Предпосылки к изобретению
Агрегация тромбоцитов является первоначальной гемостатической реакцией для сокращения кровотечения, индуцированного васкулярным повреждением. Однако патологическое распространение этого нормального гемостатического процесса может привести к образованию тромба. Конечным, обычным путем в агрегации тромбоцитов является связывание фибриногена с активированным, подвергнутого воздействию тромбоцитов гликопротеином IIb/IIIa (GPIIb/IIIa). Агенты, которые препятствуют связыванию фибриногена с GPIIb/IIIa, следовательно, ингибируют агрегацию тромбоцитов. Эти агенты, следовательно, полезны при лечении тромбоцит-опосредованных тромботических нарушений, таких как артериальный и венозный тромбоз, острый инфаркт миокарда, нестабильная стенокардия, реокклюзия после тромболитической терапии и пластической операции на сосудах, воспаление и различные вазо-окклюзивные нарушения. Рецептор фибриногена (GPIIb/IIIa) активируется стимулами, такими как ADP, коллаген и тромбин, подвергающими домены связывания действию двух различных пептидных областей фибриногена: α-цепь Arg-Gly-Asp (RGD) и γ-цепь His-His-Leu-Gly-Gly-Ala-Lys-Gln-Ala-Gly-Asp-Val (HHLGGAKQAGDV, γ400-411). Поскольку было показано, что эти пептидные фрагменты сами ингибируют связывание фибриногена с GPIIb/IIIa, миметик этих фрагментов может служить также в качестве антагониста. На самом деле, до настоящего изобретения были открыты сильнодействующие антагонисты на основе RGD, которые ингибируют как связывание фибриногена с GPIIb/IIIa, так и агрегацию тромбоцитов, например, Ro-438857 (L. Alig, J. Med. Chem., 1992, 35, 4393) имеет IC50 0,094 мкМ против индуцированной тромбином агрегации тромбоцитов in vitro. Некоторые из этих агентов показали также эффективность in vivo в качестве антитромботических агентов и в некоторых случаях были также использованы вместе с фибринолитической терапией, например t-PA или стрептокиназой (J.A. Zablocki, Current Pharmaceutical Design 1995, 1, 533). Как показано результатами фармакологических исследований, описанных далее, соединения настоящего изобретения проявляют способность блокировать связывание фибриногена с выделенным GPIIb/IIIa (IC50 0,0002-1,39 мкМ), ингибировать агрегацию тромбоцитов in vitro в присутствии различных стимулов тромбоцитов (0,019-65,0 мкМ против тромбина) и, кроме того, ингибировать ех vivo агрегацию тромбоцитов в моделях животных. Кроме того, было показано, что эти агенты проявляют эффективность в моделях тромбоза животных в качестве их предшественников ("Nipecotic Acad Derivatives As Antithrombotic Compounds", серийный номер заявки 08/213772, подача 16 марта 1994). Соединения настоящего изобретения проявляют эффективность в качестве антитромботических агентов благодаря их способности предотвращать агрегацию тромбоцитов. Кроме того, поскольку соединения данного изобретения ингибируют интегрин-опосредованную межклеточную адгезию или адгезию клетка-матрица, они могут быть также полезными против воспаления, резорбции костей, метастазов опухолевых клеток и так далее (D. Сох, Drug News and Perspectives 1995, 8, 197).
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, представленным следующей общей формулой (I):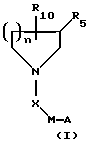
где А, X, М, R5, R10 и п такие, как определено ниже. Эти ингибиторы агрегации тромбоцитов полезны при лечении тромбоцит-опосредованных тромботических нарушений, таких как артериальный и венозный тромбоз, острый инфаркт миокарда, реокклюзия после тромболитической терапии и пластической операции на сосудах, воспаление, нестабильная стенокардия и различные вазо-окклюзивные нарушения. Эти соединения также полезны в качестве антитромботических средств, используемых в сочетании с фибринолитической терапией (например, t-PA или стрептокиназой). Фармацевтические композиции, содержащие такие соединения, также являются частью настоящего изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Более конкретно, настоящее изобретение относится к соединениям следующей формулы (I):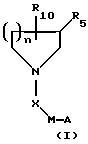
где М представляет (СН2)m или пиперидин-1-ил;
где А выбирают из любого из пиперидин-2-ила, пиперидин-3-ила, пиперидин-4-ила, пиперазин-1-ила, пирролидин-2-ила, пирролидин-3-ила, NHR2 или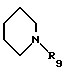
где R9 выбирают из любого из Н, алкила, СН(NН), CMe(NH) или ацила, предпочтительно R9 представляет водород;
где R10 представляет Н или C(O)N(R1)YZ;
где R1 выбирают из Н или циклоалкила;
где R2 выбирают из любого из Н, алкила или ацила. Предпочтительно R2 представляет водород;
где R5 представляет Н или С(О)NHQ(CHW)rCO2R8; где Q выбирают из СН2, СН-арил, СН-гетероарил, СН-замещенный гетероарил или СН-алкил; предпочтительно, Q представляет CH2, СН-замещенный гетероарил или СН-гетероарил; W выбирают из Н или N(R6)Т-R7, предпочтительно, W представляет Н, когда Q представляет СН, и N(R6)-T-R7, когда Q представляет СН2; где R6 выбирают из любого из Н, алкила или ацила; предпочтительно R6 представляет водород, Т выбирают из С(О), C(N-CN) или SO2, предпочтительно, Т представляет С(О) и R7 выбирают из любого из алкила, арила, аралкила, алкокси или аминоалкила; и R8 выбирают из Н, алкила или аралкила; предпочтительно, R8 представляет Н;
где m равно целому числу 1, 2 или 3. Предпочтительно, m равно 1 или 2;
где Х выбирают из любого из С(О), С(О)О, C(O)NH, CH2 или SO2;
где n равно целому числу 1, 2 или 3;
где r равно 0 или 1;
где R1 выбирают из Н или циклоалкила;
где Y выбирают из любого из (СН2)р, CH(R3)(CH2)q, (CH2)qCH(R3), (CH(COR4)CH2)q, (CH2)qCHOH или пиперидин-3-карбоновой кислоты, при условии, что, когда Y представляет (СН2)р и р равно 2, Х отличен от С(O), или, когда Х представляет С(О), то либо R1 отличен от Н, либо R2 отличен от Н, и при условии, что, когда Y представляет (CH(CO2R4)CH2)q, Х отличен от С(О) или СН2;
где р равно 2 или 3;
где q равно 1, 2 или 3. Предпочтительно, q равно 1;
где R3 представляет алкил, С2-С8-алкенил, С2-С8-алкинил, арил, аралкил или гетероарил;
где R4 представляет Н или алкил или циклоалкил. Предпочтительно, R4 представляет водород;
где Z представляет СO2Н, СО2-алкил, SО3Н, РО3Н2 или 5-тетразол; при условии, что по меньшей мере один из R5 и R10 представляет водород;
или их энантиомеру или фармацевтически приемлемой соли.
Предпочтительно, группа C(O)N(R1)YZ присоединена к углероду кольца центрального азацикла в 3- или 4-положении (4-положении, когда кольцо более чем пятичленное), и наиболее предпочтительно, в 3-положении.
Используемый здесь, если не указано иное, термин алкил и алкокси, используют ли его отдельно или как часть замещающей группы, включает неразветвленные и разветвленные цепи, имеющие 1-8 углеродов. Например, алкильные радикалы включают метил, этил, пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, 3-(2-метил)бутил, 2-пентил, 2-метилбутил, неопентил, н-гексил, 2-гексил и 2-метилпентил. Алкокси-радикалы представляют кислород-простые эфиры, образованные из ранее описанных алкильных групп с неразветвленной или разветвленной цепью. Циклоалкильные группы содержат 5-8 атомов углерода в кольце и предпочтительно 6-7 атомов углерода.
Термин "арил", "гетероарил" или "замещенный гетероарил", используемый здесь отдельно или в комбинации с другими терминами, обозначает ароматические или гетероароматические группы, такие как фенил, нафтил, пиридил, тиенил, фуранил или хинолинил, где заместителем является алкильная группа. Термин "аралкил" означает алкильную группу, замещенную арильной группой.
Термин "ацил", используемый здесь, означает органический радикал, имеющий 2-6 атомов углерода, образованный из органической кислоты путем удаления гидроксильной группы.
Соединения настоящего изобретения могут присутствовать также в форме фармацевтически приемлемой соли. Фармацевтически приемлемая соль обычно имеет форму, в которой атом азота на 1-пиперидиновом (пирролидиновом, пиперазиновом) заместителе протонируется неорганической или органической кислотой. Характерные органические или неорганические кислоты включают хлористоводородную, бромистоводородную, иодистоводородную, перхлорную, серную, азотную, фосфорную, уксусную, пропионовую, гликолевую, молочную, янтарную, малеиновую, фумаровую, яблочную, винную, лимонную, бензойную, миндальную, метансульфоновую, гидроксиэтансульфоновую, бензолсульфоновую, щавелевую, памовую, 2-нафталинсульфоновую, п-толуолсульфоновую, циклогексансульфаминовую, салициловую, сахариновую или трифторуксусную.
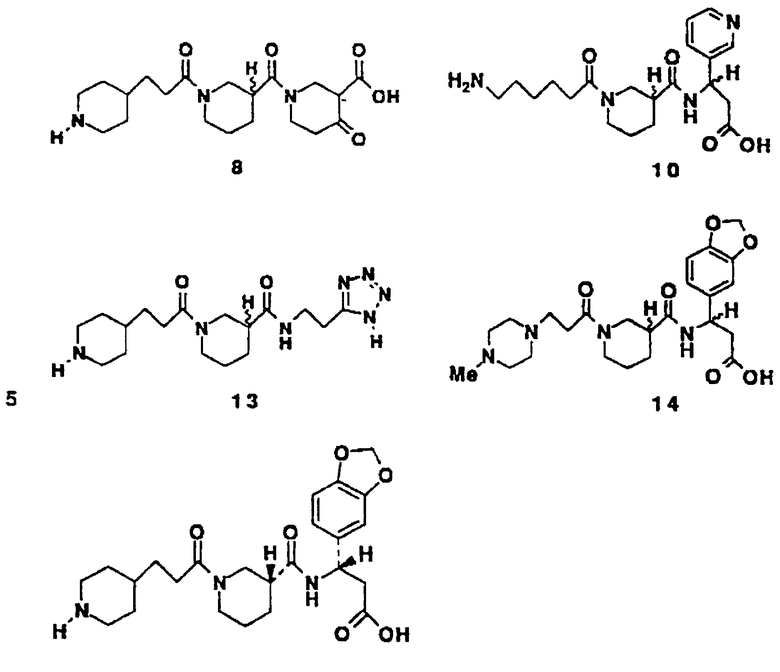
Особенно предпочтительные соединения настоящего изобретения включают те соединения, указанные в таблице I, где "Замест." обозначает положение присоединения группы С(О)N(R1) YCO2H к центральному азациклу и где буква "R" после цифры "3" обозначает абсолютную конфигурацию (правила Кана-Ингольда-Перлога). Те цифры, не имеющие никакой установленной конфигурации, представляют рацемические смеси.
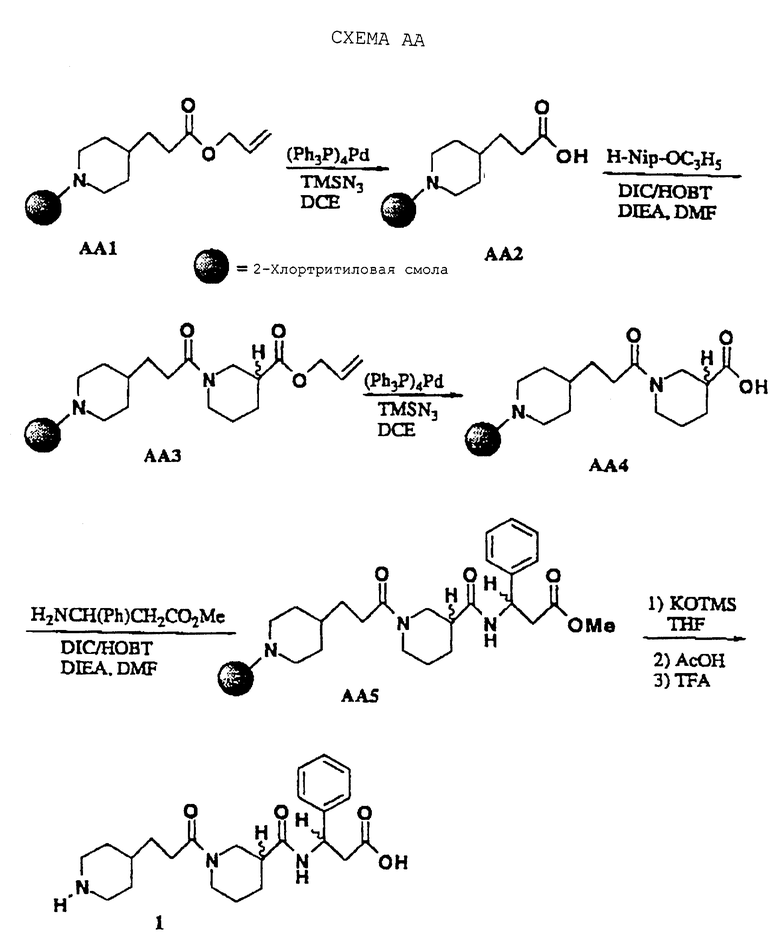
Соединения изобретения, где R5 представляет Н, R10 представляет C(O)N(R1)YZ, M представляет (СН2)m и А представляет пиперидин-2-ил, пиперидин-3-ил, пиперидин-4-ил, пиперазин-1-ил, пирролидин-2-ил, пирролидин-3-ил или NHR2, можно получить, как показано на схеме АА. В этой схеме аллиловый эфир нипекотиновой кислоты (либо рацемическая смесь, либо любой отдельный энантиомер) можно обработать связанной со смолой 4-пиперидинпропионовой кислотой в присутствии DIC/HOBT и третичного амина. Аллиловый эфир затем удяляют через палладий-опосредованный катализ и продолжают повторяющийся процесс азосочетания, получая конечный продукт после омыления триметилсиланолатом калия (например, соединение 1). По аналогии, соединения (соединения 2 и 3), у которых третичный амид заменен на мочевину и уретан, были получены реакцией амина (спирта), осажденного на твердом носителе, с п-нитрофенилхлорформиатом и затем с этилнипекотатом (S.M. Hutchins Tetrahedron Lett. 1994, 35, 4055).
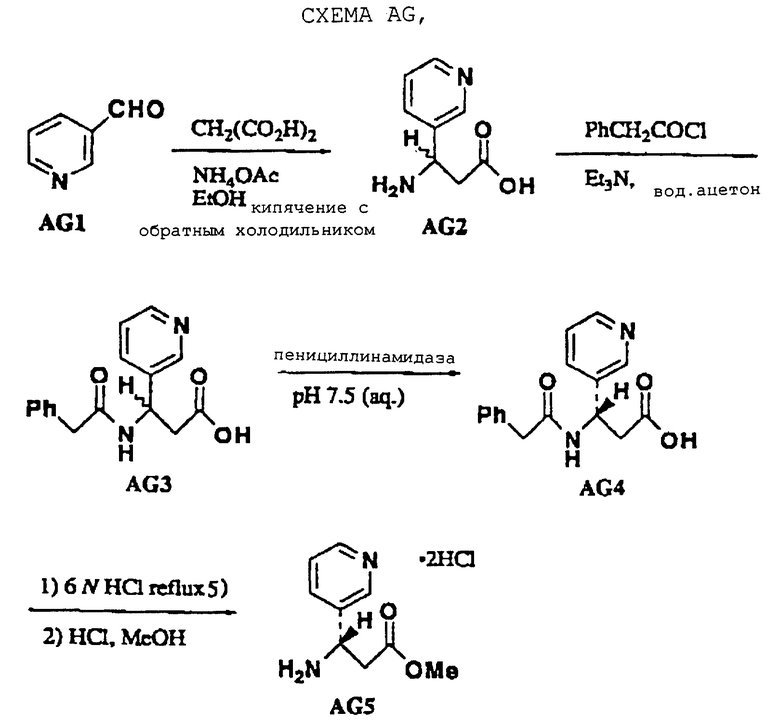
Промежуточные соединения эфиры тризамещенной 3-аминопропионовой кислоты получали с использованием модифицированной методики Кневенагеля (Knoevenagel) (Scheme AG; Т. Profft, J.Prakt Chem., 1965, 30, 18) с последующей этерификацией Фишера получаемой карбоновой кислоты (когда они коммерчески недоступны). Эти промежуточные соединения получали в энантиомерно-обогащенной форме разделением (расщеплением) пенициллинамидазой рацемических фенилацетамидов, таких как промежуточный AG3 (V.A. Soloshonok, Tetrahedron: Asymmetry 1995, 6, 1601). И здесь, нежелательный R-энантиомер гидролизуют амидазой, тогда как целевой S-энантиомер сохраняет фенилацетильную группу. Разделения можно также проводить на (-)-эфедриновых солях рацемических тризамещенных 3-N-Вос-аминопропионовых кислот, как опубликовано (J.A. Zablocki, J. Med. Chem. , 1995, 38, 2378). Этилнипекотат и этилизонипекотат являются коммерчески доступными промежуточными продуктами.
Синтез 5- и 7-членных кольцевых аналогов нипекотамидов (4 и 17, соответственно) осуществляли синтезом в твердой фазе с использованием метилпирролидин-3-карбоксилатных и метилгексагидроазепин-3-карбоксилатных промежуточных соединений для аналогичного превращения АА2 в АА3 (схема АА). Метилпирролидин-3-карбоксилат и метилгексагидроазепин-3-карбоксилат получили, как опубликовано (Н. Paroport, J. Org. Chim., 1974, 39, 893). Например, N-бензилгексагидроазепин-2-он взаимодействовал с системой диизопропиламид лития/диэтилкарбонат и этот продукт затем восстанавливали литий-алюминийгидридом, получая N-бензил-3-гидроксиметилгексагидроазепин. Бензильную группу удаляли гидрогенолизом (H2, PdC, MeOH), азот защищали (ди-трет-бутилдикарбонат/гидроксид натрия) и спирт окисляли триоксидом хрома, получая N-Boc-гексагидроазепин-3-карбоновую кислоту. Вос-группу удаляли совместно с этерификацией карбоксилата с использованием HCl/MeOH, получая метилгексагидроазепин-3-карбоксилат.
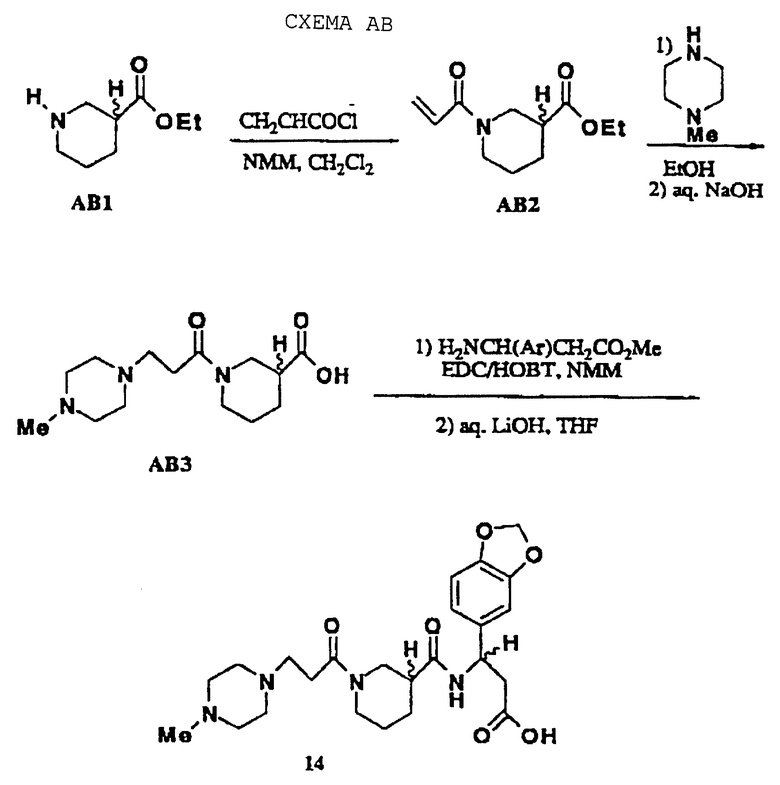
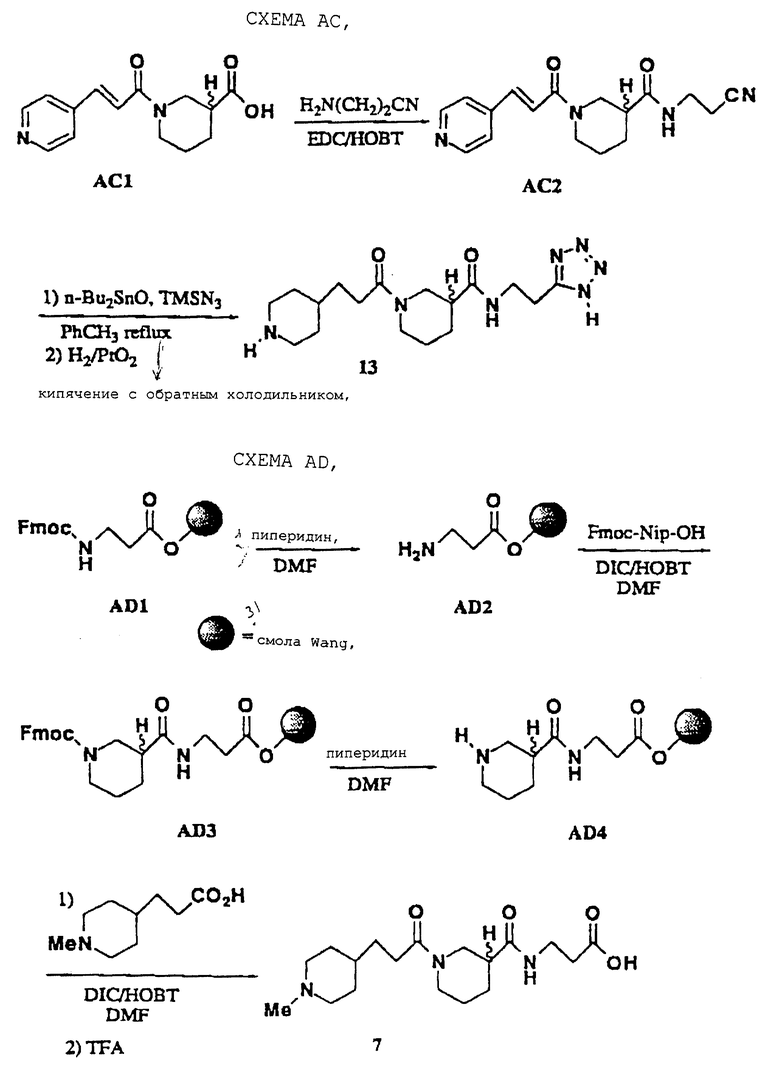
Аналоги пиперазина получали, как приводится в качестве примера в схеме АВ, как опубликовано (S.G.Gilbreath, J. Am. Chem. Soc., 1988, 110, 6172). Тетразолы (13) получали из соответствующих нитрилов с использованием системы азидотриметилсилан/оксид дибутилолова, как опубликовано (схема AC; S. J. Wittenberger, J. Org. Chem., 1993, 58, 4139). И здесь, нитрильный предшественник АС2 получали стандартным сочетанием амидной связи с 3-аминопропионитрилом и восстанавливали на конечной синтетической стадии с использованием опосредованного диоксидом платины гидрирования (W.J. Hoekstra, J. Med. Chem. , 1995, 38, 1582).
Аналоги N-метилпиперидина можно получить методиками на основе Fmoc синтезов пептидов в твердой фазе, как показано на схеме AD (P.Sieber, Tetrahedron Lett. 1987, 28, 6147). Fmoc-защитные группы расщепляли с использованием 20% пиперидин/DMF, сочетания проводили с использованием DIC/HOBT/DMF и конечные продукты удаляли из смолы 95% TFA.
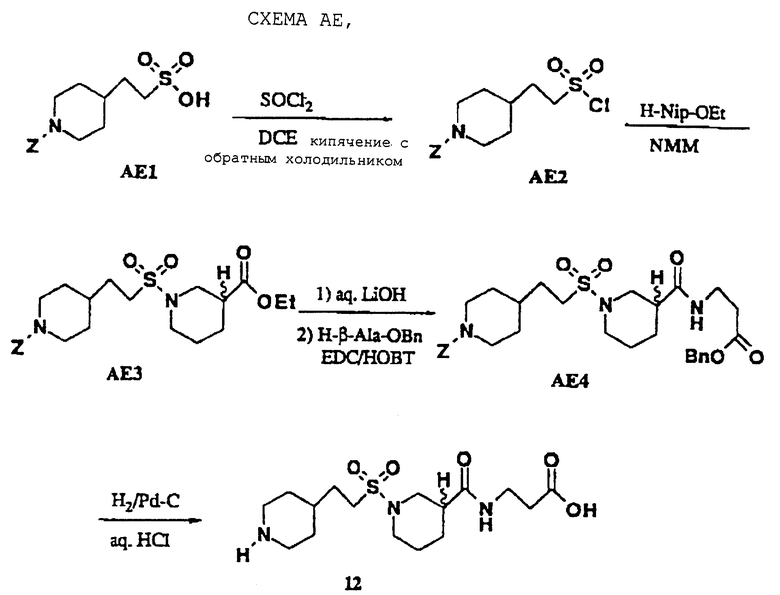
Сульфонамид 12 получали, как показано на схеме АЕ. Промежуточный АЕ1 выделяли в две стадии из 4-пиридинэтансульфоновой кислоты путем гидрирования/защиты, как описано (J. I. DeGaw, J. Heterocyclic Chem., 1966, 3, 90) и затем хлорировали с использованием стандартных условий для хлорирования тионилхлоридом (P.J. Hearst. Org. Syn. 1950, 30, 58), получая АЕ2. Промежуточный АЕ2 затем превращали в конечный продукт с использованием стандартного синтеза в фазе раствора (W.J. Hoekstra, J.Med. Chem, 1995, 38, 1582).
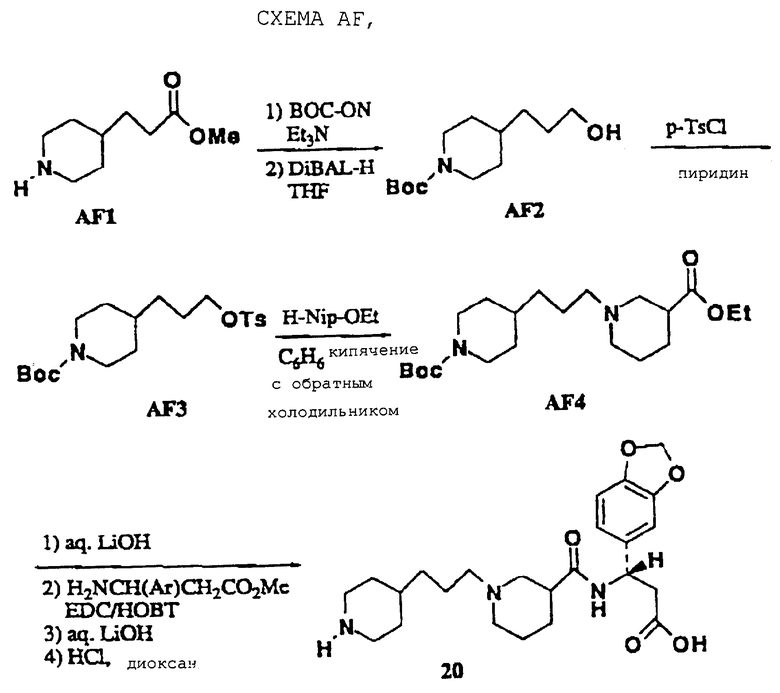
Пиперидинпропилнипекотамид 20 получали, как показано на схеме AF. Сложный эфир AF1 защищали Вос-группой с использованием стандартных Boc-ON-условий (D. S. Tarbell, Proc. Natl. Acad, Sci, USA 1972, 69, 730) и затем восстанавливали в его соответствующий первичный спирт системой DiBAL-H/THF (E. Winterfeldt, Synthesis 1975, 617), получая промежуточный AF2. Это соединение превращали в его соответствующий тозилат AF3 с использованием п-TsCl (L.F. Awad, Bull. Chem. Soc. Jpn., 1986, 59, 1578). Этилнипекотат затем алкилировали промежуточным AF3 с использованием стандартных условий (бензол/нагревание; I. Seki, Chem. Pharm. Bull. Jpn., 1970, 18, 1104).
Энантиомерно обогащенный этиловый эфир (R)-(-)-нипекотиновой кислоты выделяли хиральным разделением рацемического материала в виде его соответствующей соли D-винной кислоты (А. М. Akkerman, Rec. Trav. Chim. Pays-Bas, 1951, 70, 899).
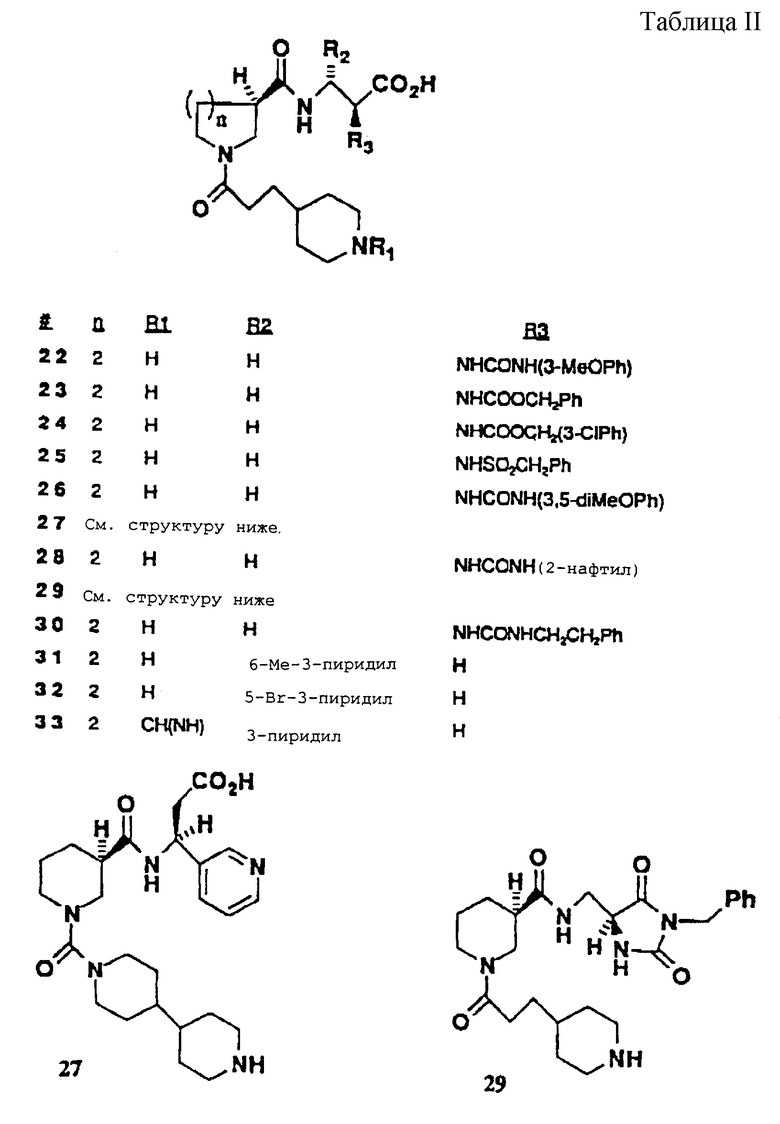
Особенно предпочтительные соединения настоящего изобретения включают те соединения, показанные в таблице I (и таблице II), где буква "R" после цифры "3" обозначает абсолютную конфигурацию (правила Кана-Ингольда-Прелога).
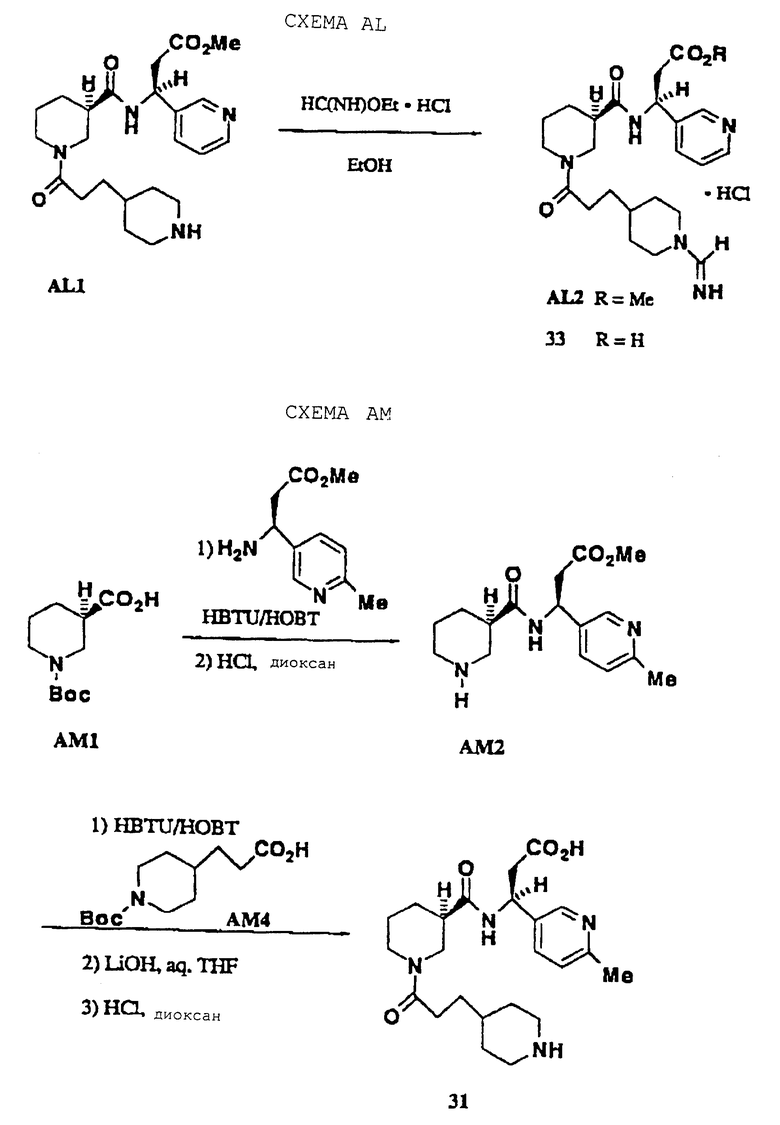
Антагонисты диаминопропионовые кислоты, где R5 представляет С(O)NHQ(CHW)rCO2R8, R10 представляет Н, М представляет пиперидин-1-ил и А представляет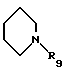
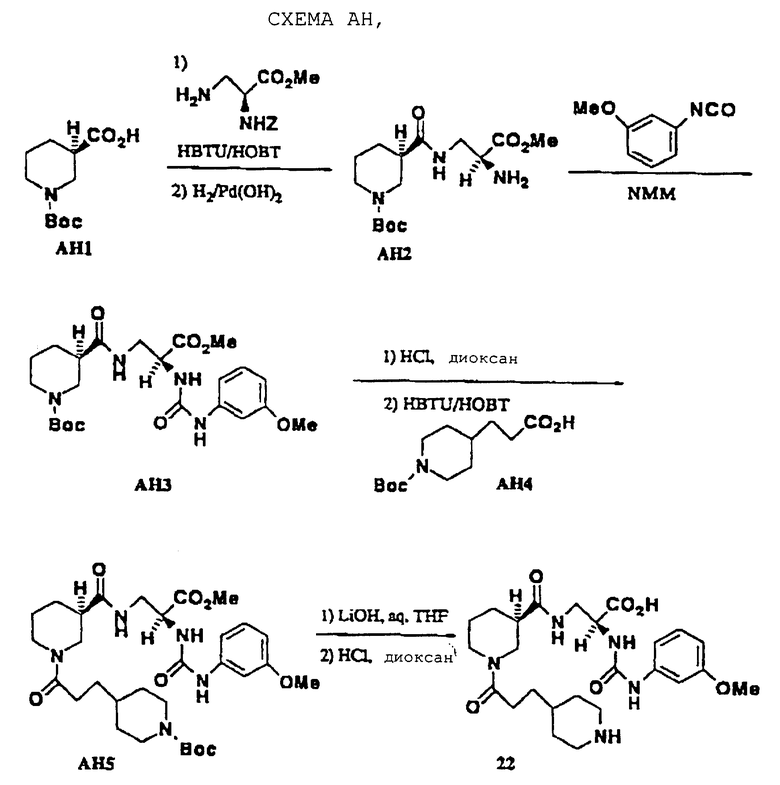
можно получить, как показано на схеме АН. Метил-N-α-Z-диаминопропионат ацилировали HBTU-активированным АН1, Z-группу удаляли гидрогенолизом для получения АН2 (для 23 Z-группу сохраняли), и затем получаемый первичный амин реагировал с требуемым изоцианатом (или алкилхлорформиатом для 24, алкилсульфонилхлоридом для 25) для получения АН3. Вос-группу промежуточного АН3 удаляли НС1 и получаемый вторичный амин ацилировали HBTU-активированным АН4 для получения АН5. Этот материал омыляли гидроксидом лития и Вос-группу удаляли НС1 для получения 22.
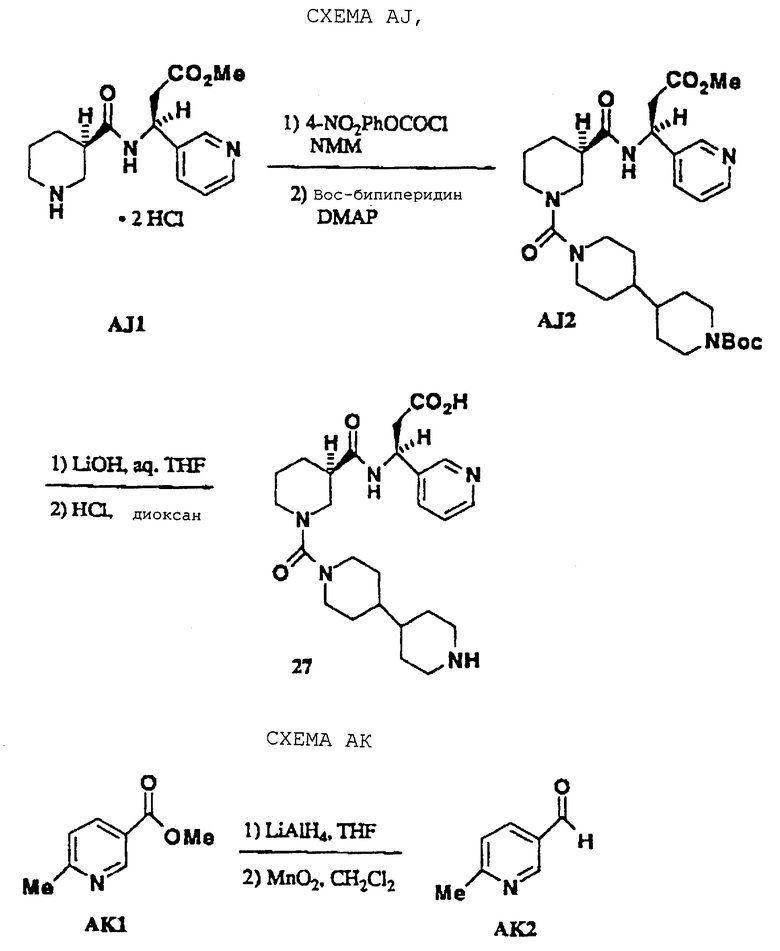
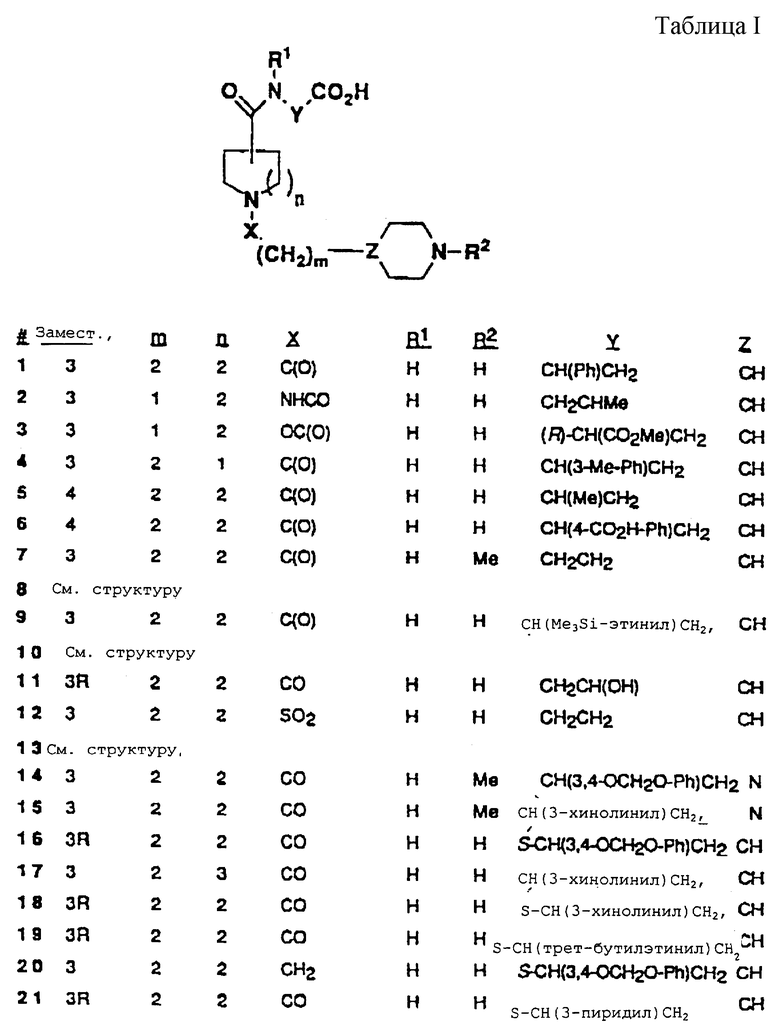
Антагонисты на основе бипиперидин-мочевины настоящего изобретения можно получить, как показано на схеме AJ. Промежуточный АJ1 получали, как описано в схеме AG. АJ1 ацилировали п-нитрофенилхлорформиатом и затем давали реагировать с Вос-бипиперидином (для синтеза, см. W. Bondinell, заявка на патент WO 94/14776). Сложный эфир AJ2 омыляли гидроксидом лития и Вос-группу удаляли НС1 для получения 27. Промежуточные соединения замещенные пиперидинальдегиды, такие как АК2, получали восстановлением литийалюминийгидридом их соответствующих метиловых эфиров никотиновых кислот (АК1) с последующим окислением диоксидом марганца (схема АК). Альдегиды затем превращали в β-аминокислоты, как показано на схеме AG. Формамидин AL3 получали, как показано на схеме AL. Амин AL1 ацилировали этилформимидатом, как описано М.К. Scott (J. Med. Chem. , 1983, 26, 534). Сложный эфир AL2 омыляли 4NHC1 (комнатная температура, 20 ч), получая 33. Антагонисты типа тризамещенных β-аминокислот были синтезированы, как показано на схеме AM. Разделенный 6-метилпиридил-β-аминоэфир ацилировали HBTU-активированным AM1 и продукт сочетания обрабатывали НС1, получая амин АМ2. Амин ацилировали HBTU-активированным АМ4, сложный эфир омыляли и Вос-группу удаляли при помощи НС1, получая 31.
Для получения фармацевтических композиций настоящего изобретения одно или более соединений формулы (I) или их солей изобретения в качестве активного ингредиента однородно смешивают с фармацевтическим носителем согласно обычным методикам приготовления фармацевтических средств, этот носитель может принимать большое число форм в зависимости от формы препарата, которая желательна для введения, например перорального или парентерального, такого как внутримышечное. При получении композиций в виде пероральной лекарственной формы можно применять любую из обычных фармацевтических сред. Так, для жидких пероральных препаратов, таких как, например, суспензии, эликсиры и растворы, подходящие носители и добавки включают воду, гликоли, масла, спирты, отдушки, консерванты, красители и тому подобное; для твердых пероральных препаратов, таких как, например, порошки, капсулы, каплеты, желатиновые капсулы и таблетки, подходящие носители и добавки включают крахмалы, сахара, разбавители, гранулирующие агенты, смазывающие вещества, связующие, дезинтегрирующие агенты и тому подобное. Вследствие легкости их введения таблетки и капсулы представляют наиболее благоприятные пероральные формы единичной дозы, в этих случаях очевидно, что применяют твердые фармацевтические носители. При желании, таблетки могут быть покрыты сахаром или покрыты энтеросолюбильной оболочкой с применением стандартных методик. Для парентерального введения носитель обычно включает стерильную воду, хотя могут быть включены другие ингредиенты, например, для таких целей, как способствование растворению или для консервирования. Можно также приготовить инъецируемые суспензии, в этом случае можно применять подходящие жидкие носители, суспендирующие агенты и тому подобное. Фармацевтические композиции будут содержать, на единичную дозу, например, таблетку, капсулу, порошок, инъекцию, чайную ложку и тому подобное, количество активного ингредиента, необходимое для доставки эффективной дозы, как описано выше. Фармацевтические композиции будут содержать, на единичную дозу, например, таблетку, капсулу, порошок, инъекцию, суппозиторий, чайную ложку и тому подобное, от примерно 0,03 мг до 100 мг/кг (предпочтительно, 0,1-30 мг/кг) и могут быть даны при дозе от примерно 0,1 до 300 мг/кг/день (предпочтительно 1-50 мг/кг/день). Дозировки, однако, могут меняться в зависимости от потребности пациентов, серьезности состояния, которое лечат, и используемого соединения. Можно использовать либо ежедневное введение, либо пост-периодическую дозировку.
БИОЛОГИЯ
Соединения настоящего изобретения препятствуют связыванию фибриногена с гликопротеином тромбоцитов IIb/IIIa (GPIIb/IIIa) и тем самым ингибируют агрегацию тромбоцитов. Такие соединения, следовательно, полезны при лечении тромбоцит-опосредованных тромботических нарушений, таких как артериальный и венозный тромбоз, острый инфаркт миокарда, реокклюзия после тромболитической терапии и пластической операции на сосудах и различные вазо-окклюзивные нарушения. Поскольку конечным обычным путем в нормальной агрегации тромбоцитов является связывание фибриногена с активированным, подвергнутым воздействию GPIIb/IIIa, ингибирование этого связывания представляет вполне состоятельный антитромботический подход. Рецептор активируется такими стимулами, как АДР, коллаген и тромбин, подвергающими домены связывания действию двух различных пептидных областей фибриногена: α-цепь Arg-Gly-Asp (RGD) и γ-цепь 400-411. Как показано результатами фармакологических исследований, описанных ниже, соединения настоящего изобретения проявляют способность блокировать связывание фибриногена с выделенным GPIIb/IIIa (IC50 0,0002-1,39 мкМ), ингибировать агрегацию тромбоцитов in vitro в присутствии различных стимулов тромбоцитов (0,019-65,0 мкМ против тромбина) и, следовательно, ингибировать ex vivo агрегацию тромбоцитов на моделях животных.
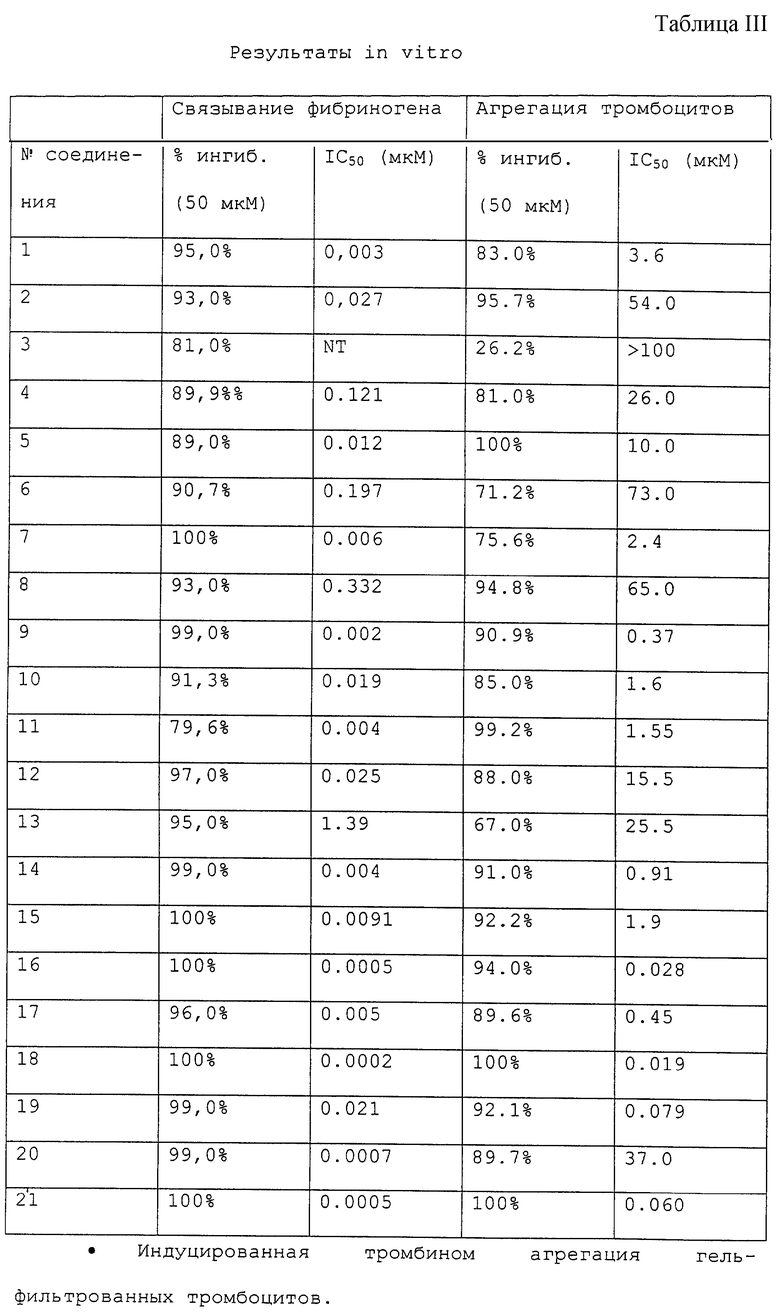
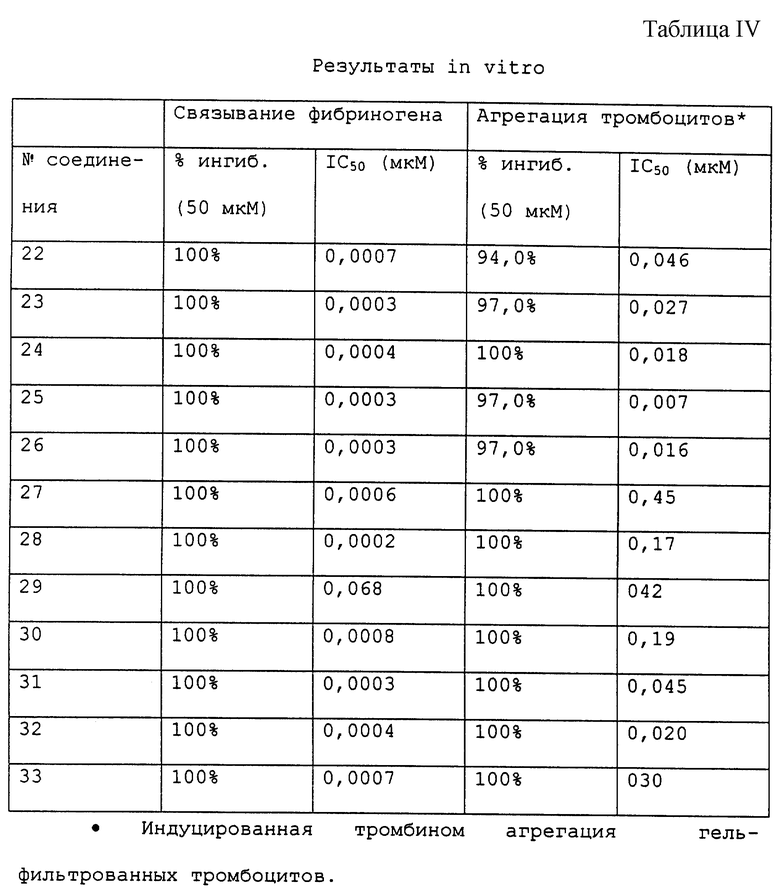
ИСПЫТАНИЯ НА СВЯЗЫВАНИЕ IN VITRO ОЧИЩЕННОГО В ТВЕРДОЙ ФАЗЕ ГЛИКОПРОТЕИНА IIB/IIIA
Пластину микротитратора с 96 лунками иммулон-2 (Dynatech-Immulon) покрывают 50 мкл/лунку RGD-подобным очищенным GPIIb/IIIa (эффективный диапазон 0,5-10 мкг/мл) в 10 мМ HEPES 150 мМ NaCl, 1 мМ при рН 7,4. Пластину покрывают и инкубируют в течение ночи при 4oС. Раствор GPIIb/IIIa выгружают и добавляют 150 мкл 5% BSA и инкубируют при комнатной температуре в течение 1-3 ч. Пластину промывают экстенсивно модифицированным буфером Туrode. В лунки, которые содержат испытуемые соединения (25 мкл/лунку), добавляют биотинилированный фибриноген (25 мкл/лунку) при удвоенной 2-х конечной концентрации. Пластину покрывают и инкубируют при комнатной температуре в течение 2-4 ч. За двадцать минут до завершения инкубации добавляют одну каплю Реагента А (набор пероксидазы из хрена АВС, Vecta Stain, Vector Laboratories, Inc.) и одну каплю Реагента В при смешивании к 5 мл модифицированной буферной смеси Tyrode и оставляют стоять. Раствор лиганда выгружают и пластину промывают (5•200 мкл/лунку) модифицированным буфером Tyrode. Добавляют реагент Vecta Stain HRP-биотин-авидин (50 мкл/лунку, как получен выше) и инкубируют при комнатной температуре в течение 15 мин. Раствор Vecta Stain выгружают и лунки промывают (5•200 мкл/лунку) модифицированным буфером Tyrode. Добавляют проявляющий буфер (10 мл 50 мМ цитрат/фосфатного буфера, рН 5,3, 6 мг o-фенилендиамина, 6 мкл 30% H2O2; 50 мкл/лунку) и инкубируют при комнатной температуре в течение 3-5 мин и затем добавляют 2 NH2SO4 (50 мкл/лунку). Считывают оптическую плотность при 490 нМ. Результаты приводятся в таблицах III и IV.
ИСПЫТАНИЕ НА ИНГИБИРОВАНИЕ IN VITRO ИНДУЦИРОВАННОЙ ТРОМБИНОМ АГРЕГАЦИИ ГЕЛЬ-ФИЛЬТРОВАННЫХ ТРОМБОЦИТОВ
Процент агрегации тромбоцитов вычисляют как увеличение в пропускании света концентрата тромбоцитов, обработанных соединением, по сравнению с концентратом контроль-обработанных тромбоцитов. Кровь человека получают от нормальных доноров без лекарственных средств в пробирки, содержащие 0,13 М цитрата натрия. Обогащенную тромбоцитами плазму (PRP) собирают центрифугированием цельной крови при 200-х г в течение 10 мин при 25oС. PRP (5 мл) гель-фильтруют через Sepharose 2B (объем слоя 50 мл) и количество тромбоцитов доводят до 2•107 тромбоцитов на образец. К силицированной кювете добавляют следующие компоненты: концентрированный тромбоцитный фильтрат и буфер Tyrode (0,14 М NaCl, 0,0027 М КСl, 0,012 М NаНСО3, 0,76 мМ Na2HPO4, 0,0055 М глюкозы, 2 мг/мл BSA и 5,0 мМ HEPES, pH 7,4) в количестве, равном 350 мкл, 50 мкл 20 мМ кальция и 50 мкл испытуемого соединения. Агрегацию контролируют в агрегометре BIODATA в течение 3 мин после добавления агониста (50 мкл тромбина, 1 единица/мл). Результаты приводятся в таблицах III и IV.
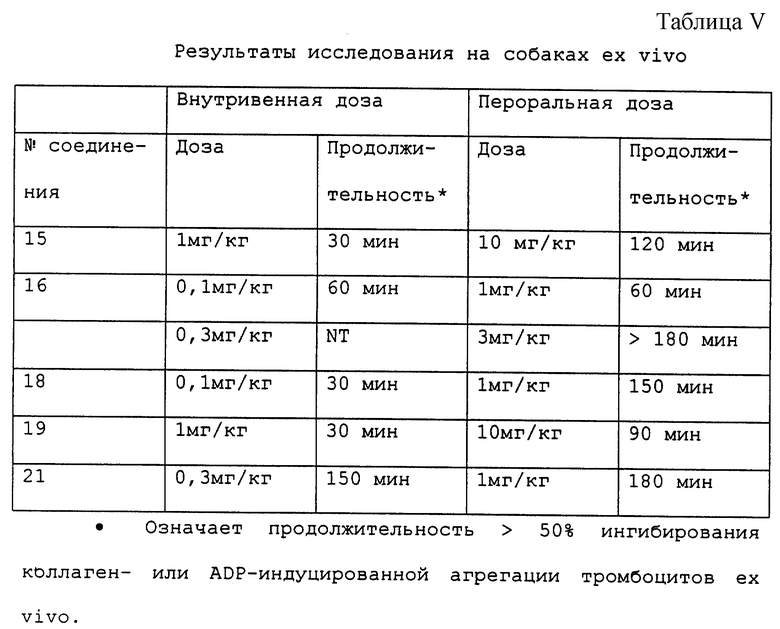
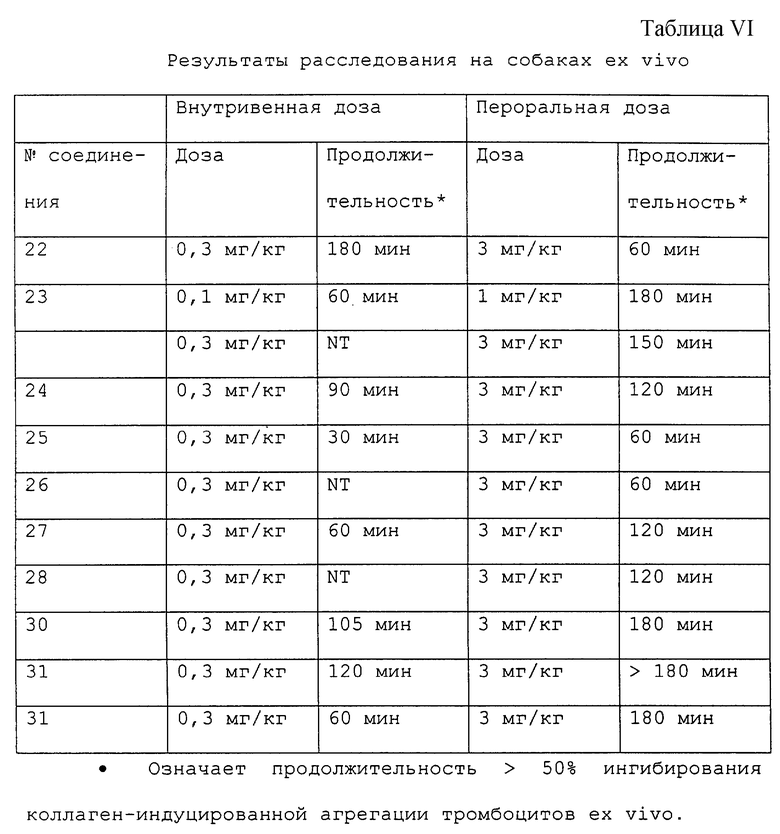
ИССЛЕДОВАНИЕ НА СОБАКАХ EX VIVO
Взрослых собак-дворняжек (8-13 кг) анестезировали пентобарбиталом натрия (35 мг/кг, внутривенно) и переводили на искусственное дыхание. Артериальное кровяное давление и частоту сердечных сокращений измеряли с использованием датчика давления Millar с катетером-наконечником, вставленным в бедренную артерию. Другой датчик Millar помещали в левый желудочек (LV) через сонную артерию для измерения конечного диастолического давления LV и показателей сократимости сердечной мышцы. Электрокардиограмму в отведении II регистрировали из электродов для конечностей. Катетеры помещали в бедренную артерию и вену для взятия проб крови и инфузионного введения лекарственных средств, соответственно. Реакции непрерывно регистрировали с использованием системы Moduar Instruments data aquisition system.
Пробы артериальной крови (5-9 мл) отбирали в пробирки, содержащие 3,8% цитрата натрия для получения плазмы, обогащенной тромбоцитами (PRP), и определения влияния на параметры коагуляции; протромбиновое время (РТ) и активированное парциальное тромбопластиновое времмя (АРРТ). Отдельные пробы крови (1,5 мл) были отобраны в EDTA (этилендиаминтетрауксусная кислота) для определения гематокрита и количества клеток (тромбоцитов, RBC и лейкоцитов). Темплатное время кровотечения определяли на внутриротовой поверхности с использованием устройства для рассечения symplate и фильтровальной бумаги Whatman.
Агрегацию PRP проводили с использованием агрегометра BioData. Для агрегации цельной крови использовали агрегометр Chronolog impedance. РТ и АРТТ определяли на анализаторе коагуляции либо BioData, либо ACL 3000+. Клетки подсчитывали при помощи Sysmex K-1000.
Соединения растворяли в небольшом объеме диметилформамида (DMF) и разбавляли солевым раствором до конечной концентрации 10% DMF. Соединения вводили внутривенным путем инфузионным насосом Harvard. Дозы вводили на протяжении интервала 15 мин при постоянной скорости 0,33 мл/мин. Данные получали после каждой дозы и с интервалами 30 мин после конца введения лекарственного средства. Пероральные дозы вводили в виде водных растворов через шприц.
Соединения вызывали заметное ингибирование реакций агрегации тромбоцитов ex vivo. Так, в цельной крови соединения ингибировали коллаген-стимулированную (или ADP) агрегацию в дозах 0,1-10 мк/кг с заметным ингибированием коллаген-стимулированного выделения АТР тромбоцитов. В PRP соединения также ингибировали стимулированную коллагеном агрегацию тромбоцитов с заметной активностью при 0,1-10 мг/кг. Соединения не имели измеримого гемодинамического действия при дозах вплоть до 1 мг/кг, внутривенно. Лекарственные средства вызывают повышение в темплатном времени кровотечения при 0,1-1 мг/кг с быстрым восстановлением после обработки. Никакого влияния на коагуляцию (РТ и АРТТ) не наблюдали во время обработки, и числа тромбоцитов, лейкоцитов и RBC не изменялись при любой дозе соединений.
Результаты показывают, что соединения являются широко эффективными ингибиторами агрегации тромбоцитов ex vivo (антагонизируя как коллагеновый, так и ADP пути) после внутривенного введения доз, в интервале от 0,1 до 1 мк/кг, или 1-10 мг/кг перорально (таблицы V и VI). Антиагрегирующие действия сопровождались повышениями времени кровотечения при более высоких дозах. Никакие другие гемодинамические или гематологические эффекты не наблюдались.
Соединения 16 и 18 проявляли эффективность в модели тромбоза с использованием артериовенозного шунтирования собак в доза-зависимом способе (способ описан в "Производные нипекотиновой кислоты в качестве антитромботических соединений", серийный заявки 08/213772, поданной 16 марта 1994). Например, соединения 16 ингибируют образование тромба при кумулятивных дозах 10, 30 и 100 мкг/кг/мин, введенных внутривенной инфузией (75%, 37%, 12% веса тромба относительно контроля с носителем соответственно). Соединение 18 ингибирует образование тромба при кумулятивных дозах 3, 10 и 30 мкг/кг/мин, введенных внутривенной инфузией (82%, 41%, 12% веса тромба относительно контроля с носителем соответственно).
Примеры
Защищенные аминокислоты были приобретены у Aldrich Chemical или Bachem Bioscience Inc. 2-Хлортритиловую смолу и смолу Wang получали от Novabiochem Corp. Энантиомерно-обогащенные этиловые эфиры циклоалкилиден-3-карбоновых кислот выделяли хиральным разделением рацемического материала, как опубликовано (A. M. Akkerman, Rec. Trav. Chim. Pays-Bas, 1951, 70, 899). Все другие химикаты были приобретены у Aldrich Chemical Company, Inc. Конечные продукты, кислотно-аддитивные соли, можно превратить в свободные основания основной ионнообменной хроматографией. Спектры высокополевого 1Н ЯМР регистрировали на спектрометре Bruker AC-360 при 360 МГц, константы взаимодействия даются в Герцах. Точки плавления определяли на устройстве для измерения точки плавления Mel-Temp II и не корректировали. Микроанализы проводили в Robertson Microlit Laboratories, Inc., Madison, New Jersey. В тех случаях, когда продукт получали в виде соли, свободное основание получали методами, известными специалистам в данной области, например основной ионообменной очисткой. В примерах и на тексте описания следующие аббревиатуры имеют значения, перечисленные ниже:
Вn или Bzl = Бензил
Воc = трет-Бутоксикарбонил
BOC-ON = 2-(трет-Бутоксикарбонилоксиимино)-2-фенилацетонитрил
ВОР-С1 = Хлорангидрид бис(2-оксо-3-оксазолидинил)фосфиновой кислоты
СР = соединение
DCE = 1,2-Дихлорэтан
DCM = Дихлорметан
DIBAL-H = Диизобутилалюминийгидрид
DIC = Диизопропилкарбодиимид
DIEA = Диизопропилэтиламин
DMAP = 4-Диметиламинопиридин
DMF = N,N-Диметилформамид
EDC = Этилдиметиламинопропилкарбодиимид
EDTA = Этилендиаминтетрауксусная кислота
Et2O = Простой эфир
HBTU = Гексафторфосфат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония
НОВТ = Гидроксибензотриазол
i-Pr = Изопропил
KOTMS = Триметилсиланолат калия
NMM = N-Метилморфолин
Nip = Нипекотил (если не оговорено особо, рацемический в 3-положении)
NT = не испытывали
РРТ = осадок
PTSA = п-Толуолсульфоновая кислота
КТ = комнатная температура
TFA = Трифторуксусная кислота
TMSN3 = Азидотриметилсилан
Z = Бензилоксикарбонил
Аллил-3-(4-пиперидин)пропионат • НСl (предшественник АД1)
К смеси 3-(4-пиридин)акриловой кислоты (10,0 г, 0,066 моль) и водного НС1 (2,0 N, 50 мл) в атмосфере азота добавляют оксид платины (IV) (0,54 г). Эту смесь гидрируют при 3,515 ат (50 psi) при КТ в течение 21 ч, фильтруют через целит (Celite) и выпаривают, получая 3-(4-пиперидин)пропионовая кислота • НС1 в виде белого порошка (12,9 г, 99%). Этот порошок обрабатывают аллиловым спиртом (50 мл) и нагревают при 50oС в течение 2 ч. Этот раствор охлаждают до КТ, выпаривают приблизительно до объема 10 мл и разбавляют Et2O (250 мл). Получаемый осадок собирают и промывают Et2O, получая белый порошок (14,5 г, 94%):
1H NMR (DMSO-d6) δ 8.7-9.1 (m, 2 Н), 5.9 (m, 1 Н), 5.25 (d, J=7.15, 2 Н), 4.53 (d, J=4, 2 Н), 3.21 (d, J=8, 2 Н), 2.74 (t, J=7, 2 Н), 2.35 (t, J= 4, 2 Н), 1.72 (d, J=8, 2 Н), 1.5 (m, 3 Н), 1,3 (m, 2 H); MS m/e 198 (МН+).
Метил-(S)-3-амино-3-(3-пиридидол)пропионат • 2НСl (AG5)
Промежуточный фенилацетамид AG3 получают с использованием стандартных методов, как показано на схеме AG (E.Profft, J. Pract. Chem., 1965, 30, 18). Смесь AG1 (0,47 моль), EtOH (100 мл), NH40ac (0,47 моль) и малоновой кислоты (0,70 моль) нагревают при кипячении с обратным холодильником в течение 6 ч, охлаждают и фильтруют. Белый твердый продукт промывают EtOH и МеОН и сушат. Этот твердый продукт растворяют в смеси 2:1 ацетон/вода (360 мл), обрабатывают триэтиламином (0,72 моль) и фенилацетилхлоридом (0,36 моль) и перемешивают в течение 22 ч. Смесь выпаривают и остаток растворяют в воде (500 мл) и рН устанавливают до 12 (IN NaOH). рН водного слоя устанавливают до 2 (концентр. НС1), экстрагируют Et2O и выпаривают до получения белой пены. Пену очищают хроматографией на силикагеле (10% MeOH/DCM), получая AG3. рН раствора соединения AG3 (0,22 моль) в воде (600 мл) при КТ устанавливают до 7,5 с использованием КОН (3,0 N) и раствор обрабатывают пенициллинамидазой (91520 единиц, Sigma). Эту смесь перемешивают в течение 47 г, подкисляют до рН 1 при помощи НС1 (концентр.) и получаемый осадок фильтруют через целит. Фильтрат экстрагируют Et2O (3•300 мл), концентрируют в вакууме и обрабатывают МеОН/концентр. NH4OH (9: 1). Этот содержащий продукт раствор очищают хроматографией на силикагеле (элюент DCM/MeOH/NH4OH, 78:18;4), получая аммониевую соль (S)-3-фенилацетамидо-3-(3-пиридил)пропионовой кислоты (19,5 г, 58%). Этот продукт обрабатывают СН1 (6,0 N, 292 мл), нагревают при кипячении с обратным холодильником в течение 5 ч, охлаждают до КТ и экстрагируют Et2O (3•200 мл). рН водного слоя устанавливают до рН 12, концентрируют в вакууме и получаемый твердый продукт растирают с МеОН (2•300 мл). Этот раствор выпаривают, получая приблизительно 14 г натриевой соли. Этот материал обрабатывают МеОН (500 мл), 2,2-диметоксипропаном (44 мл) и НС1 (4 N в диоксане, 84 мл) и перемешивают в течение 90 ч при КТ. Эту смесь фильтруют и фильтрат концентрируют в вакууме. Получаемый не совсем белый твердый продукт растирают с Et2O (2•150 мл) и сушат, получая соединение AG5 (16,7 г, 96% энантиомерного избытка, ее) в виде белого, аморфного твердого продукта.
Пример 1
N-3-(4-Пиперидинпропионил)нипекотил-(3-амино-3-фенил)пропионовая кислота • TFA (1)
В спекшийся стеклянный сосуд на 25 мл в атмосфере азота загружают 2-хлортритилхлоридную смолу (0,24 г, 0,36 ммоль, Novabiochem) и DMF (5 мл). Смолу перемешивают в
атмосфере азота в течение 5 мин для набухания и DMF удаляют. Смолу последовательно обрабатывают DMF (5 мл), DIEA (0,31 мл, 5 экв.) и аллил-3-(4-пиперидин)пропионат•НСl (0,20 г, 2,4 экв.) и перемешивают в течение 8 ч. Получаемый темно-зеленый раствор удаляют и смолу промывают DMF (3•5), водным DMF (25%, 3•5 мл), THF (3•5 мл), DCM (3•5 мл) и Et2O (5 мл). Смоле дают набухать в DCE (5 мл) и обрабатывают смесью гидрата фторида тетрабутиламмония (0,28 г, 3 экв.), азидотриметилсилана (0,38 мл, 10 экв.), тетракис(трифенилфосфин) палладия (0,084 г, 20 мол.%) и DCE (5 мл). Смолу перемешивают в течение 15 ч и оранжевый раствор удаляют. Смолу промывают DCM (3•5 мл), DMF (3•5 мл), THF (3•5 мл), THF (3•5 мл) и Et2O (5 мл). Смоле дают набухать в DMF (5 мл) и обрабатывают DIEA (0,18 мл, 3 экв.), аллилнипекотат • НСl (0,17 г, 3 экв.), DIC (0,17 мл, 3 экв.) и НОВТ (1 мг). Смолу перемешивают в течение 15 ч и затем реакционный раствор удаляют. Смолу промывают DMF ( 3•5 мл), водным DMF (25%, 3•5 мл), THF (3•5 мл), DCM (3•5 мл) и Et2O (5 мл). Смоле дают набухать в DCE (5 мл) и обрабатывают смесью гидрата фторида тетрабутиламмония (0,28 г, 3 экв. ), азидотриметилсилана (0,38 мл, 10 экв.), тетракис(трифенилфосфин)палладия (0,084 г, 20 мол.%) и DCE (5 мл). Смолу перемешивают в течение 15 ч и оранжевый раствор удаляют. Смолу промывают DCM (3•5 мл), DMF (3•5 мл), THF (3•5 мл) и Et2O (5 мл). Смоле дают набухать в DMF (5 мл) и обрабатывают DIEA (0,18 мл, 3 экв.), метил-D,L-3-амино-3-фенилпропионат • НСl (0,23 г, 3 экв.), DIC (0,17 мл, 3 экв.) и НОВТ (1 мг). Смолу перемешивают в течение 17 ч и затем реакционный раствор удаляют. Смолу промывают DMF (3•5 мл), водным DMF (25%, 3•5 мл), THF (3•5 мл), DCM (3•5 мл) и Et2O (5 мл). Смоле дают набухать в THF (5 мл) и обрабатывают раствором KOTMS (0,23 г, 10 экв.) и THF (2 мл). Смолу перемешивают в течение 18 ч и затем реакционный раствор удаляют. Смолу промывают DMF (3•5 мл), смесью уксусная кислота/THF (1: 1, два раза), водным DMF (25%, 3•5 мл), THF (3•5 мл), DCM (3•5 мл) и Et2O (5 мл). Смолу обрабатывают TFA/DCM (1:1, 10 мл), перемешивают в течение 15 мин и получаемый красный раствор собирают. Этот раствор выпаривают и получаемое масло растирают с Et2O (3•5 мл) и сушат, получая соединение 1 в виде прозрачного стекла (0,11 г):
1Н NMR (DMSO-d6) δ 8.6 (m, 1 Н), 8.42 (d, J=7, 1 Н), 8.2 (m, 1 H); 7.3 (m, 3 Н), 7.2 (m, 2 Н), 5.18 (d, J=6, 1 Н), 4.3 (m, 1 Н), 3.7 (m, 1 Н), 3.2 (m, 3 Н), 2.8 (m, 2 Н), 2.6 (m, 2 Н), 2.3 (m, 5 Н), 1.1-1.9 (m, 11 H); MS m/e 416 (МН+).
С использованием той же общей методики синтеза в твердой фазе, как описано в примере 1, соединения указанных примеров получали по схеме АА, как описано в конкретном примере.
Пример 2
N-(4-Пиперидинметиламинокарбонил)нипекотил-(З-амино-2-метил)-пропионовая кислота • TFA (2)
Соединение 2 получают, как показано на схеме АА. Связанному смолой 4-пиперидинметиламину (0,36 ммоль) дают набухать в DCE (5 мл), обрабатывают п-нитрофенилхлорформиатом (0,36 ммоль) и DIEA (0,36 ммоль), перемешивают в течение 1 ч, и растворитель удаляют. Смолу промывают (см. пример 1), дают ей набухать в DCE (5 мл), обрабатывают солью аллилнипекотат • НСl (0,36 ммоль) и DIEA (0,72 ммоль) и перемешивают в течение 16 ч. Растворитель удаляют, смолу промывают (см. пример 1) и аллиловый эфир расщепляют до соответствующей кислоты (см. пример 1). Смоле дают набухать в DMF (5 мл), кислоту сочетают с метил-3-амино-2-метилпропионатом (0,36 ммоль) и синтез завершают, как показано в примере 1. Соединение 2 выделяют в виде прозрачного стекла (0,11 г):
1H NMR (СD3ОD) δ 3.9 (m, 2 Н); 3.2 (m, 4 Н), 3.10 (d, J=7, 2 H), 2.9 (m, 3Н), 2.6 (m, 2 Н), 2.3 (m, 1 Н), 1.9 (m, 4 Н), 1.7-1.9 (m, 5 Н), 1.3-1.5 (m, 5 Н), 1.11 (d, J=7, 3 H); MS m/е 35Б (МН+).
Пример 3
α-Метиловый эфир N-(4-пиперидинметилоксикарбонил) нипекотил-D-аспарагиновой кислоты • TFA (3)
Соединение 3 получают, как показано на схеме АА. Связанному смолой 4-пиперидинметанолу (0,36 ммоль) дают набухать в DCE (5 мл), обрабатывают п-нитрофенилхлорформиатом (0,36 ммоль) и DIEA (0,36 ммоль), перемешивают в течение 1ч, и растворитель удаляют. Смолу промывают (см. пример 1), дают набухать в DCE (5 мл), обрабатывают аллилнипекотат • НСl (0,36 ммоль) и DIEA (0,72 ммоль) и перемешивают в течение 16 ч. Растворитель удаляют, смолу промывают (см. пример 1) и аллиловый эфир расщепляют до соответствующей кислоты (см. пример 1). Смоле дают набухать в DMF (5 мл), кислоту сочетают с H-D-Asp(oBn)-Оме (0,36 ммоль) и синтез завершают, как показано в примере 1. Соединение 3 выделяют в виде желтого стекла (0,019 г):
1H NMR (СD3ОD) δ 4.8 (m, 2 Н), 3.9 (m, 3 Н), 3.70 (d, J=9, 4 Н), 3.39 (s, 3 Н), 3.3 (m, 2 Н), 2.9 (m, 4 Н), 2.8 (m, 2 Н), 1.9 (m, 4 Н), 1.7 (m, 2 Н), 1.4 (m, 4 H); MS m/e 400 (МН+).
Пример 4
N-3-(4-Пиперидинпропионил)пирролидин-3-карбокси-[3-амино-3-(4-тодил)] пропионовая кислота • TFA (4)
Соединение 3 получают, как показано на схеме АА. Промежуточному АА2 (0,36 ммоль) дают набухать в DCE (5 мл), обрабатывают метилпирролидин-3-карбоксилат • НСl (0,36 ммоль), DIC (0,72 ммоль) и DIEA (0,72 ммоль) и перемешивают в течение 16 ч. Растворитель удаляют, смолу промывают (см.пример 1) и метиловый эфир расщепляют до соответствующей кислоты при помощи KOTMS (см. пример 1). Смоле дают набухать в DMF (5 мл), кислоту сочетают с метил-3-амино-3-(4-толил)пропионатом (0,36 ммоль) и затем синтез завершают, как показано в примере 1. Соединение 4 выделяют в виде прозрачного стекла (0,081 г):
1H NMR (СD3OD) δ 7.19 (d, J=5, 2 H), 7.10 (d, J=5, 2 Н), 5.31 (dd, J= 3,10; 1 H) 3.6 (m, 4 Н), 3.3 (m, 2 Н), 2.9 (m, 4 H), 2.7 (m, 2 Н), 2-3 (m, 2 Н), 2.1 (m, 3 Н), 1.9 (m, 4 Н), 1.6 (m, 4 Н), 1.3 (m, 4 H); MS m/e 416 (MH+).
Пример 5
N-3-(4-Пиперидинпропионил)изонипекотил-(3-амино-3-метил)пропионовая кислота • TFA (5)
Соединение 5 получают, как показано на схеме АА. Промежуточному АА2 (0,36 ммоль) дают набухать в ОСЕ (5 мл), обрабатывают этилизонипекотатом (0,36 ммоль), DIC (0,72 ммоль) и DIEA (0,72 ммоль) и перемешивают в течение 16 ч. растворитель удаляют, смолу промывают (см. пример 1) и этиловый эфир расщепляют до соответствующей кислоты при помощи KOTMS (см. пример 1). Смоле дают набухать в DMF (5 мл), кислоту сочетают с метил-3-амино-3-метилпропионатом (0,36 ммоль) и затем синтез завершают, как показано в примере 1. Соединение 5 выделяют в виде рыжевато-коричневого стекла (0,033 г):
1H NMR (СD3ОD) δ 4.5 (m, 1 Н), 4.2 (m, 1 Н), 3.9 (m, 1 Н), 3.3 (m, 2 H), 3,3 (m, 3 Н) 3.1 (m, 1 H), 2.9 (m, 3 Н), 2.7 (m, 2 Н), 2.4 (m, 2 Н), 2.0 (m, 2 Н), 1.7 (m, 2 Н) 1.5 (m, 6 Н), 1.3 (m, 2 Н), 1.15 (d, J=9, 3 H); MS m/e 354 (МН+).
Пример 6
N-3-(4-Пиперидинпропионил)изонипекотил-[3-амино-3-(4-карбоксифенил)пропионовая кислота • TFA (6)
Соединение 6 получают, как показано на схеме АА. Промежуточному АА2 (0,36 ммоль) дают набухать в DCE (5 мл), обрабатывают этилизонипекотатом (0,36 ммоль), DIC (0,72 ммоль) и DIEA (0,72 ммоль) и перемешивают в течение 16 ч, растворитель удаляют, смолу промывают (см. пример 1) и этиловый эфир расщепляют до соответствующей кислоты KOTMS (см. пример 1). Смоле дают набухать в DMF (5 мл), кислоту сочетают с метил-3-амино-3-(4-карбоксиметилфенил)пропионатом (0,36 ммоль) и затем синтез завершают, как показано в примере 1. Соединение 6 выделяют в виде рыжевато-коричневого стекла (0,034 г):
1Н NMR (СD3ОD) δ 7.9 (m, 3 Н), 7.43 (d, J = 5, 2 Н), 5.4 (m, 1 H), 4.5 (m, 1 Н), 4.0 (m, 1 Н), 3.3 (m, 4 Н), 3.1 (m, 1 Н), 2.9 (m, 2 Н), 2.7 (m, 2 Н), 2.7 (m, 1 Н), 2.5 (m, 4 Н), 2.0 (m, 2 Н), 1.2-1.9 (m, 10 H); MS m/e 460 (МН+).
Пример 7
N-3-(4-N-Метилпиперидинпропионил)нипекотил-3-аминопропионовая кислота • TFA (7)
Соединение 7 получают, как показано на схеме AD. Связанный смолой Fmos-β-Ala (1 ммоль) обрабатывают смесью 20% пиперидин/DMF (10 мл), перемешивают в течение 2 ч, и растворитель удаляют. Смолу промывают DMF, дают набухать в DMF (10 мл) и обрабатывают Fmos-нипекотиновой кислотой (1 ммоль), DIC (2 ммоль) и DIEA (1 ммоль). Смолу перемешивают в течение 16 ч, растворитель удаляют и смолу промывают DMF и DCM. Смолу промывают смесью 20% пиперидин/DMF (10 мл) в течение 2 ч, растворитель удаляют и смолу промывают DMF. Смоле дают набухать в DMF (10 мл), обрабатывают 4-N-метилпиперидинпропионовой кислотой (1 ммоль), DIC (2 ммоль) и DIEA (1 ммоль) и перемешивают в течение 16 ч. Растворитель удаляют и смолу промывают DMF и DCM. Смолу расщепляют 95% TFA (10 мл) и TFA выпаривают, получая 7 в виде белого порошка (0,26 г): т.пл. 172-177oС;
1H NMR (СDCl3) δ 4.4 (m, 1 Н), 3.7 (m, 1 Н), 3.4 (m, 1 Н), 3.2 (m, 1 Н), 3.1 (m, 1 Н), 2.7 (m, 2 Н), 2.3 (m, 6 Н), 2.21 (s, 3 Н), 1.9 (m, 4 Н), 1.3-1.8 (m, 10 H); MS m/e 354 (МН+).
Пример 8
N-3-(4-Пиперидинпропионил)нипеколит-4-оксонипекотиновая кислота • TFA (8)
Соединение 8 получают, как показано на схеме АА. Промежуточному АА2 (0,36 ммоль) дают набухать в DCE (5 мл), обрабатывают этилнипекотатом (0,36 ммоль), DIC (0,72 ммоль) и DIEA (0,72 ммоль) и перемешивают в течение 16 ч. Растворитель удаляют, смолу промывают (см. пример 1) и этиловый эфир расщепляют до соответствующей кислоты KOTMS (см. пример 1). Смоле дают набухать в DMF (5 мл), кислоту сочетают с метил-4-оксонипекотатом (0,36 ммоль) и затем синтез завершают, как показано в примере 1. Соединение 8 выделяют в виде прозрачного стекла (0,04 г):
1H NMR (DMSO-d6) δ 8.5 (m, 1 Н), 8.2 (m, 1 Н), 6.5 (m, 1 Н), 4.3 (m, 1 Н), 3.4 -3.8 (m 4 Н), 3.2 (m, 2 Н), 3.0 (m, 1 Н), 2.8 (m, 2 H), 2.2-2.6 (m, 6 Н), 1.8 (m, 2 Н) 1.1-1.7 (m, 11 H); MS m/e 394 (MH+).
Пример 9
N-3-(4-Пиперидинпропионил)нипекотил-[3-амино-3-(2-триметилсилилэтинил)] пропионовая кислота • TFA (9)
Соединение 9 получают, как показано на схеме АА. Промежуточному АА2 (0,36 ммоль) дают набухать в DCE (5 мл), обрабатывают этилнипекотатом (0,36 ммоль), DIC (0,72 ммоль) и DIEA (0,72 ммоль) и перемешивают в течение 16 ч. Растворитель удаляют, смолу промывают (см. пример 1) и этиловый эфир расщепляют до соответствующей кислоты KOTMS (см. пример 1). Смоле дают набухать в DMF (5 мл), кислоту сочетают с метил-3-амино-3-(2-триметилсилилэтинил) пропионатом (для получения см. J. Zablocki, J. Med. Chem., 1995, 38, 2378; 0,36 ммоль) и затем синтез завершают, как показано в примере 1. Соединение 9 выделяют в виде желтого стекла (0,12 г):
1H NMR (СD3ОD) δ 3.8 (m, 1 Н), 3.2-3.4 (m, 4 Н), 2.9 (m, 3 Н), 2.7 (m, 2 Н), 2.3-2.5 (m, 2 Н), 1.9 (m, 4 Н), 1.1-1.9 (m, 13 Н), 0.0 (s, 9 H); MS m/e 436 (МН+).
Пример 10
N-(6-Аминокапроил)нипекотил-3-амино-3-(3-пиридил) пропионовая кислота • 3TFA (10)
Соединение 10 получают, как показано на схеме АА. Связанной смолой 6-аминокапроновой кислоте (0,36 ммоль) дают набухать в DCT (5 мл), обрабатывают этилнипекотатом (0,36 ммоль), DIC (0,72 ммоль) и DIEA (0,72 ммоль) и перемешивают в течение 16 ч. Растворитель удаляют, смолу промывают (см. пример 1) и этиловый эфир расщепляют до соответствующей кислоты при помощи KOTOMS (см. пример 1). Смоле дают набухать в DMF (5 мл), кислоту сочетают с метил-3-амино-3-(3-пиридил)пропионатом (0,36 ммоль) и затем синтез завершают, как показано в примере 1. Соединение 10 выделяют в виде прозрачного стекла (0,008 г):
1H NMR (DMSO-d6) δ 8.6 (m, 2 Н), 8.1 (s, 1 Н), 7.0-7.7 (m, 5 Н), 5.15 (t, J= 3, 1 Н), 4.4 (m, 1 Н), 4.1 (m, 1 Н), 3.7 (m, 2 Н), 3.1 (m, 1 Н), 2.7 (m, 4 Н), 2.5 (m, 1 Н), 2.3 (m, 2 Н), 1.2-1.9 (m, 11 H); MS m/e 391 (МН+).
Анализ. Вычислено для С20Н30N4O4 • 3 TFA • 2Н2O (768,60): С 40,63; Н 4,85; N 7,29; F 22,25. Найдено: С 40,81; H 4,70; N 6,12; F 23,83.
Пример 11
N-3-(4-Пиперидинпропионил)-R-(-)-нипекотил-(3-амино-2-гидрокси)-пропионовая кислота • TFA (11)
Соединение 11 получают, как показано на схеме АА. Промежуточному АА2 (0,36 ммоль) дают набухать в ОСЕ (5 мл), обрабатывают этил-R-нипекотатом (0,36 ммоль), DIC (0,72 ммоль) и DIEA (0,72 ммоль) и перемешивают в течение 16 ч. Растворитель удаляют, смолу промывают (см. пример 1) и этиловый эфир расщепляют до соответствующей кислоты при помощи KOTOMS (см. пример 1). Смоле дают набухать в DMF (5 мл), кислоту сочетают с метил-3-амино-2-гидроксипропионатом (0,36 ммоль) и затем синтез завершают, как показано в примере 1. Соединение 11 выделяют в виде розового стекла (0,05 г):
1H NMR (DMSO-d6) δ 8.5 (m, 1 Н), 8.2 (m, 1 Н), 7.8 (m, 1 Н), 4.0-4.4 (m, 2 Н), 3.7 (m, 1 Н), 3.2 (m, 3 Н), 2.8 (m, 3 Н), 2.6 (m, 1 Н), 2.1-2.3 (m, 3 Н), 1.8 (m, 4 Н), 1.0-1.4 (m, 10 H); MS m/e 358 (МН+).
Пример 12
N-3-(4-Пиперидинэтансульфонил)нипекотил-3-аминопропионовая кислота • НСl (12)
Соединение 12 получали, как показано на схеме АЕ. Промежуточный АЕ1 синтезируют следующей методикой. 2-(4-Пиридин)-этансульфоновую кислоту (3,0 г, 0,016 моль) растворяют в водной НСl (2,0 N, 12 мл) и этот раствор обрабатывают диоксидом платины (0,13 г) и гидрируют при 3,515 ат (50 psi) при КТ в течение 18 ч. Смесь фильтруют через целит и выпаривают, получая 2-(4-пиперидин)этансульфоновая кислота • НСl (3,5 г, белый порошок). Этот порошок растворяют в водном THF (1:1, 70 мл) при КТ и обрабатывают NMM (3,7 мл, 2,2 экв. ) и бензилхлорформиатом (2,2 мл, 1 экв.). Эту смесь перемешивают в течение 15 ч, подкисляют водной лимонной кислотой и экстрагируют СНСl3 (2•100 мл). Органический слой сушат Na2SO4 и выпаривают, получая 2-(4-N-Z-пиперидин)этансульфоновую кислоту (2,75 г, золотистое масло). Это масло превращают в конечный продукт 12 в пять синтетических стадий (схема АЕ, W.J. Hoekstra, J. Med. Chem., 1995, 38, 1582) и выделяют в виде прозрачного стекла (0,060 г):
1H NMR (DMSO-d6) δ 8.9 (m, 1 Н), 8.6 (m, 1 Н), 3.5 (m, 2 Н), 3.1- 3.3 (m, 4 Н), 3.0 (m, 2 Н), 2.6-2.8 (m, 4 Н), 2.3 (m, 3 Н), 1.65-1.9 (m, 5 Н), 1.6 (m, 3 Н), 1.2-1.4 (m, 5 H); MS m/e 376 (МН+).
Пример 13
N-3-(4-Пиперидинпропионил)нипекотил-5Н-(2-аминоэтил) тетразол • НСl (13)
Соединение 13 получают, как показано в схеме АС. Промежуточный АСl (получен, как в W.J. Hoekstra, J. Med. Chem., 1995, 38, 1582; 1,9 ммоль) растворяют в DCM (50 мл) и обрабатывают BOP-CI (1,9 ммоль), NMM (1,9 ммоль) и 3-аминопропионитрилом (1,9 ммоль). Реакционную смесь перемешивают в течение 18 ч, разбавляют насыщенным NH4Cl и слои разделяют. Органический слой выпаривают и продукт очищают хроматографией на силикагеле (10% EtOH/DCM), получая масло. Масло растворяют в толуоле (10 мл), обрабатывают азидотриметилсиланом (2,4 ммоль) и оксидом дибутилолова (1,2 ммоль) и нагревают при кипячении с обратным холодильником в течение 16 ч. Охлаждение дает коричневый осадок, который растирают с Et2O. Этот твердый продукт гидрируют над диоксидом платины (0,08 г) в МеОН (12 мл) при 3,515 ат (50 psi) в течение 15 ч, фильтруют и выпаривают, получая 13 в виде желтой пены (0,065 г):
1H NMR (DMSO-d6) δ 8.9 (m, 1 Н), 8.6 (m, 1 Н), 8.13 (d, J=28, 1 Н), 4.2 (m, 2 Н), 3.2 (m, 3 Н), 3.0 (m, 4 Н), 2.7 (m, 4 Н), 2.31 (q, J=8, 2 Н), 1.7-1.9 (m, 3 Н), 1.4-1.6 (m, 5 Н), 1.1-1.3 (m, 4 H); MS m/e 364 (МН+).
Пример 14
N-3-(4-N-Метилпиперазинпропионил)-нипекотил-[3-амино-3-(3,4-метилендиоксифенил)]пропионовая кислота • Na (14)
Соединение 14 получают, как показано на схеме АВ. Этилнипекотат (3 ммоль) растворяют в DCM (50 мл), обрабатывают акрилоилхлоридом (3 ммоль) и NММ (3 ммоль) и перемешивают в течение 1 ч. Растворитель выпаривают, и остаток растворяют в EtOH (50 мл) и обрабатывают N-метилпиперазином (3 ммоль). Раствор нагревают при 60oС в течение 15 ч, охлаждают до КТ, и растворитель выпаривают. Остаток распределяют между DCM (100 мл) и водой (10 мл) и слои разделяют. Органический слой сушат и выпаривают, получая пену. Пену растворяют в воде, обрабатывают NaOH (3 ммоль), перемешивают в течение 1 ч и выпаривают, получая АВ3-Na. Этот синтез завершают, как иллюстрировано (W.J. Hoekstra, J. Med. Chem., 1995, 38, 1582), с использованием метил-3-амино-3-(3,4-метилендиоксифенил)пропионата (2,5 ммоль), получая 14 в виде белого аморфного твердого продукта (0,14 г):
1Н NMR (D2O) δ 6.8 (m, 3 Н), 5.91 (s, 2 Н), 5.0 (m, 1 Н), 4.0 (m, 1 H); 3.7 (m, 1 Н), 2.8-3.4 (m, 11 Н), 2.69 (s 3 Н), 2.4-2.6 (m, 7 Н), 1.9 (m, 1 Н), 1.7 (m, 2 Н), 1.5 (m, 1 H); MS m/e 475 (МН+).
Анализ. Вычислено для C24H33N4O6 • Na • Н2O (514,56): С 56,02; Н 6,86; N 10,89. Найдено: С 55,72; Н 6,78; N 10,52.
Пример 15
N-3-(4-N-Метилпиперазинпропионил)-нипекотил-[3-амино-3-(3-хинолинил)] пропионовая кислота • 3TFA (15)
Соединение 15 получают, как описано в примере 14. Синтез завершают, как иллюстрировано (W.J.Hoekstra, J. Med. Chem., 1995, 38, 1582), с использованием метил-3-амино-3-(3-хинолинил)пропионата (6 ммоль) с АВ3. Соединение 15 выделяют в виде желтого порошка (1,89 г):
1Н NMR (DMSO-d6) δ 8.94 (s 1 Н), 8.12 (s, 1 Н), 7.9 (m, 2 Н), 7.6 (m, 2 Н), 7.07 (d, J=4, 1 H), 5.2 (m, 1 Н), 4.1 (m, 1 Н), 3.7 (m, 1 Н), 3.1-3.3 (m, 2 Н), 2.9 (m, 2 Н), 2.6 (m, 2 Н), 2.43 (s, 3 Н), 1.9-2.4 (m, 12 Н), 1.2-1.5 (m, 4 H); MS m/e 482 (MH+).
Пример 16
N-3-(4-Пиперидинпропионил)-R-(-)-нипекотил-[(S)-3-амино-3-(3,4-Пиперидинпропионил)-R-(-)-нипекотил-[(S)-3-амино-3- (3, 4-метилендиоксифенил) ] пропионовая кислота • НСl (16)
К охлажденному (5oС) раствору Boc-R-нипекотиновой кислоты (9 ммоль) и метил-(S)-3-амино-3-(3,4-метилендиоксифенил)пропионата (см. пример AG5; 9 ммоль) в MeCN (100 мл) добавляют HBTU (9 ммоль), НОВТ (9 ммоль) и NMM (18 ммоль). Эту смесь перемешивают в течение 15 ч, разбавляют водой (10 мл) и выпаривают. Остаток разбавляют EtOAc (100 мл) и органический слой сушат и выпаривают, получая белую пену. Пену обрабатывают НCI (2 N в диоксане, 20 мл), перемешивают в течение 3 ч и выпаривают до пены. Пену растворяют в MeCN (100 мл) и обрабатывают Вос-пиперидинпропионовой кислотой (7 ммоль), HBTU (7 ммоль), НОВТ (7 ммоль) и NMM (14 ммоль) с перемешиванием в течение 6 ч. Смесь разбавляют водой (10 мл), выпаривают и разбавляют EtOAc (100 мл). Органический слой сушат, выпаривают и очищают хроматографией на силикагеле (7% EtOH/DCM), получая пену. К раствору, пены (4,6 моль) в THF, охлажденному на ледяной бане, по каплям добавляют LiOH • Н2O (6,9 ммоль, растворенный в 30 мл воды). Эту смесь перемешивают в течение 1,5 ч, подкисляют АсОН (1,7 мл) и нагревают до КТ. Раствор разбавляют СНCIз (75 мл), и слои разделяют. Органический слой сушат (Nа2SO4) и выпаривают, получая белую пену. Пену растворяют в диоксане (20 мл) и анизоле (0,3 мл), охлаждают на ледяной бане, обрабатывают НCI (15 мл, 4,0 N в диоксане) и перемешивают в течение 3 ч, получая осадок. Осадок фильтруют и промывают Et2O (150 мл) и MeCN (20 мл), получая 16 в виде белого порошка (1,78 г); т.пл. 190-200oС;
1H NMR (DMSO-d6) δ 8.9 (m, 1 Н), 8.6 (m, 1 Н), 8.4 (m, 1 Н), 6.83 (d, J= 5, 1 Н), 6.79 (d, J=5, 1 Н), 6.7 (m, 1 Н), 5.95 (s, 2 Н), 5.08 (dd, J=5, 11.1 Н), 4.1-4.3 (m, 1 Н), 3.7 (m, 1 Н), 3.15 (d, J=10, 2 Н), 3.0 (m, 1 Н), 2.7 (m, 2 Н), 2.8 (m, 3 H), 2.31 (d, J=7, 2 Н), 1.81 (d, J=10, 2 Н), 1.2-1.7 (m, 11 H); MS m/e 480 (МН+); [α]24D =0.478o (с 1.00. МeОН),
Пример 17
N-3-(4-Пиперидинпропионил)-гексагидроазепин-3-карбокси-[3-амино-3-(3-хинолинил)]пропионовая кислота • 2 TFA (17)
Соединение 17 получают, как показано на схеме AА. Промежуточному АА2 (0,36 ммоль) дают набухать в DCE (5 мл), обрабатывают метилгексагидроазепин-3-карбоксилат • НCI (0,36 ммоль), DIC (0,72 ммоль) и DIEA (0,72 ммоль) и перемешивают в течение 16 ч. Растворитель удаляют, смолу промывают (см. пример 1) и метиловый эфир расщепляют, превращая в соответствующую кислоту при помощи KOTMS (см. пример 1). Смоле дают набухать в DMF (5 мл), кислоту сочетают с метил-3-амино-3-(3-хинолинил)-пропионатом (0,36 ммоль) и затем синтез завершают, как показано в примере 1. Соединение 17 выделяют в виде стекла (0,10 г):
1H NMR (D2O) δ 9.06 (s, 1 Н), 8.9 (m, 1 Н), 8.2 (m, 1 Н), 8.04 (s, 1 Н), 8.0 (t, J=4, 2 Н), 7.8 (t, J=4, 2 Н), 5.6 (m, 1 Н), 3.8 (m, 1 Н), 3.3 (m, 4 Н), 3.0 (m, 2 Н), 2.7 (m, 4 Н), 2.0-2.4 (m, 6 Н), 1.7-1.9 (m, 4 Н), 1.1-1.6 (m, 8 H); MS m/e 481 (МН+).
Пример 18
N-3-(4-Пиперидинпропионил)-R-(-)-нипекотил-[(S)-3-амино-3-(3-хинолинил)] пропионовая кислота • 2 НCI (18)
Соединение 18, полученное, как описано в примере 16, исходя из Boc-R-нипекотиновой кислоты (7,1 ммоль) и метил-(S)-3-амино-3-(3-хинолинил)пропионата (см. пример AG5; 7,1 ммоль), выделяют в виде белых хлопьев (1,11 г): т.пл. 142-144oС; МС m/е 467 (МН+); [α]24D=-173o (с = 0,1, МеОН), Анализ. Вычислено для C26H34N4O4 • 2,25 НCI • Н2О (566,64): С 55,11; Н 6,80; N 9,89; Cl 14,08. Найдено: С 54,85; Н 6,62; N 10,04; Cl 13,68.
Пример 19
N-3-(4-Пиперидинпропионил)-R-(-)-нипекотил-[(S)-3-амино-3-(2-трет-бутилэтинил)]пропионовая кислота • НCI (19)
Соединение 19, полученное, как описано в примере 16, исходя из Boc-R-нипекотиновой кислоты (3,2 ммоль) и метил-(S)-3-амино-3-(2-трет-бутилэтинил)пропионата (см. J. A. Zablocki, J. Med. Chem., 1995, 38, 2378; 3,2 ммоль), выделяют в виде белого порошка (0,33 г); МС m/е 420 (МН+). Анализ. Вычислено для C23H37N3O4 • 1,07 HCl • 0,43 Н2О (468,97): С 59,21; Н 8,42; N 8,96; Cl 8,09. Найдено: С 58,92; Н 8,58; N 8,76; Cl 7,82.
Пример 20
N-3-(4-Пиперидинпропил)-нипекотил-[(S)-3-амино-3-(3,4-метилендиоксифенил)]пропионовая кислота • 2TFA (20)
Соединение 20 получают, как показано на схеме AF. Промежуточный AF3 (2,8 ммоль) растворяют в бензоле (50 мл), обрабатывают этилнипекотатом (2,8 ммоль) и нагревают при кипячении с обратным холодильником в течение 7 ч. Реакционную смесь охлаждают, распределяют между водой (15 мл) и EtOAc (70 мл) и слои разделяют. Органический слой сушат и выпаривают, получая AF4. AF4 превращают в 20, как описано ранее ((W.J. Hoekstra, J.Med. Chem., 1995, 38, 1582), и выделяют в виде белого порошка (0,33 г):
1Н NMR (СD3ОD) δ 8.6-8.8 (m, 3 Н), 6.7-6.9 (m, 3 Н), 5.91 (s, 2 Н), 5.1-5.2 (m, 1 Н), 3.3-3.5 (m, 4 Н), 2.8-3.1 (m, 6 Н), 2.6-2.7 (m, 3 Н), 1.5-2.0 (m, 11 Н), 1.2-1.4 (m, 4 Н); MS m/e 446 (МН+).
Пример 21
N-3- (4-Пиперидинпропионил) -R- (-) -нипекотил- [ (S) -3-амино-3-(3-пиридил)]пропионовая кислота • 2 TFA (21)
Соединение 21, полученное, как описано в примере 16, исходя из Boc-R-нипекотиновой кислоты (6,4 ммоль) и метил -(S)-3-амино-3-(3-пиридил)пропионата (см. пример AG5; 6,4 ммоль), выделяют в виде белого аморфного твердого продукта (1,60 г): т.пл. 74-81oС; МС m/e 417 (МН+). Анализ. Вычислено для C22H32N4O4 • 2,1 С2НF3O2 • 0,7 Н2O (668,58): С 47,07; Н 5,35; N 8,38; F 17,90; KF 1,89. Найдено: С 47,08; Н 5,31; N 8,41; F 17,68; KF 2,00.
Пример 22
N-3-(4-Пиперидинпропионил)-R-(-)-нипекотил-[(S)-2-(3-метоксианилино)карбониламино-3-амино]пропионовая кислота (22)
Метил-Вос-R-нипекотил-[(S)-2-Z-амино-3-амино] пропионат (получен из метил-N-α-Z-L-диаминопропионата и Вос-R-нипекотиновой кислоты, как показано в примере 16; 9,5 ммоль) растворяют в МеОН (40 мл) и гидрируют при 3,515 ат (50 psi) над гидроксидом палладия (0,4 г) в течение 24 ч. Смесь фильтруют и выпаривают, получая белый твердый продукт АН2. АН2 (9,1 ммоль) растворяют в DCM (100 мл), охлаждают (5oС), обрабатывают 3-метоксифенилизоцианатом (9,1 ммоль) и NMM (9,1 ммоль) и перемешивают в течение 17 ч. Раствор разбавляют насыщенным NН4Сl (10 мл), слои разделяют и органический слой сушат, выпаривают до масла и очищают хроматографией на силикагеле (4% EtOH/DCM), получая АН3. Промежуточный АН3 превращают в 22 в четыре стадии, как в примере 16, получая белый аморфный твердый продукт (1,35 г): т.пл. 72-76oС;
1H NMR (DMSO-d6) δ 8.7 (m, 3 H): 7.8 (m, 1 Н), 7.1 (m 2 Н), 6.8 (d, 1 Н), 6.5 (d 2 Н), 3.66 (s, 3 Н), 3.4 (m, 2 Н), 3.2 (d, 2 Н), 2.7 (dd, 4 Н), 2,3 (m, 3 Н), 1.6 (m, 3 Н), 1.1-1.7 (m, 11 H); MS m/e 504 (МH+).
Анализ. Вычислено для C25H37N5O6 • 1,2 HCl • 1,0 Н2O (565,37): С 53,11; Н 7,17; N 12,39; Cl 7,53. Найдено: С 53,40; Н 7,44; N 12,14; Cl 7,66.
С использованием той же самой общей методики синтеза, как описано в примере 22, соединения примеров 26, 28-30 получают по схеме АН, указанной в конкретном примере. Для карбаматных аналогов применяемым ацилирующим агентом был подходящий алкилхлорформиат (аналогичное превращение АН2 в АН3; один молярный эквивалент). Для сульфонамидов применяемым сульфирующим агентом был подходящий сульфонилхлорид (один молярный эквивалент).
Пример 23
N-3-(4-Пиперидинпропионил)-R-(-)-нипекотил-[(S)-2-бензилоксикарбониламино-3-амино]пропионовая кислота • НCI (23)
Соединение 23, полученное из метил-N-α-Z-L-диаминопропионата (8,8 ммоль) и Boc-R-нипекотиновой кислоты (8,8 ммоль), как показано в примере 16, выделяют в виде белого порошка (1,65 г): т.пл. 110-113oС; МС m/е 489 (МН+).
Анализ. Вычислено для C25H36N4O6 • 1,15 НСl • 0,5 Н2О • 0,5 диоксан (583,57): С 55,56; Н 7,41; N 9,60; CI 6,99. Найдено: С 55,23; Н 7,79; N 9,85; CI 7,01.
Пример 24
N-3-(4-Пиперидинпропионил)-R-(-)-нипекотил-[(S)-2-(3-хлорбензилокси)карбониламино-3-амино]пропионовая кислота • НCI (24)
Соединение 24, полученное реакцией 3-хлорбензилоксикарбонилхлорида (6,6 ммоль) с АН2 (6,6 ммоль), как описано в примере 22, выделяют в виде белого аморфного твердого продукта (1,33 г): т. пл. 89-96oС; МС m/е 524 (МН+). Анализ. Вычислено для C25H35N4O6 • 1,25 HCl • 0,5 Н2О • 1,0 диоксан (637,20): С 50,89; Н 7,08; N 8,78; Cl 12,52. Найдено: С 51,10; Н 6,71; N 8,38; Cl 12,20.
Пример 25
N-3-(4-Пиперидинпропионил)-R-(-)-нипекотил-[(S)-2-бензилсульфониламино-3-амино]пропионовая кислота • HC1 (25)
Соединение 25, полученное реакцией бензилсульфонилхлорида (5,2 ммоль) с АН2 (5,2 ммоль), как показано в примере 22, выделяют в виде белого порошка (0,87 г): т.пл. 145-149oС; МС m/е 509 (МН+). Анализ. Вычислено для С24Н36N4O6S • 1,3 HCl • 0,3 диоксан (568,06): С 50,75; Н 7,04; N 9,86; Cl 8,11. Найдено: С 51,03; Н 6,93; N 9,46; Cl 7,85.
Пример 26
N-3-(4-Пиперидинпропионил)-R-(-)-нипекотил-[(S)-2-(3,5-диметоксианилино)карбониламино-3-амино]пропионовая кислота • HC1 (26)
Соединение 26, полученное реакцией 3,5-диметоксифенил-изоционата (10,2 ммоль) с АН2 (10,2 ммоль), как показано в примере 22, выделяют в виде белого порошка (1,89 г): т.пл. 190-193oС; МС m/е 534 (МН+). Анализ. Вычислено для С26Н39N5O7 • 1,2 HCl • 0,2 диоксан (585,40): С 53,35; Н 7,20; N 11,96; Cl 7,27. Найдено: С 53,48; Н 7,38; N 12,05; Cl 6,97.
Пример 27
N-(4,4'-Бипипepидин-1-ил)карбонил] -R-(-)-нипекотил-[(S)-3-амино-3-(3-пиридил)]пропионовая кислота • 3НСl (27)
Промежуточный АJ1 (5,5 ммоль), полученный, как показано в примере 16, растворяют в DCM (140 мл), охлаждают (5oС), обрабатывают п-нитрофенилхлорформиатом (5,5 ммоль) и (16,5 ммоль) и перемешивают в течение 2 ч. Смесь разбавляют водой (15 мл), слои разделяют и органический слой сушат и выпаривают до масла. Масло растворяют в MeCN (70 мл), обрабатывают М-Вос-4,4'-бипиперидином (7,5 ммоль) и DMAP (5,5 ммоль) и нагревают при кипячении с обратным холодильником в течение 24 ч. Смесь охлаждают, выпаривают до получения твердого продукта и распределяют между EtOAc (150 мл) и NaOH (1 N, 20 мл). Слои разделяют и органический слой сушат, выпаривают до получения твердого продукта и очищают хроматографией на силикагеле (8% EtOH/DCM), получая AJ2 в виде зеленого стекла (1,5 ммоль). AJ2 омыляют и освобождают от защитной группы, как описано в примере 16, получая 27 в виде бледно-желтого порошка (0,73 г): т.пл. 121-125oС; МС m/е 472 (МН+). Анализ. Вычислено для C25H37N5O4 • 3,6 НСl • 1,0 диоксан (690,98): С 50,41; Н 7,09; N 10,14; Cl 18,47. Найдено: С 50,80; Н 7,31; N 10,20; Cl 18,78.
Пример 28
N-3-(4-Пиперидинпропионил)-R-(-)-нипекотил-[(S)-2-(2-нафтиламино)карбониламино-3-амино]пропионовая кислота НСl • (28)
Соединение 28, полученное реакцией 2-нафтилизоцианата (8,5 ммоль) с АН2 (8,5 ммоль), как показано в примере 22, выделяют в виде белого порошка (1,65 г): т. пл. 187-193oС; МС m/е 524 (МН+). Анализ. Вычислено для С28Н37N5О5 • 1,36 НСl • 0,72 диоксан (602,07): С 55,86; Н 7,39; N 11,63; Сl 8,01. Найдено: С 56,03; Н 7,11; N 11,23; Cl 7,97.
Пример 29
N-3-(4-Пиперидинпропионил)-R-(-)-нипекотиламинометил-5-(S)-(3-N-бензил)имидазолин-2,4-дион • НCI (29)
Гидрохлорид N-3-(4-пиперидинпропионил)-R-(-)-нипекотил-[(S)-2-(2-бензиламино)карбониламино-3-амино] пропионовой кислоты (0,15 г), полученный из промежуточного АН2 (4,4 ммоль) и бензилизоцианата (4,4 ммоль), как описано в примере 22, растворяют в водной НCI (3 N) и перемешивают в течение 18 ч при КТ. Этот раствор концентрируют в вакууме, получая белый твердый продукт. Этот продукт растирают и сушат, получая 29 в виде белой пены (0,144 г):
1Н NMR (DMSO-d6) δ 9.0 (m, 1 Н), 8.6 (m, 1 Н), 8.3 (m, 1 Н), 7.2 (m 5 Н), 4.48 (s, 2 Н), 4.2 (m, 2 Н), 3.7 (m, 1 Н), 3.4 (m, 1 Н), 3.2 (d, 3 Н), 2.7 (d, 3 Н), 2.2 (m, 3 Н), 1.7 (m, 3 Н), 1.0-1.6 (m, 10 H); MS m/e 470 (MH+).
Пример 30
N-3-(4-Пиперидинпропионил)-R-(-)-нипекотил-[(S)-2-(фенетиламино)карбониламино-3-амино]пропионовая кислота • НСO2Н (30)
Соединение 30, полученное реакцией 2-фенетилизоцианата (4,1 ммоль) с АН2 (4,1 ммоль), как показано в примере 22, выделяют в виде рыжевато-коричневой пены (0,41 г): т. пл. 65-72oС; МС m/е 502 (МН+). Анализ. Вычислено для С26Н39N5О5 • 1,2 НСО2Н • 1,0 Н2О (574,87): С 56,83; Н 7,61; N 12,18. Найдено: С 57,12; Н 7,80; N 11,85.
6-Метил-3-пиридинкарбоксальдегид (АК2)
Альдегидный предшественник АК2 получают в две стадии с использованием стандартных условий. АК1 (0,066 моль) растворяют в THF (100 мл), охлаждают (-78oС), обрабатывают LiAlH4 (0,066 моль) и перемешивают в течение 4 ч. Реакционную смесь гасят насыщенным NH4C1, нагревают, фильтруют с промыванием СНСl3 (250 мл) и слои разделяют. Органический слой сушат и выпаривают, получая светлое масло (0,054 моль). Масло растворяют в DCM (200 мл), обрабатывают MnO2 (70 г) и нагревают при кипячении с обратным холодильником в течение 6 ч. Смесь охлаждают, фильтруют и растворитель выпаривают, получая АК2 (0,052 моль) в виде коричневого масла.
Пример 31
N-3-(4-Пиперидинпропионил)-R-(-)-нипекотил-[(S)-3-амино-3-(6-метил-3-пиридил)]пропионовая кислота • 2 НСl (31)
Соединение 31 получают, как описано в примере 16, исходя из Boc-R-нипекотиновой кислоты (6,9 ммоль) и метил-(S)-3-амино-3-(6-метил-3-пиридил)пропионата (см. примеры АК5, AG5; 6,9 ммоль). Соединение 31 выделяют в виде белой пены (1,20 г): т.пл. 99-105oC; МС m/е 431 (МН+). Анализ. Вычислено для С23Н34N4O4 • 2,24 НСl • 1,0 H2O • 0,24 ацетонитрил (534,33): С 51,70; Н 7,35; N 11,11; Сl 14,82. Найдено: С 51,32; Н 7,45; N 11,23; CI 14,42.
Пример 32
N-3-(4-Пиперидинпропионил)-R-(-)-нипекотил-[(S)-3-амино-3-(5-бром-3-пиридил)]пропионовая кислота • 2 НCI (32)
Соединение 32, полученное, как описано в примере 16, исходя из Boc-R-нипекотиновой кислоты (4,8 ммоль) и метил-3-S-амино-3-(5-бром-3-пиридил)пропионата (см. примеры АК5, AG5; 4,8 ммоль), выделяют в виде белой пены (1,24 г): т.пл.98-101oС; МС m/е 496 (МН+). Анализ. Вычислено для С22Н31Вr4O4 • 2,2 НСl • 1,0 Н2O • (593,67): С 44,51; Н 5,98; N 9,44, Cl 13,14. Найдено: С 44,17; Н 6,37; N 9,81; Cl 13,10.
Пример 33
N-3-(4-Формамидинопиперидинпропионил)-R-(-)-нипекотил-[(S)-3-амино-3-(3-пиридил)]пропионовая кислота • 2НСl (33)
Формамидин 33 получают по методике M.K.Scott (J.Med. Chem., 1983, 26, 534), как показано на схеме AL. Промежуточный AL1 (см. пример 21; 2,3 ммоль) растворяют в EtOH (20 мл), обрабатывают этилформимидат • НCI (3,7 ммоль), перемешивают в течение 22 ч и фильтруют. Фильтрат обрабатывают Et2O (40 мл), охлаждают на ледяной бане и фильтруют, получая стекловидный AL2. AL2 растворяют в водной НCI (4N, 15 мл), перемешивают в течение 28 ч и выпаривают, получая 33 в виде белой пены (0,75 г): т.пл. 49-55oС.
1H NMR (DMSO-d6) δ 9.35 (s, 1 Н), 9.1 (m, 2 Н), 8.8 (m, 2 Н), 8.70 (d, 1 Н), 8.5 (m, 1 Н), 7.8 (m, 2 Н), 5.2 (dd, 1 Н), 4.2 (m, 1 Н), 3.8 (m, 2 Н), 3.2 (m, 2 Н), 2.8 (m, 2 Н), 2.6 (m, 1 Н), 2.3 (m, 2 Н), 1.8 (m, 3 Н), 1.0-1.7 (m, 12 H); MS m/e 444 (MH+).
Изобретение относится к новым производным пирролидина или пиперидина ф-лы (I), их энантиомерам и фармацевтически приемлемым солям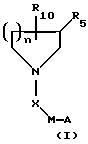
где R10 - Н или C(O)N(R1)YZ, R1 - Н, Y - (СН2)р, (CH2)qCH(R3) или CH(R3)(CH2)q, R3 - арил, аралкил или гетероарил, q = 1-3, р = 2 или 3, Z - CO2H, СО2-алкил или 5-тетразол, Х-С(O), М-(СН2)n, или пиперидин-1-ил, m = 2, n = 2, R5 - Н, А выбирают из пиперидин-2-ила, пиперидин-3-ила, пиперидин-4-ила или N-замещенного пиперидина. Соединения ф-лы (I) ингибируют агрегацию тромбоцитов и могут найти применение в медицине. 3 с. и 2 з.п. ф-лы, 6 табл.
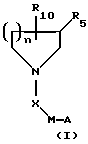
где R10 представляет Н или C(O)N(R1)YZ, где R1 представляет Н; Y представляет (СН2)р, (CH2)qCH(R3) или CH(R3) (CH2)q; где R3 представляет арил, аралкил или гетероарил, q = 1-3 и р = 2 или 3; Z представляет СО2Н, СO2-алкил или 5-тетразол;
Х представляет С(O);
М представляет (СН2)m или пиперидин-1-ил, где m = 2;
n = 2;
R5 представляет Н;
А выбирают из любого из пиперидин-2-ила, пиперидин-3-ила, пиперидин-4-ила или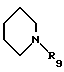
где R9 представляет Н, алкил, CH(NH), CMe(NH) или ацил,
их энантиомеры и фармацевтически приемлемые соли.
N-3-(4-пиперидинпропионил)нипекотил(3-амино-3-фенил)пропионовая кислота,
N-3-(4-пиперидинпропионил)изонипекотил-[3-амино-3-(4-карбоксифенил)] пропионовая кислота,
N-3-(4-пиперидинпропионил)нипекотил-5Н-(2-аминоэтил)тетразол,
N-3-(4-пиперидинпропионил)-R-(-)-нипекотил-[(S)-3-амино-3-(3,4-метилендиоксифенил)] пропионовая кислота,
N-3-(4-пиперидинпропионил)-R-(-)-нипекотил-[(S)-3-амино-3-(3-хинолинил)] пропионовая кислота,
N-3-(4-пиперидинпропионил)-нипекотил-[(S)-3-амино-3-(3,4-метилендиоксифенил)] пропионовая кислота,
N-3-(4-пиперидинпропионил)-R-(-)нипекотил-[(S)-3-амино-3-(3-пиридил)] пропионовая кислота,
N-[(4,4'-бипиперидин-1-ил)карбонил] -R-(-)-нипекотил-[(S)-3-амино-3-(3-пиридил)] пропионовая кислота,
N-3-(4-пиперидинпропионил)-R-(-)-нипекотил-[(S)-3-амино-3-(6-метил-3-пиридил)] пропионовая кислота,
N-3-(4-пиперидинпропионил)-R-(-)-нипекотил-[(S)-3-амино-3-(5-бром-3-пиридил)] пропионовая кислота и
N-3-(4-формамидинопиперидинпропионил)-R-(-)-нипекотил-[(S)-3-амино-3-(3-пиридил)] пропионовая кислота.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| SU 1486059 A3, 07.06.1989. | |||

