, опубликованной 23 апреля 1998 г., в частности неоплазии, образовавшийся из эпителиальных клеток. В частности, соединения по изобретению пригодны для лечения рака печени, мочевого пузыря, панкреатической железы, яичника, предстательной железы, шейки матки, легкого и молочной железы, главным образом рака желудочно-кишечного тракта, например рака толстой кишки, и рака кожи, например рака и меланомы клеток простого сквамозного эпителия или базальных клеток.
Под термином "лечение" в контексте настоящего описания следует понимать как терапевтические, так и профилактические способы лечения, например в отношении лечения неоплазии - терапию, способствующую предупреждению начала клинического или доклинического проявления неоплазии или способствующую предупреждению начала развития злокачественных клеток либо прекращению или реверсии развития предраковых клеток в раковые, а также предупреждению или ингибированию развития неоплазии или метастазов. В этом контексте следует, в частности, отметить, что под объем настоящего изобретения подпадает применение соединений по изобретению для ингибирования или предупреждения развития рака кожи, например карциномы клеток простого сквамозного эпителия или базальных клеток, вызванной УФ облучением, например, в результате постоянного облучения солнечным светом.
Соединения по изобретению обладают активностью при указанных выше показаниях и при этом практически лишены нежелательного побочного действия, такого как образование язв желудочно-кишечного тракта, что характерно для обычных ингибиторов циклооксигеназы.
Соединения по изобретению также обладают способностью абсорбировать УФ излучение и пригодны для блокирования или абсорбции УФ излучения, например для лечения или предупреждения солнечного ожога, например в виде предохраняющих от солнечного ожога композиций.
Соединения по изобретению также могут применяться при глазных болезнях, что предусматривает лечение глазных болезней, в частности глазных воспалительных болезней, глазной боли, включая боль, связанную с хирургией глаза, такой как операции по поводу PRK или катаракты, глазной аллергии, фотофобии различной этиологии, повышенного внутриглазного давления при глаукоме), за счет ингибирования продуцирования протеина, связанного с реакцией трабекулярной сети на глюкокортикоиды (TIGR), и болезни, связанной с сухостью глаз.
Вышеуказанные свойства могут быть продемонстрированы с помощью опытов in vitro и in vivo преимущественно с использованием млекопитающих, например крыс, мышей, собак, обезьян и выделенных из них клеток или препаратов ферментов. Эти соединения могут применяться in vitro в форме растворов, например водных растворов, и in vivo главным образом орально, местно или парентерально, например внутривенно. Применяемая in vitro доза может варьироваться в диапазоне молярных концентраций примерно от 10-5 до 10-9. Применяемая in vivo доза в зависимости от пути введения может варьироваться в диапазоне примерно от 1 до 100 мг/кг.
Ингибирование циклооксигеназы определяют in vitro с помощью опытов на клетках по ингибированию как циклооксигеназы-1, так и циклооксигеназы-2.
Опыты на клетках по выявлению активности ингибиторов циклооксигеназы основаны на том факте, что фермент циклооксигеназа (простагландин-Н-синтаза) катализирует скорость решающей стадии в пути синтеза простагландина из арахидоновой кислоты. В этой реакции участвуют два фермента: СОХ-1, представляющий собой конститутивную форму фермента, и СОХ-2, индуцирующийся в ответ на различные факторы роста и цитокины. Были выявлены клеточные линии, которые экспрессируют одну форму фермента, а именно, линия фибробластов кожи человека, у которой IL-1 (интерлейкин-1) может индуцировать синтез СОХ-2, и линия эпителиальных клеток почки 293, которая была стабильно трансфектирована с целью конститутивной экспрессии СОХ-1. Обе изоформы ответственны за метаболическое превращение арахидоновой кислоты в стабильный метаболит простагландин Е2. Арахидоновая кислота может быть добавлена экзогенно с целью увеличить продуцирование до легко выявляемых уровней. Уровни простагландина Е2 во внеклеточной среде определяют с помощью радиоиммунного анализа путем оценки активности фермента. С целью определения избирательной активности соединения сравнивают относительные активности каждой изоформы.
Ингибирование циклооксигеназы-1 (СОХ-1) и циклооксигеназы-2 (СОХ-2) in vitro определяют с помощью опытов на клетках с целью определения активности in vitro и избирательности в отношении ингибирования СОХ-2 с помощью радиоиммунного анализа простагландина Е2. В опытах используют клетки, представляющие собой первичные фибробласты человека, индуцированные интерлейкином-1 с целью продуцирования СОХ-2, и линию эпителиальных клеток 293 почки человека, стабильно трансфектированную с целью конститутивной экспрессии СОХ-1. Клетки высевают в лунки планшетов, в которых проводят анализ. Фибробласты стимулируют к синтезу СОХ-2 обработкой в течение ночи IL-1, при этом клетки линии 293 не нуждаются в индуцировании. Обе клеточные линии предварительно обрабатывают в течение 15 мин при 37oС соединениями с различной степенью разведения, а затем добавляют 40 мкМ арахидоновой кислоты в качестве экзогенного субстрата с целью продуцирования простагландина Е2 (PGE2), количество которого оценивают в надосадочной жидкости с помощью радиоиммунного анализа. Для определения значений IC50 соединения тестируют в 5 концентрациях, используя каждый раз по 4 повторности (наиболее высокая концентрация 30 мкМ), для каждой концентрации рассчитывают среднее значение ингибирования PGE2 (по сравнению с клетками, не обработанными соединением), для всех экспериментов строят кривую зависимости среднего % ингибирования от десятичного логарифма (log) концентрации соединения и рассчитывают общее значение IC50, используя аппроксимацию 4-параметрической логарифмической зависимостью.
Как правило, значения IС50 соединений формулы I, определенные в опыте по ингибированию СОХ-2, близки примерно к 0,005 мкМ, в то время как значения IC50, определенные в опыте по ингибированию СОХ-1, превышают 30 мкМ.
У репрезентативных соединений из примеров 6, 9 и 18 значения IC50 в отношении ингибирования СОХ-2 составляют примерно 0,13, 0,25 и 0,007 мкМ соответственно, при этом они не вызывают никакого заметного ингибирования СОХ-1 в концентрации 30 мкМ.
Ингибирование продуцирования простагландина Е2 с помощью СОХ-2 может быть определено in vivo с использованием в качестве модели крыс, которым в подкожный воздушный карман вводят липополисахарид (ЛПС) (см. "Advances in Inflammation Research", Raven Press, 1986 и J. Med. Chem. 396, 1846 (1996)).
Самок крыс линии Lewis подвергают анестезии и затем создают у них спинные воздушные карманы путем подкожной инъекции 10 мл воздуха через стерильный, снабженный фильтром шприц размером 0,45 мкм. Через 24 ч после этой процедуры в воздушные карманы инъецируют ЛПС (8 мкг/карман), суспендированный в стерильном забуференном фосфатом физиологическом растворе. Исследуемые соединения суспендируют в обогащенном кукурузном крахмале и вводят через желудочный зонд за 1 ч до инъекции ЛПС. Содержимое карманов собирают через 3 ч после инъекции ЛПС и путем ферментативного иммунного анализа оценивают уровни PGE2, присутствующие в жидкостях карманов. Значения ЕD50 в отношении ингибирования образования PGE2 рассчитывают линейной регрессией по методу наименьших квадратов. У репрезентативных соединений из примеров 6, 9, 18 и 24 значения ED50 находятся в диапазоне от примерно 0,2 мг/кг при введении перорально до примерно 0,6 мг/кг при введении перорально.
Ингибирование in vivo продуцируемого под воздействием СОХ-1 тромбоксана В2 (ТХВ2) может быть оценено ех vivo в сыворотке крыс после орального введения соединения.
В целом эта методика состоит в том, что крысам не дают корм в течение ночи, вводят через желудочный зонд соединение в носителе, представляющем собой обогащенный кукурузный крахмал, и через некоторый промежуток времени, составляющий от 30 мин до 8 ч, умерщвляют введением углекислого газа путем ингаляции. Кровь собирают путем сердечной пункции в пробирки без антикоагулянта, дают образоваться сгустку и отделяют сыворотку путем центрифугирования. Сыворотку хранят в замороженном виде до последующего определения уровня тромбоксана В2 радиоиммунным анализом. В каждом эксперименте используют следующие группы (5-6 крыс на группу): контрольную группу, обработанную носителем, и группы, обработанные либо разными дозами тестируемого соединения, либо обработанные в различные моменты времени. Данные, полученные для тромбоксана В2, выражают в виде процента относительно уровней, обнаруженных в контрольной группе, обработанной только носителем. Репрезентативные соединения I(d), l(g), 3(a) и 6(а) вызывают менее чем 50%-ное ингибирование продуцирования тромбоксана В2 в сыворотке при введении оральной дозы, которая в 50-150 раз выше значения ED50, необходимого для ингибирования COX-2 in vivo.
Противовоспалительную активность определяют в опыте, в котором у крыс вызывают отек лапки с помощью каррагинана.
Крысам линии Sprague Dawley (весом 200-225 г) не дают корм в течение ночи, а затем вводят орально соединение, суспендированное в растворе обогащенного кукурузного крахмала. Через 1 ч в подподошвенную область левой задней лапы инъецируют 0,1 мл 1%-ного каррагинана в физиологическом растворе, что вызывает воспалительную реакцию. Через 3 ч после введения каррагинана крыс безболезненно умерщвляют и отрезают обе задние лапы по линии начала волосяной части лапы и взвешивают на электронных весах. Массу отека воспаленной лапы определяют вычитанием веса невоспаленной лапы (правой) из веса воспаленной лапы (левой). Процент ингибирования соединением определяют для каждого животного в виде процента увеличения веса лапы по сравнению со средним значением в контроле. Значения ЕD30 определяют для каждой кривой зависимости реакции от дозы, используя для аппроксимации кривой следующую формулу:
100/1+тангенс угла наклона кривой (концентрация лекарства/ЕD50).
Значения ЕD30 рассчитывают как средние значения ЕD30, полученные из независимых опытов по определению зависимости реакции от дозы.
Анализ переносимости желудком используют для оценки образования язв у крыс через 4 ч после орального введения тестируемого соединения. Этот опыт проводят следующим образом.
Крысам не дают корм в течение ночи, вводят через желудочный зонд соединение в носителе, представляющем собой обогащенный кукурузный крахмал, и через 4 ч умерщвляют введением углекислого газа путем ингаляции. Удаляют желудки и подсчитывают и оценивают общие повреждения желудка, получая общую длину повреждения на особь. В каждом эксперименте используют следующие группы (5-6 крыс на группу): контрольную группу, обработанную носителем, группы, обработанные тестируемым соединением, и группы, обработанные диклофенаком в качестве эталонного соединения.
Данные рассчитывают в виде среднего количества язв в группе, средней длины язв (мм) в группе и индекса изъязвления (ИИ).
ИИ = средняя длина язв в группе х встречаемость язв, где встречаемость язв представляет собой долю животных в группе с повреждениями (100%-ная встречаемость равна 1).
Репрезентативные соединения из примеров 6, 9, 18 и 24 практически не оказывают какого-либо ульцерогенного воздействия на желудок в дозе 100 мг/кг при введении перорально.
Переносимость кишечником может быть определена путем оценки воздействия на проницаемость кишечника. Отсутствие возрастания проницаемости свидетельствует о переносимости кишечником.
Использованный для этой цели метод представляет собой модификацию метода, описанного Davis и др., Pharm. Res. 1994, 11: 1652-1656, и основан на том факте, что экскреция введенной орально 51Сr-ЭДТК, маркера проницаемости тонкого кишечника, увеличивается под воздействием НСПВЛ. Группам крыс (≥12 особей/группу) вводят однократную оральную дозу тестируемого соединения или носителя путем зондирования желудка. Немедленно после обработки соединением каждой крысе путем зондирования желудка вводят 51Сr-ЭДТК (5 мкКи/особь). Крыс помещают в отдельные клетки для измерения обмена веществ и дают пищу и воду по желанию. Мочу собирают в течение 24 ч. Через 24 ч после введения Cr-ЭДТК крыс умерщвляют. Для количественной оценки воздействия соединения на проницаемость кишечника измеренное количество 51 Сr-ЭДТК, выведенной с мочой, у обработанных соединением крыс сравнивают с количеством 51Сr-ЭДТК, выведенной с мочой, у крыс, обработанных носителем. Относительную проницаемость определяют, рассчитывая активность, присутствующую в каждом образце мочи, в виде процента от введенной дозы после коррекции на фоновый уровень излучения.
Репрезентативные соединения из примеров 6, 9, 18 и 24 не оказывают воздействия или оказывают лишь незначительное воздействие на проницаемость кишечника в дозе 30 мг/кг при введении перорально.
Аналгезирующее действие соединений по изобретению определяют, используя хорошо известный тест Рендалла-Селитто (Randall-Selitto). Согласно тесту Рендалла-Селитто по определению давления на лапу антиноцицепцию (аналгезирующее действие) в воспаленной ткани оценивают путем сравнения порогового давления на воспаленную лапу крысы после орального введения тестируемого лекарства и порогового давления на воспаленную лапу крыс, которым вводят орально носитель, т.е. кукурузный крахмал.
Группам из 10 самцов крыс линии Wistar весом 40-50 г не дают корм в течение ночи до опыта. Гипералгезию индуцируют путем инъекции 0,1 мл 20%-ной суспензии пивных дрожжей с помощью иглы номер 26 в подподошвенную область правой задней лапы. В левую лапу инъекцию не делают и ее используют в качестве контрольной лапы для определения гипералгезии. Носитель (обогащенная 3%-ная суспензия кукурузного крахмала) в дозе 10 мл/кг, эталонное соединение (диклофенак используют в каждом эксперименте в такой же дозе, что и тестируемые соединения) и тестируемые соединения в различных дозах, суспендированные в носителе, вводят в дозе 10 мл/кг орально через 2 ч после инъекции дрожжей. Пороговое давление, при котором происходит отдергивание лапы, оценивают количественно с помощью аналгезиметра типа Basile через 1 ч после орального введения тестируемых соединений.
Ноцицептивный порог определяют как силу в граммах, при которой крыса отдергивает лапу или издает звуки. И издание звуков, и отдергивание лапы считаются ответной реакцией.
Данные анализируют, сравнивая средний порог чувствительности к боли в обработанной носителем, т.е. кукурузным крахмалом, группе, в которую входят как крысы с воспаленными лапами, так и с невоспаленными лапами, с порогом чувствительности отдельных крыс, обработанных лекарством. Отдельных крыс в группах, обработанных лекарством, и группу, представляющую собой положительный контроль (обработка диклофенаком), называют "реактантами", если индивидуальный порог чувствительности к боли в каждой лапе превышает среднее значение порога в контрольной группе на два стандартных отклонения от среднего значения. Средние значения порогов чувствительности к боли воспаленной лапы в контрольной группе сравнивают с индивидуальными порогами чувствительности к боли воспаленной лапы в группе, обработанной тестируемым лекарством. Среднее значение порогового давления в контрольной группе с невоспаленными лапами сравнивают с индивидуальными значениями порогового давления в тестируемых группах. Результаты выражают в виде количества реактантов в каждой тестируемой группе (n=10) с воспаленными и невоспаленными лапами. Проценты рассчитывают делением количества реактантов на общее количество крыс, взятых для оценки каждого соединения.
Все репрезентативные соединения из примеров 6, 9, 18 и 24 увеличивают порог чувствительности к боли в воспаленной лапе при введении орально в дозе 10 мг/кг. Эти соединения избирательного увеличивают порог чувствительности к боли в воспаленной лапе и не увеличивают порог чувствительности к боли в невоспаленной лапе, что свидетельствует об их периферическом механизме действия.
Антиартритное действие соединений по изобретению может быть выявлено с помощью хорошо известного теста на крысах с индуцированным адъювантом хроническим артритом. Офтальмическое действие может быть продемонстрировано с использованием методов, хорошо известных в данной области.
Соединения формулы I могут быть получены, например,
(а) сочетанием соединения формулы II или На
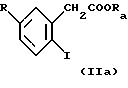
где R имеет указанные выше значения, Ra обозначает (низш.)алкил, предпочтительно изопропил и R6 и R7 обозначают (низш.)алкил или R6 и R7 вместе с атомом азота обозначают пиперидино, пирролидино или морфолино, с соединением формулы III
где R1, R2, R3, R4 и R5 имеют указанные выше значения, в присутствии меди и йодида меди с получением соединения формулы IV или IVa

и гидролизом образовавшегося соединения формулы IV или IVa с получением соединения формулы I, или
(б) для соединений, в которых R обозначает этил, конденсацией соединения формулы V
где R1-R7 имеют указанные выше значения, с реакционноспособным функционально активным производным уксусной кислоты, таким как ацетилхлорид, по реакции ацилирования Фриделя-Крафтса с получением соединения формулы VI
где R1-R7 имеют указанные выше значения, которое в свою очередь подвергают гидрогенолизу, а затем гидролизуют с получением соединения формулы I, в котором R обозначает этил, или
(в) гидролизом лактама формулы VII
где R и R1-R5 имеют указанные выше значения, в присутствии сильного основания и при необходимости осуществлением в вышеуказанных процессах временной защиты любых препятствующих реакции реакционноспособных групп с последующим выделением образовавшегося соединения по изобретению и при необходимости превращением любого образовавшегося соединения в другое соединение по изобретению и/или при необходимости превращением свободной карбоновой кислоты по изобретению в ее фармацевтически приемлемое производное в виде сложного эфира и/или при необходимости превращением образовавшейся свободной кислоты в соль или образовавшейся соли в свободную кислоту или в другую соль.
В исходных и в промежуточных продуктах, которые подвергают превращению в соединения по изобретению приведенным в настоящем описании способом, присутствующие функционально активные группы, такие как амино-, гидрокси- и карбоксильные группы, необязательно защищают общепринятыми защитными группами, известными в области препаративной органической химии. Защищенные гидрокиси-, амино- и карбоксильные группы представляют собой группы, которые могут быть превращены в мягких условиях в свободные амино-, гидрокси- и карбоксильные группы без вступления в другие нежелательные побочные реакции. Например, гидроксизащитные группы предпочтительно представляют собой бензильные или замещенные бензильные группы.
Получение соединений формулы IV согласно варианту (а) осуществляют в условиях модифицированной реакции конденсации Ульмана с получением диариламинов, например, в присутствии порошкообразной меди и йодида меди (I) и карбоната калия, в инертном растворителе с высокой температурой кипения, таком как нитробензол, толуол, ксилол или N-метилпирролидон, при повышенной температуре, например при температуре в диапазоне 100-200oС, предпочтительно при температуре дефлегмации, согласно общей методике, описанной у F. Nohara, Chem. Abstr. 94. 15402 (1951), и у Moser и др., J. Med. Chem. 33, 2358 (1990).
Промежуточные продукты формулы IV, в которых R1 или R5 обозначает метил или этил, могут быть получены из промежуточных продуктов формулы IV, в которых R1 или R5 обозначают бром, взаимодействием тетраметилолова или тетраэтилолова в условиях реакции Хека, т.е. в присутствии соли палладия (такой как Pd(OAc)2 или Pd(Cl2), триарилфосфина (такого как три(ортотолил)фосфина) и основания (такого как триэтиламин, ацетат натрия) в полярном растворителе, таком как диметилформамид.
Гидролиз образовавшихся орто-анилинофенилацетамидов формулы IV осуществляют в водном гидроксиде щелочного металла, например в 6н. NaOH, в присутствии спирта (например этанола, пропанола, бутанола) при повышенной температуре, такой как температура дефлегмации реакционной смеси.
Гидролиз сложных эфиров формулы IVa осуществляют по известным в данной области методам, например в щелочных условиях, как описано выше для соединений формулы IV, или в альтернативном варианте в кислых условиях, например с использованием метансульфоновой кислоты.
Исходные продукты формулы II или IIа обычно являются известными или могут быть получены по методам, известным в данной области, например по приведенным у F. Nohara в описании к заявке JP 78/96434 (1978).
Так, например, 5-метил- или 5-этилантраниловую кислоту превращают и производные орто-диазония с последующей обработкой йодидом щелочного металла в кислоте (например, в сульфоновой кислоте) с получением 5-алкил-2-йодбензойной кислоты. Путем восстановления с получением соответствующего бензилового спирта (например, в присутствии диборана), превращения этого спирта сначала в бромид, а затем в нитрил, гидролиза этого нитрила с получением уксусной кислоты и ее превращения в N,N-диалкиламид согласно известным в данной области методам получают исходный продукт формулы II. В альтернативном варианте исходные продукты формулы II, в которых R обозначает этил, могут быть получены по реакции Фриделя-Крафтса ацилированием оксиндола, например ацетилхлоридом, в присутствии хлорида алюминия, восстановлением образовавшегося кетона путем, например каталитического гидрогенолиза, с последующим гидролитическим расщеплением образовавшегося 5-этилоксиндола с получением 5-этил-2-аминофенилуксусной кислоты. Диазотизацией в присутствии, например, йодида калия, получают 5-этил-2-йодфенилуксусную кислоту, которую превращают в амид формулы II. Эфиры формулы IIа получают из соответствующих кислот согласно известным в данной области методам получения сложных эфиров.
Анилины формулы III либо являются известными в данной области, либо их получают по методам, хорошо известным в данной области, либо по методам, приведенным в настоящем описании.
Получение 5-этилзамещенных соединений согласно варианту (б) осуществляют в условиях реакции ацилирования Фриделя-Крафтса, например в присутствии хлорида алюминия в инертном растворителе, таком как 1,2-дихлорэтан, с последующим гидрогенолизом, например с использованием в качестве катализатора палладия на древесном угле, предпочтительно с использованием в качестве растворителя уксусной кислоты, при комнатной температуре и при давлении примерно 3 атм.
Исходные продукты формулы V обычно получают согласно описанному выше варианту (а), но используя в качестве исходного продукта амид формулы II, в котором R обозначает водород, например согласно методу, описанному в J. Med. Chem. 33, 2358(1990).
Получать соединения по изобретению согласно способу (в) можно в условиях, хорошо известных в данной области для гидролитического расщепления лактамов, предпочтительно с использованием сильного водного основания, такого как водный гидроксид натрия, необязательно в органическом смешивающемся с водой растворителе, таком как метанол, при повышенной температуре в диапазоне примерно от 50 до 100o С, как это в общем виде представлено в патенте US 3558690.
Исходные продукты, представляющие собой оксиндолы, получают N-ацилированием диариламина формулы VIII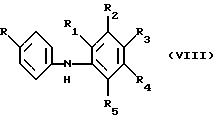
где R и R1-R5 имеют указанные выше значения, с галоацетилхлоридом, предпочтительно с хлорацетилхлоридом, предпочтительно при повышенной температуре, например близкой к 100oС, с получением соединения формулы IX
где R и R1-R5 имеют указанные выше значения. Циклизацию соединения формулы IX осуществляют в условиях реакции алкилирования Фриделя-Крафтса в инертном растворителе, таком как дихлорбензол, в присутствии катализаторов Фриделя-Крафтса, например хлорида алюминия и дихлорида этилалюминя, при повышенной температуре, например при 120-175oС.
Диариламины формулы VIII могут быть получены путем реакции конденсации Ульмана согласно приведенному в настоящем описании способу или с помощью других методов, известных в данной области, например по реакции сочетания Бухвальда.
Так, например, диариламины формулы VIII, в которых R1, R2, R4 и R5 обозначают фтор, a R3 обозначает водород, могут быть получены взаимодействием соответствующего анилина (4-этил- или 4-метиланилина) с пентафторбензолом в присутствии сильного основания, такого как амид лития или н-бутиллития, как это в общем виде представлено в J. Fluorine Chemistry 5, 323 (1975).
Эфиры карбоновых кислот формулы I получают конденсацией карбоновой кислоты в форме соли или в присутствии основания с галогенидом (бромидом или хлоридом), соответствующим участвующему в реакции этерификации спирту (такому как бензилхлорацетат), по методам, хорошо известным в данной области, например в полярном растворителе, таком как диметилформамид, и при необходимости с дополнительной модификацией образовавшегося продукта.
Так, например, если продукт этерификации сам является сложным эфиром, он может быть превращен в карбоновую кислоту, например гидрогенолизом образовавшегося бензилового эфира. Если продукт этерификации сам является галогенидом, то он может быть, например, превращен в нитроорксипроизводное взаимодействием, например, с нитратом серебра.
Например, соединения формулы Iа предпочтительно получают конденсацией соли карбоновой кислоты приведенной выше формулы I с соединением формулы
Х-СН2СООRa,
где Х обозначает уходящую группу, a Ra обозначает карбоксизащитную группу, с получением соединения формулы I с защищенной карбоксигруппой с последующим удалением защитной группы Ra.
Этерификацию можно проводить в условиях, хорошо известных в данной области, например в полярном растворителе, таком как диметилформамид, при температуре в диапазоне от комнатной до примерно 100oС, предпочтительно в диапазоне от 40 до 60oС.
Соль кислоты формулы I предпочтительно представляет собой соль щелочного металла, например соль натрия, которая может быть получена in situ.
Уходящая группа Х предпочтительно обозначает галоген, например хлор или бром, или (низш.)алкилсульфонилокси-, например метансульфонилоксигруппу.
Карбоксизащитная группа Ra предпочтительно обозначает бензил.
Образовавшиеся бензиловые эфиры могут быть превращены в свободные кислоты формулы Iа предпочтительно гидрогенолизом водородом в присутствии в качестве катализатора, например, Pd/C, в уксусной кислоте при атмосферном давлении или гидрогенизацией по Парру в диапазоне температур от комнатной до примерно 50oС.
Изобретение включает новые исходные продукты и способы их получения.
И, наконец, соединения по изобретению либо получают в свободной форме, либо в форме их соли, если в них присутствуют солеобразующие группы.
Кислотные соединения по изобретению могут быть превращены в соли металлов с помощью фармацевтически приемлемых оснований, например водного гидроксида щелочного металла, предпочтительно в присутствии растворителя, представляющего собой простой эфир или спирт, такой как (низш.)алканол. Образовавшиеся соли могут быть превращены в свободные соединения обработкой кислотами. Эти или другие соли также могут использоваться для очистки полученных соединений. Аммонийные соли получают взаимодействием с пригодным амином, например диэтиламином, и т.п.
Соединения по изобретению, имеющие основные группы, могут быть превращены в кислотно-аддитивные соли, прежде всего в фармацевтически приемлемые соли. Их получают, например, с неорганическим кислотами, такими как минеральные кислоты, например серная кислота, фосфорная кислота или галогенводородными кислотами, или с органическими карбоновыми кислотами, такими как С1-С4 алканкарбоновые кислоты, которые, например, могут быть незамещенными или могут быть замещены галогеном, например уксусная кислота, такими как насыщенные или ненасыщенные дикарбоновые кислоты, например щавелевая, янтарная, малеиновая или фумаровая кислота, такими как гидроксикарбоновые кислоты, например гликолевая, молочная, яблочная, винная или лимонная кислота, такими как аминокислоты, например аспарагиновая или глутаминовая кислота, или с органическими сульфоновыми кислотами, такими как С1-С4 алкилсульфоновые кислоты (например, метансульфоновая кислота) или арилсульфоновые кислоты, которые могут быть незамещенными или могут быть замещены (например, галогеном). Предпочтительными являются соли, полученные с соляной кислотой, метансульфоновой кислотой и малеиновой кислотой.
В связи с тем, что свободные соединения и соединения в форме их солей тесно связаны друг с другом, то всякий раз, когда в данном контексте упоминается соединение, также подразумевается соответствующая соль, при условии, что это возможно или требуется в данных обстоятельствах.
Соединения, включая их соли, также могут быть получены в форме их гидратов или могут включать другие растворители, которые используются для кристаллизации.
Фармацевтические композиции по изобретению представляют собой композиции, пригодные для энтерального, такого как оральное или ректальное, трансдермального, местного и парентерального введения млекопитающим, включая человека, для ингибирования активности СОХ-2 и для лечения зависимых от СОХ-2 нарушений и включают эффективное количество фармацевтически активного соединения по изобретению индивидуально или в сочетании с одним либо несколькими фармацевтически приемлемыми носителями.
Более конкретно фармацевтические композиции включают эффективное ингибирующее циклооксигеназу-2 количество соединения по изобретению, у которого практически отсутствует ингибирующая активность в отношении циклооксигеназы-1 и связанные с ней побочные действия.
Фармакологически активные соединения по изобретению пригодны для изготовления фармацевтических композиций, включающих эффективное количество этого соединения в сочетании или в смеси с эксципиентами или носителями, пригодными либо для энтерального, либо для парентерального введения. Предпочтительными являются таблетки и желатиновые капсулы, содержащие действующее вещество в сочетании с а) разбавителями, например с лактозой, декстрозой, сахарозой, маннитом, сорбитом, целлюлозой и/или с глицином, б) замасливателями, например с двуокисью кремния, тальком, стеариновой кислотой, ее магниевой или кальциевой солью и/или с полиэтиленгликолем, а для таблеток также в сочетании с в) связующими агентами, например с алюмосиликатом магния, крахмальной пастой, желатином, трагакантом, метилцеллюлозой, натрийкарбоксиметилцеллюлозой и/или с поливинилпирролидоном, при необходимости с г) разрыхлителями, например с крахмалами, агаром, альгиновой кислотой или с ее натриевой солью или с шипучими смесями, и/или с д) абсорбентами, красителями, корригентами и подслащивающими веществами. Инъецируемые композиции предпочтительно представляют собой водные изотонические растворы или суспензии, а суппозитории предпочтительно приготавливают с использованием жирных эмульсий или суспензий. Эти композиции могут быть стерильными и/или содержать адъюванты, такие как консерванты, стабилизаторы, смачивающие или эмульгирующие агенты, вещества, усиливающие растворимость, соли для регулирования осмотического давления и/или буферы. Кроме того, они также могут содержать другие ценные с терапевтической точки зрения вещества. Эти композиции готовят согласно общепринятым методам смешения, гранулирования или нанесения покрытия соответственно, и они содержат примерно от 0,1 до 75%, предпочтительно примерно от 1 до 50% действующего вещества.
На таблетки известными в данной области методами может быть нанесено либо пленочное, либо энтеросолюбильное покрытие.
Приемлемые композиции для трансдермального применения включают эффективное количество соединения по изобретению в сочетании с носителем. Пригодные для этой цели носители включают абсорбирующиеся фармакологически приемлемые растворители, способствующие проникновению через кожу хозяина, например приспособления для трансдермального введения могут иметь форму бандажа, имеющего каркас, резервуар, содержащий соединение необязательно в смеси с носителями, необязательно контролирующий скорость барьер для введения соединения через кожу хозяина с контролируемой и предварительно заданной скоростью в течение продолжительного периода времени, и средства для закрепления устройства на коже.
Приемлемые композиции для местного нанесения, например на кожу и в глаза, включают водные растворы, суспензии, мази, кремы, гели или распыляемые композиции, например, предназначенные для введения с помощью аэрозоля или т. п. Такие системы для местного введения могут быть предназначены, например, для кожного нанесения, например, для лечения рака кожи, например для профилактического применения в солнцезащитных кремах, разбрызгиваемых лосьонах и т. п. В этой связи следует отметить, что соединения по изобретению, например соединения из приведенных ниже примеров 2 и 9, обладают способностью абсорбировать УФ-лучи в диапазоне длин волн от 290 до 320 нм, но при этом пропускают лучи с большей длиной волны, относящиеся к желто-красной области спектра. Таким образом, они особенно пригодны для приготовления местных композиций, в том числе косметических, таких как перечисленные выше, хорошо известные в данной области композиции. Они могут содержать солюбилизаторы, стабилизаторы, повышающие тоничность агенты, буферы и консерванты. Композиции, пригодные для местного нанесения, могут быть приготовлены, например, согласно методам, описанным в патенте US 4784808. Композиции для введения в глаза могут быть приготовлены, например, согласно методам, описанным в патентах US 4829088 и US 4960799.
Фармацевтические композиции содержат эффективное в качестве ингибитора СОХ-2 количество соединения по изобретению, как оно определено выше, либо индивидуально, либо в сочетании с другим терапевтическим агентом.
Приемлемые дополнительные действующие вещества, пригодные для лечения неоплазии, включают, например, любые противоопухолевые агенты или агенты, предназначенные для радиологической защиты, перечисленные в указанной выше публикации WO 98/16227, начиная со стр. 24, строка 19.
При использовании в сочетании с другим действующим веществом соединение по изобретению может вводиться либо одновременно, до или после другого действующего вещества, либо раздельно с использованием этого же или другого пути введения или вместе в одной и той же фармацевтической композиции.
Доза вводимого действующего вещества зависит от вида теплокровных животных (млекопитающих), веса тела, возраста, индивидуального состояния и от пути введения. Стандартная доза для орального введения млекопитающему весом примерно от 50 до 70 кг может находиться в диапазоне примерно от 5 до 500 мг действующего вещества.
Настоящее изобретение также относится к способам применения соединений по изобретению и их фармацевтически приемлемых солей или содержащих их фармацевтических композиций для ингибирования СОХ-2 или для лечения зависимых от СОХ-2 состояний, указанных в данном описании, например воспаления, боли, ревматоидного артрита, остеоартрита, глазных нарушений, в частности глазных воспалительных заболеваний, глаукомы болезни, связанной с сухостью глаза, у млекопитающих.
Кроме того, изобретение относится к применению соединений по изобретению для приготовления лекарственных средств, предназначенных для лечения связанных с СОХ-2 болезней и состояний.
В частности, настоящее изобретение относится к способу избирательного ингибирования активности циклооксигеназы-2 у млекопитающего практически без ингибирования активности циклооксигеназы-1, который включает введение млекопитающему, нуждающемуся в таком лечении, эффективного в качестве ингибитора циклооксигеназы-2 количества соединения по изобретению.
Более конкретно настоящее изобретение относится к способу лечения связанных с циклооксигеназой-2 нарушений у млекопитающих, практически не сопровождающемуся нежелательными побочными действиями, связанными с ингибированием активности циклооксигеназы-1, который включает введение млекопитающему, нуждающемуся в таком лечении, эффективного в качестве ингибитора циклооксигеназы-2 количества соединения по изобретению, у которого практически полностью отсутствует ингибирующая активность в отношении циклооксигеназы-1.
В частности, это относится к способу лечения ревматоидного артрита, остеоартрита, боли или воспаления у млекопитающих, при осуществлении которого не происходит нежелательного образования язв желудочно-кишечного тракта и который включает введение млекопитающему, нуждающемуся в таком лечении, соединения по изобретению, обладающего требуемой активностью.
Ниже изобретение проиллюстрировано на примерах, не ограничивающих его объем. Температуры указаны в градуса Цельсия. Если не указано иное, то все процессы выпаривания осуществляют при пониженном давлении, предпочтительно приблизительно при давлении от 15 до 100 мм рт.ст. (что соответствует 20-133 мбар). Строение конечных продуктов, промежуточных и исходных продуктов подтверждают стандартными аналитическими методами, например микроанализом и с помощью спектроскопических характеристик (например, МС, ИК, ЯМР). Используемые сокращения соответствуют принятым в данной области.
Примеры
Пример 1
(а) N, N-диметил-5-метил-2-(2',4'-дихлор-6'-метиланилино)фенилацета-мид (1,5 г, 4,3 ммоля) гидролизуют с помощью 6 н. NaOH (70 мл) в виде двухфазного раствора с н-ВuОН (40 мл) при нагревании с обратным холодильником в течение 14 ч. После охлаждения до комнатной температуры смесь сливают на лед (100 мл). Добавляют толуол (100 мл) и смесь переносят в делительную воронку. Значение рН водной фазы доводят до 1 с помощью 3 н. НСl. Органическую фазу отделяют и водную фазу повторно экстрагируют толуолом (100 мл). Объединенный органический раствор сушат (MgSO4) и концентрируют в глубоком вакууме (35-50 мбар) на роторном испарителе, не давая температуре смеси превысить 50oС. После кристаллизации из Et2O/гексана получают 5-метил-2-(2',4'-дихлор-6'-метиланилино)фенилуксусную кислоту в виде твердого вещества темного желтовато-коричневого цвета, tпл 137-141oС.
Исходный продукт, т. е. N,N-диметил-5-метил-2-(2',4'-дихлор-6'-метиланилино)фенилацетамид, получают следующим образом.
5-метил-2-йодбензойную кислоту (100 г, 0,38 моля) растворяют в ТГФ (350 мл) и охлаждают на ледяной бане. Добавляют по каплям комплекс боран-ТГФ (380 мл 1М в ТГФ, 0,38 моля). После завершения добавления реакционную смесь нагревают до комнатной температуры и перемешивают в течение 14 ч. Смесь переносят в большую колбу Эрленмейера, охлаждают на ледяной бане и осторожно прекращают реакцию, добавляя воду (250 мл). После выпаривания ТГФ на роторном испарителе получают белую суспензию, которую обрабатывают дополнительной порцией воды (1 л) и затем фильтруют и сушат в вакуумном эксикаторе над P2O5, получая 2-йод-5-метилбензиловый спирт в виде твердого вещества белого цвета, tпл 82-85oС.
Бензиловый спирт (99,8 г, 0,38 моля) растворяют в 48%-ной НВr (500 мл) и нагревают с обратным холодильником в течение 4 ч. Образовавшийся бензилбромид выделяют в виде твердого вещества желтого цвета, сливая охлажденную смесь на большой объем воды (1,5 л), с последующей фильтрацией. Бензилбромид (осторожно: слезоточивое вещество!) растворяют в EtOH (400 мл) и перемешивают при комнатной температуре. Растворяют цианид натрия (56 г, 1,14 моля) в минимальном количестве (~100 мл) воды и затем добавляют этанольный раствор бензилбромида. Реакционную смесь нагревают с обратным холодильником в течение 3 ч и затем охлаждают до комнатной температуры. Этанол удаляют на роторном испарителе и остаток промывают большим объемом (1 л) воды. Путем фильтрации выделяют 2'-йод-5'-метилфенилацетонитрил в виде твердого вещества белого цвета, tпл 77-79oС.
Нитрил (94,5 г, 0,37 моля) растворяют в EtOH (350 мл) и обрабатывают NaOH (29,4 г, 0,74 моля), растворенным в воде (200 мл). Реакционную смесь нагревают с обратным холодильником в течение 14 ч. После охлаждения до комнатной температуры этанол удаляют на роторном испарителе и добавляют 6 н. НСl до тех пор, пока значения рН не достигнет 1. Твердую 5-метил-2-йодфенилуксусную кислоту отфильтровывают и промывают водой (2•500 мл). После сушки над Р2О5 в вакуумном эксикаторе твердую 5-метил-2-йодфенилуксусную кислоту (tпл 112-114oС, 83 г, 0,30 моля) растворяют в CH2Cl2 (450 мл), содержащем несколько капель ДМФ. К раствору добавляют тионилхлорид (32 мл, 0,450 моля) и реакционную смесь нагревают с обратным холодильником в течение ночи. После охлаждения до комнатной температуры реакционную смесь разбавляют дополнительной порцией CH2Cl2 (500 мл) и промывают водой (2•250 мл), насыщенным раствором NаНСО3 (250 мл) и соляным раствором (250 мл). Раствор сушат (MgSO4) и концентрируют на роторном испарителе, получая 5-метил-2-йодфенилацетилхлорид в виде желтоватого масла.
К раствору 5-метил-2-йодфенилацетилхлорида в Et2O (500 мл), охлажденному на ледяной бане, добавляют по каплям диметиламин (200 мл 2М раствора в ТГФ). По завершении добавления добавляют EtOAc (350 мл) и раствор промывают водой (350 мл), соляным раствором (250 мл) и сушат (MgSO4). После упаривания на роторном испарителе и растирания со смесью Et2O/гексан (1:1) получают N,N-диметил-5-метил-2-йодфенилацетамид в виде твердого вещества светлого желтовато-коричневого цвета, tпл 47-49oС.
N,N-диметил-5-метил-2-йодфенилацетамид (3,5 г, 11,5 ммоля) и 2,4-дихлор-6-метиланилин (4,1 г, 23 ммоля) перемешивают в ксилолах (100 мл) с порошкообразной медью (0,18 г, 2,9 ммоля), йодидом меди (I) (0,55 г, 2,9 ммоля) и безводным карбонатом калия (1,6 г, 11,5 ммоля). Реакционную смесь нагревают с обратным холодильником в течение 48 ч. Еще слегка теплую (40oС) суспензию коричневого цвета фильтруют через подушку из целита, которую затем промывают толуолом (75 мл). Фильтрат упаривают на роторном испарителе и подвергают экспресс-хроматографии на силикагеле (Rf составляет 0,30 в 40%-ном EtOAc/гексане), получая N, N-диметил-5-метил-2-(2', 4'-дихлор-6'-метиланилино)фенилацетамид в виде беловатого кристаллического твердого вещества, tпл 119-124oС.
Аналогичным путем получают следующие соединения формулы I, в которых R имеет значения, указанные в таблице 1.
5-этил-2-йод-N,N-диметилфенилацетамид, который является исходным продуктом в примерах 12-17, получают следующим образом.
А1С13 (303 г, 2,27 моля) помещают в трехгорлую колбу, снабженную термометром и капельной воронкой. По каплям при перемешивании добавляют ДМФ (50 мл), при этом температура повышается до 60oС. Затем смесь охлаждают до 45oС и добавляют в виде трех порций оксиндол (33 г, 0,25 моля). Еще через 10 мин добавляют ацетилхлорид (36 мл, 0,5 моля). Затем смесь перемешивают еще в течение 30 мин при комнатной температуре. Смесь сливают на лед (3000 г). Это приводит к образованию твердого вещества, которое отфильтровывают, промывают сначала водой, а затем холодным метанолом (1000 мл), и далее сушат, получая 5-ацетилоксиндол. 5-Ацетилоксиндол (54 г, 308 ммолей), уксусную кислоту (400 мл) и палладий на угле (10%-ный, 5 г) объединяют и обрабатывают водородом в течение 14 ч при давлении 55 фунтов/кв.дюйм. Катализатор удаляют фильтрацией через подушку из целита, фильтрат концентрируют при пониженном давлении и остаток обрабатывают простым эфиром, получая 5- этилоксиндол.
5-этилоксиндол (~ 54 г, ~335 ммолей), этанол (750 мл), воду (150 мл) и гидроксид калия (65 г, 1,62 моля) объединяют и нагревают с обратным холодильником в течение 3 дней. Смеси дают охладиться и затем фильтруют через слой целита. Фильтрат концентрируют при пониженном давлении, добавляют воду и значение рН доводят до 6,5. Осадок отфильтровывают, промывают водой и сушат в течение ночи, получая 5-этил-2-аминофенилуксусную кислоту.
Смесь, содержащую воду (405 мл) и концентрированную НСl (48 мл), перемешивают и охлаждают до 0oС. Медленно добавляют 5-этил-2-аминофенилуксусную кислоту (53,7 г, 300 ммолей), поддерживая температуру на уровне 0-2oС. После этого добавления по каплям в течение 30 мин добавляют раствор нитрита натрия (22,2 г, 322 ммоля) в 60 мл воды, поддерживая температуру на уровне 0-2oС. Еще через 20 мин добавляют по каплям раствор йодида калия (48 г, 290 ммолей) в 18 мл концентрированной НСl и 130 мл воды, поддерживая температуру ниже 10oС. Реакционной смеси дают нагреться до комнатной температуры и затем нагревают с обратным холодильником в течение 2 ч. Смесь экстрагируют этилацетатом и простым эфиром (смесь 1:1, 4•300 мл), после чего органический слой сначала промывают 30%-ным водным раствором тиосульфита натрия, а затем раствором гидроксида натрия (0,1М) и после этого подкисляют до рН 6 и экстрагируют этилацетатом. Этот раствор промывают насыщенным соляным раствором, сушат (над сульфатом магния), фильтруют и растворитель удаляют при пониженном давлении. Этот остаток обрабатывают гексаном, получая 5-этил-2- йодфенилуксусную кислоту.
5-этил-2-йодфенилуксусную кислоту растворяют в метиленхлориде (400 мл) и добавляют ДМФ (1 мл). Затем по каплям в течение 20 мин добавляют тионилхлорид (21 мл, 300 молей). Смесь нагревают до температуры дефлегмации и выдерживают при этой температуре в течение 3,5 ч, а затем смесь охлаждают и добавляют ледяную воду (400 мл) и метиленхлорид (300 мл). Слои разделяют, органический слой промывают раствором бикарбоната натрия, насыщенным соляным раствором, сушат (над сульфатом магния) упаривают при пониженном давлении, получая 5-этил-2-йодфенилацетилхлорид.
Хлорангидрид (46 г, 150 ммолей) растворяют в простом эфире (500 мл) и перемешивают при -35oС. По каплям при -35oС добавляют диметиламин (250 мл 2М раствора в ТГФ, 500 ммолей) и смеси дают нагреться до комнатной температуры и затем перемешивают в течение 60 ч. Добавляют воду и этилацетат и слои разделяют. Органический слой промывают насыщенным соляным раствором и объединенные водные слои промывают простым эфиром. Затем объединенные органические слои сушат (над сульфатом магния) и растворитель удаляют при пониженном давлении. Добавляют гексан и получают N,N-диметил-5-этил-2-йодфенилацетамид в виде твердого вещества.
N, N-диметил-5-этил-2-фенилацетамид, который является исходным продуктом в примерах 18 и 19, получают следующим образом.
N, N-диметил-2-йодфенилацетамид (60 г, 0,208 моля), 2',3',5',6'- тетрафторанилин (100 г, 0,606 моля), порошкообразную медь (6,6 г, 0,104 ммоля), йодид меди (I) (19,8 г, 0,104 моля) и безводный карбонат калия (28,7 г, 0,208 моля) перемешивают в 1000 мл ксилолов. Реакционную смесь нагревают с обратным холодильником в течение 48 ч. Еще слегка теплую (40oС) суспензию коричневого цвета фильтруют через подушку из целита, которую в свою очередь промывают толуолом (250 мл). Фильтрат упаривают на роторном испарителе и затем подвергают экспресс-хроматографии на силикагеле (Rf составляет 0,25 в 30%-ном растворе EtOAc в гексане). После кристаллизации из пентана/Et2O получают N, N-диметил-2-(2',3',5',6'-тетрафторанилино)фенилацетамид, tпл 109-110oС.
В атмосфере инертного газа к суспензии хлорида алюминия (51,2 г, 0,385 моля), перемешанной в 1,2-дихлорэтане (750 мл), медленно добавляют ацетилхлорид (29,1 мл, 0,385 моля). После перемешивания при комнатной температуре в течение 1 ч получают раствор желтого цвета. Раствор охлаждают на ледяной бане и добавляют N,N-диметил-2-(2',3',5,6'-тетрафторанилино)фенилацетамид (40 г, 0,123 моля). Реакционной смеси дают нагреться до комнатной температуры и затем нагревают до 80oС в течение 0,5 ч. Реакционную смесь сливают на лед и экстрагируют EtOAc (2•750 мл). Органический экстракт промывают водой (750 мл), насыщенным раствором NaHCO3 (500 мл) и соляным раствором (500 мл). После упаривания на роторном испарителе и растирания с Et2O получают N,N-диметил-5-ацетил-2 (2',3',5',6'-тeтpaфтopaнилинo)фeнилaцeтaмид в виде твердого вещества белого цвета, tпл 112-114oС.
N, N-димeтил-5-aцeтил-2-(2', 3',5',6'-тетрафторанилино)фeнилaцeтaмид (30 г, 0,802 моля) растворяют в НОАс (150 мл) и гидрируют (55 фунтов/кв.дюйм) в присутствии 10%-ного Pd/C (1,5 г) как катализатора в течение 8 ч. Катализатор удаляют фильтрацией через целит и фильтрат сливают на воду (500 мл) и EtOAc (500 мл). Органический слой промывают водой (750 мл), нейтрализуют насыщенным раствором Na2CO3 (500 мл) и промывают соляным раствором (500 мл). После упаривания на роторном испарителе с последующим растиранием с гексаном получают N, N-диметил-5-этил-2-(2',3',5',6'-тетрафторанилино)фенилацетамид, tпл 105-106oС.
N,N-диметил-5-этил-2-(4'-хлор-2'-фтор-6'-метиланилино)фенилацетамид, который является исходным продуктом в примерах 21 и 22, получают следующим образом.
По реакции конденсации Ульмана N,N-диметил-5-этил-2-йодфенилацетамида с 2-бром-4-хлор-6-фторанилином аналогично примеру 1 получают N,N- димeтил-5-этил-2-(2'-бром-4'-xлop-6'-фтopaнилинo)фeнилaцeтaмид.
N, N-диметил-5-этил-2-(2'-бром-4'-хлор-6'-фторанилино)фенилацетамид (2,5 г, 6,0 ммолей) объединяют с ДМФ (10 мл), триэтиламином (10 мл), триорто-толилфосфином (0,5 г, 1,6 ммоля), тетраметилоловом (4 мл, 5,16 г, 28,9 ммоля) и ацетатом палладия (0,25 г, 1,1 ммоля) и смесь выдерживают в запечатанной пробирке в течение 3 дней при 95oС. Пробирке дают охладиться и осторожно открывают. К реакционной смеси добавляют воду и этилацетат и смесь разделяют. Органическую фракцию промывают разбавленным раствором NaCl (2•50 мл). Объединенные водные фракции затем промывают этилацетатом и далее объединенные органические фракции сушат (над сульфатом магния). Продукт абсорбируют небольшим количеством силикагеля и очищают экспресс- хроматографией (на двуокиси кремния, этилацетат/гексан в соотношении от 1:4 до 1:1), получая N,N-диметил-5-этил-2-(4'-хлор-2'-фтор-6' - метиланилино) фенилацетамид.
2-хлор-4-пивалоилокси-6-фторанилин, который является исходным продуктом в примере 23, получают следующим образом.
К охлажденной до 0oС смеси, содержащей 7,0 г (0,045 моля) 3-фтор-4- нитрофенола и 6,7 г (0,067 моля) триэтиламина в 20 мл метиленхлорида, по каплям добавляют 6,5 г (0,054 моля) пивалоилхлорида. Реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Реакцию прекращают добавлением воды и экстрагируют этилацетатом. Органический слой последовательно промывают 1 н. соляной кислотой, насыщенным водным раствором бикарбоната натрия и насыщенным соляным раствором, а затем сушат над сульфатом магния. После фильтрации и удаления растворителей получают 10,5 г неочищенного 2-фтор-4-пивалоилоксинитробензола, который растворяют в 200 мл абсолютного этанола. К раствору добавляют 0,9 г 5%-ного палладия на угле и затем смесь гидрируют при давлении водорода 30 фунтов/кв.дюйм в течение 2 ч. Катализатор отфильтровывают и растворитель удаляют, получая 2-фтор- 4 - пивалоилоксианилин.
Смесь, содержащую 7,3 г (0,035 моля) 2-фтор-4-пивалоилоксианилина и 5,1 г (0,038 моля) N-хлорсукцинимида в 50 мл фторбензола, нагревают с обратным холодильником в атмосфере азота в течение 2 ч. После охлаждения до комнатной температуры растворитель удаляют, добавляют воду и смесь экстрагируют этилацетатом. Органический слой промывают 1 н. раствором гидроксида натрия и насыщенным соляным раствором и сушат над сульфатом магния. После фильтрации и удаления растворителей получают остаток, который очищают хроматографией на силикагеле (20%-ный раствор этилацетата в гексане), получая 2-хлор-4-пивалоилокси-6-фторанилин.
Превращение 2-хлор-4-пивалоилокси-6-фторанилина в 5-метил-2-(2'-хлор-4'-гидрокси-6'-фторанилино)фенилуксусную кислоту проводят аналогично примеру 1, при этом пивалоильную группу удаляют гидролизом на последней стадии с помощью диметиламида, получая конечный продукт.
Пример 24
5-этил-2-(2', 3', 5', 6'-тетрафторанилино)фенилуксусную кислоту (1,0 г, 3,06 ммоля) в ТГФ (100 мл) обрабатывают 1н. раствором гидроксида натрия (3,06 мл, 3,06 ммоля) в течение 1 ч. Смесь концентрируют на роторном испарителе и затем сушат упариванием сначала с ТГФ (2•100 мл), а затем с бензолом (2•100 мл). Оставшуюся беловатую натриевую соль 5-этил-2-(2',3',5', 6'-тетрафторанилино)фенилуксусной кислоты сушат в течение ночи в глубоком вакууме. 5-Этил-2-(2', 3',5',6'-тетрафторанилино)фенилацетат натрия (0,5 г, 1,43 ммоля) и бензил-2-бромацетат (272 мкл, 1,72 ммоля) перемешивают при 50oС в диметилформамиде (50 мл) в течение 14 ч. Реакционную смесь охлаждают до комнатной температуры и распределяют между EtOAc (200 мл) и водой (200 мл). Органический слой вновь промывают водой (2•200 мл), соляным раствором (100 мл), сушат (MgSO4) и концентрируют на роторном испарителе. Неочищенный бензиловый эфир подвергают экспресс-хроматографии на силикагеле (10-15%-ный раствор EtOAc в гексане), получая бензилоксикарбонилметиловый эфир 5-этил-2-(2', 3', 5', 6'-тетрафторанилино)фенилуксусной кислоты в виде бесцветного масла. Масло растворяют в НОАс (20 мл) и гидрируют (55 фунтов/кв.дюйм) в присутствии 10%-ного Pd/C (0,1 г) как катализатора в течение 1 ч. Катализатор удаляют фильтрацией через целит и фильтрат сливают на воду (200 мл) и EtOAc (200 мл). Органический слой промывают водой (250 мл) и соляным раствором (100 мл). После упаривания на роторном испарителе и растирания с Et2O/гексаном получают эфир, т.е. карбоксиметил-5-этил-2-(2',3',5',6'-тетрафторанилино)фенилацетат с tпл 151-153oС формулы (1а), где R обозначает этил, R1, R2, R4, R5 обозначают фтор и R3 обозначает Н.
Пример 34
5-этил-2-(2', 3', 5', 6'-тетрафторанилино)фенилуксусную кислоту (1,0 г, 3,06 ммоля) в ТГФ (100 мл) обрабатывают 1 н. раствором гидроксида натрия (3,06 мл, 3,06 ммоля) в течение 1 ч. Смесь концентрируют на роторном испарителе и затем остаток обрабатывают и упаривают досуха сначала с ТГФ (2•100 мл), а затем с бензолом (2х100 мл). Оставшуюся беловатую натриевую соль 5-этил-2-(2', 3', 5',6'-тетрафторанилино)фенилуксусной кислоты сушат в течение ночи в глубоком вакууме. 5-этил-2-(2',3',5',6'-тетрафторанилино)фенилацетат натрия (2,0 г, 6,2 ммоля) растворяют в ДМФ (70 мл) и обрабатывают 1-бром-4-хлорбутаном (1,2 г, 6,9 ммоля) при комнатной температуре в течение ночи. Реакционную смесь концентрируют в глубоком вакууме (35-50 мбар) на роторном испарителе. Образовавшееся масло распределяют между водой (200 мл) и Et2O (200 мл). Органический слой промывают соляным раствором (100 мл), сушат (MgSO4) и концентрируют на роторном испарителе, получая хлорбутиловый эфир в виде масла светло-коричневого цвета. Хлорбутиловый эфир растворяют в СН3CN (100 мл) и обрабатывают нитратом серебра (8,7 г, 50 ммолей) при нагревании с обратным холодильником в течение 18 ч. Реакционную смесь охлаждают до комнатной температуры и растворитель удаляют на роторном испарителе. Остаток распределяют между СНСl2 (200 мл) и водой (200 мл). Органический слой сушат (MgSO4), концентрируют и подвергают экспресс-хроматографии (5%-ный раствор EtOAc в гексане), получая нитрооксибутил-5-этил-2-(2',3',5',6'-тетрафторанилино)фенилацетат в виде прозрачного масла.
Пример 35
5-этил-2- (2',3',5',6'-тетрафторанилино)фенилацетат натрия (7,3 г, 20,9 ммоля) растворяют в ДМФ (100 мл) и обрабатывают бензил-2-метил-2-бромпропионатом (6,2 г, 24,2 ммоля) при 50oС в течение 96 ч. Реакционную смесь охлаждают до комнатной температуры и концентрируют в глубоком вакууме (35-50 мбар) на роторном испарителе. Образовавшееся масло распределяют между водой (200 мл) и Et2O (200 мл). Органический слой промывают соляным раствором (100 мл), сушат (MgSO4) и концентрируют на роторном испарителе, получая масло светло-коричневого цвета. После экспресс-хроматографии (0-10%-ный раствор EtOAc в гексане) на силикагеле получают эфир в виде светло-красного масла. Эфир (1,5 г, 3,0 ммоля) растворяют в EtOAc (150 мл) и гидрируют (55 фунтов/кв. дюйм) в присутствии 10%-ного Pd/C (0,3 г) как катализатора в течение 1 ч. Катализатор удаляют фильтрацией через целит (500 мл). После упаривания на роторном испарителе с последующим растиранием с гексаном получают 1-карбокси-1-метилэтил-5-этил-2-(2', 3', 5', 6'-тетрафторанилино)фенилацетат в виде кристаллического твердого вещества белого цвета, tпл 104-108oС.
Пример 36
Изопропил-5-метил-2-(2'-фтор-6'-трифторметиланилино)фенилацетат (2,9 г, 8,4 ммоля) растворяют в метансульфоновой кислоте (25 мл) и перемешивают при комнатной температуре в течение 8 ч. Реакционную смесь медленно добавляют к 200 мл льда в химическом стакане. После того, как лед растает, раствор перемешивают, получая твердое вещество белого цвета, которое выделяют фильтрацией. Твердое вещество подвергают экспресс-хроматографии на силикагеле, используя 35%-ный EtOAc в качестве элюента, с получением 5-метил-2-(2'-фтор-6'-трифторметиланилино)фенилуксусной кислоты в виде твердого вещества белого цвета, tпл 155-156oС.
Исходный продукт получают следующим образом.
2-йод-5-метилфенилуксусную кислоту (20,0 г, 72 ммоля) и каталитическое количество 98%-ной серной кислоты (0,2 мл) растворяют в изопропиловом спирте (200 мл) и нагревают с обратным холодильником в течение 48 ч. Растворитель удаляют на роторном испарителе и остаток в виде масла распределяют между EtOAc (500 мл) и насыщенным раствором NаНСО3 (500 мл). Органический слой отделяют, сушат (MgSO4) и концентрируют на роторном испарителе. Оставшееся масло отгоняют, используя устройство, снабженное трубкой с шаровым расширением, в результате чего получают прозрачное бесцветное масло, которое затвердевает при комнатной температуре с образованием изопропил-2-йод-5-метилфенилацетата, tпл 48-50oС.
Изопропил-2-йод-5-метилфенилацетат (10,0 г, 31 ммоль), 2-амино-3- фторбензотрифторид (20,0 г, 111 ммолей), порошкообразную медь (1,1 г, 16 ммолей), йодид меди (I) (3,1 г, 16 ммолей) и К2СО3 (4,3 г, 31 ммоль) перемешивают в ксилолах. Реакционную смесь нагревают с обратным холодильником в течение 48 ч. Еще слегка теплую (40oС) суспензию коричневого цвета фильтруют через подушку из целита, которую в свою очередь промывают толуолом (100 мл). Фильтрат упаривают на роторном испарителе и затем подвергают экспресс-хроматографии на силикагеле, используя 3-4%-ный EtOAc в гексане в качестве элюента. Выделяют продукт, т.е. изопропил-5-метил-2-(2'-фтор-6'-трифторметиланилино)фенилацетат, в виде масла светло-желтого цвета.
Пример 37
Аналогично примеру 36 получают 5-метил-2-(2',4'-дихлор-6'-трифторметиланилино)фенилуксусную кислоту, tпл 157-158oС.
Пример 38
N-(2,3,5,6-тетрафторфенил)-5-этилоксиндол (72,67 г, 0,235 моля) суспендируют в воде, содержащей небольшое количество метанола (10 об.%, 235 мл), и добавляют раствор гидроксида натрия (50 мас.%, 16,1 мл). Смесь перемешивают при 80-85oС в течение 2-4 ч, а затем охлаждают до окружающей температуры. Реакционный раствор частично концентрируют при пониженном давлении (25-30 мм рт.ст.). После удаления 50 мл растворителя смесь разбавляют водой (150 мл) и метил-трет-бутиловым эфиром (250 мл). Охлажденную смесь подкисляют до рН 6,5-7,0 водным раствором НСl (12,1 н., 19,5 мл), поддерживая температуру на уровне 0-5oС. Водный слой отбрасывают, а органический слой промывают водой (250 мл). Органический слой концентрируют при пониженном давлении (20-100 мм рт. ст.), заменяя растворитель на толуол. После удаления летучих компонентов объем смеси доводят до 400-450 мл. Эту смесь нагревают до 70oС, осветляют, концентрируют до половины объема и охлаждают до 0oС. После перемешивания при этой температуре в течение 2 ч продукт собирают и промывают толуолом/гептаном (10:90, 100 мл). Образовавшееся твердое вещество сушат при пониженном давлении и при 50-60oС в течение 4-8 ч, получая 5-этил-2-(2',3',5', 6'-тетрафторанилино)фенилуксусную кислоту из примера 18.
Исходный продукт получают следующим образом.
4-этиланилин (246,36 г, 2,00 моля) растворяют в безводном тетрагидрофуране (900 мл). В атмосфере N2 при охлаждении, поддерживая температуру реакционной смеси на уровне, не превышающем 15oС, добавляют раствор н- BuLi (2,5M в гексане, 800 мл, 2,00 моля). После перемешивания в течение 1 ч при 10oС при охлаждении смеси, поддерживая температуру на уровне 10-20oС, добавляют чистый пентафторбензол (168,06 г, 1,00 моль). Реакционную смесь перемешивают при температуре окружающей среды в течение 1,5 ч, а затем медленно добавляют при интенсивном перемешивании и охлаждении, поддерживая температуру реакционной смеси на уровне, не превышающем 35oС, водный раствор НСl (6 н., 500 мл). После прекращения реакции смесь перемешивают при температуре окружающей среды в течение 0,5-18 ч. Нижний водный слой отделяют и верхнюю органическую фазу концентрируют при пониженном давлении (30-150 мм рт.ст.) до одной четверти объема. Концентрат разбавляют гептаном (300 мл) и экстрагируют водой (300 мл). Отделенный верхний органический слой перемешивают над силикагелем (50 г) с размером частиц 230-400 меш и фильтруют. Остаток на фильтре промывают гептаном (4•50 мл). Объединенные фильтрат и смывы концентрируют при пониженном давлении (20-30 мм рт.ст.), получая твердый неочищенный продукт. Этот продукт перекристаллизовывают из горячего гептана (200 мл) и собирают при 0oС. Это твердое вещество промывают холодным гептаном (100 мл) и сушат при пониженном давлении при 40oС, получая чистый N-(2',3',5',6'-тетрафторфенил)-4-этиланилин.
Дифениламиновое производное (230 г, 0,854 моля, 1,0 экв.) обрабатывают хлорацетилхлоридом (192,96 г, 1,709 моля, 2,0 экв.) при 100-115oС в течение 2 ч (интенсивность выделения НСl контролируется скоростью нагревания). Смесь охлаждают до температуры окружающей среды, а затем концентрируют при пониженном давлении (10-12 мм рт.ст.) до 80-90% первоначального объема. Добавляют 1,2-дихлорбензол (80 мл) и разбавленную смесь концентрируют при пониженном давлении (10-12 мм рт.ст.) до тех пор, пока присутствие хлорацетилхлорида больше не выявляется методом газовой хроматографии (30-40 мл удалено путем дистилляции), с получением в растворе неочищенного N-(2',3',5',6'-тетрафторфенил)-N-хлорацетил-4-этиланилина.
Безводный AlCl3 (170,84 г, 1,281 ммоля, 1,5 экв.) суспендируют с 1,2-дихлорбензолом (480 мл) в атмосфере N2 и охлаждают до 0oС. Неочищенный продукт в виде раствора, полученного на предыдущей стадии (теоретическое содержание 295,34 г, 0,854 моля, 1,0 экв.), медленно добавляют при интенсивном перемешивании, поддерживая температуру на уровне, не превышающем 60oС. Добавляют раствор EtAlCl2 (1,8M в толуоле, 733 мл, 1,319 моля, 1,7 экв.) и реакционную смесь нагревают при интенсивном перемешивании до ~160oС, отгоняя (135-160oС) толуол при нормальном атмосферном давлении. После прекращения дистилляции (~ 690 мл) температуру реакционной смеси поддерживают на уровне 155-165oС в течение 3,5-5 ч. Смесь охлаждают до температуры окружающей среды, а затем сливают на ледяную крошку (2,5 кг) при интенсивном перемешивании в атмосфере N2. Реакционный сосуд промывают 1,2-дихлорбензолом (50 мл). Холодную реакционную смесь, в которой прекратилась реакция, в виде суспензии фильтруют и остаток на фильтре промывают последовательно смесью 10%-ного 1,2-дихлорбензола в гептане (100 мл) и гептаном (100 мл). Продукт сушат при пониженном давлении при 80-90oС в течение 12-16 ч, получая N-(2',3',5',6'-тетрафторфенил) -5-этилоксиндол.


Описаны определенные 5-алкил-2-ариламинофенилуксусные кислоты и их производные общей формулы I, где R обозначает метил, этил; R1 обозначает хлор или фтор; R2 обозначает водород или фтор; R3 обозначает водород, фтор, хлор, метил, этил, метокси, этокси или гидрокси; R4 обозначает водород или фтор и R5 обозначает хлор, фтор, трифторметил или метил, или их фармацевтически приемлемые соли, либо их фармацевтически приемлемое пролекарство в виде сложного эфира, три способа их получения, соединения, а также фармацевтические композиции, обладающие способностью избирательного ингибирования циклооксигеназы-2, способы лечения связанных с циклооксигеназой-2 заболеваний у млекопитающих, а также способ избирательного ингибирования активности циклооксигеназы-2 у млекопитающих. 9 с. и 3 з.п. ф-лы, 2 табл.

где R обозначает метил или этил;
R1 обозначает хлор или фтор;
R2 обозначает водород или фтор;
R3 обозначает водород, фтор, хлор, метил, этил, метокси, этокси или гидрокси;
R4 обозначает водород или фтор;
R5 обозначает хлор, фтор, трифторметил или метил,
или его фармацевтически приемлемая соль либо его фармацевтически приемлемое пролекарство в виде сложного эфира.
5-метил-2-(2',4'-дихлор-6'-метиланилино)фенилуксусную кислоту,
5-метил-2-(2',3',5',6'-тетрафторанилино)фенилуксусную кислоту,
5-метил-2-(2',6'-дихлоранилино)фенилуксусную кислоту,
калиевую соль 5-метил-2-(2',6'-дихлоранилино)фенилуксной кислоты,
натриевую соль 5-метил-2-(2',6'-дихлоранилино)фенилуксусной кислоты,
5-метил-2-(2'-хлор-6'-фторанилино)фенилуксусную кислоту,
5-метил-2-(2',6'-дихлор-4'-метиланилино)фенилуксусную кислоту,
5-метил-2-(2'-хлор-6'-метиланилино)фенилуксусную кислоту,
5-метил-2-(2',4'-дифтор-6'-хлоранилино)фенилуксусную кислоту,
5-метил-2-(2'-фтор-4',6'-дихлоранилино)фенилуксусную кислоту,
5-метил-2-(2'-хлор-4'-фтор-6'-метиланилино)фенилуксусную кислоту,
5-этил-2-(2'-фтор-6'-хлоранилино)фенилуксусную кислоту,
5-этил-2-(2'-хлор-6'-метиланилино)фенилуксусную кислоту,
5-этил-2-(2',3',6'-трифторанилино)фенилуксусную кислоту,
5-этил-2-(2',3',5',6'-тетрафтор-4'-этоксианилино)фенилуксусную кислоту,
5-этил-2-(2'-хлор-4',6'-дифторанилино)фенилуксусную кислоту,
5-этил-2-(2',4'-дихлор-6'-фторанилино)фенилуксусную кислоту,
5-этил-2-(2',3',5',6'-тетрафторанилино)фенилуксусную кислоту,
5-этил-2-(2',4'-дихлор-6'-метиланилино)фенилуксусную кислоту,
5-этил-2-(2'-фтор-4'-хлор-6'-метиланилино)фенилуксусную кислоту,
5-этил-2-(2',4'-дифтор-6'-метиланилино)фенилуксусную кислоту,
5-этил-2-(2'-хлор-4'-фтор-6'-метиланилино)фенилуксусную кислоту,
5-метил-2-(2'-хлор-4'-гидрокси-6'-фторанилино)фенилуксусную кислоту,
карбоксиметил-5-этил-2-(2',3',5',6'тетрафторанилино)фенилацетат,
карбоксиметил-5-этил-2-(2'-хлор-6'-метиланилино)фенилацетат,
карбоксиметил-5-этил-2-(2',6'-дихлоранилино)фенилацетат,
карбоксиметил-5-этил-2-(2',4'-дифтор-6'-хлоранилино)фенилацетат,
карбоксиметил-5-этил-2-(2,4'-дихлор-6'-фторанилино)фенилацетат,
карбоксиметил-5-этил-2-(2'-хлор-6'-фторанилино)фенилацетат,
карбоксиметил-5-метил-2-(2'-фтор-4',6'-дихлоранилино)фенилацетат,
карбоксиметил-5-метил-2-(2',6'-дихлоранилино)фенилацетат,
карбоксиметил-5-метил-2-(2'-хлор-6'-фторанилино)фенилацетат,
карбоксиметил-5-метил-2-(2',4'-дифтор-6'-хлоранилино)фенилацетат,
нитроксибутил-5-этил-2-(2',3',5',6'-тетрафторанилино)фенилацетат,
1-карбокси-1-метилэтил-5-этил-2-(2', 3', 5',6'-тетрафторанилино) фенилацетат,
5-метил-2-(2'-фтор-6'-трифторметиланилино)фенилуксусную кислоту,
5-метил-2-(2', 4'-дихлор-6'-трифторметиланилино)фенилуксусную кислоту, или
5-этил-2-(2',3',5',6'-тетрафторанилино)фенилуксусную кислоту,
или его фармацевтически приемлемая соль либо его фармацевтически приемлемое пролекарство в виде сложного эфира.
где R, R1, R2, R3, R4 и R5 имеют значения, указанные в п.1,
и его фармацевтически приемлемые соли.

где R имеет значения, указанные в п.1;
Ra обозначает (низш.)алкил;
R6 и R7 обозначают (низш. )алкил или R6 и R7, вместе с атомом азота обозначают пиперидино, пирролидино или морфолино,
с соединением формулы III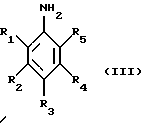
где R1, R2, R3, R4 и R5 имеют значения, указанные в п.1,
в присутствии меди и йодида меди с получением соединения формулы IV или IVa
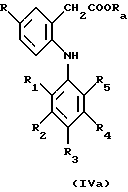
и гидролиз образовавшего соединения формулы IV или IVa с получением соединения формулы I, и при необходимости включающий осуществление в вышеуказанном процессе временной защиты любых препятствующих реакции реакционноспособных групп с последующим выделением образовавшегося соединения по изобретению и при необходимости включающий превращение любого образовавшегося соединения в другое соединение по изобретению и/или при необходимости превращение свободной карбоновой кислоты по изобретению в ее фармацевтически приемлемое производное в виде эфира и/или при необходимости превращение образовавшейся свободной кислоты в соль или образовавшейся соли в свободную кислоту или в другую соль.
где R1-R7 имеют указанные выше значения,
с реакционноспособным функционально активным производным уксусной кислоты, таким, как ацетилхлорид, в реакции ацилирования Фриделя-Крафтса с получением соединения формулы VI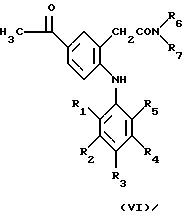
где R1-R7 имеют указанные выше значения, которое в свою очередь подвергают гидрогенолизу, а затем гидролизуют с получением соединения формулы I, в котором R обозначает этил, и при необходимости включающий осуществление в вышеуказанном процессе временной защиты любых препятствующих реакции реакционноспособных групп с последующим выделением образовавшегося соединения по изобретению и при необходимости включающий превращение любого образовавшегося соединения в другое соединение по изобретению и/или при необходимости превращение свободной карбоновой кислоты по изобретению в ее фармацевтически приемлемое производное в виде эфира и/или при необходимости превращение образовавшейся свободной кислоты в соль или образовавшейся соли в свободную кислоту или в другую соль.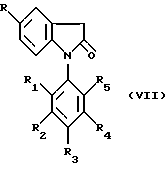
где R и R1-R5 имеют указанные выше значения,
в присутствии сильного основания и при необходимости включающий осуществление в вышеуказанном процессе временной защиты любых препятствующих реакции реакционноспособных групп с последующим выделением образовавшегося соединения по изобретению и при необходимости включающий превращение любого образовавшегося соединения в другое соединение по изобретению и/или при необходимости превращение свободной карбоновой кислоты по изобретению в ее фармацевтически приемлемое производное в виде эфира и/или при необходимости превращение образовавшейся свободной кислоты в соль или образовавшейся соли в свободную кислоту или в другую соль.
| ПРОИЗВОДНЫЕ 4-АМИНОФЕНОЛА ИЛИ ИХ N-АЛКИЛЬНЫЕ ИЛИ СОЛЕВЫЕ ПРОИЗВОДНЫЕ, ПРОЯВЛЯЮЩИЕ ПРОТИВОВОСПАЛИТЕЛЬНУЮ АКТИВНОСТЬ | 1990 |
|
RU2049779C1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| Экономайзер | 0 |
|
SU94A1 |
| DE 3445011 А1, 13.06.1985 | |||
| US 3558690 А1, 26.01.1971 | |||
| US 4548952 А1, 22.10.1985 | |||
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |

