Объектом настоящего изобретения являются производные алкиламинотиазола, их получение и их применение в терапии.
Уже известны производные алкиламинотиазола, описанные в документах WO 03/014095 A, WO 2004/009565 A и WO 2004/033439 A, являющиеся ингибиторами образования β-амилоидного пептида (β-А4).
Всегда существует потребность в поиске и разработке веществ, являющихся ингибиторами образования β-амилоидного пептида (β-А4). Соединения согласно изобретению отвечают этой цели.
Первым объектом изобретения являются соединения, отвечающие общей формуле (I):
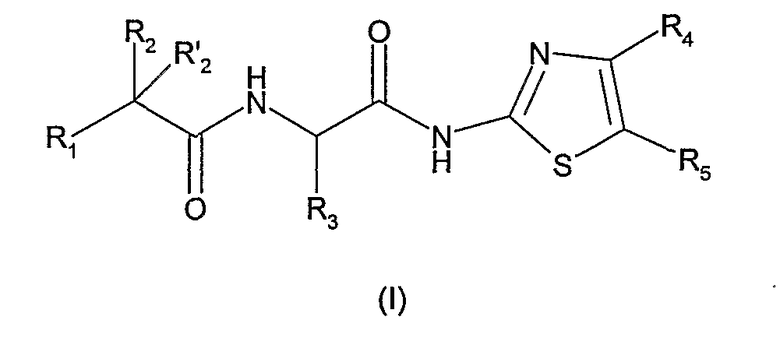
в которой:
R1 обозначает: либо С1-6-алкил, в известных случаях замещенный одним, двумя или тремя заместителями, выбранными из: галогена, трифторметила, гидроксигруппы, С1-6-алкоксигруппы, С1-6-тиоалкила, тиофена или фенила; либо С3-7-циклоалкил, тиофен, бензотиофен, пиридинил, фуранил или фенил; причем вышеупомянутые фенильные группы в известных случаях могут быть замещены одним, двумя или тремя заместителями, выбранными из: атома галогена, С1-6-алкила, С1-6-алкоксигруппы, гидроксигруппы, метилендиоксигруппы, феноксигруппы или бензилоксигруппы, или трифторметила;
R2 и R2 ' обозначают, независимо один от другого, атом водорода, атом галогена, гидроксигруппу, С1-3-алкоксигруппу, С1-3-алкил, С3-7-циклоалкил, группу О-С(О)-С1-6-алкил, или R2 и R2 ' вместе образуют оксогруппу;
R3 обозначает атом водорода, С1-6-алкил, в известных случаях замещенный гидроксильной группой, С1-6-циклоалкил или С1-3-алкоксигруппу;
один или другой из радикалов R4 и R5 представляет собой группу Z
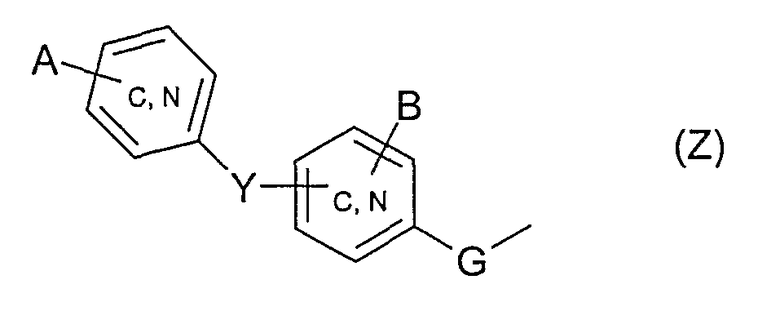
и один или другой из радикалов R4 и R5 представляет собой группу -С(Х)R6;
G обозначает простую связь или группу -СН2-;
Y обозначает простую связь, атом кислорода, атом серы, С1-4-алкиленовую группу или -N(W)-, причем С1-4-алкиленовая группа, в известных случаях, замещена гидроксигруппой или С1-3-алкоксигруппой;
W обозначает либо атом водорода, либо С1-3-алкил, в известных случаях замещенный фенилом, либо фенил;
А и В обозначают, независимо один от другого, атом водорода, атом галогена, гидроксигруппу, С1-3-алкил, С1-3-алкоксигруппу, трифторметил, трифторметоксигруппу или -О-СНF2; при условии, что если Y представляет собой простую связь или атом кислорода и если группа Z представляет собой группу типа
тогда А отлично от атома водорода;
Х обозначает атом кислорода или атом серы;
R6 обозначает С1-6-алкоксигруппу, гидроксигруппу или -NR7R8; при этом С1-6-алкоксигруппа в известных случаях замещена фенилом;
R7 и R8 обозначают, независимо один от другого, либо атом водорода; либо С1-6-алкильную группу, в известных случаях замещенную С3-7-циклоалкилом, С3-7-циклоалкенилом, С1-3-алкоксигруппой, фенилом, морфолинилом или пиридинилом; либо С3-7-циклоалкильную группу, С1-6-алкоксигруппу или фенил; причем вышеупомянутые С3-7-циклоалкильная и фенильная группы, в известных случаях, замещены одной или двумя группами, выбранными из атома галогена, гидроксигруппы, С1-3-алкила или С1-3-алкоксигруппы; или
R7 и R8 вместе с атомом азота, который их несет, образуют азиридиновый, азетидиновый, пирролидиновый, пиперидиновый, морфолиновый или бензопиперидиновый цикл.
Среди соединений общей формулы (I) первая подгруппа соединений образована соединениями, для которых R1 обозначает С1-6-алкил или фенил, в известных случаях замещенный одним, двумя или тремя атомами галогена.
Среди соединений общей формулы (I) вторая подгруппа соединений образована соединениями, для которых R2 и R2 ' обозначают, независимо один от другого, атом водорода, гидроксигруппу или R2 и R2 ' вместе образуют оксогруппу.
Среди соединений общей формулы (I) третья подгруппа соединений образована соединениями, для которых R3 обозначает С1-6-алкил.
Среди соединений общей формулы (I) четвертая подгруппа соединений образована соединениями, для которых:
один или другой из радикалов R4 и R5 представляет собой группу Z
и один или другой из радикалов R4 и R5 представляет собой группу -С(Х)R6;
G обозначает простую связь;
Y обозначает простую связь, атом кислорода, серу, С1-4-алкиленовую группу, более конкретно, метиленовую группу;
А и В обозначают, независимо один от другого, атом водорода, галоген, более конкретно, фтор, трифторметильную группу, трифторметоксигруппу; при условии, что если Y представляет собой простую связь или атом кислорода и если группа Z представляет собой группу типа
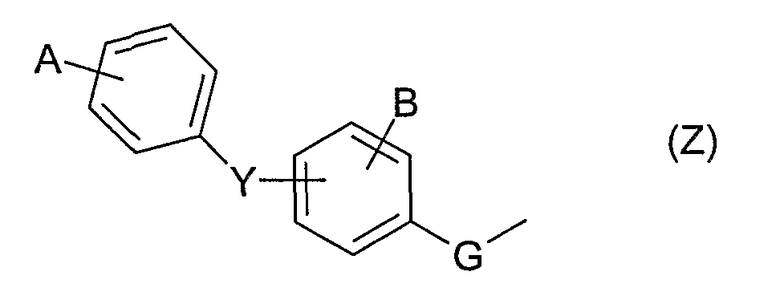
тогда А отлично от атома водорода;
Х обозначает атом кислорода или атом серы;
R6 обозначает С1-6-алкоксигруппу, более конкретно, метоксигруппу или этоксигруппу.
Соединения, для которых одновременно A, B, W, X, Y, Z, R1, R2, R2 ', R3, R4, R5, R6, R7 и R8 являются такими, которые определены в вышеупомянутых подгруппах соединений, образуют пятую подгруппу.
Среди соединений общей формулы (I) и вышеупомянутых подгрупп шестая подгруппа соединений образована соединениями, для которых:
R1 обозначает С1-4-алкил, предпочтительно, изопропил или трет-бутил, или фенил, замещенный двумя атомами фтора; и/или
R2 обозначает атом водорода или гидроксигруппу и R2 ' обозначает атом водорода; и/или
R3 обозначает С1-4-алкил, предпочтительно, метил, этил или пропил; и/или
Х обозначает атом водорода.
В рамках настоящего изобретения
- под Сt-z, где t и z принимают значения от 1 до 7, понимают углеродную цепь, способную содержать от t до z атомов углерода, например С1-3-углеродная цепь, которая может содержать от 1 до 3 атомов углерода, С3-6-углеродная цепь, которая может содержать от 3 до 6 атомов углерода,
- под алкилом понимают насыщенную алифатическую группу, линейную или разветвленную, например, С1-6-алкильная группа представляет собой углеродную цепь, содержащую от 1 до 6 атомов углерода, линейную или разветвленную, более конкретно, метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, …, предпочтительно, метил, этил, пропил или изопропил;
- под алкиленом понимают двухвалентную насыщенную алкильную группу, линейную или разветвленную, например, С1-3-алкилен представляет собой двухвалентную углеродную цепь, содержащую от 1 до 3 атомов углерода, линейную или разветвленную, более конкретно, метиленовую, этиленовую, изопропиленовую, пропиленовую;
- под циклоалкилом понимают циклическую алкильную группу, например, С3-7-циклоалкил представляет собой циклическую углеродную цепь, содержащую от 3 до 7 атомов углерода, более конкретно, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, предпочтительно, циклопентил или циклогексил;
- под циклоалкенилом понимают моно- или полиненасыщенную циклическую алкильную группу, например, С3-7-циклоалкенил представляет собой моно- или полиненасыщенную циклическую углеродную цепь, содержащую от 3 до 7 атомов углерода, более конкретно, циклопропенил, циклобутенил, циклопентенил, циклогексенил, циклогептенил, предпочтительно, циклопентенил или циклогексенил;
- под тиоалкилом понимают группу S-алкила с насыщенной алифатической цепью, линейная или разветвленная;
- под алкоксигруппой понимают группу -О-алкила с насыщенной алифатической цепью, линейной или разветвленной;
- под атомом галогена понимают фтор, хлор, бром или йод;
- под «R2 и R2 ' вместе образуют оксогруппу» понимают такую группу, что:
и
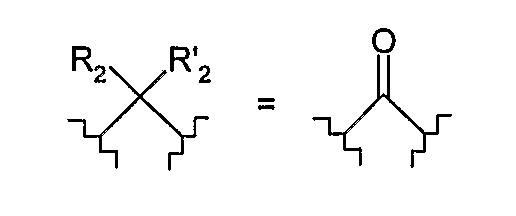
и
- в группе Z ароматическая группа 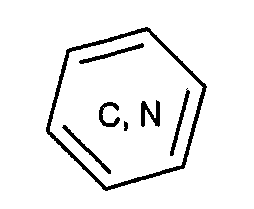 такова, что один из атомов углерода ароматического цикла может быть заменен атомом азота в положении, в котором нет заместителя А или В.
такова, что один из атомов углерода ароматического цикла может быть заменен атомом азота в положении, в котором нет заместителя А или В.
Соединения общей формулы (I) могут содержать один или несколько асимметрических атомов углерода. Следовательно, они могут существовать в форме энатиомеров или диастереоизомеров. Упомянутые энатиомеры, диастереоизомеры, а также их смеси, включая рацемические смеси, входят в изобретение. Когда атом углерода, несущий R2 и R2 ', и/или атом углерода, несущий R3, являются асимметрическими, предпочитают соединения общей формулы (I), в которых атом углерода, несущий R2 и R2 ', имеет конфигурацию (S) и/или атом углерода, несущий R3, имеет конфигурацию (S).
Соединения формулы (I) могут существовать в виде оснований или солей присоединения кислот. Такие аддитивные соли входят в изобретение.
Вышеупомянутые соли получают, преимущественно, с фармацевтически приемлемыми кислотами, но соли других кислот, используемые, например, для очистки или выделения соединений формулы (I), также входят в изобретение.
Соединения общей формулы (I) могут находиться в форме гидратов или сольватов, а именно, в форме ассоциатов или соединений с одной или несколькими молекулами воды, или с растворителем. Такие гидраты и сольваты равным образом входят в изобретение.
Вторым объектом настоящего изобретения являются способы получения соединений формулы (I).
Так, эти соединения могут быть получены способами, иллюстрированными в схемах, следующих ниже, операционные условия которых являются обычными для специалиста.
Под термином «защитная группа» подразумевают группу, дающую возможность препятствовать реакционной способности функциональной группы или положения во время химической реакции, которая может их затрагивать и которая воссоздает молекулу после разрыва химической связи согласно методам, известным специалисту. Примеры защитных групп, а также способы защиты и снятия защиты даны, между прочим, в монографии Protective groups in Organic Synthesis, Greene et coll., 2nd Ed. (John Wiley & Sons, Inc., New York).
Значения A, B, W, X, Y, Z, R1, R2, R2 ', R3, R4, R5, R6, R7 и R8 в соединениях формул (II)-(XVIII), приведенных ниже, являются такими, которые определены для соединений формулы (I), если только другое определение не будет уточнено.
Согласно схеме 1, приведенной ниже, соединение формулы (I) может быть получено пептидным связыванием 2-аминотиазола формулы (III) с ациламинокислотой формулы (II) в операционных условиях, известных специалисту, например, в присутствии гексафторфосфата бензотриазол-1-илокси-трис(пирролидино)фосфония (РуВОР) или гексафторфосфата бензотриазол-1-илокси-трис(диметиламино)фосфония (ВОР) и N-этилморфолина или N-метилморфолина в инертном растворителе, таком как N,N-диметилформамид, ацетонитрил или дихлорметан при температуре, которая может варьировать от 0°С до комнатной температуры.
Соединение формулы (II) может быть получено пептидным связыванием соединения формулы (IV) с защищенной кислотой формулы (V), в которой Pg представляет собой защитную группу, например, бензильную, согласно способам, известным специалисту, таким, как описанные выше. Соединение, полученное таким образом, затем освобождают от защиты. В случае, когда защитная группа является бензильной, для получения соединения формулы (II) соединение предварительно гидрируют, предпочтительно, в присутствии палладия на углероде в абсолютном этаноле при атмосферном давлении водорода при комнатной температуре.
СХЕМА 1
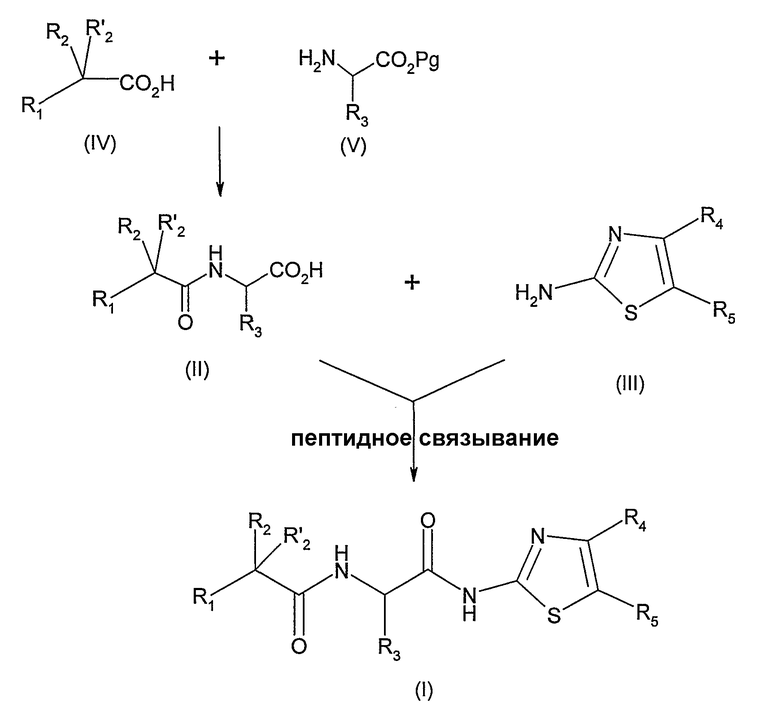
Альтернативно, соединение формулы (I) может быть получено согласно схеме 2.
Согласно схеме 2, следующей ниже, соединение формулы (I) может быть получено пептидным связыванием соединения формулы (IV) с амином формулы (VI) согласно способам, известным специалисту, как, например, в присутствии гидрата гидроксибензотриазола ГБТ (НОBt) и хлоргидрата 1-этил-3-(3-диметиламинопропил)карбодиимида (ЭДАК, HCl) (EDAK, HСl).
СХЕМА 2
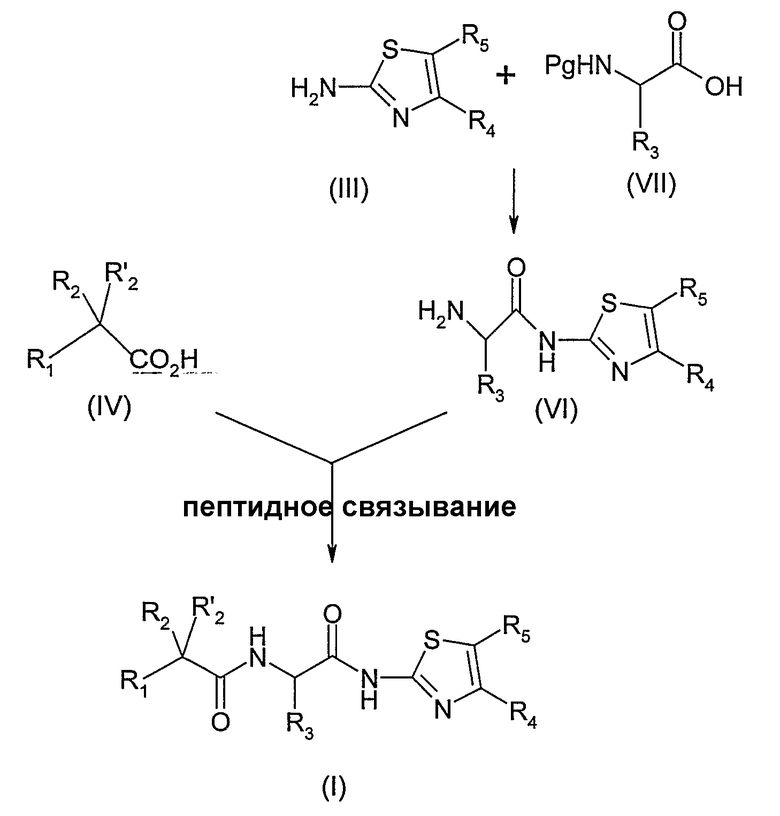
Соединение формулы (VI) может быть получено пептидным связыванием 2-аминотиазола формулы (III) с защищенным амином формулы (VII), в которой Pg обозначает защитную группу, например, N-трет-бутилоксикарбонильную (БОК)(Boc), согласно способам, известным специалисту, таким, как описанные выше. Соединение, полученное таким образом, затем освобождают от защиты. В случае, когда защитная группа представляет собой БОК, для того, чтобы получить соединение формулы (VI), снятие защиты осуществляется кислотным гидролизом в присутствии газообразной соляной кислоты, растворенной в безводном растворителе, или трифторуксусной кислоты.
Соединения формулы (I), в которой R2 и R2 ' образуют оксогруппу, могут быть получены окислением соединения формулы (I), в которой R2 или R2 ' представляет собой гидроксигруппу. Реакция может быть осуществлена в условиях, известных специалисту, например, с реактивом Десса Мартина (Dess Martin). Равным образом, указанные соединения могут быть получены прямым связыванием кетокислоты формулы IV), в которой R2 и R2 ' вместе образуют оксогруппу, с амином формулы (VI) в условиях, известных специалисту. Способы получения таких кетокислот известны специалисту.
Соединения формулы (III), в которой R4=-C(O)-R6, при этом R6 представляет собой С1-6-алкоксигруппу, может быть получено согласно схеме 3, следующей ниже.
СХЕМА 3
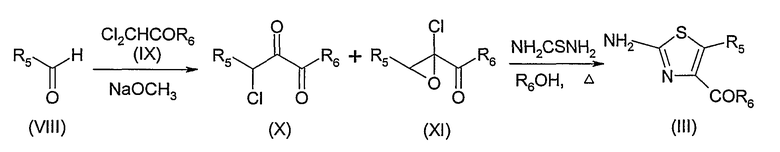
Согласно схеме 3, соединение формулы (III) может быть получено реакцией альдегида формулы (VIII), в которой R5 является таким, который определен выше, с метилдихлорацетатом формулы (IX), в которой R6 представляет собой С1-6-алкоксигруппу, в известных случаях, замещенную фенилом, и, например, метилатом или этилатом натрия при 0°С согласно адаптации способа, описанного Такедой (Takeda) (Bull. Chem. Soc. Jp., 1970, p. 2997). Для получения соединения формулы (III), полученную смесь продуктов (X) и (XI) обрабатывают тиомочевиной в присутствии, например, метанола или этанола с обратным холодильником в течение 4 или 8 часов.
Соединения формулы (III), в которой R4=-C(O)-R6, при этом R6 представляет собой гидроксигруппу, может быть получено гидролизом вышеупомянутых соединений, для которых R6 представляет собой С1-6-алкоксигруппу, в известных случаях, замещенную фенилом, в условиях, известных специалисту.
Соединение формулы (III), в которой R5=-C(O)-R6, при этом R6 представляет собой С1-6-алкоксигруппу, может быть получено согласно схеме 4, следующей ниже.
СХЕМА 4
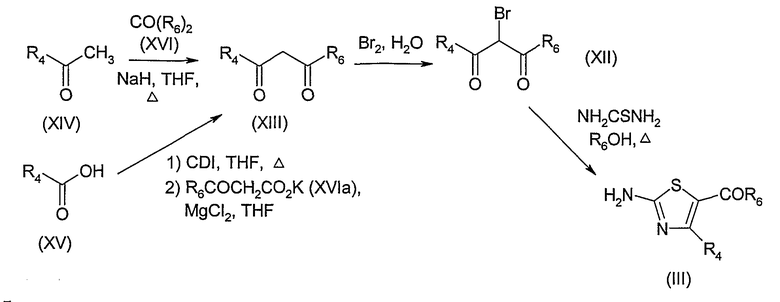
Согласно схеме 4, соединение формулы (III) может быть получено бромированием сложного β-кетоэфира формулы (XIII), в которой R6 представляет собой С1-6-алкоксигруппу, в известных случаях, замещенную фенилом, с получением соединения формулы (XII), и последующей реакцией с тиомочевиной согласно адаптации способа, описанного Бартоном с коллегами (Barton et coll.) (J.C.S. Perkin I, 1982, p.159).
Сложный β-кетоэфир формулы (XIII) может быть получен реакцией кетона формулы (XIV) с диалкилкарбонатом формулы (XVI), в которой R6 представляет собой С1-6-алкоксигруппу, в известных случаях, замещенную фенилом, согласно адаптации способа, описанного Кромби с коллегами (L. Crombie et coll.) (J.C.S. Perkin Trans I, 1987, p.323). Равным образом, сложный β-кетоэфир формулы (XIII) может быть получен реакцией кислоты формулы (XV), активированной карбонилдиимидазолом (КДИ) (CDI), с малонатом формулы (XVIa), в которой R6 представляет собой С1-6-алкоксигруппу, в известных случаях, замещенную фенилом, согласно адаптации способа, описанного, например, Бруксом с коллегами (D.W. Brooks et coll.) (Angew. Chem. Int. Ed., 1979, p.72).
Соединение формулы (III) в которой R5=-C(O)-R6, при этом R6 представляет собой гидроксигруппу, может быть получено гидролизом вышеупомянутых соединений, для которых R6 представляет собой С1-6-алкоксигруппу, в условиях, известных специалисту.
Соединение формулы (III) в которой R4 или R5 представляет собой -C(O)-NR7R8, может быть получено согласно схеме 5.
СХЕМА 5
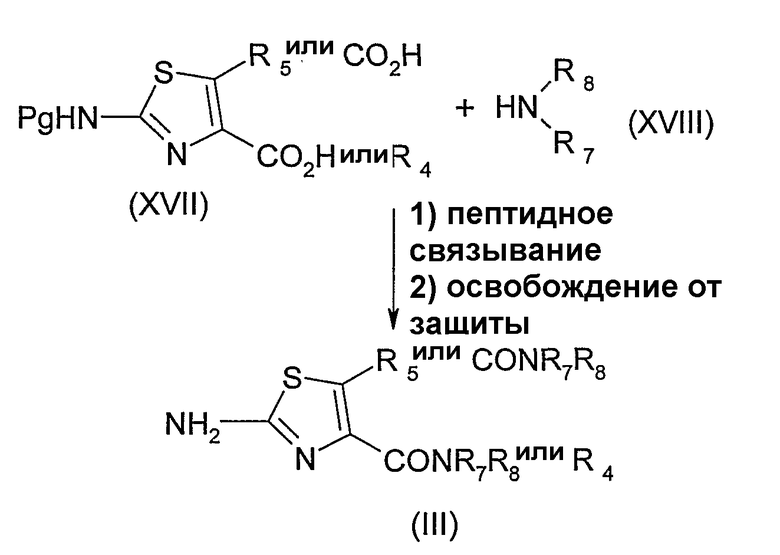
Согласно схеме 5, соединение формулы (III) получают пептидным связыванием соединения формулы (XVII), в которой R5 или R4 представляет собой карбоксильную группу и Pg - защитную группу, такую как БОК, с соединением формулы (XVIII) в присутствии, например, ГБТ и (ЭДАК, HСl). Соединение, полученное таким образом, освобождают от защиты в условиях, известных специалисту. Соединение формулы (XVII), в которой Pg представляет собой БОК, может быть получено при помощи защиты соединения формулы (III), в которой R4 или R5 представляет собой группу -С(О)R6 и R6 представляет собой С1-6-алкоксигруппу, в известных случаях, замещенную фенилом, воздействием ди-трет-бутилдикарбоната в безводном терагидрофуране в присутствии диметиламинопиридина при комнатной температуре и последующим гидролизом карбоксилата в условиях, известных специалисту, например, с гидроксидом лития в смеси тетрагидрофуран/вода 7:3 (об./об.) при температуре 60°С.
Соединения общей формулы (I), в которой R4 или R5 представляет собой группу -C(Х)R6 и Х=S, может быть получено, исходя из соответствующих соединений общих формул (I) или (III), в которых R4 или R5 представляет собой группу -C(Х)R6 и Х=О, превращением группы С(О) в группу СS, например, при помощи реактива Лэвессона (Lawesson) согласно способу, аналогичному способу, описанному Кава с коллегами (M.P. Cava et coll) в Tetrahedron, 1985, p.5061.
В схемах 1-5 исходные соединения и реагенты, в частности, соединения формул (III), (IV), (V), (VII), (VIII), (IX), (XIV), (XV), (XVI), (XVIa), (XVII) и (XVIII), когда способ их получения не описан, являются доступными в торговле или описаны в литературе, или могут быть получены способами, которые описаны в литературе или которые известны специалисту.
Например, соединения формулы (IV), в которой R2 или R2 ' представляет собой гидроксигруппу, могут быть получены присоединением триметилсилилцианида к альдегиду согласно адаптации способа, описанного Эвансом с коллегами (D.A. Evans et coll.) (J. C. S., Chem. Comm. 1973, p.55) или действием нитрита натрия на альфа-аминокислоту согласно адаптации способа, описанного Шинном с коллегами (I. Shinn et coll.) (J. Org. Chem., 2000, p.7667).
Например, соединения формулы (XV), в которой Y=O, могут быть получены согласно адаптации способа, описанного Синделом с коллегами (Sindel et coll.) (Collect. csech. Tchecosl., 1982, p.72) или Аткисоном с коллегами (Atkison et coll.) (J. Med. Chem., 1983, p.1353).
Например, соединения формулы (XV), в которой Y представляет собой группу типа С1-4-алкиленовой, могут быть получены, например, согласно адаптации способа, описанного Кроу с коллегами (Crow et coll.) (Austral. J. Chem., 1981, p.1037), или, альтернативно, реакцией Судзуки (Suzuki) согласно адаптации способа, описанного Шахеном с коллегами (Chahen et coll.) (Synlett, 2003, p.1668).
Например, соединения формулы (XV), в которой Y=S, могут быть получены согласно способу, описанному Голдбергом (Goldberg) (Chem. Ber., 1994, p.4526).
Например, соединения формулы (XV), в которой Y=N(W), могут быть получены согласно способу, описанному Шаном с коллегами (Chane et coll.) (Tetrahedron Letters, 1998, p.2933) или Шамэном (Chamain) (Tetrahedron Letters, 1998, p.4179) или Хьювом с коллегами (Huwe et coll.) (Tetrahedron Letters, 1999, p.683).
Например, соединения формулы (XV), в которой Y представляет собой простую связь, могут быть получены реакцией Судзуки (Suzuki) в условиях, известных специалисту, например, согласно способу, описанному Денгом с коллегами (Deng et coll.) Synthesis, 2003, p.337 или Мейером с коллегами (Meier et coll.) Synthesis, 2003, p.551.
Например, соединения формулы (VIII) могут быть получены восстановлением соединений формулы (XV) в условиях, известных специалисту.
Когда функциональная группа соединения является реакционноспособной, например, когда R1 содержит гидроксигруппу, она может требовать предварительной защиты перед реакцией. Специалист сможет легко определить необходимость предварительной защиты.
Следующие примеры описывают получение некоторых соединений согласно изобретению. Эти примеры не являются ограничительными и только иллюстрируют изобретение.
Номера соединений, приведенных в качестве примеров, соответствуют номерам, приведенным в таблице, следующей ниже. Элементные микроанализы и анализы методами ЯМР, ИК-спектрометрии или ЖХ-МС (LC-MS) (жидкостная хроматография в сочетании с масс-спектрометрией) подтверждают структуры полученных соединений.
Пример 1: 2-{2-(S)-[2-(3,5-дифторфенил)ацетиламино]пентаноил}амино-5-[2-(4-фторфенокси)фенил]тиазол-4-метилкарбоксилат
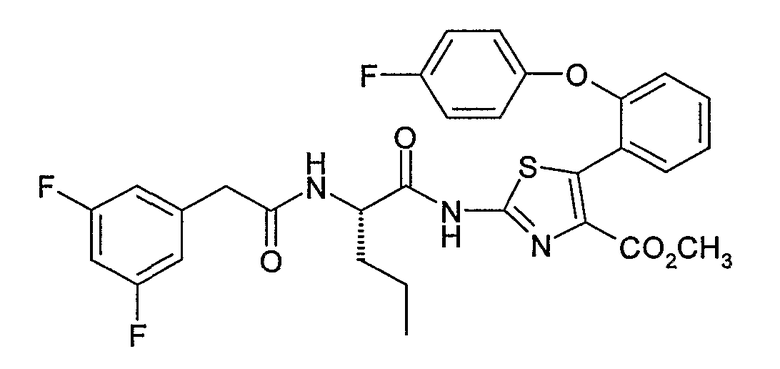
Пример 1.1: 2-(4-фторфенокси)бензойная кислота
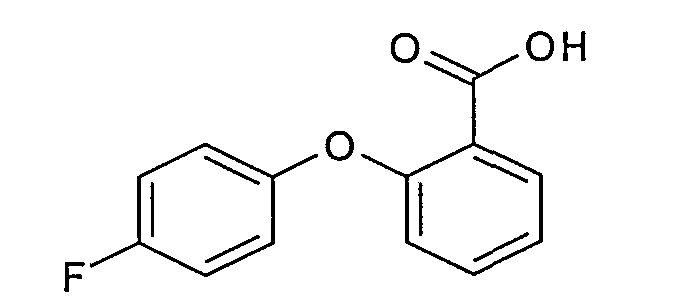
К смеси 120 г 2-йодбензойной кислоты, 1 г порошкообразной меди и 54,4 г 4-фторфенола в 200 мл N,N-диметилформамида медленно добавляют 100 г карбоната калия. Греют при 160°С в течение 4 часов, затем дают остыть перед выпариванием. Остаток извлекают дистиллированной водой, подкисляют водным раствором 1 н. соляной кислоты, затем экстрагируют этилацетатом. Органическую фазу промывают насыщенным раствором хлорида натрия, сушат над безводным сульфатом натрия, затем концентрируют. Целевой продукт кристаллизуют из смеси диэтиловый эфир/пентан. Получают 50 г белого твердого вещества.
ЖХ/МС: МН+=233
Пример 1.2: 2-(4-фторфенокси)-O,N-диметилбензамид
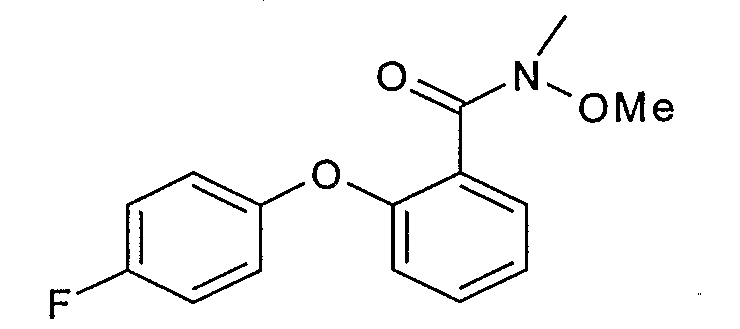
К раствору 50 г 2-(4-фторфенокси)бензойной кислоты, полученной на стадии 1.1, в 450 мл N,N-диметилформамида добавляют 29,7 г гидрата гидроксибензотриазола, затем 37 г (ЭДАК, HСl), 19 г (O,N-диметилгидроксиламин, HСl) и 19,4 г N-метилморфолина. Перемешивают при комнатной температуре в течение 16 часов. Выпаривают, извлекают этилацетатом и промывают 2 раза насыщенным водным раствором хлорида натрия, 1 раз дистиллированной водой, 1 раз водным раствором 1 М гидросульфата калия, затем насыщенным водным раствором хлорида натрия. Сушат над безводным сульфатом натрия, затем концентрируют. Получают 49 г окрашенного масла, которое используют в данном состоянии впоследствии.
ЖХ/МС: МН+=276
Пример 1.3: 2-(4-фторфенокси)бензальдегид
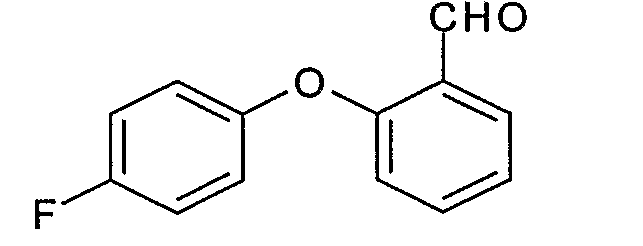
К 100 мл раствора 1М литийалюминийгидрида в тетрагидрофуране при 0°С по каплям добавляют 48,5 г 2-(4-фторфенокси)-O,N-диметилбензамида, полученного на стадии 1.2, в 300 мл безводного тетрагидрофурана. Перемешивают при 0°С в течение 1 часа, затем гидролизуют по каплям 40 мл водного раствора 1М гидросульфата калия. Выпаривают, извлекают этилацетатом, промывают 2 раза водным раствором 1М гидросульфата калия, затем насыщенным водным раствором хлорида натрия. Сушат над безводным сульфатом натрия, затем концентрируют. Получают 35 г окрашенного масла.
ЖХ/МС: МН+=217
ЯМР, 300 МГц (CDCl3), δ (м.д.): 6,70-7,00 (м, 4Н); 7,20-7,35 (м, 2Н); 7,55 (т, 1Н); 7,95 (д, 1Н); 10,43 (с, 1Н).
Пример 1.4: 2-амино-5-[2-(4-фторфенокси)фенил]тиазол-4-метилкарбоксилат
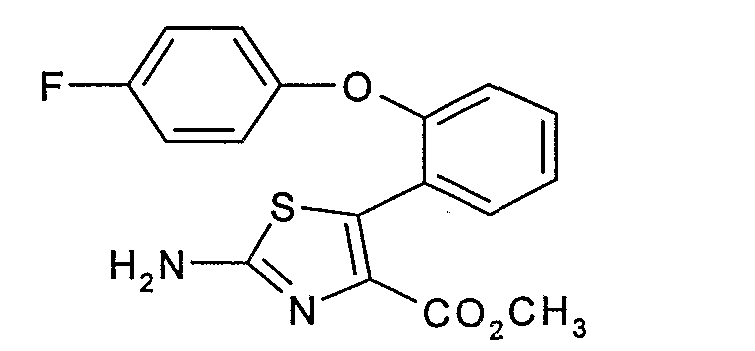
К раствору 35 г 2-(4-фторфенокси)бензальдегида, полученного на стадии 1.3, в 400 мл диэтилового эфира добавляют при 0°С 30 г метилдихлорацетата, затем по каплям 325 мл раствора метилата натрия (0,5 М) в метаноле. После 1 ч при 0°С выпаривают только диэтиловый эфир, сохраняя метанол, добавляют 11 г тиомочевины и нагревают с обратным холодильником в течение 6 часов. Выпаривают реакционную смесь досуха, извлекают ее этилацетатом и промывают ее 10%-ным водным раствором гидроксида аммония, затем насыщенным водным раствором хлорида натрия. Сушат органическую фазу над безводным сульфатом натрия, затем ее концентрируют. Извлекают остаток 100 мл диэтилового эфира и фильтруют его через стеклянный фильтр. Получают 30 г белого твердого вещества.
ЖХ/МС: МН+=345
ЯМР, 300 МГц (CDCl3), δ (м.д.): 3,70 (с, 3Н); 5,55 (шир.с, 2Н); 6,55-6,80 (м,4Н); 7,00 (д, 1Н); 7,20 (т, 1Н); 7,35-7,45 (м, 2Н).
Пример 1.5: 2-[2-(S)-пентаноиламино]амино-5-[2-(4-фторфенокси)фенил]тиазол-4-метилкарбоксилат
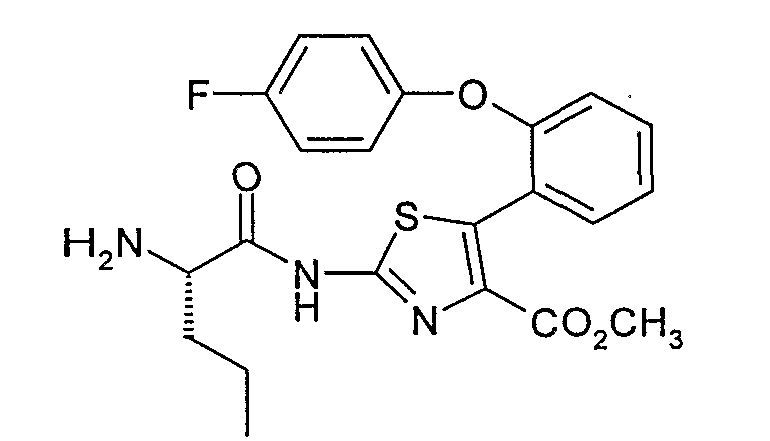
К 8,6 г 2-амино-5-[2-(4-фторфенокси)фенил]тиазол-4-метилкарбоксилата, полученного на стадии 1,4, в растворе в 200 мл N,N-диметилформамида при 0°С добавляют 2,75 г N-метилморфолина, 14,30 г PyBOP, затем 5,97 г (S)-БОК-норвалина. Дают реакционной среде возвратиться к комнатной температуре, затем перемешивают ее в течение 16 ч. После выпаривания извлекают осадок этилацетатом и промывают его 2 раза насыщенным водным раствором бикарбоната натрия, 2 раза водой, 1 раз водным раствором 1М гидросульфата калия, затем насыщенным водным раствором хлорида натрия. Сушат органическую фазу над безводным сульфатом натрия, затем ее концентрируют. Хроматографируют остаток на колонке с силикагелем, элюируя смесью этилацетата и петролейного эфира 3:7 (об./об.). Получают 8,5 г белого твердого вещества.
ЖХ/МС: МН+=544
ЯМР, 300 МГц (CDCl3), δ (м.д.): 0,88 (т, 3Н); 1,38 (с, 9Н); 1,39-1,55 (2м, 2Н); 1,75 (м, 2Н); 3,35 (шир.с, 1Н); 3,68 (с, 3Н); 4,28 (м, 1Н); 5,65 (д, 1Н); 6,80-6,90 (м, 5Н); 7,10 (т, 1Н); 7,20-7,32 (м, 2Н).
Перемешивают 6,5 г продукта, полученного выше, в растворе в 60 мл трифторуксусной кислоте при комнатной температуре в течение 30 мин, затем его упаривают, осадок извлекают этилацетатом и промывают его 2 раза насыщенным водным раствором карбоната натрия, затем насыщенным водным раствором хлорида натрия. Сушат органическую фазу над безводным сульфатом натрия, затем ее выпаривают с получением 3,9 г белого твердого вещества.
ЖХ/МС: МН+=444
Пример 1.6: 2-{2-(S)-[2-(3,5-дифторфенил)ацетиламино]пентаноил}амино-5-[2-(4-фторфенокси)фенил]тиазол-4-метилкарбоксилат
К 0,7 г 2-амино-2-[2-(S)-пентаноиламино]-5-[2-(4-фторфенокси)фенил]тиазол-4-метилкарбоксилата, полученного согласно примеру 1.5, в растворе в 30 мл N,N-диметилформамида при 0°С добавляют 0,20 г N-метилморфолина, 0,99 г PyBOP, затем 0,33 г 3,5-дифторфенилуксусной кислоты. Дают реакционной среде возвратиться к комнатной температуре и перемешивают ее в течение 18 ч. Выпаривают реакционную среду. Осадок извлекают этилацетатом и промывают его 2 раза насыщенным водным раствором бикарбоната натрия, 2 раза водой, 1 раз водным раствором 1М гидросульфата калия, затем насыщенным водным раствором хлорида натрия. Сушат органическую фазу над безводным сульфатом натрия, затем ее концентрируют. Хроматографируют остаток на колонке с диоксидом кремния, элюируя смесью петролейный эфир/этилацетат 1:1 (об./об.) с получением 0,56 г белого твердого вещества.
ЖХ/МС: МН+=558
Спектр ЯМР описан в таблице (соединение № 13).
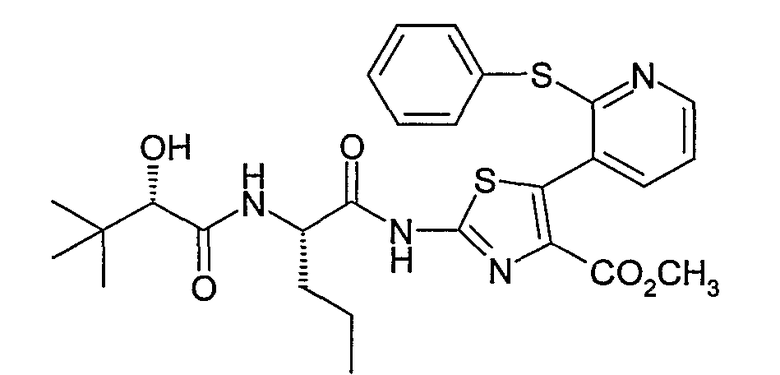
Пример 2: 2-{2-(S)-[2-(S)-гидрокси-(3,3-диметил)бутириламино]пентаноил}амино-5-[2-(фенилтио)-3-пиридил]тиазол-4-метилкарбоксилат (соединение № 19)
Пример 2.1: (2-фенилтио)-O,N-диметилникотинамид
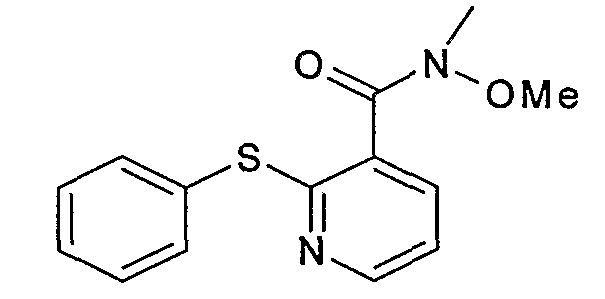
(2-фенилтио)-O,N-диметилникотинамид может быть получен согласно способу, аналогичному описанному на стадии 1.2 примера 1. Исходя из 20 г 2-фенилтионикотиновой кислоты получают 21,9 г бесцветного масла, которое используют в данном состоянии впоследствии.
ЖК/МС: МН+=275
ЯМР, 300 МГц (CDCl3), δ (м.д.): 3,35 (шир.с, 3Н); 3,58 (шир.с, 3Н); 7,35 (м, 3Н); 7,50 (м, 2Н); 7,60 (д, 2Н); 8,40 (д, 1Н).
Пример 2.2: (2-фенилтио)никотинальдегид
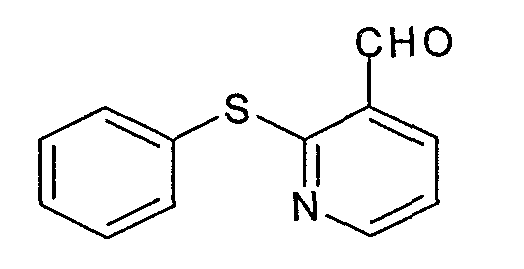
(2-фенилтио)никотинальдегид может быть получен согласно способу, аналогичному описанному на стадии 1.3 примера 1. Исходя из 21,9 г (2-фенилтио)-O,N-диметилникотинамида (амид Вайнреба (Weinreb)), полученного на стадии 2.1, и 48 мл раствора 1М литийалюминийгидрида в 300 мл тетрагидрофурана, получают 16,6 г белого твердого вещества.
ЯМР, 300 МГц (CDCl3), δ (м.д.): 7,18 (м, 1Н); 7,42 (м, 3Н); 7,57 (м, 2Н); 8,05 (д, 1Н); 8,46 (д, 1Н); 10,35 (с, 1Н).
Пример 2.3: 2-амино-5-[2-(фенилтио)-3-пиридил]тиазол-4-метилкарбоксилат (соединение № 19)
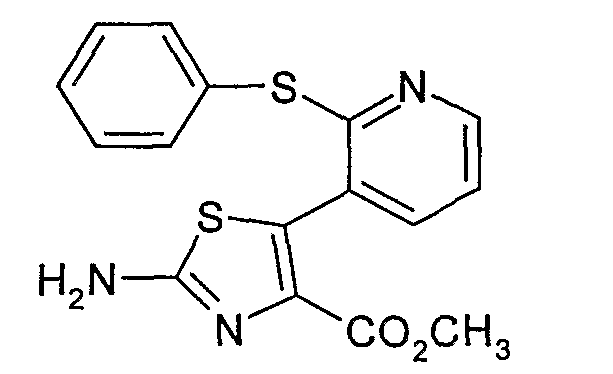
2-Амино-5-[2-(фенилтио)-3-пиридил]тиазол-4-метилкарбоксилат может быть получен согласно методу, аналогичному описанному на стадии 1.4 примера 1. Исходя из 16,5 г (2-фенилтио)никотинальдегида, полученного на стадии 2.2, 11,2 г метилдихлорацетата и 150 мл 0,5 М метилата натрия в 300 мл диэтилового эфира, получают 19 г бледно-желтого твердого вещества.
ЖК/МС: МН+=344
ЯМР, 300 МГц (ДМСОd6), δ (м.д.): 3,41 (шир.с, 3Н); 3,60 (с, 3Н); 7,20 (шир.д, 1Н); 7,38-7,48 (м, 5Н); 7,65 (шир.д, 1Н); 8,30 (шир.д, 1Н).
Пример 2.4: 2-[2-(S)-пентаноиламино]амино-5-[2-(фенилтио)-3-пиридил]тиазол-4-метилкарбоксилат
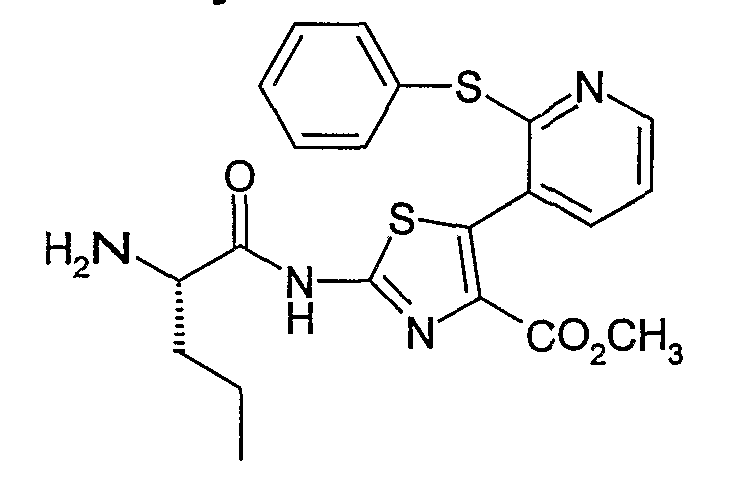
2-[2-(S)-пентаноиламино]амино-5-[2-(фенилтио)-3-пиридил]тиазол-4-метилкарбоксилат может быть получен согласно методу, аналогичному описанному на стадии 1.5 примера 1. Исходя из 5,14 г 2-амино-5-[2-(фенилтио)-3-пиридил]тиазол-4-метилкарбоксилата, полученного в примере 2.3, и 3,58 г (S)-БОК-норвалина в присутствии 8,58 г PyBOP и 1,66 г N-метилморфолина в N,N-диметилформамиде при 0°С, получают, после хроматографирования, 3,5 г бледно-желтого твердого вещества.
ЖК/МС: МН+=542
ЯМР, 300 МГц (ДМСОd6), δ (м.д.): 0,87 (т, 3Н); 1,45 (с, 9Н); 1,70 (м, 2Н); 1,97 (м, 2Н); 3,72 (с, 3Н); 4,45 (м, 1Н); 5,23 (м, 1Н); 7,10 (м, 1Н); 7,30 (м, 3Н); 7,42 (м, 2Н); 7,50 (д, 1Н); 8,39 (д, 1Н).
Растворяют 3,5 г амина, полученного выше, в 150 мл раствора 4М соляной кислоты в 1,4-диоксане и 20 мл метанола. Перемешивают 1 ч 30 мин при комнатной температуре, затем выпаривают. Получают 3,2 г бледно-желтого твердого вещества.
ЖК/МС: МН+=442
Пример 2.5: 2-{2-(S)-[2-(S)-гидрокси-(3,3-диметил)бутириламино]пентаноил}амино-5-[2-(фенилтио)-3-пиридил]тиазол-4-метилкарбоксилат
2-{2-(S)-[2-(S)-гидрокси-(3,3-диметил)бутириламино]пентаноил}амино-5-[2-(фенилтио)-3-пиридил]тиазол-4-метилкарбоксилат может быть получен согласно методу, аналогичному описанному на стадии 1.6 примера 1. Исходя из 0,9 г 2-[2-(S)-пентаноиламино]амино-5-[2-(фенилтио)-3-пиридил]тиазол-4-метилкарбоксилата, полученного на стадии 2.4, и 0,25 г (S)-2-гидрокси-3,3-диметилмасляной кислоты в присутствии 1 г PyBOP и 0,59 г N-метилморфолина в 90 мл N,N-диметилформамида при 0°С, получают, после хроматографирования на колонке с силикагелем, элюируемой смесью этилацетат/петролейный эфир 7:3 (об./об.), 0,5 г белого порошка.
ЖК/МС: МН+=557
Спектр ЯМР описан в таблице (соединение № 19).
Пример 3: 2-{2-(S)-[2-(3,5-дифторфенил)ацетиламино]пентаноил}амино-5-[2-(4'-трифторметил)дифенил]тиазол-4-метилкарбоксилат (соединение № 5)
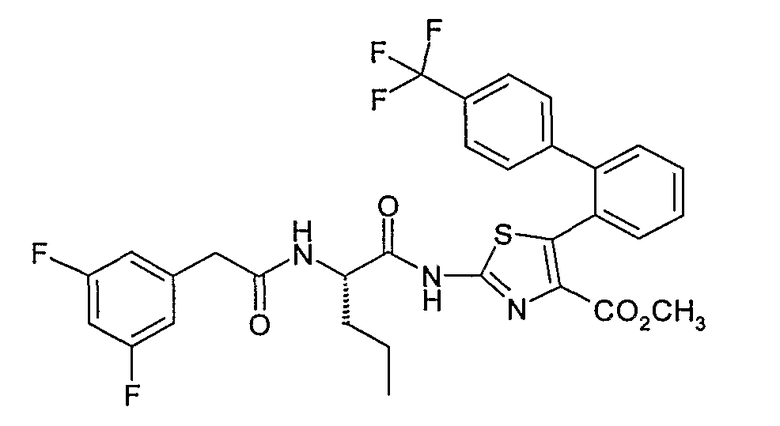
Пример 3.1: 2-амино-5-[2-(4'-трифторметил)дифенил]тиазол-4-метилкарбоксилат
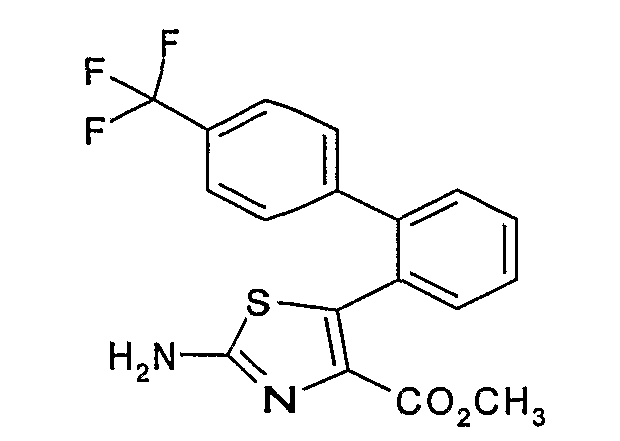
2-Амино-5-[2-(4'-трифторметил)дифенил]тиазол-4-метилкарбоксилат может быть получен согласно способу, аналогичному описанному на стадиях 1.2-1.4 примера 1, исходя из 26,6 г 4-трифторметил-2-дифенилкарбоновой кислоты. Амид Вейнреба (Weinreb) данной кислоты восстанавливают в альдегид литийалюминийгидридом с получением 18,7 г прозрачного желтого масла. Альдегид (18 г) вводят в реакцию с 10,3 г метилдихлорацетата в присутствии 144 мл 0,5 М метилата натрия, затем с 4,7 г тиомочевины в метаноле при нагревании с обратным холодильником. Получают 16 г бледно-желтого твердого вещества.
ЖХ/МС: МН+=379
Пример 3.2: 2-[2-(S)-Пентаноиламино]амино-5-[2-(4'-трифторметил)дифенил]тиазол-4-метилкарбоксилат
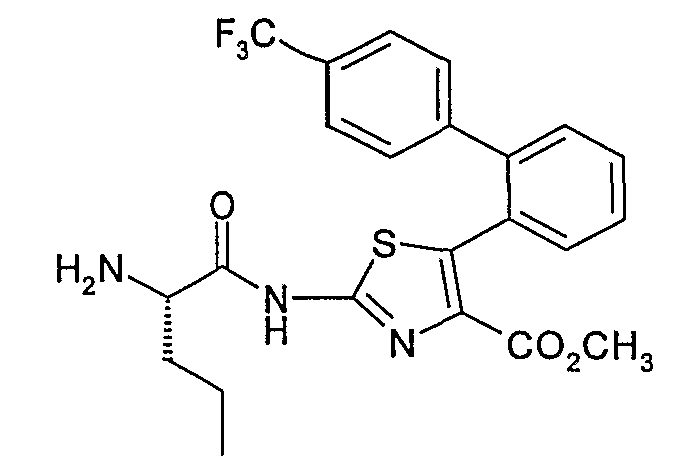
2-[2-(S)-Пентаноиламино]амино-5-[2-(4'-трифторметил)дифенил]тиазол-4-метилкарбоксилат может быть получен согласно способу, аналогичному описанному на стадии 1.5 примера 1. Исходя из 2,26 г 2-амино-5-[2-(4'-трифторметил)дифенил]тиазол-4-метилкарбоксилата, полученного по примеру 3.1, и 1,43 г (S)-БОК-норвалина, в присутствии 3,43 г РуВОР и 0,66 г N-метилморфолина в 120 мл N,N-диметилформамида при °С, получают, после хроматографии на колонке с силикагелем, элюируемой смесью петролейный эфир/этилацетат 8:2 (об./об.), 2 г бледно-желтого твердого вещества.
ЖХ/МС: МН+=578
ЯМР 300 МГц (CDCl3) δ (м.д.): 0,92 (т, 3Н); 1,45 (с, 9Н); 1,70 (м, 2Н); 1,80 (м, 2Н); 3,69 (с, 3Н); 4,40 (м, 1Н); 5,10 (м, 1Н); 6,98 (м, 4Н); 7,27 (м, 2Н); 7,32 (д, 2Н).
Затем полученное соединение освобождают от защиты 50 мл трифторуксусной кислоты, следуя способу, описанному в примере 1.5. Получают 1 г белой пены.
ЖХ/МС: МН+=478
Пример 3.3: 2-{2(S)-[2-(3,5-Дифторфенил)ацетиламино]пентаноил}амино-5-[2-(4'-трифторметил)дифенил]тиазол-4-метилкарбоксилат
2-{2(S)-[2-(3,5-Дифторфенил)ацетиламино]пентаноил}амино-5-[2-(4'-трифторметил)дифенил]тиазол-4-метилкарбоксилат может быть получен согласно способу, аналогичному описанному на стадии 1.6 примера 1. Исходя из 0,94 г 2-[2-(S)-пентаноиламино]амино-5-[2-(4'-трифторметил)дифенил]тиазол-4-метилкарбоксилата, полученного на стадии 3.2, и 0,18 г 3,5-дифторфенилуксусной кислоты в присутствии 0,55 г РуВОР и 0,11 г N-метилморфолина в 50 мл N,N-диметилформамида при 0°С, получают, после хроматографии на колонке с силикагелем, элюируемой смесью этилацетат/ петролейный эфир 7:3 (об./об.), 0,45 г белого порошка.
ЖХ/МС: МН+=632
Спектр ЯМР описан в таблице (соединение №5).
Таблица, следующая ниже, иллюстрирует химические структуры и физические свойства нескольких примеров соединений согласно изобретению.
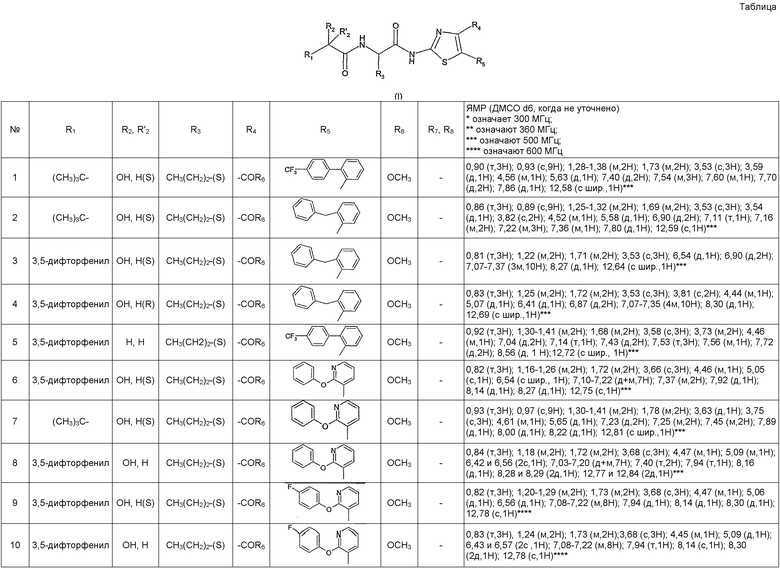
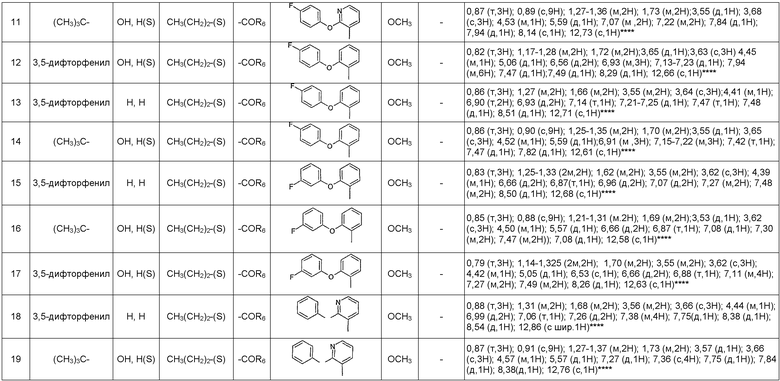
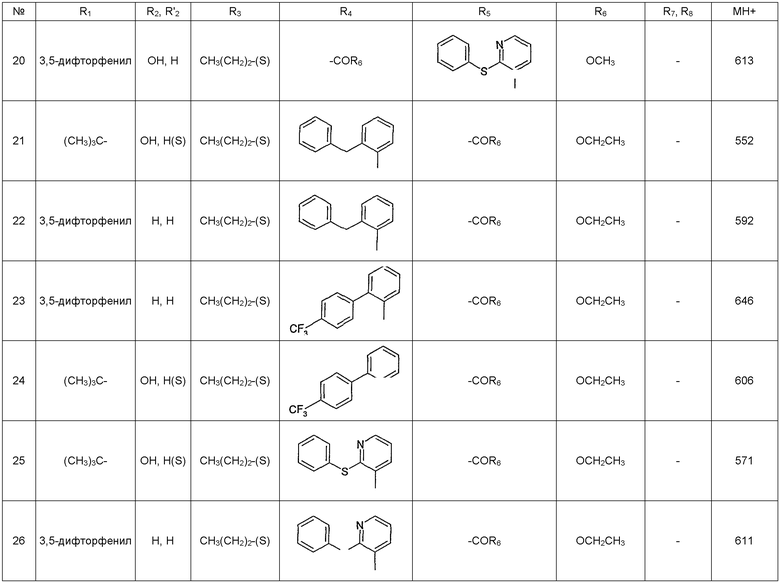
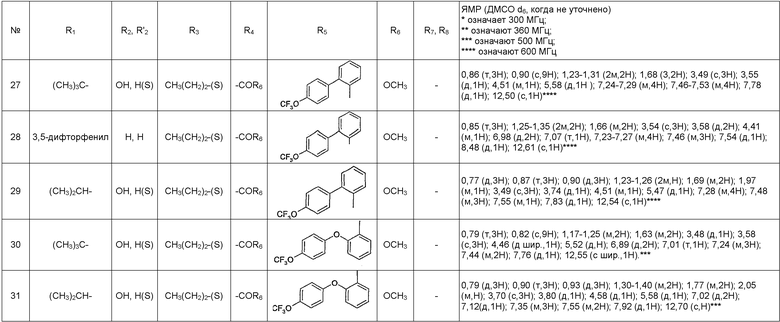
В таблице:
- (S) или (R) в колонках «R3» и «R2, R2' » указывают стереохимию асимметрического атома углерода, несущего R3 или R2, в формуле (I). Для атома углерода, несущего R2, указание (S) или (R) не касается случая, в котором R2 и R2' вместе образуют оксогруппу;
- МН+ представляет собой значение массы соединения, протонированного атомом водорода (масса соединения + 1), определенное методом ЖХ/МС.
Соединения согласно изобретению составили предмет фармакологических испытаний, которые показали их пользу как терапевтически активных веществ.
В частности, они были испытаны относительно их ингибирующего действия на продукцию β-амилоидного пептида (β-А4).
β-Амилоидный пептид (β-А4) представляет собой фрагмент более важного белка-предшественника, называемого БПА (белок-предшественник амилоида) (APP (Amyloid Precursor Protein)). Этот последний продуцируется и присутствует в различных клетках ткани животного или человека. Однако его расщепление в мозговой ткани ферментами типа протеазы, приводит к образованию пептида β-А4, который накапливается в форме амилоидных бляшек. Две протеазы, ответственные за продукцию амилоидного пептида, известны под названиями бета-секретаза и гамма-секретаза (Wolf MS, Secretase targets for Alzheimer's disease: identification and therapeutic potential, J. Med. Chem., 2001, 44(13), 2039-60).
Было показано, что упомянутое постепенное отложение пептида β-А4 является нейротоксичным и может играть важную роль в болезни Альцгеймера.
Таким образом соединения согласно настоящему изобретению в качестве ингибитора продуцирования β-амилоидного пептида (β-А4) за счет ингибирования гамма-секретазы могут быть использованы при лечении патологий, таких как старческое слабоумие, болезнь Альцгеймера, синдром Дауна, болезнь Паркинсона, амилоидная ангиопатия, и/или цереброваскулярные нарушения, лобно-височные деменции и болезнь Пика, посттравматические деменции, патологии, связанные с воспалительными процессами в нервной системе, болезнь Гентингтона и корсаковский синдром.
Испытания были проведены согласно протоколу, описанному ниже.
Для клеточного испытания β-амилоида используют линию клеток СНО-К1, совместно экспрессирующую СТ100 из БПА (АРР) и PS1 M146L клон 30-12. Клеточная линия нацелена на ингибирование гамма-секретазы. Пресенилин связан с активностью гамма-секретазы (Wolf MS, Haass C., The role of presenilins in gamma-secretase activity, (J. Biol. Chem., 2001, 276(8):5413-6)) и его совместная экспрессия с амилоидным белком или его N-концевым фрагментом приводит к увеличению секреции пептида А1-42 (β-А4), генерируя таким образом фармакологический инструмент, позволяющий оценить игибирование соединениями формулы (I) продуцирования пептида β-А4. Засев 96-тилуночных планшетов для культивирования осуществляют из расчета 1×105 клеток на лунку в 150 мкл инкубационной среды. Присутствие минимального процентного содержания (1,3% конечное) сыворотки дает возможность клеточной адгезии к пластику после 2-3 часов инкубирования при 37°С в присутствии 5% СО2. Продукты (15 мкл) тестируют 10 мкМ 1% ДМСО (конечное содержание) и инкубируют в течение 24-25 ч при 37°С в присутствии 5% СО2 при 100%-ной влажности. После упомянутого 24-25-часового инкубирования клеточные супернатанты (100 мкл) переносят в планшеты ELISA, обработанные захватывающими антителами 6Е10 (6Е10, эпитоп: аа1-17, INTERCHIM/SENETEK 320-10), чтобы определить содержание амилоидных пептидов, секретированных клетками в присутствии соединений согласно изобретению. Параллельно обрабатывают гамму контрольного синтетического пептида, «пептид 1-40», с концентрациями 5 и 10 нг/мл. Планшеты ELISA инкубируют в течение ночи при 4°С.
Количество связанного пептида детектируют косвенным способом в присутствии конкурента, соответствующего усеченному пептиду, пептид 1-28, связанный с биотином, который затем детектируют при помощи стрептавидина, связанного со щелочной фосфатазой. Субстрат, п-нитрофенилфосфат (pNPP FAST p-Nitrophenyl Phosphate, Sigma N2770), дает четкий растворимый желтый продукт реакции при 405 нм. Реакцию останавливают раствором 0,1М ЭДТК (EDTA). Для этого, после фиксации амилоидного пептида в планшете ELISA, 50 мкл биотинилированного пептида 1-28 добавляют к 100 мкл отстоявшейся клеточной жидкости и инкубируют 30 минут при комнатной температуре. Затем планшеты ELISA промывают 3 раза. После сушки перевертыванием на фильтровальную бумагу в лунку добавляют 100 мкл стрептавидин-щелочной фосфатазы (Interchim/Jackson Immunoresearch Laboratories 016-050-084) и инкубируют 1 час при комнатной температуре. Планшеты вновь промывают, затем добавляют субстрат щелочной фосфатазы (pNPP 1 мг/мл) из расчета 100 мкл на лунку. После инкубирования в течение 30 минут при комнатной температуре реакцию останавливают добавлением 100 мкл 0,1М ЭДТК на лунку и осуществляют считывание на 405 нм.
Наиболее активные соединения формулы (I) согласно настоящему изобретению имеют СЕ50 (концентрация, соответствующая эффективности 50%) меньше 500 нМ, более конкретно меньше 100 нМ. Например, соединение №13 таблицы имеет СЕ50 6нМ.
Результаты биологических испытаний показывают, что соединения являются ингибиторами образования β-амилоидного пептида (β-А4).
Следовательно, эти соединения могут быть использованы для лечения патологий, при которых ингибитор образования β-амилоидного пептида (β-А4) приносит терапевтическую пользу. В частности, такими патологиями являются старческое слабоумие, болезнь Альцгеймера, синдром Дауна, болезнь Паркинсона, амилоидная ангиопатия, цереброваскулярные нарушения, лобно-височные деменции и болезнь Пика, посттравматические деменции, патологии, связанные с воспалительными процессами в нервной системе, болезнь Гентингтона и корсаковский синдром.
Применение соединений согласно изобретению для получения лекарства, предназначенного для лечения вышеупомянутых патологий, является неотъемлемой частью изобретения.
Равным образом, предметом изобретения являются лекарственные средства, которые содержат соединение формулы (I), или соль присоединения этого последнего с фармацевтически приемлемой кислотой, или гидрат или сольват соединения формулы (I). Эти лекарственные средства находят применение в терапии, в частности, при лечении вышеупомянутых патологий.
Согласно другому из его аспектов настоящее изобретение касается фармацевтических композиций, содержащих в качестве действующего начала по меньшей мере одно соединение согласно изобретению. Эти фармацевтические композиции содержат эффективную дозу соединения согласно изобретению, или фармацевтически приемлемой соли, гидрата или сольвата вышеупомянутого соединения и, в известных случаях, один или несколько фармацевтически приемлемых эксципиентов.
Вышеупомянутые эксципиенты выбирают сообразно фармацевтической форме и желаемому способу введения из обычных эксципиентов, известных специалисту.
В фармацевтических композициях по изобретению для перорального, сублингвального, подкожного, внутримышечного, внутривенного, топического, местного, интратрахеального, интраназального, чрескожного или ректального введения вышеупомянутое действующее начало формулы (I), его соль, его сольват или его возможный гидрат могут быть введены в разовой форме введения, в смеси с классическими фармацевтическими эксципиентами, животным и людям для профилактики или лечения расстройств или болезней, упомянутых выше.
Соответствующие разовые формы введения включают в себя формы для перорального приема, такие как таблетки, мягкие или твердые желатиновые капсулы, порошки, гранулы, жевательные резинки и растворы или суспензии для перорального приема; формы для сублингвального, буккального, интратрахеального, внутриглазного, интраназального, ингаляционного введения, формы для подкожного, внутримышечного или внутривенного введения и формы для ректального или вагинального введения. Для топического нанесения можно использовать соединения согласно изобретению в форме кремов, помад или лосьонов.
В качестве примера, разовая форма введения соединения согласно изобретению в виде таблетки может содержать следующие компоненты:
- соединение согласно изобретению 50 мг;
- манит 223,75 мг;
- натрийсодержащая кроскарамеллоза 6,0 мг;
- кукурузный крахмал 15,0 мг;
- гидроксипропилметилцеллюлоза 2,25 мг;
- стеарат магния 3,0 м.
Чтобы получить желаемый профилактический или терапевтический эффект, доза действующего начала может варьировать от 0,1 до 200 мг на кг массы тела в день. Хотя указанные дозировки будут являться примерами для ситуации в среднем, могут быть особые случаи, в которых применяют более высокие или более низкие дозировки, такие дозировки равным образом принадлежат изобретению. Согласно обычной практике, дозировка, соответствующая каждому пациенту, определяется врачом сообразно способу введения, массе и ответной реакции вышеупомянутого пациента.
Каждая разовая доза может содержать от 0,1 до 1000 мг, предпочтительно от 0,1 до 500 мг действующего начала в сочетании с одним или несколькими фармацевтическими эксципиентами. Указанная разовая доза может вводиться от 1 до 5 раз в день таким образом, чтобы ввести дневную дозу от 0,5 до 5000 мг, предпочтительно от 0,5 до 2500 мг.
Согласно другому своему аспекту, настоящее изобретение касается также способа лечения патологий, указанных выше, который заключается во введении соединения согласно изобретению, фармацевтически приемлемой соли или гидрата вышеупомянутого соединения.
Настоящее изобретение относится к соединениям общей формулы (I) в форме основания или фармацевтически приемлемой соли присоединения с кислотой. Соединения настоящего изобретения обладают свойствами ингибитора образования β-амилоидного пептида (β-А4). В формуле (I)
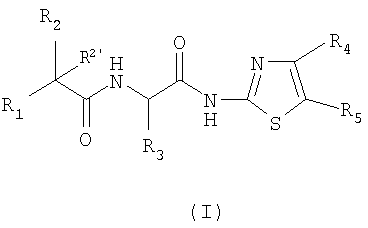
R1 обозначает: C1-6-алкил или фенил; причем указанные фенильные группы замещены двумя заместителями, выбранными из атомов галогена; R2 и R2' обозначают, независимо один от другого, атом водорода или гидроксигруппу; R3 обозначает C1-6-алкил; один или другой из
радикалов R4 и R5 представляет собой группу Z
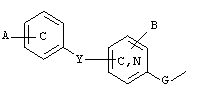
и один или другой из радикалов R4 и R5 представляет собой группу -С(Х)R6; G обозначает простую связь; Y обозначает простую связь, атом кислорода, атом серы, C1-4-алкиленовую группу; А и В обозначают, независимо один от другого, атом водорода, галоген, трифторметил, трифторметоксигруппу; при условии, что если Y представляет собой простую связь или атом кислорода и если группа Z представляет
собой группу типа 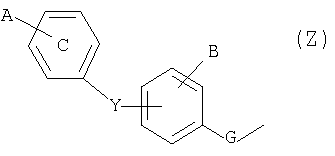 тогда А отлично от атома водорода; Х обозначает атом кислорода; R6 обозначает C1-6-алкоксигруппу. Изобретение также относится к способу получения соединений формулы (I), к лекарственному средству и фармацевтической композиции на их основе, и к применению соединений формулы (I) для получения лекарственного средства. 5 н. и 1 з.п. ф-лы, 1 табл.
тогда А отлично от атома водорода; Х обозначает атом кислорода; R6 обозначает C1-6-алкоксигруппу. Изобретение также относится к способу получения соединений формулы (I), к лекарственному средству и фармацевтической композиции на их основе, и к применению соединений формулы (I) для получения лекарственного средства. 5 н. и 1 з.п. ф-лы, 1 табл.
1. Соединение, отвечающее общей формуле (I)
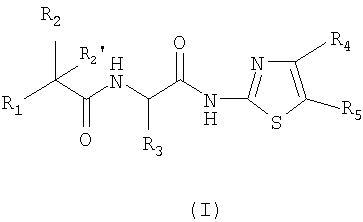
в которой R1 обозначает C1-6-алкил или фенил; причем указанные фенильные группы замещены двумя заместителями, выбранными из атомов галогена;
R2 и R2' обозначают, независимо один от другого, атом водорода или гидроксигруппу;
R3 обозначает C1-6-алкил;
один или другой из радикалов R4 и R5 представляет собой группу Z
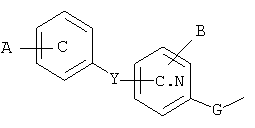
и один или другой из радикалов R4 и R5 представляет собой группу -С(Х)R6;
G обозначает простую связь;
Y обозначает простую связь, атом кислорода, атом серы, С1-4-алкиленовую группу;
А и В обозначают, независимо один от другого, атом водорода, галоген, трифторметил, трифторметоксигруппу; при условии, что, если Y представляет собой простую связь или атом кислорода и если группа Z представляет собой группу типа
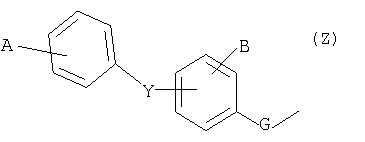
тогда А отлично от атома водорода;
Х обозначает атом кислорода;
R6 обозначает C1-6-алкоксигруппу;
в форме основания или фармацевтически приемлемой соли присоединения с кислотой.
2. Способ получения соединения формулы (I) по п.1, включающий стадию, заключающуюся в осуществлении пептидного связывания соединения формулы (IV)
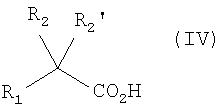
с амином формулы (VI)
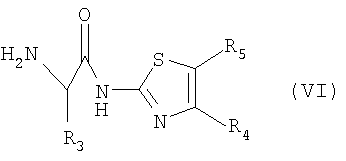
в которых R1, R2, R2' R3, R4, R5 являются такими, как определенные в формуле (I) по п.1.
3. Лекарственное средство, обладающее свойствами ингибитора образования β-амилоидного пептида (β-А4), отличающееся тем, что оно содержит соединение формулы (I) по п.1 в форме фармацевтически приемлемых основания или соли.
4. Фармацевтическая композиция, обладающая свойствами ингибитора образования β-амилоидного пептида (β-А4), содержащая по меньшей мере одно соединение формулы (I) по п.1 в форме фармацевтически приемлемых основания или соли и, в известных случаях, один или несколько фармацевтически приемлемых эксципиентов.
5. Соединение формулы (I) по п.1 в форме фармацевтически приемлемых основания или соли, предназначенное для получения лекарственного средства, обладающего свойствами ингибитора образования β-амилоидного пептида (β-А4).
6. Применение соединения формулы (I) по п.1 в форме фармацевтически приемлемых основания или соли для получения лекарственного средства, предназначенного для лечения старческого слабоумия, болезни Альцгеймера, синдрома Дауна, болезни Паркинсона, амилоидной ангиопатии, цереброваскулярных нарушений, лобно-височных деменций и болезни Пика, посттравматических деменций, патологий, связанных с воспалительными процессами в нервной системе, болезни Гентингтона и/или корсаковского синдрома.
| Wolfe M.S | |||
| "Therapeutic strategies for Alzheimer's disease" NATURE REVIEWS | |||
| DRUG DISCOVERY, 1(11), 2002, 859-866 | |||
| ПРОИЗВОДНЫЕ ГИДРОКСАМОВОЙ ИЛИ КАРБОНОВОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКАЯ ИЛИ ВЕТЕРИНАРНАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2136657C1 |
| WO 03014095 A1, 20.02.2003 | |||
| EA 200200247 A1, 29.08.2002 | |||
| WO 9822433 A1, 28.05.1998. | |||

