ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к органическим соединениям, используемым в терапии и/или профилактике у млекопитающих, и, в частности, относится к ингибиторам белков IAP, используемым для лечения злокачественных опухолей.
ОСНОВА ИЗОБРЕТЕНИЯ
Апоптоз, или запрограммированная гибель клеток, представляет собой генетически и биохимически регулируемый механизм, который играет важную роль в развитии и гомеостазе у беспозвоночных, так же как и у позвоночных. Нарушения апоптоза, которые приводят к преждевременной гибели клеток, связывают с множеством болезней развития. Дефицит апоптоза, который приводит к тому, что клетки не погибают, связывают со злокачественными опухолями и хроническими вирусными инфекциями (Thompson et al., (1995) Science 267, 1456-1462).
Одними из ключевых эффекторных молекул в апоптозе являются каспазы (цистеин-содержащие аспартат-специфические протеиназы). Каспазы представляют собой сильные протеиназы, производящие расщепление [пептидной связи] сразу после остатка аспарагиновой кислоты, и, будучи однажды активированы, они осуществляют расщепление клеточных белков внутри живых клеток. Поскольку каспазы являются весьма сильными протеиназами, то для предотвращения преждевременной гибели клеток необходим строгий контроль этого семейства белков. Обычно каспазы синтезируются в виде крупных неактивных зимогенов, которым для активации необходим протеолитический процессинг. Протеолитический процессинг является одним из путей, который осуществляет регуляцию каспаз. Второй механизм реализуется посредством семейства белков, которые связывают и осуществляют регуляцию каспазы.
Семейство молекул, которые ингибируют активность каспаз, представляет собой ингибиторы апоптоза (IAP) (Deveraux et al., J Clin Immunol (1999), 19:388-398). IAP первоначально были обнаружены в бакуловирусах по их функциональной способности замещать белок P35, антиапоптотический ген (Crook et al. (1993) J. Virology 67, 2168-2174). IAP были описаны в организмах, начиная от дрозофилы до человека. Независимо от их происхождения структурно IAP содержат один из трех повторяющихся доменов IAP бакуловирусов (BIR), и большинство из них также содержат карбокси-концевой «мотив безымянных пальцев» (RING finger motif). Домен BIR как таковой представляет собой цинк-связывающий домен, приблизительно состоящий из 70 остатков, содержащий 4 альфа-спирали и 3 бета-цепи, с цистеиновыми и гистидиновыми остатками, которые координируют ион цинка (Hinds et al.,(1999) Nat. Struct. Biol. 6, 648-651). Считается, что именно домен BIR вызывает антиапоптотический эффект путем ингибирования каспаз и, таким образом, ингибирования апоптоза. В качестве примера показано, что связанный с X-хромосомой человека IAP (XIAP) ингибирует каспазу 3, каспазу 7 и Apaf-1-цитохром C-опосредованную активацию каспазы 9 (Deveraux et al., (1998) EMBO J. 17, 2215-2223). Каспазы 3 и 7 ингибируются BIR2- доменом XIAP, в то время как BIR3-домен XIAP ответственен за ингибирование активности каспазы 9. XIAP экспрессирован повсеместно в большинстве эмбриональных тканей и тканей взрослых (Liston et al, Nature, 1996, 379(6563): 349), и уровень его экспрессии повышен в целом ряде линий опухолевых клеток панелей клеточных линий NCI 60 (Fong et al, Genomics, 2000, 70: 113; Tamm et al, Clin. Cancer Res. 2000, 6(5): 1796). Было показано, что повышенная экспрессия XIAP в опухолевых клетках осуществляет защиту против различных проапоптотических стимулов и стимулирует резистентность к химиотерапии (LaCasse et al, Oncogene, 1998, 17(25): 3247). В соответствии с этим была выявлена жесткая корреляция между уровнями белка XIAP и выживаемостью пациентов с острой миелогенной лейкемией (Tamm et al., выше). Снижение регуляции экспресии XIAP под действием антисмысловых олигонуклеотидов, как было показано, делает опухолевые клетки чувствительными к гибели, индуцируемой обширным разнообразием про-апоптотических агентов, как in vitro, так и in vivo (Sasaki et al, Cancer Res., 2000, 60(20): 5659; Lin et al., Biochem J., 2001, 353: 299; Hu et al., Clin. Cancer Res., 2003, 9(7): 2826). Было показано также, что Smac/DIABLO-полученные пептиды тоже повышают чувствительность целого ряда линий опухолевых клеток к апоптозу, индуцируемому различными про-апоптотическими лекарственными средствами (Arnt et al., J. Biol. Chem., 2002, 277(46): 44236; Fulda et al., Nature Med., 2002, 8(8): 808; Guo et al., Blood, 2002, 99(9): 3419; Vucic et al., J. Biol. Chem., 2002, 277(14): 12275; Yang et al., Cancer Res., 2003, 63(4): 831).
Меланомный IAP (МL-IAP) представляет собой IAP, который не детектируется в большинстве тканей взрослых, но уровень которого сильно повышен в меланомах (Vucic et al., (2000) Current Bio 10: 1359-1366). Определение структуры белка выявило значительную гомологию ML-IAP BIR и «мотива безымянных пальцев» (RING finger motif) с соответствующими доменами, присутствующими в человеческих XIAP, C-IAP1 и C-IAP2. BIR-домен ML-IAP имеет наибольшее сходство с BIR2- и BIR3-доменами XIAP, C-IAP1 и C-IAP2 и, по-видимому, является ответственным за ингибирование апоптоза, как было определено с помощью метода делеционного анализа. Кроме того, Vucic et al. показали, что ML-IAP может ингибировать апоптоз, индуцируемый химиотерапевтическим агентом. Агенты, такие как адриамицин и 4-(трет-бутил)фенол (4-TBP), были тестированы в системе культуры клеток меланомы, в повышенном количестве экспрессирующих ML-IAP, и химиотерапевтические агенты были значительно менее эффективны в киллерной активности в отношении этих клеток по сравнению с их активностью в отношении нормальных меланоцитов в контроле. Механизм, с помощью которого ML-IAP продуцирует антиапоптотическую активность, частично обусловлен ингибированием каспазы 3 и 9. ML-IAP оказался не способен к эффективному ингибированию каспаз 1, 2, 6 или 8.
Поскольку апоптоз является путем, строго контролируемым множеством взаимодействующих факторов, обнаружение того факта, что можно обеспечить регуляцию IAP как таковых, не было столь уж необычным. В плодовой мушке дрозофиле белки Reaper (rpr), Head Involution Defective (hid) и GRIM физически взаимодействуют с семейством IAP дрозофилы и ингибируют их антиапоптотическую активность. У животных SMAC/DIABLO действуют таким образом, что блокируют белки IAP и дают возможность реализации апоптоза. Было показано, что в процессе нормального апоптоза SMAC в результате процессинга превращается в активную форму и высвобождается из митохондрий в цитоплазму, где он физически связывается с белками IAP и предотвращает связывание IAP с каспазой. Такое ингибирование IAP позволяет каспазе оставаться активной и, таким образом, продолжать участие в апоптозе. Любопытно, что гомология между последовательностями ингибиторов IAP показывает, что на N-конце подвергнутых процессингу активных белков существует мотив из четырех аминокислот. Этот тетрапептид, по-видимому, связывается внутри гидрофобного кармана в BIR-домене и нарушает связывание BIR-домена с каспазами (Chai et al., (2000) Nature 406:855-862, Liu et al., (2000) Nature 408:1004-1008, Wu et al., (2000) Nature 408 1008-1012).
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном из аспектов настоящего изобретения предусмотрены новые ингибиторы белков IAP, имеющие общую формулу (I)
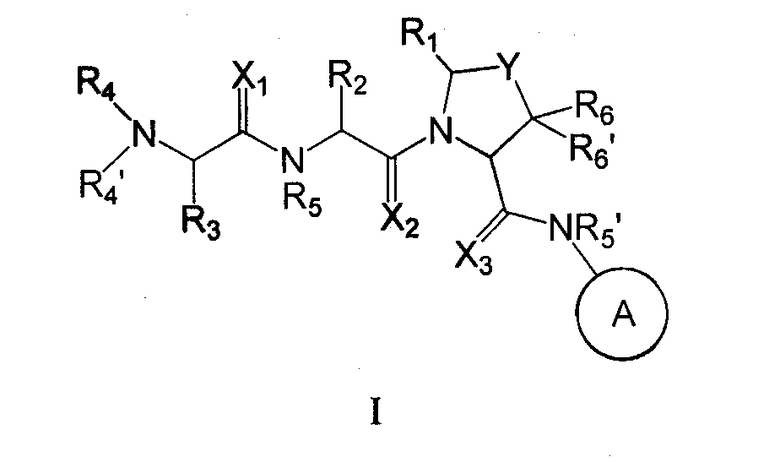
где
каждый X1, X2 и X3 независимо представляет собой O или S;
Y представляет собой (CHR7)n, O или S; где n - это 1 или 2, и R7 представляет собой H, галоген, алкил, арил, аралкил, амино, ариламино, алкиламино, аралкилaмино, алкокси, арилокси или аралкилокси;
A представляет собой 5-членный гетероцикл, содержащий от 1 до 4 гетероатомов, необязательно замещенных амино, гидроксилом, меркапто, галогеном, карбоксилом, амидино, гуанидино, алкилом, алкокси, арилом, арилокси, ацилом, ацилокси, ациламино, алкоксикарбониламино, циклоалкилом, алкилтио, алкилсульфинилом, алкилсульфонилом, аминосульфонилом, алкиламиносульфонилом, алкилсульфониламино или гетероциклом; где каждое алкильное, алкокси-, арильное, арилокси-, ацильное, ацилокси-, ациламино-, циклоалкильное и гетероциклическое замещение необязательно замещено гидроксилом, галогеном, меркапто, карбоксилом, алкилом, алкокси, галогенoалкилом, амино, нитро, циано, циклоалкилом, арилом или гетероциклом;
R1 представляет собой H, или R1 и R2 вместе образуют 5-8 членное кольцо;
R2 представляет собой алкил, циклоалкил, циклоалкилалкил, арил, аралкил, гетероцикл или гетероциклический алкил; каждый из которых необязательно замещен гидроксилом, меркапто, галогеном, амино, карбоксилом, алкилом, галогеналкилом, алкокси или алкилтио;
R3 представляет собой H или алкил;
каждый из R4 и R4' независимо представляет собой H, гидроксил, амино, алкил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероарил или гетероарилалкил, где каждый алкил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероарил и гетероарилалкил необязательно замещены галогеном, гидроксилом, меркапто, карбоксилом, алкилом, алкокси, амино и нитро;
каждый из R5 и R5' независимо представляет собой H или алкил;
каждый из R6 и R6' независимо представляет собой H, алкил, арил или аралкил; и соли и их сольваты.
В другом аспекте изобретения предусмотрены композиции, включающие в себя соединения формулы I и носитель, растворитель или эксципиент.
В другом аспекте изобретения предусмотрен способ индуцирования апоптоза в клетке, включающий в себя введение в указанную клетку соединения формулы I.
В другом аспекте изобретения предусмотрен способ сенсибилизации клетки к апоптотическому сигналу, включающий в себя введение в указанную клетку соединения формулы I.
В другом аспекте изобретения предусмотрен способ ингибирования связывания белка IAP с белком каспазы, включающий в себя приведение в контакт указанного белка IAP с соединением формулы I.
В другом аспекте изобретения предусмотрен способ лечения заболевания или состояния, ассоциированного с повышенной экспрессией у млекопитающего белка IAP, включающий в себя введение указанному млекопитающему эффективного количества соединения формулы I.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВОПЛОЩЕНИЙ.
Термин "алкил" означает разветвленную или неразветвленную, насыщенную или ненасыщенную (т.е. алкенил, алкинил) алифатическую углеводородную группу, имеющую до 12 атомов углерода, если не указано иначе. В случае, когда "алкил" является частью другого термина, например "алкиламино", тогда алкил предпочтительно означает насыщенную углеводородную цепь, однако включающую также ненасыщенные углеводородные цепи, такие как "алкениламино" и "алкиниламино”. Примеры предпочтительных алкильных групп включают в себя метил, этил, н-пропил, изопропил, н-бутил, изо-бутил, втор-бутил, трет-бутил, н-пентил, 2-метилбутил, 2,2-диметилпропил, н-гексил, 2-метилпентил, 2,2-диметилбутил, н-гептил, 3-гептил, 2-метилгексил и пр. Термин "низший алкил", "C1-C4 алкил" и "алкил с 1-4 атомами углерода" являются синонимами и используются взаимозаменяемо и означают метил, этил, 1-пропил, изопропил, циклопропил, 1-бутил, втор-бутил или трет-бутил. Если не указано иначе, замещенные алкильные группы могут содержать один (предпочтительно), два, три или четыре заместителя, которые могут быть одинаковыми или различными. Примеры таких замещенных алкильных групп включают в себя, без ограничения, цианометил, нитрометил, гидроксиметил, тритилоксиметил, пропионилоксиметил, аминометил, карбоксиметил, карбоксиэтил, карбоксипропил, алкилоксикарбонилметил, алкилоксикарбониламинометил, карбамоилоксиметил, метоксиметил, этоксиметил, трет-бутоксиметил, ацетоксиметил, хлорметил, бромметил, йодметил, трифторметил, 6-гидроксигексил, 2,4-дихлор(н-бутил), 2-амино(изопропил), 2-карбамоилоксиэтил и пр. Алкильная группа может быть также замещена группой углеродного кольца. Примеры включают в себя циклопропилметильные, циклобутилметильные, циклопентилметильные и циклогексилметильные группы, а также соответствующие этильные, пропильные, бутильные, пентильные, гексильные группы и т.д. Предпочтительными замещенными алкилами являются замещенные метилы, например метильная группа, замещенная теми же заместителями, что и "замещенная Cn-Cm-алкильная" группа. Примеры замещенной метильной группы включают в себя такие группы, как гидроксиметильная, защищенная гидроксиметильная (например, тетрагидропиранилоксиметильная), ацетоксиметильная, карбамоилоксиметильная, трифторметильная, хлорметильная, карбоксиметильная, бромметильная и йодметильная.
Термин "амидин" означает группу -C(NH)-NHR, где R представляет собой H, или алкил, или аралкил. Предпочтительным амидином является группа -NH-C(NH)-NH2.
Термин "амино" означает первичные (т.е. -NH2), вторичные (т.е. -NRH) и третичные (т.е. -NRR) амины. Предпочтительными вторичными и третичными аминами являются алкиламин, диалкиламин, ариламин, диариламин, аралкиламин и диаралкиламин. Особенно предпочтительными вторичными и третичными аминами являются метиламин, этиламин, пропиламин, изопропиламин, фениламин, бензиламин, диметиламин, диэтиламин, дипропиламин и диизопропиламин.
Используемый здесь термин "амино-защитная группа" относится к производному групп, обычно используемых для блокировки или защиты аминогруппы в процессе реакции с другими функциональными группами соединения. Примеры таких защитных групп включают в себя карбаматы, амиды, алкильные и арильные группы, имины, так же как и многие производные N-гетероатома, которые могут быть удалены для восстановления необходимой амино-группы. Предпочтительными амино-защитными группами являются Boc, Fmoc и Cbz. Другие примеры таких групп можно найти в публикации T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", 2<nd> ed., John Wiley & Sons, Inc., New York, NY, 1991, chapter 7; E. Haslam, "Protective Groups in Organic Chemistry", J. G. W. McOmie, Ed., Plenum Press, New York, NY, 1973, Chapter 5, and T.W. Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, New York, NY, 1981. Термин "защищенный амино" относится к аминогруппам, замещенным одной из упомянутых выше амино-защитных групп.
Термин "арил", используемый отдельно или как часть другого термина, означает карбоциклическую ароматическую группу, конденсированную или не конденсированную, имеющую указанное количество атомов углерода или, если это количество не указано, то вплоть до 14 атомов углерода. Предпочтительные арильные группы включают в себя фенил, нафтил, бифенил, фенантренил, нафтаценил и пр. (см., например, Lang's Handbook of Chemistry (Dean, J. A., ed.) 13<th> ed., Таблицы 7-2 [1985]), и наиболее предпочтительным является фенил. Замещенный фенил или замещенный арил означает фенильную группу или арильную группу, замещенную одним, двумя, тремя, четырьмя или пятью, предпочтительно 1-2, 1-3 или 1-4, заместителями, выбранными, если не указано иначе, из галогена (F, Cl, Br, I), гидрокси, защищенной гидрокси-группы, циано, нитро, алкила (предпочтительно C1-C6-алкила), алкокси-группы (предпочтительно C1-C6-алкокси), бензилокси-группы, карбокси-группы, защищенной карбокси-группы, карбоксиметила, защищенного карбоксиметила, гидроксиметила, защищенного гидроксиметила, аминометила, защищенного аминометила, трифторметила, алкилсульфониламино, арилсульфониламино, гетероциклилсульфониламино, гетероциклила, арила или других указанных групп. Одна или более метиновая (CH) и/или метилeновая (CH2) группы в этих заместителях, в свою очередь, могут быть замещены подобной группой, как определено выше. Примеры, связанные с термином "замещенный фенил", включают в себя, но без ограничения, моно- или ди(галоген)фенильную группу, такую как 2-хлорфенильную, 2-бромфенильную, 4-хлорфенильную, 2,6-дихлорфенильную, 2,5-дихлорфенильную, 3,4-дихлорфенильную, 3-хлорфенильную, 3-бромфенильную, 4-бромфенильную, 3,4-дибромфенильную, 3-хлор-4-фторфенильную, 2-фторфенильную и пр.; моно- или ди(гидрокси)фенильную группу, такую как 4-гидроксифенильную, 3-гидроксифенильную, 2,4-дигидроксифенильную, их защищенные гидрокси-производные и пр.; нитрофенильную группу, такую как 3- или 4-нитрофенильную; цианофенильную группу, например 4-цианофенильную; моно- или ди(низший алкил)фенильную группу, такую как 4-метилфенильную, 2,4-диметилфенильную, 2-метилфенильную, 4-(изопропил)фенильную, 4-этилфенильную, 3-(н-пропил)фенильную и пр.; моно или ди(алкокси)фенильную группу, например 3,4-диметоксифенильную, 3-метокси-4-бензилоксифенильную, 3-метокси-4-(1-хлорметил)бензилоксифенильную, 3-этоксифенильную, 4-(изопропокси)фенильную, 4-(трет-бутокси)фенильную, 3-этокси-4-метоксифенильную и пр.; 3- или 4-трифторметилфенильную; моно- или дикарбоксифенильную или (защищенный карбокси)фенильную группу, такую как 4-карбоксифенильную; моно- или ди(гидроксиметил)фенильную группу или (защищенный гидроксиметил)фенильную группу, такую как 3-(защищенный гидроксиметил)фенильную группу или 3,4-ди(гидроксиметил)фенильную группу; моно- или ди(аминометил)фенильную или (защищенный аминометил)фенильную группу, такую как 2-(аминометил)фенильную или 2,4-(защищенный аминометил)фенильную группу; или моно- или ди(N-(метилсульфониламино))фенильную группу, такую как 3-(N-метилсульфониламино))фенильную группу. Термин "замещенный фенил" также означает двузамещенные фенильные группы, где заместители различны, например 3-метил-4-гидроксифенильная, 3-хлор-4-гидроксифенильная, 2-метокси-4-бромфенильная, 4-этил-2-гидроксифенильная, 3-гидрокси-4-нитрофенильная, 2-гидрокси-4-хлорфенильная и прочие группы, а также трехзамещенные фенильные группы, где заместители различны, например 3-метокси-4-бензилокси-6-метилсульфониламино, 3-метокси-4-бензилокси-6-фенил-сульфониламино-группы, и четырехзамещенные фенильные группы, где заместители различны, например такую как 3-метокси-4-бензилокси-5-метил-6-фенилсульфониламино-группу. Предпочтительные замещенные фенильные группы включают в себя 2-хлорфенильную, 2-аминофенильную, 2-бромфенильную, 3-метоксифенильную, 3-этоксифенильную, 4-бензилоксифенильную, 4-метоксифенильную, 3-этокси-4-бензилоксифенильную, 3,4-диэтоксифенильную, 3-метокси-4-бензилоксифенильную, 3-метокси-4-(1-хлорметил)бензилоксифенильную, 3-метокси-4-(1-хлорметил)бензилокси-6-метилсульфонил-аминофенильную группы. Конденсированные арильные кольца могут также быть замещены какими-нибудь заместителями, предпочтительно 1, 2 или 3 заместителями, подробно описанными здесь, подобно тому, как и для замещенных алкильных групп.
Термины "карбоциклил", "карбоциклический", "карбоцикл" и "карбоцикло", отдельно и в случае, когда используются как фрагмент сложной группы, такой как карбоциклоалкильная группа, относятся к моно-, би- или трициклическому алифатическому кольцу, имеющему от 3 до 14 атомов углерода, и предпочтительно от 3 до 7 атомов углерода, которые могут быть насыщенными или ненасыщенными, ароматическими или неароматическими. Предпочтительные насыщенные карбоциклические группы включают в себя циклопропильную, циклобутильную, циклопентильную и циклогексильную группы, а более предпочтительными являются циклопропильная и циклогексильная группы, и наиболее предпочтительной является циклогексильная группа. Предпочтительными ненасыщенными карбоциклами являются ароматические, например арильные группы, описанные ранее, наиболее предпочтительной является фенильная группа. Термины "замещенный карбоциклил", "карбоцикл" и "карбоцикло" означают такие группы, которые замещены теми же заместителями, что и группа "замещенный алкил".
Термин "карбокси-защитная группа" относится здесь к одному из производных сложного эфира группы карбоновой кислоты, обычно используемого для блокировки или защиты группы карбоновой кислоты в процессе реакции с другими функциональными группами соединения. Примеры таких защитных групп карбоновой кислоты включают в себя 4-нитробензил, 4-метоксибензил, 3,4-диметоксибензил, 2,4-диметоксибензил, 2,4,6-триметоксибензил, 2,4,6-триметилбензил, пентаметилбензил, 3,4- метилендиоксибензил, бензгидрил, 4,4'-диметоксибензгидрил, 2,2',4,4'-тетраметоксибензгидрил, алкильные группы, такие как трет-бутил или трет-амил, тритил, 4-метокситритил, 4,4'-диметокситритил, 4,4',4"-триметокситритил, 2-фенилпроп-2-ил, триметилсилил, трет-бутилдиметилсилил, фенацил, 2,2,2-трихлорэтил, бета-(триметилсилил)этил, бета-(ди(н-бутил)метилсилил)этил, пара-толуолсульфонилэтил, 4-нитробензилсульфонилэтил, аллил, циннамил, 1-(триметилсилилметил)проп-1-ен-3-ил и прочие фрагменты. Используемые разновидности карбокси-защитной группы не столь важны, так как дериватизированная карбоновая кислота стабильна в условиях последующих реакций на других фрагментах молекулы и может быть удалена на соответствующем участке без деструктурирования остатка молекулы. В частности, важно не подвергать карбокси-защищенную молекулу воздействию сильных нуклеофильных оснований, таких как гидроксид лития или NaOH, или не создавать восстановительных условий с использованием сильно активированных гидридов металла, таких как LiAlH4. (Таких жестких условий удаления надо избегать также в случае удаления амино-защитных групп и гидрокси-защитных групп, упомянутых выше.) Подходящими защитными группами карбоновой кислоты являются алкильные (например, метил, этил, трет-бутил), аллильные, бензильные и пара-нитробензильные группы. Подобные карбокси- защитные группы, используемые также в областях цефалоспорина, пенициллина и пептида, также можно использовать для защиты заместителей карбокси-группы. Другие примеры таких групп можно найти в публикации T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", 2<nd> ed., John Wiley & Sons, Inc., New York, N. Y., 1991, chapter 5; E. Haslam, "Protective Groups in Organic Chemistry", J. G. W. McOmie, Ed., Plenum Press, New York, N. Y., 1973, Chapter 5, and T. W. Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, New York, NY, 1981, Chapter 5. Термин "защищенный карбокси" относится к карбокси-группе, замещенной одной или более карбокси-защитной группами.
Термин "гуанидин" означает группу -NH-C(NH)-NHR, где R представляет собой H, или алкил, или аралкил. Предпочтительной гуанидиновой группой является группа -NH-C(NH)-NH2.
Используемый здесь термин "гидрокси-защитная группа" относится к производному гидрокси-группы, обычно используемому для блокировки или защиты гидрокси-группы в процессе реакции с другими функциональными группами соединения. Примеры таких защитных групп включают в себя тетрагидропиранилокси-, бензоильную, ацетокси-, карбамоилокси-, бензильную группы и группы силильных эфиров (например, TBS, TBDPS). Другие примеры таких групп можно найти в публикации T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", 2<nd> ed., John Wiley & Sons, Inc., New York, NY, 1991, chapters 2-3; E. Haslam, "Protective Groups in Organic Chemistry", J. G. W. McOmie, Ed., Plenum Press, New York, NY, 1973, Chapter 5, and T. W. Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, New York, NY, 1981. Термин "защищенный гидрокси" относится к гидрокси-группе, замещенной одной или более из указанных выше гидрокси-защитных групп.
Термины "гетероциклическая группа", "гетероциклический", "гетероцикл", "гетероциклил" или "гетероцикло", отдельно и в случае, когда используются как фрагмент сложной группы, такой как гетероциклоалкильная группа, используются взаимозаменяемо и относятся к некоторому моно-, би- или трициклическому, насыщенному или ненасыщенному, ароматическому (гетероарил) или неароматическому кольцу, имеющему некоторое установленное количество, обычно от 5 до 14, кольцевых атомов, причем атомами кольца являются атомы углерода и по меньшей мере один гетероатом (азот, сера или кислород), но предпочтительно 1-4 гетероатома. Обычно 5-членное кольцо имеет 0-2 двойные связи, и 6- или 7- членное кольцо имеет 0-3 двойные связи, и гетероатомы азота или серы могут быть необязательно оксидированы (например, SO, SO2), и некоторые гетероатомы азота могут быть необязательно кватернизированы. Предпочтительные неароматические гетероциклы включают в себя морфолинил (морфолино), пирролидинил, оксиранил, оксетанил, тетрагидрофуранил, 2,3-дигидрофуранил, 2H-пиранил, тетрагидропиранил, тииранил, тиетанил, тетрагидротиетанил, азиридинил, азетидинил, 1-метил-2-пирролил, пиперазинил и пиперидинил. Группа "гетероциклоалкил" является гетероциклической группой, определяемой выше, имеющей ковалентную связь с алкильной группой, которая определена выше. Предпочтительные 5-членные гетероциклы, содержащие атомы серы или кислорода и от одного до трех атомов азота, включают в себя тиазолил, в частности тиазол-2-ил и тиазол-2-ил-N-оксид, тиадиазолил, в частности 1,3,4-тиадиазол-5-ил и 1,2,4-тиадиазол-5-ил, оксазолил, предпочтительно оксазол-2-ил, и оксадиазолил, такой как 1,3,4-оксадиазол-5-ил и 1,2,4-оксадиазол-5-ил. Предпочтительные гетероциклические 5-членные кольца, содержащие от 2 до 4 атомов азота, включают в себя имидазолил, предпочтительно имидазол-2-ил; триазолил, предпочтительно 1,3,4-триазол-5-ил; 1,2,3-триазол-5-ил, 1,2,4-триазол-5-ил и тетразолил, предпочтительно 1H-тетразол-5-ил. Предпочтительными бензо-конденсированными 5-членными гетероциклами являются бензоксазол-2-ил, бензтиазол-2-ил и бензимидазол-2-ил. Предпочтительные 6-членные гетероциклы содержат от 1 до 3 атомов азота и необязательно атомы серы или кислорода, например пиридил, такой как пирид-2-ил, пирид-3-ил и пирид-4-ил; пиримидил, предпочтительно пиримид-2-ил и пиримид-4-ил; триазинил, предпочтительно 1,3,4-триазин-2-ил и 1,3,5-триазин-4-ил; пиридaзинил, в частности пиридазин-3-ил и пиразинил. Пиридин-N-оксидные и пиридaзин-N-оксидные и пиридильные, пиримид-2-ильные, пиримид-4-ильные, пиридaзинильные и 1,3,4-триазин-2-ильные группы являются предпочтительными. Примеры заместителей для необязательно замещенных гетероциклов и другие примеры 5- и 6-членных систем колец, описанных выше, можно найти в публикации W. Druckheimer et al., U.S. Patent No. 4278793.
Термин "гетероарил" используемый отдельно или как фрагмент сложной группы, такой как гетероаралкильная группа, относится к некоторой моно-, би- или трициклической ароматической системе колец, имеющей установленное число атомов, где хотя бы одно кольцо является 5-, 6- или 7-членным кольцом, содержащим от одного до четырех гетероатомов, выбранных из азотной, кислородной или серной групп, и предпочтительно, чтобы хотя бы один гетероатом являлся азотом (Lang's Handbook of Chemistry, выше). В определение включены также некоторые бициклические группы, в которых некоторые из вышеупомянутых гетероарильных колец слиты с бензольным кольцом. Предпочтительны гетероарилы, в которых гетероатомами являются азот или кислород. Следующие системы колец являются примерами гетероарильных (замещенных или незамещенных) групп, обозначенных термином "гетероарил": тиенил, фурил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, изоксазолил, триазолил, тиадиазолил, оксадиазолил, тетразолил, тиатриазолил, оксатриазолил, пиридил, пиримидил, пиразинил, пиридaзинил, тиазинил, оксазинил, триазинил, тиадиазинил, оксадиазинил, дитиазинил, диоксазинил, оксатиазинил, тетразинил, тиатриазинил, оксатриазинил, дитиадиазинил, имидазолинил, дигидропиримидил, тетрагидропиримидил, тетразолo[1,5-b]пиридaзинил и пуринил, так же как и бензо-конденсированные производные, например, бензоксазолил, бензофурил, бензотиазолил, бензотиадиазолил, бензотриазолил, бензоимидазолил и индолил. Особенно предпочтительна группа "гетероарила", которая включает в себя 1,3-тиазол-2-ил, 4-(карбоксиметил)-5-метил-1,3-тиазол-2-ил, 4-(карбоксиметил)-5-метил-1,3-тиазол-2-ил соль натрия, 1,2,4-тиадиазол-5-ил, 3-метил-1,2,4-тиадиазол-5-ил, 1,3,4-триазол-5-ил, 2-метил-1,3,4-триазол-5-ил, 2-гидрокси-1,3,4-триазол-5-ил, 2- карбокси-4-метил-1,3,4-триазол-5-ил соль натрия, 2-карбокси-4-метил-1,3,4-триазол-5-ил, 1,3-оксазол-2-ил, 1,3,4-оксадиазол-5-ил, 2-метил-1,3,4-оксадиазол-5-ил, 2-(гидроксиметил)-1,3,4-оксадиазол-5-ил, 1,2,4-оксадиазол-5-ил, 1,3,4-тиадиазол-5-ил, 2-тиол-1,3,4-тиадиазол-5-ил, 2-(метилтио)-1,3,4-тиадиазол-5-ил, 2-амино-1,3,4-тиадиазол-5-ил, 1H-тетразол-5-ил, 1-метил-1H-тетразол-5-ил, 1-(1-(диметиламино)эфир-2-ил)-1H-тетразол-5-ил, 1-(карбоксиметил)-1H-тетразол-5-ил, 1-(карбоксиметил)-1H-тетразол-5-ил соль натрия, 1-(метилсульфоновая кислота)-1H-тетразол-5-ил, 1-(метилсульфоновая кислота)-1H-тетразол-5-ил соль натрия, 2-метил-1H-тетразол-5-ил, 1,2,3-триазол-5-ил, 1-метил-1,2,3-триазол-5-ил, 2-метил-1,2,3-триазол-5-ил, 4-метил-1,2,3-триазол-5-ил, пирид-2-ил-N-оксид, 6-метокси-2-(н-оксид)-пиридаз-3-ил, 6-гидроксипиридаз-3-ил, 1-метилпирид-2-ил, 1-метилпирид-4-ил, 2-гидроксипиримид-4-ил, 1,4,5,6-тетрагидро-5,6-диоксо-4-метил-ас-триазин-3-ил, 1,4,5,6-тетрагидро-4-(формилметил)-5,6-диоксо-ас-триазин-3-ил, 2,5-дигидро-5-оксо-6-гидрокси-ас-триазин-3-ил, 2,5-дигидро-5-оксо-6-гидрокси-ас-триазин-3-ил соль натрия, 2,5-дигидро-5-оксо-6-гидрокси-2-метил-ас-триазин-3-ил соль натрия, 2,5-дигидро-5-оксо-6-гидрокси-2-метил-ас-триазин-3-ил, 2,5-дигидро-5-оксо-6-метокси-2-метил-ас-триазин-3-ил, 2,5-дигидро-5-оксо-ас-триазин-3-ил, 2,5-дигидро-5-оксо-2-метил-ас-триазин-3-ил, 2,5-дигидро-5-оксо-2,6-диметил-ас-триазин-3-ил, тетразолo[1,5-b]пиридaзин-6-ил и 8-аминотетразолo[1,5-b]пиридaзин-6-ил. Альтернативная "гетероарильная" группа включает в себя; 4-(карбоксиметил)-5-метил-1,3-тиазол-2-ил, 4-(карбоксиметил)-5-метил-1,3-тиазол-2-ил соль натрия, 1,3,4-триазол-5-ил, 2-метил-1,3,4-триазол-5-ил, 1H-тетразол-5-ил, 1-метил-1H-тетразол-5-ил, 1-(1-(диметиламино)эфир-2-ил)-1H-тетразол-5-ил, 1-(карбоксиметил)-1H-тетразол-5-ил, 1-(карбоксиметил)-1H-тетразол-5-ил соль натрия, 1-(метилсульфоновая кислота)-1H-тетразол-5-ил, 1-(метилсульфоновая кислота)-1H-тетразол-5-ил соль натрия, 1,2,3-триазол-5-ил, 1,4,5,6-тетрагидро-5,6-диоксо-4-метил-ас-триазин-3-ил, 1,4,5,6-тетрагидро-4-(2-формилметил)-5,6-диоксо-ас-триазин-3-ил, 2,5-дигидро-5-оксо-6-гидрокси-2-метил-ас-триазин-3-ил соль натрия, 2,5-дигидро-5-оксо-6-гидрокси-2-метил-ас-триазин-3-ил, тетразолo[1,5-b]пиридaзин-6-ил и 8-аминотетразолo[1,5-b]пиридaзин-6-ил.
Термин "ингибитор" означает соединение, которое уменьшает или предотвращает связывание белков IAP с белками каспазы или которое уменьшает или предотвращает ингибирование апоптоза белком IAP. В альтернативном варианте термин "ингибитор" означает соединение, которое предотвращает связывание взаимодействия X-IAP с каспазой или связывание взаимодействия МL-IAP с SMAC.
Термин "фармацевтически приемлемые соли" включает в себя как аддитивные соли кислот, так и аддитивные соли оснований. Термин "фармацевтически приемлемые аддитивные соли кислот" относится к таким солям, которые сохраняют биологическую эффективность и свойства свободных оснований и которые не являются биологически или в каком-нибудь другом отношении нежелательными, образованными с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, угольная кислота, фосфорная кислота и пр., и органические кислоты, которые могут быть выбраны из алифатических, циклоалифатических, ароматических, аралифатических, гетероциклических, карбоновых и сульфоновых классов органических кислот, таких как муравьиная кислота, уксусная кислота, пропионовая кислота, гликолевая кислота, глюконовая кислота, молочная кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, аспарагиновая кислота, аскорбиновая кислота, глутаминовая кислота, антраниловая кислота, бензойная кислота, коричная кислота, миндальная кислота, памовая кислота, фенилуксусная кислота, метансульфоновая кислота, этансульфоновая кислота, пара-толуолсульфоновая кислота, салициловая кислота и пр.
Термин "фармацевтически приемлемые аддитивные соли оснований" включает в себя производные неорганических оснований, таких, например, как соли натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди, марганца, алюминия и пр. Особенно предпочтительными являются соли аммония, калия, натрия, кальция и магния. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают в себя соли первичных, вторичных и третичных аминов, замещенных аминов, включая природно образующиеся замещенные амины, циклические амины и ион основания, замещенный полимерами, такими как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, 2-диэтиламиноэтанол, триметамин, дициклогексиламин, лизин, аргинин, гистидин, кофеин, процаин, гидрабамин, хлорин, бетаин, этилендиамин, глюкозамин, метилглюкамин, теобромин, пурины, пиперазин, пиперидин, N-этил пиперидин, полиамины и пр. Особенно предпочтительными органическими нетоксичными основаниями являются изопропиламин, диэтиламин, этаноламин, триметамин, дициклогексиламин, хлорин и кофеин.
Настоящее изобретение связано с новыми соединениями общей формулы
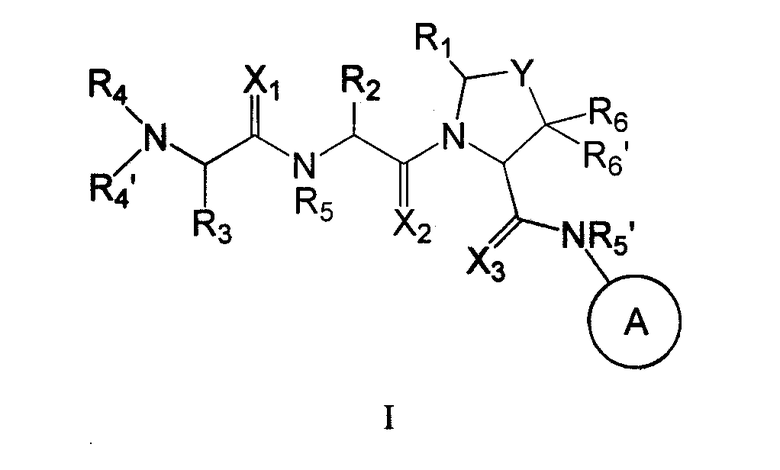
где X, Y, A, R1, R2, R3, R4, R4', R5, R5', R6 и R6' - такие, как здесь описано,
каждый из X1 и X2 независимо представляет собой O или S. В другом предпочтительном воплощении оба, X1 и X2, представляют собой O. В другом предпочтительном воплощении оба, X1 и X2, представляют собой S. В другом предпочтительном воплощении X1 представляет собой S, тогда как X2 представляет собой O. В другом предпочтительном воплощении X1 представляет собой O, тогда как X2 представляет собой S.
Y представляет собой (CHR7)n, O или S; где n представляет собой 1 или 2, и R7 представляет собой H, галоген, алкил, арил, аралкил, амино, ариламино, алкиламино, аралкиламино, алкокси, арилокси или аралкилокси. В отдельном воплощении Y представляет собой CH2. В отдельном воплощении n представляет собой 1. В отдельном воплощении n представляет собой 1, и Y представляет собой CHR7, где R7 представляет собой аралкилокси, например бензилокси. В отдельном воплощении n представляет собой 1, и Y представляет собой CHR7, где R7 представляет собой F. В отдельном воплощении n представляет собой 1, и Y представляет собой CHR7, где R7 представляет собой аралкиламино, например бензиламино. В другом отдельном воплощении Y представляет собой O. В другом частном воплощении Y представляет собой S.
Кольцо 'A' представляет собой 5-членный гетероцикл, содержащий 1-4 гетероатома, необязательно замещенных амино, гидроксилом, меркапто, галогеном, карбоксилом, амидино, гуанидином, алкилом, алкокси, арилом, арилокси, ацилом, ацилокси, ациламино, алкоксикарбониламино, циклоалкилом, алкилтио, алкилсульфинилом, алкилсульфонилом, аминосульфонилом, алкиламиносульфонилом, алкилсульфониламино или гетероциклом; где каждое алкильное, алкокси-, арильное, арилокси-, ацильное, ацилокси-, ациламино-, циклоалкильное и гетероциклическое замещение необязательно замещено гидроксилом, галогеном, меркапто, карбоксилом, алкилом, алкокси, галогеналкилом, амино, нитро, циано, циклоалкилом, арилом или гетероциклом. В другом воплощении группы 5-членного гетероциклического кольца А необязательно замещены амино, гидроксилом, меркапто, галогеном, карбоксилом, амидино, гуанидином, алкилом, алкокси, арилом, арилокси, ацилом, ацилокси, ациламино, циклоалкилом или гетероциклом, где каждое алкильное, алкокси-, арильное, арилокси-, ацильное, ацилокси-, ациламино-, циклоалкильное и гетероциклическое замещение необязательно замещено гидроксилом, галогеном, меркапто, карбоксилом, алкилом, галогеналкилом, амино, нитро, циклоалкилом, арилом или гетероциклом. В отдельном воплощении кольцо A является ароматическим. В другом отдельном воплощении кольцо A имеет формулу IIa или IIb:
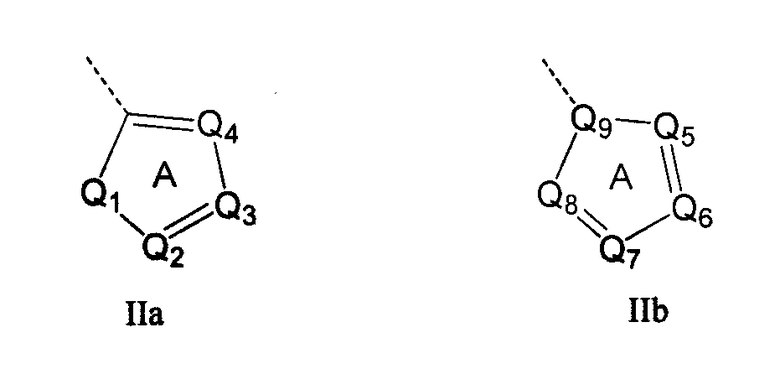
где Q1 представляет собой NR8, O или S; каждый из Q2, Q3, Q4, Q5, Q6, Q7 и Q8 независимо представляет собой CR9 или N; где R9 представляет собой H, амино, гидроксил, меркапто, галоген, карбоксил, амидино, гуанидино, алкил, алкокси, арил, арилокси, ацил, ацилокси, ациламино, циклоалкил или гетероцикл; где каждое алкильное, алкокси-, арильное, арилокси-, ацильное, ацилокси-, ациламино-, циклоалкильное и гетероциклическое замещение необязательно замещено гидроксилом, галогеном, меркапто, карбоксилом, алкилом, галогеналкилом, амино, нитро, циклоалкилом, арилом или гетероциклом; R8 представляет собой H, алкил, ацил, арил, циклоалкил или гетероцикл; где каждый алкил, арил, циклоалкил и гетероцикл необязательно замещены гидроксилом, галогеном, меркапто, карбоксилом, алкилом, галогеналкилом, амино, нитро, циклоалкилом, арилом или гетероциклом; и Q9 представляет собой CH или N. В отдельном воплощении кольцо A представляет собой группу формулы II. В отдельном воплощении кольцо A представляет собой группу формулы II, в которой Q4 представляет собой CR9, где R9 представляет собой арил или гетероарил, необязательно замещенный, как описано выше. В отдельном воплощении кольцо A представляет собой группу формулы II, в которой Q4 представляет собой CR9, где R9 представляет собой фенил. В отдельном воплощении кольцо A представляет собой группу формулы II, в которой Q4 представляет собой CR9, где R9 представляет собой фенил, и Q3 представляет собой CH или CF. В другом воплощении кольцо A представляет собой группу формулы II, в которой Q4 представляет собой CR9, где R9 представляет собой пиридин-2-ил. В другом воплощении кольцо A представляет собой группу формулы II, в которой Q4 представляет собой CR9, где R9 представляет собой пиридин-2-ил, и Q3 представляет собой C-Me.
В другом воплощении кольцо A согласно формуле IIа или IIb представляет собой пиррольное кольцо, необязательно замещенное алкилом, арилом, аралкилом, циклоалкилом, циклоалкилалкилом, гетероциклом или гетероциклоалкилом, необязательно замещенным галогенгидроксилом, меркапто, карбоксилом, алкилом, галогеналкилом, амино, нитро, арилом или гетероарилом. В отдельном воплощении кольцо A замещено арильной или гетероарильной группой. В другом воплощении кольцо A выбрано из группы, состоящей из следующего:
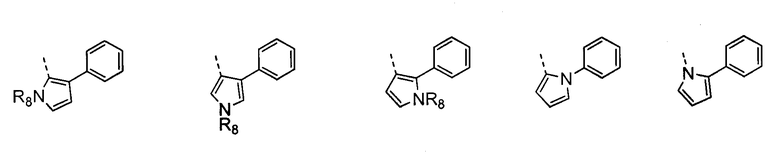
где R8 представляет собой H, алкил (например, метил, этил или пропил) или ацил (например, ацетил). В отдельном воплощении R8 представляет собой H.
В другом воплощении кольцо A представляет собой фуран, необязательно замещенный алкилом, арилом, аралкилом, циклоалкилом, циклоалкилалкилом, гетероциклом или гетероциклоалкилом, необязательно замещенным галогенгидроксилом, меркапто, карбоксилом, алкилом, галогеналкилом, амино, нитро, арилом или гетероарилом. В другом воплощении кольцо A замещено арильной или гетероарильной группой. В отдельном воплощении кольцо A выбрано из группы, состоящей из
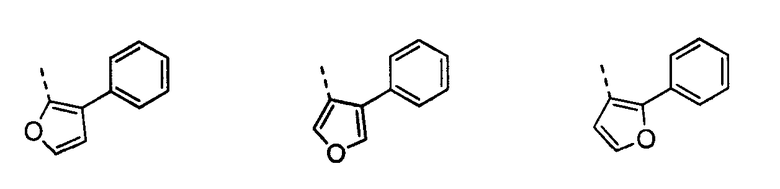
В другом воплощении кольцо A представляет собой тиофен, необязательно замещенный алкилом, арилом, аралкилом, циклоалкилом, циклоалкилалкилом, гетероциклом или гетероциклоалкилом, необязательно замещенным галогенгидроксилом, меркапто, карбоксилом, алкилом, галогеналкилом, амино, нитро, арилом или гетероарилом. В другом воплощении кольцо A замещено арильной или гетероарильной группой. В отдельном воплощении кольцо A выбрано из группы, состоящей из следующего:
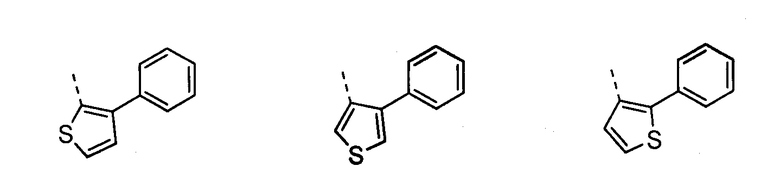
В другом воплощении кольцо A представляет собой пиразол, необязательно замещенный алкилом, арилом, аралкилом, циклоалкилом, циклоалкилалкилом, гетероциклом или гетероциклоалкилом, необязательно замещенным галогенгидроксилом, меркапто, карбоксилом, алкилом, галогеналкилом, амино, нитро, арилом или гетероарилом. В другом воплощении кольцо A замещено арильной или гетероарильной группой. В отдельном воплощении кольцо A выбрано из группы, состоящей из следующего:
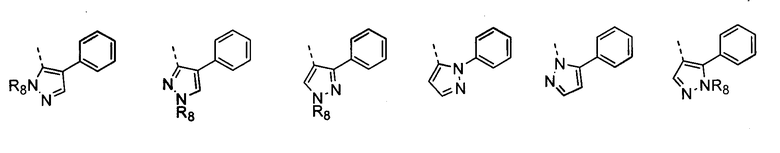
где R8 представляет собой H, алкил (например, метил, этил или пропил) или ацил (например, ацетил). В отдельном воплощении R8 представляет собой H.
В другом воплощении кольцо A представляет собой имидазол, необязательно замещенный алкилом, арилом, аралкилом, циклоалкилом, циклоалкилалкилом, гетероциклом или гетероциклоалкилом, необязательно замещенным галогенгидроксилом, меркапто, карбоксилом, алкилом, галогеналкилом, амино, нитро, арилом или гетероарилом. В другом воплощении кольцо A замещено арильной или гетероарильной группой. В отдельном воплощении кольцо A выбрано из группы, состоящей из следующего:
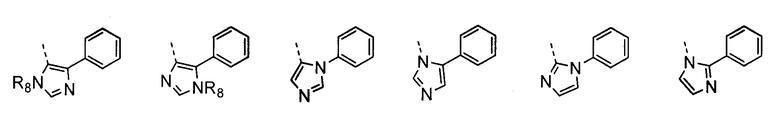
где R8 представляет собой H, алкил (например, метил, этил или пропил) или ацил (например, ацетил). В отдельном воплощении R8 представляет собой H.
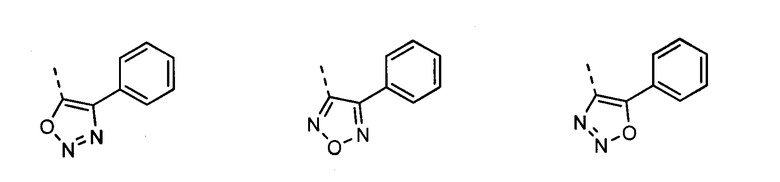
В другом воплощении кольцо A представляет собой оксазол, необязательно замещенный алкилом, арилом, аралкилом, циклоалкилом, циклоалкилалкилом, гетероциклом или гетероциклоалкилом, необязательно замещенным галогенгидроксилом, меркапто, карбоксилом, алкилом, галогеналкилом, амино, нитро, арилом или гетероарилом. В другом воплощении кольцо A замещено арильной или гетероарильной группой. В отдельном воплощении кольцо A выбрано из группы, состоящей из следующего:
В другом воплощении кольцо A представляет собой изоксазол, необязательно замещенный алкилом, арилом, аралкилом, циклоалкилом, циклоалкилалкилом, гетероциклом или гетероциклоалкилом, необязательно замещенным галогенгидроксилом, меркапто, карбоксилом, алкилом, галогеналкилом, амино, нитро, арилом или гетероарилом. В другом воплощении кольцо A замещено арильной или гетероарильной группой. В отдельном воплощении кольцо A выбрано из группы, состоящей из следующего:
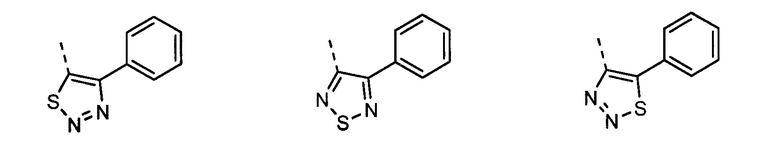
В другом воплощении кольцо A представляет собой тиазол, необязательно замещенный алкилом, арилом, аралкилом, циклоалкилом, циклоалкилалкилом, гетероциклом или гетероциклоалкилом, необязательно замещенным галогенгидроксилом, меркапто, карбоксилом, алкилом, галогеналкилом, амино, нитро, арилом или гетероарилом. В другом воплощении кольцо A замещено арильной или гетероарильной группой. В отдельном воплощении кольцо A выбрано из группы, состоящей из следующего:

В другом воплощении кольцо A представляет собой изотиазол, необязательно замещенный алкилом, арилом, аралкилом, циклоалкилом, циклоалкилалкилом, гетероциклом или гетероциклоалкилом, необязательно замещенным галогенгидроксилом, меркапто, карбоксилом, алкилом, галогеналкилом, амино, нитро, арилом или гетероарилом. В другом воплощении кольцо A замещено арильной или гетероарильной группой. В отдельном воплощении кольцо A выбрано из группы, состоящей из следующего:
В другом воплощении кольцо A представляет собой 1,2,3-триазол, необязательно замещенный алкилом, арилом, аралкилом, циклоалкилом, циклоалкилалкилом, гетероциклом или гетероциклоалкилом, необязательно замещенным галогенгидроксилом, меркапто, карбоксилом, алкилом, галогеналкилом, амино, нитро, арилом или гетероарилом. В другом воплощении кольцо A замещено арильной или гетероарильной группой. В отдельном воплощении кольцо A выбрано из группы, состоящей из следующего:
где R8 представляет собой H, алкил (например, метил, этил или пропил) или ацил (например, ацетил). В отдельном воплощении R8 представляет собой H.
В другом воплощении кольцо A представляет собой 1,2,4-триазол, необязательно замещенный алкилом, арилом, аралкилом, циклоалкилом, циклоалкилалкилом, гетероциклом или гетероциклоалкилом, необязательно замещенным галогенгидроксилом, меркапто, карбоксилом, алкилом, галогеналкилом, амино, нитро, арилом или гетероарилом. В другом воплощении кольцо A замещено арильной или гетероарильной группой. В отдельном воплощении кольцо A выбрано из группы, состоящей из следующего:
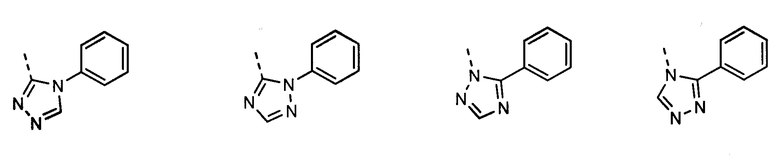
В другом воплощении кольцо A представляет собой оксадиазол, необязательно замещенный алкилом, арилом, аралкилом, циклоалкилом, циклоалкилалкилом, гетероциклом или гетероциклоалкилом, необязательно замещенным галогенгидроксилом, меркапто, карбоксилом, алкилом, галогеналкилом, амино, нитро, арилом или гетероарилом. В другом воплощении кольцо A замещено арильной или гетероарильной группой. В отдельном воплощении кольцо A выбрано из группы, состоящей из следующего:
В другом воплощении кольцо A представляет собой тиадиазол, необязательно замещенный алкилом, арилом, аралкилом, циклоалкилом, циклоалкилалкилом, гетероциклом или гетероциклоалкилом, необязательно замещенным галогенгидроксилом, меркапто, карбоксилом, алкилом, галогеналкилом, амино, нитро, арилом или гетероарилом. В другом воплощении кольцо A замещено арильной или гетероарильной группой. В отдельном воплощении кольцо A выбрано из группы, состоящей из следующего:
В другом воплощении кольцо A представляет собой тетразол, необязательно замещенный алкилом, арилом, аралкилом, циклоалкилом, циклоалкилалкилом, гетероциклом или гетероциклоалкилом, необязательно замещенным галогенгидроксилом, меркапто, карбоксилом, алкилом, галогеналкилом, амино, нитро, арилом или гетероарилом. В другом воплощении кольцо A замещено арильной или гетероарильной группой. В отдельном воплощении кольцо A выбрано из группы, состоящей из следующего:

В отдельном воплощении кольцо A представляет собой следующее:
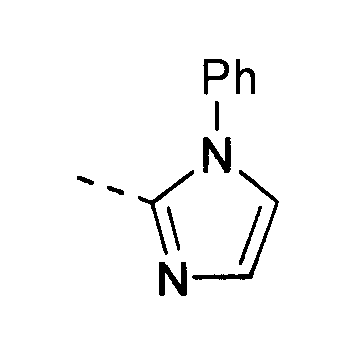
В отдельном воплощении кольцо A представляет собой следующее:
R1 представляет собой H, или R1 и R2 вместе образуют 5-8-членное кольцо. В отдельном воплощении R1 представляет собой H. В отдельном воплощении R1 и R2 вместе образуют 6-членное кольцо. В отдельном воплощении R1 и R2 вместе образуют 7-членное кольцо. В другом отдельном воплощении R1 и R2 вместе образуют 8-членное кольцо. В другом отдельном воплощении R1 и R2 вместе образуют 7-членное кольцо, тогда как Y представляет собой S. В другом отдельном воплощении R1 представляет собой H, тогда как Y представляет собой CH2. В другом отдельном воплощении R1 представляет собой H, тогда как Y представляет собой S. В другом отдельном воплощении R1 представляет собой H, тогда как Y представляет собой O.
R2 представляет собой алкил, циклоалкил, циклоалкилалкил, арил, аралкил, гетероцикл или гетероциклилалкил. В предпочтительном воплощении R2 представляет собой алкил или циклоалкил. В одном из воплощений каждая группа R2 необязательно замещена гидроксилом, меркапто, галогеном, амино, карбоксилом, алкилом, галогеналкилом, алкокси или алкилтио. В одном из воплощений изобретения R2 представляет собой трет-бутил, изопропил, циклогексил, циклопентил или фенил. В отдельном воплощении R1 представляет собой циклогексил. В другом отдельном воплощении R2 представляет собой тетрагидропиран-4-ил. В другом отдельном воплощении R2 представляет собой изопропил (т.е. боковую цепь аминокислоты валина). В другом отдельном воплощении R2 представляет собой трет-бутил. В отдельном воплощении R2 ориентировано таким образом, что аминокислота или аналог аминокислоты, включенные в R2, находятся в L-конфигурации.
R3 представляет собой H или алкил. В предпочтительном воплощении R3 представляет собой H или метил, этил, пропил или изопропил. В отдельном предпочтительном воплощении R3 представляет собой H или метил. В наиболее предпочтительном воплощении R3 представляет собой метил. В другом отдельном воплощении R3 представляет собой трет-бутил. В предпочтительном воплощении R3 ориентировано таким образом, что аминокислота или аналог аминокислоты, включенные в R2, находятся в L-конфигурации.
Каждый из R4 и R4' независимо представляет собой H, гидроксил, амино, алкил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероарил, или гетероарилалкил, где каждый алкил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероарил и гетероарилалкил необязательно замещены галогеном, гидроксилом, меркапто, карбоксилом, алкилом, алкокси, амино и нитро. В отдельном воплощении оба, R4 и R4', представляют собой H. В другом отдельном воплощении R4 представляет собой метил, и R4' представляет собой H. В отдельном воплощении один из R4 и R4' представляет собой гидроксил (OH), тогда как другой представляет собой H. В другом отдельном воплощении один из R4 и R4' представляет собой амино, например NH2, NHMe и NHEt, тогда как другой представляет собой H. В отдельном воплощении R4' представляет собой H, и R4 представляет собой H, алкил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероарил гетероарилалкил. В отдельном воплощении R4 представляет собой группу, выбранную из группы, состоящей из следующего:
Каждый из R5 и R5' независимо представляет собой H или алкил. В предпочтительном воплощении R5 и R5' представляют собой H или метил. В отдельном воплощении R5 представляет собой H, и R5' представляет собой метил. В другом отдельном воплощении R5 представляет собой метил, и R5' представляет собой H. В другом отдельном воплощении оба, R5 и R5', представляют собой метил. В другом отдельном воплощении оба, R5 и R5', представляют собой H.
Каждый из R6, и R6' независимо представляет собой H, алкил, арил или аралкил. В отдельном воплощении R6 представляет собой алкил, например метил. В другом отдельном воплощении R6 представляет собой арил, например фенил. В другом отдельном воплощении R6 представляет собой аралкил, например бензил. В отдельном воплощении оба, R6 и R6', одинаковы, например, оба являются алкилом, например метилом. В другом отдельном воплощении R6 представляет собой метил, и R6' представляет собой H.
Соединения, используемые в изобретении, содержат один или более асимметричных атомов углерода. Соответственно, соединения существуют в виде диастереомеров, энантиомеров или их смесей. В процессе синтеза соединений могут быть использованы рацематы, диастереомеры или энантиомеры в качестве исходных материалов или промежуточных соединений. Диастереомерные соединения могут быть выделены хроматографическим методом и методом кристаллизации. Аналогично энантиомерные смеси могут быть выделены с использованием таких же и других известных в этой области методов. Каждый из асимметричных атомов углерода может находиться в R- или S-конфигурации, и обе эти конфигурации входят в объем настоящего изобретения. Соединения согласно изобретению предпочтительно имеют следующую стереохимическую конфигурацию формулы I'
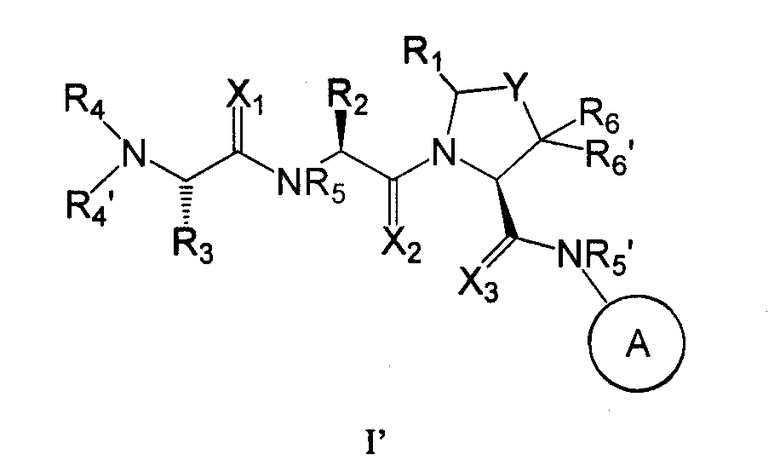
где X, Y, A, R1, R2, R3, R4, R4', R5, R5', R6 и R6' имеют значения, которые описаны выше.
Изобретение также охватывает пролекарства соединений, описанных выше. Подходящими пролекарствами являются те, которые приемлемым образом включают в себя известные амино-защитные и карбокси-защитные группы, которые отделяют, например, посредством гидролиза, с получением на выходе исходного соединения в физиологических условиях. Предпочтительным классом пролекарств являются соединения, в которых атом азота в амино-, амидино-, аминоалкиленамино-, иминоалкилeнамино- или гуанидино- группе замещен гидрокси (OH) группой, алкилкарбонильной (-CO-R) группой, алкоксикарбонильной (-CO-OR), ацилоксиалкил-алкоксикарбонильной (-CO-O-R-O-CO-R) группой, где R представляет собой моновалентную или дивалентную группу, определяемую выше, или группу, имеющую формулу -C(O)-O-CP1P2-галогеналкил, где P1 и P2 - одинаковы или различны и представляют собой H, низший алкил, низший алкокси, циано, галоген-замещенный низший алкил или арил. Предпочтительно атом азота представляет собой один из атомов азота амидино-группы соединений согласно изобретению. Эти пролекарственные соединения получают посредством реакции соединений, описанных выше в данном изобретении, с активированным ацильным соединением для связывания атома азота в соединении согласно изобретению с карбонилом активированного ацильного соединения. Подходящие активированные карбонильные соединения содержат хорошую уходящую группу, связанную с карбонильным углеродом и включающую в себя ацилгалогениды, ацильные амины, ацильные соли пиридиния, ацильные алкоксиды, в частности ацильные феноксиды, такие как пара-нитрофеноксиацил, динитрофеноксиацил, фторфеноксиацил и дифторфеноксиацил. Эти реакции обычно являются экзотермическими и протекают в инертных растворителях при пониженных температурах, таких как -78 - 50°C. Реакции обычно протекают в присутствии неорганического основания, такого как карбонат калия, бикарбонат натрия, или органического основания, такого как амин, включая пиридин, триэтиламин и т.д. Один из способов получения пролекарств описан в USSN 08/843369, поданном 15 апреля, 1997 (соответствующей публикации РСТ WO9846576), описание которого включено в настоящее описание в виде ссылки во всей его полноте.
Отдельные соединения формулы I включают в себя следующее:
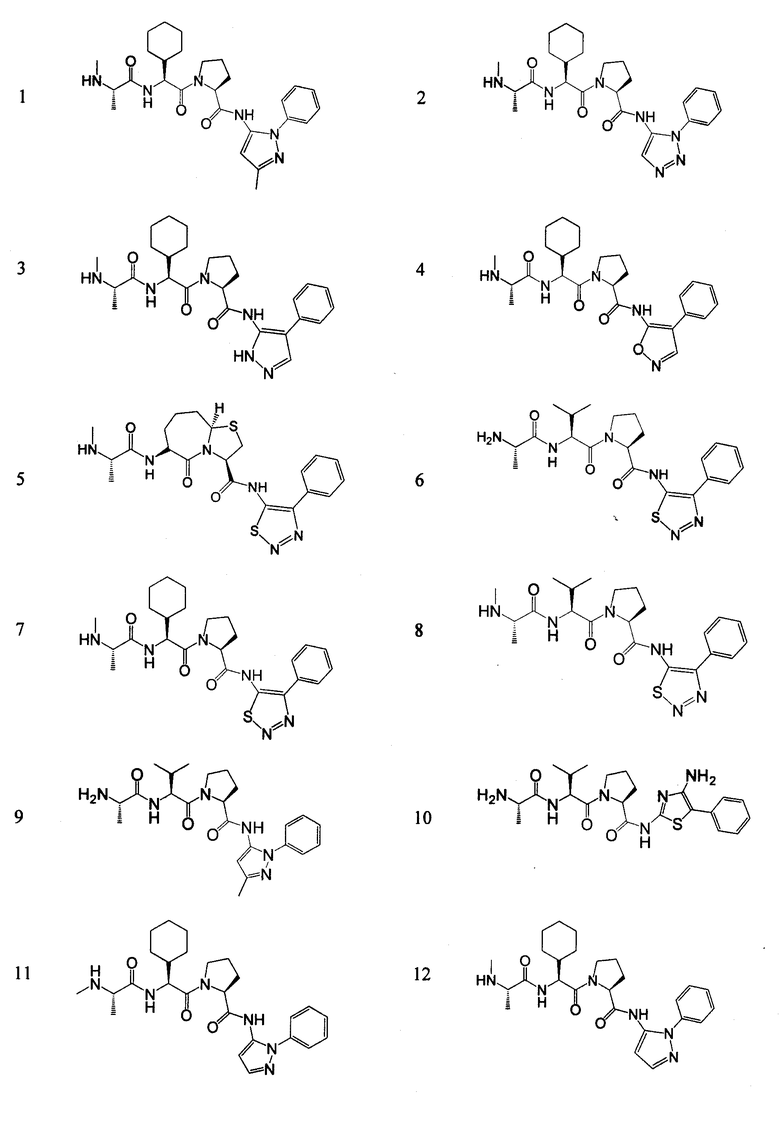
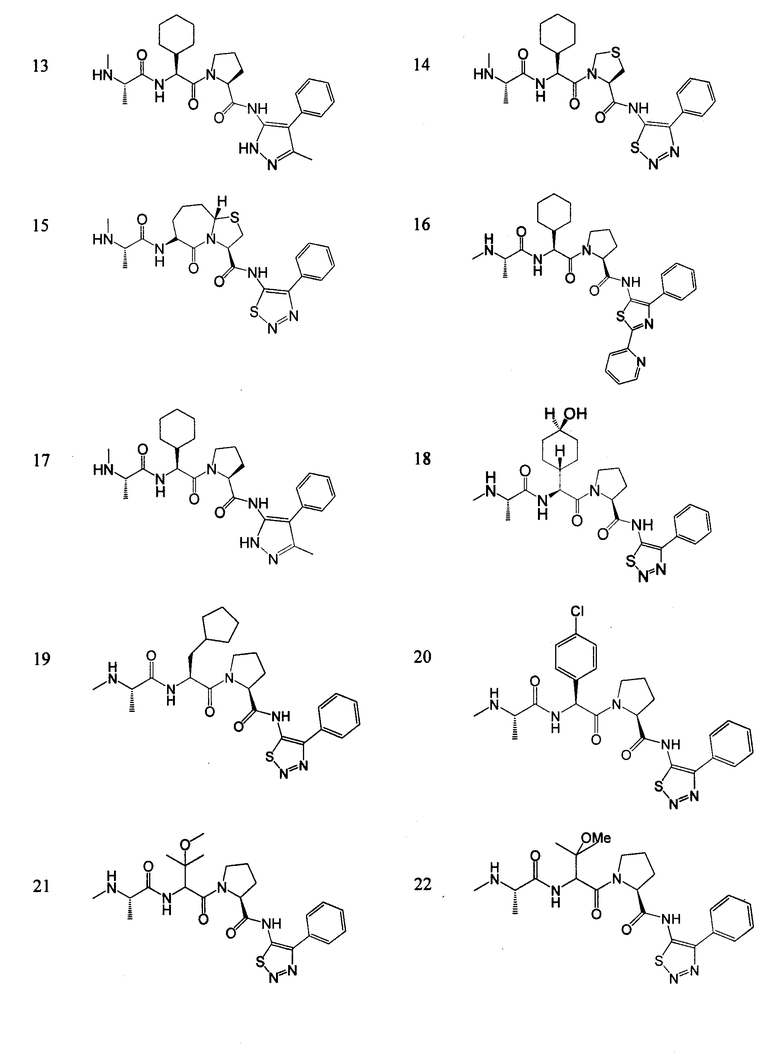

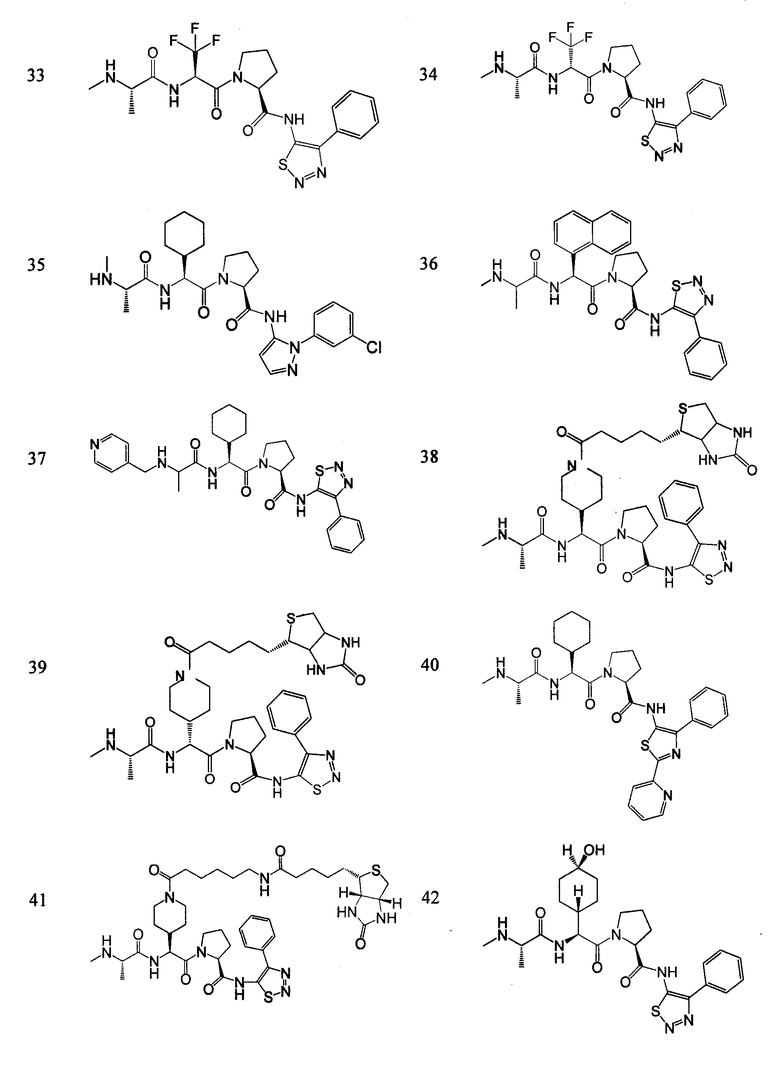
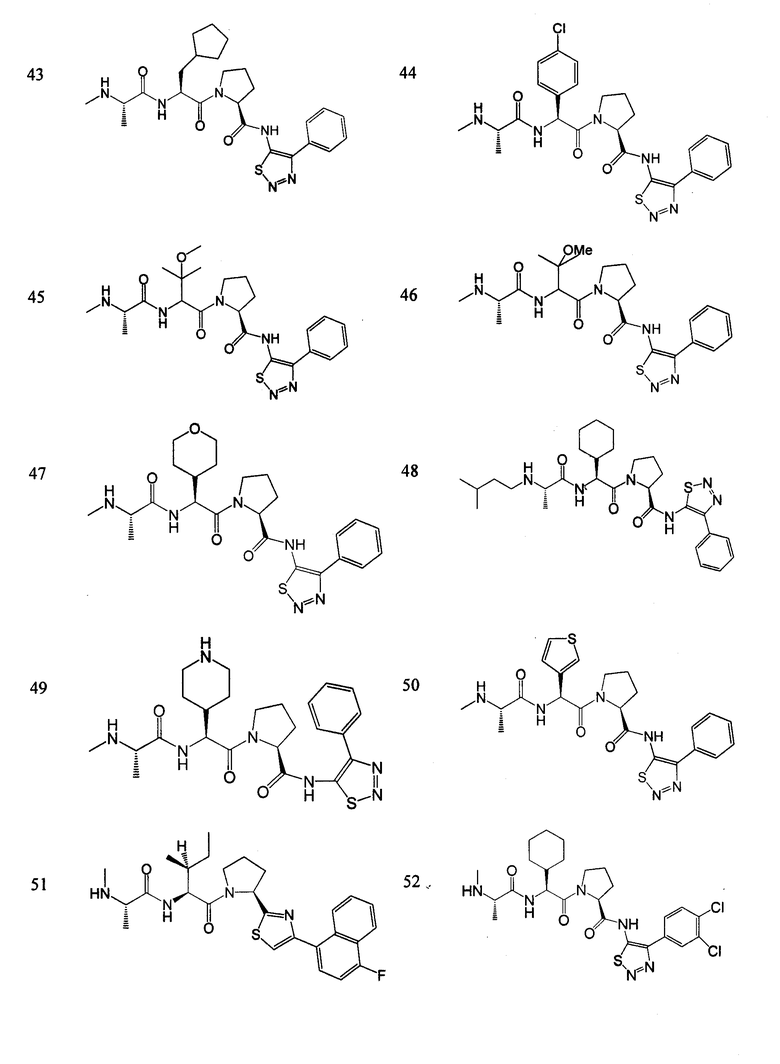

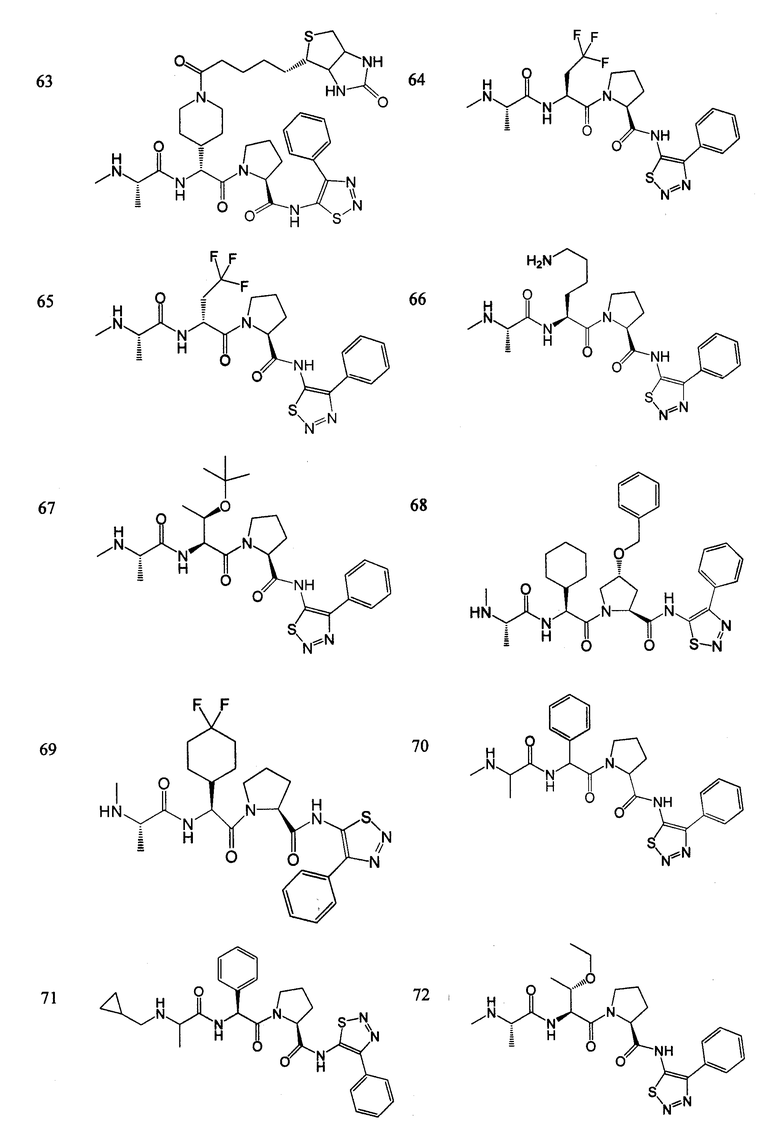
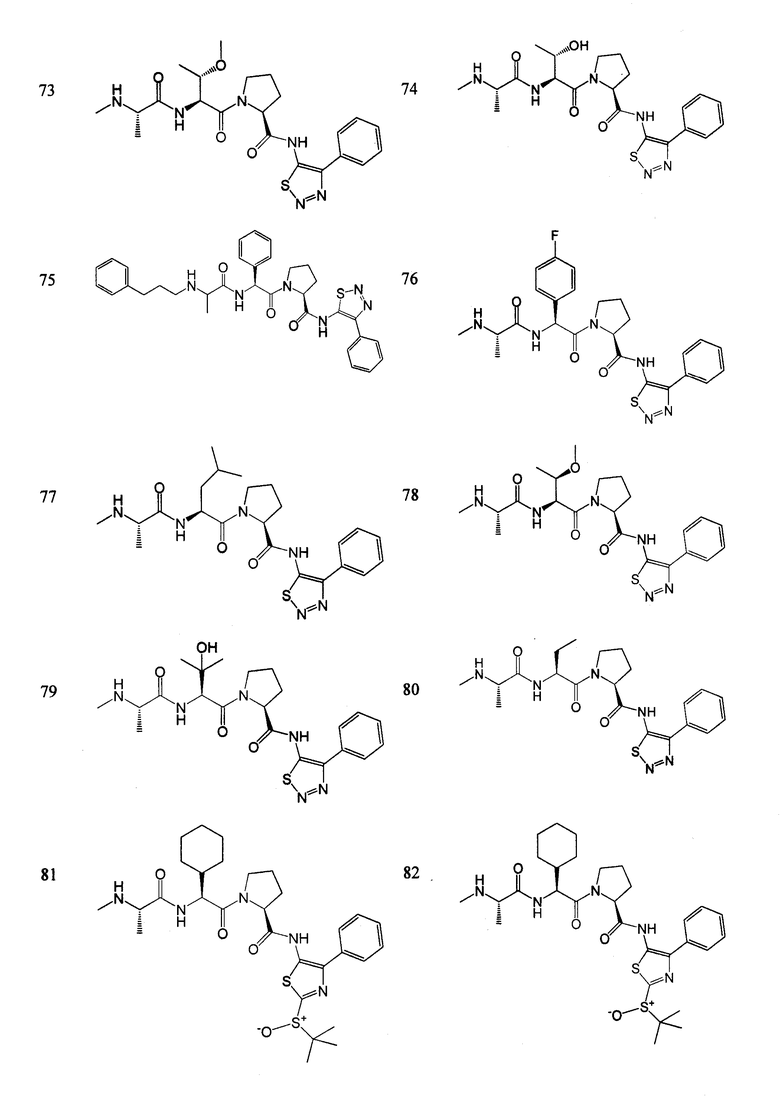
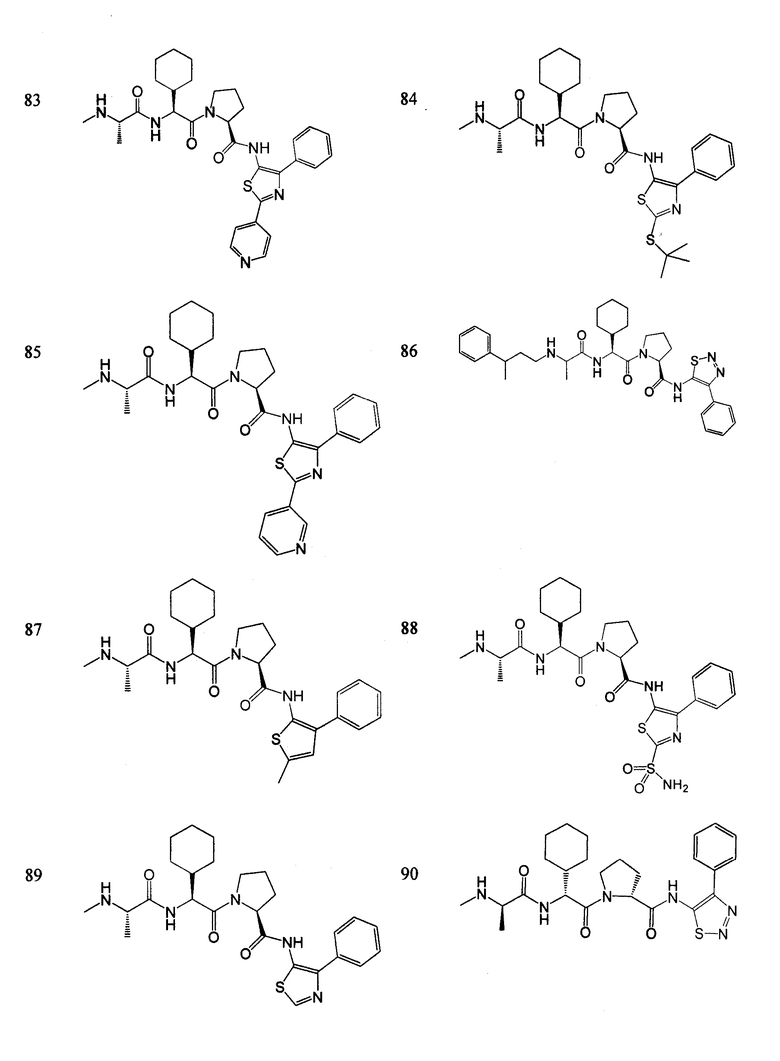
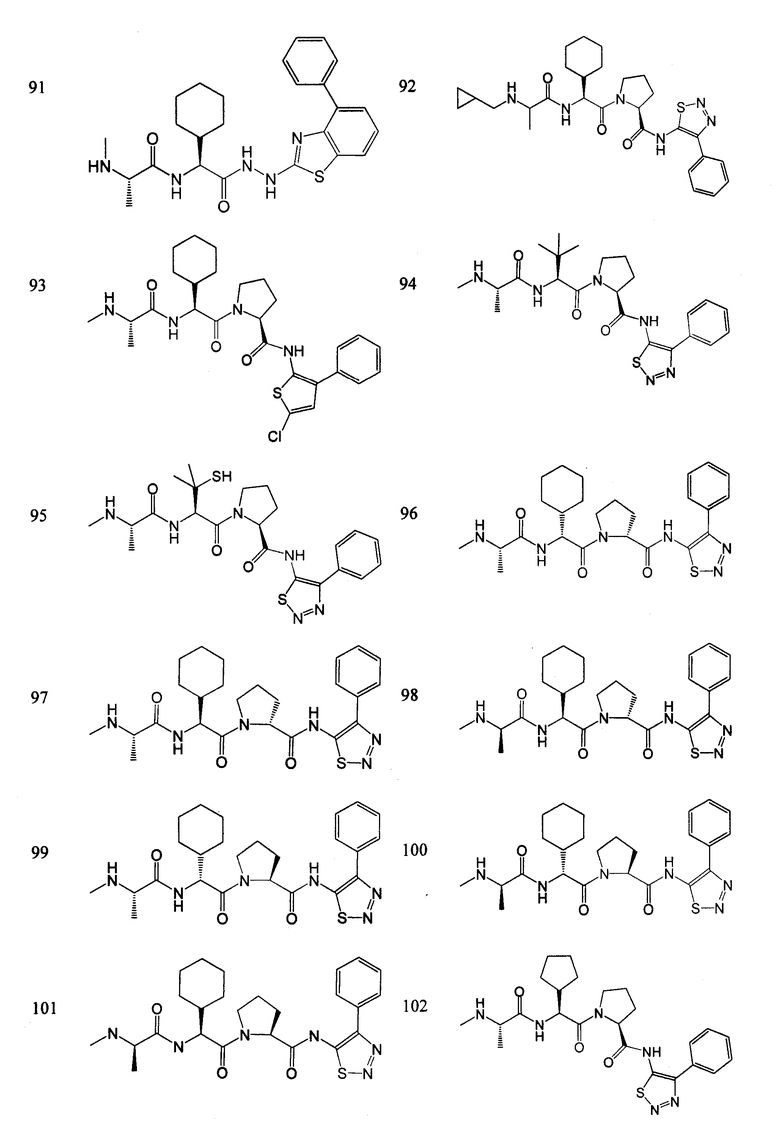
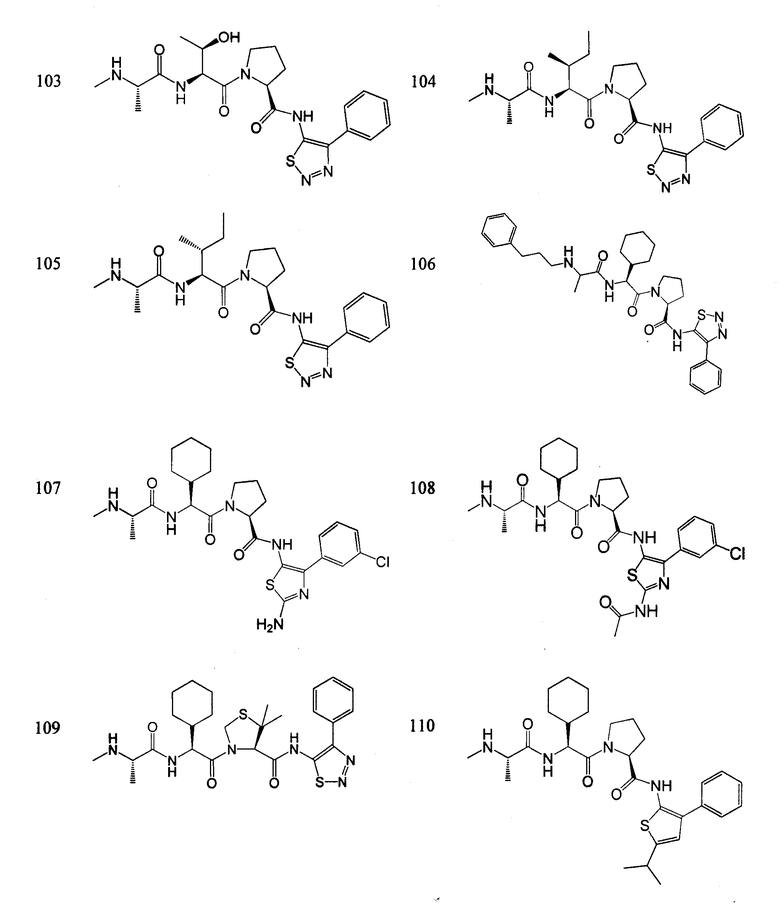
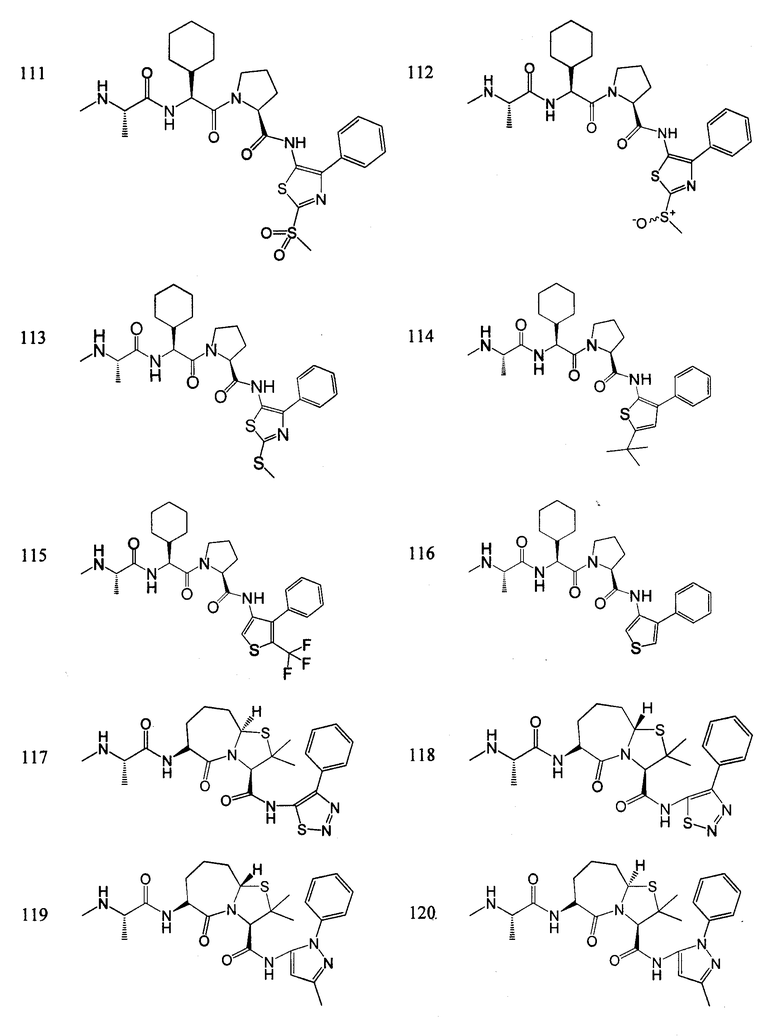

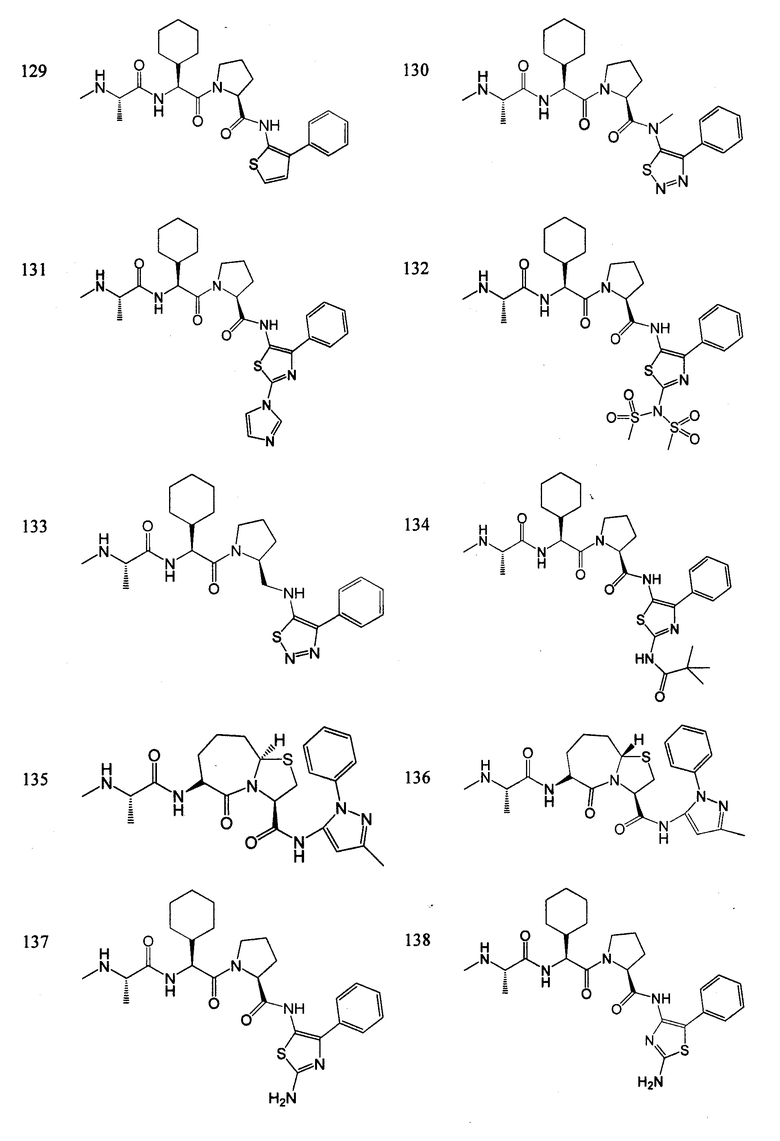
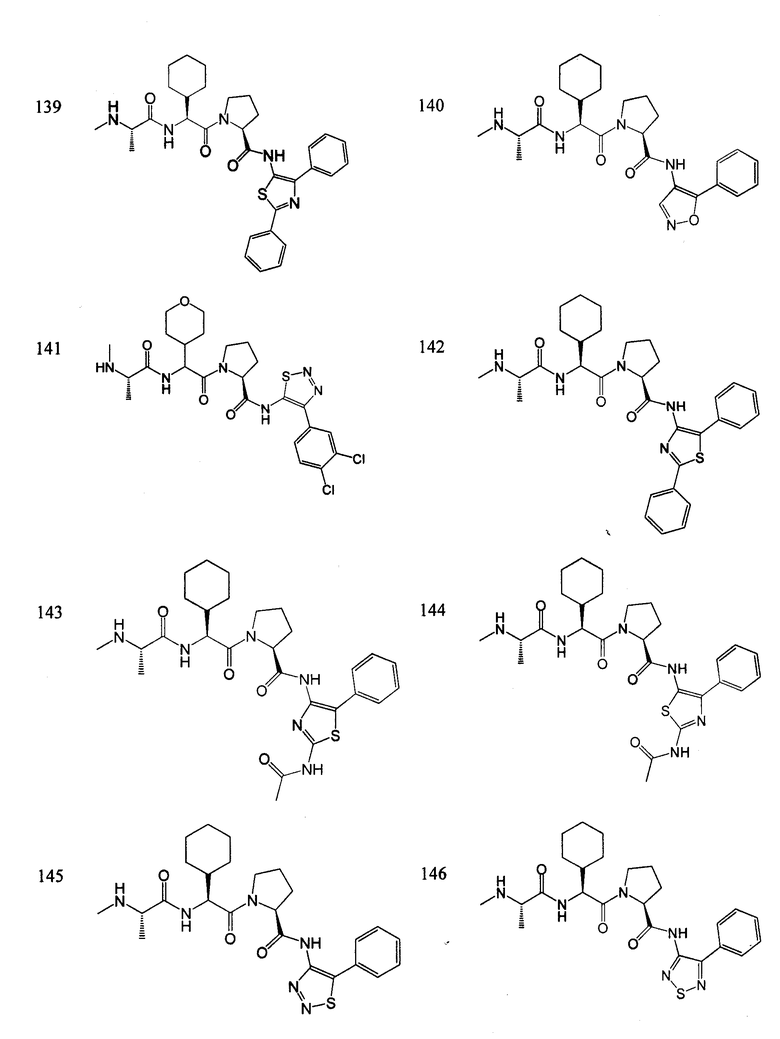
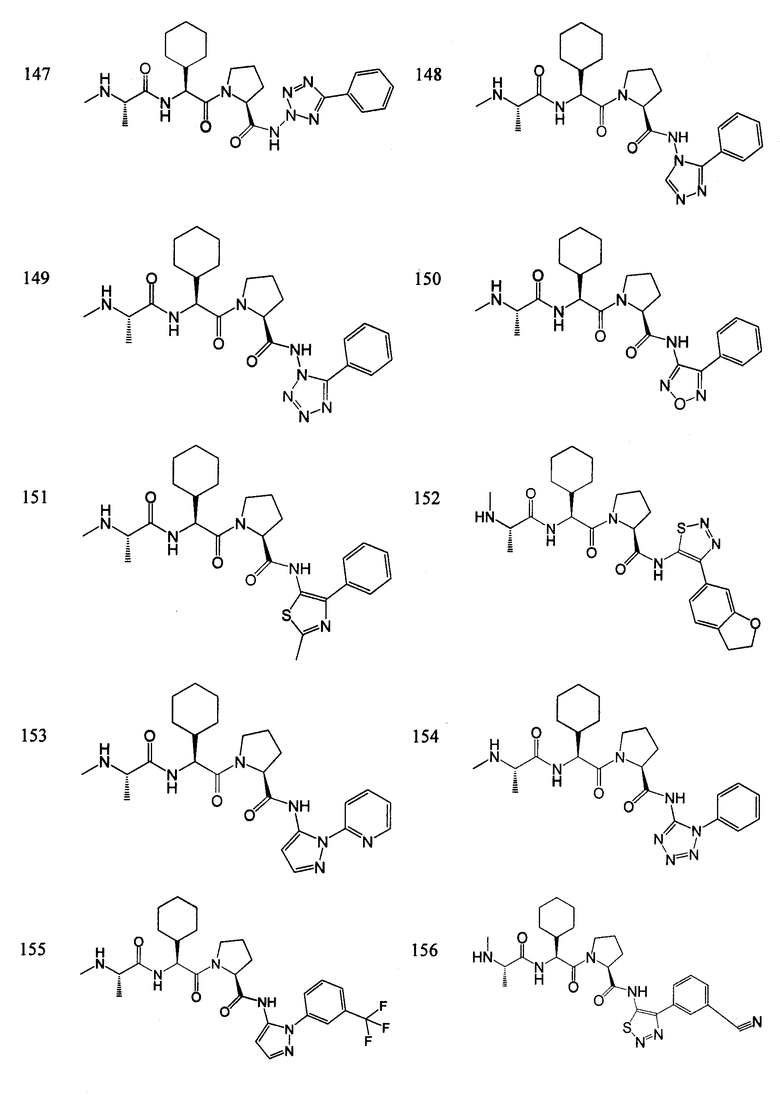
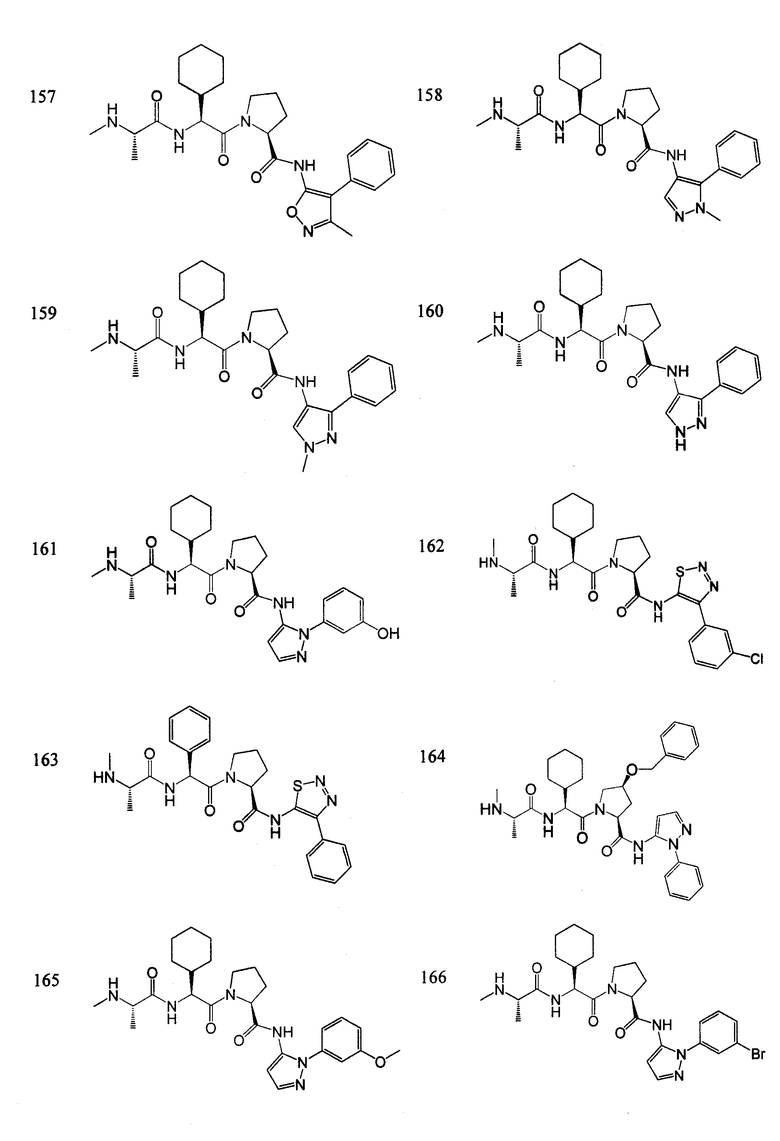
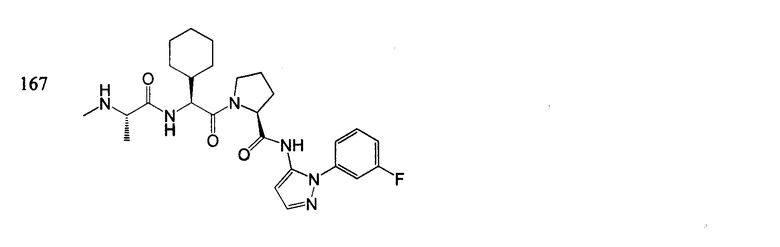
ПРОЦЕДУРЫ СИНТЕЗА
Соединения согласно изобретению получены с использованием стандартных способов органического синтеза из коммерчески доступных веществ и реагентов. Важно, что процедуры синтеза, используемые в получении соединений согласно изобретению, будут зависеть от конкретных заместителей, присутствующих в соединении, и что могут потребоваться защита и снятие защиты, как и в стандартных процедурах в органическом синтезе. В основной схеме синтеза соединения согласно изобретению могут быть получены с использованием методов, типичных для области химии пептидов, посредством связывания аналогов аминокислотного остатка с использованием типичных процедур связывания амидо. На схеме 1 аналоги амино-защищенного аминокислотного остатка последовательно связываются, и с них снимается защита, с получением конечных соединений.

Важно также и то, чтобы аналоги аминокислоты могли связываться друг с другом в любом порядке и могли быть получены с использованием твердофазной подложки, которая широко используется в данной области.
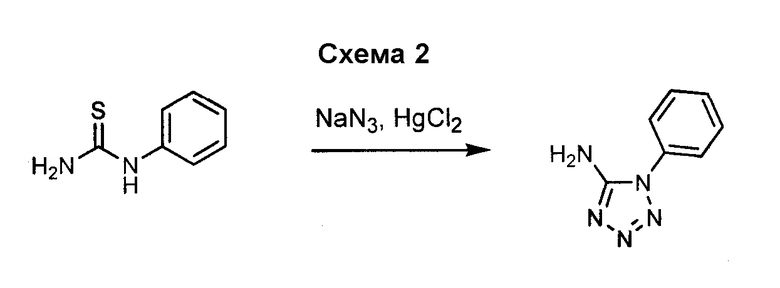
Амино-замещенное кольцо A, которое служит промежуточным соединением для получения соединений согласно изобретению, является коммерчески доступным или, кроме того, может быть получено из коммерчески доступных реагентов с использованием стандартных в органической химии методов. Например, 1-арил-5-аминотетразол, такой как фенил-5-аминотетразол, может быть получен согласно схеме 2 из коммерчески доступной фенилтиомочевины посредством реакции с азидом натрия и хлоридом ртути.
3-Арил-5-амино-1,2,3-триазол, такой как 3-фенил-3H-[1,2,3]триазол-4-иламин, может быть получен согласно процедуре, описанной в J. Org. Chem, 1981, 46:856-9 и показанной ниже на схеме 3, посредством реакции фениламина с аминоацетонитрилом.
Аналогично 5-амино-1-фенил-1H-[1,2,3]триазол-4-карбонитрил может быть получен посредством реакции фениламина с 2-амино-малонитрилом, как показано на схеме 4.
4-Арил-5-амино-1,2,5-оксадиазол, такой как 4-фенил-фуразан-3-иламин, может быть получен согласно процедурам, описанным в публикации Lakhan et al. (Indian Journal of Chemistry, Section B: Organic Chemistry Including Medicinal Chemistry (1987), 26B(7), 690-2) и показанным на схеме 5, посредством реакции бензоилцианида с гидроксиламином.
4-Арил-3-амино-1,2,4-триазол, такой как 4-фенил-4H-[1,2,4]триазол-3-иламин, может быть получен посредством реакции фенилизотиоцианата с гидразинкарбоксимидамидом, с получением 5-амино-4-фенил-4H-[1,2,4]триазол-3-тиола, в котором тиоловая группа может быть удалена с помощью катализатора никеля Ренея, как показано на схеме 6.

4-Арил-5-амино-1,2,3-триазол, такой как 3,5-дифенил-3H-[1,2,3]триазол-4-иламин, может быть получен согласно процедурам, описанным в публикации J. Org. Chem., 1990, 55:3351-62, и, как показано на схеме 7, посредством реакции бензолацетонитрила с азидобензолом (или, в альтернативном варианте, триметилсилилазидом, TMS-N3).
4-Арил-3-аминопиразол, такой как 4-фенил-2H-пиразол-3- иламин, может быть получен согласно процедурам, описанным в патенте EP 269859 и показанным на схеме 8, посредством реакции бензолацетонитрила со сложным триэтиловым эфиром ортомуравьиной кислоты, с получением 3-оксо-2-фенилпропионитрила, который может вступать в реакцию с гидразином.
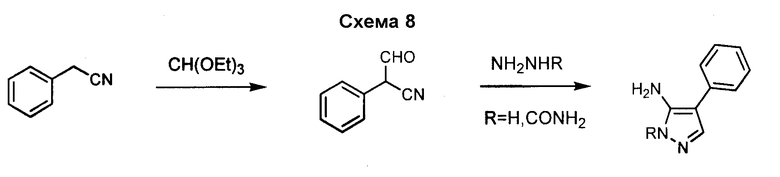
Различные гидразины и производные бензолацетонитрила могут быть использованы для получения замещенного-4-арил-3-аминопиразола, как показано на схеме 9.
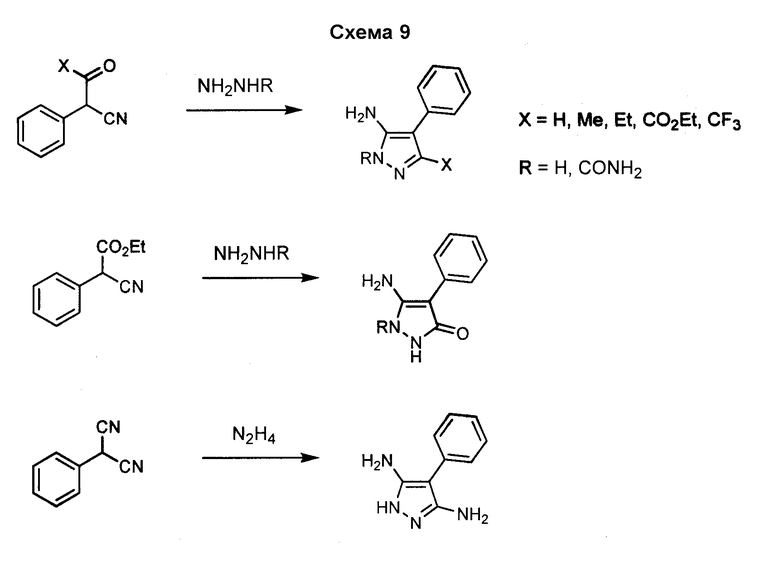
1-Арил-5-аминопиразол, такой как 2-фенил-2H-пиразол-3-иламин, может быть получен посредством реакции фенилгидразина с 3-оксо-пропионитрилом. Могут использоваться различные нитрилы для осуществления замещения пиразольного кольца в положении 3, как показано на схеме 10.
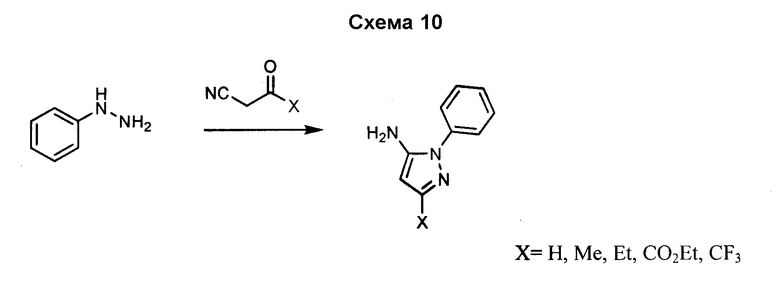
3-Арил-4-аминоимидазол, такой как 3-фенил-3H-имидазол-4-иламин, может быть получен посредством реакции фениламина с аминоацетонитрилом и сложным триэтиловым эфиром ортомуравьиной кислоты, как показано на схеме 11. Замещение имидазола в положении 2 может быть осуществлено с использованием аналогов сложного триэтилового эфира ортомуравьиной кислоты, как показано ниже.
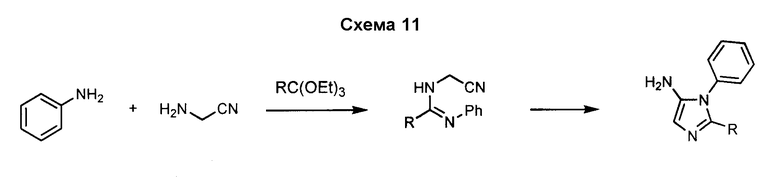
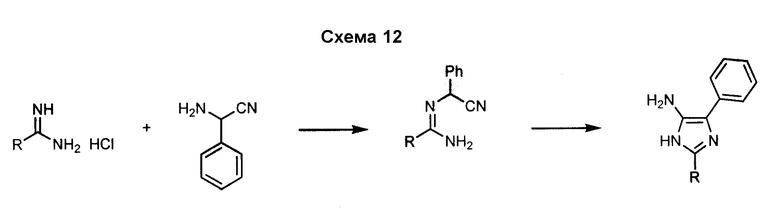
5-Арил-4-аминоимидазол, такой как 5-фенил-3H-имидазол-4-иламин, может быть получен посредством реакции формамида с аминофенилацетонитрилом, как показано на схеме 12. Замещение в положении 2 имидазольного кольца может быть осуществлено с использованием аналогов формамида.
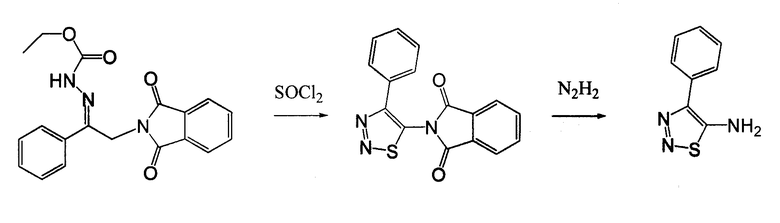
4-Арил-[1,2,3]тиадиазол-5-иламин, такой как 4-фенил-[1,2,3]тиадиазол-5-иламин, может быть получен согласно процедуре, показанной на схеме 13. 2-Бром-1-фенил-этанон вступает в реакцию с фталимидом лития, и продукт замещения вступает в реакцию со сложным этиловым эфиром гидразинкарбоксилата. Полученный сложный этиловый эфир гидразинкарбоксилата циклизован с образованием тиадиазола посредством реакции с тионилхлоридом, с последующим удалением группы фталимида с помощью гидразина.

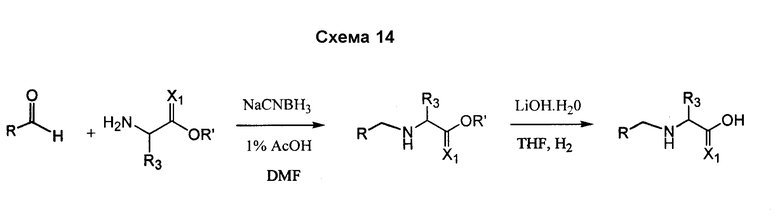
Соединения согласно изобретению, в которых R4 или R4' не являются H, могут быть получены согласно стандартным методам органической химии, например посредством восстановительного аминирования, где исходный аналог аминокислотного остатка, например NH2-CH(R3)-C(O)-OH, вступает в реакцию с подходящим альдегидом или кетоном, с получением требуемых заместителей R4 и R4'. См. схему 14. Полученная промежуточная R4/R4'-замещенная аминокислота далее может быть сопряжена со следующей промежуточной аминокислотой или с остатком соединения, с использованием стандартных процедур спаривания пептидов.
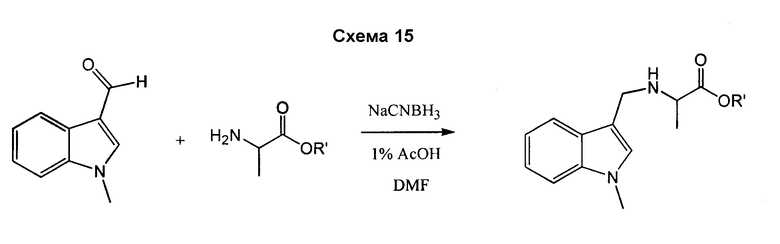
В отдельном воплощении аланин взаимодействует с 1-метилиндол-2-карбоксальдегидом и восстанавливается цианоборогидридом натрия, растворенным в 1% HOAc/DMF, с получением N-замещенного аланинового остатка, который может быть использован для получения соединений согласно изобретению. См. схему 15.
В альтернативном варианте процедура восстановительного аминирования для получения R4/R4'-заместителей является конечной стадией в получении соединения.
В случае, когда соединения согласно изобретению включают в себя R4- или R4'- заместители, отличные от H, они также могут быть получены посредством замещения подходящей промежуточной кислотой, которая включает в себя уходящую группу с требуемым амином. Например, Br-CH(R3)-C(O)-OH замещается амином R4-NH2 или R4-NH-R4', согласно схеме 16.
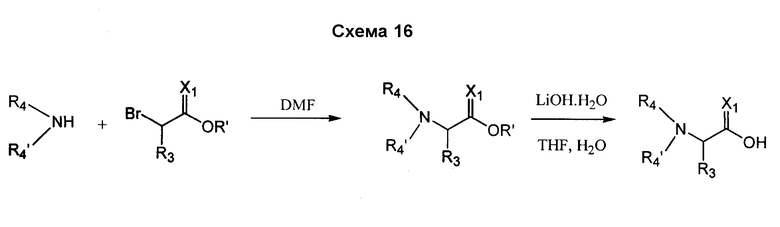
В альтернативном варианте процедура замещения для получения R4- или R4'- заместителей может выполняться как конечная стадия в получении соединения, как показано на схеме 17.

В отдельном воплощении 2-бромпропионовая кислота взаимодействует со следующими аминами, растворенными в DMF, и процесс кипения продолжается до завершения процедуры замещения и образования N-замещенного аланинового остатка:

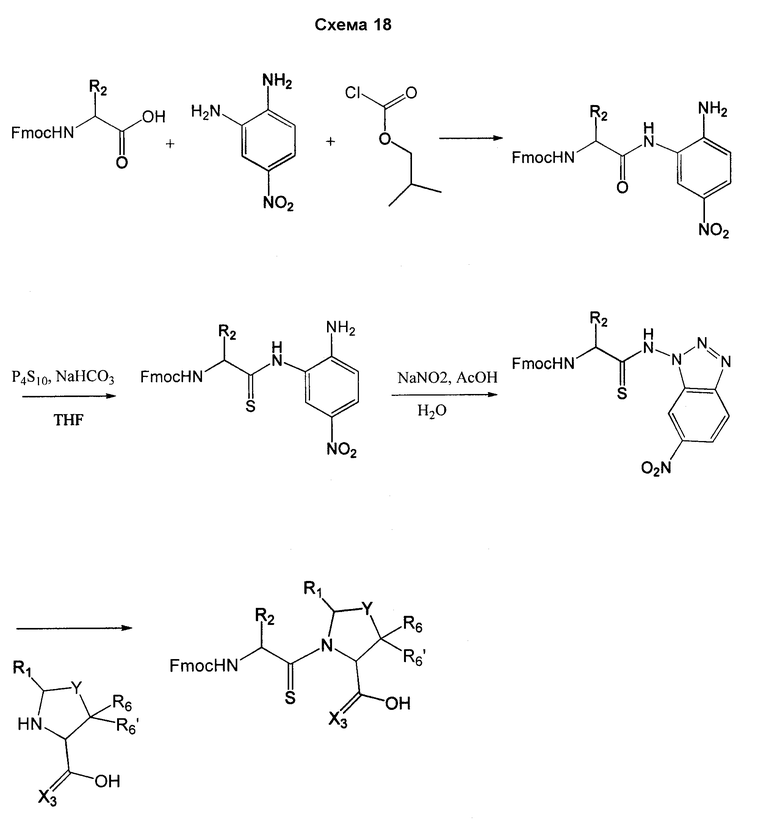
Соединения согласно изобретению, в которых один или более из X1, X2 и X3 представляют собой серу, т.е соединение включает в себя тиоамид, могут быть получены согласно установленным способам органической химии. Например, соединения, в которых X2 представляет собой серу, могут быть получены согласно схеме 18, где в качестве исходного материала использовали Fmoc-аналог аминокислотного остатка NH2-CH(R2)-COOH, который растворяли в THF и охлаждали до -25°C с добавлением DIPEA, далее добавляли изобутилхлорформат. Через 10 минут добавляли диамин, 4-нитробензол-1,2-диамин и реакционную смесь непрерывно перемешивали при температуре -25°C в течение 2 часов, далее непрерывно перемешивали при комнатной температуре в течение ночи. THF удаляли под вакуумом и смесь подвергали флэш-хроматографии, используя 50% EtOAc/гексан, получая на выходе требуемый продукт. Производную Fmoc-аланина, фосфор, пентасульфид и кабонат натрия смешивали в THF и перемешивали в течение ночи. Раствор концентрировали и, применяя флэш-хроматографию с использованием 80% EtOAc/гексана, получали на выходе активированный тиоаланин. Активированный тиоаланин и нитрит натрия далее смешивали в уксусной кислоте и разбавляли H2O. Полученный осадок фильтровали и сушили, получая на выходе продукт. Тиоаланин соединяли с аналогом OH-защищенного пролинового аминокислотного остатка, растворяя оба в DMF. Далее снимали защиту с тиоамида посредством воздействия 20% PIP/DMA в течение 15 минут и использовали для сопряжения с аналогом аминокислотного остатка R4/R4'-N-CH(R3)-COOH, с последующим снятием защиты OH и соединением с замещенным промежуточным кольцом A. В альтернативном варианте сначала Fmoc-защищенный тиоамид соединяли с амино-замещенным промежуточным кольцом A, далее снимали защиту с Fmoc и затем соединяли с аналогом аминокислотного остатка R4/R4'-N-CH(R3)-COOH.
ПРИМЕНИМОСТЬ
Соединения согласно изобретению ингибируют связывание белков IAP с каспазами, в частности взаимодействие связывания X-IAP с каспазами 3 и 7. Эти соединения ингибируют связывание ML-IAP с белком Smac. Соответственно, соединения согласно изобретению используются для индуцирования апоптозов в клетках или для повышения чувствительности клеток, в частности злокачественных клеток, к апоптотическим сигналам. Соединения согласно изобретению используются для индуцирования апоптозов в клетках, в которых наблюдается повышенная экспрессия белков IAP. Альтернативно соединения согласно изобретению используются для индуцирования апоптозов в клетках, в которых митохондриальный путь апоптоза разрушен, так что высвобождение белков Smac из белков ML-IAP ингибируется путем повышения регуляции Bcl-2 или понижения регуляции Bax/Bak. В более широком плане соединения могут быть использованы для лечения всех типов злокачественных опухолей, при которых клетки не подвержены апоптозу. Примеры злокачественных опухолей такого типа включают в себя нейробластому, злокачественную опухоль кишечника, например злокачественную опухоль прямой кишки, злокачественную опухоль толстой кишки, семейную аденоматозно-полипозную карциному и наследственную неполипозную злокачественную опухоль прямой кишки, эзофагеальную карциному, губную злокачественную опухоль, злокачественную опухоль гортани, злокачественную опухоль подглоточника, злокачественную опухоль языка, злокачественную опухоль слюнных желез, злокачественную опухоль желудка, аденокарциному, медуллярную карциному щитовидной железы, сосочковую карциному щитовидной железы, почечную карциному, карциному почечной паренхимы, карциному яичника, карциному шейки матки, карциному тела матки, карциному эндометрия, карциному хориона, карциному поджелудочной железы, карциному предстательной железы, карциному яичка, карциному молочной железы, карциному мочевого пузыря, меланому, опухоли мозга, такие как глиобластома, астроцитома, менингиома, медуллобластома и периферические, нейроэктодермальные опухоли, лимфому Ходжкина, неходжкинскую лимфому, лимфому Бэркитта, острую лимфатическую лейкемию (ALL), хроническую лимфатическую лейкемию (CLL), острую миелоидную лейкемию (AМL), хроническую миелоидную лейкемию (CМL), лимфому при T-клеточной лейкемии у взрослых, гепатоклеточную карциному, карциному желчного пузыря, бронхиальную карциному, мелкоклеточную карциному легких, немелкоклеточную карциному легких, множественную миелому, базалиому, тератому, ретинобластому, меланому сосудистой оболочки глаза, семиному, рабдомиосаркому, краниофарингеому, остеосаркому, хондросаркому, миосаркому, липосаркому, фибросаркому, саркому Эвинга и плазмоцитому.
Соединения согласно изобретению используются для повышения чувствительности клеток к апоптотическим сигналам. Соответственно, эти соединения могут быть введены до, одновременно или после радиционной терапии или цитостатической либо антинеопластической химиотерапии. Подходящие цитостатические химиотерапевтические соединения включают в себя, не ограничиваясь ими, (i) антиметаболиты, такие как цитарабин, флударабин, 5-фтор-2'-дезоксиуиридин, гемцитабин, гидроксимочевина или метотрексат; (ii) ДНК-фрагментирующие агенты, такие как блеомицин, (iii) поперечно-сшивающие ДНК агенты, такие как хлорамбуцил, цисплатин, циклофосфамид или азотистый иприт; (iv) интеркалирующие агенты, такие как адриамицин (доксорубицин) или митоксантрон; (v) ингибиторы белкового синтеза, такие как L-аспарагиназа, циклогексимид, пуромицин или дифтерийный токсин; (vi) яды топоизомеразы I, такие как камптотецин или топотекан; (vii) яды топоизомеразы II, этопозид (VP-16) или тенипозид; (viii) агенты, направленные на микротрубочки, такие как колцемид, колхицин, паклитаксел, винбластин или винкристин; (ix) ингибиторы киназы, такие как флавопиридол, стауроспорин, STI571 (CPG 57148B) или UCN-01 (7-гидроксистауроспорин); (x) разнообразные агенты для исследовательской деятельности, такие как тиоплатин, PS-341, фенилбутират, ET-18-OCH3 или ингибиторы фарнезилтрансферазы (L-739749, L-744832); полифенолы, такие как кверцетин, резвератрол, пицеатаннол, эпигаллокатехин-галлат, теафлавины, флаванолы, процианидины, бетулиновая кислота и ее производные; (xi) гормоны, такие как глюкокортикоиды или фенретинид; (xii) антагонисты гормонов, такие как тамоксифен, финастерид или антагонисты LHRH. В предпочтительном воплощении соединения согласно изобретению вводят совместно с цитостатическим соединением, выбранным из группы, состоящей из цисплатина, доксорубицина, таксола, таксотера и митомицина C. Наиболее предпочтительным цитостатическим соединением является доксорубицин.
Другим классом активных соединений, которые могут быть использованы в настоящем изобретении, являются такие соединения, которые способны повысить чувствительность к апоптозу или индуцировать апоптоз путем связывания с рецепторами гибели клеток ("агонисты рецепторов гибели клеток"). Такие агонисты рецепторов гибели клеток включают в себя лиганды рецепторов, такие как фактор некроза опухоли (TNF-α), фактор некроза опухоли β (TNF-β, лимфотоксин-α), LT-β (лимфотоксин-β), TRAIL (Apo2L, лиганд DR4), CD95 (Fas, APO-1)лиганд, TRAMP (DR3, Apo-3) лиганд, DR6-лиганд, а также фрагменты и производные любого из лигандов. Предпочтительно, чтобы лиганд рецептора гибели клеток представлял собой TNF-α. Более предпочтительно, чтобы лиганд рецептора гибели клеток представлял собой Apo2L/TRAIL. Кроме того, агонисты рецепторов гибели включают в себя агонистические антитела к рецепторам гибели, такие как анти-CD95-антитело, анти-TRAIL-R1(DR4)-антитело, анти-TRAIL-R2(DR5)-антитело, анти- TRAIL-R3-антитело, анти-TRAIL-R4-антитело, анти-DR6-антитело, анти-TNF-R1-антитело и анти-TRAMP(DR3)-антитело, а также фрагменты и производные любого из указанных антител.
С целью повышения чувствительности клеток к апоптозу соединения согласно изобретению могут быть использованы также в сочетании с радиационной терапией. Фраза "радиационная терапия" относится к применению электромагнитного излучения или корпускулярного излучения при лечении неоплазии. Радиационная терапия основана на том принципе, что высокая доза радиоактивного излучения, направленного на область-мишень, будет приводить к гибели репродуцирующихся клеток как в опухолевых, так и в нормальных тканях. Режим дозирования радиоактивного излучения обычно определяется в терминах дозы поглощенной радиации (rad), времени и частоты облучения и должен быть тщательно подобран онкологом. Количество радиации, которое получает пациент, будет зависеть от разных обстоятельств, но два наиболее важных из них связаны с локализацией опухоли по отношению к остальным критическим структурам или органам организма и со степенью распространения опухоли в организме. Примеры радиоактивных терапевтических агентов включают в себя, без ограничения, радиотерапевтические агенты, известные в данной области (Hellman, Principles of Radiation Therapy, Cancer, in Principles I and Practice of Oncology, 24875 (Devita et al., 4th ed., vol. 1, 1993). Современные успехи в радиационной терапии включают в себя трехмерное конформное направленное наружное излучение, рентгенотерапию с модуляцией интенсивности потока излучения (IMRT), стереотактическую радиохирургию и брахитерапию (интерстициальная радиационная терапия), где в последнем случае источник радиоактивного излучения помещают непосредственно внутри опухоли в виде имплантированных "зерен". Указанные новые способы терапевтического воздействия позволяют действовать на опухоль более высокими дозами радиоактивного излучения, чем и объясняется их повышенная эффективность по сравнению со стандартной рентгенотерапией с модуляцией интенсивности потока излучения.
Ионизирующая радиация бета-излучающими радионуклидами считается наиболее эффективной среди рентгенотерапевтических воздействий в связи с умеренной линейной потерей энергии (LET) ионизирующей частицы (электрона) и ее промежуточной дальностью проникновения (обычно на несколько миллиметров вглубь ткани). Значительно более низкие уровни гамма-облучения способны обеспечить необходимую дозу облучения на значительно больших расстояниях. Альфа-частицам свойственна другая крайность, они способны доставлять очень высокую дозу энергии (LET), однако имеют исключительно ограниченную область воздействия, и, следовательно, они должны находиться в непосредственном контакте с клетками той ткани, которая подвергается терапевтическому воздействию. Кроме того, альфа-излучателями обычно являются тяжелые металлы, что ограничивает возможности химии и связано с избыточным риском утечки радионуклида из той области, которая должна быть подвергнута обработке. В зависимости от опухоли, которая должна быть подвергнута лечению, в объем настоящего изобретения входит использование любых возможных типов излучающих частиц.
Кроме того, настоящее изобретение охватывает различные виды неионизирующего излучения, такие, например, как ультрафиолетовое (УФ) излучение, видимый свет высоких энергий, микроволновое излучение (гипертермическая терапия), инфракрасное (ИК) излучение и лазерное излучение. В особом воплощении настоящего изобретения применяется УФ-излучение.
Настоящее изобретение включает в себя также фармацевтические композиции или лекарственные средства, содержащие соединения согласно изобретению и терапевтически инертный носитель, разбавитель или наполнитель, а также способы применения соединений согласно изобретению для получения таких композиций и лекарственных средств. Обычно соединения формулы I, используемые в способах согласно изобретению, обычно составляют [в композиции] в результате смешивания при комнатной температуре и при соответствующем значении pH, при соблюдении требуемой степени чистоты, с физиологически приемлемыми носителями, например носителями, которые не являются токсичными для реципиентов в тех дозах и концентрациях, которые используются в галеновых лекарственных формах для внутреннего введения. Значение pH композиции зависит главным образом от конкретного применения и от концентрации соединения, однако оно предпочтительно заключено в интервале приблизительно от 3 до 8. Композиция в ацетатном буфере при pH 5 соответствует предпочтительному воплощению настоящего изобретения.
Ингибирующее соединение для применения в настоящем изобретении предпочтительно должно быть стерильным. Обычно такое соединение должно храниться в виде твердой композиции, хотя приемлемыми являются также и лиофилизированные композиции или водные растворы.
Составление композиции согласно изобретению, определение ее дозировки и пути введения производятся в соответствии с общепринятой медицинской практикой. Факторы, которые в этой связи принимаются во внимание, включают в себя конкретное заболевание, которое предстоит подвергнуть лечению, конкретное млекопитающее, которое будет подвергнуто лечению, клиническое состояние пациента, причину расстройства, область организма, в которую должно быть доставлено лекарственное средство, способ введения, режим введения, а также другие факторы, известные практикующим врачам. "Эффективное количество" соединения, которое следует вводить, обусловлено перечисленными выше факторами и составляет минимальное количество, необходимое для ингибирования взаимодействия IAP с каспазами, для индукции апоптоза или повышения чувствительности злокачественных клеток к апоптотическому сигналу. Предпочтительно, чтобы такое количество составляло ниже той дозы, которая является токсичной для нормальных клеток или для млекопитающего в целом.
Обычно первоначальное фармацевтически эффективное количество соединения согласно изобретению вводят парентерально в дозе, составляющей приблизительно от 0,01 до 100 мг/кг, предпочтительно приблизительно от 0,1 до 20 мг/кг веса тела пациента в день, при этом обычная область первоначально используемых доз соединения составляет приблизительно от 0,3 до 15 мг/кг/день. Доза лекарственных форм для перорального введения, таких как таблетки и капсулы, предпочтительно содержит приблизительно от 25 до 1000 мг соединения согласно изобретению.
Соединение согласно изобретению можно вводить любым из подходящих способов, включая пероральный, местный, трансдермальный, парентеральный, подкожный, внутрибрюшинный, внутрилегочный и интраназальный способы введения, а также, если это необходимо для локальной обработки, допустимо введение внутрь пораженной ткани. Парентеральные инфузии включают в себя внутримышечное, внутривенное, внутриартериальное, внутрибрюшинное или подкожное введение. Примером подходящей пероральной лекарственной формы является таблетка, содержащая приблизительно 25 мг, 50 мг, 100 мг, 250 мг или 500 мг соединения согласно изобретению, смешанного приблизительно с 90-30 мг безводной лактозы, приблизительно с 5-40 мг натрий-кросскармеллозы, приблизительно с 5-30 мг поливинилпирролидона (PVP) K30 и приблизительно с 1-10 мг стеарата магния. Измельченные в порошок ингредиенты сначала смешивают друг с другом, а затем смешивают с раствором PVP. Полученная в результате композиция может быть высушена, гранулирована, смешана со стеаратом магния и спрессована в таблетки, с использованием соответствующего оборудования. Аэрозольная композиция может быть получена путем растворения соединения согласно изобретению, например 5-400 мг соединения, в подходящем буферном растворе, например фосфатном буфере, в случае необходимости, с добавлением вещества, повышающего тоничность раствора, например, соли, такой как хлористый натрий. Обычно раствор фильтруют, например, через фильтр с размером пор 0,2 микрона для удаления загрязнений и примесей.
ПРИМЕРЫ
Настоящее изобретение описывается более наглядно с помощью следующих примеров. Эти примеры не предназначены для ограничения объема настоящего изобретения. Аббревиатуры, используемые в описании примеров, следующие:
ACN: ацетонитрил;
Chg: циклогексилглицин;
DCM: дихлорметан;
DIPEA: диизопропилэтиламин;
DMAP: 4-диметиламинопиридин;
DME: 1,2-диметоксиэтан;
DMF: диметилформамид;
DMSO: диметилсульфоксид;
EDC: 1-этил-3-(3-диметиламинопропил)карбодиимид;
EEDQ: 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин;
LCMS: жидкостная хроматографическая масс-спектометрия;
HATU: O-(7-азобензотриазол-1-ил)-1,1,3,3-тетраметилурониумгексафторфосфат;
HOBt: N-гидроксибензотриазол;
HBTU: 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний-гексафторфосфат;
HPLC: высокоэффективная жидкостная хроматография;
NBS: N-бромосукцинамид;
TASF: трис(диметиламино)сульфоний-дифтортриметилсиликат;
TEA: триэтиламин;
TFA: трифторацетат;
THF: тетрагидрофуран;
Пример 1. Сложный этиловый эфир 6-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)-5-оксо-октагидротиазолo[3,2-a]азепин-3-карбоновой кислоты
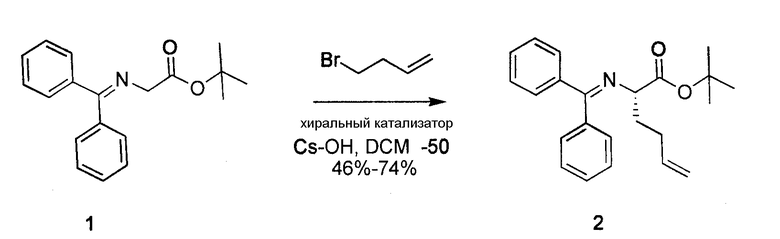
К перемешиваемому раствору сложного трет-бутилового эфира N-(дифенилметилен) глицина 1 (3,0 г, 10,1 ммоль) и хирального катализатора O-аллил-N-(9-антрасенилметил)-цихонидинбромида (613 мг, 1,0 ммоль) в сухом DCM (30 мл) добавляли гидроксид цезия (17 г, 101 ммоль). Реакционную смесь охлаждали до -78°C в бане из сухого льда с ацетоном и добавляли покапельно 4-бром-1-бутен. Далее реакционную смесь энергично перемешивали в атмосфере азота при температуре -48°C в течение 48 часов. Далее последовательно добавляли этиловый эфир и H2O. Органический слой отделяли, дважды промывали H2O, один раз соляным раствором, сушили над MgSO4 и концентрировали. Полученный продукт очищали посредством SiO2-хроматографии в градиенте 0-10% EtOAc в гексане, получая на выходе 65% соединения 2.
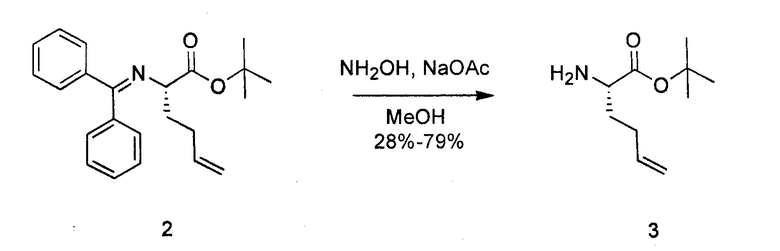
К перемешиваемому раствору соединения 2 (1,52 г, 4,3 ммоль) в сухом MeOH (50 мл) добавляли NaOAc (720 мг, 8,6 ммоль) и NH2OH·HCl (540 мг, 7,6 ммоль). Реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 2 часов. Далее добавляли DCM и 0,1 н. NaOH. Водный слой отделяли и трижды экстрагировали с использованием DCM, сушили над Na2SO4, объединяли фракции DCM и концентрировали. Полученный продукт очищали посредством SiO2-хроматографии в градиенте 0-10% MeOH в DCM с 0,05% TEA, получая на выходе 70% соединения 3.
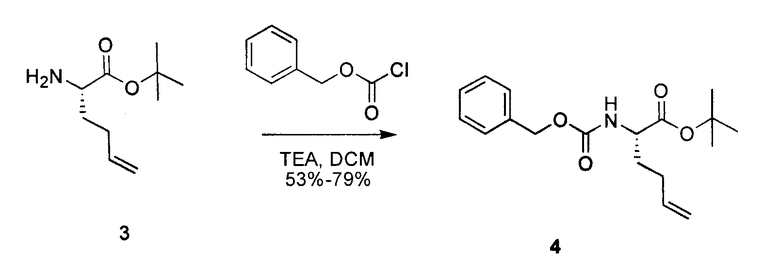
К раствору 3 (610 мг, 3,3 ммоль) в сухом DCM (20 мл) добавляли триэтиламин (550 мкл, 3,9 ммоль) и бензилхлорформат (550 мкл, 3,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Раствор концентрировали и очищали посредством SiO2-хроматографии в градиенте 0-30% EtOAc в гексане, получая на выходе 66% соединения 4.
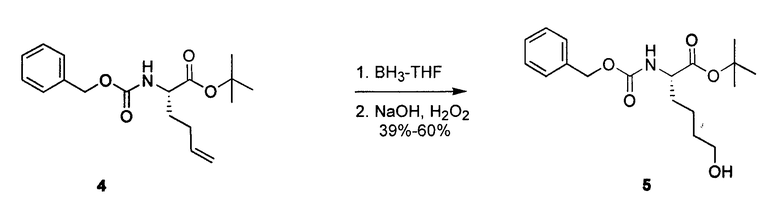
К перемешиваемому раствору 4 (577 мг, 1,8 ммоль) в THF (20 мл) в атмосфере азота добавляли BH3·THF. По истечении 1 часа добавляли 3 н. NaOH (300 мкл, 0,9 ммоль) и H2O2 (306 мкл, 2,7 ммоль). Реакционную смесь перемешивали в течение ночи и впоследствии разбавляли H2O, дважды экстрагировали этиловым эфиром, сушили над MgSO4 и концентрировали. Полученный продукт очищали посредством SiO2-хроматографии с градиентом 10-45% EtOAc в гексане, получая на выходе 50% соединения 5.

К перемешиваемому раствору 5 (71 мг, 0,21 ммоль) в MeOH (2 мл) в атмосфере 1 атм. H2 добавляли 10% гидроксидпалладия на угле (30 мг). Реакция завершалась через 30 минут. Реакционную смесь фильтровали через целит и концентрировали, получая соединение 6 с количественным выходом.
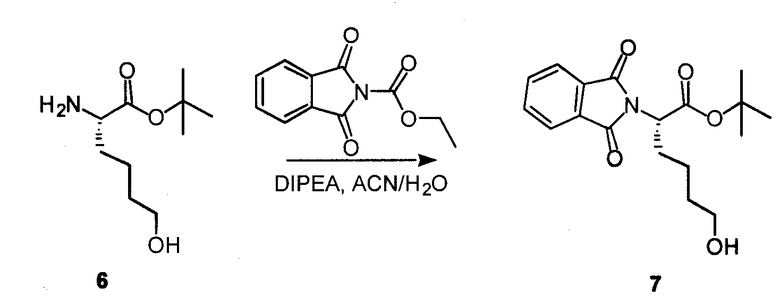
К раствору соединения 6 (42 мг, 0,21 ммоль) в ACN (2 мл) добавляли карбэтоксифталимид (50 мг, 0,23 ммоль) с DEPEA (40 мкл, 0,23 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Добавляли H2O (1 мл) и перемешивали дополнительно в течение 10 минут. ACN выпаривали и добавляли DCM и 10% лимонную кислоту. Водный слой отделяли и трижды экстрагировали с DCM, фракции DCM комбинировали, сушили над Na2SO4 и концентрировали, получая на выходе 95% соединения 7.
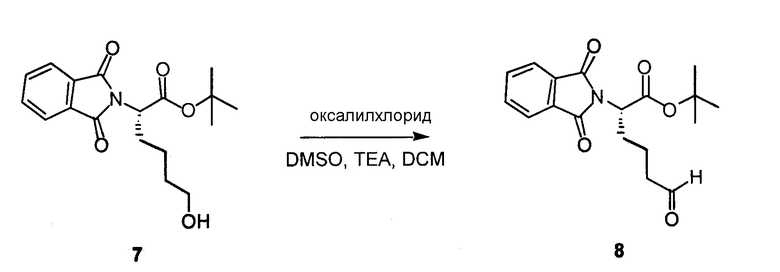
Оксалилхлорид (561 мкл, 6,60 ммоль) растворяли в DCM (35 мл), охлаждали до -78°C, перемешивали в течение 5 минут, далее добавляли раствор диметилсульфоксида (870 мкл, 12,3 ммоль) в DCM (2,5 мл). После перемешивания в течение 5 минут соединения 7 (1,05 г, 3,15 ммоль) в дихлорметане (20 мл) добавляли триэтиламин (2,37 мл, 17,0 ммоль). Температуру реакционной смеси медленно доводили до комнатной температуры. Добавляли DCM и H2O, водный слой отделяли и дважды экстрагировали DCM. Фракции DCM комбинировали, фильтровали через Na2SO4 и концентрировали, получая на выходе 95% соединения 8.
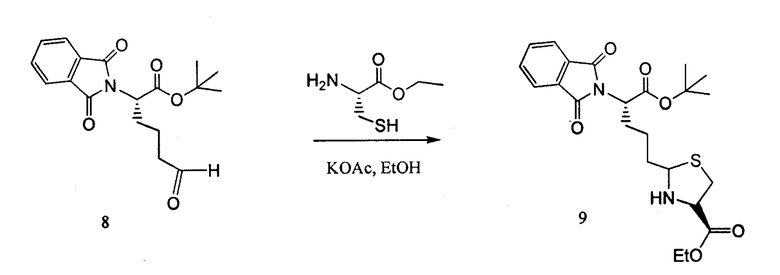
Сложный этиловый эфир L-цистеин-гидрохлорида (643 мг, 3,5 ммоль) и ацетат калия (343 мг, 3,5 ммоль) растворяли в перемешиваемом EtOH (13 мл) и охлаждали до 0°C в ледяной бане. Соединение 8 растворяли в EtOH (13 мл) и добавляли к реакционной смеси. Реакционную смесь перемешивали при 0°C в течение 4 часов, методами LCMS определяли превращение соединения 8 в два диастереомерных продукта. Реакционную смесь фильтровали, выпаривали EtOH, снова растворяли в DCM и промывали соляным раствором, сушили над MgSO4 и концентрировали, получая количественный выход смеси диастереомеров 9 1:1.
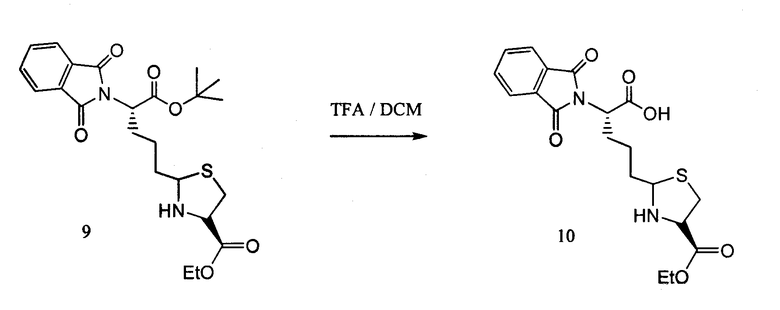
Диастереомеры растворяли в смеси TFA:DCM 1:1 (10 мл) и перемешивали в течение 1 часа при комнатной температуре. Методы LCMS подтвердили полное превращение в соединение 10. Реакционную смесь концентрировали, получая на выходе 95% для двух диастереомеров соединения 10.

К перемешиваемому раствору 10 (675 мг, 1,67 ммоль) в THF (20 мл) добавляли EEDQ (619 мг, 2,50 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение двух дней. THF удаляли при пониженном давлении, полученный продукт снова растворяли в EtOAc. Органический слой промывали 0,5 н. HCl, 0,5% NaHCO3, H2O, соляным раствором. Раствор EtOAc сушили над MgSO4 и концентрировали. Полученный продукт очищали посредством HPLC с обращенной фазой с использованием 10-70% ACN в H2O, получая два диастереомера 11, с выходом 20% для диастереомера 1 и 18% диастереомера 2.
Пример 2. 1-[2-Циклогексил-2-(2-метиламинопропиониламино)-ацетил]пирролидин-2-карбоновая кислота(2-фенил-2H-пиразол-3-ил)-амид
Раствор Boc-MeAla-Chg-Pro-OH (47,0 мг, 0,107 ммоль) и пиридина (26 мкл, 0,32 ммоль) в безводном дихлорметане (300 мкл) охлаждали до 0°C и покапельно добавляли раствор оксалилхлорида в дихлорметане (54 мкл, 2,0 M, 0,11 ммоль) в течение 10 минут. Раствор перемешивали при температуре 0°C в течение 15 минут, затем при температуре окружающей среды - в течение 45 минут, затем добавляли раствор 5-амино-1-фенилпиразола (15,9 мг, 0,100 ммоль; TCI America catalog # A0174) и пиридина (15,5 мкл, 0,191 ммоль) в дихлорметане (0,5 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 16 часов, разбавляли дихлорметаном до объема 20 мл и промывали 0,2 н. водным гидроксидом натрия (20 мл). Органическую фазу сушили над MgSO4 и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали посредством колоночной хроматографии (силикагель, 60% этилацетат в гексане, затем 100% этилацетат), получая на выходе желтое масло: m/z 581 (M+H+). Полученное масло обрабатывали 5% трифторуксусной кислотой в дихлорметане (2 мл) и по истечении 18 часов растворитель удаляли в вакууме. Полученное в результате масло (29,3 мг, выход 57% через 2 стадии) далее очищали посредством HPLC с обращенной фазой, получая на выходе продукт (TFA соль, 9,6 мг, 15% выход):
m/z 481 (M+H+), 503 (M+Na+).
Пример 3. 4-Фенил-[1,2,3]тиадиазол-5-иламин
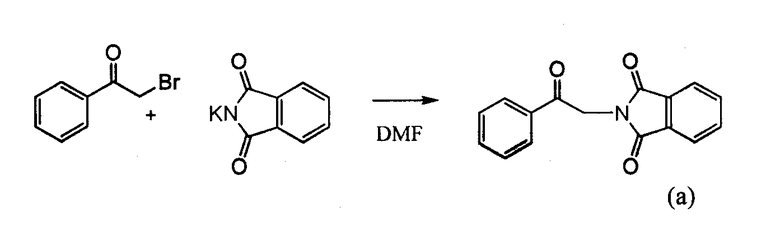
2-Бромацетофенон растворяли в DMF (3 об.) и добавляли фталимид калия (1,1 экв.). Реакционную смесь, исходно слабо экзотермическую, перемешивали в течение ночи при комнатной температуре. DMF удаляли в вакууме и реакционную смесь разбавляли DCM (~3 об.), далее разбавляли 0,1 н. NaOH (~3 об.; 1:1 вод./орг.) и энергично перемешивали, затем экстрагировали. Органический слой, содержащий некоторое количество твердого вещества, концентрировали в вакууме и полученное твердое вещество суспендировали в диэтиловом эфире и собирали посредством всасывающей фильтрации, получая на выходе 95% соединения (a) в виде белого кристаллического твердого вещества.
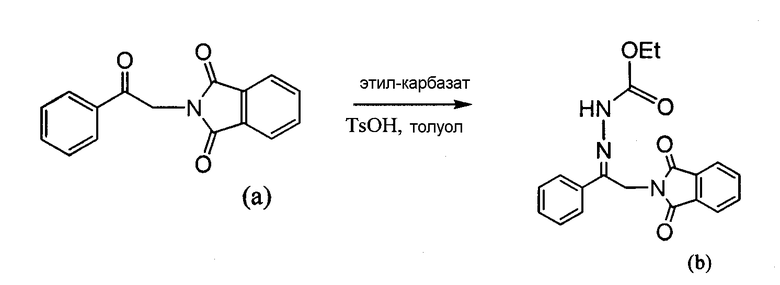
Соединение (a), этил-карбазат (1,5 экв.) и TsOH-H,O (0,1 экв.) соединяли в толуоле (5 об.) и кипятили с обратным холодильником, используя сепаратор Dean-Stark для удаления воды. Раствор приобретал темно-красный цвет, и реакцию завершали посредством TLC в течение ~2 часов. Приблизительно половину толуола удаляли путем дистилляции, температуру раствора доводили до комнатной температуры и концентрировали в вакууме. Полученное твердое вещество суспендировали в EtOH (минимальный объем, необходимый для перемешивания), кипятили с обратным холодильником в течение 30 минут и охлаждали льдом для обеспечения выпадения в осадок обоих изомеров. Твердое вещество собирали посредством всасывающей фильтрации, промывали холодным EtOH и сушили над вакуумом, получая оба изомера соединения (b) в виде беловатого твердого вещества, с выходом ~90%.
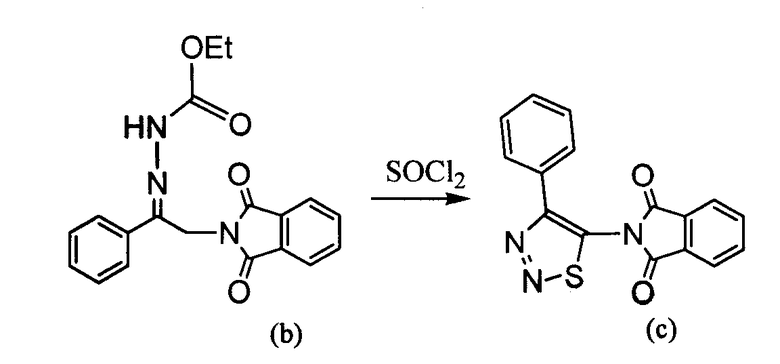
К охлажденному льдом тионилхлориду (4 экв., -0,85 об.) добавляли порционно (для контроля экзотермы) смесь изомеров (b). Ледяную баню удаляли и температуру реакционной смеси доводили до комнатной температуры и перемешивали в течение ночи. Тионилхлорид удаляли в вакууме, добавляли DCM (1 об.) и реакционную смесь перемешивали с 0,1 M NaOH (1 об.; 1:1 вод./орг.). Суспензию экстрагировали и органические фазы концентрировали в вакууме, суспендировали в кипящем EtOАс (минимальный объем, необходимый для удобного перемешивания) в течение 30 минут, температуру реакционной смеси доводили до комнатной температуры, собирали посредством всасывающей фильтрации, промывали минимальным количеством холодного EtOАс и сушили в вакууме, получая (c) в виде беловатого кристаллического твердого вещества, с выходом ~80%.
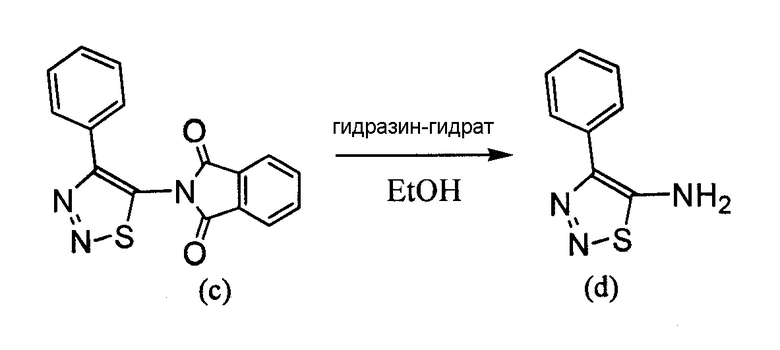
Раствор гидразин-гидрата (2,4 экв.) в EtOH (1 об.) добавляли покапельно к кипящему раствору (c) в EtOH (8 об.). Немедленно образовывался осадок, и реакцию завершали посредством TLC в течение ~3 часов. Раствор охлаждали до комнатной температуры и побочный продукт расщепления фталимида удаляли путем фильтрации и затем промывали DCM. Фильтрат EtOH/DCM концентрировали в вакууме до получения кристаллического образования. Эту суспензию перемешивали в течение ночи и кристаллическую твердую смесь собирали посредством всасывающей фильтрации и промывали холодным EtOH до удаления цветных примесей, получая тиадиазоламин (d) в виде беловатого кристаллического твердого вещества, с выходом ~75%.
Пример 4. 1-[2-Циклогексил-2-(2-метиламинопропиониламино)-ацетил]пирролидин-2-карбоновая кислота(4-фенил-[1,2,3]тиадиазол-5-ил)-амид

Boc-L-Pro (2 экв.), HOBt (1,9 экв.), EDC-HCl (1,9 экв.) DIPEA (5 экв.) растворяли в DMF (10-15 об.). К этой смеси далее добавляли тиадиазоламин (d). Реакционную смесь, исходно слабо экзотермическую, нагревали до 75°C и перемешивали в течение ночи, температуру смеси доводили до комнатной температуры и DMF частично удаляли в вакууме. Реакционную смесь разбавляли EtOAc (10-15 об.), далее дважды промывали 1 M HCl, единожды - NaHCO3 и единожды - соляным раствором (1:1 вод./орг.). Органический слой концентрировали в вакууме и полученный твердый продукт суспендировали в кипящем MeCN (минимальный объем, необходимый для удобного перемешивания) в течение 30 минут, температуру реакционной смеси доводили до комнатной температуры. Посредством всасывающей фильтрации получали Boc-защищенный сопряженный продукт в виде беловатого кристаллического твердого вещества, с выходом ~77%. Boc-защищенный продукт суспендировали в 4 M растворе HCl/диоксан (4-5 экв. кислоты) и MeCN (1 об. экв. к раствору диоксана) и перемешивали при комнатной температуре, до тех пор пока методы LCMS не показали полное снятие защиты, ~1 час. Реакционную смесь концентрировали в вакууме и полученное твердое вещество энергично суспендировали в кипящем MeCN (минимальный объем, необходимый для удобного перемешивания), температуру реакционной смеси доводили до комнатной температуры, твердое вещество собирали посредством всасывающей фильтрации, промывали холодным MeCN до удаления цвета осадка из брикета, получая соль HCl (e) в виде беловатого твердого вещества, с приблизительным количественным выходом.
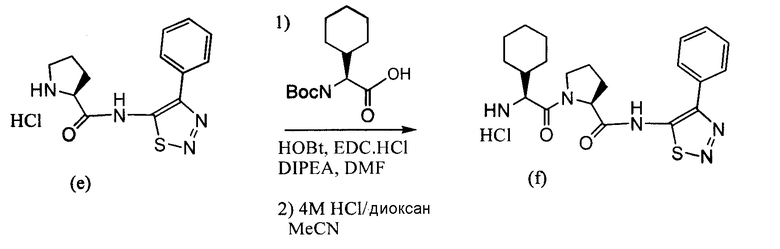
Соль HCl (e) растворяли в DMF (10-15 об.) и DIPEA (5 экв.). К этому раствору добавляли Boc-L-Chg (1,5 экв.), HOBt (1,4 экв.) и EDC-HCl (1,4 экв.). Завершение процесса взаимодействия по истечении ~2 часов определяли методами LCMS. Реакционную смесь разбавляли EtOAc (15 об.) и дважды промывали 1 M HCl, единожды -NaHCO3 и единожды соляным раствором (1:1 вод./орг.). Органический экстракт сушили над сульфатом натрия и концентрировали в вакууме. Полученное твердое вещество суспендировали в EtOH/Гексан (20:80) (минимальный объем, необходимый для удобного перемешивания) и фильтровали, получая Boc-защищенный сопряженный продукт в виде беловатого твердого вещества, с выходом -80%. Boc-защищенный продукт растворяли в растворе 4 M HCl/диоксан (4-5 экв. кислоты) и MeCN (0,25 экв. объема к раствору диоксана) и перемешивали при комнатной температуре, пока методами LCMS не выявляли полное снятие защиты, ~1 час. Реакционную смесь дважды концентрировали досуха посредством толуола (такой же объем, как и в случае снятия защиты), получая соль HCl (f) в виде белого кристаллического твердого вещества, с приблизительным количественным выходом.

Соль HCl (f) растворяли в DMF (10-15 об.) и DIPEA (5 экв.). К этому раствору добавляли Boc-L-N-метил Ala (1,5 экв.), HOBt (1,4 экв.) и EDC-HCl (1,4 экв.). Завершение процесса взаимодействия по истечении ~1 часа определяли методами LCMS. Реакционную смесь разбавляли EtOAc (15 об.) и дважды промывали 1 M HCl, единожды - NaHCO3 и единожды - соляным раствором (1:1 вод./орг.). Органический экстракт сушили над сульфатом натрия и концентрировали в вакууме, получая Boc-защищенный сопряженный продукт в виде бежевого, пенистого твердого вещества, с выходом 85%. Boc-защищенный сопряженный продукт растворяли в растворе 4 M HCl/диоксан (4-5 экв. кислоты) и MeCN (0,25 экв. объема к раствору диоксана) и перемешивали при комнатной температуре, пока методами LCMS не выявляли полное снятие защиты, ~1 час. Реакционную смесь дважды концентрировали досуха посредством толуола (такой же объем, как и в случае снятия защиты) и полученное твердое вещество суспендировали в MTBE/EtOAc (70:30) (минимальный объем, необходимый для удобного перемешивания), фильтровали и собирали, получая неочищенный продукт (g) в виде беловатого сыпучего твердого вещества. Неочищенную соль HCl (g) суспендировали в MeOH (4 об. минимум) и растворяли, перемешивая при температуре 65°C. Добавляли двумя порциями теплый изопропилацетат (6-8 об.), сохраняя при этом температуру приблизительно 60°C, и давали раствору остыть в процессе перемешивания. Кристаллизация происходила быстро, суспензию перемешивали при комнатной температуре в течение нескольких часов, затем перемешивали при температуре 0°C в течение 1 часа, после чего твердое вещество собирали посредством всасывающей фильтрации, промывали MeOH/iPrOAc (1:4, 2 об.) и сушили, получая конечный продукт в виде белого/беловатого крусталлического твердого вещества, с выходом ~80% от (f).
Пример 5. 2-[трет-бутоксикарбонил-(1H-пиррол-2-илметил)-амино]пропионовая кислота

Аланин-этиловый сложный эфир (5 г, 32,5 ммоль), пиррол-2-карбоксальдегид (3,1 г, 32,5 ммоль), цианоборгидрид натрия (2,04 г, 32,5 ммоль) и AcOH (1%) соединяли в DMF и перемешивали в течение ночи. Реакцию гасили водой и DMF выпаривали. Реакционную смесь разбавляли EtOAc, промывали 0,1 н. NaOH, сушили и концентрировали, получая на выходе 2,5 г продукта. Полученный сложный эфир (2,5 г, 12,8 ммоль), ди-трет-бутилдикарбонат (3,06 г, 14 ммоль) смешивали в THF, H2O с NaHCO3 и перемешивали в течение ночи. THF выпаривали и смесь разбавляли EtOAc, промывали 1 н. NaOH, насыщенной NH4Cl и солевым раствором. Далее сушили, смесь концентрировали, получая на выходе Boc-защищенный сложный эфир 3,3 г. Boc-защищенный сложный эфир (1,67 г, 5,6 моль), моногидрат гидроксида лития (284 мг, 6,77 ммоль) смешивали в THF и H2O при температуре 0°C. THF удаляли в вакууме и раствор подкисляли, разбавляя H2SO4, дважды экстрагировали EtOAc. Органические слои комбинировали, сушили и выпаривали.
Пример 6. Тетрагидропиранилглицин
Тетрагидропиранилглицин приобретали в NovaBiochem или синтезировали согласно процедурам, описанным в литературе: Ghosh, A. K.; Thompson, W. J.; holloway, M. K.; McKee, S. P.; Duong, T. T.; Lee, H. Y.; Munson, P. M.; Smith, A. M.; Wai, J. M; Darke, P. L.; Zugay, J. A.; Emini, E. A.; Schleife, W. A.; Huff, J. R.; Anderson, P. S. J. Med. Chem, 1993, 36, 2300-2310.
Пример 7. Пиперидинилглицин
Пиперидинилглицин синтезировали согласно процедурам, описанным в литературе: Shieh, W-C; Xue, S.; Reel, N.; Wu, R.; Fitt, J.; Repic, O. Tetrahedron: Asymmetry, 2001, 12, 2421-2425.
Пример 8. 4,4-Дифторциклогексилглицин
4,4-Дифторциклогексилглицин получали согласно процедурам, описанным в US 2003/0216325.
Пример 9. Boc(S)-2-амино-2-(4-гидроксициклогексил)уксусная кислота

Следуя процедуре Sheih (Tetrahedron: Asymmetry, 2001, 12, 2421-2425), раствор кетона а (8,4 г) и EtOAc (30 мл) добавляли к раствору сложного метилового эфира N-Cbz-фосфоноглицина b, TMG (4,5 мл) и EtOAc (30 мл). Раствор выдерживали при комнатной температуре в течение 48 часов, затем промывали 1 н. HCl (3×50 мл), соляным раствором (1×50 мл), сушили над Na2SO4, фильтровали и концентрировали. Осадок абсорбировали на целите и очищали посредством хроматографии, затем дополнительно очищали посредством рекристаллизации из EtOAc/гексанов, получая 5,2 г продукта c.
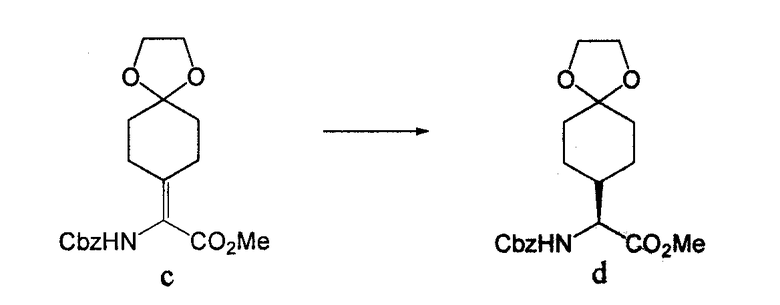
Следуя процедуре Sheih (Tetrahedron: Asymmetry, 2001, 12, 2421-2425), раствор енамида c (5,0 г), (S,S)-Me-BPE-Rh(I) (1,5 г, Strem Chemicals, Newburyport, MA) и MeOH (100 мл) энергично встряхивали в атмосфере H2, 70 пси, в течение 48 часов. Растворитель удаляли при пониженном давлении. Осадок растворяли в EtOAc и фильтровали через SiO2 с EtOAc. Растворитель удаляли при пониженном давлении, получая 4,0 г продукта d в виде бесцветного твердого вещества.
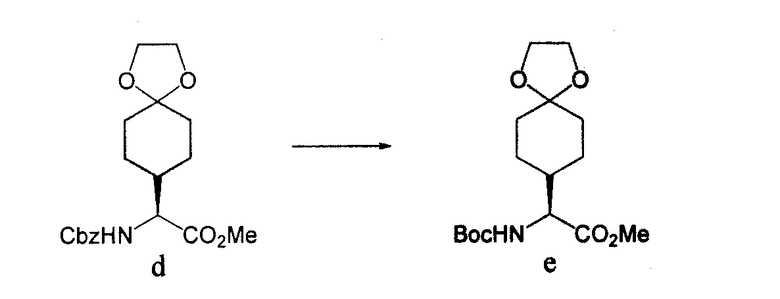
Смесь Cbz-карбамата d, (4,0 г), Boс2O, (2,9 г), 20% Pd(OH)2·C (1,0 г) и MeOH (30 мл) выдерживали в атмосфере H2 в течение 6 часов. Смесь фильтровали через целит с использованием MeOH. Растворитель удаляли при пониженном давлении, получая 4,5 г остатка e, который непосредственно и использовали в дальнейшем.
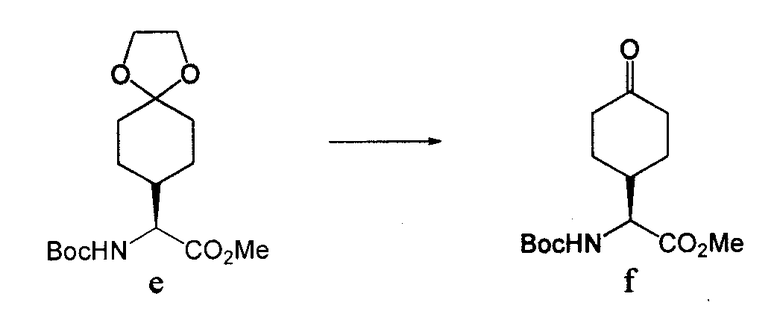
Полученный выше осадок e растворяли в H2O (10 мл), AcOH (30 мл), THF (5 мл) и дихлоруксусной кислоте (3 мл) и выдерживали при комнатной температуре в течение ночи. Добавляли воду (5 мл) и раствор выдерживали до полного завершения процесса гидролиза, определяемого методами HPLC-MS. Осторожно добавляли твердый Na2CO3 до прекращения выделения газа, смесь разбавляли водным NaHCO3 и экстрагировали 10%EtOAc/DCM. Комбинированные органические фазы единожды промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали. Осадок очищали посредством хроматографии, получая 2,9 г продукта f.
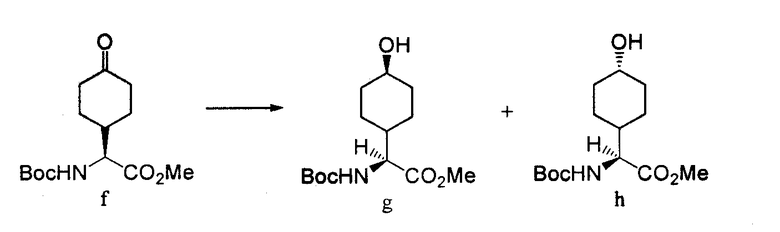
Смесь кетона f (1,5 г) и MeOH (50 мл) обрабатывали NaBH4 (290 мг) при температуре 0°C в течение 20 минут. Смесь подкисляли до ~pH 1 посредством 10% водной лимонной кислоты и удаляли MeOH при пониженном давлении. Осадок разбавляли водой и экстрагировали 20% EtOAc/DCM. Объединенные органические фазы промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали. Осадок очищали посредством хроматографии, получая 1,17 г продукта g и 0,23 г продукта h.
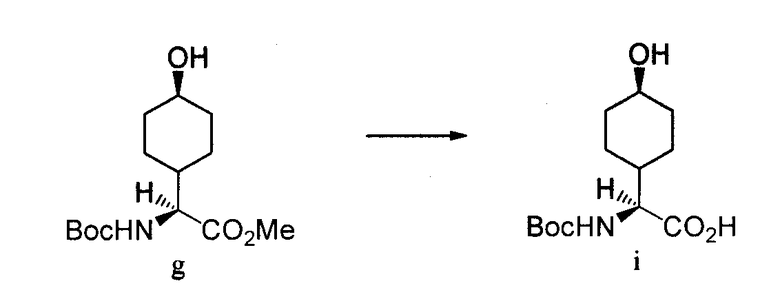
Смесь сложного эфира g (1,17 г), LiOH·H2О (160 мг), THF (3 мл) и воды (4,5 мл) энергично перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли соляным раствором и полностью экстрагировали EtOAc. Объединенные органические фазы промывали единожды соляным раствором, сушили над Na2SO4, фильтровали и концентрировали, получая кислоту i (525 мг).
Пример 10. Соединение 29
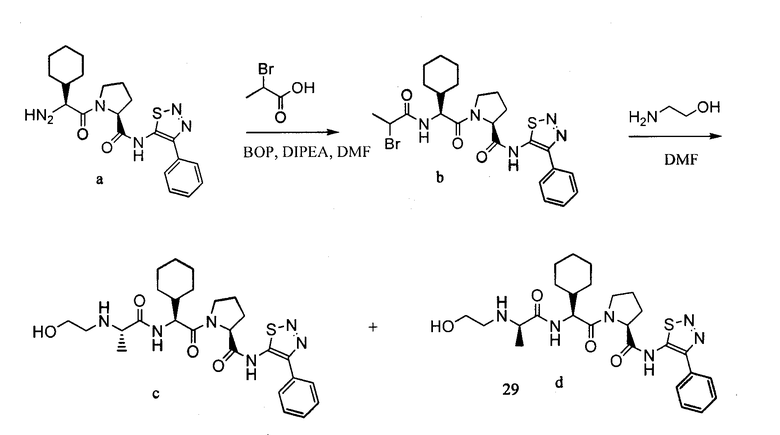
Смесь амина a (1,56 ммоль), 2-бромпропионовой кислоты (0,72 г, 4,68 ммоль), BOP (2,1 г, 4,68 ммоль) и DIPEA (1,6 мл, 9,36 ммоль) в 10 мл DMF перемешивали при комнатной температуре в течение 2 часов. Методами LCMS-анализа определяли завершение реакции. Добавляли 100 мл EtOAc к реакционной смеси и органический слой промывали насыщенным NaHCO3, затем солевым раствором, сушили над Na2SO4 и концентрировали досуха. Неочищенный материал очищали посредством хроматографии, используя 50% EtOAc/гексан, получая соединение b.
Соединение b (0,832 г, 1,5 ммоль) обрабатывали этаноламином (200 мкл, 2,73 ммоль) в 3 мл DMF и перемешивали в течение ночи до завершения реакции. Реакционную смесь очищали посредством HPLC с обращенной фазой, получая два диастереомера, c (53 мг) и d (соединение 29) (150 мг).
Пример 11. N-Boc-N-циклопропилметил-L-аланин
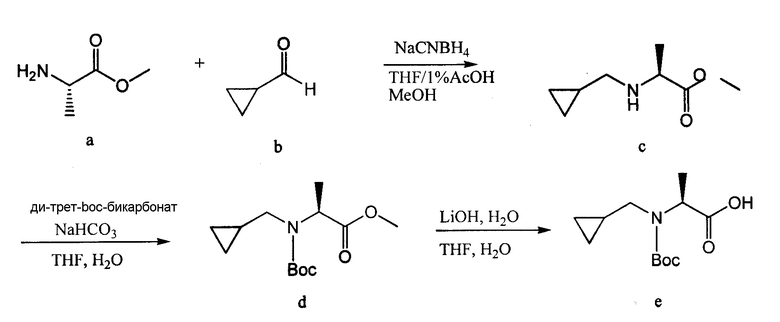
Сложный метиловый эфир L-аланин-гидрохлорида a (5 г, 35,8 ммоль) и циклопропанкарбоксальдегид b (2,67 мл, 35,8 ммоль) суспендировали в 50 мл THF w/1% AcOH. При добавлении 5 мл CH3OH мутный раствор становился прозрачным. Добавляли NaCNBH4 (2,25 г, 35,8 ммоль) и реакционную смесь перемешивали в течение ночи. Реакцию гасили, добавляя 1 н. водный NaOH, дважды экстрагировали EtOAc, органические слои сушили над Na2SO4 и концентрировали досуха. Неочищенный материал очищали посредством хроматографии, используя 30% EtOAc/гексан (окрашенный нингидрином), с получением соединения c (1 г, 18%).
Соединение c (1 г, 6,37 ммоль) и ди-трет-boc-дикарбонат (2,1 г, 9,55 ммоль) разбавляли THF (20 мл) и добавляли H2O (20 мл), NaHCO3 (1,3 г, 15,9 ммоль). Реакционную смесь перемешивали в течение ночи до завершения реакции. THF удаляли при пониженном давлении, водный слой трижды экстрагировали EtOAc. Комбинированные органические слои промывали 1 н. NaOH, насыщенным NH4Cl, далее солевым раствором, концентрировали досуха. Boc-защищенное соединение d (1,39 г, 5,40 ммоль) перемешивали с LiOH-H2O (1,14 г, 27 ммоль) в THF (20 мл) и H2O (20 мл) в течение ночи при комнатной температуре. THF удаляли и подкисляли водный слой, добавляя 10% лимонную кислоту, до получения pH 4, затем трижды экстрагировали EtOAc. Комбинированные органические слои промывали соляным раствором и концентрировали. Полученный неочищенный продукт очищали на колонке C-18 с обращенной фазой, элюировали 0-50% ацетонитрил/H2O, получая чистое соединение e в виде белого твердого вещества (794 мг).
Пример 12. Процедура связывания кислоты с фторидом
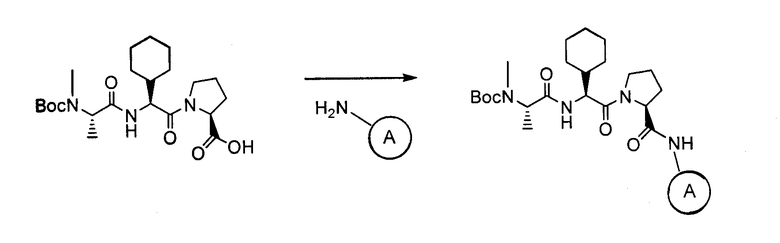
Раствор Boc-MeAla-Chg-Pro-OH (2,3 ммоль) и пиридина (6,9 мкмоль) в безводном дихлорметане (23 мл) охлаждали до 0°C и покапельно добавляли цианурфторид (2,3 ммоль) с интервалом в 30 секунд. Полученную смесь перемешивали при температуре 0°C в течение 15 минут, при температуре окружающей среды - в течение 5 часов и затем гасили водой. Смесь трижды экстрагировали дихлорметаном (общий объем 100 мл) и комбинированные органические фазы промывали раствором соли и сушили над безводным сульфатом натрия. Далее фильтровали и концентрировали в вакууме, получая на выходе пептид фторангидрида в виде чистого бесцветного масла, которое использовали в дальнейшем без предварительной очистки.
Раствор неочищенного фторангидрида (0,50 ммоль) и пиридина (1,5 ммоль) в дихлорметане (2,5 мл) добавляли к твердому амину (0,50 ммоль) и реакционную смесь перемешивали при температуре окружающей среды или при температуре 50°C (в запечатанном сосуде). Смесь вливали в водный бикарбонат натрия и трижды экстрагировали дихлорметаном (общий объем 100 мл). Комбинированные органические фазы промывали раствором соли и сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный пептид-амид использовали непосредственно без предварительной очистки.
Пример 13. 1-Фенил-1H-пиразол-5-амин
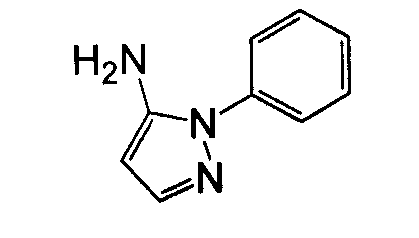
1-Фенил-1H-пиразол-5-амин коммерчески доступен из TCI America (каталог# A0174).
Пример 14. 3-Метил-1-фенил-1H-пиразол-5-амин
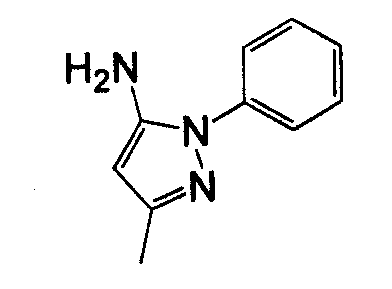
3-Метил-1-фенил-1H-пиразол-5-амин коммерчески доступен из TCI America (каталог# A1311).
Пример 15. 5-Фенилтиазол-2,4-диамин
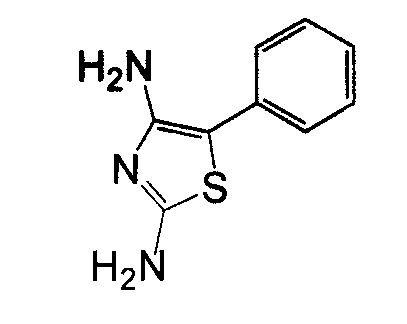
5-Фенилтиазол-2,4-диамин коммерчески доступен из Acros Organics (catalog# 11234-0010).
Пример 16. 5-(Трифторметил)-4-фенилтиофен-3-амин
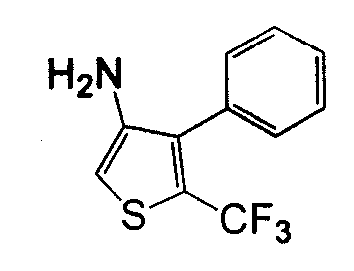
5-(Трифторметил)-4-фенилтиофен-3-амин коммерчески доступен из Acros Organics (каталог# SEW03133DA).
Пример 17. 4-Фенил-1H-пиразол-3-амин
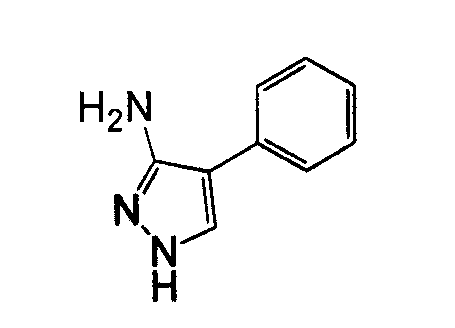
4-Фенил-1H-пиразол-3-амин получали согласно процедурам, описанным в публикации E. L. Anderson et al.; J. Med. Chem., 1964, 7, 259-268.
Пример 18. 5-Метил-4-фенил-1H-пиразол-3-амин
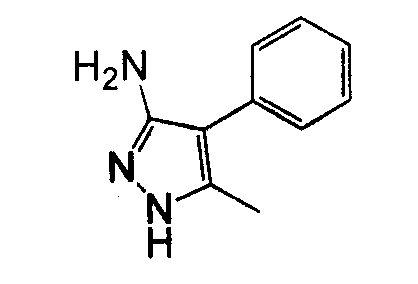
5-Метил-4-фенил-1H-пиразол-3-амин получали согласно процедурам, описанным в публикации E. L. Anderson et al.; J. Med. Chem., 1964, 7, 259-268.
Пример 19. 3-Фенил-3H-1,2,3-триазол-4-амин

3-Фенил-3H-1,2,3-триазол-4-амин получали согласно процедурам, описанным в публикации K. M. Baines, T. W. Rourke, K. Vaughan; J. Org. Chem., 1981, 46, 856-859.
Пример 20. 4-Фенилизоксазол-5-амин
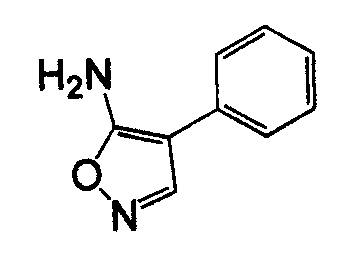
4-Фенилизоксазол-5-амин получали согласно процедурам, описанным в публикации H. Peeters, W. Vogt; EP 43024.
Пример 21. 3-Фенил-1H-пиразол-4-амин
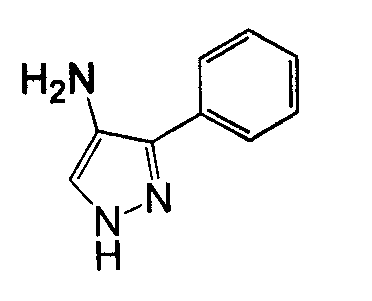
3-Фенил-1H-пиразол-4-амин получали согласно процедурам, описанным в публикации C. Chen, K. Wilcoxen, J. R. McCarthy; Tetrahedron Lett., 1988, 39, 8229-8232.
Пример 22. 1-Метил-3-фенил-1H-пиразол-4-амин

1-Метил-3-фенил-1H-пиразол-4-амин получали согласно процедурам, описанным в публикации C. Chen, K. Wilcoxen, J. R. McCarthy; Tetrahedron Lett., 1988, 39, 8229-8232.
Пример 23. 1-Метил-5-фенил-1H-пиразол-4-амин
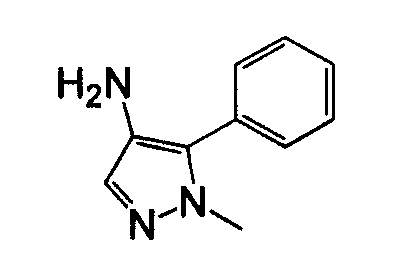
1-Метил-5-фенил-1H-пиразол-4-амин получали согласно процедурам, описанным в публикации C. Chen, K. Wilcoxen, J. R. McCarthy; Tetrahedron Lett, 1988, 39, 8229-8232.
Пример 24. 3-Метил-4-фенилизоксазол-5-амин
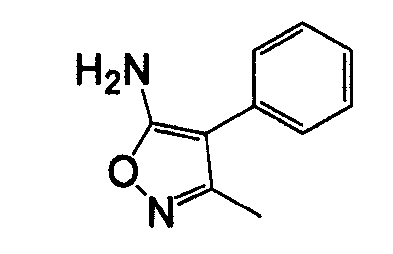
3-Метил-4-фенилизоксазол-5-амин получали согласно процедурам, описанным в публикации H. Peeters, W. Vogt; EP 43024.
Пример 25. 1-Фенил-1H-тетразол-5-амин
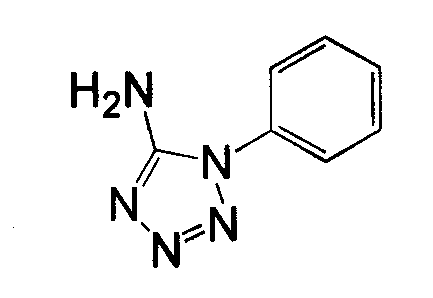
1-Фенил-1H-тетразол-5-амин получали согласно процедурам, описанным в публикации R. A. Batey, D. A. Powell; Org. Lett., 2000, 2, 3237-3240.
Пример 26. 4-Фенил-1,2,5-оксадиазол-3-амин
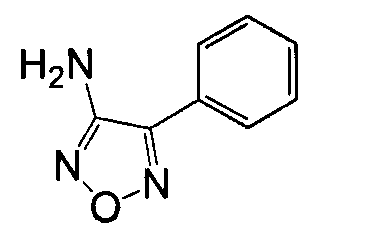
4-Фенил-1,2,5-оксадиазол-3-амин получали согласно процедурам, описанным в публикации R. Lakhan, O. P. Singh; Ind. J. Chem., 1987, 26B, 690-692.
Пример 27. 1-Амино-5-фенил-1H-тетразол
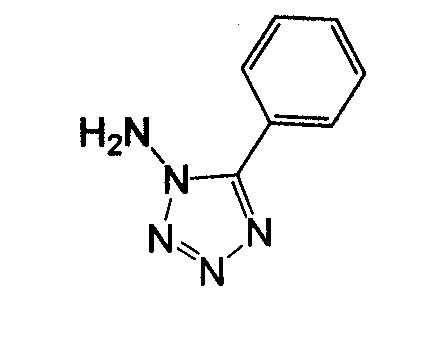
1-Амино-5-фенил-1H-тетразол получали согласно процедурам, описанным в публикации T. L. Gilchrist, G. E. Gymer, C. W. Rees; J. Chem. Soc., Perkin Trans. 1, 1975, 1747-1750.
Пример 28. 4-Амино-3-фенил-4H-1,2,4-триазол
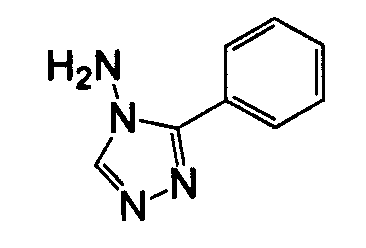
4-Амино-3-фенил-4H-1,2,4-триазол получали согласно процедурам, описанным в публикации A. А. Ikizler, N. Yildirim; J. Heterocyclic Chem., 1998, 35, 377-380.
Пример 29. 3-Фенилтиофен-2-амин
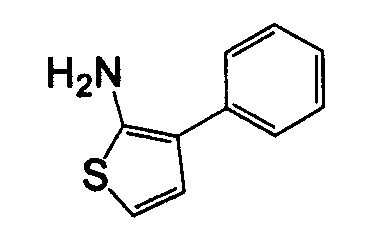
3-Фенилтиофен-2-амин получали согласно процедурам, описанным в публикации Y. Yoshikawa et al.; EP 737682 (US 5747518).
Пример 30. 2-Фенилтиофен-3-амин
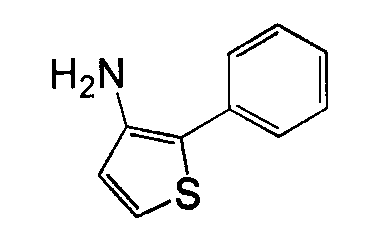
2-Фенилтиофен-3-амин получали согласно процедурам, описанным в публикации Y. Yoshikawa et al.; EP 737682 (US 5747518).
Пример 31. 4-Фенилтиофен-3-амин

4-Фенилтиофен-3-амин получали согласно процедурам, описанным в публикации G. Kirsch, D. Cagniant, P. Cagniant; J. Heterocyclic Chem., 1982, 19, 443-445.
Пример 32. 5-Амино-4-фенилтиазол-2-тиол
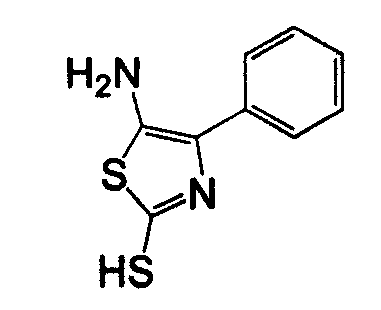
5-Амино-4-фенилтиазол-2-тиол получали согласно процедурам, описанным в публикации A. H. Cook, I. Heilbron, A. L. Levy; J.Chem. Soc., 1947, 1598-1609.
Пример 33. 2-(Метилтио)-4-фенилтиазол-5-амин
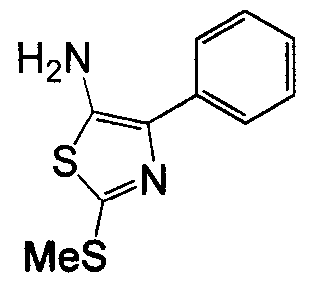
2-(Метилтио)-4-фенилтиазол-5-амин получали согласно процедурам, описанным в публикации A. H. Cook, I. Heilbron, A. L. Levy; J. Chem. Soc., 1947, 1598-1609.
Пример 34. 5-Амино-2-(метилсульфинил)-4-фенилтиазол

К раствору 5-амино-2-(метилсульфанил)-4-фенилтиазола (305 мг, 1,37 ммоль) в уксусной кислоте (3,0 мл) покапельно добавляли водную перекись водорода (660 мкл, 30% вес, 6,9 ммоль) при температуре окружающей среды. Через 4 часа смесь распределяли между дихлорметаном (60 мл) и водой (60 мл). Органическую фазу отделяли, промывали раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали чистый 5-амино-2-(метилсульфинил)-4-фенилтиазол (285 мг, 87%).
Пример 35. 5-Амино-2-(метилсульфонил)-4-фенилтиазол
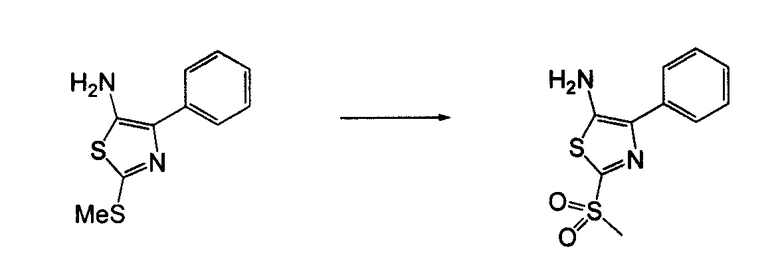
К раствору 5-амино-2-(метилсульфанил)-4-фенилтиазола (302 мг, 1,36 ммоль) в дихлорметане (5,0 мл) порционно добавляли 3-хлорпербензойную кислоту (638 мг, 77% вес., 2,9 ммоль), охлаждая в процессе до температуры 0°C. Смесь разбавляли дихлорметаном (3,0 мл), выдерживали смесь в течение 5 минут и оставляли смесь, позволяя ей нагреться до температуры окружающей среды. Через 3 часа порционно добавляли следующее количество 3-хлорпербензойной кислоты (305 мг, 77% вес, 1,4 ммоль). Через 20 часов смесь обрабатывали тиосульфатом натрия (2 мл, 1,0 M), вливали в насыщенный водный бикарбонат натрия и трижды экстрагировали дихлорметаном (общий объем 100 мл). Комбинированные органические фазы промывали насыщенным водным бикарбонатом натрия и соляным раствором, сушили над сульфатом натрия и концентрировали в вакууме, получая темно-коричневое пенистое вещество. Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали чистый 5-амино-2-(метилсульфонил)-4-фенилтиазол (90 мг, 26%).
Пример 36. 5-Амино-2-(аминосульфонил)-4-фенилтиазол и 5-амино-4-фенилтиазол
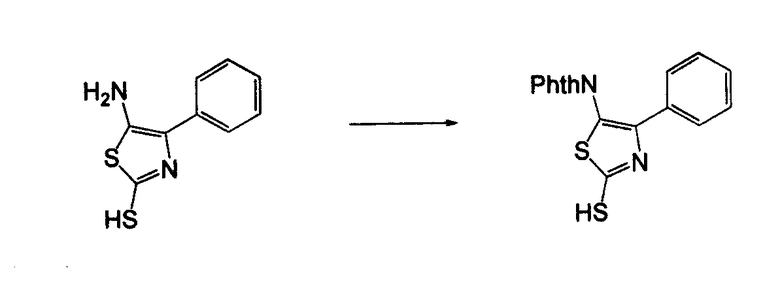
Смесь 5-амино-2-меркапто-4-фенилтиазола (1,01 г, 4,83 ммоль) и фталевого ангидрида (716 мг, 4,84 ммоль) в уксусной кислоте (20 мл) нагревали до 100°C в течение 64 часов и давали остыть. Смесь разбавляли в холодной воде (150 мл) и осадок собирали путем фильтрации, промывали водой (50 мл) и сушили под сильным вакуумом (1,46 г, 90%). Хотя фталимид и загрязнялся незначительным количеством дисульфида, но его использовали без дальнейшей очистки.
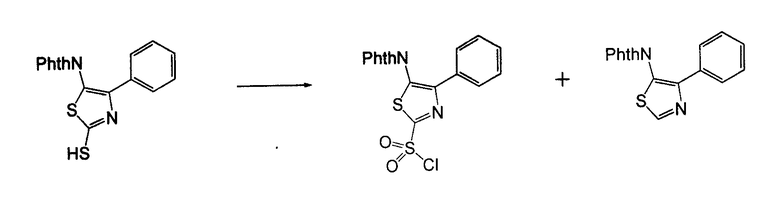
Раствор 2-меркапто-4-фенил-5-фталимидотиазола (203 мг, 600 мкмоль) в уксусной кислоте (4,5 мл) и воде (0,5 мл) при температуре 0°C обрабатывали N-хлорсукцинимидом (243 мг, 1,82 ммоль), одной порцией. Смесь перемешивали при температуре 0°C в течение 10 минут, оставляли на 1 час, позволяя смеси достичь температуры окружающей среды, и затем распределяли между дихлорметаном (50 мл) и водой (50 мл). Водную фазу дважды экстрагировали дихлорметаном (2 × 25 мл) и комбинированные органические фазы промывали соляным раствором и сушили над сульфатом натрия. Фильтровали и концентрировали в вакууме, получая смесь (231 мг) 2-(хлорсульфонил)-4-фенил-5-фталимидотиазола (больший компонент) с 4-фенил-5-фталимидотиазолом (2: 1), которую использовали в дальнейшем без очистки.
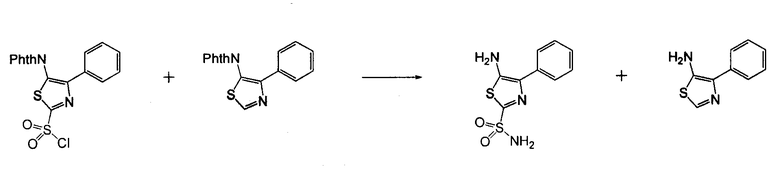
Неочищенную смесь сульфонилхлорида с 4-фенил-5-фталимидотиазолом (231 мг) в дихлорметане (10 мл) покапельно обрабатывали раствором аммония в метаноле (900 мкл, 2,0 M) при температуре окружающей среды. Через 10 минут смесь концентрировали в вакууме. Осадок суспендировали в этаноле (10 мл), обрабатывали этанолгидразином (660 мкл, 1,0 M, 660 мкмоль) и кипятили с обратным холодильником. Через 1,5 часа добавляли следующую порцию этанолгидразина (660 мкл, 1,0 M, 660 мкмоль) и кипятили с обратным холодильником в течение 15 часов. Охлажденный раствор фильтровали и концентрировали в вакууме. Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали чистый 5-амино-2-(аминосульфонил)-4-фенилтиазол (56 мг, 36% в три стадии) и 5-амино-4-фенилтиазол (17 мг, 16% в три стадии).
Пример 37. 5-Амино-2-(трет-бутилсульфанул)-4-фенилтиазол
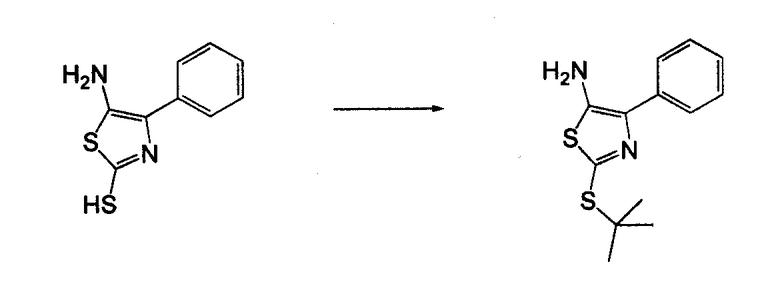
К суспензии 5-амино-2-меркапто-4-фенилтиазола (210 мг, 1,01 ммоль) в воде (1,0 мл) и трет-бутаноле (82 мг, 1,1 ммоль) добавляли концентрированную серную кислоту (3,0 мл), охлаждая при этом до ~ 20°C. Через 1,5 часа добавляли следующую порцию трет-бутанола (300 мкл, 1,0 M, 300 мкмоль) в воде при температуре окружающей среды. Через 1,5 часа смесь вливали в избыточно водный бикарбонат натрия и трижды экстрагировали дихлорметаном (общий объем 120 мл). Комбинированные органические фазы промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали 5-амино-2-(трет-бутилсульфанил)-4-фенилтиазол (220 мг, 82%).
Пример 38. 5-Амино-2-(трет-бутилсульфинил)-4-фенилтиазол
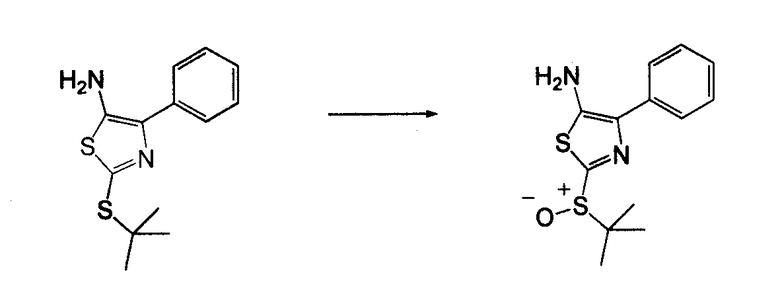
К 5-амино-2-(трет-бутилсульфанил)-4-фенилтиазол (102 мг, 385 мкмоль) в уксусной кислоте (5,0 мл) покапельно добавляли водный раствор перекиси водорода (218 мкл, 30% вес, 1,9 ммоль) при температуре окружающей среды. Через 5 часов смесь распределяли между дихлорметаном (50 мл) и водой (50 мл). Водную часть отделяли и экстрагировали дихлорметаном (20 мл). Комбинированные органические фазы промывали насыщенным водным бикарбонатом натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая фактически чистый 5-амино-2-(трет-бутилсульфинил)-4-фенилтиазол (110 мг, колич.).
Пример 39. 5-Амино-4-фенил-2-(трифторметилсульфанил)тиазол

Суспензию 5-амино-2-меркапто-4-фенилтиазола (503 мг, 2,41 ммоль) в уксусной кислоте (5,0 мл) обрабатывали гексан-2,5-дионом (290 мкл, 2,47 ммоль) в течение 14 часов при температуре окружающей среды, затем кипятили с обратным холодильником в течение 3 часов. Смесь гомогенизировали при кипячении с обратным холодильником, и при охлаждении образовывался осадок, который восстанавливали путем фильтрации, промывали уксусной кислотой (3 × 1,0 мл) и сушили в вакууме, получая чистый пирролидинотиазол (624 мг, 90%) в виде ярко-желтого микрокристаллического твердого вещества.
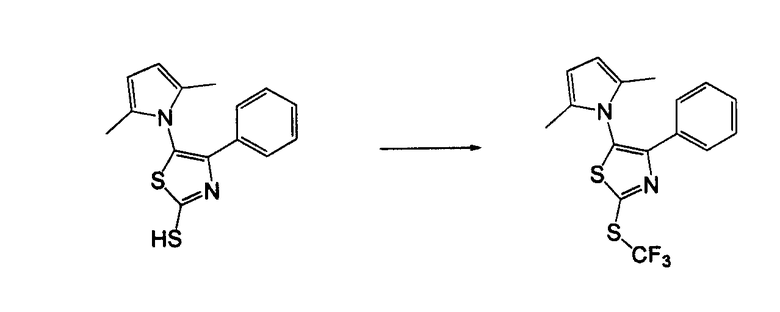
Раствор меркапто-тиазола (201 мг, 701 мкмоль) и карбоната калия (291 мг, 2,11 ммоль) в DMF (2,0 мл) насыщали путем барботирования трифторметилйодидом в течение 5 минут и в запечатанном сосуде нагревали до 50°C в течение 30 минут. Охлажденную смесь снова насыщали трифторметилйодидом и нагревали до 1000C в течение 1,5 часов. Смесь еще раз насыщали трифторметилйодидом и нагревали до 100°C (общий период 24 часа) и давали остыть. Смесь вливали в воду и трижды экстрагировали этилацетатом (общий объем 100 мл). Комбинированные органические фазы промывали водой и соляным раствором, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали чистый (трифторметилсульфанил)тиазол (72 мг, 29%) в виде бесцветной кристаллической пленки.
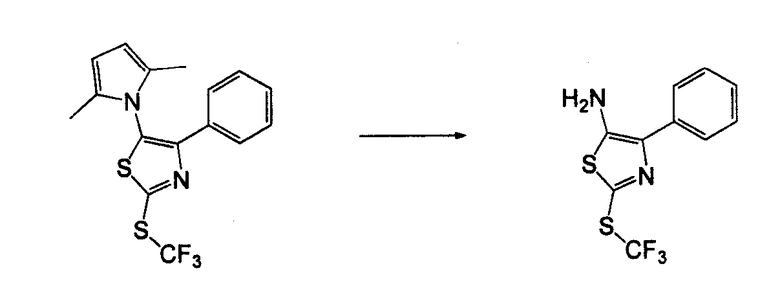
Суспензию тиазола (72 мг) и гидроксиламингидрохлорида (71 мг, 1,0 ммоль) в этаноле (5,0 мл) кипятили с обратным холодильником в течение 17 часов, разбавляли уксусной кислотой (3 мл), снова кипятили с обратным холодильником в течение дальнейших 2 часов и концентрировали до объема ~ 3 мл. Охлажденную смесь обрабатывали водным гидроксиламином (1,0 мл, 50% вес) и снова кипятили с обратным холодильником в течение 42 часов. Смесь обрабатывали водой (50 мл) и насыщенным водным бикарбонатом натрия (50 мл) и трижды экстрагировали в дихлорметане (общий объем 100 мл). Комбинированные органические фазы промывали соляным раствором, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали чистый 5-амино-4-фенил-2-(трифторметилсульфанил)тиазол (12,5 мг, 22%).
Пример 40. 5-Амино-4-фенил-2-(трифторметил)тиазол

α-Аминофенилацетамид (2,00 г, 13,3 ммоль) в метаноле (50 мл) обрабатывали этилтрифторацетатом (3,2 мл, 27 ммоль) при температуре 0°C в течение 30 минут и оставляли реакционную смесь, позволяя ей достичь температуры окружающей среды в течение 18 часов. Смесь концентрировали в вакууме, гомогенизировали метанолом и снова концентрировали, получая на выходе чистый трифторацетамид (3,27 г, колич.).
Трифторацетамид (881 мг, 3,58 ммоль) и реактив Лавессона (1,45 г, 3,59 ммоль) вместе обрабатывали пиридином (7,2 мл) и смесь нагревали до 100°C в течение 20 часов. Охлажденную смесь вливали в насыщенный водный бикарбонат натрия и трижды экстрагировали в хлороформе (общий объем 120 мл). Комбинированные органические фазы промывали водой, содержащей насыщенный водный бикарбонат натрия (10:1), соляным раствором и сушили над сульфатом натрия. Фильтровали и концентрировали в вакууме, получая на выходе красно-коричневое масло (829 мг). Полученный неочищенный продукт обрабатывали водным гидроксидом натрия (25 мл, 1,0 н.) в течение 15 минут и трижды экстрагировали в дихлорметане (общий объем 100 мл). Комбинированные органические фазы промывали водным гидроксидом натрия (25 мл, 1,0 н.) и соляным раствором и сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали чистый 5-амино-4-фенил-2-(трифторметил)тиазол (65 мг, 7,5%).
Пример 41. 3-Амино-4-фенил-1,2,5-тиадиазол

К раствору монохлорида серы (24,0 г, 178 ммоль) в DMF (30 мл) добавляли порционно каждые 20 минут α-аминофенилацетонитрил гидрохлорид (10,0 г, 59,3 ммоль) при температуре 0°C. Через 40 минут раствор оставляли на 20 минут, позволяя ему нагреться до температуры окружающей среды, разбавляли DMF (20 мл) и перемешивали в течение последующих 20 часов, далее вливали в ледяную воду. Полученную смесь экстрагировали эфиром (200 мл), фильтровали и еще дважды экстрагировали эфиром (2 × 50 мл). Комбинированные органические фазы промывали соляным раствором, сушили над сульфатом магния и концентрировали в вакууме, получая 3-хлор-4-фенил-1,2,5-тиадиазол в виде подвижного оранжевого масла (10,1 г, 87%). Посредством ускоренной дистилляции этого масла (9,35 г) при пониженном давлении получали на выходе чистое бесцветное масло (7,75 г, 83%), которое кристаллизовалось в процессе охлаждения.

3-Хлор-4-фенил-1,2,5-тиадиазол (3,19 г, 16,2 ммоль) в THF (32 мл) покапельно обрабатывали раствором литий-бис(триметилсилил)амида в THF (17,0 мл, 1,0 M, 17,0 ммоль) при температуре 0°C. Через 10 минут смесь оставляли на 1,5 часа, позволяя ей нагреться до температуты окружающей среды, далее обрабатывали 1 н. соляной кислотой и трижды экстрагировали в эфире (общий объем 300 мл). Комбинированные органические фазы промывали насыщенным водным бикарбонатом натрия и соляным раствором, сушили над сульфатом магния и концентрировали в вакууме. Полученный осадок растворяли в метаноле (50 мл) и триэтиламине (0,5 мл), кипятили с обратным холодильником в течение 15 часов и снова концентрировали в вакууме. Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали 3-амино-4-фенил-1,2,5-тиадиазол (1,96 г, 68%) в виде бесцветного твердого вещества.
Пример 42. 5-Амино-2-метил-4-фенилтиазол
К суспензии α-аминофенилацетонитрил-гидрохлорида (3,37 г, 20,0 ммоль) и измельченной в пудру серы (641 мг, 20,0 ммоль) в этаноле (20 мл) при температуре 0°C добавляли триэтиламин (4,18 мл, 30,0 ммоль), затем добавляли уксусный альдегид (2,3 мл, 41 ммоль). Реакционную смесь помещали в запечатанный сосуд и сосуд нагревали до температуры 60-70°C в течение 1 часа. Охлажденную смесь фильтровали и концентрировали в вакууме, полученный осадок обрабатывали этанолом (20 мл) и соляной кислотой (20 мл, 1 н.) в течение 15 часов. Далее смесь обрабатывали водным карбонатом натрия и трижды экстрагировали в этилацетате (общий объем 300 мл). Комбинированные органические фазы промывали соляным раствором, сушили над сульфатом натрия и концентрировали в вакууме, получая темно-коричневое масло. Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали 5-амино-2-метил-4-фенилтиазол (1,31 г, 34%), которое кристаллизовали из толуола.
Пример 43. 5-Амино-2-метил-4-фенилтиазол

Суспензию α-аминофенилацетонитрил-гидрохлорида (1,69 г, 10,0 ммоль), измельченную в пудру серу (321 мг, 10,0 ммоль) и 4-пиридинкарбоксальдегид (1,91 мл, 20,0 ммоль) в этаноле (10 мл) обрабатывали триэтиламином (2,09 мл, 15,0 ммоль) и реакционную смесь перемешивали при температуре 50°C в течение 80 минут. Охлажденную смесь разбавляли этанолом (5 мл) и обрабатывали водным гидроксиламином (700 мкл, 50% вес, 11 ммоль) при температуре окружающей среды в течение 15 часов, далее разбавляли дихлорметаном (50 мл). Добавляли насыщенный водный бикарбонат натрия и отделенные водные фазы дважды экстрагировали дихлорметаном (общий объем 100 мл). Комбинированные органические фазы сушили над сульфатом натрия и концентрировали в вакууме, получая темно-коричневое маслянистое пенистое вещество (3,23 г). Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали 5-амино-2-(4-пиридил)-4-фенилтиазол (1,41 г, 56%).
Пример 44. 2,4-Дифенилтиазол-5-амин
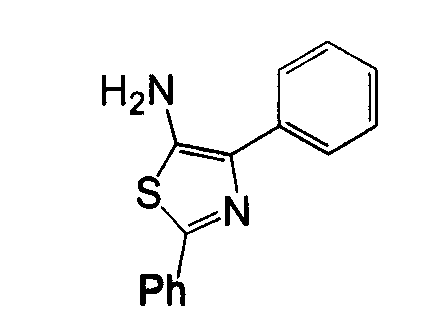
2,4-Дифенилтиазол-5-амин получали согласно процедурам, описанным в публикации K. Gewald, H. Schonfelder, U. Hain; J. Prakt. Chem., 1974, 361, 299-303.
Пример 45. 4-Фенил-2-(пиридин-2-ил)тиазол-5-амин
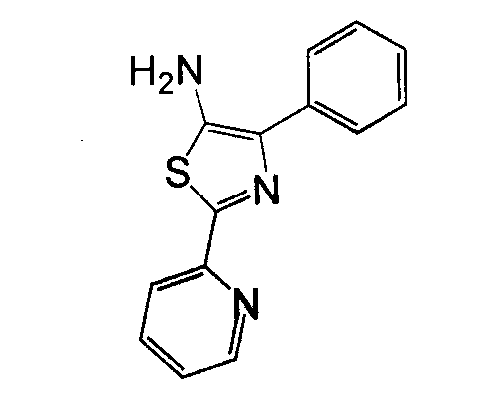
4-Фенил-2-(пиридин-2-ил)тиазол-5-амин получали согласно процедурам, описанным в публикации K. Gewald, H. Schonfelder, U. Hain; J. Prakt. Chem., 1974, 361, 299-303.
Пример 46. 4-Фенил-2-(пиридин-3-ил)тиазол-5-амин
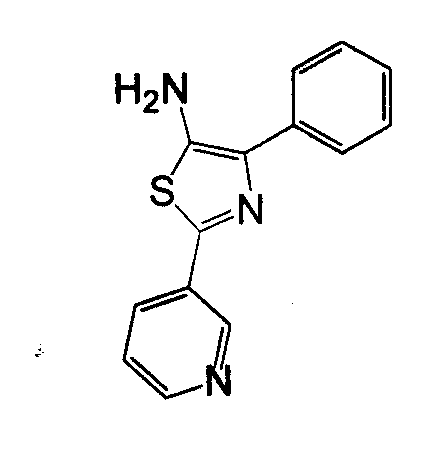
4-Фенил-2-(пиридин-3-ил)тиазол-5-амин получали согласно процедурам, описанным в публикации K. Gewald, H. Schonfelder, U. Hain; J. Prakt. Chem., 1974, 361, 299-303.
Пример 47. 5-Амино-2-(Fmoc-амино)-4-фенилтиазол
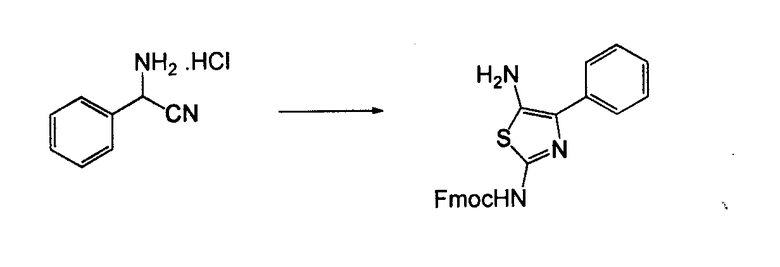
Суспензию α-аминофенилацетонитрил-гидрохлорида (3,19 г, 18,9 ммоль) и Fmoc-изотиоцианата (5,31 г, 18,9 ммоль) в DCM обрабатывали этилдиизопропиламином (3,62 мл, 20,8 ммоль) при температуре 0°C в течение 1 часа и затем при температуре окружающей среды в течение 3 часов. Далее смесь вливали в водный насыщенный бикарбонат натрия и трижды экстрагировали в этилацетате. Комбинированные органические фазы промывали водой, насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали в вакууме. Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали 5-амино-2-(Fmoc-амино)- 4-фенилтиазол (3,75 г, 48%).
Пример 48. N-(5-амино-4-фенилтиазол-2-ил)ацетамид
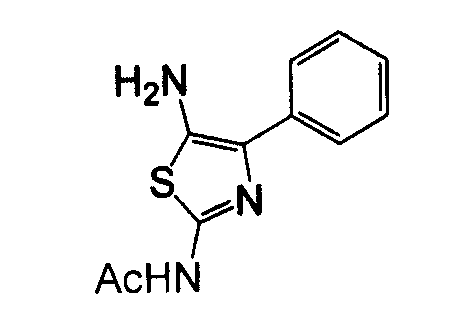
N-(5-амино-4-фенилтиазол-2-ил)ацетамид получали согласно процедурам, аналогичным процедурам, описанным в примере 47.
Пример 49. N-(5-амино-4-фенилтиазол-2-ил)бензамид
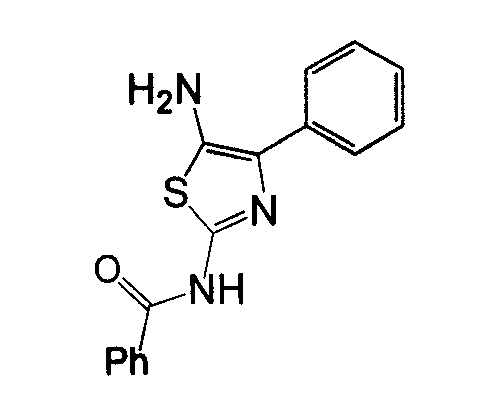
N-(5-амино-4-фенилтиазол-2-ил) получали согласно процедурам, аналогичным процедурам, описанным в примере 47.
Пример 50. Этил 5-амино-4-фенилтиазол-2-илкарбамат
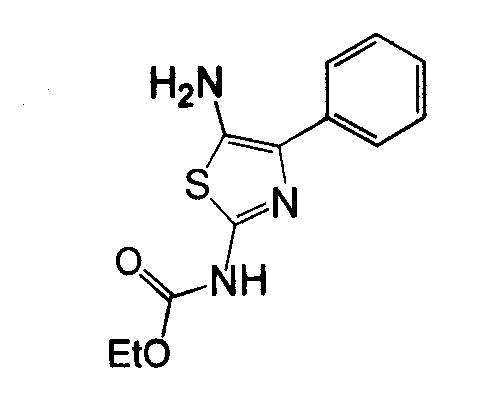
Этил 5-амино-4-фенилтиазол-2-илкарбамат получали согласно процедурам, аналогичным процедурам, описанным в примере 47.
Пример 51. N-(5-Амино-4-(2-хлорфенил)тиазол-2-ил)ацетамид
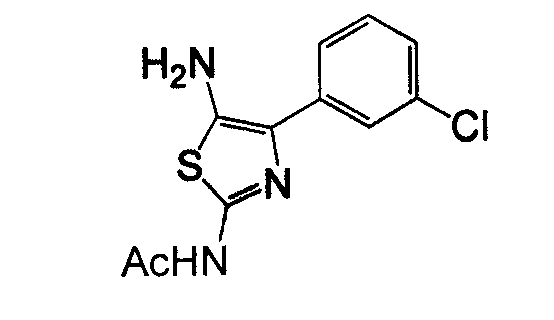
N-(5-амино-4-(2-хлорфенил)тиазол-2-ил)ацетамид получали согласно процедурам, аналогичным процедурам, описанным в примере 47.
Пример 52. (9H-флуорен-9-ил)метил 5-амино-4-(2-хлорфенил)тиазол-2-илкарбамат
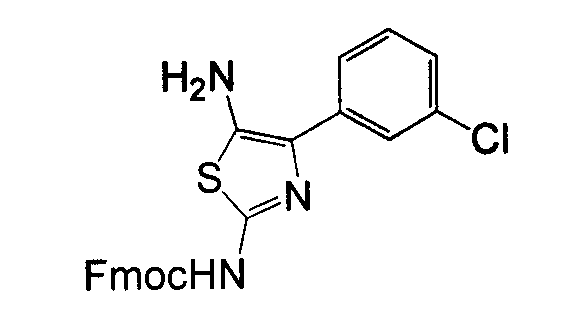
(9H-флуорен-9-ил)метил 5-амино-4-(2-хлорфенил)тиазол-2-илкарбамат получали согласно процедурам, аналогичным процедурам, описанным в примере 47.
Пример 53. 5-Амино-2-(1-имидазолил)-4-фенилтиазол

Суспензию α-аминофенилацетонитрил-гидрохлорида (5,01 г, 29,7 ммоль) и тиокарбонилдиимидазола (5,30 г, 29,7 ммоль) в DCM (100 мл) обрабатывали этилдиизопропиламином (5,69 мл, 32,7 ммоль) при температуре 0°C в течение 15 минут и затем при температуре окружающей среды в течение 3 часов. Далее смесь вливали в водный насыщенный бикарбонат натрия (50 мл) и воду (150 мл) и трижды экстрагировали в дихлорметане (общий объем 300 мл). Комбинированные органические фазы промывали насыщенным солевым раствором, сушили над сульфатом магния и концентрировали в вакууме, получая темно-коричневое масло (8,18 г). Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали 5-амино-2-(1-имидазолил)-4-фенилтиазол (2,47 г, 34%).
Пример 54. Амид, образованный пептидом MeAla-Chg-Pro и 2,5-диамино-4-фенилтиазолом
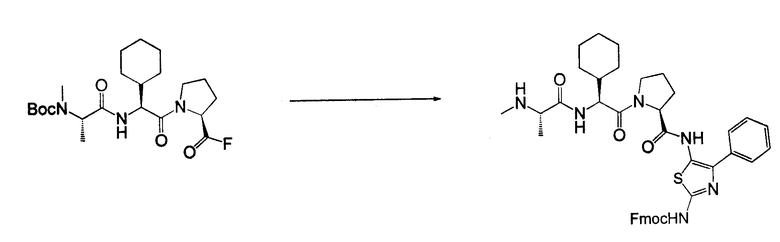
5-Амино-2-(Fmoc-амино)-4-фенилтиазол (250 мг, 605 мкмоль) обрабатывали фторангидридом (730 мкмоль; производная форма Boc-MeAla-Chg-Pro-OH, как описано ранее) и пиридином (147 мкл, 1,82 ммоль) в дихлорметане (2,0 мл) при температуре окружающей среды в течение 6 дней. Далее смесь вливали в водный насыщенный бикарбонат натрия и трижды экстрагировали в дихлорметане (общий объем 100 мл). Комбинированные органические фазы промывали соляным раствором, сушили над сульфатом магния и концентрировали в вакууме, получая неочищенный пептид амида в виде желтого масла (525 мг), которое впоследствии использовали без предварительной очистки.
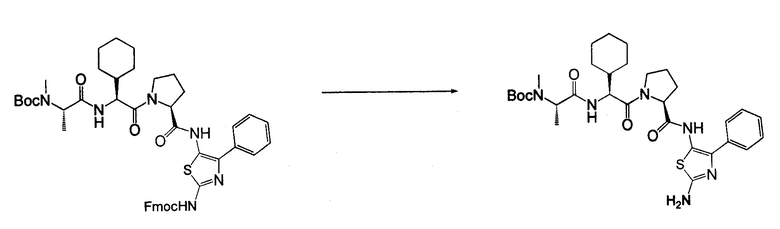
Неочищенный пептид амида в DMF (9,0 мл) обрабатывали пиперидином (1,0 мл) при температуре окружающей среды в течение 20 минут и затем концентрировали в вакууме. Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали 2,5-диамино-4-фенилтиазол-пептид амида (228 мг, 61% в две стадии).
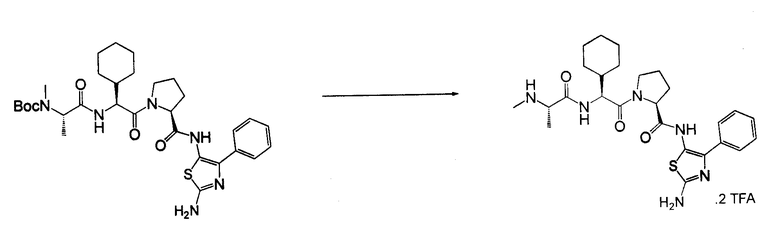
Неочищенный пептид амида (48 мг, 78 мкмоль) в дихлорметане (2,0 мл) обрабатывали трифторуксусной кислотой (2,0 мл) при температуре окружающей среды в течение 30 минут. Реакционную смесь концентрировали в вакууме, гомогенизировали дихлорметаном и снова концентрировали. Осадок очищали посредством препаративной HPLC (ацетонитрил/вода) с обращенной фазой, получая соль амида, образованного пептидом и трифторуксусной кислотой, с полным снятием защиты (42 мг, 73%), в виде белого аморфного твердого вещества.
Пример 55. Амид, образованный пептидом MeAla-Chg-Pro и 2,5-диамино-4-(3-хлорфенил)тиазолом
Амид, образованный пептидом MeAla-Chg-Pro и 2,5-диамино-4- (3-хлорфенил)тиазолом, получали, используя процедуры, описанные в примере 55.
Пример 56. Амид, образованный MeAla-Chg-Pro и 5-амино-2-(пивалоиламино)-4-фенилтиазолом
Boc-пептид-амино-тиазол (48 мг, 78 мкмоль) и этилдиизопропиламин (140 мкл, 0,80 ммоль) в дихлорметане (2,0 мл) обрабатывали пивалоилхлоридом (50 мкл, 0,40 ммоль) при температуре окружающей среды в течение 3 часов и затем насыщенным водным бикарбонатом натрия, далее трижды экстрагировали в дихлорметане (общий объем 60 мл). Комбинированные органические фазы промывали соляным раствором, сушили над сульфатом магния и концентрировали в вакууме. Полученное неочищенное масло обрабатывали трифторуксусной кислотой (5,0 мл) в дихлорметане (5,0 мл) при температуре окружающей среды в течение 20 минут. Реакционную смесь концентрировали в вакууме, гомогенизировали дихлорметаном и снова концентрировали. Осадок растворяли в водной уксусной кислоте (50%) для дальнейшего очищения посредством препаративной HPLC (ацетонитрил/вода) с обращенной фазой, получая на выходе чистую соль амида, образованного пептидом и трифторуксусной кислотой (38 мг, 68% в две стадии) в виде белого аморфного твердого вещества.
Пример 57. Амид, образованный MeAla-Chg-Pro и 5-амино-2-(пивалоиламино)-4-фенилтиазолом
Boc-пептид-амино-тиазол (38 мг, 62 мкмоль) и этилдиизопропиламин (107 мкл, 0,61 ммоль) в дихлорметане (2,0 мл) обрабатывали метансульфонилхлоридом (24 мкл, 0,31 ммоль) при температуре окружающей среды в течение 20 минут, затем насыщенным водным бикарбонатом натрия, далее трижды экстрагировали в дихлорметане. Комбинированные органические фазы промывали соляным раствором, сушили над сульфатом магния и концентрировали в вакууме. Полученное неочищенное масло обрабатывали трифторуксусной кислотой (4,0 мл) в дихлорметане (4,0 мл) при температуре окружающей среды в течение 20 минут. Реакционную смесь концентрировали в вакууме, гомогенизировали дихлорметаном и снова концентрировали. Осадок растворяли в водной уксусной кислоте (50%) для дальнейшего очищения посредством препаративной HPLC (ацетонитрил/вода) с обращенной фазой, получая на выходе чистую соль амида, образованного пептидом и трифторуксусной кислотой (11 мг, 23% в две стадии), в виде белого аморфного твердого вещества.
Пример 57. 2-(Ацетиламино)-4-амино-5-фенилтиазол
α-Бромфенилацетонитрил (1,08 г, 5,48 ммоль) в этаноле (10 мл) обрабатывали N-ацетилтиомочевиной (649 мг, 5,49 ммоль) при температуре окружающей среды в течение 4 часов, затем кипятили с обратным холодильником в течение 3,5 часов. Охлажденную смесь концентрировали в вакууме и затем распределяли между дихлорметаном и насыщенным водным бикарбонатом натрия. Органическую фазу промывали соляным раствором, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали 2-(ацетиламино)-4-амино-5-фенилтиазол (295 мг, 23%).
Пример 58. 2,5-Дифенилтиазол-4-амин

2,5-Дифенилтиазол-4-амин получали, используя процедуры, описанные в примере 57.
Пример 59. 5-Фенил-2-(пиразин-2-ил)тиазол-4-амин
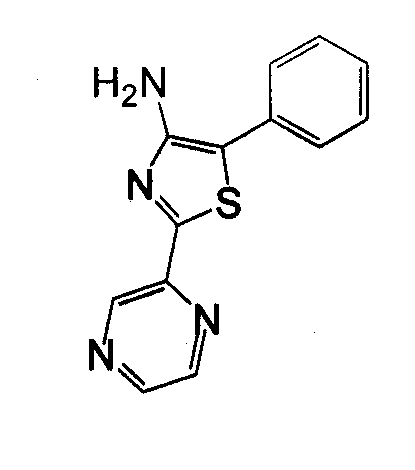
5-Фенил-2-(пиразин-2-ил)тиазол-4-амин получали, используя процедуры, описанные в примере 57.
Пример 60. 5-Амино-1-(3'-нитрофенил)пиразол

3-Нитрофенилгидразин-гидрохлорид (7,03 г, 36,3 ммоль), диизопропилэтиламин (9,5 мл, 54,5 ммоль) и этанол (60 мл) перемешивали в атмосфере азота при комнатной температуре в течение 2 часов. Далее добавляли этоксиметиленмалононитрил (4,52 г, 36,3 ммоль), после чего реакционную смесь кипятили с обратным холодильником в течение 1 часа. Температуру реакционной смеси доводили до комнатной температуры. Растворитель удаляли при пониженном давлении до тех пор, пока не выпадал осадок. Полученное твердое вещество фильтровали, получая 6,54 г циклизованного продукта (выход 78%).

5-Амино-1-(3'-нитрофенил)-4-цианопиразол (559 мг, 2,44 ммоль) и фосфорную кислоту (86%, 6 мл) кипятили с обратным холодильником при температуре 170°C в течение 15 часов. Температуру реакционной смеси доводили до комнатной температуры и нейтрализовали ее гидроксидом аммония. Органические фазы трижды экстрагировали диэтиловым эфиром (общий объем 40 мл), промывали соляным раствором и сушили над сульфатом магния. После удаления растворителя получали 5-амино-1-(3'-нитрофенил)пиразол в виде желтого порошка (398 мг, выход 80%).
Пример 61. 1-(2-Фторфенил)-1H-пиразол-5-амин

1-(2-Фторфенил)-1H-пиразол-5-амин получали, используя процедуры, описанные в примере 60.
Пример 62. 1-(3-Хлорфенил)-1H-пиразол-5-амин

1-(3-Хлорфенил)-1H-пиразол-5-амин получали, используя процедуры, описанные в примере 60.
Пример 63. 1-(3-Фторфенил)-1H-пиразол-5-амин

1-(3-Фторфенил)-1H-пиразол-5-амин получали, используя процедуры, описанные в примере 60.
Пример 64. 1-(3-Бромфенил)-1H-пиразол-5-амин
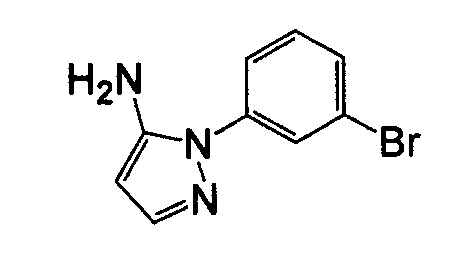
1-(3-Бромфенил)-1H-пиразол-5-амин получали, используя процедуры, описанные в примере 60.
Пример 65. 1-(3-Трихлорметилфенил)-1H-пиразол-5-амин
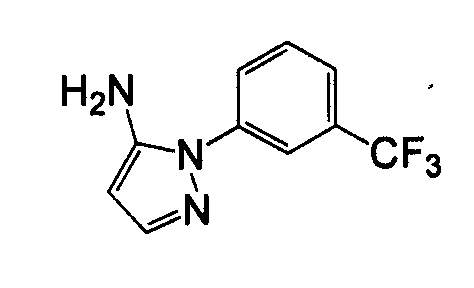
1-(3-Трихлорметилфенил)-1H-пиразол-5-амин получали, используя процедуры, описанные в примере 60.
Пример 66. 1-(Пиридин-2-ил)-1H-пиразол-5-амин
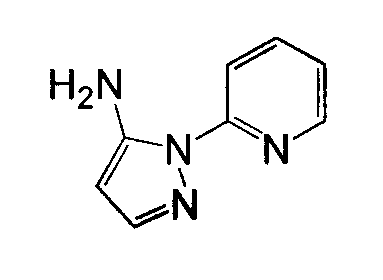
1-(Пиридин-2-ил)-1H-пиразол-5-амин получали, используя процедуры, описанные в примере 60.
Пример 67. 1-(3-Метоксифенил)-1H-пиразол-5-амин
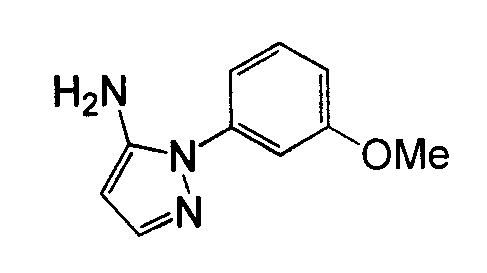
1-(3-Метоксифенил)-1H-пиразол-5-амин выделяли посредством дальнейшего децианилирования 5-амино-4-циано-1-(3'-метоксифенил)пиразола, полученного в примере 60.
Пример 67. 1-(3-Гидроксифенил)-1H-пиразол-5-амин
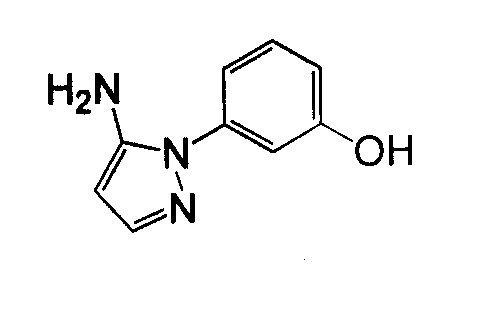
1-(3-Гидроксифенил)-1H-пиразол-5-амин получали, используя процедуры, описанные в примере 60.
Пример 68. 4-Амино-5-фенил-1,2,3-тиадиазол

Фенил-пировиноградную кислоту (25 г, 149 ммоль) и этилкарбазат (16 г, 149 ммоль) кипятили в бензоле (225 мл) с обратным холодильником в течение 2 часов и полученную смесь концентрировали в вакууме. Полученный неочищенный продукт растворяли в минимально теплом дихлорметане, получая на выходе гидразон в виде желтого осадка, который охлаждали до температуры окружающей среды, выделяли посредством фильтрации (30,4 г, 81%) и использовали без дальнейшей очистки.
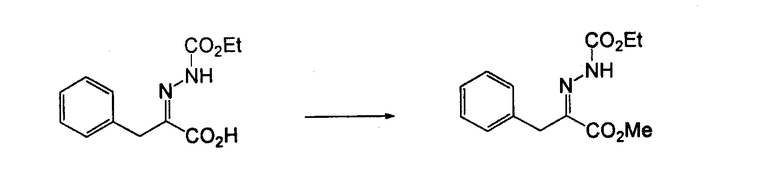
Диазометан получали, покапельно, в течение 45 минут добавляя раствор Diazald (N-метил-N-нитрозо-пара-толуолсульфонамида (18,6 г, 86,9 ммоль) в диэтиловом эфире (180 мл) к раствору гидроксида калия (18,2 г, 325 ммоль) в воде (37 мл) и 2-(2-этоксиэтокси)-этанола (37 мл) при температуре 65°C. В результате дистилляции получали эфирный раствор диазометана, который немедленно добавляли к перемешиваемому раствору гидразона (10,9 г, 43,5 ммоль) в метаноле (150 мл) при температуре 0°C. Эту систему промывали избыточным диэтиловым эфиром до тех пор, пока дистиллят не становился чистым, далее смесь обрабатывали уксусной кислотой (1 мл) и концентрировали в вакууме. Полученное масло распределяли между этилацетатом (200 мл) и бикарбонатом натрия (200 мл) и органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая на выходе сложный метиловый эфир в виде желтого твердого вещества (10,2 г, 89%).

Сложный гидразон-метиловый эфир (10,2 г, 38,6 ммоль) обрабатывали тионилхлоридом (25 мл, 343 ммоль) при температуре окружающей среды в течение 24 часов и смесь концентрировали в вакууме. Кристаллизация из гексана давала на выходе тиадиазол-сложный метиловый эфир (4,81 г, 56%).

Сложный тиадиазол-метиловый эфир (2,79 г, 12,7 ммоль) обрабатывали гидратом гидразина (1,09 мл, 93,9 ммоль) в метаноле (50 мл) при температуре окружающей среды в течение 24 часов и полученный белый осадок собирали посредством фильтрации. Повторная кристаллизация из изопропанола давала на выходе тиадиазол-гидразид (3,99 г, 83%).

Раствор тиадиазол-гидразида (3,99 г, 18,1 ммоль) в воде (40 мл) и концентрированную соляную кислоту (1,8 мл, 21,9 ммоль) покапельно обрабатывали раствором нитрита натрия (1,52 г, 21,3 ммоль) в воде (15 мл) при температуре 0°C в течение 2 часов. Полученный осадок собирали фильтрованием, получая на выходе азид тиадиазолсодержащей кислоты в виде беловатого твердого вещества (3,95 г, 94%).

Согласно процедурам, описанным в публикации K. Masuda et al.; Chem. Pharm. Bull., 1981, 29, 1743-1747, азид тиадиазол-содержащей кислоты (3,95 г, 17,1 ммоль) кипятили с обратным холодильником в этаноле (40 мл) в течение 45 минут, далее смесь концентрировали в вакууме. Кристаллизация из бензола давала на выходе этилкарбамат (3,37 г, 74%).

Этилкарбамат (399 мг, 1,60 ммоль) и бромистый водород в уксусной кислоте (3 мл, 30% вес) нагревали в запечатанном сосуде при температуре 80°C в течение 18 часов. Охлажденную смесь распределяли между этилацетатом (15 мл) и водой (15 мл) и органическую фазу концентрировали в вакууме. Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали на выходе 4-амино-5-фенил-1,2,3-тиадиазол (136 мг, 49%).
Пример 69. 4-Амино-5-фенилизоксазол

5-Фенил-4-изоксазолкарбоновую кислоту (460 мг, 2,36 ммоль) и тионилхлорид (1,71 мл, 23,6 ммоль) кипятили с обратным холодильником в течение 3 часов и реакционную смесь концентрировали в вакууме, получая на выходе хлорид кислоты, который использовали без очистки.

Неочищенный хлорид кислоты в ацетоне (7 мл) обрабатывали раствором азида натрия (165 мг, 2,62 ммоль) в воде (2 мл) при температуре 0°C в течение 1,5 часов, далее температуру реакционной смеси доводили до температуры окружающей среды и концентрировали смесь в вакууме. Полученное белое твердое вещество промывали водой и сушили над вакуумом и использовали в дальнейшем без очистки.

Азид кислоты (409 мг, 1,91 ммоль) кипятили с обратным холодильником в метаноле в течение 6 часов, далее смесь концентрировали в вакууме, получая на выходе метилкарбамат в виде белого твердого вещества, которое использовали в дальнейшем без очистки.

Метилкарбамат (378 мг, 1,73 ммоль) обрабатывали гидробромной кислотой (13 мл, 48% вес, 115 ммоль), гомогенизировали уксусной кислотой (2 мл), нагревали при температуре 65°C в течение 48 часов и давали остыть. Смесь нейтрализовывали водным гидроксидом натрия и экстрагировали с этилацетатом (2 × 125 мл). Комбинированные органические фазы сушили над сульфатом натрия и концентрировали в вакууме, получая на выходе 4-амино-5-фенилизоксазол в виде белого твердого вещества (193 мг, 70%).
Пример 70. Синтез 5-алкил-2-амино-3-фенилтиофена

Бензилцианид (2,33 мл, 20 ммоль) обрабатывали основанием Verkade (2,8,9-триметил-2,5,8,9-тетрааза-1-фосфабицикло[3.3.3]ундекан; 441 мг, 2,0 ммоль) и 3,3-диметилбутиральдегидом (2,64 мл, 200 ммоль) в метаноле (4 мл) и реакционную смесь нагревали в запечатанном сосуде при температуре 45°C в течение 16 часов. Охлажденный раствор концентрировали в вакууме, получая на выходе ненасыщенный нитрил в виде бесцветного масла, которое использовали без очистки.

Нитрил (10,0 ммоль), карбонат калия (2,34 г, 23,4 ммоль) и измельченную в пудру серу (330 мг, 10,3 ммоль) в этаноле (2 мл) нагревали в запечатанном сосуде при температуре 160°C в течение 24 часов. Охлажденный раствор разбавляли водой, дважды экстрагировали в диэтиловом эфире и комбинированные органические фазы концентрировали в вакууме. Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали на выходе 5-амино-2-трет-бутил-4-фенилтиазол (75%).
Пример 71. 5-Метил-3-фенилтиофен-2-амин
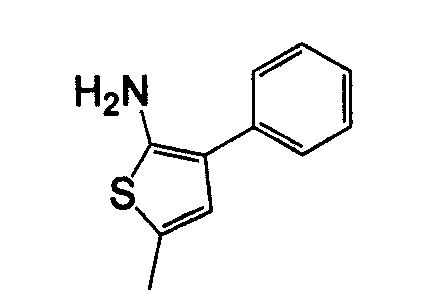
5-Метил-3-фенилтиофен-2-амин получали, используя процедуры, описанные в примере 70.
Пример 72. 5-Изопропил-3-фенилтиофен-2-амин
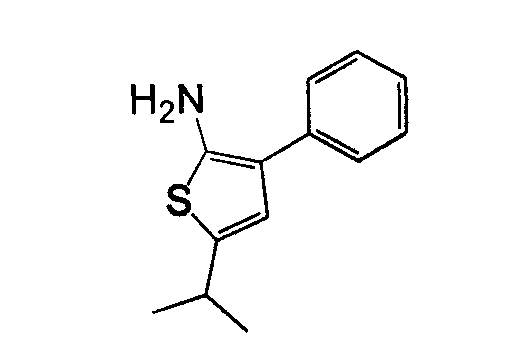
5-Изопропил-3-фенилтиофен-2-амин получали, используя процедуры, описанные в примере 70.
Пример 73. 2-Амино-5-хлор-3-фенилтиофен

2-Амино-3-фенилтиофен (12,0 ммоль) в THF (7 мл) обрабатывали ди-трет-бутилдикарбонатом (2,97 г, 13,3 ммоль) и диизопропилэтиламином (3,15 мл, 18,1 ммоль) при температуре окружающей среды в течение 60 часов и реакционную смесь концентрировали в вакууме. Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали на выходе 2-(N-Boc-амино)-3-фенилтиофен (1,98 г, 59%).

К 2-(N-Boc-амино)-3-фенилтиофену (89 мг, 0,32 ммоль) в дихлорметане (4 мл) при температуре 0°C медленно добавляли N-хлорсукцинимид (48 мг, 0,36 ммоль), затем давали смеси нагреться до температуры окружающей среды в течение 16 часов. Далее смесь разбавляли дихлорметаном, промывали водой и органические фазы концентрировали в вакууме. Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали на выходе 2-(N-Boc-амино)-5-хлор-3-фенилтиофен (66 мг, 66%).

2-(N-Boc-амино)-5-хлор-3-фенилтиофен (66 мг, 0,21 ммоль) обрабатывали трифторуксусной кислотой (1 мл) в дихлорметане (3 мл) при температуре окружающей среды в течение 1 часа. Реакционную смесь разбавляли DMF (1 мл) и летучие материалы удаляли при пониженном давлении. Полученный раствор 2-амино-5-хлор-3-фенилтиофена в DMF использовали в последующей стадии связывания без очистки.
Пример 74. 1-Метил-4-(метиламино)-3-фенилпиразол

К 1-метил-4-амино-3-фенилпиразолу (572 мг, 3,30 ммоль) и ди-трет-бутилдикарбонату (799 мг, 3,66 ммоль) в THF (10 мл) и воде (3 мл) покапельно добавляли насыщенный водный бикарбонат натрия (3 мл, 1,2 M, 3,6 ммоль). Полученную смесь перемешивали при температуре окружающей среды в течение 7 часов и затем вливали в водную лимонную кислоту (0,5 M) и трижды экстрагировали в эфире (общий объем 100 мл). Комбинированные органические фазы промывали насыщенным водным бикарбонатом натрия и соляным раствором, сушили над сульфатом магния и концентрировали в вакууме, получая на выходе неочищенный карбамат в виде коричневого масла (920 мг), которое использовали в дальнейшем без очистки.

Суспензию гидрита натрия в минеральном масле (327 мг, 60% вес, 8,18 ммоль) промывали THF (2 × 5 мл) и суспендировали в THF (3,0 мл) при температуре 0°C. К этой смеси добавляли покапельно пиразол (744 мг, 2,72 ммоль) в THF (5,0 мл) и через 15 минут добавляли метилйодид (187 мкл, 3,00 ммоль). Далее выдерживали смесь при температуре 0°C следующие 30 минут, затем позволяли смеси нагреться до температуры окружающей среды в течение 18 часов и далее обрабатывали реакционную смесь насыщенным водным хлоридом аммония и достаточным для растворения твердых веществ количеством воды. Смесь трижды экстрагировали в эфире (общий объем 120 мл) и комбинированные органические фазы промывали соляным раствором, сушили над сульфатом магния и концентрировали в вакууме, получая на выходе неочищенный N-метилкарбамат в виде янтарного масла (750 мг, 96%), которое использовалось в дальнейшем без очистки.
Неочищенный N-метилкарбамат в DCM (1,0 мл) обрабатывали трифторуксусной кислотой (1,0 мл) при температуре окружающей среды в течение 40 минут. Реакционную смесь концентрировали в вакууме, гомогенизировали дихлорметаном и снова концентрировали, получая на выходе в основном чистый 1-метил-4-(метиламино)-3- фенилпиразол (150 мг, колич.) в виде коричневого масла.
Пример 75. N-метил-4-фенил-1,2,3-тиадиазол-5-амин
N-метил-4-фенил-1,2,3-тиадиазол-5-амин получали, используя процедуры, описанные в примере 74.
Пример 76. 1-трет-бутил-4-амино-3-фенилпиразол и 1-трет-бутил-4-амино-5-фенилпиразол

Раствор 2-бромацетофенона (30,0 г, 151 ммоль) в DMF (120 мл) порционно обрабатывали фталимидом калия (30,8 г, 166 ммоль) при температуре окружающей среды, затем нагревали до температуры 40°C в течение 3,5 часов. Охлажденную смесь вливали в воду (600 мл) и экстрагировали хлороформом (300 мл затем 100 мл). Комбинированные органические фазы промывали гидроксидом натрия (200 мл, 0,2 н.), водой (2 × 100 мл), соляным раствором (100 мл), сушили над сульфатом магния и концентрировали в вакууме. Полученное пастообразное твердое вещество суспендировали в эфире (100 мл), собирали посредством фильтрации, промывали эфиром (100 мл) и сушили в вакууме, получая на выходе чистый 2-фталимидоацетофенон в виде белого твердого вещества (34,3 г, 86%).

Согласно процедурам, описанным у C. Chen, K. Wilcoxen, J. R. McCarthy; Tetrahedron Lett, 1988, 39, 8229-8232, суспензию 2- фталимидоацетофенона (13,3 г, 50,0 ммоль) в диметилформамид-диметилацетале (26,7 мл, 200 ммоль) кипятили с обратным холодильником в течение 28 часов и концентрировали в вакууме. Полученное янтарное масло кристаллизовали из изопропанола (100 мл) и промывали изопропанолом (2 × 5 мл), получая на выходе 3-(диметиламино)-1-фенил-2-фталимид-2-пропен-1-он в виде желтых игл (13,7 г, 85%).
Смесь 3-(диметиламино)-1-фенил-2-фталимид-2-пропен-1-она (3,00 г, 9,38 ммоль) и трет-бутилгидразингидрохлорида (1,29 г, 10,3 ммоль) в этаноле (94 мл) и воде (9,4 мл) перемешивали при температуре окружающей среды в течение 64 часов и затем кипятили с обратным холодильником в течение 24 часов. Охлажденную смесь обрабатывали гидразином (590 мкл, 18,8 ммоль) и снова кипятили с обратным холодильником в течение 75 минут. Образование осадка происходило в процессе охлаждения при температуре окружающей среды. Смесь фильтровали, твердое вещество промывали смесью этанола (5 мл) и воды (0,5 мл) и фильтрат концентрировали в вакууме. Полученный осадок распределяли между эфиром (250 мл) и насыщенным водным бикарбонатом натрия (50 мл), разбавляли водой (100 мл) и водную фазу дважды экстрагировали эфиром (2 × 50 мл). Комбинированные органические фазы промывали соляным раствором, сушили над сульфатом натрия и концентрировали в вакууме, получая блекло-белое твердое вещество (1,92 г). Применяя флэш-хроматографию на силикагеле (этилацетат/гексан), получали на выходе 1-трет-бутил-4-амино-3-фенилпиразол (1,52 г, 75% в 2 стадии) и 1-трет-бутил-4-амино-5-фенилпиразол (114 мг, 6% в 2 стадии).
Пример 77. 1-(2,2,2-Трифторэтил)-3-фенил-1H-пиразол-4-амин и 1-(2,2,2-трифторэтил)-5-фенил-1H-пиразол-4-амин
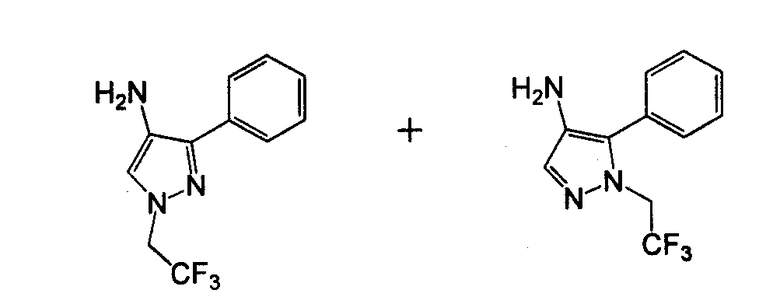
1-(2,2,2-Трифторэтил)-3-фенил-1H-пиразол-4-амин и 1-(2,2,2-трифторэтил)-5-фенил-1H-пиразол-4-амин получали аналогично из 2,2,2-трифторэтилгидразина согласно процедурам, описанным в примере 76.
Пример 78. Анализы ингибирования IAP
В следующих экспериментах используется химерный домен BIR, обозначаемый MLXBIR3SG, в котором 11 из 110 остатков соответствуют остаткам, обнаруженным в XIAP-BIR3, тогда как остальные соответствуют ML-IAP-BIR. Было показано, что химерный белок MLXBIR3SG значительно лучше связывает и ингибирует каспазу-9, чем любой из природных доменов BIR, за исключением связывания пептидов на основе Smac и связывания зрелого Smac, аффинности которых были подобны аффинностям нативного ML-IAP-BIR. Улучшенное ингибирование каспазы-9 химерным BIR-доменом белка MLXBIR3SG коррелировало с повышенным ингибированием доксорубицин-индуцированного апоптоза при трансфекции в клетках MCF7.
Последовательность MLXBIR3SG:
MGSSHHHHHHSSGLVPRGSHMLETEEEEEEGAGATLSRGPAFPGMGSEELRLASFYDWP LTAEVPPELLAAAGFFHTGHQDKVRCFFCYGGLQSWKRGDDPWTEHAKWFPGCQFLLR SKGQEYINNIHLTHSL (SEQ ID NO: 1)
TR-FRET-анализ связывания пептидов
Эксперименты по конкурентному резонансному переносу энергии флуоресценции с временным разрешением (TR-FRET) были предприняты на установке Wallac Victor2 Multilabeled Counter Reader (Perkin Elmer Life and Analytical Sciences, Inc.) в соответствии с методикой, описанной Kolb et al. (Journal of Biomolecular Screening, 1996, 1(4):203). Смесь реагентов, содержащую 300 нM his-меченного MLXBIR3SG; 200 нM биотинилированного пептида SMAC (AVPI); 5 мкг/мл анти-his-аллофикоцианина (XL665) (CISBio International); и 200 нг/мл стрептавидин-европия (Perkin Elmer), готовили в реагентном буфере (50 мM Tris [pH 7,2], 120 мM NaCl, 0,1% бычьих глобулинов, 5 мM DTT и 0,05% октилглюкозида). (Альтернативно эту смесь можно приготовить, используя меченный европием анти-His (Perkin Elmer) и стрептавидин-аллофикоцианин (Perkin Elmer) в концентрациях порядка 6,5 нM и 25 нM соответственно). Реагентную смесь инкубировали при комнатной температуре в течение 30 минут. После инкубации смесь добавляли к серийным разведениям 1:3 соединения-антагониста (начальная концентрация порядка 50 мкM) в 384-луночных черных планшетах FIA (Greiner Bio-One, Inc.). После 90-минутной инкубации при комнатной температуре измеряли флуоресценцию, используя фильтры [в области длины волны] возбуждения европия (340 нм) и [в области длины волны] испускания европия (615 нм) и аллофикоцианина (665 нм). Данные по антагонистической [активности] вычисляли в виде отношения сигнала эмиссии (испускания) аллофикоцианина при 665 нм к величине сигнала европия при 615 нм (эти отношения умножали на коэффициент 10000 для облегчения расчетов с полученными данными). Полученные в результате значения наносили на график в виде функции концентрации антагониста и подставляли в четырехпараметрическое уравнение, используя компьютерную программу Kaleidograph (Synergy Software, Reading, PA). Показатели уровня антагонистической активности определяли из значений IC50. Было обнаружено, что соединения согласно изобретению обладают ингибиторной активностью IAP, которая была продемонстрирована в настоящем анализе.
Анализ связывания пептидов методом поляризации флуоресценции
Поляризационные эксперименты были предприняты на установке Analyst HT 96-384 (Molecular Devices Corp.) в соответствии с методикой, описанной Keating, S.M., Marsters, J, Beresini, M., Ladner, C, Zioncheck, K., Clark, K., Arellano, F., and Bodary., S.(2000) in Proceedings of SPIE: In Vitro Diagnostic Instrumentation (Cohn, G.E., Ed.) pp 128-137, Bellingham, WA. Образцы для измерения аффинности поляризации флуоресценции были получены путем добавления серийных разведений 1:2, начиная с конечной концентрации 5 мкM MLXBIR3SG в поляризационном буфере (50 мM Tris [pH 7,2], 120 мM NaCl, 1% бычьих глобулинов, 5 мM DTT и 0,05% октилглюкозида), к 5-карбоксифлуоресцеин-конъюгированному AVPdi-Phe-NH2 (AVP-diPhe-FAM) в конечной концентрации 5 нM.

Образец AVP-diPhe-FAM
Реакции считывали после 10-минутной инкубации при комнатной температуре, используя стандартные отсекающие фильтры для флуоресцеин-флуорофора (λex = 485 нм; λem = 530 нм) в 96-луночных черных планшетах HE96 (Molecular Devices Corp.). Значения флуоресценции наносили на график в виде функции концентрации белка и значения IC50 получали, подставляя полученные данные в четырехпараметрическое уравнение, используя компьютерную программу Kaleidograph (Synergy software, Reading, PA). Эксперименты по конкурентному анализу были предприняты путем добавления MLXBIR3SG при 30 нM к лункам, содержащим 5 нM образца AVP-diPhe-FAM, а также путем серийных разведений 1:3 соединений антагониста, начиная с концентрации в 300 мкM в поляризационном буфере. Считывание образцов производили после 10-минутной инкубации. Значения поляризации флуоресценции наносили на график в виде функции концентрации антагониста, и значения IC50 были получены в результате подстановки полученных данных в четырехпараметрическое уравнение с использованием компьютерной программы Kaleidograph (Synergy software, Reading, PA). Константы ингибирования (K i) для антагонистов определяли из значений IC50. Было обнаружено, что соединения согласно изобретению обладают ингибиторной активностью IAP, которая была продемонстрирована в настоящем анализе.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
| МОДУЛИРУЮЩИЕ JAK КИНАЗУ ХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2529019C2 |
| ИНГИБИТОРЫ IAP | 2013 |
|
RU2728789C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| Бициклические гетероциклические соединения и их применения в терапии | 2012 |
|
RU2662827C2 |
| 19-НОР C3, 3-ДИЗАМЕЩЕННЫЕ C21-N-ПИРАЗОЛИЛЬНЫЕ СТЕРОИДЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2675855C2 |
| ИНГИБИТОРЫ IAP | 2013 |
|
RU2593259C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ОБЕСПЕЧИВАЮЩИЕ РАЗРУШЕНИЕ БЕЛКА IKAROS И БЕЛКА AIOLOS | 2020 |
|
RU2833608C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| СПОСОБ ЛЕЧЕНИЯ АЛЛЕРГИИ С ИСПОЛЬЗОВАНИЕМ ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2001 |
|
RU2277909C2 |

Настоящее изобретение относится к новым ингибиторам IAP, которые могут использоваться в качестве терапевтических агентов для лечения злокачественных опухолей, где соединения имеют общую формулу I,
в которой X, Y, A, R1, R2, R3, R4, R4', R5, R5', R6 и R6' имеют такие же значения, которые указаны в описании изобретения. 2 н. и 14 з.п. ф-лы.
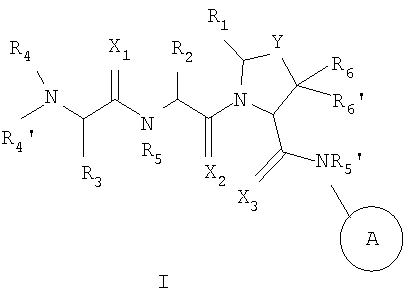
1. Соединение формулы I:
где каждый из X1, X2 и Х3 независимо представляет собой О;
Y представляет собой (CHR7)n или S; где n=1, и R7 представляет собой Н или С6-С10арилС1-С6алкилокси;
А представляет собой 5-членный гетероцикл, содержащий от 1 до 4 гетероатомов, выбранных из азота, серы или кислорода, необязательно замещенный амино, галогеном, насыщенным C1-С6алкилом, С6-С10арилом, ациламино, C1-С6алкоксикарбониламино, C1-С6алкилтио, C1-С6алкилсульфинилом, C1-С6алкилсульфонилом, аминосульфонилом, С1-С6алкилсульфониламино или гетероциклом, представляющим собой моно или бициклическое, насыщенное или ненасыщенное, ароматическое или неароматическое кольцо, имеющее 5-10 кольцевых атомов, и содержащее 1-2 гетероатома, выбранных из N и О; где каждое алкильное замещение необязательно замещено галогеном, и арильное замещение необязательно замещено гидроксилом, галогеном, галогеналкилом, алкокси, нитро, циано;
R1 представляет собой Н, или R1 и R2 вместе образуют 5-8 членное насыщенное кольцо;
R2 представляет собой C1-С6алкил, необязательно замещенный гидроксилом, меркапто, галогеном, амино, C1-С6алкилом, галогеналкилом, C1-С6алкокси или C1-С6алкилтио; С3-С6циклоалкил, необязательно замещенный гидроксилом, галогеном; С3-С6циклоалкилС1-С6алкил; С6-С10арил, необязательно замещенный галогеном; гетероцикл, представляющий собой тиофен; или насыщенный гетероциклический алкил, представляющий собой 6-ти членное кольцо, содержащее 1-2 гетероатома, выбранных из N и О, который необязательно замещен карбоксилом;
R3 представляет собой Н или насыщенный C1-С6алкил;
каждый из R4 и R4' независимо представляет собой Н;
насыщенный C1-С6алкил, необязательно замещенный гидроксилом;
С10арилС1-С6алкил; С3-С6циклоалкилС1-С6алкил; гетероарилалкил, где гетероарильная группа представляет собой ароматическое 6-ти членное кольцо, содержащее 1-2 атомов азота;
каждый из R5, и R5' независимо представляет собой Н или C1-С6алкил;
каждый из R6, и R6' независимо представляет собой Н, C1-С6алкил;
а также их фармацевтически приемлемые соли.
2. Соединение по п.1, имеющее следующую стереохимическую конфигурацию формулы I'
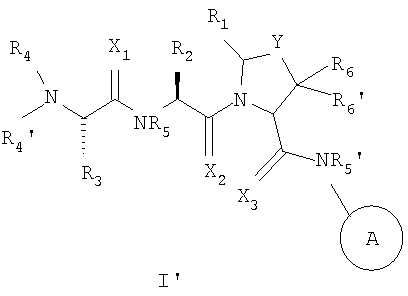
где X, Y, A, R1, R2, R3, R4, R4', R5, R5', R6 и R6' имеют значения, определенные в п.1.
3. Соединение по п.2, где кольцо А имеет формулу IIа или IIb:
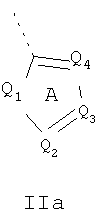
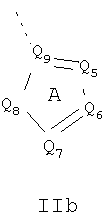
где Q1 представляет собой NR8, О или S; каждый из Q2, Q3, Q4, Q5, Q6, Q7, и Q8 независимо представляет собой CR9 или N; где R9 представляет собой Н, амино, галоген, насыщенный C1-С6алкил, С6-С10арил, ациламино или гетероцикл, представляющий собой моно или бициклическое, насыщенное или ненасыщенное, ароматическое или неароматическое кольцо, имеющее 5-10 кольцевых атомов, и содержащее 1-2 гетероатома, выбранных из N и О; где каждое алкильное замещение необязательно замещено галогеном, и арильное замещение необязательно замещено гидроксилом, галогеном, галогеналкилом, алкокси, нитро;
R8 представляет собой Н, насыщенный C1-С6алкил, С6-С10арил, ациламино или гетероцикл, представляющий собой моно или бициклическое, насыщенное или ненасыщенное, ароматическое или неароматическое кольцо, имеющее 5-10 кольцевых атомов, и содержащее 1-2 гетероатома, выбранных из N и О; где каждое алкильное замещение необязательно замещено галогеном, и арильное замещение необязательно замещено гидроксилом, галогеном, галогеналкилом, алкокси, нитро; и Q9 представляет собой СН или N.
4. Соединение по п.2, где кольцо А выбрано из группы, состоящей из следующего:
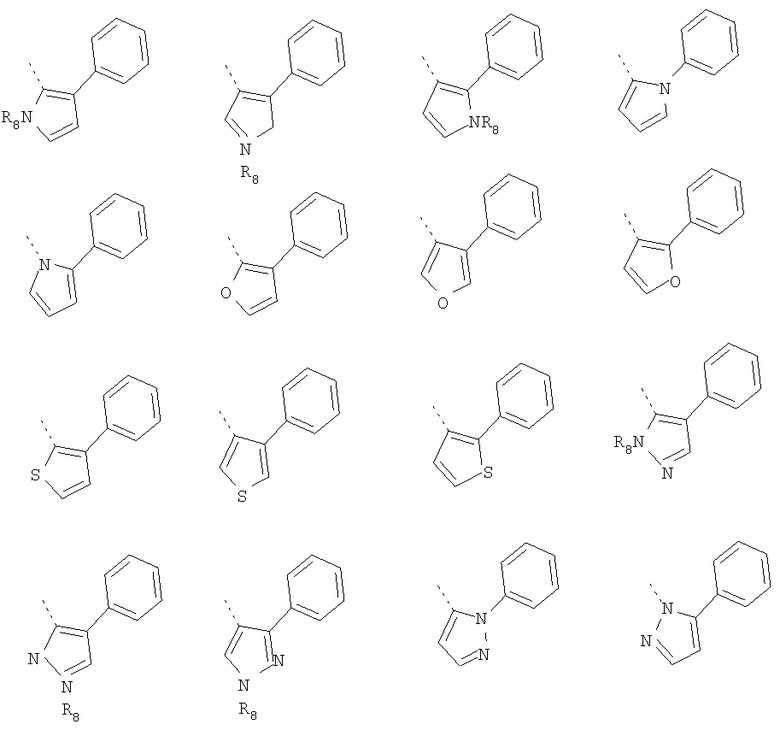
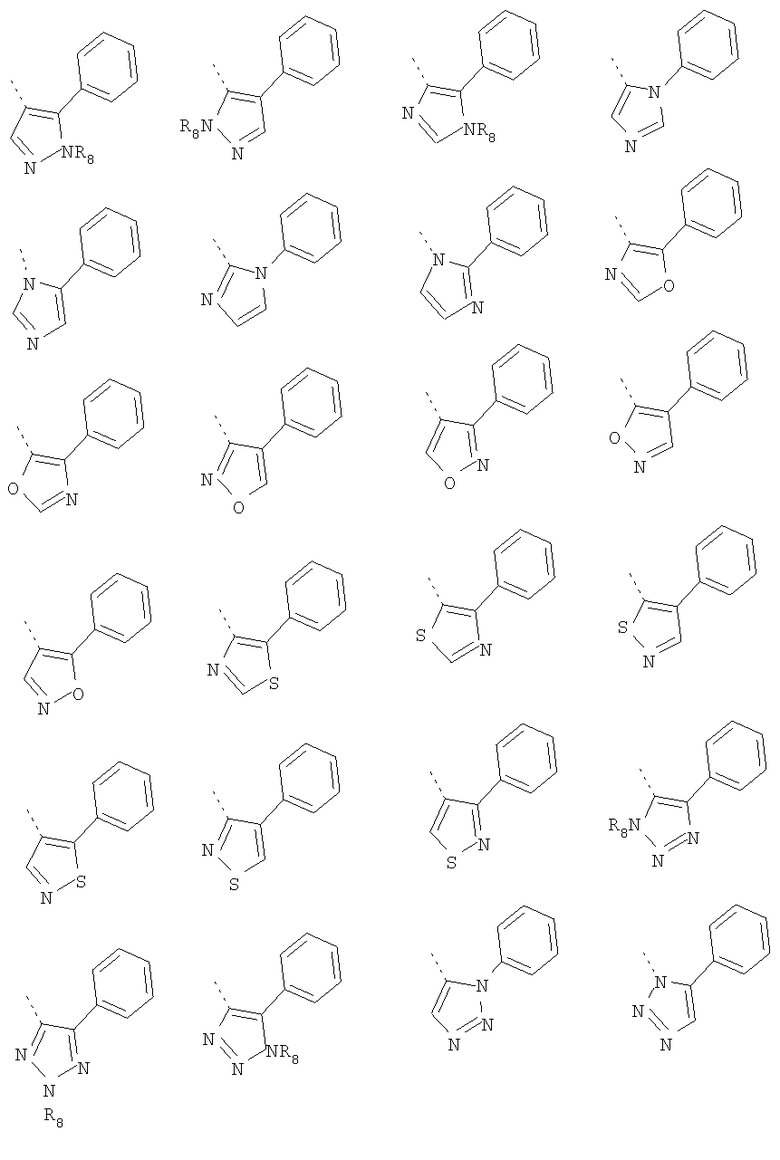
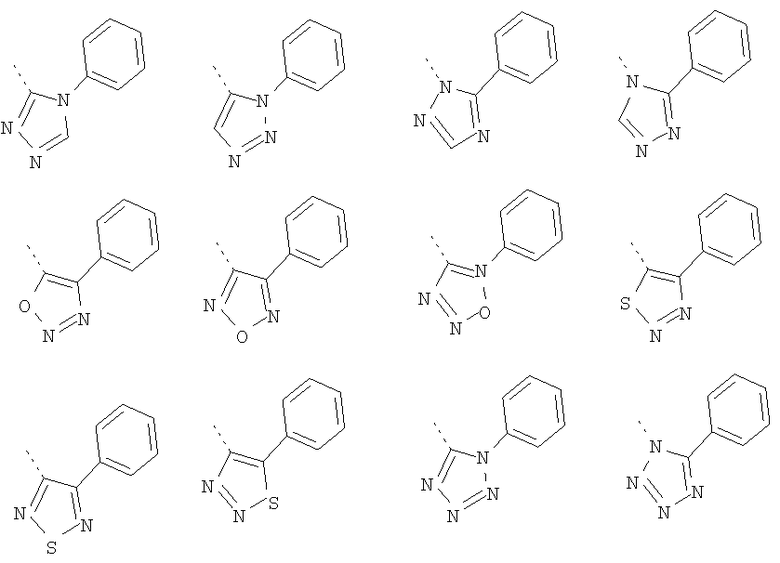
где R8 представляет собой Н, C1-С6алкил.
5. Соединение по п.2, где R8 представляет собой Н.
6. Соединение по п.2, где R1 и R2 вместе образуют 5-8 членное насыщенное кольцо.
7. Соединение по п.2, где R1 представляет собой Н.
8. Соединение по п.2, где R2 представляет собой C1-С6алкил или С3-С6циклоалкил.
9. Соединение по п.2, где R2 представляет собой изопропил, трет-бутил, циклогексил, фенил или тетрагидропиран-4-ил.
10. Соединение по п.2, где R3 представляет собой метил.
11. Соединение по п.2, где R4 представляет собой Н или метил, и R4' представляет собой Н.
12. Соединение по п.2, где каждый из R5 и R5' независимо представляет собой Н или метил.
13. Соединение по п.2, где каждый из R6 и R6' независимо представляет собой Н или метил.
14. Соединение по п.2, где каждый из X1, Х2 и Х3 представляет собой О.
15. Соединение по п.2, где R1 представляет собой Н; R2 представляет собой изопропил, трет-бутил, циклогексил, фенил или тетрагидропиран-4-ил; R3 представляет собой метил; R4 представляет собой Н или метил, и R4' представляет собой Н, R5 и R5' представляют собой Н или метил; X1, Х2 и Х3 представляют собой О.
16. Соединение представляет собой:

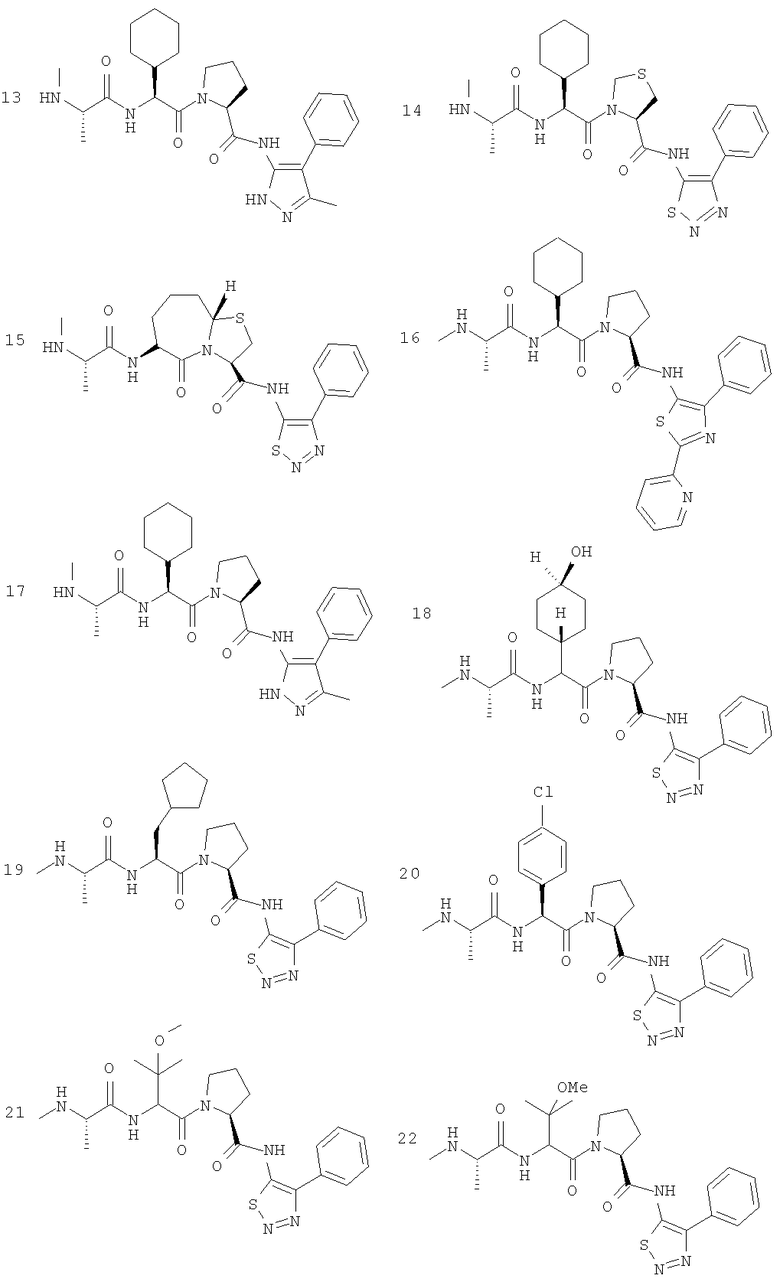

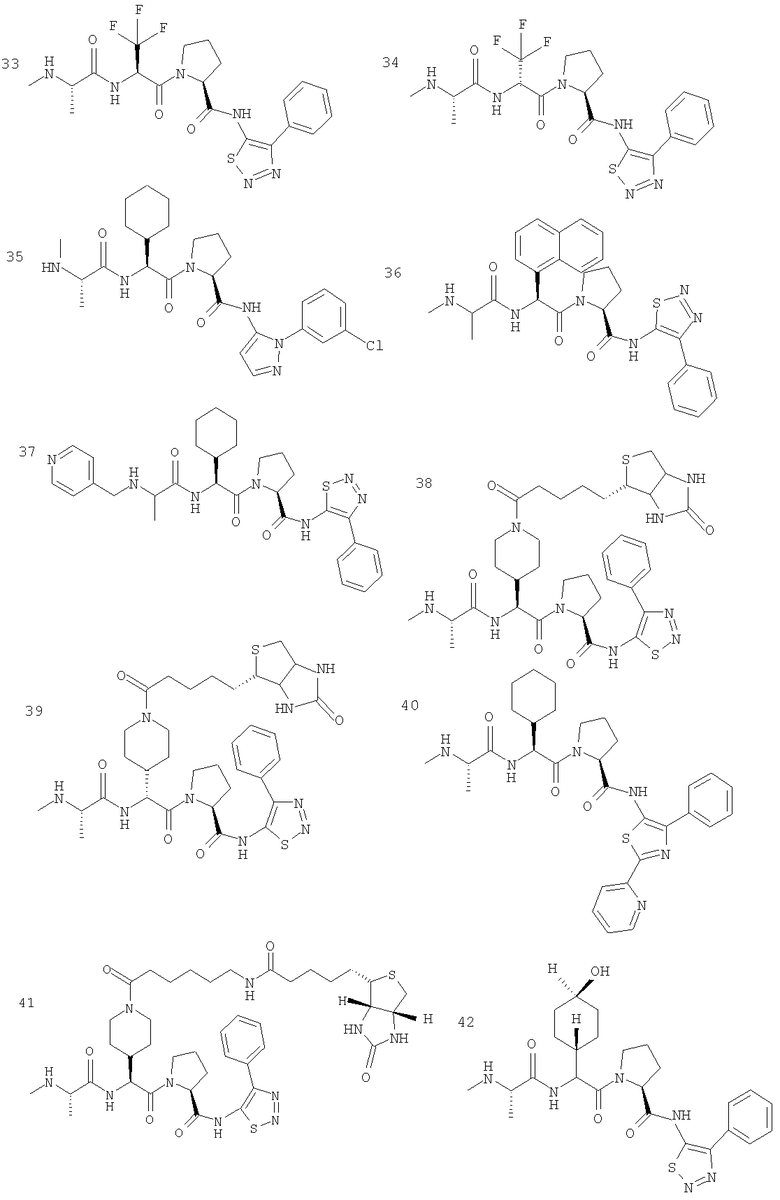
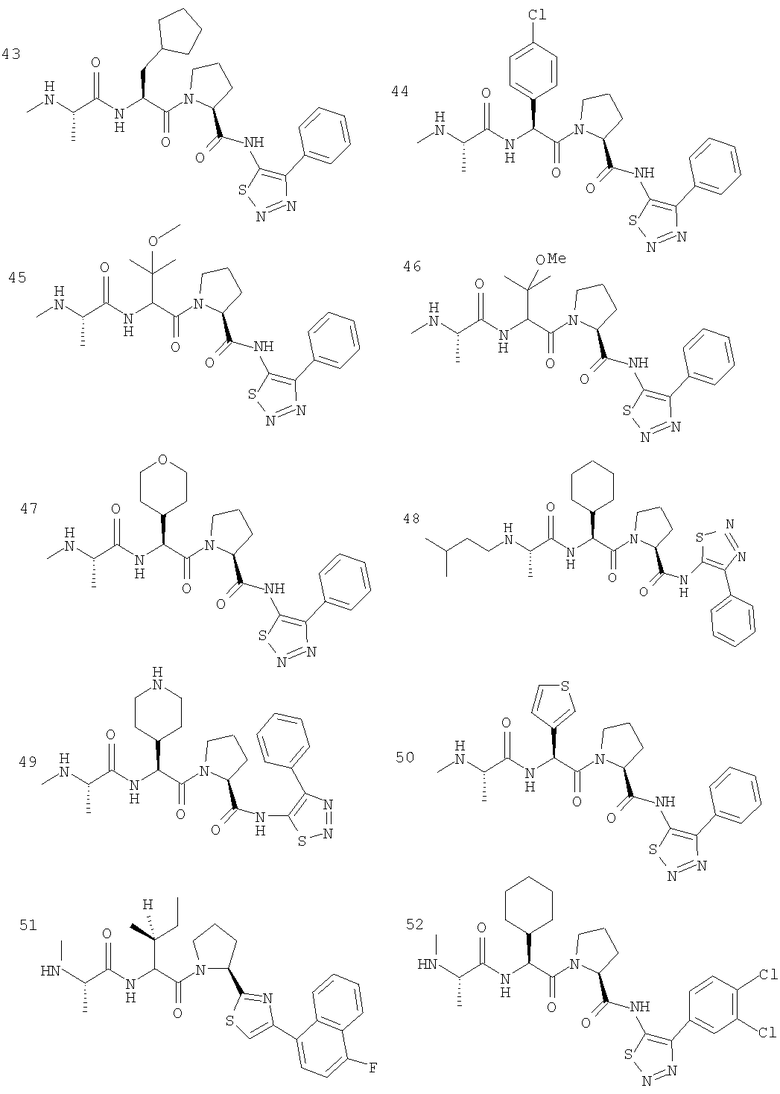
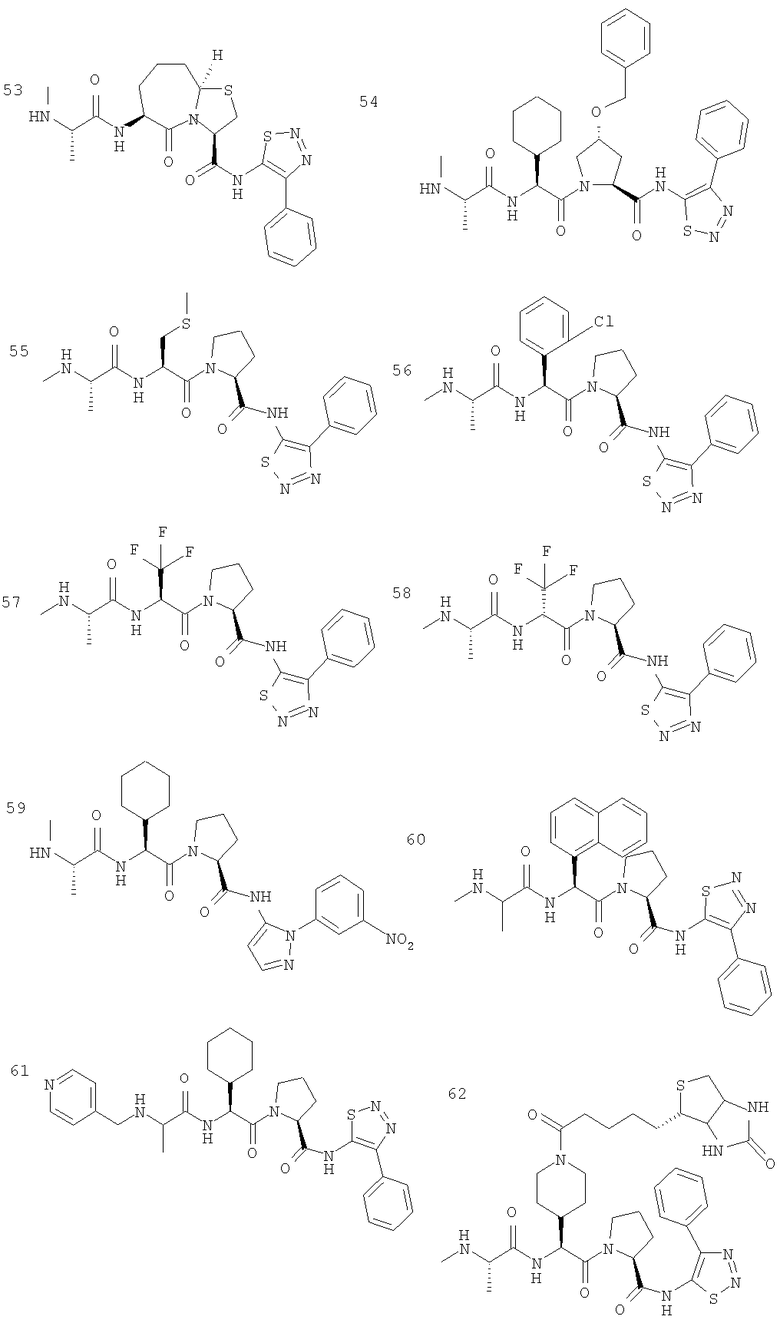

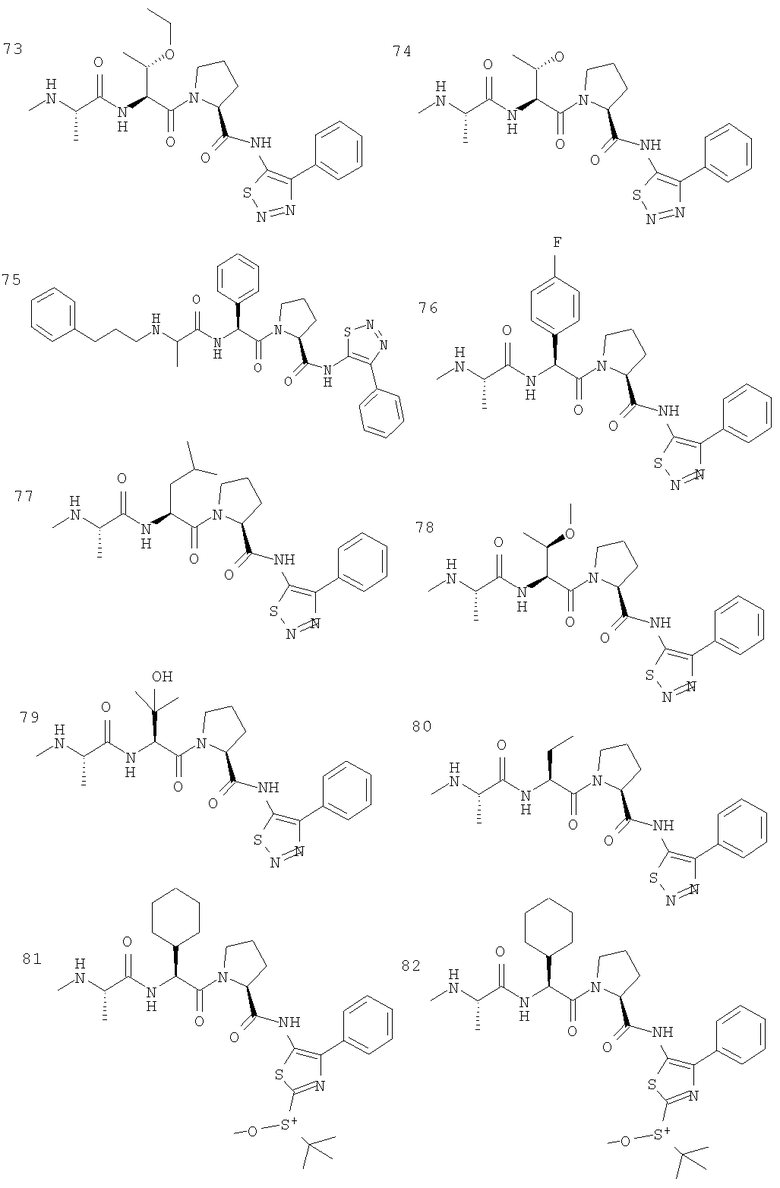
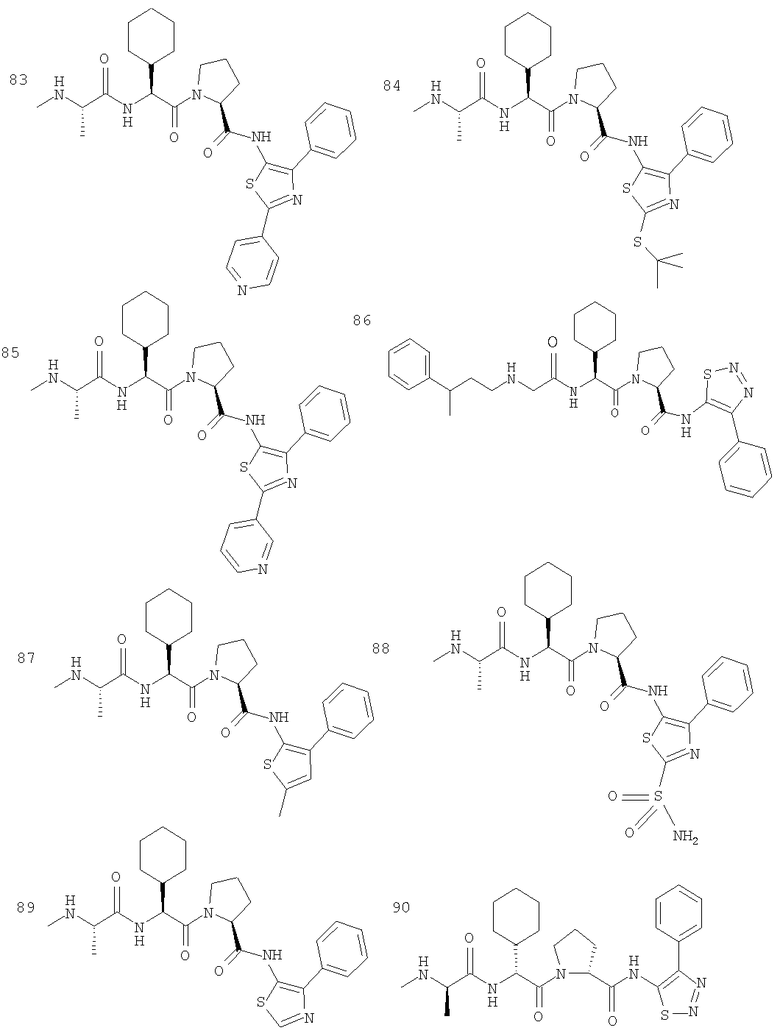
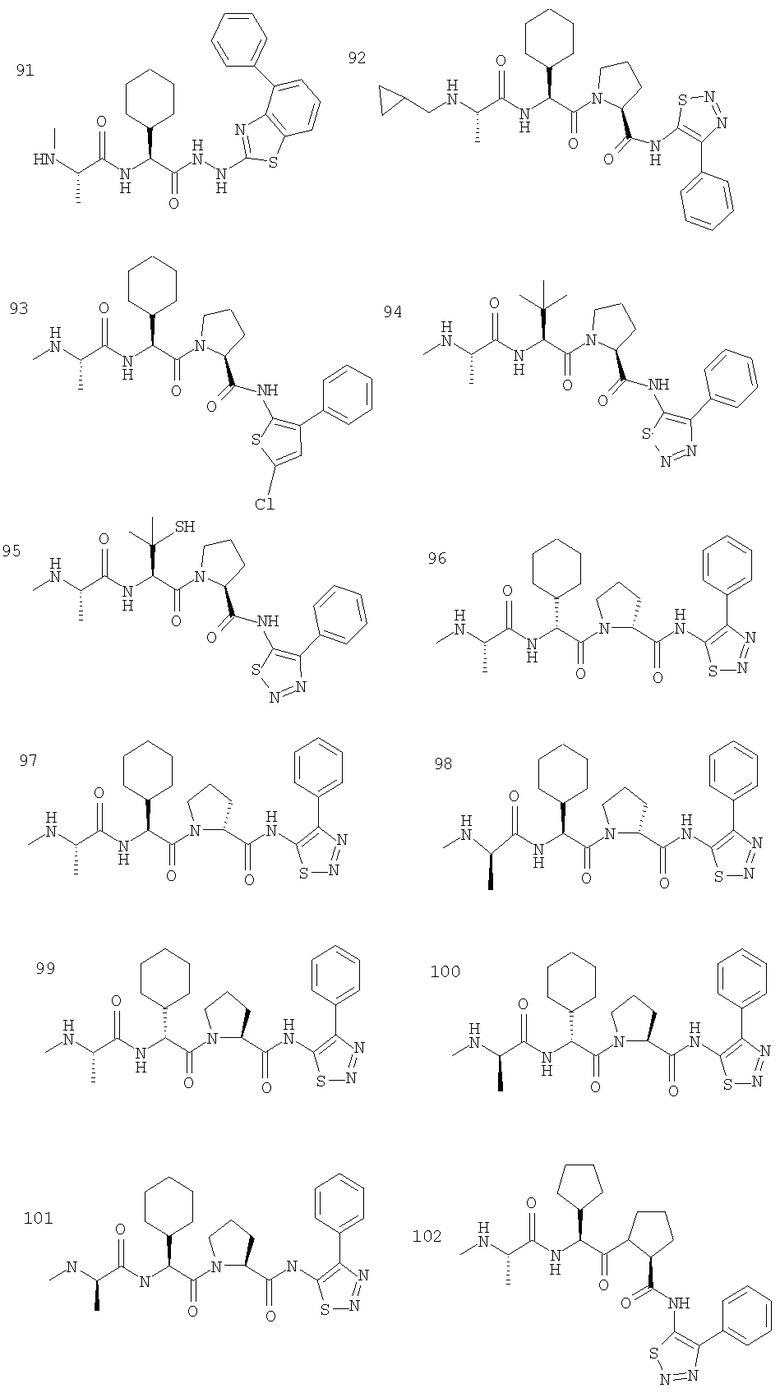

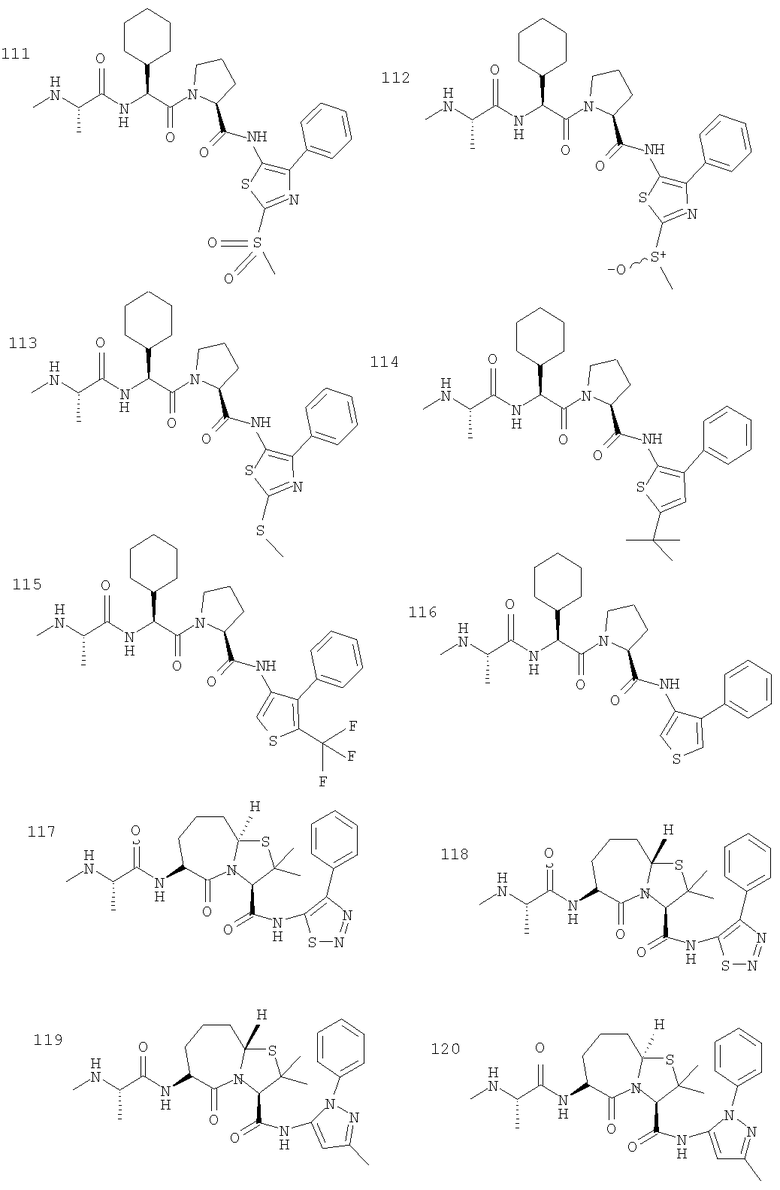
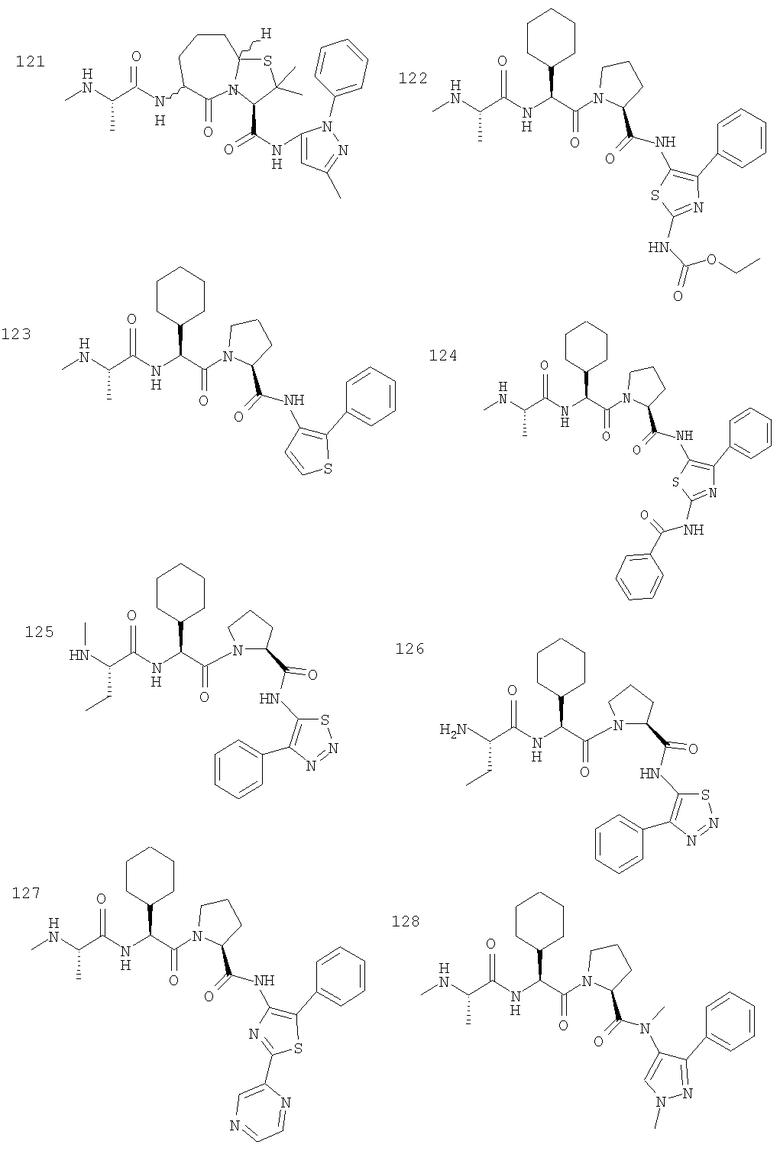
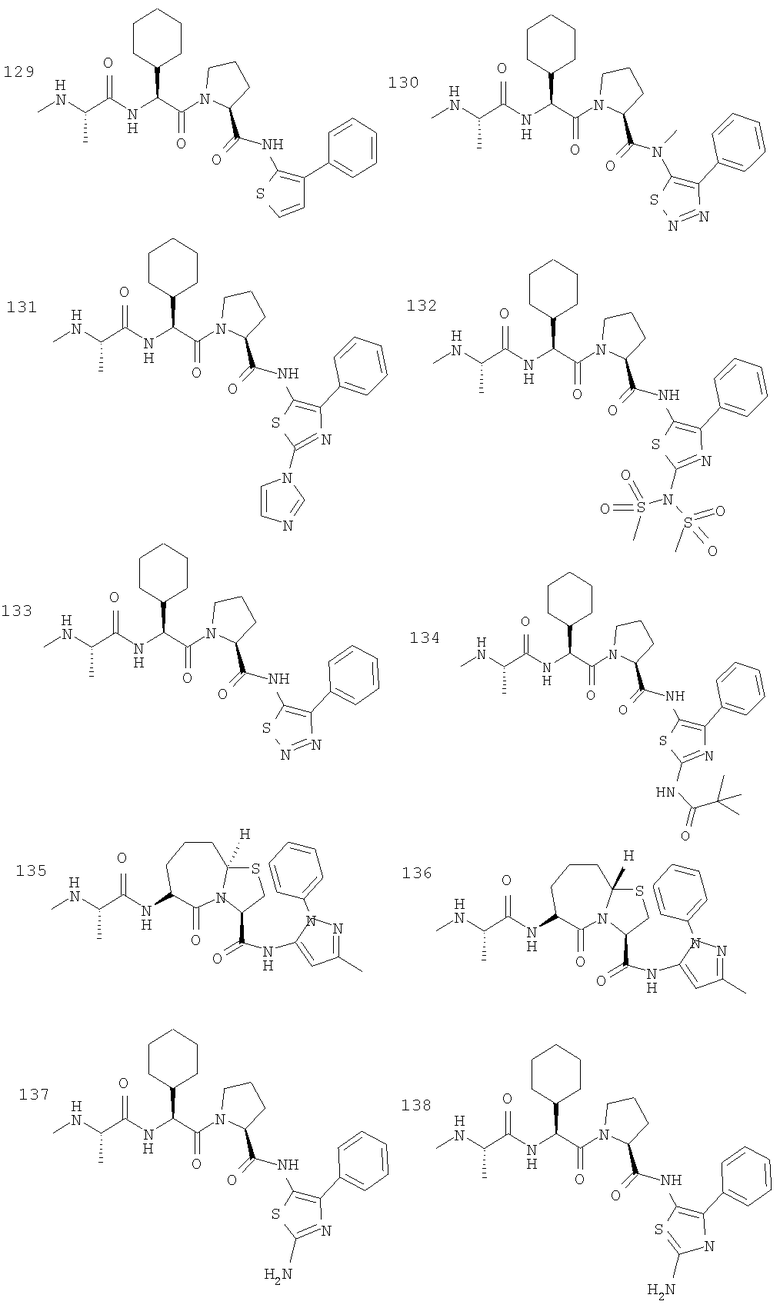
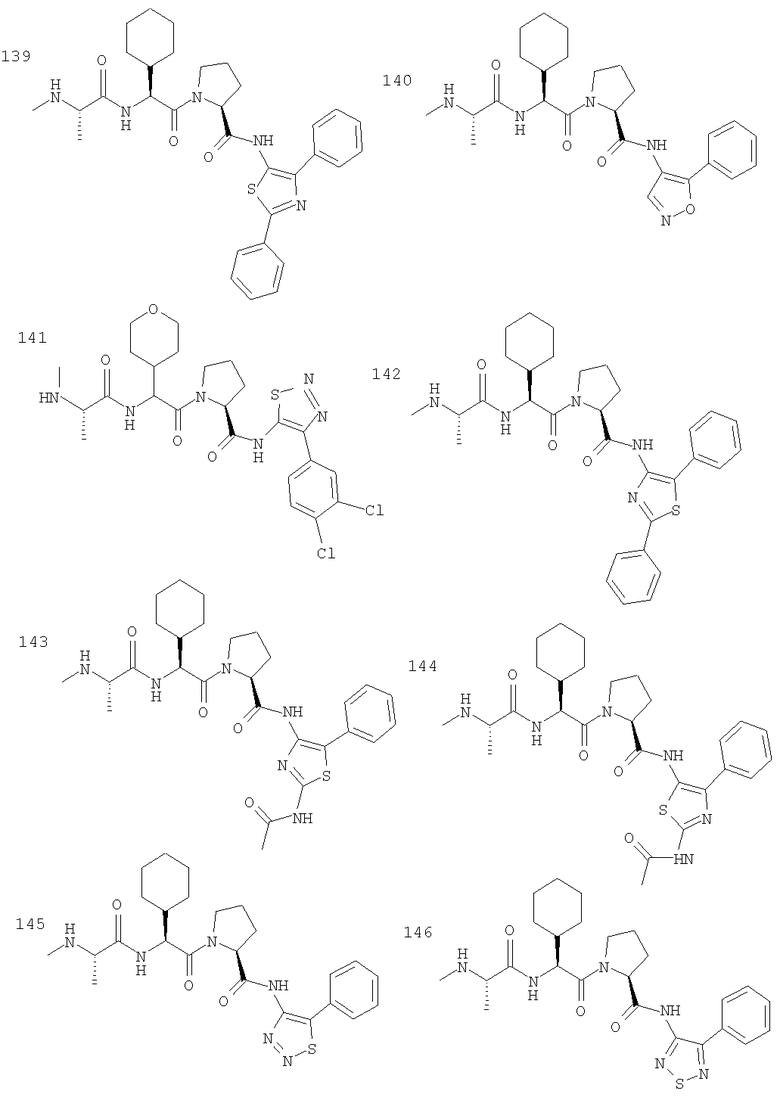
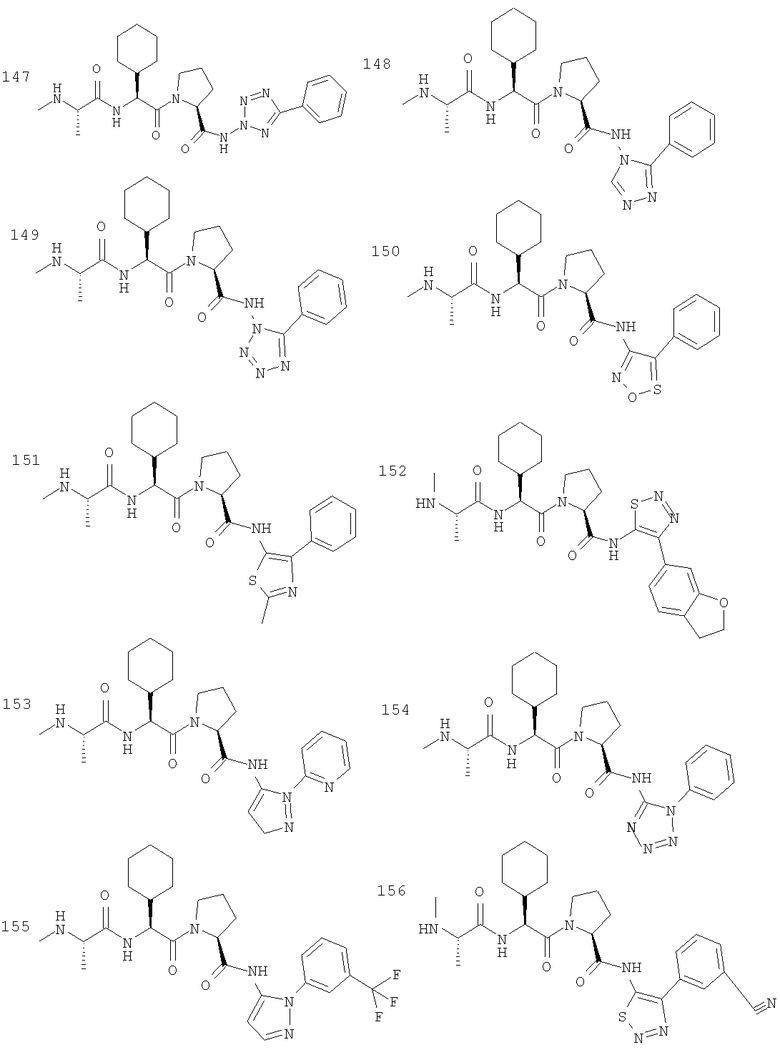
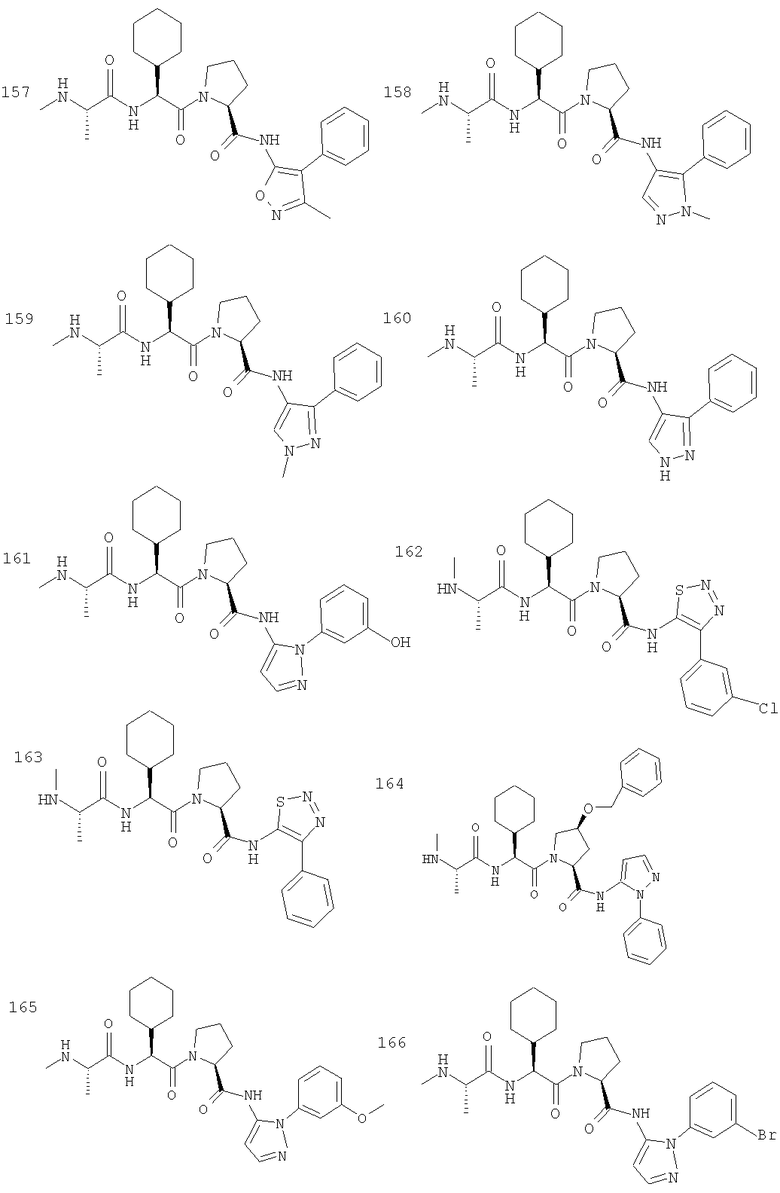
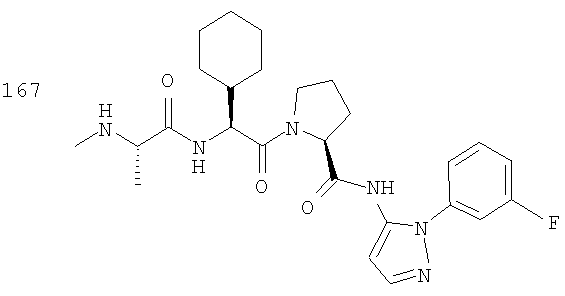
а также их фармацевтически приемлемые соли.
| RU 2000126298 А, 27.10.2002 | |||
| CHAI JIJIE ET AL "STRUCTURAL AND BIOCHEMICAL BASIS OF | |||
| APOPTOTIC ACTIVATION BY SMAC/DIABLO", NATURE, NATURE | |||
| PUBLISHING GROUP, 2000, v.406, no 6798, 855-862 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

