Объектом изобретения являются производные ациламинотиазола, их получение и их применение в терапии.
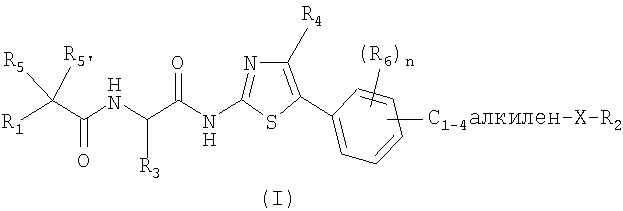
Первым объектом изобретения являются соединения, отвечающие общей формуле (I):
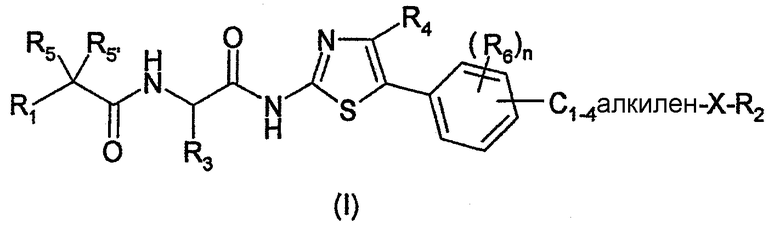
в которой:
X обозначает атом кислорода или серы;
R1 обозначает С1-10-алкил, в случае необходимости замещенный C3-7-циклоалкилом, фенилом, тиенилом; или R1 обозначает C3-7-циклоалкил, тиенил, пиридинил, пиримидинил;
тиенильные группы в случае необходимости могут быть замещены 1-3 С1-3-алкильными группами; фенил может быть в случае необходимости замещен 1-5 атомами галогена или C1-3-алкильными группами, C1-3-алкокси-группами, С1-3-фторалкильными группами, С1-3-фторалкокси-группами;
R2 обозначает C1-6-алкил, в случае необходимости замещенный C3-7-циклоалкилом, фенилом, C1-3-алкоксилом, гидроксилом; или R2 обозначает С3-7-циклоалкил, пиперидинил, фенил или пиридинил;
C3-7-циклоалкил и пиперидинил в случае необходимости могут быть замещены одной или несколькими группами C1-3-алкила, C1-3-алкоксила, гидроксила, C1-3-фторалкила, C1-3-фторалкоксила;
фенил и пиридинил в случае необходимости могут быть замещены одним или несколькими атомами галогена или группами CN, C1-3-алкила, C1-3-алкоксила, гидроксила, Cl-3-фторалкила, C1-3-фторалкоксила;
R3 обозначает атом водорода или С1-6-алкил, который может быть замещен С3-7-циклоалкилом;
R4 обозначает атом водорода или С1-6-алкил;
R5 и R5' обозначают, независимо один от другого, атом водорода, галогена, гидроксил, C1-3-алкил; или R5 и R5' вместе образуют оксо-группу или оксимную группу, такую как:
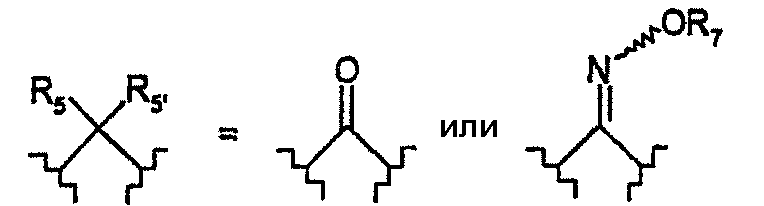
где R7 обозначает атом водорода или C1-3-алкил;
n обозначает целое число от 0 до 3; и
R6 обозначает, независимо один от другого, когда n=2 или 3, атом водорода, галогена, гидроксил, C1-3-алкил, С1-3-алкоксил, C1-3-фторалкил или C1-3-фторалкоксил.
Из соединений общей формулы (I), подгруппа предпочтительных соединений представлена соединениями, в которых:
- X обозначает атом кислорода или серы; и/или
- R1 обозначает C1-5-алкил, предпочтительно, метил, этил, 1-метилэтил, 1,1-диметилэтил, пропил, 1-метилпропил, 2-метилпропил, 1-этилпропил, которые могут быть замещены фенилом, тиенилом; или R1 обозначает C3-7-циклоалкил, предпочтительно, циклогексил, тиенил, пиридинил; тиенильные группы могут быть замещены одним или двумя C1-3-алкильными, предпочтительно, метильными группами; фенил может быть замещен одним или двумя атомами галогена, предпочтительно, хлора или фтора; и/или
- R2 обозначает C1-6-алкил, предпочтительно, этил, 1-метилэтил; или R2 обозначает C3-7-циклоалкил, предпочтительно, циклогексил, фенил или пиридинил; причем фенил может быть замещен 1-3 группами CN, C1-3-алкилами, предпочтительно, метилом, этилом, C1-3-алкоксилами, предпочтительно, метокси, этокси, гидроксилами, фторалкоксилами, предпочтительно, трифторметокси, или атомами галогена, предпочтительно хлора, фтора; и/или
- R3 обозначает C1-6-алкил, предпочтительно, метил, этил, пропил; и/или
- R4 обозначает атом водорода или C1-6-алкил, предпочтительно, метил или 4-метилпентил; и/или
- R5 и R5' обозначают, независимо один от другого, атом водорода, гидроксил; или R5 и R5' вместе образуют оксо-группу; и/или
- R6 обозначает атом водорода, галогена, предпочтительно, хлора или фтора, C1-3-алкил, предпочтительно, метил, C1-3-алкоксил, предпочтительно, метокси или этокси; и/или
- n=0 или 1.
Соединения, в которых одновременно X, R1, R2, R3, R4, R5, R5', R6 и n имеют значения, определенные выше для подгруппы предпочтительных соединений, являются особенно предпочтительными, и наиболее предпочтительными среди них являются соединения, в которых:
X обозначает атом кислорода; и/или
C1-4-алкилен является метиленом; и/или
атом углерода, несущий группу R3 имеет конфигурацию (S).
В качестве примера предпочтительных соединений можно назвать следующие соединения:
1. (2S)-2-{[(2R)-2-циклогексил-2-гидроксиацетил]амино}-N-(5-{2-[(циклогексилокси)метил]фенил}-1,3-тиазол-2-ил)пентанамид;
2. (2S)-2-{[(2S)-2-циклогексил-2-гидроксиацетил]амино}-N-(5-{2-[(циклогексилокси)метил]фенил}-1,3-тиазол-2-ил)пентанамид;
3. (2S)-N-(5-{2-[(циклогексилокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[2-(3-пиридинил)ацетил]амино}пентанамид;
4. N-((1S)-1-{[(5-{2-[(циклогексилокси)метил]фенил}-1,3-тиазол-2-ил)амино]карбонил}бутил)-2-гидрокси-4-метилпентанамид;
5. (2S)-N-{5-[2-(изопропоксиметил)фенил]-4-метил-1,3-тиазол-2-ил}-2-{[2-(3-тиенил)ацетил]амино}пентанамид;
6. (2S)-N-{5-[2-(изопропоксиметил)фенил]-1,3-тиазол-2-ил}-2-{[2-(3-тиенил)ацетил]амино}пентанамид;
7. (2S)-N-{5-[2-(изопропоксиметил)фенил]-1,3-тиазол-2-ил}-2-{[3-(3-тиенил)пропаноил]амино}пентанамид;
8. (2S)-N-{5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил}-2-{[2-(3-тиенил)ацетил]амино}пентанамид;
9. (2S)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}-N-{5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил}пентанамид;
10. (2S)-N-{5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил}-2-{[2-(2-тиенил)ацетил]амино}бутанамид;
11. (2S)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}-N-{5-[2-(изопропоксиметил)фенил]-1,3-тиазол-2-ил}пентанамид;
12. (2S)-N-{5-[2-(изопропоксиметил)фенил]-1,3-тиазол-2-ил}-2-{[2-(2-тиенил)ацетил]амино}пентанамид;
13. (2S)-2-[(3,3-диметилбутаноил)амино]-N-{5-[2-(изопропоксиметил)фенил]-1,3-тиазол-2-ил}пентанамид;
14. N-{(1S)-1-[({5-[2-(изопропоксиметил)фенил]-1,3-тиазол-2-ил}амино)карбонил]бутил}-3-метил-2-оксопентанамид;
15. (2S)-N-{5-[2-(этоксиметил)фенил]-1,3-тиазол-2-ил}-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
16. (2S)-2-{[2-(2,5-диметил-3-тиенил)ацетил]амино}-N-{5-[2-(этоксиметил)фенил]-1,3-тиазол-2-ил}пентанамид;
17. (2S)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}-N-[5-(2-{[3-(трифторметокси)фенокси]метил}фенил)-1,3-тиазол-2-ил]пентанамид;
18. (2S)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}-N-{5-[4-метокси-2-(феноксиметил)фенил]-1,3-тиазол-2-ил}пентанамид;
19. (2S)-N-(5-{2-[(2-фторфенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
20. (2S)-N-(5-{2-[(2-этилфенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
21. (2S)-2-{[3-(2,4-дихлорфенил)пропаноил]амино}-N-{5-[2-{феноксиметил)фенил]-1,3-тиазол-2-ил}пентанамид;
22. (2S)-2-{[2-(5-метил-2-тиенил)ацетил]амино}-N-{5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил}пентанамид;
23. (2S)-N-{5-{2-[(2,3-диметоксифенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
24. (2S)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}-N-[5-(2-{[2-(трифторметокси)фенокси]метил}фенил)-1,3-тиазол-2-ил]пентанамид;
25. (2S)-N-(5-{2-[(3,5-диметоксифенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
26. (2S)-N-(5-{2-[(2,3-диметилфенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
27. (2S)-N-(5-{2-[(3,4-диметилфенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
28. (2S)-N-(5-{2-[(2,6-диметилфенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
29. (2S)-N-(5-{2-[(3-хлорфенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
30. (2S)-N-(5-{2-[(3,4-диметоксифенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
31. (2S)-N-(5-{2-[(2,6-диметоксифенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
32. (2S)-N-(5-{2-[(2,4-диметилфенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
33. (2S)-N-(5-{2-[(2,5-диметилфенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
34. (2S)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}-N-{4-метил-5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил}пентанамид;
35. (2R)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}-N-{5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил}пентанамид;
36. (2S)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}-N-(5-{2-[(2-метилфенокси)метил]фенил}-1,3-тиазол-2-ил}пентанамид;
37. (2S)-2-{[(2S)-2-гидрокси-3-метилбутаноил]амино}-N-{5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил}пентанамид;
38. (2S)-2-[(2-гидрокси-3,3-диметилбутаноил)амино]-N-(5-{2-[(2-метоксифенокси)метил]фенил}-1,3-тиазол-2-ил)пентанамид;
39. (2S)-N-(5-{2-[(2-этоксифенокси)метил]фенил}-1,3-тиазол-2-ил)-2-[(2-гидрокси-3,3-диметилбутаноил)амино]пентанамид;
40. (2S)-2-[(2-гидрокси-3-фенилпропаноил)амино]-N-{5-[2-(феноксиметил)-фенил]-1,3-тиазол-2-ил}пентанамид;
41. (2S)-N-(5-{2-[(2,6-дихлорфенокси)метил]фенил}-1,3-тиазол-2-ил)-2-[(2-гидрокси-3,3-диметилбутаноил)амино]пентанамид;
42. (2R)-3-этил-2-гидрокси-N-{(1S)-1-[({5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил}амино)карбонил]бутил}пентанамид;
43. (2S)-3-этил-2-гидрокси-N-{(1S)-1-[({5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил}амино)карбонил]бутил}пентанамид;
44. (2S)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}-N-{5-[2-(изопропоксиметил)-4-метоксифенил]-1,3-тиазол-2-ил}пентанамид;
45. (2S)-N-(5-{2-[(2-(хлор-6-метилфенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
46. (2S)-N-(5-{2-[(2,6-дифторфенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
47. (2S)-N-{5-[4-хлор-2-(феноксиметил)фенил]-1,3-тиазол-2-ил}-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
48. (2S)-N-{5-[4-фтор-2-(феноксиметил)фенил]-1,3-тиазол-2-ил}-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
49. (2S)-2-{[(2S)-2-гидрокси-3-метилбутаноил]амино}-N-{5-[4-метокси-2-(феноксиметил)фенил]-1,3-тиазол-2-ил}пентанамид;
50. (2S)-N-(5-{2-[(3,4-дихлорфенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
51. (2S)-N-((1S)-1-{[(5-{2-[(циклогексилокси)метил]фенил}-1,3-тиазол-2-ил}амино]карбонил}бутил)-2-гидрокси-4-метилпентанамид;
52. (2S)-N-{5-[4-этокси-2-(феноксиметил)фенил]-1,3-тиазол-2-ил}-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
53. (2S)-N-{5-[4-этокси-2-(феноксиметил)фенил]-1,3-тиазол-2-ил}-2-{[(2S)-2-гидрокси-3-метилбутаноил]амино}пентанамид;
54. (2S)-N-{5-[5-фтор-2-(феноксиметил)фенил]-1,3-тиазол-2-ил}-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
55. (2S)-N-{5-[5-хлор-2-(феноксиметил)фенил]-1,3-тиазол-2-ил}-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
56. (2S)-N-{5-[5-фтор-2-(феноксиметил)фенил]-1,3-тиазол-2-ил}-2-{[(2S)-2-гидрокси-3-метилбутаноил]амино}пентанамид;
57. (2S)-N-(5-{2-[(циклогексилокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
58. (2S)-2-гидрокси-3,3-диметил-N-[(1S)-1-метил-2-оксо-2-({5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил}амино)этил]бутанамид;
59. (2S)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}-N-{5-[4-метил-2-(феноксиметил)фенил]-1,3-тиазол-2-ил}пентанамид;
60. (2S)-2-{[(2S)-2-гидрокси-3-метилбутаноил]амино}-N-{5-[5-метил-2-(феноксиметил)фенил]-1,3-тиазол-2-ил}пентанамид;
61. (2S)-N-(5-{2-[(3-цианофенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
62. (2S)-N-(5-{2-[(3-фторфенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
63. (2S)-N-(5-{2-[(3-фторфенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3-метилбутаноил]амино}пентанамид;
64. (2S)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}-N-{5-[5-метил-2-(феноксиметил)фенил]-1,3-тиазол-2-ил}пентанамид;
65. (2S)-2-{[(2S)-2-гидрокси-3-метилбутаноил]амино}-N-{5-[4-метил-2-(феноксиметил)фенил]-1,3-тиазол-2-ил}пентанамид;
66. (2S)-N-((1S)-2-{[(5-{2-[(циклогексилокси)метил]фенил}-1,3-тиазол-2-ил}-амино]-1-метил-2-оксоэтил}-2-гидрокси-3,3-диметилбутанамид;
67. (2S)-N-(5-{2-[(2-хлор-5-метилфенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
68. (2S)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}-N-(5-{2-[(3-метилфенокси)метил)фенил}-1,3-тиазол-2-ил)пентанамид
69. (2S)-N-(5-{2-[(2-цианофенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
70. (2S)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}-N-(5-{2-[(4-пиридинилокси)метил]фенил}-1,3-тиазол-2-ил)пентанамид;
71. (2S)-N-(5-{2-[(2-хлор-4,5-диметилфенокси))метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
72. (2S)-N-(5-{2-[(4-хлор-3-метилфенокси))метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
73. (2S)-N-(5-{2-[(2,3-дихлорфенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
74. (2S)-N-(5-{2-[(2,3-дифторфенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид;
75. (2S)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}-N-{4-(4-метилпентил)-5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил)пентанамид.
Объектом изобретения также являются, среди соединений общей формулы (I), соединения общей формулы (I'):

(I')
в которой:
X обозначает атом кислорода или серы;
R1 обозначает C1-10-алкил, в случае необходимости замещенный C3-7-циклоалкилом, фенилом, тиенилом; или R1 обозначает C3-7-циклоалкил, тиенил, пиридинил, пиримидинил;
тиенильные группы в случае необходимости могут быть замещены 1-3 С1-3-алкильными группами; фенил может быть в случае необходимости замещен 1-5 атомами галогена или C1-3-алкильными группами, C1-3-алкокси-группами, С1-3-фторалкильными группами, С1-3-фторалкокси-группами;
R2 обозначает C1-6-алкил, в случае необходимости замещенный C3-7-циклоалкилом, фенилом, C1-3-алкоксилом, гидроксилом; или R2 обозначает С3-7-циклоалкил, пиперидинил, фенил или пиридинил;
C3-7-циклоалкил и пиперидинил в случае необходимости могут быть замещены одной или несколькими группами C1-3-алкила, C1-3-алкоксила, гидроксила, C1-3-фторалкила, C1-3-фторалкоксила;
фенил и пиридинил в случае необходимости могут быть замещены одним или несколькими атомами галогена или группами C1-3-алкила, C1-3-алкоксила, гидроксила, C1-3-фторалкила, C1-3-фторалкоксила;
R3 обозначает атом водорода или С1-6-алкил, который может быть замещен С3-7-циклоалкилом;
R4 обозначает атом водорода или С1-4-алкил;
R5 и R5' обозначают, независимо один от другого, атом водорода, галогена, гидроксил, C1-3-алкил; или R5 и R5' вместе образуют оксо-группу или оксимную группу, такую как:

где R7 обозначает атом водорода или C1-3-алкил;
n обозначает целое число от 0 до 3; и
R6 обозначает, независимо один от другого, когда n=2 или 3, атом водорода, галогена, гидроксил, C1-3-алкил, C1-3-алкоксил, C1-3-фторалкил или C1-3-фторалкоксил.
Из соединений общей формулы (I') подгруппа предпочтительных соединений представлена соединениями, в которых:
- X обозначает атом кислорода или серы; и/или
- R1 обозначает C1-5-алкил, предпочтительно, метил, этил, 1-метилэтил, 1,1-диметилэтил, пропил, 1-метилпропил, 2-метилпропил, 1-этилпропил, которые могут быть замещены фенилом, тиенилом; или R1 обозначает C3-7-циклоалкил, предпочтительно, циклогексил, тиенил, пиридинил; тиенильные группы могут быть замещены одним или двумя C1-3-алкильными, предпочтительно, метильными группами; фенил может быть замещен одним или двумя атомами галогена, предпочтительно, хлора или фтора; и/или
- R2 обозначает C1-6-алкил, предпочтительно, этил, 1-метилэтил; или R2 обозначает C3-7-циклоалкил, предпочтительно, циклогексил, или фенил;
причем фенил может быть замещен одним или двумя C1-3-алкильными, предпочтительно, метильными, этильными, C1-3-алкокси-, предпочтительно, метокси-, этокси-, гидроксильными, фторалкокси-, предпочтительно, трифторметокси-, группами или одним или двумя атомами галогена, предпочтительно хлора, фтора; и/или
- R3 обозначает C1-6-алкил, предпочтительно, этил, пропил; и/или
- R4 обозначает атом водорода или C1-4-алкил, предпочтительно, метил; и/или
- R5 и R5' обозначают, независимо один от другого, атом водорода, гидроксил; или R5 и R5' вместе образуют оксо-группу; и/или
- R6 обозначает атом водорода, галогена, предпочтительно, хлора или фтора, C1-3-алкоксил, предпочтительно, метокси; и/или
- n=0 или 1.
В рамках изобретения понимают под:
- Ct-z, где t и z могут принимать значения от 1 до 10, углеродную цепь, которая может содержать от t до z атомов углерода, например, С1-3-углеродную цепь, которая может содержать от 1 до 3 атомов углерода, С3-6-углеродную цепь, которая может содержать от 3 до 6 атомов углерода, …;
- алкилом, прямую или разветвленную насыщенную алифатическую группу, например, С1-6-алкил обозначает прямую или разветвленную углеродную цепь из 1-6 атомов углерода, в частности, метил, этил, пропил, 1-метилэтил, бутил, 1-метилпропил, 2-метилпропил, 1,1-диметилэтил, …, предпочтительно, метил, этил, пропил или 1-метилэтил;
- алкиленом, двухвалентную алкильную группу;
- циклоалкилом, циклическую алкильную группу, например, C3-7-циклоалкил обозначает углеродный цикл из 3-7 атомов углерода, в частности, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, предпочтительно, циклопентил или циклогексил;
- алкоксилом, -О-алкильную группу, где алкил имеет указанные выше значения;
- фторалкилом, алкильную группу, в которой один или несколько атомов водорода замещены атомом фтора;
- фторалкоксилом, алкокси-группу, в которой один или несколько атомов водорода замещены атомом фтора; и
- атомом галогена, фтор, хлор, бром или иод.
Соединения общей формулы (I) могут содержать один или несколько асимметрических атомов углерода. Таким образом, они могут существовать в форме энантиомеров или диастереоизомеров. Эти энантиомеры, диастереоизомеры, а также их смеси, включая рацемические смеси, составляют часть изобретения.
Соединения формулы (I) могут существовать в виде оснований или солей присоединения с кислотами. Такие соли присоединения составляют часть изобретения.
Эти соли предпочтительно получают с фармацевтически приемлемыми кислотами, однако, соли с другими кислотами, пригодными, например, для очистки или выделения соединений формулы (I), также составляют часть изобретения.
Соединения общей формулы (I) могут находиться в форме гидратов или сольватов, а именно, в форме ассоциаций или комбинаций с одной или несколькими молекулами воды или с растворителем. Такие гидраты и сольваты также составляют часть изобретения.
В дальнейшем описании под отделяемой группой понимают группу, которая может быть легко отщеплена от молекулы с удалением электронной пары, путем разрыва гетеролитической связи. Эта группа может, таким образом, быть легко заменена на другую группу, например, в ходе реакции замещения. Такими отделяемыми группами являются, например, галогены или активированная гидроксильная группа, такая как мезилатная, тозилатная, трифлатная, ацетильная группы… и т.д. Примеры отделяемых групп, а также источники, в которых описано их получение, приведены в Advanced Organic Chemistry, J. March, 3rd Edition, Wiley Interscience, p 310-316.
Под защитной группой понимают группу, которая позволяет устранить реакционоспособность функциональной группы или положения в ходе химической реакции, которая может ее (его) затронуть, и которая приводит к исходной молекуле после отщепления известными специалисту методами. Примеры защитных групп, а также методы установления и снятия защиты, приведены, в числе прочих, Protective groups in Organic Synthesis, Greene et al., 2nd Ed. (John Wiley & Sons, Inc., New York).
Вторым объектом изобретения являются способы получения соединений формулы (I).
Так, эти соединения могут быть получены способами, иллюстрируемыми нижеследующими схемами, рабочие условия которых являются обычными известными специалисту.
Схема 1
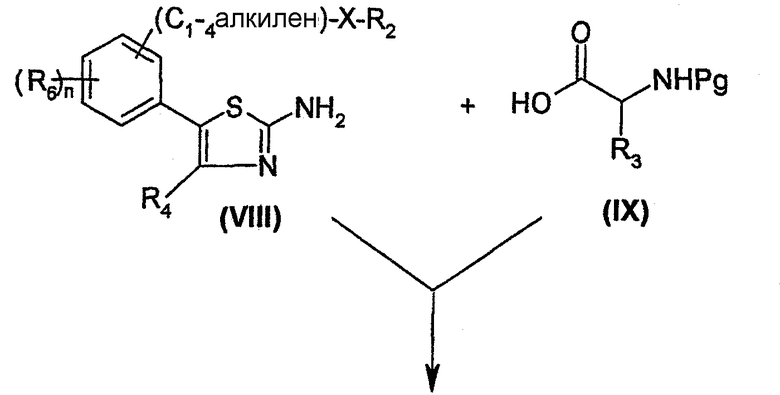
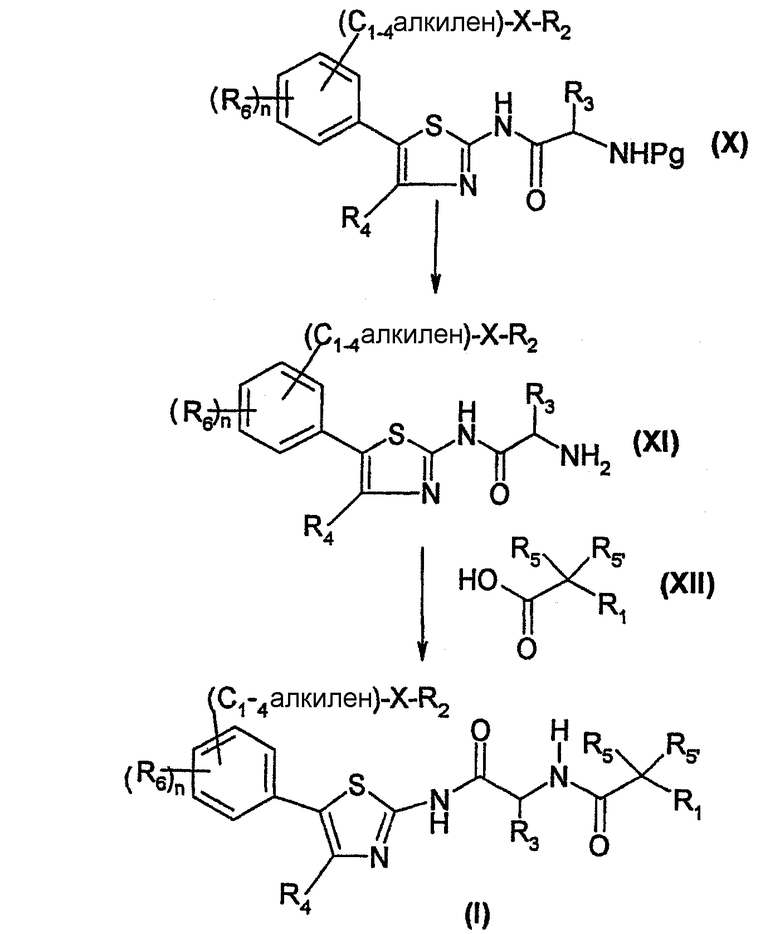
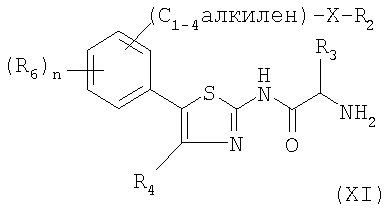
Согласно схеме 1, соединение формулы (I) может быть получено пептидным связыванием амина формулы (XI) с кислотой формулы (XII) в условиях, известных специалисту, например, в присутствии гексафторфосфата бензотриазол-1-илокси-трис(диметиламино) фосфония (ВОР) и N-этилморфолина или N-метилморфолина в инертном растворителе, таком как диметилформамид, ацетонитрил или дихлорметан при температуре в интервале от 0°C до комнатной температуры.
Амин формулы (XI) получают пептидным связыванием амина формулы (VIII) с аминокислотой формулы (IX), в которой Pg обозначает защитную группу, в условиях, описанных выше, с получением соединения формулы (X). Аминокислоту формулы (IX) защищают, например, при помощи N-трет.-бутилоксикарбонила (Boc). Затем удаляют защиту с соединения (X) известными специалисту методами, получая амин формулы (XI). Например, если используемой защитной группой является Boc, ее удаляют с помощью кислотного гидролиза в присутствии безводной газообразной соляной кислоты.
Соединение формулы (VIII) может быть получено согласно схеме 2.
Схема 2
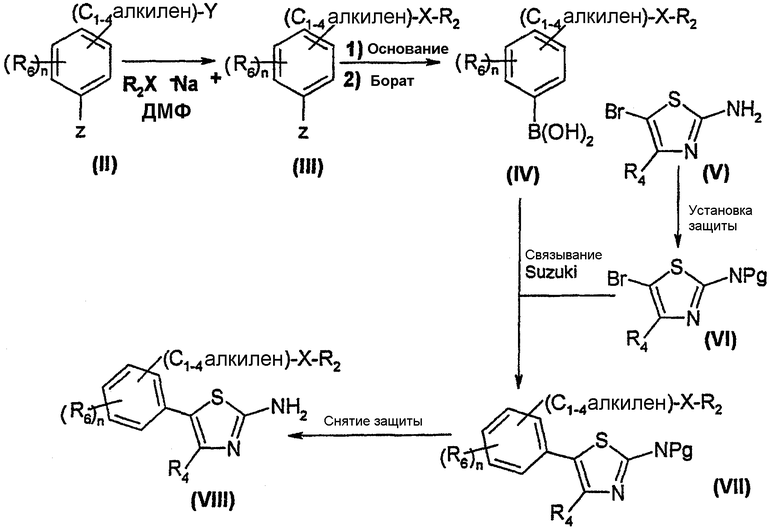
Согласно этой схеме, аралкил формулы (II), в которой Y обозначает отделяемую группу, предпочтительно, атом галогена, такой как атом брома, и Z обозначает атом галогена, такой как атом брома, конденсируют с алкоголятом или тиолатом щелочного металла, например, формулы R2X-Na+, где Х обозначает атом кислорода или серы. Реакцию проводят в инертном растворителе, таком как диметилформамид, при температуре от 0°C до 50°C, получая соединение формулы (III). Арил формулы (III) превращают в бороновую кислоту формулы (IV) способом, являющимся адаптацией способа, описанного Schoevaars, J. Am. Chem. Soc., 1999, 121, 9550-9561. Это превращение может, например, быть осуществлено путем предварительного образования аниона соединения (III), например, действием сильного основания, такого как бутиллитий, в эфирном растворителе, таком как тетрагидрофуран, при температурах от -50°C до -80°C. Этот анион затем вводят в реакцию с боратом, таким как триметилборат, с получением, в результате гидролиза, бороновой кислоты формулы (IV).
Связывание бороновой кислоты (IV) с 5-бромтиазолом формулы (VI), в которой Pg обозначает защитную группу, такую как иминогруппа, например, дифенилкетонимин, может быть осуществлено в соответствии с реакцией по Suzuki, с помощью способа, являющегося адаптацией способа, описанного Wolfe, J. Org. Chem., 1997, 62, 4943-4948, с получением 5-фенилтиазола формулы (VII). Связывание осуществляют, например, в эфирном растворителе, таком как диоксан, в присутствии тригидрата фосфата трикалия и катализатора, такого как тетракис(трифенилфосфин) палладия (0), при температуре в интервале от комнатной температуры до температуры кипения растворителя. С полученного таким образом 5-фенилтиазола формулы (VII) удаляют защитную группу известными специалисту методами, получая 5-фенил-2-аминотиазол формулы (VIII).
5-бромтиазол формулы (VI) получают, устанавливая защиту аминогруппы соответствующего соединения формулы (V). Предпочтительно, ее защищают в форме дифенилкетонимина в известных специалисту условиях.
Исходные соединения, в частности соединения формул (II), (V), (IX) и (XII), если способ их получения не описан, доступны на рынке или известны из литературы, или могут быть получены описанными в ней способами, или известны специалисту.
Например, 5-бром-2-амино-тиазол (V) может быть получен бромированием соответствующего 2-амино-тиазола согласно способу, являющемуся адаптацией способа, описанного Kaye, J. Chem. Soc. Perkins I, 1981, 2335-2339.
Например, соединение формулы (XII) может быть получено способом, являющимся адаптацией способа, описанного Middleton et al., J. Org. Chem., 45, 14, 1980, 2883-2887 и Miyamoto et al., J. Amer. Chem. Soc., 114, 15, 1992, 6256-6257.
Значения X, R1, R2, R3, R4, R5, R5', R6 и n в соединениях формул (II)-(XII) соответствуют значениям, определенным для соединений формулы (I).
В следующих примерах описано получение некоторых соединений согласно изобретению. Эти примеры не являются ограничительными и служат только для иллюстрации изобретения. Номера соединений, представленных в примерах, соответствуют номерам в следующей далее таблице. Микроанализы элементарного состава и спектры ЯМР, ИК или масс-спектры подтверждают структуру полученных соединений.
Пример 1 (соединение 9)
(2S)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}-N-{5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил}пентанамид
1.1. 1-бром-2-(феноксиметил)бензол
К 20,2 г фенола в растворе в 150 мл диметилформамида добавляют порциями при 5°C 1,2 г гидрида натрия (в виде 50%-й суспензии в масле). Перемешивают при комнатной температуре и вводят при 5°C 37,2 г 2-бромбензилбромида в растворе в 15 мл диметилформамида. Через 2 часа при 20°C реакционную среду выливают на смесь воды со льдом и экстрагируют этилацетатом. Органическую фазу высушивают над безводным сульфатом натрия и концентрируют, получая 36 г масла.
ЯМР 1H δ в ч.млн (ДМСО d6): 5,22 (с, 2H); 7,09-7,67 (мультиплет, 9H).
1.2. 2-(феноксиметил)фенилбороновая кислота
К 36 г 1-бром-2-(феноксиметил)бензола, полученного на стадии 1.1, в растворе в 150 мл тетрагидрофурана, добавляют по каплям при -70°C, 90 мл n-бутиллития (1,6 M) в растворе в гексане. Через 2 часа при -70°C по каплям добавляют 16 мл триметилбората. Дают температуре среды подняться до -30°C. Осуществляют гидролиз с помощью насыщенного раствора хлорида аммония, затем экстрагируют этилацетатом и высушивают органическую фазу безводным сульфатом натрия. После упаривания получают 33 г твердого вещества белого цвета.
ЯМР 1H δ в ч.млн (ДМСО d6): 5,25 (с, 2H); 6,85-7,67 (мультиплет, 11H).
1.3. 5-бром-N-(дифенилметилен)-1,3-тиазол-2-амин
К 34 г 5-бром-1,3-тиазол-2-аминбромгидрата в суспензии в 300 мл 1,2-дихлорэтана добавляют 26 г бензофенонимина. Смесь кипятят в течение 18 часов с обратным холодильником. Отфильтровывают сформировавшийся осадок и концентрируют фильтрат, получая 37,2 г твердого вещества.
Т.пл.=109°C.
ЯМР 1H δ в ч.млн (ДМСО d6): 7,34 (м, 2H); 7,50-7,76 (мультиплет, 9H).
1.4. 5-{2-[(фенокси)метил]фенил}-N-(дифенилметилен)-1,3-тиазол-2-амин
В 14,8 г 2-(феноксиметил)фенилбороновой кислоты, полученной на стадии 1.2, в растворе 250 мл 1,4-диоксана последовательно вводят 15 г дигидрата фосфата трикалия, 10,5 г 5-бром-N-(дифенилметилен)-1,3-тиазол-2-амина, полученного на стадии 1.3, 1,5 г тетракис(трифенилфосфина) палладия (0) и кипятят 1 час с обратным холодильником. Реакционную среду упаривают досуха, помещают в этилацетат и промывают водой. Органическую фазу высушивают над безводным сульфатом натрия и концентрируют растворители. Остаток хроматографируют на колонке с силикагелем, используя в качестве элюента дихлорметан, и получают 35 г масла желтого цвета.
ЯМР 1H δ в ч.млн: 4,81 (с, 2H); 7,17-7,83 (мультиплет, 20H).
1.5. 5-{2-[(фенокси)метил]фенил}-1,3-тиазол-2-амин
К 35 г 5-{2-[(фенокси)метил]фенил}-N-(дифенилметилен)-1,3-тиазол-2-амина, полученного на стадии 1.4, в растворе в 250 мл метанола, добавляют 150 мл водного раствора соляной кислоты (1 M) и перемешивают 18 часов при 20°C. Упаривают досуха, помещают остаток в диэтиловый эфир и промывают водным раствором гидроксида натрия (0,5 M). Органическую фазу высушивают над безводным сульфатом натрия и концентрируют. Остаток хроматографируют на колонке с силикагелем, используя в качестве элюента смесь дихлорметан/метанол 98/2 (об./об.), получая 15 г твердого вещества бежевого цвета.
Т.пл.=154°C.
ЯМР 1H δ в ч.млн (ДМСО d6): 5,07 (с, 2H); 6,98-7,65 (мультиплет, 10H).
1.6. трет.-бутил-(1S)-1-[({5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил}-амино)карбонил]бутилкарбамат
К 3,35 г (2S)-2-[(трет.-бутилоксикарбонил)амино]пентановой кислоты в растворе в 35 мл диметилформамида добавляют при 0°C 7,1 г гексафторфосфата бензотриазол-1-илокси-трис(диметиламино)фосфония, затем, по каплям, 2,1 мл N-метилморфолина. Через 15 минут при 0°C вводят 4 г 5-{2-[(фенокси)метил]фенил}-1,3-тиазол-2-амина, полученного на стадии 1.5, и перемешивают 18 часов при комнатной температуре. Среду помещают в этилацетат и промывают 2 раза водой. Органическую фазу высушивают над безводным сульфатом натрия и концентрируют. Остаток хроматографируют на колонке с силикагелем, используя в качестве элюента смесь дихлорметан/метанол 98/2 (об./об.), получая 5,2 г бесцветного масла.
ЯМР 1H δ в ч.млн (ДМСО d6): 0,88 (т, 3H); 1,22-1,65 (мультиплет, 13H); 4,24 (кв, 1H); 5,09 (с, 2H); 6,94-7,67 (мультиплет, 10H) 12,23 (с, 1H).
1.7. гидрохлорид (2S)-2-амино-N-(5-{2-[(фенокси)метил]фенил}-1,3-тиазол-2-ил)пентанамида
К 5 г трет.-бутил-(1S)-2-[(5-{2-[(фенокси)метил]фенил}-1,3-тиазол-2-ил)амино]-1-пропил-2-оксоэтилкарбамата, полученного на стадии 1.6, в растворе в 60 мл этилацетата, добавляют по каплям при 0°C 25 мл раствора газообразной соляной кислоты (4,5 М) в этилацетате. Среду перемешивают 18 часов при 20°C. Сформировавшийся осадок отфильтровывают, промывают 2 раза диэтиловым эфиром и высушивают, получая 3 г твердого вещества белого цвета.
Т.пл.=148°C.
ЯМР 1H δ в ч.млн (ДМСО d6): 0,90 (т, 3H); 1,39 (м, 2H); 1,85 (м, 2H); 4,18 (кв, 1H); 5,08 (с, 2H); 6,94-7,68 (мультиплет, 10H); 8,65 (с, 3H).
1.8. (2S)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}-N-{5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил}пентанамид
К 0,32 г (2S)-2-гидроксил-3,3-диметилбутановой кислоты в растворе в 25 мл диметилформамида при 0°C последовательно добавляют 1,36 г гексофторфосфата бензотриазол-1-илокситрипирролидинфосфония и 0,7 мл N-этилморфолина. Через 20 минут при 0°C вводят 0,88 г гидрохлорида (2S)-2-амино-N-(5-{2-[(фенокси)метил]фенил}-1,3-тиазол-2-ил)пентанамида, полученного на стадии 1.7, и перемешивают в течение 18 часов при комнатной температуре. Реакционную среду помещают в этилацетат и промывают водой. Органическую фазу высушивают над безводным сульфатом натрия и концентрируют. Остаток хроматографируют на колонке с силикагелем, используя в качестве элюента смесь дихлорметан/метанол 99/1 (об./об.), получая после кристаллизации в изопропиловом эфире 0,83 г твердого вещества белого цвета.
Т.пл.=84°C.
ЯМР 1H δ в ч.млн (ДМСО d6): 0,89 (т, 3H); 0,93 (с, 9H); 1,33 (м, 2H); 1,71 (кв, 21H); 3,57 (д, 1H); 4,61 (кв, 1H); 5,09 (с, 2H); 5,61 (д, 1H); 6,97-7,02 (мультиплет, 3H); 7,28-7,67 (мультиплет, 7H); 7,81 (д, 1H); 12,28 (с, 1H).
[α]D 20=-81,8 (c=1/CH3OH).
Пример 2 (соединение 16)
(2S)-2-{[2-(2,5-диметил-3-тиенил)ацетил]амино}-N-{5-[2-(этоксиметил)-фенил]-1,3-тиазол-2-ил}пентанамид
2.1. 2-(2,5-диметил-3-тиенил)-1-(4-морфолинил)-1-этантион
К 5 г 2,5-диметил-3-ацетилтиофена добавляют 1,68 г серы, 6,5 мл морфолина и кипятят 10 часов с обратным холодильником. Охлаждают до 20°C и выливают в водный раствор соляной кислоты (1 н.). Среду экстрагируют этилацетатом. Органическую фазу высушивают над безводным сульфатом натрия и концентрируют. Остаток очищают хроматографией на колонке с силикагелем, используя в качестве элюента смесь циклогексан/этилацетат 8/2 (об./об.), получая 6,8 г масла оранжевого цвета.
ЯМР 1H δ в ч.млн (ДМСО d6): 2,30 (с, 3H); 2,34 (с, 3H); 3,47 (т, 2H); 3,65 (м, 4H); 4,07 (с, 2H); 4,20 (т, 2H); 6,56 (с, 1H).
2.2. 2-(2,5-диметил-3-тиенил)уксусная кислота
2-(2,5-диметил-3-тиенил)уксусную кислоту получают способом, описанным в Heterocycl. Chem; EN; 25; 1988; 1571-1581.
К 6,7 г 2-(2,5-диметил-3-тиенил)-1-(4-морфолинил)-1-этантиона, полученного на стадии 2.1, в растворе в 70 мл метанола добавляют 21 мл водного раствора гидроксида натрия (50 мас.%) и кипятят 6 часов с обратным холодильником. После выпаривания метанола остаток разбавляют водой и подкисляют водным раствором соляной кислоты (6 н.). Сформировавшийся осадок отфильтровывают, затем хроматографируют на колонке с силикагелем, используя в качестве элюента дихлорметан, и получают 3,6 г кристаллов бежевого цвета.
Т.пл.=65°C.
ЯМР 1H δ в ч.млн (ДМСО d6): 2,26 (с, 3H); 2,34 (с, 3H); 3,39 (с, 2H); 6,56 (с, 1H).
2.3. (2S)-2-{[(2,5-диметил-3-тиенил)ацетил]амино}-N-{5-[2-(этоксиметил)фенил]-1,3-тиазол-2-ил}пентанамид
Действуют аналогично стадии 1.8 примера 1, заменяя (2S)-2-гидрокси-3,3-диметилбутановую кислоту 2-(2,5-диметил-3-тиенил) уксусной кислотой, полученной на стадии 2.2. Получают 0,67 г кристаллов белого цвета.
Т.пл.=84°C.
ЯМР 1H δ в ч.млн (ДМСО d6): 0,89 (т, 3H); 1,16 (т, 3H); 1,29 (м, 2H); 1,65 (м, 2H); 2,25 (с, 3H); 2,30 (с, 3H); 3,33 (м, 2H); 3,47 (кв, 2H); 4,44 (с, 2H); 4,49 (кв, 1H); 4,55 (с, 1H); 7,35-7,55 (мультиплет, 5H); 8,32 (д, 1H); 12,28 (с, 1H).
[α]D 20=-103 (c=1/CH3OH).
Пример 3 (соединение 22)
(2S)-2-{[2-(5-метил-2-тиенил)ацетил]амино}-N-{5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил}пентанамид
3.1. 2-(5-метил-2-тиенил)-1-(4-морфолинил)-1-этантион
2-(5-метил-2-тиенил)-1-(4-морфолинил)-1-этантион получают способом, аналогичным способу, описанному в примере 2.1.
ЯМР 1H δ в ч.млн (ДМСО d6): 2,40 (с, 3H); 3,49 (т, 2H); 3,64 (т, 2H); 3,81 (т, 2H); 4,19 (т, 2H); 4,40 (с, 2H); 6,49 (д, 1H); 6,77 (д, 1H).
3.2. 2-(5-метил-2-тиенил)уксусная кислота
2-(5-метил-2-тиенил)уксусную кислоту получают способом, аналогичным способу, описанному в примере 2.2.
Т.пл.=54°C.
ЯМР 1H δ в ч.млн (ДМСО d6): 2,38 (с, 3H); 3,72 (с, 3H); 6,61 (д, 1H); 6,69 (д, 1H).
3.3. (2S)-2-{[2-(5-метил-2-тиенил)ацетил]амино}-N-{5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил}пентанамид
Действуют аналогично стадии 1.8 примера 1, заменяя (2S)-2-гидрокси-3,3-диметилбутановую кислоту 2-(5-метил-2-тиенил) уксусной кислотой, полученной на стадии 3.2. Получают 0,73 г кристаллов бежевого цвета.
Т.пл.=81°C.
ЯМР 1H δ в ч.млн (ДМСО d6): 0,87 (т, 3H); 1,32 (м, 2H); 1,64 (м, 2H); 2,36 (с, 3H); 3,62 (кв, 2H); 4,48 (кв, 1H); 5,07 (с, 2H); 6,59 (д, 1H); 6,67 (д, 1H); 6,95-7 (мультиплет, 3H); 7,27-7,73 (мультиплет, 7H); 8,41 (д, 1H); 12,30 (с, 1H).
[α]D 20=-91,7 (c=1/CH3OH).
Пример 4 (соединение 7)
(2S)-N-{5-[2-(изопропоксиметил)фенил]-1,3-тиазол-2-ил}-2-{[3-(3-тиенил)пропаноил]амино}пентанамид
4.1. (E)-3-(3-тиенил)-2-пропеновая кислота
К 25 г 3-тиенальдегида в растворе 100 мл пиридина добавляют 46 г малоновой кислоты, 2 мл пиперидина и нагревают при 100°C в течение 4 часов. Реакционную среду охлаждают до 30°C и выливают в водный раствор соляной кислоты (2 н.). Сформировавшийся осадок отфильтровывают и промывают изопропиловым эфиром, получая после высушивания 30 г твердого вещества белого цвета.
Т.пл.=152°C.
ЯМР 1H δ в ч.млн (ДМСО d6): 6,36 (д, 1H); 7,51-7,62 (мультиплет, 3H); 7,93 (д, 1H); 12,27 (с, 1H).
4.2. Этил-(E)-3-(3-тиенил)-2-пропеноат
К 11 г (E)-3-(3-тиенил)-2-пропеновой кислоты, полученной на стадии 4.1, в растворе в 50 мл диметилформамида добавляют 11,5 г карбоната калия, 6,8 мл иодэтана и перемешивают в течение 48 часов при 20°C. Среду помещают в этилацетат и промывают водой. Органическую фазу высушивают над безводным сульфатом натрия и концентрируют, получая 12,5 г масла.
ЯМР 1H δ в ч.млн (ДМСО d6): 1,26 (т, 3H); 4,17 (кв, 2H); 6,47 (д, 1H); 7,57-7,71 (мультиплет, 3H); 8,01 (д, 1H).
4.3. Этил-3-(3-тиенил)-2-пропаноат
К 12,5 г этил-(E)-3-(3-тиенил)-2-пропеноата, полученного на стадии 4.2, в растворе в 100 мл этанола, добавляют 4 г 10%-ого палладия на угле и перемешивают в течение 24 часов при 60°C под давлением водорода 5 бар. Отфильтровывают катализатор и концентрируют фильтрат, получая 11 г масла.
ЯМР 1H δ в ч.млн (ДМСО d6): 1,20 (т, 3H); 2,62 (т, 2H); 2,88 (т, 2H); 4,07 (кв, 2H); 7,02 (д, 1H); 7,18 (м, 1H); 7,45 (м, 1H).
4.4. 3-(3-тиенил)пропановая кислота
К 11 г этил-3-(3-тиенил)-2-пропаноата, полученного на стадии 4.3, в растворе в 100 мл этанола добавляют 75 мл водного раствора гидроксида натрия (2 н.). Перемешивают в течение 18 часов при 20°C. После выпаривания растворителей остаток подкисляют. Сформировавшийся осадок отфильтровывают и высушивают в вакууме, получая 6,3 г твердого вещества бежевого цвета.
Т.пл.=59°C.
ЯМР 1H δ в ч.млн (ДМСО d6): 2,56 (т, 2H); 2,85 (т, 2H); 7,02 (т, 1H); 7,18 (с, 1H); 7,45 (м, 1H); 12,14 (с, 1H).
4.5. (2S)-N-{5-[2-изопропоксиметил)фенил]-1,3-тиазол-2-ил}-2-{[3-(3-тиенил)пропаноил]амино}пентанамид
Действуют аналогично стадии 1.8 примера 1, заменяя (2S)-2-гидрокси-3,3-диметилбутановую кислоту 3-(3-тиенил)пропановой кислотой, полученной на стадии 4.4. Получают 0,75 г кристаллов бежевого цвета.
Т.пл.=101°C.
ЯМР 1H δ в ч.млн (ДМСО d6): 0,87 (т, 3H); 1,13 (д, 6H); 1,33 (м, 2H); 1,62 (м, 2H); 2,48 (т, 2H); 2,82 (т, 2H); 3,67 (м, 1H); 4,45 (с, 2H); 4,54 (кв, 1H); 6,98 (д, 1H); 7,12 (д, 1H); 7,35-7,57 (мультиплет, 6H); 8,21 (д, 1H); 12,26 (с, 1H).
[α]D 20=-71(c=1/CH3OH).
Пример 5
(2R)-3-этил-2-гидрокси-N-{(1S)-1-[({5-[2-(феноксиметил) фенил]-1,3-тиазол-2-ил}амино)карбонил]бутил}пентанамид (соединение n 42) и
(2S)-3-этил-2-гидрокси-N-{(1S)-1-[({5-[2-(феноксиметил) фенил]-1,3-тиазол-2-ил}амино)карбонил]бутил}пентанамид (соединение 43)
5.1. 3-этил-2-гидроксипентановая кислота
К раствору 1,24 мл 2-этилбутиральдегида в 18 мл безводного дихлорметана осторожно добавляют 1,5 мл триметилсилилцианида, затем каталитическое количество иодида цинка. Реакционную среду перемешивают в течение 2 часов при комнатной температуре, затем в течение 3,5 часов при 60°C. Реакционную среду охлаждают до 0°C и добавляют 3,5 мл концентрированной соляной кислоты. Реакционную среду перемешивают в течение 18 часов при комнатной температуре, затем 1 час при кипячении с обратным холодильником. После охлаждения реакционную смесь выливают в воду и экстрагируют дважды с помощью 50 мл этилацетата. Объединенные органические фазы экстрагируют с помощью 100 мл гидроксида натрия (7,5 н.) при 4°C. После разделения водную фазу промывают 3 раза с помощью 50 мл этилацетата. Водную фазу подкисляют с помощью 70 мл соляной кислоты (12 н.) и экстрагируют трижды с помощью 50 мл этилацетата. Объединенные органические фазы высушивают и выпаривают растворитель.
Т.пл.=84°C.
5.2. (2R)-3-этил-2-гидрокси-N-{(1S)-1-[({5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил}амино)карбонил]бутил}пентанамид и
(2S)-3-этил-2-гидрокси-N-{(1S)-1-[({5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил}амино)карбонил]бутил}пентанамид
Действуют аналогично стадии 1.8 примера 1, заменяя (2S)-2-гидрокси-3,3-диметилбутановую кислоту 3-этил-2-гидроксипентановой кислотой, полученной на стадии 5.1. Получают 0,78 г твердого вещества белого цвета.
Соединение 42 (SR):
Т.пл.=67,4°C.
ЯМР 1H δ в ч.млн (ДМСО d6): 0,74 (т, 3H); 0,84-0,90 (мультиплет, 6H); 1,26-1,71 (мультиплет, 9H); 3,94 (м, 1H); 4,57 (кв, 1H); 5,07 (с, 2H); 5,40 (с, 1H); 6,94-6,99 (мультиплет, 3H); 7,26-7,93 (мультиплет, 7H); 7,91 (д, 1H); 12,27 (с, 1H).
[α]D 20=-41,5(c=1/CH3OH).
Соединение 43 (SS):
Т.пл.=122,5°C.
ЯМР 1H δ в ч.млн (ДМСО d6): 0,78-0,89 (мультиплет, 9H); 1,18-1,38 (мультиплет, 6H); 1,55 (м, 1H); 1,68 (кв, 2H); 3,92 (м, 1H); 4,60 (кв, 1H); 5,07 (с, 2H); 5,49 (д, 1H); 6,95-7 (мультиплет, 3H); 7,26-7,62 (мультиплет, 7H); 7,87 (д, 1H); 12,27 (с, 1H).
[α]D 20=-72,6 (c=1/CH3OH).
Пример 6 (соединение 40)
(2S)-2-[(2-гидрокси-3-фенилпропаноил)амино]-N-{5-[2-(феноксиметил)-фенил]-1,3-тиазол-2-ил}пентанамид
6.1. 3-фенил-2-гидроксипропионовая кислота
К суспензии 1,6 г фенилаланина в 5,3 мл серной кислоты (2,5 н.) добавляют по каплям при 0°C раствор 0,829 г нитрита натрия в 4,2 мл воды. Реакционную смесь перемешивают в течение 2 часов при 0°C, затем 17 часов при комнатной температуре. Реакционную смесь экстрагируют дважды с помощью 100 мл этилацетата. Объединенные органические фазы промывают 100 мл насыщенного раствора хлорида натрия в воде. После высушивания получают 1,2 г кристаллов желтого цвета.
Т.пл.=97°C.
6.2. 2-[(2-гидрокси-3-фенилпропаноил)амино]-N-{5-[2-(феноксиметил)фенил]-1,3-тиазол-2-ил}пентанамид
Действуют аналогично стадии 1.8 примера 1, заменяя (2S)-2-гидрокси-3,3-диметилбутановую кислоту 3-фенил-2-гидроксипропионовой кислотой, полученной на стадии 6.1. Получают 0,8 г твердого вещества белого цвета.
Т.пл.=86°C.
ЯМР 1H δ в ч.млн (ДМСО d6): 0,85 (т, 3H); 1,24 (м, 2H); 1,63 (м, 2H); 2,70 (м, 1H); 2,57 (м, 1H); 4,17 (м, 1H); 4,56 (кв, 1H); 5,08 (с, 2H); 6,94-7,63 (мультиплет, 15H); 8,02 (м, 1H); 12,25 (с, 1H).
[α]D 20=-28 (c=1/CH3OH).
Пример 7 (соединение 70)
(2S)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}-N-(5-{2-[(4-пиридинилокси)метил]фенил}-1,3-тиазол-2-ил)пентанамид
7.1. 2-(2-амино-1,3-тиазол-5-ил)фенилметанол
К 29,86 г 5-[2-(трет.-бутоксиметил)фенил]-N-дифенилметилен)-1,3-тиазол-2-амина, полученного способом, аналогичным способу, описанному на стадиях 1.1-1.4 примера 1, в растворе в 140 мл метанола добавляют 70 мл водного 3М раствора соляной кислоты и оставляют на 18 часов при комнатной температуре, затем кипятят с обратным холодильником в течение 4 часов. Метанол выпаривают. Остаток помещают в водный 6М раствор соляной кислоты и экстрагируют диэтиловым эфиром. рН водной фазы доводят до основного значения при одновременном охлаждении и экстрагируют этилацетатом. Этилацетатную фазу высушивают над безводным сульфатом натрия и концентрируют. Остаток отверждают диизопропиловым эфиром и получают 6 г твердого вещества бежевого цвета.
Т.пл.=145°C.
7.2. трет.-бутил-5-[2-(гидроксиметил)фенил]-1,3-тиазол-2-илкарбамат
К 6 г 2-(2-амино-1,3-тиазол-5-ил)фенилметанола, полученного на стадии 7.1, в растворе в 80 мл 1,4-диоксана последовательно добавляют 1,17 г оксида магния, 29 мл водного 2М раствора гидроксида натрия, затем при 0°C, порциями - 7,6 г ди-трет.-бутилкарбоната (ВОС2О). Оставляют на 48 часов при комнатной температуре, затем среду концентрируют, помещают в воду и экстрагируют этилацетатом. Органическую фазу промывают 5%-м раствором гидросульфата калия, высушивают над безводным сульфатом натрия и концентрируют. Остаток хроматографируют на колонке с силикагелем, используя в качестве элюента смесь дихлорметан/метанол 99/1 (об./об.), и получают 2,3 г масла, которое отверждают диизопропиловым эфиром.
Т.пл.=180,7°C.
7.3. трет.-бутил-5-{2-[(4-пиридинилокси)метил]фенил}-1,3-тиазол-2-илкарбамат
К 5,24 г трифенилфосфина в растворе в 60 мл тетрагидрофурана добавляют порциями при 0°C 4,15 г диизопропилазодикарбоксилата (DIAD). Через 30 минут при приблизительно 10°C добавляют порциями 1,96 г 4-гидроксипиридина, оставляют на 30 минут при приблизительно 10°C и вводят 4,2 г трет.-бутил-5-[2-(гидроксиметил)фенил]-1,3-тиазол-2-илкарбамата, полученного на стадии 7.2. Оставляют на 4 дня при комнатной температуре. Среду концентрируют, помещают в насыщенный раствор карбоната натрия и экстрагируют дихлорметаном. Органическую фазу высушивают над безводным сульфатом натрия и концентрируют. Остаток хроматографируют на колонке с силикагелем, используя в качестве элюента смесь с возрастающей полярностью от дихлорметан/метанол 99/1 (об./об.) до дихлорметан/метанол 90/10 (об./об.), и получают 1 г масла.
ЯМР 1H δ в ч.млн (ДМСО d6): 1,52 (с, 9H); 5,27 (с, 2H); 6,10 (кв, 2H); 7,05 (м, 1H); 7,41-7,54 (мультиплет, 6H); 11,60 (с, 1H).
7.4. 5-{2-[(4-пиридинилокси)метил]фенил}-1,3-тиазоламин
К 1 г трет.-бутил-5-{2-[(4-пиридинилокси)метил]фенил}-1,3-тиазол-2-илкарбамата, полученного на стадии 7.3, в 20 мл дихлорметана добавляют 20 мл 4M раствора газообразной соляной кислоты в этилацетате. Оставляют на 4 часа при комнатной температуре. Реакционную среду концентрируют и доводят рН до основного значения с помощью 5%-ого раствора гидросульфата натрия. Сформировавшийся осадок отфильтровывают и хроматографируют на колонке с силикагелем, используя в качестве элюента смесь дихлорметан/метанол 95/5 (об./об.), и получают 0,58 г пены бежевого цвета.
ЯМР 1H δ в ч.млн (ДМСО d6): 5,25 (с, 2H); 6,10 (д, 2H); 6,98-7,55 (мультиплет, 9H).
7.5. (2S)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}-N-(5-{2-[(4-пиридинилокси)метил]фенил}-1,3-тиазол-2-ил)пентанамид
Действуют аналогично стадиям 1.6-1.8 примера 1, заменяя 5-{2-[(фенокси)метил]фенил}-1,3-тиазол-2-амин 5-{2-[(4-пиридинилокси)метил]фенил}-1,3-тиазоламином, полученным на стадии 7.4. Получают 0,4 г в виде кристаллов.
Т.пл.=112,7°C.
ЯМР 1H δ в ч.млн (ДМСО d6): 0,89 (т, 3H); 0,92 (с, 9H); 1,34 (м, 2H); 1,68 (кв, 2H); 3,58 (д, 1H); 4,61 (кв, 1H); 5,24 (с, 2H); 5,61 (д, 1H); 6,09 (д, 2H); 7,04 (м, 1H); 7,41-7,53 (мультиплет, 6H); 7,80 (д, 1H); 12,36 (с, 1H).
Соединения n 61 ((2S)-N-(5-{2-[(3-цианофенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид) и n 69 ((2S)-N-(5-{2-[(2-цианофенокси)метил]фенил}-1,3-тиазол-2-ил)-2-{[(2S)-2-гидрокси-3,3-диметилбутаноил]амино}пентанамид) могут быть получены способом, описанным в примере 7, с заменой 4-гидроксипиридина соответственно 3-цианофенолом или 2-цианофенолом.
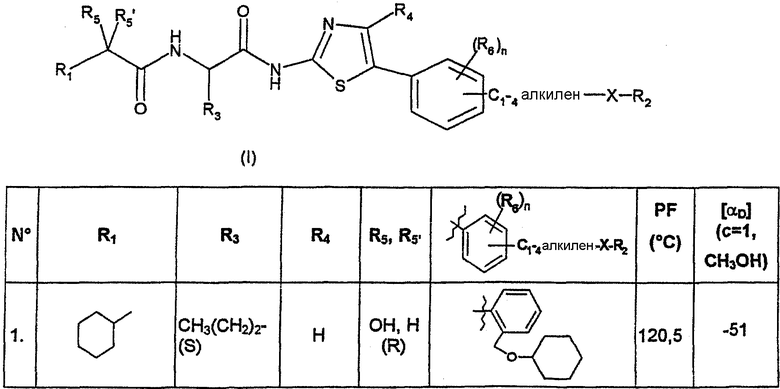
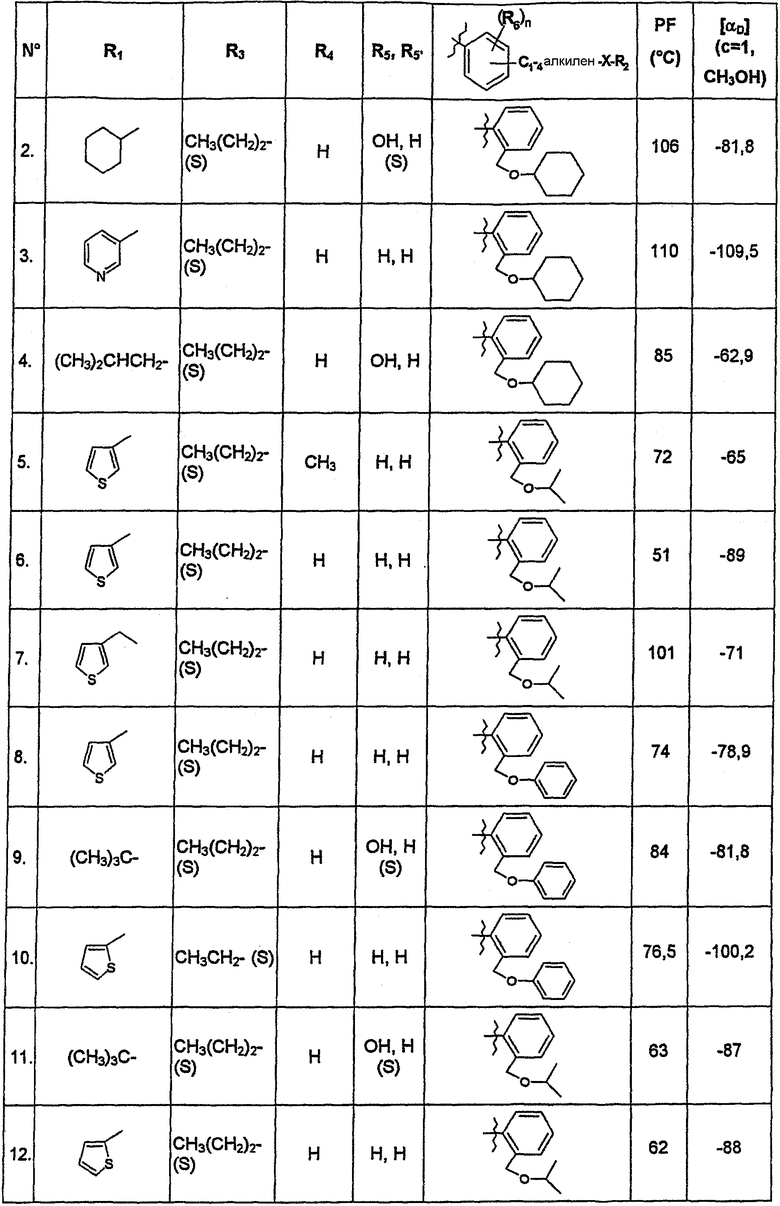
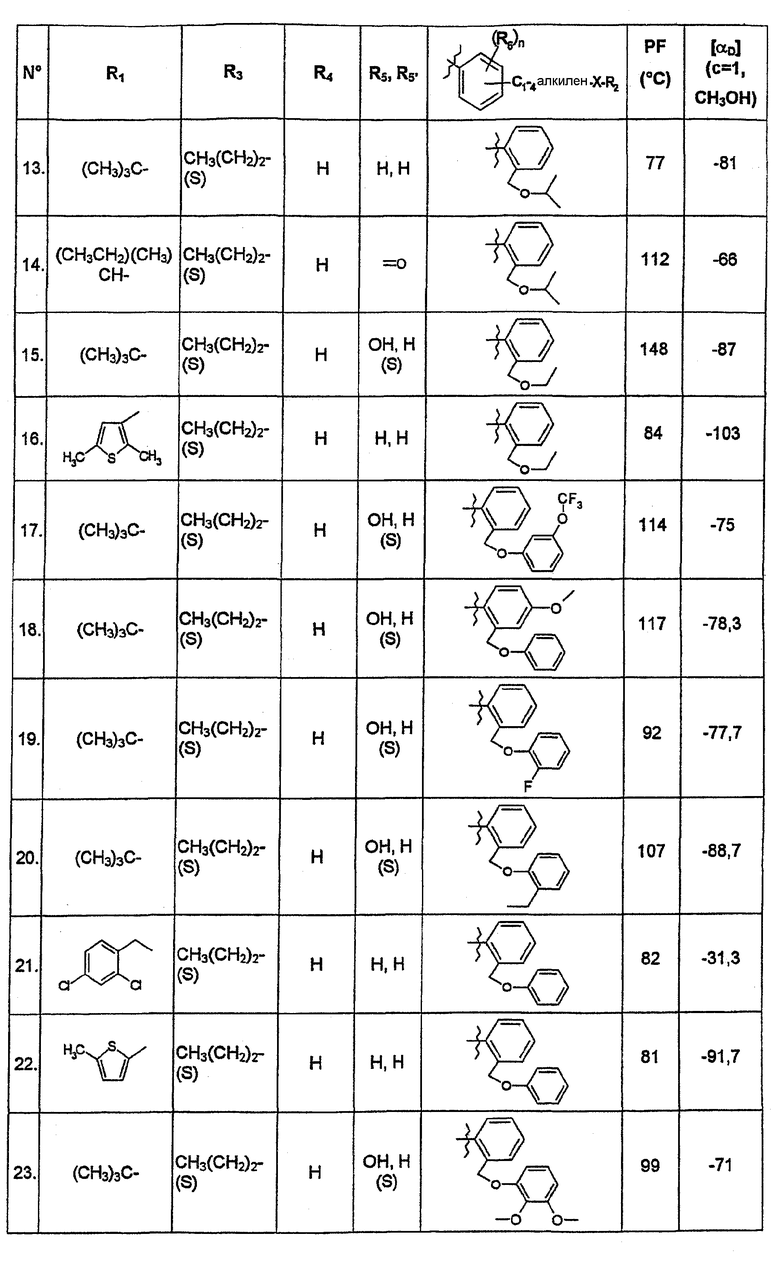
В нижеследующей таблице представлены химические структуры и физические свойства некоторых соединений по изобретению.
В этой таблице:
- Т.пл. (°C) обозначает точку плавления соединения, в градусах Цельсия;
- [αD] (c=1, CH3OH) обозначает вращающую способность соединения в концентрации 1 г/л в метаноле;
- (S) или (R) в столбцах «R3» и «R5, R5'» указывают на стереохимию асимметрических атомов углерода, несущих соответственно R3 или R5, в формуле (I).
Для атома углерода, несущего R5, обозначение (S) или (R) не относится к случаю, когда R5 и R5' вместе образуют оксо-группу или оксимную группу.
Соединения, описанные в этой таблице, были получены способами, описанными выше.
Таблица

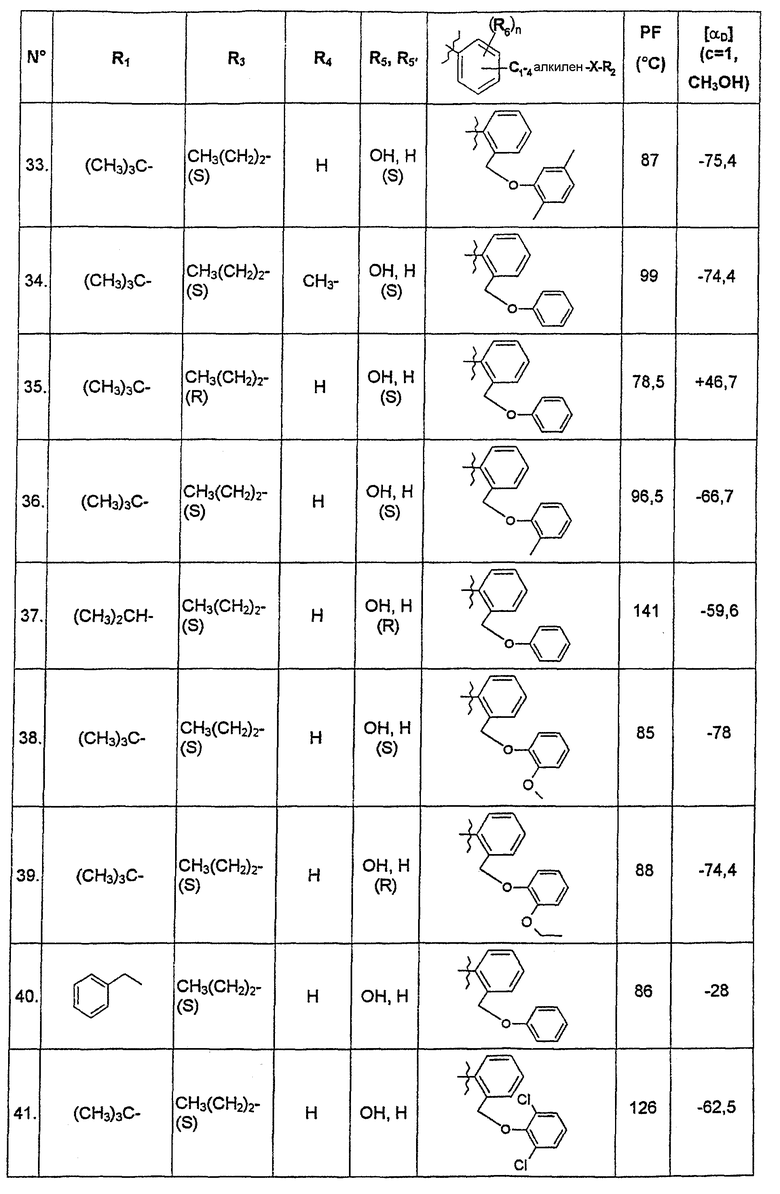

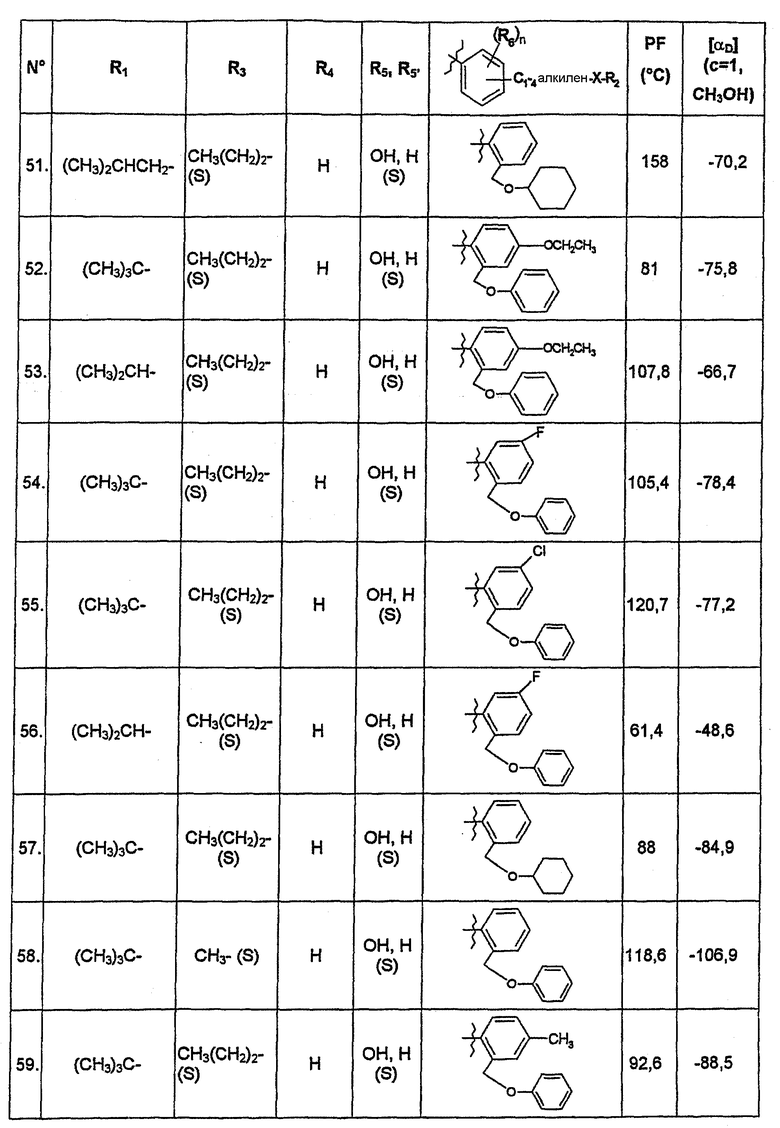
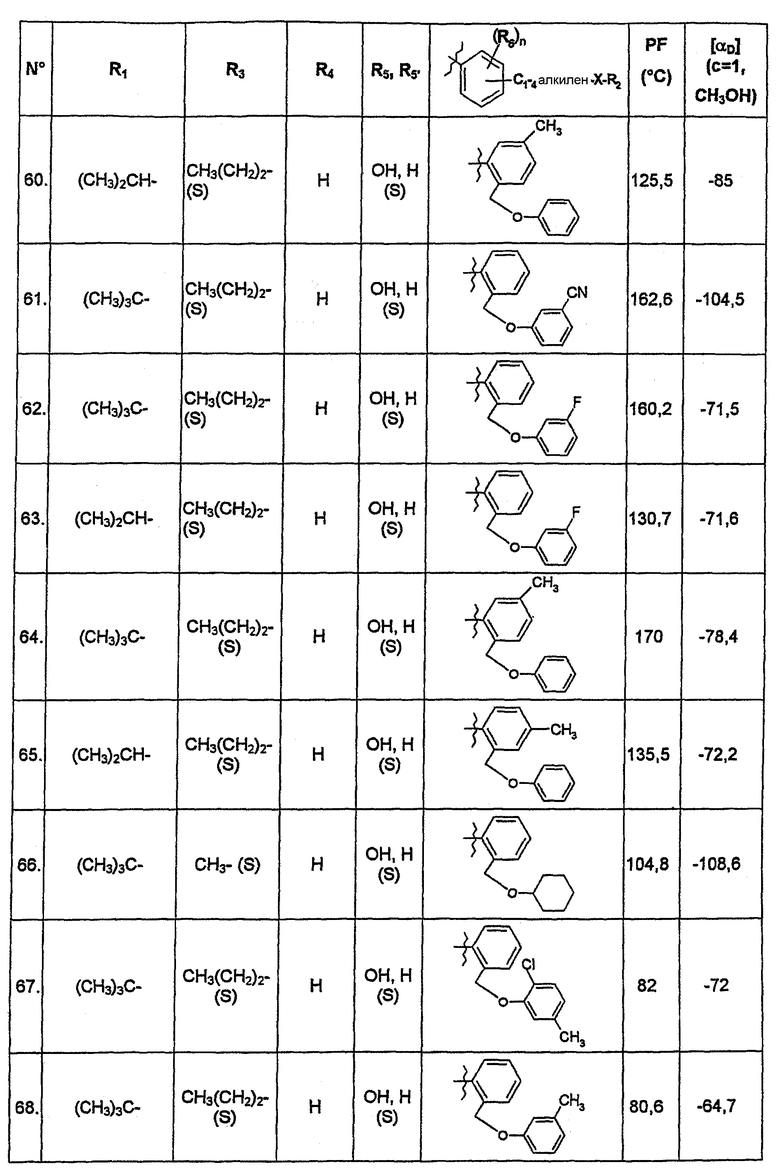
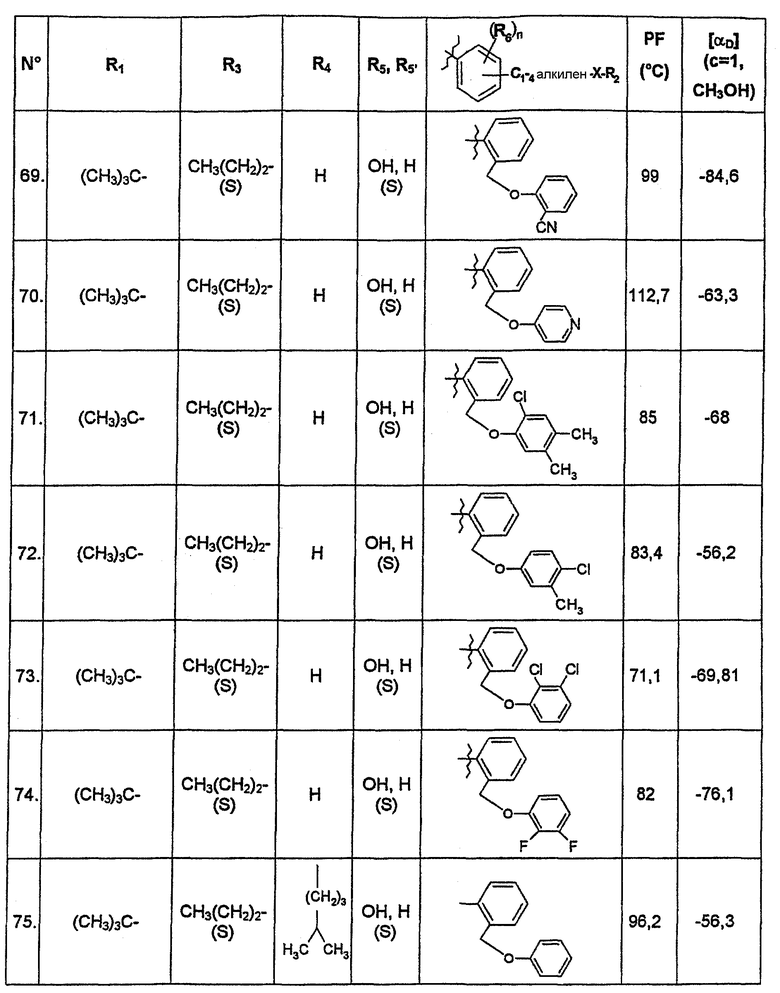
Соединения по изобретению были объектами фармакологических исследований, которые показали их ценность в качестве терапевтически активных веществ.
В частности, их тестировали в отношении их ингибирующего действия на продукцию β-амилоидного пептида (β-А4).
β-амилоидный пептид (β-А4) представляет собой фрагмент более крупного белка-предшественника, называемого APP (Amyloid Precursor Protein). Последний продуцируется и присутствует в различных клетках тканей животных или человека. На уровне мозга его расщепление ферментами типа протеаз приводит к образованию пептида β-А4, который аккумулируется в виде амилоидных бляшек. Две протеазы, отвечающие за продукцию амилоидного пептида, известны под названиями бета- и гамма-секретаз (Wolfe MS, Secretase targets for Alzheimer's disease : identification and therapeutic potential, J.Med. Chem., 2001 Jun 21; 44 (13), 2039-60).
Однако было показано, что постепенно накапливающийся пептид β-А4 нейротоксичен и может играть важную роль в развитии болезни Альцгеймера.
Таким образом, соединения по изобретению, будучи ингибиторами продукции β-амилоидного пептида (β-А4) за счет ингибирования гамма-протеаз, могут быть использованы в лечении таких патологий, как старческая деменция, болезнь Альцгеймера, синдром Дауна, болезнь Паркинсона, амилоидная ангиопатия, сосудисто-мозговые расстройства, лобно-височные деменции и болезнь Пика, посттравматические деменции, патологии, связанные с нервно-воспалительными процессами, болезнь Гентингтона и Корсаковский синдром.
Тестирования проводили согласно приведенному ниже протоколу.
Для β-амилоидного клеточного исследования использовали линию CHO-K1, коэкспрессирующую CT100 APP PS1 M146L клон 30-12. Целью этой линии является ингибирование гамма-секретазы. Пресенилин связан с гамма-секретазной активностью (Wolfe MS, Haass C., The Role of presenilins in gamma-secretase activity, J. Biol. Chem., 2001 Feb 23, 276 (8), 5413-6) и его совместная экспрессия с амилоидным белком или его N-концевым фрагментом приводит к увеличению секреции пептида А1-42 (β-А4), что создает фармакологический инструмент, позволяющий оценить ингибирование соединениями формулы (I) продукции пептида β-А4. Засеивание 96-лунковых культуральных планшетов осуществляли из расчета 1х105 клеток на лунку в 150 мкл инкубационной среды. Присутствие минимального количества (конечное 1,3%) сыворотки делает возможной адгезию клеток на пластике после 2-3 часов инкубации при 37°C в присутствии 5% СО2. Тестируемые продукты (15 мкл) тестируют при конечной концентрации 10 мкМ ДМСО 1% и инкубируют в течение 24-25 ч при 37°C в присутствии 5% СО2 и при 100% влажности. После этой 24-25-часовой инкубации, клеточные супернатанты (100 мкл) переносят на планшеты ELISA, обрабатывают антителами захвата 6Е10 (6E10, эпитоп :aa1-17, INTERCHIM/SENETEK 320-10) для определения количества амилоидных пептидов, секретированных клетками в присутствии соединений по изобретению. Параллельно обрабатывают гамму контрольного синтетического пептида, «пептида 1-40», в концентрациях 5 и 10 нг/мл. Планшеты ELISA инкубируют в течение ночи при 4°C.
Количество фиксированного пептида определяют косвенно в присутствии конкурента, соответствующего усеченному пептиду, пептиду 1-28, связанному с биотином, который затем детектируют с помощью стрептавидина, связанного со щелочной фосфатазой. Субстрат, п-Нитрофенил Фосфат (pNPP FAST p-Nitrophényl Phosphate, Sigma N2770), приводит к растворимому продукту реакции желтого цвета, видимому при 405 нм. Реакцию останавливают с помощью 0,1М раствора ЭДТУ. Для этого после фиксации амилоидного пептида на планшете ELISA к 100 мкл клеточного супернатанта добавляют 50 мкл биотинилированного пептида 1-28 и инкубируют 30 минут при комнатной температуре. Затем планшеты ELISA промывают 3 раза. После высушивания на абсорбирующей бумаге добавляют стрептавидин-Щелочную Фосфатазу (Interchim/Jackson ImmunoResearch Laboratories 016-050-084) в количестве 100 мкл на лунку и инкубируют 1 час при комнатной температуре. Планшеты снова промывают, затем добавляют субстрат для щелочной фосфатазы (pNPP 1 мг/мл) из расчета 100 мкл на лунку. После инкубации в течение 30 минут при комнатной температуре реакцию останавливают добавлением 100 мкл 0,1М ЭДТУ на лунку и осуществляют считывание при 405 нм.
Соединения формулы (I) по изобретению демонстрируют CE50 (эффективную концентрацию при 50%) ниже 500 нМ. В частности, соединение n 70 согласно таблице демонстрирует CE50, равное 295 нМ. Соединения формулы (I) по изобретению демонстрируют CE50, в частности, ниже 100 нМ.
Результаты биологических тестов показывают, что эти соединения являются ингибиторами образования β-амилоидного пептида (β-А4).
Таким образом, эти соединения могут быть использованы в лечении патологий, в которых ингибитор образования β-амилоидного пептида (β-А4) приносит терапевтическую пользу. Такими патологиями, в частности, являются старческая деменция, болезнь Альцгеймера, синдром Дауна, болезнь Паркинсона, амилоидная ангиопатия, сосудисто-мозговые расстройства, лобно-височные деменции и болезнь Пика, посттравматические деменции, патологии, связанные с нервно-воспалительными процессами, болезнь Гентингтона и Корсаковский синдром.
Применение соединений по изобретению в виде фармацевтически приемлемых основания, соли, гидрата или сольвата для получения лекарственного средства, предназначенного для лечения вышеназванных патологий, составляет часть изобретения.
Объектом изобретения также являются лекарственные средства, содержащие соединение формулы (I) или его соль присоединения с фармацевтически приемлемой кислотой, или гидрат или сольват соединения формулы (I). Эти лекарственные средства находят применение в терапии, в частности, в лечении вышеназванных патологий.
Согласно другому аспекту, настоящее изобретение относится к фармацевтическим композициям, содержащим в качестве действующего начала по меньшей мере одно соединение по изобретению. Эти фармацевтические композиции содержат эффективную дозу соединения по изобретению или фармацевтически приемлемой соли, гидрата или сольвата указанного соединения и, в случае необходимости, один или несколько фармацевтически приемлемых эксципиентов. Эти эксципиенты выбирают, в зависимости от используемой фармацевтической формы и желаемого пути введения, из обычных известных специалисту эксципиентов.
В фармацевтических композициях согласно настоящему изобретению для перорального, сублингвального, подкожного, внутримышечного, внутривенного, топического, местного, интратрахеального, интраназального, чрескожного или ректального введения вышеуказанное действующее начало формулы (I) или, в случае необходимости, его соль, сольват или гидрат могут быть введены в виде стандартной лекарственной формы, в смеси с обычными фармацевтическими эксципиентами, животным или людям для профилактики или лечения вышеназванных расстройств или заболеваний.
Подходящие стандартные лекарственные формы для введения включают формы для перорального введения, такие как таблетки, мягкие или твердые желатиновые капсулы, порошки, гранулы, жевательные резинки и растворы или суспензии для перорального введения, формы для сублингвального, буккального, интратрахеального, внутриглазного, интраназального введения, введения путем ингаляции, формы для подкожного, внутримышечного или внутривенного введения и формы для ректального или вагинального введения. Для топического нанесения соединения по изобретению можно использовать в составе кремов, помад или лосьонов.
В качестве примера, стандартная лекарственная форма для введения соединения по изобретению в виде таблетки может содержать следующие компоненты:
Для получения желаемого профилактического или терапевтического эффекта доза действующего начала может варьировать от 0,1 мг до 200 мг на кг массы тела в сутки. Хотя указанные дозы являются примерами средней ситуации, в особых случаях могут оказаться приемлемыми более высокие или более низкие дозы, которые также входят в рамки изобретения. Согласно обычной практике дозу, подходящую для каждого пациента, определяет врач в зависимости от способа введения, массы тела и индивидуальной чувствительности указанного пациента.
Каждая разовая доза может содержать от 0,1 до 1000 мг, предпочтительно, от 0,1 до 500 мг действующего начала в комбинации с одним или несколькими фармацевтическими эксципиентами. Эта разовая доза может вводиться от 1 до 5 раз в сутки таким образом, чтобы суточная доза составила от 0,5 до 5000 мг, предпочтительно, от 0,5 до 2500 мг.
Настоящее изобретение также относится, согласно другому аспекту, к способу лечения вышеназванных патологий, который включает введение соединения по изобретению, фармацевтически приемлемой соли, сольвата или гидрата указанного соединения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ УЛУЧШЕНИЯ КОГНИТИВНЫХ ФУНКЦИЙ | 2012 |
|
RU2665021C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ УЛУЧШЕНИЯ КОГНИТИВНЫХ ФУНКЦИЙ | 2012 |
|
RU2792010C2 |
| УЛУЧШЕННЫЕ АГОНИСТЫ АПЕЛИНОВОГО РЕЦЕПТОРА (APJ) И ИХ ИСПОЛЬЗОВАНИЕ | 2016 |
|
RU2766148C1 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| НОВОЕ ФОСФОРАМИДАТНОЕ ПРОИЗВОДНОЕ НУКЛЕОЗИДА И ЕГО ПРИМЕНЕНИЕ | 2014 |
|
RU2621709C2 |
| АНТИБАКТЕРИАЛЬНЫЕ АГЕНТЫ | 2000 |
|
RU2269525C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| ЗАМЕЩЕННЫЕ ДИКЕТОПИПЕРАЗИНЫ КАК АНТАГОНИСТЫ ОКСИТОЦИНА | 2002 |
|
RU2303032C2 |
| ПРОИЗВОДНЫЕ ГИДРОКСАМОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ ИЛИ ВЕТЕРИНАРНАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2126791C1 |
| КОМБИНАЦИЯ ИНГИБИТОРА ВИЧ-ПРОТЕАЗЫ С ДРУГИМИ АНТИВИРУСНЫМИ СОЕДИНЕНИЯМИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ | 1994 |
|
RU2139052C1 |
Изобретение относится к новым соединениям формулы (I). Соединения настоящего изобретения могут найти применение в качестве лекарственного средства для лечения патологии, в которой является полезным ингибитор образования β-амилоидного пептида β-А4. В общей формуле (I):
Х обозначает атом кислорода; R1 обозначает С1-10-алкил, в случае необходимости замещенный фенилом, тиенилом; или R1 обозначает С3-7-циклоалкил, тиенил, пиридинил; тиенильные группы в случае необходимости могут быть замещены 1-2 C1-3-алкильными группами; фенил может быть в случае необходимости замещен 1-2 атомами галогена; R2 обозначает C1-6-алкил; или R2 обозначает С3-7-циклоалкил, фенил или пиридинил; фенил в случае необходимости может быть замещен одним или несколькими атомами галогена или группами CN, C1-3-алкила, C1-3-алкоксила, C1-3-фторалкоксила; R3 обозначает C1-6-алкил; R4 обозначает атом водорода или C1-6-алкил; R5 и R5' обозначают, независимо один от другого, атом водорода, гидроксил; или R5 и R5' вместе образуют оксо-группу; n обозначает целое число от 0 до 3; и R6 обозначает, независимо один от другого, атом водорода, галогена, C1-3-алкил, C1-3-алкоксил. Изобретение также относится к способу получения соединений, фармацевтической композиции, применению соединений для получения лекарственного средства. 6 н. и 2 з.п. ф-лы, 1 табл.
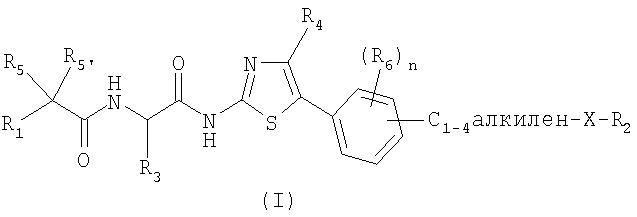
1. Соединения, отвечающие общей формуле (I)
в которой
Х обозначает атом кислорода;
R1 обозначает C1-10-алкил, в случае необходимости замещенный фенилом, тиенилом; или R1 обозначает С3-7-циклоалкил, тиенил, пиридинил;
тиенильные группы в случае необходимости могут быть замещены 1-2 C1-3-алкильными группами; фенил может быть в случае необходимости замещен 1-2 атомами галогена;
R2 обозначает C1-6-алкил; или R2 обозначает С3-7-циклоалкил, фенил или пиридинил;
фенил в случае необходимости может быть замещен одним или несколькими атомами галогена или группами CN, C1-3-алкила, C1-3-алкоксила, С1-3-фторалкоксила;
R3 обозначает C1-6-алкил;
R4 обозначает атом водорода или C1-6-алкил;
R5 и R5' обозначают, независимо один от другого, атом водорода, гидроксил; или R5 и R5' вместе образуют оксогруппу;
n обозначает целое число от 0 до 3; и
R6 обозначает, независимо один от другого, атом водорода, галогена, C1-3-алкил, C1-3-алкоксил,
в виде основания или в виде фармацевтически приемлемой соли присоединения с кислотой.
2. Соединение формулы (I) по п.1, отличающееся тем, что
Х обозначает атом кислорода;
R1 обозначает C1-5-алкил, в случае необходимости замещенный фенилом, тиенилом; или R1 обозначает С3-7-циклоалкил, тиенил, пиридинил; тиенильные группы могут быть замещены одной или двумя C1-3-алкильными группами; фенил может быть замещен одним или двумя атомами галогена;
R2 обозначает C1-6-алкил, С3-7-циклоалкил, фенил или пиридинил;
причем фенил может быть замещен 1-2 группами CN, C1-3-алкил, C1-3-алкоксил,
С1-3-фторалкоксил или атомами галогена;
R3 обозначает C1-6-алкил;
R4 обозначает атом водорода или C1-6-алкил;
R5 и R5' обозначают, независимо один от другого, атом водорода, гидроксил; или R5 и R5' вместе образуют оксогруппу;
R6 обозначает атом водорода, галогена, C1-3-алкил, C1-3-алкоксил; и
n=0 или 1,
в виде основания или в виде фармацевтически приемлемой соли присоединения с кислотой.
3. Соединение формулы (I) по п.1 или 2, отличающееся тем, что
Х обозначает атом кислорода;
R1 обозначает метил, этил, 1-метилэтил, 1,1-диметилэтил, пропил, 1-метилпропил, 2-метилпропил или 1-этилпропил, которые могут быть замещены фенилом или тиенилом; или R1 обозначает циклогексил, тиенил или пиридинил; тиенильные группы могут быть замещены одной или двумя метильными группами; фенил может быть замещен одним или двумя атомами хлора или фтора;
R2 обозначает этил, 1-метилэтил, циклогексил, фенил или пиридинил;
причем фенил может быть замещен 1-2 группами CN, метильными, этильными, метокси, этокси, трифторметоксигруппами или атомами хлора или фтора;
R3 обозначает метил, этил или пропил;
R4 обозначает атом водорода, метил или 4-метилпентил;
R5 и R5' обозначают, независимо один от другого, атом водорода, гидроксил; или R5 и R5' вместе образуют оксогруппу;
R6 обозначает атом водорода, хлора или фтора, метил, метокси или этокси;
n=0 или 1; и
C1-4-алкилен представляет собой метилен;
в виде основания или в виде фармацевтически приемлемой соли присоединения с кислотой.
4. Способ получения соединений формулы (I) по одному из пп.1-3 путем пептидного связывания амина формулы (XI)
с кислотой формулы (XII)
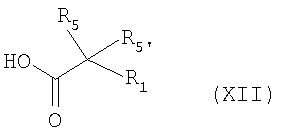
в которых X, R1, R2, R3, R4, R5, R5', R6 и n имеют значения, определенные в формуле (I) по п.1.
5. Фармацевтическая композиция для лечения патологии, в которой является полезным ингибитор образования β-амилоидного пептида β-А4, содержащая по меньшей мере одно соединение формулы (I) по одному из пп.1-3, в виде основания или в виде фармацевтически приемлемой соли присоединения с кислотой, в эффективной дозе, и, в случае необходимости, один или несколько фармацевтически приемлемых эксципиентов.
6. Соединение формулы (I) по одному из пп.1-3 в виде основания или в виде фармацевтически приемлемой соли, для применения в качестве лекарственного средства для лечения патологии, в которой является полезным ингибитор образования β-амилоидного пептида β-А4.
7. Применение соединения формулы (I) по одному из пп.1-3 в виде основания или в виде фармацевтически приемлемой соли присоединения с кислотой для получения лекарственного средства, предназначенного для лечения патологии, в которой является полезным ингибитор образования β-амилоидного пептида β-А4.
8. Применение соединения формулы (I) по одному из пп.1-3 в виде основания или в виде фармацевтически приемлемой соли присоединения с кислотой для получения лекарственного средства, предназначенного для лечения старческой деменции, болезни Альцгеймера, синдрома Дауна, болезни Паркинсона, амилоидной ангиопатии, сосудисто-мозговых расстройств, лобно-височных деменции и болезни Пика, посттравматических деменции, патологий, связанных с нервно-воспалительными процессами, болезни Гентингтона и/или Корсаковского синдрома.
| ПРОИЗВОДНЫЕ ГИДРОКСАМОВОЙ ИЛИ КАРБОНОВОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКАЯ ИЛИ ВЕТЕРИНАРНАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2136657C1 |
| WO 9822433 A1, 28.05.1998 | |||
| Способ передела желтого фосфора в красный | 1929 |
|
SU24392A1 |
| EA 200200247 A1, 29.08.2002. | |||

